

Summary:
Autoimmune (or rheumatic) diseases are increasing in prevalence but selecting the best therapy for each patient proceeds in trial-and-error fashion. This strategy can lead to ineffective therapy resulting in irreversible damage and suffering; thus, there is a need to bring the promise of precision medicine to patients with autoimmune disease. While host factors partially determine the therapeutic response to immunosuppressive drugs, these are not routinely used to tailor therapy. Thus, non-host factors likely contribute. Here, we consider the impact of the human gut microbiome in the treatment of autoimmunity. We propose that the gut microbiome can be manipulated to improve therapy and to derive greater benefit from existing therapies. We focus on the mechanisms by which the human gut microbiome impacts treatment response, provide a framework to interrogate these mechanisms, review a case study of a widely-used anti-rheumatic drug, and discuss challenges with studying multiple complex systems: the microbiome, the human immune system, and autoimmune disease. We consider open questions that remain in the field and speculate on the future of drug-microbiome-autoimmune disease interactions. Finally, we present a blue-sky vision for how the microbiome can be used to bring the promise of precision medicine to patients with rheumatic disease.
Keywords: microbiome, autoimmunity, pharmacomicrobiomics, host-microbe interactions, drug-microbiota interactions
INTRODUCTION
The trillions of microbes living in and on us produce a dizzying array of metabolites and proteins that our immune system cannot ignore. These microbial communities, or microbiota, include bacteria, viruses, archaea, and fungi. The gut microbiome, which consists of these microbes and their collective genomes living in the gut, encodes 150-fold more genes than the human genome and harbors a vast metabolic repertoire that extends the capabilities of the host genome1. Because gut microbial communities are shaped by many factors2 and are often the “first responders” to microbial and xenobiotic perturbations, the gut microbiota provides a rich source of information to the host. This information is used to calibrate multiple organ systems3, including the immune system4. Thus, the human microbiome exerts a sustained and powerful impact on the host immune system from the moment we are born5. These diverse microbes are with us in sickness and in health, including autoimmunity. While much of our current research in microbiome studies has sought to characterize healthy individuals6, increasing focus has been directed towards disease7. However, the exact mechanisms by which microbes impact the host, and vice versa, are still being elucidated. The role microbes play in autoimmunity is multifactorial and manifests as an emergent property of multiple metabolic pathways, both host and microbial. Given this complex ecosystem that spans multiple phylogenetic kingdoms, studies of the microbiome-host interaction will take us into a vast unknown with the potential for uncovering novel mechanisms in biology and autoimmunity.
Here, we consider the impact of the human gut microbiome in the treatment of rheumatic disease. Others have reviewed the impact of the microbiome on disease pathogenesis in multiple autoimmune conditions8–13. Here, we focus on treatment and argue that the gut microbiome can be manipulated to improve therapy for patients with rheumatic disease and to derive greater benefit from existing therapies. In this review, we will focus on the mechanisms by which the human gut microbiome impacts treatment response, provide a framework to interrogate these mechanisms, dive into a case study of a widely-used anti-rheumatic drug (methotrexate), and review the challenges with studying a triumvirate of complex systems, each with many “unknowns:” the microbiome, the human immune system, and clinical rheumatology.
Autoimmune (also called rheumatic) diseases such as rheumatoid arthritis, systemic lupus erythematosus, and spondyloarthritis, affect over 3% of the American14 and global population15, with recent estimates suggesting that as many as 8% of the US population is living with autoimmune disease16. Alarmingly, recent indicators suggest that autoimmune disease is on the rise, prompting U.S. scientific institutions like the National Institutes of Health (NIH) to launch the Office of Autoimmune Disease Research in 202117. These chronic diseases result in significant financial costs to patients and reduced economic gross domestic product. Further, autoimmune diseases cause irreversible joint and organ damage if patients are not expeditiously diagnosed and treated. Because there is a “window of opportunity” for treatment, it is imperative to find the right therapy for each patient at the time of diagnosis.
However, selecting immune-suppressing therapies generally proceeds in trial-and-error fashion. This is due to an “embarrassment of riches:” patients now have many more options for treatment because of advances in molecular medicine and pharmacology. Over a short timespan (e.g., 40 years), there has been an explosion of biologic therapies, including those that target specific molecules in the host immune system18. This has led to multiple options for patients and providers alike. But guidance on choosing a specific therapy is limited. Thus, to ensure that limited financial resources are distributed effectively, funders of therapy (i.e., insurance companies, Medicare, Medicaid, and sometimes patients themselves) require less-expensive therapies to be tried first.
While this strategy may reduce overall cost by using cost-effective therapies initially, it causes some patients to be initiated on ineffective therapy, thereby resulting in irreversible damage and suffering. Thus, there is a need to identify, at the time of diagnosis, which therapies will be effective or ineffective for a particular patient. In essence, there is a need to bring the promise of precision medicine to patients with rheumatic disease.
Many investigators have looked to patient characteristics, particularly those that are modifiable, to identify host factors that determine therapeutic response19. In the case of the common disease rheumatoid arthritis (RA), host factors such as genetics, age, disease duration and extent of damage at the time of therapy, and smoking are all factors that may influence treatment response to the first-line therapy called methotrexate (MTX). Patients themselves often inquire whether timing of therapy (e.g., “Should I take the medicine at night or in the morning?”), ingestion with food, and diet may impact treatment response. While providers give guidance, we lack strong predictors of therapeutic response that are based on host-associated factors alone. Thus, non-host factors likely contribute, which is why in recent years, our group and others have turned their attention to the impact of the microbiome on treatment of autoimmunity.
Technology paves the way for discovery, allowing us to approach an old question with a new lens. While many of the current drugs used to treat autoimmunity were developed over 50 years ago, at a time when drug targets were not known20, we are now equipped with vastly improved tools and technologies to study the human gut microbiome. These tools allow us to understand the impact of drugs on microbes, to better define the transformations these microbes carry out on drugs, and to better test host-drug-microbiota interactions in vivo. Because of advances in tools and technologies, we can revisit past studies with a new perspective that focuses on the microbiota and how it shapes treatment response to autoimmune disease.
1. The microbiome in the treatment of disease
The gut microbiota shapes host physiology, and therefore is a prime candidate for impacting therapeutic outcomes. Indeed, gut microbiota help us harness energy from the foods we eat, synthesize essential vitamins, and produce metabolites that are critical for the development of the host immune system21. Microbial metabolites may act at sites beyond the intestinal tract, influencing host neurodevelopment and cardiovascular and renal health22. However, many of the metabolic activities of our gut microbiota remain to be discovered, and ~50% of bacterial genes are of unknown function, representing the “dark matter” of the gut microbiome23.
The composition and function of the gut microbiome is highly personalized, leading researchers to postulate that variation in the gut microbiome contributes to variation in host phenotypes, including treatment response24. The gut microbiota is dynamic and responsive to multiple factors, including diet and antibiotics24. But non-antibiotic host-targeted drugs can have off-target effects on the microbiota, as demonstrated in a study showing that >200 drugs affect microbial growth in vitro25. Additionally, the microbiota can metabolize a significant number of host-targeted drugs26–29.
Evidence from multiple clinical disciplines suggest that, in addition to host and environmental factors, the gut microbiota contributes to treatment outcomes in patients (Figure 1). A notable example comes from the field of Endocrinology. Metformin is an antidiabetic drug that exerts, in part, its antidiabetic effects through the gut microbiota. In a double-blind randomized controlled trial, 18 to 22 patients with type 2 diabetes were treated with either metformin or placebo for 4 months30. Fecal and plasma samples were collected at 0, 2 and 4 months of treatment. The microbiota of metformin-treated patients was altered relative to placebo controls at 2 and 4 months, suggesting that metformin acts on the gut microbiome. Further, the researchers performed a fecal microbiota transplant (FMT) of microbial communities from patients into gnotobiotic mice. Compared to mice transplanted with placebo-exposed microbial communities, mice with metformin-exposed microbial communities had improved glucose tolerance. These results support the hypothesis that metformin-induced changes to the gut microbiota led to antidiabetic effects in the host. Subsequent studies in mice revealed multiple underlying mechanisms31 by which metformin acts on the microbiota to exert antidiabetic effects, which are comprehensively summarized in a recent review32.
Figure 1.

Variation in patient response to therapies is governed by multiple factors. These include genetics, modifiable environmental factors (like smoking and diet), immune activation at the time of drug initiation, and concurrent therapies. Emerging evidence implicates the human gut microbiome (right side) is also a determinant of patient response to therapies and thus an important consideration in efforts to advance precision medicine.
The field of oncology provides additional examples showing that gut microbes influence treatment outcomes33. Observational cohort studies in humans have shown that pre-treatment microbial community composition and function is associated with cancer therapy response, and studies in mice have suggested that microbes contribute to drug outcomes34–36. In three back-to-back papers in 2018, multiple investigators showed that the gut microbiome differed in patients that responded to checkpoint blockade with anti-PD134–36. Patients with melanoma (N=53 in one study and N=42 in a second study)35,36, non-small cell lung cancer (N=60), or renal cell carcinoma (n=40)34 were studied. Between ~40–50% of patient responded to therapy in each study. All three groups found that clinical response to PD1 blockade was associated with pre-treatment microbial composition and function. For example, Routy et al34 showed that non-small cell lung cancer patients (N=140) and renal cell carcinoma patients (N=67) given antibiotics in the months preceding or shortly after the first dose of checkpoint blockade therapy (with anti PD-1) had worse overall survival than those not receiving antibiotics. In mouse studies, the investigators found that broad-spectrum antibiotics worsened survival in mice bearing melanoma and sarcoma tumors. Further, after assessing the microbial communities of responder vs. non-responder patients with either non-small cell lung cancer or renal cell carcinoma (N=42 responders and N=36 non-responders), they found that microbial taxa differed between responders and non-responders prior to treatment. Multiple taxa differed, but one prominent taxon that was replicated in additional patient cohorts was Akkermansia muciniphila, which was enriched in responders. FMT of responder microbial communities into mice bearing tumors resulted in better outcomes to PD-1 blockade, which was not seen when mice were transplanted with communities from non-responders. Further, oral gavage with A. muciniphila conferred improved response to PD1 blockade in germ-free mice and mice colonized with fecal microbial communities from non-responders. These studies, in which specific modulation of the microbiota in animal models mediated improved efficacy, demonstrate that the microbiota contributes to treatment response in these models.
While these initial results are exciting, more remains to be uncovered, including deciphering and targeting the underlying microbial mechanisms. In the aforementioned cancer studies, all three groups went on to find specific taxa that were implicated in survival in patients or in response in murine studies. Interestingly, among the three studies, no common taxa or microbial pathways were identified that might suggest a universal mechanism that could be leveraged clinically. The microbial effectors of treatment response remain to be determined. On the host side, the immune mechanisms associated with microbial modulation of anti-PD1 efficacy remained to be elucidated, though increased immune cell infiltration into tumors by CD8+ T cells, CD4+ T cells, and innate immune cells were reported. Some initial small clinical trials suggest that modulating the microbiome might be effective in patients37,38. Larger clinical trials are ongoing to determine whether microbiome-directed therapies in patients may improve checkpoint blockade efficacy39,40. Thus, in these examples within endocrinology and oncology, gut microbes shape response by acting on host molecular pathways implicated in metabolism or immunity.
In the next section, we review the different methodologies employed to study the effect of the microbiome on drug therapy and tools to decipher underlying mechanisms.
2. Development of tools to study the impact of the microbiome on the treatment of rheumatic disease
Advances in technologies have enabled a higher resolution understanding of the diversity and functionality of gut microbial communities. These advances include next-generation sequencing, high-resolution analytical chemistry platforms, expanded microbial catalogues, bioinformatic tools, and the refinement of “culturomics” (Figure 2). Below, we review these key advances, offering limitations and strengths of each, and highlighting how a combination of orthogonal approaches is needed to understand the impact of the microbiota on treatment outcomes in autoimmunity.
Figure 2.

Advances in culture-dependent and culture-independent techniques have accelerated microbiome research. (A) Historically, knowledge of microbes was limited to those that we could culture (culture-dependent techniques). Once microbes were cultured in vitro (top), they could be introduced into gnotobiotic animals to investigate host-microbe interactions (bottom). (B) Advances in sequencing and analytical chemistry platforms have accelerated our knowledge of the human gut microbiome. Next-generation sequencing technologies have enabled cost-effective, culture-independent methods to determine the identity of microbes present in gut samples, the genes they bring with them, and whether these genes are transcribed. Advanced analytical chemistry platforms such as MALDI-TOF and LC-MS have enabled identification of the proteins and small molecule metabolites that microbes produced. NMR technologies can be used to validate the identity of novel compounds. Abbreviations: 16S, 16S ribosomal RNA gene sequencing; MALDI-TOF, matrix assisted laser desorption ionization-time of flight; LC-MS, liquid chromatography-mass spectrometry; NMR, nuclear magnetic resonance.
2.1. Culture-dependent Techniques
Microbial culture.
Historically, microbiologists studied microbes using “culture-based” techniques; we could only study organisms that we could grow in a petri dish. Successful in vitro growth enabled further careful and laborious study of the physiological and biochemical properties of microbes: colony morphologies, gram staining, nutritional requirements, biochemical reactions mediated by the microbes, among other properties. It is remarkable to remember that the identity of microbes was based on a constellation of these properties, whereas today, species identity is largely based on nucleotide sequencing (of one or more genomic regions or by whole-genome sequencing). Once microbes were cultured and identified in vitro, the direct effects of drugs, nutrients, and vitamins on microbial growth and physiology could be characterized. Further, lysates from bacteria could be used to characterize drug-protein interactions41. Thus, a key strength of in vitro microbiology model systems is that they show that drugs can act directly on microbes or interact with microbial proteins.
Gnotobiotic animals.
A key limitation of in vitro experimental systems is that they do not shed light on drug-microbiota interactions in vivo; for this, gnotobiotic animal model systems are used. Gnotobiology is the study of animals colonized with defined communities of microbes. Gnotobiotic animals are re-derived in germ-free conditions. Rederivation involves a complex breeding and fostering workflow42. Because germ-free animals are vulnerable to colonization, utmost care is required when food, supplies, and waste are moved into and out of the isolators. Thus, maintenance of germ-free animals is time-intensive, laborious, and expensive. Given this, gnotobiotic facilities are an uncommon and precious resource across research institutions. Gnotobiotic studies are indispensable for understanding the contribution of the microbiome to the treatment of autoimmune disease. Gnotobiotic animals enable us to test how drugs impact the human gut microbiota in vivo under controlled settings, which is difficult to do in humans. While similar studies can be carried out in conventional mouse facilities, the mouse microbiome differs significantly from the human, and strains that are found in mice are absent in humans. Importantly, a key strength of gnotobiotic studies is that they uniquely enable us to decipher how specific microbes and their genes, proteins, or metabolites directly contribute to drug pharmacology in vivo or how drugs reconfigure microbial communities in vivo to shape host immunity.
Rederiving animals in germ-free conditions enables us to determine whether the microbiome is required for a phenotype of interest. For example: is the commensal microbiome essential for life? Given that germ-free animals can be rederived and raised to old age, even exceeding the lifespan of colonized counterparts43, the commensal microbiome is not essential in the same way that many of our human genes are. However, germ-free animals are not normal. They are deficient in multiple ways compared to their colonized counterparts. They weigh less, tend to exhibit more anxiety and hyperactivity, and are more susceptible to infections21. The immune system of germ-free mice is dramatically impacted, with lower numbers of B cells, smaller Peyer’s patches, fewer macrophages, and lower levels of antibodies44,45. Further, the blood and organs of colonized mice are awash in microbiota-produced metabolites, many of which are absent in germ-free mice3,46,47. These small molecule metabolites bind receptors on immune cells and modulate host immunity48. This interaction between metabolites and the host immune system is just one of multiple mechanisms by which gut microbes shape host immunity (see Section 3.2). These findings suggest that while a microbiome is not essential for survival, microbes and the genes they encode are functionally important to the host and extend the capabilities of the host genome.
Beyond assessing the qualitative impact of microbiota on host phenotypes, gnotobiotic studies are a powerful tool to quantify the impact of specific microbes or microbial products on host phenotypes, such as immunity and autoimmunity. This can be achieved by colonizing mice with a single microbial strain and testing its effect on a host phenotype. Additionally, mice can be colonized with a defined consortium of bacteria, to mimic more complex microbial communities, and the addition or removal of a particular microbe or microbial gene can be assessed (Figure 3). Advances in genetic manipulation of many human gut microbes allows for us to test for the impact of specific microbial genes on host physiology. Thus, the armamentarium of gnotobiotic and anaerobic microbiology tools has expanded to allow us to dissect the uniquely microbial contributions to host immunity and drug pharmacology. For example, gnotobiotic studies have revealed that (1) the microbiome is required for the development of autoimmune arthritis in mice49, (2) the addition of specific microbes can alter the host blood metabolome50, (3) microbial metabolism of inflammatory compounds like dietary uric acid51 impacts host uric acid levels, (4) gut microbes can invade mesenteric adipose tissue and contribute to inflammation52, and invading microbes are targeted by the host immune system53. Thus, gnotobiotic studies have revealed several mechanisms by which microbes contribute to host immunity, inflammation, and autoimmunity.
Figure 3.
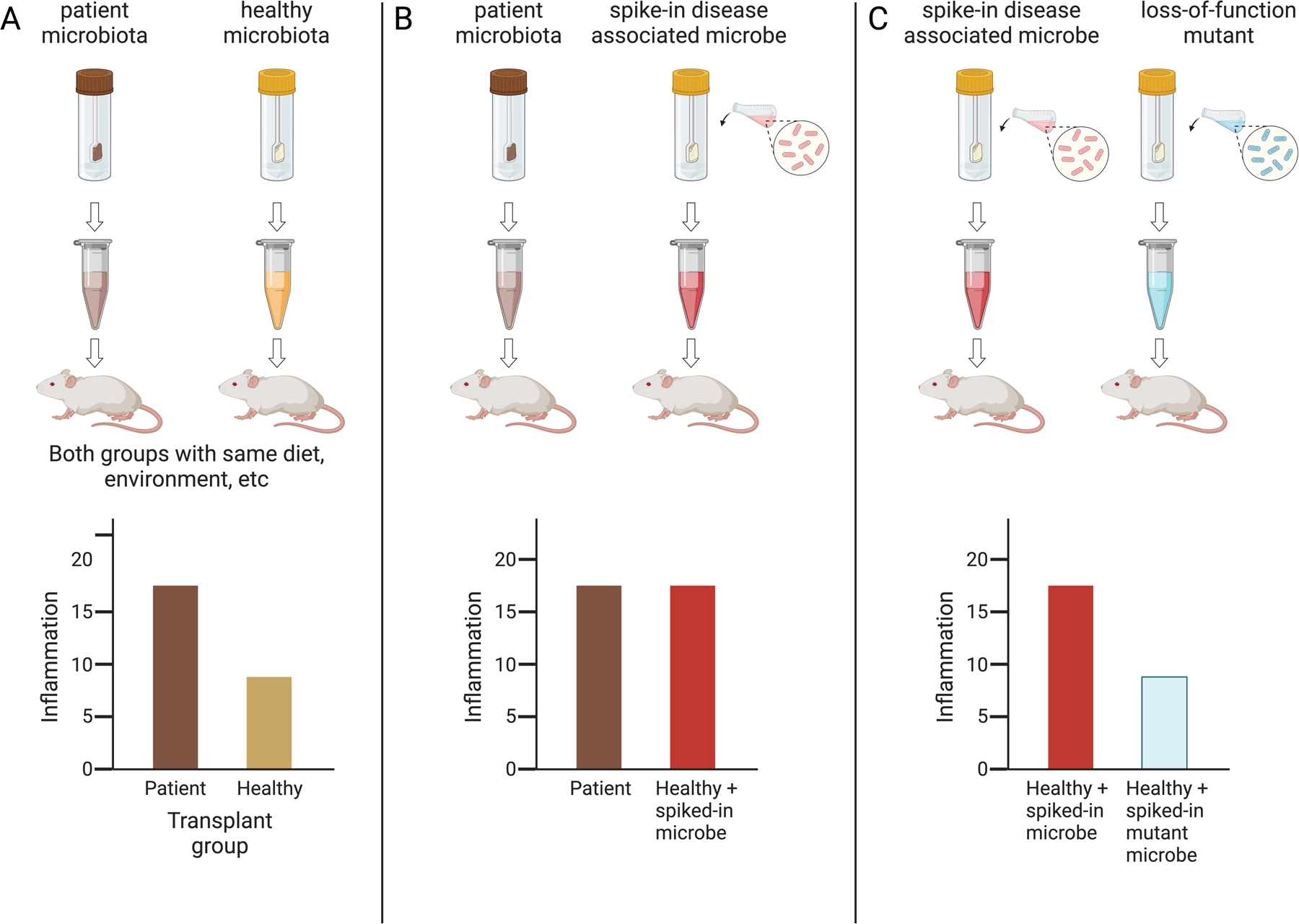
Animal models enable us to test if microbial communities, specific microbes, or microbial genes impact host phenotypes. In this example, the relationship between host inflammation and the microbiome is investigated. The underlying hypothesis is that a disease-associated microbe carrying a specific microbial gene contributes to host inflammation.
A) Microbial communities from diseased patients leads to increased inflammation in gnotobiotic mice.
B) Addition (“spike-in”) of a candidate microbe from disease-associated microbiota into healthy microbial communities increases inflammation induced by healthy microbial communities, thereby implicating the specific candidate microbe.
C) Genetic ablation of a candidate microbial gene abrogates the microbe’s ability to induce inflammation when added to a healthy microbial community.
Gnotobiotic animal studies enable us to examine bidirectional interactions between drugs and microbes in vivo. They enable us to ask how drug a might affect human gut microbes in vivo, especially when combined with sequencing technologies (described in the next section). By inducing disease in germ-free animals, we can ask whether a drug alleviates disease in the absence of a microbiome, or whether microbial communities are needed for the drug’s mechanism of action. Furthermore, microbial communities from patients that have been exposed to a drug vs. not can be transplanted into germ-free mice, and the effect of the “drug-exposed” microbial community on disease can be tested30 (akin to Figure 3A). Such fecal microbiota transplant (FMT) studies provide evidence for the hypothesis that a drug acts on the microbiota to alleviate inflammation and therefore the therapeutic effects of a drug are, at least partially, mediated through microbes. Additionally, we can examine the impact of microbial communities on drug pharmacokinetics and ask whether specific microbial genes impact drug levels in the host. Thus, gnotobiotic animal studies are a critical tool for demonstrating a causal role for the microbiome in shaping the treatment of autoimmune disease.
2.2. Culture-independent Techniques
Next-generation sequencing.
Up until recently, a major limitation of culture-based techniques was that many microbes could not be cultured using the media preparations and culture conditions employed in the past. This realization arrived on the heels of advances in nucleic acid analysis and sequencing in the 1970s. When Carl Woese, Norman Pace, and colleagues reported that the 16S rRNA gene could be used to identify bacterial species54, and this technique became more widely accessible55, we gained a greater appreciation for the immense diversity of the microbes residing in an environment. With the development of next-generation sequencing technologies, we learned that the majority of microbes living in the gut, soil, and other environment were not yet cultured at the time of discovery56.
Next-generation sequencing is a high throughput “culture-independent” technique that enables researchers to ask multiple types of questions about microbial communities (reviewed in57). Researchers can interrogate which microbes are present in a community using 16S rRNA gene amplicon sequencing (bacterial species) and internal transcribed spacer (ITS) sequencing (fungal species) (Figure 2B). Shotgun (or metagenomic) sequencing profiles all the DNA in a specimen, and thus provides information on which microbial genes are present in a sample. RNA-sequencing enables identification of the transcripts that microbial communities produce. Such sequencing-based technologies are used to determine how drugs impact microbial community structure and function in vivo or ex vivo.
Interestingly, culture-independent techniques have led to improvements in culture-dependent techniques. Next-generation sequencing enabled the growth of “culturomics,” defined as leveraging multiple culturing conditions, analytical chemistry, and sequencing for the identification of bacterial species58,59. These sequencing-based technologies contributed to the many catalogues that were developed for studying microbial genomes. From these cataloged microbial genomes, researchers were able to pin down the nutritional requirements or culture conditions required for the growth of previously fastidious microbes58,60. Microbiome researchers have made great strides in the past 10 years in cultivating hundreds of new species that were previously thought to be non-cultivatable58,59,61,62. This is because of the recognition that these species require specialized conditions to grow58. These conditions include (1) growth under anaerobic conditions, (2) use of rich media (typically containing brain, heart, or meat tissue) with multiple required trace elements and supplements, and (3) promotion of germination with bile acids or enrichment with antibiotic treatment. With these modifications, we are now able to culture the majority of microbes that are present in the human gut62. For example, about 80% of all described bacterial species to date are archived at DSMZ62. Additionally, Lau et al found that 95% of species that were present at a relative abundance >0.1% could be cultured using at least 1 of 66 different culture conditions59. Thus, it is possible to develop and study representative panels of anaerobic gut microbes in vitro that recapitulate of the diversity of microbes found in the guts of patients in vivo. However, microbes that are rare in abundance or prevalence continue to remain difficult to culture; but advanced robotics and machine learning has facilitated the evolution of high-throughput methods to lessen the burden of culturing rare species63. Thus, the microbiome community has developed methods of culturing most microbes that are prevalent and abundant in the human gut.
Analytical Chemistry.
A second major “culture-independent” technology that has facilitated studies of the microbiome, and therefore drug-microbiota interactions, has been advances in analytical chemistry platforms64,65. These platforms enable the study of microbial proteomes and small-molecule metabolites. Platforms for studying small-molecule metabolites have been instrumental in identifying which microbes metabolize therapeutic drugs and for mapping the transformations. This has only been possible with high-resolution liquid chromatography mass spectrometry (LC-MS) platforms that can distinguish between compounds with very small mass differences and transitions, and nuclear magnetic resonance (NMR) platforms that can be used to verify the identity of novel substrates.
Analytical chemistry platforms have enabled high-throughput studies of how human gut microbes transform drugs. In a landmark study, investigators incubated 271 drugs with 76 different microbial drugs and found that 176 drugs were depleted or transformed by human gut bacteria in vitro26. Such studies reveal that microbes produce drug metabolites that are similar to those that are produced by the host, but some products of microbial metabolism are unique to microbes and are novel. Thus, advanced analytical chemistry platforms have revolutionized our understanding of the diversity of biochemical activities that performed by microbes66, both on endogenous compounds and xenobiotics, such as drugs used to treat autoimmune disease.
Advanced analytical chemistry platforms combined with gnotobiotic studies allow us to better understand how microbes metabolize drugs in vivo in the host. Use of gnotobiotic animals enables investigators to control for many factors that confound microbiome studies in humans67. In these studies, mice are colonized with microbial communities of interest and drug pharmacokinetics is quantified. Most studies of drug pharmacokinetics look in the host circulation because that is where drugs act on host cells. However, examining the stool may provide insights into microbial products produced by bacteria that are either largely retained in the gut or are produced there and enter into circulation. Thus, combinations of these different technologies can be used to reveal the interplay between drugs and microbes in shaping the treatment of autoimmune disease.
2.3. Taking stock of the evidence implicating the microbiome in the treatment of autoimmune disease.
While many studies report that the microbiome is associated with multiple diseases, these snapshot studies are just the first step in understanding if the microbiome may affect treatment response. A common refrain from such cross-sectional human studies is, “Which came first – changes in the microbiome or disease?” Some of the tools described above, such as gnotobiotics studies, address such questions because they provide evidence that microbes or their products are causal agents that produce the phenotypes we observe in animal models. But, because there is still so much to learn about the impact of microbes on the treatment of autoimmune disease in humans, it is important to consider what lines of evidence exist and what studies have yet to be done (if deemed useful and feasible) to help us advance microbiome-based therapies for patients with autoimmune disease. One framework we use to determine the state of evidence for microbiome-drug interactions is provided in Table 1. This table of questions allows us to assess the extent to which experimental or epidemiological data exists in humans and model organisms to support the hypothesis that the microbiome matters for treatment outcomes in an autoimmune disease of interest. This framework is largely a guide to think broadly about the different types of data that provide evidence for the microbiome in disease and treatments; a relevant adage is “The absence of evidence is not evidence of absence.” Nevertheless, finding reproducible signatures using orthogonal tools and techniques will be critical in translating microbiome research to the clinic.
Table 1.
Questions to consider when assessing our knowledge bank on how the microbiome impacts treatment response†.
| Humans | Animal Models | |
|---|---|---|
| Pathogenesis |
|
|
| Treatment |
|
|
This table provides general queries to consider when assessing our knowledge bank of the impact of the microbiome on treating autoimmunity. We do not mean to imply that all these questions must be answered to confirm that the microbiota matters. At times, it may not be technically, cost-effectively, or ethically possible to ascertain the answers to all these questions. Currently, many of the questions remain “open” for many autoimmune diseases.
In summary, a combination of culture-dependent and culture-independent techniques are required to advance microbiome-based medicines to treat autoimmunity. In vitro and ex vivo (communities taken from the host and grown in a culture in the laboratory) microbiology studies are reductionist but are indispensable in determining whether a drug directly affects microbes or whether microbes directly metabolize a drug. Gnotobiotic animal studies are expensive and may not fully recapitulate human disease and phenotypes, but gnotobiology is indispensable for showing that microbial communities in vivo can causally impact host phenotypes. By comparison, demonstrating a causal role for specific microbes or microbial products in humans is achieved through randomized clinical trials and/or epidemiological studies; these are even more expensive and less common, but preclinical studies in gnotobiotic model systems can help guide human studies. Mechanistic studies looking at specific microbial genes, proteins, or metabolites can be performed in vitro, ex vivo and in gnotobiotic animals, especially with the development of novel methods to genetically manipulate non-model bacterial species68. Multiple “omic” technologies are used individually or in combination with culture-dependent techniques to decipher the broad impacts of drugs on the microbiome community composition, structure, and function in humans and in model systems. Advanced analytical chemistry platforms are used to determine how drugs impact the metabolome or proteome of the microbiome and are instrumental in identifying how microbes metabolize drugs. Thus, advances in these technologies have greatly expanded our understanding of drug-microbiota interactions, but as we will see in the subsequent sections, much more remains to be uncovered.
3. Mechanisms by which microbes impact treatment response
3.1. Microbes directly metabolize drugs or affect their pharmacology.
One mechanism by which the microbiome impacts the treatment of autoimmune disease is that gut microbes affect the pharmacokinetic profiles of immunosuppressive drugs. They do so through two broad mechanisms: (1) by altering the drug directly or (2) by affecting the host. In the first case, drugs are “biotransformed” or “metabolized” by microbial enzymes that reduce, hydrolyze, or otherwise structurally modify the drug. These modifications may result in drug metabolites with altered bioactivity: more toxic, less effective, more effective, less absorbed, etc. and thereby affect drug efficacy. In the second case, microbes induce changes to host proteins and enzymes that influence drug pharmacokinetics: for example, microbes alter host intestinal cell drug-transporters, liver metabolizing enzymes, renal channels responsible for excretion, and expression of other host enzymes that act on therapeutic drugs to alter their pharmacology (e.g., absorption, distribution, metabolism, excretion). Thus, microbes may exert powerful effects on a drug’s pharmacological properties. One term to encapsulate this notion is pharmacomicrobiomics69. Recent comprehensive reviews have summarized what is known about how microbes shape drug pharmacology70,71.
Why might microbes transform therapeutic drugs? One possibility is that microbes evolved these mechanisms to act on natural compounds (for example, to harvest energy and nutrients from them). Therapeutic drugs that mimic endogenous compounds may therefore be “innocent bystanders” of microbial enzymes that act on endogenous compounds. Evidence to support this possibility comes from studies showing that microbial enzymes act on a range of structurally similar compounds with specific common motifs72. In a recent large-scale analysis of 438 drugs that were incubated with human gut bacteria73, investigators compared metabolism of synthetic drugs vs. drugs found in nature or derived from natural compounds (those with slight structural modifications from the natural compound). In this study, 10% of synthetic drugs were metabolized, while 21% of natural or naturally-derived compounds were metabolized by microbes. Steroids dominated the list of natural compounds metabolized by human gut bacteria, but non-steroidal drugs included: azathioprine, mycophenolate mofetil, and allopurinol. All of these are “antimetabolites” used in the treatment of autoimmune or rheumatic disease. Similarly, additional natural compounds that are known to be metabolized by gut bacteria include levodopa (dopamine is an endogenous neurotransmitter), digoxin (from Digitalis purpurea plant, or foxglove), lovastatin (produced by fungi), sorivudine (nucleotide analog), and 5-fluorouracil (nucleotide analog)70. Thus, therapeutic drugs with chemical motifs that closely approximate endogenous compounds are likely to be impacted by human gut microbial metabolism, and many current therapies used to treat autoimmune disease possess this property.
3.2. Therapies act on microbes to impact immunity and inflammation.
While microbes affect drugs, drugs may affect microbes: drugs designed to target human pathways have off-target effects on microbial pathways. The extent to which these off-target microbial effects impact human immunity and physiology is not well defined. Since under homeostatic conditions, microbes directly impact host immunity74,75, it is possible that off-target effects of drugs on commensal microbes may affect human immunity and physiology in multiple ways (Figure 4). Thus, understanding the impact of drugs on gut commensal communities can shed light on microbiota-immune interactions.
Figure 4.

Interactions between drugs, microbes, and host immunity. Historically, we focused on how drugs directly impact the host immune system when considering autoimmunity treatment (dashed arrow). However, emerging evidence suggests that gut microbiota modify drugs and drugs act on microbes to shape host immunity (solid arrows).
Human-targeted drugs have off-target effects on microbial pathways. This was demonstrated several decades ago, in which investigators examined the effects of drugs on microbial growth in vitro76,77. These studies were undertaken to understand the mechanism of action of many of these drugs, since the targets were unknown at the time76. These initial drug-microbiota studies were also undertaken to help evolve drugs that had greater specificity for microbial proteins than human proteins78. Many of these early studies focused on one or a few drugs and usually tested activity against infectious microbes. But recently, a landmark study expanded our knowledge of drug impacts on commensal microbes. The extent to which therapeutic drugs could have off-target effects on human gut microbes was reported in study in which 1,197 drugs were tested at a single concentration against 40 representative human gut microbes25. This large and quantitative study demonstrated that 203 (24%) of the host-targeted drugs also inhibited growth of at least one human gut commensal strain in vitro. Thus, a significant percent of host-targeted drugs may have off-target growth inhibitory effects on gut microbes.
In addition to inhibiting growth, host-targeted drugs can affect the transcriptomes and metabolomes of gut commensals. Thus, the percentage of drugs affecting microbes is likely an underestimate because the investigators only screened the effects of drugs on growth25. But host-targeted drugs may change microbial physiology, transcription, and metabolite production without leading to a change in growth. For example, in a recent study looking at 14 common human gut isolates incubated with 19 top-prescribed therapeutics79, all tested drugs resulted in differential gene expression in at least one bacterial isolate. Notably, the investigators tested drug concentrations that had minimal impacts on growth, supporting the idea that host-targeted drugs can have off-target effects on the transcriptome, and likely downstream products, of gut microbes. Thus, studies looking at the effects of host-targeted drugs on in vitro growth or community composition (16S) may miss important transcriptomic or metabolomic effects that drugs have on the microbiota.
Under homeostatic conditions, the microbiome shapes host physiology, including host immunity, and drugs may act on microbes and affect these homeostatic mechanisms (Figure 4). Microbes can influence homeostatic host immunity in several ways. Commensal microbes exert tonic signals that modulate the host immune tone4, induce tolerance80, produce antigens that are recognized by the immune system10, and produce proteins, glycoproteins, lipid-sugars (LPS), and outer membrane vesicles81 that the host immune system responds to in a non-diseased state.
In the setting of disease, microbes and their products are implicated in autoimmune disease pathogenesis, and therapeutic drugs may do “double-duty” by targeting both host immune cells and pathogenic microbial mechanisms to alleviate disease. Mechanisms by which microbes are thought to contribute to autoimmune pathogenesis are multitudinous (Figure 5):
Molecular mimicry/cross-reactivity: Commensals may produce protein peptides with homology to self-peptides; thus, an appropriate immune response may also result in “friendly fire” in which immune cells attack self-organs10,82–84. Similarly, antigens directed against commensals may result in “antigen spreading” that targets self-peptides.
Immunomodulatory metabolites: Commensals produce small molecule metabolites that set the immunologic tone for multiple immune cell populations85,86; and in doing so, these commensals may “embolden” immune cells that were previously weakly responsive to self-peptides to become strongly responsive. Small molecules produced by microbes can traverse the epithelial barrier and reach high levels in the circulation47.
Immunomodulatory cell wall components: Commensals have been shown to produce lipid, protein, and carbohydrate molecules that act more locally to incite (or dampen) inflammation in immune cell populations in the lamina propria87,88. These immune cell populations then migrate to other tissues and may contribute to enhanced inflammation at gut-distal sites89.
Compromised barrier permeability: Microbes themselves may traverse the epithelial barrier if it is compromised (i.e., “leaky”), which is often the case in autoimmunity. These microbes may then unleash strong immune responses that trigger or exacerbate autoimmunity90.
Figure 5.

Microbes may shape host immunity in multiple ways. They may produce immunomodulatory small-molecule metabolites that easily traverse epithelial barriers, thereby resulting in effects on organs that are distal from gut microbes (“immunomodulatory metabolites”). Microbes also produce bulkier protein, lipid, or carbohydrate products (flagellin or LPS) that tend to affect the local immunological milieu (“cell wall components”). Microbial proteins with homology to human proteins may spur autoimmunity because an appropriate immune response may be accidentally unleashed on human tissues and organs (“cross-reactivity with autoantigens” or “molecular mimicry”). Finally, microbes may translocate into the circulation when the epithelial barrier is impaired, leading to microbial colonization of sites that were historically thought be sterile. Abbreviations: SCFA, short chain fatty acids; ATP, adenosine triphosphate; LPS, lipopolysaccharide; PSA, polysaccharide A.
How, then, do commonly used immunosuppressive or immunomodulatory drugs modulate the microbiome to exert effects on host immunity? The possibilities include:
Reducing levels of an autoimmunity-inducing microbe: Immunosuppressive drugs, in addition to suppressing the host immune system, also change the abundance of inflammatory microbes, particularly ones that trigger molecular-mimicry and autoimmunity. If we can identify a commensal that initiates, incites, or aggravates disease which is already “accidentally” targeted by existing immunomodulatory drugs, then we can develop specific therapies targeting microbiota instead of broadly immunosuppressing the host. Proof-of-principle studies showcasing that specific microbes can be targeted to alleviate autoimmunity come from studies of murine models of lupus and inflammatory bowel disease: specifically targeting Enterococcus gallinarum or Klebsiella pneumoniae strains using vaccines90 or phage therapy91, respectively, resulted in improved outcomes in mouse models. Though causality is difficult to ascertain in patients, cross-sectional patient studies also support the hypothesis that successful drug therapy is associated with reductions in specific microbial species92,93.
Reducing levels of autoimmunity-inducing microbial products: Immunosuppressive drugs may change the physiology of existing microbes, such that immune modulatory proteins, lipids, sugars, and small molecule metabolites are altered in a way that reduces host inflammation. In this case, studies profiling gut community composition using 16S or shotgun sequencing may not uncover changes that are likely happening at the transcriptomic or metabolomic level. In one example, investigators generated an experimental system to interrogate microbiota-immune interactions and how diet impacts expression of immunomodulatory antigens on a commensal microbe94. They found diet could reduce expression of an antigen on Bacteroides thetaiotaomicron that promoted activation of T cells specific for B. theta. This exemplifies how exogenous perturbations (e.g., diet) can impact the microbiota (e.g., peptide expression) with downstream consequences for host immunity (e.g., T cell activation).
Reshaping microbial communities so that they are less immune-stimulating: Immunosuppressive drugs, such as MTX, may change the community composition of microbes or emergent community dynamics. A recent study showed MTX-exposed microbial communities, showing reduced levels of Bacteroidetes phylum, elicited less immune activation (lower levels of B cells, myeloid cells, Th1 cells, and T cell activation in the spleen) when mice were exposed to DSS95.
Above, we presuppose that commensals incite inflammation, but commensals may also suppress inflammation. In this case, drugs may act on the microbiota to increase anti-inflammatory microbes, microbial products, or microbial community configurations. Elucidating these anti-inflammatory mechanisms could be critical for developing novel therapies.
The general principle underlying all these mechanisms is that immunosuppressive or immunomodulatory drugs that act on microbes can propagate effects through existing microbiota-immune pathways to lead to disease alleviation. If we can uncover the key or central microbiota-immune pathways that trigger or exacerbate disease, then we can develop drugs to target specific microbes or microbial products without inducing iatrogenic immune suppression.
4. Ode to methotrexate (or “owed to methotrexate”): rheumatology edition
Methotrexate is considered one of the first rationally-designed molecular medicines developed in the modern era96. The success of MTX and modern-day chemotherapy was born out of an erroneous (and possibly deadly) hypothesis: initially, oncologists hypothesized that leukemia was due to folate deficiency because the large size of leukemic cells resembled that of enlarged red blood cells (megaloblasts) seen in pernicious anemia, which is due to vitamin deficiency (folate or B12)97. However, clinical studies revealed that folate supplementation was not effective and possibly accelerated disease98. Instead, depriving cancer cells of folate seemed effective: diet-induced folate deficiency resulted in decreased circulating leukemic cells97. These findings spurred the development of folate antagonists for the treatment of leukemia. MTX was synthesized in the 1940s by an unsung scientific hero, Yellapragada Subbarao, shortly after the synthesis of a close analogue of MTX, aminopterin99. Aminopterin and MTX were designed to structurally mimic and antagonize folate activity. In a landmark study, aminopterin was shown by Sidney Farber and colleagues to be effective in curing pediatric leukemia100. While aminopterin was used in the initial studies, MTX replaced aminopterin as a chemotherapy due to a better therapeutic profile in murine studies97.
This was a huge success for molecular medicine and for the treatment of cancer97,101. It was the first time that a compound had been synthesized to mimic the newly discovered vitamin, folate, to treat disease. However, many things were not known at that time, including the enzymes involved in folate metabolism and the precise target of MTX102. Interestingly, in this instance, a drug was developed with little knowledge of the pathway or enzyme that was being targeted; this contrasts with the modern paradigm of drug development, in which key genes or enzymes are first implicated in disease, and drugs are developed (or screened for their ability) to target those specific proteins (e.g. anti-TNF, anti-IL17, PCSK9 inhibitors)20. Multiple scientists lauded the research teams that led to the discovery of methotrexate97, its target dihydrofolate reductase (DHFR), and the folate pathway. Some have written perspectives reminiscing about those discoveries, with Frank Heunnekens writing, “The Enzyme Game was open to everyone. Starting materials were tissues available from laboratory animals and local abattoirs or from cells provided by accommodating bacteria. The necessary reagents (often homemade or, when all else failed, purchased from Sigma) were relatively few and simple”101. Indeed, Joseph Bertino, when asked to give the 1993 Karnofsky Memorial Lecture to the American Society of Clinical Oncology, gave an “Ode to Methotrexate” to highlight all the therapeutic successes that were “owed to methotrexate”97, including cancer cure or remission for the first time in history.
MTX use and popularity has only grown since those early days. MTX was subsequently found to be effective in the treatment of multiple cancers as well as non-malignant diseases, including psoriasis, psoriatic arthritis, and rheumatoid arthritis. It supplanted therapies such as gold and aspirin based on clinical trials in multiple patient populations, especially after FDA approval for its use in treating RA in 1988103. Despite the development of other therapies similarly targeting broad metabolic pathways (e.g., leflunomide, azathioprine, cyclophosphamide) or specific cytokines or immune receptors (e.g., anti-TNF, anti-IL6, anti-IL1 receptor, etc.), MTX has remained an anchor therapy in RA.
Methotrexate’s longevity in the rheumatologic armamentarium is a testament to its multiple beneficial therapeutic properties, with many studies focusing on in the treatment of rheumatoid arthritis. First, clinicians learned that MTX reduced all-cause and cardiovascular mortality104,105, and that this was not a property shared by other orally-administered disease-modifying antirheumatic drugs like hydroxychloroquine or sulfasalazine106. Second, MTX was shown to be more tolerable than other oral disease-modifying antirheumatic drugs (DMARDS)107. Third, when biologic therapy was developed, MTX was found to reduce the production of anti-drug neutralizing antibodies, thereby increasing the efficacy of biologics (e.g., TNF inhibitors, uricase)108. Most recently, it has been shown to make gout treatment with pegloticase more effective by reducing antidrug antibody development to pegloticase109. Consequently, MTX acts synergistically with multiple biologics to alleviate disease110,111. Fourth, unlike other oral drugs, it is taken just once weekly instead of daily, which patients prefer. Fifth, because it is not under patent, oral and injectable forms are more affordable than the newer biologics. Thus, over the years, MTX has shown its worth in multiple clinical trials and studies. Expert guidelines recommend MTX as first-line therapy for RA112, and many rheumatologists use it as first-line therapy more broadly to treat inflammatory arthritis in diseases such as lupus, dermatomyositis, and sarcoidosis.
Instead of being relegated to the dustbin of rheumatologic history (as has been the fate of gold and aspirin), MTX has repeatedly shown itself to be a powerful tool in our therapeutic arsenal beyond the clinical discipline of rheumatology. It is not just used in the treatment of rheumatologic disease but has found widespread use in other clinical disciplines as well, including in the treatment of psoriasis113, mycosis fungoides113, inflammatory eye disease (uveitis)114, inflammatory bowel disease115, vasculitis116, and other immune-mediated conditions. It can act as a stand-alone therapy for many patients, and when it is insufficient, other therapies are added-on to MTX because it can act synergistically with other drugs. No other drug in rheumatology possesses so many favorable therapeutic properties. It is among the top 300 drugs prescribed in the US (#132 in 2021), with an estimated 4.4 million prescriptions each year provided to over 900,000 patients117. Thus, MTX is used to treat multiple common conditions, and many advances in autoimmune therapy are “owed to methotrexate.”
5. The microbiome vs. methotrexate: how bugs affect drugs
Although MTX has been used for multiple decades, not every patient responds to MTX therapy. Many patients experience inadequate or no response to MTX, and thus MTX has limited efficacy in these patients19. Additionally, some patients that benefit from MTX also experience significant side-effects; in these latter cases, the toxicity of MTX limits use of this cost-effective and mortality-improving drug118.
This inter-individual variation in MTX response has been heavily studied over the past 50–60 years and multiple host factors have been investigated119. Oncology investigators have found that cancer patients evolve resistance to MTX because tumor cells amplify the target of MTX, DHFR, resulting in multiple copies120. Or they mutate the target enzyme, DHFR, such that it no longer binds MTX. Additionally, MTX is actively transported into cells, and cancer cells evolve resistance by mutating transporters. Additionally, SNPs or mutations in folate pathway genes have been implicated in MTX response19. These previous investigations looking at various host factors that might govern MTX response have been reviewed in detail by others19,118,121.
Microbial metabolism of MTX was first discovered several decades ago, but most clinicians are surprised to learn this since it is not reported in clinical reviews focused on MTX113,115,122. The initial discoveries of microbial metabolism of MTX occurred in the 1960s, in which investigators found that soil72 and mouse123 bacterial strains could metabolize MTX into an inactive metabolite. It was surmised that human gut bacterial strains similarly metabolized MTX, but investigators were not able to show this directly124. This lead to controversy about whether the inactive metabolites were truly formed by microbes or whether, instead, they were already present in the infusion bag as a result of impurities in the initial synthesis of MTX124. Early pharmacokinetic studies were completed in small populations of cancer patients given MTX by the intravenous (IV) route instead of the oral (PO) route. These early studies led to the conclusion that only a small amount (~5%) of MTX is metabolized by bacteria125. Given that nowadays most MTX prescriptions are prescribed to treat autoimmunity and administered via the oral route, in which the drug is more likely to encounter microbes, these early studies in cancer patients may not be generalizable to the current cohort of patients taking MTX. Indeed, these early studies may have led to premature conclusions about the extent and impact of microbial metabolism of MTX. Our group revisited this line of investigation and found that human gut bacterial strains could directly metabolize MTX, and that this metabolism was associated with clinical response in RA patients. Below, we review this history of microbial metabolism of MTX in greater depth.
5.1. Identification of environmental microbes that metabolize MTX.
Given the growing success of MTX in the 1960’s, there was high interest in characterizing MTX and its metabolites, many of which were still largely unknown because the field of clinical pharmacology was just being developed126. Early studies noted that some bacteria inactivate MTX127. In 1967, Levy and Goldman working at NIH sought to define the metabolites of MTX, because, as they wrote “Although the biological degradation of methotrexate has been examined in several systems, in none have the degradation products been completely characterized”128. Their strategy was to find a soil microbe that could metabolize MTX and to identify products of this metabolism in the lab. They used a culture enrichment technique to facilitate finding such a microbe: they prepared a medium that selected for microbes that could use MTX as the sole source of carbon and nitrogen. They screened microbes from the mud at Rock Creek woods on minimal media containing MTX and salts. Remarkably, they found a single microbe, characterized only as a “pseudomonad”, that could survive on MTX-based minimal media. The authors deduced that the “crystalline orange pigment” left behind was deoxyaminopteroic acid (DAMPA) (Figure 6). Further, the authors found that the candidate enzyme could act on folic acid and aminopterin. They went on to show that the purified enzyme could metabolize multiple compounds ending with a glutamate and thus named the enzyme carboxypeptidase G (CPG)72, noting129 “the study with methotrexate illustrates the usefulness of microorganisms in elucidating biochemical reactions on compounds with potential interest as drugs.”
Figure 6.
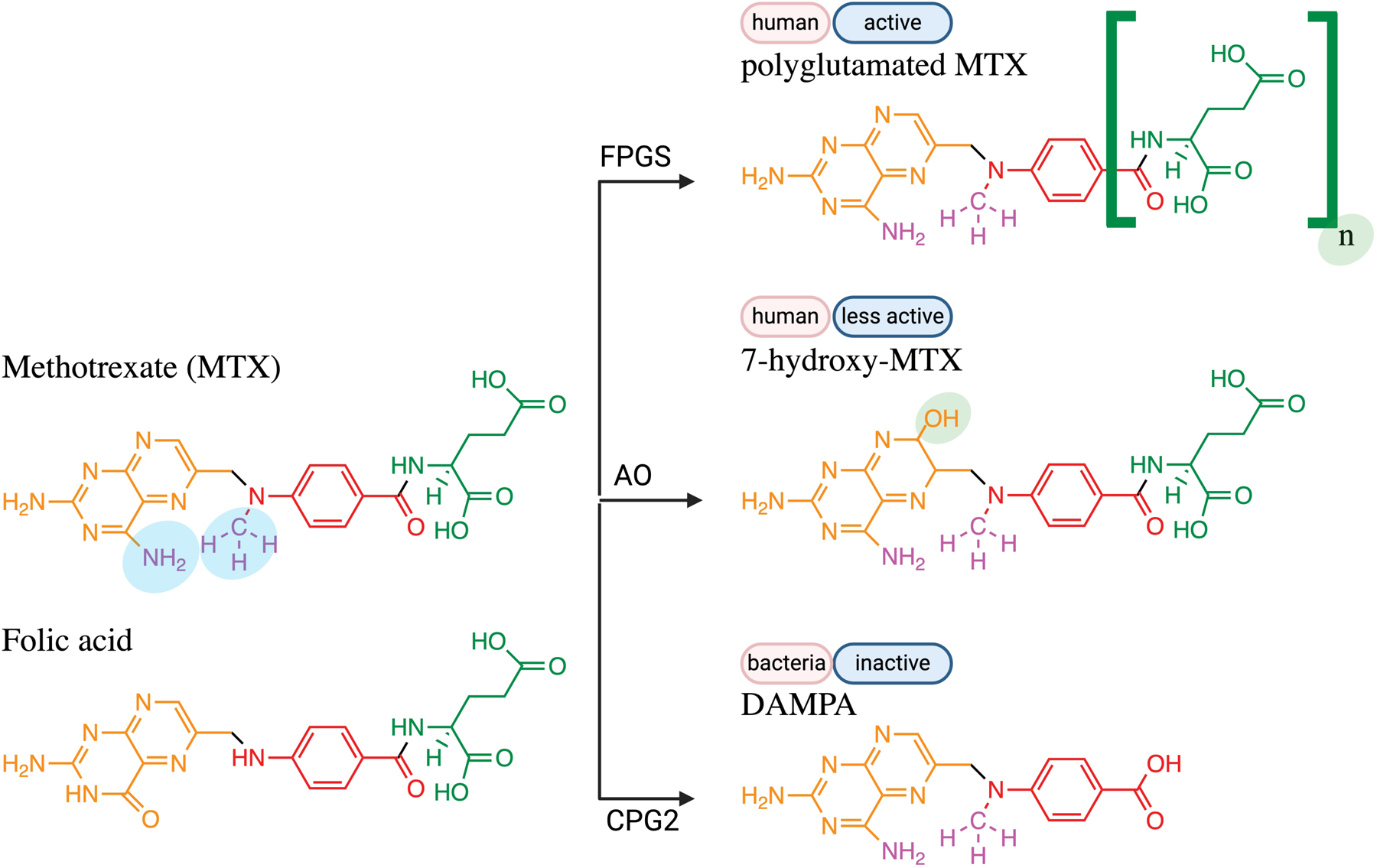
Two human enzymes and one bacterial enzyme family are known to metabolize MTX (and a related vitamin, folic acid). All eukaryotic cells possess FPGS, which makes MTX more active by adding two to seven glutamates. The liver hydroxylates MTX (or folic acid) using AO, which makes MTX less active. The first enzyme discovered to metabolize MTX was a carboxypeptidase (CPG), which removes the glutamate moiety (colored green). Abbreviations: FPGS, folyl polyglutamate synthase; AO, aldehyde oxidase; CPG2, carboxypeptidase G2.
With evidence that targeting the folate pathway was effective in curing cancer, investigators also sought to identify folate degrading enzymes that might be used therapeutically in vivo. Bacterial sources of such enzymes could be generated in large quantities. In 1971, McCullough and Chabner sought to find soil bacteria that utilize folates as the sole source of energy130. They isolated Pseudomonas stutzeri from mud obtained along the Long Island Sound, which could survive on minimal media with leucovorin or folic acid. The investigators isolated and purified carboxypeptidase G1 (CPG1) from this strain, which was similar to CPG in that it removed a terminal glutamate. They found that CPG1 was induced by the presence of folates. Further, folate analogs like MTX were substrates of CPG1, which produced DAMPA.
Once it became clear that MTX could cause life-threatening toxicity, enzymes that cleave MTX in vivo were sought, and once again, researchers turned to bacteria to identify such enzymes. This is because humans lack an enzyme that converts MTX into the inactive metabolite DAMPA. A Flavobacterium species isolated from water was found to encode a carboxypeptidase (not further named, and simply designated as “carboxypeptidase”) with the ability to metabolize MTX, with higher affinity for MTX than endogenous natural folates. These findings suggested that it might be effective in reversing the toxicity of high-dose MTX seen in cancer patients131,132. It was not until carboxypeptidase G2 (CPG2) was identified in a strain of Pseudomonas RS-16133 and cloned into E. coli134 that this strategy came to fruition in reversing toxicity in patients, and continues to be used in clinical practice135. Taken together, these studies demonstrated the power of using microbes to learn about MTX and leveraging this information to improve patient care.
Thus, the first MTX metabolite identified (DAMPA) was a bacteria-produced metabolite, not a human one. It was not until almost 3 decades after the development of MTX that the first human-produced MTX metabolites, polyglutamated MTX and 7-hydroxy-MTX, were identified and reported (Figure 6). Investigators first uncovered that MTX was polyglutamated in human red blood cells136 and human liver cells137 (Figure 6). A similar type of transformation happens to folates, which are polyglutamated once they enter the cell, but these polyglutamated forms were hard to discover initially because they were rapidly deglutamated by endogenous enzymes138. Produced by the enzyme folylpolyglutamate synthase (FPGS), polyglutamated folates are retained intracellularly and have higher affinity for enzymes like DHFR138. Similarly, polyglutamated MTX is retained intracellularly and is more effective at inhibiting DHFR and other enzymes, leading researchers to consider MTX to be a “prodrug”139. The second human-produced MTX metabolite discovered was 7-hydroxy-methotrexate, which was identified by investigators at the National Cancer Institute (NCI at NIH) studying cancer patients that were receiving high dose MTX140. In this study of 5 cancer patients treated with MTX, about 7–33% of the drug excreted in urine between 18–24 hours was 7-hydroxy-MTX. The enzyme that produces 7-hydroxy-MTX is aldehyde oxidase (AO).
Thus, all together there are three major MTX metabolites: DAMPA, polyglutamated MTX, and 7-hydroxy-MTX. The ability to detect and distinguish between these three major MTX metabolites was challenging, and many early studies suffered from insufficient sensitivity and specificity. The identity of these compounds was determined by their retention times and patterns on DEAE cellulose and thin layer chromatography techniques. Indeed, the ability to disambiguate parent and metabolite compounds was not always possible, making it challenging to make strong conclusions. And the methods were low-throughput compared to current day methods. Thus, sample sizes were small for many studies.
5.2. Studies suggesting that gut microbes metabolize MTX in animal models.
While initial studies in the 1960s suggested that soil microbes could metabolize MTX in vitro, the first hints that MTX is metabolized by gut microbes in vivo came from studies of antibiotic-treated and germ-free mice by investigators at NCI. In 1969, investigators found that when comparing normal vs. antibiotic-treated CDF1 male mice (N=3 per treatment group) treated with radioactive MTX 3 mg intraperitoneally, more MTX was recovered in the stool of antibiotic-treated mice; additionally, there was less “pre-MTX” metabolites, which were metabolites that eluted before MTX on DEAE cellulose columns141. This suggested that microbes may metabolize MTX. However, the investigators noted that in antibiotic-treated mice given neomycin and sulfathiazole, less MTX was recovered in the urine. This was counter to what was expected: if antibiotics reduce microbial metabolism, more MTX should be delivered to the circulation and excreted in the urine. The investigators then studied DBA/2 germ-free mice treated with subcutaneous MTX (dose and timepoint not explicitly reported) and found that more MTX was recovered in the urine, thereby surmising that antibiotics (e.g., neomycin) likely reduced MTX transport into the circulation. There were a few limitations to this early study. The amount of MTX and its metabolites in the blood circulation was not assessed and the identity of the “pre-MTX” metabolites was not known. Of note, the only reported metabolite to have been discovered at this point was DAMPA128; the discovery of PG-MTX and 7-OH-MTX was not accomplished until 1973 and 1976. Additionally, the investigators experienced technical challenges in distinguishing between MTX and its metabolites in the feces of germ-free animals. They were therefore unable to confirm if MTX was increased or its metabolites were decreased in germ-free fecal samples, as might be expected from the results in antibiotic-treated mice.
This same group further characterized the kinetics by which MTX metabolites (still unidentified) were observed in the urine and feces of rats and mice142. They compared the amount of MTX, “pre-MTX,” and “post-MTX” metabolites in mice and rats (N=3 per treatment group) given tritiated MTX 21 mg/m2 intraperitoneally. They used a DEAE ion-exchange column and quantified excretion at “early” and “later” time points (“early” being 0–6 hours; “late” being 6–12 hours in rats). When considering the urine, 40% of the radioactive dose (comprising both MTX and its metabolites) was excreted in the urine in the early period and another 10% in the late period. Interestingly, in the early period, most of the dose was MTX but in the late period, most of the excreted dose was a MTX metabolite. When considering the feces collected over both the early and late time periods, most of the MTX was excreted in a metabolized form. While these studies provided initial information on the kinetics of MTX metabolite production in vivo, still missing was the identity of the metabolites, the levels of MTX and its metabolites in circulation, and whether the metabolites were produced by the host or the microbiota. Additionally, these in vivo studies had not yet demonstrated that microbes directly metabolize MTX or whether the microbes induce host enzymes that metabolize MTX.
To distinguish between these possibilities, ex vivo fecal communities from mice were used to show that murine gut microbiota can directly metabolized MTX. Investigators tested MTX metabolism by ex vivo cecal communities from CDF1 mice123. MTX was metabolized into three distinct compounds, one of which was DAMPA based on multiple chemical properties. DAMPA was less abundant or absent in cecal communities from antibiotic treated mice. These ex vivo studies provided direct evidence that murine microbiota metabolize MTX into DAMPA, but whether human gut microbes were also capable of this function was unknown.
5.3. MTX pharmacokinetics in patients: hints at microbial metabolism of MTX by human microbes.
Pharmacokinetic studies in humans hinted at the possibility that microbes metabolize MTX, but without specific assays to disambiguate and quantify the metabolites (which were only more precisely determined after 1973), it was challenging to determine the source of these metabolites. In 1973, investigators at the University of Kansas Medical Center profiled radiolabeled MTX levels in the plasma, urine, and feces of 22 cancer patients given intravenous (not oral) MTX143. They found that intravenously-administered MTX was recovered in the feces, suggesting that MTX is enterohepatically circulated in humans. They also found that about 20% of the MTX excreted in the urine was a MTX metabolite, suggesting that MTX is metabolized in vivo in humans, but whether it was microbiota- or host-produced could not be determined. Thus, these early studies revealed that even MTX given intravenously encounters gut microbes.
Shortly thereafter investigators reported that MTX taken orally undergoes more metabolism than MTX administered intravenously; again, whether this was due to microbial or host enzymes was not known and the identity of the metabolites was also unknown. These investigators sought to determine how the route of administration affects MTX pharmacokinetics144. They examined 13 cancer patients, some with malignant infusions. All 13 received IV therapy. Six also received PO therapy. Both routes were tested at 2 concentrations: 30 mg and 80 mg per meter squared (body surface area-based dosing). Plasma concentrations were lower in patients given oral MTX and reflected 47% of the IV dose. While only 6% of the IV dose was excreted as metabolites, 35% of the oral dose was excreted as metabolites in the urine. This suggests that orally administered MTX undergoes more metabolism, and the authors hypothesize that this metabolism could have been either microbial or from first-pass metabolism by the liver. It was not possible to distinguish between these two possibilities in this experiment, though the authors favored microbial metabolism in their discussion.
Further, it became clear that the current tools to measure MTX and its metabolites were not sufficiently specific, and a controversy arose as to whether the metabolites pre-existed in the pill bottle itself (i.e., “impurities”) instead of being produced by host or microbes. Donehower and colleagues noted that many investigators had previously found multiple MTX impurities125. They compared two assays for quantifying MTX and tested their specificities against MTX and known MTX impurities, including DAMPA. Testing the blood and urine of 5 cancer patients receiving 6-hour IV infusions of MTX, they noted that between 3–6% of the total urinary excreted dose was excreted as DAMPA. The authors concluded that this was more than would be expected from the concentration of impurities found in the bottle (ranging between 1.1 to 2.9% DAMPA), and that a large percentage of DAMPA was produced endogenously (and not due to impurities). But the field focused on the fact that only 3–6% of the excreted dose was DAMPA. Thus, from this finding, in cancer patients taking intravenous MTX and quantifying urinary levels of DAMPA, it was extrapolated that microbial metabolism plays a minor role, either noted to be less than 5% of the excreted dose in urine145 or 5% of the administered dose146. But sometimes, what one finds (or does not find) depends on where one looks. One limitation of these studies is that microbial metabolism was not assessed in patients given oral methotrexate and levels of DAMPA in the stool (presumably where it is produced) were not examined. Given that MTX was known to be enterohepatically circulated at that time143, examining DAMPA levels in the feces might have recovered a greater contribution of microbial metabolism to MTX pharmacokinetics. In support of this, Donehower and colleagues also go on to note that DAMPA is less soluble than MTX; thus, if it is produced in the gut, it might remain there and not reach the circulation or urine in high concentrations.
The death knell for studies examining microbial impacts on MTX metabolism likely came in a study published by Stewart et al124. Given the aforementioned limitations, there was some suspicion that the amount of microbial metabolites was very little (in humans and mice given IP or IV MTX) and some suspected that the “metabolites” were actually pre-existing impurities in the initial MTX preparation/infusion bag147. Indeed, Stewart et al in 1986 tested the abundance of DAMPA in 3 patients receiving MTX as a chemotherapy given IV124. They found that only some patients had DAMPA circulating in their blood and only during some infusions (each patient received multiple cycles of MTX). This variability was thought to arise from variability in the MTX preparation. Variability in microbiota composition or the impact of chemotherapy on the microbiota was not known at the time, and therefore not considered. The investigators went on to test 5 strains of human gut microbial species under aerobic conditions for metabolism but did not observe direct bacterial metabolism. The authors note, “These findings lead us to the conclusion that the source of DAMPA in patients treated with high dose MTX is the infusion fluid itself, although we are unable to say whether the contaminant is present as an initial impurity or as a breakdown product.” After that publication, few investigators continued to report on microbial metabolism of MTX or whether it plays a role in patients taking oral MTX for treatment of autoimmunity.
5.4. Hiatus in research studies examining microbial metabolism of MTX.
In contrast to the growing use of MTX, the idea that microbial metabolism could impact MTX pharmacokinetics was largely neglected; as mentioned above, the fact that microbes metabolize MTX is known by few clinicians. There was little take-up by the scientific and medical community of these findings, perhaps because of the small sample size of mice, the limited primary data presented in the manuscripts, and the challenges with characterizing the metabolites and accurately distinguishing them from one another (DAMPA, polyglutamated MTX, and 7-OH-MTX) with sufficient specificity. Further, we now know that many MTX metabolites co-elute on column-based chromatography systems, and that higher-resolution platforms, such as mass spectrometry are required to distinguish between the multiple MTX metabolites148. These challenges with early studies of MTX likely contributed to conflicting results.
Thus, in the intervening years, many review articles reported that MTX is metabolized by the intestinal microbiota but that this metabolism is negligible. For example, Grim report “The drug is metabolized by intestinal bacteria to 4-amino-deoxy-N10-methylpteroic acid. The metabolite usually accounts for less than 5% of the administered dose, and is rarely detectable in human plasma and urine.”146 However, these studies were largely based on studies in cancer patients receiving IV or IP drug and who likely already had been treated with antibiotics, given that cancer and chemotherapy increase the likelihood of infections requiring antibiotics. The extent and impact of microbial metabolism of MTX in patients with rheumatic disease taking oral MTX was not known.
5.5. Viva la microbe: revisiting the impact of microbes on MTX metabolism.
After a ~30-year hiatus in studies looking at microbial metabolism of MTX, we and others began to revive this line of inquiry. Studying the rat microbiome, investigators149 treated male Sprague Dawley rats with MTX 0, 10, 40 or 100 mg/kg via the IP route. Profiling over 48 hours, they found MTX in both urine and stool. One technological advantage this study had over early studies (which used DEAE cellulose141, immunoassays125, or HPLC124) was the use of ultra-high performance liquid chromatography (UHPLC) coupled to mass spectrometry (MS) to find DAMPA and 7-hydroxy-MTX in the stool, suggesting that intraperitoneal MTX undergoes enterohepatic circulation. Metabolites were not found in the urine. These studies confirmed that MTX is metabolized into DAMPA and suggest that DAMPA in retained in the gut, at least in rats and at the concentrations tested in this study.
Given our observations in rheumatology clinic that patients vary in their response to oral MTX and that we do not have good ways to predict response, our group asked if and which human gut microbes were capable of metabolizing MTX. With multiple advances in microbiome research, we were able bring multiple model systems to bear on this question, but many questions still remain.
Because mouse and rat microbial communities are significantly different from human microbial communities, we sought to determine whether human gut bacteria metabolize MTX. First, we showed that human gut microbial communities could directly metabolize MTX ex vivo150. We incubated fecal samples from 22 treatment-naïve RA patients with MTX in an anaerobic chamber150. Taking aliquots of the supernatant between 0 through 48 hours, we observed that some microbial communities quickly depleted MTX from the supernatant and other microbial communities did not, as quantified by UHPLC-MS and NMR. These studies showed that microbial communities from different patients vary in their ability to metabolize MTX. Further, we tested for an association between ex vivo metabolism and clinical response to MTX: patients who tended to clinically improve with MTX were more likely to harbor microbial communities that slowly metabolized the drug in our ex vivo assay. Thus, we showed for the first time that complex human gut microbial communities could deplete MTX, that this activity varied across patients, and that this variation in ex vivo cultures correlated with clinical response in patients. This might have been the first time that microbial metabolism of a drug was correlated with a complex composite clinical outcome in patients, as opposed to drug levels or single serological markers of disease.
In a subsequent paper, Bustion et al, we screened 45 gut bacterial isolates that were representative of the diversity of the human gut microbiota and identified 10 that could deplete MTX from the culture media151. All were from the Firmicutes phylum, of which we tested 17, suggesting that at least in this small sample, about ~50% of Firmicutes can metabolize MTX. Interestingly, this taxon’s predominant members tend to be strictly anaerobic and therefore may not have been easy to study outside of an anaerobic culture chamber/jar. These studies highlight the challenges experienced by researchers in the 1980s when they were unable to detect microbial metabolism by human gut isolates grown under aerobic conditions124. Our studies showed that microbial metabolism by human gut microbes was likely prevalent across members of the Firmicutes phylum, which is one of the most abundant phyla in the human gut microbiome6.
However, many questions remain and are the focus of current studies in our lab. We did not report on the MTX metabolites produced by human microbial communities and/or whether these metabolites were found in the blood and stool of patients. Recently, we developed UHPLC-MS based methods to test and quantify MTX and metabolites in patient blood and stool. Thus, our initial findings may be the tip of the iceberg, and future studies uncovering the impact of the microbiome on MTX pharmacology are needed to understand how to tailor MTX therapy in patients.
6. Methotrexate vs. the microbiome: how drugs affect bugs
As noted above, human-targeted drugs may act on microbes, and there is now ample evidence to suggest that this is the case for MTX. While MTX may be metabolized by microbes, suggesting that microbes “see” the drug and unleash enzymes to act on the drug, it is also possible that MTX acts on microbial machinery to alter microbial physiology. And given that the microbiome is a potent modulator of host immunity, MTX may exert some of its therapeutic effects via the microbiome. Below, we consider studies supporting this hypothesis.
6.1. In vitro studies reveal that MTX inhibits bacterial growth.
Studies conducted in the 1940s suggested that MTX acts on bacterial folate pathways to inhibit growth. A group from Lederle Lab (the major synthesizer and distributor of MTX) found that MTX (ranging from 1 – 50 mg/ml) inhibited growth of Streptococcus faecalis R as assessed by optical density76. Folic acid rescued MTX-induced growth inhibition. Another group demonstrated that Bdellovibrio bacteriovorus (an environmental obligate aerobe that preys on Gram negative bacteria) growth was inhibited by MTX152. These studies were undertaken at a time when the target of MTX was still not known and when its spectrum of activity in all three domains of life was still being determined (although back then, only two domains were known).
However, some bacterial species were resistant to the growth inhibitory effects of MTX, and multiple resistance mechanisms were implicated. Strains of Lactobacillus casei were variably sensitive to MTX; those that were resistant produced large quantities of DHFR153, which scientists then leveraged as a source for enzyme purification and further study. Indeed, purified bacterial DHFR was instrumental in elucidating the structure of MTX-DHFR interactions154. Additionally, multiple laboratory and clinical isolates of microbes were found to be resistant to MTX. Examples include E. coli, in which resistance was conferred by efflux pumps that shuttled the drug out155. Thus, some of the same mechanisms by which cancer cells evade MTX were also present in bacteria: shuttle the drug out155, amplify the target enzyme153, mutate the target so that it can no longer be inhibited by MTX156, upregulate compensatory pathways (like folic acid)95, and metabolize the drug127.
6.2. Human clinical studies suggest that MTX acts on pathogens and commensals.
Epidemiological observations revealed that MTX, in addition to inhibiting growth in vitro, could treat or prevent infections in patients, similar to an antibiotic. A group from St. Jude Children’s Research Hospital noted that “children undergoing therapy for leukemia acquired infections due to Group A beta hemolytic streptococci (GABHS) less frequently than normal children. This observation appeared contrary to what might be expected from the immunosuppressed hosts with leukemia since these individuals are known to be highly susceptible to a variety of infectious disease”157. They tested the in vitro growth inhibitory activity of multiple concentrations of MTX (ranging between 0.49 μg/ml to 250 μg/ml) against 10 GABHS isolates as well as common infectious agents, including Pseudomonas aeruginosa, E. coli, Candida albicans, Staphylococcus aureus, Serratia marcescens, and a Klebsiella-Enterobacter species. While most strains were resistant to MTX 250 μg/ml, 10 GABHS strains were sensitive, suggesting that at least 1 taxon was sensitive to MTX in vitro. In vivo, they also showed that MTX delayed or prevented GABHS-induced mortality in BALB/c female mice (N=5 mice per treatment group), whereas penicillin did not. These initial findings suggested that MTX can act on infectious pathogens in vitro and in vivo, and at least in this one instance, have better efficacy than penicillin.
When full-length 16S Sanger sequencing became widely available, investigators used it to test the impact of drugs on gut microbes. Many of these studies were done in humans or in mice, making it challenging to ascertain whether the effect of the drug was directly on microbes or on the host (with subsequent indirect effects on microbes). For example, investigators158 looked at the effects of high-dose MTX chemotherapy on the DNA content of fecal samples collected from 36 pediatric patients with acute lymphoblastic leukemia. DNA content was decreased compared to 36 age-matched pediatric controls. They also quantified DNA belonging to three taxa (Bifidobacteria, Lactobacillus, and Escherichia coli) and found that high-dose MTX reduced abundance after 3 days after therapy. Seven days after therapy, there was partial rebound. These studies suggest that intravenous high-dose MTX acts on the abundance of at least 3 taxa present in the gut of pediatric cancer patients.
Many of these early studies demonstrated that MTX could act on human gut microbes, but the full spectrum of activity was not explored or known. Limitations of the early studies (i.e., before ~2010) were that they largely focused on a few experimental micro-organismal strains, in part because the ability to study anaerobic microbes was not widely available or easy at the time.
6.3. Advanced tools reveal broad impacts of MTX on rodent microbiota.
In more recent years, 16S amplicon sequencing has enabled hundreds of bacterial species to be profiled in drug-response studies. Mouse models of MTX toxicity, which primarily study high doses of MTX meant to model cancer therapy, have suggested that drug-induced microbiome changes may mediate drug toxicity. Further, modulation of the microbiome by dietary restriction159 or supplementation with probiotics160,161 can alleviate mucositis induced by high-dose MTX. For example, MTX administered intraperitoneally at 1 mg/kg every 3 days for 2 weeks to wild-type specific pathogen free (SPF) BALB/c mice resulted in monocytic inflammation of the small and large intestine, increased expression of inflammatory cytokines (TNFα, IFNγ, and IL-1β), and shifts to the microbiota, with decreases in the Bacteroidiales order160. Among these, Bacteroides fragilis was significantly decreased at day 14, and gavage of mice with B. fragilis reduced mucositis by histology. Similarly, investigators treated wild-type BALB/c mice with MTX 50 mg/kg every 3 days by oral gavage for 21 days161. Mice were supplemented with leucovorin or vehicle control in drinking water. Leucovorin was found to rescue MTX-induced changes to the gut microbiota composition. In these studies, Bifidobacterium longum was decreased by MTX and rescued by leucovorin, and gavage of B. longum to mice receiving MTX rescued mice from MTX-induced weight loss and mucositis. A third study149 evaluated the effects of MTX on community composition of rat microbial communities and found that the Bacteroidetes phylum was reduced when rats were treated with MTX 10 mg/kg; notably, the opposite occurred in rats given 40 mg/kg or 100 mg/kg. Further, the authors examined urine and fecal metabolomic changes in response to MTX and found a large number (>1000 in urine and >500 in stool) of metabolites that were altered at 48 hours after treatment. The findings demonstrate the pervasive effects of MTX on mouse and rat gut microbiota.
While knowledge that MTX may act on some microbes has been established, the hypothesis that MTX may act on the microbiota to modulate host immunity had not been specifically tested. This is interesting, given that historically, RA and other autoimmune diseases were thought to be infectious in etiology. This was supported by the fact that drugs like minocycline, sulfasalazine, hydroxychloroquine, and cotrimoxazole (trimethoprim/sulfamethoxazole) worked to alleviate RA symptoms. However, this hypothesis fell out of favor when an infectious agent could not be identified and the disease did not fulfill Koch’s postulates162. However, in more recent years, microbiome-focused studies in RA populations found associations of gut microbial changes in response to therapy in RA, but whether these were causal or correlative (i.e., reduced inflammation in the host leading to changes in microbiota composition) could not be determined from observational patient cohorts92,93. Thus, few studies tested whether the therapeutic effects of MTX were mediated by the microbiota.
6.4. Multiple model systems reveal broad impacts of MTX on human microbiota.
Because rodent microbial strains differ from those found in humans, our group sought to understand how human gut microbial communities respond to MTX and how this might impact host immunity95. We found that MTX variably and directly perturbs the growth of human gut bacteria at physiological concentrations. To test this and to determine which specific human gut bacteria are directly affected by MTX, we individually treated 45 diverse bacterial species that are commonly found in the human gut with 10 concentrations of MTX in vitro and measured growth by optical density95. One common measure of microbial drug sensitivity is the minimal inhibitory concentration (MIC), which is defined as the lowest concentration of MTX needed to inhibit >90% growth. We found that this varied among species and ranged across the full gradient, with 11 isolates resistant to the maximum concentration tested. Relative to other phyla, members of the Bacteroidetes phylum tended to be sensitive to MTX. The estimated concentration of MTX in the proximal gut (10–100 μg/ml) would be sufficient to inhibit 11–33% of the tested isolates95. Thus, MTX directly affects growth of bacteria at physiological concentrations, and sensitivity to MTX varies among bacterial species. Further, when we examined effects on the transcriptomes and metabolomes of bacterial species in vitro, we found that MTX induced profound transcriptomic and metabolomic changes, even in isolates that did not show any growth defects. These studies revealed that MTX acts on purine and pyrimidine pathways in human gut bacterial species, but many other pathways were affected as well.
In addition to these direct effects in vitro, we found that, in vivo, MTX altered community composition of human gut microbial communities in humanized mice (i.e., mice colonized with human gut microbiota)95. In germ-free mice colonized with microbial communities with a healthy human donor, MTX caused dose-dependent shifts in community composition. These controlled studies in non-diseased mice suggest that changes are due to MTX and not disease or other factors. Among the 80 taxa that were differentially abundant upon MTX treatment, there was a propensity for the Bacteroidetes phylum to be sensitive to MTX. The Bacteroidetes phylum is one that encompasses hundreds of species and that often represents 40–50% of the species in the human gut microbiome. This finding was in alignment with what we observed in vitro, where Bacteroidetes tended to be sensitive to MTX. We tested this effect in multiple additional mouse studies with small variations (i.e., microbiota from different human donors including RA patients, altered housing conditions, different route of administration). In all experiments, MTX altered community composition. The bacterial taxa that were differentially abundant partially depended on the donor community, emphasizing the highly personalized nature of the gut microbiota, but we also found >50 bacterial species detected in multiple donors that were reproducibly altered by MTX, including prevalent and abundant species such as Bacteroides thetaiotaomicron and Bacteroides ovatus.95
When we next turned to RA patient cohorts (N=23), we found that MTX altered gut microbial community composition and that these shifts were associated with clinical response95. We found that patients with new-onset RA starting MTX treatment exhibited shifts over time (between 0 and 1 month after therapy). When comparing those that clinically responded to MTX (MTX responders, or MTX-R) to those that did not (MTX-non-responders, MTX-NR), we found that a favorable clinical response was associated with a decrease in the Bacteroidetes phylum, similar to what we observed in vitro and in vivo in mice.95 These findings suggest that MTX response is associated with specific shifts in the human gut microbiota. This led us to test the novel hypothesis that MTX acts on gut microbiota to alleviate host inflammation.
Using fecal microbiota transplants in gnotobiotic mouse models of inflammation, we found that MTX reduces the inflammatory potential of gut microbiota. Given the above multiple findings that human gut microbes are directly affected by MTX, we sought to determine whether MTX exposure reduces the inflammatory potential of gut microbial communities95. To do this, we obtained MTX-exposed and MTX-non-exposed (pre-treatment) microbial communities from RA patient donors; these communities were transplanted into germ-free mice and host immunity was quantified in unchallenged (“non-inflamed”) and challenged (“inflamed”) states. Germ-free mice were colonized with pre- and post-treatment stool samples from 3 MTX-responders with the largest decrease in Bacteroidetes phylum levels, given the association of this phylum with clinical response. Each colonization group was split into unchallenged and challenged subsets, using dextran sodium sulfate (DSS) as an inflammatory challenge. Immune populations in the spleen, small intestine, and colon were examined. In unchallenged mice, 3 immunocyte populations were lower in the post-MTX relative to pre-MTX recipients (B cells and Tregs) and 1 population was higher (myeloid cells). In mice challenged with DSS, 7 immunocyte populations were significantly lower (B, myeloid, activated T, Th1, and Th17 cells) and 1 higher in post-MTX recipients95. Taken together, these results suggest that MTX exposure in patients reduces the inflammatory potential of gut microbes.
In summary, drugs used to treat autoimmunity can have effect on gut microbial communities. These off-target effects on microbes may mediate some of the therapeutic properties of drugs used to treat autoimmunity. Multiple RA patient studies further support this hypothesis, demonstrating that MTX response is predicted by gut microbiota composition and function93,150,163. Elucidating the underlying mechanisms will pave the way for microbiota-based therapies.
7. Variations on a theme: many drugs used to treat autoimmunity interact with microbes
MTX is just one of many anti-rheumatic drugs that interact with human gut microbes. In study profiling 1,197 drugs25, the following common rheumatology drugs were tested for growth inhibitory activity against 40 human gut species: MTX, azathioprine, leflunomide, sulfasalazine, allopurinol, and cyclophosphamide. Of these, MTX affected growth of 12 species, azathioprine affected 7, and leflunomide affected 2. While the others did not affect growth at the single concentration tested, perhaps trials with higher concentrations might reveal effects. Often, we do not know the drug concentrations experienced by microbes in the gut164. Another study tested multiple concentrations of MTX, sulfasalazine, and aurothiomalate against 12 oral-microbiota associated species, and found that all three affected species in vitro, albeit some at high concentrations165. These studies reveal that multiple anti-rheumatic drugs can act on human gut microbes to affect growth and likely other finer-grained phenotypes, such as transcription and metabolism.
Human gut microbes also metabolize multiple anti-rheumatic drugs. In a study profiling 271 drugs tested against 76 human gut bacterial species, the following drugs were depleted from the media in the presence of microbes: sulfasalazine (as expected, since this drug was designed to be metabolized by bacteria), mycophenolate, febuxostat, colchicine, betamethasone, dexamethasone, indomethacin, ketorolac, and etodolac26. Another study testing 438 drugs incubated with complex human gut microbial communities ex vivo revealed that allopurinol and azathioprine were depleted by gut microbes73. Hydrocortisone and mycophenolate mofetil were also transformed by gut microbiota. Thus, multiple oral immunosuppressive drugs are metabolized by human gut microbes.
However, knowledge of drug-microbiota interactions in autoimmunity is still at its infancy and many outstanding questions remain. Many anti-rheumatic drugs are given in combination to patients, and few studies have examined combinatorial effects of drugs. Previous studies26,73 laudably tested multiple drugs for metabolism, but the precise metabolic products and their bioactivity remain open questions. Importantly, these studies largely focused on drug-microbiota interactions, but for many of these, the clinical impact on the host remains largely unknown. Thus, more studies likely are warranted to understand drug-microbiota interactions in autoimmunity.
8. Challenges with studying multiple complex systems
Dissecting the impacts of the microbiome in treating autoimmunity is complex because much remains unknown about the microbiome, the host immune system, and clinical autoimmunity. As for the microbiome, advances in high throughput screening of bacterial genes166,167, genetic manipulation of microbes68, comparative genomics168, and metabolomics169 are beginning to uncover unknown genes, proteins, and microbial products. The same is true for immunity – advances in single cell technologies reveal novel cell states, suggesting that the classic distinctions between Th1, Th2, immunocyte populations are hazier than previously surmised170–172. And clinical rheumatology grapples with disease heterogeneity, lack of well-defined biomarkers to quantify disease activity, a workforce that needs expansion to keep pace with the growth in autoimmune disease, and a fragmented patient population that lacks access to academic centers to participate in research173. Moreover, unlike diabetes or cancer, there is no single diagnostic test that can be used to diagnose rheumatologic disease or to monitor its progress, severity, and improvement. Instead, rheumatologists use composite scores or indices to quantify disease174–176. These composites include both physician- and patient-reported measures, such as tender joints (reported by the patient) and swollen joints (assessed by the physician). Such patient-reported measures represent symptoms that we, as a scientific community, have not yet figured out how to quantify longitudinally using lab tests. Thus, there is the opportunity to find microbiota-based features that are highly correlated with patient outcomes or physician scores; these biomarkers may be easier to systematically leverage in future clinical studies. They may also provide mechanistic insight into problematic symptoms that we currently lack blood markers for.
These challenges serve as a clarion call: we need a well-trained scientific workforce that is bold enough to tackle these challenges; we need institutional stakeholders that are committed to facilitating the development of microbiome-based medicines. There is a need for such discovery because the number of patients with autoimmune disease is growing and impacts all ages, races, and cultures.
9. Microbiome Medicine
While mounting evidence suggests that the microbiome may impact the onset, progression, and treatment of disease in humans, essentially none of our current therapies intentionally target the microbiota. Thus, we have an opportunity to develop substantively novel therapies if we elucidate the mechanisms by which the human gut microbiota contributes to the treatment of autoimmune disease. We posit that, in addition to acting on the host immune system, immunosuppressive drugs act on gut microbiota to alleviate autoimmunity. Deciphering the underlying mechanisms of how current therapeutics act on the microbiota to alleviate disease may lead to the identification and targeting of microbes, their proteins, or metabolites that contribute to or exacerbate autoimmunity. Thus, instead of broadly immunosuppressing patients, we can develop therapies targeting facets of the microbiota that contribute to disease.
There are multiple microbiome-based therapies that can be employed. These include diet177, supplements, probiotics, fecal microbiota transplant, vaccination targeting specific microbes90, CRISPR editing to target specific microbial genes178, and small-molecule therapies targeting microbes179. Reducing microbial metabolism of existing drugs might be easily achieved by providing supplements that regulate microbial anabolic pathways. Further, if we understand how microbes “talk” to the immune system to incite inflammation, we may be able to develop drugs targeting microbial effectors mediating this communication180.
10. Concluding Remarks
Here, we reviewed the impact of the gut microbiome on the treatment of autoimmunity, and the many mechanisms by which microbes may modify therapeutic outcomes. Microbes interact with therapeutic drugs, either by affecting them (i.e., their pharmacology) or by being affected by them. Microbes shape host immunity and likely contribute to autoimmunity. Many drugs used to treat autoimmunity are taken orally and interact with human gut microbes; now we have the tools to look at these interactions with a new lens. We took an in-depth look at a widely used immunosuppressive drug, methotrexate (MTX), which revealed how an important drug that has been studied for decades continues to provide new insights into drug-microbiota-autoimmunity interactions that have strong implications for advancing precision medicine.
Finally, though there is still much to learn, the outlook for microbiome medicine is bright and holds great promise. While studying three complex systems with many unknowns seems daunting, advances in technology make this an opportune moment in history to understand the mechanisms that underly drug-microbiota-autoimmunity interactions. Understanding these interactions represents an opportunity to (1) derive greater benefit from existing therapies in autoimmunity, (2) target immune-inciting microbial products without broadly immunosuppressing patients, (3) uncover microbiota-mediated mechanisms of autoimmunity, (4) and tailor therapies for patients with autoimmune disease. Studies aimed at addressing these goals will advance care for the millions of patients globally suffering from autoimmune disease.
Acknowledgements
R.R.N was supported by the following: K08AR073930; R03AR082036 (NIAMS), I01CX002557 (VA ORD), R35GM151349 (NIGMS), Russell/Engleman Rheumatology Research Center, and the Arthritis National Research Foundation. D.A.O. was supported by: 3K08AR073930-05S1 (NIAMS/OD); ImmunoDiverse SRA Fellowship; 5R01AR074500 (NIAMS).
We would like to thank Mary Nakamura, Peter Turnbaugh, and members of the Nayak Lab (Mohana Mukherjee, Vanya Sofia Villa Soto) for their helpful comments and suggestions on the manuscript.
Footnotes
Conflict of Interest Statement
The authors have no conflict of interest.
References
- 1.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352(6285):560–564. [DOI] [PubMed] [Google Scholar]
- 3.Quinn RA, Melnik AV, Vrbanac A, et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature. 2020;579(7797):123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long SS, Swenson RM. Development of anaerobic fecal flora in healthy newborn infants. J Pediatr 1977;91(2):298–301. [DOI] [PubMed] [Google Scholar]
- 6.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lloyd-Price J, Mahurkar A, Rahnavard G, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550(7674):61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holers VM, Demoruelle MK, Kuhn KA, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol 2018;14(9):542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill T, Rosenbaum JT. Putative pathobionts in HLA-B27-associated spondyloarthropathy. Front Immunol 2020;11:586494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silverman GJ, Azzouz DF, Gisch N, Amarnani A. The gut microbiome in systemic lupus erythematosus: lessons from rheumatic fever. Nat Rev Rheumatol 2024. [DOI] [PubMed] [Google Scholar]
- 11.Ma L, Morel L. Loss of gut barrier integrity In lupus. Front Immunol 2022;13:919792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan D, Issa N, Afifi L, Jeon C, Chang HW, Liao W. The role of the skin and gut microbiome in psoriatic disease. Curr Dermatol Rep 2017;6(2):94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruff WE, Greiling TM, Kriegel MA. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol 2020;18(9):521–538. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol 1997;84(3):223–243. [DOI] [PubMed] [Google Scholar]
- 15.Conrad N, Misra S, Verbakel JY, et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: a population-based cohort study of 22 million individuals in the UK. Lancet 2023;401(10391):1878–1890. [DOI] [PubMed] [Google Scholar]
- 16.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol 2008;173(3):600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinse GE, Parks CG, Weinberg CR, et al. Increasing prevalence of antinuclear antibodies in the United States. Arthritis Rheumatol 2020;72(6):1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung SM, Kim WU. Targeted immunotherapy for autoimmune disease. Immune Netw 2022;22(1):e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romao VC, Canhao H, Fonseca JE. Old drugs, old problems: where do we stand in prediction of rheumatoid arthritis responsiveness to methotrexate and other synthetic DMARDs? BMC Med 2013;11:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature. 2020;580(7803):329–338. [DOI] [PubMed] [Google Scholar]
- 21.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glowacki RWP, Martens EC. In sickness and health: Effects of gut microbial metabolites on human physiology. PLoS Pathog 2020;16(4):e1008370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas AM, Segata N. Multiple levels of the unknown in microbiome research. BMC Biol 2019;17(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zmora N, Zeevi D, Korem T, Segal E, Elinav E. Taking it personally: personalized utilization of the human microbiome in health and disease. Cell Host Microbe 2016;19(1):12–20. [DOI] [PubMed] [Google Scholar]
- 25.Maier L, Pruteanu M, Kuhn M, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555(7698):623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570(7762):462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330(6005):831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341(6143):295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klatt NR, Cheu R, Birse K, et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science. 2017;356(6341):938–945. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Esteve E, Tremaroli V, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 2017;23(7):850–858. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Xie C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 2018;24(12):1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foretz M, Guigas B, Viollet B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinol 2023;19(8):460–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanahan D Hallmarks of cancer: new dimensions. Cancer Discov 2022;12(1):31–46. [DOI] [PubMed] [Google Scholar]
- 34.Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. [DOI] [PubMed] [Google Scholar]
- 35.Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371(6529):595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baruch EN, Youngster I, Ben-Betzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371(6529):602–609. [DOI] [PubMed] [Google Scholar]
- 39.Baruch EN, Wang J, Wargo JA. Gut microbiota and antitumor immunity: potential mechanisms for clinical effect. Cancer Immunol Res 2021;9(4):365–370. [DOI] [PubMed] [Google Scholar]
- 40.Villemin C, Six A, Neville BA, Lawley TD, Robinson MJ, Bakdash G. The heightened importance of the microbiome in cancer immunotherapy. Trends Immunol 2023;44(1):44–59. [DOI] [PubMed] [Google Scholar]
- 41.Matthews DA, Alden RA, Bolin JT, et al. Dihydrofolate reductase from Lactobacillus casei. X-ray structure of the enzyme methotrexate.NADPH complex. J Biol Chem 1978;253(19):6946–6954. [PubMed] [Google Scholar]
- 42.Arvidsson C, Hallen A, Backhed F. Generating and analyzing germ-free mice. Curr Protoc Mouse Biol 2012;2(4):307–316. [DOI] [PubMed] [Google Scholar]
- 43.Thevaranjan N, Puchta A, Schulz C, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017;21(4):455–466 e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol 1963;42(4):471–483. [PMC free article] [PubMed] [Google Scholar]
- 45.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009;9(5):313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Claus SP, Ellero SL, Berger B, et al. Colonization-induced host-gut microbial metabolic interaction. mBio 2011;2(2):e00271–00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Treuren W, Dodd D. Microbial contribution to the human metabolome: implications for health and disease. Annu Rev Pathol 2020;15:345–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skelly AN, Sato Y, Kearney S, Honda K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol 2019;19(5):305–323. [DOI] [PubMed] [Google Scholar]
- 49.Rehaume LM, Mondot S, Aguirre de Carcer D, et al. ZAP-70 genotype disrupts the relationship between microbiota and host, leading to spondyloarthritis and ileitis in SKG mice. Arthritis Rheumatol 2014;66(10):2780–2792. [DOI] [PubMed] [Google Scholar]
- 50.Han S, Van Treuren W, Fischer CR, et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature. 2021;595(7867):415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Jarman JB, Low YS, et al. A widely distributed gene cluster compensates for uricase loss in hominids. Cell. 2023;186(16):3400–3413 e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ha CWY, Martin A, Sepich-Poore GD, et al. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell. 2020;183(3):666–683 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vujkovic-Cvijin I, Welles HC, Ha CWY, et al. The systemic anti-microbiota IgG repertoire can identify gut bacteria that translocate across gut barrier surfaces. Sci Transl Med 2022;14(658):eabl3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woese CR, Stackebrandt E, Macke TJ, Fox GE. A phylogenetic definition of the major eubacterial taxa. Syst Appl Microbiol 1985;6:143–151. [DOI] [PubMed] [Google Scholar]
- 55.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 1985;82(20):6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Durack J, Lynch SV. The gut microbiome: relationships with disease and opportunities for therapy. J Exp Med 2019;216(1):20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lagier JC, Khelaifia S, Alou MT, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 2016;1:16203. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Lau JT, Whelan FJ, Herath I, et al. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med 2016;8(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tripp HJ, Kitner JB, Schwalbach MS, Dacey JW, Wilhelm LJ, Giovannoni SJ. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature. 2008;452(7188):741–744. [DOI] [PubMed] [Google Scholar]
- 61.Browne HP, Forster SC, Anonye BO, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533(7604):543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lagkouvardos I, Overmann J, Clavel T. Cultured microbes represent a substantial fraction of the human and mouse gut microbiota. Gut Microbes 2017;8(5):493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Y, Sheth RU, Zhao S, et al. High-throughput microbial culturomics using automation and machine learning. Nat Biotechnol 2023;41(10):1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ren JL, Zhang AH, Kong L, Wang XJ. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv 2018;8(40):22335–22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Plumb RSG LA; Rainville PD; Isaac G; Trengove R;King AM; Wilson ID Advances in high throughput LC/MS based metabolomics: A review. Trends Analyt Chem 2023;160:116954-. [Google Scholar]
- 66.Dodd D, Cann I. Tutorial: Microbiome studies in drug metabolism. Clin Transl Sci 2022;15(12):2812–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y. Host variables confound gut microbiota studies of human disease. Nature. 2020;587(7834):448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin WB, Li TT, Huo D, et al. Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell. 2022;185(3):547–562 e522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scher JU, Nayak RR, Ubeda C, Turnbaugh PJ, Abramson SB. Pharmacomicrobiomics in inflammatory arthritis: gut microbiome as modulator of therapeutic response. Nat Rev Rheumatol 2020;16(5):282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 2016;14(5):273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verdegaal AA, Goodman AL. Integrating the gut microbiome and pharmacology. Sci Transl Med 2024;16(732):eadg8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goldman P, Levy CC. Carboxypeptidase G: purification and properties. Proc Natl Acad Sci U S A 1967;58(4):1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Javdan B, Lopez JG, Chankhamjon P, et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell. 2020;181(7):1661–1679 e1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geva-Zatorsky N, Sefik E, Kua L, et al. Mining the human gut microbiota for immunomodulatory organisms. Cell. 2017;168(5):928–943 e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schirmer M, Smeekens SP, Vlamakis H, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167(4):1125–1136 e1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franklin AL, Belt M, et al. Biological studies with 4-amino-10-methylpteroylglutamic acid. J Biol Chem 1949;177(2):621–629. [PubMed] [Google Scholar]
- 77.Simon EJ. Inhibition of bacterial growth by drugs of the morphine series. Science. 1964;144(3618):543–544. [DOI] [PubMed] [Google Scholar]
- 78.Bushby SR, Hitchings GH. Trimethoprim, a sulphonamide potentiator. Br J Pharmacol Chemother 1968;33(1):72–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ricaurte D, Huang Y, Sheth RU, Gelsinger DR, Kaufman A, Wang HH. High-throughput transcriptomics of 409 bacteria-drug pairs reveals drivers of gut microbiota perturbation. Nat Microbiol 2024;9(2):561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wannemuehler MJ, Kiyono H, Babb JL, Michalek SM, McGhee JR. Lipopolysaccharide (LPS) regulation of the immune response: LPS converts germfree mice to sensitivity to oral tolerance induction. J Immunol 1982;129(3):959–965. [PubMed] [Google Scholar]
- 81.Hong M, Li Z, Liu H, et al. Fusobacterium nucleatum aggravates rheumatoid arthritis through FadA-containing outer membrane vesicles. Cell Host Microbe 2023;31(5):798–810 e797. [DOI] [PubMed] [Google Scholar]
- 82.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 2005;11(1):85–89. [DOI] [PubMed] [Google Scholar]
- 83.Chervonsky AV. Influence of microbial environment on autoimmunity. Nat Immunol 2010;11(1):28–35. [DOI] [PubMed] [Google Scholar]
- 84.Lanz TV, Brewer RC, Ho PP, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. 2022;603(7900):321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. [DOI] [PubMed] [Google Scholar]
- 86.Hang S, Paik D, Yao L, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature. 2019;576(7785):143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown EM, Kenny DJ, Xavier RJ. Gut microbiota regulation of T Cells during inflammation and autoimmunity. Annu Rev Immunol 2019;37:599–624. [DOI] [PubMed] [Google Scholar]
- 88.Alexander M, Ang QY, Nayak RR, et al. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022;30(1):17–30 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galvan-Pena S, Zhu Y, Hanna BS, Mathis D, Benoist C. A dynamic atlas of immunocyte migration from the gut. Sci Immunol 2024;9(91):eadi0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Manfredo Vieira S, Hiltensperger M, Kumar V, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359(6380):1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Federici S, Kredo-Russo S, Valdes-Mas R, et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell. 2022;185(16):2879–2898 e2824. [DOI] [PubMed] [Google Scholar]
- 92.Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang X, Zhang D, Jia H, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 2015;21(8):895–905. [DOI] [PubMed] [Google Scholar]
- 94.Wegorzewska MM, Glowacki RWP, Hsieh SA, et al. Diet modulates colonic T cell responses by regulating the expression of a Bacteroides thetaiotaomicron antigen. Sci Immunol 2019;4(32). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nayak RR, Alexander M, Deshpande I, et al. Methotrexate impacts conserved pathways in diverse human gut bacteria leading to decreased host immune activation. Cell Host Microbe 2021;29(3):362–377 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev 2005;57(2):163–172. [DOI] [PubMed] [Google Scholar]
- 97.Bertino JR. Karnofsky memorial lecture. Ode to methotrexate. J Clin Oncol 1993;11(1):5–14. [DOI] [PubMed] [Google Scholar]
- 98.Farber S, Cutler EC, Hawkins JW, Harrison JH, Peirce EC 2nd, Lenz GG. The action of pteroylglutamic conjugates on man. Science. 1947;106(2764):619–621. [DOI] [PubMed] [Google Scholar]
- 99.Bagri NK, Ramanan A, Ramanan AV. Dr Yellapragada SubbaRow: the forgotten figure in the history of methotrexate. Rheumatology (Oxford). 2023;62(4):1364–1365. [DOI] [PubMed] [Google Scholar]
- 100.Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 1948;238(23):787–793. [DOI] [PubMed] [Google Scholar]
- 101.Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Adv Enzyme Regul 1994;34:397–419. [DOI] [PubMed] [Google Scholar]
- 102.Huennekens FM. In search of dihydrofolate reductase. Protein Sci 1996;5(6):1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weinblatt ME. Methotrexate in rheumatoid arthritis: a quarter century of development. Trans Am Clin Climatol Assoc 2013;124:16–25. [PMC free article] [PubMed] [Google Scholar]
- 104.Krause D, Schleusser B, Herborn G, Rau R. Response to methotrexate treatment is associated with reduced mortality in patients with severe rheumatoid arthritis. Arthritis Rheum 2000;43(1):14–21. [DOI] [PubMed] [Google Scholar]
- 105.Xu J, Xiao L, Zhu J, Qin Q, Fang Y, Zhang JA. Methotrexate use reduces mortality risk in rheumatoid arthritis: A systematic review and meta-analysis of cohort studies. Semin Arthritis Rheum 2022;55:152031. [DOI] [PubMed] [Google Scholar]
- 106.Choi HK, Hernan MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet 2002;359(9313):1173–1177. [DOI] [PubMed] [Google Scholar]
- 107.Maetzel A, Wong A, Strand V, Tugwell P, Wells G, Bombardier C. Meta-analysis of treatment termination rates among rheumatoid arthritis patients receiving disease-modifying anti-rheumatic drugs. Rheumatology (Oxford). 2000;39(9):975–981. [DOI] [PubMed] [Google Scholar]
- 108.Krieckaert CL, Nurmohamed MT, Wolbink GJ. Methotrexate reduces immunogenicity in adalimumab treated rheumatoid arthritis patients in a dose dependent manner. Ann Rheum Dis 2012;71(11):1914–1915. [DOI] [PubMed] [Google Scholar]
- 109.Botson JK, Saag K, Peterson J, et al. A Randomized, Placebo-Controlled Study of Methotrexate to Increase Response Rates in Patients with Uncontrolled Gout Receiving Pegloticase: Primary Efficacy and Safety Findings. Arthritis Rheumatol 2023;75(2):293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54(1):26–37. [DOI] [PubMed] [Google Scholar]
- 111.Witte T Methotrexate as combination partner of TNF inhibitors and tocilizumab. What is reasonable from an immunological viewpoint? Clin Rheumatol 2015;34(4):629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res (Hoboken) 2021;73(7):924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen S, O’Brien T, Yap LM, Prince HM, McCormack CJ. The use of methotrexate in dermatology: a review. Australas J Dermatol 2012;53(1):1–18. [DOI] [PubMed] [Google Scholar]
- 114.Angeles-Han ST, Ringold S, Beukelman T, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the screening, monitoring, and treatment of juvenile idiopathic arthritis-associated uveitis. Arthritis Care Res (Hoboken). 2019;71(6):703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Herfarth HH, Kappelman MD, Long MD, Isaacs KL. Use of methotrexate in the treatment of inflammatory bowel diseases. Inflamm Bowel Dis 2016;22(1):224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol 2021;73(8):1366–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kane S The Top 300 of 2021, ClinCalc DrugStats Database, Version 2024.01. ClinCalc: https://clincalc.com/DrugStats/Top300Drugs.aspx. 2024. Accessed February 8, 2024.
- 118.Romao VC, Lima A, Bernardes M, Canhao H, Fonseca JE. Three decades of low-dose methotrexate in rheumatoid arthritis: can we predict toxicity? Immunol Res 2014;60(2–3):289–310. [DOI] [PubMed] [Google Scholar]
- 119.Kremer JM. Toward a better understanding of methotrexate. Arthritis Rheum 2004;50(5):1370–1382. [DOI] [PubMed] [Google Scholar]
- 120.Jolivet J, Cowan KH, Curt GA, Clendeninn NJ, Chabner BA. The pharmacology and clinical use of methotrexate. N Engl J Med 1983;309(18):1094–1104. [DOI] [PubMed] [Google Scholar]
- 121.Hoekstra M, van Ede AE, Haagsma CJ, et al. Factors associated with toxicity, final dose, and efficacy of methotrexate in patients with rheumatoid arthritis. Ann Rheum Dis 2003;62(5):423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol 2020;16(3):145–154. [DOI] [PubMed] [Google Scholar]
- 123.Valerino DM, Johns DG, Zaharko DS, Oliverio VT. Studies of the metabolism of methotrexate by intestinal flora. I. Identification and study of biological properties of the metabolite 4-amino-4-deoxy-N 10 -methylpteroic acid. Biochem Pharmacol 1972;21(6):821–831. [DOI] [PubMed] [Google Scholar]
- 124.Stewart MJ, Watson ID, Farid YY, Skellern GG. An investigation into the source of the deglutamated metabolites of methotrexate in patients treated with high dose infusions. Ann Clin Biochem 1986;23 ( Pt 2):210–215. [DOI] [PubMed] [Google Scholar]
- 125.Donehower RC, Hande KR, Drake JC, Chabner BA. Presence of 2,4-diamino-N10-methylpteroic acid after high-dose methotrexate. Clin Pharmacol Ther 1979;26(1):63–72. [DOI] [PubMed] [Google Scholar]
- 126.Wagner JG. History of pharmacokinetics. Pharmacol Ther 1981;12(3):537–562. [DOI] [PubMed] [Google Scholar]
- 127.Webb M Inactivation of analogues of folic acid by certain non-exacting bacteria. Biochim Biophys Acta 1955;17(2):212–225. [DOI] [PubMed] [Google Scholar]
- 128.Levy CC, Goldman P. The enzymatic hydrolysis of methotrexate and folic acid. J Biol Chem 1967;242(12):2933–2938. [PubMed] [Google Scholar]
- 129.Goldman P, Levy CC. The enzymatic hydrolysis of folate analogues. Biochem Pharmacol 1968;17(11):2265–2270. [DOI] [PubMed] [Google Scholar]
- 130.McCullough JL, Chabner BA, Bertino JR. Purification and properties of carboxypeptidase G 1. J Biol Chem 1971;246(23):7207–7213. [PubMed] [Google Scholar]
- 131.Albrecht AM, Boldizsar E, Hutchison DJ. Carboxypeptidase displaying differential velocity in hydrolysis of methotrexate, 5-methyltetrahydrofolic acid, and leucovorin. J Bacteriol 1978;134(2):506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chabner BA, Johns DG, Bertino JR. Enzymatic cleavage of methotrexate provides a method for prevention of drug toxicity. Nature. 1972;239(5372):395–397. [DOI] [PubMed] [Google Scholar]
- 133.Sherwood RF, Melton RG, Alwan SM, Hughes P. Purification and properties of carboxypeptidase G2 from Pseudomonas sp. strain RS-16. Use of a novel triazine dye affinity method. Eur J Biochem 1985;148(3):447–453. [DOI] [PubMed] [Google Scholar]
- 134.Minton NP, Atkinson T, Sherwood RF. Molecular cloning of the Pseudomonas carboxypeptidase G2 gene and its expression in Escherichia coli and Pseudomonas putida. J Bacteriol 1983;156(3):1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Widemann BC, Balis FM, Murphy RF, et al. Carboxypeptidase-G2, thymidine, and leucovorin rescue in cancer patients with methotrexate-induced renal dysfunction. J Clin Oncol 1997;15(5):2125–2134. [DOI] [PubMed] [Google Scholar]
- 136.Baugh CM, Krumdieck CL, Nair MG. Polygammaglutamyl metabolites of methotrexate. Biochem Biophys Res Commun 1973;52(1):27–34. [DOI] [PubMed] [Google Scholar]
- 137.Jacobs SA, Derr CJ, Johns DG. Accumulation of methotrexate diglutamate in human liver during methotrexate therapy. Biochem Pharmacol 1977;26(23):2310–2313. [DOI] [PubMed] [Google Scholar]
- 138.Covey JM. Polyglutamate derivatives of folic acid coenzymes and methotrexate. Life Sci 1980;26(9):665–678. [DOI] [PubMed] [Google Scholar]
- 139.Chabner BA, Allegra CJ, Curt GA, et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J Clin Invest 1985;76(3):907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jacobs SA, Stoller RG, Chabner BA, Johns DG. 7-Hydroxymethotrexate as a urinary metabolite in human subjects and rhesus monkeys receiving high dose methotrexate. J Clin Invest 1976;57(2):534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zaharko DS, Bruckner H, Oliverio VT. Antibiotics alter methotrexate metabolism and excretion. Science. 1969;166(3907):887–888. [DOI] [PubMed] [Google Scholar]
- 142.Zaharko DS, Oliverio VT. Reinvestigation of methotrexate metabolism in rodents. Biochem Pharmacol 1970;19(11):2923–2925. [DOI] [PubMed] [Google Scholar]
- 143.Huffman DH, Wan SH, Azarnoff DL, Hogstraten B. Pharmacokinetics of methotrexate. Clin Pharmacol Ther 1973;14(4):572–579. [DOI] [PubMed] [Google Scholar]
- 144.Wan SH, Huffman DH, Azarnoff DL, Stephens R, Hoogstraten B. Effect of route of administration and effusions on methotrexate pharmacokinetics. Cancer Res 1974;34(12):3487–3491. [PubMed] [Google Scholar]
- 145.Widemann BC, Sung E, Anderson L, et al. Pharmacokinetics and metabolism of the methotrexate metabolite 2, 4-diamino-N(10)-methylpteroic acid. J Pharmacol Exp Ther 2000;294(3):894–901. [PubMed] [Google Scholar]
- 146.Grim J, Chladek J, Martinkova J. Pharmacokinetics and pharmacodynamics of methotrexate in non-neoplastic diseases. Clin Pharmacokinet 2003;42(2):139–151. [DOI] [PubMed] [Google Scholar]
- 147.Chatterji DC, Frazier AG, Gallelli JF. Identification and quantitation of impurities in methotrexate. J Pharm Sci 1978;67(5):622–624. [DOI] [PubMed] [Google Scholar]
- 148.Rubino FM. Separation methods for methotrexate, its structural analogues and metabolites. J Chromatogr B Biomed Sci Appl 2001;764(1–2):217–254. [DOI] [PubMed] [Google Scholar]
- 149.Letertre MPM, Munjoma N, Wolfer K, et al. A two-way interaction between methotrexate and the gut microbiota of male Sprague-Dawley rats. J Proteome Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Artacho A, Isaac S, Nayak R, et al. The pretreatment gut microbiome is associated with lack of response to methotrexate in new-onset rheumatoid arthritis. Arthritis Rheumatol 2021;73(6):931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bustion AE, Nayak RR, Agrawal A, Turnbaugh PJ, Pollard KS. SIMMER employs similarity algorithms to accurately identify human gut microbiome species and enzymes capable of known chemical transformations. Elife. 2023;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pritchard MA, Langley D, Rittenberg S. Effects of methotrexate on intraperiplasmic and axenic growth of Bdellovibrio bacteriovorus. J Bacteriol 1975;121(3):1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dann JG, Ostler G, Bjur RA, et al. Large-scale purification and characterization of dihydrofolate reductase from a methotrexate-resistant strain of Lactobacillus casei. Biochem J 1976;157(3):559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bolin JT, Filman DJ, Matthews DA, Hamlin RC, Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J Biol Chem 1982;257(22):13650–13662. [PubMed] [Google Scholar]
- 155.Kopytek SJ, Dyer JC, Knapp GS, Hu JC. Resistance to methotrexate due to AcrAB-dependent export from Escherichia coli. Antimicrob Agents Chemother 2000;44(11):3210–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sirotnak FM, Donati GJ, Hutchison DJ. Genetic modification of the structure and amount of dihydrofolate reductase in amethopterin-resistant Diplococcus pneumoniae. J Biol Chem 1964;239:4298–4302. [PubMed] [Google Scholar]
- 157.Metcalfe D, Hughes WT. Effects of methotrexate on group A beta hemolytic streptococci and streptococcal infection. Cancer. 1972;30(2):588–593. [DOI] [PubMed] [Google Scholar]
- 158.Huang Y, Yang W, Liu H, et al. Effect of high-dose methotrexate chemotherapy on intestinal Bifidobacteria, Lactobacillus and Escherichia coli in children with acute lymphoblastic leukemia. Exp Biol Med (Maywood). 2012;237(3):305–311. [DOI] [PubMed] [Google Scholar]
- 159.Tang D, Zeng T, Wang Y, et al. Dietary restriction increases protective gut bacteria to rescue lethal methotrexate-induced intestinal toxicity. Gut Microbes 2020;12(1):1714401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou B, Xia X, Wang P, et al. Induction and amelioration of methotrexate-induced gastrointestinal toxicity are related to immune response and gut microbiota. EBioMedicine. 2018;33:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huang X, Fang Q, Rao T, et al. Leucovorin ameliorated methotrexate induced intestinal toxicity via modulation of the gut microbiota. Toxicol Appl Pharmacol 2020;391:114900. [DOI] [PubMed] [Google Scholar]
- 162.Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nat Rev Rheumatol 2011;7(10):569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Qiao J, Zhang SX, Chang MJ, et al. Specific enterotype of gut microbiota predicted clinical effect of methotrexate in patients with rheumatoid arthritis. Rheumatology (Oxford). 2023;62(3):1087–1096. [DOI] [PubMed] [Google Scholar]
- 164.Dressman JB, Amidon GL, Fleisher D. Absorption potential: estimating the fraction absorbed for orally administered compounds. J Pharm Sci 1985;74(5):588–589. [DOI] [PubMed] [Google Scholar]
- 165.Kussmann M, Obermueller M, Spettel K, Winkler S, Aletaha D. In vitro evaluation of disease-modifying antirheumatic drugs against rheumatoid arthritis associated pathogens of the oral microflora. RMD Open. 2021;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cain AK, Barquist L, Goodman AL, Paulsen IT, Parkhill J, van Opijnen T. A decade of advances in transposon-insertion sequencing. Nat Rev Genet 2020;21(9):526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dantas G, Sommer MO, Degnan PH, Goodman AL. Experimental approaches for defining functional roles of microbes in the human gut. Annu Rev Microbiol 2013;67:459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bisanz JE, Soto-Perez P, Lam KN, et al. Illuminating the microbiome’s dark matter: a functional genomic toolkit for the study of human gut Actinobacteria. bioRxiv 2018:304840. [Google Scholar]
- 169.Zuffa S, Schmid R, Bauermeister A, et al. microbeMASST: a taxonomically informed mass spectrometry search tool for microbial metabolomics data. Nat Microbiol 2024;9(2):336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schnell A, Littman DR, Kuchroo VK. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat Immunol 2023;24(1):19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kiner E, Willie E, Vijaykumar B, et al. Gut CD4(+) T cell phenotypes are a continuum molded by microbes, not by T(H) archetypes. Nat Immunol 2021;22(2):216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rao DA, Gurish MF, Marshall JL, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542(7639):110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Al Maini M, Adelowo F, Al Saleh J, et al. The global challenges and opportunities in the practice of rheumatology: white paper by the World Forum on Rheumatic and Musculoskeletal Diseases. Clin Rheumatol 2015;34(5):819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fries JF, Bruce B, Cella D. The promise of PROMIS: using item response theory to improve assessment of patient-reported outcomes. Clin Exp Rheumatol 2005;23(5 Suppl 39):S53–57. [PubMed] [Google Scholar]
- 175.van Riel PL, Renskers L. The Disease Activity Score (DAS) and the Disease Activity Score using 28 joint counts (DAS28) in the management of rheumatoid arthritis. Clin Exp Rheumatol 2016;34(5 Suppl 101):S40–S44. [PubMed] [Google Scholar]
- 176.Aletaha D, Nell VP, Stamm T, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7(4):R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alexander M, Turnbaugh PJ. Deconstructing mechanisms of diet-microbiome-immune interactions. Immunity 2020;53(2):264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lam KN, Spanogiannopoulos P, Soto-Perez P, et al. Phage-delivered CRISPR-Cas9 for strain-specific depletion and genomic deletions in the gut microbiome. Cell Rep 2021;37(5):109930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bhatt AP, Pellock SJ, Biernat KA, et al. Targeted inhibition of gut bacterial beta-glucuronidase activity enhances anticancer drug efficacy. Proc Natl Acad Sci U S A 2020;117(13):7374–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Woo AYM, Aguilar Ramos MA, Narayan R, et al. Targeting the human gut microbiome with small-molecule inhibitors. Nat Rev Chem 2023;7(5):319–339. [DOI] [PubMed] [Google Scholar]