

Abstract

Serine/threonine-protein kinase B-Raf (BRAF; RAF = rapidly accelerated fibrosarcoma) plays an important role in the mitogen-activated protein kinase (MAPK) signaling cascade. Somatic mutations in the BRAF gene were first discovered in 2002 by Davies et al., which was a major breakthrough in cancer research. Subsequently, three different classes of BRAF mutants have been discovered. This class includes class I monomeric mutants (BRAFV600), class II BRAF homodimer mutants (non-V600), and class III BRAF heterodimers (non-V600). Cancers caused by these include melanoma, thyroid cancer, ovarian cancer, colorectal cancer, nonsmall cell lung cancer, and others. In this study, we have highlighted the major binding pockets in BRAF protein, their active and inactive conformations with inhibitors, and BRAF dimerization and its importance in paradoxical activation and BRAF mutation. We have discussed the first-, second-, and third-generation drugs approved by the Food and Drug Administration and drugs under clinical trials with all four different binding approaches with DFG-IN/OUT and αC-IN/OUT for BRAF protein. We have investigated particular aspects and difficulties with all three generations of inhibitors. Finally, this study has also covered recent developments in synthetic BRAF inhibitors (from their discovery in 2002 to 2022), their unique properties, and importance in inhibiting BRAF mutants.
1. Introduction
The rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/mitogen extracellular kinase (MEK)/extracellular signal-regulated kinase (ERK) or mitogen-activated protein kinase (MAPK) signaling pathway is a crucial intracellular signaling pathway,1 responsible for intracellular signal transduction, namely, acute hormone responses, embryogenesis, cell differentiation, and apoptosis involved in the control of cell growth, proliferation, migration, and survival.2 By acting downstream of receptor tyrosine kinases, the MAPK pathway regulates cell fate decisions. Under physiological conditions, MAPK activation occurs via extracellular binding of growth factors to receptor tyrosine kinases,3 at the cell surface, leading to phosphorylation and activation of RAS proteins by guanosine-5′-triphosphate (GTP)-bound.4 RAF, a downstream effector of RAS, is a serine-threonine-specific protein kinase that stimulates MEK, which in turn activates ERK.5 Activation of ERK results in a growth-promoting and transforming signal (Figure 1).6,7
Figure 1.

BRAF signaling pathway. (green arrow) Normal BRAF pathway. (red arrow) Oncogenic BRAF pathway.
The RAF kinase family includes three members of the MAPK pathway: ARAF, BRAF, and CRAF. BRAF is the major activating kinase,8 and its activation occurs by binding to its RAS-binding domain.9 It encodes a cytoplasmic serine/threonine kinase and is a major regulator of the RAS pathway (RAS → RAF → MEK → ERK → MAP kinase).10,11 BRAF is essential for regulation of a variety of cellular processes, including growth, proliferation, survival, and migration.12,13
The discovery of BRAF mutations was a major breakthrough in cancer research. Somatic mutations in BRAF were first discovered in 2002 by Davies et al.14 BRAF is among the most frequently mutated oncogenes in human cancers.15 One of the most studied pathways, with over 90% mutations, is the RAS > RAF > MEK > ERK (MAPK) pathway.16 BRAF mutations occur to varying degrees in human cancers, including approximately 70–90% in melanoma, 5–20% in colon cancer,17,18 30–50% in thyroid cancer,19 5–30% in ovarian cancer,20 1–4% in nonsmall cell lung cancer, and 1–3% in hairy cell leukemia and Langerhans cell histiocytosis (Figure 2).21
Figure 2.

Global cancer statistics for new cases and deaths for 2020 and BRAFV600E (%) mutation in different cancer types.
All BRAF mutations are located in the kinase domain, either in the glycine-rich loop encoded by exon 11 or in the activation sequence encoded by exon 15.14,22 The BRAF gene has a variety of activating mutations that have been discovered and classified into three classes. The monomeric BRAFV600 codon mutation is classified as class I. In the RAS/RAF/MEK/ERK pathway, this mutation occurs in an RAS-independent manner.23 The BRAFV600 gene mutation BRAFV600E is the most common mutation 90%24 at position 600 with a single point mutation (Val600 → Glu) in which the polar, hydrophilic glutamic acid replaces the hydrophobic valine.25 This breaks the hydrophobic link between the A-loop and the P-loop, resulting in an active conformation.26 The constitutive kinase activity of BRAFV600E is 500 times higher than that of wild-type BRAF kinase.27 Another type of mutation is BRAFV600WT, with BRAFV600K (10%) being the second most common mutation, where valine is replaced by lysine.28 Rarer codon BRAFV600 mutations are BRAFV600R, BRAFV600D (6–3%), and BRAFV600E2,V600M,V600G (<1%).29 The class II includes non-V600 mutants and is regulated by mutant BRAF homodimers without involvement of RAS. Here, L597 and K601 were found in the activation segment, whereas G464 and G469 were found in the glycine-rich region of BRAF. Class III involves non-V600 mutants and is regulated by mutant BRAF heterodimers involving the RAS mutant. In this, D287, V459L, G466 V, S467, G469, N581, D594, F555, and G596 were detected. In BRAF gene mutations, the classes II and III are very rare codon mutations.23,30
Statistics of new cases and deaths in all cancers worldwide for 2020 and details of specific cancers affected by the BRAFV600E mutation are shown in (Figure 2).
2. Current Insight BRAF Protein
2.1. BRAF’s Skeletal System and Binding Pocket
The BRAF protein consists of 766 amino acid sequences.31,32 It has two lobes, the N-terminal and the C-terminal lobes. These two lobes consist of three conserved regions (CRs): CR1 (R150–290), CR2 (R360–375), and CR3 (R457–717). CR1 and CR2 are regulatory domains.33 CR1 contains the RAS-binding domain (RBD) (R155–227) and the cysteine-rich domain (CRD) (R234–280).34,35 CR1 interacts with both RAS and membrane phospholipids. CR1 acts as an autoinhibitor of the BRAF kinase domain (CR3) and ensures that signal transduction is controlled. CR2 is a serine/threonine-rich domain that serves as a flexible hinge between CR1 and CR3 and contains a binding site for the 14–3–3 protein.35,36 CR3 is the catalytic protein kinase domain located near the C-terminus [Figure 3a]. The N-terminus of CR3 is involved in adenosine triphosphate (ATP) binding. It primarily comprises an antiparallel β-sheet structure, anchors, and a glycine-rich ATP-phosphate binding loop (P-loop) (R464–471) and an αC-helix (R492–504). Through the activation segment, the P-loop region is critical for stabilizing ATP binding and maintaining an inactive BRAF conformation. The C-terminal of CR3 contains a hinge region/adenine region (R530–535), catalytic loop (R574–581), DFG motif [Asp-Phe-Gly(R594–595–596)], and an activation section (R594–623). In the CR3 domain, the ATP-phosphate binding loop, DFG motif, and activation segment are the major active binding sites.37−39 The ATP pocket has several binding regions that interact with inhibitors. These are an adenine region (similar to the hinge region), a sugar region (ATP ribose pocket), type I and II hydrophobic regions, and a solvent-accessible region that is also part of the ATP region but is not occupied. At the C-terminus, the dimerization interface (DIF) (R504–511) is another binding site for the 14–3–3 protein.36 The end of CR3 facilitates the binding of substrate proteins. This occurs through its catalytic loop, which facilitates the transfer of phosphate from ATP to BRAF substrates [Figure 3b,c].32,40 The gatekeeper (catalytic cleft between the N- and C-termini) plays an important role by controlling the access of ligands to a hydrophobic pocket deep in the active site that is not in contact with ATP, which affects the binding and specificity of inhibitors.41
Figure 3.

BRAF protein structure and binding pocket. (a) Regulatory (CR1 & CR2) and kinase (CR3) domain between N and C-terminal in BRAF kinase protein. (b) BRAF protomer (PDB ID:6P7G) with different orientations. (c) BRAF dimer (PDB ID:2FB8). Different binding pockets (p-loop, αC-helix, DIF, DFG, catalytic loop, activation segment, and hinge region) are labeled in specific colors.
2.2. Importance of BRAF Dimerization and Its Challenges
Recent studies have shown that Raf dimerization is required for normal Ras-dependent activation of Raf kinase in various cellular activities.42 The active conformation of BRAF is facilitated by dimerization.43 It was discovered that dimerization of BRAF kinases involves insights into a core cluster of residues known as the dimer interface (DIF) [(R504–511) in BRAF], which plays a critical role in dimer formation.8 DIF is located at the tail end of the αC-helix in the BRAF kinase protein. Many of the ATP-competitive inhibitors promote BRAF dimerization in an RAS-dependent manner. BRAF dimerization can be homo- or heterodimerized.44 The 14–3–3 domain protein in DIF is responsible for BRAF dimerization. The two-protein structure of BRAF–BRAF in the 14–3–3 complex in DIF is responsible for homodimer, and BRAF-MEK1–14–3–3 complex is heterodimer. Two BRAF serine sites pS729 and Ps365 open monomers in the 14–3–3 protein complex, promote formation of active BRAF dimers, and link them.36 Dimerization is enhanced by Ras and is subject to negative feedback regulation by ERK.45
The structure of an active BRAF kinase in dimerization is that of a side-by-side dimer, in which only one partner needs to be catalytically active.45 Drugs that inhibit BRAF kinase can bind to a protomer of the dimer complex (homodimers and heterodimers of BRAF) and inhibit its ATP-binding site; however, this binding enhances the activity of the protomer (in the absence of drug), leading to the induction of an active conformation.46 Although recent BRAF dimer structures have shown that the 14–3–3 dimer can bind to two kinase domain protomers simultaneously, it is unclear whether both protomers are catalytically active.47
The RBD:14–3–3 interface has a dual function, contributing first to RAF autoinhibition and second to the full spectrum of RAS-RBD interactions.47 There are two types of autoinhibitory mechanisms that must be overcome in BRAF activation. The first is 14–3–3-assisted locking of the N- and C-terminal portions, and the second is destabilization of the inactive conformation of the kinase domain itself.46 BRAF dimerization is also associated with multiple phosphorylation events. RAF dimers may have an allosteric function in facilitating activation loop autophosphorylation. The autophosphorylation of the P-loop of RAF is inextricably linked to the catalytic activity of RAF.48 The dimer-promoting potential of BRAF inhibitors may be associated with some “paradoxical” activation. Paradoxical triggers may enhance concerted movements of the αC-helix and A-loop regions.49 As mentioned earlier, the class II mutation is responsible for the homodimer, and the class III mutation is responsible for the heterodimer. Rajakulendran et al. suggested that BRAF mutations G450W, F488A, M489W, and Y538F at the dimerization interface inhibit dimer formation.46 BRAF dimerization contributes to the pathogenic effect of disease-associated mutant Raf proteins and, when activated, exhibits similar activity to the constitutively active V600E mutant. It has also been shown to influence treatment response and disease progression in individuals receiving BRAF inhibitors.50 Raf dimerization and its role in normal and oncogenic states in cell signaling is shown in Figure 4.
Figure 4.

Dimerization of Raf in cell signaling. Note: (A) In the normal RAS-dependent signaling pathway, Raf dimerization is required for Raf kinase activation and signaling to MEK for further cell proliferation and regulation. (B) In oncogenic states, it is required for MEK/ERK signaling, which is upregulated by Raf dimerization. These include: (1) mutant c-Raf protein with b-Raf in dimerization impaired kinase activity from normal to oncogenic, (2) RTKs and RasGTPases induced by mutation, (3) in the context of active Ras, treatment with ATP-competitive Raf inhibitors, and (4) Raf inhibitor resistance is mediated by self-homodimerizing BRAFV600E splice variants.
2.3. Concept of Inactive and Active Conformation of BRAF
BRAF protein switches from inactive to active conformation and is also important for kinase activation. Several regulatory elements play key roles in this process. The inactive conformation of BRAF is characterized by the position DFG-OUT/αC-OUT and is also known as the closed conformation. The active conformation is characterized by the position DFG-IN/αC-IN and is also referred to as the open conformation.51−53 In BRAF protein kinases, the conformation of the conserved DFG motif containing a catalytic [Asp-Phe-Gly (594–595–596)] residue is known to be critical for selective binding of many kinase inhibitors.54
In the DFG-OUT conformation, BRAF shows a reverse orientation. The DFG residue [Asp (594)] oscillates and is displaced from the active site, occupying part of the ATP-binding site. The Mg2+ of the DFG motif is not bound to ATP, facilitating ATP c-phosphate transfer as the catalytic cleft of BRAF becomes inaccessible to ATP. The glycine-rich loop and activation segment are close to each other due to hydrophobic interactions.55,56 The DFG-OUT conformation has created an ATP-binding site and an adjacent hydrophobic pocket. In this conformation, the DFG residue moves from the ATP-binding site to the hydrophobic pocket, which is known as allosteric movement extending outward from the ATP-binding site. In the DFG-OUT conformation, the inhibitors bind to the allosteric site (hydrophobic pocket, near the ATP-binding pocket).54
In the DFG-IN conformation, the DFG residue [Asp(594)] is in the active site, and the catalytic cleft of BRAF becomes accessible to ATP, so that the Mg2+ of the DFG motif is bound to and occupies part of the ATP-binding site. Disruption of the hydrophobic interaction occurs between the glycine-rich loop and the activation segment (due to phosphorylation in the activation segment).55,56 The DFG-IN conformation has no allosteric movement and no allosteric binding site. In this conformation, inhibitors or agonists form interactions with the hinge region by forming ∼1–3 hydrogen bonds and the adenine region (ATP binding site) by hydrophobic interaction and also interact with other parts of the ATP binding site.54
The αC-helix near the ATP-binding site is called the αC-IN position. In this catalytic position, the Glu501 residue of the αC-helix causes the formation of a salt bridge with the conserved K483 residue. The αC-helix away from the ATP site is referred to as the αC-OUT position.57,58
3. Current Insight into BRAF Inhibitors
BRAF binding modes are classified into four basic categories based on the conformation of the binding pocket of the DFG motif and the αC-helix.59 These conformations classify inhibitors as follows and are shown in (Figure 5).
Figure 5.

On the basis of DFG (purple color) and αC (cyan color) movement, four different binding conformations are shown. (a) PDB ID: 4MNF (GDC-0879); type I inhibitors (αC-IN/DFG-IN) (b) PDB ID: 5HI2 (Sorafenib); type II inhibitors (αC-IN/DFG-OUT) (c) PDB ID: 4RZV (Vemurafenib); type I1/2 or type III (αC-OUT/DFG-IN) (d) PDB ID: 5CSX (BI 882370); typeI/II or type IV (αC-OUT/DFG-OUT).
1. Type I inhibitors (αC-IN/DFG-IN) 2. Type II inhibitors (αC-IN/DFG-OUT) 3. Type I1/2 or Type III (αC-OUT/DFG-IN) 4. TypeI/II or Type IV (αC-OUT/DFG-OUT).60−63
3.1. First-Generation BRAF Inhibitors: (αC-IN)
Originally, this class was designed to target the RAS (CRAF) mutant in the MAPK pathway before the BRAF mutation was discovered.58,64 The first-generation inhibitors (Figure 6) are designed as ATP-competitive small molecules with αC-IN conformation. They are BRAF monomer inhibitors and bind in dimers to the active site of a protomer within an RAF. They are not selective enough to block mutant RAF dimers.65,66
Figure 6.

First-generation BRAF inhibitors.
Sorafenib (formally known as BAY43–9006) is a “multitargeted” RAF kinase inhibitor that inhibits both BRAF and CRAF. It is a nonselective RAF kinase inhibitor. It was the first Food and Drug Administration (FDA)-approved oral drug in 2005, targeting monomeric BRAF tumors (wild-type and V600E mutant) but ineffective against dimeric mutant.67−71 In addition to sorafenib, many small molecules have been developed as BRAF inhibitors, but most are still in the preclinical phase. L-779,450 is a selective BRAF kinase inhibitor, but it lacks relative therapeutic efficacy due to poor bioavailability.72−75 GW-5074 (GlaxoSmithKline (GSK)) is an RAF inhibitor with stronger (10-fold) inhibitory activity on RAF1 than on BRAF. A phase I study demonstrated the safety and antitumor efficacy of GW-5074 (MG–005) in combination with sorafenib, providing a unique method of antitumor activity that targets cancer cell necroptosis caused by mitochondrial dysfunction and differing from conventional Raf inhibitor therapies.76−80 Clinical studies have shown that GDC-0879 is a potent and selective BRAFV600E inhibitor in melanoma and colorectal cancer, inhibiting the RAF/MEK/ERK pathway.81−83 ZM-336372 is a pan-RAF inhibitor that was the first BRAF inhibitor investigated but simultaneously hyperactivates and inhibits CRAF, resulting in paradoxical CRAF activity without pathway activation.78,84−86 SB-590885 is a triarylimidazole moiety that has greater potency and activity for BRAFV600E mutant than for CRAF in RAF kinases.87−89
3.2. Second-Generation BRAF Inhibitors: (αC-OUT)
Following the identification of BRAF mutations in 2002, second-generation drugs were found by screening inhibitors for the BRAFV600E48 mutant. The second generation (Figure 7) has an αC-out binding conformation with specific selectivity for the tumor driven by the BRAF monomer to target the BRAFV600E mutant with a broad therapeutic window. As monotherapies or in combination with other targeted treatments, these inhibitors significantly improve clinical outcomes for patients with BRAFV600 mutation-driven melanoma and some solid malignancies.53,75,90
Figure 7.

Second-generation BRAF inhibitors.
Dabrafenib is an ATP-competitive inhibitor of BRAF kinases was first developed under the name GSK2118436 by GlaxoSmithKline (GSK). It suppresses BRAFV600E more effectively than BRAFV600WT. The Biopharmaceutics Categorization System (BCS) classifies dabrafenib as a class II.91−95 Vemurafenib is the first drug approved by the FDA in 2011 for the treatment of patients with advanced BRAF exon 15 V600E mutation metastatic melanoma.96−99 In June 2018, the FDA approved encorafenib (LGX-818), a pyrazolo-pyrimidine-based phenylsulfonamide derivative, for the treatment of BRAFV600E/K-mutated cancer. Encorafenib, a selective inhibitor of BRAF kinase, is being paired with binimetinib, an MEK inhibitor, to treat metastatic and advanced malignant melanoma. Compared with other second-generation BRAF inhibitors, encorafenib has a longer duration of action.100−103 BI882370 inhibited the growth of human BRAF-mutated melanoma cells with 100-fold greater efficacy (1–10 nmol/L) than vemurafenib. BI-882370 is a potent and selective inhibitor of both the BRAFV600E oncogenic mutant and BRAF wild-type.104−106 XL-281 is an orally bioavailable RAF inhibitor that is generally well-tolerated. XL-281 has a lower rate of keratoacanthoma and squamous cell carcinoma (4%) than BRAF inhibitors such as dabrafenib (6–10%) and vemurafenib (18–26%).107−109
3.3. Third-Generation BRAF Inhibitors: (αC-IN with Allosteric Binding Site)
Third-generation drugs mainly have the αC-IN/DFG-OUT conformation (Figure 8). They were developed to overcome the problems with BRAF dimerization and paradoxical activation. These inhibitors with αC-IN conformation have specific selectivity for tumors driven by BRAF monomers and RAS-dependent BRAF dimers. They have target specificity for BRAF monomers and RAF dimers with a narrow therapeutic window. These inhibitors with αC-OUT conformation have specific selectivity for tumor-driven BRAF monomers and RAS-independent BRAF dimers. They have target specificity for BRAF homodimers and RAF monomers with a broad therapeutic window. The DFG-OUT conformation exposes an additional hydrophobic binding site directly adjacent to the ATP-binding site, commonly referred to as the “allosteric site.” Some third-generation drugs are pan-RAF inhibitors that inhibit paradoxical activation and are also not activated during RAF dimerization.64,66,110,111
Figure 8.

Third-generation BRAF inhibitors.
AZ-628 is more effective against the BRAFV600E mutation. AZ-628 compared with sorafenib has shown excellent preclinical results in terms of catalytic inhibition of BRAF protomers and dimers.112−115 MLN-2480 has a wider cerebral distribution. MLN-2480 is an oral, experimental phase I BRAFV600E inhibitor with a delayed inactivation rate. The allosteric kinase inhibitor MLN-2480 is used to treat melanoma, colorectal, lung, and pancreatic cancer.116−119 CCT-241161 and CCT-196969 are equally effective against BRAFV600E, CRAF, and SFKs (Src family kinase). The development of BRAFV600E skin tumors was inhibited by CCT-241161 and CCT-196969. The efficacy of CCT-241161 and CCT-196969 was observed in PDXs derived from patients exhibiting innate resistance to vemurafenib.78,120−122 INU-152 only partially activates the paradoxical pathway in melanoma cells with mutated RAS.120,123 LY-3009120 inhibits the isoforms of ARAF, BRAF, and CRAF with comparable affinity. LY-3009120 stimulates dimerization of BRAF-CRAF and causes paradoxical ERK activation.120,124−126 CEP-32496 is an orally bioavailable potent BRAF inhibitor. It is also known as agerafenib or RXDX-105. CEP-32492 shows high affinity against BRAFV600E. Agerafenib (CEP-32496) inhibited activation of the ERK, MAPK pathway in neuroblastoma cells.127−131
BAL–3833 (CCT-3833) works well in in KRAS-mutated tumors because RAF and SRC are important junctions in them. It inhibits both the monomeric and dimeric forms of BRAF.78,112,132,133 LSN-307453, which inhibits dimerization of all RAF isoforms, is currently in preclinical testing. LSN-3074753 is more active against all ten mutations than BRAFV600E.120,134−136 BGB-283 (lifirafenib) inhibits the BRAF dimer with potent reversible inhibition of all RAFs as well as EGFR.120,137,138 RAF-709 inhibits monomers and dimers alike.120,139,140 PLX-8394 inhibits paradoxical formation of RAF dimers and is more potent than vemurafenib. It is a BRAF inhibitor that can be taken orally and does not lead to paradoxical activation of MAPK.141−144 Compared to vemurafenib, PLX-7904 inhibits MAPK signaling for a longer period of time, resulting in greater blockade of proliferation and lower viability. In BRAFV600E melanoma cells, PLX-7904 was found to effectively suppress RAF signaling, whereas it had no paradoxical effects on wild-type cells.120,145−147 RAF-265 is a dual inhibitor of BRAF and VEGFR2, preventing both osteoclast development and resorption. RAF-265 shows synergistic antitumor activity with ZSTK-474 in medullary thyroid cancer.117,147−149 BGB-659 is able to inhibit class I and class II BRAF mutations because it can bind both monomeric BRAFs and both protomers of an RAF dimer. BGB659 showed higher activity against BRAFWT kinase.117,136,150,151
3.4. Challenges with BRAF Inhibitors
3.4.1. Challenges with First- and Second-Generation BRAF Inhibitors
First-generation BRAF inhibitors (αC-IN) have a lack of selectivity. They are ineffective against malignancies caused by II and III BRAF mutants due to homo/heterodimerization, leading to a paradoxical outcome.66,152
Second-generation αC-OUT BRAF inhibitors are extremely selective for cancers that rely on monomeric BRAF species, resulting in a broad treatment window. Resistance to first- and second-generation BRAF kinase inhibitors is mediated by BRAF dimerization. However, they have negative allostery and paradoxical activation and are ineffective in cancers based on dimer BRAF mutations. Second-generation BRAF inhibitors (αC-OUT/DFG-IN) exhibit paradoxical activation and allosteric transactivation of BRAF dimers. As a result of treatment with these inhibitors, healthy (BRAFWT) cells may be stimulated, leading to secondary malignancies. These drugs are highly effective in BRAFV600E tumors; however, non-V600 mutated malignancies are resistant.153
Secondary skin lesions such as hyperkeratosis, keratoacanthomas, and squamous cell carcinomas may occur in patients on BRAF inhibitors. Secondary melanomas, gastric and colonic polyps, and recurrences of previous cancers have also been observed in patients on BRAF inhibitors.154 Combinations of BRAF inhibitors and MEK inhibitors increase response rates and prolong progression-free survival (PFS) and overall survival (OS) in patients with BRAFV600 mutated metastatic melanoma. Although long-term effects have been documented, many patients develop acquired resistance to these drugs.153
3.4.2. Challenges with Third Generation BRAF Inhibitors
The ability of inhibitors to efficiently and persistently target both protomers is critical for effective inhibition of such dimers. The goal is to develop third-generation BRAF inhibitors that stabilize BRAF in the αC-IN conformation that can bind to both protomers in dimeric structures while inhibiting cellular CRAF inhibits both monomeric and dimeric forms of RAFs or does not allow dimer formation. The ability of both αC-OUT and αC-IN inhibitors to promote trans-activating dimerization of RAFs, as well as their general mechanism of inhibition, results in a paradoxical stimulation of ERK signaling. αC-IN inhibitors are more potent than αC-OUT inhibitors in suppressing dimeric RAF activity. αC-OUT and αC-IN inhibitors both stimulate dimerization; however, αC-IN inhibitors often do so more strongly because they greatly increase RAF binding to the active RAS. αC-IN inhibitors promote paradoxical activation more strongly than αC-OUT inhibitors.39,66
Paradoxical Activation by BRAF Inhibitors
Oncogenic BRAF dimers show resistance to BRAF inhibitors and cause paradoxical activation. Paradoxical activation in dimerizing cells by BRAF inhibitors can be explained by two different mechanistic explanations (Figure 9). The first hypothesis states that BRAF is autoinhibited. Another proposed mechanism involves BRAF and CRAF conformational changes induced by physical binding of the RAF inhibitor that promote dimer formation between an uninhibited CRAF protomer and BRAF or CRAF bound to the inhibitor. The phenomenon of paradoxical activation is mostly unknown and hypothetical.49,154
Figure 9.

Paradoxical activation. Note: Mechanism of autoinhibition: (1) In this case, inhibition of BRAF in the presence of a mutant or growth factor-activated RAS leads to abrogation of BRAF autoinhibition, so that it homodimerizes with BRAF and becomes hyperactivated. Conformational changes: (2, 3) At low doses, the drug binds only one RAF protomer and causes the other to transactivate. (4) At high doses, it binds to and inhibits both RAF dimers, effectively knocking down the signaling complex.
4. Recent Advancements
Following the discovery of BRAF mutations in 2002 and their importance in various cancers, many academic scientists/researchers started to work on it to solve the problem related to different BRAF mutations, dimerization, and paradoxical activation. In this context, various scaffold derivatives, such as pyrazine, imidazole, pyridine, pyrazole, pyrimidine, quinoxaline, etc., and their hybrids were synthesized. Their inhibitory activity against various cell lines such as A375, WM266–4, B16BL6, LOX-IMVI, SK-MEL-5 (melanoma), T-29RKO (colorectal wild type), A549 (lung adenocarcinoma), cancer cell lines, etc., and enzyme kinase assay BRAFV600E and BRAFWT were investigated, and their results were published. A summary of the various synthesized scaffolds and their BRAF inhibitory activity and binding conformation in relation to the existing difficulties (from the discovery of BRAF mutant in 2002 to 2022) is reported in Figure 10 and Table 1.
Figure 10.
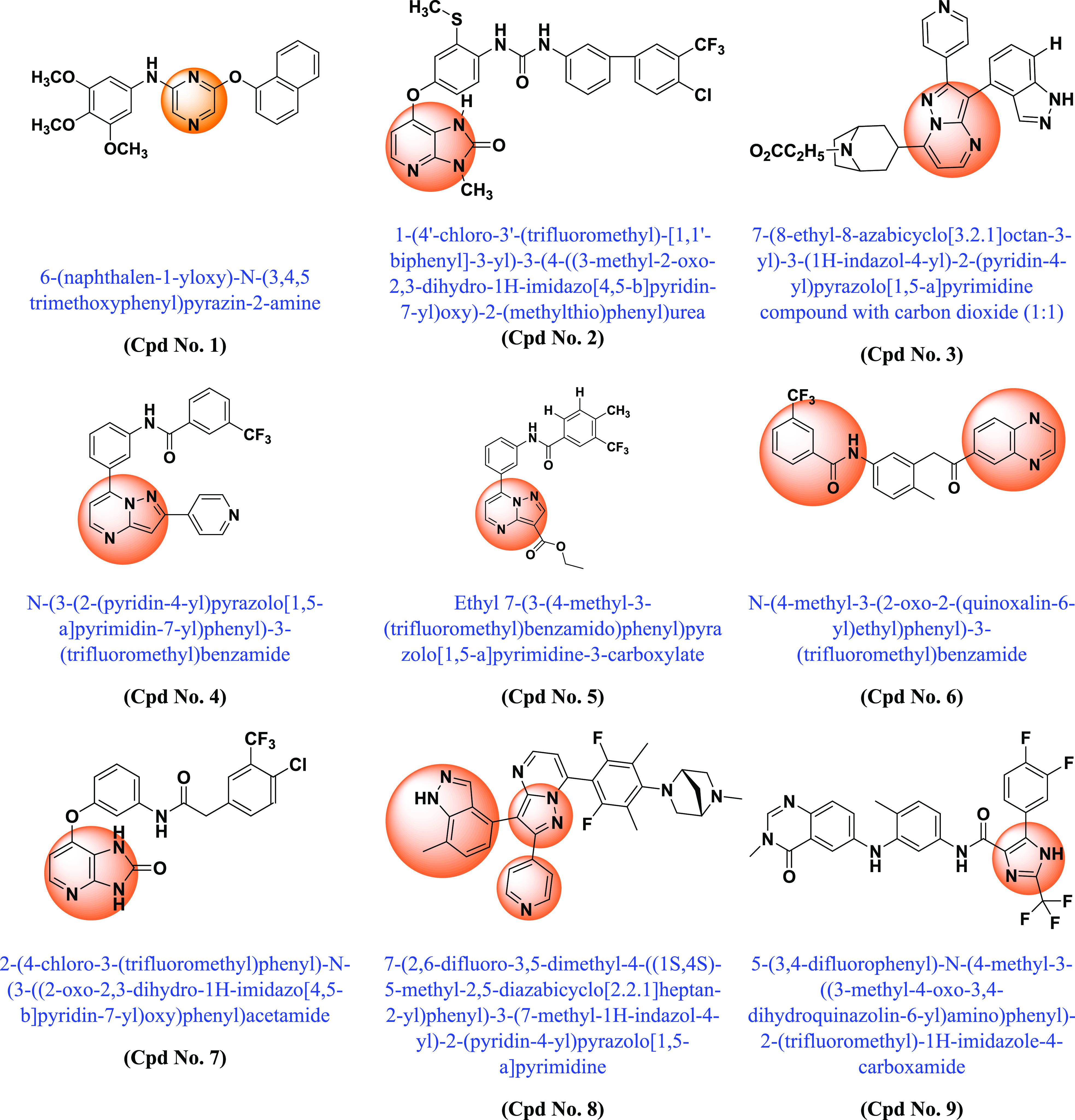
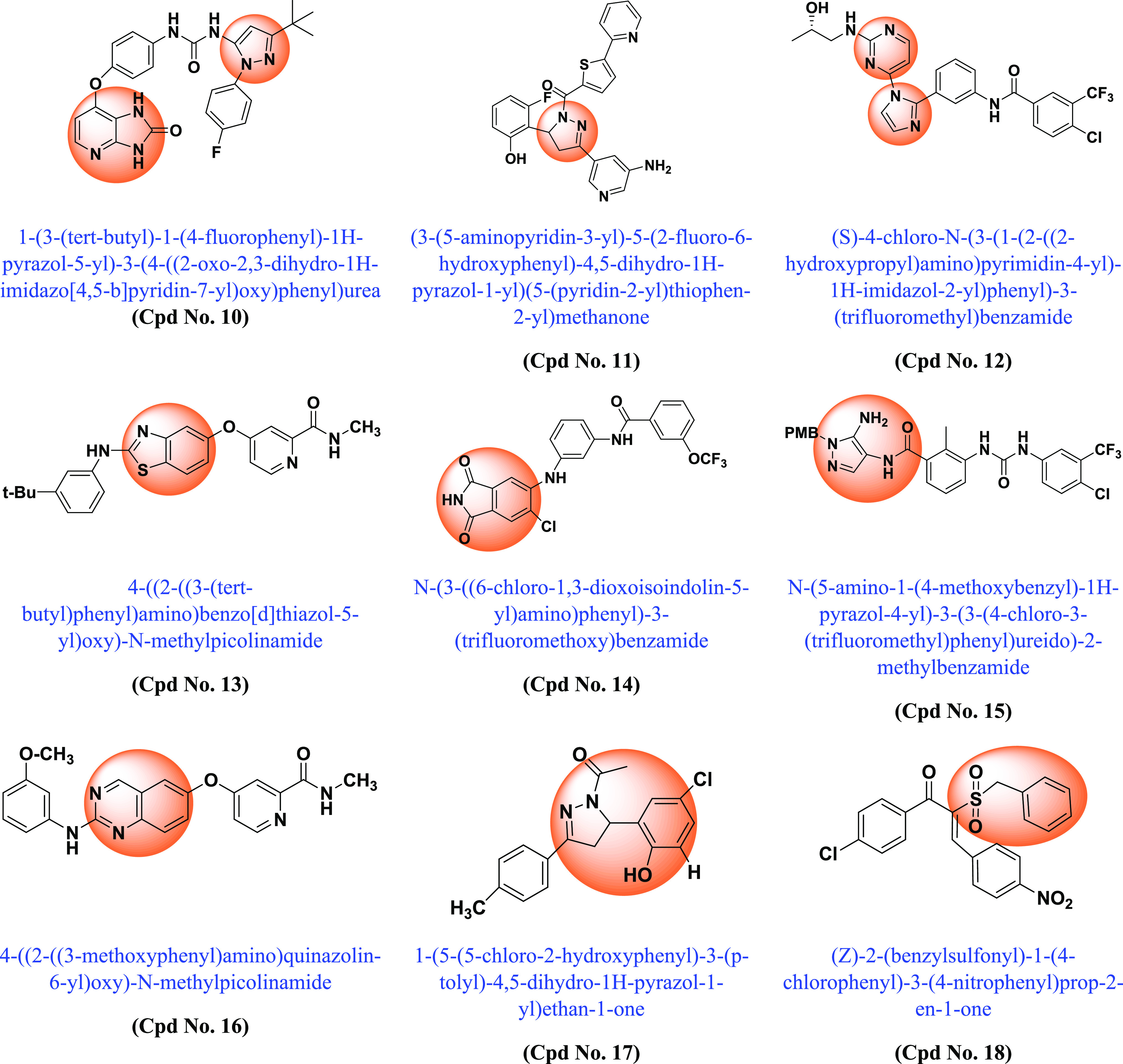
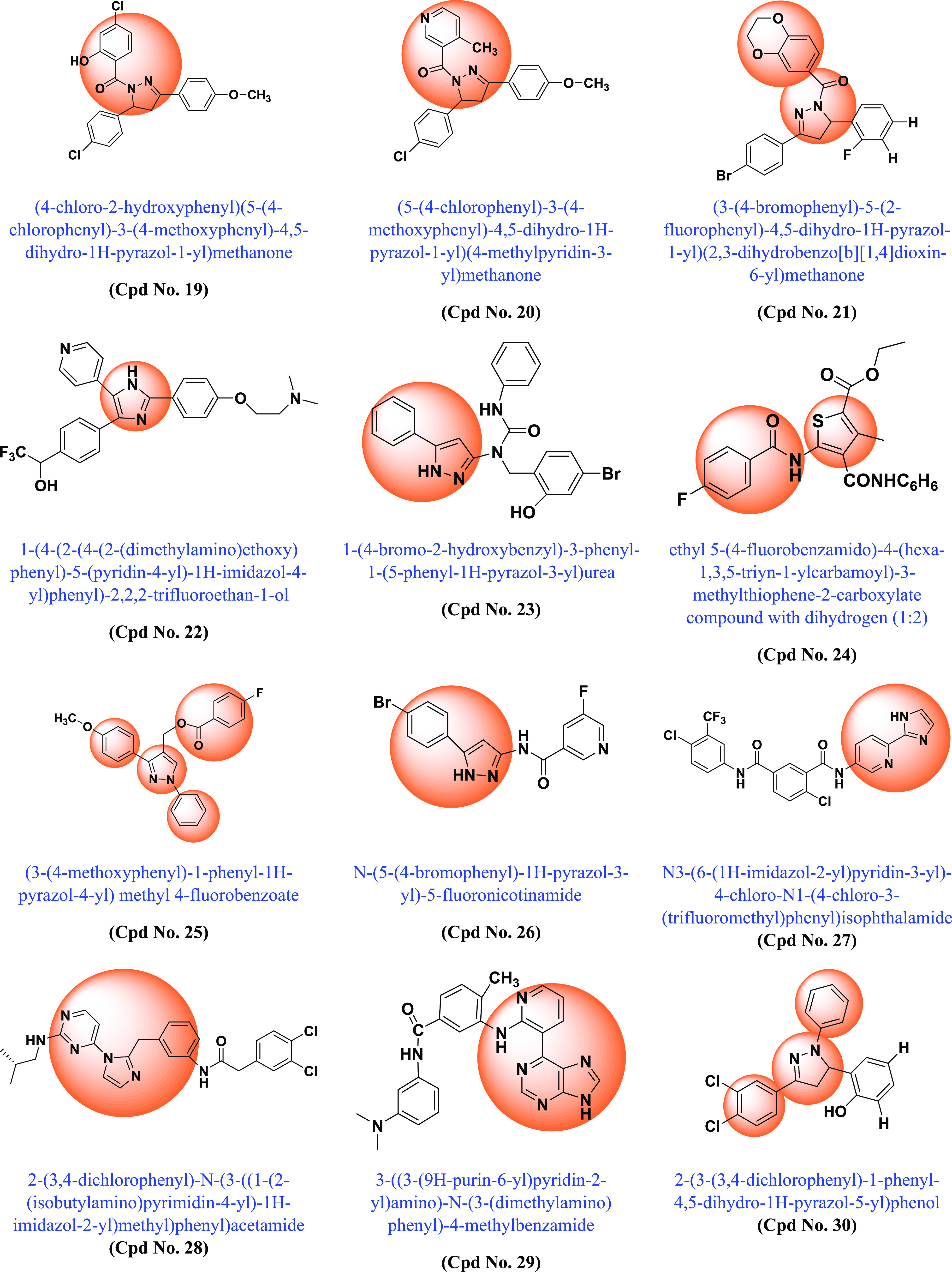
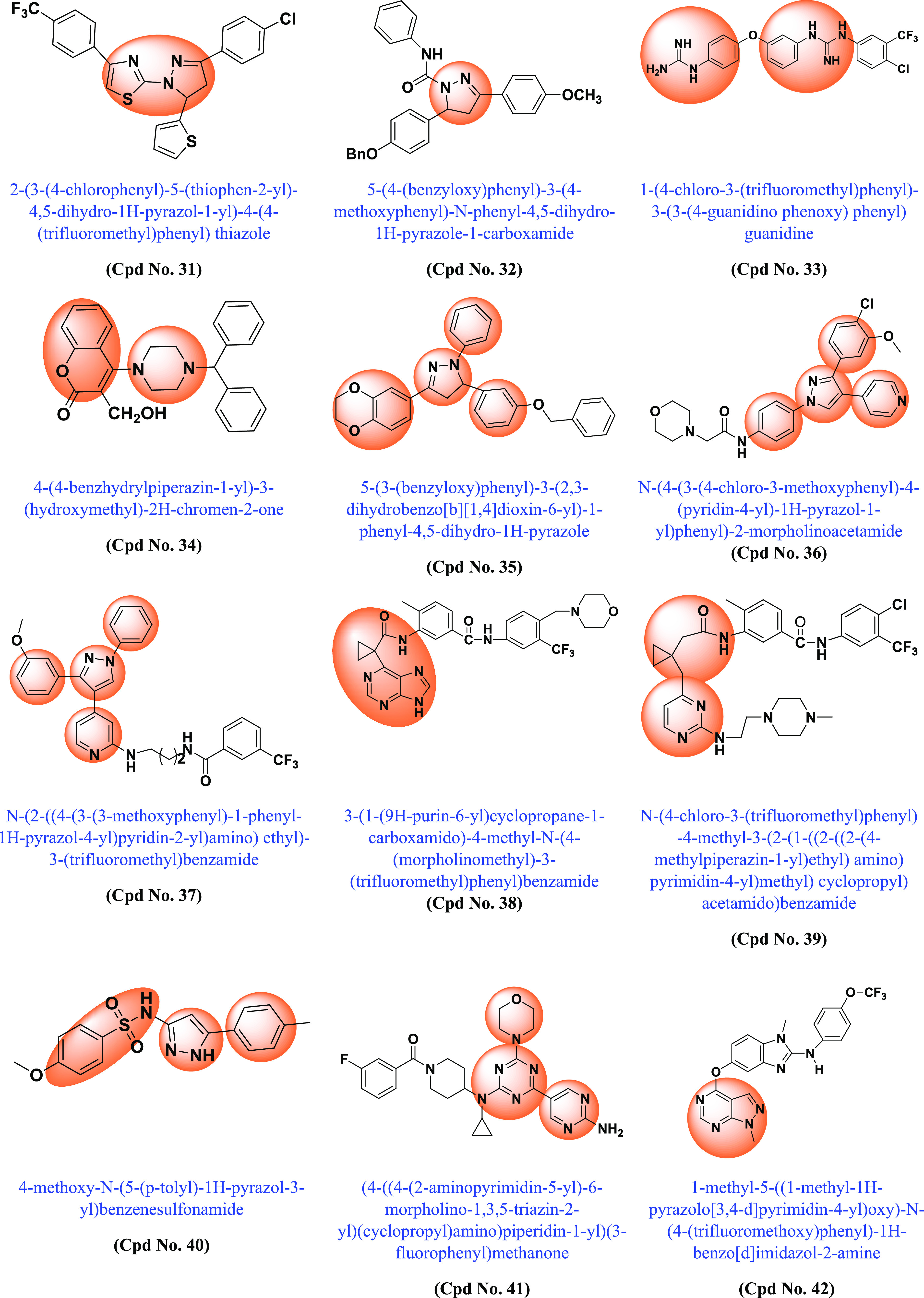
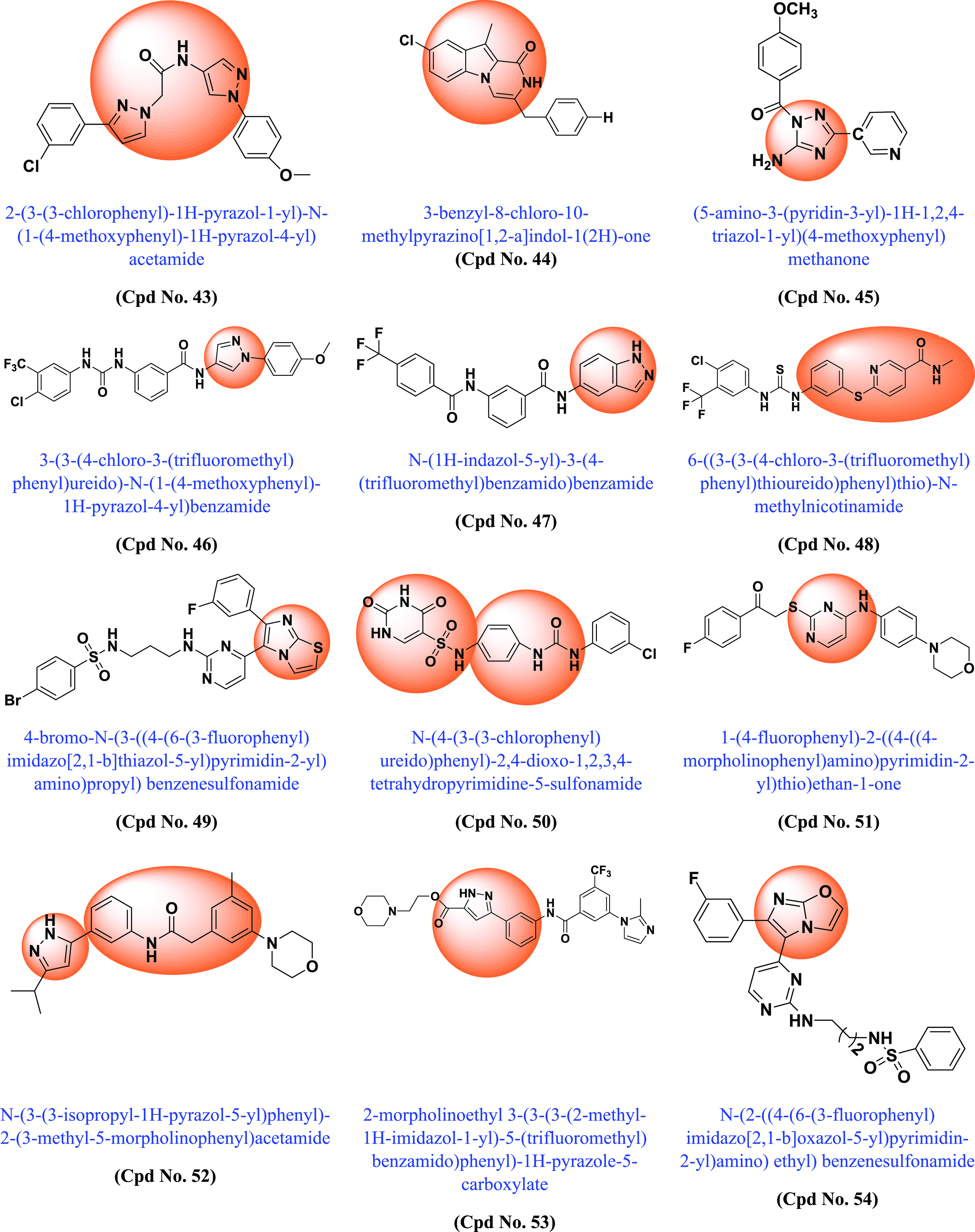
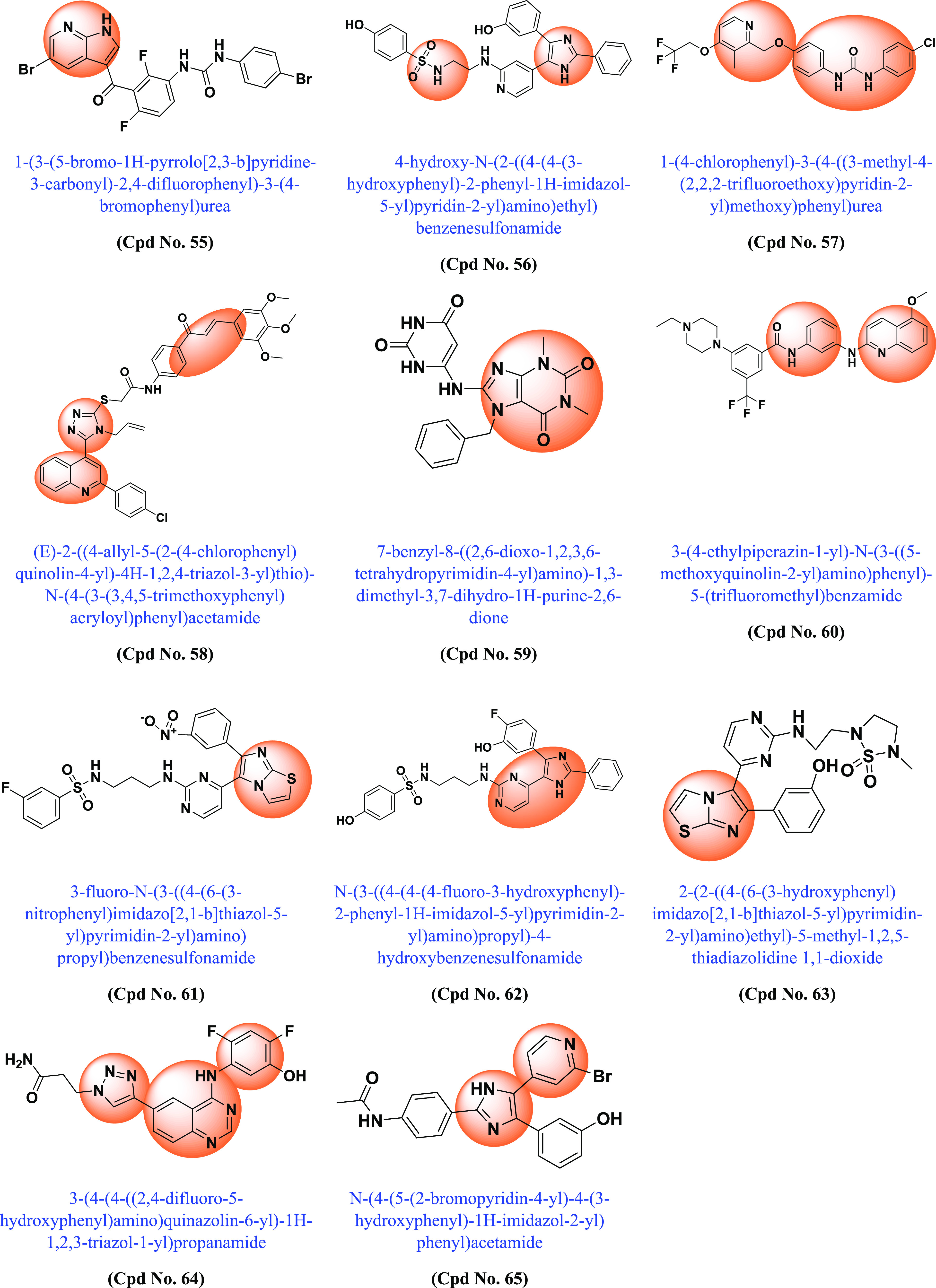
Various synthesized compounds with parents’ scaffolds as BRAF mutant inhibitors. Note: (*) Cpd No.: Compound number; (**) Parent scaffold: Different derivatives of the parent scaffold were synthesized, but only the most potent compounds based on cell line or enzyme kinase activity were chosen.
Table 1. Different Scaffolds and Their Key Aspects in BRAF Mutant.
| Compound No. | Parent scaffold* | BRAFV600E(Enzyme Kinase)/Cell line | IC50(μM) | Key points | Refs |
|---|---|---|---|---|---|
| 1 | Pyrazine | BRAFV600E | 1.2 | Pyrazine scaffold shows strong selectivity against BRAFV600E mutant compared with CRAF | (155) |
| 2 | Imidazo-pyridine | BRAFV600E | 0.001 | Demonstrated interaction with the DFG pocket | (156) |
| 3 | Pyrazolo-pyrimidine | WM266–4 | 0.30 | Bound to the active conformation of BRAF. Fusion of the tropane moiety with the pyrimidine core led to novel, potent BRAF inhibitors that unfortunately did not show good efficacy in cells. | (157) |
| 4 | Pyrazolo-pyrimidine | A375 | 0.28 | Introduction of groups interacting with the hinge region of the kinase in the 2-position of the scaffold with potent BRAF inhibitors | (158) |
| 5 | Pyrazolo-pyrimidine | BRAFV600E | 0.5 | Replacement of esters with amides was tolerated with a slight increase in enzyme potency | (159) |
| 6 | Amidoheteroaryls (quinoxaline-benzamide) | BRAFV600E | 0.001 | Association with DFG-out allosteric inhibitors of BRAF kinase | (160) |
| 7 | Imidazo-pyridinone | BRAFV600E | 0.015 | Inhibitors of the type II BRAF, in which the central phenyl ring is linked to the hinge binder and 1,3 substitutions bind to the allosteric pocket of BRAF | (161) |
| 8 | Indazolylpyrazolopyrimidine | A375 | 0.024 | Type I BRAF inhibitors. Induced dimerization/activation mechanism. In BRAF mutant xenograft models, it shows good inin vivo good anticancer activity | (162) |
| 9 | Imidazole | BRAFV600E | 0.3 | Allosteric DFG-OUT inhibitors of BRAF kinase. The quinazoline moiety binds to the hinge region, the phenyl ring binds to the gatekeeper residue, and the imidazole moiety binds to the selectivity site. | (163) |
| 10 | Imidazopyridin -Imidazole | BRAF | 0.142 | Type II kinase inhibitors with DFG-OUT pocket conformation. Substituted phenyl groups as C-rings (adjacent to imidazole) resulted in a significant improvement in cellular potency | (164) |
| 11 | Pyrazoline | BRAFV600E | 4.0 | Chemical modification of the pyrazoline scaffold led to the development of a potent and selective BRAFV600E inhibitor. (2-Fluoro,6-hydroxy) substitution enhances cellular potency | (165) |
| 12 | Pyrimidin-4-yl-1H-imidazole | WM3629 | 0.62 | Superior antiproliferative activities compared to sorafenib. Selective and potent CRAF inhibitor. | (166) |
| 13 | Benzothiazole | BRAFV600E | 0.004 | DFG-OUT pocket conformation in which substituted anilines replace Phe593, which swings out and interacts with aromatic systems in the hinge region and selectivity pocket. | (167) |
| 14 | Isoindoline-1,3-diones | BRAFV600E/WT | 0.006 | DFG-OUT pocket conformation. Structure-based drug design technology was used to develop both type I and type II BRAF inhibitors with unique chemical scaffolds and hinge binders. | (168) |
| 15 | Imidazolopyrazole | A375 | 0.27 | The compound showed competitive antiproliferative activity to sorafenib. Potential and selective BRAFV600E and CRAF inhibitor | (169) |
| 16 | Quinazoline | BRAFV600E | 0.07 | DFG-OUT conformation | (170) |
| 17 | 4,5-Dihydro-2H-pyrazole 2-hydroxyphenyl | BRAFV600E | 0.22 | Compound showed potent and strong anti-BRAFV600E activity | (171) |
| 18 | Benzylsulfonyl chalcone | BRAFV600E | 0.17 | More potent and selective BRAFV600E inhibitor compared to BRAFWT | (172) |
| 19 | Salicylamide moiety containing 3,5-diarylpyrazoline derivatives | BRAFV600E | 0.16 | Selective inhibition of proliferation of BRAFV600E and BRAFWT mutants | (173) |
| 20 | 4,5-Dihydro-1H-pyrazole niacinamide | BRAFV600E | 0.20 | Compound has highest bioactivity compared to positive control sorafenib. | (174) |
| 21 | 2,3-Dihydrobenzo [b][1,4]dioxin-containing 4,5-dihydro-1H-pyrazole | BRAFV600E | 0.11 | More potent than erlotinib and compound with selective BRAFV600E inhibitor activity | (175) |
| 22 | Imidazole | BRAFV600E | >10 | The novel entity benzo[b]thiophen-3-yl)-2,2,2-trifluoroethanol interacts with the BPII pocket of BRAF, resulting in potent BRAF kinase inhibition in vitro and in cells. | (176) |
| 23 | 5-Phenyl-1H-pyrazol | WM266.4 | 1.50 | Compound showed high antiproliferative activity comparable to positive control vemurafenib | (177) |
| 24 | N-(thiophen-2-yl) benzamide | BRAFV600E | 0.63 | Developed compound proves to be a potent BRAFV600E kinase inhibitor | (178) |
| 25 | (1,3-Diphenyl-1H-pyrazol-4-yl) methyl benzoate | WM266.4 | 0.94 | High antiproliferative effect comparable to positive control vemurafenib and highly selective BRAFV600E in vitro inhibitory activity | (179) |
| 26 | 5-Phenyl-1H-pyrazole | WM266.4 | 2.63 | Compound exhibits highly selective BRAFV600E in vitro inhibitory activity comparable to positive control vemurafenib | (180) |
| 27 | 2-(1H-Imidazol-2-yl) pyridine | A375 | 2.93 | The nitrogen of the imidazole ring forms a critical hydrogen bond with Cys531 in the hinge region, while the amide moiety forms two additional hydrogen bonds with Glu500 and Asp593 in the DFG region. | (181) |
| 28 | Benzyl 2-(1H-imidazole-1-yl) pyrimidine | A375 | 1.82 | Conformation with two different BRAFV600E/V599E mutants and CRAF inhibitor activity | (182) |
| 29 | 2-Amino-3-purinylpyridine | A375 | 1.839 | DFG-OUT conformation with BRAFV600E inhibitory activity. The compounds decreased the growth of melanoma cells A375 (BRAFV600E) via the ERK pathway, paradoxical activation was not observed in ERK melanoma cells SKMEL-2 (BRAFWT) | (183) |
| 30 | Diaryl-pyrazol | BRAFV600E | 0.15 | Addition of ortho-hydroxyl to the 4,5-dihydro-1H-pyrazole scaffold enhanced antitumor activity, while expansion of the group at N-1 of pyrazoline was similarly beneficial. The compound is more potent than vemurafenib and erlotinib. | (184) |
| 31 | 4,5-Dihydro-1H-pyrazole thiazole | WM266.4 | 0.12 | The entire BRAFV600E complex was characterized as a receptor, and the sphere was selected based on the ligand binding position of SB–590885. | (185) |
| 32 | Pyrazole | BRAFV600E | 0.63 | Potent inhibitory activities against BRAFV600E and BRAFWT | (186) |
| 33 | Guanidinium-diaryl | T-29 | 0.9 | DFG-OUT conformations. Sorafenib binds to the adenine region of the ATP-binding site while penetrating deeper into the hydrophobic pocket. | (187) |
| 34 | Benzo-α-pyrone containing piperazine | BRAFV600E | 0.37 | The drug induces cell apoptosis and marked DNA fragmentation. Cell cycle arrest in G0/G1 phase in melanoma cells. Compound tightly binds to the crystal structure of BRAFV600E at the active site. | (188) |
| 35 | Dioxin-containing triaryl pyrazoline | BRAFV600E | 0.04 | Selective for BRAFV600E against BRAFWT, CRAF and EGFR and low in toxicity | (189) |
| 36 | 1,3,4-Triaryl pyrazole | A375 | 1.82 | The compound was tested against BRAFV600E, BRAFWT, and C-RAF. Its type 2-kinase inhibitors bind to the inactive form of kinases (DFG-OUT). | (190) |
| 37 | 1,3,4-Triaryl pyrazole | SK-MEL-5 | 1.54 | The compound was tested against seven kinases: BRAFWT, BRAFV600E, RAF1, EGFR, P38a/MAPK14, ABL1, and ABL1 (T315l). Good inhibitory activity against mutant BRAFV600E and RAF1 kinases | (191) |
| 38 | Purine based cyclopropyl formamide fragment | BRAFV600E | 3.49 | Inhibits proliferation of SK-MEL-2 cell lines without paradoxical activation of ERK. DFG-OUT conformation inhibits all subtypes of Raf proteins. More potent than vemurafenib and sorafenib | (192) |
| 39 | Pyrimidine based cyclopropyl formamide fragment | BRAFV600E | 12.6 | The compound was tested against seven kinases: BRAFWT, BRAFV600E, A-RAF, C-RAF. DFG-OUT, BRAFV600E inhibitors inhibited A375 cell line proliferation via the ERK pathway without paradoxical activation of ERK in SK | (193) |
| 40 | N-(5-Phenyl-1H-pyrazol-3-yl) benzenesulfonamide | WM266.4 | 1.58 | The binding mode of the developed compound is similar to that of the BRAFV600E inhibitor. It consists of a terminal aromatic group (phenyl or pyrazole) filling the allosteric pocket formed by the displacement of the DFG loop and an amide linker connecting an aryl group in the hydrophobic pocket to a hinge-binding heterocycle. The use of an amide linker would allow rapid and efficient screening of many possible hinge-binding groups. | (194) |
| 41 | 2-(Aminopyrimidin-5-yl)-4-(morpholin-4-yl)-6-substituted triazine | A375 | 0.12 | Compound was tested against PI3K(α,β,γ), mTOR BRAFV600E BRAFWT and CRAF kinase activity, proving to be a PI3Kα and BRAFV600E dual inhibitor. | (195) |
| 42 | Pyrazolo[3,4-d]pyrimidine | A375 | 1.74 | DFG-OUT-type pan-RAF inhibitors. High inhibitory activities against both BRAFV600E and VEGFR-2 kinase, comparable to sorafenib act as dual BRAFV600E and VEGFR-2 inhibitor | (196) |
| 43 | Pyrazole | A375 | 0.96 | Compound has potential as a BRAFV600E inhibitor compared to vemurafenib | (197) |
| 44 | Pyrazino[1,2-a]indole | A-549 | 0.8 | Compound showed both BRAFV600E and antioxidant activity. | (198) |
| 45 | 1,2,4-triazole | H-460 | 1.6 | Compound showed potent BRAFV6OOE, EGFR and tubulin inhibitory activity | (199) |
| 46 | Pyrazole | A375 | 0.39 | Type II BRAFV600E inhibitors in this hit compound showed hydrogen bonding with Cys532 in the hinge region, Asp594 in the DFG motif and Glu501 in the αC-helix. The compound showed no significant effect on phosphorylation of ERK and MEK and is more potent than vemurafenib | (200) |
| 47 | Indazole | A375 | 1.97 | The compound does not induce paradoxical effect in normal cells. Shows good inhibitory BRAFV600E activities and antiproliferation activities. | (201) |
| 48 | Thioether and nicotinamide containing sorafenib analogs | B16BL6 | 19.75 | DFG-OUT conformation: the synthesized compound hasBRAF, BRAFV600E and VEGFR-2-muti kinase inhibitor | (202) |
| 49 | Imidazo[2,1-b]thiazole | BRAFV600E | 9.30 | The developed compound was tested against leukemia, nonsmall cell lung cancer, colon, melanoma, ovarian, kidney, prostate, and breast cancer and obtained moderate results. The compound showed the greatest potential inhibitory effect against BRAFV600E compared to BRAFWT and CRAF. | (203) |
| 50 | N-phenyl-(2,4-dihydroxypyrimidine-5-sulfonamido) phenylurea | A549 | 0.67 | The compound outperformed BRAF kinase inhibition and EGFR kinase inhibition | (44) |
| 51 | 4-Amino-2-thiopyrimidines | BRAFWT | 0.11 | Dual VEGFR-2 and BRAF inhibitors. In the designed compound, the 4-amino-2-thiopyrimidine core scaffold is to be located in the central gate region of the inactive DFG-OUT conformations of both enzymes. The hydrophobic substituent at the 4-amino group occupies the hydrophobic posterior pocket on one side. The substituent on the sulfide group, on the other hand, expands to fit into the hinge region. | (204) |
| 52 | N-(3-(3-Alkyl-1H-pyrazol-5-yl) phenyl)-aryl amide | BRAFV600E | 0.70 | Superior selectivity over BRAFV600E and CRAF over BRAFWT kinases | (205) |
| 53 | 3-Carbonyl-5-phenyl-1H-pyrazole | BRAFV600E | 0.10 | Paradoxical activation can be avoided by inhibiting BRAFV600E and CRAF more selectively than BRAFWT. In silico studies have shown that 3-carbonyl-5-phenyl-1H-pyrazole (3) has three hydrogen bonds near the hinge site. In addition, the pyrazole and middle phenyl rings of the compound interlock with Phe595 of the DFG motif. | (206) |
| 54 | Imidazo[2,1-b]oxazole | BRAFV600E | 0.034 | The developed compound was tested against leukemia, nonsmall cell lung cancer, CNS, colon, melanoma, ovarian, renal, prostate, and breast cancers and obtained moderate results. Most effective against BRAFV600E compared to BRAFWT and RAf1 | (207) |
| 55 | Pyrrolo[2,3-b]pyridine | BRAFV600E | 0.080 | Compound with potent BRAFV600E inhibitor | (208) |
| 56 | Imidazole-sulphonamides | BRAFV600E | 32.90 | The developed compound was tested against leukemia, nonsmall cell lung cancer, CNS, colon, melanoma, ovarian, kidney, prostate and breast cancer, achieving moderate results. Compound with potent BRAFV600E inhibitor with active binding site | (209) |
| 57 | 1-Aryl-3-[4-(pyridin-2-ylmethoxy)phenyl]urea | A549 | 2.39 | Using the colorimetric MTT technique, compounds were tested in vitro for their antiproliferative activity against cancer cell lines A549, HCT-116, PC–3, and HL7702, a normal human liver cell line. Compound more effective than sorafenib | (210) |
| 58 | Quinoline/chalcone/1,2,4-triazole | BRAFV600E | 1.1 | The compound was tested against pancreatic (Panc-1), breast (MCF–7), colon (HT–29), and epithelial cell lines (A-549). The compound showed binding affinity to the (ATP) active site of BRAFV600E and was comparable to that of vemurafenib. They have potent inhibitory effects on BRAFV600E and EGFR. | (211) |
| 59 | 7,8-Disubstituted-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione | BRAFWT | 115.1 | Molecular docking in the active site of BRAFWT revealed that C-8 and C-7 substitutions for the N-7 benzyl and tricyclic derivatives, respectively, were responsible for binding interactions with the DFG loop amino acid Asp593 and the αC-helical amino acid Glu500. The compounds showed promising multikinase activity against PI3Kα, BRAFV600E and BRAFWT | (212) |
| 60 | 2-Anilinoquinoline-based arylamides | BRAFV600E | 0.888 | The developed compound was tested against leukemia, nonsmall cell lung cancer, CNS, colon, melanoma, ovarian, renal, prostate and breast cancer and obtained moderate results. Inhibitory activity against BRAFWT, BRAFV600E (DFG-OUT) and C-RAF kinases | (213) |
| 61 | Imidazothiazole | BRAFV600E | 0.021 | The developed compound was tested against leukemia, nonsmall cell lung cancer, CNS, colon, melanoma, ovarian, kidney, prostate and breast cancer and achieved moderate results Compound with BRAFV600E and BRAFWT inhibitory activity. | (214) |
| 62 | Imidazol-5-yl-pyrimidine | LOX-IMVI | 13.90 | Association with interactions with RAS, ribose, hydrophobic pockets and hinge region, additional binding with allosteric pocket of BRAF kinase. With BRAFV600E/p38α inhibitors. | (215) |
| 63 | Imidazo[2,1-b]thiazole | BRAFV600E | 0.07 | Activity against mutant BRAF was higher than for CRAF and BRAFWT. The compounds are more potent than vemurafenib in terms of inhibition of MEK and ERK phosphorylation. | (216) |
| 64 | 4-(3-Hydroxy anilino)-6-(1H-1,2,3-triazol-4-yl)quinazolines | BRAFV600E | 0.051 | The compound was tested against BRAFV600E, CRAF EGFR, EGFRT790M, VEGFR-2, and PDGFR-β. It interacts with the DFG-OUT conformation with and strongly inhibits BRAFV600E at low dose compared to CRAF, EGFRT790 M and VEGFR-2. | (217) |
| 65 | Imidazol-5-yl-pyridine | BRAFV600E | 0.530 | The designed compound has been tested against leukemia, nonsmall cell lung cancer, CNS, colon, melanoma, ovarian, renal, prostate and breast cancer and moderate results were obtained. Dual BRAFV600E/p38α inhibitors with high affinity in kinase pockets. | (218) |
5. Conclusion
Currently, three types of BRAF mutations are reported, i.e., class I: monomeric mutants (BRAFV600); class II: BRAF homodimer mutants (non-V600); and class III: non-V600 BRAF heterodimers. Currently, FDA-approved targeted therapy specifically targets BRAFV600E mutant monomers but is insufficiently effective against non-V600E dimers. Third-generation pan-RAF inhibitors are particularly intriguing because they were designed as “paradox breakers” that do not cause paradoxical BRAF activation but, rather, perform activation in a specific manner that leads to the development of secondary cancers. The mobility of the DFG motif is critical for the selectivity of BRAF inhibitors. Many inhibitors are designed based on the αC-helix and DFG conformation; they are ineffective for current problems such as dimerization and paradoxical activation. Therefore, a structure-based design is strongly recommended to design BRAF inhibitors that exploit the properties of BRAF binding sites, conformational changes, and DFG rearrangement.
In recent years, researchers have synthesized various BRAF inhibitors, and after compiling the data, among these inhibitors, a few target the second and third classes of BRAF mutations as well as have problems like dimerization and paradoxical activation. They should design inhibitors that have binding affinity in the core active site [the nucleotide binding site (ADP or ATP), the DFG motif, the phospho-acceptor site (activation segment) adjacent to the DFG motif, and the helix] and an allosteric binding pocket. In cases of BRAF dimerization, new molecules should aim to bind to both protomers instead of one. These efforts could reduce the number of paradoxical breakers, which would provide new hope for a fourth-generation drug to overcome the current challenges of BRAF.
6. Future Perspective
Many questions regarding the BRAF protein and its regulation remain unanswered. The structure of BRAF kinase is very flexible; different ligands can induce different states of the kinase. The differences in structures may explain the selectivity of BRAF inhibitors for active and inactive BRAF. BRAF monomer, homodimer, and heterodimer protein structures have different mutations. Therefore, based on the above-mentioned mutations and the change of their conformations from active to inactive, further studies are needed.
Because of paradoxical activation, alternative methods of inhibiting Raf (BRAF) signaling using ATP-competitive inhibitors are needed. Little is known about the regulatory mechanisms controlling the formation of BRAF homodimers to heterodimers. In the long term, a better knowledge of RAF regulation would help to develop more effective and better tolerated therapies for BRAF-related malignancies.
Acknowledgments
The authors are thankful to DST-FIST, Central University of Punjab, Bathinda, for providing the necessary facilities to execute this manuscript.
Glossary
List of Abbreviations
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- BRAF
v-RAF murine sarcoma viral homologue B1
- CR
Conserved regions
- CRD
Cysteine-rich domain
- DIF
Dimerization interface
- EGFR
Epidermal growth factor receptors
- ERK
Extracellular-signal regulated kinase
- FDA
Food and drug administration
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen extracellular Kinase
- RAF
Rapidly Accelerated Fibrosarcoma
- RAS
Rat sarcoma
- VEGFR-2
Vascular endothelial growth factor receptor-2
Author Contributions
Conceptualization: Pradeep Kumar; Data collection: Adarsh Kumar, Harshwardhan Singh; Writing the manuscript: Ankit Kumar Singh and Pankaj Sonawane; Sketching of figures and data interpretation: Vladislav Naumovich, Prateek Pathak; Writing, review, and final editing of the manuscript: Habibullah Khalilullah, Mariusz Jaremko, Abdul-Hamid Emwas, Amita Verma, Maria Grishina, and Pradeep Kumar.
The APC was funded by King Abdullah University of Science and Technology, Thuwal, Jeddah, Saudi Arabia.
The authors declare no competing financial interest.
References
- Palmieri G.; Colombino M.; Cristina M.; Antonio P.; Lissia A.; Cossu A. Targeted therapies in melanoma: Successes and pitfalls. IntechOpen 2013, 29–58. 10.5772/53624. [DOI] [Google Scholar]
- Amaral T.; Sinnberg T.; Meier F.; Krepler C.; Levesque M.; Niessner H.; Garbe C. The mitogen-activated protein kinase pathway in melanoma part I–activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 85–92. 10.1016/j.ejca.2016.12.010. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer V.; Wellbrock C.; Marais R. Melanoma biology and new targeted therapy. Nature 2007, 445 (7130), 851–857. 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Palmieri G.; Rozzo C.; Gentilcore G.; Ascierto P. A.. Melanoma pathophysiology and drug targets. Futur. Med. 2012.6. 10.2217/ebo.11.29 [DOI] [Google Scholar]
- Dahl C.; Guldberg P. The genome and epigenome of malignant melanoma. APMIS 2007, 115 (10), 1161–1176. 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- Sullivan R. J.; Flaherty K. T. BRAF in melanoma: pathogenesis, diagnosis, inhibition, and resistance. J. Skin Cancer 2011, 2011, 1. 10.1155/2011/423239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri G.; Capone M.; Ascierto M. L.; Gentilcore G.; Stroncek D. F.; Casula M.; Sini M. C.; Palla M.; Mozzillo N.; Ascierto P. A. Main roads to melanoma. J. Transl. Med. 2009, 7 (1), 1–17. 10.1186/1479-5876-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneker C. M.; Rovoli M.; Kontopidis G.; Röring M.; Galda S.; Braun S.; Brummer T.; McInnes C. Design and synthesis of type-IV inhibitors of BRAF kinase that block dimerization and overcome paradoxical MEK/ERK activation. J. Med. Chem. 2019, 62 (8), 3886–3897. 10.1021/acs.jmedchem.8b01288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb N. J.; Dilworth S. M.; Mol C. D. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat. Rev. Cancer 2004, 4 (9), 718–727. 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- Houghton A. N.; Polsky D. Focus on melanoma. Cancer cell 2002, 2 (4), 275–278. 10.1016/S1535-6108(02)00161-7. [DOI] [PubMed] [Google Scholar]
- Hawryluk E. B.; Tsao H. Melanoma: clinical features and genomic insights. Cold Spring Harbor Perspect. Med. 2014, 4 (9), a015388. 10.1101/cshperspect.a015388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyle K. D.; Guldberg P. Genetic risk factors for melanoma. Hum. Genet. 2009, 126 (4), 499–510. 10.1007/s00439-009-0715-9. [DOI] [PubMed] [Google Scholar]
- Subbiah V.; Baik C.; Kirkwood J. M. Clinical development of BRAF plus MEK inhibitor combinations. Trends cancer 2020, 6 (9), 797–810. 10.1016/j.trecan.2020.05.009. [DOI] [PubMed] [Google Scholar]
- Frisone D.; Friedlaender A.; Malapelle U.; Banna G.; Addeo A. A BRAF new world. Crit. Rev. Oncol./Hematol. 2020, 152, 103008. 10.1016/j.critrevonc.2020.103008. [DOI] [PubMed] [Google Scholar]
- Han X.-R.; Chen L.; Wei Y.; Yu W.; Chen Y.; Zhang C.; Jiao B.; Shi T.; Sun L.; Zhang C.; et al. Discovery of selective small molecule degraders of BRAF-V600E. J. Med. Chem. 2020, 63 (8), 4069–4080. 10.1021/acs.jmedchem.9b02083. [DOI] [PubMed] [Google Scholar]
- Kiełbik A.; Wawryka P.; Chwiłkowska A.; Saczko J.; Kulbacka J. Signaling pathways in melanoma biology and new targeted therapeutic approaches. Med. Res. J. 2019, 4 (3), 184–188. 10.5603/MRJ.a2019.0033. [DOI] [Google Scholar]
- Saldanha G.; Potter L.; DaForno P.; Pringle J. H. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin. Cancer Res. 2006, 12 (15), 4499–4505. 10.1158/1078-0432.CCR-05-2447. [DOI] [PubMed] [Google Scholar]
- Man R.-J.; Zhang Y.-L.; Jiang A.-Q.; Zhu H.-L. A patent review of RAF kinase inhibitors (2010–2018). Expert Opin. Ther. Pat. 2019, 29 (9), 675–688. 10.1080/13543776.2019.1651842. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Lu B.; Liu D.; Shen R.; Yan Y.; Yang L.; Zhang M.; Zhang L.; Cao G.; Cao H.; et al. EBI-907, a novel BRAFV600E inhibitor, has potent oral anti-tumor activity and a broad kinase selectivity profile. Cancer Biol. Ther. 2016, 17 (2), 199–207. 10.1080/15384047.2016.1139231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim P. N.; Zhang J.; Zhang C.; Bollag G. Case History: Vemurafenib, a Potent, Selective, and First-in-Class Inhibitor of Mutant BRAF for the Treatment of Metastatic Melanoma. Annu. Rep. Med. Chem. 2013, 48, 435–449. 10.1016/B978-0-12-417150-3.00026-0. [DOI] [Google Scholar]
- Chen S.-H.; Zhang Y.; Van Horn R. D.; Yin T.; Buchanan S.; Yadav V.; Mochalkin I.; Wong S. S.; Yue Y. G.; Huber L.; et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer discovery 2016, 6 (3), 300–315. 10.1158/2159-8290.CD-15-0896. [DOI] [PubMed] [Google Scholar]
- de Snoo F. A.; Hayward N. K. Cutaneous melanoma susceptibility and progression genes. Cancer Lett. 2005, 230 (2), 153–186. 10.1016/j.canlet.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Śmiech M.; Leszczyński P.; Kono H.; Wardell C.; Taniguchi H. Emerging BRAF mutations in cancer progression and their possible effects on transcriptional networks. Genes 2020, 11 (11), 1342. 10.3390/genes11111342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbottom M. W.; Faraoni R.; Chao Q.; Campbell B. T.; Lai A. G.; Setti E.; Ezawa M.; Sprankle K. G.; Abraham S.; Tran L.; et al. Identification of 1-(3-(6, 7-dimethoxyquinazolin-4-yloxy) phenyl)-3-(5-(1, 1, 1-trifluoro-2-methylpropan-2-yl) isoxazol-3-yl) urea hydrochloride (CEP-32496), a highly potent and orally efficacious inhibitor of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF) V600E. J. Med. Chem. 2012, 55 (3), 1082–1105. 10.1021/jm2009925. [DOI] [PubMed] [Google Scholar]
- Dhomen N.; Marais R. BRAF signaling and targeted therapies in melanoma.. Hematol./Oncol. Clin 2009, 23 (3), 529–545. 10.1016/j.hoc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Richtig G.; Hoeller C.; Kashofer K.; Aigelsreiter A.; Heinemann A.; Kwong L.; Pichler M.; Richtig E. Beyond the BRAFV 600E hotspot: biology and clinical implications of rare BRAF gene mutations in melanoma patients. Br. J. Dermatol. 2017, 177 (4), 936–944. 10.1111/bjd.15436. [DOI] [PubMed] [Google Scholar]
- Leonardi G. C.; Falzone L.; Salemi R.; Zanghì A.; Spandidos D. A.; Mccubrey J. A.; Candido S.; Libra M. Cutaneous melanoma: From pathogenesis to therapy. Int. J. Oncol. 2018, 52 (4), 1071–1080. 10.3892/ijo.2018.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dratkiewicz E.; Simiczyjew A.; Pietraszek-Gremplewicz K.; Mazurkiewicz J.; Nowak D. Characterization of melanoma cell lines resistant to vemurafenib and evaluation of their responsiveness to EGFR-and MET-inhibitor treatment. Int. J. Mol. Sci. 2020, 21 (1), 113. 10.3390/ijms21010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.; Ficili B.; Candido S.; Pezzino F. M.; Guarneri C.; Biondi A.; Travali S.; McCubrey J. A.; Spandidos D. A.; Libra M. Emerging targeted therapies for melanoma treatment. Int. J. Oncol. 2014, 45 (2), 516–524. 10.3892/ijo.2014.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviano M.; Giunta E. F.; Tortora M.; Curvietto M.; Attademo L.; Bosso D.; Cardalesi C.; Rosanova M.; De Placido P.; Pietroluongo E. BRAF gene and melanoma: Back to the future. Int. J. Mol. Sci. 2021, 22 (7), 3474. 10.3390/ijms22073474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Y.-C.; Tang H.-C. Insight into molecular dynamics simulation of BRAF (V600E) and potent novel inhibitors for malignant melanoma. Int. J. Nanomed. 2015, 10, 3131. 10.2147/IJN.S80150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankner M.; Rose A. A.; Rajkumar S.; Siegel P. M.; Watson I. R. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 2018, 37 (24), 3183–3199. 10.1038/s41388-018-0171-x. [DOI] [PubMed] [Google Scholar]
- Vasef M. A.; Auerbach A.. Diagnostic Pathology: Molecular Oncology E-Book ;Elsevier Health Sciences, 2019. [Google Scholar]
- Vulpetti A.; Bosotti R. Sequence and structural analysis of kinase ATP pocket residues. Il farmaco 2004, 59 (10), 759–765. 10.1016/j.farmac.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Niihori T.; Aoki Y.; Narumi Y.; Neri G.; Cavé H.; Verloes A.; Okamoto N.; Hennekam R.; Gillessen-Kaesbach G.; Wieczorek D.; et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006, 38 (3), 294–296. 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Wang W. Structures of BRAF–MEK1–14–3-3 sheds light on drug discovery. Signal Transduction Targeted Ther. 2019, 4 (1), 1–2. 10.1038/s41392-019-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Nassan H. B. Recent progress in the identification of BRAF inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2014, 72, 170–205. 10.1016/j.ejmech.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Van Linden O. P.; Kooistra A. J.; Leurs R.; De Esch I. J.; De Graaf C. KLIFS: a knowledge-based structural database to navigate kinase–ligand interaction space. J. Med. Chem. 2014, 57 (2), 249–277. 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- Agianian B.; Gavathiotis E. Current insights of BRAF inhibitors in cancer: miniperspective. J. Med. Chem. 2018, 61 (14), 5775–5793. 10.1021/acs.jmedchem.7b01306. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr RAF protein-serine/threonine kinases: structure and regulation. Biochem. Biophys. Res. Commun. 2010, 399 (3), 313–317. 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- Angiolini M. The Role of Structural Biology in Kinase Inhibitor Drug Discovery Success. Struct. Biol. Drug Discovery 2020, 363–393. 10.1002/9781118681121.ch16. [DOI] [Google Scholar]
- Freeman A. K.; Ritt D. A.; Morrison D. K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4 (3), 180–185. 10.4161/sgtp.26117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoulia Z.; Gavathiotis E.; Poulikakos P. I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17 (11), 676–691. 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderwala A. Y.; Nimbvikar A. A.; Cope N. J.; Li Z.; Wang Z. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem. Biol. 2019, 14 (7), 1471–1480. 10.1021/acschembio.9b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto P. A.; Kirkwood J. M.; Grob J.-J.; Simeone E.; Grimaldi A. M.; Maio M.; Palmieri G.; Testori A.; Marincola F. M.; Mozzillo N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10 (1), 1–9. 10.1186/1479-5876-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummer T.; McInnes C. RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogen 2020, 39 (21), 4155–4169. 10.1038/s41388-020-1263-y. [DOI] [PubMed] [Google Scholar]
- Martinez Fiesco J. A.; Durrant D. E.; Morrison D. K.; Zhang P. Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat. Commun. 2022, 13 (1), 1–14. 10.1038/s41467-022-28084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M.; Nagel T.; Stuart D. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br. J. Cancer 2014, 111 (4), 640–645. 10.1038/bjc.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse A.; Verkhivker G. M. Exploring molecular mechanisms of paradoxical activation in the BRAF kinase dimers: atomistic simulations of conformational dynamics and modeling of allosteric communication networks and signaling pathways. PloS one 2016, 11 (11), e0166583 10.1371/journal.pone.0166583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau N. P.; Venkatanarayan A.; Quinn J. G.; Phung W.; Malek S.; Hymowitz S. G.; Sudhamsu J. Dimerization induced by C-terminal 14–3–3 binding is sufficient for BRAF kinase activation. Biochemistry 2020, 59 (41), 3982–3992. 10.1021/acs.biochem.0c00517. [DOI] [PubMed] [Google Scholar]
- Simanshu D. K.; Morrison D. K. A Structure Is Worth a Thousand Words: New Insights for RAS and RAF RegulationStructural Insights for RAS and RAF Regulation. Cancer discovery 2022, 12, OF1–OF14. 10.1158/2159-8290.CD-21-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso M.; Estrada M. A.; Ventocilla C.; Samanta M.; Maksimoska J.; Villanueva J.; Winkler J. D.; Marmorstein R. Chemically linked vemurafenib inhibitors promote an inactive BRAFV600E conformation. ACS Chem. Biol. 2016, 11 (10), 2876–2888. 10.1021/acschembio.6b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Kumar A.; Thareja S.; Kumar P. Current insights into the role of BRAF inhibitors in treatment of melanoma. Anti-Cancer Agents Med. Chem. 2023, 23, 278. 10.2174/1871520622666220624164152. [DOI] [PubMed] [Google Scholar]
- Zambon A.; Niculescu-Duvaz I.; Niculescu-Duvaz D.; Marais R.; Springer C. J. Small molecule inhibitors of BRAF in clinical trials. Bioorg. Med. Chem. Lett. 2012, 22 (2), 789–792. 10.1016/j.bmcl.2011.11.060. [DOI] [PubMed] [Google Scholar]
- Rahman M. A.; Salajegheh A.; Smith R. A.; Lam A.-Y. BRAF inhibitors: from the laboratory to clinical trials. Crit. Rev. Oncol./Hematol. 2014, 90 (3), 220–232. 10.1016/j.critrevonc.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Wellbrock C.; Hurlstone A. BRAF as therapeutic target in melanoma. Biochem. Pharmacol. 2010, 80 (5), 561–567. 10.1016/j.bcp.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Maloney R. C.; Zhang M.; Jang H.; Nussinov R. The mechanism of activation of monomeric B-Raf V600E. Comput. Struct. Biotechnol. J. 2021, 19, 3349–3363. 10.1016/j.csbj.2021.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.-h.; Zhou H.; Zhu S.-b.; Huang J.-l.; Zhao X.-x.; Ding H.; Pan Y.-l. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manage. Res. 2018, 10, 2289. 10.2147/CMAR.S170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36 (7), 422–439. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery?. ACS Chem. Biol. 2014, 9 (6), 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker S.; Kirk R.; Hayward R.; Zambon A.; Viros A.; Cantarino N.; Affolter A.; Nourry A.; Niculescu-Duvaz D.; Springer C.; et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2010, 2 (35), 35ra41–35ra41. 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- Waizenegger I. C.; Baum A.; Steurer S.; Stadtmüller H.; Bader G.; Schaaf O.; Garin-Chesa P.; Schlattl A.; Schweifer N.; Haslinger C. A Novel RAF Kinase Inhibitor with DFG-Out–Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol. Cancer Ther. 2016, 15 (3), 354–365. 10.1158/1535-7163.MCT-15-0617. [DOI] [PubMed] [Google Scholar]
- Verkhivker G. Molecular dynamics simulations and modelling of the residue interaction networks in the BRAF kinase complexes with small molecule inhibitors: probing the allosteric effects of ligand-induced kinase dimerization and paradoxical activation. Mol. BioSyst. 2016, 12 (10), 3146–3165. 10.1039/C6MB00298F. [DOI] [PubMed] [Google Scholar]
- Alqathama A. BRAF in malignant melanoma progression and metastasis: potentials and challenges. Am. J. Cancer Res. 2020, 10 (4), 1103. [PMC free article] [PubMed] [Google Scholar]
- Franovic A.; Miller N.; Severson P.; Kanouni T.; Timple N.; Jiang P.; Murphy E.; Martin E. The next-generation pan-RAF inhibitor, KIN-2787, is active in class II and class III BRAF mutant models. J. Clinical Oncology 2021, 39, 3116. [Google Scholar]
- Cotto-Rios X. M.; Agianian B.; Gitego N.; Zacharioudakis E.; Giricz O.; Wu Y.; Zou Y.; Verma A.; Poulikakos P. I.; Gavathiotis E. Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 2020, 11 (1), 1–16. 10.1038/s41467-020-18123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasskarl J. Sorafenib. Small Mol. Oncol. 2010, 184, 61–70. 10.1007/978-3-642-01222-8_5. [DOI] [PubMed] [Google Scholar]
- Plaza-Menacho I.; Mologni L.; Sala E.; Gambacorti-Passerini C.; Magee A. I.; Links T. P.; Hofstra R. M.; Barford D.; Isacke C. M. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J. Biol. Chem. 2007, 282 (40), 29230–29240. 10.1074/jbc.M703461200. [DOI] [PubMed] [Google Scholar]
- Adnane L.; Trail P. A.; Taylor I.; Wilhelm S. M. Sorafenib (BAY 43–9006, Nexavar®), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- Wilhelm S.; Carter C.; Lynch M.; Lowinger T.; Dumas J.; Smith R. A.; Schwartz B.; Simantov R.; Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery 2006, 5 (10), 835–844. 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Wilhelm S.; Carter C.; Lynch M.; Lowinger T.; Dumas J.; Smith R. A.; Schwartz B.; Simantov R.; Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discovery 2006, 5 (10), 835–844. 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Smith R. A.; Dumas J.; Adnane L.; Wilhelm S. M. Recent advances in the research and development of RAF kinase inhibitors. Curr. Top. Med. Chem. 2006, 6 (11), 1071–1089. 10.2174/156802606777812077. [DOI] [PubMed] [Google Scholar]
- Miura K.; Satoh M.; Kinouchi M.; Yamamoto K.; Hasegawa Y.; Philchenkov A.; Kakugawa Y.; Fujiya T. The preclinical development of regorafenib for the treatment of colorectal cancer. Expert Opin. Drug Discovery 2014, 9 (9), 1087–1101. 10.1517/17460441.2014.924923. [DOI] [PubMed] [Google Scholar]
- Stone E. A.; Mercado B. Q.; Miller S. J. Structure and Reactivity of Highly Twisted N-Acylimidazoles. Org. Lett. 2019, 21 (7), 2346–2351. 10.1021/acs.orglett.9b00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner D. E.; Eck M. J. A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Sci. 2021, 30 (8), 1535–1553. 10.1002/pro.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovolskaite A.; Madan M.; Pandey V.; Altomare D. A.; Phanstiel O. IV The discovery of indolone GW5074 during a comprehensive search for non-polyamine-based polyamine transport inhibitors. Int. J. Biochem. Cell Biol. 2021, 138, 106038. 10.1016/j.biocel.2021.106038. [DOI] [PubMed] [Google Scholar]
- Ibrahim P. N.; Zhang J.; Zhang C.; Bollag G. Case History: Vemurafenib, a Potent, Selective, and First-in-Class Inhibitor of Mutant BRAF for the Treatment of Metastatic Melanoma. Annu. Rep. Med. Chem. 2013, 48, 435–449. 10.1016/B978-0-12-417150-3.00026-0. [DOI] [Google Scholar]
- Karoulia Z.; Gavathiotis E.; Poulikakos P. I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer. 2017, 17 (11), 676–691. 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban-Burgos L.; Wang H.; Nieto P.; Zheng J.; Blanco-Aparicio C.; Varela C.; Gómez-López G.; Fernández-García F.; Sanclemente M.; Guerra C.; et al. Tumor regression and resistance mechanisms upon CDK4 and RAF1 inactivation in KRAS/P53 mutant lung adenocarcinomas. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (39), 24415–24426. 10.1073/pnas.2002520117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C.-C.; Ho C.-L.; Yang M.-H.; Tsai Y.-T.; Liu S.-Y.; Chang P.-Y.; Wu Y.-Y.; Chen J.-H.; Huang T.-C.; Yehn R.-H.; et al. Phase I Targeted Combination Trial of Sorafenib and GW5074 in Patients with Advanced Refractory Solid Tumors. J. Clin. Med. 2022, 11 (8), 2183. 10.3390/jcm11082183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vido M. J.; Le K.; Hartsough E. J.; Aplin A. E. BRAF splice variant resistance to RAF inhibitor requires enhanced MEK association. Cell Rep. 2018, 25 (6), 1501–1510. 10.1016/j.celrep.2018.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deeb I. M.; Lee S. H. Design and synthesis of new potent anticancer pyrazoles with high FLT3 kinase inhibitory selectivity. Bioorg. Med. Chem. 2010, 18 (11), 3961–3973. 10.1016/j.bmc.2010.04.029. [DOI] [PubMed] [Google Scholar]
- Sieber J.; Wieder N.; Clark A.; Reitberger M.; Matan S.; Schoenfelder J.; Zhang J.; Mandinova A.; Bittker J. A.; Gutierrez J.; et al. GDC-0879, a BRAFV600E inhibitor, protects kidney podocytes from death. Cell Chem. Biol. 2018, 25 (2), 175–184. 10.1016/j.chembiol.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saei A.; Eichhorn P. J. A. Adaptive responses as mechanisms of resistance to BRAF inhibitors in melanoma. Cancers. 2019, 11 (8), 1176. 10.3390/cancers11081176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gompel J. J.; Kunnimalaiyaan M.; Holen K.; Chen H. ZM336372, a Raf-1 activator, suppresses growth and neuroendocrine hormone levels in carcinoid tumor cells. Mol. Cancer Ther. 2005, 4 (6), 910–917. 10.1158/1535-7163.MCT-04-0334. [DOI] [PubMed] [Google Scholar]
- Gunderwala A.; Cope N.; Wang Z. Mechanism and inhibition of BRAF kinase. Curr. Opin. Chem. Biol. 2022, 71, 102205. 10.1016/j.cbpa.2022.102205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon A.; Niculescu-Duvaz I.; Niculescu-Duvaz D.; Marais R.; Springer C. J. Small molecule inhibitors of BRAF in clinical trials. Bioorg. Med. Chem. Lett. 2012, 22 (2), 789–792. 10.1016/j.bmcl.2011.11.060. [DOI] [PubMed] [Google Scholar]
- Aldaghi S. A.; Jalal R. Concentration-Dependent Dual Effects of Ciprofloxacin on SB-590885-Resistant BRAFV600E A375 Melanoma Cells. Chem. Res. Toxicol. 2019, 32 (4), 645–658. 10.1021/acs.chemrestox.8b00335. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Gong Q.; Wang W.; Liu F.; Kong Q.; Pan F.; Zhang X.; Yu C.; Hu S.; Fan F.; et al. The combination of Biochanin A and SB590885 potentiates the inhibition of tumour progression in hepatocellular carcinoma. Cancer Cell Int. 2020, 20 (1), 1–11. 10.1186/s12935-020-01463-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Novak J.; Kumar A.; Singh H.; Thareja S.; Pathak P.; Grishina M.; Verma A.; Yadav J. P.; Khalilullah H.; et al. Gaussian field-based 3D-QSAR and molecular simulation studies to design potent pyrimidine–sulfonamide hybrids as selective BRAF V600E inhibitors. RSC Adv. 2022, 12 (46), 30181–30200. 10.1039/D2RA05751D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S.-B.; Henry J. R.; Kaufman M. D.; Lu W.-P.; Smith B. D.; Vogeti S.; Rutkoski T. J.; Wise S.; Chun L.; Zhang Y.; et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer cell. 2015, 28 (3), 384–398. 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- King A. J.; Arnone M. R.; Bleam M. R.; Moss K. G.; Yang J.; Fedorowicz K. E.; Smitheman K. N.; Erhardt J. A.; Hughes-Earle A.; Kane-Carson L. S.; et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PloS one. 2013, 8 (7), e67583. 10.1371/journal.pone.0067583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne A. D.; Garnock-Jones K. P. Dabrafenib: First Global Approval. Drugs 2013, 73 (12), 1367–1376. 10.1007/s40265-013-0095-2. [DOI] [PubMed] [Google Scholar]
- Gibney G. T.; Zager J. S. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin. Drug Metab. Toxicol. 2013, 9 (7), 893–9. 10.1517/17425255.2013.794220. [DOI] [PubMed] [Google Scholar]
- Rai S. K.; Gunnam A.; Mannava M. K. C.; Nangia A. K. Improving the Dissolution Rate of the Anticancer Drug Dabrafenib. Cryst. Growth Des. 2020, 20 (2), 1035–1046. 10.1021/acs.cgd.9b01365. [DOI] [Google Scholar]
- Yao H.; Sun Q.; Zhu J. Identification and characterization of small-molecule inhibitors to selectively target the DFG-in over the DFG-out conformation of the B-Raf kinase V600E mutant in colorectal cancer. Arch Pharm. 2016, 349 (10), 808–815. 10.1002/ardp.201600184. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Heinzmann D.; Grippo J. F. Clinical pharmacokinetics of vemurafenib. Clin Pharmacokinet. 2017, 56 (9), 1033–1043. 10.1007/s40262-017-0523-7. [DOI] [PubMed] [Google Scholar]
- Dumaz N.; Jouenne F.; Delyon J.; Mourah S.; Bensussan A.; Lebbé C. Atypical BRAF and NRAS mutations in mucosal melanoma. Cancers 2019, 11 (8), 1133. 10.3390/cancers11081133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.; McKee A. E.; Ning Y.-M.; Hazarika M.; Theoret M.; Johnson J. R.; Xu Q. C.; Tang S.; Sridhara R.; Jiang X.; He K.; Roscoe D.; McGuinn W. D.; Helms W. S.; Russell A. M.; Miksinski S. P.; Zirkelbach J. F.; Earp J.; Liu Q.; Ibrahim A.; Justice R.; Pazdur R. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAFV600E Mutation. Clin. Cancer Res. 2014, 20 (19), 4994–5000. 10.1158/1078-0432.CCR-14-0776. [DOI] [PubMed] [Google Scholar]
- Adamopoulos C.; Ahmed T. A.; Tucker M. R.; Ung P. M.; Xiao M.; Karoulia Z.; Amabile A.; Wu X.; Aaronson S. A.; Ang C. Exploiting allosteric properties of RAF and MEK inhibitors to target therapy-resistant tumors driven by oncogenic BRAF signaling. Cancer Discovery 2021, 11, 1716. 10.1158/2159-8290.CD-20-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Jr Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- NIDDKD . Encorafenib. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury ;National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, 2012. [Google Scholar]
- Sun J.; Zager J. S.; Eroglu Z. Encorafenib/binimetinib for the treatment of BRAF-mutant advanced, unresectable, or metastatic melanoma: design, development, and potential place in therapy. OncoTargets Ther. 2018, 11, 9081–9089. 10.2147/OTT.S171693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger I. C.; Baum A.; Steurer S.; Stadtmüller H.; Bader G.; Schaaf O.; Garin-Chesa P.; Schlattl A.; Schweifer N.; Haslinger C.; et al. A Novel RAF Kinase Inhibitor with DFG-Out–Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol. Cancer Ther. 2016, 15 (3), 354–365. 10.1158/1535-7163.MCT-15-0617. [DOI] [PubMed] [Google Scholar]
- He M.; Lv W.; Rao Y., Opportunities and Challenges of Small Molecule Induced Targeted Protein Degradation. Front Cell Dev Biol. 2021, 9. 10.3389/fcell.2021.685106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger I. C.; Baum A.; Steurer S.; Stadtmüller H.; Bader G.; Schaaf O.; Garin-Chesa P.; Schlattl A.; Schweifer N.; Haslinger C.; Colbatzky F.; Mousa S.; Kalkuhl A.; Kraut N.; Adolf G. R. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol. Cancer Ther. 2016, 15 (3), 354–65. 10.1158/1535-7163.MCT-15-0617. [DOI] [PubMed] [Google Scholar]
- Ribas A.; Flaherty K. T. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat. Rev. Clin Oncol. 2011, 8 (7), 426–433. 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- Callahan M. K.; Masters G.; Pratilas C. A.; Ariyan C.; Katz J.; Kitano S.; Russell V.; Gordon R. A.; Vyas S.; Yuan J.; et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014, 2 (1), 70–79. 10.1158/2326-6066.CIR-13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson M. A.; Gordon M. S.; Edelman G.; Bendell J. C.; Kudchadkar R. R.; LoRusso P. M.; Johnston S. H.; Clary D. O.; Schwartz G. K. Phase I study of XL281 (BMS-908662), a potent oral RAF kinase inhibitor, in patients with advanced solid tumors. Invest. New Drugs 2015, 33 (2), 349–56. 10.1007/s10637-014-0191-5. [DOI] [PubMed] [Google Scholar]
- Ducreux M.; Chamseddine A.; Laurent-Puig P.; Smolenschi C.; Hollebecque A.; Dartigues P.; Samallin E.; Boige V.; Malka D.; Gelli M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol 2019, 11, 175883591985649. 10.1177/1758835919856494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.-F.; Chen Y.; Rao S.-S.; Chen X.-M.; Liu H.-C.; Qin J.-H.; Tang W.-F.; Yue-Wang; Zhou X.; Lu T. Recent advances in the research and development of B-Raf inhibitors. Curr. Med. Chem. 2010, 17 (16), 1618–1634. 10.2174/092986710791111242. [DOI] [PubMed] [Google Scholar]
- Karoulia Z.; Gavathiotis E.; Poulikakos P. I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17 (11), 676–691. 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto-Rios X. M.; Agianian B.; Gitego N.; Zacharioudakis E.; Giricz O.; Wu Y.; Zou Y.; Verma A.; Poulikakos P. I.; Gavathiotis E. Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 2020, 11 (1), 1–16. 10.1038/s41467-020-18123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-Q.; Teng Q.-X.; Lei Z.-N.; Ji N.; Cui Q.; Fu H.; Lin L.; Yang D.-H.; Fan Y.-F.; Chen Z.-S., Reversal of Cancer Multidrug Resistance (MDR) Mediated by ATP-Binding Cassette Transporter G2 (ABCG2) by AZ-628, a RAF Kinase Inhibitor. Front. Cell Dev. Biol. 2020, 8. 10.3389/fcell.2020.601400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto-Rios X. M.; Agianian B.; Gitego N.; Zacharioudakis E.; Giricz O.; Wu Y.; Zou Y.; Verma A.; Poulikakos P. I.; Gavathiotis E. Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 2020, 11 (1), 4370. 10.1038/s41467-020-18123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouladi M.; Pfister S. M. MEK and RAF inhibitors: time for a paradigm shift in the treatment of pediatric low-grade gliomas?. Neuro-Oncology 2017, 19, 741–743. 10.1093/neuonc/nox039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agianian B.; Gavathiotis E. Current insights of BRAF inhibitors in cancer: miniperspective. J. Med. Chem. 2018, 61 (14), 5775–5793. 10.1021/acs.jmedchem.7b01306. [DOI] [PubMed] [Google Scholar]
- Gampa G.; Kim M.; Mohammad A. S.; Parrish K. E.; Mladek A. C.; Sarkaria J. N.; Elmquist W. F. Brain Distribution and Active Efflux of Three panRAF Inhibitors: Considerations in the Treatment of Melanoma Brain Metastases. J. Pharmacol. Exp. Ther. 2019, 368 (3), 446–461. 10.1124/jpet.118.253708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadnover V.-I.; Mariana V.-V.; Maria B.-M.. Regulation of MAPK ERK1/2 Signaling by Phosphorylation: Implications in Physiological and Pathological Contexts. In Post-Translational Modifications in Cellular Functions and Diseases ;Shibo Y., Ed.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar]
- Pan J.-h.; Zhou H.; Zhu S.-b.; Huang J.-l.; Zhao X.-x.; Ding H.; Pan Y.-l. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manage Res. 2018, 10, 2289. 10.2147/CMAR.S170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti G.; Andrini E.; Sisi M.; Rizzo A.; Parisi C.; Di Federico A.; Gelsomino F.; Ardizzoni A. Beyond EGFR, ALK and ROS1: current evidence and future perspectives on newly targetable oncogenic drivers in lung adenocarcinoma. Crit Rev. Oncol/Hematol. 2020, 156, 103119. 10.1016/j.critrevonc.2020.103119. [DOI] [PubMed] [Google Scholar]
- Alqathama A. BRAF in malignant melanoma progression and metastasis: potentials and challenges. Am. J. Cancer Res. 2020, 10 (4), 1103–1114. [PMC free article] [PubMed] [Google Scholar]
- Hong S. P.; Ahn S. K. Discovery of a novel pan-RAF inhibitor with potent anti-tumor activity in preclinical models of BRAFV600E mutant cancer. Life Sciences 2017, 183, 37–44. 10.1016/j.lfs.2017.06.021. [DOI] [PubMed] [Google Scholar]
- Kortum R. L.; Morrison D. K. Path forward for RAF therapies: inhibition of monomers and dimers. Cancer cell. 2015, 28 (3), 279–281. 10.1016/j.ccell.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S.-B.; Henry J.; Kaufman M.; Lu W.-P.; Smith B.; Vogeti S.; Rutkoski T.; Wise S.; Chun L.; Zhang Y.; et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer cell 2015, 28 (3), 384–398. 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Kohtamäki L.; Arjama M.; Mäkelä S.; Ianevski P.; Välimäki K.; Juteau S.; Ilmonen S.; Ungureanu D.; Kallioniemi O.; Murumägi A.; Hernberg M. High-throughput ex vivo drug testing identifies potential drugs and drug combinations for NRAS-positive malignant melanoma. Transl. Oncol. 2022, 15 (1), 101290. 10.1016/j.tranon.2021.101290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehling D. E.; Harris P. A. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (19), 4047–4056. 10.1016/j.bmcl.2015.07.093. [DOI] [PubMed] [Google Scholar]
- James J.; Ruggeri B.; Armstrong R. C.; Rowbottom M. W.; Jones-Bolin S.; Gunawardane R. N.; Dobrzanski P.; Gardner M. F.; Zhao H.; Cramer M. D.; et al. CEP-32496: a novel orally active BRAFV600E inhibitor with selective cellular and in vivo antitumor activity. Mol. Cancer Ther. 2012, 11 (4), 930–941. 10.1158/1535-7163.MCT-11-0645. [DOI] [PubMed] [Google Scholar]
- Friedman J. A.; Hewit T.; Bruckheimer E.; Trusko S.; Dorsey B.; Ruggeri B. Antitumor activity of CEP-32496, a novel orally active BRAFV600E inhibitor, in a panel of Champions TumorGraft models of melanoma and colorectal cancer with B-Raf V600E mutations. Cancer Res. 2012, 72 (8_Supplement), 3755–3755. 10.1158/1538-7445.AM2012-3755. [DOI] [Google Scholar]
- Jiang C.; Xie L.; Zhang Y.; Fujinaga M.; Mori W.; Kurihara Y.; Yamasaki T.; Wang F.; Zhang M.-R. Pharmacokinetic evaluation of [11C] CEP-32496 in nude mice bearing BRAFV600E mutation-induced melanomas. Mol. Imaging 2018, 17, 153601211879595. 10.1177/1536012118795952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Yu Y.; Zhao Y.; Wu D.; Yu X.; Lu J.; Chen Z.; Zhang H.; Hu Y.; Zhai Y.; Su J.; Aheman A.; De las Casas A.; Jin J.; Xu X.; Shi Z.; Woodfield S. E.; Vasudevan S. A.; Agarwal S.; Yan Y.; Yang J.; Foster J. H. Small molecule inhibitor agerafenib effectively suppresses neuroblastoma tumor growth in mouse models via inhibiting ERK MAPK signaling. Cancer Lett. 2019, 457, 129–141. 10.1016/j.canlet.2019.05.011. [DOI] [PubMed] [Google Scholar]
- Saturno G.; Lopes F.; Niculescu-Duvaz I.; Niculescu-Duvaz D.; Zambon A.; Davies L.; Johnson L.; Preece N.; Lee R.; Viros A.; et al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann. Oncol. 2021, 32 (2), 269–278. 10.1016/j.annonc.2020.10.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saturno G.; Lopes F.; Niculescu-Duvaz I.; Niculescu-Duvaz D.; Zambon A.; Davies L.; Johnson L.; Preece N.; Lee R.; Viros A.; Holovanchuk D.; Pedersen M.; McLeary R.; Lorigan P.; Dhomen N.; Fisher C.; Banerji U.; Dean E.; Krebs M. G.; Gore M.; Larkin J.; Marais R.; Springer C. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Annals of oncology: official journal of the European Society for Medical Oncology 2021, 32 (2), 269–278. 10.1016/j.annonc.2020.10.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man R.-J.; Zhang Y.-L.; Jiang A.-Q.; Zhu H.-L. A patent review of RAF kinase inhibitors (2010–2018). Expert Opin Ther Pat. 2019, 29 (9), 675–688. 10.1080/13543776.2019.1651842. [DOI] [PubMed] [Google Scholar]
- Esteban-Burgos L.; Wang H.; Nieto P.; Zheng J.; Blanco-Aparicio C.; Varela C.; Gómez-López G.; Fernández-García F.; Sanclemente M.; Guerra C.; Drosten M.; Galán J.; Caleiras E.; Martínez-Torrecuadrada J.; Fajas L.; Peng S. B.; Santamaría D.; Musteanu M.; Barbacid M. Tumor regression and resistance mechanisms upon CDK4 and RAF1 inactivation in KRAS/P53 mutant lung adenocarcinomas. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (39), 24415–24426. 10.1073/pnas.2002520117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaguri S. C.; Akula S.; Ashireddygari V. R.; Sahoo P. S.; Burra V. L. S. P.; Silveri R.; Mupparapu V.; Korikani M.; Amanchi N. R.; Subramanian J.; Kancha R. K. Estimated sensitivity profiles of lung cancer specific uncommon BRAF mutants towards experimental and clinically approved kinase inhibitors. Toxicol. Appl. Pharmacol. 2022, 453, 116213. 10.1016/j.taap.2022.116213. [DOI] [PubMed] [Google Scholar]
- Halaban R.; Bacchiocchi A.; Straub R.; Cao J.; Sznol M.; Narayan D.; Allam A.; Krauthammer M.; Mansour T. S. A novel anti-melanoma SRC-family kinase inhibitor. Oncotarget. 2019, 10 (23), 2237. 10.18632/oncotarget.26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai J.; Gan H.; Barrow C.; Jameson M.; Atkinson V.; Haydon A.; Millward M.; Begbie S.; Brown M.; Markman B.; Patterson W.; Hill A.; Horvath L.; Nagrial A.; Richardson G.; Jackson C.; Friedlander M.; Parente P.; Tran B.; Wang L.; Chen Y.; Tang Z.; Huang W.; Wu J.; Zeng D.; Luo L.; Solomon B. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2020, 38 (19), 2140–2150. 10.1200/JCO.19.02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi G. A.; Rico A.; Tanner H.; Aversa R. J.; Taft B. R.; Subramanian S.; Setti L.; Burger M. T.; Wan L.; Tamez V.; et al. Design and Discovery of N-(2-Methyl-5′-morpholino-6′-((tetrahydro-2 H-pyran-4-yl) oxy)-[3, 3′-bipyridin]-5-yl)-3-(trifluoromethyl) benzamide (RAF709): A Potent, Selective, and Efficacious RAF Inhibitor Targeting RAS Mutant Cancers. J. med chem. 2017, 60 (12), 4869–4881. 10.1021/acs.jmedchem.6b01862. [DOI] [PubMed] [Google Scholar]
- Shao W.; Mishina Y. M.; Feng Y.; Caponigro G.; Cooke V. G.; Rivera S.; Wang Y.; Shen F.; Korn J. M.; Mathews Griner L. A.; et al. Antitumor Properties of RAF709, a Highly Selective and Potent Inhibitor of RAF Kinase Dimers, in Tumors Driven by Mutant RAS or BRAFAntitumor Properties of Novel RAF Inhibitor RAF709. Cancer Res. 2018, 78 (6), 1537–1548. 10.1158/0008-5472.CAN-17-2033. [DOI] [PubMed] [Google Scholar]
- Röck R.; Mayrhofer J.; Torres-Quesada O.; Enzler F.; Troppmair J.; Stefan E.. Abstract B45: Surveillance of RAS-RAF dynamics in vivo: Tracking activity conformations and drug-induced interactions. In Proceedings of the AACR Special Conference on Targeting RAS-Driven Cancers , December 9–12, 2018, San Diego, California, American Association for Cancer Research, 2020; Vol. 18, (5), 10.1158/1557-3125.RAS18-B45 [DOI]
- Durrant D. E.; Morrison D. K. Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br J. Cancer. 2018, 118 (1), 3–8. 10.1038/bjc.2017.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles O. J.; Drozd A.; Tee L.; Beggs A. D.; Middleton G. W. Paradox breaker BRAF inhibitors have comparable potency and MAPK pathway reactivation to encorafenib in BRAF mutant colorectal cancer. Oncotarget 2020, 11 (34), 3188–3197. 10.18632/oncotarget.27681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutuka C. S. A.; Andrews M. C.; Mariadason J. M.; Ioannidis P.; Hudson C.; Cebon J.; Behren A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16 (1), 112. 10.1186/s12943-017-0684-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Wang Z.; Guo P.; Ding N. Electrostatic mechanism of V600E mutation-induced B-Raf constitutive activation in colorectal cancer: molecular implications for the selectivity difference between type-I and type-II inhibitors. Eur. Biophys J. 2019, 48 (1), 73–82. 10.1007/s00249-018-1334-y. [DOI] [PubMed] [Google Scholar]
- Koumaki K.; Kontogianni G.; Kosmidou V.; Pahitsa F.; Kritsi E.; Zervou M.; Chatziioannou A.; Souliotis V. L.; Papadodima O.; Pintzas A. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Ε CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim Biophys Acta Mol. Basis Dis 2021, 1867 (4), 166061. 10.1016/j.bbadis.2020.166061. [DOI] [PubMed] [Google Scholar]
- Basile K. J.; Le K.; Hartsough E. J.; Aplin A. E. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigm. Cell Melanoma Res. 2014, 27 (3), 479–84. 10.1111/pcmr.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Chen Y.; Zhou X.; Gu Y.; Qian W.; Zhang F.; Han W.; Lu T.; Tang W. Design, synthesis and biological evaluation of bis-aryl ureas and amides based on 2-amino-3-purinylpyridine scaffold as DFG-out B-Raf kinase inhibitors. Eur. J. Med. Chem. 2015, 89, 581–596. 10.1016/j.ejmech.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Bertazza L.; Barollo S.; Radu C. M.; Cavedon E.; Simioni P.; Faggian D.; Plebani M.; Pelizzo M. R.; Rubin B.; Boscaro M.; Pezzani R.; Mian C. Synergistic antitumour activity of RAF265 and ZSTK474 on human TT medullary thyroid cancer cells. J. Cell. Mol. Med. 2015, 19 (9), 2244–52. 10.1111/jcmm.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummer T.; McInnes C. RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene. 2020, 39 (21), 4155–4169. 10.1038/s41388-020-1263-y. [DOI] [PubMed] [Google Scholar]
- Huo K.-G.; Notsuda H.; Fang Z.; Liu N. F.; Gebregiworgis T.; Li Q.; Pham N.-A.; Li M.; Liu N.; Shepherd F. A.; Marshall C. B.; Ikura M.; Moghal N.; Tsao M.-S. Lung Cancer Driven by BRAFG469V Mutation Is Targetable by EGFR Kinase Inhibitors. J. Thorac. Oncol. 2022, 17 (2), 277–288. 10.1016/j.jtho.2021.09.008. [DOI] [PubMed] [Google Scholar]
- Wang P.-F.; Qiu H.-Y.; Zhu H.-L. A patent review of BRAF inhibitors: 2013–2018. Expert Opin. Ther. Pat. 2019, 29 (8), 595–603. 10.1080/13543776.2019.1640680. [DOI] [PubMed] [Google Scholar]
- Eroglu Z.; Ozgun A. Updates and challenges on treatment with BRAF/MEK-inhibitors in melanoma. Expert Opin. Orphan Drugs 2018, 6 (9), 545–551. 10.1080/21678707.2018.1512402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney G. T.; Messina J. L.; Fedorenko I. V.; Sondak V. K.; Smalley K. S. Paradoxical oncogenesis—the long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10 (7), 390–399. 10.1038/nrclinonc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu-Duvaz I.; Roman E.; Whittaker S. R.; Friedlos F.; Kirk R.; Scanlon I. J.; Davies L. C.; Niculescu-Duvaz D.; Marais R.; Springer C. J. Novel inhibitors of the v-raf murine sarcoma viral oncogene homologue B1 (BRAF) based on a 2,6-disubstituted pyrazine scaffold. J. Med. Chem. 2008, 51 (11), 3261–74. 10.1021/jm070776b. [DOI] [PubMed] [Google Scholar]
- Ménard D.; Niculescu-Duvaz I.; Dijkstra H. P.; Niculescu-Duvaz D.; Suijkerbuijk B. M.; Zambon A.; Nourry A.; Roman E.; Davies L.; Manne H. A.; Friedlos F.; Kirk R.; Whittaker S.; Gill A.; Taylor R. D.; Marais R.; Springer C. J. Novel potent BRAF inhibitors: toward 1 nM compounds through optimization of the central phenyl ring. J. Med. Chem. 2009, 52 (13), 3881–91. 10.1021/jm900242c. [DOI] [PubMed] [Google Scholar]
- Di Grandi M. J.; Berger D. M.; Hopper D. W.; Zhang C.; Dutia M.; Dunnick A. L.; Torres N.; Levin J. I.; Diamantidis G.; Zapf C. W.; Bloom J. D.; Hu Y.; Powell D.; Wojciechowicz D.; Collins K.; Frommer E. Novel pyrazolopyrimidines as highly potent B-Raf inhibitors.. Bioorg. Med. Chem. Lett. 2009, 19 (24), 6957–61. 10.1016/j.bmcl.2009.10.058. [DOI] [PubMed] [Google Scholar]
- Gopalsamy A.; Ciszewski G.; Shi M.; Berger D.; Hu Y.; Lee F.; Feldberg L.; Frommer E.; Kim S.; Collins K.; Wojciechowicz D.; Mallon R. Hit to lead optimization of pyrazolo[1,5-a]pyrimidines as B-Raf kinase inhibitors.. Bioorg. Med. Chem. Lett. 2009, 19 (24), 6890–2. 10.1016/j.bmcl.2009.10.074. [DOI] [PubMed] [Google Scholar]
- Gopalsamy A.; Ciszewski G.; Hu Y.; Lee F.; Feldberg L.; Frommer E.; Kim S.; Collins K.; Wojciechowicz D.; Mallon R. Identification of pyrazolo[1,5-a]pyrimidine-3-carboxylates as B-Raf kinase inhibitors.. Bioorg. Med. Chem. Lett. 2009, 19 (10), 2735–8. 10.1016/j.bmcl.2009.03.129. [DOI] [PubMed] [Google Scholar]
- Lyne P. D.; Aquila B.; Cook D. J.; Dakin L. A.; Ezhuthachan J.; Ioannidis S.; Pontz T.; Su M.; Ye Q.; Zheng X.; Block M. H.; Cowen S.; Deegan T. L.; Lee J. W.; Scott D. A.; Custeau D.; Drew L.; Poondru S.; Shen M.; Wu A. Identification of amidoheteroaryls as potent inhibitors of mutant (V600E) B-Raf kinase with in vivo activity. Bioorg. Med. Chem. Lett. 2009, 19 (3), 1026–9. 10.1016/j.bmcl.2008.10.053. [DOI] [PubMed] [Google Scholar]
- Nourry A.; Zambon A.; Davies L.; Niculescu-Duvaz I.; Dijkstra H. P.; Ménard D.; Gaulon C.; Niculescu-Duvaz D.; Suijkerbuijk B. M.; Friedlos F.; Manne H. A.; Kirk R.; Whittaker S.; Marais R.; Springer C. J. BRAF inhibitors based on an imidazo[4,5]pyridin-2-one scaffold and a meta substituted middle ring. J. Med. Chem. 2010, 53 (5), 1964–78. 10.1021/jm901509a. [DOI] [PubMed] [Google Scholar]
- Wang X.; Berger D. M.; Salaski E. J.; Torres N.; Dutia M.; Hanna C.; Hu Y.; Levin J. I.; Powell D.; Wojciechowicz D.; Collins K.; Frommer E.; Lucas J. Indazolylpyrazolopyrimidine as highly potent B-Raf inhibitors with in vivo activity. J. Med. Chem. 2010, 53 (21), 7874–8. 10.1021/jm1007566. [DOI] [PubMed] [Google Scholar]
- Dietrich J.; Gokhale V.; Wang X.; Hurley L. H.; Flynn G. A. Application of a novel [3 + 2] cycloaddition reaction to prepare substituted imidazoles and their use in the design of potent DFG-out allosteric B-Raf inhibitors. Bioorg. Med. Chem. Lett. 2010, 18 (1), 292–304. 10.1016/j.bmc.2009.10.055. [DOI] [PubMed] [Google Scholar]
- Suijkerbuijk B. M.; Niculescu-Duvaz I.; Gaulon C.; Dijkstra H. P.; Niculescu-Duvaz D.; Ménard D.; Zambon A.; Nourry A.; Davies L.; Manne H. A.; Friedlos F.; Ogilvie L. M.; Hedley D.; Lopes F.; Preece N. P.; Moreno-Farre J.; Raynaud F. I.; Kirk R.; Whittaker S.; Marais R.; Springer C. J. Development of novel, highly potent inhibitors of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF): increasing cellular potency through optimization of a distal heteroaromatic group. J. Med. Chem. 2010, 53 (7), 2741–56. 10.1021/jm900607f. [DOI] [PubMed] [Google Scholar]
- Duffey M. O.; Adams R.; Blackburn C.; Chau R. W.; Chen S.; Galvin K. M.; Garcia K.; Gould A. E.; Greenspan P. D.; Harrison S.; Huang S. C.; Kim M. S.; Kulkarni B.; Langston S.; Liu J. X.; Ma L. T.; Menon S.; Nagayoshi M.; Rowland R. S.; Vos T. J.; Xu T.; Yang J. J.; Yu S.; Zhang Q. Discovery and optimization of pyrazoline compounds as B-Raf inhibitors. Bioorg. Med. Chem. Lett. 2010, 20 (16), 4800–4. 10.1016/j.bmcl.2010.06.113. [DOI] [PubMed] [Google Scholar]
- Lee J.; Kim H.; Yu H.; Chung J. Y.; Oh C. H.; Yoo K. H.; Sim T.; Hah J. M. Discovery and initial SAR of pyrimidin-4-yl-1H-imidazole derivatives with anti-proliferative activity against melanoma cell lines. Bioorg. Med. Chem. Lett. 2010, 20 (5), 1573–7. 10.1016/j.bmcl.2010.01.064. [DOI] [PubMed] [Google Scholar]
- Ramurthy S.; Aikawa M.; Amiri P.; Costales A.; Hashash A.; Jansen J. M.; Lin S.; Ma S.; Renhowe P. A.; Shafer C. M.; Subramanian S.; Sung L.; Verhagen J. Design and synthesis of 5,6-fused heterocyclic amides as Raf kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21 (11), 3286–9. 10.1016/j.bmcl.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Wang X.; Salaski E. J.; Berger D. M.; Powell D.; Hu Y.; Wojciechowicz D.; Collins K.; Frommer E. Structure-based design of isoindoline-1,3-diones and 2,3-dihydrophthalazine-1,4-diones as novel B-Raf inhibitors.. Bioorg. Med. Chem. Lett. 2011, 21 (23), 6941–4. 10.1016/j.bmcl.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Kim M. H.; Kim M.; Yu H.; Kim H.; Yoo K. H.; Sim T.; Hah J. M. Structure based design and syntheses of amino-1H-pyrazole amide derivatives as selective Raf kinase inhibitors in melanoma cells. Bioorg. Med. Chem. 2011, 19 (6), 1915–23. 10.1016/j.bmc.2011.01.067. [DOI] [PubMed] [Google Scholar]
- Ramurthy S.; Costales A.; Jansen J. M.; Levine B.; Renhowe P. A.; Shafer C. M.; Subramanian S. Design and synthesis of 6, 6-fused heterocyclic amides as raf kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22 (4), 1678–1681. 10.1016/j.bmcl.2011.12.112. [DOI] [PubMed] [Google Scholar]
- Liu J. J.; Zhang H.; Sun J.; Wang Z. C.; Yang Y. S.; Li D. D.; Zhang F.; Gong H. B.; Zhu H. L. Synthesis, biological evaluation of novel 4,5-dihydro-2H-pyrazole 2-hydroxyphenyl derivatives as BRAF inhibitors. Bioorg. Med. Chem. 2012, 20 (20), 6089–96. 10.1016/j.bmc.2012.08.020. [DOI] [PubMed] [Google Scholar]
- Li Q. S.; Li C. Y.; Lu X.; Zhang H.; Zhu H. L. Design, synthesis and biological evaluation of novel (E)-α-benzylsulfonyl chalcone derivatives as potential BRAF inhibitors. Eur. J. Med. Chem. 2012, 50, 288–95. 10.1016/j.ejmech.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Li Q. S.; Lv X. H.; Zhang Y. B.; Dong J. J.; Zhou W. P.; Yang Y.; Zhu H. L. Identification of novel 3,5-diarylpyrazoline derivatives containing salicylamide moiety as potential anti-melanoma agents. Bioorg. Med. Chem. lett. 2012, 22 (21), 6596–601. 10.1016/j.bmcl.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Li C. Y.; Li Q. S.; Yan L.; Sun X. G.; Wei R.; Gong H. B.; Zhu H. L. Synthesis, biological evaluation and 3D-QSAR studies of novel 4,5-dihydro-1H-pyrazole niacinamide derivatives as BRAF inhibitors. Bioorg. Med. Chem. 2012, 20 (12), 3746–55. 10.1016/j.bmc.2012.04.047. [DOI] [PubMed] [Google Scholar]
- Yang Y. S.; Li Q. S.; Sun S.; Zhang Y. B.; Wang X. L.; Zhang F.; Tang J. F.; Zhu H. L. Design, modification and 3D QSAR studies of novel 2,3-dihydrobenzo[b][1,4]dioxin-containing 4,5-dihydro-1H-pyrazole derivatives as inhibitors of B-Raf kinase. Bioorg. Med. Chem. 2012, 20 (20), 6048–58. 10.1016/j.bmc.2012.08.043. [DOI] [PubMed] [Google Scholar]
- Niculescu-Duvaz D.; Niculescu-Duvaz I.; Suijkerbuijk B. M.; Ménard D.; Zambon A.; Davies L.; Pons J. F.; Whittaker S.; Marais R.; Springer C. J. Potent BRAF kinase inhibitors based on 2,4,5-trisubstituted imidazole with naphthyl and benzothiophene 4-substituents. Bioorg. Med. Chem. 2013, 21 (5), 1284–304. 10.1016/j.bmc.2012.12.035. [DOI] [PubMed] [Google Scholar]
- Dong J. J.; Li Q. S.; Wang S. F.; Li C. Y.; Zhao X.; Qiu H. Y.; Zhao M. Y.; Zhu H. L. Synthesis, biological evaluation and molecular docking of novel 5-phenyl-1H-pyrazol derivatives as potential BRAF(V600E) inhibitors. Org. Biomol. Chem. 2013, 11 (37), 6328–37. 10.1039/c3ob40776d. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Chen X.; Qin J.; Kong X.; Ye F.; Jiang Y.; Liu H.; Jiang H.; Marmorstein R.; Luo C. Identification and synthesis of N-(thiophen-2-yl) benzamide derivatives as BRAF(V600E) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23 (8), 2306–12. 10.1016/j.bmcl.2013.02.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y.-J.; Xing M.; Zhang Y.-L.; Makawana J. A.; Jiang A.-Q.; Zhu H.-L. Design, synthesis and biological evaluation of (1, 3-diphenyl-1 H-pyrazol-4-yl) methyl benzoate derivatives as potential BRAF V600E inhibitors. RSC Adv. 2014, 4 (95), 52702–52711. 10.1039/C4RA08708A. [DOI] [Google Scholar]
- Wang S. F.; Zhu Y. L.; Zhu P. T.; Makawana J. A.; Zhang Y. L.; Zhao M. Y.; Lv P. C.; Zhu H. L. Design, synthesis and biological evaluation of novel 5-phenyl-1H-pyrazole derivatives as potential BRAF(V600E) inhibitors. Bioorg. Med. Chem. 2014, 22 (21), 6201–8. 10.1016/j.bmc.2014.08.029. [DOI] [PubMed] [Google Scholar]
- Jiao Y.; Xin B. T.; Zhang Y.; Wu J.; Lu X.; Zheng Y.; Tang W.; Zhou X. Design, synthesis and evaluation of novel 2-(1H-imidazol-2-yl) pyridine Sorafenib derivatives as potential BRAF inhibitors and anti-tumor agents. Eur. J. Med. Chem. 2015, 90, 170–83. 10.1016/j.ejmech.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Kim M.; Lee J.; Jung K.; Kim H.; Aman W.; Ryu J. S.; Hah J. M. Design, synthesis and biological evaluation of benzyl 2-(1H-imidazole-1-yl) pyrimidine analogues as selective and potent Raf inhibitors. Bioorg. Med. Chem. 2014, 24 (15), 3600–4. 10.1016/j.bmcl.2014.05.030. [DOI] [PubMed] [Google Scholar]
- Yang W.; Chen Y.; Zhou X.; Gu Y.; Qian W.; Zhang F.; Han W.; Lu T.; Tang W. Design, synthesis and biological evaluation of bis-aryl ureas and amides based on 2-amino-3-purinylpyridine scaffold as DFG-out B-Raf kinase inhibitors. Eur. J. Med. Chem. 2015, 89, 581–96. 10.1016/j.ejmech.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Yang Y. S.; Zhang F.; Tang D. J.; Yang Y. H.; Zhu H. L. Modification, biological evaluation and 3D QSAR studies of novel 2-(1,3-diaryl- 4,5-dihydro-1H-pyrazol-5-yl)phenol derivatives as inhibitors of B-Raf kinase. PloS one 2014, 9 (5), e95702. 10.1371/journal.pone.0095702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M. Y.; Yin Y.; Yu X. W.; Sangani C. B.; Wang S. F.; Lu A. M.; Yang L. F.; Lv P. C.; Jiang M. G.; Zhu H. L. Synthesis, biological evaluation and 3D-QSAR study of novel 4,5-dihydro-1H-pyrazole thiazole derivatives as BRAF(V600E) inhibitors. Bioorg. Med. Chem. 2015, 23 (1), 46–54. 10.1016/j.bmc.2014.11.029. [DOI] [PubMed] [Google Scholar]
- Li Q. S.; Lü X. H.; Yang Y.; Ruan B. F.; Yao R. S.; Liao C. Z. Discovery and pharmacophore studies of novel pyrazole-based anti-melanoma agents. Chem. Biodiversity 2015, 12 (1), 116–32. 10.1002/cbdv.201400143. [DOI] [PubMed] [Google Scholar]
- Diez-Cecilia E.; Carson R.; Kelly B.; van Schaeybroeck S.; Murray J. T.; Rozas I. Probing a 3,4’-bis-guanidinium diaryl derivative as an allosteric inhibitor of the Ras pathway. Bioorg. Med. Chem. Lett. 2015, 25 (19), 4287–92. 10.1016/j.bmcl.2015.07.082. [DOI] [PubMed] [Google Scholar]
- Chen L. W.; Wang Z. F.; Zhu B.; Man R. J.; Liu Y. D.; Zhang Y. H.; Wang B. Z.; Wang Z. C.; Zhu H. L. Design, synthesis and biological evaluation of novel benzo-α-pyrone containing piperazine derivatives as potential BRAF(V600E) inhibitors. Bioorg. Med. Chem. Lett. 2016, 26 (20), 4983–4991. 10.1016/j.bmcl.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Yang Y. S.; Yang B.; Zou Y.; Li G.; Zhu H. L. Design, biological evaluation and 3D QSAR studies of novel dioxin-containing triaryl pyrazoline derivatives as potential B-Raf inhibitors. Bioorg. Med. Chem. 2016, 24 (13), 3052–3061. 10.1016/j.bmc.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Khan M. A.; El-Gamal M. I.; Tarazi H.; Choi H. S.; Oh C. H. Design and synthesis of a new series of highly potent RAF kinase-inhibiting triarylpyrazole derivatives possessing anti-proliferative activity against melanoma cells. Future Med. Chem. 2016, 8 (18), 2197–2211. 10.4155/fmc-2016-0057. [DOI] [PubMed] [Google Scholar]
- Gamal El-Din M. M.; El-Gamal M. I.; Abdel-Maksoud M. S.; Yoo K. H.; Oh C. H. Design, synthesis, broad-spectrum anti-proliferative activity, and kinase inhibitory effect of triarylpyrazole derivatives possessing arylamides or arylureas moieties. Eur. J. Med. Chem. 2016, 119, 122–31. 10.1016/j.ejmech.2016.04.048. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wang L.; Zhang Q.; Zhu G.; Zhang Z.; Zhou X.; Chen Y.; Lu T.; Tang W. Potent Pan-Raf and Receptor Tyrosine Kinase Inhibitors Based on a Cyclopropyl Formamide Fragment Overcome Resistance. J. Chem. Inf. Model. 2017, 57 (6), 1439–1452. 10.1021/acs.jcim.6b00795. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhu G.; Zhang Q.; Duan C.; Zhang Y.; Zhang Z.; Zhou Y.; Lu T.; Tang W. Rational design, synthesis, and biological evaluation of Pan-Raf inhibitors to overcome resistance. Org. Biomol. Chem. 2017, 15 (16), 3455–3465. 10.1039/C7OB00518K. [DOI] [PubMed] [Google Scholar]
- Gong Z.-H.; Yao J.; Ji J.-F.; Yang J.; Xiang T.; Zhou C.-K. Synthesis and biological evaluation of novel N-(5-phenyl-1H-pyrazol-3-yl) benzenesulfonamide derivatives as potential BRAFV600E inhibitors. Med. Chem. Res. 2017, 26 (10), 2583–2591. 10.1007/s00044-017-1957-z. [DOI] [Google Scholar]
- Wang H. Y.; Shen Y.; Zhang H.; Hei Y. Y.; Zhao H. Y.; Xin M.; Lu S. M.; Zhang S. Q. Discovery of 2-(aminopyrimidin-5-yl)-4-(morpholin-4-yl)-6- substituted triazine as PI3K and BRAF dual inhibitor. Future Med. Chem. 2018, 10 (20), 2445–2455. 10.4155/fmc-2018-0145. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wan S.; Li Z.; Fu Y.; Wang G.; Zhang J.; Wu X. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d]pyrimidine derivatives as BRAF(V600E) and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2018, 155, 210–228. 10.1016/j.ejmech.2018.05.054. [DOI] [PubMed] [Google Scholar]
- Wang C. R.; Wang Z. F.; Shi L.; Wang Z. C.; Zhu H. L. Design, synthesis, and biological evaluation of pyrazole derivatives containing acetamide bond as potential BRAF(V600E) inhibitors. Bioorg. Med. Chem. Lett. 2018, 28 (14), 2382–2390. 10.1016/j.bmcl.2018.06.028. [DOI] [PubMed] [Google Scholar]
- Youssif B. G. M.; Abdelrahman M. H.; Abdelazeem A. H.; Abdelgawad M. A.; Ibrahim H. M.; Salem O. I. A.; Mohamed M. F. A.; Treambleau L.; Bukhari S. N. A. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem. 2018, 146, 260–273. 10.1016/j.ejmech.2018.01.042. [DOI] [PubMed] [Google Scholar]
- El-Sherief H. A. M.; Youssif B. G. M.; Bukhari S. N. A.; Abdel-Aziz M.; Abdel-Rahman H. M. Novel 1,2,4-triazole derivatives as potential anticancer agents: Design, synthesis, molecular docking and mechanistic studies. Bioorg. Chem. 2018, 76, 314–325. 10.1016/j.bioorg.2017.12.013. [DOI] [PubMed] [Google Scholar]
- Wang P. F.; Wang Z. F.; Qiu H. Y.; Huang Y.; Hu H. M.; Wang Z. C.; Zhu H. L. Identification and biological evaluation of novel type II B-RafV600E inhibitors. ChemMedChem. 2018, 13 (23), 2558–2566. 10.1002/cmdc.201800574. [DOI] [PubMed] [Google Scholar]
- Wang P. F.; Zhang Y. J.; Wang D.; Hu H. M.; Wang Z. C.; Xu C.; Qiu H. Y.; Zhu H. L. Design, synthesis, and biological evaluation of new B-Raf(V600E) kinase inhibitors. Bioorg. Med. Chem. 2018, 26 (9), 2372–2380. 10.1016/j.bmc.2018.03.038. [DOI] [PubMed] [Google Scholar]
- Sun S.; He Z.; Huang M.; Wang N.; He Z.; Kong X.; Yao J. Design and discovery of thioether and nicotinamide containing sorafenib analogues as multikinase inhibitors targeting B-Raf, B-Raf(V600E) and VEGFR-2. Bioorg. Med. Chem. 2018, 26 (9), 2381–2391. 10.1016/j.bmc.2018.03.039. [DOI] [PubMed] [Google Scholar]
- Abdel-Maksoud M. S.; Ammar U. M.; Oh C. H. Anticancer profile of newly synthesized BRAF inhibitors possess 5-(pyrimidin-4-yl)imidazo[2,1-b]thiazole scaffold. Bioorg. Med. Chem. 2019, 27 (10), 2041–2051. 10.1016/j.bmc.2019.03.062. [DOI] [PubMed] [Google Scholar]
- Abdel-Mohsen H. T.; Omar M. A.; El Kerdawy A. M.; Mahmoud A. E. E.; Ali M. M.; El Diwani H. I. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur. J. Med. Chem. 2019, 179, 707–722. 10.1016/j.ejmech.2019.06.063. [DOI] [PubMed] [Google Scholar]
- Jung H.; Kim J.; Im D.; Moon H.; Hah J. M. Design, synthesis, and in vitro evaluation of N-(3-(3-alkyl-1H-pyrazol-5-yl) phenyl)-aryl amide for selective RAF inhibition. Bioorg. Med. Chem. Lett. 2019, 29 (4), 534–538. 10.1016/j.bmcl.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Kim J.; Choi B.; Im D.; Jung H.; Moon H.; Aman W.; Hah J. M. Computer-aided design and synthesis of 3-carbonyl-5-phenyl-1H-pyrazole as highly selective and potent BRAFV600E and CRAF inhibitor. J. Enzyme Inhib. Med. Chem. 2019, 34 (1), 1314–1320. 10.1080/14756366.2019.1599366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Maksoud M. S.; Ammar U. M.; El-Gamal M. I.; Gamal El-Din M. M.; Mersal K. I.; Ali E. M. H.; Yoo K. H.; Lee K. T.; Oh C. H. Design, synthesis, and anticancer activity of imidazo[2,1-b]oxazole-based RAF kinase inhibitors. Bioorg. Chem. 2019, 93, 103349. 10.1016/j.bioorg.2019.103349. [DOI] [PubMed] [Google Scholar]
- Abdel-Maksoud M. S.; Ali E. M. H.; Ammar U. M.; Mersal K. I.; Yoo K. H.; Oh C. H. Design and synthesis of novel pyrrolo[2,3-b]pyridine derivatives targeting (V600E)BRAF. Bioorg. Med. Chem. 2020, 28 (11), 115493. 10.1016/j.bmc.2020.115493. [DOI] [PubMed] [Google Scholar]
- Ali E. M. H.; Abdel-Maksoud M. S.; Ammar U. M.; Mersal K. I.; Ho Yoo K.; Jooryeong P.; Oh C. H. Design, synthesis, and biological evaluation of novel imidazole derivatives possessing terminal sulphonamides as potential BRAF(V600E)inhibitors. Bioorg. Chem. 2021, 106, 104508. 10.1016/j.bioorg.2020.104508. [DOI] [PubMed] [Google Scholar]
- Feng J.; Li T.; Liang S.; Zhang C.; Tan X.; Ding N.; Wang X.; Liu X.; Hu C. Synthesis and biological evaluation of a new series of 1-aryl-3-[4-(pyridin-2-ylmethoxy)phenyl]urea derivatives as new anticancer agents. Med Chem Res 2020, 29 (8), 1413–1423. 10.1007/s00044-020-02554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohassab A. M.; Hassan H. A.; Abdelhamid D.; Gouda A. M.; Youssif B. G. M.; Tateishi H.; Fujita M.; Otsuka M.; Abdel-Aziz M. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent anti-proliferative agent targeting EGFR and BRAF(V600E) kinases. Bioorg. Chem. 2021, 106, 104510. 10.1016/j.bioorg.2020.104510. [DOI] [PubMed] [Google Scholar]
- Mohamed A. R.; El Kerdawy A. M.; George R. F.; Georgey H. H.; Abdel Gawad N. M. Design, synthesis and in silico insights of new 7,8-disubstituted-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione derivatives with potent anticancer and multi-kinase inhibitory activities. Bioorg. Chem. 2021, 107, 104569. 10.1016/j.bioorg.2020.104569. [DOI] [PubMed] [Google Scholar]
- El-Damasy A. K.; Haque M. M.; Park J. W.; Shin S. C.; Lee J. S.; EunKyeong Kim E.; Keum G. 2-Anilinoquinoline based arylamides as broad spectrum anticancer agents with B-RAF(V600E)/C-RAF kinase inhibitory effects: Design, synthesis, in vitro cell-based and oncogenic kinase assessments. Eur. J. Med. Chem. 2020, 208, 112756. 10.1016/j.ejmech.2020.112756. [DOI] [PubMed] [Google Scholar]
- Ammar U. M.; Abdel-Maksoud M. S.; Ali E. M. H.; Mersal K. I.; Ho Yoo K.; Oh C. H. Structural optimization of imidazothiazole derivatives affords a new promising series as B-Raf V600E inhibitors; synthesis, in vitro assay and in silico screening. Bioorg. Chem. 2020, 100, 103967. 10.1016/j.bioorg.2020.103967. [DOI] [PubMed] [Google Scholar]
- Ali E. M. H.; El-Telbany R. F. A.; Abdel-Maksoud M. S.; Ammar U. M.; Mersal K. I.; Zaraei S. O.; El-Gamal M. I.; Choi S. I.; Lee K. T.; Kim H. K.; Lee K. H.; Oh C. H. Design, synthesis, biological evaluation, and docking studies of novel (imidazol-5-yl)pyrimidine-based derivatives as dual BRAF(V600E)/p38α inhibitors. Eur. J. Med. Chem. 2021, 215, 113277. 10.1016/j.ejmech.2021.113277. [DOI] [PubMed] [Google Scholar]
- Abdel-Maksoud M. S.; El-Gamal M. I.; Lee B. S.; Gamal El-Din M. M.; Jeon H. R.; Kwon D.; Ammar U. M.; Mersal K. I.; Ali E. M. H.; Lee K. T.; Yoo K. H.; Han D. K.; Lee J. K.; Kim G.; Choi H. S.; Kwon Y. J.; Lee K. H.; Oh C. H. Discovery of New Imidazo[2,1-b]thiazole Derivatives as Potent Pan-RAF Inhibitors with Promising In Vitro and In Vivo Anti-melanoma Activity. J. Med. Chem. 2021, 64 (10), 6877–6901. 10.1021/acs.jmedchem.1c00230. [DOI] [PubMed] [Google Scholar]
- Lee C. I.; Liao C. B.; Chen C. S.; Cheng F. Y.; Chung Y. H.; Wang Y. C.; Ciou S. Y.; Hsueh W. Y.; Lo T. H.; Huang G. R.; Huang H. Y.; Tsai C. S.; Lu Y. J.; Chuang S. H.; Huang J. J. Design and synthesis of 4-anilinoquinazolines as Raf kinase inhibitors. Part 1. Selective B-Raf/B-Raf(V600E) and potent EGFR/VEGFR2 inhibitory 4-(3-hydroxyanilino)-6-(1H-1,2,3-triazol-4-yl)quinazolines. Bioorg. Chem. 2021, 109, 104715. 10.1016/j.bioorg.2021.104715. [DOI] [PubMed] [Google Scholar]
- Ali E. M. H.; Mersal K. I.; Ammar U. M.; Zaraei S. O.; Abdel-Maksoud M. S.; El-Gamal M. I.; Haque M. M.; Das T.; Kim E. E.; Lee J. S.; Lee K. H.; Kim H. K.; Oh C. H. Structural optimization of 4-(imidazol-5-yl)pyridine derivatives affords broad-spectrum anticancer agents with selective B-RAF(V600E)/p38α kinase inhibitory activity: Synthesis, in vitro assays and in silico study. Eur. J. Pharm. Sci. 2022, 171, 106115. 10.1016/j.ejps.2022.106115. [DOI] [PubMed] [Google Scholar]