

ABSTRACT
The circadian clock is a crucial regulator of mammalian physiology, controlling daily oscillations in key biological processes, such as cell proliferation, apoptosis, and DNA damage repair. Disruption of circadian rhythms has been identified as a significant risk factor for cancer development and progression, yet the specific molecular mechanisms linking circadian dysfunction to cancer remain poorly understood. Recent studies have increasingly focused on the role of diet in modulating circadian rhythms, highlighting the potential for dietary interventions in cancer management. However, how dietary factors like glucose restriction interact with circadian rhythms to influence cancer cell behavior remains an open question. Here, we investigate the mechanisms underlying glucose restriction‐induced apoptosis in non‐small cell lung cancer (NSCLC) cells, with a focus on the role of circadian clock genes. Analysis of the GEPIA database revealed that the circadian gene Bmal1 is highly expressed in normal tissues and associated with better prognosis in lung adenocarcinoma patients. In NSCLC cells, Bmal1 expression correlated with proapoptotic gene activity. In a tumor xenograft model using severe combined immunodeficiency (SCID) mice, a glucose‐restricted (ketogenic) diet significantly delayed tumor growth and increased the expression of Bmal1 and proapoptotic genes. These findings suggest that glucose restriction promotes apoptosis in NSCLC cells through a Bmal1‐mediated pathway, providing novel insights into the intersection between circadian regulation and cancer biology. Targeting core circadian clock genes like Bmal1 may represent a promising therapeutic strategy for managing lung cancer, broadening our understanding of how circadian rhythms can be harnessed for cancer prevention and treatment.
Keywords: AMPK, apoptosis, Bmal1, glucose restriction, NSCLC
In this study we investigate the role of the core circadian gene BMAL1 in regulating mechanisms within non‐small cell lung cancer cells, revealing that circadian rhythm disruption may represent another significant factor triggering apoptosis.

Abbreviations
- GR
glucose restriction
- KD
ketogenic diet
- NSCLC
non‐small cell lung cancer
- PMSF
phenylmethylsulfonyl fluoride
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- SCID
severe combined immune deficiency
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
1. Introduction
In the worldwide perspective, lung cancer stands out as a prevalent form of cancer [1]. Even with advancements in radiotherapy, chemotherapy, and surgical methods, cancer continues to be a significant contributor to global mortality [2, 3]. Presently, increasing research is directed toward dietary measures as a supplementary treatment in tumor progression and prognosis. Such interventions could provide numerous advantages for cancer patients undergoing treatment, such as mitigating drug side effects, boosting chemotherapy effectiveness, and regulating cell growth and death processes [4, 5, 6, 7].
The disruption of circadian rhythms is closely related to tumor occurrence [8]. Stable circadian rhythms play a role in tumor suppression, and when the steady state imposed by the biological clock is inhibited, it promotes the occurrence and development of tumors. During this process, cells exhibit uncontrolled rapid proliferation, increased metabolic demands, and resistance to apoptosis [9, 10]. Circadian rhythm is an endogenous oscillation with a cycle of about 24 h [11]. At the molecular level, the circadian clock consists of different transcription factors, such as Bmal1 (brain and muscle ARNT‐like protein 1), Clock (circadian locomotor output cycles kaput), Cry (Cryptochrome), and Per (Period) [12]. These core transcription elements create a transcription‐translation negative feedback loop, which controls several signaling pathways in the cell, ranging from cell proliferation, apoptosis, to DNA damage repair [13, 14, 15].
Recent studies have shown that disruption of circadian rhythms leads to shortened lifespan, dysregulated gene expression, and hepatic metabolic dysfunction in mice [16], ultimately resulting in tumorigenesis. Both physiological disruption of circadian rhythms and genetic disruption (mutations in Per2 and Bmal1) have been found to exacerbate pulmonary tumorigenesis [17]. However, the molecular mechanisms underlying how circadian clock gene expression influences tumor progression remain unclear. With growing insights into circadian genes in cancer progression, modulating the expression of these circadian genes may help ameliorate circadian rhythm disruptions in cancer, thereby reducing tumor growth and disease progression.
Bmal1 forms a heterodimer with another circadian clock protein, clock, driving the transcription of downstream genes that control various biological functions, including cell cycle regulation, DNA damage repair, and metabolism essential for maintaining circadian rhythms, but it also plays a crucial role in regulating metabolic processes [18, 19, 20]. Studies have shown that diet can directly influence circadian rhythms by modulating the expression of Bmal1 and other clock genes. For instance, time‐restricted feeding or specific dietary compositions, such as ketogenic diets, can enhance Bmal1 expression, aligning metabolic cycles with circadian rhythms [17, 21]. The connection to cancer has garnered increasing attention in recent years. Disruption of Bmal1 expression has been linked to tumorigenesis, as Bmal1 regulates genes involved in cell proliferation, apoptosis, and immune responses [22, 23, 24]. Reduced Bmal1 expression is associated with poor prognosis in various cancers [25], including lung adenocarcinoma, whereas restoration of circadian function through Bmal1 has been shown to suppress tumor growth [26]. Therefore, Bmal1 represents a critical link between circadian rhythms and cancer, positioning it as a promising therapeutic target in cancer prevention and management [27, 28].
Research suggests that certain dietary habits can either promote or inhibit tumor growth [29]. By consciously selecting our food intake, we may lower the likelihood of developing particular tumors [30]. Dietary patterns significantly influence the internal clock [31]. According to the Warburg effect, tumor cells largely rely on glucose for energy, unlike normal cells that can derive energy from ketone bodies when glucose is limited. This is one of the main characteristics that distinguish tumor cells from normal cells [32, 33]. Meal timing, nutrient intake, and food composition have substantial impacts on circadian rhythms [34]. Furthermore, circadian rhythms play an essential role in coordinating apoptosis. Apoptosis, or programmed cell death, is a natural process that eliminates damaged or unnecessary cells [16, 35]. Dysregulation of apoptosis can lead to tumor formation and progression. Caspases (cysteinyl aspartate‐specific protease), crucial enzymes in the apoptosis pathway [36, 37]. The process of apoptosis is essentially a cascading amplification reaction of caspase‐mediated irreversible limited substrate hydrolysis [38].
Our previous studies have demonstrated that glucose restriction can inhibit the progression of non‐small cell lung cancer by regulating AMPK‐SIRT1 [39]. This study primarily explores the mechanism by which glucose restriction induces apoptosis in non‐small cell lung cancer cells from the perspective of energy metabolism and circadian clock genes. In this study, we found that a low‐carbohydrate ketogenic diet can inhibit the growth of xenograft tumors in mice and restore disrupted circadian rhythms. These findings can provide a more profound insight into the internal mechanisms of dietary changes on tumorigenesis, thereby facilitating the development of more effective new treatment regimens.
2. Materials and Methods
2.1. Cell Culture and Reagents
A549 and NCI‐H460 cells were sourced from the Chinese Academy of Sciences Committee on Type Culture Collection Cell Bank in Shanghai, China. These cells were maintained in RPMI‐1640 medium obtained from Procell Life Science & Technology Co. Ltd. (Wuhan, China), supplemented with 10% heat‐inactivated fetal bovine serum (CHUANQIU Biotechnology Co. Ltd., Shanghai, China), 100 U/mL penicillin, and 100 mg/mL streptomycin. The cultures were incubated at 37°C in an atmosphere containing 5% CO2. To create cell cultures with varying glucose levels, anhydrous D‐glucose (purity > 99.8%; YIFEIXUE BIOTECH Co. Ltd., Nanjing, China) was added to glucose‐free RPMI‐1640 medium (Procell).
2.2. GEPIA Database and Kaplan–Meier Plotter Database Analysis
The Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer‐pku.cn/index.html) is an interactive web‐based tool that utilizes data from 9736 tumor samples and 8587 normal tissues, which are drawn from both the TCGA and GTEx databases. GEPIA enables the comparison of gene expression patterns across various cancer types. Meanwhile, the Kaplan–Meier Plotter (http://kmplot.com/analysis/) is a robust and comprehensive web application designed for survival analysis. The most extensively studied cancers in this database include breast, ovarian, lung, and gastric cancers. Its data sources consist of GEO, EGA, and TCGA datasets, as well as high‐throughput platforms such as gene chips and RNA‐seq. The platform's main function is to identify and evaluate survival markers using meta‐analysis, making it an ideal tool for survival studies, clinical feature correlation, and immunoassay analysis.
2.3. Quantitative Real‐Time PCR
Total RNA was extracted using an RNAiso Plus kit (YIFEIXUE Biotech. Co. Ltd.) following the manufacturer's instructions. Total RNA was reverse transcribed with HiScript II Q RT SuperMix for qPCR (R222; Vazyme Biotech Co. Ltd.). Quantitative real‐time polymerase chain reaction (qRT‐PCR) was performed using the AceQ Universal SYBR qPCR Master Mix (Q511; Vazyme Biotech Co. Ltd.) and a Roche Light Cycler 96 real‐time PCR system (Roche, Basel, Switzerland). The sequences of the PCR primers used in this study are provided in Table 1. GAPDH was used as an internal mRNA control. The fold change was calculated using the method.
TABLE 1.
Primer sequences used for quantitative real‐time PCR.
| Species | Gene | Forward primer | Reverse primer |
|---|---|---|---|
| Human | AMPKα1 | 5′‐GTAGTAAAAACAGGCTCCACGAA‐3′ | 5′‐CACCAGAAAGGATCTGTTGGA‐3′ |
| Human | GAPDH | 5′‐CAAGGTCATCCATGACAACTTTG‐3′ | 5′‐GTCCACCACCCTGTTGCTGTAG‐3′ |
| Human | Bmal1 | 5′‐GGGCAGCAGATGGATGGATTTTTG‐3′ | 5′‐GGATCTTGAAGACAGACTCTGAGACA‐3′ |
| Human | Bax | 5′‐CCCGAGAGGTCTTTTTCCGAG‐3′ | 5′‐CCAGCCCATGATGGTTCTGAT‐3′ |
| Human | Bcl‐2 | 5′‐GAGGATTGTGGCCTTCTTTG‐3′ | 5′‐GCCGGTTCAGGTACTCAGTC‐3′ |
| Human | Caspase‐3 | 5′‐TGGAACAAATGGACCTGTTGACC‐3′ | 5′‐AGGACTCAAATTCTGTTGCCACC‐3′ |
| Human | Caspase‐8 | 5′‐CAAATGCAAACTGGATGATGAC‐3′ | 5′‐AGCAGGCTCTTGTTGATTTGG‐3′ |
| Mice | GAPDH | 5′‐GCAAAGTGGAGATTGTTGCCAT‐3′ | 5′‐CCTTGACTGTGCCGTTGAATTT‐3′ |
| Mice | Bmal1 | 5′‐ACAGTCAGATTGAAAAGAGGCG‐3′ | 5′‐GCCATCCTTAGCACGGTGAG‐3′ |
| Mice | Bax | 5′‐AGGATGCGTCCACCAAGAA‐3′ | 5′‐CAAAGTAGAAGAGGGCAACCAC‐3′ |
| Mice | Bcl‐2 | 5′‐ACGGTGGTGGAGGAACTCTTCAG‐3′ | 5′‐GGTGTGCAGATGCCGGTTCAG‐3′ |
| Mice | Caspase‐3 | 5′‐ATGGAGAACAACAAAACCTCAGT‐3′ | 5′‐TTGCTCCCATGTATGGTCTTTAC‐3′ |
| Mice | Caspase‐8 | 5′‐GGACTACATCCCACACAAGAAGCAC‐3′ | 5′‐GTTGCAGTCTAGGAAGTTGACCAGC‐3′ |
2.4. Western Blot Analysis
Proteins were extracted using RIPA buffer with PMSF (protease and phosphatase inhibitors), and concentrations were determined via the BCA protein assay (Beyotime Biotechnology Co. Ltd., Shanghai, China). After separation by 10% SDS‐PAGE (Invitrogen, Life Technologies Corp, Carlsbad, CA), proteins were transferred onto nitrocellulose membranes (GE Healthcare UK Ltd., Bucks, UK). Membranes were blocked with 5% skim milk, incubated overnight at 4°C with primary antibodies, and subsequently incubated for 1 h at room temperature with HRP‐conjugated secondary antibodies, facilitating protein detection. Primary antibodies were as follows: anti‐AMPKα1, anti‐Bmal1, anti‐Bax, anti‐Bcl‐2, anti‐β‐actin (ZENBIO, Chengdu, China); anti‐Caspase‐8, anti‐Cleaved Caspase‐3 (ABclonal Technology Co. Ltd.). The secondary antibody is HRP‐Conjugated AffiniPure Goat Anti‐Mouse IgG (ZSGB‐BIO, Beijing, China).
2.5. Plasmid Transfection
NSCLC cells were transfected with the human Bmal1 plasmid or the control plasmid using Lipofectamine 2000, as per the manufacturer's protocol; the plasmid was constructed by the Corues Biotechnology Company (Nanjing, China). Similarly, the human AMPKα1 plasmid or its control plasmid was transfected into NSCLC cells using Lipofectamine 2000 in accordance with the provided instructions.
2.6. Flow Cytometry Analysis
Cells were harvested and washed twice with chilled PBS, followed by resuspension in binding buffer. To evaluate apoptosis, an Annexin V‐FITC/PI apoptosis detection kit (BD Biosciences, NJ, USA) was used. The cells were incubated with Annexin V‐FITC for 15 min at room temperature, kept in the dark, and propidium iodide (PI) was subsequently added for 10 min. Apoptosis levels were then measured using flow cytometry (A00‐1‐1102; Beckman, USA).
2.7. Xenograft Studies
Ten male SCID mice (4 weeks old, 18–20 g) were housed at Nanjing Medical University under controlled temperature (22°C ± 1°C) and humidity (55% ± 5%). Each mouse was subcutaneously injected with 5 × 106 A549 spheroid cells. After 2 weeks, the mice were divided into ketogenic diet (90% fat, 1.6% carbohydrate, 8.4% protein) [40] and control diet groups (10% fat, 80% carbohydrate, 10% protein). After 2 weeks of treatment, the mice were euthanized and tissues collected. Another cohort of 36 mice, including 24 injected with A549 cells, was divided similarly into ketogenic, control, and blank groups. After 1 month of treatment, the mice were euthanized, and tissues collected at 6:00 a.m., 12:00 p.m., 6:00 p.m., and 12:00 a.m.
2.8. Statistical Analysis
The experimental data were presented as mean values ± standard deviation, based on results from at least three separate experiments. Statistical analysis of quantitative variables was carried out using both Student's t‐test and one‐way analysis of variance (ANOVA). For comparisons across multiple groups, one‐way ANOVA was employed, followed by Tukey's post hoc test to pinpoint specific differences between groups. Various software tools, including SPSS version 22.0 (SPSS Inc., Chicago, IL, USA), GraphPad Prism version 8.0 (GraphPad Software Inc., La Jolla, CA, USA), and FlowJo V10, were utilized to perform all statistical analyses. p < 0.05 was considered to indicate statistical significance.
3. Results
3.1. Glucose Restriction Enhanced the Expression of Apoptotic Genes in NSCLC Cells
Our previous research has shown that glucose restriction plays a significant role in promoting apoptosis in non‐small cell lung cancer cells [39]. Previous cellular phenotype experiments have demonstrated that glucose restriction can induce apoptosis in tumor cells. The NSCLC cell lines A549 and NCI‐H460 were treated with different concentrations of glucose (11, 5.5, 2.5 mM) in cell culture medium for 12 h to examine changes in apoptosis‐related genes. The results indicate that both tumor cells exhibit an apoptosis trend after glucose restriction. The expression of proapoptotic genes Bax, Caspase‐8, and Cleaved Caspase‐3 increased, whereas the expression of the antiapoptotic gene Bcl‐2 decreased (Figure 1A,B). Similarly, Western Blot experiments showed a similar trend in the protein expression levels of apoptosis‐related genes (Figure 1C,D). These experimental results confirm that glucose restriction can influence the expression of apoptosis‐related genes, thereby enhancing the apoptotic levels of NSCLC cells.
FIGURE 1.

Glucose restriction promotes apoptosis in tumor cells. (A, B) The mRNA levels of Bax, Bcl‐2, Caspase‐3 and Caspase‐8 in cells were detected by qRT‐PCR after treated with different concentrations of glucose (11, 5.5, 2.5 mM) for 12 h. (C, D) Western Blot analysis of Bax, Bcl‐2, Cleaved Caspase‐3, Caspase‐8 in cells cultured with different concentrations of glucose for 24 h.*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.2. Glucose Restriction Induced Apoptosis in NSCLC Cells via AMPK
It is known that glucose intake is a major nutritional input determining AMPK activity [41]. Our previous studies have shown that AMPK expression is positively regulated by ROS when glucose is restricted. We now focus primarily on the role of AMPK in the apoptosis process. By overexpressing AMPK levels through plasmid transfection in A549 and NCI‐H460 cells, we investigate the impact of AMPK on downstream apoptotic regulatory genes. As seen in Figure 2A, we successfully overexpressed AMPK in lung cancer cells. Subsequently, we examine the expression trends of relevant apoptotic genes through qRT‐PCR and Western blot experiments. Figure 2C,E, demonstrate an upregulation in the expression of Bax, Caspase‐8, and Cleaved Caspase‐3 genes, whereas the expression of Bcl‐2 is reduced. This suggests that following AMPK overexpression, the cells exhibit an overall proapoptotic state. The results in A549 and NCI‐H460 cells are consistent.
FIGURE 2.

Glucose restriction promotes apoptosis in NSCLC cells through AMPK involvement. (A) mRNA expression of AMPK after transfected with AMPKα1 overexpression plasmid. (B) mRNA expression of AMPK after AMPK inhibitor compound C (40 μM) used. (C) mRNA expression of Bax, Bcl‐2, Caspase‐3 and Caspase‐8 after AMPK overexpression. (D) mRNA expression of Bax, Bcl‐2, Caspase‐3 and Caspase‐8 after AMPK inhibition. Protein expression of Bax, Bcl‐2, Cleaved Caspase‐3 and Caspase‐8 after AMPK overexpression (E) and AMPK inhibition (F).
When we inhibit AMPK activity with the AMPK inhibitor Dorsomorphin (Compound C), the apoptosis process in tumor cells is significantly suppressed (Figure 2B). The expression of the proapoptotic gene Bax shows a decreasing trend, whereas the antiapoptotic gene Bcl‐2 exhibits an increasing trend (Figure 2D,F). These results indicate that AMPK plays a crucial role in promoting the cell apoptosis process.
3.3. High Expression of the Circadian Clock Gene Bmal1 Was Associated With Better Prognosis in NSCLC
Analyzing the expression of circadian clock genes in non‐small cell lung cancer using a database revealed interesting findings. Comparing tumor tissue samples and adjacent normal tissue samples from lung adenocarcinoma patients in the GEPIA database, it was observed that the expression of circadian clock genes Bmal1, Clock, and Cry1 showed relatively small differences between tumor and normal tissues, with only Per1 showing significant differential expression, higher in normal tissue than in tumor tissue (Figure 3A). Further comparison of tumor tissue samples from lung adenocarcinoma patients in the GEPIA database with normal lung tissue samples from the GTEx database indicated that while Clock and Cry1 gene expressions remained nonsignificant between tumor and normal tissues, Bmal1 exhibited a trend similar to Per1, with higher expression in normal lung tissue than in tumor tissue (Figure 3B). Survival analysis of lung adenocarcinoma patients based on circadian clock gene expression in the Kaplan–Meier database unveiled that patients with high expression of the Clock gene had lower overall survival compared to those with low expression; Cry1 showed no significant difference; patients with high expression of Bmal1 and Per1 had better prognoses, with significant differences (Figure 3C). Those research findings suggest that Bmal1 exhibits a significant difference in expression between NSCLC patients and healthy individuals. Notably, in NSCLC patients, elevated Bmal1 expression is correlated with a more favorable prognosis. This highlights the potential prognostic value of Bmal1 in lung cancer and suggests that it may play a critical role in influencing patient outcomes.
FIGURE 3.

Expression levels of circadian clock genes in lung adenocarcinoma and normal tissue. (A) Analysis of circadian gene expression in lung adenocarcinoma tumor samples and adjacent noncancerous tissues from the GEPIA database. (B) Analysis of circadian gene expression in tumor tissue samples patients from the GEPIA database compared to normal lung tissue samples from the GTEx database. (C) Surviavl analysis of differential expression levels of clock genes. *p < 0.05.
3.4. Bmal1 Participated in Regulating the Apoptosis Process of NSCLC Cells
The findings presented in Section 3.3 raise an intriguing question: could the elevated expression of Bmal1 be attributed to its proapoptotic effect, thereby contributing to the improved prognosis observed in patients? To further explore the role of Bmal1 in the apoptosis process of NSCLC cells, we chose to overexpress Bmal1 in A549 cells through plasmid transfection and successfully constructed cells with overexpressed Bmal1 gene (Figure 4A). Based on this, we examined the expression trends of relevant apoptotic genes. The experimental results show a significant increase in mRNA expression levels of proapoptotic markers, such as Bax, Caspase‐3, and Caspase‐8, whereas the mRNA expression level of the antiapoptotic gene Bcl‐2 significantly decreased, and these differences are statistically significant (Figure 4D). Furthermore, through Western Blot analysis, we observed a significant increase in the expression level of Cleaved Caspase‐3 protein (Figure 4B). In addition, we used the same method to construct A549 and NCI‐H460 cells with overexpressed Bmal1 gene and detected the level of cell apoptosis using flow cytometry (Figure 4E). The results indicate that in both cell lines, the level of cell apoptosis in the Bmal1 overexpression group significantly increased, and the difference is significant. In conclusion, these results indicate that Bmal1 plays an important regulatory role in the apoptosis process of NSCLC cells. More importantly, these findings support the notion that the reason for the better prognosis associated with high expression of Bmal1 may be due to its proapoptotic effect.
FIGURE 4.
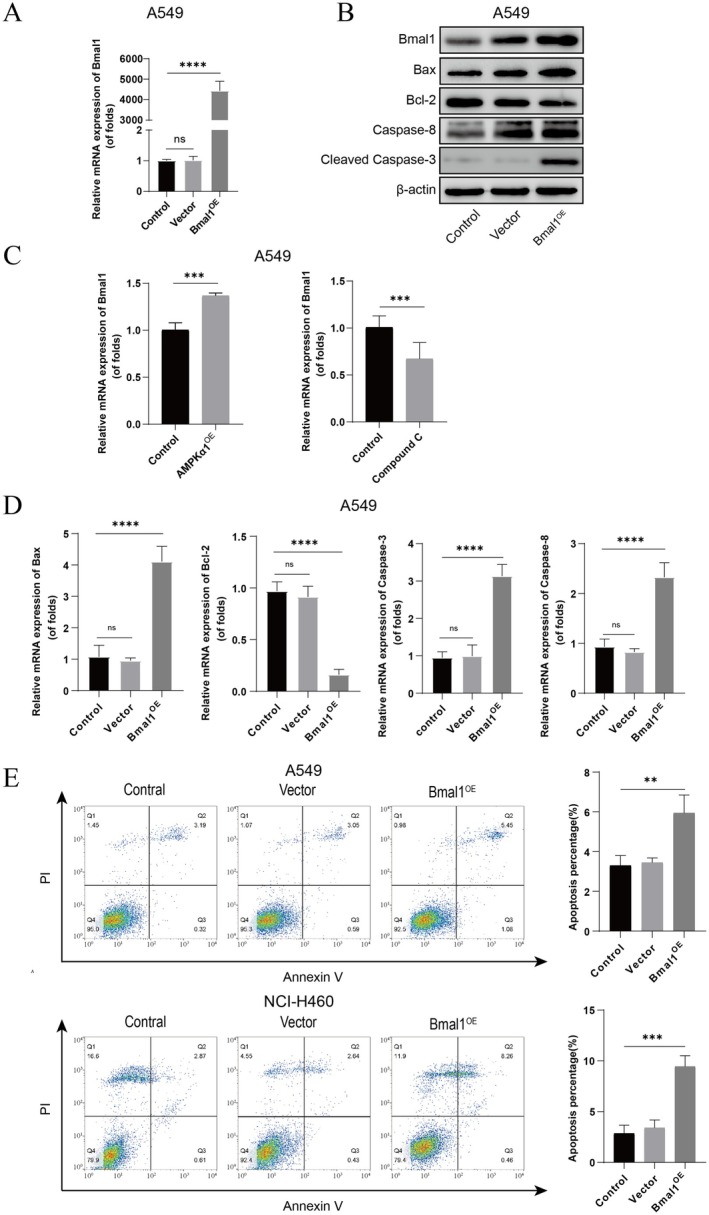
Regulation of apoptosis gene expression by Bmal1 in NSCNC cells. (A, D) mRNA expression of Bmal1, Bax, Bcl‐2, Caspase‐3 and Caspase‐8 after transfected with Bmal1 overexpression plasmid. (B) Protein expression of Bmal1, Bax, Bcl‐2, Cleaved Caspase‐3 and Caspase‐8 after transfected with Bmal1overexpression plasmid. (C) The impact of AMPK overexpression or inhibition on Bmal1 expression. (E) Flow cytometry measured after Bmal1 overexpression. **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.5. AMPK Is Involved in the Regulation of Bmal1 in NSCLC Cells
AMPK and Bmal1 play key roles in energy metabolism and circadian rhythm regulation, respectively. Recent studies have shown a close connection between the two. In tumor cells, disrupted energy metabolism and circadian rhythm are prominent features. The interaction between AMPK and Bmal1 plays a significant role in the growth and apoptosis of tumor cells. Our research has found that AMPK can regulate the expression of Bmal1, thereby controlling the apoptosis of tumor cells. When AMPK is activated, both the mRNA and protein levels of Bmal1 significantly increase. Conversely, when we use an AMPK inhibitor, the expression levels of AMPK decrease, and the mRNA and protein levels of Bmal1 also decrease significantly (Figure 4C).
3.6. Glucose Restriction Promoted the Progression of Apoptosis In Vivo
To investigate whether glucose restriction affects tumor growth, we used a mouse xenograft model. Figure 5A indicates that we divided the mice into three groups and shows the proportions of the three macronutrients in a normal diet and a ketogenic diet. Figure 5B shows that the mRNA expression levels of the proapoptotic gene Bax, Caspase‐3, and Caspase‐8 in the ketogenic diet group were significantly higher than those in the normal diet group. Protein expression also follows the same trend (Figure 5D). This suggests that the ketogenic diet plays a crucial role in promoting apoptosis in tumor cells. Figure 5C demonstrates that, compared to the normal diet group, the ketogenic diet group exhibited a marked recovery in circadian rhythms with an increased oscillation amplitude, indicating that glucose restriction can restore circadian rhythm disruptions caused by tumors. We next investigated the effects of a ketogenic diet on normal mice. The experimental results showed that in normal mice, the ketogenic diet did not alter apoptosis levels in vivo (Figure S1), indicating that the proapoptotic effects induced by the ketogenic diet were tumor‐dependent.
FIGURE 5.

Glucose restriction increased the expression of apoptosis‐related genes in vivo. (A) Grouping of xenograft tumor mice (by Figdraw) and the proportions of the three macronutrients in a normal diet and a ketogenic diet. (B) mRNA expression of Bax, Bcl‐2, Caspase‐3, Caspase‐8 on normal diet and ketogenic diet groups. (C) The rhythmic expression levels of Bmal1 on blank control, normal diet and ketogenic diet groups. (D) Protein expression of Bax, Bcl‐2, Caspase‐8 on normal diet and ketogenic diet groups. *p < 0.05, ***p < 0.001.
4. Discussion
Recent research has highlighted that circadian disruption has been linked to a range of health problems, with mounting evidence indicating a strong connection between disturbances in circadian rhythms and cancer development [42, 43]. Specifically, mutations in key circadian genes, such as Clock and Bmal1 have been associated with heightened risks of various cancers, including pancreatic, prostate, and breast cancer, in both humans and animal models [44, 45, 46]. Additionally, emerging studies suggest that dietary modifications, particularly glucose deprivation, can influence tumor progression by delaying cancer development and selectively inducing apoptosis in cancer cells while sparing normal cells [47, 48]. In this study, we have revealed the crucial role of glucose restriction in inhibiting the progression of NSCLC. At a mechanistic level, glucose restriction significantly increases the expression levels of AMPK, a key signaling molecule that further induces the upregulation of the core circadian gene BMAL1. This process effectively restores the normal day–night rhythmic expression pattern, ultimately promoting the activation of the cancer cell apoptosis program, leading to a potent inhibitory effect on tumor growth trends.
Cancer cells often require more glucose supply due to their high energy demands [49]. The Warburg effect, where tumor cells rely on glycolysis for energy, plays a crucial role in the growth advantage of tumors [32, 50]. Compared to normal cells, tumor cells have a metabolic vulnerability that relies more on glucose, making glucose restriction an effective method to inhibit tumor progression [51, 52]. Several clinical studies have explored the potential benefits of a ketogenic diet, a low glucose diet, for cancer patients [52]. Glucose restriction can inhibit tumor progression by inducing apoptosis in tumor cells [53]. This study found that glucose restriction can upregulate the expression of the energy‐responsive factor AMPK, subsequently increasing BMAL1 expression and ultimately promoting apoptosis. These findings offer new insights into the mechanisms of glucose restriction‐induced apoptosis for a better understanding. Despite the promising research prospects, several key issues require attention, such as addressing patient compliance in implementing glucose restriction within clinical settings, as well as conducting comprehensive evaluations of the long‐term effects and safety of prolonged glucose deprivation in cancer patients.
The CLOCK/BMAL‐mediated transcriptional activation constitutes the core of circadian rhythm regulation, and the disruption of this system is closely associated with cancer progression [54]. Despite extensive evidence establishing the link between circadian rhythm disruption and tumorigenesis, the specific details of its underlying molecular mechanisms remain to be further elucidated [55]. Rhythmic abnormalities severely impact cell cycle regulation, promote cell proliferation, alter tumor microenvironment characteristics, and interfere with DNA damage repair mechanisms, promoting tumorigenesis [56, 57]. Directly targeting the human CLOCK/BMAL transcriptional complex poses significant challenges, with current drug development strategies focusing on regulating clock negative feedback elements (such as REV‐ERB agonists, ROR agonists, and CRY stabilizers) in an attempt to restore rhythmic balance [58]. Unfortunately, these candidate drugs commonly exhibit insufficient specificity and significant side effects, hindering their clinical progress. This study demonstrates that adopting a glucose restriction strategy as a novel and practical approach not only effectively enhances clock gene expression, restoring a normal circadian rhythm pattern, but also is safe and reliable, offering a promising therapeutic pathway for inhibiting tumor development. This approach highlights the broad prospects of metabolic interventions in regulating chronobiology and in the field of anticancer therapies.
Apoptosis, a widespread form of programmed cell death, is a process meticulously regulated by various intrinsic and extrinsic signaling pathways [59]. These pathways activate in response to several triggers, such as cellular stress, DNA damage, and immune surveillance [60]. This highly ordered mechanism of cell death serves as a crucial strategy for eliminating abnormal or harmful cells, including tumor cells, particularly through the coordinated action of intrinsic and extrinsic pathways [61]. Consequently, inducing apoptosis emerges as a powerful approach against tumors, aimed at effectively clearing tumor cells by precisely modulating these key signaling pathways. In this study, we investigate the role of the core circadian gene BMAL1 in regulating mechanisms within non‐small cell lung cancer cells, revealing that circadian rhythm disruption may represent another significant factor triggering apoptosis.
In summary, we have revealed glucose restriction as a promising therapeutic intervention by activating the AMPK‐BMAL1 signaling pathway, promoting apoptosis in non‐small cell lung cancer cells, effectively restraining disease progression. Although the precise regulation of BMAL1 activity by AMPK and the molecular mechanisms by which BMAL1 mediates apoptosis remain to be further explored, our research findings have clearly indicated the critical role of circadian rhythm disruption in the progression of non‐small cell lung cancer. These novel insights not only provide a theoretical basis for chronotherapy strategies but also pave the way for the development of glucose restriction‐based adjunctive therapies aimed at optimizing cancer treatment efficacy and safety, thus opening new research avenues.
Author Contributions
Tao Wei: data curation, formal analysis, methodology, validation, writing – original draft. Ying Cheng: data curation, validation. Jierong Ge: data curation, validation. Manting Zhu: data curation. Hong Chen: data curation. Qing Feng: methodology, supervision, writing – review and editing.
Ethics Statement
Approval of the research protocol by an Institutional Review Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: Animal experiments have been approved by the Experimental Animal Welfare Ethics Committee of Nanjing Medical University.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. The ketogenic diet does not affect apoptosis in normal mice (C57BL/6J).
Funding: This work was Supported by the Key Project of Jiangsu Commission of Health (grant no. ZD2022012) and the Top‐notch Innovative Talent Project of Nanjing Medical University (grant no. NJMUTY20230094).
Tao Wei and Ying Cheng should be considered joint first author.
References
- 1. Kratzer T. B., Bandi P., Freedman N. D., et al., “Lung Cancer Statistics, 2023,” Cancer 130 (2024): 1330–1348. [DOI] [PubMed] [Google Scholar]
- 2. Li Y., Yan B., and He S., “Advances and Challenges in the Treatment of Lung Cancer,” Biomedicine & Pharmacotherapy 169 (2023): 115891. [DOI] [PubMed] [Google Scholar]
- 3. Hirsch F. R., Scagliotti G. V., Mulshine J. L., et al., “Lung Cancer: Current Therapies and New Targeted Treatments,” Lancet 389 (2017): 299–311. [DOI] [PubMed] [Google Scholar]
- 4. Chen J., Liu X., Zou Y., et al., “A High‐Fat Diet Promotes Cancer Progression by Inducing Gut Microbiota‐Mediated Leucine Production and PMN‐MDSC Differentiation,” Proceedings of the National Academy of Sciences of the United States of America 121 (2024): e2306776121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y., Yamamoto T., Takahashi Y., et al., “Metabolic Intervention by Low Carbohydrate Diet Suppresses the Onset and Progression of Neuroendocrine Tumors,” Cell Death & Disease 14 (2023): 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeong Y. J., Rogers T. J., Anderson C. E., and Lien E. C., “Tumor Lipid Metabolism: A Mechanistic Link Between Diet and Cancer Progression,” Current Opinion in Biotechnology 84 (2023): 102993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weng M. L., Chen W. K., Chen X. Y., et al., “Fasting Inhibits Aerobic Glycolysis and Proliferation in Colorectal Cancer via the Fdft1‐Mediated AKT/mTOR/HIF1α Pathway Suppression,” Nature Communications 11 (2020): 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Masri S. and Sassone‐Corsi P., “The Emerging Link Between Cancer, Metabolism, and Circadian Rhythms,” Nature Medicine 24 (2018): 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rabinovich‐Nikitin I., Lieberman B., Martino T. A., and Kirshenbaum L. A., “Circadian‐Regulated Cell Death in Cardiovascular Diseases,” Circulation 139 (2019): 965–980. [DOI] [PubMed] [Google Scholar]
- 10. Qu M., Zhang G., Qu H., et al., “Circadian Regulator BMAL1::CLOCK Promotes Cell Proliferation in Hepatocellular Carcinoma by Controlling Apoptosis and Cell Cycle,” Proceedings of the National Academy of Sciences of the United States of America 120 (2023): e2214829120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang W., Ramsey K. M., Marcheva B., and Bass J., “Circadian Rhythms, Sleep, and Metabolism,” Journal of Clinical Investigation 121 (2011): 2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bass J. and Takahashi J. S., “Circadian Integration of Metabolism and Energetics,” Science 330 (2010): 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang R., Lahens N. F., Ballance H. I., Hughes M. E., and Hogenesch J. B., “A Circadian Gene Expression Atlas in Mammals: Implications for Biology and Medicine,” Proceedings of the National Academy of Sciences of the United States of America 111 (2014): 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ikeda R., Tsuchiya Y., Koike N., et al., “REV‐ERBα and REV‐ERBβ Function as Key Factors Regulating Mammalian Circadian Output,” Scientific Reports 9 (2019): 10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bouchard‐Cannon P., Mendoza‐Viveros L., Yuen A., Kærn M., and Cheng H. Y., “The Circadian Molecular Clock Regulates Adult Hippocampal Neurogenesis by Controlling the Timing of Cell‐Cycle Entry and Exit,” Cell Reports 5 (2013): 961–973. [DOI] [PubMed] [Google Scholar]
- 16. Kettner N. M., Voicu H., Finegold M. J., et al., “Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis,” Cancer Cell 30 (2016): 909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papagiannakopoulos T., Bauer M. R., Davidson S. M., et al., “Circadian Rhythm Disruption Promotes Lung Tumorigenesis,” Cell Metabolism 24 (2016): 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi J. S., “Molecular Architecture of the Circadian Clock in Mammals,” in A Time for Metabolism and Hormones, ed. Sassone‐Corsi P. and Christen Y. (Springer, 2016), 13–24. [PubMed] [Google Scholar]
- 19. Partch C. L., Green C. B., and Takahashi J. S., “Molecular Architecture of the Mammalian Circadian Clock,” Trends in Cell Biology 24 (2014): 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Acosta‐Rodríguez V., Rijo‐Ferreira F., Izumo M., et al., “Circadian Alignment of Early Onset Caloric Restriction Promotes Longevity in Male C57BL/6J Mice,” Science 376 (2022): 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sherman H., Genzer Y., Cohen R., Chapnik N., Madar Z., and Froy O., “Timed High‐Fat Diet Resets Circadian Metabolism and Prevents Obesity,” FASEB Journal 26 (2012): 3493–3502. [DOI] [PubMed] [Google Scholar]
- 22. Pick R., Wang C., Zeng Q., Gül Z. M., and Scheiermann C., “Circadian Rhythms in Anticancer Immunity: Mechanisms and Treatment Opportunities,” Annual Review of Immunology 42 (2024): 83–102. [DOI] [PubMed] [Google Scholar]
- 23. Tang Q., Cheng B., Xie M., et al., “Circadian Clock Gene Bmal1 Inhibits Tumorigenesis and Increases Paclitaxel Sensitivity in Tongue Squamous Cell Carcinoma,” Cancer Research 77 (2017): 532–544. [DOI] [PubMed] [Google Scholar]
- 24. Gery S. and Koeffler H. P., “Circadian Rhythms and Cancer,” Cell Cycle 9 (2010): 1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beker M. C., Caglayan B., Caglayan A. B., et al., “Interaction of Melatonin and Bmal1 in the Regulation of PI3K/AKT Pathway Components and Cellular Survival,” Scientific Reports 9 (2019): 19082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Battaglin F., Chan P., Pan Y., et al., “Clocking Cancer: The Circadian Clock as a Target in Cancer Therapy,” Oncogene 40 (2021): 3187–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gangitano E., Gnessi L., Lenzi A., and Ray D., “Chronobiology and Metabolism: Is Ketogenic Diet Able to Influence Circadian Rhythm?,” Frontiers in Neuroscience 15 (2021): 756970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Genzer Y., Dadon M., Burg C., Chapnik N., and Froy O., “Ketogenic Diet Delays the Phase of Circadian Rhythms and Does Not Affect AMP‐Activated Protein Kinase (AMPK) in Mouse Liver,” Molecular and Cellular Endocrinology 417 (2015): 124–130. [DOI] [PubMed] [Google Scholar]
- 29. Gonzalez C. A. and Riboli E., “Diet and Cancer Prevention: Where We Are, Where We Are Going,” Nutrition and Cancer 56 (2006): 225–231. [DOI] [PubMed] [Google Scholar]
- 30. Key T. J., Schatzkin A., Willett W. C., Allen N. E., Spencer E. A., and Travis R. C., “Diet, Nutrition and the Prevention of Cancer,” Public Health Nutrition 7 (2004): 187–200. [DOI] [PubMed] [Google Scholar]
- 31. Patterson R. E., Laughlin G. A., LaCroix A. Z., et al., “Intermittent Fasting and Human Metabolic Health,” Journal of the Academy of Nutrition and Dietetics 115 (2015): 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Warburg O., “On the Origin of Cancer Cells,” Science 123 (1956): 309–314. [DOI] [PubMed] [Google Scholar]
- 33. Seyfried T. N. and Shelton L. M., “Cancer as a Metabolic Disease,” Nutrition & Metabolism 7 (2010): 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panda S., “Circadian Physiology of Metabolism,” Science (New York, N.Y.) 354, no. 6315 (2016): 1008–1015, 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elmore S., “Apoptosis: A Review of Programmed Cell Death,” Toxicologic Pathology 35 (2007): 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lowe S. W. and Lin A. W., “Apoptosis in Cancer,” Carcinogenesis 21 (2000): 485–495. [DOI] [PubMed] [Google Scholar]
- 37. Fan T. J., Han L. H., Cong R. S., and Liang J., “Caspase Family Proteases and Apoptosis,” Acta Biochimica et Biophysica Sinica 37 (2005): 719–727. [DOI] [PubMed] [Google Scholar]
- 38. Degterev A. and Yuan J., “Expansion and Evolution of Cell Death Programmes,” Nature Reviews. Molecular Cell Biology 9 (2008): 378–390. [DOI] [PubMed] [Google Scholar]
- 39. Li B., Chen Q., Feng Y., et al., “Glucose Restriction Induces AMPK‐SIRT1‐Mediated Circadian Clock Gene Per Expression and Delays NSCLC Progression,” Cancer Letters 576 (2023): 216424. [DOI] [PubMed] [Google Scholar]
- 40. Allen B. G., Bhatia S. K., Buatti J. M., et al., “Ketogenic Diets Enhance Oxidative Stress and Radio‐Chemo‐Therapy Responses in Lung Cancer Xenografts,” Clinical Cancer Research 19 (2013): 3905–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hardie D. G., Ross F. A., and Hawley S. A., “AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis,” Nature Reviews. Molecular Cell Biology 13 (2012): 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dallmann R., Okyar A., and Lévi F., “Dosing‐Time Makes the Poison: Circadian Regulation and Pharmacotherapy,” Trends in Molecular Medicine 22 (2016): 430–445. [DOI] [PubMed] [Google Scholar]
- 43. Sancar A. and Van Gelder R. N., “Clocks, Cancer, and Chronochemotherapy,” Science 371 (2021): eabb0738. [DOI] [PubMed] [Google Scholar]
- 44. Hoffman A. E., Yi C. H., Zheng T., et al., “CLOCK in Breast Tumorigenesis: Genetic, Epigenetic, and Transcriptional Profiling Analyses,” Cancer Research 70 (2010): 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou L., Zhang Z., Nice E., Huang C., Zhang W., and Tang Y., “Circadian Rhythms and Cancers: The Intrinsic Links and Therapeutic Potentials,” Journal of Hematology & Oncology 15 (2022): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aiello I., Fedele M. M., Román F., et al., “Circadian Disruption Promotes Tumor‐Immune Microenvironment Remodeling Favoring Tumor Cell Proliferation,” Science Advances 6 (2020): eaaz4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boyd P., O'Connor S. G., Heckman‐Stoddard B. M., and Sauter E. R., “Time‐Restricted Feeding Studies and Possible Human Benefit,” Journal of the National Cancer Institute 6 (2022): pkac032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pan X., Taylor M. J., Cohen E., Hanna N., and Mota S., “Circadian Clock, Time‐Restricted Feeding and Reproduction,” International Journal of Molecular Sciences 21 (2020): 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vander Heiden M. G., Cantley L. C., and Thompson C. B., “Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation,” Science 324 (2009): 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pavlova N. N. and Thompson C. B., “The Emerging Hallmarks of Cancer Metabolism,” Cell Metabolism 23 (2016): 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zuccoli G., Marcello N., Pisanello A., et al., “Metabolic Management of Glioblastoma Multiforme Using Standard Therapy Together With a Restricted Ketogenic Diet: Case Report,” Nutrition & Metabolism (London) 7 (2010): 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Allen B. G., Bhatia S. K., Anderson C. M., et al., “Ketogenic Diets as an Adjuvant Cancer Therapy: History and Potential Mechanism,” Redox Biology 2 (2014): 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Klement R. J. and Champ C. E., “Calories, Carbohydrates, and Cancer Therapy With Radiation: Exploiting the Five R's Through Dietary Manipulation,” Cancer Metastasis Reviews 33 (2014): 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sulli G., Lam M. T. Y., and Panda S., “Interplay Between Circadian Clock and Cancer: New Frontiers for Cancer Treatment,” Trends Cancer 5 (2019): 475–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pariollaud M. and Lamia K. A., “Cancer in the Fourth Dimension: What Is the Impact of Circadian Disruption?,” Cancer Discovery 10 (2020): 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fu L. and Lee C. C., “The Circadian Clock: Pacemaker and Tumour Suppressor,” Nature Reviews. Cancer 3 (2003): 350–361. [DOI] [PubMed] [Google Scholar]
- 57. Perrin L., Loizides‐Mangold U., Chanon S., et al., “Transcriptomic Analyses Reveal Rhythmic and CLOCK‐Driven Pathways in Human Skeletal Muscle,” eLife 7 (2018): e34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Solt L. A., Wang Y., Banerjee S., et al., “Regulation of Circadian Behaviour and Metabolism by Synthetic REV‐ERB Agonists,” Nature 485 (2012): 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Youle R. J. and Strasser A., “The BCL‐2 Protein Family: Opposing Activities That Mediate Cell Death,” Nature Reviews. Molecular Cell Biology 9 (2008): 47–59. [DOI] [PubMed] [Google Scholar]
- 60. Carneiro B. A. and El‐Deiry W. S., “Targeting Apoptosis in Cancer Therapy,” Nature Reviews Clinical Oncology 17 (2020): 395–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fuchs Y. and Steller H., “Programmed Cell Death in Animal Development and Disease,” Cell 147 (2011): 742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The ketogenic diet does not affect apoptosis in normal mice (C57BL/6J).