

SUMMARY
Recurrent Clostridioides difficile infection (CDI) results in significant morbidity and mortality. We previously established that CDI in mice does not protect against reinfection and is associated with poor pathogen-specific B cell memory (Bmem), recapitulating our observations with human Bmem. Here, we demonstrate that the secreted toxin TcdB2 is responsible for subversion of Bmem responses. TcdB2 from an endemic C. difficile strain delayed immunoglobulin G (IgG) class switch following vaccination, attenuated IgG recall to a vaccine booster, and prevented germinal center formation. The mechanism of TcdB2 action included increased B cell CXCR4 expression and responsiveness to its ligand CXCL12, accounting for altered cell migration and a failure of germinal center-dependent Bmem. These results were reproduced in a C. difficile infection model, and a US Food and Drug Administration (FDA)-approved CXCR4-blocking drug rescued germinal center formation. We therefore provide mechanistic insights into C. difficile-associated pathogenesis and illuminate a target for clinical intervention to limit recurrent disease.
In brief
Norman et al. use mouse models of C. difficile vaccination and infection to delineate a CXCR4-dependent mechanism by which the secreted toxin TcdB2 suppresses germinal center formation and antibody recall responses. Germinal centers were rescued by a CXCR4-blocking drug, suggesting a therapeutic avenue for prevention of recurrent C. difficile infection.
Graphical Abstract

INTRODUCTION
The spore-forming bacterium Clostridioides difficile is the largest cause of nosocomial infection globally, surpassing that caused by methicillin-resistant Staphylococcus aureus.1–3 C. difficile infection (CDI) survivors commonly suffer from long-lasting complications due to high rates of disease recurrence, with the chances of relapse increasing after each infection.4,5 Prolonged dysbiosis caused by antibiotic therapy and pathogen re-exposure likely contribute to CDI recurrence. However, recent evidence, albeit from a few laboratories, indicates that poor B cell memory responses following infection leave both murine and human hosts vulnerable to CDI recurrence.6–9 Despite these key observations, mechanistic insights into how C. difficile prevents protective B cell memory following infection are lacking.
C. difficile spores are transmitted via the fecal-oral route and remain viable on surfaces under diverse environmental conditions.10,11 Germination of ingested spores is facilitated by dysbiosis of the host microbiome, often induced by broad-spectrum antibiotic therapy.12,13 CDI establishes in the colon, and symptoms range from mild diarrhea to severe pseudomembranous colitis, sepsis, and death.14–16 C. difficile secretes large single-subunit toxins known as toxin A (TcdA) and toxin B (TcdB), both of which are significant virulence factors.17 Analysis of clinical C. difficile strains and studies in animal models establish TcdB as the main driver of disease pathology.18,19 TcdB enters host cells, primarily epithelial cells, by receptor mediated endocytosis.17,20–22 Cytosol-localized TcdB glucosylates small guanosine triphosphatase (GTPase) family members, including Rho, Rac, and Cdc42, leading to altered signaling and cytoskeletal function.17,21,22 TcdB-induced glucosylation of small GTPases in epithelial cells results in cell rounding and, ultimately, cell death by apoptosis.21,22 This diminishes the structural integrity of the colonic lining, allowing tissue infection and dissemination of C. difficile and its toxins.23–26 Critically, disease severity has increased in recent years, concomitant with the emergence of C. difficile strains such as R20291 (ribotype 027), which express a more virulent version of TcdB known as TcdB2.27,28
CDI can be successfully cleared in animal models and patients by the innate immune response.28 MyD88-mediated activated neutrophils29 are necessary for clearance, as are type 1, 2, and 3 innate-like lymphocytes (ILC1s, ILC2s, and ILC3s, respectively), which have been reported to mitigate disease severity.30–33 Additionally, interleukin-25 (IL-25)-regulated eosinophils contribute to protection.34 While innate mechanisms can resolve disease, there is a failure to establish adequate adaptive immune memory following infection. TcdB-specific immunoglobulin G (IgG) constitutes the clearest correlate of protection against recurrent CDI.28,35,36 Furthermore, analysis of the TcdB-specific memory B cell compartment in individuals who have recovered from CDI shows an apparent deficiency in Ig class switch and poor TcdB-neutralizing capability of the limited IgG that is produced.8 These observations are recapitulated in mouse models of recurrent CDI, wherein infection poorly stimulates TcdB-specific IgG responses, B cell memory, or expansion of T follicular helper (Tfh) cells.7
The limited class-switched B cell memory following infection suggests that CDI impacts mechanisms underpinning Ig class switch and/or germinal center (GC) reactions. For an effective GC reaction, exposure to antigen needs to result in altered lymphocyte migration and reorganization of secondary lymphoid organ architecture, wherein antigen-experienced B cells undergo CD40L-dependent class switch, somatic hypermutation, and affinity maturation.37 The chemokine receptors CXCR4 and CXCR5 play a critical role in establishing GC function and consequent adaptive immune memory.38,39 CXCR4 is responsible for lymphocyte migration toward CXCL12 present near the border of light zones of GCs within lymph nodes.38,39 Once the lymphocyte has reached the edge of the light zone, CXCR4 is downregulated and CXCR5 is upregulated, inducing cell migration into the light zone of GCs. It is in the light zone where antigen selection and class switching take place.38,39 If this balance between CXCR4 and CXCR5 is disrupted, then lymphocytes are unable to properly migrate, and GC formation is disrupted.38,39
With the knowledge that TcdB2 is a critical virulence factor, we tested the hypothesis that TcdB2 exerts a deleterious impact on the mechanisms essential for the establishment of host B cell memory. Using a mouse model in which immunization with an inactivated TcdB2 vaccine antigen was preceded by active TcdB2 treatment, it was observed that TcdB2 delayed IgG class switch while blocking IgG recall responses and GC formation in secondary lymphoid organs. Analysis of differentially expressed genes (DEGs) revealed an increased expression of the chemokine receptor CXCR4. An increase in migration of B cells in response to the CXCL12 was also observed, explaining the lack of GC formation and the suppression of B cell memory. Effects of TcdB2 were recapitulated in a CDI challenge model, and GC formation was rescued by the US Food and Drug Administration (FDA)-approved CXCR4-blocking drug AMD3100.40 We therefore provide mechanistic insights into C. difficile-associated pathogenesis and illuminate CXCR4 as a target for clinical intervention to boost host humoral immunity and limit recurrent disease.
RESULTS
TcdB2 inhibits IgG recall responses
B6 mice were treated with TcdB2 before immunization to evaluate its impact on humoral immunity to the C. difficile vaccine antigen B2Δ, a TcdB2 mutant incapable of entering and intoxicating host cells (Figure 1A). TcdB2 treatment attenuated B2-Δ-specific IgG recall responses (Figure 1B). Sera were collected on day 67 (pre-boost) and day 81 (post-boost) from mice treated with TcdB2 lacking the increases in B2Δ-specific IgG1, IgG2b, and IgG2c titers observed in controls (Figure 1B). To measure the impact of TcdB2 on antibody (Ab) function, sera from vehicle -or TcdB2-treated B2Δ-immunized mice were tested for their ability to prevent in vitro intoxication of the Chinese hamster ovary (CHO) cell line with TcdB2 (Figure 1C). Recall sera collected from TcdB2-treated mice had a significantly decreased ability to neutralize TcdB2 in vitro as compared to the immunized control sera (Figure 1C). In contrast, the affinity of sera from control and TcdB2-treated mice was not significantly different (Figure S1A), indicating that the amounts of B2-Δ-specific IgG in the sera accounted for differences in TcdB2 neutralization rather than differences in affinity.
Figure 1. Inhibited IgG recall responses following TcdB2 treatment.
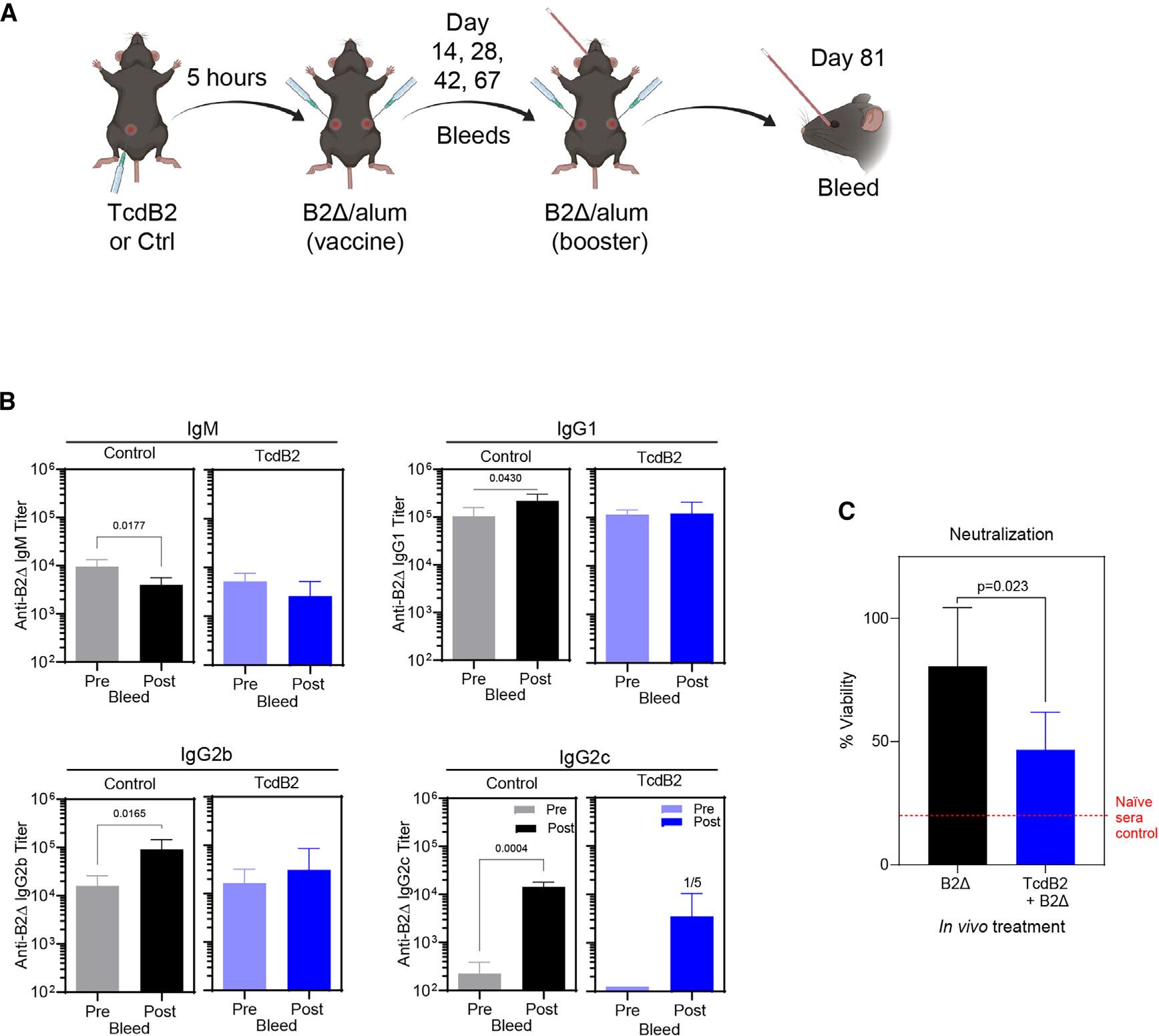
(A) Female B6 mice (n = 5 per group) were given PBS vehicle or 1 ng TcdB2 (i.p.) and then immunized with 10 μg of B2Δ/Alum (subcutaneously [s.c.]) after 5 h. Mice were bled on days 14, 28, 42, and 67 (primary). On day 67, mice were boosted with 10 μg of B2Δ in PBS (s.c.) and then bled on day 81 (recall).
(B) Serum B2Δ-specific IgM, IgG1, IgG2b, and IgG2c endpoint recall titers were determined by ELISA. All control mice had an IgG2c booster response, while 1 of 5 TcdB2-treated mice responded. Graphs show comparison of mean ± SD endpoint titers pre and post booster vaccine administration, and significant differences were determined by a matched-pairs t test. Further, Mann-Whitney U tests were used to compare post-booster vaccine titers between control and TcdB2-treated groups (for IgG1, p = 0.036).
(C) CHO cells were incubated with TcdB2, sera, or sera and TcdB2 for 24 h. Cell viability was determined using the CCK-8 assay as a measure of TcdB2 neutralization. The graph shows mean ± SD cell viability using recall sera from control (n = 5) and TcdB2-treated (n = 6) groups, and significance was determined by unpaired t test.
Other data related to this figure are depicted in Figure S1.
TcdB2 had no demonstrable effect on B2Δ-specific IgM titers but delayed production of IgG1 and IgG2b during the primary response (Figure S1B). TcdB2 neutralization in primary bleed sera reflected the primary B2Δ-specific IgG titers (Figure S1C). To determine whether enzymatic activity of TcdB2 was required for the impact on primary and recall responses, mice were treated with TcdB2 or an equivalent amount of a TcdB2 mutant containing a D270N point mutation, rendering it glucosyltransferase null (subsequently referred to as D270N) (Figures S1D and S1E). Mice treated with bioactive TcdB2 displayed lower primary and recall IgG responses than those treated with D270N (Figures S1D and S1E). However, differences in recall titers were not significant. Taken together, this suggests the possibility that TcdB2 can exert a low-level glucosyl transferase-independent effect on Ab production.
Our results demonstrate that a single bolus of TcdB2 delayed IgG class switching, blocked IgG recall responses, and limited production of TcdB2-neutralizing Abs.
CD40 activation restores and enhances IgG recall responses in TcdB2-treated mice
Given the inhibitory action of TcdB2 on recall IgG titers, the effects of a higher dose of the B2Δ/alum vaccine and supplemental B cell stimulation were tested (Figure 2). This was achieved by administration of an agonistic anti-CD40 monoclonal antibody (mAb) clone, FGK4.5., that mimics the effects of CD40L ligation of CD40 in vivo.41,42 When using twice the vaccine dose as in the previous experiment, TcdB2 administration resulted in minimal effects on IgM titers, as expected (Figure 2A). Primary and recall IgG1 titers were not significantly influenced by TcdB2, but CD40 activation resulted in a 31% increase in endpoint recall titers as compared to untreated controls (Figure 2B). In contrast, TcdB2 exerted strong effects on IgG2b, reducing recall IgG2b titers by 84% (Figure 2C). Administration of the anti-CD40 monoclonal Ab (mAb) restored and enhanced recall IgG2b titers so that they were 330% higher than that those observed in control mice (Figure 2C). A more pronounced effect was observed when measuring IgG2c responses in that TcdB2 completely abrogated the response, which was rescued by CD40 activation (Figure 2D). The data were also expressed as fold change between primary and recall titers, and each experimental group was compared (Figure 2E). TcdB2 had no significant effect on the magnitude of the IgM or IgG1 recall response, likely due to the higher dose of B2Δ than in Figure 1. In contrast, TcdB2 inhibited the IgG2b response and eliminated IgG2c recall responses. Activation of CD40 rescued the IgG2b and IgG2c recall titers and enhanced them to levels above those observed in controls.
Figure 2. CD40 activation-associated restoration of IgG recall responses in TcdB2-treated mice.
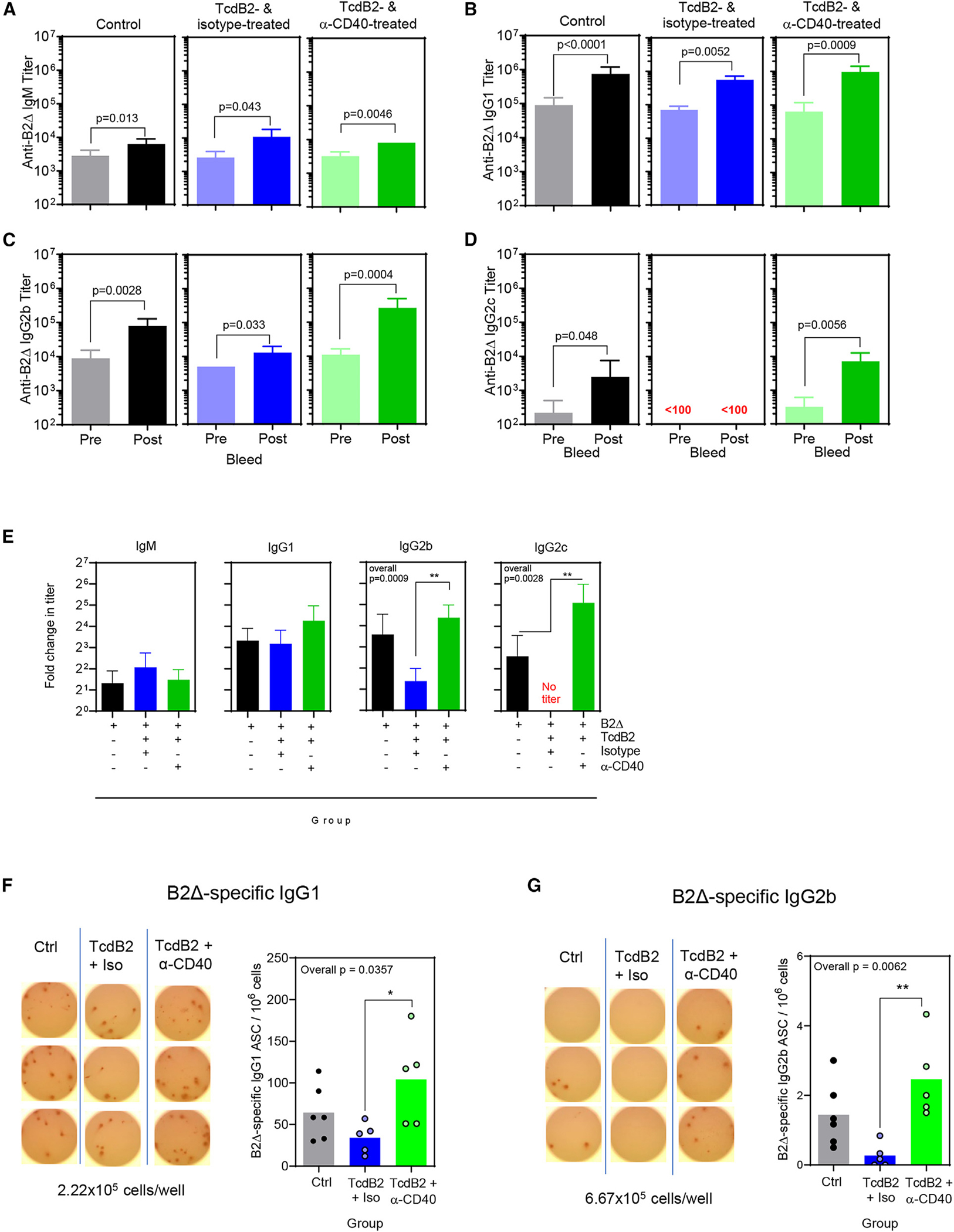
Female B6 mice were given PBS vehicle (left, gray/black, n = 6) or 1 ng TcdB (i.p.) (center and right, n = 10 in total) and then immunized after 5 h with 20 μg of B2Δ/Alum (s.c.). TcdB2-treated mice were injected s.c. with 100 μg isotype control mAb (center, blue n = 5) or 100 μg anti-CD40 mAb (right, green, n = 5) on days 1 and 8. A booster vaccine was administered on day 60 and consisted of 20 μg of B2Δ in PBS. Blood samples were collected before (day 60) and after (day 74) the booster.
(A–D) Data show (A) IgM, (B) IgG1, (C) IgG2b, and (D) IgG2c B2Δ-specific endpoint titers ± SD. Matched-pairs t-tests were used to measure significance.
(E) Data from (A)–(D) were re-analyzed by calculating fold change (mean ± SD) in endpoint IgM, IgG1, IgG2b, and IgG2c titers following booster vaccine administration and comparing the three experimental groups. Additional ANOVAs with Kruskal-Wallace post-test were performed to compare post-booster titers in all groups. IgG2b overall p = 0.0009, IgG2c overall p = 0.0028. For post-test, **p < 0.01.
(F and G) Representative images of ELISPOT wells with spots attributable to B2Δ-specific IgG1 and IgG2b. Graphs depict the number of B2Δ-specific spots per million cells. Each symbol represents an individual mouse. *p < 0.05, **p < 0.01.
At the final time point, bone marrow cells were harvested and analyzed by enzyme-linked immunosorbent spot (ELISPOT) assay to enumerate memory B cell-derived long-lived plasma cells (Figures 2F and 2G). The numbers of B2Δ-specific IgG1- and IgG2b-secreting plasma cells were consistent with serum titers (Figures 2F and 2G). Total plasma cells (of all specificities) did not differ significantly between experimental groups (Figures S2A and S2B). The assay background was zero spots (Figure S2C), and TcdB2 did not alter total numbers of cells recovered from bone marrow (Figure S2D). These results demonstrate that TcdB2-exposed B cells were intrinsically capable of IgG class switch provided alternative Th cell-type signals were provided and that TcdB2 had a long-term impact on the establishment of a long-lived plasma cell compartment following immunization.
TcdB2 results in non-lethal lymphocyte intoxication
TcdB2 glucosylates small GTPases and induces apoptosis in host epithelia.17 We determined that TcdB2 administered in vivo had no effect on the number of recoverable cells from lymphoid organs (Figure S3). As dead cells could be cleared in vivo, splenocytes were isolated from naive B6 mice and cultured in vitro with or without TcdB2. TcdB2 did not induce necrosis or apoptosis in B cells or CD4+ T cells above the background level in the cultures (Figures S4A and S4B). We considered whether a TcdB2-induced stress response could impact B cells, but at the doses administered, norepinephrine responses were minimal (Figure S4C). The lack of necrotic or apoptotic B cells observed following TcdB2 treatment suggested the possibility of non-lethal effects. Since the mechanism of action of TcdB2 requires glucosylation of small GTPases, including Rac1, the impact on amounts of non-glucosylated Rac1 were measured using a capillary-based immunoblot. TcdB2 resulted in a loss of non-glucosylated Rac1 in splenocytes and isolated B cells in a glucosyltransferase-dependent manner (Figures S4D–S4F). These results demonstrate that TcdB2 glucosylates Rac1 in B cells and appears to cause non-lethal intoxication.
TcdB2 blocks immunization-induced GC formation
The effects of TcdB2 on immunization-induced reorganization of lymphoid architecture were determined. B6 mice were treated with TcdB2 or the inactive D270N mutant and then immunized with B2Δ/alum. After 21 days, inguinal lymph nodes (iLNs) were collected and sections mounted on slides for analysis (Figure 3). Sections were stained with hematoxylin and eosin (H&E) (Figure 3A). It was observed that there were significantly less numerous or undetectable GCs in sections from TcdB2-treated and immunized mice than in sections from immunized controls (treated with vehicle or D270N) (Figures 3A and 3B). Those detectable GCs in sections from TcdB2-treated mice were significantly smaller than those in controls (Figure 3C). To confirm the results, draining lymph node (iLN) sections were stained with fluorochrome-conjugated anti-B220 and anti-Ki67 to examine GC structures (Figure 3D). These data demonstrate that TcdB2 inhibited GC formation, consistent with inhibited Ig class switch and recall responses.
Figure 3. TcdB2 blockade of immunization-induced GCs.
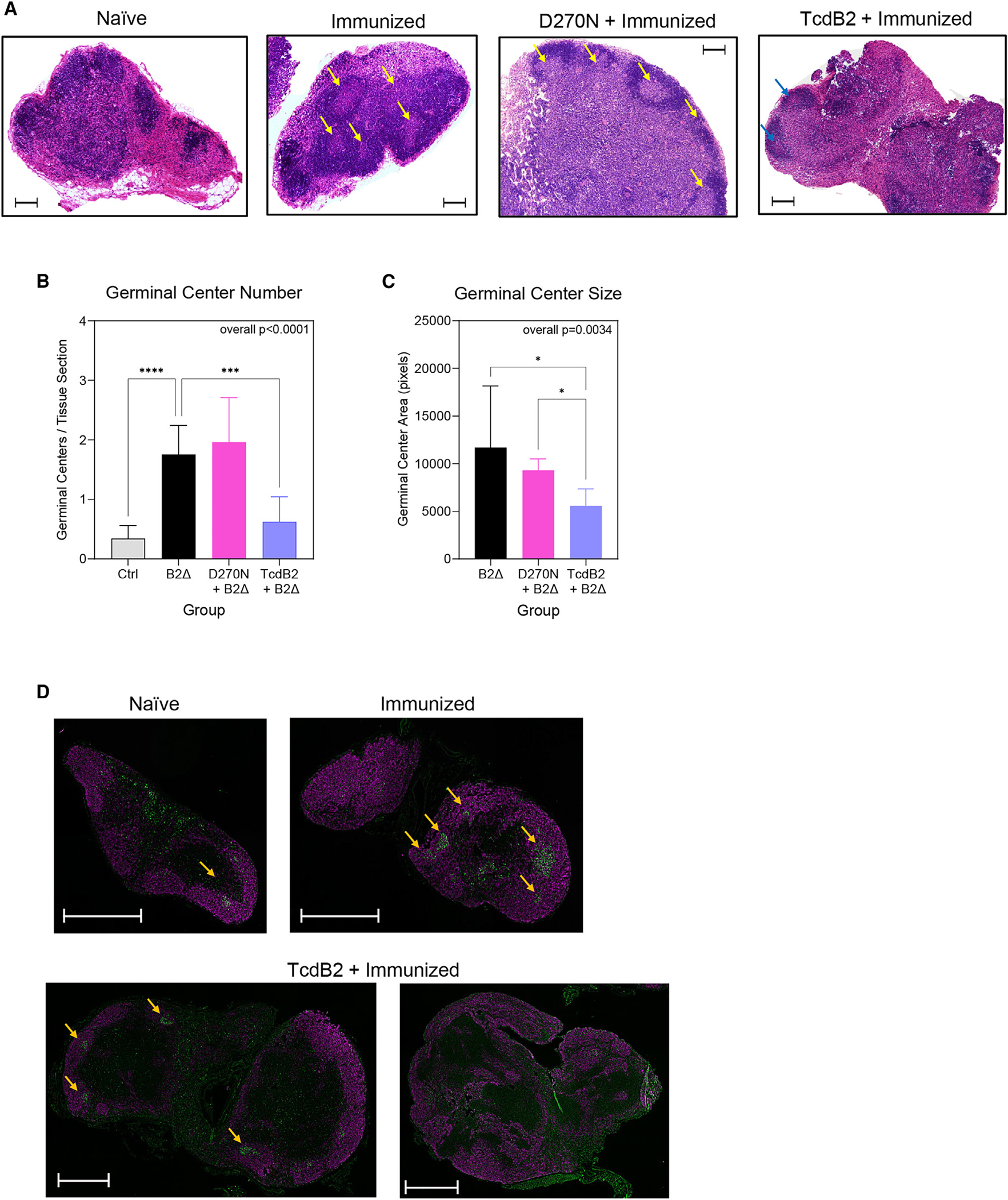
Mice were treated as follows: immunized s.c. with PBS vehicle control (n = 7), immunized s.c. with 10 μg of B2Δ/Alum (n = 7), injected i.p. with 1 ng TcdB2 and then immunized s.c. (n = 7), or injected with 1 ng D270N and then immunized s.c. (n = 6).
(A) Representative H&E sections of iLNs. Arrows indicate GCs. The scale bar depicts 500 μm.
(B) Mean ± SD GC count.
(C) Mean ± SD GC area per mouse.
(B and C) Data are from three pooled experiments. Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparisons post-test. For post-tests: *p < 0.05, ***p < 0.001, ****p < 0.0001.
(D) Representative immunofluorescent sections from mice described in (A). B220+ total B cells (purple) and Ki67+ proliferating GC B cells (green) are shown.
Arrows indicate GCs. The scale bar depicts 500 μm. Two examples are shown for TcdB2 treatment, as GCs were either small or undetectable.
TcdB2 exposure increases CXCR4 gene expression and cell surface expression by lymphocytes
To identify a potential mechanism by which TcdB2 inhibits GC formation, differentially expressed genes (DEGs) in draining lymph node cells were profiled following TcdB2 or D270N treatment (Figures 4A and 4B). DEGs were curated and represented as volcano plots (Figure 4A). CXCR4 gene expression was significantly upregulated when comparing TcdB2 to PBS and TcdB2 to D270N but not when comparing D270N to PBS, showing that TcdB2 must be enzymatically active to alter CXCR4 expression (Figure 4B). Analysis of CXCR5 and CCR7 expression and their corresponding ligands did not reveal any significant alteration following TcdB2 or D270N treatment (Figure 4B). Analysis of all chemokine receptor and ligand genes in the gene panel also did not reveal any significant changes in their expression (Table S1). The Msc gene encoding musculin was significantly downregulated, and the Cepbp gene encoding CCAAT enhancer binding protein beta (CEBPB) was significantly upregulated in response to TcdB2. Msc, also referred to as activated B cell factor 1 (ABF-1) inhibits transactivation of the E47 E2A protein involved in cell commitment and differentiation43 while CEBPB regulates BLIMP1 expression by post-GC B cells.44 As results of the Nanostring DEG analyses were obtained using whole mixed lymph node cells, B cells were isolated before confirmation of the results by qPCR (Figure 4C). These data demonstrate that enzymatically active TcdB2 selectively increases CXCR4-encoding mRNA expression in B cells. Given our focus on the lack of GCs following TcdB2 treatment, we focused the remaining studies on CXCR4.
Figure 4. Differentially expressed genes following TcdB2 exposure include CXCR4.
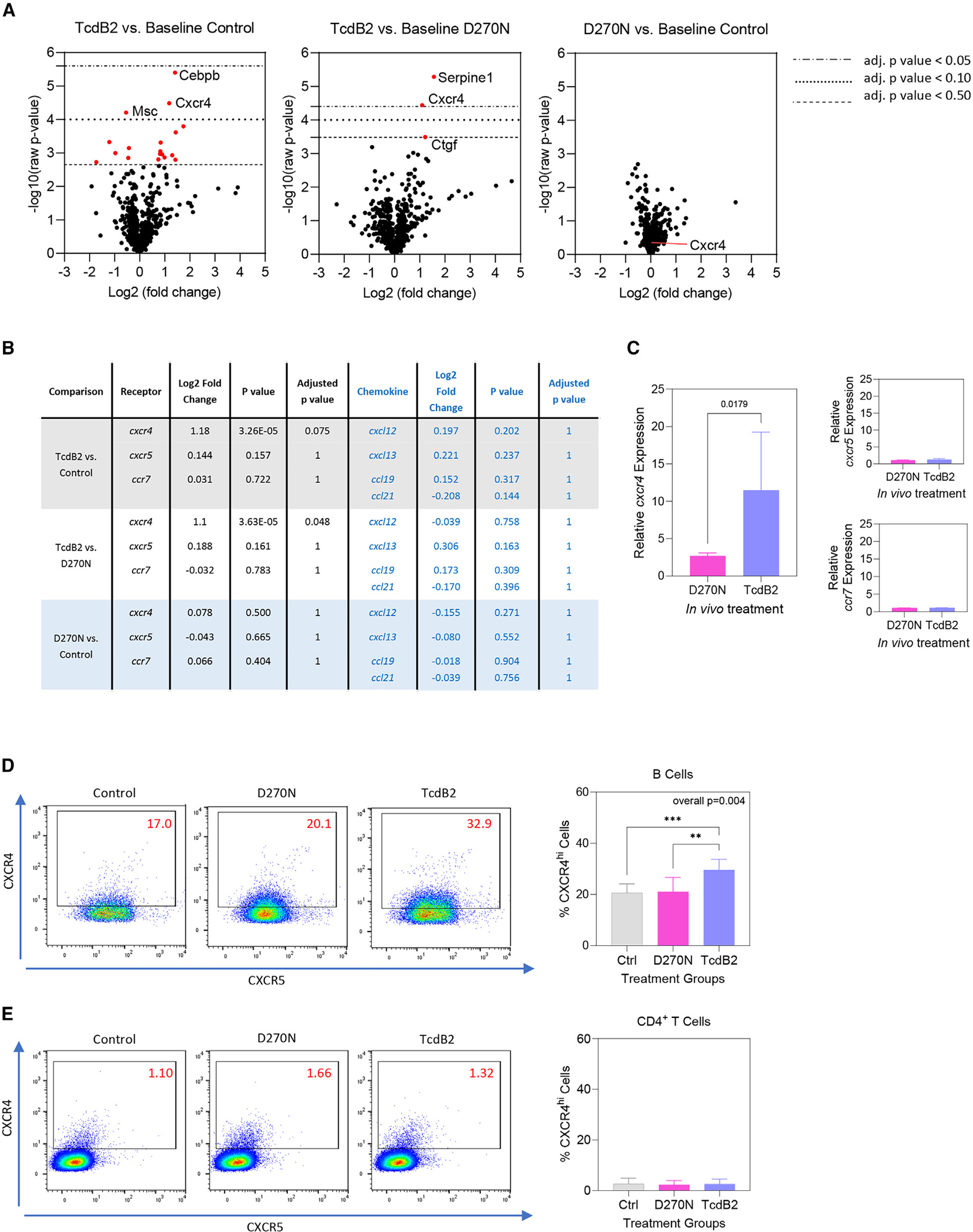
Female mice (n = 4 per group) were given 200 μL PBS vehicle control, 10 ng D270N, or 10 ng TcdB2 in PBS by the s.c. route. Seven days post treatment, RNA was purified from axillary and inguinal lymph nodes (aLNs and iLNs). Gene expression was quantified using the Nanostring nCounter SPRINT profiler platform.
(A) Differentially expressed genes (DEGs) comparing TcdB2 to PBS (left), TcdB2 to D270N (center), or D270N to PBS (right).
(B) Summary of the log2 fold change, raw p values, and adjusted p values (Benjamin-Yekutieli method) for each two-way comparison in the experiment. Values for cxcr4, cxcr5, ccr7, and their ligands are depicted. A full list of chemokines and their receptors is shown in (Table S1).
(C) Relative expression of cxcr4, cxcr5, and ccr7 in isolated B cells as determined by qPCR. Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the ΔΔCT method. Data show mean ± SD for 5 mice per group.
(D and E) B cell (D) and CD4+ T cell (E) CXCR4 and CXCR5 expression was measured by flow cytometry. Flow plots show CXCR4 versus CXCR5 expression, while graphs depict the mean ± SD percentage of each cell type expressing high levels of CXCR4. Data in are pooled from 3 experiments (n = 9 per group). Statistical significance was determined by one-way ANOVA with Kruskal-Wallace post-test (**p < 0.01, ***p < 0.001).
Using flow cytometry, it was observed that TcdB2 significantly increased CXCR4 cell surface expression by B cells, whereas the D270N mutant had no effect on expression, consistent with DEG and qPCR data (Figure 4D). The increase in CXCR4 expression was not observed in CD4+ T cells (Figure 4E). TcdB2 did not significantly affect CXCR5 cell surface expression by B cells or CD4+ T cells (Figures S5A and S5B).
TcdB2 leads to increased migration of B cells to the CXCR4 ligand CXCL12
Transwell migration assays were performed to measure responsiveness to the CXCR4 ligand CXCL12 after in vivo treatment with vehicle, D270N, or TcdB2 (Figure 5A). Background levels of migration increased in splenocytes from TcdB2-treated mice, but the effect was not statistically significant (Figure 5B). Cells from vehicle control mice showed a statistically significant response to the CXCL12 ligand, as did cells from D270N- and TcdB2-treated mice (Figure 5B). However, the highest level of migration was observed in splenocytes from TcdB2-treated mice, which was significantly different from all other experimental conditions (Figure 5B).
Figure 5. Increased B cell migration toward the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
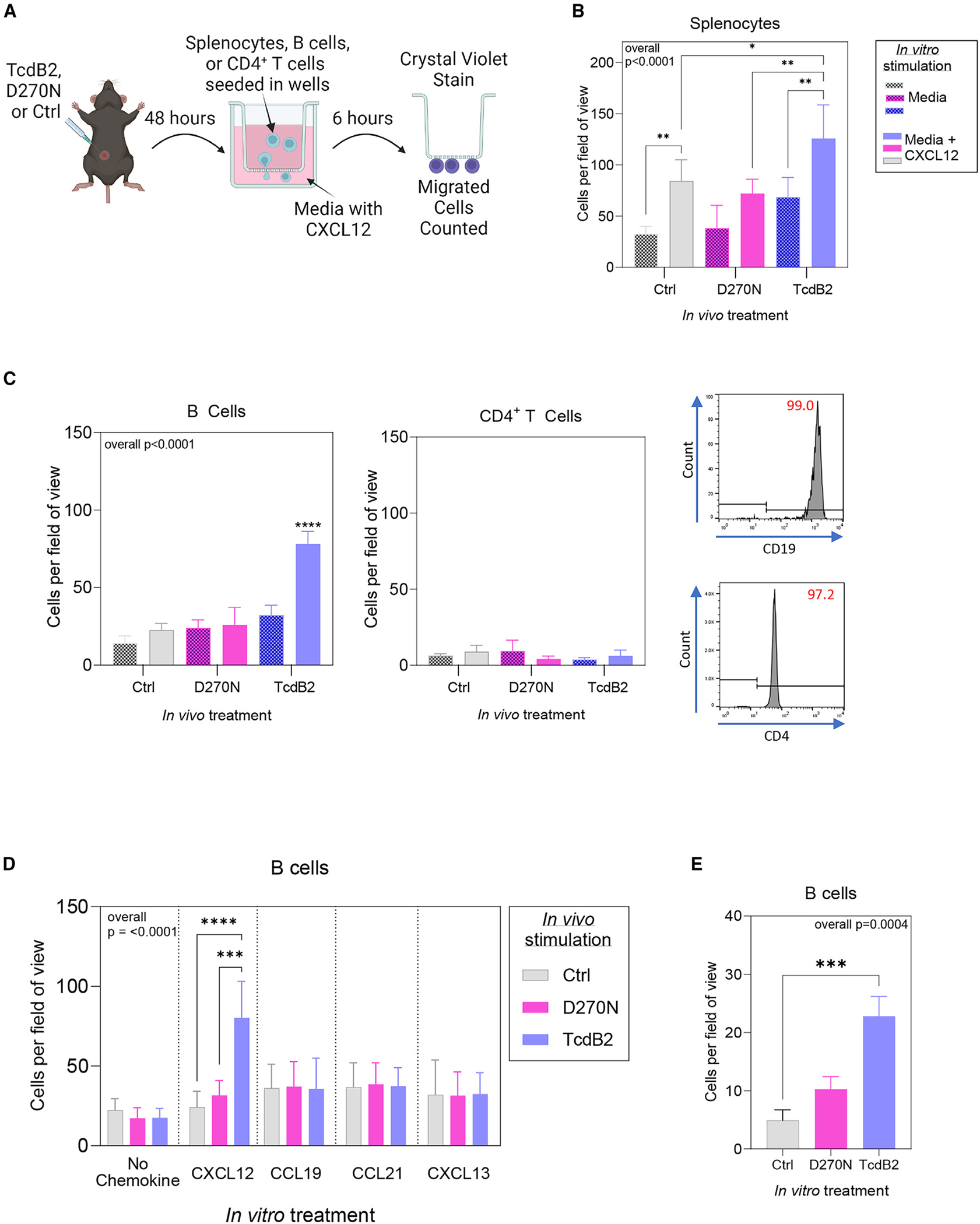
(A) Female B6 mice (n = 4 per group) were given 1 ng TcdB2, 1 ng D270N, or PBS vehicle control by the s.c. route. After 48 h, splenocytes, B cells, or CD4+ T cells were isolated (B and CD4+ T cells by magnetic separation) and seeded into the top of a Transwell. The bottom of the Transwell contained serum-free medium with or without CXCR12. Cells were incubated for 6 h, and then migratory cells were fixed and stained with crystal violet and counted.
(B) Quantification of migratory splenocytes averaged from 4 fields of view from each Transwell membrane (mean ± SD, n = 4 per group). Data are representative of two independent experiments.
(C) Representative flow cytometry plots for isolated B cells and CD4+ T cells. Graphs depict quantification of migratory B cells and CD4+ T cells (mean ± SD, n = 4 per group). Data are representative of two independent experiments.
(D) Isolated B cells from vehicle-, D270N-, and TcdB2-treated mice were stimulated in vitro with ligands for CXCR4, CXCR5, and CCR7 (CXCL12, CCL19/CCL21, and CXCL13, respectively). Data are pooled from two independent experiments (mean ± SD, n = 8 per group).
(E) Isolated splenic B cells were cultured with vehicle, D270N, or TcdB2 for 6 h (n = 3). Graph shows mean ± SD, and data are representative of 2 similar experiments. Statistical significance was determined by one-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Isolated B cells from TcdB2-treated mice showed a prominent and significant increase in migration as compared to all other experimental conditions (Figure 5C). In contrast, naive CD4+ T cell migration was minimal and not affected by TcdB2 treatment (Figure 5C). The ligands for CCR7 (CCL19 and CCL21) and CXCR5 (CXCL13) stimulated a low level of B cell migration. TcdB2 had no effect on those responses, demonstrating selective effects on CXCR4-mediated migration (Figure 5D). In vitro treatment of B cells with TcdB2 also revealed a glucosyltransferase-dependent and direct effect on CXCR4-mediated migration (Figure 5E). These results demonstrate that enzymatically active TcdB2 increases CXCR4 expression by B cells but not CD4+ T cells and increases their migration toward the CXCL12 chemo-attractant. These results are consistent with inhibited GC formation and IgG recall responses by TcdB2.
CDI increases B cell CXCR4 expression and cell migration
To determine whether TcdB2 effects on CXCR4 expression and cell migration were recapitulated in a CDI model, B6 mice were infected with TcdB2-secreting C. difficile R20291 spores (Figure 6A). Successful infection was confirmed by measuring weight loss (Figure 6B), C. difficile colony-forming units (CFUs) in fecal samples (Figure 6C), and measurement of cecum and colon length (Figure 6D). After 2 days, spleen and axillary lymph nodes (aLNs), iLNs, and mesenteric lymph nodes (mLN) were collected for CXCR4 and CXCR5 cell surface expression and cell migration (Figures 6E–6J and S5C–S5F). Lymphocytes from proximal (mLN) (Figure 6F) and regional (iLN, spleen) (Figures 6G and 6H) lymphoid organs had an increase in CXCR4 cell surface expression from the CDI group compared to the uninfected control group, while lymphocytes from distal lymphoid organs (aLN) (Figure 6I) did not have a change in CXCR4 expression. CXCR5 expression was not altered in CDI versus the control group (Figures S5C–S5F).
Figure 6. Increased lymphocyte migration toward the CXCR4 chemoattractant CXCL12 following CDI.
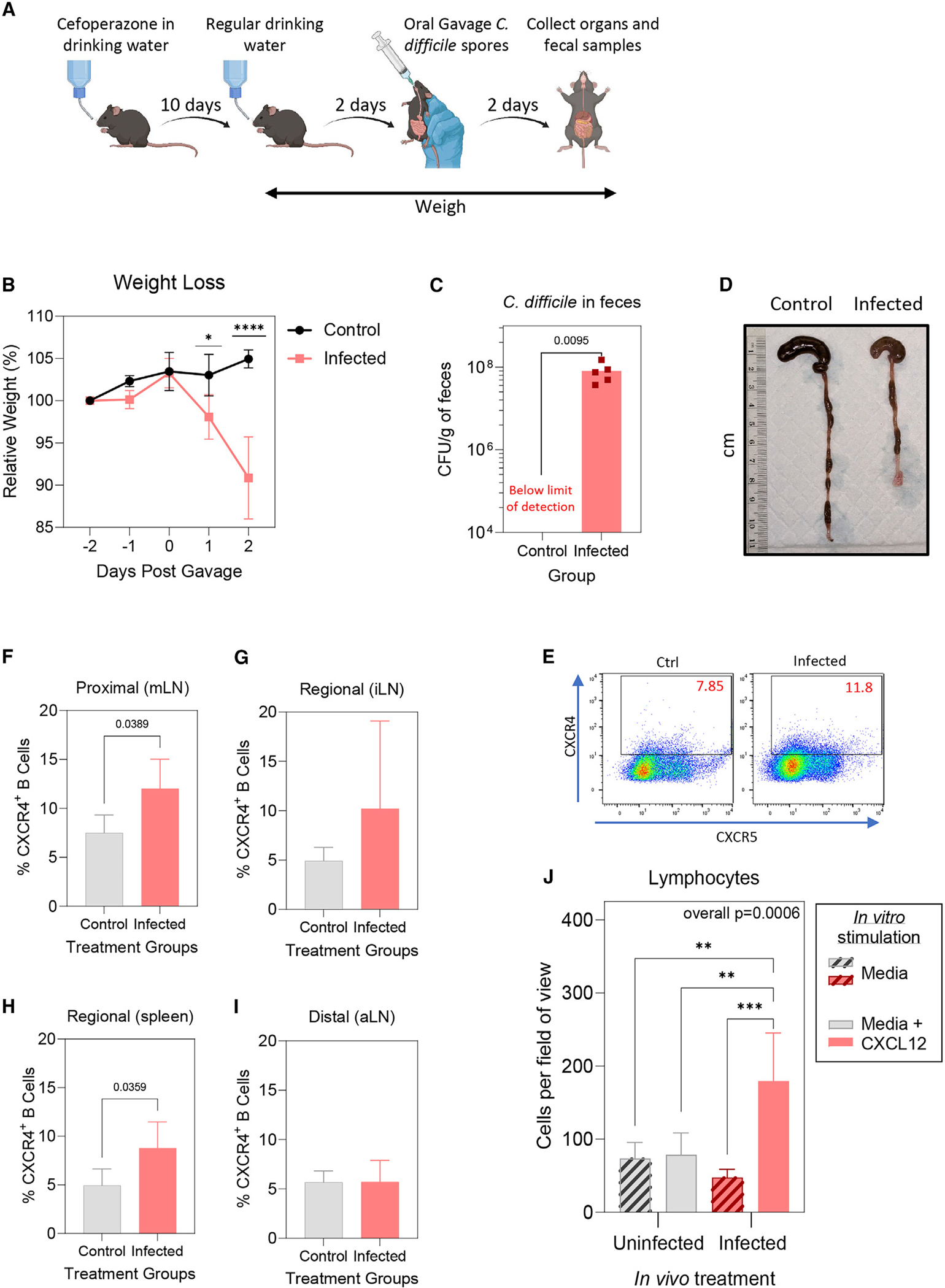
(A) Female B6 mice (n = 5 infected, n = 4 control) were given cefoperazone for 10 days, then distilled drinking water for 2 days. Mice were then given heat-treated C. difficile R20291 spores or distilled water via oral gavage and lymphatic organs, colon, and cecum, and fecal samples were collected 2 days post gavage.
(B) Mean ± SD weights (relative to the starting weight obtained 2 days before gavage).
(C) Mean ± SD C. difficile CFUs from fecal samples collected 2 days post gavage.
(D) Representative image of cecum and colon from a control mouse (left) and infected mouse (right).
(E) Representative flow cytometry plot of the CXCR4 versus CXCR5 gating strategy (left, control; right, infected).
(F–I) Mean ± SD percentage of CXCR4hi B cells in (F) mLNs, (G) iLNs, (H) spleen, and (I) aLNs, determined by flow cytometry.
(J) Mean ± SD numbers of migratory lymphocytes from mLNs averaged from 4 fields of view from each Transwell membrane.
Statistical significance was determined by one-way ANOVA with Tukey’s multiple-comparisons post-test or two-tailed t test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Cells from mice infected with C. difficile showed significantly higher CXCL12-dependent migration than mLN cells from uninfected control mice (Figure 6J). The data demonstrate that the upregulation of CXCR4 on B cells and its effect on migration was recapitulated in a CDI mouse model. This implicates C. difficile-secreted TcdB2 in humoral immune suppression during CDI.
The CXCR4 antagonist AMD3100 rescues GC formation
The FDA-approved drug AMD3100 is a CXCR4 antagonist that prevents binding of its ligand CXCL12.40 To determine whether the effects of TcdB2 on CXCR4 function could be blocked in vivo, mice were administered TcdB2 with or without AMD3100 treatment (Figure 7A). A standard (10 μg/g (mouse weight)) and a lower tonic dose (1 μg/g)45,46 of AMD3100 significantly decreased TcdB2-induced lymphocyte migration (Figure 7A). AMD3100 resulted in a significant increase in GC size and number following TcdB2 administration and B2Δ/alum vaccination (Figure 7B). In contrast, AMD3100 did not reverse TcdB2-suppressed IgG1, IgG2b, or IgG2c recall responses (Figure 7C), showing that correction of CXCR4 migration is insufficient to restore humoral immunity. Mice were also infected with C. difficile and treated with vehicle or AMD3100 (Figure S6A). AMD3100-treated mice had significantly attenuated disease (Figure S6B), increased survival (Figure S6C), and a lower bacterial burden (Figure S6D). Thirty days after the initial infection, mice were immunized with a reduced dose of the B2Δ/alum vaccine. Serum B2Δ-specific IgG1 was increased in mice that had previously been treated with AMD3100, but IgG1 titers were low (Figure S6E) and insufficient to translate to significantly elevated fecal IgG1 (Figure S6F) or IgA (Figure S6G). This suggests the potential for AMD3100 to prime a stronger humoral immune response following infection, but optimization is required.
Figure 7. The CXCR4 antagonist AMD3100 rescues TcdB2-suppressed GC formation but not IgG recall.
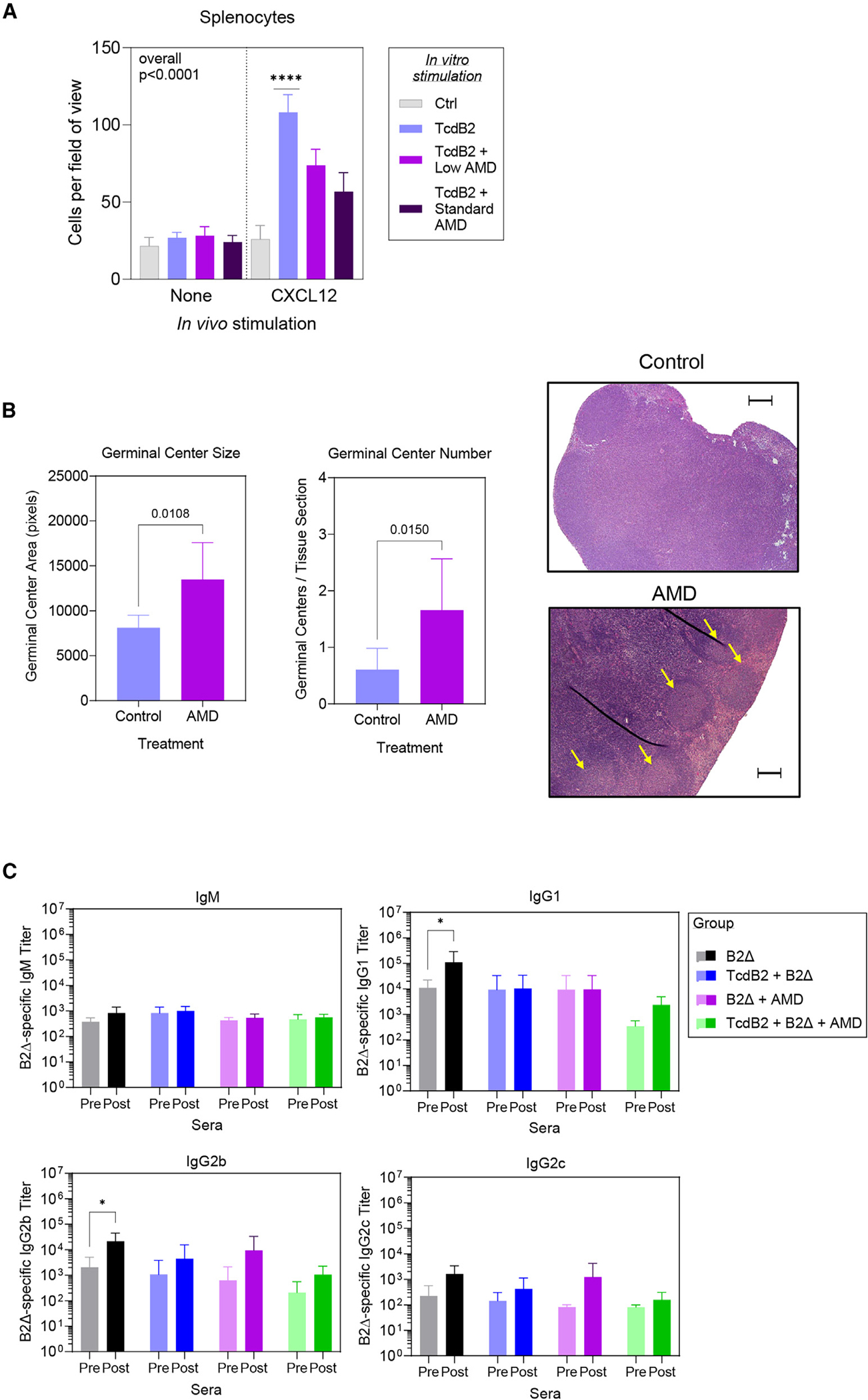
(A) Female B6 mice were given 1 ng TcdB2 or PBS vehicle control (i.p.) and then given either PBS vehicle or AMD3100 (1 or 10 μg/g of body weight) by the s.c. route. After 48 h, splenocytes were isolated, and migration toward CXCL12 was measured as described in Figure 5. The graph depicts mean ± SD numbers of migratory splenocytes averaged from 4 fields of view from each Transwell membrane. Data are from 2 pooled experiments (n = 7 mice per group). Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparison post-test; ****p < 0.0001.
(B) Mice (n = 7 per group from 2 pooled experiments) were given 1 ng TcdB2 or PBS vehicle control (i.p.) and then immunized s.c. with 10 μg of B2Δ/Alum after 5 h. At 0, 24, and 48 h post PBS or TcdB2 treatment, mice were treated with AMD3100 or PBS vehicle control (i.p.). Graphs depict the mean ± SD area and number of GCs in iLNs collected 21 days post treatment. Statistical significance was determined by two-tailed t test. Images show representative H&E sections from lymph nodes. Yellow arrows indicate GCs. Thin dark lines were due to a crease in the section. The scale bar depicts 500 μm.
(C) Mice were treated with PBS vehicle and then immunized with B2Δ/alum (B2Δ), treated with TcdB2 and then immunized (TcdB2 + B2Δ), immunized and then treated with AMD3100 (AMD + B2Δ), or treated with TcdB2, immunized, and treated with AMD3100 (AMD + TcdB2 + B2Δ). Sera were collected on day 60 (pre-boost), and a booster vaccine was administered before collection of sera on day 74 (post-boost). Data show mean titers ± SD for 5 mice per group. Significance was determined by two-way ANOVA with Sidak’s multiple comparison post-test. *p < 0.05.
DISCUSSION
We have taken significant steps toward understanding the mechanisms responsible for disease recurrence following CDI. Here, we report the discovery that the secreted toxin TcdB2, the main driver of disease pathology in CDI,25,47,48 exerts a profound and deleterious effect on the humoral immune response. TcdB2 was shown to delay IgG class switch, block GC formation, and curtail IgG recall responses. The mechanism includes increased CXCR4 expression by B cells, resulting in altered migration, and the effects of TcdB2 on GC formation could be blocked by the FDA-approved drug known as AMD3100. The Cebpb gene, reported to promote post-GC BLIMP1 expression and plasma cell development, was also significantly upregulated.44 This suggests another avenue for exploration of the impact of TcdB2 on humoral immunity beyond CXCR4-regulated GC formation.
Serum TcdB2-neutralizing IgG titers remain the best-known correlate of protection against recurrent CDI,28,49 but it has remained unclear why infection fails to stimulate an immune response that adequately prevents CDI recurrence. Several factors likely contribute to recurrence, including continued antibiotic therapy-maintained dysbiosis, continued C. difficile exposure in the environment, and possible germination of spores that are resident in host epithelial cells.4,11,50 However, we propose that insufficiently protective B cell memory following infection likely contributes to the overall risk of recurrence. This study provides a framework for understanding how protective humoral immunity is subverted by C. difficile.
We observed that TcdB2 administration prior to a standard immunization protocol resulted in delayed IgG class switch and severely abrogated IgG recall responses following administration of a booster vaccine. As a result, IgG titers were lower in TcdB2-treated mice, and this was reflected in the reduced ability of that IgG to neutralize TcdB2. However, the average affinity of the IgG, measured indirectly by ELISA, suggested no significant difference in affinity maturation of the IgG molecules that were successfully produced. Although our results showed that TcdB2 blocked GC formation, there is precedent for extra-GC affinity maturation. For example, GC-independent affinity maturation occurred in lymphotoxin-α-deficient mice when immunized with (4-hydroxy-3-nitrophenyl)acetyl-ovalbumin (intraperitoneally [i.p.]).51 Our data suggests that B cells under conditions of TcdB2 intoxication are still intrinsically able to undergo GC-independent affinity maturation.
This observation parallels what has been seen in patients following recovery from CDI. TcdB-specific memory B cells (Bmem) were isolated from patients following recovery from CDI. The Bmem-encoded Abs were fully sequenced and produced as IgG1 mAbs.8 Analysis of the curated Ig gene sequences revealed affinity maturation with significant numbers of somatic mutations in the Ig complementarity-determining 3 (CDR3) regions.8 The IgG1 mAbs were able to bind TcdB1 and TcdB2, but very few were able to neutralize toxin in vitro.8 The results are also consistent with the reported lack of IgG class switch and B cell memory following CDI of naive B6 mice,7 and suggestive of some GC-independent affinity maturation in patients.
In the present study, the effects of TcdB2 on recall responses were exerted to a greater extent on IgG2b and IgG2c than on IgG1. This is suggestive of selective effects of TcdB2 on the cytokine environment, possibly transforming growth factor β (TGF-β) and interferon γ (IFNγ) production and warrants further investigation. There is some relevance to this observation in human subjects, since previous infection with C. difficile was associated with selective effects on TcdA-specific IgG subclasses, including IgG2 subclass.52
The delay in IgG class switch and the abrogated IgG recall response were consistent with a deficiency in class switch signals. This could be reflective of the B cells’ ability to respond to class switch signals such as CD40 engagement by CD40L (expressed by CD4+ T helper [Th] cells), of Th cells to provide those signals, or insufficient communication between B cells and Th cells. In this study, stimulation of CD40 using an agonistic mAb to mimic CD40L interaction prevented the effects of TcdB2 on IgG recall responses and boosted the Ab titers. Furthermore, while TcdB2 reduced the number of post-GC, long-lived, TcdB2-specific bone marrow-resident plasma cells, CD40 activation exerted a rescue effect, showing that CD40 activation pathways remained intact after TcdB2 exposure.
Apart from demonstrating that TcdB2-exposed B cells retain an intrinsic capacity to respond to Th cell-derived IgG class switch signals, the data suggest a potential therapeutic approach for CDI. Arguably, targeted CD40 activation during an initial incidence of CDI could be used to boost IgG class switch, B cell memory, and the establishment of a long-lived plasma cell compartment in patients. Given the mortality rate associated with recurrent CDI, further consideration of a CD40-based therapeutic strategy may be considered.
The D270N mutant that renders TcdB2 unable to glucosylate small GTPases53 did not have a clear impact on IgG class switch and recall, suggesting the possibility of low-level toxicity that is not readily detected by in vivo immunization experiments. However, the data provide clues to the mechanisms by which toxin impacts IgG class switch and recall responses. Wild-type TcdB2 did not elevate cell death in cultured B or CD4+ T cells, but immunoblotting analysis revealed that the small GTPase Rac1 was glucosylated by TcdB2. This implies a direct effect of TcdB2 on B cells, leading to altered function, although we acknowledge that indirect effects of TcdB2 on B cell function could also occur. Consistent with the hypothesis of direct effects, the small GTPases, including Rac1 and Rac2 are known to affect IgG class switch.54 Conditional deletion of Rac1 in B cells of a Rac2−/− mouse tempered IgG2b production following sheep red blood cell immunization, but increased IgG2b production when B cells were stimulated with an IgG2b-promoting cocktail (lipopolysaccharide, IL-4, IL-5, and TGF-β).54 These data suggest a possible relationship between Rac1, Rac2, and γ2b gene transcription, which is disrupted by TcdB2 but also influenced by the nature of the B cell stimulation and the type of antigen used. Examining other small GTPases that TcdB2 glucosylates, such as Rho and Cdc42, and a study to examine the signaling pathways impacted by TcdB2 in B cells are therefore of interest.
The inhibition of IgG class switch and recall responses by TcdB2 was consistent with the disrupted GC reactions that were also observed. The observation that immunization-induced GC formation was blocked by TcdB2 but not the D270N mutant was particularly striking and isolates the glucosyltransferase activity of the toxin to impact on GC reactions. The effects of TcdB2 were apparent both in GC numbers and in their size; fewer, smaller GCs were observed. Given that GCs have inherent complexity, such as dark and light zones, the latter being the location of affinity maturation,55 it is reasonable to suggest that TcdB2 could differentially impact light and dark zone formation. Since high CXCR4 expression denotes B and T cells that are not resident in GC light zones, we suggest that TcdB2 subverts B and CD4+ T cells interactions in the light zone of GCs, resulting in blockade of light zone formation within GCs.
A targeted transcriptomic approach followed by confirmatory qPCR identified the chemokine receptor CXCR4 as being upregulated in draining lymph node cells following TcdB2 administration. CXCR4 protein expression was confirmed by flow cytometry to increase following TcdB2 treatment, while CXCR5 expression did not change. The conformation of the 7-transmembrane receptor CXCR4 and, thus, its ligand-binding and signaling properties are regulated by Rac1.56 By facilitating GTP binding activity, Rac1 is involved in several important processes, including cellular migration (Rac1 Rac family small GTPase 1 [Mus musculus (house mouse)]; https://www.ncbi.nlm.nih.gov/gene/19353). Rac1 is responsible for regulating conformational changes in CXCR4 that alter receptor activation and migration toward the chemoattractant CXCL12.56 Considering that TcdB2 targets Rac1, these data suggest a potential link between TcdB2 and CXCR4 activation on B cells. TcdB2 may enhance CXCR4 activation, explaining why the substantial increase in cell migration is not explained fully by increases in CXCR4 protein expression.
CXCR5 is synonymous with a follicular (or GC) localization, while CXCR4 is typical of extra-follicular localization.39 CXCR4 downregulation is required for migration of activated B cells from the dark to the light zone of the GC, where selection and affinity maturation occur.39,55 Indeed, a study by Balabanian et al. showed that a gain-of-function mutation in CXCR4 left IgG responses intact but resulted in disorganized lymphoid structure, small GC-like structures, and an increase in the ratio of light-zone to dark-zone cells,57 much in line with our observations. Cell migration assays revealed that TcdB2 treatment resulted in increased migration of B cells, but not CD4+ T cells, to the ligand CXCL12. While TcdB2 appeared to have a broad impact on B cell migration but no detectable effect on CD4+ T cells, this does not preclude an impact on antigen-specific B and T cells in the context of vaccination. Suitable B cell and Th cell reporter mice to track antigen-specific B and Th cells are likely needed to address that question. However, our data firmly implicate altered CXCR4 B cell migration as a mechanism underpinning the TcdB2-subverted humoral immune response.
CDI of mice increased CXCR4 cell surface expression on B cells, and migration to the CXCL12 ligand was profoundly increased. The data recapitulated results from TcdB2 treatment alone, demonstrating the potential of TcdB2 to suppress the humoral immune response during CDI. It was also observed that B cell CXCR4 expression increases in mLNs, iLNs, and the spleen, but not aLNs, suggesting that the effects are local and regional rather than systemic. However, the humoral immune suppression caused by CDI may be more regional and only systemic during the most severe disease. Arguably, individuals experiencing CDI are unlikely to seek vaccines until they have recovered. The duration and breadth of impact of TcdB2 on vaccine-induced GC reactions is worth further consideration. In this study, CDI had no effects on the distal aLNs but increased CXCR4 expression and cell migration in mLNs. If an individual were to receive a vaccination while experiencing CDI, we posit that the vaccine may still have efficacy, since it would be delivered intramuscularly in the deltoid muscle, distal from the peritoneal cavity. However, further work would be needed to make firm conclusions in this regard.
To mitigate the effects TcdB2 has on the upregulation of CXCR4 on B cells, the FDA-approved drug AMD3100 was utilized. AMD3100 treatment decreased the effects of TcdB2 to a remarkable degree, rescuing GC formation in TcdB2-intoxicated mice. When AMD3100 was used in the CDI model, symptoms were less severe and the mortality rate decreased, likely a consequence of neutrophil recruitment.58 Interestingly, AMD3100 treatment appeared to “prime” the mice to respond more effectively to vaccination, as indicated by low TcdB2-specific IgG titers. This approach may therefore be developed to determine how to improve vaccine responses or to reduce the severity of recurrent disease. AMD3100 may also have potential to be used in parallel with other CDI treatments in patients.
Our work adds to the growing body of observations that bacterial toxins can impact host immunity utilizing a diversity of mechanisms. A notable example is Bacillus anthracis lethal toxin, which alters the function of several immune cell types, including dendritic cells, B cells, T cells, and invariant natural killer T cells.59–62 Another example is streptococcal pyrogenic exotoxin A, which stimulates Tfh cells to kill B cells in a granzyme B-dependent manner.63 It has also been reported that other C. difficile toxins have specific effects on the immune system. Binary toxin can suppress eosinophilia,64 while TcdB1 activates ILC3s.65 Therefore, many bacterial toxins are not limited to killing target cells but, because of co-evolution with their hosts, deploy sophisticated mechanisms to alter the function of specific cellular targets and cell behavior. In CDI, TcdB2 appears to scramble B-to-Th cell communication so that adaptive immune memory is subverted. This study provides a framework for several lines of investigation into how TcdB2 impacts humoral immunity to C. difficile and may inform therapeutic interventions.
Limitations of the study
Our study unequivocally identifies altered CXCR4-mediated B cell migration that is dependent on the glucosyltransferase activity of TcdB2. However, the study does not address potentially glucosyltransferase-independent effects on other aspects of humoral immunity. Further study is needed to address other immune-altering functions of TcdB2 and to identify mechanisms that may be targeted in combination with the CXCR4 mechanism for therapy. A deeper understanding of the effects of TcdB2 on GCs is also desirable. Arguably, there could be specific effects on dark versus light zone formation as opposed to a complete GC blockade by TcdB2 which this study did not address. Although we did not detect a strong stress response to the doses of TcdB2 used, the impact of sustained TcdB2 during infection should be examined in future studies for impacts on humoral immunity. Finally, as reagents become available, a full analysis of the impact of TcdB2 on C. difficile-specific CD4+ T cells is warranted, as this was not possible for the present study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mark L. Lang (mark-lang@ouhsc.edu).
Materials availability
Requests for resources and reagents should be directed to and will be fulfilled by the lead contact Mark L. Lang (mark-lang@ouhsc.edu).
Data and code availability
Raw data files corresponding to the results reported herein have been deposited in Mendeley (https://doi.org/10.17632/m2v6dj33s5.2).
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Female C57BL/6 mice (C57BL/6NCr) at 8 weeks of age were purchased from Charles River Laboratories (Bethesda, MD, Cat # model 027). All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center (OUHSC) (original protocol 20–020-AHI, renewed as 22–082-CHI). Procedures were performed under inhalational anesthesia using a 4% isoflurane/96% medical air mixture dispersed through a precision vaporizer. This study was performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
METHOD DETAILS
Antibodies and fluorochromes
Horseradish peroxidase (HRP)-conjugated anti-mouse IgA, IgM, IgG1, IgG2b, and IgG2c were purchased from Southern Biotech (Birmingham, AL). Biotin-conjugated anti-CXCR5 (2G8), FITC-conjugated anti-B220 (RA3–6B2), FITC-conjugated anti-CD21/23 (7G6), and APC-conjugated anti-CD5 (53–7.3) monoclonal antibodies (mAbs) and PE-conjugated streptavidin were from BD Biosciences (San Jose, CA). APC-Cy7-conjugated anti-CD19 (6D5), Alexa 488-conjugated anti-Ki67 (11f6), and Biotin-conjugated anti-CXCR5 (L138D7) mAbs were purchased from Biolegend (San Diego, CA). APC-conjugated anti-CXCR4 (2B11) and PE-conjugated anti-CD19 (1D3) were purchased from eBiosciences (San Diego, CA). VioletFluor 450-conjugated anti-CD4 (GK 1.5) was purchased from Tonbo (Cytek, San Diego, CA). Alexa Fluor 750-conjegated anti-CD93 (223427) was purchased from R&D Systems (Minneapolis, MN). PE-conjugated anti-Annexin V and 7AAD were from Stemcell Technologies (Cambridge, MA). Invitrogen, (Carlsbad, CA). FcR-blocking mAb 2.4G2 was from BioXCell (Lebanon, NH).
Purification of TcdB2, B2Δ, and D270N
TcdB2, B2Δ, and D270N were expressed in Bacillus megaterium (MoBiTec, Göttingen, Germany) and purified by nickel affinity chromatography (GE Life Sciences, Boston, MA) as previously described.66 Purity and integrity were confirmed by SDS-PAGE and each batch of TcdB2 was tested for toxicity using a CHO cell killing assay.67
Immunization and bleed schedule
Mice were administered 1 ng TcdB2 in sterile phosphate-buffered saline (PBS) intraperitoneal (i.p.) injection. Control mice were injected with PBS or 1 ng of an enzymatically inactivated TcdB mutant known as D270N.53 After 5 h, mice were immunized subcutaneously (s.c.) with 10 μg B2Δ, a TcdB2 mutant incapable of entering host cells.66 Unless stated otherwise, the B2Δ was adsorbed to a 2% w/v Alhydrogel alum adjuvant suspension (Invivogen, San Diego, CA) in PBS and is referred to as B2Δ/alum herein. Vaccines were divided equally over both flanks. The mice were immunized with B2Δ/alum on day 0 and then boosted on day 67 with B2Δ in PBS. Retro-orbital blood was collected using heparin capillary tubes on days 14, 28, 42, 67 (pre-boost), and 81 (14 days postboost), unless stated otherwise. Blood samples were incubated for 2 h at room temperature, then centrifuged at 13,000 rcf for 15 min. Sera were collected, aliquoted, and stored at −20°C. Where indicated, TcdB2-treated and B2Δ-immunized mice were injected s.c. with 100 μg anti-CD40 (rat IgG2a, clone FGK4.5) or isotype control mAbs (rat IgG2a, clone 2A3) formulated for in vivo use (InVivoPlus, BioXCell, Lebanon, NH). The mAbs were injected on days 1 and 8.
ELISA
Nunc MaxiSorp Enzyme-linked Immunosorbent Assay (ELISA) 96-well plates (Thermo Scientific, Waltham, MA) were coated overnight at 4°C with phosphate coating buffer (0.1 M Na2HPO4, pH 9.0) containing B2Δ at a final concentration of 10 μg/mL. Wells were washed 4 times with PBS-T (1x PBS, 0.05% Tween) and blocked with 1% bovine serum albumin (BSA) in PBS-T for 2 h at room temperature. This was followed by washing 4 times with PBS-T. Wells were incubated overnight at 4°C with mouse sera (serially diluted 2-fold) or fecal supernatant and PBS-T, then washed with PBS-T 4 times. Wells were incubated with 0.2 μg/mL HRP-conjugated IgG1, IgG2b, IgG2c, or IgM for 1 h at 4°C. Wells were washed with PBS-T and developed for 5 min at room temperature by addition of 90 μL of 2,2′azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS) substrate to each well (SeraCare, Milford, MA). To stop the reaction, 110 μL of 10% w/v sodium dodecyl sulfate (SDS) solution was added to each well. Optical density (OD) of the samples at an absorbance of 405 nm was measured using the Spectrostar Nano spectrophotometer (BMG Labtech, Cary, NC) within 30 min.
Cell culture and in vitro neutralization
The hamster epithelial cell line CHO-K1 (American Type Culture Collection, Manassas, VA) was cultured in F12-K media (Gibco, Life Technologies Corporation, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA), 100 units/ml Antibiotic / Anti-mycotic solution (Corning, Manassas, VA). Cells were cultured at 37°C in 5% CO2 and passed every 48 h using tryptic digestion. Ninety-six-well plates (Falcon, Corning, Durham, NC) were seeded with CHO-K1 cells at a density of 3 × 104 cells/well and incubated overnight at 37°C in 5% CO2. Sera were diluted to 1:100 or 1:2000, then incubated for 1 h at 37°C with F12-K media containing TcdB2 at a final concentration of 250 nM. The CHO cell culture media was replaced with the sera-TcdB2-media solution (or media lacking TcdB2 or sera). Cells were incubated for 24 h at 37C before addition of 100 μL media containing 10 μL Cell Counting Kit-8 (CCK-8) (Sigma-Aldrich, Millipore Sigma, Saint Louis, MO). Cells were incubated for a further 2–3 h at 37°C until the absorbance at 450 nm associated with untreated control cells reached 3.0. Background absorbance was near zero and subtracted from all samples. The absorbance associated with the TcdB-treated control cells was then subtracted. The percent viability was calculated as absorbance (experimental sample)/absorbance (untreated controls) × 100.
Murine cell preparation
Murine spleens, lymph nodes (axillary (aLN), inguinal (iLN), and mesenteric (mLN)), and bone marrow cells were isolated from B6 mice. Cells from spleen and lymph nodes were isolated by mechanical disruption. Bone marrow cells were obtained by trimming the ends of the femurs and tibias and flushing with media using a 27-gauge syringe needle. Erythrocytes were removed by hypotonic lysis with Tris-buffered ammonium-chloride (ACT) for 5 min then washed with PBS. Final cell pellets were resuspended in RMPI 1640 media containing 10% FBS, 100 units/ml penicillin, and 100 μg/mL streptomycin.
ELISPOTS
Multiscreen High-throughput Satellite (HTS) enzyme-linked immunosorbent spot (ELISPOT) wells (Millipore, Bedford, MA) were prepared for antigen coating by incubating with 35% v/v ethanol for 30 s and washed twice with PBS. The plates were coated overnight with anti-mouse Ig or B2Δ (10 μg/mL final concentration) at 4°C. Plates were washed 3 times with PBS and blocked with RPMI 1640 containing 10% FBS for 2 h at room temperature. Isolated bone marrow cells (3×106 cells per well) were added then subjected to a 1:3 serial dilution such that the wells contained 2.00 × 106, 6.67 × 105, 2.22 × 105, or 7.41 × 104 cells. The plates were then incubated in 5% CO2 at 37°C for 4.5 h. The cells were lysed and removed by 4 washes with PBS-T. Plates were incubated overnight at 4°C with 5% v/v FBS in PBS containing HRP-goat anti-mouse IgG1 and IgG2b mAb at a final concentration of 1.0 μg/mL (Southern Biotech, Birmingham, AL). The plates were then washed with PBS-T and colorimetric detection performed. The developing solution was prepared by dissolving one tablet of 3-amino-9-ethyl-carbazole (AEC) (Sigma Chemical Co., St. Louis, MO) in 2.5 mL dimethylformamide (Sigma Chemical Co., St. Louis, MO) and mixing with 47.5 mL of a 0.0075 N acetic acid and 0.0175 M sodium acetate buffer. The solution was passed through a 0.2 μm syringe filter before adding hydrogen peroxide to a final concentration of 0.0005% v/v. One hundred microliters of developing solution was added to each well and incubated at room temperature for 10 min. The reaction was stopped by addition of deionized water. The plates were then washed 20 times and allowed to air dry at room temperature under light-protected conditions for 24 h. Antibody secreting cells (spots) were enumerated using an ImmunoSpot analyzer (Cellular Technology Limited, Shaker Heights, OH).
Flow cytometry
Splenocytes and lymph node cells (3 × 106) were incubated with anti-FcR-blocking Ab 2.46G at a final concentration of 20 μg/mL for 5 min. Cells were then stained with fluorochrome-conjugated mAb cocktails to detect CD4+ T cells (anti-CD4) and B cell (anti-CD19 or anti-B220) populations. After incubating at 4°C for 1 h, cells were washed with PBS 3 times by centrifugation at 250 rcf for 5 min. Cells were then fixed with 1% w/v paraformaldehyde in PBS. Data were collected using a Stratedigm S1200Ex flow cytometer (Stratedigm, San Jose, CA) and analyzed with FlowJo software (Version 2.0.1, Tree Star, Ashland, OR).
Cell viability assay
Three million splenocytes in RPMI 1640 media were added to each well of a 48-well tissue culture plate. Vehicle (media), TcdB2, or D270N was added to a final concentration of 0.1, 1, or 10 pM. Cells were incubated at 37°C for 30 min, 6, 24, or 48 h before removal of TcdB2 or D270N by washing. Cells were re-suspended in RPMI 1640 media containing 1% v/v FBS and incubated with an anti-FcR-blocking Ab (2.4G2) at a final concentration of 20 μg/mL for 5 min. Cell were stained with anti-B220 and anti-CD4 fluorochrome-conjugated Abs for 30 min then with anti-Annexin and 7AAD in Annexin V Binding Buffer (Stemcell Technologies, Cambridge, MA) for 15 min at room temperature. The cells were washed with 1 mL of Annexin V Binding Buffer (250 rcf for 5 min), then re-suspended in 200 μL of Annexin V Binding Buffer. The cells were analyzed immediately by flow cytometry.
Capillary electrophoresis and western blot
Splenocytes or splenic B cells isolated with the STEMCELL Pan B cell Isolation Kit (Stemcell Technologies, Cambridge, MA) were incubated for 4 h at 37°C in RPMI 1640 with 5% v/v FBS in the presence or absence of TcdB2 or D270N at a final concentration of 10 pM. Cells were washed twice in PBS and re-suspended at 1 × 108 cells/ml in Mammalian Protein Extraction Reagent (MPER) (Thermo Scientific, Rockford, IL) containing 1X protease inhibitor cocktail set 1 (EMD Millipore, Billerica, MA). Cells were shaken for 10 min on ice and then centrifuged at 14,000 rcf for 15 min. Supernatants were collected and aliquots stored at −80°C. The protein concentration in the lysates was calculated using a Bradford Protein Assay (Bio-Rad, Mountain View, CA).
The Jess Simple Western machine (Protein Simple, San Jose CA) is an automated capillary-based protein separation and immunoblotting system. The manufacturer’s standard protocol for the 12–230-kDa Jess separation module was followed. Cell lysates were prepared in 0.1X Sample buffer and Fluorescent 5X master mix containing dithiothreitol (400 mM final concentration) to achieve a final loading of 1.5–5 μg of protein per capillary. Samples were denatured at 95°C for 5 min before being loaded into each well. The 12–230 kDa ladder and cellular lysates were separated by electrophoresis in their respective capillaries and then fixed. Capillary bound proteins were washed, blocked and incubated with a 1:100 dilution of anti-Rac1 (Clone 102/Rac1, Becton Dickinson, San Diego, CA) and a 1:500 dilution of anti-GAPDH (Clone 6C5, Abcam, Cambridge, MA) Abs. Capillaries were washed again and incubated with a 1:100 dilution of HRP-conjugated anti-mouse IgG2b (Rac1) or 1:200 IgG1 (GAPDH) (Southern Biotech, Birmingham, AL). The HRP-labeled Abs were detected using peroxide/luminol-S reagent (Protein Simple, San Jose, CA). Imaging of the chemiluminescence from the capillary bound proteins was performed using Compass for Simple Western software (Version 6.1.0, Protein Simple, San Jose, CA). An internal system control was included in each run.
Norepinephrine assay
Noradrenaline (Norepinephrine) was measured following the Noradrenaline High Sensitive ELISA Assay Kit protocol from Eagle Biosciences according to manufacturers instructions (Amherst, NH). All steps were performed at room temperature unless stated otherwise. Plasma samples were prepared and stored at −80°C until use. Ten microliters of plasma or standards in duplicates were mixed with 100 μL Extraction Buffer in an Extraction Plate and subject to shaking (high speed) for 1 h before decanting and washing with 1 mL wash buffer (5 min while shaking slowly), then decanted. This was followed by addition of 150 μL of Acylation Buffer and 50 μL of Acylation Reagent to each well and incubation for 20 min while shaking (medium speed). The plate was then decanted and washed twice. 1 mL of Wash Buffer was then incubated with 125 μL 0.025 M HCl per well under light-protected conditions for 20 min while shaking at medium speed. One hundred microliters of samples from each well was then transferred from the Extraction Plate to the Enzyme Plate. Twenty microliters of Enzyme Mix was added to each well, followed by shaking (medium) for 1 min under light-protected conditions and a further 90 min incubation at 37°C without shaking. One hundred microliters of sample from each well was then transferred from the Enzyme Plate to a well of the MT-strips (wells precoated with Noradrenaline). Twenty microliters of Noradrenaline Antiserum was added to each well. The MT-strips were incubated overnight at 4°C under light-protected conditions. Wells were decanted then washed with 250 μL of Wash Buffer 3 times (without shaking). Wash buffer was removed then samples were then incubated with 100 μL POD-Conjugate per well at for 60 min while shaking (medium) and washed 4 times (decanting the wells between each wash). After adding 100 μL Substrate to each well, samples were incubated for 40 min while shaking (medium). This was followed by addition of 100 μL of Stop Solution. The colorimetric change was detected at an absorbance of 450 nm using a Spectrostar Nano plate reader (BMG Labtech, Cary, NC), with a reference wavelength of 570 nm. Noradrenaline concentrations were calculated by interpolating from a standard curve (sigmoidal curve – 4 Parameter Logistic), where X is the concentration of each sample.
Histology and immunohistochemical (IHC) staining
Mice were injected s.c. with 1 ng TcdB2 or D270N in sterile PBS and controls were given sterile PBS. Mice were then immunized with B2Δ/Alum following the previously described method. The iLNs were isolated and fixed in Excalibur’s Alcoholic Z-Fix (containing formaldehyde). Paraffin sections (5 μm thick) were mounted on slides by Excalibur Pathology Inc. (Norman, OK). A portion of the slides were stained with hematoxylin and eosin (H&E) and imaged. The remaining slides were used for immunohistochemical (IHC) staining. For IHC staining, slides were deparaffinized by incubating for 3 min in xylene, 1:1 xylene:ethanol, then 95% and 50% ethanol concentrations. Slides were then submerged in sodium citrate buffer (10 nM NA Citrate, 0.05% Tween 20, pH 6.0) and microwaved on a low power setting for 1 h. Slides were washed 3 times with PBS for 5 min and incubated with 0.3% Triton X-100 in PBS for 15 min. After washing with PBS, slides were blocked for 30 min in a humidity chamber by incubation with 10% v/v normal goat serum (NGS) in 1% w/v BSA. Tissue sections were stained with fluorochrome-conjugated mAbs to detect B cells and proliferating cells to identify germinal centers (GCs). Slides were washed with PBS and mounted in prolong gold medium (Invitrogen, Carlsbad, CA), dried for 24 h, then visualized using the Leica M205-MFC THUNDER microscope (Leica, Deer-field, IL).
Germinal center image quantification
Bright field images of H&E-stained lymph node sections at a 10X magnification were analyzed using ImageJ (NIH, distributed by Fiji, Bethesda, MD). The area of a GC was measured by drawing around the boundary of the structure and calculated as pixel number by the ImageJ software. For each GC 6–8 consecutive 5 μm sections were analyzed and the average area of a given GC calculated. Where more than one GC was observed in a section, the average size of a GC within the lymph node was calculated and used to obtain a single data point.
Multi-targeting mRNA profiling
Mice were injected s.c. with 10 ng TcdB2 in PBS (controls were given PBS or 10 ng D270N in PBS). The aLNs and iLNs were isolated after 7 days. Lymph nodes were placed immediately in DNA/RNA Shield (Zymo Research, Irvine, CA). RNA was purified from the lymph nodes using Zymo Quick-RNA Miniprep Plus Kit (Zymo Research, Irvine, CA). Purified RNA was stored at −80°C until needed. RNA analysis was performed by the OUHSC core facility using a NanoString nCounter SPRINT profiler (NanoString, Seattle, WA) in conjunction with the nCounter Myeloid Innate Immunity Panel. Data were analyzed with nSolver 4.0 Advanced Analysis Software (Version 2.0.134, NanoString, Seattle, WA). Analysis was performed by normalizing the raw transcript counts utilizing negative synthetic sequences to account for background noise and positive synthetic sequences to account for technical variations.65 Raw data supporting the analyses herein are available at Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi, Accession # GSE263792).
cDNA preparation
B cells were isolated from aLNs and iLNs as described, resuspended in 500 μL of DNA/RNA Shield (Zymo Research; Irvine, CA), before immediate RNA purification. RNA was purified from B cells using the Quick-RNA MiniPrep Plus Kit (Zymo Research; Irvine, CA). Upon purification, RNA concentration was measured using a NanoDrop 2000 Spectrophotometer (Thermo Scientific; Waltham, MA). RNA was converted to cDNA using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen; Carlsbad, CA) following the manufacturers protocol and stored at −20°C until ready for use. Five micrograms of RNA in a 1 μL volume was added to 1 μL Oligo(dT) primer (50 μM), 1 μL dNTP mix (10 mM), and 7 μL DEPC-treated water for each sample into 0.2 mL TempAssure PCR tube strips (USA Scientific; Ocala, FL). Samples were incubated at 65°C for 5 min in an Applied Biosystems 2720 Thermal Cycler (Waltham, MA), then on ice for 1 min. Ten microliters of cDNA Synthesis Mix (2 μL 10X RT buffer, 4 μl 25 mM MgCl2, 2 μL 0.1 M DTT, 1 μL RNaseOUT, and 1 μL SuperScript III RT) was added to each sample. Additionally, no-RT controls were made by adding 10 μL of cDNA Synthesis Mix to the control but replacing the SuperScript III RT with 1 μL of DEPC-treated water. Samples were pulsed on VWR mini centrifuge (Radnor, PA), then incubated in the thermal cycler for 50 min at 50°C followed by 85°C for 5 min, then on ice for 1 min. After brief centrifugation, 1 μL RNase H was added to each sample and incubated in the thermal cycler at 37°C for 20 min. The cDNA concentration was then measured.
qPCR
For each sample, the following was added into 96-well PCR plate (Thermo Scientific; Waltham, MA): 10 μL Power SYBR Greeen PCR Master Mix (2X) (Applied Biosystems; Waltham, MA), 100nM in 1μL primer mix, 100 ng in 1 μL cDNA template, and 8 μL nuclease-free water. Primers used were cxcr4, cxcr5, ccr7, and gapdh (housekeeping) (OriGene; Rockville, MD). Samples were run on an Applied Biosystems QuantStudio Thermal Cycler (Waltham, MA) with the following conditions: Hold for 10 min at 95°C and cycled 40X for 15 s at 95°C then 60 s at 60°C. The ΔΔCT method of analysis was used wherein expression of a given gene was normalized to the gapdh housekeeping control gene.
Transwell migration assays
B cells and CD4+ T cells were isolated from splenocytes using magnetic bead separation following the manufacturer’s protocol for the EasySep Mouse Pan-B Cell and EasySep Mouse Naive CD4+ T cell Isolation Kits (STEMCELL Technologies, Cambridge, MA). Five hundred microliters of serum free media (RPMI 1640) or media containing CXCL12, CXCL13, CCL19 or CCL21 (Sino Biological, Wayne, PA) at a final concentration of 100 ng/mL was added to the lower chamber of a 24-well transwell plate (Corning, Manassas, VA). Thirty thousand cells in a 300 μL volume, consisting of either total splenocytes, isolated B cells, or isolated CD4+ T cells in serum free media were added to the upper chamber of the transwell plates (6.5 mm diameter insert with 8.0 μm pore size). Cells were then incubated for 6 h at 37°C in 5% CO2. The media was carefully aspirated from the transwell inserts and the interior membrane gently swabbed with a sterile cotton swab to remove any non-migratory cells. Transwell inserts were then placed in plate wells containing 700 μL of 70% ethanol for 10 min and allowed to air dry for 10 min to fix migratory cells on the underside of the membrane as cells do not drop into the well below the membrane.68 Once dry, transwell inserts were placed in plate wells containing 600 μL of a 0.02% w/v crystal violet solution for 10 min to stain migratory cells. The inserts were gently dipped in distilled water to remove excess crystal violet and left to dry overnight. The membrane inserts were imaged using a light microscope at 20X magnification selecting four different randomized fields of view per well. Migratory cells in each field were counted and the average value used to obtain each data point.68,69
C. difficile spore preparation
C. difficile R20291 spores were prepared and isolated as previously described.70 Briefly, a single colony grown on Brain Heart Infusion (BHI) + Taurocholic acid (TCA) was streaked onto a pre-reduced 70:30 agar plate and incubated at 37°C for 5 days in an anaerobic chamber. Spores were then harvested from the agar surface by adding 1.5 mL sterile water and scraping the cell/spore material into a 1.5 mL microcentrifuge tube. Cell/spore material was stored at 4°C for 7 days and then separated in a 50% w/v sucrose gradient (centrifuged at 250 rcf for 20 min). Finally, the spore pellet was washed 5 times with sterile distilled water and placed in a glass vial at 4°C. Before infection of mice, C. difficile spores were enumerated by plating on TCCFA (Taurocholate Cycloserine Cefoxitin Fructose Agar). A pre-calculated concentration of spore inoculum was heated at 65°C for 20 min, then allowed to cool for 5 min at room temperature.71
C. difficile infection
Mice were housed in sterile cages with sterile food. The animals were provided with Cefoperazone sodium salt (Millipore Sigma, St. Louis, MO) in distilled drinking water (0.5 g/L) for 10 days, followed by 2 days of distilled water (Thermofisher, Waltham, MA). Mice were infected by oral gavage with 1 × 107 heat-treated C. difficile R20291 spores or distilled water to control for the gavage procedure. C. difficile-associated pathology was assessed by monitoring daily weights, and other clinical signs such as lethargy, hunched posture, and diarrhea. Animals were euthanized if the weight loss reached 20.0% or the mice were moribund.71 Bacteria were quantified on day 2 post-gavage by homogenizing fecal pellets with 1X PBS, serially diluted, plated on TCCFA and cultured under anaerobic conditions at 37°C. CFUs were counted within 48 h.71
AMD3100 drug treatment
Mice were injected via the i.p. route with AMD3100 (Millipore Sigma) (1 or 10 μg/g body weight) in sterile PBS at hrs 0, 24, and 48 post intoxication or oral gavage.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental design
All experiments were designed with prior consideration of statistical power and were repeated to demonstrate reproducibility. Typically ‘n’ refers to the number of individual mice in an experimental group. Figure legends provide information on group size and number of experimental replicates and whether data shown are representative or pooled. Where indicated, blinded analyses were performed, specifically for histology images and for cell migration assays.
Statistics
Data were analyzed using GraphPad Prism (Version 9.1.1, La Jolla, CA). A two-tailed t test or a Mann-Whitney U test, and one-way ANOVA with Tukey’s or Dunnett’s multiple comparison test was used for statistical analysis between two and multiple experimental groups respectively. Where indicated, paired t-tests were used. A Two-way Repeated Measure ANOVA with Tukey’s multiple comparisons test was used to determine statistical significance in weight loss measured at multiple time points. A p value of 0.05 or less was considered statistically significant. Differential gene expression analysis used the Nanostring platform in conjunction with the nCounter analysis package. This system reports the log2 fold change in gene expression relative to a baseline or comparison sample. A raw p value is reported as well as an adjusted p value. The adjusted p value is obtained using the Benjamini-Yekutieli method which assumes a 0.05 False Discovery Rate (FDR).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| HRP anti-mouse IgM | Southern Biotech | Cat# 1020–05 RRID: AB_2794201 |
| HRP anti-mouse IgG1 | Southern Biotech | Cat# 1070–05 RRID_2650509 |
| HRP anti-mouse IgG2b | Southern Biotech | Cat# 1090–05 RRID: AB_2794521 |
| HRP anti-mouse IgG2c | Southern Biotech | Cat# 1079–05 RRID: AB_2794466 |
| HRP anti-mouse IgA | Southern Biotech | Cat # 1040–05 RRID# AB_2714213 |
| Anti-mouse/rat/human GAPDH, clone 6C5 | Abcam | Cat # Ab8245 RRID: AB_2107448 |
| Biotin anti-mouse CXCR5, clone 2G8 | BD Biosciences | Cat # 551960 RRID: AB_394301 |
| FITC anti-mouse B220, clone RA3–6B2 | BD Biosciences | Cat # 553088, RRID: AB_394618 |
| FITC anti-mouse CD21/23, clone 7G6 | BD Biosciences | Cat# 553818 RRID: AB_395070 |
| APC anti-mouse CD5, clone 53–7.3 | BD Biosciences | Cat# 550035 RRID: AB_398457 |
| Anti-Rac1 (multi species), clone 102/Rac1 | BD Biosciences | Cat# 610651 RRID: AB_397978 |
| APC-Cy7 anti-mouse CD19, clone 6D5 | Biolegend | Cat# 152411 RRID: AB_2922473 |
| Alexa 488 anti-mouse/human-Ki67, clone 17F6 | Bioegend | Cat# 151204 RRID# AB_2566800 |
| PE anti-mouse CD19, clone 1D3 | eBiosciencess | Cat# 12–0193-82 RRID: AB_657659 |
| APC anti-mouse CXCR4, clone 2B11 | eBioscience | Cat# 17–9991-82 RRID AB_10670878 |
| V450 anti-mouse CD4, clone GK1.5 | Tonbo | Cat # 75–0041 RRID: AB_2621927 |
| Alexa 750 anti-mouse CD93, clone 223437 | R&D Systems | Cat# FAB1 696S RRID: AB_2935869 |
| Anti-mouse FcR, clone 2.4G2 | BioXCell | Cat# BE0307 RRID: AB_2736987 |
| InVivoPlus™ isotype control mAb, clone 2A3 | BioXCell | Cat# BP0089 RRID: AB_1107769 |
| InVivoPlus™ anti-mouse CD40 mAb, clone FGK4.5 | BioXCell | Cat# BP0016–2 RRID: AB_1107647 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| C. difficile R20291 strain | N/A | |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TcdB2: wild type recombinant C. difficile toxin | Bacillus megaterium expression system, in house | N/A |
| TcdB2 D270N: glucosyltransferase-null recombinant C. difficile toxin | Bacillus megaterium expression system, in house | N/A |
| B2Δ: TcdB2Δ1769–1787, receptor-binding-null recombinant C. difficile toxin | Bacillus megaterium expression system, in house | N/A |
| Brain Heart Infusion (BHI) | Millipore Sigma | Cat # 1104930500 |
| Bacto proteose peptone No. 3 | BD Biosciences | Cat # 211693 |
| Select Agar | Invitrogen | Cat# 30191–023 |
| Fructose | Fisher Chemical | Cat #L95–500 |
| 2% w/v Alhydrogel suspension | Invivogen | Cat # vac-alu-250 |
| Cefoperazone sodium salt | Millipore Sigma | Cat #C4292 |
| Distilled drinking water | Corning | Cat # 25–055-CM |
| Phycoerythrin-conjugated streptavidin | BD Biosciences | Cat# 554061 |
| APC streptavidin | BD Biosciences | Cat# 554067 |
| RPMI1640 media | Corning | Cat # 10–104-CV |
| FBS | Atlanta Biologicals | Cat #S11150 |
| F12-K media | Corning | Cat # 21127–022 |
| Antibiotic Antimycotic Solution | Corning | Cat # 30–004-C1 |
| 2,2’azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS) | SeraCare | Cat# 5120–0042 |
| Sodium Dodecyl Sulfate (SDS) | J.T. Baker | Cat #L050–7 |
| 3-amino-9-ethyl-carbazole (AEC) | Millipore Sigma | Cat# A6926 |
| Protease Inhibitor Cocktail Set 1 | EMD Millipore | Cat # 539131 |
| Mouse CXCL12 recombinant chemokine | Sino Biological | Cat # 50025-MNAE |
| Mouse CXCL13 recombinant chemokine | R&D Systems | Cat # AF470 |
| Mouse CCL19 recombinant chemokine | R&D Systems | Cat # 440-M3 |
| Mouse CCL21 recombinant chemokine | R&D Systems | Cat # AF457 |
| Power SYBR® Green PCR Master Mix | Applied Biosystems | Cat # 4367659 |
| SuperScript® III First-Strand Synthesis System for RT-PCR | Invitrogen | Cat # 18080–051 |
| Cxcr4 Mouse qPCR Primer Pair | OriGene | Cat # MP202423 |
| Cxcr5 Mouse qPCR Primer Pair | OriGene | Cat # MP202369 |
| Ccr7 Mouse qPCR Primer Pair | OriGene | Cat # MP202402 |
| Gapdh Mouse qPCR Primer Pair | OriGene | Cat # MP205604 |
| Excalibur’s Alcoholic Z-Fix | Excalibur Pathology | N/A |
| AMD3100 | Millipore Sigma | Cat # A5602 |
|
| ||
| Critical commercial assays | ||
|
| ||
| EasySep™ Mouse Pan-B Cell Isolation Kit | STEMCELL Technologies | Cat # 19844 |
| EasySep™ Mouse Naive CD4+ T cell Isolation Kit | STEMCELL Technologies | Cat # 19765 |
| PE-anti-Annexin V | STEMCELL Technologies | Cat # 100–0331 |
| 7-AAD | STEMCELL Technologies | Cat # 75001 |
| Annexin V binding buffer | STEMCELL Technologies | Cat #: 100–0334 |
| DNA/RNA Shield | Zymo Research | Cat #R1100–50 |
| Zymo Quick-RNA Miniprep Plus Kit | Zymo Research | Catt #R1057 |
| Bradford Protein Assay | Bio-Rad | Cat # 500002 |
| nCounter Myeloid Innate Immunity Panel | NanoString | Cat # XT-CSO-MMII2–12 |
| 12–230-kDa Jess separation module | Protein Simple | Cat # SM-WOO1 |
| Cell Counting Kit 8 (CCK-8) | Sigma-Aldrich | Cat # 96992 |
| Noradrenaline (Norepinephrine) High Sensitive ELISA | Eagle Biosciences Inc. | Cat # EA633/96 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data for manuscript | Mendeley | https://doi.org/10.17632/m2v6dj33s5.2 |
| Raw data for Nanostring analysis | Gene Expression Omnibus (Geo) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi | Accession #: GSE263792 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| CHO-K1 cell line | ATCC | Cat # CCL-61 RRID: CVCL_0214 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mice, C57Bl/6NCr | Charles River | Cat # model 027 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo v10.8.1 | Tree Star | https://www.flowjo.com/solutions/flowjo RRID: SCR_008520 |
| GraphPad prism v9.5.1 | GraphPad Software LLC | http://www.graphpad.com RRID: SCR_002798 |
| ImageJ | NIH, Fiji distributors | https://imagej.net SCR_003070 |
| nSolver 4.0 Advanced Analysis Software | NanoString | http://www.nanostring.com/products/nSolver RRID: SCR_003420 |
| Compass for Simple Western Software | Protein Simple | https://www.bio-techne.com/resources/instrument-software-download-center/compass-software-simple-western RRID: SCR_022930 |
|
| ||
| Other | ||
|
| ||
| Mammalian Protein Extraction Reagent (M-PER) | Thermo Scientific | Cat # 78501 |
| Peroxide/Luminol-S reagent | Protein Simple | Cat # PS-CS01 |
Highlights.
C. difficile toxin B (TcdB2) suppresses antibody recall responses
TcdB2 blocks germinal center formation
TcdB2 alters B cell CXCR4-mediated cell migration
A CXCR4-blocking drug rescues germinal center formation
ACKNOWLEDGMENTS
This research was funded by NIH awards RO1 AI134719, RO1 AI119048, and U19 AI174994. We thank Ms. J. Gipson and Mr. J. Henthorn of the OUHSC Institutional Research Core Facility for assistance with mRNA profiling and flow cytometry, respectively. We thank Ms. C. Katarzyna (OUHS) for assistance with immunohistochemistry and Dr. S. Chiliveru (OU Stephenson Cancer Center) for assistance with ELISPOT analysis. We thank Ms. D.A. Doyle for assistance with harvesting intact colons and acknowledge preliminary observations by Dr. S. Amadou Amani that TcdB could limit IgG production. Schematics were created using BioRender.
Footnotes
DECLARATION OF INTERESTS
K.M.N., M.L.L., and J.D.B. are listed as inventors on provisional US patent OKLA.P0023US.P1 filed on 09/13/23, which is related to the work described here.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114245.
REFERENCES
- 1.Magill SS, O’Leary E, Janelle SJ, Thompson DL, Dumyati G, Nadle J, Wilson LE, Kainer MA, Lynfield R, Greissman S, et al. (2018). Changes in Prevalence of Health Care-Associated Infections in U.S. Hospitals. N. Engl. J. Med. 379, 1732–1744. 10.1056/NEJMoa1801550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartlett JG (2010). Clostridium difficile: progress and challenges. Ann. N. Y. Acad. Sci. 1213, 62–69. 10.1111/j.1749-6632.2010.05863.x. [DOI] [PubMed] [Google Scholar]
- 3.Kaslow DC, and Shiver JW (2011). Clostridium difficile and methicillin-resistant Staphylococcus aureus: emerging concepts in vaccine development. Annu. Rev. Med. 62, 201–215. 10.1146/annurev-med-051109-101544. [DOI] [PubMed] [Google Scholar]
- 4.Kelly CP, and LaMont JT (2008). Clostridium difficile–more difficult than ever. N. Engl. J. Med. 359, 1932–1940. 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 5.Olsen MA, Yan Y, Reske KA, Zilberberg MD, and Dubberke ER (2015). Recurrent Clostridium difficile infection is associated with increased mortality. Clin. Microbiol. Infect. 21, 164–170. 10.1016/j.cmi.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Monaghan TM, Robins A, Knox A, Sewell HF, and Mahida YR (2013). Circulating Antibody and Memory B-Cell Responses to C. difficile Toxins A and B in Patients with C. difficile-Associated Diarrhoea, Inflammatory Bowel Disease and Cystic Fibrosis. PLoS One 8, e74452. 10.1371/journal.pone.0074452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amadou Amani S, Shadid T, Ballard JD, and Lang ML (2020). Clostridioides difficile Infection Induces an Inferior IgG Response to That Induced by Immunization and Is Associated with a Lack of T Follicular Helper Cell and Memory B Cell Expansion. Infect. Immun. 88, e00829–19. 10.1128/iai.00829-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah HB, Smith K, Scott EJ 2nd, Larabee JL, James JA, Ballard JD, and Lang ML (2020). Human C. difficile toxin-specific memory B cell repertoires encode poorly neutralizing antibodies. JCI Insight 5, e138137. 10.1172/jci.insight.138137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees WD, and Steiner TS (2018). Adaptive immune response to Clostridium difficile infection: A perspective for prevention and therapy. Eur. J. Immunol. 48, 398–406. 10.1002/eji.201747295. [DOI] [PubMed] [Google Scholar]
- 10.Fawley WN, and Wilcox MH (2001). Molecular epidemiology of endemic Clostridium difficile infection. Epidemiol. Infect. 126, 343–350. 10.1017/s095026880100557x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen SH, Tang YJ, Rahmani D, and Silva J Jr. (2000). Persistence of an endemic (toxigenic) isolate of Clostridium difficile in the environment of a general medicine ward. Clin. Infect. Dis. 30, 952–954. 10.1086/313807. [DOI] [PubMed] [Google Scholar]
- 12.Bignardi GE (1998). Risk factors for Clostridium difficile infection. J. Hosp. Infect. 40, 1–15. 10.1016/S0195-6701(98)90019-6. [DOI] [PubMed] [Google Scholar]
- 13.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, and Young VB (2015). Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect. Immun. 83, 934–941. 10.1128/iai.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, and Burdon DW (1978). Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br. Med. J. 1, 695. 10.1136/bmj.1.6114.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobson G, Hickey C, and Trinder J (2003). Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 29, 1030. 10.1007/s00134-003-1754-7. [DOI] [PubMed] [Google Scholar]
- 16.Gerding DN, Johnson S, Peterson LR, Mulligan ME, and Silva J Jr. (1995). Clostridium difficile-associated diarrhea and colitis. Infect. Control Hosp. Epidemiol. 16, 459–477. 10.1086/648363. [DOI] [PubMed] [Google Scholar]
- 17.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, and Minton NP (2010). The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713. 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 18.Alfa MJ, Kabani A, Lyerly D, Moncrief S, Neville LM, Al-Barrak A, Harding GK, Dyck B, Olekson K, and Embil JM (2000). Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 38, 2706–2714. 10.1128/jcm.38.7.2706-2714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, et al. (2009). Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179. 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hippenstiel S, Schmeck B, N’Guessan PD, Seybold J, Krüll M, Pre-issner K, Eichel-Streiber CV, and Suttorp N (2002). Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L830–L838. 10.1152/ajplung.00467.2001. [DOI] [PubMed] [Google Scholar]
- 21.Chandrasekaran R, and Lacy DB (2017). The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 41, 723–750. 10.1093/femsre/fux048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voth DE, and Ballard JD (2005). Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263. 10.1128/cmr.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Bella S, Ascenzi P, Siarakas S, Petrosillo N, and di Masi A (2016). Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins 8, 134. 10.3390/toxins8050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, and Feng H(2012). Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J. Infect. Dis. 205, 384–391. 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamm EE, Voth DE, and Ballard JD (2006). Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. Proc. Natl. Acad. Sci. USA 103, 14176–14181. 10.1073/pnas.0604725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaban L, Chen Y, Fasciano AC, Lin Y, Kaplan DL, Kumamoto CA, and Mecsas J (2018). A 3D intestinal tissue model supports Clostridioides difficile germination, colonization, toxin production and epithelial damage. Anaerobe 50, 85–92. 10.1016/j.anaerobe.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanis JM, Barua S, and Ballard JD (2010). Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog. 6, e1001061. 10.1371/journal.ppat.1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steele J, Mukherjee J, Parry N, and Tzipori S (2013). Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207, 323–330. 10.1093/infdis/jis669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarchum I, Liu M, Shi C, Equinda M, and Pamer EG (2012). Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect. Immun. 80, 2989–2996. 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, Ling L, Leiner I, and Pamer EG (2015). Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe 18, 27–37. 10.1016/j.chom.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fachi JL, Pral LP, Dos Santos JAC, Codo AC, de Oliveira S, Felipe JS, Zambom FFF, Câmara NOS, Vieira PMMM, Colonna M, and Vinolo MAR (2021). Hypoxia enhances ILC3 responses through HIF-1alpha-dependent mechanism. Mucosal Immunol. 14, 828–841. 10.1038/s41385-020-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fachi JL, Secca C, Rodrigues PB, Mato FCP, Di Luccia B, Felipe JS, Pral LP, Rungue M, Rocha VM, Sato FT, et al. (2020). Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med. 217. 10.1084/jem.20190489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frisbee AL, Saleh MM, Young MK, Leslie JL, Simpson ME, Abhyankar MM, Cowardin CA, Ma JZ, Pramoonjago P, Turner SD, et al. (2019). IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 10, 2712. 10.1038/s41467-019-10733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buonomo EL, Cowardin CA, Wilson MG, Saleh MM, Pramoonjago P, and Petri WA Jr. (2016). Microbiota-Regulated IL-25 Increases Eosinophil Number to Provide Protection during Clostridium difficile Infection. Cell Rep. 16, 432–443. 10.1016/j.celrep.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kink JA, and Williams JA (1998). Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 66, 2018–2025. 10.1128/iai.66.5.2018-2025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, et al. (2010). Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28, 965–969. 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]
- 37.De Silva NS, and Klein U (2015). Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 15, 137–148. 10.1038/nri3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barinov A, Luo L, Gasse P, Meas-Yedid V, Donnadieu E, Arenzana-Seisdedos F, and Vieira P (2017). Essential role of immobilized chemokine CXCL12 in the regulation of the humoral immune response. Proc. Natl. Acad. Sci. USA 114, 2319–2324. 10.1073/pnas.1611958114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, and Cyster JG (2004). Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 5, 943–952. 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 40.Donzella GA, Schols D, Lin SW, Esté JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, and Moore JP (1998). AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 4, 72–77. 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 41.Laman JD, Claassen E, and Noelle RJ (2017). Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 37, 371–420. 10.1615/CritRevImmunol.v37.i2-6.100. [DOI] [PubMed] [Google Scholar]
- 42.Lang GA, Exley MA, and Lang ML (2006). The CD1d-binding glycolipid alpha-galactosylceramide enhances humoral immunity to T-dependent and T-independent antigen in a CD1d-dependent manner. Immunology 119, 116–125. 10.1111/j.1365-2567.2006.02413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massari ME, Rivera RR, Voland JR, Quong MW, Breit TM, van Dongen JJ, de Smit O, and Murre C (1998). Characterization of ABF-1, a novel basic helix-loop-helix transcription factor expressed in activated B lymphocytes. Mol. Cell Biol. 18, 3130–3139. 10.1128/mcb.18.6.3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee G, Jang E, and Youn J (2020). CCAAT/enhancer binding protein β Induces Post-Switched B Cells to Produce Blimp1 and Differentiate into Plasma Cells. Immune Netw. 20, e42. 10.4110/in.2020.20.e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Tannous BA, Poznansky MC, and Chen H (2020). CXCR4 antagonist AMD3100 (plerixafor): From an impurity to a therapeutic agent. Pharmacol. Res. 159, 105010. 10.1016/j.phrs.2020.105010. [DOI] [PubMed] [Google Scholar]
- 46.Majumdar S, Pontejo SM, Jaiswal H, Gao JL, Salancy A, Stassenko E, Yamane H, McDermott DH, Balabanian K, Bachelerie F, and Murphy PM (2023). Severe CD8+ T Lymphopenia in WHIM Syndrome Caused by Selective Sequestration in Primary Immune Organs. J. Immunol. 210, 1913–1924. 10.4049/jimmunol.2200871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT, et al. (1995). Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J. Clin. Invest. 95, 2004–2011. 10.1172/jci117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carter GP, Chakravorty A, Pham Nguyen TA, Mileto S, Schreiber F, Li L, Howarth P, Clare S, Cunningham B, Sambol SP, et al. (2015). Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections. mBio 6, e00551. 10.1128/mBio.00551-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta SB, Mehta V, Dubberke ER, Zhao X, Dorr MB, Guris D, Molrine D, Leney M, Miller M, Dupin M, and Mast TC (2016). Antibodies to Toxin B Are Protective Against Clostridium difficile Infection Recurrence. Clin. Infect. Dis. 63, 730–734. 10.1093/cid/ciw364. [DOI] [PubMed] [Google Scholar]
- 50.Castro-Córdova P, Mora-Uribe P, Reyes-Ramírez R, Cofré-Araneda G, Orozco-Aguilar J, Brito-Silva C, Mendoza-León MJ, Kuehne SA, Minton NP, Pizarro-Guajardo M, and Paredes-Sabja D (2021). Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 12, 1140. 10.1038/s41467-021-21355-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, and Chaplin DD (1996). Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature 382, 462–466. 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 52.Katchar K, Taylor CP, Tummala S, Chen X, Sheikh J, and Kelly CP (2007). Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin. Gastroenterol. Hepatol. 5, 707–713. 10.1016/j.cgh.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 53.Bilverstone TW, Garland M, Cave RJ, Kelly ML, Tholen M, Bouley DM, Kaye P, Minton NP, Bogyo M, Kuehne SA, and Melnyk RA (2020). The glucosyltransferase activity of C. difficile Toxin B is required for disease pathogenesis. PLoS Pathog. 16, e1008852. 10.1371/journal.ppat.1008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerasimčik N, He M, Dahlberg CIM, Kuznetsov NV, Severinson E, and Westerberg LS (2017). The Small Rho GTPases Rac1 and Rac2 Are Important for T-Cell Independent Antigen Responses and for Suppressing Switching to IgG2b in Mice. Front. Immunol. 8, 1264. 10.3389/fimmu.2017.01264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakagawa R, and Calado DP (2021). Positive Selection in the Light Zone of Germinal Centers. Front. Immunol. 12, 661678. 10.3389/fimmu.2021.661678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zoughlami Y, Voermans C, Brussen K, van Dort KA, Kootstra NA, Maussang D, Smit MJ, Hordijk PL, and van Hennik PB (2012). Regulation of CXCR4 conformation by the small GTPase Rac1: implications for HIV infection. Blood 119, 2024–2032. 10.1182/blood-2011-06-364828. [DOI] [PubMed] [Google Scholar]
- 57.Balabanian K, Brotin E, Biajoux V, Bouchet-Delbos L, Lainey E, Fenneteau O, Bonnet D, Fiette L, Emilie D, and Bachelerie F (2012). Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmentalization in mice. Blood 119, 5722–5730. 10.1182/blood-2012-01-403378. [DOI] [PubMed] [Google Scholar]
- 58.Liu Q, Li Z, Gao JL, Wan W, Ganesan S, McDermott DH, and Murphy PM (2015). CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice. Eur. J. Immunol. 45, 1855–1867. 10.1002/eji.201445245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, and Pulendran B (2003). Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 424, 329–334. 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- 60.Fang H, Xu L, Chen TY, Cyr JM, and Frucht DM (2006). Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J. Immunol. 176, 6155–6161. 10.4049/jimmunol.176.10.6155. [DOI] [PubMed] [Google Scholar]
- 61.Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D’Elios MM, Tang WJ, Montecucco C, and Baldari CT (2005). Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 201, 325–331. 10.1084/jem.20041557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joshi SK, Lang GA, Larabee JL, Devera TS, Aye LM, Shah HB, Ballard JD, and Lang ML (2009). Bacillus anthracis lethal toxin disrupts TCR signaling in CD1d-restricted NKT cells leading to functional anergy. PLoS Pathog. 5, e1000588. 10.1371/journal.ppat.1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dan JM, Havenar-Daughton C, Kendric K, Al-Kolla R, Kaushik K, Rosales SL, Anderson EL, LaRock CN, Vijayanand P, Seumois G, et al. (2019). Recurrent group A Streptococcus tonsillitis is an immunosusceptibility disease involving antibody deficiency and aberrant T(FH) cells. Sci. Transl. Med. 11, eaau3776. 10.1126/scitranslmed.aau3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cowardin CA, Buonomo EL, Saleh MM, Wilson MG, Burgess SL, Kuehne SA, Schwan C, Eichhoff AM, Koch-Nolte F, Lyras D, et al. (2016). The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat. Microbiol. 1, 16108. 10.1038/nmicrobiol.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pope RL, Chitrakar A, Sah P, Shadid T, Ballard JD, and Zenewicz LA (2022). Clostridioides difficile Toxin B Activates Group 3 Innate Lymphocytes. Infect. Immun. 90, e0007322. 10.1128/iai.00073-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bland SJ, Larabee JL, Shadid TM, Lang ML, and Ballard JD (2019). Deletion of a 19-Amino-Acid Region in Clostridioides difficile TcdB2 Results in Spontaneous Autoprocessing and Reduced Cell Binding and Provides a Nontoxic Immunogen for Vaccination. Infect. Immun. 87, e00210–19. 10.1128/IAI.00210-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ehrich M, Van Tassell RL, Libby JM, and Wilkins TD (1980). Production of Clostridium difficile antitoxin. Infect. Immun. 28, 1041–1043. 10.1128/iai.28.3.1041-1043.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Justus CR, Marie MA, Sanderlin EJ, and Yang LV (2023). Transwell In vitro Cell Migration and Invasion Assays. Methods Mol. Biol. 2644, 349–359. 10.1007/978-1-0716-3052-5_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Justus CR, Leffler N, Ruiz-Echevarria M, and Yang LV (2014). In vitro cell migration and invasion assays. J. Vis. Exp. 51046. 10.3791/51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weldy M, Evert C, Dosa PI, Khoruts A, and Sadowsky MJ (2020). Convenient Protocol for Production and Purification of Clostridioides difficile Spores for Germination Studies. STAR Protoc. 1, 100071. 10.1016/j.xpro.2020.100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winston JA, Thanissery R, Montgomery SA, and Theriot CM (2016). Cefoperazone-treated mouse model of clinically-relevant Clostridium difficile strain R20291. JoVE. JoVE, e54850. 10.3791/54850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data files corresponding to the results reported herein have been deposited in Mendeley (https://doi.org/10.17632/m2v6dj33s5.2).
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.