

Abstract
The endoplasmic reticulum (ER) is an important organelle that controls the intracellular and extracellular environments. The ER is responsible for folding almost one‐third of the total protein population in the eukaryotic cell. Disruption of ER‐protein folding is associated with numerous human diseases, including metabolic disorders, neurodegenerative diseases, and cancer. During ER perturbations, the cells deploy various mechanisms to increase the ER‐folding capacity and reduce ER‐protein load by minimizing the number of substrates entering the ER to regain homeostasis. These mechanisms include signaling pathways, degradation mechanisms, and other processes that mediate the reflux of ER content to the cytosol. In this review, we will discuss the recent discoveries of five different ER quality control mechanisms, including the unfolded protein response (UPR), ER‐associated‐degradation (ERAD), pre‐emptive quality control, ER‐phagy and ER to cytosol signaling (ERCYS). We will discuss the roles of these processes in decreasing ER‐protein load and inter‐mechanism crosstalk.
Keywords: COPII, ERAD, ERCYS, ER‐Phagy, ER‐pQC, UPR
During ER stress, an accumulation of misfolded proteins within the ER disrupts proper ER‐protein folding. Thus, there is a need for ER quality control mechanisms to decrease ER‐protein load until the stress is resolved. In this review, we highlight five mechanisms that aim to decrease ER‐protein load and increase ER‐protein folding capacity, and we discuss the crosstalk between them. Graphical abstract created with Biorender.com.

Abbreviations
- AGR2
anterior gradient 2
- ARE
antioxidant response element–dependent
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- bZIP
basic‐leucine zipper motif
- COPII
Coat Protein Complex II
- DR5
Death Receptor 5
- EIF2α
Eukaryotic translation Initiation Factor 2‐alpha
- EIF2αK
Eukaryotic translation Initiation Factor 2‐alpha Kinase
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum‐associated degradation
- ERAS
endoplasmic reticulum‐associated RNA silencing
- ERCYS
endoplasmic reticulum to cytosol signaling
- ERGIC
ER‐Golgi intermediate compartment
- ERLAD
endoplasmic reticulum to lysosomes‐associated degradation
- ER‐pQC
endoplasmic reticulum pre‐emptive quality control
- GCN2
general control nonderepressible 2
- GRP78
glucose‐regulating protein 78
- HRI
heme‐regulated inhibitor
- IRE1
Inositol requiring enzyme‐1
- ISR
integrated stress response
- NRF2
nuclear factor erythroid 2–related factor 2
- PDI
protein disulfide isomerase
- PERK
protein kinase R (PKR)‐like endoplasmic reticulum kinase
- RIDD
Regulated IRE1‐Dependent mRNA Decay
- SRP
signal recognition particle
- UPR
unfolded protein response
- UPS
ubiquitin proteasome system
- XBP1
X‐Box binding protein
Introduction
Secreted and membrane proteins are targeted to the ER for proper folding and post‐translational modifications. Proteins in the secretory pathway possess a signal sequence in their N‐terminal that is recognized by the signal recognition particle (SRP); this sequence is highly variable and can reach up to 30 positively charged amino acids that contain a core of hydrophobic residues [1, 2, 3]. The polypeptides of the ER‐resident proteins are translated on ribosomes localized to the cytosolic face of the ER membrane. Once there, the nascent polypeptides are co‐translationally translocated to the ER lumen through a channel composed of several proteins, including the sec family called the translocon, and SRP [3, 4]. Upon translocation to the ER lumen, the signal peptide is cleaved off at −1 and −2 positions containing short‐side chains [1, 5, 6, 7]. The ER‐resident enzymes and chaperones ensure that the translocated polypeptides gain proper and productive protein folding. Correctly folded proteins are then moved to the Golgi apparatus to be sorted and directed to reach their final destination [3]. Proteins that fail to fold correctly are retrieved and degraded by the ER‐associated degradation machinery (ERAD) [8, 9].
A number of intrinsic and extrinsic factors affect ER homeostasis, accumulating misfolded proteins, and causing ER stress. Initially, cells attempt to correct this by inducing a signaling pathway, called the unfolded protein response (UPR), to regain homeostasis by increasing ER‐folding capacity. In addition, cells have developed several mechanisms to restore ER homeostasis by decreasing the ER‐protein load and increasing ER‐folding capacity. These work by (a) degrading mRNAs encoding for proteins destined to the ER lumen, (b) inhibiting global protein translation, (c) eliminating misfolded and damaged proteins from the ER to be degraded in the cytosol, (d) Autophagy of the ER subdomains that contain damaged and misfolded proteins, and (e) reflux of ER‐localized proteins to allow rapid restoration of the ER. These processes work together during perturbation of the ER homeostasis, with much inter‐mechanism crosstalk being reported.
Here, we summarize the different signaling pathways and ER‐protein quality control systems that aim to restore ER homeostasis by decreasing the ER‐protein load.
ER quality control mechanisms that reduce ER‐protein load
The unfolded protein response (UPR)
Cells are exposed to different physiological and chemical factors that affect protein folding in the ER, leading to ER stress. ER stress occurs when the ER‐protein folding demand exceeds capacity. To handle this situation, cells activate a conserved intracellular signaling pathway known as the unfolded protein response (UPR) [10, 11, 12]. In mammalian cells, the UPR is initiated by three ER‐resident sensors: inositol requiring enzyme 1 (IRE1), protein kinase R‐like ER kinase (PERK), and activating transcription factor 6 (ATF6). Initially, the UPR aims to restore ER homeostasis by increasing the ER‐protein folding capacity through inducing the expression of ER‐chaperones and enzymes and decreasing the number of client proteins in the ER. In addition, it was demonstrated that the UPR activation induces caspases, Death Receptor 5 (DR5), proapoptotic BH3‐only BCL2 family members BIM and NOXA, TXNIP, and other proapoptotic proteins to instigate cell death [13, 14, 15, 16, 17, 18, 19, 20, 21, 22].
Inositol requiring enzyme‐1 (IRE1)
Inositol requiring enzyme‐1 is the only conserved UPR arm from yeast to humans and is a major UPR component. In yeast cells, there is only one form of IRE1 that plays an adaptive role, while mammalian cells have two isoforms of IRE1 (IRE1α and IRE1β) [12, 15]. In mammalian cells, IRE1 signaling contributes to the progression of different human diseases, including inflammatory and neurodegenerative diseases, diabetes, and cancer [23, 24, 25, 26, 27, 28]. According to current dogma, IRE1, PERK and ATF6 activation depends on the ER HSP70 chaperone BiP (also known as Glucose‐regulating protein 78 ‐GRP78). BiP binds and inhibits IRE1, ATF6 and PERK in controlled and unstressed conditions. Unfolded and misfolded proteins in the ER compete with BiP binding, which dissociates BiP from the UPR sensors to cause their activation. Upon activation of IRE1, BiP levels increase, restoring homeostasis and negatively autoregulating activation [29]. Moreover, misfolded proteins can directly associate with IRE1—independently of BiP—n order to activate it [30, 31]. Once activated, IRE1 trans‐autophosphorylates at serine 73 and auto‐dimerizes, activating its RNAse domain in an unconventional cleavage of 26‐nt from the X‐Box binding protein‐1 (Xbp1) mRNA encoding for ER stress‐responsive b‐ZIP family activating transcription factor (ATF). Spliced Xbp1 (Xbp1s) is then translated and translocated to the nucleus to induce the expression of a large number of ER‐resident chaperones and glycosylation enzymes aimed to increase ER‐folding capacity [24, 32, 33, 34, 35]. On the other hand, Xbp1s increases ERAD activity by increasing the expression of HRD1, Herpud1 and other ERAD‐specific genes to trigger misfolded protein degradation by the proteasome. It is evident that the IRE1/xbp1s axis initially attempts to reduce ER‐protein load by decreasing the amount of unfolded/misfolded proteins and increasing ER‐folding capacity and ERAD activity, thereby eliminating misfolded proteins [36, 37] (Fig. 1A).
Fig. 1.
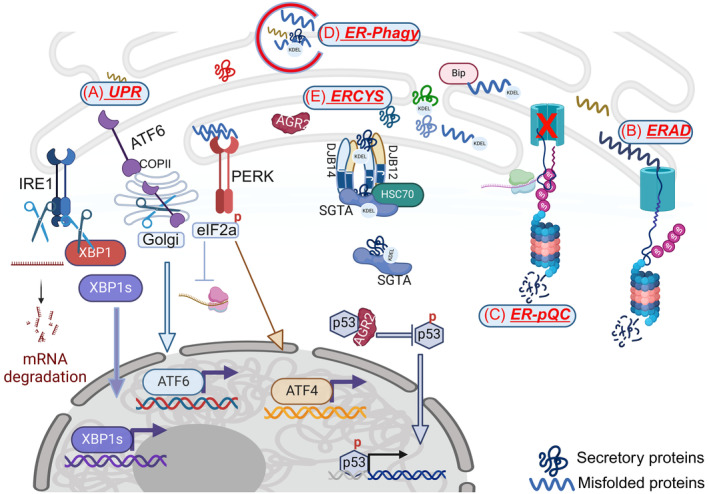
Schematic showing various mechanisms to relieve the ER of its content during stress. (A) The UPR: (a) RIDD results in degradation of mRNAs encoding for proteins in the secretory pathway and induces XBP1s target genes, (b) PERK inhibits global protein translation by phosphorylation of eIF2α and increases ATF4 target genes, and (c) ATF6 induces genes involved in increasing the ER‐protein folding capacity; (B) the ERAD eliminates misfolded proteins from the ER by directing their degradation to the proteasome. (C) The pre‐emptive quality control mechanism (ER‐pQC) reroutes synthesized proteins to the cytosol for proteasomal degradation. (D) ER‐phagy removes part of the damaged ER‐containing misfolded proteins (E) ERCYS induces reflux of soluble ER‐resident proteins to the cytosol, where they gain new function.
Another output of IRE1 RNAse domain activation is the degradation of a subset of mRNAs during ER stress [38]. This mechanism is called Regulated IRE1‐Dependent mRNA Decay, or RIDD, and is independent of Xbp1s activity. RIDD was discovered in drosophila and was shown to be conserved in many other organisms, including fission yeast and mammals. RIDD targets selective mRNAs encoding for proteins in the secretory pathway destined to enter the ER [39, 40]. Despite that, not all mRNAs encoding for ER‐targeted proteins are degraded by RIDD, and some meet the opposite fate, where the molecular chaperone BiP escapes RIDD and gets truncated and stabilized [40]. RIDD also affects gene expression and cell fate during ER stress by degrading selective miRNAs to stabilize the specific genes. This was shown by the degradation of miRNA17 that targets the pro‐inflammatory protein TXNIP [18, 19]. On the other hand, RIDD plays an adaptive response by decreasing the number of proteins entering the ER. Instead of increasing the ER‐folding capacity, RIDD reduces the number of substrate proteins that need to be folded by the ER‐localized chaperones during ER stress (Fig. 1A).
Another example is the degradation of the mRNA encoding for BLOS1 (biogenesis of lysosome‐related organelles 1 subunit 1), which protects against ER stress and the accumulation of ubiquitinated protein aggregates through repositioning of late endosomes (LEs)/lysosomes to the microtubule‐organizing center. Thus, RIDD activity towards BLOS1 protects against protein aggregation by promoting the late endosome/lysosome‐mediated microphagy.
Similarly, a new conserved mechanism called ERAS (ER‐associated RNA silencing), which promotes ER‐associated RNA turnover, has recently been discovered [41]. In ERAS, the Argonaute protein RDE‐1/AGO2 mediates ERAS and synergizes with ERAD to restore ER homeostasis and function [41].
Activating IRE1/XBP1 aims to restore homeostasis in different ways. XBP1s increases the capacity of ER‐protein folding to increase the folding and processing of the substrates and increases ERAD activity to eliminate misfolded proteins. IRE1‐RIDD activity results in the degradation of thousands of mRNAs encoding for proteins in the secretory pathways, thereby decreasing the amount of substrates in the ER. Altogether, IRE1 increases ER‐protein folding capacity and reduces the overall load in the ER lumen.
Protein kinase R‐like ER kinase (PERK)
PERK, or EIF2αK3 (Eukaryotic translation Initiation Factor 2‐alpha Kinase), is less conserved than IRE1 and is an essential ER homeostasis regulator. PERK is a type‐I transmembrane protein kinase located on the ER membrane with one lumenal and one cytosolic domain (the kinase domain). The ER lumenal domain of PERK is very similar to that of the IRE1 that binds BiP when inactive. The cytoplasmic kinase domain of PERK is very similar to other kinases known to phosphorylate the Eukaryotic translation Initiation Factor 2A (eIF2α). When PERK is active, it phosphorylates eIF2α to phos‐eIF2α at residue number 51 and inhibits global protein translation to decrease the overall substrates entering the ER [10, 16]. eIF2α is part of the integrated stress response (ISR) and can be phosphorylated by different kinases besides PERK. Osmotic and oxidative stresses activate the heme‐regulated inhibitor (HRI), also known as EIF2αK1, that phosphorylates eIF2α [42, 43, 44]. General control nonderepressible 2 (GCN2), also known as EIF2αK4, phosphorylates eIF2α in response to nutrient starvation and lack of amino acids [45, 46]. Finally, the protein kinase R (PKR), also known as EIF2αK2 and activated by viruses and other stimuli, triggers the ISR by phosphorylating eIF2α and inhibiting global protein translation [42, 47]. Thus, phosphorylation of eIF2α is an escape strategy that occurs during different cellular stresses, reducing global protein translation and allowing cells to resolve stress by decreasing proteotoxicity through a decrease in translation [48, 49] (Fig. 1A).
The inhibition of cap‐dependent protein translation spares cap‐independent translation of the activating transcription factor 4 (ATF4) [50, 51]. ATF4 transcriptionally activates many genes to help reduce protein load from the ER. These mainly encode ER‐chaperones and ERAD genes to reduce misfolded proteins. Moreover, ATF4 regulates a large number of genes encoding for antioxidant proteins, and genes regulating autophagy and thus help in eliminating damaged and misfolded proteins [52, 53].
Another PERK direct substrate is the nuclear factor erythroid 2–related factor 2 (NRF2). NRF2 controls the expression of numerous antioxidant response element–dependent (ARE) genes encoding for antioxidants and proteins to eliminate oxidatively damaged proteins. In resting cells, NRF2 binds a protein called Keap1 that prevents its nuclear translocation. Upon phosphorylation by PERK, NRF2 dissociates from Keap1, leading to its stability and nuclear accumulation [54, 55]. In summary, eIF2α phosphorylation by PERK and other kinases decreases the ER‐protein load by (a) inhibiting global protein translation and helping reduce the misfolded proteins in the ER, and (b) inducing cap‐independent translation of ATF4 to eliminate misfolded proteins from the ER, by either folding or directing the proteins to ERAD (Fig. 1A).
Activating transcription factor 6 (ATF6)
In mammalian cells, the ATF6 pathway consists of two isoforms, ATF6α and ATF6β. ATF6α is the one that carries most of the induction of ER stress response genes [17, 56]. ATF6 is a type II transmembrane protein activated similarly to IRE1 and PERK, but differing from those sensors in that it lacks a kinase domain. Moreover, ATF6 has a transcriptional activation domain and a DNA‐binding domain containing the basic‐leucine zipper motif (bZIP). Upon activation, ATF6 acts as a transcription factor that regulates the various genes needed for ER proteostasis [17]. During ER stress, BiP dissociates from ATF6, causing its activation. Activated ATF6 interacts with a protein complex called COPIII that is needed for vesicular protein trafficking from the ER to the Golgi [57]. In the Golgi, ATF6 is activated and proteolytically cleaved by two proteases, S1P and S2P, to release the cytosolic fragment of ATF6 and form ATF6f. ATF6f then translocates to the nucleus to regulate the transcription of genes involved in ER proteostasis and ER quality control, including ERAD and autophagy components [58, 59]. ATF6f also enhances ER‐folding capacity by inducing increases in the mRNA levels of XBP1 itself, which is then cleaved by IRE1. Moreover, many of the XBP1s target genes are regulated (and coregulated) by ATF6, including lipid biosynthesis and ER‐chaperones such as GRP78, GRP94, protein disulfide isomerases and others [59, 60]. Finally, ATF6 and XBP1 were also found to be involved in activating PERK [61]. These data, together, further emphasize that ATF6 activation is an important adaptive response for the reduction of the ER‐protein load by (a) increasing ER‐folding capacity, (b) increasing the ERAD and autophagy activities to reduce the number of misfolded proteins in the ER, and (c) crosstalk with the other two UPR branches, IRE1 and PERK, to further regain homeostasis (Fig. 1A).
ER‐associated degradation (ERAD)
A large number of intrinsic and extrinsic factors cause the accumulation of misfolded proteins in the ER, leading to ER stress. For instance, tumor cells are exposed to internal and environmental factors (hypoxia, oxidative stress, insufficient amino acids, glucose deprivation and lactic acidosis) that affect ER proteostasis, accumulation of misfolded proteins, and activation of the UPR. Misfolded and damaged proteins are retrieved from the ER to the cytosol, ubiquitylated by complex cytosolic machinery, and degraded by the 26S proteasome to ensure that only the correctly folded proteins are secreted. This process, which is part of the ubiquitin proteasome system, or the UPS, starts with the recognition of the misfolded proteins in the ER and ends with their destruction in the cytosol, termed ER‐associated degradation (ERAD) [62, 63, 64] (Fig. 1B).
Upon accumulation of misfolded proteins in the ER, one or more ERAD branches are activated to remove and degrade these misfolded proteins. The process starts by recognition of the misfolded proteins, either in the lumen of the ER or on the ER membrane, generated by a complex on the ER membrane consisting mainly of ubiquitin ligases [64]. Several E3 ubiquitin ligase complexes in mammalian cells specialize in recognizing different substrates [65]. Once the substrate is identified, the E3 ligase recruits the E2 ubiquitin‐conjugating enzyme that transfers ubiquitin to the substrates [66]. The next step is the transfer of the proteins from the ER to the cytosol, which requires energy. To this end, the ATPase complex p97/VCP (orthologue of the yeast CDC48) is recruited to pull the substrates to the cytosol for proteasomal degradation [67] (Fig. 1B).
The ERAD not only plays an important role in the ER‐associated degradation of misfolded proteins, but also controls the abundance of some of the UPR sensors, especially IRE1, a target of SEL1‐HRD1, and its degradation depends on p97 [68]. On the other hand, the UPR regulates a large number of genes involved in ERAD. This crosstalk between ERAD and the UPR is important for maintaining proper ER‐protein folding, clearing the ER of misfolded proteins, and regaining homeostasis. The ERAD is an essential component for removal of misfolded proteins from the ER, thereby decreasing the load on the ER‐folding machinery. In addition, the ERAD ensures the maintenance of ER proteostasis and regeneration of the ER lumen through its interaction with other signaling and quality control systems on the ER membrane.
ER pre‐emptive‐quality control (ER‐pQC)
The ER‐pQC is another adaptive UPS‐dependent mechanism that aims to decrease the burden of substrates entering the ER during stress. During ER stress, the ER‐pQC ensures a transient and rapid decrease in the translocation of protein in the secretory pathway to the ER [69]. During ER‐pQC, the attenuation in protein translocation is very specific and targets ER‐resident non‐chaperone proteins. This specificity is important because it targets substrate proteins that need folding, but spares chaperones that are required to fold proteins in the ER. The attenuated proteins are then captured by the Derlin family proteins that interact with the SRP, and rerouted to the UPS system [70]. This process requires the activity of Bag6 and VP97 and chaperones and cochaperones from the ER and the cytosol. DNAJC3, which is associated with the sec61 translocon, recruits cytosolic HSP70 chaperones to pull the proteins out of the ER for proteasomal degradation [69, 70, 71]. Moreover, other components of the E3‐ligases have been implicated in this process, including RNF149. Deletion of RNF149 causes a massive flux of substrates into the ER and increases the load on ER‐protein folding machinery [72]. Thus, during ER stress, it is important to decrease the translocation of more substrates to the ER in order to decrease ER load. On the other hand, it is important to degrade those proteins to prevent their aggregation and cytotoxicity. While ERAD targets misfolded proteins to reduce the E‐ protein load, the ER‐pQC targets the newly synthesized proteins and prevents translocation (Fig. 1C).
Autophagic degradation of the endoplasmic reticulum (ER‐Phagy)
Autophagy is an important housekeeping process needed to eliminate damaged proteins (aggregated, mutated, and misfolded) and organelles such as the mitochondria, ER, and peroxisomes [73]. The eliminated materials are encapsulated by vesicles, which then fuse to the lysosome for lysosomal degradation of their content. Thus, autophagy is an adaptive process that aims to clear misfolded and damaged proteins and organelles and degrade them, using the degradation as another reusable energy source.
As discussed above, misfolded proteins in the ER and proteins that fail to mature are routed to ERAD for degradation. Some of those are ERAD‐resistant proteins that the proteasome cannot degrade for several reasons, including aggregation status (too large to cross the ER membranes) or those that the ERAD machinery does not recognize [74]. Thus, there is a need for new mechanisms to identify and eliminate those proteins. ERLAD or ER to lysosome‐associated degradation is a mechanism that recognizes and targets those proteins for lysosomal degradation [75]. ER‐phagy (Eating the ER) is part of ERLAD and aims to remove damaged proteins from the ER for degradation in the cytosol as a means to regain ER homeostasis. ER‐phagy relies on the induction of specific receptors (regulatory proteins) called ER‐phagy receptors, that are induced during stress to facilitate the swelling process. These receptors are usually lumenal, ER‐bound and cytosolic proteins, recruited during ER‐phagy. At least 11 ER‐phagy receptors in mammalian cells have ATG Interacting motifs to interact with ATG8 family proteins under stress, and target specific ER proteins and ER subdomains for degradation [73]. In addition, it was shown that ubiquitination of the reticulon‐like protein FAM134B (one of the ER‐phagy receptors) increases its oligomerization and cluster size, and promotes binding to LC3B to stimulate ER‐phagy. These data further strengthen the crosstalk between ER‐phagy and the UPS in mammalian cells [76] (Fig. 1D).
Another dominant player in ER‐phagy is the Coat Protein Complex II (COPII). COPII is a complex of proteins that facilitate the anterograde transport of proteins from the ER to the Golgi or ER to the ER‐Golgi intermediate compartment (ERGIC). COPII was initially discovered in yeast and was shown to be essential for cell viability [77, 78, 79, 80]. In yeast, the COPII comprises a minimal machinery comprising Sar1p ‐small GTPase‐ and two protein complexes, the inner coat proteins Sec23/24p and the outer coat proteins Sec13/31p [78, 80]. Initially, Sar1p allows the recruitment of the sec23/24p complex, leading to the binding and recruitment of Sec13/31p complexes to induce coat polymerization and membrane budding and vesicle formation [79, 81]. In addition, this complex also consists of two other proteins, Sec12p and Sec16p. Sec16p is essential in yeast and is required for COPII‐dependent vesicle formation through interaction with several COPII subunits. It is believed to have a role as an organizer. At the same time, Sec12p functions as an ER transmembrane guanine nucleotide exchange factor of Sar1p [77, 82].
COPII is essential for the sequential export of materials from the ER to Golgi and plays an important role in autophagosome formation to direct ER cargos to selective ER‐phagy [83]. Lst1‐Sec23, which sorts proteins into vesicles to Golgi, also has an important role in ER‐phagy through association with Atg40. This process is required for ER‐phagy but not for bulk autophagy and aims to reduce misfolded proteins from the ER and reduce their abundance in the ER to regain homeostasis. Moreover, it was shown that ER stress induces the expression of Atg40 and increases its association with ER‐phagy. And on the other hand, deletion of Atg40 or Lst1 (atg40Δ and lst1Δ mutants.) results in ATZ aggregates accumulation. In mammalian cells, SEC24C, the mammalian homolog, is also required for the delivery of FAM134B and RTN3 to lysosomes for degradation. [84]. Moreover, it was recently shown that the misfolded α‐1‐antitrypsin Z (ATZ) mutants are cleared of the ER to lysosome by COPII. The COPII subunit SEC24C and p24 family proteins, including TMP21 and TMED9, promote ATZ clearance (but not the WT) through direct interaction with FAM134B [81].
Thus, the proper activity of the COPII decreases the number of ER proteins from the ER by mediating anterograde ER‐Golgi trafficking and directing misfolded proteins for lysosomal degradation through ER‐phagy.
ER to cytosol signaling (ERCYS)
Numerous studies have reported that ER‐resident proteins could be found in non‐ER locations, including the plasma membrane, nucleus and cytosol [85, 86, 87, 88, 89]. BiP, which primarily functions as a master chaperone within the endoplasmic reticulum, was also reported to be present in various non‐ER locations, including the cytosol, nucleus cell surface, and extracellular [87, 90, 91, 92, 93, 94]. Since the year 1986 [89], the PDI family of proteins has been extensively reported to localize to the cytosol. However, in recent years, more and more pieces of evidence show that PDIA3 not only localizes to the cytosol, but has also been reported to interact with cytosolic proteins, including HIF1α, STAT3, NLRP3, and caspase3‐7 [95, 96, 97, 98, 99]. Proteomics also shows that, in apoptotic cells, PDI colocalizes with caspase‐7 and is cleaved by caspase 3–7 to gain new antiapoptotic actions [95]. Another member of the PDI family that was shown to be localized to the cytosol is AGR2, a known inhibitor of p53, that stabilizes the p65 protein and contributes to cell metastasis [100]. As with other members of the PDI family, high expression of AGR2 was also shown to be associated with inactivation/activation of STAT3, p53, and p38 in the cytosol [101, 102, 103, 104, 105].
Despite all the evidence of non‐ER localizations of ER proteins, the mechanism and complete identity of those proteins have not yet been discovered. Recently, it was shown that, in yeast experiencing ER stress conditions, a subset of the ER proteins are rerouted to the cytosol [106, 107]. This mechanism is chaperone‐mediated, depends on the activity of ER and cytosolic chaperones, and proceeds more vigorously when the ERAD is crippled. Moreover, using a photo‐converted version of the fluorescent mEOS protein indicates that the refluxed proteins are rerouted to the cytosol in their folded and functional state [106, 107]. This suggests that the reflux of the ER proteins to the cytosol is independent of the ERAD, which targets misfolded proteins and may work as a parallel mechanism during stress. Such a mechanism can benefit yeast cells by debulking to reduce the ER of its content (Fig. 1E).
The ER stress‐induced protein reflux mechanism is also conserved in higher eukaryotes [108, 109]. In mammalian cells, this mechanism targets a wide range of proteins in different cell lines experiencing ER stress. In isolated tumors, from Glioblastoma multiform (GBM) patients and from GBM mouse models, this mechanism is constitutively active and reroutes a large number of ER proteins to the cytosol. In tumors, the reflux of ER proteins plays an important pathophysiological role by relieving the ER of its content and gain‐of‐ function of the refluxed proteins in the cytosol. This was demonstrated by the inhibition of p53 protein by the refluxed AGR2 [109]. ERCYS (ER to CYtosol Signaling) may constitute a novel mechanism conserved from yeast to humans to relieve the ER of its content. Other ER‐refluxed proteins may gain new functions in the cytosol, as demonstrated earlier by the interaction between PDI proteins and other cytosolic proteins such as STAT3, p53, and caspase3‐7 [95, 96, 97, 98]. Cancer cell dependency on ERCYS makes it a promising target for anticancer drugs (Fig. 1E).
In yeast, ERCYS depends on the activity of the DNAJ protein HLJ1, an HSP40 cochaperone localized to the ER membrane. Moreover, a subset of the yeast cytosolic HSP70 machinery was also needed [106, 107]. In mammalian cells, HLJ1 has five putative orthologues, including DNAJB12, DNAJB14, DNAJC14, DNAJC18 and DNAJC30 [110, 111, 112, 113, 114, 115]. Moreover, ERCYS requires that the HSC70‐SGTA machinery from the cytosol interact with the ER membrane‐localized HSP40 cochaperones [115]. DNAJB12 and DNAJB14 are both required for this mechanism, while DNAJB12 is also sufficient to promote ERCYS by overexpression and to inhibit wt‐p53 under genotoxic stress [115]. The Reflux of ER proteins is similar to the escape of polyomaviruses from the ER to the cytosol during infection [116]. This activity depends on BiP, DNAJB12, DNAJB14, HSC70, SGTA and other ER and cytosolic components [116], suggesting that viruses have hijacked this conserved mechanism – from yeast to human – to escape the ER and invade the host cell (Fig. 1E).
Despite this indication, the cytosolic function of those proteins that are usually active in an oxidizing environment in the ER is not fully understood. This is important, especially for the PDI family of proteins that rely on their thioredoxin domains and active cysteine to carry out many redox‐sensitive activities in the ER. Those activities are believed to be crippled in the highly reduced environment of the cytosol, that contains a millimolar range of the reductant Glutathione (GSH).
We should note here that another mechanism was also reported to reflux protein from the ER to the cytosol through permeabilization of the ER membrane. This mechanism depends on the activity of IRE1 and is mediated by Bax and Bak. In the permeabilization model, Bnip3 (BH3‐domain‐containing protein) triggers the oligomerization of Bax and Bak on the ER membrane to induce permeabilization [87]. Unlike ERCYS, the permeabilization process is chaperone‐independent. It occurs during high levels of ER stress regimes that induce apoptosis, while ERCYS proceeds at low to mild ER stresses in the adaptive range and seems transient.
Finally, the dual functionality of protein in different compartments is not unique to the ER. In unstressed cells, cytochrome c is localized to the mitochondrial intermembrane space (IMS), which is an electron shuttle. Upon apoptotic stimuli, cytochrome c leaves the IMS and enters the cytosol to activate caspase‐9 and caspase‐3 by forming a complex with Apaf‐1 (apoptosome) [117, 118]. Another example is the mitochondrial protein HSPDI (HSP60), which plays an antiapoptotic function in the mitochondria while playing both anti‐ and proapoptotic functions in the cytosol [119, 120, 121].
These processes are highly interconnected and aim to reflux many ER proteins to the cytosol during stress. ERCYS targets correctly folded proteins that reside in the ER during stress to reduce the load on the ER. In summary, ERCYS increases cell fitness by maintaining proper secretory functions and cytosolic gain‐of‐function of the refluxed proteins and by decreasing the amount of proteins inside the ER.
The interconnection between the ER disposal mechanisms
Eukaryotic cells developed several ER disposal mechanisms that are activated during certain conditions and aim to regain ER homeostasis. Although each mechanism seems to act differently and more specialized than others, those mechanisms have many interconnection nodes. As discussed above, the UPR reduces ER‐protein load by RIDD and inhibits global protein translation. It also regulates genes and pathways to increase the ER‐folding capacity and remove misfolded proteins from the ER. In addition, the UPR regulates the UPS activity, including the pre‐emptive quality control and ERAD systems. The UPR regulates genes involved in ER‐phagy, ERAD, and the pre‐emptive quality control system. IRE1‐XBP1 regulates the expression of HRD1, Herpud1, and other ERAD‐specific genes to trigger misfolded protein degradation by the proteasome. ATF6 on the other hand regulates, Derlin‐3, Herp, SEL1L, HRD1 and EDEM1 [59]. Moreover, PERK inhibition disrupts ER‐Golgi anterograde and impairs ERAD by preventing ER to cytosol retro‐translocation by increasing the levels of Erp72 [122]. Interestingly, UPS can regulate the UPR as well. Under normal and unstressed conditions, IRE1 is a substrate of the Sel1L‐Hrd1 ERAD complex in a mechanism that requires BiP and the OS9 (Lectin protein) [68]. In addition, ATF6 is an ERAD substrate and is regulated by the Sel1L‐Hrd1 ERAD complex [123].
In yeast, IRE1‐HAC1 stimulates COPII‐vesicle formation and suppresses the sec12‐4 mutants that are defective in vesicle budding from the ER [124, 125]. PERK/ATF4 also regulates COPII gene expression and facilitates ATF6 transport to the Golgi. Those genes include GTPase activating proteins, COPII vesicle coat proteins, and genes associated with vesicle formation and fusion [126]. These later results emphasize a highly complex interconnection between the UPR that regulates the COPII component and, at the same time, the ATF6 branch of the UPR is regulated by a proper activity of COPII [57].
ER‐phagy receptors are induced by various stressors, including ER stress, hypoxia, and nutrient starvation [127, 128]. These stressors cause the UPR induction to induce gene expression and regain homeostasis. Recent articles have shown that the UPR signaling pathway regulates the ER‐phagy genes. For instance, the induction of CCPG1 gene encoding for cell‐cycle progression gene1 proteins is an important component of ER‐phagy that interacts with core autophagy proteins, depending on the induction of the UPR. In Schizosaccharomyces pombe, IRE1 promotes ER‐phagy by upregulating the expression of Epr1 – soluble autophagy receptor [129]. Moreover, BiP forms a complex with ER‐phagy receptor FAM134B, and loss of FAM134B increases ER stress and reduces cell proliferation [127]. This UPR/ER‐phagy node further strengthens the crosstalk between the different quality control systems to clear the ER of its content during stress and to regain homeostasis by decreasing ER‐protein load.
Yet the role of the UPR in regulating ERCYS is not established, and future studies are required to unravel this node between the unfolded protein response and the exit of correctly folded proteins from the ER.
Conclusions and future perspectives
During evolution, numerous mechanisms have evolved to reduce stress within the endoplasmic reticulum. These, such as the UPR, can act as signaling pathways to induce the expression of different factors (enzymes, chaperones, transcription factors, and others) to increase ER‐folding capacity. Moreover, some of those signaling pathways induce the expression of genes that regulate the elimination of misfolded and unfolded proteins from the ER, including ERAD and ER‐phagy. Hence, protein folding capacity is increased, and misfolded proteins decreased, in the ER. IRE1 and PERK can also actively reduce the amount of substrates translocating to the ER by degrading mRNA encoding for ER proteins or inhibiting global protein translation.
On the other hand, ERAD, ER‐Phagy, ER‐pQC, and ERCYS help reduce the ER load by targeting the pre‐existing or newly synthesized proteins. ERAD targets misfolded proteins; ER‐phagy targets misfolded and damaged proteins that are ERAD‐resistant to lysosomal degradation; and ER‐pQC marks stalled polypeptides on the ribosomes and targets them for degradation.
ERCYS is a novel mechanism recently discovered in yeast, and conserved in mammalian cells and isolated human tumors. ERCYS differs from ERAD, ER‐phagy, and pre‐QC in that it neither targets misfolded proteins nor causes their degradation by the UPS. ERCYS consists of a transient and quick debulking of the ER to relieve it from its content and allow cells to regain homeostasis.
Some of the mechanisms discussed above have been extensively studied. However, we still lack substantial information on the other pathways, and have only recently begun to understand their contribution to ER homeostasis and molecular mechanism of action. There is insufficient understanding of those molecular mechanisms and their role in human diseases.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
SD and GT wrote the sections on the UPR, ERCYS, pre‐QC and contributed to other sections. AI, planned the concept and the design of the review, wrote the section on ER‐Phagy, made the figure with SD and GT and wrote the conclusion. SB, wrote the section on COPII and the Interconnection between the different pathways.
Acknowledgements
This work was funded by grants from the Israeli Science Foundation (ISF‐977/21), The Israel Cancer Research Fund (ICRF). Salam Dabsan is funded by the Israel Council for Higher Education (VATAT). Figures and graphical abstract created with Biorender.com.
Salam Dabsan and Gal Twito contributed equally to this article.
References
- 1. Walter P & Blobel G (1981) Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence‐dependent and site‐specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walter P, Ibrahimi I & Blobel G (1981) Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in‐vitro‐assembled polysomes synthesizing secretory protein. J Cell Biol 91, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rapoport TA (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669. [DOI] [PubMed] [Google Scholar]
- 4. Deshaies RJ, Sanders SL, Feldheim DA & Schekman R (1991) Assembly of yeast sec proteins involved in translocation into the endoplasmic reticulum into a membrane‐bound multisubunit complex. Nature 349, 806–808. [DOI] [PubMed] [Google Scholar]
- 5. Wickner W (1979) The assembly of proteins into biological membranes: the membrane trigger hypothesis. Annu Rev Biochem 48, 23–45. [DOI] [PubMed] [Google Scholar]
- 6. Martoglio B & Dobberstein B (1998) Signal sequences: more than just greasy peptides. Trends Cell Biol 8, 410–415. [DOI] [PubMed] [Google Scholar]
- 7. von Heijne G (1985) Signal sequences. The limits of variation. J Mol Biol 184, 99–105. [DOI] [PubMed] [Google Scholar]
- 8. Bonifacino JS, Suzuki CK, Lippincott‐Schwartz J, Weissman AM & Klausner RD (1989) Pre‐Golgi degradation of newly synthesized T‐cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol 109, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lippincott‐Schwartz J, Bonifacino JS, Yuan LC & Klausner RD (1988) Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell 54, 209–220. [DOI] [PubMed] [Google Scholar]
- 10. Bertolotti A, Zhang Y, Hendershot LM, Harding HP & Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded‐protein response. Nat Cell Biol 2, 326–332. [DOI] [PubMed] [Google Scholar]
- 11. Sidrauski C & Walter P (1997) The transmembrane kinase Ire1p is a site‐specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. [DOI] [PubMed] [Google Scholar]
- 12. Mori K, Ma W, Gething MJ & Sambrook J (1993) A transmembrane protein with a cdc2+/CDC28‐related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. [DOI] [PubMed] [Google Scholar]
- 13. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D & Kaufman RJ (2006) Adaptation to ER stress is mediated by differential stabilities of pro‐survival and pro‐apoptotic mRNAs and proteins. PLoS Biol 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kozutsumi Y, Segal M, Normington K, Gething MJ & Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose‐regulated proteins. Nature 332, 462–464. [DOI] [PubMed] [Google Scholar]
- 15. Cox JS, Shamu CE & Walter P (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- 16. Harding HP, Zhang Y & Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397, 271–274. [DOI] [PubMed] [Google Scholar]
- 17. Haze K, Yoshida H, Yanagi H, Yura T & Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10, 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M et al. (2012) IRE1alpha induces thioredoxin‐interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16, 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H et al. (2014) Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fribley AM, Evenchik B, Zeng Q, Park BK, Guan JY, Zhang H, Hale TJ, Soengas MS, Kaufman RJ & Wang CY (2006) Proteasome inhibitor PS‐341 induces apoptosis in cisplatin‐resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem 281, 31440–31447. [DOI] [PubMed] [Google Scholar]
- 21. Urano F, Bertolotti A & Ron D (2000) IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci 113(Pt 21), 3697–3702. [DOI] [PubMed] [Google Scholar]
- 22. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL & Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12, 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C & Chevet E (2017) Endoplasmic reticulum proteostasis in glioblastoma‐from molecular mechanisms to therapeutic perspectives. Sci Signal 10, eaal2323. [DOI] [PubMed] [Google Scholar]
- 24. Obacz J, Avril T, Rubio‐Patino C, Bossowski JP, Igbaria A, Ricci JE & Chevet E (2019) Regulation of tumor‐stroma interactions by the unfolded protein response. FEBS J 286, 279–296. [DOI] [PubMed] [Google Scholar]
- 25. Chevet E, Hetz C & Samali A (2015) Endoplasmic reticulum stress‐activated cell reprogramming in oncogenesis. Cancer Discov 5, 586–597. [DOI] [PubMed] [Google Scholar]
- 26. Cabral‐Miranda F, Tamburini G, Martinez G, Ardiles AO, Medinas DB, Gerakis Y, Hung MD, Vidal R, Fuentealba M, Miedema T et al. (2022) Unfolded protein response IRE1/XBP1 signaling is required for healthy mammalian brain aging. EMBO J 41, e111952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee AH, Heidtman K, Hotamisligil GS & Glimcher LH (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci USA 108, 8885–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martinez‐Turtos A, Paul R, Grima‐Reyes M, Issaoui H, Krug A, Mhaidly R, Bossowski JP, Chiche J, Marchetti S, Verhoeyen E et al. (2022) IRE1alpha overexpression in malignant cells limits tumor progression by inducing an anti‐cancer immune response. Onco Targets Ther 11, 2116844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okamura K, Kimata Y, Higashio H, Tsuru A & Kohno K (2000) Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun 279, 445–450. [DOI] [PubMed] [Google Scholar]
- 30. Gardner BM & Walter P (2011) Unfolded proteins are Ire1‐activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karagoz GE, Acosta‐Alvear D, Nguyen HT, Lee CP, Chu F & Walter P (2017) An unfolded protein‐induced conformational switch activates mammalian IRE1. elife 6, e30700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clauss IM, Chu M, Zhao JL & Glimcher LH (1996) The basic domain/leucine zipper protein hXBP‐1 preferentially binds to and transactivates CRE‐like sequences containing an ACGT core. Nucleic Acids Res 24, 1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG & Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- 34. Yoshida H, Matsui T, Yamamoto A, Okada T & Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- 35. Cox JS & Walter P (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. [DOI] [PubMed] [Google Scholar]
- 36. Chiang WC, Kroeger H, Sakami S, Messah C, Yasumura D, Matthes MT, Coppinger JA, Palczewski K, LaVail MM & Lin JH (2015) Robust endoplasmic reticulum‐associated degradation of rhodopsin precedes retinal degeneration. Mol Neurobiol 52, 679–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chiang WC, Messah C & Lin JH (2012) IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol Biol Cell 23, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hollien J & Weissman JS (2006) Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science 313, 104–107. [DOI] [PubMed] [Google Scholar]
- 39. Hollien J, Lin JH, Li H, Stevens N, Walter P & Weissman JS (2009) Regulated IRE1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimmig P, Diaz M, Zheng J, Williams CC, Lang A, Aragon T, Li H & Walter P (2012) The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. elife 1, e00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Efstathiou S, Ottens F, Schutter LS, Ravanelli S, Charmpilas N, Gutschmidt A, Le Pen J, Gehring NH, Miska EA, Boucas J et al. (2022) ER‐associated RNA silencing promotes ER quality control. Nat Cell Biol 24, 1714–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR & Hovanessian AG (1990) Molecular cloning and characterization of the human double‐stranded RNA‐activated protein kinase induced by interferon. Cell 62, 379–390. [DOI] [PubMed] [Google Scholar]
- 43. Lu L, Han AP & Chen JJ (2001) Translation initiation control by heme‐regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21, 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen JJ, Throop MS, Gehrke L, Kuo I, Pal JK, Brodsky M & London IM (1991) Cloning of the cDNA of the heme‐regulated eukaryotic initiation factor 2 alpha (eIF‐2 alpha) kinase of rabbit reticulocytes: homology to yeast GCN2 protein kinase and human double‐stranded‐RNA‐dependent eIF‐2 alpha kinase. Proc Natl Acad Sci USA 88, 7729–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF & Hinnebusch AG (1992) Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene‐specific translational control of GCN4 in yeast. Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- 46. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR et al. (2002) The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22, 6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levin DH, Petryshyn R & London IM (1980) Characterization of double‐stranded‐RNA‐activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF‐2 alpha) in reticulocyte lysates. Proc Natl Acad Sci USA 77, 832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Costa‐Mattioli M & Walter P (2020) The integrated stress response: from mechanism to disease. Science 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szaruga M, Janssen DA, de Miguel C, Hodgson G, Fatalska A, Pitera AP, Andreeva A & Bertolotti A (2023) Activation of the integrated stress response by inhibitors of its kinases. Nat Commun 14, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu PD, Harding HP & Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vattem KM & Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG & Bell JC (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24, 7469–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P & Bruhat A (2013) The eIF2alpha/ATF4 pathway is essential for stress‐induced autophagy gene expression. Nucleic Acids Res 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ & Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol 23, 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Suzuki T, Takahashi J & Yamamoto M (2023) Molecular basis of the KEAP1‐NRF2 signaling pathway. Mol Cells 46, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M & Mori K (2001) Endoplasmic reticulum stress‐induced formation of transcription factor complex ERSF including NF‐Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol Cell Biol 21, 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schindler AJ & Schekman R (2009) In vitro reconstitution of ER‐stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci USA 106, 17775–17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gade P, Ramachandran G, Maachani UB, Rizzo MA, Okada T, Prywes R, Cross AS, Mori K & Kalvakolanu DV (2012) An IFN‐gamma‐stimulated ATF6‐C/EBP‐beta‐signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc Natl Acad Sci USA 109, 10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A & Mori K (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33, 75–89. [DOI] [PubMed] [Google Scholar]
- 60. Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ et al. (2009) ATF6alpha induces XBP1‐independent expansion of the endoplasmic reticulum. J Cell Sci 122, 1626–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spaan CN, Smit WL, van Lidth de Jeude JF, Meijer BJ, Muncan V, van den Brink GR & Heijmans J (2019) Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis 10, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS & Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER‐associated degradation. Cell 101, 249–258. [DOI] [PubMed] [Google Scholar]
- 63. Friedlander R, Jarosch E, Urban J, Volkwein C & Sommer T (2000) A regulatory link between ER‐associated protein degradation and the unfolded‐protein response. Nat Cell Biol 2, 379–384. [DOI] [PubMed] [Google Scholar]
- 64. Hiller MM, Finger A, Schweiger M & Wolf DH (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin‐proteasome pathway. Science 273, 1725–1728. [DOI] [PubMed] [Google Scholar]
- 65. Christianson JC, Jarosch E & Sommer T (2023) Mechanisms of substrate processing during ER‐associated protein degradation. Nat Rev Mol Cell Biol 24, 777–796. [DOI] [PubMed] [Google Scholar]
- 66. Ciechanover A, Finley D & Varshavsky A (1984) Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 37, 57–66. [DOI] [PubMed] [Google Scholar]
- 67. Bodnar N & Rapoport T (2017) Toward an understanding of the Cdc48/p97 ATPase. F1000Res 6, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, Ma H, Inoue T, Gao B, Kim H et al. (2015) IRE1alpha is an endogenous substrate of endoplasmic‐reticulum‐associated degradation. Nat Cell Biol 17, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J & Hegde RS (2006) Substrate‐specific translocational attenuation during ER stress defines a pre‐emptive quality control pathway. Cell 127, 999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kadowaki H, Nagai A, Maruyama T, Takami Y, Satrimafitrah P, Kato H, Honda A, Hatta T, Natsume T, Sato T et al. (2015) Pre‐emptive quality control protects the ER from protein overload via the proximity of ERAD components and SRP. Cell Rep 13, 944–956. [DOI] [PubMed] [Google Scholar]
- 71. Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL et al. (2006) Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 126, 727–739. [DOI] [PubMed] [Google Scholar]
- 72. Legesse A, Kaushansky N, Braunstein I, Saad H, Lederkremer G, Navon A & Stanhill A (2023) The role of RNF149 in the pre‐emptive quality control substrate ubiquitination. Commun Biol 6, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Reggiori F & Molinari M (2022) ER‐phagy: mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum. Physiol Rev 102, 1393–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P & Tsai B (2019) Cells deploy a two‐pronged strategy to rectify misfolded proinsulin aggregates. Mol Cell 75, 442–456.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bernales S, McDonald KL & Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4, e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gonzalez A, Covarrubias‐Pinto A, Bhaskara RM, Glogger M, Kuncha SK, Xavier A, Seemann E, Misra M, Hoffmann ME, Brauning B et al. (2023) Ubiquitination regulates ER‐phagy and remodelling of endoplasmic reticulum. Nature 618, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Salama NR, Chuang JS & Schekman RW (1997) Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol Biol Cell 8, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schekman RW (1994) Regulation of membrane traffic in the secretory pathway. Harvey Lect 90, 41–57. [PubMed] [Google Scholar]
- 79. Rexach MF, Latterich M & Schekman RW (1994) Characteristics of endoplasmic reticulum‐derived transport vesicles. J Cell Biol 126, 1133–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M & Schekman R (1994) COPII: a membrane coat formed by sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907. [DOI] [PubMed] [Google Scholar]
- 81. Roberts BS, Mitra D, Abishek S, Beher R & Satpute‐Krishnan P (2024) The p24‐family and COPII subunit SEC24C facilitate the clearance of alpha1‐antitrypsin Z from the endoplasmic reticulum to lysosomes. Mol Biol Cell 35, ar45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Watson P, Townley AK, Koka P, Palmer KJ & Stephens DJ (2006) Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 7, 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cui Y, Parashar S & Ferro‐Novick S (2020) A new role for a COPII cargo adaptor in autophagy. Autophagy 16, 376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cui Y, Parashar S, Zahoor M, Needham PG, Mari M, Zhu M, Chen S, Ho HC, Reggiori F, Farhan H et al. (2019) A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 365, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Akagi S, Yamamoto A, Yoshimori T, Masaki R, Ogawa R & Tashiro Y (1988) Distribution of protein disulfide isomerase in rat hepatocytes. J Histochem Cytochem 36, 1533–1542. [DOI] [PubMed] [Google Scholar]
- 86. Akagi S, Yamamoto A, Yoshimori T, Masaki R, Ogawa R & Tashiro Y (1988) Localization of protein disulfide isomerase on plasma membranes of rat exocrine pancreatic cells. J Histochem Cytochem 36, 1069–1074. [DOI] [PubMed] [Google Scholar]
- 87. Kanekura K, Ma X, Murphy JT, Zhu LJ, Diwan A & Urano F (2015) IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci Signal 8, ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Turano C, Gaucci E, Grillo C & Chichiarelli S (2011) ERp57/GRP58: a protein with multiple functions. Cell Mol Biol Lett 16, 539–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lewis MJ, Mazzarella RA & Green M (1986) Structure and assembly of the endoplasmic reticulum: biosynthesis and intracellular sorting of ERp61, ERp59, and ERp49, three protein components of murine endoplasmic reticulum. Arch Biochem Biophys 245, 389–403. [DOI] [PubMed] [Google Scholar]
- 90. Zhang Y, Liu R, Ni M, Gill P & Lee AS (2010) Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem 285, 15065–15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu Z, Liu G, Ha DP, Wang J, Xiong M & Lee AS (2023) ER chaperone GRP78/BiP translocates to the nucleus under stress and acts as a transcriptional regulator. Proc Natl Acad Sci USA 120, e2303448120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T & Helenius A (2011) BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol 13, 1305–1314. [DOI] [PubMed] [Google Scholar]
- 93. Shim SM, Choi HR, Sung KW, Lee YJ, Kim ST, Kim D, Mun SR, Hwang J, Cha‐Molstad H, Ciechanover A et al. (2018) The endoplasmic reticulum‐residing chaperone BiP is short‐lived and metabolized through N‐terminal arginylation. Sci Signal 11, eaan0630. [DOI] [PubMed] [Google Scholar]
- 94. Duriez M, Rossignol JM & Sitterlin D (2008) The hepatitis B virus precore protein is retrotransported from endoplasmic reticulum (ER) to cytosol through the ER‐associated degradation pathway. J Biol Chem 283, 32352–32360. [DOI] [PubMed] [Google Scholar]
- 95. Na KS, Park BC, Jang M, Cho S, Lee DH, Kang S, Lee CK, Bae KH & Park SG (2007) Protein disulfide isomerase is cleaved by caspase‐3 and ‐7 during apoptosis. Mol Cells 24, 261–267. [PubMed] [Google Scholar]
- 96. Kobayashi Y, Oguro A, Hirata Y & Imaoka S (2021) The regulation of hypoxia‐inducible Factor‐1 (HIF‐1alpha) expression by protein disulfide isomerase (PDI). PLoS One 16, e0246531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Eufemi M, Coppari S, Altieri F, Grillo C, Ferraro A & Turano C (2004) ERp57 is present in STAT3‐DNA complexes. Biochem Biophys Res Commun 323, 1306–1312. [DOI] [PubMed] [Google Scholar]
- 98. Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD & Sehgal PB (2002) Association of the chaperone glucose‐regulated protein 58 (GRP58/ER‐60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interf Cytokine Res 22, 555–563. [DOI] [PubMed] [Google Scholar]
- 99. Rosarda JD, Stanton CR, Chen EB, Bollong MJ & Wiseman RL (2023) Pharmacologic targeting of PDIA1 inhibits NLRP3 Inflammasome assembly and activation. Isr J Chem e202300125. doi: 10.1002/ijch.202300125 [DOI] [Google Scholar]
- 100. Jia M, Guo Y, Zhu D, Zhang N, Li L, Jiang J, Dong Y, Xu Q, Zhang X, Wang M et al. (2018) Pro‐metastatic activity of AGR2 interrupts angiogenesis target bevacizumab efficiency via direct interaction with VEGFA and activation of NF‐kappaB pathway. Biochim Biophys Acta Mol basis Dis 1864, 1622–1633. [DOI] [PubMed] [Google Scholar]
- 101. Hrstka R, Bouchalova P, Michalova E, Matoulkova E, Muller P, Coates PJ & Vojtesek B (2016) AGR2 oncoprotein inhibits p38 MAPK and p53 activation through a DUSP10‐mediated regulatory pathway. Mol Oncol 10, 652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang Z, Li H, Deng Y, Schuck K, Raulefs S, Maeritz N, Yu Y, Hechler T, Pahl A, Fernandez‐Saiz V et al. (2021) AGR2‐dependent nuclear import of RNA polymerase II constitutes a specific target of pancreatic ductal adenocarcinoma in the context of wild‐type p53. Gastroenterology 161, 1601–1614.e23. [DOI] [PubMed] [Google Scholar]
- 103. Takabatake K, Konishi H, Arita T, Kataoka S, Shibamoto J, Furuke H, Takaki W, Shoda K, Shimizu H, Yamamoto Y et al. (2021) Anterior gradient 2 regulates cancer progression in TP53‐wild‐type esophageal squamous cell carcinoma. Oncol Rep 46, 260. [DOI] [PubMed] [Google Scholar]
- 104. Chevet E, Fessart D, Delom F, Mulot A, Vojtesek B, Hrstka R, Murray E, Gray T & Hupp T (2013) Emerging roles for the pro‐oncogenic anterior gradient‐2 in cancer development. Oncogene 32, 2499–2509. [DOI] [PubMed] [Google Scholar]
- 105. Bouchalova P, Sommerova L, Potesil D, Martisova A, Lapcik P, Koci V, Scherl A, Vonka P, Planas‐Iglesias J, Chevet E et al. (2022) Characterization of the AGR2 Interactome uncovers new players of protein disulfide isomerase network in cancer cells. Mol Cell Proteomics 21, 100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Igbaria A, Merksamer PI, Trusina A, Tilahun F, Johnson JR, Brandman O, Krogan NJ, Weissman JS & Papa FR (2019) Chaperone‐mediated reflux of secretory proteins to the cytosol during endoplasmic reticulum stress. Proc Natl Acad Sci USA 116, 11291–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lajoie P & Snapp EL (2019) Size‐dependent secretory protein reflux into the cytosol in association with acute endoplasmic reticulum stress. bioRxiv 573428. 10.1101/573428 [PREPRINT] [DOI] [PMC free article] [PubMed]
- 108. Dreno L, Tiraboschi C, Lacoste S, Gomez SC, Garrido MF, Bertrand M, Tannoury M, Jacquet E, Nhiri N, Loriot Y et al. (2022) Abstract 400: functional, structural and binding studies of the atypical ER‐resident protein FKBP7, a potential target in chemoresistant prostate cancer. Cancer Res 82, 400. [Google Scholar]
- 109. Sicari D, Centonze FG, Pineau R, Le Reste PJ, Negroni L, Chat S, Mohtar MA, Thomas D, Gillet R, Hupp T et al. (2021) Reflux of endoplasmic reticulum proteins to the cytosol inactivates tumor suppressors. EMBO Rep 22, e51412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Piette BL, Alerasool N, Lin ZY, Lacoste J, Lam MHY, Qian WW, Tran S, Larsen B, Campos E, Peng J et al. (2021) Comprehensive interactome profiling of the human Hsp70 network highlights functional differentiation of J domains. Mol Cell 81, 2549–2565.e8. [DOI] [PubMed] [Google Scholar]
- 111. Malinverni D, Zamuner S, Rebeaud ME, Barducci A, Nillegoda NB & De Los Rios P (2023) Data‐driven large‐scale genomic analysis reveals an intricate phylogenetic and functional landscape in J‐domain proteins. Proc Natl Acad Sci USA 120, e2218217120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yamamoto YH, Kimura T, Momohara S, Takeuchi M, Tani T, Kimata Y, Kadokura H & Kohno K (2010) A novel ER J‐protein DNAJB12 accelerates ER‐associated degradation of membrane proteins including CFTR. Cell Struct Funct 35, 107–116. [DOI] [PubMed] [Google Scholar]
- 113. Grove DE, Fan CY, Ren HY & Cyr DM (2011) The endoplasmic reticulum‐associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3‐dependent degradation of nascent CFTRDeltaF508. Mol Biol Cell 22, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Purificacao ADD, Debbas V, Tanaka LY, Gabriel GVM, Wosniak Junior J, De Bessa TC, Garcia‐Rosa S, Laurindo FRM & Oliveira PVS (2023) DNAJB12 and DNJB14 are non‐redundant Hsp40 redox chaperones involved in endoplasmic reticulum protein reflux. Biochim Biophys Acta Gen Subj 1868, 130502. [DOI] [PubMed] [Google Scholar]
- 115. Dabsan S, Zur G, Gilad A & Igbaria A (2023) Chaperone complexes from the endoplasmic reticulum (ER) and the cytosol inhibit wt‐p53 by activation the ER to cytosol signaling. bioRxiv 2023.08.01.551134. 10.1101/2023.08.01.551134 [DOI]
- 116. Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards AP, Van Goor KE, Paton AW, Paton JC, Atwood WJ, Tsai B et al. (2011) BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio 2, e00101‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zou H, Henzel WJ, Liu X, Lutschg A & Wang X (1997) Apaf‐1, a human protein homologous to C. Elegans CED‐4, participates in cytochrome c‐dependent activation of caspase‐3. Cell 90, 405–413. [DOI] [PubMed] [Google Scholar]
- 118. Liu X, Kim CN, Yang J, Jemmerson R & Wang X (1996) Induction of apoptotic program in cell‐free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. [DOI] [PubMed] [Google Scholar]
- 119. Lai HC, Liu TJ, Ting CT, Yang JY, Huang L, Wallace D, Kaiser P & Wang PH (2007) Regulation of IGF‐I receptor signaling in diabetic cardiac muscle: dysregulation of cytosolic and mitochondria HSP60. Am J Physiol Endocrinol Metab 292, E292–E297. [DOI] [PubMed] [Google Scholar]
- 120. Chun JN, Choi B, Lee KW, Lee DJ, Kang DH, Lee JY, Song IS, Kim HI, Lee SH, Kim HS et al. (2010) Cytosolic Hsp60 is involved in the NF‐kappaB‐dependent survival of cancer cells via IKK regulation. PLoS One 5, e9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chandra D, Choy G & Tang DG (2007) Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro‐apoptotic or pro‐survival functions involve differential interactions with caspase‐3. J Biol Chem 282, 31289–31301. [DOI] [PubMed] [Google Scholar]
- 122. Gupta S, McGrath B & Cavener DR (2010) PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes 59, 1937–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Horimoto S, Ninagawa S, Okada T, Koba H, Sugimoto T, Kamiya Y, Kato K, Takeda S & Mori K (2013) The unfolded protein response transducer ATF6 represents a novel transmembrane‐type endoplasmic reticulum‐associated degradation substrate requiring both mannose trimming and SEL1L protein. J Biol Chem 288, 31517–31527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Higashio H & Kohno K (2002) A genetic link between the unfolded protein response and vesicle formation from the endoplasmic reticulum. Biochem Biophys Res Commun 296, 568–574. [DOI] [PubMed] [Google Scholar]
- 125. Sato M, Sato K & Nakano A (2002) Evidence for the intimate relationship between vesicle budding from the ER and the unfolded protein response. Biochem Biophys Res Commun 296, 560–567. [DOI] [PubMed] [Google Scholar]
- 126. Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG & Wek RC (2011) The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 22, 4390–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chipurupalli S, Ganesan R, Martini G, Mele L, Reggio A, Esposito M, Kannan E, Namasivayam V, Grumati P, Desiderio V et al. (2022) Cancer cells adapt FAM134B/BiP mediated ER‐phagy to survive hypoxic stress. Cell Death Dis 13, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lim Y, Kim S & Kim EK (2021) Palmitate reduces starvation‐induced ER stress by inhibiting ER‐phagy in hypothalamic cells. Mol Brain 14, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhao D & Du LL (2020) Epr1, a UPR‐upregulated soluble autophagy receptor for reticulophagy. Autophagy 16, 2112–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]