

Abstract
Cell death can be a highly regulated process. A large and growing number of mammalian cell death mechanisms have been described over the past few decades. Major pathways with established roles in normal or disease biology include apoptosis, necroptosis, pyroptosis and ferroptosis. However, additional non-apoptotic cell death mechanisms with unique morphological, genetic, and biochemical features have also been described. These mechanisms may play highly specialized physiological roles or only become activated in response to specific lethal stimuli or conditions. Understanding the nature of these emerging and understudied mechanisms may provide new insight into cell death biology and suggest new treatments for diseases such as cancer and neurodegeneration.
Keywords: Apoptosis, ferroptosis, necrosis, pyroptosis, necroptosis, non-apoptotic cell death, ROS
1. Cell death can be executed by diverse mechanisms
Shortly after the emergence of cell theory in the 19th century came early reports of what we now recognize as cell death[1–3]. Over a century later, it became clear that cell death could be executed in a stereotypical manner by a genetically regulated mechanism – apoptosis[4–6]. Apoptotic cells have unique morphological characteristics: nuclear and cytoplasmic condensation, fragmentation of the cell into bleb-like bodies, and clearance of these bodies by other cells[4]. The activation of cysteine-aspartic proteases (“caspases”) during apoptosis generates characteristic protein fragments (e.g., cleaved poly (adenosine diphosphate-ribose) polymerase, or PARP) that uniquely define this process biochemically[7,8].
Apoptosis accounts for most cell death that is developmentally regulated and homeostatic[4]. However, cell death events with morphologies inconsistent with apoptosis have long been observed in mammalian cells and systems[9,10]. Among early paradoxical mechanistic observations that pointed towards non-apoptotic mechanisms of cell death, caspase inhibition can in some cells result in more cell death, not less, when combined with a canonical pro-apoptotic ligand like tumor necrosis factor alpha (TNF-α) [11]. Furthermore, the deletion of the key pro-apoptotic genes B cell lymphoma 2 (BCL2) antagonist/killer 1 (Bak1), BCL2 associated X (Bax), and BCL2-related ovarian killer (Bok) is not entirely incompatible with early embryonic development or even the subsequent development of a small percentage of mice into adults [12,13]. Assuming that developmental cell death is not entirely dispensable, this implies that non-apoptotic cell death mechanisms must exist and contribute to normal physiology. This raises many questions. How many ways can a mammalian cell die? Can non-apoptotic mechanisms contribute to disease, or be useful targets for modulation to treat disease[14,15]? These and related mechanistic questions remain important and only partially answered.
In this review, we survey non-apoptotic cell death mechanisms, especially those discovered in recent years to operate in mammalian cells. We will not consider here unusual forms of cell death that to date have been observed only in non-mammalian model organisms. Even restricting this discussion to mammalian systems, there is still extensive complexity and disagreement. For example, cell death following cell detachment from a substratum is referred to by a specific name – anoikis – but this process can be executed by either apoptotic or non-apoptotic mechanisms[16,17]. Likewise, mitotic catastrophe may either represent a distinct cell death mechanism or a sub-branch of apoptosis[18,19]. There are also examples of cell death processes that use elements of the apoptosis pathway to induce non-apoptotic cell death[20,21]. We will bypass these thorny issues and instead attempt to focus mostly on cell death mechanisms that are thought to be predominately non-apoptotic in nature.
2. Classifying different forms of non-apoptotic cell death
Mammalian cells can engage the core apoptosis machinery in response to pro-death ligands, removal of pro-survival ligands, or specific perturbations like DNA damage or proteasome inhibition[22–31]. However, other signaling events and stresses do not engage the core apoptosis pathway and instead yield various forms of non-apoptotic cell death. These non-apoptotic mechanisms have been identified and characterized over the past twenty years (Fig. 1). Compared to apoptosis, non-apoptotic cell death mechanisms appear to have unique morphological, genetic, and biochemical features. However, in many instances these features are only partly elucidated.
Figure 1. Cell death mechanism citations over time.
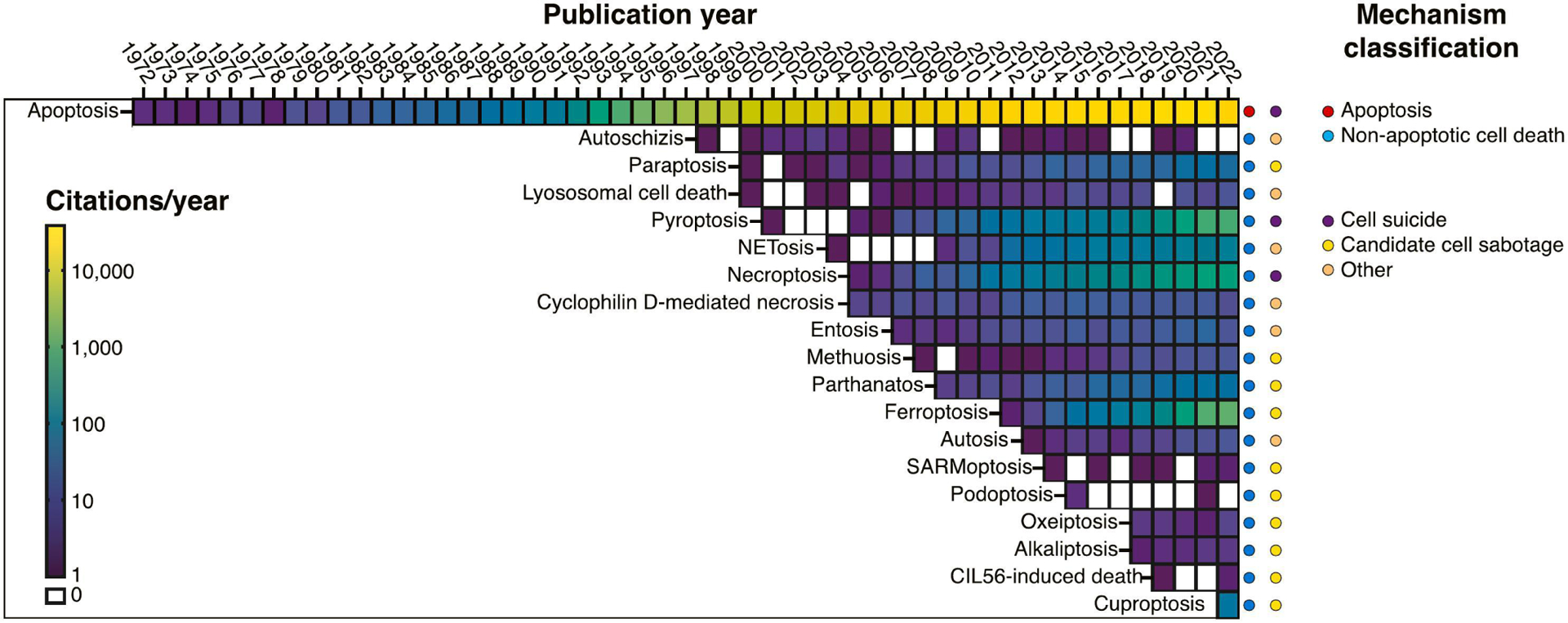
Heatmap representing the number of citations reported in PubMed for different cell death mechanisms discussed in the review, arranged by year of first mention. Note that the first mention of a named pathway in PubMed in some cases comes years after the first observations of the mechanism, before it is recognized as unique and named. Each cell in the heatmap represents the number of PubMed citations mentioning each mechanism per calendar year. Years without any citations are white. On the right, cell death mechanisms are categorized as apoptotic or non-apoptotic, and sub-categorized as suicide, sabotage, or mixed/other mechanisms. This sub-classification is somewhat arbitrary and may shift over time as more is learned.
There are several ways to organize our understanding of apoptotic and non-apoptotic cell death mechanisms. It is intuitive that extremes of pressure, heat, cold, pH, mechanical stress, or detergent treatment could directly rupture the membrane of mammalian cells in a manner that is entirely unregulated[32]. These effects are not of great interest here. By contrast, subtler stresses that trigger mechanisms of cell death whose execution can be enhanced or suppressed by specific molecular processes (e.g., gene deletion, small molecule treatment), yielding evidence of regulation, are of more interest to this discussion[32–34]. A key point of distinction is therefore between unregulated and regulated cell death. Apoptosis is the original example of a regulated cell death process (Fig. 2). A major conceptual advance over the past twenty years has been the recognition that non-apoptotic cell death can also be highly regulated.
Figure 2. Cell suicide mechanisms.
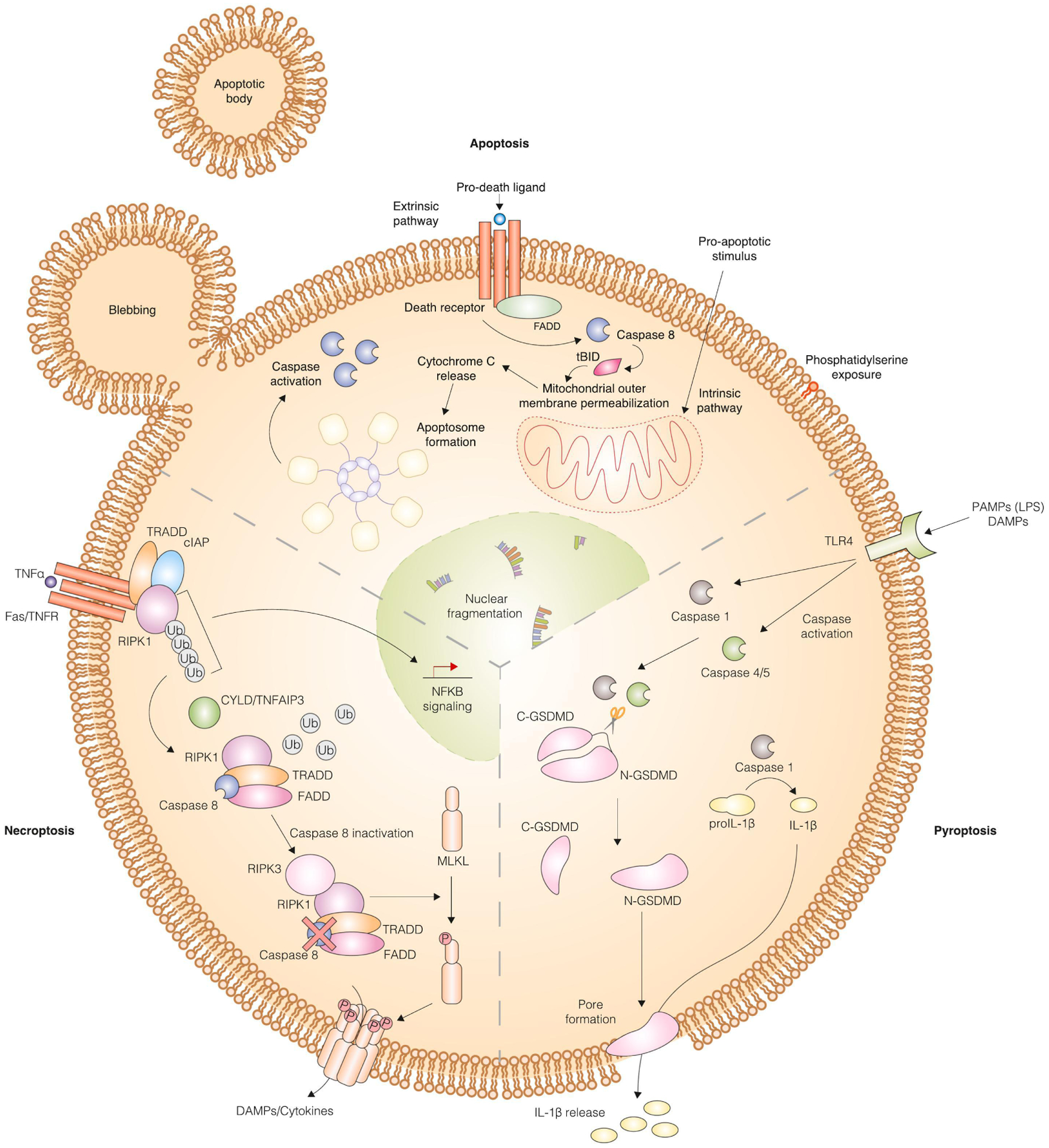
Mechanism of apoptosis, necroptosis and pyroptosis. Apoptosis can be triggered by the extrinsic pathway involving binding of a ligand to its corresponding death receptor, caspase 8 activation, BH3 interacting domain death agonist (BID) cleavage into truncated BID (tBID), mitochondrial outer membrane permeabilization (MOMP), cytochrome C release, and apoptosome formation. Alternatively, the intrinsic pathway involves incorporation of signals that converge on the B cell lymphoma 2 (BCL2) family of proteins, which is comprised of pro- and anti-apoptotic proteins. When the pro-apoptotic proteins overcome inhibition by the anti-apoptotic proteins, MOMP occurs leading to formation of the apoptosome. Executioner caspase activation occurs following both extrinsic and intrinsic pathways and causes hallmarks of apoptosis including nuclear fragmentation, phosphatidyl serine exposure, and apoptotic body formation through blebbing occur. The mechanism of pyroptosis is distinct. Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) bind to toll-like receptor 4 (TLR4) to activate specific caspases (1 and 4/5), which initiate the pyroptotic cascade. These caspases cleave gasdermin D into two fragments, with the N-terminal fragment translocating to the plasma membrane to create a pore through which processed interleukin 1 beta (IL-1β) can pass. The mechanism of necroptosis is likewise distinct from apoptosis and pyroptosis. Tumor necrosis factor alpha (TNFα) or other pro-death ligands bind to cell surface receptors to activate “complex I”, which includes TNF receptor type 1-associated death domain (TRADD), cellular inhibitor of apoptosis protein 1 (cIAP), and receptor interacting protein kinase 1 (RIPK1). Cylindromatosis lysine 63 deubiquitase (CYLD)/TNF alpha-induced protein 3 (TNFAIP3) catalyzes the deubiquitylation of RIPK1, which enables the formation of “complex II”. Complex II includes RIPK1, TRADD, Fas-associated death domain (FADD), and caspase 8. When caspase 8 is inactive, RIPK3 associates with RIPK1 to promote phosphorylation and subsequent polymerization of mixed lineage kinase domain like pseudokinase (MLKL). Polymerized MLKL localizes to the plasma membrane where it forms a pore through which DAMPs and cytokines are released.
Under the umbrella of regulated cell death, another potentially useful sub-classification scheme is between cell suicide and cell sabotage mechanisms[35]. Cell suicide processes have dedicated biochemical effectors that promote cell death, and these forms of cell death serve specific physiological roles. Apoptosis can be a form of cell suicide, clearly, with dedicated inducers and effectors, and a defined role in many physiological cell death events. Additionally, the non-apoptotic processes of pyroptosis and necroptosis may be forms of cell suicide. Both have dedicated signal transduction mechanisms and lethal pore-forming effector proteins (gasdermin D and mixed lineage kinase domain like pseudokinase, or MLKL, respectively)[14,36–42] (Fig. 2). These lethal mechanisms can also both be activated in response to physiological signals, such as infection, and help maintain organismal homeostasis[43,44]. These three cell suicide mechanisms (apoptosis, necroptosis, and pyroptosis) have been extensively studied and reviewed, and so will not be a focus of this review.
Cell sabotage mechanisms differ from cell suicide mechanisms in the way they use the ongoing normal metabolic, signaling, or cell biological activity of the cell to drive the execution of cell death under certain conditions[35]. For example, mammalian metabolic activity normally results in the generation of oxidized lipid species (i.e., lipid peroxides) at levels that are non-toxic to the cell; cell membranes are normally protected from lipid peroxidation by a series of endogenous defense mechanisms[45]. However, inactivation of these defenses results in toxic accumulation of high levels of lipid peroxides and subsequent plasma membrane rupture, hallmarks of the non-apoptotic process of ferroptosis[46–48] (Fig. 3a). Here, it is the normal formation of membrane lipid peroxides that drives death forward. In other cases, cell sabotage can involve hyperactivation of a given enzyme or process, resulting in cell death under certain circumstances.
Figure 3. Mechanisms of cell death that fit into the cell suicide/sabotage dichotomy.
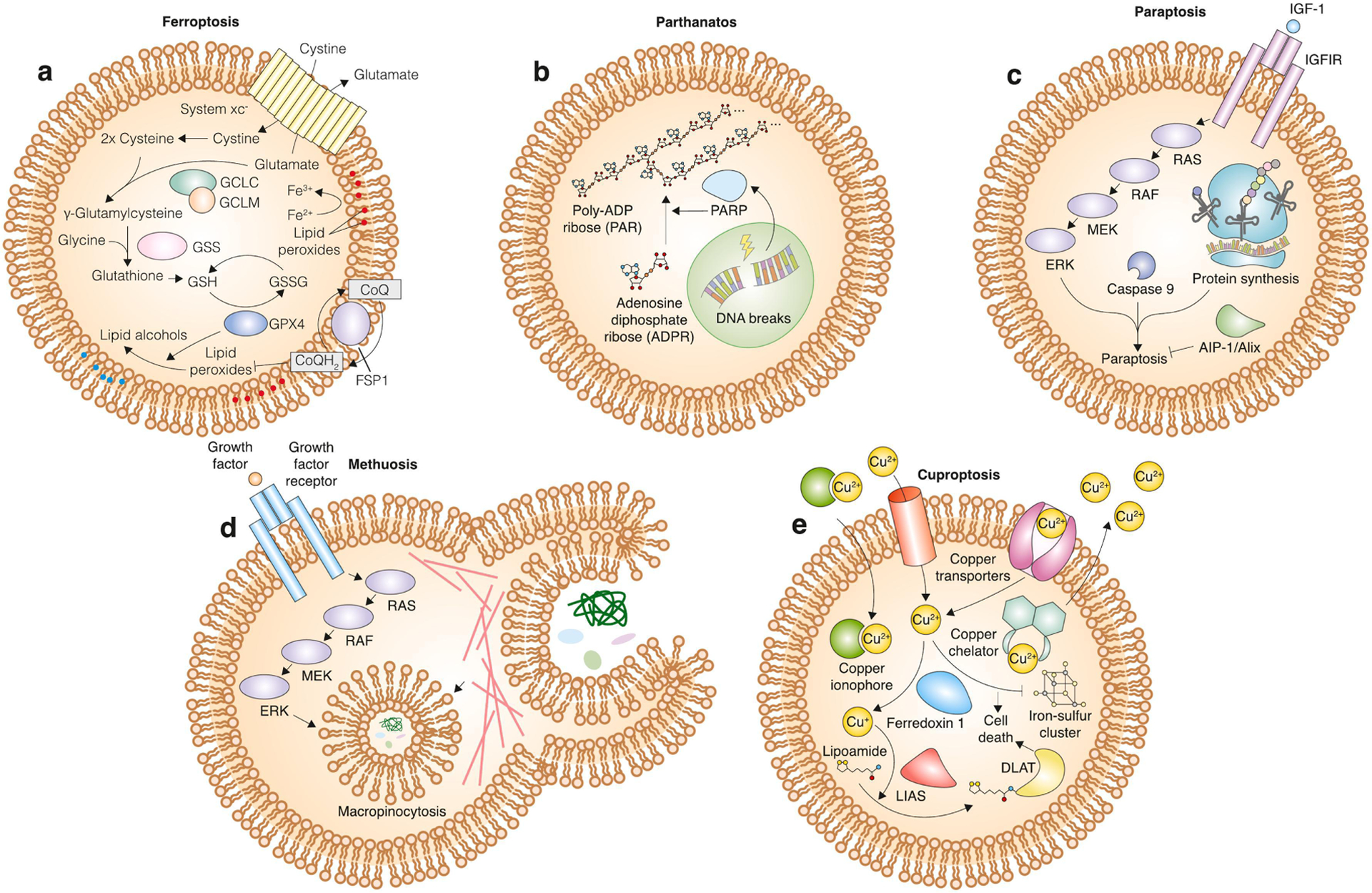
a, mechanism of ferroptosis. The cystine/glutamate antiporter system xc− normally allows for entry of cystine into the cell, where it can be reduced into cysteine and incorporated into the antioxidant tripeptide glutathione. Glutathione is a cofactor for the phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4), which catalyzes the conversion of potentially toxic lipid peroxides into non-toxic lipid alcohols. In parallel, the oxidoreductase ferroptosis suppressor protein 1 (FSP1) can catalyze the synthesis of reduced electron carriers such as coenzyme Q10 (CoQ10-H2), which in turn inhibit lipid reactive oxygen species (ROS) spread at the plasma membrane. Inhibition of these lipid peroxide defenses leads to ferroptosis. b, mechanism of parthanatos. DNA breaks lead to activation of poly (adenosine diphosphate-ribose) polymerase (PARP), which then catalyzes the polymerization of adenosine diphosphate (ADP) ribose into poly (adenosine diphosphate-ribose) (PAR), which leads to death induction. c, mechanism of paraptosis. Activation of insulin-like growth factor receptor 1 (IGF1R) by insulin-like growth factor (IGF-1) leads to downstream activation of the mitogen-activated protein kinase (MAPK) pathway. The MAPK pathway activates paraptosis in a protein synthesis- and caspase 9-dependent manner. This death pathway is opposed by actin-integrating protein 1 (AIP-1)/apoptosis-linked gene 2-interacting protein X (Alix). d, mechanism of methuosis. Growth factors bind to their respective receptors to activate the MAPK pathway. Increased signaling through this pathway induces micropinocytosis of extracellular material. Excessive micropinocytosis leads to dramatic vacuolization that physically detaches the cell from its substrate and leads to death induction. e, mechanism of cuproptosis. Intracellular copper levels are modulated by a variety of factors. Copper transporters shuttle copper in either direction across the membrane. Copper ionophores bind to and bring copper ions into the cell across the membrane, and copper chelators sequester copper and decrease cytosolic levels. Intracellular copper is oxidized by ferredoxin 1 and then serves as a co-factor for lipoic acid synthase (LIAS)-mediated lipoylation of target proteins. Lipoylation of dihydrolipoamide S-acetyltransferase (DLAT) and copper-dependent inhibition of iron-sulfur cluster formation leads to cuproptosis.
Cell sabotage mechanisms are non-apoptotic, do not have dedicated biochemical effectors like pore-forming proteins, and do not appear to have a role in normal development or homeostasis[49]. However, cell sabotage mechanisms may be activated by specific pathological stresses and could therefore be therapeutic targets to activate for cancer therapy or to inhibit to prevent degenerative diseases. Relatively little attention has been placed on cell sabotage mechanisms as a group, and it remains unclear which mechanisms belong in this category. Below, we will focus on examples of emerging and understudied non-apoptotic cell death mechanisms that may represent examples of cell sabotage, along with other non-apoptotic mechanisms that appear to share features of both cell sabotage and cell suicide phenotypes.
3. Candidate cell sabotage mechanisms
Here, we consider non-apoptotic cell death mechanisms that appear best described as forms of cell sabotage, focusing on key elements of regulation.
Ferroptosis
Ferroptosis was reported in 2012, but is likely identical to the process of oxytosis described some years prior[50]. There also are observations in the literature of what we can now recognize as ferroptosis dating back several decades[51,52]. This process contributes to pathological cell death in the nervous system, kidney, and other tissues in response to both acute and chronic stresses[53–56]. Ferroptosis may also play a physiological role as an endogenous tumor suppressor mechanism[57,58]. Molecules that can induce ferroptosis are being developed for cancer treatment and there is therefore much interest in the fundamental regulation of this process[59].
Ferroptosis is a prototypic example of a cell sabotage mechanism[35,49]. The iron-dependent accumulation of toxic lipid peroxides during ferroptosis results from a loss of balance between pro-oxidant and anti-oxidant forces in the cell[60] (Fig. 3a). Apoptotic caspases 3 and 7 are not activated during ferroptosis[61], and, unlike necroptosis or pyroptosis, specific pro-ferroptotic pore-forming proteins have not yet been identified. Instead, the O2, iron, and peroxidizable membrane phospholipids present in mammalian cells combine to drive the cell towards death when one or more endogenous lipid peroxide defense systems are inactivated. The pro-death lipid peroxidation can be enzyme-catalyzed[62] or result from non-specific Fenton chemistry[63,64]. Several enzymes and pathways normally operate to keep ferroptotic lipid peroxidation in check, including the glutathione-dependent phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4)[65–68], the ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10)/vitamin K pathway[69–71], the guanosine triphosphate (GTP) cyclohydrase 1 (GCH1)/tetrahydrobiopterin (BH4) pathway[72,73], and possibly a dihydroorotate dehydrogenase (DHODH)/nicotinamide adenine dinucleotide phosphate (NADPH) pathway[74]. When one or more of these mechanisms is inactivated, lipid peroxidation runs rampant and destroys plasma membrane integrity[75].
Beyond lipid peroxidation, distinguishing features of ferroptosis have been difficult to ascertain, possibly because cells undergoing ferroptosis can die very quickly. For example, the morphology of ferroptotic cells is largely unremarkable and may involve only slightly altered mitochondrial structure before the onset of membrane permeabilization[46]. Increased cell surface exposure of the transferrin receptor may be a molecular marker of ferroptotic cells[76], but cell surface expression of this receptor per se is not specific to ferroptosis. More work is needed[77], and newer techniques such as machine learning[78], redox lipidomics[79,80], or ferroptosis-specific antibodies[81] may yet unveil unique markers of ferroptosis that can be useful in basic and translational studies.
Parthanatos
Parthanatos is a form of non-apoptotic cell death linked to diseases including Parkinson’s and Alzheimer’s and therefore studied most often in a neuronal context[82]. There is no evidence that parthanatos plays any developmental or homeostatic role – it appears to be purely a mechanism of pathological cell death. Biochemically and morphologically, parthanatos is characterized by depolarization of mitochondria and rupture of the plasma membrane in the absence of membrane blebbing or swelling[83]. In cultured cells, parthanatos can be triggered by DNA damage (e.g., using N-methyl-N-nitroso-N′-nitroguanidine, or MNNG)[84–86]. Extensive DNA damage activate PARP-family enzymes[87]. PARPs catalyze the synthesis of PAR chains from adenosine diphosphate ribose (ADPR) monomers[88]. ADPR can be polymerized into poly-ADPR (pADPR or PAR) by PAR polymerase, and target proteins are poly-ADP ribosylated (or PARylated) to promote DNA repair and cellular survival. However, PAR can also bind directly to the apoptosis-inducing factor (AIF/AIFM1) and disrupt its attachment to the mitochondria[89]. AIF forms a complex with macrophage migration inhibitory factor (MIF), which then translocates to the nucleus and fragments DNA to induce cell death[90] (Fig. 3b).
The induction of parthanatos involves effects on metabolism that are consistent with and help further illustrate the concepts of regulated non-apoptotic cell death and cell sabotage. Nicotinamide adenine dinucleotide (NAD+) is required for parthanatos since PARP uses NAD+ to synthesize PAR. Excess PARP activation promotes death, in part, by depleting intracellular NAD+ stores through PAR-mediated hexokinase inhibition and subsequent glycolytic collapse[91]. NAD+ is a central co-factor to several metabolic pathways at the mitochondria, and NAD+ depletion favors AIF nuclear translocation by disrupting mitochondrial integrity[92]. This mechanism of cell death execution can therefore be blocked by small molecule PARP inhibitors[91], which prevents NAD+ consumption. AIF has an essential role in normal mitochondrial physiology[93]; the AIF protein presumably moonlights as a pro-death effector in cells experiencing extreme levels of metabolic stress.
Paraptosis
Paraptosis was initially described in 2000, making it one of the first non-apoptotic mechanisms to be proposed[94]. Paraptosis can be triggered in cultured cells by overexpression of various proteins including the intracytoplasmic domain of the insulin-like growth factor 1 receptor, toxicity and c-Jun N-terminal kinase inducer (TAJ)/TNF receptor superfamily member 19 (TROY), and programmed cell death 5 (PDCD5)[94–97] (Fig. 3c). However, paraptosis is not merely an artefact of protein overexpression, as several natural products with anti-cancer properties (e.g., curcumin, paclitaxel, and celastrol) are also proposed to induce paraptosis in some contexts[98]. Inhibition of pancreatic endoplasmic reticulum (ER) kinase (PKR)-like ER kinase (PERK) in cancer cells can also stimulate a tumor-clearing immune response by inducing paraptosis, suggesting that this mechanism could be a target for certain cancers[99].
Paraptotic cells do not exhibit telltale characteristics of apoptosis such as nuclear fragmentation, caspase 3 processing, or the formation of apoptotic bodies[94]. Rather, the defining feature of paraptosis is the swelling of intracellular organelles, particularly the ER and mitochondria, without any other morphological signs consistent with apoptosis[100]. Early studies indicated that paraptosis requires de novo protein synthesis, providing evidence of regulation[94]. This also suggests that ongoing protein synthesis and possibly protein accumulation drives paraptosis induction in a cell sabotage-like manner. Supporting the physiological relevance of paraptosis, the morphological alterations characteristic of paraptosis are observed in vivo[101]. However, organelle swelling reminiscent of paraptosis can also occur without cell death in some contexts (e.g., organelle swelling caused by altered vesicle trafficking or protein secretion)[102,103]. It is therefore possible that the observed morphological changes in paraptotic cells are not themselves lethal. Rather, these changes could correlate with some other lethal biochemical change or that unique characteristics of paraptotic organellar swelling that remain to be elucidated.
Methuosis
Methuosis is a form of non-apoptotic cell death characterized by high levels of macropinocytosis[104] (Fig. 3d). Macropinocytosis is a process by which cells ingest extracellular material into large, membrane-bound vesicles - the macropinosomes[105]. Macropinocytosis can be triggered by hyperactive rat sarcoma virus (RAS) protein activation or other signaling mechanisms[106–109]. Methuosis induced by transient transfection of oncogenic RAS involves the accumulation of large vacuoles that are not acidic, do not concentrate microtubule-associated protein 1A/1B-light chain 3-II (LC3-II), and are not double membrane-bound, indicating that the death is not autophagic[104]. The intracellular accumulation of large numbers of pinocytic vesicles triggers detachment of the cell from its substrate and, eventually, lethal rupture of the plasma membrane[104]. Inhibition of macropinocytosis using a small molecule Rac family small GTPase 1 (RAC1) inhibitor can prevent cell death, indicating that methuosis fits the definition of a regulated form of cell sabotage[110].
RAS is frequently mutated in cancer. Consequently, it might seem that RAS-mutant cancer cells should readily undergo methuosis[111–116]. In fact, RAS-driven macropinocytosis often seems to enhance cancer cell viability and proliferation in vitro and in vivo[107,108]. Why RAS hyperactivation induces methuosis in some contexts but enhances cancer cell viability in others is unclear. Methuosis has also been observed to occur in response to methamphetamine and nerve growth factor, suggesting that this pathway may also occur in other physiological contexts but this remains understudied[111].
Cuproptosis
Copper is a cofactor for diverse enzymes in the cell that have functions spanning processes as diverse as blood clotting and energy production, making this metal essential for sustaining life[117]. Free copper is normally maintained at sub-picomolar concentration within the cell[118,119], and tight homeostatic regulation of free copper is necessary because high levels of this metal can be cytotoxic[119]. Indeed, mutations in copper transporters, resulting in intracellular copper accumulation, can lead to a rare but devastating disease, Wilson’s syndrome[120,121]. Copper ionophores such as elesclomol have also been explored as novel anti-cancer agents up to human clinical trials[122], and there is therefore much interest in the mechanisms of copper toxicity.
Various lethal mechanisms have been proposed to explain copper toxicity including paraptosis[122] (see above) or the induction of oxidative stress[123]. Indeed, it is possible that different lethal mechanisms are involved depending upon the dose of copper. Recently, a different non-apoptotic mechanism of copper toxicity was identified and termed cuproptosis[124,125]. Here, elesclomol and other copper ionophores trigger cell death by shuttling copper into the mitochondria, where Cu2+ is reduced to Cu+ by ferredoxin 1[124]. Cu+ then promotes the aberrant lipoylation (i.e., the addition of lipoamide to target proteins) of several tricarboxylic acid (TCA) cycle proteins, including dihydrolipoamide S-acetyltransferase (DLAT)[124]. Copper directly binds and oligomerizes DLAT in a lipoylation-dependent manner. This accumulation of lipoylated TCA cycle proteins disrupts iron-sulfur cluster biogenesis, which in turn leads to proteotoxic stress and cell death[124] (Fig. 3e). Cuproptosis is exacerbated by increased mitochondrial oxidative phosphorylation and can be attenuated by reduced levels of this process. Thus, akin to ferroptosis, the ongoing metabolic activity of the cell drives it towards the precipice of death – a hallmark of cell sabotage.
Oxeiptosis
The lipid peroxides that characterize ferroptosis are not the only potentially lethal forms of oxidizing species in the cell. A form of soluble reactive oxygen species (ROS), hydrogen peroxide, can be toxic to cells[126,127]. Soluble ROS are already proposed to contribute to necroptosis[128], pyroptosis[129], and autophagic cell death[130]. However, ozone and hydrogen peroxide were recently suggested to trigger a specific and unique non-apoptotic cell death mechanism termed oxeiptosis[131] (Fig. 4a). In this form of cell death, ROS accumulation is sensed by the oxidation-sensitive protein kelch like ECH associated protein 1 (KEAP1). KEAP1 normally binds to phosphoglycerate mutase 5 (PGAM5), sequestering it at the mitochondrial outer membrane. Upon ROS increase, the KEAP1-PGAM5 complex is proposed to dissociate, allowing PGAM5 to enter into the mitochondria, dephosphorylate AIFM1 (also called AIF), and cause cell death[131]. The involvement of the endogenous cellular machinery renders this mechanism a potential example of cell sabotage, although to date there is no evidence that disruption of a specific intracellular defense mechanism (e.g., catalase) can unleash the lethal process.
Figure 4. Mechanisms of cell death that fit into the cell suicide/sabotage dichotomy (continued).
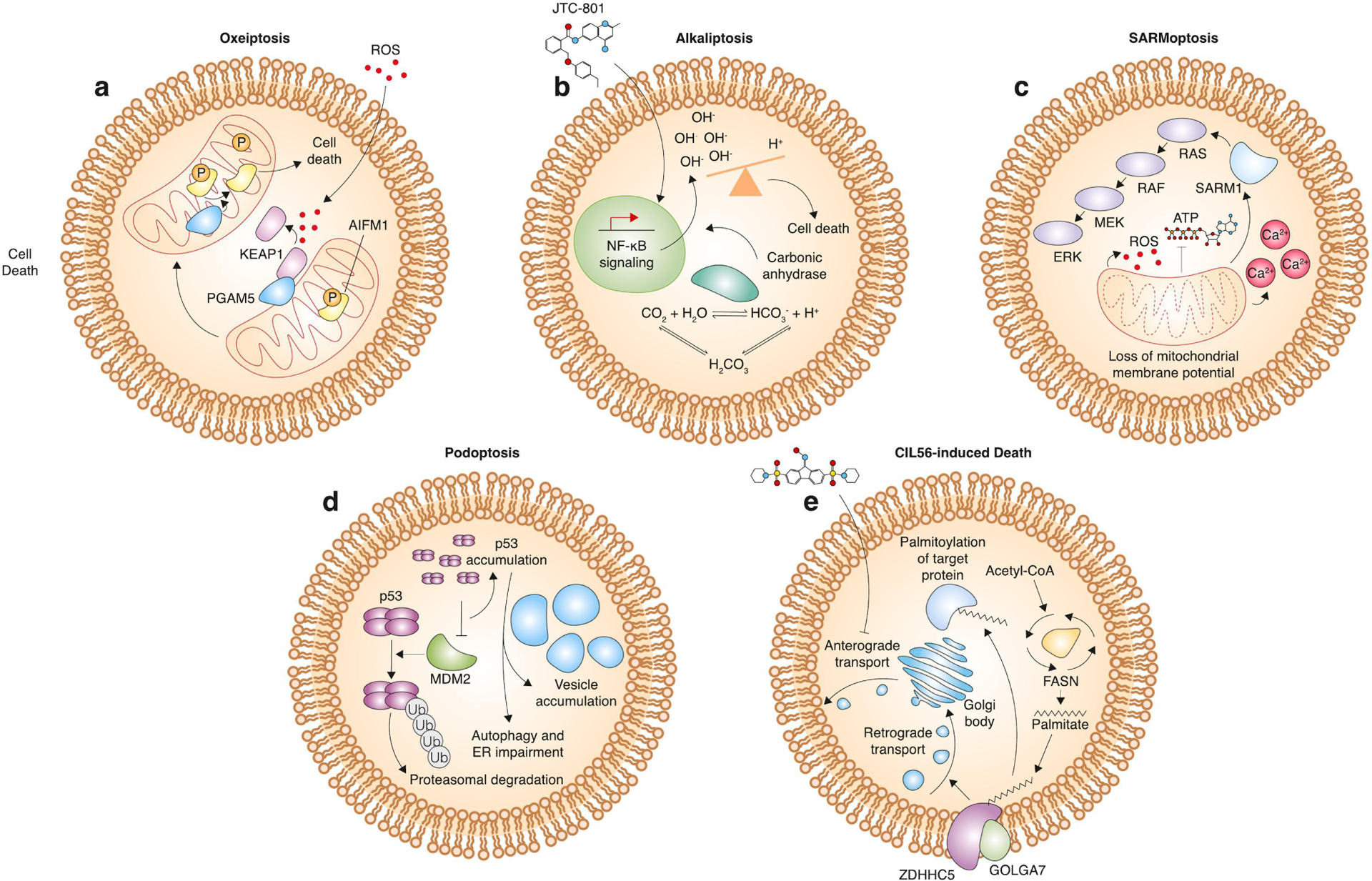
a, mechanism of oxeiptosis. Excessive ROS leads to dissociation of kelch like ECH associated protein 1 (KEAP1) from phosphoglycerate mutase 5 (PGAM5) and subsequent release of PGAM5 from the mitochondria. PGAM5 can then act as a phosphatase to remove phosphates from apoptosis-inducing factor (AIFM1), which then leads to death induction. b, mechanism of alkaliptosis. JTC-801 leads to increased NF-κB pathway activity, which decreases expression of carbonic anhydrase. This leads to an increase in intracellular pH and cell death. c, mechanism of SARMoptosis. Compromising of mitochondrial membrane potential leads to ROS accumulation, depletion of intracellular ATP, accumulation of calcium, and sterile alpha and Toll/interleukin receptor (TIR) motif containing 1 (SARM) activation. SARM can activate the MAPK pathway, which in turn results in cell death. d, mechanism of podoptosis. Inhibition of the E3 ubiquitin ligase mouse double minute 2 (MDM2) leads to accumulation of the p53 tetramer. p53 accumulation causes vesicle accumulation and impairment of autophagy and normal endoplasmic reticulum homeostasis, leading to cell death. e, Caspase-independent lethal (CIL56)-induced cell death. CIL56 inhibits anterograde transport from the Golgi body to the plasma membrane. Zinc finger DHHC-type palmitoyltransferase 5 (ZDHHC5) and golgin A7 (GOLGA7) form a protein S-acylation complex that promotes retrograde transport from the plasma membrane to the Golgi body. This process is dependent upon palmitate, a 16-carbon saturated fatty acid.
Oxeiptosis can be triggered by viral infection, which leads to toxic ROS accumulation in the host cell. Certain viral proteins bind protein members of the host antioxidant response system to prevent death and thereby promote viral reproduction[131]. Oxeiptosis may also be important in clinical contexts: ROS-inducing treatments could kill cancer cells in an AIFM1-dependent manner[132–134] and oxeiptosis may also contribute to vitiligo due via induction of melanocyte cell death[135]. While ROS-induced cell death has been observed for decades, the concept of a specific PGAM5 and AIFM1-dependent mechanism - oxeiptosis - is novel and further investigation of how this process may intersect with other forms of non-apoptotic, AIFM1-dependent cell death such as parthanatos is an open question.
Alkaliptosis
Cells must maintain pH within a narrow range to ensure survival[136]. Low intracellular pH (also known as acidosis or acidification) can kill cells via apoptosis[136] or necroptosis[137]. On the other hand, increased intracellular pH (or alkalinization) may induce a form of cell death distinct from other mechanisms, termed alkaliptosis[138] (Fig. 4b). At present, a single stimulus has been shown to trigger this form of cell death, the small molecule opioid analgesic JTC-801[139]. JTC-801 was originally developed for pain management[140] but was subsequently found to exhibit anti-cancer properties[138]. While some evidence indicates that JTC-801 induces apoptosis by inhibiting the phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB) axis[141,142], other results indicate that it induces caspase-independent cell death[143]. While nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling can inhibit apoptosis[144], this transcription factor can promote alkaliptosis in response to JTC-801 by suppressing carbonic anhydrase 9 (CA9) expression and thereby reducing intracellular pH[138]. The direct target(s) of JTC-801 that result in this effect is presently unknown. Furthermore, little is known about the defining morphological characteristics, physiological inducers or potential disease-relevance of alkaliptosis. Tumors tend to maintain a relatively low pH due to the Warburg effect and constant production of lactic acid[145]. The ability to exploit alkaliptosis for cancer therapy may vary considerably depending on the metabolic status of the cell. Whether this mechanism can be activated by stimuli other than JTC-801 remains to be established.
SARMoptosis
In general, cells of the nervous system appear especially susceptible to unique forms of non-apoptotic cell death[9]. Wallerian degeneration is a neuron-specific degenerative phenotype caused by axonal crush and related injuries[146]. This lethal process appears to engage a unique non-apoptotic cell death mechanism termed SARMoptosis. Here, neuronal cell death in response to axonal injury relies on sterile alpha and Toll/interleukin receptor (TIR) motif containing 1 (SARM1) and other NAD+-related enzymes[147] (Fig. 4c). Neuronal injury disrupts mitochondrial inner membrane transmembrane potential, and ongoing metabolism drives ROS accumulation, loss of adenosine triphosphate (ATP), and an increase in calcium, which in turn activate SARM1[147]. SARM1 then enzymatically depletes the cell of NAD+ and also triggers a mitogen-activated protein kinase (MAPK) signaling cascade that further disrupts energy homeostasis and causes axonal degradation[148,149]. In the absence of SARM1-dependent NAD+ depletion, neurons are better able to withstand Wallerian degeneration[150]. While altered NAD+ metabolism connects SARMoptosis and parthanatos, SARMoptosis alone results in the production of cyclic ADP-ribose (cADPR) which could be used as a unique biomarker for this process[151]. Interestingly, in addition to axonal injury, SARMoptosis may be induced via viral infection and is thus a potential point of intervention for the treatment of multiple diseases[147,151,152].
Podoptosis
Podocytes are glomerular kidney cells adjacent to Bowman’s capsule that enwrap capillaries[153]. Mouse double minute 2 (MDM2) is an E3-ubiquitin ligase that normally degrades p53 to keep its basal level low. MDM2 knockdown or inactivation in podocytes leads to p53 stabilization and a novel non-apoptotic type of cell death called podoptosis[154,155] (Fig. 4d). In podoptosis, p53 activation results in endoplasmic reticulum stress and cytosolic vacuole formation, presenting a unique biochemical/morphological signature[154]. Podoptosis also exhibits dysregulation of autophagy, which indicates that it may share some similarities with paraptosis[154]. Whether these morphological correlates of cell death contribute directly to lethal membrane permeabilization is, however, unclear. Of note, in cancer cells p53 stabilization can lead to activation of pro-death BCL2 family members to cause apoptosis[26]. However, there are examples in cells other than podocytes where MDM2 depletion and p53 stabilization induces non-apoptotic cell death[156], so this concept is not entirely unprecedented. Regardless, it is interesting that p53 stabilization appears capable of inducing either apoptosis or non-apoptotic cell death depending on the cellular context.
Diseases affecting the glomerulus are severe and, in some cases, lethal to the organism. The loss of podocytes is a major contributor to these morbidities[157]. As such, preventing podoptosis could serve an important role in treating kidney disease. However, it remains controversial whether podocyte loss is indeed due to a programmed cell death mechanism, physical detachment of viable cells, or a combination of both[158].
CIL56-induced death
Small molecule phenotypic screening can identify small molecules that induce novel cell death mechanisms[159]. A large-scale small molecule screen identified 451 caspase-independent lethal (CIL) compounds that killed cancer cells without activating the pro-apoptotic caspases 3 and 7[34]. Out of 56 compounds with high potency, the lethality of ten molecules was modulated by genetic perturbations and/or pharmacological agents, indicating that these lethal molecules triggered cell death in a regulated manner. This screen identified CIL56 as one novel lethal agent. CIL56 was initially thought to trigger ferroptosis at low doses and non-ferroptotic cell death at higher doses. However, in most cells it appears that the latter, non-ferroptotic mechanism is responsible for death induction[160,161]. CIL56-induced cell death requires zinc finger DHHC-type palmitoyltransferase 5 (ZDHHC5) and golgin A7 (GOLGA7)[160,161]. These two proteins form a mutually stabilizing protein S-acylation (i.e., “protein palmitoylation”) complex that promotes retrograde vesicle transport from the plasma membrane. This process is dependent upon palmitate, a sixteen carbon saturated fatty acid[160]. CIL56 inhibits anterograde transport from the Golgi body to the plasma membrane, suggesting that a perturbation of vesicle trafficking between the Golgi apparatus and the plasma membrane may be involved in cell killing upon CIL56 treatment[160] (Fig. 4e).
CIL56 offers a striking example of cell sabotage where a normally homeostatic mechanism (i.e., ZDHHC5/GOLGA7 complex-driven retrograde membrane trafficking) is converted into a death-promoting mechanism upon inactivation of a different mechanism (i.e., anterograde transport). CIL56 (also called CA3) can also inhibit the yes-associated protein 1 (YAP)-transcriptional enhancer factor domain family member 1 (TEAD) axis and may be useful as a potential cancer therapy in diseases where this pathway is active[162]. How YAP-TEAD modulation may relate to vesicle trafficking is, however, not clear. Defining morphological features of this lethal mechanism, if any exist, are presently unknown. Like JTC-801-induced alkaliptosis, it is also an open question whether other molecules or endogenous stimuli can induce this process.
4. More complex non-apoptotic cell death mechanisms
Certain regulated cell death mechanisms are not easily classified as cell suicide or cell sabotage processes. Like a cell suicide mechanism, they may serve important physiological roles and not obviously be caused by ongoing or hyperactivated intracellular signaling or metabolic network activity, and yet like a cell sabotage mechanism they may employ the normal machinery of the cell to execute cell death and not rely on any dedicated lethal effectors. Furthermore, for certain processes, the line between apoptosis and non-apoptotic cell death (i.e., necrosis) is blurry, suggesting that our conception of non-apoptotic cell death mechanisms may need to expand to include mechanisms that elude simple classification.
NETosis
Neutrophils are the most common type of granulocyte and an essential part of the innate immune system. Neutrophils are activated by microbial compounds such as pro-inflammatory cytokines and lipopolysaccharides (LPS). These stimuli trigger a vast structural transformation of the cell that yield the formation of neutrophil extracellular traps (NETs). NETs are physical structures, composed of fragments of chromatin and granule proteins, that can act to capture and degrade microbes[163]. The process of NET formation not only induces death of the target microbe, but it also occurs as part of a non-apoptotic cell death cascade within neutrophils called NETosis[164]. In some cases, activation of the rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway activates ROS production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2)[165]. NOX-derived ROS induce the release from cytosolic granules of neutrophil elastase (NE), which translocate to the nucleus to promote chromatin decondensation[166]. This translocation process is dependent on myeloperoxidase (MPO) and results in degradation of cytosolic F-actin[167]. In other cases, NETosis can be induced in a NOX-independent manner by calcium ionophores which activate mitochondrial ROS to trigger NET formation[168]. In both cases, degradation of the cytoskeleton coupled with chromatin decondensation results in extrusion of the newly formed NET out of the cell, compromising the plasma membrane and killing the neutrophil (Fig. 5a).
Figure 5. Mechanisms of cell death that do not fit into the cell suicide/sabotage dichotomy.
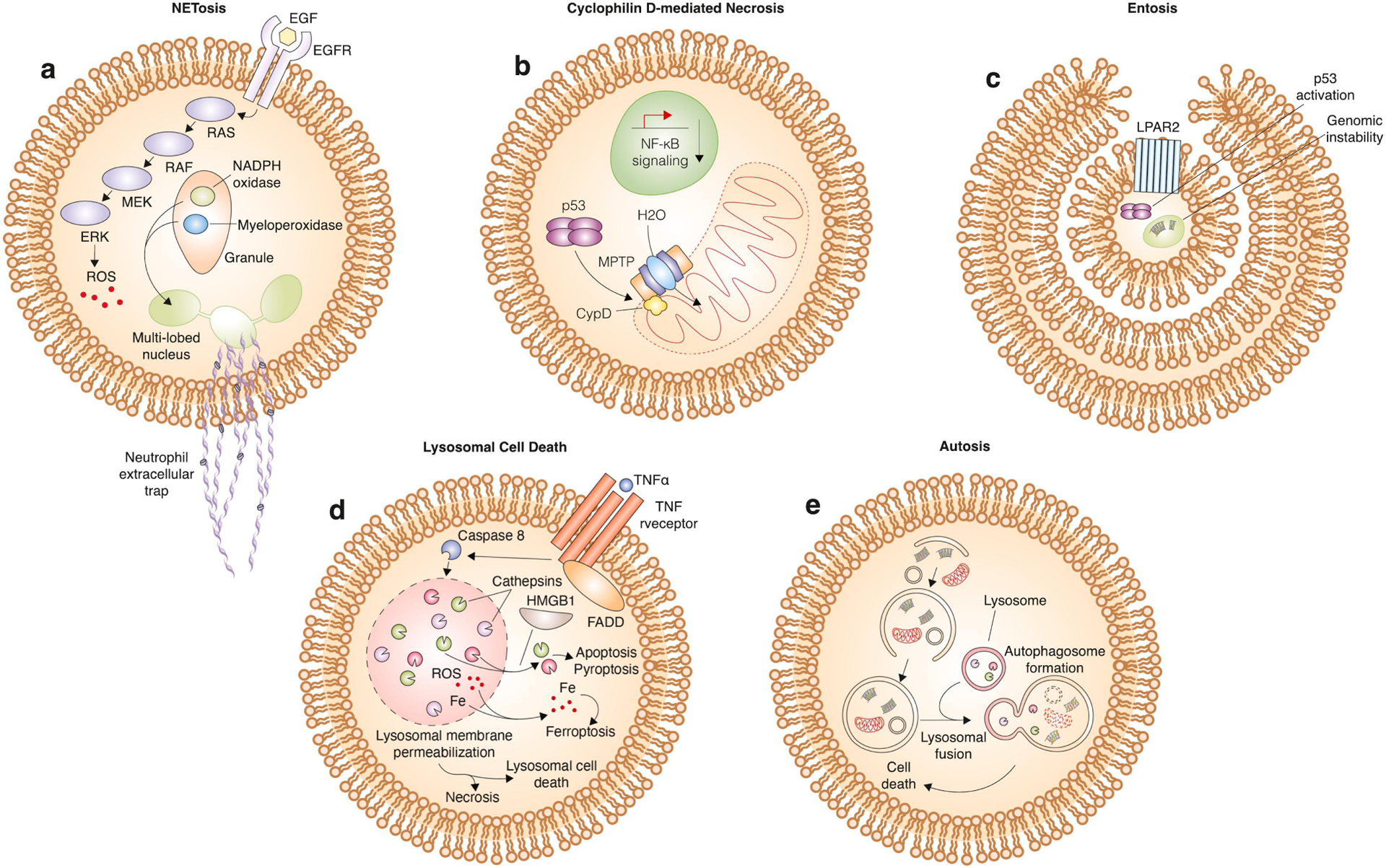
a, mechanism of NETosis. Activation of the MAPK pathway by binding of epidermal growth factor (EGF) to its associated receptor leads to ROS accumulation. ROS stimulates the release of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and myeloperoxidase from granules within the neutrophil. Translocation of these proteins to the multi-lobed nucleus results in release of nuclear contents, including DNA and histones, into the extracellular space where they can interact with target cells to induce cell death. b, mechanism of Cyclophilin D-mediated necrosis. The mitochondria permeability transition pore (MPTP), upon activation involving binding of CypD, results in permeabilization of the outer mitochondrial membrane. This releases water into the mitochondria that causes it to burst open and induce cell death. This form of cell death is associated with decreased nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB signaling). c, mechanism of entosis. Cell cannibalism or the cell-in-cell phenotype is activated by close contact of cells and formation of adherens junctions. In entosis, one cell is completely engulfed within another, and the process is activated by G-protein coupled lysophosphatidic acid receptor 2 (LPAR2) activation, p53 activation, and genomic instability. The cell can be recycled back to the plasma membrane or killed by enzymes within the lysosome upon fusion of the cell-containing vesicle with the lysosome. d, mechanism of lysosomal cell death. TNFα binds to its receptor to activate FADD and caspase 8. Caspase 8 induces lysosomal membrane permeabilization and subsequent release of cathepsins, ROS, and iron into the cytosol. High mobility group box 1 (HMGB1) facilitates the release of cathepsins specifically, which leads to induction of apoptosis or pyroptosis. Alternatively, cytosolic ROS accumulation can lead to the induction of ferroptosis. In addition, cell death by necrosis or an independent lysosomal cell death pathway can ensue. e, mechanism of autosis. Activation of autophagy leads to recycling of cellular components including proteins, nucleic acids, and organelles. Fusion with the lysosome leads to degradation of these components, and excessive activation of this pathway leads to cell death.
The main physiological role of NETosis is the elimination of pathogens: NETosis can mediate host protection from the bacteria, viruses, fungi, and protozoans[169,170]. This does not always come without a cost though. For example, in severe sepsis cases, toll-like receptor 4 (TLR4) activates platelets and neutrophils to eliminate bacteria, but they also damage normal surrounding tissue[171]. Beyond infectious diseases, NETosis has also been shown to promote autoimmune disorders such as Lupus as well as metastasis and thrombosis in cancer patients, prompting the development of NETosis-inhibiting drugs for treatment of these conditions[172,173]. Like a cell sabotage process, NETosis appears to use only the cells normal signaling machinery to execute cell death. However, it uniquely configures this machinery to generate a specific lethal effector mechanism – the defining extracellular traps.
Cyclophilin D-mediated necrosis
Opening of the mitochondrial permeability transition (mPT) pore defines a classic form of regulated non-apoptotic cell death. This mechanism requires cyclophilin D (CypD, encoded by the gene PPIF) as a core component of the mPT pore complex[174,175] (Fig. 5b). Mice lacking CypD are resistant to ischemia-reperfusion injury[174,175], and numerous studies attest to the physiological relevance of this form of death to pathology in vivo. Interestingly, CypD overexpression inhibits apoptosis yet promotes necrosis[176]. In line with this, mice lacking CypD are no longer able to undergo necrosis whereas apoptosis still occurs[175]. As such, the mPT pore promotes a form of necrotic cell death distinct from apoptosis. Similar to how p53 can directly bind and activate BAX to promote apoptosis[177], p53 can also directly bind the mPT pore to promote necrosis in an ATP synthase inhibitory factor 1 (ATP5IF1)-dependent manner, opening the pore and enabling water influx that leads to mitochondrial rupture and necrosis[20,178]. CypD activation is associated with decreased NF-κB signaling[179], which can potentially be used as a marker as this type of cell death.
The cyclophilin family derives its name from its members’ inhibition by the immunosuppressive transplant drug cyclosporine A[180]. The main physiological role of CypD outside of its regulation of the mPT is unclear. It belongs to the peptidyl-prolyl cis-trans isomerase protein family, which plays a role in protein folding, yet which proteins CypD may act on and how this impacts cellular physiology is still somewhat unclear[181]. It is therefore possible that CypD is a specific regulator of cell death, like BAX or BAK1 for apoptosis. However, the incident stimuli which specifically trigger this form of cell death specifically have not been clearly isolated and in some cases (e.g., ROS) appear to overlap with distinct cell death mechanisms [174,175].
Entosis
Entosis is a distinct mechanism, leading to cell death, that involves engulfment and destruction of one cell by another[182]. It has long been known that epithelial cells can phagocytize apoptotic cells[183], and instances of cell-in-cell morphologies have been observed for over a century[184]. While detachment of cells from matrix typically results in a specific type of apoptosis termed anoikis, in the case of entosis, the engulfed cell does not activate apoptotic caspases or exhibit morphological characteristics of apoptosis. The engulfment process is dependent upon E- and P-cadherin and results from physical compaction of cells that drives formation of adherens junctions. Since cancer cells are highly proliferative and compact, they may be particularly sensitive to this form of cell death [182,185]. The cell, once engulfed, becomes encapsulated in the lysosome and destroyed in a cathepsin-dependent manner[182] (Fig. 5c). Alternatively, the engulfed cell can be recycled back to the plasma membrane and be released from the engulfing cell without being killed[182].
Cells with highly mutated DNA are preferentially targeted for engulfment and clearance by entosis, suggesting that this form of cell death may be part of a genome surveillance and quality control mechanism[186]. p53 can also activate entosis by sensing genome instability via prolonged mitosis and aneuploidy[187]. Other triggers for entosis include glucose starvation[188], lysophosphatidic acid (LPA)-mediated activation of G-protein coupled LPA receptor 2 (LPAR2)[189], and tumor necrosis factor-related apoptosis-inducing ligand signaling[190]. These triggers can be activated in cancer and motivate cannibalistic activity of tumor cells, promoting entosis, recycling of nutrients, and tumor progression[184]. Entosis could represent a homeostatic mechanism of cell non-autonomous destruction and may therefore be best classified as a form of cell suicide. However, this destructive non-apoptotic mechanism appears to rely entirely upon known signaling processes and lysosomal enzymes to trigger cell death.
Lysosomal cell death
Lysosomes lie at the nexus of several distinct cell death mechanisms. Lysosomal membrane permeabilization (LMP) occurs in response to various stresses such as ROS or lyosomotropic agents[191]. LMP may promote or contribute to the execution of necrotic[192], apoptotic[193], pyroptotic[194], ferroptotic[195,196], or entotic[182] cell death. Further, caspase 8 and the pro-apoptotic protein BID can induce LMP upon activation by upstream pro-apoptotic receptor signaling through TNF-α receptor (TNFR), linking classic pro-apoptotic stimuli potentially to the induction of non-apoptotic cell death[197].
However, lysosomes are also involved in a unique non-apoptotic process called lysosomal cell death. This is a vesiculate cytoplasmic cell death mechanism where membrane-bound organellar structures form and expand in number and size until they are contiguous with the extracellular space[198]. This process is distinct from autophagy[199]. LMP and cathepsin leakage is necessary for the induction of this form of cell death[200] (Fig. 5d). LMP can be induced by various pharmacological and physiological factors, including quinolone antibiotics and oleic acid, an abundant monounsaturated fatty[201]. Lysosomal cell death exhibits some of the hallmarks of apoptosis including BAX and BAK oligomerization yet death is induced in a caspase-independent manner[200]. Lysomotropic agents, or weak bases that become protonated and trapped in the lysosome, can accumulate and induce LMP to trigger cell death[202]. Lysosomal cell death has very recently been termed lysoptosis following further elucidation of its mechanism, which includes reliance on cathepsins and inhibition by serpins[203].
Autosis
Autophagy is a scavenging process that involves recycling of cellular components in conditions of low nutrients to prevent demise of a cell. In some cases, the stresses are too severe, and autophagy is incapable of preventing cell death. Autophagy has long been observed as a feature of dying cells; however, whether autophagy plays an active role in the execution of cell death or instead acts as a passive bystander or scavenger of dying debris is often unclear and has led to confusion around the term “autophagic cell death” (ACD)[204]. To further understand specific modalities of cell death involving autophagy, an autophagy-inducing peptide resembling a domain of Beclin, an autophagy protein, was developed by investigating the interactions between viruses and host enzymes[205]. This peptide was lethal to cells in a dose-dependent manner, and the death was suppressed by inhibitors of autophagy but not other forms of cell death, suggesting that autosis is a unique form of autophagy-dependent cell death[206] (Fig. 5e). Autosis may be clinically relevant as it was previously known that autophagy plays an important role in the cell death observed during ischemia-reperfusion injury[207–209]. In addition, some cancer therapeutic agents may induce autosis in cancer cells. For example, a ginger extract induces autosis in pancreatic cancer cells[210]. Indeed, it remains somewhat unclear whether autosis should be considered distinct from autophagy-dependent/associated forms of cell death that have been described historically.
5. Conclusions and Future Directions
The cell death field has gone through several evolutions over the past sixty years, from the discovery that cell death could be regulated, to the cellular and molecular characterization of apoptosis, to the explosive growth in our understanding of non-apoptotic cell death mechanisms. Here, we have highlighted examples of emerging and under-studied non-apoptotic mechanisms. Some of these processes are best described as cell suicide mechanisms whereas others fit the concept of cell sabotage mechanisms. Yet other processes do not seem to fit in either category, indicating that the ways we classify cell death remains a ripe area for the development of new concepts. Below, we highlight four additional areas where important questions require further elucidation.
For most non-apoptotic cell death mechanisms there are large gaps in our basic understanding or ability to manipulate these processes. For example, we have excellent pharmacological tools to activate or inhibit ferroptosis and can perceive how oxidative damage to membrane phospholipids is fatal; however, defining morphological features and biochemical markers that can be used to track this process in vivo, akin to nuclear condensation and cleaved caspase 3 for apoptosis, are elusive. By contrast, for methuosis, macropinocytosis seems to be important for this lethal mechanism, and this cell biological process also provides a defining morphological feature, but there are few specific pharmacological modulators of this process that could be used to investigate this process in vivo. For other processes, like paraptosis, we have some useful means to induce this process and a solid understanding of the defining morphological features, but the precise explanation for cell death following ER swelling remains elusive. Ultimately, if apoptosis is any guide, it will take much further investigation to fully characterize any of the above cell death mechanisms.
Whether and how different cell death mechanisms connect to one another is under-explored. A key discovery lying at the origin of the non-apoptotic cell death field highlights the potential importance of these connections: inhibition of caspase activity in some cells can convert a pro-apoptotic stimulus into a pro-necroptotic stimulus[11,14]. Several stimuli are reported to induce both paraptosis and apoptosis, suggesting that paraptosis may be primarily a morphological manifestation of ER stress that occasionally causes death before the apoptosis mechanism can be fully engaged[211,212]. How or why cells die via paraptosis instead of apoptosis under different conditions remains unclear. Likewise, methuosis occurs with caspase activation, but inhibition of caspases does not prevent death[213]. For copper, accumulation of this metal is linked to both paraptosis[214] and cuproptosis[124]. It is unclear whether and why cells engage different forms of non-apoptotic cell death in response to the same lethal stimulus. One hypothesis is that the kinetics of stress accumulation could be important for determining whether a cell succumbs via apoptosis or is more quickly overwhelmed in a non-apoptotic fashion[215–217]. Even between different non-apoptotic mechanisms there may be underappreciated cross-talk. NAD+ serves as a building block for both pADPR (involved in parthanatos), cADPR (involved in SARMoptosis), and NADPH (a key regulator of ferroptosis)[218]. Consequently, perturbation of NAD+ metabolic pathways might therefore alter sensitivity to all three mechanisms in parallel. Further, certain proteins are vital for multiple forms of cell death. For example, caspase 8 activity or lack thereof can determine the tendency of a cell towards apoptosis, necroptosis, or pyroptosis[219], and AIF family members are involved in parthanatos, oxeiptosis, and ferroptosis[84,131,220]. More work is needed to elucidate how a single protein contributes to multiple cell death mechanisms, and how this interplay impacts organismal physiology. Finally, the role of different oxidizing species in cell death remains a subject of controversy. The accumulation of oxidative species is implicated in ferroptosis, NETosis, oxeiptosis, lysosomal cell death and other mechanisms. While lipid peroxidation may be a uniquely defining feature of ferroptosis[221], whether soluble ROS species (H2O2, superoxide, singlet oxygen) always trigger a specific form of non-apoptotic cell death remains unclear.
The concept of cell sabotage rests on the notion that ongoing cellular activity drives a cell towards death when another balancing or protective process is inhibited. However, stressors such as proteasome inhibition and microtubule disruption cause apoptotic cell death in a manner that is promoted by ongoing cellular activity (e.g., protein synthesis or cell division, in the cases or proteasome inhibition or microtubule disruption, respectively)[51]. Thus, why certain stresses engage the apoptosis machinery and others induce various forms of non-apoptotic cell death emerges as a key open question. We may infer that cells have evolved sensors and transducers for certain stresses that allow for the core apoptotic machinery to be engaged in some cases, but why these exist for some stresses and not others is unclear.
Finally, the physiological relevance of different non-apoptotic cell death mechanisms and therefore whether it is appropriate or perhaps counter-productive to bestow specific names on these processes remains an open question. For example, an early critique of the importance of ferroptosis was that this process could only be induced by synthetic small molecules, such as erastin. However, once it was understood that erastin inhibited the cell surface system xc− cystine/glutamate antiporter it became clearer how a physiological stimulus encountered in the nervous system, like high concentration of extracellular glutamate, could engage this process in a pathological setting[46,222]. In turn, it became easier to imagine that this cell sabotage mechanism played important roles in vivo. For other processes, specific physiological stimuli are lacking, the bridge to normal or disease physiology remains to be established, and therefore the importance of the mechanism is less clear. For example, the small molecule CIL56 clearly induces a unique form of cell death, but whether this is related to any form of cell death that can be activated by physiological stimuli is presently unknown. As another example, the process of autoschizis was proposed over twenty years ago as a unique form of oxidative necrosis induced by treatment with the redox-cycling agents vitamin C, vitamin K3, or the combination of both agents[223,224]. These agents may be useful for the treatment of cancer, which alone makes this process notable. However, whether this exact mechanism can be induced by any configuration of endogenous stimuli is unclear. When and whether it is appropriate to name unique cell death mechanisms that can only be caused by highly specific and often non-physiological stimuli is an open question in the field[33].
Highlights:
Mammalian cells can be killed by several distinct cell death mechanisms
Some non-apoptotic cell death mechanisms correspond to cell sabotage
Crosstalk between non-apoptotic cell death mechanisms is common
The complete set of cell death mechanisms that can be activated is unclear
Acknowledgements
This work was supported by awards to S.J.D. from the NIH (1R01GM122923) and the American Cancer Society (RSG-21-017-01-CCG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
S.J.D. is a co-founder of Prothegen Inc., a member of the scientific advisory board for Ferro Therapeutics and Hillstream BioPharma, and an inventor on patents related to ferroptosis.
References
- [1].Mazzarello P, A unifying concept: the history of cell theory, Nat Cell Biol, 1 (1999) E13–E15. [DOI] [PubMed] [Google Scholar]
- [2].Vogt C, Untersuchungen über die Entwicklungsgeschichte der Geburtshelferkrœte (Alytes obstetricans), Jent & Gassmann, 1842. [Google Scholar]
- [3].Majno G, Joris I, Apoptosis, oncosis, and necrosis An overview of cell death, Am J Pathol, 146 (1995) 3–15. [PMC free article] [PubMed] [Google Scholar]
- [4].Kerr JFR, Wyllie AH, Currie AR, Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics, Br J Cancer, 26 (1972) 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tata JR, Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture, Developmental Biology, 13 (1966) 77–94. [DOI] [PubMed] [Google Scholar]
- [6].Lockshin RA, Williams CM, Programmed cell death—II Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths, Journal of Insect Physiology, 10 (1964) 643–649. [Google Scholar]
- [7].Kaufmann SH, Induction of Endonucleolytic DNA Cleavage in Human Acute Myelogenous Leukemia Cells by Etoposide, Camptothecin, and Other Cytotoxic Anticancer Drugs: A Cautionary Note1, Cancer Research, 49 (1989) 5870–5878. [PubMed] [Google Scholar]
- [8].Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR, The C elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme, Cell, 75 (1993) 641–652. [DOI] [PubMed] [Google Scholar]
- [9].Yuan J, Lipinski M, Degterev A, Diversity in the mechanisms of neuronal cell death, Neuron, 40 (2003) 401–413. [DOI] [PubMed] [Google Scholar]
- [10].Yuan J, Kroemer G, Alternative cell death mechanisms in development and beyond, Genes Dev, 24 (2010) 2592–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P, Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor, J Exp Med, 187 (1998) 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, Strasser A, Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK, Cell, 173 (2018) 1217–1230.e17. [DOI] [PubMed] [Google Scholar]
- [13].Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, et al. , The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues, Mol Cell, 6 (2000) 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J, Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury, Nat Chem Biol, 1 (2005) 112–119. [DOI] [PubMed] [Google Scholar]
- [15].Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J, Identification of RIP1 kinase as a specific cellular target of necrostatins, Nat Chem Biol, 4 (2008) 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Frisch S, Francis H, Disruption of epithelial cell-matrix interactions induces apoptosis | Journal of Cell Biology | Rockefeller University Press, Journal of Cell Biology, 124 (1994) 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ishikawa F, Ushida K, Mori K, Shibanuma M, Loss of anchorage primarily induces non-apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling, Cell Death Dis, 6 (2015) e1619–e1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].King KL, Cidlowski JA, Cell cycle and apoptosis: Common pathways to life and death, Journal of Cellular Biochemistry, 58 (1995) 175–180. [DOI] [PubMed] [Google Scholar]
- [19].Nakahata K, Miyakoda M, Suzuki K, Kodama S, Watanabe M, Heat shock induces centrosomal dysfunction, and causes non-apoptotic mitotic catastrophe in human tumour cells, Int J Hyperthermia, 18 (2002) 332–343. [DOI] [PubMed] [Google Scholar]
- [20].Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM, p53 opens the mitochondrial permeability transition pore to trigger necrosis, Cell, 149 (2012) 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L, TNF Induces Pathogenic Programmed Macrophage Necrosis in Tuberculosis through a Mitochondrial-Lysosomal-Endoplasmic Reticulum Circuit, Cell, 178 (2019) 1344–1361.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nagata S, Apoptosis by Death Factor, Cell, 88 (1997) 355–365. [DOI] [PubMed] [Google Scholar]
- [23].Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S-I, Sameshima M, Hase A, Seto Y, Nagata S, The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis, Cell, 66 (1991) 233–243. [DOI] [PubMed] [Google Scholar]
- [24].Smith CA, Farrah T, Goodwin RG, The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death, Cell, 76 (1994) 959–962. [DOI] [PubMed] [Google Scholar]
- [25].Trauth BC, Klas C, Peters AMJ, Matzku S, Möller P, Falk W, Debatin K-M, Krammer PH, Monoclonal Antibody-Mediated Tumor Regression by Induction of Apoptosis, Science, 245 (1989) 301–305. [DOI] [PubMed] [Google Scholar]
- [26].van Delft MF, Huang DC, How the Bcl-2 family of proteins interact to regulate apoptosis, Cell Res, 16 (2006) 203–213. [DOI] [PubMed] [Google Scholar]
- [27].Bredesen DE, Ye X, Tasinato A, Sperandio S, Wang JJ, Assa-Munt N, Rabizadeh S, p75NTR and the concept of cellular dependence: seeing how the other half die, Cell Death Differ, 5 (1998) 365–371. [DOI] [PubMed] [Google Scholar]
- [28].Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE, Induction of apoptosis by the low-affinity NGF receptor, Science, 261 (1993) 345–348. [DOI] [PubMed] [Google Scholar]
- [29].Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, Bredesen DE, The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis, Nature, 395 (1998) 801–804. [DOI] [PubMed] [Google Scholar]
- [30].Welihinda AA, Tirasophon W, Kaufman RJ, The cellular response to protein misfolding in the endoplasmic reticulum, Gene Expr, 7 (1999) 293–300. [PMC free article] [PubMed] [Google Scholar]
- [31].Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP, 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer, Journal of Cellular Biochemistry, 82 (2001) 110–122. [DOI] [PubMed] [Google Scholar]
- [32].Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe’er D, Shaik F, Bulinski JC, Stockwell BR, Modulatory profiling identifies mechanisms of small molecule-induced cell death, Proc Natl Acad Sci U S A, 108 (2011) E771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, et al. , Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ, 25 (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR, Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis, Nat Chem Biol, 12 (2016) 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Green DR, The Coming Decade of Cell Death Research: Five Riddles, Cell, 177 (2019) 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].He W, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, Han J, Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion, Cell Res, 25 (2015) 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, Molecular mechanisms of necroptosis: an ordered cellular explosion, Nat Rev Mol Cell Biol, 11 (2010) 700–714. [DOI] [PubMed] [Google Scholar]
- [38].Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang J-G, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RCJ, Hilton DJ, Babon JJ, Nicola NA, et al. , The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism, Immunity, 39 (2013) 443–453. [DOI] [PubMed] [Google Scholar]
- [39].Cookson BT, Brennan MA, Pro-inflammatory programmed cell death, Trends in Microbiology, 9 (2001) 113–114. [DOI] [PubMed] [Google Scholar]
- [40].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling, Nature, 526 (2015) 666–671. [DOI] [PubMed] [Google Scholar]
- [41].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, Nature, 526 (2015) 660–665. [DOI] [PubMed] [Google Scholar]
- [42].Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, Shao F, Pore-forming activity and structural autoinhibition of the gasdermin family, Nature, 535 (2016) 111–116. [DOI] [PubMed] [Google Scholar]
- [43].Pasparakis M, Vandenabeele P, Necroptosis and its role in inflammation, Nature, 517 (2015) 311–320. [DOI] [PubMed] [Google Scholar]
- [44].Bergsbaken T, Fink SL, Cookson BT, Pyroptosis: host cell death and inflammation, Nat Rev Microbiol, 7 (2009) 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gutteridge JM, Lipid peroxidation and antioxidants as biomarkers of tissue damage, Clinical Chemistry, 41 (1995) 1819–1828. [PubMed] [Google Scholar]
- [46].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR, Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death, Cell, 149 (2012) 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rush GF, Gorski JR, Ripple MG, Sowinski J, Bugelski P, Hewitt WR, Organic hydroperoxide-induced lipid peroxidation and cell death in isolated hepatocytes, Toxicology and Applied Pharmacology, 78 (1985) 473–483. [DOI] [PubMed] [Google Scholar]
- [48].Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D, Ferroptosis: process and function, Cell Death Differ, 23 (2016) 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Green DR, Victor B, The pantheon of the fallen: why are there so many forms of cell death?, Trends Cell Biol, 22 (2012) 555–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lewerenz J, Ates G, Methner A, Conrad M, Maher P, Oxytosis/Ferroptosis—(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System, Frontiers in Neuroscience, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cao JY, Dixon SJ, Mechanisms of ferroptosis, Cell Mol Life Sci, 73 (2016) 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hirschhorn T, Stockwell BR, The development of the concept of ferroptosis, Free Radic Biol Med, 133 (2019) 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz P-S, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Bräsen JH, Kunzendorf U, Anders H-J, et al. , Synchronized renal tubular cell death involves ferroptosis, Proceedings of the National Academy of Sciences, 111 (2014) 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li J, Cao F, Yin H, Huang Z, Lin Z, Mao N, Sun B, Wang G, Ferroptosis: past, present and future, Cell Death Dis, 11 (2020) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang H-T, Linkermann A, Gu W, Min J, Wang F, Ferroptosis as a target for protection against cardiomyopathy, Proceedings of the National Academy of Sciences, 116 (2019) 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, Li G, Yang L, Liu W, Min J, Wang F, Characterization of ferroptosis in murine models of hemochromatosis, Hepatology, 66 (2017) 449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, Baer R, Gu W, Ferroptosis as a p53-mediated activity during tumour suppression, Nature, 520 (2015) 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, Zhuang L, Chen G, Xiao Z-D, Hung M-C, Chen J, Huang P, Li W, Gan B, BAP1 links metabolic regulation of ferroptosis to tumour suppression, Nat Cell Biol, 20 (2018) 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen X, Kang R, Kroemer G, Tang D, Broadening horizons: the role of ferroptosis in cancer, Nat Rev Clin Oncol, 18 (2021) 280–296. [DOI] [PubMed] [Google Scholar]
- [60].Stockwell BR, Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications, Cell, 185 (2022) 2401–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR, RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels, Nature, 447 (2007) 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV, Vladimirov YA, Shvedova AA, Philpott CC, Bayir H, Kagan VE, Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction?, Free Radical Biology and Medicine, 133 (2019) 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zielinski ZAM, Pratt DA, Lipid Peroxidation: Kinetics, Mechanisms, and Products, J. Org. Chem, 82 (2017) 2817–2825. [DOI] [PubMed] [Google Scholar]
- [64].Conrad M, Pratt DA, The chemical basis of ferroptosis, Nat Chem Biol, 15 (2019) 1137–1147. [DOI] [PubMed] [Google Scholar]
- [65].Seibt TM, Proneth B, Conrad M, Role of GPX4 in ferroptosis and its pharmacological implication, Free Radical Biology and Medicine, 133 (2019) 144–152. [DOI] [PubMed] [Google Scholar]
- [66].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR, Regulation of Ferroptotic Cancer Cell Death by GPX4, Cell, 156 (2014) 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ursini F, Maiorino M, Gregolin C, The selenoenzyme phospholipid hydroperoxide glutathione peroxidase, Biochimica et Biophysica Acta (BBA) - General Subjects, 839 (1985) 62–70. [DOI] [PubMed] [Google Scholar]
- [68].Maiorino M, Roveri A, Ursini F, Gregolin C, Enzymatic determination of membrane lipid peroxidation, J Free Radic Biol Med, 1 (1985) 203–207. [DOI] [PubMed] [Google Scholar]
- [69].Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, Mourão A, Buday K, Sato M, Wanninger J, Vignane T, Mohana V, Rehberg M, Flatley A, Schepers A, Kurz A, et al. , FSP1 is a glutathione-independent ferroptosis suppressor, Nature, 575 (2019) 693–698. [DOI] [PubMed] [Google Scholar]
- [70].Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA, The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis, Nature, 575 (2019) 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, Shi J, Liu X, Horbath A, Das M, Li W, Poyurovsky MV, Olszewski K, Gan B, A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers, Nat Commun, 13 (2022) 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, Birsoy K, Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers, Nat Chem Biol, 16 (2020) 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kössl J, Brandner S, Daniels JD, Schmitt-Kopplin P, Hauck SM, Stockwell BR, Hadian K, Schick JA, GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling, ACS Cent Sci, 6 (2020) 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, Poyurovsky MV, Olszewski K, Gan B, DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer, Nature, 593 (2021) 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA, Dixon SJ, Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State, Cell Chemical Biology, 26 (2019) 420–432.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, Upadhyayula PS, Canoll P, Uchida K, Soni RK, Hadian K, Stockwell BR, Transferrin Receptor Is a Specific Ferroptosis Marker, Cell Reports, 30 (2020) 3411–3423.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hadian K, Stockwell BR, A roadmap to creating ferroptosis-based medicines, Nat Chem Biol, 17 (2021) 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jin J, Schorpp K, Samaga D, Unger K, Hadian K, Stockwell BR, Machine Learning Classifies Ferroptosis and Apoptosis Cell Death Modalities with TfR1 Immunostaining, ACS Chem Biol, 17 (2022) 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bayır H, Anthonymuthu TS, Tyurina YY, Patel SJ, Amoscato AA, Lamade AM, Yang Q, Vladimirov GK, Philpott CC, Kagan VE, Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis, Cell Chem Biol, 27 (2020) 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, et al. , Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nat Chem Biol, 13 (2017) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Homma T, Nishino Y, Fujii J, Yokoyama C, Flow cytometric determination of ferroptosis using a rat monoclonal antibody raised against ferroptotic cells, J Immunol Methods, 510 (2022) 113358. [DOI] [PubMed] [Google Scholar]
- [82].Wang X, Ge P, Parthanatos in the pathogenesis of nervous system diseases, Neuroscience, 449 (2020) 241–250. [DOI] [PubMed] [Google Scholar]
- [83].Aki T, Funakoshi T, Uemura K, Regulated necrosis and its implications in toxicology, Toxicology, 333 (2015) 118–126. [DOI] [PubMed] [Google Scholar]
- [84].Andrabi SA, Dawson TM, Dawson VL, Mitochondrial and Nuclear Cross Talk in Cell Death, Annals of the New York Academy of Sciences, 1147 (2008) 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Virág L, Szabó C, The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors, Pharmacol Rev, 54 (2002) 375–429. [DOI] [PubMed] [Google Scholar]
- [86].Roos WP, Thomas AD, Kaina B, DNA damage and the balance between survival and death in cancer biology, Nat Rev Cancer, 16 (2016) 20–33. [DOI] [PubMed] [Google Scholar]
- [87].Amé J-C, Spenlehauer C, de Murcia G, The PARP superfamily, BioEssays, 26 (2004) 882–893. [DOI] [PubMed] [Google Scholar]
- [88].Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC, Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38, Science, 262 (1993) 1056–1059. [DOI] [PubMed] [Google Scholar]
- [89].Wang Y, Kim NS, Haince J-F, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM, Poly(ADP-Ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical for PAR Polymerase-1–Dependent Cell Death (Parthanatos), Science Signaling, 4 (2011) ra20–ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, Chen R, Wang JE, Kam T-I, Jeong JS, Xie Z, Neifert S, Qian J, Andrabi SA, Blackshaw S, Zhu H, et al. , A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1, Science, 354 (2016) aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagné J-P, Poirier GG, Dawson VL, Dawson TM, Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis, Proc Natl Acad Sci U S A, 111 (2014) 10209–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA, NAD+ Depletion Is Necessary and Sufficient forPoly(ADP-Ribose) Polymerase-1-Mediated Neuronal Death, J. Neurosci, 30 (2010) 2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Joza N, Pospisilik JA, Hangen E, Hanada T, Modjtahedi N, Penninger JM, Kroemer G, AIF: Not Just an Apoptosis-Inducing Factor, Annals of the New York Academy of Sciences, 1171 (2009) 2–11. [DOI] [PubMed] [Google Scholar]
- [94].Sperandio S, de Belle I, Bredesen DE, An alternative, nonapoptotic form of programmed cell death, Proc Natl Acad Sci U S A, 97 (2000) 14376–14381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, Bredesen DE, Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix, Cell Death Differ, 11 (2004) 1066–1075. [DOI] [PubMed] [Google Scholar]
- [96].Pehar M, O’Riordan KJ, Burns-Cusato M, Andrzejewski ME, Del Alcazar CG, Burger C, Scrable H, Puglielli L, Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death, Aging Cell, 9 (2010) 174–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang Y, Li X, Wang L, Ding P, Zhang Y, Han W, Ma D, An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression, J Cell Sci, 117 (2004) 1525–1532. [DOI] [PubMed] [Google Scholar]
- [98].Lee D, Kim IY, Saha S, Choi KS, Paraptosis in the anti-cancer arsenal of natural products, Pharmacology & Therapeutics, 162 (2016) 120–133. [DOI] [PubMed] [Google Scholar]
- [99].Mandula JK, Chang S, Mohamed E, Jimenez R, Sierra-Mondragon RA, Chang DC, Obermayer AN, Moran-Segura CM, Das S, Vazquez-Martinez JA, Prieto K, Chen A, Smalley KSM, Czerniecki B, Forsyth P, Koya RC, Ruffell B, Cubillos-Ruiz JR, Munn DH, Shaw TI, et al. , Ablation of the endoplasmic reticulum stress kinase PERK induces paraptosis and type I interferon to promote anti-tumor T cell responses, Cancer Cell, 0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kim E, Lee DM, Seo MJ, Lee HJ, Choi KS, Intracellular Ca2 + Imbalance Critically Contributes to Paraptosis, Front Cell Dev Biol, 8 (2020) 607844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Petrillo S, Chiabrando D, Genova T, Fiorito V, Ingoglia G, Vinchi F, Mussano F, Carossa S, Silengo L, Altruda F, Merlo GR, Munaron L, Tolosano E, Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis, Cell Death Differ, 25 (2018) 573–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Li H, Ericsson M, Rabasha B, Budnik B, Chan SH, Freinkman E, Lewis CA, Doench JG, Wagner BK, Garraway LA, Schreiber SL, 6-Phosphogluconate Dehydrogenase Links Cytosolic Carbohydrate Metabolism to Protein Secretion via Modulation of Glutathione Levels, Cell Chem Biol, 26 (2019) 1306–1314.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lima S, Milstien S, Spiegel S, Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking, J Biol Chem, 292 (2017) 3074–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chi S, Kitanaka C, Noguchi K, Mochizuki T, Nagashima Y, Shirouzu M, Fujita H, Yoshida M, Chen W, Asai A, Himeno M, Yokoyama S, Kuchino Y, Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells, Oncogene, 18 (1999) 2281–2290. [DOI] [PubMed] [Google Scholar]
- [105].Lewis WH, Pinocytosis by Malignant Cells, The American Journal of Cancer, 29 (1937) 666–679. [Google Scholar]
- [106].Bar-Sagi D, Feramisco JR, Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins, Science, 233 (1986) 1061–1068. [DOI] [PubMed] [Google Scholar]
- [107].Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar-Sagi D, Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells, Nature, 497 (2013) 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Jayashankar V, Edinger AL, Macropinocytosis confers resistance to therapies targeting cancer anabolism, Nat Commun, 11 (2020) 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ehrkamp A, Herrmann C, Stoll R, Heumann R, Ras and Rheb Signaling in Survival and Cell Death, Cancers, 5 (2013) 639–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bhanot H, Young AM, Overmeyer JH, Maltese WA, Induction of Nonapoptotic Cell Death by Activated Ras Requires Inverse Regulation of Rac1 and Arf6, Molecular Cancer Research, 8 (2010) 1358–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Overmeyer JH, Young AM, Bhanot H, Maltese WA, A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells, Mol Cancer, 10 (2011) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Robinson MW, Overmeyer JH, Young AM, Erhardt PW, Maltese WA, Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death, J Med Chem, 55 (2012) 1940–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Chen H, Miao Y, Bian A, Ye J, Wang J, Cong X, Jian S, Yi Z, Liang L, Sun Z, Yang F, Ding T, A novel small-molecule activator of unfolded protein response suppresses castration-resistant prostate cancer growth, Cancer Lett, 532 (2022) 215580. [DOI] [PubMed] [Google Scholar]
- [114].Wu J, Hu H, Ao M, Cui Z, Zhou X, Qin J, Guo Y, Chen J, Xue Y, Fang M, Design, synthesis, and biological evaluation of 5-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)-1H-Indole-2-Carbohydrazide derivatives: the methuosis inducer 12A as a Novel and selective anticancer agent, J Enzyme Inhib Med Chem, 36 (2021) 1436–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Fang Y, Zhong T, Yang L, Luo F, Li Q, Wang D, Li Q, Fan Y, Yang X, Spiropachysine A suppresses hepatocellular carcinoma proliferation by inducing methuosis in vitro and in vivo, Phytomedicine, 102 (2022) 154151. [DOI] [PubMed] [Google Scholar]
- [116].Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, Vanden Berghe T, Cappe B, Ladik M, Riquet FB, Heyndrickx L, Bridelance J, Vuylsteke M, Vandecasteele K, Vandenabeele P, Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death, Cell Death Dis, 11 (2020) 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kim B-E, Nevitt T, Thiele DJ, Mechanisms for copper acquisition, distribution and regulation, Nat Chem Biol, 4 (2008) 176–185. [DOI] [PubMed] [Google Scholar]
- [118].Kim B-E, Nevitt T, Thiele DJ, Mechanisms for copper acquisition, distribution and regulation, Nat Chem Biol, 4 (2008) 176–185. [DOI] [PubMed] [Google Scholar]
- [119].Tapiero H, Townsend DM, Tew KD, Trace elements in human physiology and pathology Copper, Biomed Pharmacother, 57 (2003) 386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Mercer JFB, The molecular basis of copper-transport diseases, Trends in Molecular Medicine, 7 (2001) 64–69. [DOI] [PubMed] [Google Scholar]
- [121].Bandmann O, Weiss KH, Kaler SG, Wilson’s disease and other neurological copper disorders, The Lancet Neurology, 14 (2015) 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, Bussolati O, Franchi-Gazzola R, Marchiò L, Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells, J Am Chem Soc, 133 (2011) 6235–6242. [DOI] [PubMed] [Google Scholar]
- [123].Hasinoff BB, Yadav AA, Patel D, Wu X, The cytotoxicity of the anticancer drug elesclomol is due to oxidative stress indirectly mediated through its complex with Cu(II), J Inorg Biochem, 137 (2014) 22–30. [DOI] [PubMed] [Google Scholar]
- [124].Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD, Eaton JK, Frenkel E, Kocak M, Corsello SM, Lutsenko S, Kanarek N, Santagata S, Golub TR, Copper induces cell death by targeting lipoylated TCA cycle proteins, Science, 375 (2022) 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, Kocak M, Kory N, Tsherniak A, Santagata S, Whitesell L, Ghobrial IM, Markley JL, Lindquist S, Golub TR, Mitochondrial metabolism promotes adaptation to proteotoxic stress, Nat Chem Biol, 15 (2019) 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Villalpando-Rodriguez GE, Gibson SB, Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat, Oxid Med Cell Longev, 2021 (2021) 9912436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Tan S, Sagara Y, Liu Y, Maher P, Schubert D, The regulation of reactive oxygen species production during programmed cell death, J Cell Biol, 141 (1998) 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Radogna F, Cerella C, Gaigneaux A, Christov C, Dicato M, Diederich M, Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma, Oncogene, 35 (2016) 3839–3853. [DOI] [PubMed] [Google Scholar]
- [129].Zhou B, Zhang J, Liu X, Chen H, Ai Y, Cheng K, Sun R, Zhou D, Han J, Wu Q, Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis, Cell Res, 28 (2018) 1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lee YJ, Kim N-Y, Suh Y-A, Lee C, Involvement of ROS in Curcumin-induced Autophagic Cell Death, Korean J Physiol Pharmacol, 15 (2011) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, Pennemann FL, Schnepf D, Wettmarshausen J, Braun M, Leung DW, Amarasinghe GK, Perocchi F, Staeheli P, Ryffel B, Pichlmair A, Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway, Nat Immunol, 19 (2018) 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Marconett CN, Morgenstern TJ, San Roman AK, Sundar SN, Singhal AK, Firestone GL, BZL101, a phytochemical extract from the Scutellaria barbata plant, disrupts proliferation of human breast and prostate cancer cells through distinct mechanisms dependent on the cancer cell phenotype, Cancer Biology & Therapy, 10 (2010) 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Zhang J, Gao R-F, Li J, Yu K, Bi K-X, Alloimperatorin activates apoptosis, ferroptosis, and oxeiptosis to inhibit the growth and invasion of breast cancer cells in vitro, Biochem Cell Biol, 100 (2022) 213–222. [DOI] [PubMed] [Google Scholar]
- [134].Wang C-X, Chen L-H, Zhuang H-B, Shi Z-S, Chen ZC, Pan J-P, Hong Z-S, Auriculasin enhances ROS generation to regulate colorectal cancer cell apoptosis, ferroptosis, oxeiptosis, invasion and colony formation, Biochem Biophys Res Commun, 587 (2022) 99–106. [DOI] [PubMed] [Google Scholar]
- [135].Kang P, Chen J, Zhang W, Guo N, Yi X, Cui T, Chen J, Yang Y, Wang Y, Du P, Ye Z, Li B, Li C, Li S, Oxeiptosis: a novel pathway of melanocytes death in response to oxidative stress in vitiligo, Cell Death Discov, 8 (2022) 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Lagadic-Gossmann D, Huc L, Lecureur V, Alterations of intracellular pH homeostasis in apoptosis: origins and roles, Cell Death Differ, 11 (2004) 953–961. [DOI] [PubMed] [Google Scholar]
- [137].Wang Y-Z, Wang J-J, Huang Y, Liu F, Zeng W-Z, Li Y, Xiong Z-G, Zhu MX, Xu T-L, Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction, ELife, 4 (2015) e05682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D, Wang P, Ma X, Kroemer G, Bartlett DL, Billiar TR, Lotze MT, Zeh HJ, Kang R, Tang D, JTC801 Induces pH-dependent Death Specifically in Cancer Cells and Slows Growth of Tumors in Mice, Gastroenterology, 154 (2018) 1480–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shinkai H, Ito T, Iida T, Kitao Y, Yamada H, Uchida I, 4-Aminoquinolines: Novel Nociceptin Antagonists with Analgesic Activity, J. Med. Chem, 43 (2000) 4667–4677. [DOI] [PubMed] [Google Scholar]
- [140].Yamada H, Nakamoto H, Suzuki Y, Ito T, Aisaka K, Pharmacological profiles of a novel opioid receptor-like1 (ORL(1)) receptor antagonist, JTC-801, Br J Pharmacol, 135 (2002) 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Zhao B, Hu T, JTC-801 inhibits the proliferation and metastasis of the Hep G2 hepatoblastoma cell line by regulating the phosphatidylinositol 3-kinase/protein kinase B signalling pathway, Oncol Lett, 17 (2019) 1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zheng C-J, Yang L-L, Liu J, Zhong L, JTC-801 exerts anti-proliferative effects in human osteosarcoma cells by inducing apoptosis, J Recept Signal Transduct Res, 38 (2018) 133–140. [DOI] [PubMed] [Google Scholar]
- [143].Zhu S, Liu J, Kang R, Yang M, Tang D, Targeting NF-κB–dependent alkaliptosis for the treatment of venetoclax-resistant acute myeloid leukemia cells, Biochemical and Biophysical Research Communications, 562 (2021) 55–61. [DOI] [PubMed] [Google Scholar]
- [144].Van Antwerp DJ, Martin SJ, Verma IM, Green DR, Inhibition of TNF-induced apoptosis by NF-κB, Trends in Cell Biology, 8 (1998) 107–111. [DOI] [PubMed] [Google Scholar]
- [145].Swietach P, Vaughan-Jones RD, Harris AL, Hulikova A, The chemistry, physiology and pathology of pH in cancer, Philosophical Transactions of the Royal Society B: Biological Sciences, 369 (2014) 20130099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Waller A, Experiments on the Section of the Glosso-Pharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres, Edinb Med Surg J, 76 (1851) 369–376. [PMC free article] [PubMed] [Google Scholar]
- [147].Summers DW, DiAntonio A, Milbrandt J, Mitochondrial Dysfunction Induces Sarm1-Dependent Cell Death in Sensory Neurons, J Neurosci, 34 (2014) 9338–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Summers DW, Gibson DA, DiAntonio A, Milbrandt J, SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation, Proc Natl Acad Sci U S A, 113 (2016) E6271–E6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, Tessier-Lavigne M, Pathological axonal death through a MAPK cascade that triggers a local energy deficit, Cell, 160 (2015) 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou Y-J, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, et al. , dSarm/Sarm1 is required for activation of an injury-induced axon death pathway, Science, 337 (2012) 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Chen Y-H, Sasaki Y, DiAntonio A, Milbrandt J, SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration, Exp Neurol, 339 (2021) 113636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Mukherjee P, Woods TA, Moore RA, Peterson KE, Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death, Immunity, 38 (2013) 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Reiser J, Altintas MM, Podocytes, F1000Res, 5 (2016) F1000 Faculty Rev-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H, Kotb AM, Endlich N, Anders H-J, Murine Double Minute-2 Prevents p53-Overactivation-Related Cell Death (Podoptosis) of Podocytes, J Am Soc Nephrol, 26 (2015) 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Mulay SR, Thomasova D, Ryu M, Kulkarni OP, Migliorini A, Bruns H, Gröbmayr R, Lazzeri E, Lasagni L, Liapis H, Romagnani P, Anders H-J, Podocyte loss involves MDM2-driven mitotic catastrophe, J Pathol, 230 (2013) 322–335. [DOI] [PubMed] [Google Scholar]
- [156].Liu G, Terzian T, Xiong S, Van Pelt CS, Audiffred A, Box NF, Lozano G, The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development, J Pathol, 213 (2007) 360–368. [DOI] [PubMed] [Google Scholar]
- [157].Wiggins RC, The spectrum of podocytopathies: a unifying view of glomerular diseases, Kidney Int, 71 (2007) 1205–1214. [DOI] [PubMed] [Google Scholar]
- [158].Yin L, Yu L, He JC, Chen A, Controversies in Podocyte Loss: Death or Detachment?, Front Cell Dev Biol, 9 (2021) 771931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Yang WS, Stockwell BR, Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells, Chemistry & Biology, 15 (2008) 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Ko P-J, Woodrow C, Dubreuil MM, Martin BR, Skouta R, Bassik MC, Dixon SJ, A ZDHHC5-GOLGA7 Protein Acyltransferase Complex Promotes Nonapoptotic Cell Death, Cell Chemical Biology, 26 (2019) 1716–1724.e9. [DOI] [PubMed] [Google Scholar]
- [161].Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR, Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death, ACS Chem. Biol, 10 (2015) 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, Skinner HD, Johnson RL, Ding S, Ajani JA, A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma, Molecular Cancer Therapeutics, 17 (2018) 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A, Neutrophil Extracellular Traps Kill Bacteria, Science, 303 (2004) 1532–1535. [DOI] [PubMed] [Google Scholar]
- [164].Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A, Novel cell death program leads to neutrophil extracellular traps, Journal of Cell Biology, 176 (2007) 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H, Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation, Nat Chem Biol, 7 (2011) 75–77. [DOI] [PubMed] [Google Scholar]
- [166].Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A, Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps, Journal of Cell Biology, 191 (2010) 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V, A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis, Cell Reports, 8 (2014) 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Douda DN, Khan MA, Grasemann H, Palaniyar N, SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx, Proceedings of the National Academy of Sciences, 112 (2015) 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S, Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1, Cell Host & Microbe, 12 (2012) 109–116. [DOI] [PubMed] [Google Scholar]
- [170].Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T, Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality, Cell Death Differ, 18 (2011) 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, DeVinney R, Doig CJ, Green FHY, Kubes P, Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood, Nat Med, 13 (2007) 463–469. [DOI] [PubMed] [Google Scholar]
- [172].Demers M, Wagner DD, NETosis: A New Factor in Tumor Progression and Cancer-Associated Thrombosis, Semin Thromb Hemost, 40 (2014) 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M, Laybutt DR, Meikle PJ, Biden TJ, A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking, Biochem J, 435 (2011) 267–276. [DOI] [PubMed] [Google Scholar]
- [174].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death, Nature, 434 (2005) 658–662. [DOI] [PubMed] [Google Scholar]
- [175].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y, Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death, Nature, 434 (2005) 652–658. [DOI] [PubMed] [Google Scholar]
- [176].Li Y, Johnson N, Capano M, Edwards M, Crompton M, Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis, Biochem J, 383 (2004) 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chipuk JE, Maurer U, Green DR, Schuler M, Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription, Cancer Cell, 4 (2003) 371–381. [DOI] [PubMed] [Google Scholar]
- [178].Guo L, Mitochondrial ATP synthase inhibitory factor 1 interacts with the p53-cyclophilin D complex and promotes opening of the permeability transition pore, J Biol Chem, 298 (2022) 101858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Dhingra R, Guberman M, Rabinovich-Nikitin I, Gerstein J, Margulets V, Gang H, Madden N, Thliveris J, Kirshenbaum LA, Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy, Cardiovasc Res, 116 (2020) 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Walsh CT, Zydowsky LD, McKeon FD, Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction, Journal of Biological Chemistry, 267 (1992) 13115–13118. [PubMed] [Google Scholar]
- [181].Giorgio V, Soriano ME, Basso E, Bisetto E, Lippe G, Forte MA, Bernardi P, Cyclophilin D in mitochondrial pathophysiology, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1797 (2010) 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS, A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion, Cell, 131 (2007) 966–979. [DOI] [PubMed] [Google Scholar]
- [183].Monks J, Rosner D, Jon Geske F, Lehman L, Hanson L, Neville MC, Fadok VA, Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release, Cell Death Differ, 12 (2005) 107–114. [DOI] [PubMed] [Google Scholar]
- [184].Fais S, Overholtzer M, Cell-in-cell phenomena in cancer, Nat Rev Cancer, 18 (2018) 758–766. [DOI] [PubMed] [Google Scholar]
- [185].Sun Q, Cibas ES, Huang H, Hodgson L, Overholtzer M, Induction of entosis by epithelial cadherin expression, Cell Res, 24 (2014) 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Chen R, Ram A, Albeck JG, Overholtzer M, Entosis is induced by ultraviolet radiation, IScience, 24 (2021) 102902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Liang J, Niu Z, Zhang B, Yu X, Zheng Y, Wang C, Ren H, Wang M, Ruan B, Qin H, Zhang X, Gu S, Sai X, Tai Y, Gao L, Ma L, Chen Z, Huang H, Wang X, Sun Q, p53-dependent elimination of aneuploid mitotic offspring by entosis, Cell Death Differ, 28 (2021) 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Hamann JC, Surcel A, Chen R, Teragawa C, Albeck JG, Robinson DN, Overholtzer M, Entosis Is Induced by Glucose Starvation, Cell Reports, 20 (2017) 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Purvanov V, Holst M, Khan J, Baarlink C, Grosse R, G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion, ELife, 3 (2014) e02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Bozkurt E, Düssmann H, Salvucci M, Cavanagh BL, Van Schaeybroeck S, Longley DB, Martin SJ, Prehn JHM, TRAIL signaling promotes entosis in colorectal cancer, J Cell Biol, 220 (2021) e202010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Boya P, Kroemer G, Lysosomal membrane permeabilization in cell death, Oncogene, 27 (2008) 6434–6451. [DOI] [PubMed] [Google Scholar]
- [192].Yuan X, Nie W, He Z, Yang J, Shao B, Ma X, Zhang X, Bi Z, Sun L, Liang X, Tie Y, Liu Y, Mo F, Xie D, Wei Y, Wei X, Carbon black nanoparticles induce cell necrosis through lysosomal membrane permeabilization and cause subsequent inflammatory response, Theranostics, 10 (2020) 4589–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Kågedal K, Zhao M, Svensson I, Brunk UT, Sphingosine-induced apoptosis is dependent on lysosomal proteases, Biochemical Journal, 359 (2001) 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, Xiao G, Li S, Fan L, Billiar TR, Wilson MA, Fan J, Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis, Cell Death Differ, 21 (2014) 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, Dai E, Ferroptosis is a lysosomal cell death process, Biochemical and Biophysical Research Communications, 503 (2018) 1550–1556. [DOI] [PubMed] [Google Scholar]
- [196].Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, Yamada K, An essential role for functional lysosomes in ferroptosis of cancer cells, Biochemical Journal, 473 (2016) 769–777. [DOI] [PubMed] [Google Scholar]
- [197].Werneburg N, Guicciardi ME, Yin X-M, Gores GJ, TNF-α-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent, American Journal of Physiology-Gastrointestinal and Liver Physiology, 287 (2004) G436–G443. [DOI] [PubMed] [Google Scholar]
- [198].Clarke PGH, Developmental cell death: morphological diversity and multiple mechanisms, Anat Embryol, 181 (1990) 195–213. [DOI] [PubMed] [Google Scholar]
- [199].Franko J, Pomfy M, Prosbová T, Apoptosis and cell death (mechanisms, pharmacology and promise for the future), Acta Medica (Hradec Kralove), 43 (2000) 63–68. [PubMed] [Google Scholar]
- [200].Boya P, Andreau K, Poncet D, Zamzami N, Perfettini J-L, Metivier D, Ojcius DM, Jäättelä M, Kroemer G, Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion, J Exp Med, 197 (2003) 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Sargeant TJ, Lloyd-Lewis B, Resemann HK, Ramos-Montoya A, Skepper J, Watson CJ, Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization, Nat Cell Biol, 16 (2014) 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Villamil Giraldo AM, Appelqvist H, Ederth T, Öllinger K, Lysosomotropic agents: impact on lysosomal membrane permeabilization and cell death, Biochem Soc Trans, 42 (2014) 1460–1464. [DOI] [PubMed] [Google Scholar]
- [203].Luke CJ, Markovina S, Good M, Wight IE, Thomas BJ, Linneman JM, Lanik WE, Koroleva O, Coffman MR, Miedel MT, Gong Q, Andress A, Campos Guerrero M, Wang S, Chen L, Beatty WL, Hausmann KN, White FV, Fitzpatrick JAJ, Orvedahl A, et al. , Lysoptosis is an evolutionarily conserved cell death pathway moderated by intracellular serpins, Commun Biol, 5 (2022) 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Kroemer G, Levine B, Autophagic cell death: the story of a misnomer, Nat Rev Mol Cell Biol, 9 (2008) 1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, et al. , Identification of a candidate therapeutic autophagy-inducing peptide, Nature, 494 (2013) 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Liu Y, Shoji-Kawata S, Sumpter RM, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PGH, Puyal J, Levine B, Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia, Proc Natl Acad Sci U S A, 110 (2013) 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA, Cardiac autophagy is a maladaptive response to hemodynamic stress, J Clin Invest, 117 (2007) 1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J, Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion, Circulation Research, 100 (2007) 914–922. [DOI] [PubMed] [Google Scholar]
- [209].Nah J, Zablocki D, Sadoshima J, Autosis: A New Target to Prevent Cell Death, JACC: Basic to Translational Science, 5 (2020) 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Akimoto M, Iizuka M, Kanematsu R, Yoshida M, Takenaga K, Anticancer Effect of Ginger Extract against Pancreatic Cancer Cells Mainly through Reactive Oxygen Species-Mediated Autotic Cell Death, PLOS ONE, 10 (2015) e0126605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Pyrczak-Felczykowska A, Reekie TA, Jąkalski M, Hać A, Malinowska M, Pawlik A, Ryś K, Guzow-Krzemińska B, Herman-Antosiewicz A, The Isoxazole Derivative of Usnic Acid Induces an ER Stress Response in Breast Cancer Cells That Leads to Paraptosis-like Cell Death, Int J Mol Sci, 23 (2022) 1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Wang W-B, Feng L-X, Yue Q-X, Wu W-Y, Guan S-H, Jiang B-H, Yang M, Liu X, Guo D-A, Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90, J Cell Physiol, 227 (2012) 2196–2206. [DOI] [PubMed] [Google Scholar]
- [213].Maltese WA, Overmeyer JH, Methuosis: nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments, Am J Pathol, 184 (2014) 1630–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Gandin V, Pellei M, Tisato F, Porchia M, Santini C, Marzano C, A novel copper complex induces paraptosis in colon cancer cells via the activation of ER stress signalling, J Cell Mol Med, 16 (2012) 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Forcina GC, Conlon M, Wells A, Cao JY, Dixon SJ, Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells, Cell Syst, 4 (2017) 600–610.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, Wells A, Kahanu A, Magtanong L, Watts JL, Pratt DA, Dixon SJ, A compendium of kinetic modulatory profiles identifies ferroptosis regulators, Nat Chem Biol, 17 (2021) 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Richards R, Schwartz HR, Honeywell ME, Stewart MS, Cruz-Gordillo P, Joyce AJ, Landry BD, Lee MJ, Drug antagonism and single-agent dominance result from differences in death kinetics, Nat Chem Biol, 16 (2020) 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Shimada K, Hayano M, Pagano NC, Stockwell BR, Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity, Cell Chem Biol, 23 (2016) 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, Lamkanfi M, Krönke M, Pasparakis M, Kashkar H, Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis, Nature, 575 (2019) 683–687. [DOI] [PubMed] [Google Scholar]
- [220].Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D, AIFM2 blocks ferroptosis independent of ubiquinol metabolism, Biochemical and Biophysical Research Communications, 523 (2020) 966–971. [DOI] [PubMed] [Google Scholar]
- [221].Van Coillie S, Van San E, Goetschalckx I, Wiernicki B, Mukhopadhyay B, Tonnus W, Choi SM, Roelandt R, Dumitrascu C, Lamberts L, Dams G, Weyts W, Huysentruyt J, Hassannia B, Ingold I, Lele S, Meyer E, Berg M, Seurinck R, Saeys Y, et al. , Targeting ferroptosis protects against experimental (multi)organ dysfunction and death, Nat Commun, 13 (2022) 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Armenta DA, Dixon SJ, Investigating Nonapoptotic Cell Death Using Chemical Biology Approaches, Cell Chemical Biology, 27 (2020) 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Jamison JM, Gilloteaux J, Taper HS, Calderon PB, Summers JL, Autoschizis: a novel cell death, Biochemical Pharmacology, 63 (2002) 1773–1783. [DOI] [PubMed] [Google Scholar]
- [224].Gilloteaux J, Jamison JM, Arnold D, Ervin E, Eckroat L, Docherty JJ, Neal D, Summers JL, Cancer cell necrosis by autoschizis: synergism of antitumor activity of vitamin C: vitamin K3 on human bladder carcinoma T24 cells, Scanning, 20 (1998) 564–575. [DOI] [PubMed] [Google Scholar]