

ABSTRACT
Small molecules are components of fungal extracellular vesicles (EVs), but their biological roles are only superficially known. NOP16 is a eukaryotic gene that is required for the activity of benzimidazoles against Cryptococcus deuterogattii. In this study, during the phenotypic characterization of C. deuterogattii mutants expected to lack NOP16 expression, we observed a reduced EV production. Whole-genome sequencing, RNA-Seq, and cellular proteomics revealed that, contrary to our initial findings, these mutants expressed Nop16 but exhibited altered expression of 14 genes potentially involved in sugar transport. Based on this observation, we designated these mutant strains as Past1 and Past2, representing potentially altered sugar transport. Analysis of the small molecule composition of EVs produced by wild-type cells and the Past1 and Past2 mutant strains revealed not only a reduced number of EVs but also an altered small molecule composition. In a Galleria mellonella model of infection, the Past1 and Past2 mutant strains were hypovirulent. The hypovirulent phenotype was reverted when EVs produced by wild-type cells, but not mutant EVs, were co-injected with the mutant cells in G. mellonella. These results connect EV biogenesis, cargo, and cryptococcal virulence.
KEYWORDS: Cryptococcus, extracellular vesicles
INTRODUCTION
Extracellular vesicles (EVs) are membranous structures produced by all domains of life (1). In microorganisms, EVs participate in processes of immunopathogenesis, cell-to-cell communication, cellular differentiation, and antimicrobial resistance, among others [reviewed in reference (2)]. Fungal EVs were first characterized in the Cryptococcus genus (3). In this model, EVs have immunostimulatory activity (4) and vaccinal potential (5). Cryptococcal EVs are also required for fungal virulence (6) and intraspecies communication (7).
As widely discussed in the literature, several questions related to fungal EVs remain unanswered (8). The mechanisms of biogenesis of fungal EVs are still poorly known, which impairs the design of experimental models for the study of their functions. Although much progress has been made in the identification of the protein and nucleic acid components of fungal EVs (5, 9–12), their small molecule composition was only recently addressed in C. deuterogattii (13), Penicillium digitatum (14), P. chrysogenum (15), and Histoplasma capsulatum (16). In C. deuterogattii, small molecule analysis of EVs revealed the presence of a peptide controlling infection in the Galleria mellonella model (13). A possible relationship between small molecule composition and fungal virulence remains to be determined, as well as the genes regulating the formation of fungal EVs and their small molecule cargo.
NOP16 is the gene encoding nucleolar protein 16. In humans, Nop16 is expressed in 237 different tissues (17), but its functions are poorly known. In Saccharomyces cerevisiae, Nop16 is a constituent of 66S pre-ribosomal particles, with involvement in 60S ribosomal subunit biogenesis (18, 19). There is no evidence in the literature pointing to the roles of Nop16 in the formation of EVs, although ribosomal proteins are abundant in cryptococcal EVs (5, 20).
During the search for novel anti-cryptococcal agents, we found that Nop16 was involved in the antifungal activity of mebendazole by still unknown mechanisms (21). In this study, we report on the generation and characterization of two independent mutants of C. deuterogattii initially thought to lack expression of Nop16 but lately identified to have altered expression of genes involved in sugar transport. In comparison to wild-type (WT) cells, the mutants were hypovirulent in a G. mellonella model of infection, although they shared many phenotypic properties with WT cells including growth rates, ultrastructural features, capsule formation, and susceptibility to phagocytosis. The mutants were less efficient in producing EVs, in association with an altered small molecule cargo. Infection of G. mellonella with the mutant strains in the presence of EVs produced by parental cells resulted in increased virulence, suggesting a role for sugar transport in EV biogenesis and cargo. These results also suggest that the composition of EVs can be a determinant for the virulence of C. deuterogattii.
RESULTS
Generation of the C. deuterogattii mutants
We previously identified the NOP16 (CNBG_3695) gene in C. deuterogattii as a potential target for antifungal development (21) and aimed at generating null mutants by employing the Delsgate methodology, as previously described (22). Tentative NOP16 gene inactivation allele was performed by employing the vector pDONR-NAT (22). Amplification of the NOP16 gene flanking regions and the internal fragment was performed with the primers that are listed in Table 1. Two potentially null mutants were identified based on the absence of the amplification with diagnostic primers (Fig. 1A and B) and decreased sensitivity to mebendazole (Fig. 1C), based on the previously described association between the antifungal activity of benzimidazoles and NOP16 expression (21). These mutants were then denominated nop16Δ.1 and nop16Δ.2.
TABLE 1.
Primers used in this study
| Primer name | Sequence (5′−3′) | Purpose |
|---|---|---|
| NOP16_5F | AAAATAGGGATSACAGGGTAATGAAGATCCTTGAAGTGCTCTGG | Disruption construct for NOP16, 5′ flank |
| NOP16_5R | GGGGACAAGTTTGTSCAAAAAAGCAGGCTATCCTCAACGTGGAATCTGCTATC | |
| NOP16_3F | GGGGACCACTTTGTACAAGAAAGCTGGGTAACTTCTTCTGCGGTACTGTGTG | Disruption construct for NOP16, 3′ flank |
| NOP16_3R | AAAAATTACCCTGTTATCCCTACCCTAGCCAGCTGTAAACTC | |
| NOP16_IF | GAGGACTGAGGTCGTCAAGAG | Confirmation of NOP16 disruption |
| NOP16_IR | CAGTGTCGTCTCCGTATCTATC | |
| ACTF | CGGTATCGTCACAAACTGG | Amplification of ACT1 |
Fig 1.

Tentative generation of null mutants of NOP16 in C. deuterogattii. (A) NOP16 inactivation strategy. TV is the representation of the targeting vector constructed by the Delsgate methodology, with 5 NOP16 and 3 NOP16 representing the 5′ and 3′ gene flanks of the NOP16 gene, respectively. The primers used to amplify the 5′ (5F and 5R) and 3′ (3F and 3R) of NOP16 are represented as arrowheads. NATR is the cassette that confers nourseothricin resistance. (B) Nourseothricin-resistant cells were evaluated for the presence of the NOP16 gene using internal diagnostic primers (IF and IR), using the ACT gene as a loading control. (C) WT and two null mutants were evaluated for their sensitivity to mebendazole. Bars represent the average of the ratio between growth in 1 mM mebendazole normalized to the growth in a drug-free medium obtained in three independent experiments. Mutant cells display decreased sensitivity to mebendazole (**P < 0.01 and ***P < 0.0001) as revealed by analysis of variance followed by Dunnett’s multiple-comparison analysis. In contrast to what these results suggested, NOP16 deletion was not achieved. Please see Results and Fig. 2 to 8 for a detailed explanation and further correction.
We initially assumed that these mutants lacked the expression of NOP16. However, in a follow-up approach, we performed an RNA-Seq analysis to understand at the molecular level what was changed in the mutants. Total RNA was isolated from the WT strain of C. deuterogattii as well as from the two generated mutants from cultures in YPD medium (1% yeast extract, 2% peptone, and 2% dextrose). After quality trimming with fastp (23) and alignment to the C. deuterogattii R265 genome sequence using STAR (24), the transcriptome profile was observed using the Integrative Genomics Viewer (IGV) (25). Unexpectedly, we detected transcripts that span the entire NOP16 (CNBG_3695) gene in both WT cells and the generated mutants (Fig. 2A).
Fig 2.

Transcriptional profile (A) and genomic structure (B) of the NOP16 (CNBG_3695) gene in the analyzed strains. (A) IGV panel illustrating the coverage of aligned RNA-Seq reads (read cov) of the WT (upper), nop16.1 (middle), and nop16.2 (bottom) strains. The blue arrows below the histogram panel represent the gene architecture. (B) Genomic structure of the NOP16 (CNBG_3695) locus in the analyzed strains. IGV panel illustrating the coverage of aligned DNA Seq reads (read cov) of the WT (upper), nop16.1 (middle), and nop16.2 (bottom) mutants. The blue arrows below the histogram panel represent the gene architecture.
This unexpected finding guided us to a closer examination of the primer sequences used to construct the inactivation plasmid. This analysis revealed that they were based on the flanking sequences of the gene CNBG_3591, which codes for a protein predicted (by sequence comparison) to function as a peptide chain release factor. As we could not detect the presence of a segment of the NOP16 gene in the generated mutants by PCR, we hypothesized that the PCR results could be a false negative. To overcome the potential limitations of PCR diagnosis, we determined the genome sequence of the WT and two mutant strains. To confirm the results obtained with the RNA-Seq analysis, which suggests that the NOP16 (CNBG_3695) gene is present in the mutants, we determined the genome sequence of the three strains using Illumina DNA Seq. We quality trimmed the reads with fastp (23) and aligned them to the C. deuterogattii R265 genome sequence using BWA (26). Visual inspection of the aligned reads confirmed the presence of the NOP16 (CNBG_3695) gene in both the WT strain and the generated mutants (Fig. 2B).
As the plasmid used to transform the WT strain contains sequences of the CNBG_3591 locus, we also evaluated the coverage of such genomic sequence. We confirmed that the sequences used to construct the plasmid were indeed present in higher abundance in the mutant strains compared to the WT strain (Fig. 3). To investigate the molecular events that lead to the integration of extra copies of the plasmid into the genome of C. deuterogattii R265, we used the perSVade pipeline (27) to detect structural variants (SVs) in the three analyzed strains (WT and mutants) compared to the reference genomic sequence of C. deuterogattii R265 (NCBI accession GCA_002954075.1). The pipeline allows the identification of five major types of SVs: deletions, insertions, inversions, tandem duplications, and translocations. The analysis conducted with perSVade resulted in the identification of all the aforementioned alterations (Supplementary file 1). Most of the deletions are common to the three strains compared to the reference genome sequence. Some were detected as differential. However, further visual inspection of the aligned reads confirmed that such deletions were present in all strains compared to the reference genome sequence. For instance, perSVade identified a deletion present in the mutant strains only encompassing the region between 660,117 and 660,776 of the chromosome CP025769.1. Nevertheless, visual inspection confirmed that this region is absent in the DNA Seq data from all strains analyzed here (Fig. 4A), possibly due to inherent properties of the sequence (e.g., repetitions), low quality of the sequences, or low abundance of input DNA in this region.
Fig 3.

Genomic structure of CNBG_3591 locus in the analyzed strains. IGV panel illustrating the DNA Seq aligned reads to the WT (upper), nop16.1 (middle), and nop16.2 (bottom) mutants. The blue arrows below the histogram panel represent the gene architecture of CNBG_3591 (center) and the partial architecture of Fig. 3 genomic structure of the CNBG_3591 locus in the analyzed strains. IGV panel illustrating the coverage of aligned DNA Seq reads (read cov) of the WT (upper), nop16.1 (middle), and nop16.2 (bottom) mutants. The blue arrows below the histogram panel represent the CNBG_3591 gene architecture as well as the partial maps of surrounding genes CNBG_3590 (left) and CNBG_3592 (right).
Fig 4.

Predicted deleted (A), tandem-duplicated (B) loci specific to nop16.1 and nop16.2. (A) IGV panel illustrating the coverage of aligned DNA Seq reads (read cov) of the WT (upper), nop16.1 (middle), and nop16.2 (bottom) mutants. (B) IGV panel illustrating the coverage of aligned DNA Seq reads (read cov) of the WT (upper), nop16.1 (middle), and nop16.2 (bottom) mutants.
The pipeline did not identify differential SVs related to insertions or inversions (Supplementary file 1). However, a common tandem duplication was found in the region of nucleotides 1853467–1856827 of chromosome CP025759.1. This region exactly encompasses the sequence used for the construction of the plasmid, based on flanking sequences of the gene CNBG_3591 (Fig. 4B). In this way, considering all the other potential SVs, which are predicted in regions with low coverage or with low confidence based on visual inspection of results in IGV, we concluded that the key differences between the two generated mutant strains and the WT strain are due to the tandem duplication of two segments of the genome encompassing the CNBG_3591 gene.
Aiming to understand the molecular impact of such SVs on molecular processes present in the mutant strains compared to the WT strain, we returned to our RNA-Seq analysis. Total RNA was isolated in triplicate conditions and used as input for Illumina sequencing. The reads were quality trimmed with fastp (23) and aligned to the C. deuterogattii R265 genome sequence using STAR (24). The identification of differentially expressed genes (DEGs) was performed using the R package DESeq2 (28), employing stringent criteria [absolute value of log2FC ≥1 and false discovery rate (FDR)-corrected P-value <0.05). Principal component analysis (PCA) of the normalized expression allowed us to infer that the transcriptional profiles of the three strains analyzed are, in fact, distinct when comparing the two mutants with the WT strain (Fig. 5A). In addition, we detected 21 DEGs when comparing the transcriptional profile of WT vs nop16.1 and 44 DEGs when comparing the transcriptional profile of WT vs nop16.2 (Fig. 5B).
Fig 5.

Comparative analysis of gene expression in WT and mutant strains. (A) PCA of transformed data from DESeq2, highlighting the analyzed groups. (B) Venn diagram highlighting the shared and specific DEGs from the comparison between WT and nop16.1 or nop16.2 strains.
It is important to note that, despite differences in the number of DEGs, the general profile of alteration of expression is common for both strains, as revealed by the PCA (Fig. 5A) and by the analysis of volcano plots (Fig. 6A), which could also be confirmed by the full list of DEGs found (Supplementary file 2). In line with this observation, all DEGs with increased expression in WT compared to the nop16.1 mutant could also be detected as positively regulated in WT compared to the nop16.1 mutant (Fig. 6B). These 14 genes mainly code for sugar transporters (Table 2).
Fig 6.
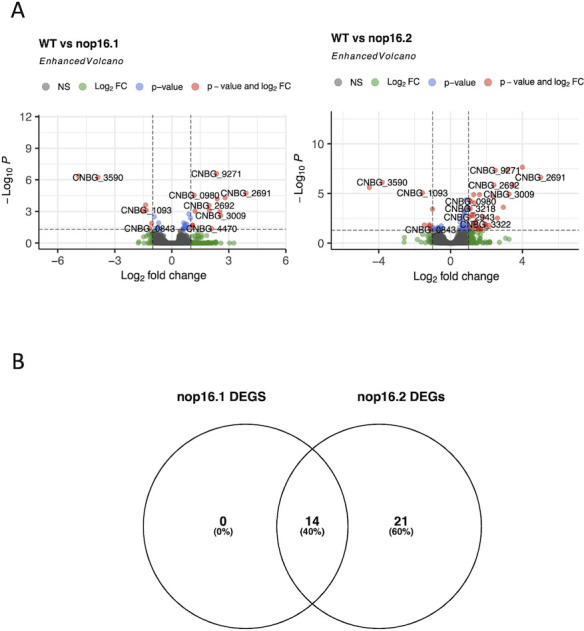
Regulation of gene expression in the mutant strains of C. deuterogattii. (A) DEGs profile presented by nop16 mutants. Data from DESeq2 were plotted using the EnhancedVolcano (7) package to evaluate the set of DEGs found between the comparison of WT and nop16.1 mutant (left) or WT and nop16.2 mutant (right). (B) Venn diagram highlighting the positively regulated genes in WT compared to nop16.1 or nop16.2 mutants.
TABLE 2.
Identity of common WT-upregulated DEGs compared in Past1 and Past2 strains
| Gene ID | Product description |
|---|---|
| CNBG_0980 | Xylitol dehydrogenase |
| CNBG_1797 | Conserved hypothetical protein |
| CNBG_2487 | Monosaccharide transporter |
| CNBG_2490 | Conserved hypothetical protein |
| CNBG_2691 | Nicotinamide mononucleotide permease |
| CNBG_2692 | Mandelate racemase/muconate lactonizing enzyme |
| CNBG_2834 | Sugar transporter |
| CNBG_3009 | Sugar transporter |
| CNBG_3725 | Sugar transporter |
| CNBG_4470 | Sugar transporter |
| CNBG_5449 | Transaldolase |
| CNBG_9239 | Unspecified product |
| CNBG_9245 | MFS domain-containing protein |
| CNBG_9271 | Glyco_hydro_79C domain-containing protein |
We also performed a proteomic analysis to gain insight into the molecular mechanisms governing the observed phenotypes in the mutants nop16.1 and nop16.2 compared to the WT strain. Total protein extracts were prepared from cells cultured in YPD and subjected to trypsinization, as previously described (29). The analysis of the extracts was performed in a nanoliquid chromatography (nLC)-tandem mass spectrometry (MS/MS) using an UltiMate 3000 RSLC system coupled to an Orbitrap Fusion Lumos mass spectrometer. Finally, the data were analyzed using PatternLab V (30, 31). While 17 proteins were detected as differentially abundant in the comparison of nop16.1 vs WT (Fig. 7; Supplementary file 3), this number is higher in the comparison between nop16.2 and WT (n = 58 – Fig. 8A; Supplementary file 4). Despite dozens of differentially abundant proteins (DAPs) being detected in both comparisons (nop16.1 vs WT and nop16.2 vs WT), a low number of them were shared between the comparisons (Fig. 8B).
Fig 7.

DAPs profiles presented by the nop16.1 mutant compared to the WT strain (A) or nop16.2 mutant compared to the WT strain (B). Data were processed using the PatternLab V pipeline and plotted after applying the TFold function. The F-stringency was set to 0.45, and the q-value cutoff value was 0.05. Blue dots represent DAPs.
Fig 8.

Venn diagrams highlighting the shared and specific DAPs from the comparison between WT and nop16.1 or nop16.2 mutants (A) and the shared and specific DAPs and DEGs from the comparison between WT and nop16.1 or nop16.2 mutants (B).
It is noteworthy that we observed no overlap between the set of DEGs and respective DAPs observed in both mutants (Fig. 8B). However, the product of the gene CNBG_3888 (ATP-dependent RNA helicase FAL1) is a DAP that is more abundant in WT cells compared to nop16.1 and nop16.2 mutants. This suggests an unbalanced ratio of transcription to translation in cells, impacting the correlation of transcripts to proteins.
Considering the above results, we assumed that the tandem duplication of the plasmid used for transformation led to a potentially altered sugar transport in the mutant cells, which ultimately could cause the phenotypic alterations observed in the so-called nop16.1 and nop16.2 mutant strains. Therefore, a major requirement for these changes is the nomenclature of the mutants. Instead of the nop16Δ.1 and nop16Δ.2 mutant strains, we adopted from this point the use of Past1 and Past2 strains, which stands for potentially altered sugar transport. The following phenotypic results were already described in the original version of this paper (32), and the conclusions originated from them have not changed.
Phenotypic characterization of C. deuterogattii mutant strains revealed a hypovirulent profile
As part of our antifungal development program (21), we tested the mutants for altered antifungal susceptibility but did not find any clear connection (data not shown). Nevertheless, we proceeded with the characterization of the mutants. Our tests included virulence potential in the G. mellonella model, determination of growth rates, observation of general cellular aspects, production of virulence factors, and characterization of EVs, as follows.
We first tested the virulence potential of the mutants by comparing their ability to kill G. mellonella with that demonstrated by parental (WT) cells. Both mutants took longer than WT cells to kill the invertebrate host (Fig. 9; P < 0.0001). In the representative experiment (one out of three) that we described in this study, all larvae were killed by WT cells on day 2 post infection, while the mutants took 7 days to kill the whole G. mellonella population. Replicates produced similar results (data not shown).
Fig 9.

C. deuterogattii mutant strains manifest a hypovirulent phenotype. Infection of G. mellonella with the independent Past mutants 1 (A) and 2 (B) resulted in a smaller efficacy in killing the animals, in comparison with wild-type (WT) cells (P < 0.0001 for both mutants). Control systems were injected with phosphate buffered saline (PBS) only. Statistical analysis was performed with the Mantel-Cox test.
We asked why the mutant strains had reduced abilities to kill G. mellonella. To address this question, we first determined the proliferation rates of the mutants and compared them to those obtained for WT cells at 37°C in three different media. In all media [Sabouraud, YPD, and Roswell Park Memorial Institute (RPMI); Fig. 10A through C], the mutants manifested growth rates that were higher than those of WT cells, which led us to discard the possibility that their decreased virulence was a consequence of reduced growth capacities. We then searched for evident cellular alterations in C. deuterogattii. WT cells and the mutant strains had similar ultrastructural aspects, as concluded from the observation of transmission electron microscopy (TEM) micrographs (Fig. 10D through F).
Fig 10.

Analysis of growth rates (A–C) and ultrastructural aspects (D–F) of wild-type (WT) and mutant cells. Independently of the use of Sabouraud (A), YPD (B), or RPMI (C) as the growth media, the mutant strains always manifested higher proliferation rates, in comparison to WT cells. Analysis of the ultrastructural features of WT (D), Past1 (E), and Past2 (F) cells revealed no evident alterations. Scale bars in D–F correspond to 500 nm.
Since the cell surface of C. deuterogattii is essential for interaction with host immune cells and, consequently, pathogenesis (33), we evaluated cell wall and capsular structures. Staining of the cell wall components chitin and chitooligomers of WT and mutant strains revealed similar characteristics (Fig. 11A through C), which were in agreement with those reported in the literature (34). As for the capsular structures of C. deuterogattii, immunofluorescence analysis of parental and mutant cells using a monoclonal antibody to the main cryptococcal capsular component, namely, glucuronoxylomannan (GXM), showed similar profiles of serological reactivity in all strains (Fig. 11A through C). The mutants tended to produce higher amounts of extracellular GXM, but the differences were very discrete (Fig. 11D). The general similarity in the capsular structures was confirmed by India ink counterstaining, and no visual evidence of altered capsules was observed (Fig. 11E through G). We then determined the capsular dimensions in the three strains. Although the capsules of the mutants were statistically smaller than those of WT cells (P < 0.0001), the average values were very close (Fig. 11H). Scanning electron microscopy (SEM) confirmed the immunofluorescence and counterstaining results, with no apparent differences between the capsules of WT and mutant cells (Fig. 11I through K). Since the susceptibility to phagocytosis, which also involves the participation of the cryptococcal capsule (35), is a determinant for the pathogenesis of Cryptococcus (36), we also compared the phagocytic rates of WT and mutant strains. We found no significant differences between the phagocytic rates of WT and mutant cells by mouse macrophages (Fig. 11L). Altogether, these results led us to the conclusion that the hypovirulent phenotypes of the mutant strains were not related to evident cellular alterations, capsule formation, or its possible interference with phagocytosis. We also tested melanin formation as another cryptococcal virulence factor (37). Once again, WT and mutant strains had similar abilities to make pigments (data not shown).
Fig 11.

Analysis of the surface architecture of C. deuterogattii and its impact on fungal phagocytosis. Fluorescence microscopy analysis of the cell surface of WT (A), Past1 (B), and Past2 (C) cells revealed similar aspects of cell wall chitin (blue fluorescence), chitooligomers (green fluorescence), and the capsule (red fluorescence) in the three strains. GXM secretion tended to be higher in the Past1 mutant (**P = 0.0053), but not the Past2 strain (D; ns, not significant). India ink counterstaining of WT (E), Past1 (F), and Past2 (G) cells suggested similar capsular dimensions, but the determination of the capsule sizes revealed lower average values for the mutant cells (H, ****P < 0.0001, with a 95% confidence level of 95.61%). SEM was also used for the observation of the capsules of WT (I), Past1 (J), and Past 2 (K) cells, revealing similar capsular structures. The phagocytosis rates of the three strains by mouse macrophages were also determined (L). No significant (ns) differences between the three strains were observed.
EV formation is affected in the mutant strains
EVs are required for virulence mechanisms in C. deuterogattii (6). We then hypothesized that EV formation could be affected in the mutant strains. The genes regulating formation of fungal EVs are not well known, but in Saccharomyces cerevisiae, EV production was influenced by the functionality of Golgi-related secretory proteins (38). Therefore, we analyzed the Golgi of WT and mutant strains after staining the cells with C6-NBD-ceramide (39).
WT cells showed the typical pattern of disperse Golgi staining previously observed in C. neoformans (40, 41) (Fig. 12A). In mutant cells, Golgi staining was clearly less intense, and most of the cells gave weak or negative fluorescent signals. This visual perception was confirmed by quantitative determination of fluorescence staining, which revealed a significantly reduced fluorescence signal in mutant cells (Fig. 12B). These results were suggestive that the mutant strains manifested defects in the Golgi. We then asked whether EV formation was also altered in the mutant cells.
Fig 12.

Cell wall (calcofluor white, CFW) and Golgi staining (C6-NBD-Cer) in WT and mutant cells. (A) Microscopic examination of stained fungal cells suggested reduced levels of Golgi staining in mutant cells. (B) Quantitative determination of fluorescent cells (100 cells for each condition) confirmed a significantly reduced detection of Golgi staining in the mutants, in comparison with WT cells. Scale bars, 10 µm.
To address this question, we isolated EVs from WT and mutant cells for characterization by TEM and nanoparticle tracking analysis (NTA) (Fig. 13A through C). TEM revealed that WT and mutant cells produced EVs with their typical morphological aspects, including cup-shaped structures and bilayered membranes. NTA demonstrated that in the three strains, the EV population was mainly distributed in the 100–300 nm range, as extensively observed for the Cryptococcus genus (5, 42, 43). Minor populations in the 300–600 nm range were also observed in the three strains. However, the quantification of EVs produced by the three strains by NTA revealed a significantly reduced formation of vesicles in the mutant strains (Fig. 13D). After normalization of the number of EVs in each sample to the number of EV-producing cells, one of the mutant strains (Past1) was approximately twice less efficient than WT cells to produce EVs, while EV production was approximately threefold higher in the parental strain than in the second (Past2) mutant. In an attempt to validate this observation, we measured extracellular urease activity in the three strains, since this enzyme is an extracellular virulence factor exported in EVs (20). Urease activity was significantly smaller in mutant cells, in comparison with the enzyme activity measured in the parental strain (Fig. 13E).
Fig 13.
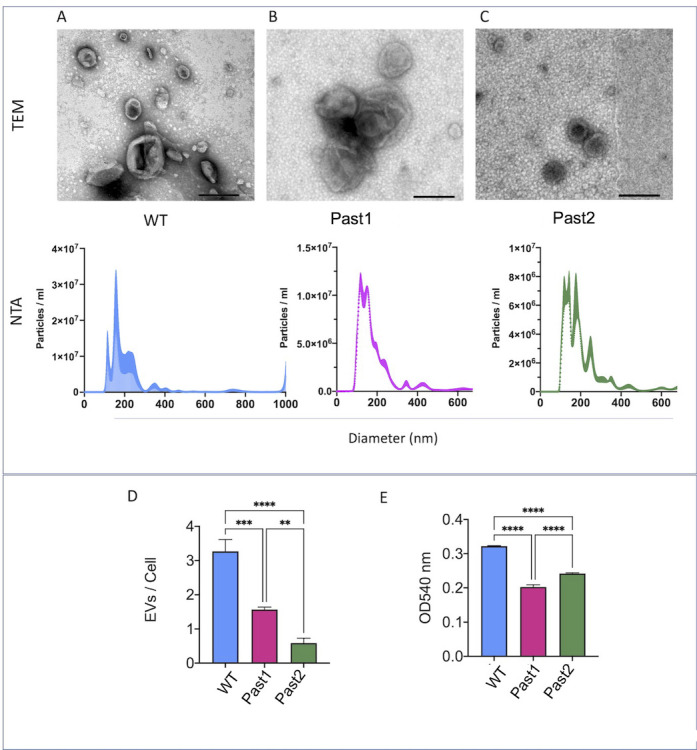
Analysis of EV formation in C. deuterogattii. EVs produced by wild-type (A), Past1 (B), and Past2 (C) cells were characterized by TEM (upper panels) and NTA (lower panels). EVs from all strains, in general, manifested similar properties. (D) Quantification of EVs produced by each strain revealed a significantly reduced number of vesicles produced by mutant cells, in comparison to the WT strain (****P < 0.0001; ***P = 0.002). EV production was smaller in the Past2 mutant than in the Past1 strain (**P = 0.0036). (E) Measurement of urease activity in the three strains revealed that the mutants had a lower ability to hydrolyze urea (****P < 0.0001). Both mutants manifested similar urease activities (ns, not significant).
Small molecule composition of EVs
The reduced formation of EVs in the mutant strains suggested a role of the affected genes in EV biogenesis. Since EV biogenesis involves the mechanisms of cargo (44), we also asked whether the composition of EVs was altered in the mutants. As a proof of concept, we focused our analysis on the 13 EV small molecules that we recently described in C. deuterogattii (13), since this number was much smaller than those found for proteins (5) and RNA (9) in cryptococcal vesicles. Of the 13 previously described small molecule components of cryptococcal EVs, two were below the detection level (Val-Leu-Pro-Val-Pro and asperphenamate) in this study, so we concentrated our analysis on the remaining 11 compounds. To compare the small molecule composition of EVs from WT and mutant cells, we used the partial least square discriminant analysis (PLS-DA). This analysis allows the comparison of data including several variables under an algorithm-supervised mode, as is the case of mass spectrometry (MS) data. Different chemical profiles of small molecule composition of EVs from WT and mutant cells were found (Fig. 14). As anticipated, the composition of the EVs from the mutant strains was similar. However, EV composition in WT cells was clearly different from that observed in mutant vesicles. Three of the annotated metabolites were classified as variables important in the projection (VIPs) identified by PLS-DA, namely, Phe-Pro, Pyro Glu-Leu, and Cyclo (Tyr-Pro). They were used for the discrimination between the three groups (small molecule composition of EVs produced by WT and mutant cells). These results proved the concept that EV formation was altered in the C. deuterogattii mutants.
Fig 14.

Multivariate data analysis of small molecule composition in WT and mutant cells of C. deuterogattii based on liquid chromatography-mass spectrometry (LC-MS) data. (A) PLS-DA of EVs LC-MS data. Each sphere in this analysis represents a single replicate. The elliptical area around the three replicates represents the confident zone of each group. In this analysis, R2 and Q2 values corresponded to 0.98 and 0.20, respectively, confirming the consistency between the original and cross-validation predicted data. (B) Three metabolites [Phe-Pro, Pyro Glu-Leu, and Cyclo (Tyr-Pro)] were classified as variables important in the projection (VIP) features indicated by PLS-DA. Only VIPs with a coefficient score above 3.0 and P < 0.01 for the permutation test were selected. Their molecular formula [M + H]+ and coefficient scores calculated for the PLS-DA model shown in A are listed.
EVs from parental cells increase the virulence of mutant cells
Since the EVs were altered in the mutant strains, we asked whether the vesicles were the elements required for virulence. We raised two hypotheses. First, the hypovirulent profile of the mutants could be a result of the reduced number of EVs. Second, the altered composition of EVs would be responsible for the hypovirulent phenotypes. To address these hypotheses, we returned to the G. mellonella model of infection using the second (Past2) mutant strain, in which the formation of EVs was more evidently reduced. To test whether reduced EV concentrations would be the reason for the hypovirulence, we infected G. mellonella with mutant cells supplementing the injecting suspension with EVs produced by this mutant strain. The amount of EVs added to this suspension was calculated on the basis that this mutant was threefold less effective in producing EVs than WT cells. Therefore, for each cryptococcal cell used for infection, we added three EVs produced by the mutant. Independently of the presence of the EVs produced by the mutant strain, all larvae died by day 8 post infection, and no significant differences were observed between systems injected with EVs plus the mutant and those receiving the mutant strain only (Fig. 15; P = 0.5488). We then speculated that the number of EVs was likely not a determinant for pathogenesis. To test whether the composition of EVs influenced virulence, we injected G. mellonella with the mutant strain in the presence of EVs produced by WT cells, using the same ratio of three EVs for each cryptococcal cell used for infection. In comparison with systems receiving no EVs or EVs from mutant cells, a clearly accelerated death curve was observed when G. mellonella was injected with the mutant in the presence of EVs produced by WT cells (Fig. 15; P = 0.0007).
Fig 15.

A role for EVs in the ability of C. deuterogattii to kill G. mellonella. All larvae survived after injection with PBS alone, EVs from wild-type (WT) cells, or EVs from the Past2 mutant. Animals infected with the Past2 mutant alone or the mutant in the presence of their own EVs had similar mortality rates (P = 0.5488). In contrast, infection of G. mellonella with the Past2 mutant in the presence of EVs produced by WT cells resulted in higher mortality rates in comparison with all other systems infected with the Past2 mutant of C. deuterogattii (P = 0.0007). Statistical analysis was performed with the Mantel-Cox test.
DISCUSSION
In this study, our results indicated a connection between regular EV production and cryptococcal virulence. In this sense, it is noteworthy that the current literature indicates that EVs play puzzling roles in cryptococcal pathogenesis. These structures were denominated “virulence bags” due to their complex composition that includes several virulence factors (20), which agrees with studies suggesting that cryptococcal EVs function in favor of disease progress. For instance, C. neoformans EVs facilitated cryptococcal traversal across the blood-brain barrier in mice, promoting an enhanced infection of the brain (45). An RNAi mutant strain of C. neoformans lacking expression of Sec6, a gene regulating conventional secretion, had attenuated virulence in mice in association with the absence of EV formation (46). More recently, long-distance communication via EVs was demonstrated to be essential for the virulence of C. deuterogattii (6). One additional factor that may be related to the connection between EVs and virulence is urease. The role of urease in fungal virulence was well established in C. neoformans and Coccidioides posadasii (47), and its activity in cryptococcal EVs was demonstrated in early studies (20). It remains unknown whether the altered urease activity derived from the misfunctioning of unknown cellular structures, but our results support the supposition that EV production, urease activity, and virulence are somehow connected in Cryptococcus. It remains unknown if these connections occur in other fungi, but C. posadasii is a natural candidate for this type of investigation.
Paradoxically, several studies indicate that cryptococcal EVs stimulate disease control. EVs stimulated the antifungal activity of macrophages in association with nitric oxide and cytokine production (4). In addition, EVs enriched with sterylglucosides induced protection against C. neoformans in a G. mellonella model of infection (48). Similarly, an EV peptide produced by C. deuterogattii induced the control of animal mortality in the same model (13). In mice, exposure to EVs produced by an acapsular strain of C. neoformans resulted in attenuated disease (5), which agrees with reports in the C. albicans model (49). The versatility of EVs in cryptococcal pathogenesis was efficiently illustrated in a recent study that demonstrated that vesicle properties changed according to the growth condition (50). EVs produced under a poor nutritional condition contained more virulence compounds and induced a more robust inflammatory pattern than those produced in a rich nutritional medium. On the other hand, EVs produced in a rich medium inhibited the expression of genes related to the inflammasome, suggesting an involvement of EVs in the pathogenic process (50). We demonstrated recently that cryptococcal EVs are highly diverse in their physical-chemical properties, suggesting that their functions can indeed show a high diversity (43).
Our results suggest that an altered composition of EVs can result in attenuated virulence, as concluded from the observation that EVs produced by wild-type cells restored, at least partially, the virulence of the mutant strain. On the basis of the previous reports demonstrating that EVs contribute to the pathogenic potential of Cryptococcus (6, 20, 45, 46, 50), we speculate that the hypovirulent phenotype of the mutants could be a result of altered EV production. Our results suggested that increasing the concentration of EVs was not sufficient to restore the pathogenic potential of the mutants, since the mortality of G. mellonella did not change in the presence of an increased amount of EVs produced by these cells. In contrast, infection with the mutant cells in the presence of EVs produced by the parental cells resulted in an increased pathogenic potential. These results strongly suggest that EV cargo is determinant for the ability of C. deuterogattii to kill G. mellonella. One limitation of our study comes from the lack of knowledge of the EV components impacting virulence, since it is very likely that the composition of the vesicles produced by the mutants was altered at multiple levels and not only in the small molecule cargo. Regardless, these results might represent a proof of concept that EV formation is connected to virulence through their composition. Future studies identifying bioactive EV components and determining their vesicular concentration will be necessary for the characterization of the molecules impacting infection, as recently described for the Ile-Pro-Ile tripeptide (13). However, we anticipate that this might be a complex analysis since we cannot anticipate whether a molecular association between different vesicle components will be required for fungal virulence.
MATERIALS AND METHODS
Strains
Fungal strains were maintained on Sabouraud agar plates (1% yeast extract, 2% peptone, 4% dextrose, and 1.5% agar) grown at 30°C for 24 h and stored at 4°C. Twenty-four hours before the experiments, the strains were transferred to liquid YPD medium and incubated at 30°C for 24 h with shaking (200 rpm). In experiments where capsule induction was necessary, the cells underwent an extra step of incubation in the capsule induction medium [Sabouraud 10% diluted in morpholinepropanesulfonic acid (MOPS) 50 mM, pH 7.4] (51) for 24 h at 37°C and 5% CO2. For the GXM detection assay by enzyme-linked immunosorbent assay (ELISA), cells grown in YPD were submitted to an incubation step in RPMI (37°C, 5% CO2 for 24 h) before the collection of the supernatants. The Delsgate methodology was used to construct the NOP16 gene inactivation allele by employing the vector pDONR-NAT, as described previously (22). Primers used to amplify the NOP16 gene flanking regions, as well as the internal fragment, are listed in Table 1. For the confirmation of NOP16 deletion, the strains were cultured in YPD for 24 h with shaking (200 rpm). The cells were washed with fresh YPD and counted in a hemacytometer. A total of 500,000 cells were inoculated into fresh YPD in the absence (control) or presence of mebendazole (1 mM) in a 96-well plate. After incubation for 24 h at 30°C, the OD600 was determined, and the relative growth was determined by the ratio of optical densities in the presence of mebendazole by the control condition. For determination of growth rates, WT and mutant strains were grown in Sabouraud broth overnight at 30°C with shaking (200 rpm). The cultures were washed three times with PBS and counted in a Neubauer chamber. The cells were suspended (2.5 × 105 cells/mL) in liquid Sabouraud, YPD, or RPMI medium supplemented with 1% glucose, buffered (pH 7.0), with 165 mM MOPS. Each suspension (200 µL, triplicates) was placed onto the wells of 96-well plates and incubated at 37°C on a Molecular Devices SpectraMax Paradigm microplate reader. Fungal growth was determined with optical density measurements at 530 nm every hour for 48 h.
Genome sequencing
For genome sequencing, we first extracted the DNA from the three isolates (WT, Past1/nop16Δ.1, and Past2/nop16Δ.2) independently as follows. The isolates were grown in YPD medium overnight at 30°C with 200 rpm agitation. For DNA recovery, 2 mL of each sample was centrifuged for 5 min at 13,000 rpm, washed in PBS, and frozen at −80°C. To the frozen pellet, we added 500 µL of extraction buffer (NaCl, Tris-HCl, pH 8, 1% SDS, Triton-X-100, and EDTA) and zirconium beads, vortex mixed in four cycles of 2-min vortexing plus 30 seconds on ice, followed by the addition of 500 µL of phenol-chloroform (vol/vol) and 30 seconds of vortexing. Samples were then centrifuged for 5 min at 13,000 rpm, and the aqueous phase was transferred to a new tube. One volume of phenol-chloroform was added, and samples were vortex mixed for 30 seconds before centrifugation for 5 min at 13,000 rpm. The aqueous phase was removed to a new tube, and DNA was precipitated by the addition of 20 µL of sodium acetate (3 M, pH 5.2) to each 100 µL of sample and 250 µL of pure ethanol. This was followed by a 15-min incubation at −70°C. Samples were then centrifuged for 15 min at 13,000 rpm, the supernatant was discarded, and the pellet was washed with 70% cold ethanol. Samples were air dried at room temperature and resuspended in 100 µL of Tris-ethylene-diamine-tetraacetic acid (TE) buffer with 50µg/mL of RNAse (Thermo Fisher Scientific) for 45 min at 37°C. RNAse was inactivated in a 100-µL phenol-chloroform (vol/vol) solution and centrifuged for 2 min at 15,000 rpm, and the DNA precipitation step was repeated as described above before DNA quantification in a nanodrop system. The absorbance ratio 260/280 was superior to 1.8, and the A260/230 was higher than 2, confirming the absence of contaminants. Genome sequencing was performed on an Illumina NextSeq 2000 instrument after constructing a DNA library using the Illumina DNA Prep kit, quantification by quantitative PCR, clustering, and sequencing (NextSeq 2 × 100 bp, average coverage of 10 million clusters or 20 million paired reads per sample). The genome sequencing service was provided by the Centre for Functional Genomics of the Luiz de Queiroz College of Agriculture at the University of São Paulo (ESALQ-USP, https://sites.usp.br/cgf/).
Bioinformatic analysis of DNA Seq data
Raw reads recovered from Illumina BaseSpace were quality filtered using fastp (23) with default options. The resulting files were used for further analysis. Alignment of the paired-end clean reads to C. deuterogattii R265 (NCBI accession GCA_002954075.1) was performed using the BWA aligner v. 0.7.17 (24) with default options. Generation of BAM and indexed BAM files was performed using samtools (52) v. 1.18. Visualization of aligned reads was carried out using IGV 2.16.2 (25).
The detection of SVs was performed using perSVade (27) v1.02.6, essentially as described in the GitHub manual, available at https://github.com/Gabaldonlab/perSVade/wiki/6.-Examples#running-persvade-modules-persvade-module-args-recommended.
RNA-Seq analysis
First, RNA from the three strains was extracted in independent triplicates using the Trizol-Qiagen protocol (Ambion, Life Technologies, USA). Cells were grown in YPD medium overnight, and 106 cells were recovered by centrifugation, washed three times in sterile water, and stored at −80°C. Cell lysis was performed by adding 750 µL of Trizol and zirconium beads to the frozen pellet. Samples were agitated using the FastPrep-24 (MP Biomedical) at 4.0 ms for 20 seconds and 1 min on ice in nine cycles. The mixture was then incubated at room temperature for 5 min, and 200 µL of iced chloroform was added to each tube, which was hand shaken for 15 seconds, followed by a 3-min incubation at room temperature and a 12,000 × g centrifugation at 4°C for 15 min. The aqueous phase was transferred to a new tube, and 500 µL of 100% isopropanol was added, followed by a 10-min incubation at room temperature and centrifugation at 12,000 × g for 10 min at 4°C. The RNA pellet was washed with 1 mL of 75% cold ethanol, vortex mixed, and centrifuged at 7,500 × g for 5 min at 4°C. Samples were vacuum dried and resuspended in PCR water for Nanodrop quantification, and the RNA quality was measured on the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The RNA extracted from WT and mutant cells was prepared for RNA-Seq analysis according to the Illumina Stranded mRNA Prep Ligation protocol (Illumina, USA), following its instructions (Illumina, Document #1000000124518 v 02). The RNA sample input was adjusted to a final concentration of 130 ng in distilled water with a final volume of 25 µL per well. Messenger RNA (mRNA) was captured using oligo (dT) magnetic beads in a magnetic separation rack (New England Biolabs, Inc., USA), eluted in the elution buffer, and cleaned up using fragmentation master mix, bead binding buffer, and bead washing buffer before fragmentation and denaturation of the mRNA using mixes and thermal cycles indicated by the manufacturer. To the purified mRNA, the first-strand synthesis mix and the reverse transcriptase were added to synthesize the first cDNA strand using the thermal cycler, followed by the synthesis of the second cDNA strand with the appropriate mix. After cDNA generation, the strands were cleaned up using Agencourt AMPure XP and washing beads with fresh 80% ethanol. The 3′ ends of the cDNA strands were adenylated with the A-Tailing Mix to prevent self-ligation between the fragments and assure adapter complementarity by the thymine nucleotide located at the 3′ end of the adapters. After adenylation, the samples were ligated to the index anchors and PCR amplified. The fragments were captured with Agencourt AMPure XP, washed with 80% ethanol, and resuspended in resuspension buffer. The library was then selectively amplified in an enhanced PCR mix for the anchor-ligated DNA fragments, and indexes and primer sequences were added to form clusters in a dual-indexed library. The library was finally checked for concentration and quality and then sequenced on the Illumina NextSeq 500 (Illumina, USA).
Bioinformatic analysis of RNA-Seq data
Raw reads were quality filtered using fastp (23) with default options. The resulting files were used for alignment to C. deuterogattii R265 (NCBI accession GCA_002954075.1) using STAR v. 2.7.10b (24) with the following parameters: –quantMode GeneCounts –outFilterType BySJout –alignIntronMin 10 –alignIntronMax 3000 –outFilterIntronMotifs RemoveNoncanonical. A count table from each paired-end library was fed into R 4.3.1 for differential expression analysis using DESeq2 v. 3.18 (28), referring to the vignettes associated with the package. Volcano plots were prepared using the EnhancedVolcano package (53) in R.
Protein extraction from fungal cells
Fungal cells were washed three times with PBS and subsequently collected. The samples were instantly frozen at −80°C in an ultra-freezer. Following freezing, they underwent lyophilization at −50°C and 0.04 mBar for 18 h. Next, the process of fungal cell lysis was initiated. This involved adding 0.1-mm zirconium beads to the dried pellets, followed by 10 cycles of vortex shaking for 1 min and then resting for 1 min on ice. Afterward, the pellets were treated with a 0.1% RapiGest SF solution (wt/vol) in 50 mM ammonium bicarbonate (NH4HCO3). This step was followed by five more cycles of vortex shaking for 1 min and 1 min of ice incubation. Post-lysis, the samples were centrifuged at 18,000 rpm for 15 min. The supernatants, which contained the proteins, were collected. Finally, the protein concentration in each sample was determined using fluorometric quantification (Qubit, Thermo Fisher Scientific, Inc.).
Sample preparation for mass spectrometry
For each sample, 100 µg of proteins was initially subjected to reduction. This was achieved using dithiothreitol (DTT) at a final concentration of 10 mM, with the samples being incubated at 60°C for 30 min. After the reduction process, the samples were allowed to cool to room temperature. Following this, alkylation was performed using iodoacetamide (IAA), where the samples were treated with a final concentration of 25 mM IAA for 25 min at room temperature, ensuring exposure to light was avoided. The samples then underwent a second incubation with DTT (final concentration 10 mM) at room temperature. The next step involved digestion with trypsin, executed at a 1/50 enzyme-to-substrate ratio, and allowed to proceed overnight. Following digestion, the peptides were desalted and concentrated using Stage-Tips (54). Finally, for peptide quantification, a fluorimetric assay (Qubit, Thermo Fisher Scientific, Inc.) was utilized, adhering to the manufacturer’s guidelines.
Mass spectrometry analysis
The mass spectrometry analysis was conducted in technical duplicates. Peptides obtained from the desalting process were loaded onto reverse-phase columns, which were in-house packed with ReprosilPur C18 Acqua stationary phase. These columns had 3-µm-diameter beads, 120 Å pore size, were 30 cm long, and had a 75-µm internal diameter. The peptide loading was performed using the UltiMate 3000 nanochromatography system from Thermo Fisher Scientific, Inc.
For elution, a chromatographic gradient was employed over a duration of 145 min. The gradient initiation was at 5% and gradually increased to 50% of phase B (consisting of 95% acetonitrile in 0.1% formic acid) over 100 min. This was followed by a 20-min gradient increase from 50% to 95% of the same mobile phase, with a maintained flow rate of 250 µL/min. The eluate was then directly fed into the Orbitrap Fusion Lumos mass spectrometer, located at the Proteomics and Mass Spectrometry facility of the Carlos Chagas Institute, Fiocruz-PR. Mass spectra were acquired using the Orbitrap Fusion Lumos in data-dependent acquisition mode. This mode involved alternating automatically between full-scan MS and MS/MS acquisition, with a dynamic exclusion time of 45 seconds. The initial MS scan was set at a resolution of 60,000 at 200 m/z, and every 2 seconds, the most intense ions with 2+ and 3+ charges were isolated and subjected to high-energy collision dissociation with normalized collision energies of 30. The resulting fragment ions were analyzed at a resolution of 15,000 at 200 m/z. Operational parameters for the mass spectrometer included a spray voltage of 2.6 kV, no flow of sheath and auxiliary gas, a heated capillary temperature of 250°C, enabled predictive automatic gain control, and a radiofrequency level of 70% in the S-lens. The scan functions and solvent gradients in the nLC were controlled and managed using the Xcalibur 4 software from Thermo Fisher Scientific.
Peptide spectral matching (PSM)
We utilized PatternLab for Proteomics V for peptide spectrum matching due to its proven reliability and accessibility; the software is freely available at http://patternlabforproteomics.org. Data analysis followed the software’s protocol, with the feature that enables multiplexed spectra identification through the Y.A.D.A. 3.0 deconvolution algorithm (30, 31, 55). The database was prepared using the “target-decoy” approach to provide a set of reliable identifications. This set included sequences of Cryptococcus deuterogattii downloaded on 18 October 2023, obtained from FungiDB (https://fungidb.org/), including the reverse version of each sequence found in the database and sequences of 127 common contaminants in mass spectrometry (e.g., keratin, trypsin). The Comet search tool (56) was used to compare the experimentally obtained tandem mass spectra with the theoretical spectra generated from the database, selecting the most likely candidates. In summary, the search was limited to tryptic candidates with up to two cleavage failures, and carbamidomethylation of cysteine and oxidation of methionine were considered as fixed modification and variable modification, respectively.
PSM validation
Validation was performed by the Search Engine Processor (57). Identifications were grouped by charge state (+2 and >+3), resulting in two distinct subgroups. For each result, the cross correlation, DeltaCN, and spectrum count score resulting from Comet were used to generate a Bayesian discriminator. Identifications were ranked in ascending order according to discriminator values. Minimum values (cut-offs) were established for identifications to accept a 1% FDR at the protein level, based on the number of labeled decoys (58). Additionally, a restriction to sequences with six or more amino acid residues was imposed. Results were post-processed to accept PSMs with less than 6 ppm, a minimum cross-correlation value of 2.0, and proteins with one or more independent pieces of evidence in the identification. This final filter was applied to ensure that all search results achieved a 0.1% FDR (59).
Relative protein quantification
Quantification was performed using extracted ion chromatograms. The TFold module of PatternLab V (60) was used to identify differentially abundant proteins between the different groups. Our proteomic comparison considered only proteins identified with two or more unique peptides (i.e., peptides that map to a single sequence in the database) and a q-value ≤0.
Ultrastructure of C. deuterogattii
For the analysis of possible cellular alterations, fungal cells were processed for TEM (42). The cells were fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, for 1 h at room temperature. The fixed samples were washed three times with cacodylate buffer by centrifugation (5,000 × g for 2 min). The pellet was treated with 1% osmium tetroxide and 5 mM potassium ferrocyanide diluted in 0.1 M cacodylate buffer (1:1 ratio) for 1 h at room temperature. The samples were then washed three times with cacodylate buffer and then sequentially dehydrated with 30%, 50%, 70%, 90%, and twice with 100% acetone. In each step, the samples were incubated with acetone for 30 min. The cell pellet was covered with the EMbed 812 resin (EMS) diluted in acetone. The initial dilution was one part resin to two parts 100% acetone, and this proportion was changed daily for 3 days (one part resin to one part acetone on the second day; two parts resin to one part acetone on the third day). On the fourth day, the solution was replaced with 100% resin. This process was repeated, and the cell pellet covered with resin was incubated at 60°C for 48 h until complete polymerization of the resin was achieved. Ultra-sections were prepared with an ultramicrotome (Leica EM UC6; thickness of 70–80 nm) and mounted on microscopic grids. The observation of the samples was performed using a JEOL 1400Plus microscope with an acceleration of 90 kV. The images were obtained using a digital camera with an 8-megapixel CCD coupled to the equipment.
Analysis of the polysaccharide capsule
Fungal cells were first counterstained with India ink for visualization of the capsule by light microscopy. As the polysaccharide structure is not permeable to India ink, counterstaining generates a high-contrast zone that allows the capsular structure to be visualized. C. deuterogattii cells were incubated in 10% Sabouraud in 50 mM MOPS, pH 7.4 (51), for 24 h at 37°C and 5% CO2. After capsule induction, the cells were recovered by centrifugation at 3,000 × g, washed with PBS, and fixed with 4% paraformaldehyde. The cell suspension (3 µL) was placed onto a glass slide and supplemented with 2 µL of Indian ink. The sample was finally covered with a glass coverslip and observed with an inverted microscope (Leica DMi8). The images obtained in this process were analyzed using the ImageJ software (NIH). The capsule size was estimated as the cell body diameter subtracted from the total capsule diameter. For serological detection of capsular GXM, cryptococcal cells grown in YPD for 24 h were centrifuged, and the cell pellet was washed with PBS. The cells were transferred to the capsule induction medium under the conditions described above. The cells were then centrifuged at 3,000 × g for 3 min, and the pellet was washed three times with sterile PBS. The cells were suspended in 150 µL of 4% paraformaldehyde in PBS and incubated at room temperature for 30 min. After fixation, the cell suspension was centrifuged again at 3,000 × g for 3 min. The cell pellet was subjected to three washes with PBS and blocked with 1 mL of blocking buffer (1% bovine serum albumin [BSA] in PBS), followed by incubation for 1 h at 37°C. The suspension was centrifuged, the supernatant was discarded, and 150 µL of 25 µM calcofluor was added to the cell pellet, followed by incubation for 30 min at 37°C. Then, three washes were performed with PBS, and 120 µL of an anti-GXM antibody 18B7 (donated by Dr. Arturo Casadevall, Johns Hopkins University, Baltimore, USA) at 10 µg/mL in blocking buffer was added to the cell suspension, for subsequent incubation for 1 h at 37°C. The cells were washed three times with PBS and suspended in a solution of 120 µL of a secondary antibody (goat to mouse immunoglobulin antibody) conjugated to Alexa 546 diluted in blocking buffer (1:1,500). The cells were incubated for 1 h in the dark at room temperature. The cells were washed again with PBS and stained with fluorescein-isothiocyanate (FITC)-labeled wheat germ agglutinin (WGA) for detection of chitin oligomers in the cell wall (34). This step was carried out by suspending the cell pellet in 600 µL of WGA at 5 µg/mL, followed by incubation for 30 min at 37°C. The cells were washed with PBS and observed with an inverted fluorescence microscope DMi8 (Leica Microsystems). Extracellular GXM was measured by ELISA as previously described (61) with minor modifications (3).
The surface of fungal cells was also analyzed by SEM (42). The C. deuterogattii cells grown in YPD were centrifuged at 3,000 × g, and the cell pellet was washed with PBS. The sample was fixed with 2.5% glutaraldehyde in cacodylate buffer (0.1 M sodium cacodylate, pH 7.2). The cells were washed three times with a post fixative solution (0.2 M sucrose, 0.1 M sodium cacodylate buffer, 2 mM MgCl2). The washed samples (150 µL) were allowed to adhere to 0.01% poly-L-lysine (type I)-coated coverslips for 30 min at room temperature. The coverslips were sequentially dehydrated with 30%, 50%, and 70% ethanol solutions (5 min for each concentration), followed by 90% ethanol and two rounds of 100% ethanol, with these three steps lasting 10 min each. The cells were dried on a critical point chamber (Leica EM CPD300) and coated with gold particles on a metallizer (Leica EM ACE200). The visualization of the samples was performed using a JEOL JSM-6010 Plus/LA microscope with an acceleration of 5 kV.
Golgi staining
We followed the protocol originally described by Pagano and colleagues (39) and adapted by our group to the analysis of the Golgi in cryptococci (40, 41). Briefly, yeast cells (107) were fixed with 4% paraformaldehyde in PBS, followed by washing with PBS and incubation with C6-NBD-ceramide (10 µM) for 16 h at 4°C. The cells were then incubated with fetal calf serum (10%) at 4°C for 1 h to remove the excess of C6-NBD-ceramide. The cell wall was stained with calcofluor white (0.1 mg/mL) for 30 min at room temperature, followed by washing with PBS and analysis by fluorescence microscopy as previously described (40, 41). Virtually, all cells were efficiently stained with calcofluor white, and the percentage of cells giving positive signals for C6-NBD-ceramide-derived fluorescence was manually determined in populations of 100 cells for each tested condition.
Isolation of EVs
The isolation of EVs produced by mutant and WT C. deuterogattii cells followed our recently described protocol (42). Fungal cultures had their cell densities determined in a Neubauer chamber and adjusted to 3.5 × 107 cells/mL. Aliquots of each inoculum (300 µL) were spread with a Drigalski loop onto Petri dishes containing 25 mL of solid YPD. Three plates per strain were used. The plates were incubated at 30°C for 24 h to reach confluence, and then, the fungal cells were recovered by gently scraping with an inoculation loop and transferred to a centrifuge tube containing 30 mL of sterile PBS. Sequential centrifugation was used to remove the cells. In the first centrifugation step, the samples were centrifuged at 5,000 × g for 15 min at 4°C. The supernatants from the first centrifugation step were transferred to sterile tubes. These tubes were submitted to a second centrifugation step at 15,000 × g for 15 min at 4°C for sedimentation of the cellular debris. After this step, the supernatants were filtered through 0.45-µm membranes and ultracentrifuged at 100,000 × g for 1 h at 4°C. The pelleted EVs were suspended in a final volume of 300 µL of PBS and stored at 4°C.
Quantification and morphological analysis of EVs
The quantification of EVs was performed by nanoparticle tracking analysis (NTA) using LM10 nanoparticle analysis system coupled to a 488-nm laser, equipped with a camera and flow pump (Malvern Panalytical, Malvern, United Kingdom) and the NTA 3.0 software (Malvern Panalytical) (42). The samples were diluted 200-fold in PBS to achieve the optimal readout range of 9 × 107 to 2.9 × 109 particles/mL. The samples were injected with 1-mL syringes attached to a continuous flow injection pump. Three 60-second videos (camera level at 15, gain at 3) were obtained per sample after the passage of the samples through the light beam. The viscosity of the samples was indicated as the same as that of water. For data analysis, the camera gain was changed to 10–15, and the detection limit used was three for all samples. TEM was used to visualize the EVs (42). The samples were homogenized with a vortex for 2 min to break up possible aggregates. The EV suspensions (50 µL) were adhered to Formvar-coated grids for 60 min at room temperature. The grids were then washed with 30 µL of sterile PBS. The excess buffer was removed by applying filter paper to the bottom of each grid. The grids were then incubated with 30 µL of the Karnowski solution for 10 min, washed three times with cacodylate buffer, and finally dried with filter paper. The samples were counterstained with 5% uranyl acetate for 2 min. The grids were washed once with H2O, dried with filter paper, and transferred to a metallizer (Leica EM ACE200), where they were covered with carbon particles for later visualization with a JEOL 1400Plus microscope with beam acceleration at 90 kV.
Urease activity
Cell suspensions at 1 × 108 cells/mL were prepared in urea broth/Robert’s medium (final volume of 1 mL). The systems were incubated overnight at 30°C with shaking. The cells were pelleted by centrifugation and the supernatants (200 µL) were transferred to the wells of 96-well plates. Optical densities at 540 nm were determined on a SpectraMax PARADIGM microplate reader (Molecular Devices).
C. deuterogattii phagocytosis
Murine macrophage cells (RAW 264.7) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% innactivated fetal bovine serum (FBS) at 37°C in 5% CO2. On the day before the experiment, a suspension containing 5 × 105 cells/mL was prepared in DMEM supplemented with 10% FBS. Two hundred microliters of this suspension were added to each well of 96-well plates. The plates were incubated at 37°C and 5% CO2 for 24 h. Fungal cells were adjusted to a density of 1 × 106 cells/mL in YPD, washed once with PBS, and then incubated with 0.5 mg/mL FITC in PBS at room temperature for 15 min in the dark. The cells were washed three times with PBS, and then, the inoculum was adjusted to 5 × 105 cells/mL in DMEM. The 18B7 anti-GXM antibody was added to a final concentration of 10 µg/mL, and opsonization was carried out by incubating these suspensions for 1 h at 37°C in a 5% CO2 atmosphere. Opsonized cells were used to infect macrophages previously seeded in 96-well plates, using the MOI (multiplicity of infection) of 1:1. The systems were incubated for 3 h at 37°C in 5% CO2. After incubation, the wells were washed three times with PBS to remove free fungal cells, and 200 µL of DMEM + 10% FBS was added to each well. The plates were then transferred to an Operetta high-content imaging system (PerkinElmer) (62). The equipment was adjusted to a temperature of 37°C with a 5% CO2 atmosphere and programmed to automatically photograph the infected cells. The images were obtained in the Alexa 488 channels to detect the fluorescence emitted by the FITC and also under the bright field mode. The images were processed using the Harmony high-content imaging and analysis software (PerkinElmer) and ImageJ (NIH). Phagocytosis indices consisted of the percentage of cells infected with the fungus in each field.
Galleria mellonella infection
To assess the pathogenic potential of the WT and mutant strains, the invertebrate model of infection in Galleria mellonella, which was validated for the study of cryptococcal pathogenesis (63), was used. For acclimatization, G. mellonella larvae weighing between 0.10 and 0.15 g were divided into groups of 15 animals in Petri dishes and incubated overnight at 37°C before infection. Suspensions containing 1 × 108 cells/mL were prepared in sterile PBS for each of the strains to be analyzed. Ten microliters of these suspensions, containing 1 × 106 fungi, were used to infect the larvae of G. mellonella using a Hamilton syringe as previously described (13). The plates were then incubated at 37°C to monitor the survival of the animals over the days. The groups analyzed were (i) animals infected with the WT, R265 strain; (ii) animals infected with the mutant cells; and (iii) control group inoculated only with PBS. Larvae mortality was observed daily, as evidenced by the lack of movement after stimulation with forceps.
G. mellonella mortality was also the model used to evaluate the role of EVs on fungal pathogenesis. In these assays, all infection conditions were similar to those described above. However, we only used one of the mutant strains (Past2), based on its lower ability to produce EVs. Six groups were analyzed, including non-infected controls [larvae injected with (i) PBS alone, (ii) EVs from WT cells, or (iii) EVs from the mutant] and infected systems [infection with (i) mutant cells alone, (ii) mutant cells with EVs from WT cells, or (iii) the mutant strain with its own EVs]. The amount of externally supplemented EVs corresponded to three vesicles per each of the 1 × 106 fungi used to infect G. mellonella. Monitoring of mortality was performed as described above.
Small molecule analysis
C. deuterogattii EVs were prepared and analyzed as recently described by our group (13, 14). Briefly, the samples were vacuum dried, extracted with methanol, filtered through 0.22-µm membranes, dried under a N2 flux, and stored at −20°C. The vesicular extracts were suspended in MeOH and submitted to ultra-high-performance liquid chromatography coupled to mass spectrometry (UHPLC-MS) on a Thermo Scientific QExactive hybrid Quadrupole-Orbitrap mass spectrometer. The parameters used for this analysis, including those used for MS/MS, were recently detailed by our group. UHPLC-MS operation and spectra analyses were performed using Xcalibur software (version 3.0.63). A molecular network was created using the online workflow (https://ccms-ucsd.github.io/GNPSDocumentation/) on the GNPS website (http://gnps.ucsd.edu) as reported in our recent study (13).
LC-MS data analysis for multivariate data analysis
After molecular networking analysis on the GNPS database, the online visualization option was selected to process the original files. The LC-MS chromatograms were analyzed in the Feature finding option using MZMine2 (Dashboard online GNPS version). The following parameters were used: precursor tolerance of 10 ppm; noise level of 10E4; minimum and maximum peak width, 0.05–1.5 min; and a retention time tolerance of 0.3 min. The job can be accessed online at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=395c343397384433998e001434bedfed. The quantification table (.csv) was submitted to MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/) for statistical analysis, and the data were processed using sum normalization and Pareto Scaling. PCA and PLS-DA were performed on the data set. For the individual metabolite quantification, the interesting features (annotated m/z) were searched in the Xcalibur software (version 3.0.63), through extracted ion chromatogram analysis and integrated.
Statistical analysis
Statistical analyses were performed using the GraphPad Prism software 9.3 (GraphPad Software, Inc., La Jolla, USA). The results were analyzed using a one-way analysis of variance and Tukey’s post hoc tests, except for the survival assay with G. mellonella, where the analysis with Mantel-Cox was used. The differences found were considered significant when the P-values were less than 0.05.
ACKNOWLEDGMENTS
M.L.R. is supported by grants from the Brazilian Ministry of Health (grant 440015/2018-9), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grants 405520/2018-2 and 301304/2017-3), and Fiocruz (grants PROEP-ICC 442186/2019-3, VPPCB-007-FIO-18, and VPPIS-001-FIO18). M.L.R. also acknowledges the support from the Instituto Nacional de Ciência e Tecnologia de Inovação em Doenças de Populações Negligenciadas (INCT-IDPN). H.C.O. received scholarships from the Inova Program of Fiocruz. F.C.G.R. received a scholarship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil, Finance Code 001). T.P.F. acknowledges the support from FAPESP, grant number 2021/00728-0. O.Z. is funded by grant PID2020-114546RB from MCIN/AEI/10.13039/501100011033. The funders had no role in the decision to publish or preparation of the manuscript.
We thank Dr. Nuria Trevijano Contador for the help with RNA-Seq and Michel Batista and his team for the help with proteomic analysis. The authors are thankful to the mass spectrometry technological platform of the Carlos Chagas Institute, which is supported by the Program for Technological Development in Tools for Health of Fiocruz (RPT-FIOCRUZ). The authors are grateful to the Program for Technological Development in Tools for Health-RPT- FIOCRUZ for using the microscopy facility, RPT07C, Carlos Chagas Institute, Fiocruz- Paraná.
M.L.R. is currently on leave from the position of associate professor at the Microbiology Institute of the Federal University of Rio de Janeiro, Brazil.
Contributor Information
Marcio L. Rodrigues, Email: marcio.rodrigues@fiocruz.br.
Andreas J. Bäumler, University of California, Davis, Davis, California, USA
DATA AVAILABILITY
The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE (64) partner repository with the dataset data set identifier PXD048601. Raw data are available at NCBI under Bioproject code PRJNA1059231.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/iai.00037-24.
Structural variants (SVs) in the three analyzed strains.
Differentially expressed genes in WT and mutant cells.
Proteins differentially detected in WT and mutant cells.
Proteins differentially detected in WT and mutant cells.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Couch Y, Buzàs EI, Di Vizio D, Gho YS, Harrison P, Hill AF, Lötvall J, Raposo G, Stahl PD, Théry C, Witwer KW, Carter DRF. 2021. A brief history of nearly EV-erything – the rise and rise of extracellular vesicles. J Extracell Vesicles 10:e12144. doi: 10.1002/jev2.12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coelho C, Casadevall A. 2019. Answers to naysayers regarding microbial extracellular vesicles. Biochem Soc Trans 47:1005–1012. doi: 10.1042/BST20180252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodrigues ML, Nimrichter L, Oliveira DL, Frases S, Miranda K, Zaragoza O, Alvarez M, Nakouzi A, Feldmesser M, Casadevall A. 2007. Vesicular polysaccharide export in Cryptococcus neoformans is a eukaryotic solution to the problem of fungal trans-cell wall transport. Eukaryot Cell 6:48–59. doi: 10.1128/EC.00318-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oliveira DL, Freire-de-Lima CG, Nosanchuk JD, Casadevall A, Rodrigues ML, Nimrichter L. 2010. Extracellular vesicles from Cryptococcus neoformans modulate macrophage functions. Infect Immun 78:1601–1609. doi: 10.1128/IAI.01171-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rizzo J, Wong SSW, Gazi AD, Moyrand F, Chaze T, Commere P-H, Novault S, Matondo M, Péhau-Arnaudet G, Reis FCG, Vos M, Alves LR, May RC, Nimrichter L, Rodrigues ML, Aimanianda V, Janbon G. 2021. Cryptococcus extracellular vesicles properties and their use as vaccine platforms. J Extracell Vesicles 10:e12129. doi: 10.1002/jev2.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bielska E, Sisquella MA, Aldeieg M, Birch C, O’Donoghue EJ, May RC. 2018. Pathogen-derived extracellular vesicles mediate virulence in the fatal human pathogen Cryptococcus gattii. Nat Commun 9:1556. doi: 10.1038/s41467-018-03991-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bitencourt TA, Hatanaka O, Pessoni AM, Freitas MS, Trentin G, Santos P, Rossi A, Martinez-Rossi NM, Alves LL, Casadevall A, Rodrigues ML, Almeida F. 2022. Fungal extracellular vesicles are involved in intraspecies intracellular communication. mBio 13:e0327221. doi: 10.1128/mbio.03272-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodrigues ML, Nimrichter L. 2022. From fundamental biology to the search for innovation: the story of fungal extracellular vesicles. Eur J Cell Biol 101:151205. doi: 10.1016/j.ejcb.2022.151205 [DOI] [PubMed] [Google Scholar]
- 9. Peres da Silva R, Puccia R, Rodrigues ML, Oliveira DL, Joffe LS, César GV, Nimrichter L, Goldenberg S, Alves LR. 2015. Extracellular vesicle-mediated export of fungal RNA. Sci Rep 5:7763. doi: 10.1038/srep07763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rayner S, Bruhn S, Vallhov H, Andersson A, Billmyre RB, Scheynius A. 2017. Identification of small RNAs in extracellular vesicles from the commensal yeast Malassezia sympodialis. Sci Rep 7:39742. doi: 10.1038/srep39742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peres da Silva R, Longo LGV, Cunha J da, Sobreira TJP, Rodrigues ML, Faoro H, Goldenberg S, Alves LR, Puccia R. 2019. Comparison of the RNA content of extracellular vesicles derived from Paracoccidioides brasiliensis and Paracoccidioides lutzii. Cells 8:765. doi: 10.3390/cells8070765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodrigues ML, Nakayasu ES, Almeida IC, Nimrichter L. 2014. The impact of proteomics on the understanding of functions and biogenesis of fungal extracellular vesicles. J Proteomics 97:177–186. doi: 10.1016/j.jprot.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reis FCG, Costa JH, Honorato L, Nimrichter L, Fill TP, Rodrigues ML. 2021. Small molecule analysis of extracellular vesicles produced by Cryptococcus gattii: identification of a tripeptide controlling cryptococcal infection in an invertebrate host model. Front Immunol 12:654574. doi: 10.3389/fimmu.2021.654574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Costa JH, Bazioli JM, Barbosa LD, Dos Santos Júnior PLT, Reis FCG, Klimeck T, Crnkovic CM, Berlinck RGS, Sussulini A, Rodrigues ML, Fill TP. 2021. Phytotoxic tryptoquialanines produced in vivo by Penicillium digitatum are exported in extracellular vesicles. mBio 12:e03393-20. doi: 10.1128/mBio.03393-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samuel AZ, Horii S, Nakashima T, Shibata N, Ando M, Takeyama H. 2022. Raman microspectroscopy imaging analysis of extracellular vesicles biogenesis by filamentous fungus Penicilium chrysogenum. Adv Biol (Weinh) 6:e2101322. doi: 10.1002/adbi.202101322 [DOI] [PubMed] [Google Scholar]
- 16. Cleare LG, Zamith D, Heyman HM, Couvillion SP, Nimrichter L, Rodrigues ML, Nakayasu ES, Nosanchuk JD. 2020. Media matters! alterations in the loading and release of Histoplasma capsulatum extracellular vesicles in response to different nutritional milieus. Cell Microbiol 22:e13217. doi: 10.1111/cmi.13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. UniProt . 2022. UniProtKB - Q9Y3C1 (NOP16_HUMAN). Available from: https://www.uniprot.org/uniprot/Q9Y3C1#expression
- 18. Horsey EW, Jakovljevic J, Miles TD, Harnpicharnchai P, Woolford JL. 2004. Role of the yeast Rrp1 protein in the dynamics of pre-ribosome maturation. RNA 10:813–827. doi: 10.1261/rna.5255804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harnpicharnchai P, Jakovljevic J, Horsey E, Miles T, Roman J, Rout M, Meagher D, Imai B, Guo Y, Brame CJ, Shabanowitz J, Hunt DF, Woolford JL. 2001. Composition and functional characterization of yeast 66S ribosome assembly intermediates. Mol Cell 8:505–515. doi: 10.1016/s1097-2765(01)00344-6 [DOI] [PubMed] [Google Scholar]
- 20. Rodrigues ML, Nakayasu ES, Oliveira DL, Nimrichter L, Nosanchuk JD, Almeida IC, Casadevall A. 2008. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot Cell 7:58–67. doi: 10.1128/EC.00370-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joffe LS, Schneider R, Lopes W, Azevedo R, Staats CC, Kmetzsch L, Schrank A, Del Poeta M, Vainstein MH, Rodrigues ML. 2017. The anti-helminthic compound mebendazole has multiple antifungal effects against Cryptococcus neoformans. Front Microbiol 8:535. doi: 10.3389/fmicb.2017.00535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schneider R de O, Fogaça N de S, Kmetzsch L, Schrank A, Vainstein MH, Staats CC. 2012. Zap1 regulates zinc homeostasis and modulates virulence in Cryptococcus gattii. PLoS One 7:e43773. doi: 10.1371/journal.pone.0043773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. doi: 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schikora-Tamarit MÀ, Gabaldón T. 2022. PerSVade: personalized structural variant detection in any species of interest. Genome Biol 23:175. doi: 10.1186/s13059-022-02737-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crestani J, Carvalho PC, Han X, Seixas A, Broetto L, Fischer J de S da G, Staats CC, Schrank A, Yates JR, Vainstein MH. 2012. Proteomic profiling of the influence of iron availability on Cryptococcus gattii. J Proteome Res 11:189–205. doi: 10.1021/pr2005296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clasen MA, Santos MDM, Kurt LU, Fischer J, Camillo-Andrade AC, Sales LA, de Arruda Campos Brasil de Souza T, Lima DB, Gozzo FC, Valente RH, Duran R, Barbosa VC, Carvalho PC. 2023. Patternlab V handles multiplex spectra in shotgun proteomic searches and increases identification. J Am Soc Mass Spectrom 34:794–796. doi: 10.1021/jasms.3c00063 [DOI] [PubMed] [Google Scholar]
- 31. Santos MDM, Lima DB, Fischer JSG, Clasen MA, Kurt LU, Camillo-Andrade AC, Monteiro LC, de Aquino PF, Neves-Ferreira AGC, Valente RH, Trugilho MRO, Brunoro GVF, Souza T, Santos RM, Batista M, Gozzo FC, Durán R, Yates JR, Barbosa VC, Carvalho PC. 2022. Simple, efficient and thorough shotgun proteomic analysis with PatternLab V. Nat Protoc 17:1553–1578. doi: 10.1038/s41596-022-00690-x [DOI] [PubMed] [Google Scholar]
- 32. Castelli RF, Pereira A, Honorato L, Valdez A, de Oliveira HC, Bazioli JM, Garcia AWA, Klimeck TDF, Reis FCG, Staats CC, Nimrichter L, Fill TP, Rodrigues ML. 2022. Extracellular vesicle formation in Cryptococcus deuterogattii impacts fungal virulence and requires the NOP16 gene. Infect Immun 90:e0023222. doi: 10.1128/iai.00232-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaragoza O. 2019. Basic principles of the virulence of Cryptococcus. Virulence 10:490–501. doi: 10.1080/21505594.2019.1614383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fonseca FL, Nimrichter L, Cordero RJB, Frases S, Rodrigues J, Goldman DL, Andruszkiewicz R, Milewski S, Travassos LR, Casadevall A, Rodrigues ML. 2009. Role for chitin and chitooligomers in the capsular architecture of Cryptococcus neoformans. Eukaryot Cell 8:1543–1553. doi: 10.1128/EC.00142-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zaragoza O, Rodrigues ML, De Jesus M, Frases S, Dadachova E, Casadevall A. 2009. The capsule of the fungal pathogen Cryptococcus neoformans. Adv Appl Microbiol 68:133–216. doi: 10.1016/S0065-2164(09)01204-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esher SK, Zaragoza O, Alspaugh JA. 2018. Cryptococcal pathogenic mechanisms: a dangerous trip from the environment to the brain. Mem Inst Oswaldo Cruz 113:e180057. doi: 10.1590/0074-02760180057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eisenman HC, Casadevall A. 2012. Synthesis and assembly of fungal melanin. Appl Microbiol Biotechnol 93:931–940. doi: 10.1007/s00253-011-3777-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oliveira DL, Nakayasu ES, Joffe LS, Guimarães AJ, Sobreira TJP, Nosanchuk JD, Cordero RJB, Frases S, Casadevall A, Almeida IC, Nimrichter L, Rodrigues ML. 2010. Characterization of yeast extracellular vesicles: evidence for the participation of different pathways of cellular traffic in vesicle biogenesis. PLoS One 5:e11113. doi: 10.1371/journal.pone.0011113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pagano RE, Sepanski MA, Martin OC. 1989. Molecular trapping of a fluorescent ceramide analogue at the Golgi apparatus of fixed cells: interaction with endogenous lipids provides a trans-Golgi marker for both light and electron microscopy. J Cell Biol 109:2067–2079. doi: 10.1083/jcb.109.5.2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rizzo J, Oliveira DL, Joffe LS, Hu G, Gazos-Lopes F, Fonseca FL, Almeida IC, Frases S, Kronstad JW, Rodrigues ML. 2014. Role of the Apt1 protein in polysaccharide secretion by Cryptococcus neoformans. Eukaryot Cell 13:715–726. doi: 10.1128/EC.00273-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kmetzsch L, Joffe LS, Staats CC, de Oliveira DL, Fonseca FL, Cordero RJB, Casadevall A, Nimrichter L, Schrank A, Vainstein MH, Rodrigues ML. 2011. Role for Golgi reassembly and stacking protein (GRASP) in polysaccharide secretion and fungal virulence. Mol Microbiol 81:206–218. doi: 10.1111/j.1365-2958.2011.07686.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reis FCG, Borges BS, Jozefowicz LJ, Sena BAG, Garcia AWA, Medeiros LC, Martins ST, Honorato L, Schrank A, Vainstein MH, Kmetzsch L, Nimrichter L, Alves LR, Staats CC, Rodrigues ML. 2019. A novel protocol for the isolation of fungal extracellular vesicles reveals the participation of a putative scramblase in polysaccharide export and capsule construction in Cryptococcus gattii. mSphere 4:e00080-19. doi: 10.1128/mSphere.00080-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reis FCG, Gimenez B, Jozefowicz LJ, Castelli RF, Martins ST, Alves LR, de Oliveira HC, Rodrigues ML. 2021. Analysis of cryptococcal extracellular vesicles: experimental approaches for studying their diversity among multiple isolates, kinetics of production, methods of separation, and detection in cultures of titan cells. Microbiol Spectr 9:e0012521. doi: 10.1128/spectrum.00125-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Colombo M, Raposo G, Théry C. 2014. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289. doi: 10.1146/annurev-cellbio-101512-122326 [DOI] [PubMed] [Google Scholar]
- 45. Huang SH, Wu CH, Chang YC, Kwon-Chung KJ, Brown RJ, Jong A. 2012. Cryptococcus neoformans-derived microvesicles enhance the pathogenesis of fungal brain infection. PLoS One 7:e48570. doi: 10.1371/journal.pone.0048570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Panepinto J, Komperda K, Frases S, Park YD, Djordjevic JT, Casadevall A, Williamson PR. 2009. Sec6-dependent sorting of fungal extracellular exosomes and laccase of Cryptococcus neoformans. Mol Microbiol 71:1165–1176. doi: 10.1111/j.1365-2958.2008.06588.x [DOI] [PubMed] [Google Scholar]
- 47. Rutherford JC. 2014. The emerging role of urease as a general microbial virulence factor. PLoS Pathog 10:e1004062. doi: 10.1371/journal.ppat.1004062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colombo AC, Rella A, Normile T, Joffe LS, Tavares PM, de S Araújo GR, Frases S, Orner EP, Farnoud AM, Fries BC, Sheridan B, Nimrichter L, Rodrigues ML, Del Poeta M. 2019. Cryptococcus neoformans glucuronoxylomannan and sterylglucoside are required for host protection in an animal vaccination model. mBio 10:e02909-18. doi: 10.1128/mBio.02909-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vargas G, Honorato L, Guimarães AJ, Rodrigues ML, Reis FCG, Vale AM, Ray A, Nosanchuk JD, Nimrichter L. 2020. Protective effect of fungal extracellular vesicles against murine candidiasis. Cell Microbiol 22:e13238. doi: 10.1111/cmi.13238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marina CL, Bürgel PH, Agostinho DP, Zamith-Miranda D, Las-Casas L de O, Tavares AH, Nosanchuk JD, Bocca AL. 2020. Nutritional conditions modulate C. neoformans extracellular vesicles’ capacity to elicit host immune response. Microorganisms 8:1815. doi: 10.3390/microorganisms8111815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zaragoza O, Casadevall A. 2004. Experimental modulation of capsule size in Cryptococcus neoformans. Biol Proced Online 6:10–15. doi: 10.1251/bpo68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. 2021. Twelve years of SAMtools and BCFtools. Gigascience 10:giab008. doi: 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blighe K, Rana S, Lewis M. 2023. Publication-ready volcano plots with enhanced colouring and labeling. Bioconductor. doi: 10.18129/B9.bioc.EnhancedVolcano [DOI] [Google Scholar]
- 54. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670. doi: 10.1021/ac026117i [DOI] [PubMed] [Google Scholar]
- 55. Carvalho PC, Lima DB, Leprevost FV, Santos MDM, Fischer JSG, Aquino PF, Moresco JJ, Yates JR, Barbosa VC. 2016. Integrated analysis of shotgun proteomic data with PatternLab for proteomics 4.0. Nat Protoc 11:102–117. doi: 10.1038/nprot.2015.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eng JK, Jahan TA, Hoopmann MR. 2013. Comet: an open-source MS/MS sequence database search tool. Proteomics 13:22–24. doi: 10.1002/pmic.201200439 [DOI] [PubMed] [Google Scholar]
- 57. Carvalho PC, Fischer JSG, Xu T, Cociorva D, Balbuena TS, Valente RH, Perales J, Yates JR, Barbosa VC. 2012. Search engine processor: filtering and organizing peptide spectrum matches. Proteomics 12:944–949. doi: 10.1002/pmic.201100529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barboza R, Cociorva D, Xu T, Barbosa VC, Perales J, Valente RH, França FMG, Yates JR, Carvalho PC. 2011. Can the false‐discovery rate be misleading? Proteomics 11:4105–4108. doi: 10.1002/pmic.201100297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yates JR, Park SKR, Delahunty CM, Xu T, Savas JN, Cociorva D, Carvalho PC. 2012. Toward objective evaluation of proteomic algorithms. Nat Methods 9:455–456. doi: 10.1038/nmeth.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carvalho PC, Yates JR, Barbosa VC. 2012. Improving the TFold test for differential shotgun proteomics. Bioinformatics 28:1652–1654. doi: 10.1093/bioinformatics/bts247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Casadevall A, Mukherjee J, Scharff MD. 1992. Monoclonal antibody based ELISAs for cryptococcal polysaccharide. J Immunol Methods 154:27–35. doi: 10.1016/0022-1759(92)90209-c [DOI] [PubMed] [Google Scholar]
- 62. de Oliveira HC, Joffe LS, Simon KS, Castelli RF, Reis FCG, Bryan AM, Borges BS, Soares Medeiros LC, Bocca AL, del Poeta M, Rodrigues ML. 2020. Fenbendazole controls in vitro growth, virulence potential, and animal infection in the Cryptococcus model. Antimicrob Agents Chemother 64:e00286-20. doi: 10.1128/AAC.00286-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fuchs BB, O’Brien E, Khoury JBE, Mylonakis E. 2010. Methods for using Galleria mellonella as a model host to study fungal pathogenesis. Virulence 1:475–482. doi: 10.4161/viru.1.6.12985 [DOI] [PubMed] [Google Scholar]
- 64. Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, Walzer M, Wang S, Brazma A, Vizcaíno JA. 2022. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50:D543–D552. doi: 10.1093/nar/gkab1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural variants (SVs) in the three analyzed strains.
Differentially expressed genes in WT and mutant cells.
Proteins differentially detected in WT and mutant cells.
Proteins differentially detected in WT and mutant cells.
Data Availability Statement
The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE (64) partner repository with the dataset data set identifier PXD048601. Raw data are available at NCBI under Bioproject code PRJNA1059231.