

Abstract
The gastrointestinal tract is colonized by trillions of commensal microorganisms that collectively form the microbiome and make essential contributions to organism homeostasis. The intestinal immune system must tolerate these beneficial commensals, whilst preventing pathogenic organisms from systemic spread. Humoral immunity plays a key role in this process, with large quantities of immunoglobulin (Ig)A secreted into the lumen on a daily basis, regulating the microbiome and preventing bacteria from encroaching on the epithelium. However, there is an increasing appreciation of the role of IgG antibodies in intestinal immunity, including beneficial effects in neonatal immune development, pathogen and tumour resistance, but also of pathological effects in driving chronic inflammation in inflammatory bowel disease (IBD). These antibody isotypes differ in effector function, with IgG exhibiting more proinflammatory capabilities compared with IgA. Therefore, the process that leads to the generation of different antibody isotypes, class‐switch recombination (CSR), requires careful regulation and is orchestrated by the immunological cues generated by the prevalent local challenge. In general, an initiating signal such as CD40 ligation on B cells leads to the induction of activation‐induced cytidine deaminase (AID), but a second cytokine‐mediated signal determines which Ig heavy chain is expressed. Whilst the cytokines driving intestinal IgA responses are well‐studied, there is less clarity on how IgG responses are generated in the intestine, and how these cues might become dysfunctional in IBD. Here, we review the key mechanisms regulating class switching to IgA vs IgG in the intestine, processes that could be therapeutically manipulated in infection and IBD.
Keywords: antibodies, B cells, intestinal immunity
1. INTRODUCTION
Humoral immunity plays a critical role in the gastrointestinal tract, an organ colonized by trillions of commensal microorganisms, collectively known as the microbiome. The microbiome contributes to physiological processes, including nutrient absorption and barrier protection, and occupies a tolerogenic nutrient‐rich niche that enables a symbiotic relationship with the host. 1 , 2 Given the large density of microorganisms, particularly in the lower gastrointestinal tract, the intestinal immune system must strike a balance, tolerating commensals whilst preventing the invasion of pathogens. Disruption of this balance leads to dysbiosis and inflammation, and is associated with a variety of intestinal and systemic disorders. 3 In this context, immunoglobulins (Ig) are produced in the gut mucosa and secreted into the gut lumen, mediating tolerance to commensals and shaping the composition of the microbiome through a non‐inflammatory process primarily known as immune exclusion. 4 , 5 IgA is the dominant antibody isotype at mucosal surfaces, with large quantities (3–5 g per day in humans) 6 secreted into the gut lumen. In addition to IgA, other antibody isotypes also contribute to intestinal humoral immunity. IgM is transcytosed and secreted into the gut lumen in humans with similar efficiency to IgA, and it participates in immune exclusion. 7 Furthermore, despite the limited presence of IgG+ plasma cells in the healthy gut, elevated IgG‐expressing cells have long been noted in the mucosa of patients with intestinal inflammation. 8 , 9 , 10 , 11 Recently, there has been renewed interest in the role of IgG in intestinal host‐microbe interactions both in health and disease. 4 , 12 However, the signals that regulate class switching of naïve B cells to IgG‐producing plasma cells and memory B cells in the intestine are not fully understood. Here, we consider the differing effector functions of IgA vs IgG, review the molecular processes underpinning class‐switch recombination (CSR) and discuss the factors that determine class switching to IgA vs IgG in the intestine in health and disease.
2. B CELL SUBSETS AND ACTIVATION
B cell antibody production is shaped by the nature of the stimulating antigen and the environmental context in which that antigen is encountered. Protein antigens induce high‐affinity antibody responses within secondary lymphoid organs (SLOs), such as lymph nodes, spleen and gut‐associated lymphoid tissue (GALT), and require cognate interactions with antigen‐specific CD4 T cells, a process termed as T cell–dependent (TD) antibody production. Following T cell interaction, a subset of activated B cells generate short‐lived extrafollicular plasmablasts. Others form germinal centres (GCs), where they undergo class switching from IgM to IgG, IgA or IgE expression and affinity maturation, culminating in the emergence of long‐lived plasma cells and memory B cells. T cell–independent (TI) responses are mounted against carbohydrate/polysaccharide antigens in areas largely, but not completely, devoid of germinal centres. 13 These microscopically visible solitary isolated lymphoid tissues (SILTs) include cryptopatches as well as immature and mature isolated lymphoid follicles (ILFs). 14 , 15 Here, activated B cells and plasma cells exhibit lower levels of somatic hypermutation (SHM), indicative of GC independence. TI B cell responses can be broadly divided into two classes based on the antigen in question: TI type 1 responses are induced by antigens that polyclonally activate B cells, such as potent TLR or coreceptor signalling, and TI type 2 responses mediated by B cell receptor (BCR) recognition of multivalent epitopes. 16 In addition to BCR activation, TI type 2 responses require accessory signals to promote the development of antigen‐specific plasma cells. TI responses by marginal zone B cells in the spleen can also be induced in both a contact‐dependent and contact‐independent manner by a broad range of innate cells, including neutrophils, 17 dendritic cells (DCs) 18 and mast cells. 19 These innate ‘B helper cells’ have been shown to be important (in mice) for protection from a range of pathogens including S typhimurium 20 and West Nile Virus. 21 The reliance on innate cells in these responses has led to the proposal that they may constitute a “TI‐3” response distinct from TI type 1 and TI type 2 responses. 22
Further complexity arises when considering B cells themselves. In mice, the B cell lineage is broadly divided into two cell types–B1 and B2 cells–annotated according to their ontology, with B1 cells evident in the early prenatal period prior to the development of bone marrow, from which B2 cells arise. Post‐natally, B2 cells form the major B cell population, giving rise to follicular B cells that reside within SLOs that participate in both TD and TI responses, as well as marginal zone (MZ) B cells–a specialized splenic B cell subset that produces TI antibody in response to blood‐borne encapsulated bacteria.
Innate‐like B1 cells, along with MZ B cells, mainly express germ line–encoded antigen receptors with limited diversity and also participate in TI responses. 23 In mice, they are further subdivided into B1a and B1b cells, based on the presence or absence of CD5 expression, respectively, and are principally located in the peritoneal and pleural cavities, and to a lesser extent, in lymphoid tissues. 24 B1a cells produce low‐affinity polyreactive natural antibodies, mainly IgM and IgG3 in mice, with reactivity against self and foreign carbohydrate antigens, whilst B1b cells contribute to adaptive antibody responses to TI antigens, for example, in response to infection. 25 Their presence and phenotype in humans are debated, 26 with a CD20+ CD27+ CD43+ CD70− population initially identified as the human counterpart to murine B1 cells subsequently contested as representing preplasmablasts. 27 , 28 , 29 However, CD5 is not a specific marker for B1 cells in humans, and their existence in humans is still debated.
3. ANTIBODY EFFECTOR FUNCTION–IgA VS IgG
The major output of B cell activation is an antibody‐secreting cell. Each antibody isotype exhibits distinct effector functions; these include variable domain‐dependent functions such as bacterial/viral/toxin neutralization and Fc domain‐dependent functions, including complement activation, Fc receptor engagement and epithelial cell transcytosis. Therefore, the generation of specific Ig isotypes is tightly regulated to enable a humoral response optimized for the current, prevalent immune challenge. In considering why IgA dominates at mucosal surfaces, and why IgG, although the major circulating antibody, may be expressed in the gut in health and disease, it is instructive to consider the differing effector functions of IgA vs subclasses of IgG.
3.1. IgA antibody structure and effector function
Mice possess a single IgA subclass, whilst humans and other great apes have two subclasses, IgA1 and IgA2. IgA1 dominates in human serum, but in the intestine, the relative abundance of IgA1 vs IgA2 varies along the length of the digestive tract, with the IgA1:IgA2 ratio decreasing from 3:1 in the proximal small intestine to 1:3 in the colon. 4 , 30 IgA2 possess a shorter hinge region than IgA1, which makes it less susceptible to Streptococcus‐derived proteases, 31 and this may increase its durability in the microbe‐rich colon. 32 , 33 Polymeric IgA, consisting of predominantly dimeric IgA covalently connected by the joining or J chain, binds to the polymeric Ig receptor (pIgR) on the basolateral side of intestinal epithelial cells, before being internalized and transcytosed to the apical surface. 4 Here, it is proteolytically cleaved and released as secretory IgA (sIgA) into the gut lumen 6 (Figure 1). Class‐switched IgA+ B cells are mainly generated in the gut‐associated lymphoid tissue (GALT), including Peyer's patches and mesenteric lymph nodes, with long‐lived IgA+ plasma cells seeding from there to the intestinal lamina propria.
FIGURE 1.

Intestinal IgA and IgG in health and disease: During homeostasis (left), sIgA is the predominant antibody present in the gut mucosa. IgA+ plasma cells, quiescent MNPs and FoxP3‐expressing regulatory T cells form an immunotolerant triad in the tract, which directs IgA responses mostly against commensal microbes. sIgA is transported from the basolateral side of epithelial cells by the polymeric immunoglobulin receptor (pIgR), which attaches a secretory component to the IgA, releasing it into the intestinal lumen as sIgA. Here, sIgA mediates homeostasis by preventing commensal activation of epithelial cells and warding off potential pathobionts, including by enchaining bacteria. There is a relative paucity of anti‐commensal IgG during homeostasis, some of which is maternallyderived, and functions to prevent systemic spread of potential pathogens. In conditions such as UC and CD (right), IgG‐secreting B cells are induced and appear in large numbers. When gross epithelial barrier breach occurs, IgG can bind both pathogenic and commensal bacteria, causing crosslinking and activation of resident MNPs via FcγR interactions, induction of proinflammatory IL‐1β, and engagement of RORγt‐expressing Th17 cells
IgA mainly mediates its effector functions by binding to microbes and inhibiting epithelial invasion, as well as trapping or enchaining bacteria within the mucus layer, thereby promoting tolerance of intestinal commensals 34 (Table 1). Fc‐mediated effector functions (particularly in the gut) are limited; IgA weakly activates complement, it does not mediate NK cell–mediated cytotoxicity, and overall, it has weak opsonising capability. In humans, there is a specific IgA Fc receptor (FcαRI (CD89)), which is expressed on myeloid cells, particularly neutrophils, but also on monocytes and some macrophage subsets. 35 However, mice do not express FcαRI, but other IgA‐binding receptors include the transferrin receptor (CD71), asialoglycoprotein (ASGP)‐R, FcαμR and the polymeric IgR. 35 Of note, the induction of systemic protective IgA antibodies has been described following vaccination and in a number of bacterial and viral infections. 36 , 37 In this context, IgA does have some capacity to opsonize viruses or bacteria for uptake by phagocytes and may induce TRIM21‐dependent proteasomal destruction of intracellular virus. 36 , 37 , 38 , 39
TABLE 1.
Antibody effector functions (human)
| Effector functions | IgG1 | IgG2 | IgG3 | IgG4 | IgA | |
|---|---|---|---|---|---|---|
| Fc dependent | Complement activation | ++ | + | +++ | − | + |
| Opsonization | +++ | +/− | ++ | − | + | |
| ADCC | ++ | − | ++ | − | − | |
| V‐region–dependent | Neutralization | ++ | ++ | ++ | ++ | ++ |
ADCC = NK cell–mediated antibody‐dependent cellular cytotoxicity.
3.2. IgG antibody structure and effector function
There are four IgG subclasses in humans (IgG1‐4) and mice (IgG1, IgG2a/c, IgG2b and IgG3) (Table 1). Human IgG1 is the most abundant and predominantly targets soluble protein antigens and membrane proteins. 40 The generation of IgG1 is largely TD, and it exhibits potent effector functions, including complement activation and antibody‐mediated cellular cytotoxicity. 41 In mice, the effector profile of IgG2a and IgG2b is most similar to human IgG1 and also show strong effector function in vivo. 42 Human IgG2 responses (IgG3 in mice) are almost completely restricted to TI bacterial capsular carbohydrates, although anticarbohydrate IgG antibodies of other subclasses do exist. 43 IgG2 and IgG4 antibodies have a short, rigid hinge region compared with IgG1 and 3, resulting in impaired antibody flexibility, and this influences affinity for IgG Fc receptors (FcγRs) and C1q. Human IgG3 antibodies are the most effective subclass in terms of their activating effector functions, with avid complement‐activating capacity and affinity for activating FcγRs, but they exhibit a lower half‐life than other IgG subtypes due to impaired recycling via the neonatal Fc receptor (FcRn). 44 Finally, IgG4 is associated with induction by long‐term exposure to antigens in a non‐infectious setting, as observed in immune responses to allergens or parasitic infection. 40 IgG4 has relatively high affinity for the inhibitory receptor FcγRIIB, does not fix complement, exhibits an ability to spontaneously dissociate and form bispecific antibodies 45 and has the capacity to compete with IgE for allergens. It is therefore proposed to act as an inhibitor of effector responses. 46 Beyond subclass, post‐translational modification of the Fc region of IgG, most notably via N‐linked glycosylation, fine tunes FcγR affinity and complement activity. 47 , 48 , 49 , 50 Each IgG heavy chain carries a single covalently attached biantennary N‐glycan at the asparagine 297 residue of the Fc fragment Cγ2 domains, with over 900 IgG glycoforms possible. 51 Biantennary complexes can contain additional bisecting N‐acetylglucosamine (GlcNAc), core fucose, galactose and sialic acid residues. 52
Although IgA dominates in the intestine, commensal‐reactive IgG2b and IgG3 have been identified in GALT in healthy mice. 53 In homeostatic, non‐inflamed human colon, scRNA sequencing has shown that CD38‐expressing IgG+ plasma cells are enriched in the distal sigmoid colon compared with the more proximal caecum and transverse colon 33 (Figure 2). This spatial segregation of IgG plasma cells in the colon was coincident with an increased bacterial diversity, Th1:Th17 ratio, IgA plasma cells and CD4+ T cell clonal expansion in the distal colon. IgG plasma cells were predominantly IgG1 and IgG2; however, memory B cells predominately expressed IgG1 but little IgG2, indicating that IgG isotype expression is distinct between plasma or memory B cell fates. Although this study did not address IgG binding to commensal bacteria, distal colon‐resident commensal microbes were more likely to be bound by IgA than proximal colon‐resident microbes, suggesting that spatial differences in plasma cell abundances are functionally relevant and likely to have specific effects on the local predominance of antibody‐bound commensal microbes. 33
FIGURE 2.
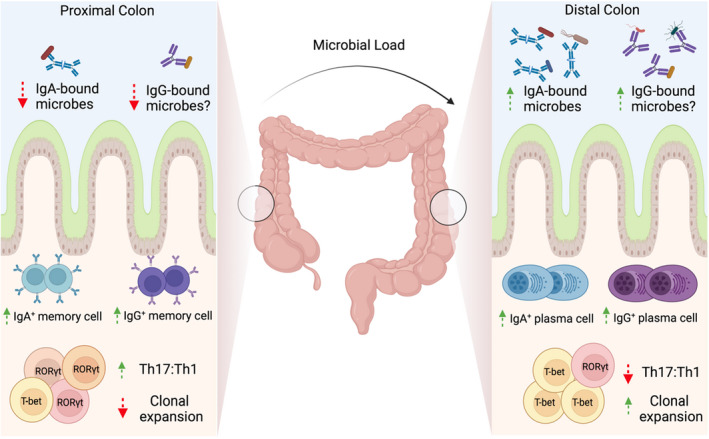
IgG and IgA cell profiles in the colon: The microbial load of the colon is known to increase from proximal to distal in both mice and man. Memory B cells predominate in the proximal colon, with more plasma cells of both the IgA and IgG variety in the distal colon. This plasma cell–rich profile of the distal colon is accompanied by a decreased Th17:Th1 ratio and increased clonal expansion of CD4+ T cells vs the proximal colon. More IgA‐bound microbes can also be found in the lumen of the distal colon, with potentially more IgG‐bound microbes to be found here also
Overall, relative to IgA, IgG is a far more proinflammatory immunoglobulin. As noted above, most IgG subclasses can activate complement, but many immune activating effects of IgG are mediated by binding to FcγRs. In humans, there are several activating receptors (FcγRIIA, FcγRIIC, FcγRIIIA and FcγRIII/) and a single inhibitory receptor, FcγRIIB, which plays a critical role in suppressing IgG‐mediated inflammation. 54 , 55 FcγRs are expressed on almost every immune cell type, including neutrophils, monocytes, macrophages, DCs, mast cells, natural killer (NK) cells and B cells, and this pattern of expression enables IgG to engage almost every facet of the immune system, underpinning its potent proinflammatory potential. Most FcγRs are low‐to‐medium affinity for IgG, requiring cross‐linking of several receptors into signalling synapses on the cell surface in order to initiate productive signalling. This is achieved through the formation of immune complexes (IC) between antigen and antigen‐specific IgG or by IgG‐opsonised cells. The inhibitory receptor, FcγRIIB, acts as an additional regulatory mechanism to suppress IgG‐mediated inflammation, although its expression is heterogeneous across cells of the immune system and subject to regulation by various stimuli, particularly by the cytokine milieu. 56 , 57 The ratio of activating to inhibitory FcγRs on any given cell is known as the activating/inhibitory (A/I) ratio, and its context‐specific modulation allows for appropriate immune responses to be raised. 54 , 58 Genetic polymorphisms in human FCGR genes that alter receptor expression or function are frequently associated with differential susceptibility to both infection and autoimmunity, including inflammatory bowel disease. 55 , 58 , 59 Differences in IgG glycosylation can also alter affinity for activating vs inhibitory FcγRs 60 , 61 , 62 , 63 ; for example, defucosylation increases the binding affinity of IgG for activating FcγRIIIA (but not FcγRIIB) 10–50 fold. 64
IgG functions in the intestine in homeostasis include protection against infectious challenge 65 , 66 , 67 , 68 and allergic intolerance, 69 neonatal immune development 53 , 70 and tumour resistance. 71 Conversely, chronic inflammation of the intestine in inflammatory bowel disease (IBD), a clinically heterogenous group of disorders, may be driven by the proinflammatory effects of IgG, activating local FcγR‐expressing cells 72 , 73 , 74 , 75 (Figure 1).
4. CLASS‐SWITCH RECOMBINATION
The ability to change the antibody isotype produced, in‐line with the nature and context of the immunological challenge, is a key feature of humoral immunity. The process of selecting the heavy chain which will confer the most appropriate effector function profile for the current challenge is central to achieve an effective response.
4.1. Molecular processes underpinning CSR
CSR occurs by an intrachromosomal deletion recombination event with the replacement of the default expressed Cμ exon cluster in naïve B cells with Cγ, Cϵ or Cα, for IgG, IgE and IgA, respectively (Figure 3). 76 The exact molecular mechanisms governing the intrachromosomal DNA recombination events have been reviewed extensively elsewhere. 77 , 78 , 79 Briefly, the exons for the different constant heavy chains that specify isotype class are located downstream of the heavy chain VDJ sequences. All of the constant genes, with the exception of δ, are preceded by switch (S) regions, which vary in length depending on the constant gene in question, but are characterized by the presence of highly repetitive nucleotide sequences. 80 , 81 CSR begins when intronic promoters' upstream of the targeted S region begin transcribing germ‐line transcripts (GLTs), untranscribed mRNA units consisting of the S region promoter, S region and constant region gene. The unique repetitive nucleotide sequences in the S region lead to the formation of bubble‐like structures called R loops in the DNA as transcription proceeds, whereby the non‐template DNA strand is displaced, forming single‐stranded DNA, which is the substrate of activation‐induced cytidine deaminase (AID). 82 , 83 Accessory proteins then allow for the formation of double‐stranded breaks through either base excision repair or mismatch repair depending on the proximity of the AID‐induced single‐stranded breaks to one another. 84 The donor region, which is invariably the Sμ region, is then joined to the acceptor S region of the constant gene being switched in, and the intervening DNA sequences of other constant genes are excised (Figure 3). The somatic editing nature of class‐switch recombination makes it irreversible.
FIGURE 3.
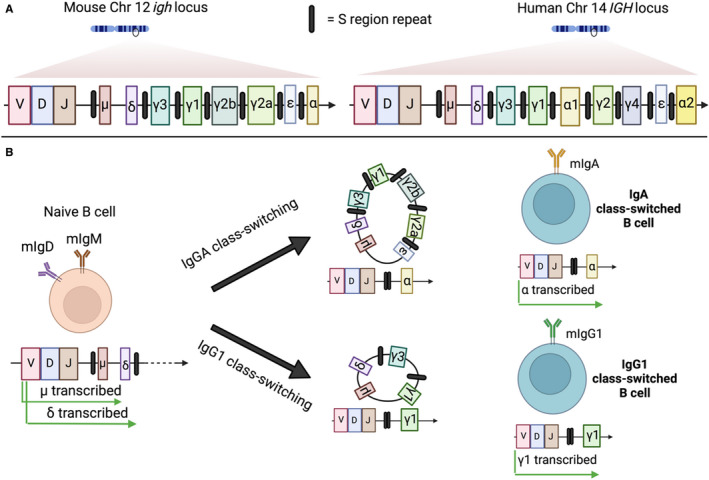
Class‐switch recombination: (A) The locus for the heavy chain constant region for both mouse and human DNA is depicted. The genes encoding the heavy chain constant regions are arrayed alongside each other downstream of the VDJ segments, which defines the antigen specificity of the BCR. All the heavy chain constant regions, with the exception of the δ gene, are flanked at their 5′ end by switch (S) regions, which are responsible for governing the AID‐mediated recombination events required to switch isotype class during activation. (B) Naïve B cells that have not encountered their cognate antigen (left) express both membrane‐bound IgM and IgD. Upon antigen encounter and T cell help, class switching begins, substituting these existing transcribed constant domains of the BCR for other isotypes. Mouse B cell isotype switching to IgA or IgG1 is presented as an example of class‐switch recombination. Depending on the cytokine and T cell signals delivered to the B cell during activation, it will preferentially excise intervening constant region genes by a recombination event mediated by S region joining (middle), resulting in the placement immediately downstream of the VDJ segment of the desired isotype (right)
4.2. Signals determining CSR
A variety of immune‐ and pathogen‐derived signals regulate the initiation and isotype selection of CSR, playing a critical role in determining the dominant antibody isotype produced in different contexts. Broadly, B cell–intrinsic CD40 and/or Toll‐like receptor signalling act as critical primary signals in CSR involved in the induction of the enzyme AID and other CSR factors, 79 whilst T cell‐ and DC‐derived cytokines provide secondary signals that determine the fate of CSR, namely the isotype selected.
4.2.1. Primary signals
CD40 ligand (CD40L) and/or TLR stimulation provide essential signals to initiate B cell proliferation and CSR. 79 , 85 In TD antibody responses, antigen‐activated B cells migrate to the T‐B border in secondary lymphoid organs where they form cognate interactions with DC‐primed T cells destined to become T follicular helper (Tfh) cells, 86 as well as CD40L expression; these Tfh cells are essential providers of B cell–activating cytokines, particularly IL‐4 and IL‐21, that direct GC activity, CSR and the emergence of high‐affinity plasma cells and memory B cells. 87 , 88 Recently, Tfh cells were found to progressively secrete IL‐21 and IL‐4 for the induction of high‐affinity BCR clones and the development of Blimp‐1–dependent plasma cells, respectively. 89 Moreover, additional stimuli that induce B cell proliferation can be delivered from antigen‐capturing stromal follicular dendritic cells (FDCs), including CXCL13, 90 B cell–activating factor (BAFF) 90 and cholesterol metabolites. 91
In TI antibody responses, PAMPs acting through TLRs can induce AID expression in an NFkB‐dependent manner, with TLR signalling involved in polysaccharide‐specific IgG generation and immune responses to encapsulated bacteria. TLR1‐2, TLR4, TLR7 and TLR8 can synergise with the BCR to induce class switching to IgG3 and other isotypes in the absence of T cell help, with murine IgG3 levels relatively unaffected by the absence of CD40 or T cells. 23 , 92 , 93 Lipopolysaccharide (LPS), found in the outer membrane of Gram‐negative bacteria, is the only known microbial product that can directly induce CSR through simultaneous TLR4 ligation and BCR cross‐linking in murine B cells, although not in human B cells, and can potently activate B1 and MZ B cells. 94
In the absence of T cells, B cells participating in TI type 2 responses may receive additional proactivation and survival signals that promote B cell activation and proliferation, such as BAFF and a proliferation‐inducing ligand (APRIL). 16 , 17 These tumour necrosis factor (TNF) ligand superfamily members engage the BAFF receptor (BAFF‐R), B cell maturation antigen (BCMA) and transmembrane activator and CAML interactor (TACI) to stimulate AID expression. 92 , 95 BAFF and APRIL are released in response to TLR stimulation by several cell subsets, including mononuclear phagocytes, neutrophils, eosinophils, ILCs, DCs, FDCs and epithelial cells. 17 , 90 , 96 , 97 , 98 , 99 , 100
4.2.2. Secondary signals
Naïve B cells can switch to any isotype, an event that is controlled by immune cells, such as T cells and DCs, through the secretion of specific cytokines. These cytokines dictate the isotype most appropriate for eliciting pathogen/antigen‐tailored responses in the context of type 1, 2 or 17 immunity. In mice, Th2‐associated IL‐4 supports class switching to IgG1 and IgE in the presence of CD40L, whilst TGF‐β plays a crucial role in the induction of IgA. TGF‐β can induce histone modification at the Sα region that makes it more amenable to the CSR machinery. 101 In addition to TGFβ, BAFF, APRIL, IL‐10 and IL‐6 have also been documented to support IgA CSR. 87 , 102 In contrast, Th1‐associated IFNγ induces class switching to IgG2a in vitro and in vivo. 103 , 104 , 105 More recently, Th17 cells have been demonstrated to support IgG CSR in vivo: IL‐17A and IL‐21 promote IgG2a/IgG3 and IgG1/2b CSR, respectively, in mice. 106
Beyond class‐switching itself, T cell–derived cytokines can also influence IgG glycosylation patterns at the asparagine 297 residue on each IgG heavy chain. A recent elegant study demonstrated that in response to IL‐23, Th17‐derived IL‐22 and IL‐21 could regulate IgG sialylation and augment IgG inflammatory activity in a murine model of rheumatoid arthritis (RA). 107 Therefore, cytokines play a crucial role in determining both Ig isotype CSR and the inflammatory potential of IgG. The relevance of these pathways to intestinal IgA and IgG class switching will be discussed below.
5. INTESTINAL CLASS SWITCHING IN HEALTH
IgA class switching in the gut requires the coordinated interaction of intestinal epithelium, DCs, macrophages and regulatory T cells, which enables commensal microbes and antigens to be sampled and to subsequently induce B cell class switching in the context of a homeostatic milieu rich in IL‐10 and TGFβ 4 , 6 , 87 , 108 (Figure 4). The signals regulating IgA class switching in the gut have been extensively studied and reviewed. 4 , 6 , 108 , 109 , 110 Here, we discuss the major principles of intestinal IgA CSR and how these observations may be relevant to understanding the regulation of intestinal IgG response in health and disease.
FIGURE 4.
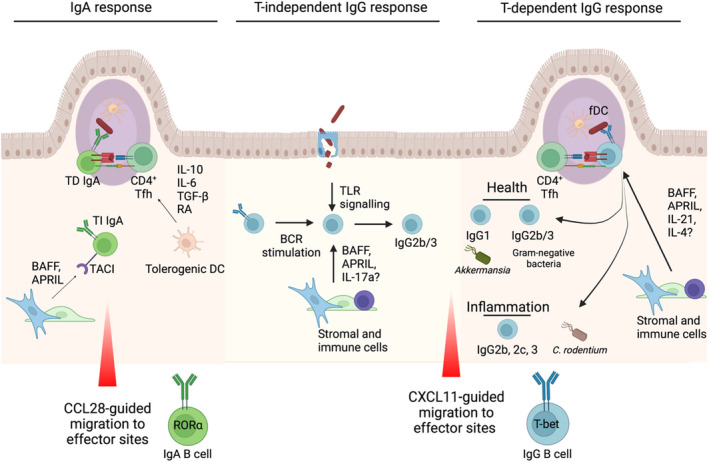
IgA vs. IgG generation and maintenance in the intestinal tract: IgA responses, which can be both T cell–dependent and –independent. In T‐dependent responses in the Payer's patches, a variety of cytokines including IL‐10, IL‐6, TGF‐β and retinoic acid (RA) from DCs drive IgA induction via CD4+ Tfh cells. BAFF and APRIL can mediate T‐independent responses outside the Peyer's patches via TACI binding on B cells. Once generated, IgA+ B cells can migrate from their inductive sites to effector sites via chemokines including epithelial‐derived CCL28, where they express the transcription factor RORα. In mice, intestinal IgG2b/3 CSR in health (middle) has been shown to be largely independent of T cells and dependent on B cell–intrinsic TLR signalling. BAFF and APRIL, released by haematopoietic and stromal cells following microbial stimulation, can promote TI AID expression in B cells, while IL‐17A can support IgG3 CSR. The species of microbes targeted by mucosal IgG responses differ between experimental conditions, suggesting a plastic response highly dependent on the composition of the microbiota. In healthy mice, T cell–dependent IgG responses towards the microbiota that have been identified include IgG1 targeting of Akkermansia muciniphila, as well as cross‐reactive IgG2b/3 responses towards Gram‐negative bacteria that arise following dissemination of intestinal bacteria and provide protection against systemic infection. Inflammatory T cell–dependent IgG responses are also critical for elimination of C rodentium in mice, with similar TD mechanisms likely to be involved in inflammatory bowel disease in humans and mice
5.1. Microbial antigens and T cell help in IgA vs IgG responses
GALT, in particular Peyer's patches of the small intestine, are the major site of IgA+B cell generation in the gut. 4 , 6 , 108 TD responses arising from B2 cells are directed towards protein antigens, occur within Peyer's patches and mesenteric lymph nodes (MLNs) and rely on CD40L‐CD40 interactions with Tfh cells. In contrast, TI IgA responses occur within the GALT and MLN, as well as in non‐lymphoid tissues and can arise from both B1b and B2 cells in the presence of BAFF and APRIL. 108 , 111 , 112 , 113 Many enteric pathogens elicit TD high‐affinity IgA responses, whilst commensals can elicit both TD and TI responses.
Peyer's patches are composed of several B cell follicles on a network of follicular dendritic cells, with interfollicular regions of DCs and T cells directly underlying the follicle‐associated epithelium and subepithelial dome. 114 Germinal centres are constitutively present in Peyer's patches, 6 resulting from continuous exposure to microbial antigens that are trafficked through M cells. GC formation in Peyer's patches and the MLN is strictly dependent on T cells, with Tfh cells potentially arising from other CD4+ T cell subsets due to their plasticity. 115 , 116 , 117 , 118 However, non‐cognate T‐B interactions have been shown to be sufficient for GC formation and SHM in Peyer's patches. B cells expressing surrogate Ig receptors can become activated to form GCs in Peyer's patches, in the presence of non‐cognate T cell help and microbiota‐derived signals. 119 Therefore, under certain circumstances, BCR‐independent B cell activation can support T cell–dependent IgA production in Peyer's patches.
GALT GCs and IgA+ plasma cell formation are severely impaired in CD40‐ and T cell–deficient mice. 115 , 120 , 121 , 122 Strikingly, however, total levels of IgA‐opsonised commensal microbes remain largely unaffected in these mice, although they exhibit impaired antigen‐specific IgA responses to protein antigens, such as cholera toxin. 121 , 122 , 123 Whilst the microbial species targeted in T cell–deficient mice differs significantly from WT mice, including an inability to target segmented filamentous bacteria (SFB), this was found to be independent of CSR or SHM. In a series of elegant experiments, commensal reactive IgA was found to derive from T cell–independent B1b and T cell–responsive B2 cells, with both responses covering overlapping and diverse bacterial taxa, 115 although the majority of IgA+ plasma cells showed signs of SHM. Therefore, whilst T‐B interactions are not required for low‐affinity TI IgA responses, they are required to mount specific IgA responses to microbes and orally administered TD antigens.
Although dominated by IgA, intestinal B cell IgG class switching occurs at a low level during homeostasis. Koch et al demonstrated commensal‐reactive IgG2b and IgG3 responses within MLN and Peyer's patches in mice. 53 These antibodies were largely generated independently of T cells but required B cell–intrinsic TLR signalling. This is consistent with the known requirements for TI IgG3 responses in mice, which predominantly target carbohydrate antigens. 93 Furthermore, IgG2b/IgG3+ B cells exhibited cell‐surface expression phenotype consistent with plasma cells and B1 cells, including CD138 and CD43, respectively. In a complementary study, gut microbiota‐induced IgG antibodies (predominantly IgG2b and IgG3) were shown to mediate systemic protection against E coli and Salmonella challenge, 66 as well as B cell–intrinsic TLR signalling; IgG production was dependent on T cells in this study, attributed to the spread of Gram‐negative commensal bacteria to systemic sites, including the spleen.
Although smaller in magnitude than IgG2b and IgG3 levels, a subset of microbiota is also targeted by a T cell–dependent IgG1 response (Figure 4), shown to be enriched for Akkermansia muciniphila, 124 a widely‐studied commensal bacterium with potential therapeutic applications. 125 A muciniphila‐specific TCR‐transgenic T cells demonstrated preferential T cell differentiation towards a Tfh phenotype within Peyer's patches when transferred into colonized mice, as well as a mixed Th phenotype within the gut lamina propria, including Th1, Th17 cells and Tregs. This T cell response was heavily microbiota‐dependent, with T cell fate differing substantially between altered Schaedler flora and SPF mice. 124 Therefore, homeostatic mucosal IgG1 responses are linked to a subset of microbial species capable of inducing antigen‐specific adaptive immunity, unlike broadly polyreactive and T cell–independent IgA responses. 126
Reductive experiments in which germ‐free mice were challenged with an auxotrophic non‐replicating E coli commensal strain enabled additional features of antigens that stimulate IgA vs IgG responses to be determined, beyond the nature of the bacterial strain, without the experimental interference of a pre‐existing host microbiome and antibody response. 127 , 128 In this model, recombinant dimeric monoclonal IgA antibodies derived from mucosal plasma cells exclusively bound plasma membrane bacterial antigens, and not cytoplasmic or ribosomal bacterial proteins. 129 Intriguingly, another study using the same auxotrophic E coli strain confirmed that mucosal IgA is predominantly targeted to cell‐surface antigens, whilst IgG can target both intracellular and plasma membrane antigens. 128 This suggests differing bacterial antigen processing may be required to generate IgA vs IgG responses. Therefore, although mucosal IgA and IgG have some overlapping antigen targets, there are clearly distinct mechanisms of action and signals required to mount mucosal IgA responses vs intestinal IgG responses.
5.2. Cytokine signals in IgA vs IgG responses
Several cytokines and soluble factors, including TGFβ, IL‐10, IL‐6 and retinoic acid, can promote IgA CSR in GALT. Of these, the most critical cytokine in Peyer's patches is TGFβ (Figure 4). Mice deficiency in TGFβRII exhibit abrogated IgA levels in Peyer's patches and elevated local and systemic IgG responses. 130 Numerous cellular sources of TGFβ have been identified, such as T and B cells, FDCs and DCs, and it acts to induce expression of germ‐line α‐transcripts in B cells. 6 , 131 Recently, DCs within the subepithelial dome (SED) of Peyer's patches were found to play a critical role in IgA CSR through αvβ8‐mediated activation of TGFβ. Following activation, CCR6+ pre‐GC B cells migrate towards CCL20‐expressing follicle‐associated epithelial cells enabling interactions with DCs located in the SED that direct IgA CSR. 132 Nitric oxide–producing DCs also support TD and TI IgA CSR through TGFβ receptor induction in B cells and DC‐intrinsic BAFF/APRIL expression, respectively. Beyond TGFβ, GALT DCs can also promote B cell gut tropism and IgA secretion through the production of RA in the absence of T cells. 133 BAFF and APRIL are expressed within GALT and the lamina propria and can promote TI IgA CSR, 100 , 134 , 135 although their primary role is suggested to be plasma cell maintenance. 4 , 96 , 100 , 110 , 136 Their importance is highlighted by common variable immunodeficiency (CVID) and selective IgA deficiency (SIgAD) linked to mutations in TNFRSF13B, encoding TACI. 137
It is notable that SIgAD is the most common primary immunodeficiency and remains largely asymptomatic in the majority of individuals. Compensation of other Ig subclasses, including IgG2, is required to prevent severe infections and complications. 138 However, SIgAD patients are at increased risk of IgG‐associated disorders linked to a defective mucosal barrier, including coeliac disease, UC and autoimmunity. 137 , 138 , 139 Therefore, intestinal penetrance of microbial and other environmental antigens seems to play an important role in mucosal and systemic IgG responses. However, little is known about the cytokine‐mediated signals that directly promote IgG class switching in the gut, although BAFF and APRIL can support IgG1 CSR in vitro. 134 , 135 Given the preferential induction of Peyer's patch‐resident Tfh cells by a subsets of the microbiota, IL‐4 and IL‐21 may play a role in mucosal IgG1 CSR in mice. 124 However, very few commensal microbes have been identified that induce antigen‐specific IgA or IgG responses. Indeed, T cells appear dispensable for the majority of anticommensal IgA responses in health. 53 Notably, circulating B cells and serum IgG/IgM levels are significantly reduced in humans with BAFF‐R deficiency, whilst these patients exhibit normal or high levels of IgA. 140 However, further work is needed to identify the additional factors that promote homeostatic IgG responses in GALT.
6. IgG CLASS SWITCHING IN INFLAMMATORY BOWEL DISEASE
IBD is a chronic relapsing inflammatory disease of the gastrointestinal tract driven by an aberrant immune response against the microbiota. There are two major subtypes of IBD, Crohn's disease (CD) and ulcerative colitis (UC), which differ in their clinical presentations, genetic associations and determinant pathological immunity. CD may affect any part of the GI tract, most commonly the terminal ileum and colon, with inflammation occurring segmentally and transmural in nature. 141 Genetic susceptibility to CD is associated with defects in microbial sensing and Th17 function (NOD2, ATG16L1, LRRK2, IL23R and STAT3). 142 In contrast, UC targets the colon, with continuous superficial inflammation, and is genetically linked to alterations in barrier integrity (HNF4A) and the major histocompatibility complex region, Th17 function and FCGR2A polymorphisms. 143
Beyond genetic susceptibility, microbial dysbiosis occurs in patients with IBD, with strong evidence indicating a role for the intestinal microbiota in triggering disease. 144 In particular, lower bacterial diversity, a reduction in Bacteroides and Firmicutes bacteria, and an increase in Proteobacteria and Actinobacteria, is observed in CD, with similar changes reported in UC. 141 , 143 , 145 Approximately, a third of CD patients have an increase in adherent‐invasive Escherichia coli, 146 which have been shown to promote Th17 inflammation in vivo, 147 whilst the presence of short‐chain fatty acid (SCFA)‐producing bacteria, such as Bifidobacterium, in CD patients is associated with quiescent disease and anti‐TNFα treatment response. This demonstrates a significant impact of microbial communities on the underlying immune response. 144 , 145
Although the pathogenic role of T cells and the IL‐23 pathway has been delineated, both clinically and in murine models of colitis, 142 , 148 , 149 , 150 , 151 the role B cells and antibodies in IBD is much less well understood. The reported ineffectiveness of a grossly underpowered randomized controlled trial of rituximab (anti‐CD20 IgG) in the treatment of UC, 152 which also represents a suboptimal strategy to deplete IgG‐producing plasma cells that do not express CD20, 153 as well as case reports of de novo Crohn's disease following rituximab administration, 154 , 155 has led to a general conception that humoral immunity is unimportant in IBD. However, a combination of genetic, 142 , 156 single‐cell RNA sequencing 75 , 157 , 158 and functional human and murine studies 72 , 73 , 74 , 159 support a pathogenic role for IgG in the pathogenesis of IBD. In particular, attention has centred on Fcγ receptors, given that a low‐affinity variant of the activating receptor FcγRIIA is linked to protection from UC and leads to attenuated myeloid cell responses to IgG. 72 , 142 , 156 The genetic association of an IgG receptor and IBD is on the surface, counterintuitive, given the dominance of IgA in the intestine in health. However, we and others identified a marked increase in luminal, commensal‐binding IgG in UC, 8 , 9 , 10 , 11 suggesting a shift in the class‐switching signals encountered by intestinal B cells in IBD. Using two mouse models of intestinal inflammation, Citrobacter rodentium, a model of human attaching‐effacing Escherichia coli infection in humans, and dextran sodium sulphate (DSS) administration, we found a strong induction of IgG antibodies directed against the microbiota and enteropathogens. 66 , 72 , 74 In these models, epithelial barrier breach induces local and systemic IgG that controls bacterial dissemination but may promote colitis through the activation of local FcγR‐expressing cells. 72 , 73 , 74 , 75 Specifically, FcyR‐expressing intestinal macrophages activation by commensal‐IgG immune complexes results in IL‐1β production, which in turn stimulates Th17 activation. 63 Given the timescale of de novo IgG induction (typically beyond day 7) and the well‐known ability of DSS to induce disease in Rag2‐deficient mice, IgG is likely to predominantly contribute to chronic phases of inflammation in this model, although circulating anticommensal IgG is present in healthy mice that may be involved at disease onset. 53 , 66 Beyond bone fide infection models and chemically induced colitis, antimicrobial IgG is observed in a variety of spontaneous colitis models in immune‐replete mice, including Il10 −/−, 160 C3H/HeJBir 161 and Nod2 −/− Cybb −/− mice, 162 as well as mice strains that exhibit increased microbial penetrance, such as Myd88 −/− Ticam −/− and Nos2 −/− Cybb −/− double‐deficient mice. 163 However, it remains to be determined whether IgG promotes intestinal inflammation across different murine models. Indeed, in Nod2/Cybb and Nos2/Cybb double‐deficient mice, the induction of microbiota‐targeting IgA and IgG antibodies is protective against microbial dissemination. 162 , 163
Whether the emergence of antimicrobial IgG is secondary to epithelial barrier dysfunction in IBD patients is not understood. It is noteworthy that anti‐microbial IgG is elevated in Crohn's disease patients several years prior to disease diagnosis, 164 suggesting significant adaptive humoral immune dysregulation during very early disease. Moreover, IgG+ plasma cells are enriched in terminal ileum biopsies of newly diagnosed, early‐onset paediatric Crohn's disease patients. 75 Therefore, it is likely that mucosal IgG responses exert differing effector functions across the timecourse of IBD, with variation in their detrimental contribution to inflammation.
Overall, the genetic signal, the prominent induction of intestinal IgG observed in IBD and subsequent functional experiments proving a causative role suggest that agents that could block IgG binding to activating FcγRs, or promote engagement or expression of the inhibitory FcyRIIB, may be useful therapeutic targets in IBD. In addition, the signals regulating the class switch to IgG observed in IBD may also represent a potential therapeutic target to prevent IgG generation and its subsequent inflammatory effector functions within the mucosa. Given their abundance in active disease, it is likely that T cells are involved in the IgG class‐switch response in IBD (Figure 4). Notably, clearance of C rodentium is critically dependent on B cells and IgG, 68 , 165 , 166 with mice lacking IgG, but not IgM or secretory IgA, developing exacerbated intestinal pathology and succumb to systemic spread of infection. The IgG response specifically targets bacterial virulence factors and promotes pathogen eradication through activation of local myeloid cells, 165 as well as IgG; CD4+ T cells are required for sterilizing immunity and C rodentium‐specific IgG responses are severely impaired in T cell–deficient mice. 166 Nod2‐deficient animals also exhibit impaired IgG and IFNγ responses following C rodentium challenge. 167 Given the role of IFNγ in promoting IgG CSR in vivo, this suggests a potential role for Th1 cells in intestinal IgG CSR.
T cells can also participate in shaping IgG glycosylation. As mentioned previously, IL‐23–mediated inflammation in mice has been shown to result in IgG desialylation that promotes inflammatory activity in models of rheumatoid arthritis. 107 Indeed, IgG glycome profiling has identified decreased IgG sialylation in CD patients. 168 Whether this directly impacts inflammatory IgG activity in IBD is not known but highlights a potential link between a major IBD‐associated risk pathway and local IgG induction. Furthermore, the impact of treatments, such as anti‐TNFα and anti‐IL‐23 monoclonal antibodies, on the frequency and inflammatory nature of IgG will be of great interest.
In addition to T cells, other immune cells and cytokines may impact the local IBD IgG response. In particular, BAFF expression is significantly increased in mucosal biopsies from IBD patients 169 and represents a promising target in B cell–mediated diseases, such as lupus erythematosus and antibody‐mediated rejection. 170 BAFF promotes B cell survival and augments B cell proliferation and Ig secretion following BCR engagement. 171 , 172 , 173 BAFF also acts as a costimulatory factor in T cell activation, 169 suggesting a potential role in targeting BAFF for dual T cell/B cell suppression. The nature of the IBD‐associated microbiota is likely to be critical in dictating B cell class switching. Certain bacterial species, such as adherent‐invasive E coli, which are prevalent in IBD, may influence T cell–dependent IgG responses in a manner analogous to invasive bacteria in mice. Furthermore, B cell responses may be directly or indirectly regulated by microbiota‐derived metabolites, such as SCFAs and aryl hydrocarbon receptor (AHR) ligands, which are known to be disrupted in IBD. 145 , 174 , 175
The T‐box transcription factor family member T‐bet has traditionally been associated with initiating and directing effector Th1 responses and controlling IFN‐y secretion in viral infection, as well as in dictating Type 1 identity in ILC1s and a subset of mucosal ILC3s. 176 , 177 More recently, an atypical B cell subset expressing T‐bet and secreting IgG has received much attention due to their presence in both mice and humans in a variety of diseases, including autoimmune disorders such as multiple sclerosis and SLE, as well as infectious diseases including Hepatitis C, HIV and rhinovirus. 178 , 179 , 180 , 181 , 182 B cell–specific T‐bet‐deficient mice demonstrated decreased class switching to IgG2a, and to a lesser extent, IgG2b and IgG3, in response to IFN‐y, as well as impaired IgG autoantibody production in a mouse model of lupus. 183 Enforced expression of T‐bet ex‐vivo in T‐bet‐deficient B cells rescued germ‐line IgG2a transcription, suggesting a B cell–intrinsic role for T‐bet in IgG2a class switching. 105 In mice immunized with the hapten NP‐KLH (nitrophenylacetyl‐keyhole limpet hemocyanin) following transfer of WT and Tbx21 −/− splenic B cells, IgG2a+ memory B cell and plasma cells were reduced in the absence of T‐bet. 184 A variety of overlapping factors have been proposed to promote T‐bet expression in B cells, mostly Type 1 cytokines including IL‐12, IL‐18, IFN‐y, IL‐27, as well as TLR7 and TLR9 agonists. 185 , 186 , 187 , 188 The presence of T‐bet‐expressing B cells is at least partially T cell contact‐ and antigen‐dependent, as they do not develop in the absence of MHC‐II or CD40 expression in B cells. 189 Crucially, T‐bet+ B cells have been reported to be increased in frequency in the intestinal mucosa of CD, where their presence correlated with increased disease severity, suggestive of a proinflammatory contribution in CD. 190 Moreover, these atypical T‐bet+ B cells were predominantly IgG+ and expressed higher amounts of IFN‐y than their IgA+ and IgM+ counterparts. 190 IgA+ memory B cells do not rely on T‐bet for class switching and rather rely on the transcription factor RORα. 184 This suggests that T‐bet, or the factors that drive its upregulation, may be a therapeutic target in IBD, as it is required for the generation of IgG+ B cells and CD4+ Th1 cells, both of which have pathogenic roles in IBD.
Recently, concurrent studies have identified the importance of anti‐fungal antibody responses in the gastrointestinal tract, expanding the role of humoral intestinal immunity into the eukaryotic kingdom. The presence of specific serum IgG against fungal species has been well‐documented in Crohn's disease patients, specifically anti‐Saccharomyces cerevisiae antibodies (ASCAs). 191 , 192 However, mucosal Ig responses to fungal species are distinct from those in the serum, 193 predominantly targeting Candida species, particularly the pathobiont C albicans, which may be pathogenic, causing fatal extraintestinal diseases, including meningoencephalitis in some contexts. 194 Intriguingly, parallel studies investigating adaptive immunity‐dependent mucosal responses in mice against C albicans (mostly IgA with some IgG1) have shown they are preferentially targeted against antigens expressed only on the tissue‐invasive hyphal morphotype of the fungus and not the less virulent circular yeast form. 195 , 196 These hyphae‐expressing C albicans are associated with worse colitis and extraintestinal diseases. In a cohort of 12 patients with Crohn's disease and 9 healthy controls, the targeting of these hyphal‐associated virulence factors by intestinal antibodies was decreased in CD patients, leading to increased hyphal forms of fungi. 195 Thus, by selectively targeting for pathogenic forms of the same fungus over commensal forms, the intestinal humoral immune response can promote homeostasis and ward off distinct morphotypes that are pathogenic if they gain access to the systemic system. 197 Similar disparate antibody responses to distinct genetic phase variations of a single bacterium species have also been reported. 129
Although, at baseline, intestinal fungi are bound by only low amounts of mucosal IgG, this can be increased rapidly by adding ex vivo of serum IgG in both humans and mice. 193 This IgG is antigen‐specific, dependent on T cell presence for its generation and significantly lowered in germ‐free mice. IgG2b and IgG3 constitute the majority of serum IgG capable of binding to commensal gut fungi in mice. This IgG‐bound mycobiota targets predominantly Candida albicans, with no other strains, including S cerevisiae, capable of generating serum IgG responses in germ‐free mice. Intriguingly, the site of B cell expansion and IgG isotype switching was found to be not the Peyer's patches or the mesenteric lymph nodes, but the spleen, which accumulated more Fas+GL‐7+ germinal centre B cells and IgG+ B cells following C albicans colonization. 193 This suggest that controlled movement of gut‐resident antigens and/or B cells to extraintestinal lymphoid tissues can educate extraintestinal humoral immunity and induce systemic mucosal‐educated IgG antibodies with targeted specificity for gut fungi, in a similar manner has been reported for IgA gut‐educated antibodies. 198 , 199 , 200 Depletion of intestinal CXC3R1+ MNPs in mice disrupts systemic anti‐C albicans IgG and polymorphisms in the coding region of CXC3R1 in humans have been associated with increased antifungal systemic IgG in CD patients. 201 Whether these systemic IgG antibodies can subsequently gain access to the intestinal mucosa during periods of inflammation is unknown.
7. MUCOSAL PLASMA CELL NICHES
Beyond IgG CSR, the gut‐trophic factors and distinct mucosal niches occupied by IgG+ and IgA+ plasma cells, may represent another therapeutic target in intestinal disease. For example, IgG+ plasma cells are enriched in inflamed intestinal tissue across a range of disorders (HIV infection, chronic granulomatous disease and CD) and exhibit unique chemokine receptor expression, with reduced CCR10 and increased CXCR4 expression relative to IgA plasma cells. 202 , 203
Broadly speaking, the chemokine CCL25 is required for the recruitment of IgA+ plasma cells into the lamina propria of the small intestine, whilst CCL28 is more important for localizing plasma cells in the large intestine, 204 , 205 , 206 with eosinophils implicated as a niche component for IgA+ plasma cells, 136 and this differs from the niche requirements of IgG plasma cells. For example, Salmonella‐specific IgA plasmablasts generated by oral vaccination in humans display robust migration towards the mucosal‐associated cytokine CCL28 in exvivo transwell assays, in contrast to Salmonella‐specific IgG plasmablasts, which showed little CCL28‐dependent chemotaxis. 207 Consistent with this, CCL28 is abundantly expressed in both intestinal and extraintestinal mucosal tissues, and its receptor CCR10 is highly expressed by IgA+ plasmblasts. 204 , 208 Intestinal IgG+ B cells preferentially migrated towards CXCL11 209 expressed by monocytes in response to microbial stimulation and overexpressed in inflamed tissue in IBD patients. 210 , 211 Collectively, these studies suggest that distinct chemokine cues orchestrate isotype‐specific B cell and plasmablast movement and residency to specific intestinal niches in health and disease.
In the human colon, IgG+ plasma cells are enriched in the distal sigmoid colon, 33 which suggests that niches for these cells could be influenced by bacterial diversity or the local Th1:Th17 ratio. In addition, the distribution of commensal‐sensing luminal‐facing epithelial cell receptors shows non‐uniform distribution along the intestine, highlighting areas where host‐microbiota immunological interfaces are more likely to occur. 212 Local IgG‐commensal immune complexes may also stimulate mononuclear phagocytes to provide IgG plasma cell niche factors, as myeloid cells have been well‐described as plasma cell niche participants in the bone marrow, via surface CD80/86 expression and cytokines such as IL‐6. 213 Single‐cell transcriptomic studies have identified cellular modules associated with IgG plasma niches in CD patients refractory to anti‐TNF therapy. 158 Further delineation of the specific factors, which initiate and maintain intestinal IgG+ plasma cell niches, could enable therapeutic targeting to disrupt the production of pathogenic IgG+ in IBD whilst sparing beneficial homeostatic IgA‐producing cells.
8. CONCLUSION
Despite the renewed interest in recent years in mucosal IgG, much work is needed to elucidate the mechanisms that regulate the induction of IgG responses in health, enteric infection and chronic inflammatory disease within the gastrointestinal tract. Whilst IgA is clearly the dominant isotype during homeostasis, low‐level IgG production occurs continuously against certain constituents of the microbiome, 53 , 124 providing systemic protection against infection, 66 and is significantly induced during infection and colitis. 33 , 68 , 72 Continued dissection of the molecular mechanisms regulating IgG class switching in the GALT and mucosa will help identify potential targets to manipulate the course of intestinal infection or chronic inflammation.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENT
AF and T.C‐D. were supported by studentships from the Wellcome Trust Infection, Immunity and Inflammation PhD programme at Cambridge University (102163/B/13/Z). T.C‐D. and MRC were also supported by a Medical Research Council New Investigator Research Grant (MR/N024907/01). MRC is supported by the National Institute for Health Research (NIHR), the NIHR Blood and Transplant Research Unit (NIHR BTRU‐2014‐10027) and a Wellcome Trust Investigator Award (220268/Z/20/Z). Figures were created with BioRender.com.
Fleming A, Castro‐Dopico T, Clatworthy MR. B cell class switching in intestinal immunity in health and disease. Scand J Immunol. 2022;95:e13139. doi: 10.1111/sji.13139
Aaron Fleming and Tomas Castro‐Dopico equal contribution.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159‐169. doi: 10.1038/nri2710 [DOI] [PubMed] [Google Scholar]
- 2. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268‐1273. doi: 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17:219‐232. doi: 10.1038/nri.2017.7 [DOI] [PubMed] [Google Scholar]
- 4. Bunker JJ, Bendelac A. IgA responses to microbiota. Immunity. 2018;49:211‐224. doi: 10.1016/j.immuni.2018.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal‐Vonarburg SC. IgA function in relation to the intestinal microbiota. Annu Rev Immunol. 2018;36:359‐381. doi: 10.1146/annurev-immunol-042617-053238 [DOI] [PubMed] [Google Scholar]
- 6. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell‐dependent and T cell‐independent IgA synthesis. Annu Rev Immunol. 2010;28:243‐273. doi: 10.1146/annurev-immunol-030409-101314 [DOI] [PubMed] [Google Scholar]
- 7. Brandtzaeg P, Johansen F‐E. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev. 2005;206:32‐63. doi: 10.1111/j.0105-2896.2005.00283.x [DOI] [PubMed] [Google Scholar]
- 8. Rüthlein J, Ibe M, Burghardt W, Mössner J, Auer IO. Immunoglobulin G (IgG), IgG1, and IgG2 determinations from endoscopic biopsy specimens in control, Crohn's disease, and ulcerative colitis subjects. Gut. 1992;33:507‐512. doi: 10.1136/gut.33.4.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi K, Asakura H, Hamada Y, et al. T lymphocyte subpopulations and immunoglobulin‐containing cells in the colonic mucosa of ulcerative colitis; a morphometric and immunohistochemical study. J Clin Lab Immunol. 1988;25:63‐68. [PubMed] [Google Scholar]
- 10. Helgeland L, Tysk C, Jarnerot G, et al. IgG subclass distribution in serum and rectal mucosa of monozygotic twins with or without inflammatory bowel disease. Gut. 1992;33:1358‐1364. doi: 10.1136/gut.33.10.1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scott MG, Nahm MH, Macke K, Nash GS, Bertovich MJ, MacDermott RP. Spontaneous secretion of IgG subclasses by intestinal mononuclear cells: differences between ulcerative colitis, Crohn's disease, and controls. Clin Exp Immunol. 1986;66:209‐215. [PMC free article] [PubMed] [Google Scholar]
- 12. Castro‐Dopico T, Clatworthy MR. IgG and Fcγ receptors in Intestinal Immunity and Inflammation. Front Immunol. 2019;10. doi: 10.3389/fimmu.2019.00805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fenton TM, Jørgensen PB, Niss K, et al. Immune profiling of human gut‐associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region‐specific immunity. Immunity. 2020;52:557‐570.e556. doi: 10.1016/j.immuni.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsuji M, Suzuki K, Kitamura H, et al. Requirement for lymphoid tissue‐inducer cells in isolated follicle formation and T cell‐independent immunoglobulin A generation in the gut. Immunity. 2008;29:261‐271. doi: 10.1016/j.immuni.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 15. Hamada H, Hiroi T, Nishiyama Y, et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol. 2002;168:57‐64. doi: 10.4049/jimmunol.168.1.57 [DOI] [PubMed] [Google Scholar]
- 16. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177:524‐540. doi: 10.1016/j.cell.2019.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puga I, Cols M, Barra CM, et al. B cell‐helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2012;13:170‐180. doi: 10.1038/ni.2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balázs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T‐independent immune responses. Immunity. 2002;17:341‐352. doi: 10.1016/s1074-7613(02)00389-8 [DOI] [PubMed] [Google Scholar]
- 19. Merluzzi S, Frossi B, Gri G, et al. Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA‐secreting plasma cells. Blood. 2010;115:2810‐2817. doi: 10.1182/blood-2009-10-250126 [DOI] [PubMed] [Google Scholar]
- 20. Kuley R, Draves KE, Fuller DH, et al. B cell activating factor (BAFF) from neutrophils and dendritic cells is required for protective B cell responses against Salmonella typhimurium infection. PLoS One. 2021;16:e0259158. doi: 10.1371/journal.pone.0259158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giordano D, Kuley R, Draves KE, et al. BAFF produced by neutrophils and dendritic cells is regulated differently and has distinct roles in antibody responses and protective immunity against West Nile Virus. J Immunol. 2020;204:1508‐1520. doi: 10.4049/jimmunol.1901120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vinuesa CG, Chang PP. Innate B cell helpers reveal novel types of antibody responses. Nat Immunol. 2013;14:119‐126. doi: 10.1038/ni.2511 [DOI] [PubMed] [Google Scholar]
- 23. Rawlings DJ, Schwartz MA, Jackson SW, Meyer‐Bahlburg A. Integration of B cell responses through Toll‐like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282‐294. doi: 10.1038/nri3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baumgarth N. A Hard(y) look at B‐1 cell development and function. J Immunol. 2017;199:3387‐3394. doi: 10.4049/jimmunol.1700943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paul WE. Fundamental Immunology. 7th ed. Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 26. Tangye SG. To B1 or not to B1: that really is still the question! Blood. 2013;121:5109‐5110. doi: 10.1182/blood-2013-05-500074 [DOI] [PubMed] [Google Scholar]
- 27. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70. J Exp Med. 2011;208:67‐80. doi: 10.1084/jem.20101499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Covens K, Verbinnen B, Geukens N, et al. Characterization of proposed human B‐1 cells reveals pre‐plasmablast phenotype. Blood. 2013;121:5176‐5183. doi: 10.1182/blood-2012-12-471953 [DOI] [PubMed] [Google Scholar]
- 29. Li W, Batliwalla F, Rothstein TL. Human B‐1 cells are not preplasmablasts: analysis of microarray data and other issues. Blood. 2013;122:3691‐3693. doi: 10.1182/blood-2013-08-520031 [DOI] [PubMed] [Google Scholar]
- 30. Lin M, Du L, Brandtzaeg P, Pan‐Hammarström Q. IgA subclass switch recombination in human mucosal and systemic immune compartments. Mucosal Immunol. 2014;7:511‐520. doi: 10.1038/mi.2013.68 [DOI] [PubMed] [Google Scholar]
- 31. Plaut AG, Wistar R Jr, Capra JD. Differential susceptibility of human IgA immunoglobulins to streptococcal IgA protease. J Clin Invest. 1974;54:1295‐1300. doi: 10.1172/jci107875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667‐685. doi: 10.1038/nri3738 [DOI] [PubMed] [Google Scholar]
- 33. James KR, Gomes T, Elmentaite R, et al. Distinct microbial and immune niches of the human colon. Nat Immunol. 2020;21:343‐353. doi: 10.1038/s41590-020-0602-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fagarasan S. Evolution, development, mechanism and function of IgA in the gut. Curr Opin Immunol. 2008;20:170‐177. doi: 10.1016/j.coi.2008.04.002 [DOI] [PubMed] [Google Scholar]
- 35. Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annu Rev Immunol. 2003;21:177‐204. doi: 10.1146/annurev.immunol.21.120601.141011 [DOI] [PubMed] [Google Scholar]
- 36. Duchemin M, Khamassi M, Xu L, Tudor D, Bomsel M. IgA targeting human immunodeficiency virus‐1 envelope gp41 triggers antibody‐dependent cellular cytotoxicity cross‐clade and cooperates with gp41‐specific IgG to increase cell lysis. Front Immunol. 2018;9:244. doi: 10.3389/fimmu.2018.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wills S, Hwang K‐K, Liu P, et al. HIV‐1‐specific IgA monoclonal antibodies from an HIV‐1 Vaccinee mediate Galactosylceramide blocking and phagocytosis. J Virol. 2018;92: doi: 10.1128/JVI.01552-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mazanec MB, Kaetzel CS, Lamm ME, Fletcher D, Nedrud JG. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci U S A. 1992;89:6901‐6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bidgood SR, Tam JC, McEwan WA, Mallery DL, James LC. Translocalized IgA mediates neutralization and stimulates innate immunity inside infected cells. Proc Natl Acad Sci U S A. 2014;111:13463‐13468. doi: 10.1073/pnas.1410980111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:1‐17. doi: 10.3389/fimmu.2014.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bruhns P, Iannascoli B, England P, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716‐3725. doi: 10.1182/blood-2008-09-179754 [DOI] [PubMed] [Google Scholar]
- 42. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34‐47. doi: 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 43. von Gunten S, Smith DF, Cummings RD, et al. Intravenous immunoglobulin contains a broad repertoire of anticarbohydrate antibodies that is not restricted to the IgG2 subclass. J Allergy Clin Immunol. 2009;123:1‐16. doi: 10.1016/j.jaci.2009.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stapleton NM, Andersen JT, Stemerding AM, et al. Competition for FcRn‐mediated transport gives rise to short half‐life of human IgG3 and offers therapeutic potential. Nat Commun. 2011;2:599. doi: 10.1038/ncomms1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van der Neut Kolfschoten M, Schuurman J, Losen M, et al. Anti‐inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554‐1557. doi: 10.1126/science.1144603 [DOI] [PubMed] [Google Scholar]
- 46. Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105:9‐19. doi: 10.1046/j.0019-2805.2001.01341.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti‐inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670‐673. doi: 10.1126/science.1129594 [DOI] [PubMed] [Google Scholar]
- 48. Li T, DiLillo DJ, Bournazos S, et al. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci. 2017;114:3485‐3490. doi: 10.1073/pnas.1702173114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pincetic A, Bournazos S, DiLillo DJ, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15:707‐716. doi: 10.1038/ni.2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karsten CM, Pandey MK, Figge J, et al. Anti‐inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin‐1. Nat Med. 2012;18:1401‐1406. doi: 10.1038/nm.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lauc G, Huffman JE, Pučić M, et al. Loci associated with N‐glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013;9:e1003225. doi: 10.1371/journal.pgen.1003225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Theodoratou E, Campbell H, Ventham NT, et al. The role of glycosylation in IBD. Nat Rev Gastroenterol Hepatol. 2014;11:588‐600. doi: 10.1038/nrgastro.2014.78 [DOI] [PubMed] [Google Scholar]
- 53. Koch M, Reiner G, Lugo K, et al. Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell. 2016;165:827‐841. doi: 10.1016/j.cell.2016.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nimmerjahn F, Ravetch JV. Fc‐receptors as regulators of immunity. Adv Immunol. 2007;96:179‐204. doi: 10.1016/S0065-2776(07)96005-8 [DOI] [PubMed] [Google Scholar]
- 55. Smith KGC, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328‐343. doi: 10.1038/nri2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pricop L, Redecha P, Teillaud JL, et al. Differential modulation of stimulatory and inhibitory Fc gamma receptors on human monocytes by Th1 and Th2 cytokines. J Immunol. 2001;166:531‐537. [DOI] [PubMed] [Google Scholar]
- 57. Liu Y, Masuda E, Blank MC, et al. Cytokine‐mediated regulation of activating and inhibitory Fc gamma receptors in human monocytes. J Leukoc Biol. 2005;77:767‐776. doi: 10.1189/jlb.0904532 [DOI] [PubMed] [Google Scholar]
- 58. Willcocks LC, Smith KG, Clatworthy MR. Low‐affinity Fcgamma receptors, autoimmunity and infection. Expert Rev Mol Med. 2009;11:e24. doi: 10.1017/S1462399409001161 [DOI] [PubMed] [Google Scholar]
- 59. Clatworthy MR. In: Ackerman ME, Nimmerjahn F, eds. Antibody Fc. Linking Adaptive and Innate Immunity. Ch. 12, Academic Press, Elsevier. 2014:217‐238. [Google Scholar]
- 60. Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N‐linked oligosaccharide improves binding to human Fcgamma RIII and antibody‐dependent cellular toxicity. J Biol Chem. 2002;277:26733‐26740. doi: 10.1074/jbc.M202069200 [DOI] [PubMed] [Google Scholar]
- 61. Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The carbohydrate at FcgammaRIIIa Asn‐162. An element required for high affinity binding to non‐fucosylated IgG glycoforms. J Biol Chem. 2006;281:5032‐5036. doi: 10.1074/jbc.M510171200 [DOI] [PubMed] [Google Scholar]
- 62. Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21‐50. doi: 10.1146/annurev.immunol.25.022106.141702 [DOI] [PubMed] [Google Scholar]
- 63. Lux A, Nimmerjahn F. Impact of differential glycosylation on IgG activity. Adv Exp Med Biol. 2011;780:113‐124. doi: 10.1007/978-1-4419-5632-3_10 [DOI] [PubMed] [Google Scholar]
- 64. Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510‐1512. doi: 10.1126/science.1118948 [DOI] [PubMed] [Google Scholar]
- 65. Caballero‐Flores G, Sakamoto K, Zeng MY, et al. Maternal immunization confers protection to the offspring against an attaching and effacing pathogen through delivery of IgG in breast milk. Cell Host Microbe. 2019;1‐11. doi: 10.1016/J.CHOM.2018.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zeng MY, Cisalpino D, Varadarajan S, et al. Gut microbiota‐induced immunoglobulin g controls systemic infection by symbiotic bacteria and pathogens. Immunity. 2016;44:647‐658. doi: 10.1016/j.immuni.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Masuda A, Yoshida M, Shiomi H, et al. Fcgamma receptor regulation of Citrobacter rodentium infection. Infect Immun. 2008;76:1728‐1737. doi: 10.1128/IAI.01493-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maaser C, Housley MP, Iimura M, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. 2004;72:3315‐3324. doi: 10.1128/IAI.72.6.3315-3324.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ohsaki A, Venturelli N, Buccigrosso TM, et al. Maternal IgG immune complexes induce food allergen‐specific tolerance in offspring. J Exp Med. 2018;215(1):91‐113. doi: 10.1084/jem.20171163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gomez de Agüero M, Ganal‐Vonarburg SC, Fuhrer T, et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351:1296‐1302. doi: 10.1126/science.aad2571 [DOI] [PubMed] [Google Scholar]
- 71. Baker K, Rath T, Flak M, et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity. 2013;39:1095‐1107. doi: 10.1016/j.immuni.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Castro‐Dopico T, Dennison TW, Ferdinand JR, et al. Anti‐commensal IgG drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity. 2019;50:1099‐1114.e1010. doi: 10.1016/j.immuni.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Uo M, Hisamatsu T, Miyoshi J, et al. Mucosal CXCR4+ IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcγR‐mediated CD14 macrophage activation. Gut. 2013;62:1734‐1744. doi: 10.1136/gutjnl-2012-303063 [DOI] [PubMed] [Google Scholar]
- 74. Kobayashi K, Qiao S, Yoshida M, et al. An FcRn‐dependent role for anti‐flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology. 2009;137:1746‐1756.e1741. doi: 10.1053/j.gastro.2009.07.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Elmentaite R, Ross ADB, Roberts K, et al. Single‐cell sequencing of developing human gut reveals transcriptional links to childhood Crohn's disease. Dev Cell. 2020;55:771‐783.e775. doi: 10.1016/j.devcel.2020.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class‐switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12:517‐531. doi: 10.1038/nri3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Methot SP, Di Noia JM. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol. 2017;133:37‐87. doi: 10.1016/bs.ai.2016.11.002 [DOI] [PubMed] [Google Scholar]
- 78. Yu K, Lieber MR. Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit Rev Biochem Mol Biol. 2019;54:333‐351. doi: 10.1080/10409238.2019.1659227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261‐292. doi: 10.1146/annurev.immunol.26.021607.090248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825‐1828. doi: 10.1126/science.7892607 [DOI] [PubMed] [Google Scholar]
- 81. Zarrin AA, Tian M, Wang J, Borjeson T, Alt FW. Influence of switch region length on immunoglobulin class switch recombination. Proc Natl Acad Sci U S A. 2005;102:2466‐2470. doi: 10.1073/pnas.0409847102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R‐loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442‐451. doi: 10.1038/ni919 [DOI] [PubMed] [Google Scholar]
- 83. Muramatsu M, Kinoshita K, Fagarasan S, et al. Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553‐563. doi: 10.1016/s0092-8674(00)00078-7 [DOI] [PubMed] [Google Scholar]
- 84. Guikema JEJ, Linehan EK, Tsuchimoto D, et al. APE1‐ and APE2‐dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017‐3026. doi: 10.1084/jem.20071289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tarlinton D. B cells still front and centre in immunology. Nat Rev Immunol. 2019;19:85‐86. doi: 10.1038/s41577-018-0107-2 [DOI] [PubMed] [Google Scholar]
- 86. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. 2015;15:137‐148. doi: 10.1038/nri3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8:421‐434. doi: 10.1038/nri2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly ‐ TFH cells in human health and disease. Nat Rev Immunol. 2013;13:412‐426. doi: 10.1038/nri3447 [DOI] [PubMed] [Google Scholar]
- 89. Weinstein JS, Herman EI, Lainez B, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17:1197‐1205. doi: 10.1038/ni.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Suzuki K, Maruya M, Kawamoto S, et al. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity. 2010;33:71‐83. doi: 10.1016/j.immuni.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 91. Trindade BC, Ceglia S, Berthelette A, et al. The cholesterol metabolite 25‐hydroxycholesterol restrains the transcriptional regulator SREBP2 and limits intestinal IgA plasma cell differentiation. Immunity. 2021;54:2273‐2287.e2276. doi: 10.1016/j.immuni.2021.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. He B, Santamaria R, Xu W, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836‐845. doi: 10.1038/ni.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pone EJ, Zan H, Zhang J, et al. Toll‐like receptors and B‐cell receptors synergize to induce immunoglobulin class‐switch DNA recombination: relevance to microbial antibody responses. Crit Rev Immunol. 2010;30:1‐29. doi: 10.1615/CritRevImmunol.v30.i1.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fagarasan S, Honjo T. T‐Independent immune response: new aspects of B cell biology. Science. 2000;290:89‐92. doi: 10.1126/science.290.5489.89 [DOI] [PubMed] [Google Scholar]
- 95. Grasset EK, Chorny A, Casas‐Recasens S, et al. Gut T cell‐independent IgA responses to commensal bacteria require engagement of the TACI receptor on B cells. Sci Immunol. 2020;5: doi: 10.1126/sciimmunol.aat7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chu VT, Fröhlich A, Steinhauser G, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151‐159. doi: 10.1038/ni.1981 [DOI] [PubMed] [Google Scholar]
- 97. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298‐306. doi: 10.1038/nature10208 [DOI] [PubMed] [Google Scholar]
- 98. Magri G, Miyajima M, Bascones S, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol. 2014;15:354‐364. doi: 10.1038/ni.2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tezuka H, Abe Y, Iwata M, et al. Regulation of IgA production by naturally occurring TNF/iNOS‐producing dendritic cells. Nature. 2007;448:929‐933. doi: 10.1038/nature06033 [DOI] [PubMed] [Google Scholar]
- 100. He B, Xu W, Santini PA, et al. Intestinal bacteria trigger T cell‐independent immunoglobulin A2 class switching by inducing epithelial‐cell secretion of the cytokine APRIL. Immunity. 2007;26:812‐826. doi: 10.1016/j.immuni.2007.04.014 [DOI] [PubMed] [Google Scholar]
- 101. Kaminski DA, Stavnezer J. Stimuli that enhance IgA class switching increase histone 3 acetylation at S alpha, but poorly stimulate sequential switching from IgG2b. Eur J Immunol. 2007;37:240‐251. doi: 10.1002/eji.200636645 [DOI] [PubMed] [Google Scholar]
- 102. Koscsó B, Kurapati S, Rodrigues RR, et al. Gut‐resident CX3CR1(hi) macrophages induce tertiary lymphoid structures and IgA response in situ. Sci Immunol. 2020;5:eaax0062. doi: 10.1126/sciimmunol.aax0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Park S‐R, Zan H, Pal Z, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class‐switch DNA recombination and somatic hypermutation. Nat Immunol. 2009;10:540‐550. doi: 10.1038/ni.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mohr E, Cunningham AF, Toellner KM, et al. IFN‐{gamma} produced by CD8 T cells induces T‐bet‐dependent and ‐independent class switching in B cells in responses to alum‐precipitated protein vaccine. Proc Natl Acad Sci USA. 2010;107:17292‐17297. doi: 10.1073/pnas.1004879107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Peng SL, Szabo SJ, Glimcher LH. T‐bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci USA. 2002;99:5545‐5550. doi: 10.1073/pnas.082114899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mitsdoerffer M, Lee Y, Jager A, et al. Proinflammatory T helper type 17 cells are effective B‐cell helpers. Proc Natl Acad Sci USA. 2010;107:14292‐14297. doi: 10.1073/pnas.1009234107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pfeifle R, Rothe T, Ipseiz N, et al. Regulation of autoantibody activity by the IL‐23 – T H 17 axis determines the onset of autoimmune disease. Nat Immunol. 2016;18:1‐7. doi: 10.1038/ni.3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pabst O. New concepts in the generation and functions of IgA. Nat Rev Immunol. 2012;12:821‐832. doi: 10.1038/nri3322 [DOI] [PubMed] [Google Scholar]
- 109. Lycke NY, Bemark M. The role of Peyer's patches in synchronizing gut iga responses. Front Immunol. 2012;3:1‐9. doi: 10.3389/fimmu.2012.00329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lycke NY, Bemark M. The regulation of gut mucosal IgA B‐cell responses: recent developments. Mucosal Immunol. 2017;10:1361‐1374. doi: 10.1038/mi.2017.62 [DOI] [PubMed] [Google Scholar]
- 111. Kato LM, Kawamoto S, Maruya M, Fagarasan S. Gut TFH and IgA: key players for regulation of bacterial communities and immune homeostasis. Immunol Cell Biol. 2014;92:49‐56. doi: 10.1038/icb.2013.54 [DOI] [PubMed] [Google Scholar]
- 112. Milpied PJ, McHeyzer‐Williams MG. High‐affinity IgA needs TH17 cell functional plasticity. Nat Immunol. 2013;14:313‐315. doi: 10.1038/ni.2567 [DOI] [PubMed] [Google Scholar]
- 113. Masahata K, Umemoto E, Kayama H, et al. Generation of colonic IgA‐secreting cells in the caecal patch. Nat Commun. 2014;5:3704. doi: 10.1038/ncomms4704 [DOI] [PubMed] [Google Scholar]
- 114. Reboldi A, Cyster JG. Peyer's patches: organizing B‐cell responses at the intestinal frontier. Immunol Rev. 2016;271:230‐245. doi: 10.1111/imr.12400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bunker J, Flynn T, Koval J, et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity. 2015;43:541‐553. doi: 10.1016/j.immuni.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kawamoto S, Maruya M, Kato L, et al. Foxp3+ T cells regulate immunoglobulin A selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41:152‐165. doi: 10.1016/j.immuni.2014.05.016 [DOI] [PubMed] [Google Scholar]
- 117. Hirota K, Turner J‐E, Villa M, et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell‐dependent IgA responses. Nat Immunol. 2013;14:372‐379. doi: 10.1038/ni.2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tsuji M, Komatsu N, Kawamoto S, et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science. 2009;323:1488‐1492. doi: 10.1126/science.1169152 [DOI] [PubMed] [Google Scholar]
- 119. Casola S, Otipoby KL, Alimzhanov M, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317‐327. doi: 10.1038/ni1036 [DOI] [PubMed] [Google Scholar]
- 120. Kawamoto S, Tran TH, Maruya M, et al. The inhibitory receptor PD‐1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485‐489. doi: 10.1126/science.1217718 [DOI] [PubMed] [Google Scholar]
- 121. Benckert J, Schmolka N, Kreschel C, et al. The majority of intestinal IgA + and IgG + plasmablasts in the human gut are antigen‐specific. J Clin Invest. 2011;121:1946‐1955. doi: 10.1172/JCI44447DS1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bergqvist P, Gardby E, Stensson A, Bemark M, Lycke NY. Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J Immunol. 2006;177:7772‐7783. doi: 10.4049/jimmunol.177.11.7772 [DOI] [PubMed] [Google Scholar]
- 123. Macpherson AJ, Gatto D, Sainsbury E, et al. A primitive T Cell‐independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222‐2226. doi: 10.1126/science.288.5474.2222 [DOI] [PubMed] [Google Scholar]
- 124. Ansaldo E, Slayden LC, Ching KL, et al. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science. 2019;364:1179‐1184. doi: 10.1126/science.aaw7479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Depommier C, Everard A, Druart C, et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof‐of‐concept exploratory study. Nat Med. 2019;25:1096‐1103. doi: 10.1038/s41591-019-0495-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bunker JJ, Erickson SA, Flynn TM, et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science. 2017;358:eaan6619. doi: 10.1126/science.aan6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59‐69. doi: 10.1016/j.smim.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 128. Li H, Limenitakis JP, Greiff V, et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature. 2020;584:274‐278. doi: 10.1038/s41586-020-2564-6 [DOI] [PubMed] [Google Scholar]
- 129. Rollenske T, Burkhalter S, Muerner L, et al. Parallelism of intestinal secretory IgA shapes functional microbial fitness. Nature. 2021;598:657‐661. doi: 10.1038/s41586-021-03973-7 [DOI] [PubMed] [Google Scholar]
- 130. Cazac BB, Roes J. TGF‐β receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443‐451. doi: 10.1016/S1074-7613(00)00044-3 [DOI] [PubMed] [Google Scholar]
- 131. Gros MJ, Naquet P, Guinamard RR. Cell intrinsic TGF‐beta 1 regulation of B cells. J Immunol. 2008;180:8153‐8158. doi: 10.4049/jimmunol.180.12.8153 [DOI] [PubMed] [Google Scholar]
- 132. Reboldi A, Arnon TI, Rodda LB, et al. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer's patches. Science. 2016;352:1‐10. doi: 10.1126/science.aaf4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mora JR, Iwata M, Eksteen B, et al. Generation of gut‐homing IgA‐secreting B cells by intestinal dendritic cells. Science. 2006;314:1157‐1160. doi: 10.1126/science.1132742 [DOI] [PubMed] [Google Scholar]
- 134. Castigli E, Scott S, Dedeoglu F, et al. Impaired IgA class switching inn APRIL‐deficient mice. Proc Natl Acad Sci USA. 2004;101:3903‐3908. doi: 10.1073/pnas.0307348101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Castigli E, Wilson SA, Scott S, et al. TACI and BAFF‐R mediate isotype switching in B cells. J Exp Med. 2005;201:35‐39. doi: 10.1084/jem.20032000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chu V, Beller A, Rausch S, et al. Eosinophils promote generation and maintenance of immunoglobulin‐A‐expressing plasma cells and contribute to gut immune homeostasis. Immunity. 2014;40:582‐593. doi: 10.1016/j.immuni.2014.02.014 [DOI] [PubMed] [Google Scholar]
- 137. Yel L. Selective IgA deficiency. J Clin Immunol. 2010;30:10‐16. doi: 10.1007/s10875-009-9357-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA deficiency: epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Scand J Immunol. 2017;85:3‐12. doi: 10.1111/sji.12499 [DOI] [PubMed] [Google Scholar]
- 139. Cunningham‐Rundles C, Brandeis WE, Pudifin DJ, Day NK, Good RA. Autoimmunity in selective IgA deficiency: relationship to anti‐bovine protein antibodies, circulating immune complexes and clinical disease. Clin Exp Immunol. 1981;45:299‐304. [PMC free article] [PubMed] [Google Scholar]
- 140. Smulski CR, Eibel H. BAFF and BAFF‐receptor in B cell selection and survival. Front Immunol. 2018;9:1‐10. doi: 10.3389/fimmu.2018.02285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Torres J, Mehandru S, Colombel J‐F, Peyrin‐Biroulet L. Crohn's disease. Lancet. 2017;389:1741‐1755. doi: 10.1016/S0140-6736(16)31711-1 [DOI] [PubMed] [Google Scholar]
- 142. Jostins L, Ripke S, Weersma RK, et al. Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119‐124. doi: 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel J‐F. Ulcerative colitis. Lancet. 2017;389:1756‐1770. doi: 10.1016/S0140-6736(16)32126-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Blumberg R, Powrie FM. Disease, and back to health: a metastable journey. Sci Transl Med. 2012;4:137rv137. doi: 10.1126/scitranslmed.3004184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yilmaz B, Juillerat P, Øyås O, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med. 2019;25:1. doi: 10.1038/s41591-018-0308-z [DOI] [PubMed] [Google Scholar]
- 146. Darfeuille‐Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent‐invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. 2004;127:412‐421. doi: 10.1053/j.gastro.2004.04.061 [DOI] [PubMed] [Google Scholar]
- 147. Manukian G, Kivolowitz C, DeAngelis T, et al. IgA‐coated E coli enriched in Crohn's disease spondyloarthritis promote TH17‐dependent inflammation. Sci Transl Med. 2017;9:eaaf9655. doi: 10.1126/scitranslmed.aaf9655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Powrie F, Leach MW, Mauze S, et al. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553‐562. doi: 10.1016/1074-7613(94)90045-0 [DOI] [PubMed] [Google Scholar]
- 149. Ahern PP, Schiering C, Buonocore S, et al. Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279‐288. doi: 10.1016/j.immuni.2010.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Duerr RH, Taylor KD, Brant SR, et al. A genome‐wide association study identifies IL23R as an inflammatory bowel disease. Gene. 2006;13:8‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Sandborn WJ, Gasink C, Gao L‐L, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012;367:1519‐1528. doi: 10.1056/NEJMoa1203572 [DOI] [PubMed] [Google Scholar]
- 152. Leiper K, Martin K, Ellis A, et al. Randomised placebo‐controlled trial of rituximab (anti‐CD20) in active ulcerative colitis. Gut. 2011;60:1520‐1526. doi: 10.1136/gut.2010.225482 [DOI] [PubMed] [Google Scholar]
- 153. Uzzan M, Ko HM, Rosenstein AK, Pourmand K, Colombel JF, Mehandru S. Efficient long‐term depletion of CD20 + B cells by rituximab does not affect gut‐resident plasma cells. Ann N Y Acad Sci. 2018;1415:5‐10. doi: 10.1111/nyas.13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Morita K, Shibano T, Maekawa K, et al. Crohn's disease following rituximab treatment in a patient with refractory nephrotic syndrome. CEN Case Rep. 2019;8:55‐60. doi: 10.1007/s13730-018-0364-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Varma P, Falconer J, Aga A, Prince HM, Pianko S. Rituximab‐induced Crohn’s disease. Scand J Gastroenterol. 2017;52:606‐608. doi: 10.1080/00365521.2017.1280530 [DOI] [PubMed] [Google Scholar]
- 156. Asano K, Matsushita T, Umeno J, et al. A genome‐wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325‐1329. doi: 10.1038/ng.482 [DOI] [PubMed] [Google Scholar]
- 157. Smillie CS, Biton M, Ordovas‐Montanes J, et al. Intra‐ and inter‐cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178:714‐730.e722. doi: 10.1016/j.cell.2019.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Martin JC, Chang C, Boschetti G, et al. Single‐cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti‐TNF therapy. Cell. 2019;178:1493‐1508.e20. doi: 10.1016/j.cell.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Rengarajan S, Vivio EE, Parkes M, et al. Dynamic immunoglobulin responses to gut bacteria during inflammatory bowel disease. Gut Microbes. 2020;11(3):405‐420. doi: 10.1080/19490976.2019.1626683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ahn J, Son S, Oliveira SC, Barber GN. STING‐dependent signaling underlies IL‐10 controlled inflammatory colitis. Cell Rep. 2017;21:3873‐3884. doi: 10.1016/j.celrep.2017.11.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Brandwein SL, McCabe RP, Cong Y, et al. Spontaneously colitic C3H/HeJBir mice demonstrate selective antibody reactivity to antigens of the enteric bacterial flora. J Immunol. 1997;159:44‐52. [PubMed] [Google Scholar]
- 162. Caruso R, Mathes T, Martens EC, et al. A specific gene‐microbe interaction drives the development of Crohn’s disease–like colitis in mice. Sci Immunol. 2019;4:eaaw4341. doi: 10.1126/sciimmunol.aaw4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Slack E, Hapfelmeier S, Stecher B, et al. Innate and adaptive immunity cooperate flexibly to maintain host‐microbiota mutualism. Science. 2009;325:617‐620. doi: 10.1126/science.1172747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Choung RS, Princen F, Stockfisch TP, et al. Serologic microbial associated markers can predict Crohn's disease behaviour years before disease diagnosis. Aliment Pharmacol Ther. 2016;43:1300‐1310. doi: 10.1111/apt.13641 [DOI] [PubMed] [Google Scholar]
- 165. Kamada N, Sakamoto K, Seo S‐U, et al. Humoral immunity in the gut selectively targets phenotypically virulent attaching‐and‐effacing bacteria for intraluminal elimination. Cell Host Microbe. 2015;17:617‐627. doi: 10.1016/j.chom.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Simmons CP, Clare S, Ghaem‐Maghami M, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2003;71:5077‐5086. doi: 10.1128/IAI.71.9.5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kim Y‐G, Kamada N, Shaw M, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2‐dependent recruitment of inflammatory monocytes. Immunity. 2011;34:769‐780. doi: 10.1016/j.immuni.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Miyoshi E, Shinzaki S, Fujii H, Iijima H, Kamada Y, Takehara T. Role of aberrant IgG glycosylation in the pathogenesis of inflammatory bowel disease. Proteomics Clin Appl. 2016;10:384‐390. doi: 10.1002/prca.201500089 [DOI] [PubMed] [Google Scholar]
- 169. Uzzan M, Colombel JF, Cerutti A, Treton X, Mehandru S. B Cell‐Activating Factor (BAFF)‐targeted B cell therapies in inflammatory bowel diseases. Dig Dis Sci. 2016;61:3407‐3424. doi: 10.1007/s10620-016-4317-9 [DOI] [PubMed] [Google Scholar]
- 170. Banham GD, Flint SM, Torpey N, et al. Belimumab in kidney transplantation: an experimental medicine, randomised, placebo‐controlled phase 2 trial. Lancet. 2018;391:2619‐2630. doi: 10.1016/S0140-6736(18)30984-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260‐263. doi: 10.1126/science.285.5425.260 [DOI] [PubMed] [Google Scholar]
- 172. Hsu BL, Harless SM, Lindsley RC, Hilbert DM, Cancro MP. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J Immunol. 2002;168:5993‐5996. doi: 10.4049/jimmunol.168.12.5993 [DOI] [PubMed] [Google Scholar]
- 173. Do RKG, Hatada E, Lee H, et al. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192:953‐964. doi: 10.1084/jem.192.7.953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Takeuchi T, Miyauchi E, Kanaya T, et al. Acetate differentially regulates IgA reactivity to commensal bacteria. Nature. 2021;595:560‐564. doi: 10.1038/s41586-021-03727-5 [DOI] [PubMed] [Google Scholar]
- 175. Suzuki M, Sujino T, Chiba S, et al. Host‐microbe cross‐talk governs amino acid chirality to regulate survival and differentiation of B cells. Sci Adv. 2021;7. doi: 10.1126/sciadv.abd6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445‐489. doi: 10.1146/annurev-immunol-030409-101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41:366‐374. doi: 10.1016/j.immuni.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 178. Knox JJ, Myles A, Cancro MP. T‐bet(+) memory B cells: generation, function, and fate. Immunol Rev. 2019;288:149‐160. doi: 10.1111/imr.12736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chang LY, Li Y, Kaplan DE. Hepatitis C viraemia reversibly maintains subset of antigen‐specific T‐bet+ tissue‐like memory B cells. J Viral Hepat. 2017;24:389‐396. doi: 10.1111/jvh.12659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wang S, Wang J, Kumar V, et al. IL‐21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T‐bet(+) B cells in SLE. Nat Commun. 2018;9:1758. doi: 10.1038/s41467-018-03750-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Couloume L, Ferrant J, Le Gallou S, et al. Mass cytometry identifies expansion of T‐bet(+) B cells and CD206(+) monocytes in early multiple sclerosis. Front Immunol. 2021;12:653577. doi: 10.3389/fimmu.2021.653577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Eccles JD, Turner RB, Kirk NA, et al. T‐bet+ memory B cells link to local cross‐reactive IgG upon human rhinovirus infection. Cell Rep. 2020;30:351‐366.e357. doi: 10.1016/j.celrep.2019.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rubtsova K, Rubtsov AV, Thurman JM, et al. B cells expressing the transcription factor T‐bet drive lupus‐like autoimmunity. J Clin Invest. 2017;127:1392‐1404. doi: 10.1172/jci91250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wang NS, McHeyzer‐Williams LJ, Okitsu SL, et al. Divergent transcriptional programming of class‐specific B cell memory by T‐bet and RORα. Nat Immunol. 2012;13:604‐611. doi: 10.1038/ni.2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Rubtsov AV, Rubtsova K, Fischer A, et al. Toll‐like receptor 7 (TLR7)‐driven accumulation of a novel CD11c⁺ B‐cell population is important for the development of autoimmunity. Blood. 2011;118:1305‐1315. doi: 10.1182/blood-2011-01-331462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Harris DP, Goodrich S, Gerth AJ, Peng SL, Lund FE. Regulation of IFN‐gamma production by B effector 1 cells: essential roles for T‐bet and the IFN‐gamma receptor. J Immunol. 2005;174:6781‐6790. doi: 10.4049/jimmunol.174.11.6781 [DOI] [PubMed] [Google Scholar]
- 187. Larousserie F, Charlot P, Bardel E, et al. Differential effects of IL‐27 on human B cell subsets. J Immunol. 2006;176:5890‐5897. doi: 10.4049/jimmunol.176.10.5890 [DOI] [PubMed] [Google Scholar]
- 188. Rivera‐Correa J, Guthmiller JJ, Vijay R, et al. Plasmodium DNA‐mediated TLR9 activation of T‐bet(+) B cells contributes to autoimmune anaemia during malaria. Nat Commun. 2017;8:1282. doi: 10.1038/s41467-017-01476-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Russell Knode LM, Naradikian MS, Myles A, et al. Age‐associated B cells express a diverse repertoire of V(H) and Vκ genes with somatic hypermutation. J Immunol. 2017;198:1921‐1927. doi: 10.4049/jimmunol.1601106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang Z, Wang Z, Wang J, et al. T‐bet‐expressing B cells are positively associated with Crohn's disease activity and support Th1 inflammation. DNA Cell Biol. 2016;35:628‐635. doi: 10.1089/dna.2016.3304 [DOI] [PubMed] [Google Scholar]
- 191. McKenzie H, Main J, Pennington CR, Parratt D. Antibody to selected strains of Saccharomyces cerevisiae (baker's and brewer's yeast) and Candida albicans in Crohn's disease. Gut. 1990;31:536‐538. doi: 10.1136/gut.31.5.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Annese V, Andreoli A, Andriulli A, et al. Familial expression of anti‐Saccharomyces cerevisiae Mannan antibodies in Crohn's disease and ulcerative colitis: a GISC study. Am J Gastroenterol. 2001;96:2407‐2412. doi: 10.1111/j.1572-0241.2001.04043.x [DOI] [PubMed] [Google Scholar]
- 193. Doron I, Leonardi I, Li XV, et al. Human gut mycobiota tune immunity via CARD9‐dependent induction of anti‐fungal IgG antibodies. Cell. 2021;184:1017‐1031.e1014. doi: 10.1016/j.cell.2021.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Drummond RA, Collar AL, Swamydas M, et al. CARD9‐dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 2015;11:e1005293. doi: 10.1371/journal.ppat.1005293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Doron I, Mesko M, Li XV, et al. Mycobiota‐induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn's disease. Nat Microbiol. 2021;6:1493‐1504. doi: 10.1038/s41564-021-00983-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ost KS, O’Meara TR, Stephens WZ, et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature. 2021;596:114‐118. doi: 10.1038/s41586-021-03722-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Dambuza IM, Brown GD. Managing the mycobiota with IgA. Nat Microbiol. 2021;6:1471‐1472. doi: 10.1038/s41564-021-01006-7 [DOI] [PubMed] [Google Scholar]
- 198. Fitzpatrick Z, Frazer G, Ferro A, et al. Gut‐educated IgA plasma cells defend the meningeal venous sinuses. Nature. 2020;587:472‐476. doi: 10.1038/s41586-020-2886-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Rojas OL, Pröbstel A‐K, Porfilio EA, et al. Recirculating intestinal IgA‐producing cells regulate neuroinflammation via IL‐10. Cell. 2019;176:610‐624.e618. doi: 10.1016/j.cell.2018.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Pröbstel AK, Zhou X, Baumann R, et al. Gut microbiota‐specific IgA(+) B cells traffic to the CNS in active multiple sclerosis. Sci Immunol. 2020;5. doi: 10.1126/sciimmunol.abc7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Leonardi I, Li X, Semon A, et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science. 2018;359:232‐236. doi: 10.1126/science.aao1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Buckner CM, Moir S, Kardava L, et al. CXCR4/IgG‐expressing plasma cells are associated with human gastrointestinal tissue inflammation. J Allergy Clin Immunol. 2014;133:1676‐1685.e1675. doi: 10.1016/j.jaci.2013.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Uo M, Hisamatsu T, Miyoshi J, et al. Mucosal CXCR4 + IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcγR‐mediated CD14 macrophage activation. Gut. 2013;62:1734‐1744. doi: 10.1136/gutjnl-2012-303063 [DOI] [PubMed] [Google Scholar]
- 204. Lazarus NH, Kunkel EJ, Johnston B, et al. A common mucosal chemokine (mucosae‐associated epithelial chemokine/CCL28) selectively attracts IgA plasmablasts. J Immunol. 2003;170:3799‐3805. doi: 10.4049/jimmunol.170.7.3799 [DOI] [PubMed] [Google Scholar]
- 205. Hieshima K, Kawasaki Y, Hanamoto H, et al. CC chemokine ligands 25 and 28 play essential roles in intestinal extravasation of IgA antibody‐secreting cells. J Immunol. 2004;173:3668‐3675. [DOI] [PubMed] [Google Scholar]
- 206. Mora JR, von Andrian UH. Differentiation and homing of IgA‐secreting cells. Mucosal Immunol. 2008;1:96‐109. doi: 10.1038/mi.2007.14 [DOI] [PubMed] [Google Scholar]
- 207. Sundström P, Lundin SB, Nilsson LA, Quiding‐Järbrink M. Human IgA‐secreting cells induced by intestinal, but not systemic, immunization respond to CCL25 (TECK) and CCL28 (MEC). Eur J Immunol. 2008;38:3327‐3338. doi: 10.1002/eji.200838506 [DOI] [PubMed] [Google Scholar]
- 208. Kunkel EJ, Kim CH, Lazarus NH, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab‐secreting cells. J Clin Invest. 2003;111:1001‐1010. doi: 10.1172/jci17244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Johansson C, Ahlstedt I, Furubacka S, et al. Differential expression of chemokine receptors on human IgA+ and IgG+ B cells. Clin Exp Immunol. 2005;141:279‐287. doi: 10.1111/j.1365-2249.2005.02843.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Schroepf S, Kappler R, Brand S, et al. Strong overexpression of CXCR3 axis components in childhood inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1882‐1890. doi: 10.1002/ibd.21312 [DOI] [PubMed] [Google Scholar]
- 211. Liu Z, Chen X, Wang X, et al. Chemokine CXCL11 links microbial stimuli to intestinal inflammation. Clin Exp Immunol. 2011;164:396‐406. doi: 10.1111/j.1365-2249.2011.04382.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Price AE, Shamardani K, Lugo KA, et al. A map of toll‐like receptor expression in the intestinal epithelium reveals distinct spatial, cell type‐specific, and temporal patterns. Immunity. 2018;49:560‐575.e566. doi: 10.1016/j.immuni.2018.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Koorella C, Nair JR, Murray ME, et al. Novel regulation of CD80/CD86‐induced phosphatidylinositol 3‐kinase signaling by NOTCH1 protein in interleukin‐6 and indoleamine 2,3‐dioxygenase production by dendritic cells. J Biol Chem. 2014;289:7747‐7762. doi: 10.1074/jbc.M113.519686 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.