

Abstract
p53 is a tetrameric protein with a thermodynamically unstable deoxyribonucleic acid (DNA)‐binding domain flanked by intrinsically disordered regulatory domains that control its activity. The unstable and disordered segments of p53 allow high flexibility as it interacts with binding partners and permits a rapid on/off switch to control its function. The p53 tetramer can exist in multiple conformational states, any of which can be stabilized by a particular modification. Here, we apply the allostery model to p53 to ask whether evidence can be found that the “activating” C‐terminal phosphorylation of p53 stabilizes a specific conformation of the protein in the absence of DNA. We take advantage of monoclonal antibodies for p53 that measure indirectly the following conformations: unfolded, folded, and tetrameric. A double antibody capture enzyme linked‐immunosorbent assay was used to observe evidence of conformational changes of human p53 upon phosphorylation by casein kinase 2 in vitro. It was demonstrated that oligomerization and stabilization of p53 wild‐type conformation results in differential exposure of conformational epitopes PAb1620, PAb240, and DO12 that indicates a reduction in the “unfolded” conformation and increases in the folded conformation coincide with increases in its oligomerization state. These data highlight that the oligomeric conformation of p53 can be stabilized by an activating enzyme and further highlight the utility of the allostery model when applied to understanding the regulation of unstable and intrinsically disordered proteins.
Keywords: p53, CK2, allosteric regulation, conformational change, oligomerization, phosphorylation, protein conformation, protein folding
Introduction
Human p53 is a 393‐amino acid tetrameric tumor suppressor protein involved in the transcriptional regulation of several genes involved in the cell cycle and differentiation. The protein is activated by a variety of stress signals such as deoxyribonucleic acid (DNA) damage, hypoxia/anoxia, microtubule inhibitors, telomere erosion, metabolic changes, heat shock, pH changes, and oncogenic activation. In response, p53 triggers differentiation, DNA repair, senescence, cell cycle arrest at low levels of p53 and/or apoptosis at high levels of p53 to prevent cells from undergoing tumorigenic alterations.1 The p53 protein can be divided into four functional regions: (i) an acidic N‐terminal domain (aa 1–101) that contains a transactivation domain (aa 1–42) responsible for transactivation and MDM2 oncoprotein interaction;2 (ii) a central core domain (aa 102–290) that is involved in sequence‐specific DNA binding;3, 4 (iii) a C‐terminal domain that contains a tetramerization domain (aa 320–356);5 (iv) and a highly basic C‐terminal regulatory domain (aa 363–393).6
Solving the intra‐ and interdomain interactions between these isolated regions on p53 has paralleled new developments in the protein science field over the past ten to 15 years on the structure‐function paradigm. These include concepts such as (i) a large proportion of amino acid sequences in higher eukaryotes are intrinsically disordered,7 (ii) a large number of protein–protein interactions occur through small peptide linear motifs that can be embedded within structural domains or in disordered domains;8 (iii) a large number of regulatory proteins are thermodynamically unstable, evolving presumably to allow a rapid on/off switch in signal transduction;9 and (iv) a new model of allostery that incorporates disorder and instability—the ensemble model.10
P53 protein has been a model for such concepts including (i) the N‐ and C‐termini of p53, containing most binding interactions as well as phosphorylation, acetylation, and ubiquitin‐like covalent modifications, are within intrinsically disordered linear motifs;11 (ii) the precise protein–protein interactions occur through relatively small and specific amino acid stretches whereby even the same C‐terminal peptide motifs can exist in different conformations—extended conformation, an alpha‐helix, or mixed beta‐sheet, depending upon the binding partner;12 (iii) the p53 DNA‐binding domain is thermodynamically unstable;13 and (iv) hundreds of different conformations of p53 are required in theory to adopt the hundreds of sequence‐specific protein–protein interactions on the multivalent p53 scaffold.7
The nature of allostery in p53 is controversial and is based on differing methods used to access its conformational dynamics. Originally, an allosteric model in p53 DNA‐binding conformations was noted based on the stoichiometry of an “activating” full‐length monoclonal antibody that binds to the C‐terminal regulatory domain, compared to the stoichiometry of its FAB fragment, indicating that negative cooperativity exists in the tetramer and that allosteric changes presumably propagate its activation as a tetramer.14 This was supported by data using ultracentrifugation sizing methods showing that the tetramerization domain fragment of p53 is stabilized by C‐terminal phosphorylation at Ser39215 and that a recombinant p53 protein with an S392E mutation is more thermostable than the wild‐type protein and is in a more folded conformation as defined using conformational‐specific monoclonal antibodies.16 The importance of Ser392 for proper p53 function was also demonstrated in mouse models bearing mutation in the homologous phosphosite at Ser389. Mice carrying the p53 S389A mutation showed increased sensitivity to ultraviolet (UV)‐induced skin tumor development, signifying the importance of serine 389 phosphorylation for the tumor‐suppressive function of p53.17, 18
In another allosteric model,19 p53 is structurally flexible and can reversibly adopt a latent conformation or an active conformation that binds DNA in a sequence‐specific manner. Additionally, in the latent conformation, p53 binds DNA nonsequence‐specifically and independently of the sequence‐specific DNA binding central domain. Together, these data suggest that C‐terminal modifications shift the conformation of p53 into a high affinity DNA‐binding state. However, structural biology has failed to detect “two” stable conformations of p53 (low affinity and high affinity20), thus, the actual nature of the low affinity and the high affinity DNA‐binding conformations of p53 are not defined. Indeed, other regulatory models exist in which p53 is not structurally flexible, the C‐terminus regulates nonspecific DNA binding or sliding on DNA fragments and acetylation stimulates DNA binding through stabilizing protein–protein interactions.21 However, the sequence‐specific DNA‐dependence in acetylation of the p53 tetramer22, 23 provides additional evidence that acetylation can also occur through a coupling of its conformational or allosteric dynamics. Thus, the ensemble model of allostery can be applied, in part, to solve what classic structural biology has not been able to divulge concerning the conformational basis of the low and high affinity DNA‐binding conformations of the unstable p53 tetramer, whereby kinetic and biophysical approaches will be required to understand the basis for the intrinsically unstable conformations that p53 can exist in.
Multisite phosphorylation by different cellular kinases on C‐terminal serine residues is the predominant mechanism for activating p53 sequence‐specific DNA binding.14 Phosphorylation sites on human p53 include Ser378 by protein kinase C, Ser315 by cyclin‐dependent kinase 2/cyclin A and Ser392 by casein kinase 2 (CK2).15, 24 These phosphorylation events may also be mutually exclusive, acting as a switch between stress signaling pathways.25, 26 The role of Ser392 phosphorylation as a universal switch for different signaling pathways was also documented by Cox and Meek who showed Ser392 phosphorylation by other kinases in response to different stimuli.24 Mutation of the CK2 p53 phosphorylation site to alanine in mouse transgenes can stimulate carcinogen‐induced cancer development, suggesting that CK2‐mediated phosphorylation stimulates the tumor suppressor function of p53.17, 18, 27 CK2 is a serine/threonine protein kinase with α2β2 tetrameric structure and is conserved in all eukaryotes. CK2 interacts with the C‐termini of transcription factors such as c‐myc, NF‐κB, and p53 and has a global role in transcription‐related chromatin remodeling and modification. Ser392 p53 phosphorylation by CK2 has been shown to: (i) cause accumulation of p53,15 (ii) enhance the p53‐DNA interaction,6 (iii) induce transcriptional activity of p5328 and (iv) induce tetramer formation in p53 and intrinsic DNA binding affinity by removing the inhibitory effect of C‐terminal p53 sequences.29 Biophysical studies on phosphopeptides have previously shown that Ser392 phosphorylation increases the association constant for reversible tetramer formation by nearly 10‐fold, in comparison to a small effect produced by Ser315 and Ser378 phosphorylation.15 In this study, we have set up assays to determine whether full length p53 oligomerizational states can be altered by phosphorylation. This would provide additional insights into the allosteric control of p53 and also provide new in vitro assays in which to develop small molecules that can stabilize p53 oligomerization.
Results
p53 forms oligomers upon CK2 phosphorylation
p53 is phosphorylated when both kinase and adenosine triphosphate (ATP) phosphate donor are present. Phosphorylation was detected by Western blotting [Fig. 1(A)] and enzyme linked‐immunosorbent assay (ELISA) [Fig. 1(B)]. The p53 + CK2 control shows that any trace amounts of endogenous ATP do not cause phosphorylation, while the p53 + ATP control shows only trace immunoreactivity with the phospho‐Ser392 specific antibody FP3 in the absence of CK2.
Figure 1.
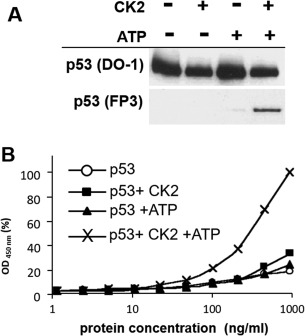
Samples of purified p53 protein were phosphorylated in vitro by CK2 and ATP. (A) Samples were phosphorylated in vitro, subjected to electrophoresis on a 4–16% (w/v) SDS‐polyacrylamide gel, and visualized by immuno‐blotting with DO‐1 and FP3 monoclonal antibodies at 1 μg/mL. (B) Samples were phosphorylated in vitro and titrated 10 times in a NP40 buffer for two‐site ELISA detection. Samples were captured by DO‐1 monoclonal antibody and subsequently detected with FP3‐HRP conjugated antibody. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (○) p53 only; (◼) p53 + CK2; (▲) p53 + ATP; (×) p53 + CK2/ATP.
In Figure 2(A), we describe a method to detect oligomers (presumably tetramers), using DO‐1 to capture p53 and DO‐1‐horseradish peroxidase (HRP) to detect the captured protein. In theory, any monomeric p53 captured by DO‐1 will have its N‐terminal domain blocked. If p53 exists as a higher oligomer, exposed N‐terminal domains of the higher oligomer can be detected by DO‐1‐HRP. As a control, we used a nonspecific mouse monoclonal antibody as capture, and we obtained no signal, indicating the assay's specificity for detecting p53. We show that phosphorylated p53 can form oligomers in the presence of CK2 and ATP [Fig. 2(B,C)].
Figure 2.
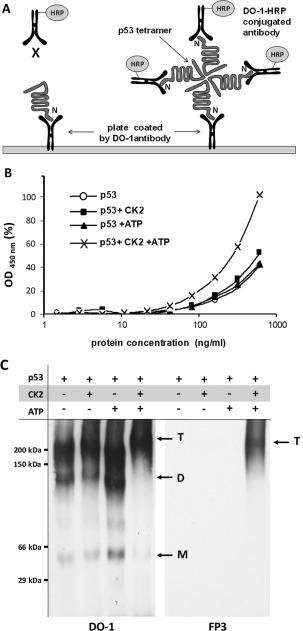
Phosphorylated p53 forms oligomers and can be detected by two‐site ELISA. (A) Schematic of two‐site ELISA detection of p53 tetramers. In the monomeric state, the N‐terminal region of p53 is captured and blocked by DO‐1. The addition of the kinase and phosphate donor pair causes p53 to form tetramers. These oligomers have exposed N‐terminal regions which can be detected by DO‐1‐HRP conjugated antibodies. (B) Samples were phosphorylated in vitro and serially diluted 10 times in NP40 buffer. Samples were captured by DO‐1 monoclonal antibody and subsequently detected with DO‐1‐HRP conjugate. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (○) p53 only; (◼) p53 + CK2; (▲) p53 + ATP; (×) p53 + CK2/ATP. (C) The samples were analyzed using blue native electrophoresis. The proteins transferred on membranes by western blotting were detected by DO1 antibody, phosphorylated Ser392 was detected by FP3 monoclonal antibody. Arrows labeled M, D, T indicate monomers dimers and tetramers, respectively.
CK2 phosphorylation enhances the stability of p53 oligomer
CK2 phosphorylation stabilizes p53 protein structure from mutant to wild‐type conformation resulting in increased reactivity with monoclonal antibody PAb1620 (Fig. 3) and reduced reactivity with monoclonal antibodies PAb240 and DO12 [Fig. 4(A,B)]. In addition, we added a control where p53 protein was denatured in the presence of an ionic detergent, sodium dodecyl sulfate (SDS). The absorbance at 450 nm gives the total amount of p53 in each sample and shows that only 9.1 to 12.7% of p53 exists in the mutant conformation originally [Fig. 4(C)]. Our evidence demonstrates that PAb1620 binding correlates with the phosphorylated form of p53. This indicates that conformation changes of the C‐terminal domain can have a profound effect on tetramer stability and these data can be explained by the ensemble model of allostery.
Figure 3.
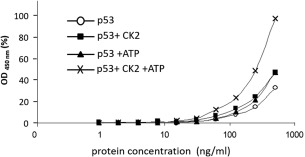
Phosphorylated p53 shows increased reactivity with conformation‐specific PAb1620 monoclonal antibodies. Samples were phosphorylated in vitro and serially diluted in NP40 buffer. Samples were captured with PAb1620 monoclonal antibodies and subsequently detected with DO‐1‐HRP conjugate antibody. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (○) p53 only; (◼) p53 + CK2; (▲) p53 + ATP; (×) p53 + CK2/ATP.
Figure 4.
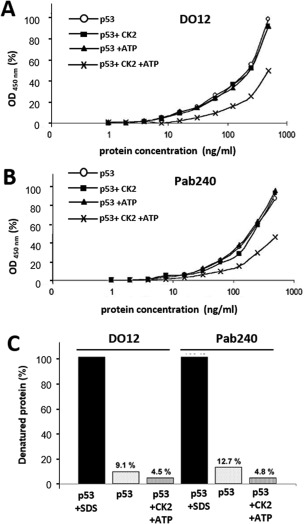
Detection of p53 denaturation. (A) Samples were phosphorylated in vitro and serially diluted for two‐site ELISA detection. Samples were captured with DO12 monoclonal antibody and subsequently detected with DO‐1‐HRP conjugate. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against FP3‐HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (○) p53 only; (◼) p53 + CK2; (▲) p53 + ATP; (×) p53 + CK2/ATP. (B) Same conditions as (A) except that capture antibody used was PAb240. (C) Phosphorylated p53 was denatured and tested for reactivity with two conformation‐specific monoclonal antibodies, DO12 and PAb240. Samples were phosphorylated in vitro and serially diluted 10 times for two‐site ELISA detection. One sample was denatured by 15 mM of SDS for 30 min at 65°C. Samples were captured with DO12 or PAb240 monoclonal antibodies and subsequently detected with DO‐1‐HRP conjugate. In the bar chart, HRP activity (absorbance at 450 nm) is represented as a percentage value on the y‐axis. The absorbance values of the p53 + SDS samples are taken as 100% denatured p53 protein for reference.
CK2 associates with p53 to form a stable complex
We observed that increasing concentrations of CK2 (5 to 500 units) give rise to increasing levels of phosphorylation [Fig. 5(A)]. Since we did not observe the same increase in phosphorylation in ELISA [Fig. 5(B)], we propose that binding of phospho‐specific antibody is blocked in the presence of higher amounts of CK2 in ELISA, possibly due to the formation of p53‐CK2 complexes in the native state that are abolished following SDS‐polyacrylamide gel electrophoresis (PAGE) and Western blotting. The level of p53 oligomerization [Fig. 6(A)] is also dependent on the concentration of CK2 added. Correspondingly, if the monoclonal antibody to the CK2 regulatory β subunit was used to detect potential p53‐CK2 complexes captured by DO‐1, we obtained a similar profile [Fig. 6(B)]. Overall, these data suggest that CK2‐p53 complexes have been formed. This is not without precedent; it has been shown that CK2 ß subunits bind stably to the kinase docking region (aa 325–344) on p53.30 Also, mammalian CK2‐p53 complexes are formed in response to UV‐induced DNA damage. These complexes contain CK2, p53 and the chromatin transcriptional elongation factor FACT (hSpt16 and SSRP1 heterodimer). In vitro and in vivo studies show that FACT alters CK2 conformation and binding specificity to phosphorylate p53 preferentially over other substrates.28
Figure 5.
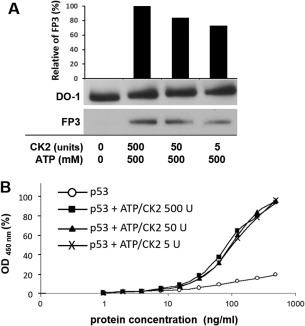
Different CK2 concentrations affect p53 phosphorylation status. (A) Samples were phosphorylated in vitro, subjected to electrophoresis on a 4–16% (w/v) SDS‐polyacrylamide gel and visualized by immunoblotting with DO‐1 and FP3 monoclonal antibodies at 1 μg/mL. The graph shows the relative density of bands detected by phospho‐specific antibody FP3. (B) Samples were phosphorylated in vitro and serially diluted for two‐site ELISA detection. Samples were captured with DO‐1 monoclonal antibody and subsequently detected with FP3‐HRP conjugate. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (○) p53 only; (◼) p53 + ATP/CK2 500 U; (▲) p53 + ATP/CK2 50 U; (×) p53 + ATP/CK2 5 U.
Figure 6.
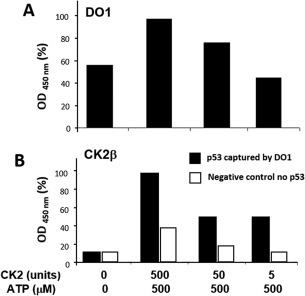
CK2 concentration affects p53 oligomerization. CK2 forms complexes with phosphorylated p53. (A) Samples were phosphorylated in vitro and serially diluted for two‐site ELISA detection. Samples were captured with DO‐1 monoclonal antibody and subsequently detected with DO‐1‐HRP conjugate. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as percentage of the maximal value obtained. (B) Samples were prepared identically and were detected by anti‐CK2 β rabbit monoclonal antibody and secondary swine‐anti‐rabbit polyclonal antibodies conjugated to HRP.
CK2 binds stably to p53 and blocks the Bp53‐10 epitope on p53 only when ATP is present
The CK2 ß subunit docking region (aa 325–344) is situated close to the Bp53‐10 binding site (aa 371–380). The Bp53‐10 epitope is masked when CK2 is present at high concentrations [Fig. 7(A)] in comparison to low concentrations [Fig. 7(B)]. It is unusual that CK2 requires the presence of ATP to form higher order structures. This is shown in Figure 7(B), where the Bp53‐10 epitope cannot be masked in p53 + CK2 samples in the absence of ATP. It can be postulated that every time a p53 monomer is phosphorylated, the conformation of its C‐terminus is altered, allowing CK2 to bind cooperatively in an ATP‐dependent manner as the p53 monomer oligomerizes. Interestingly, Bp53‐10 has the same effect as CK2 phosphorylation of the C‐terminal region of p53.31 The binding of CK2 may block the Bp53‐10 antibody from accessing a newly formed cryptic region. The new cryptic region (aa 371–380) contains phosphorylation, ubiquitination, neddylation and acetylation sites.32 Modification events at these sites may lead to an increased degradation of p53, and may be a reason why phosphorylated p53 protein exhibits increased stability (Fig. 3).
Figure 7.
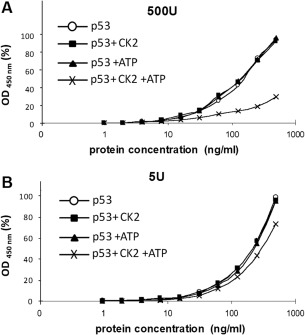
CK2 binding to p53 blocks the Bp53‐10 epitope within the C‐terminal region of p53. p53 was incubated with either 500 units (A) or 5 units (B) of CK2 and ATP. Samples were phosphorylated in vitro and serially diluted ten times in NP40 buffer. Samples were captured with DO‐1 monoclonal antibody and subsequently detected with Bp53‐10‐HRP conjugate. Protein concentration on the x‐axis is plotted on a semilogarithmic scale against HRP activity (absorbance at 450 nm) on the y‐axis, plotted as a percentage of the maximal value obtained.
Discussion
In this paper, we test whether C‐terminal phosphorylation of p53 changes the oligomeric conformation of full‐length p53 using an indirect assay that measures p53 oligomerization. We have used a quantitative and reproducible in vitro two‐site ELISA technique on purified human p53 protein from bacteria. We studied the oligomerization of p53 during activation by CK2 phosphorylation at Ser392 in the presence of an ATP phosphate donor. We showed that oligomerization gives increased levels of the epitope of PAb1620 and masks the cryptic epitopes of PAb240 and DO12, signifying an increase in native conformation and reduction in mutant conformation. Also, we found evidence that stable physical complexes of CK2 and p53 are formed only in the presence of ATP, and these complexes mask the Bp53‐10 monoclonal antibody epitope on the p53 protein. All this was done without the addition of DNA, as the p53 protein forms tetramers and complex oligomers in the absence and presence of DNA.33 Our data show that in the absence of DNA, the p53 oligomeric conformation is altered by phosphorylation, suggesting that the dynamics of the flexible and unstable tetramer can be altered kinetically. This view of p53 regulation that cannot necessarily be solved using classic structural biology can be approached by applying the ensemble model of allostery. The change in p53 oligomerization provides biochemical evidence to explain mechanisms responsible for why murine transgenes with alanine‐mutations at the Ser392 equivalent on mouse p53 have enhanced cancer development after carcinogen exposure and reduced transactivation potential after DNA damage.27 In addition, a recent transgenic mouse model has demonstrated an embryonic lethality from deleting the C‐terminal regulatory domain of p53, indicating a highly penetrant negative regulatory function for this motif of p53 during mouse development.34 As many regulatory domains on eukaryotic proteins are themselves unstable thermodynamically and also destabilize adjacent structural domains, the ensemble view of allostery will shed light on allosteric regulation of other regulatory eukaryotic proteins.
Materials and Methods
Expression of p53 protein in bacteria and purification of p53 protein
Full length human p53 was expressed from pEXP14 vector (Invitrogen) in Escherichia coli BL21 Star (DE3). Expression was induced by addition of 1 mM Isopropyl β‐D‐1‐thiogalactopyranoside at optical density 0.5 at 25°C. Cells were harvested 3 h postinduction by centrifugation (6,000 g at 4°C). Cell pellets from 1 L culture were suspended in 50 mL lysis buffer (15% [v/v] glycerol, 15 mM HEPES/KOH, 200 mM KCl) supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 mg/mL lysozyme and 50 U/mL benzonase nuclease (Sigma Aldrich). Lysates from E. coli were centrifuged at 15,000 g for 30 min and the supernatant was diluted fivefold in low salt purification buffer (15% [v/v] glycerol, 15 mM HEPES/KOH, pH 8.0, 0.04% Triton X‐100, 5 mM dithiothreitol, 2 mM benzamidine, and 1 mM β‐glycerophosphate), filtered and loaded on to a 5 mL heparin–sepharose column (GE Healthcare). The p53 protein was eluted by a 0–1 M KCl gradient, and the peak fractions that eluted at approximately 0.5–0.6 M KCl were pooled, dialyzed against low‐salt purification buffer for 12 h at 4°C and loaded on to an anion exchange HQ column (GE Healthcare).
In vitro phosphorylation of p53 protein
Purified human p53 protein was phosphorylated in vitro by CK2 (New England Biolabs). The kinase reaction buffer (New England Biolabs) contained 20 mM Tris‐HCl pH 7.5, 50 mM KCl and 10 mM MgCl2. ATP of 500 µM was utilized as a phosphate donor and the reaction was performed at 30°C for 30 min. Unless indicated otherwise, we used 50 U of CK2 (corresponding to 58 ng of enzyme) for phosphorylation of 1 μg p53 protein. Samples were analyzed by Western blotting and ELISA.
Western‐blot analysis and antibodies
The protein was analyzed using SDS gradient‐PAGE according to standard protocol. Monoclonal antibodies were purified from either cell culture supernatants or ascites using Protein A columns.35 The antibodies used in this assay were as follows:
DO‐1 monoclonal antibody, directed toward the epitope S20DLWKL25 within the p53 N‐terminal region.36
Bp53‐10 monoclonal antibody, directed toward the epitope S371KKGQSTSRH380 within the p53 C‐terminal region.37
FP3 phospho‐specific monoclonal antibody, which recognizes the C‐terminus of p53 protein phosphorylated on Ser392.38
PAb1620 monoclonal antibody, directed toward three conformation‐specific epitopes of DNA binding domain.39 These epitopes are present only when wild‐type p53 is in the native form.40
DO12 monoclonal antibody, directed toward the epitope T256LEDSSGNLLGRNSF270 and is specific for mutant or denatured p53.41
PAb240 monoclonal antibody, directed toward the epitope R213 HSVV217 and is specific for mutant or denatured p53.42
DO‐1, Bp53‐10, and FP3 were coupled to HRP through amine linkages (Pierce EZ‐Link Activated Peroxidase and Kit). Monoclonal antibodies conjugated to HRP were used for detection of captured p53 protein in ELISA experiments.
Blue native PAGE
p53 oligomers were analyzed using blue native PAGE as described by Wittig et al.43 The sample loading buffer composed of glycerol (40%), Coomassie Brilliant Blue G250 (0.4%) and cathode buffer was added to the samples in a one‐to‐one ratio and then the samples were loaded on a native gel consisting of a 3.5% stacking and a 7% separating polyacrylamide gel. In contrast to the original protocol, the cathode buffer was prepared without adding Coomassie Brilliant Blue G250 to ensure compatibility with western blotting.
Enzyme linked‐immunosorbent assay
Investigation of p53 conformation was carried out using two‐site ELISA. Each well on a 96‐well plate was incubated with 50 µL of 15 µg/mL monoclonal antibodies in carbonate buffer (0.1 M CO3, HCO3 pH 9–9.8) at 4°C overnight. The wells were blocked with 3% bovine serum albumin (BSA A7030, Sigma) in phosphate‐buffered saline (PBS) at room temperature for 2 h. The protein was phosphorylated in vitro and titrated in NP40 buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP40) with additional protease and phosphate inhibitors (Sigma). Each sample of 50 µL was plated into wells and incubated at 4°C for 3 h. The plates were washed three times in 0.1% Tween 20 in PBS before addition of the required monoclonal HRP‐conjugated antibody diluted in 1% BSA. Plates were incubated at room temperature for 2 h. Analysis of bound peroxidase was performed by colorimetric detection with a tetramethylbenzidine peroxidase EIA substrate kit (Bio‐Rad) and absorbance was measured with an automatic ELISA plate reader at 450 nm.44
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We would like to thank Dr. P. J. Coates for critical reading of the manuscript.
Contributor Information
Petr Muller, Email: muller@mou.cz.
Borivoj Vojtesek, Email: vojtesek@mou.cz.
References
- 1. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408:307–310. [DOI] [PubMed] [Google Scholar]
- 2. Fields S, Jang SK (1990) Presence of a potent transcription activating sequence in the p53 protein. Science 249:1046–1049. [DOI] [PubMed] [Google Scholar]
- 3. Bargonetti J, Manfredi JJ, Chen X, Marshak DR, Prives C (1993) A proteolytic fragment from the central region of p53 has marked sequence‐specific DNA‐binding activity when generated from wild‐type but not from oncogenic mutant p53 protein. Genes Dev 7:2565–2574. [DOI] [PubMed] [Google Scholar]
- 4. Halazonetis TD, Kandil AN (1993) Conformational shifts propagate from the oligomerization domain of p53 to its tetrameric DNA binding domain and restore DNA binding to select p53 mutants. EMBO J 12:5057–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang P, Reed M, Wang Y, Mayr G, Stenger JE, Anderson ME, Schwedes JF, Tegtmeyer P (1994) P53 domains: Structure, oligomerization, and transformation. Mol Cell Biol 14:5182–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hupp TR, Meek DW, Midgley CA, Lane DP (1992) Regulation of the specific DNA binding function of p53. Cell 71:875–886. [DOI] [PubMed] [Google Scholar]
- 7. Tompa P (2012) On the supertertiary structure of proteins. Nat Chem Biol 8:597–600. [DOI] [PubMed] [Google Scholar]
- 8. Neduva V, Linding R, Su‐Angrand I, Stark A, de Masi F, Gibson TJ, Lewis J, Serrano L, Russell RB (2005) Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol 3:e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: Re‐assessing the protein structure‐function paradigm. J Mol Biol 293:321–331. [DOI] [PubMed] [Google Scholar]
- 10. Hilser VJ (2010) Biochemistry. An ensemble view of allostery. Science 327:653–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maslon MM, Hrstka R, Vojtesek B, Hupp TR (2010) A divergent substrate‐binding loop within the pro‐oncogenic protein anterior gradient‐2 forms a docking site for reptin. J Mol Biol 404:418–438. [DOI] [PubMed] [Google Scholar]
- 12. Uversky VN, Oldfield CJ, Dunker AK (2008) Intrinsically disordered proteins in human diseases: Introducing the d2 concept. Annu Rev Biophys 37:215–246. [DOI] [PubMed] [Google Scholar]
- 13. Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor MR, Lane DP, Fersht AR (1997) Thermodynamic stability of wild‐type and mutant p53 core domain. Proc Natl Acad Sci USA 94:14338–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hupp TR, Lane DP (1994) Allosteric activation of latent p53 tetramers. Curr Biol 4:865–875. [DOI] [PubMed] [Google Scholar]
- 15. Sakaguchi K, Sakamoto H, Lewis MS, Anderson CW, Erickson JW, Appella E, Xie D (1997) Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 36:10117–10124. [DOI] [PubMed] [Google Scholar]
- 16. Nichols NM, Matthews KS (2002) Human p53 phosphorylation mimic, s392e, increases nonspecific DNA affinity and thermal stability. Biochemistry 41:170–178. [DOI] [PubMed] [Google Scholar]
- 17. Bruins W, Zwart E, Attardi LD, Iwakuma T, Hoogervorst EM, Beems RB, Miranda B, van Oostrom CT, van den Berg J, van den Aardweg GJ, Lozano G, van Steeg H, Jacks T, de Vries A (2004) Increased sensitivity to uv radiation in mice with a p53 point mutation at ser389. Mol Cell Biol 24:8884–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoogervorst EM, Bruins W, Zwart E, van Oostrom CT, van den Aardweg GJ, Beems RB, van den Berg J, Jacks T, van Steeg H, de Vries A (2005) Lack of p53 ser389 phosphorylation predisposes mice to develop 2‐acetylaminofluorene‐induced bladder tumors but not ionizing radiation‐induced lymphomas. Cancer Res 65:3610–3616. [DOI] [PubMed] [Google Scholar]
- 19. Yakovleva T, Pramanik A, Kawasaki T, Tan‐No K, Gileva I, Lindegren H, Langel U, Ekstrom TJ, Rigler R, Terenius L, Bakalkin G (2001) p53 latency. C‐terminal domain prevents binding of p53 core to target but not to nonspecific DNA sequences. J Biol Chem 276:15650–15658. [DOI] [PubMed] [Google Scholar]
- 20. Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE, Arrowsmith CH (2001) Latent and active p53 are identical in conformation. Nat Struct Biol 8:756–760. [DOI] [PubMed] [Google Scholar]
- 21. McKinney K, Mattia M, Gottifredi V, Prives C (2004) P53 linear diffusion along DNA requires its c terminus. Mol Cell 16:413–424. [DOI] [PubMed] [Google Scholar]
- 22. Dornan D, Shimizu H, Burch L, Smith AJ, Hupp TR (2003) The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA‐dependent acetylation of p53. Mol Cell Biol 23:8846–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Češková P, Chichger H, Wallace M, Vojtesek B, Hupp TR (2006) On the mechanism of sequence‐specific DNA‐dependent acetylation of p53: The acetylation motif is exposed upon DNA binding. J Mol Biol 357:442–456. [DOI] [PubMed] [Google Scholar]
- 24. Cox ML, Meek DW (2010) Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal 22:564–571. [DOI] [PubMed] [Google Scholar]
- 25. Pospisilova S, Brazda V, Kucharikova K, Luciani MG, Hupp TR, Skladal P, Palecek E, Vojtesek B (2004) Activation of the DNA‐binding ability of latent p53 protein by protein kinase c is abolished by protein kinase ck2. Biochem J 378:939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meek DW, Cox M (2011) Induction and activation of the p53 pathway: A role for the protein kinase ck2?. Mol Cell Biochem 356:133–138. [DOI] [PubMed] [Google Scholar]
- 27. Bruins W, Bruning O, Jonker MJ, Zwart E, van der Hoeven TV, Pennings JL, Rauwerda H, de Vries A, Breit TM (2008) The absence of ser389 phosphorylation in p53 affects the basal gene expression level of many p53‐dependent genes and alters the biphasic response to uv exposure in mouse embryonic fibroblasts. Mol Cell Biol 28:1974–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H (2001) A DNA damage‐induced p53 serine 392 kinase complex contains ck2, hspt16, and ssrp1. Mol Cell 7:283–292. [DOI] [PubMed] [Google Scholar]
- 29. Hupp TR (1999) Regulation of p53 protein function through alterations in protein‐folding pathways. Cell Mol Life Sci 55:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gotz C, Scholtes P, Prowald A, Schuster N, Nastainczyk W, Montenarh M (1999) Protein kinase ck2 interacts with a multi‐protein binding domain of p53. Mol Cell Biochem 191:111–120. [PubMed] [Google Scholar]
- 31. Jagelska E, Brazda V, Pospisilova S, Vojtesek B, Palecek E (2002) New ELISA technique for analysis of p53 protein/DNA binding properties. J Immunol Methods 267:227–235. [DOI] [PubMed] [Google Scholar]
- 32. Appella E, Anderson CW (2000) Signaling to p53: Breaking the posttranslational modification code. Pathol Biol 48:227–245. [PubMed] [Google Scholar]
- 33. Friedman PN, Chen X, Bargonetti J, Prives C (1993) The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc Natl Acad Sci USA 90:3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamard PJ, Barthelery N, Hogstad B, Mungamuri SK, Tonnessen CA, Carvajal LA, Senturk E, Gillespie V, Aaronson SA, Merad M, Manfredi JJ (2013) The C terminus of p53 regulates gene expression by multiple mechanisms in a target‐ and tissue‐specific manner in vivo. Genes Dev 27:1868–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harlow E, Lane D (2006) Antibody purification on protein a or protein g columns. CSH Protoc 2006:pdb.prot4283. [DOI] [PubMed] [Google Scholar]
- 36. Vojtĕsek B, Bártek J, Midgley CA, Lane DP (1992) An immunochemical analysis of the human nuclear phosphoprotein p53. New monoclonal antibodies and epitope mapping using recombinant p53. J Immunol Methods 151:237–244. [DOI] [PubMed] [Google Scholar]
- 37. Dolezalova H, Vojtesek B, Kovarik J (1997) Epitope analysis of the human p53 tumour suppressor protein. Folia Biol 43:49–51. [PubMed] [Google Scholar]
- 38. Pospisilova S, Kankova K, Svitakova M, Nenutil R, Vojtesek B (2001) New monoclonal antibodies recognizing p53 protein phosphorylated by casein kinase ii at serine 392. Folia Biol 47:148–151. [PubMed] [Google Scholar]
- 39. Wang PL, Sait F, Winter G (2001) The ‘wildtype’ conformation of p53: Epitope mapping using hybrid proteins. Oncogene 20:2318–2324. [DOI] [PubMed] [Google Scholar]
- 40. Milner J, Cook A, Sheldon M (1987) A new anti‐p53 monoclonal antibody, previously reported to be directed against the large t antigen of simian virus 40. Oncogene 1:453–455. [PubMed] [Google Scholar]
- 41. Vojtesek B, Dolezalova H, Lauerova L, Svitakova M, Havlis P, Kovarik J, Midgley CA, Lane DP (1995) Conformational changes in p53 analysed using new antibodies to the core DNA binding domain of the protein. Oncogene 10:389–393. [PubMed] [Google Scholar]
- 42. Gannon JV, Greaves R, Iggo R, Lane DP (1990) Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J 9:1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wittig I, Braun HP, Schagger H (2006) Blue native page. Nat Protoc 1:418–428. [DOI] [PubMed] [Google Scholar]
- 44. Harlow E, Lane D (2006) Indirect detection using horseradish peroxidase‐labeled reagents. CSH Protoc 2006:pdb.prot4296. [DOI] [PubMed] [Google Scholar]