


Abstract
PHF13 is a H3K4me3 epigenetic reader that modulates key chromatin processes including transcription, DNA damage response, and chromatin architecture. PHF13 is found aberrantly regulated in different cancers and its misexpression alters the epigenetic landscape of key transcription factors that regulate epithelial-to-mesenchymal transition. In this study, we sought to understand how PHF13’s chromatin affinity and diverse chromatin functions are intrinsically regulated. Our results show that PHF13 can oligomerize via conserved ordered regions in its N- and C- terminus increasing its chromatin valence and avidity, promoting polymer–polymer phase separation (PPPS) and chromatin inaccessibility. Impressively, a ∼3- to 5-fold overexpression of PHF13 was sufficient to globally compact chromatin visible by optical microscopy, dependent on its ordered dimerizing regions and oligomerization potential. Unexpectedly, we discovered that PHF13 can self-associate independent of its ordered domains via intrinsically disordered regions, which conversely reduced PHF13’s chromatin affinity, formed liquid–liquid phase separated (LLPS) condensates, and differentially impacted gene expression. Our findings support that there is an intrinsic balance between PHF13’s ordered and disordered regions and that PHF13 can phase transition between polymer–polymer and liquid–liquid phase separation states to impact chromatin structure and function.
Graphical Abstract
Graphical Abstract.

Introduction
PHF13 also known as SPOC1 (survival-time associated PHD finger protein in ovarian cancer 1) is a highly conserved epigenetic reader/effector protein [1] across vertebrates. PHF13 plays a role in diverse chromatin processes, including transcription [1–4], replication [2–4], DNA damage response [5], and cell division [6], implying its importance for cellular homeostasis. Consistently, aberrant PHF13 expression has been reported to promote epithelial mesenchymal transition (EMT) -like phenotypes and correlates with a poor prognosis in different cancers [7, 8]. Further emphasizing the essential nature of PHF13, mosaic knockout of PHF13 in mice leads to non-Mendelian genetics due to late embryonic (∼E14) and early post-natal lethality as well as a block in spermatogonia stem cell differentiation [9]. These observations all underline that PHF13 has indispensable chromatin regulatory roles that need to be tightly regulated to maintain genome integrity.
Previously, we have demonstrated that PHF13 interacts with chromatin in a multivalent fashion via a centrally localized nonspecific DNA-binding domain and a C-terminal PHD domain which specifically binds to methylated lysine 4 on histone H3 [1]. We and others have shown that PHF13 binds at active and bivalent promoters and that PHF13 interacts with key epigenetic regulators such as Polycomb [1, 3, 7], K9 methyltransferases [5], and RNA polymerase II [1, 3], to modulate the epigenetic landscape and transcriptome of its target genes [1, 7]. However, what regulates PHF13 chromatin association and affinity has not been previously explored. Here we uncover that PHF13 has the ability to self-associate and oligomerize via two different and independent mechanisms: (i) via its intrinsically disorder regions (IDRs) and (ii) via ordered aggregation domains located in its N- and C-terminus. Interestingly, however, PHF13 oligomerization via these different mechanisms has distinct effects on PHF13 localization, transcriptional regulation, and chromatin structure. Utilizing in silico predictions and genetic deletions, we explore the intricate balance between these oligomerization mechanisms in regulating PHF13 chromatin function.
IDRs are highly prevalent in chromatin and transcriptional regulators, implying an important function of these disordered regions in regulating chromatin activity [10–14]. IDRs are reported to mediate protein–protein and/or protein–nucleic acid interactions [10, 15], weak multivalent oligomerization into higher order complexes [13, 16], to regulate subcellular localization [17] and protein stability [10, 18]. Many nuclear proteins form dynamic and selective liquid–liquid phase separated (LLPS) condensates, via their IDRs, enabling the local spatial accumulation of related proteins. LLPS condensates are spherical in shape and characterized by dynamic and weak multivalent interactions, the ability to fuse, they increase in size as the external concentration increases, and may or may not involve nucleic acid interactions [19]. Some prominent examples include the nucleolus [20] transcription and replication factories [11], splicing speckles [21], and DNA damage foci [22]. However, depending on their molecular properties, IDRs can also form stronger and less dynamic interactions. For example, IDRs containing cationic stretches of amino acids, such as in histone H1 [23–25] or H5 [26, 27], neutralize DNA’s negative charge and invoke chromatin compaction and gene repression [28]. This phenomenon is not only true for linker histones but also for nonhistone proteins such as protamine [29], TDG [30], PRC1 [31], and HP1 [32].
PHF13 is composed of five IDR regions and is 55% disordered. PHF13’s polycationic nonspecific DNA binding region is an intrinsically disordered region (IDR2) that localizes PHF13 to the nucleus (NLS) and to chromatin via its DNA-binding affinity [1, 6]. In addition to this, PHF13 contains a putative RNA-binding region and nucleolar localizing sequence (IDR5) in its C-terminus. Our data indicate that PHF13’s DNA and putative RNA binding IDRs are involved in driving chromatin structural changes. Further stabilizing this interaction, we demonstrate that PHF13 can oligomerize via its ordered globular domains, creating a polyvalent chromatin binding protein, with alternating DNA- and histone-binding domains, which greatly increases its chromatin avidity and promotes PHF13 spreading along the chromatin fiber. Impressively, we find that a ∼3- to5-fold overexpression of PHF13 is sufficient to promote global chromatin compaction, dependent on its oligomerization via its N- and C- terminal ordered domains. Moreover, molecular dynamics (MD) simulations recapitulated these in vivo observations, indicating that PHF13 can induce chromatin phase transitions dependent on its polyvalent chromatin association. These features more closely resemble PPPS, namely a protein polymer acting on chromatin polymer, or chromatin associated proteins that crosslink different chromatin segments, that tend to compact the chromatin fiber. While PPPS may or may not involve multivalent chromatin interactions, this does not rely on IDR mediated homotypic multivalent interactions, and PPPS may or may not be spherical [33, 34].
Conversely, PHF13 has two intrinsically disordered PEST domains that were previously shown to regulate PHF13 stability [6]. Here we show that PEST1 (IDR1) and PEST2 (IDR3) contribute to IDR mediated oligomerization, which antagonizes PHF13’s chromatin affinity and PPPS state and promotes LLPS. ATAC- and mRNA-sequencing confirmed that PHF13’s LLPS and PPPS states differentially influenced chromatin structure and gene expression, demonstrating that PHF13 phase transitions regulate its chromatin impact and functions. These findings uncover an innate cross-talk between PHF13’s intrinsically disordered and ordered regions, in governing PHF13 chromatin valency and function. Furthermore, they reveal PHF13’s ability to phase transition between LLPS and PPPS states and suggest this as a potential general mechanism in governing chromatin architectural and transcriptional changes.
Materials and methods
Reagents and materials availability
The following plasmids generated in this study will be deposited to Addgene. The antibodies used in this study are listed below in Table 1.
Table 1.
Antibodies used in this study
| Antibody name | Clone number | Company | Catalog | Method |
|---|---|---|---|---|
| Actin | AC-15 | Sigma | A5441 | WB |
| Flag | M2 | Sigma | F1804 | WB |
| GAPDH | 6C5 | Santa Cruz | SC-32233 | WB |
| GFP | 7.1 and 13.1 | Roche | 11814460001 | WB |
| GST | Mouse mAb | Thermo Fisher | MA4-004 | WB |
| Histone H3 | E173-58 | Epitomics | 1326-1 | WB |
| Histone H3K4me3 | MC315 | Millipore | 04-745 | WB |
| Histone H3S10phos | 6G3 | Cell Signaling | 9706 | IF |
| Ki67 | Rb mAb EPR3610 | Abcam | ab92742 | IF |
| NoH61 | Rb pAb | Gift Schmidt-Zachmann | IF | |
| Nucleolin | Mouse mAb | Santa Cruz | Sc-8031 | IF |
| PHF13 | Rb pAb CR53 | gift Hans Will | IF | |
| PHF13 | Rb pAb S173 | gift Hans Will | IP | |
| PHF13 | Rb pAb CR56 | gift Hans Will | IP | |
| PHF13 m11 | Rat mAb 1D3 | Elizabeth Kremmer | WB, IF | |
| PHF13 m33 | Rat mAb 7F8 | Elizabeth Kremmer | IF | |
| PHF13 m34 | Rat mAb 4B2 | Elizabeth Kremmer | IF | |
| PHF13 m45 | Rat mAb 6F6 | Elizabeth Kremmer | IF, WB | |
| SMC3 | Rabbit | Abcam | Ab2693 | WB |
| Topoisomerase 1 | Rb pAb | Biozol | Ab3825 | IF |
| Tubulin | B-5-1-2 | Sigma | T5168 | WB |
| Vinculin | Mouse mAb | Abcam | Ab18058 | WB |
pEGFP-C1-PHF13
pFlag-CMV4-PHF13
pFlag-CMV4-PHF13(100-200)
pFlag-CMV4-PHF13 (1-150)
pFlag-CMV4-PHF13 (150-300)
pEYFP-C1-PHF13
pEYFP-C1-PHF13_NTD (21-70)
pEYFP-C1-PHF13ΔNTD (del21-70)
pEYFP-C1-PHF13 Δ24–40
pEYFP-C1-PHF13 (150–300)
PEYFP-C1-PHF13ΔPEST1
PEYFP-C1-PHF13ΔPEST2
pEYFP-C1-PHF13 Δ24–40Δ232–238
pEYFP-C1-PHF13 Δ24–40Δ272–280
pECFP-C1-PHF13
pECFP-C1-PHF13ΔNTD (del21-70)
pECFP-C1-PHF13ΔPEST2 (del141-190)
pECFP-C1-PHF13(1–150)
pECFP-C1-PHF13(150–300)
pECFP-C1-PHF13(100–200)
pECFP-C1-PHF13Δ24–40
pECFP-C1-PHF13_NTD (21-70)
pGEX-4T3-PHF13
pGEX-4T3-PHF13ΔNTD (del21-70)
pGEX-4T3-PHF13ΔPHD
pCDNA-4TO-PHF13
pCDNA-4TO-PHF13ΔNTD (del21-70)
pCDNA-4TO-PHF13Δ24–40
pCDNA-4TO-PHF13ΔPEST1(del52-88)
pCDNA-4TO-PHF13ΔPEST2(del141-190)
pCDNA-4TO-PHF13M246A
pCDNA-4TO-PHF13W255A
pCDNA-4TO-GAL4DDPHF13ΔNTD
Biological resources
Mammalian cell lines: The cell lines used in this study were not authenticated. All cell lines tested negative for mycoplasmas.
U2OS
HTB-96; Human epithelial Osteo-Sarcoma cells (female); wild-type p53 and Rb; p16-negative: were grown under standard culture conditions at 37°C and 5% CO2. Cells were grown in DMEM (Gibco) supplemented with 10% FBS (PanBio), 1× Penicillin/Streptomycin, 1× Hepes, and 1× sodium pyruvate.
U2OS PHF13 clone 5
U2OS cells were genetically modified with Tetracycline repressor and Tet-operated PHF13. This cell line was grown as described for U2OS cells, except that PHF13 was induced by the addition of doxycycline (1 μg/ml) to the medium. This cell line was verified for PHF13 expression using monoclonal antibodies against PHF13.
293T
Human Embryonic Kidney cells (fetal) were grown under standard culture conditions at 37°C and 5% CO2. Cells were grown in DMEM (Gibco) supplemented with 10% FBS (PanBio), 1× Penicillin/Streptomycin, 1× Hepes, and 1× sodium pyruvate.
H1299 cells
Human lung cell carcinoma cell line: P53 negative, were grown under standard culture conditions at 37°C and 5% CO2. Cells were grown in DMEM (Gibco) supplemented with 10% FBS (PanBio), 1× Penicillin/Streptomycin, 1× Hepes, and 1× sodium pyruvate.
Method details
Cell lysis and immunoblotting
Fractionation of lysate was performed by lysing the pellet for 10 min on ice in CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.1% NP-40) supplemented with 1× complete protease inhibitor cocktail (Roche), followed by 5 min centrifugation at 2000 × g. Supernatant containing the cytoplasmic proteins was discarded and the pellet resuspended in mild chromatin buffer (100 mM NaCl, 20 mM Tris–HCl pH 7.5, 0.5% Triton-X, 2 mM CaCl2, 2.5 mM MgCl2) plus complete protease inhibitors (Roche) for 10 min on ice, followed by 5 min centrifugation at 5000 × g. Supernatant was collected and stored as nucleoplasmic soluble fraction and the pellet was resuspended in mild chromatin buffer plus complete protease inhibitors and then sonicated on a Bioruptor Plus (Diagenode) for 10 cycles (30s on/ 30s off) at high intensity. Following sonication, 1 μl (250U) of Benzonase (Novagene) was added, and RNA/DNA was allowed to degrade on ice for 30 min, followed by centrifugation at 14 000 × g for 5 min. Supernatant was collected (chromatin fraction) and any residual pellet was discarded. Total cell lysis was performed by lysing cells in RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% NaDoc, 0.1% Sodium dodecyl sulfate (SDS)) supplemented with 1× complete protease inhibitor cocktail for 10 min on ice, followed by sonication on a Bioruptor Plus (Diagenode) for 10 cycles (30s on/ 30s off) at high intensity and then treatment with 1 μl (250U) of Benzonase (Novagene) on ice for 30 min, followed by centrifugation at 14 000 × g for 5 min. Supernatant (total cell lysis) was collected and residual pellet was discarded. Laemmli buffer was added and the lysates were denatured for 10 min at 99°C and then loaded on 4%–15% precast polyacrylamide gels (Bio-Rad) and transferred to polvinylidene fluoride (PVDF) membranes (Transblot Turbo-Biorad) using a Trans Blot Turbo Transfer System (Bio-Rad) and the mixed molecular weight program. Membranes were then blocked for 2 h at RT in 5% milk (in TBST) and then probed overnight with primary antibody. Membranes were then washed three times in 1× TBST followed by 1 h incubation in secondary antibody in 5% milk (in TBST) at RT. Membranes were then washed three times in 1× Tris-buffer saline with Tween 20 (TBST) and then developed using Pico, Dura (Pierce) or Western Lightning Ultra (Perking Elmer) on a Biorad ChemiDoc XRS.
G2 synchronization
U2OS cells were cultured for 21 h in medium supplemented with a Ro3066 inhibitor at a concentration of 10 μM, maintained at 37°C with 5% CO2. After the 21-h incubation period, the cells were collected and further processed.
Cell fractionation
Cells were harvested, counted in a cell counter (Eve Automated Cell Counter) pelleted in even numbers (1–5 × 106 cells) and washed 1× in Phosphase buffered saline (PBS). Cell pellets were resuspended in 198 μl Hypotonic Swelling Buffer (20 mM Tris–HCl pH7.4, 10 mM KCl, 2 mM MgCl2, 1 mM Ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA), 0.5 mM dithiothreitol (DTT), 0.5 mM Phenylmethylsulfonyl fluoride (PMSF), supplemented with fresh 1× Complete protease inhibitor from Roche) for 3 min on ice. Lysates were supplemented with 0.1% NP-40 (2 μl) and incubated for 3 min on ice and then pelleted at 1000 × g at 4°C for 5 min. Supernatant (cytoplasm) was removed and the pellet resuspended in 200 μl of Nucleoplasmic Buffer (20 mM Tris–HCl pH 7.4, 100 mM NaCl, 0.5% Triton X, supplemented with fresh 1× Complete protease inhibitor from Roche) for 10 min on ice followed by centrifugation at 2000 × g for 5 min. The supernatant (nucleoplasmic fraction) was collected and the pellet resuspended in 200 μl of Chromatin Buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 2 mM MgCl2, 0.1% SDS, 10% sodium deoxycholate-DoC, 1% NP-40) supplemented with 1× complete protease inhibitor (Roche) and 1 μl of -25U of Benzonase (Merck), sonicated in a Bioruptor for 10 cycles (30 on/30 off) on high intensity, briefly centrifuged, and incubated on ice for 30 min. Lysates were then centrifuged at 10 000 × g for 5 min and supernatant (chromatin fraction) retained.
Cell fractionation of EYFP expressing cells and G2 synchronized cells
Cells were trypsinized and pelleted by centrifugation at 500 × g for 4 min. The cell pellets were washed with ice-cold DPBS and centrifuged again at 500 × g for 4 min at 4°C. Cells were lysed with hypotonic buffer [20 mM Hepes (pH 8.0), 10 mM KCl, 1 mM MgCl2, 0.1% Triton X-100 (v/v), 20% glycerol (v/v), and 1× protease inhibitors] on ice for 10 min. Lysates were centrifuged at 2300 × g for 2 min at 4°C, and the cytoplasmic fractions (supernatant) were collected. The nuclear pellets were resuspended in hypertonic buffer [20 mM Hepes (pH 8.0), 150 mM NaCl or 400 mM NaCl, 1 mM EDTA, 20% glycerol (v/v), 0.1% Triton X-100 (v/v), and 1× protease inhibitors] and lysed on ice for 20 min with brief vortexing. Lysates were centrifuged at 20 400 × g for 5 min at 4°C, and the soluble nuclear fraction (supernatant) was collected. The pellets were resuspended in insoluble buffer [2 mM Tris, pH 8.0, 150 mM NaCl, 2 mM MgCl2, 1% SDS (wt/vol), 1% NP-40 (v/v), 10 mM iodoacetamide, and 1× protease inhibitors] with Benzonase, then shaken on a vortex genie for 50 min at 4°C. Lysates were centrifuged at 20 400 × g for 5 min at 4°C, and the insoluble nuclear fraction (supernatant) was collected.
Immunoblot signal quantification
Chemiluminescence signals were quantified using Image Lab 6.1 software (Bio-Rad). The volume tool function was employed by delineating a fixed area per blot, utilizing the subtraction method “local” to extract the adjusted volume values. For each transfection experiment, the adjusted volume values of fractions (cytoplasmic, soluble nuclear, and insoluble nuclear) were summed, and the percentage of each fraction was calculated accordingly. Any negative adjusted volume values were adjusted to zero. Data visualization and plotting were performed using GraphPad Prism software.
Immunoprecipitation
Immunoprecipitations were performed with 2 × 106 cells per IP condition and 25 μl of Flag-M2 agarose (Sigma) or 10 μl of magnetic GFP-Trap (Chromtek) beads. IPs were either performed from chromatin fraction or RIPA lysates (see above) which were diluted 1:5 in dilution buffer (20 mM Tris–HCl pH 7.4, 100 mM NaCl, supplemented with 1× complete protease inhibitor and 2× PhosStop) or (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA supplemented with 1× complete protease inhibitor and 2× PhosStop), respectively, and then rotated overnight at 4°C. The next day the beads were washed 4× with the 1:5 diluted IP buffer (5 min each) and then resuspended in the lysis buffer supplemented with 1× Laemmli buffer, heated for 10 min at 99°C, and loaded onto 4%–15% gradient gels (Biorad).
Luciferase reporter assays
Luciferase assays were performed in H1299 or U2OS cells by polyethylenimine (PEI) co-transfection of equivalent amounts (0.5 μg/12-well) of a luciferase reporter (pGalTK-Luc), Renilla-luciferase (pRL-TK), and Gal4-tagged Ad5-E1B55K, PHF13, or PHF13 mutants (in pM2 vector) or by co-transfection with pcDNA4TO-PHF13 or PHF13 deletion constructs with pCyclinG-Luc or pMdm2-Luc and human p53 (0.015 μg) for 24 h. Luciferase activity was measured on an automated luminometer (Lumat LB9510; Berthold Technologies) using Dual Luciferase kit (Promega). All samples were normalized for transfection efficiency by measuring Renilla-luciferase activity.
GST protein production
BL21 ecoli carrying pGEX4T3 -PHF13 Glutathione-S-transferase (GST) fusion proteins were grown in 50 ml of LB-medium supplemented with 100 μg/ml ampicillin and 3.4 μg/ml chloramphenicol under rotation (200 rpm) at 35°C, overnight. 200 ml of YT medium was inoculated with 5 ml of the overnight culture and incubated under rotation (200 rpm) at 37°C until an OD600 of 0.5 was reached. Expression of GST-fusion proteins was induced for 4 h at 32°C with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). After induction, the bacterial suspension was centrifuged for 15 min at 5000 rcf (4°C). Cell pellets were stored at −80°C overnight and then resuspended in 10 ml of ice-cold lysis buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 5 mM EDTA, 1% Triton-X-100, 30 mg of Lysozyme, and 1 complete protease tablet) and incubated for 10 min on ice. The lysate was then sonicated five times for 10 s and then centrifuged at 16 400 rcf for 30 min at 4°C. The clarified supernatant was then incubated with 500 μl of 50% glutathione agarose suspension in GST purification buffer (50 mM Tris–HCl pH 8, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40) and incubated under rotation for 2–4 h at 4°C. GST beads were then washed 4× in 10 ml of GST purification buffer and briefly centrifuged (300 × g, 1 min, 4°C) in between. Finally, the beads were resuspended in 900 μl of GST purification buffer and stored in aliquots at −80°C. The purity and quantity of prepared GST-fusion proteins bound to glutathione agarose was analyzed by sodium sodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE).
GST pulldowns
Cells were lysed on ice for 10 min with a CSK buffer [10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.1% NP-40, 1× complete protease inhibitor cocktail (Roche), and 1 mM PMSF] and then centrifuged for 5 min at 2000 × g. The cytoplasm was discarded and the pellet was then lysed for 10 min on ice in a mild chromatin buffer [20 mM Tris–HCl pH 7.5, 100 mM NaCl, 0.5% Triton-X 100, 2 mM CaCl2, 2.5 mM MgCl2, and 1× complete protease inhibitor cocktail (Roche)] to generate nucleoplasmic fractions. The cells were then centrifuged for 5 min at 10 000 × g. The supernatant was discarded and the pellet was then lysed in the mild chromatin buffer supplemented with 1× complete protease inhibitor cocktail (Roche) and benzonase (1 μl/3 × 106 cells; Novagene 250 U/μl 2 698 486) sheared for 10 min on a Bioruptor (30 s on 30 s off at high intensity) and incubated on ice for 30 min. The cells were then centrifuged for 5 min at 10 000 × g, and the chromatin supernatant was collected. GST pull downs were performed using 1, 2.27, 1.92, and 1.92 μg of recombinant GST, GST-PHF13, GST-ΔPHD, and GST-ΔNTD proteins bound to Glutathione sepharose beads, respectively, and nuclease digested chromatin lysate dilute 1:5 in dilution buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, and 1× complete protease inhibitor cocktail). The beads were incubated with the lysate at 4°C for 4 h and then washed several times in the diluted chromatin buffer, prior to denaturation in Laemmli buffer and loading on an SDS–PAGE gel.
In vitro turbidity and droplet formation assays
For the in vitro turbidity assay and droplet formation, recombinant fusion proteins were measured for concentration, and then diluted or mixed to equimolar final concentrations in storage buffer (50 mM Tris pH 7.5, 125 mM NaCl, 1 mM DTT, and 10% glycerol) and then imaged for the turbidity assay. For the condensate formation assay, these solutions were further diluted 1:1 in PEG-8000 in de-ionized water (w/v). Ten microliters of this suspension was pipetted onto chambered coverslips, and immediately imaged using a LSM880 confocal microscope equipped with a ×63/1.40 oil DIC objective. All images were acquired from within the solution interface and performed before droplets settled onto the bottom of the coverslip [35].
In vitro transcription/translation
In vitro translation was performed using a human cell free in vitro translation kit (Thermo Scientific; Cat.: 88855) according to the manufacturer’s instructions. Briefly, PHF13 and PHF13ΔNTD were cloned into the provided pT7-CFE1-CHis vector. A stop codon was inserted prior to the CHis to prevent the C-terminal fusion of PHF13 to the hexaHis tag. To obtain higher protein yields, the translation reaction was performed for 6–8 h and 2 h after the translation reaction was initiated additional 2 μl of transcription product were added. As a positive control, pT7-CFE1-CHis expressing eGFP was transcribed and translated in parallel and analyzed by fluorescence microscopy and western blot. Expression of PHF13 and PHF13ΔNTD was verified by Western blot.
Chromatography
Chromatography was performed using recombinant proteins cleaved of their GST-tags (PreScission protease; GE Healthcare) on a Superose6 10/300 GL column (GE Healthcare) run in HMG250 buffer [25 mM HEPES (pH 7.5), 2.5 mM MgCl2, 0.5 mM EDTA, 10% glycerol, 0.05% IGEPAL, and 250 mM KCl) with a flow rate of 0.3 ml/min and a fraction size of 0.5 ml/fraction. Size-exclusion chromatography of in vitro translated proteins (generated with a human cell free in vitro translation kit according to the manufacturer’s instructions; Thermo Scientific, Cat. no. 88 855) was performed on a Superdex 200 5/150 GL column (GE Healthcare) run in 50 mM phosphate buffer (25 mM KH2PO4, 25 mM Na2HPO4 * 2H2O, 150 mM NaCl). Five hundred microliters of fractions were collected with a flow rate of 0.3 ml/min.
Transfection
Condensation experiments were performed in U2OS cells by transfection with Fugene 6, Fugene HD or Fugene 4K according to manufactures instructions using 0.5–1 μg of PHF13 expression plasmid per 6-well for 24 h.
FACS FRET
Fluorescence activated cell sorting (FACS) -based fluorescence resonance energy transfer (FRET) analysis were performed as previously described [36]. In brief, 293T cells were transfected with 1 μg of CFP and YFP constructs per 12-well using standard calcium-phosphate transfection for 24 h. Cells were resuspended in FACS buffer (1× PBS, supplemented with 1% FBS and 0.5 mM EDTA), and double positive cells were analyzed for occurrence of FRET by fluorescence cell sorting with a BD-FACS Canto 2.
Indirect immunofluorescence
Cells were grown on 10-mm round coverslips or on ibidi eigth-well dishes. Cells were fixed for 10 min with 4% paraformaldehyde at RT and then washed once with 1× PBS prior to permeabilization with 0.5% Triton-X for 10 min at RT, followed by three washes (5 min) with 1× PBS and 2 h blocking in 5% BSA/PBS. Blocked cells were then incubated for 1 h at RT with primary antibodies (using supplier recommended concentrations; or 1:200 PHF13 rabbit polyclonal antibodies or undiluted PHF13 rat monoclonal antibodies), followed by three washes (5 min) with 1× PBS and 1 h incubation with secondary antibodies and DAPI or Hoechst (Molecular Probes/Abcam 1:1000) and a final by three washes (5 min) with 1× PBS. Coverslips were then mounted on glass slides using Mowiol (Sigma) and allowed to harden before imaging. Pre-extraction IF experiments were performed using the same protocol except that the coverslips were first treated with CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 0.1% NP-40) for 20 min at RT and washed twice in PBS prior to 4% paraformaldehye (PFA) fixation and 0.5% Triton-X permeabilization. To look at the impact of transcriptional inhibition or GSK3β on PHF13 cellular localization, cells were treated either with 40 mM LiCl for 6h to inhibit GSK-3β activity or with 0.5 μg/ml Actinomycin D (AMD) for 4 h to block RNA polymerase activity, prior to fixation and preparation for imaging. Images were obtained by light microscopy using an Axiophot microscope (Zeiss), Observer (Zeiss), and Confocal microscope (Confocor 2; Zeiss).
Microscopy
To ensure, quantifiable linear signal values all image acquisitions followed strict rules to prevent over/under exposed pixels/used. All used signal intensities were adjusted by exposure time, dwell time, and laser/led power to achieve at least 95% of pixels within the lower 30% of the maximal detector’s digitalization ability. Furthermore, all images were digitalized in 8-bit. Mean Intensities are interpreted as densities, while sum intensities exhibit a total of stained structures/molecules. The standard deviation utilized as hetero/homogeneity-factor of a given signal within regions of interest (ROIs).
Quantification of phenotypes
To assess the levels of PHF13 protein and the chromosome condensation phenotype, we utilized an image data analysis approach. Image quantification analysis was performed using the ZEN 3.4 Image Analysis module. Nuclei were identified within tile series images via mild smoothing, fixed DAPI intensity thresholds, closing operations, and watershed segmentation. Resulting masks were filtered by area and circularity. The analysis provided output parameters for different channels, including mean intensity, total intensity, and intensity standard deviation. Mean intensity values were interpreted as density measurements, while total intensity reflected the overall amount of stained structures or molecules. The standard deviation was used to assess the heterogeneity or homogeneity of the signal within ROIs. Data visualization was performed using GraphPad Prism and R package ggplot2, with plot refinements made in Adobe Illustrator.
The data were gated based on the total intensity of PHF13 signal, dividing the cells into four groups: nontransfected, low overexpression, high overexpression, and outliers. For example, the gating strategy for Figs 4I and 5H is shown below.
Figure 4.

PHF13 drives global chromatin condensation. (A–H) IF: Phase contrast (A–C and F) and DIC imaging (D) of U2OS (A and C–H) or U2OS H2B-GFP (B) cells transfected with PHF13 (A–F), YFP-PHF13 (G), or CFP-PHF13 (H) for 24 h and paraformaldehyde fixed (A–E and G–H) or pre-extracted prior to fixation (F). PHF13 was detected with rabbit peptide Ab CR53 (A and F) or rat mAb 6F6 (B–E) and DNA was stained with Dapi (A, B, and F) or with DRAQ5 (D). Images were captured on a Zeiss Axiophot Phase Fluorescence microscope (A, B, andF), a Zeiss Z1 Observer (E, G, and H) or a Zeiss ConfoCor 2 LSM5 microscope (D). (I) Graphical depiction of the penetrance of the chromosome condensation phenotype for low (1- to 5-fold) and high (5- to 15-fold) PHF13-expressing cells in comparison to nontransfected endogenous PHF13-expressing cells. Dotted line represents background signal in nontransfected cells.
Figure 5.

PHF13 oligomerization is required for chromatin compaction. (A–G and I) IF staining of paraformaldehyde fixed U2OS cells, transfected with PHF13 (A), PHF13ΔNTD (B), PHF13Δ24-40 (C), PHF13ΔPEST1 (D), PHF13ΔPHD (E), PHF13-M246A (F), PHF13-W225A (G), and PHF13-GAL4DDΔNTD (I). PHF13 was stained with rat mAb 6F6 (A and E–G) or 1D3 (B–D and I) and DNA was stained with DRAQ5. Nucleoli were detected with NOH61 or Ki67 antibodies. Images were captured on a Zeiss Confocor 2 LSM5 microscope. (H) Graphical depiction of the penetrance of the chromosome condensation phenotype for PHF13 full-length, PHF13ΔNTD, PHF13Δ24-40, PHF13ΔPHD, PHF13-M246A, PHF13-W225A, and PHF13ΔPEST1 overexpressing mutants in U2OS cells. Black dotted line represents background signal in nontransfected cells.
Cells were categorized into these four groups based on total PHF13 signal intensity. The intensity range for the nontransfected group was empirically determined using the PHF13 total intensity distribution from the control (nontransfected well). After establishing this baseline, the low overexpression group was defined as having PHF13 total intensity levels from baseline up to 5-fold higher than the nontransfected group, while the high overexpression group was defined as having PHF13 total intensity levels between 5- and 15-fold higher than the nontransfected group. Cells with PHF13 intensity levels that did not fit within these three defined groups were classified as outliers and excluded.
Each cell within these expression groups was assigned a chromatin compaction phenotype status (yes = chromatin compaction or no = no chromatin compaction) based on the standard deviation of bright field intensity. Cells exhibiting a standard deviation of bright field intensity ≥4 were classified as having the “yes” phenotype, while those with values below 4 were designated as “no” phenotype. The penetrance of the chromatin compaction phenotype in each PHF13 expression group was calculated as the ratio of cells with a bright field intensity standard deviation (au) ≥ 4 to the total number of cells in that group.
Finally, we calculated the penetrance of the phenotype (YES or NO) as the ratio between the number of cells with standard deviation of bright field intensity ≥ 4 over the number of cells with standard deviation of bright field intensity < 4. GraphPad Prism and the ggplot2 package of R were used to plot the data.
The same parameters were used to calculate the penetrance of PHF13 versus, ΔNTD, Δ24-40, ΔPHD, M246A, W255A, ΔPEST1, ΔPEST2, Δ24-40Δ232-238, Δ24-40Δ272-280, and Δ8 condensation phenotype.
Flow cytometry
YFP + cells were sorted and collected into ice-cold FACS medium (DMEM phenol-free (Gibco, catalog no. 21 063) supplemented with 2% FBS (Pan Biotech, catalog no. P30-2002), 1% Penicillin–Streptomycin (Gibco, catalog no. 15 140 122) and 3.4 mM EDTA (PanReac AppliChem, catalog no. A4892). One to two million YFP + sorted cells, per condition were then used for RNA extraction step for downstream RNA-seq and 100 000–500 000 YFP + sorted cells, per condition were used for ATAC-seq experiments.
RNA-sequencing
Following sorting, nuclei were isolated using the Nuclei EZ Prep Kit (Sigma–Aldrich, catalog no. NUC101) according to the manufacturer’s instructions for cell suspensions. The isolated nuclei were stored at -80°C in RTL buffer (QIAGEN, catalog no. 79216) supplemented with 2 M DTT. RNA was extracted from the nuclei employing the RNeasy Plus Mini Kit (QIAGEN, catalog no. 74134), following the manufacturer’s guidelines. RNA concentration was assessed using the Qubit™ RNA High Sensitivity (HS) Assay (Thermo Fisher, catalog no. Q32852) while fragment size distribution was determined using the Agilent RNA 6000 Nano Kit (Agilent Technologies, catalog no. 5067-1511) on the 2100 Bioanalyzer (Agilent Technologies). Library preparation was conducted using 300 ng of RNA per condition with the Illumina Stranded mRNA Prep Kit (Illumina, catalog no. 20040893, 20040897, 20040895, 20040899, and 20091649) per the manufacturer’s protocol. Libraries concentration was assessed using the Qubit™ DNA HS Assay (Thermo Fisher, catalog no. Q32851) while fragment size distribution was determined using the Agilent HS DNA Kit (Agilent Technologies, catalog no. 5067-4626) on the 2100 Bioanalyzer (Agilent Technologies). Sequencing of the libraries was performed on the AVITI platform (Element Biosciences).
ATAC-sequencing
A total of 100 000 sorted cells per condition were used in an optimized ATAC-seq protocol as described by Corces et al. [37]. Briefly, all samples were spiked-in with 7500 Drosophila nuclei (Active Motif, catalog no. 53154) and resuspended in 50 μl of Illumina resuspension buffer supplemented with 1.25 μl of Nextera Tn5 Transposase (TDE1; Illumina, catalog no. FC-121-1030). The reaction was incubated at 37°C for 30 min in a thermomixer (1000 rpm) prior to stopping the reaction. DNA was purified using Zymo DNA Clean & Concentrator Kit (Zymo Research, catalog no. D4011) and eluted with 20 μl of elution buffer. The resulting DNA was amplified using NEBNext High-Fidelity 2× PCR Master Mix (NEB, catalog no. M0541) and a unique combination of Barcoded PCR Primer [38] and purified with AMPure XP beads (Beckman Coulter, catalog no. A63881) and paired end sequenced using the AVITI platform (Element Biosciences).
RNA-seq data processing
RNA-seq data from U2OS cells were filtered and trimmed for adapters using TrimGalore v0.6.10 and then mapped to a custom human genome hg38, including the cloned mYFP sequence, using STAR aligner v2.7.11b with default parameters [39]. Count-read tables were generated by using the same program. Differential expression analysis was performed using the DEseq2 package in R version 4.4 [40]. Differentially expressed genes were defined as having a fold-change >1, adjusted P-value smaller than or equal to ≤0.01, and a minimum mean read count across the experiment samples of 30 reads. Principal component analysis (PCA) was carried out using the PCAPlot function from the DEseq2 package on the normalized read matrix that was transformed using the variance stabilizing transformation (VST) function from the DEseq2 package and plotted using ggplot2. Volcano plots were created using ggplot2. Heatmaps were plotted with the aid of seaborn v0.13.2 in Python 3.10, and cluster analysis was performed using the clustermap function using Euclidean distance and Ward linkage. Functional analysis of differentially expressed genes and clusters was performed using g:Profiler with the default parameters [41]. Bigwig files for visualization were obtained with deeptools bamCoverage using 50 bp bins and CPM normalization [42]. Genome tracks visualization was done with pygenometracks tool set [43].
ATAC-seq data processing
ATAC-seq data from U2OS cells were filtered and trimmed for adapters using BWA v0.7.17 [44]. Samples were sequenced in two runs, and both fastq files were used as inputs. Samples were mapped to the human genome hg38 and Drosophila melanogaster dmel r6.61 genome for spike-in normalization. Samtools v1.20 [45] was used for SAM-to-BAM conversion, sorting, indexing, and counting mapped reads for normalization. Picard v3.3.0 was used to remove duplicated reads (http://broadinstitute.github.io/picard/). Bedtools v2.31.1 was used to create bedgraph files with genomecov function [46]. Coverage was then normalized by spike-in factor and normalized bedgraph files were converted to bigwig for visualization using UCSC bedGraphToBigWig tool.Peak calling was performed using MACS3 v3.1. using broadpeak parameters [47]. Analysis and differential peak calling were done with DiffBind v3.6.5. Genome track visualization was performed using the PyGenometracks tool set. The count table for the peak was created using featureCounts v2.0.8 using bedtools to obtain a common set of peaks from all replicates [48]. Heatmaps were plotted as described above. All other plots were created using ggplot2.
Reprocessing of published ChIP-seq data (mouse)
To estimate genomic binding of PHF13 for MD simulations, ChIP-seq analysis from fastq files of PHF13, SMC3, RAD21, CTCF, H3K4me3, H3K4me1, H3K27ac, H3K27me3, and H3K9me3 were downloaded via Array Express (https://www.ebi.ac.uk/arrayexpress/ for PHF13) or fastq-dump (other experiments). Reads were mapped to mm10 reference genome using Bowtie2 [49] (–very-sensitive), and if applicable, mapped reads from the same experiments but different sequencing runs were first merged and then filtered (-h -b -F 282 -q 10) with SAMtools [44]. Reads mapping to blacklisted regions (ENCFF547MET) (Dunham et al., 2012) were filtered out with SAMtools as well. Signal tracks were computed with bamCoverage (-of bigwig -bs 10 -e 300 –normalizeUsing RPKM –ignoreDuplicates) [42]. Regions with significant enrichment over input sample were identified using MACS2 (-g mm –keep-dup auto –bw 300 -q 0.05 -f BAM) [47] using the –broad option for SMC3, RAD21, and PHF13 ChIP data. Heatmaps (computeMatrix - reference-point mode and plotHeatmap tool of the deepTools package) [42], correlation analysis (multiBigwigSummary and plotCorrelation [42], and visual inspections of the tracks (IGV and USCS genome browsers) were then used to depict normalized ChIP-seq signal distribution and was the basis of our MDs model.
Molecular dynamics simulations details
The system composed by polymer beads and PHF13 molecules experiences thermal fluctuations at temperature T and the particles obey to the Langevin equation (Allen and Tildesley, 1989). For sake of simplicity, all monomers have same diameter σ and mass m, both set to 1 in dimensionless units [50], unless explicitly stated. To account for excluded volume effects, we use between any two particles a repulsive Lennard-Jones (LJ) potential, with length scale σ and energy scale ϵ (KBT units). Consecutive beads of the polymer are linked by a finitely extensible nonlinear elastic spring (FENE; [50]), with standard parameter [51, 52] (length constant 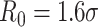 and spring constant
and spring constant  ). Polymer stiffness is modeled through a standard three body interaction:
). Polymer stiffness is modeled through a standard three body interaction:  , where
, where  is the angle formed by three consecutive beads and
is the angle formed by three consecutive beads and 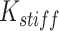 is set to
is set to 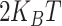 . Bonds between monomers of PHF13 are modeled as harmonic springs:
. Bonds between monomers of PHF13 are modeled as harmonic springs: 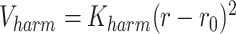 , with
, with 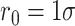 and spring constant
and spring constant  ).
).
All attractive interactions, i.e. protein–protein and protein–chromatin interactions, are modelled by a short-range, shifted attractive LJ potential  :
:
 |
for distances below the interaction threshold 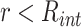 (
(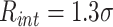 ), 0 otherwise. The interaction affinity
), 0 otherwise. The interaction affinity  between two generic types A and B is given by the minimum of
between two generic types A and B is given by the minimum of 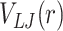 and is controlled by ϵ and
and is controlled by ϵ and 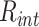 . In our simulations, we consider specific affinities taken from the following ranges, which ensure the coil–globule transition of the polymer [52]:
. In our simulations, we consider specific affinities taken from the following ranges, which ensure the coil–globule transition of the polymer [52]:  ,
, 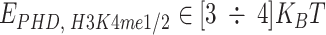 ,
, 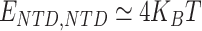 and
and 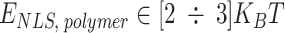 . Note that all the affinities are taken in the weak biochemical range, i.e. as typical protein–DNA and protein–protein interactions investigated in similar polymer models of chromatin [51, 52]. Values reported in table of Supplementary Fig. S6B indicate the exact affinities used for the simulations reported throughout the paper and figures. Simulations are performed with the software LAMMPS [53]. The parameters defining the Langevin equation are set to standard values [50]: friction coefficient
. Note that all the affinities are taken in the weak biochemical range, i.e. as typical protein–DNA and protein–protein interactions investigated in similar polymer models of chromatin [51, 52]. Values reported in table of Supplementary Fig. S6B indicate the exact affinities used for the simulations reported throughout the paper and figures. Simulations are performed with the software LAMMPS [53]. The parameters defining the Langevin equation are set to standard values [50]: friction coefficient 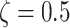 , temperature
, temperature  and integration time step
and integration time step 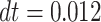 [54], expressed in dimensionless units. The system is confined in a cubic simulation box with periodic boundary conditions, with edge size
[54], expressed in dimensionless units. The system is confined in a cubic simulation box with periodic boundary conditions, with edge size  , in order to minimize finite size effects. Each simulation starts with the polymer initialized to a random Self-Avoiding-Walk (SAW) configuration [52]. Analogously, when two polymers are simulated (Supplementary Fig. S6F, central panel), they start in a SAW configuration and are separated within the simulations box. PHF13 molecules are uniformly distributed in the box, with a monomer concentration per volume unit
, in order to minimize finite size effects. Each simulation starts with the polymer initialized to a random Self-Avoiding-Walk (SAW) configuration [52]. Analogously, when two polymers are simulated (Supplementary Fig. S6F, central panel), they start in a SAW configuration and are separated within the simulations box. PHF13 molecules are uniformly distributed in the box, with a monomer concentration per volume unit  sampled in the range
sampled in the range  , where
, where 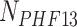 is the total number of PHF13 molecules and
is the total number of PHF13 molecules and 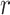 the radius of the monomer. Note that c is related to the molar concentration cm through the equation cm = c/
the radius of the monomer. Note that c is related to the molar concentration cm through the equation cm = c/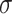 3NA, NA Avogadro number and
3NA, NA Avogadro number and 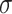 mapped in physical units [52]. For each parameter choice, we perform 10 independent simulations, which are equilibrated up to 3 × 107 timesteps, so to ensure the coil–globule phase transition. Polymer configurations are sampled every 106 timesteps after the transition.
mapped in physical units [52]. For each parameter choice, we perform 10 independent simulations, which are equilibrated up to 3 × 107 timesteps, so to ensure the coil–globule phase transition. Polymer configurations are sampled every 106 timesteps after the transition.
To quantitatively evaluate the structural differences observed in the different models, we computed the sphericity of the equilibrium polymer configurations, using the standard geometrical expression π1/3(6V)2/3/A, where V is the volume and the surface of the structure respectively, estimated as a convex hull computed from the position of the polymer beads with the ConvexHull function from the Python package scipy.spatial (Supplementary Fig. S6G). Since this approximation tend to provide regular and smoothed surfaces, the returned sphericities are generally high (>0.7). Using a-sphericity as shape descriptor computed from the gyration tensor returned similar results (data not shown).
Simulation of PHF13 mutations mutations mutations and model variants
To simulate the effect of mutations on PHF13 molecules we act in general on the interaction potential between the PHF13 monomers involved in the mutation and chromatin binding sites, keeping the rest of the system mostly unchanged. Therefore, deletion of the PhD domain is modeled by silencing all the attractive interactions involving PhD, i.e. by setting  with
with  and
and  .
.
Analogously, deletion of the NTD domain is modeled by silencing the attractive interactions involving NTD, i.e. by setting  , with
, with  and
and  . Here, phase separation of PHF13 is no longer observed, while residual interactions due to PhD-H3K4me3 persist. Upon reduction of
. Here, phase separation of PHF13 is no longer observed, while residual interactions due to PhD-H3K4me3 persist. Upon reduction of  such interactions disappear, compaction is no longer observed and the polymer remains in an open, randomly folded configuration (Fig. 6).
such interactions disappear, compaction is no longer observed and the polymer remains in an open, randomly folded configuration (Fig. 6).
Figure 6.

Molecular dynamic simulations and model of PHF13 induced condensation. (A) PHF13 is modeled as 3 beads representing the NTD, NLS, and PHD, which mediates PHF13 self-interaction and the interactions with chromatin and with chromatin interaction partners. (B and C) IP:WB GFP-TRAP from U2OS cells expressing YFP-tagged proteins and probed for the co-precipitation of endogenous cohesin proteins (B) or endogenous PRC1 proteins (C). (D and E) Polymer folding dynamics from initially randomly open configurations is monitored by the gyration radius (D). When all PHF13 interactions are enabled and no indirect C-terminal dimerization with a chromatin partner is considered, the polymer stably folds into compacted structures (E) and the gyration radius sharply decreases (PHF13, red curve; D). Deletion of the PHD (ΔPHD) or the NTD domain (ΔNTD) prevent polymer compaction, which remains in an open conformation (D and E). (F and G) Polymer model end states of cohesin/PRC1-only (F) or PHF13 + cohesin/PRC1 (G). (H) Model: PHF13 is able to oligomerize via direct N-terminal dimers. This oligomerization creates a PHF13 polymer with alternating DNA binding (cation–π interactions) and H3K4me1/2/3 binding (PHD domain) regions allowing it to spread along chromatin fiber and facilitate linear compaction which may be coupled to cohesin or PRC1 (gray ovals) long distance looping, to promote global chromosome condensation.
Simulations of PHF13 having beads with diameter 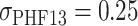 (Supplementary Fig. S6F, left panel) were performed as previously described, with length parameters (
(Supplementary Fig. S6F, left panel) were performed as previously described, with length parameters (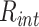 and diameters in LJ potential) rescaled to match the affinity range above reported. In this case, integration of the Langevin equation was performed with timestep
and diameters in LJ potential) rescaled to match the affinity range above reported. In this case, integration of the Langevin equation was performed with timestep 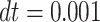 .
.
Polymer model including PHF13 and cohesin
Simulations including cohesin are performed by introducing in the above-described system an additional type of molecule, modeled as a dimer of two beads having same diameter and connected by harmonic spring. Such molecules can bind to specific sites regularly located along the polymer (density ∼0.2, two consecutive sites every 11 beads) with a very strong interaction (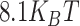 ), the other interaction affinities taken from the lower part of the above reported ranges. Concentration is taken as 10% of PHF13. Perturbations of the system are simulated as described before. Hence, depletion of cohesin is simulated by simply switching off its attractive interactions and keeping unchanged the rest of the system. Conversely, depletion of the entire PHF13 is simulated by switching off all its attractive interactions with chromatin. In this case, the observed elongated structures depend on the distribution of these binding sites, as higher binding site densities would lead to more compact configurations [55].
), the other interaction affinities taken from the lower part of the above reported ranges. Concentration is taken as 10% of PHF13. Perturbations of the system are simulated as described before. Hence, depletion of cohesin is simulated by simply switching off its attractive interactions and keeping unchanged the rest of the system. Conversely, depletion of the entire PHF13 is simulated by switching off all its attractive interactions with chromatin. In this case, the observed elongated structures depend on the distribution of these binding sites, as higher binding site densities would lead to more compact configurations [55].
Polymer model including loop extrusion
loop extrusion process (Supplementary Fig. S6F, right panel) is simulated as previously described [56]. Briefly, extruders are modelled as harmonic springs (elastic constant 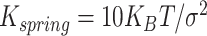 and equilibrium distance
and equilibrium distance 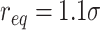 ), using extruder density ∼125 kb (i.e. 1 extruder every 5 beads) and processivity 500 kb, in line with recent computational implementations [56, 57]. Extrusion steps occurs every 500 MD timesteps.
), using extruder density ∼125 kb (i.e. 1 extruder every 5 beads) and processivity 500 kb, in line with recent computational implementations [56, 57]. Extrusion steps occurs every 500 MD timesteps.
Website/Database/in silico data
Ordered and putative interaction domains were predicted using Tango [58]. Disordered regions in PHF13 were called using PONDR [59], and inferred from AlphaFold2-advanced results. AlphaFold2-advanced [60] was used to predict if PHF13 dimerizes and to identify interacting regions. The prediction of nucleolar localization was done using the NoD -nucleolar localization sequence detector algorithm [61]. The predicted ability of PHF13 to phase separate was performed using the following algorithms PSP predictor [62], ParSe v2 [63], and FuzDrop [64].
Results
PHF13 N-terminal domain is essential for repression
PHF13 is a highly conserved (Supplementary Fig. S1A) H3K4me3 epigenetic reader, transcriptional regulator and modulator of chromatin architecture. PHF13 interacts with key epigenetic and transcriptional repressors and has been extensively reported to positively and negatively regulate gene expression, DNA damage response, chromatin structure, and cell division [1–7]. While we have previously demonstrated that PHF13 contacts chromatin in a multivalent manner via its C-terminal PHD domain and a central DNA-binding region [1], how PHF13 chromatin association is regulated is not understood. To address this question, we decided to molecularly dissect PHF13 and look at the consequence on chromatin association, transcriptional regulation, and chromatin structure.
PHF13 is composed of several regulatory domains (Fig. 1A and Supplementary Fig. S1A), including nuclear (NLS) and nucleolar (NoLS; Supplementary Fig. S1B) localizing sequences, histone and DNA-binding domains (PHD and polycationic stretch embedded in the NLS, respectively), two PEST domains which regulate its half-life, and a conserved N-terminal domain (NTD) of unknown function [1, 6]. To explore PHF13’s influence on transcription and which domains are involved in PHF13 transcriptional regulation, we utilized luciferase assays. To this end, we fused luciferase to different promoters, and measured the impact of PHF13 or mutant proteins on luciferase expression.
Figure 1.

PHF13 represses gene expression in an NTD-dependent manner. (A) Schematic overview of PHF13 domain structure. (B– E) Luciferase assays: Luciferase reporter gene expression in H1299 cells from a GAL4-responsive promoter (B and C) after transfection with GAL4 alone (pM2) or GAL4 fusion proteins (PHF13, PHF13ΔNTD, PHF13ΔPHD, or E1B) or from p53 regulated cyclin G and Mdm2 promoters (D and E) that were co-transfected with p53 and the indicated pcDNA-4TO expression vectors for 24 h. E1B served as a positive control. Immunoblots of the expressed proteins are shown above each plot (D and E). All values were normalized to Renilla expression (B–E).
PHF13 targeting to a GAL4-responsive promoter repressed luciferase expression in a dose-dependent manner and to a similar extent as GAL4-fused Ad5-E1B55K (Fig. 1B and C, and Supplementary Fig. S1C) a potent repressor [65]. Notably, we observed a loss of repression with the GAL4-tagged PHF13-ΔNTD protein whereas deletion of the PHD domain had no consequence (Fig. 1C). Similar effects were also observed for p53 dependent cyclin G and Mdm2 promoters (Fig. 1D and E, and Supplementary Fig. S1D and E). Minimal luciferase expression was observed for cyclin G and Mdm2 promotors in the absence of exogenous p53 in H1299 cells (p53 deficient). However, co-expression with p53 greatly stimulated luciferase expression from both promoters which was strongly repressed by PHF13, PHF13-ΔPHD, and Ad5-E1B-55K but not by PHF13-ΔNTD (Fig. 1D and E, and Supplementary Fig. S1D and E). These results indicate that in this assay at these promoters, PHF13 functionally represses transcription, which is dependent on its NTD but not its PHD domain (Fig. 1C–E and Supplementary Fig. S1C–E), suggesting that the conserved NTD with unknown function is required for PHF13 transcriptional repression.
PHF13’s NTD is a direct dimerization domain
To understand the importance of the NTD in transcriptional repression, we first utilized different in silico analysis to gain an understanding of its putative role (Fig. 2A and B). Cross analysis of Predictor of Naturally Disordered Regions (PONDR, red peaks) and TANGO (black peaks), which determine the propensity of a protein to be disordered (PONDR) [59] or to form ordered aggregates/self-associate (TANGO) [58], predicted that PHF13 is a predominantly disordered protein and identified two structured domains at residues 30–40 within the NTD with high aggregation potential and bookmarking the PHD domain at residues 232–238 and 272–280 with a weaker albeit above threshold aggregation probability (Fig. 2A). In addition to the strong ordered aggregation domain predicted by TANGO, PONDR indicated that the NTD contains as well an IDR, which is like-wise reported to mediate protein–protein interactions (Fig. 2A).
Figure 2.

PHF13‘s NTD mediates PHF13 homo-dimerization. (A and B) In silico analysis: Merged PONDR and Tango plots of PHF13 measuring disorder and order, respectively (A). Red and black dotted lines represent the cut offs for calling disorder and aggregation potential, respectively (A). AlphaFold2_advanced analysis of PHF13 dimers (shown as a string model and contact map). The confidence of the called structure is colour coded (B). (C and D) Schematic of constructs used in IP:WB. Flag-IP:WB of cells co-expressing EGFP-PHF13 and Flag-PHF13 full-length or 1–150, 100–200, or 150–300 deletion proteins (C). Flag IP: WB from lysates co-expressing Flag-PHF13 with YFP, YFP-PHF13, YFP-ΔNTD, YFP-NTD, or YFP-Δ24–20 (D). (E) GST-pulldowns with purified recombinant GST, GST-PHF13, GST-PHF13-ΔNTD, and GST-PHF13-ΔPHD incubated with nuclease digested chromatin lysates and immunoblotted for GST and H3K4me3. (F) WB of fractionated lysates from cells expressing PHF13, PHF13ΔNTD, and PHF13-GAL4DDΔNTD and detected with PHF13 (rat mAb ID3), Tubulin, and H3 antibodies.
To gain further structural insight into PHF13 and to address whether or not it's NTD has an intrinsic dimerization potential, we employed the AlphaFold2_advanced algorithm [60] (Fig. 2B). AlphaFold2 predicted that PHF13 can homo-dimerize via an N-terminal α-helix (aa 30–40; Fig. 2B), which overlaps with the N-terminal aggregation domain predicted by TANGO (Fig. 2A). Furthermore, AlphaFold2 confirmed that PHF13 is a predominantly disordered protein, except for a region in its NTD and the PHD domain, consistent with PONDR and TANGO (Fig. 2A).
These predictions motivated us to biochemically explore PHF13’s ability to dimerize and the role of NTD in this function. To this end, we performed classical co-immunoprecipitation experiments using differentially tagged PHF13 full length and deletion proteins (Fig. 2C and D). Flag-PHF13 (1–300 aa) or Flag-PHF13 deletion mutants (100–200 aa, 1–150 aa, and 150–300 aa) were co-expressed with full-length EGFP-PHF13 (1–300 aa) and immunoprecipitated using Flag-M2 agarose (Fig. 2C). These experiments revealed that full-length Flag-PHF13, the N-terminal half (Flag-PHF13_1–150), and the C-terminal half (Flag-PHF13_150–300) of PHF13 were all capable of co-precipitating EGFP-PHF13. In contrast, Flag-PHF13_100–200, which contains PHF13’s DNA binding domain and the majority of IDRs did not co-precipitate EGFP-PHF13 (Fig. 2C). These findings indicate that PHF13 can self-associate via N- and C- terminal interactions, located in the first (1–100) and last (200–300) 100 aa, consistent not only with homo-dimerization indicated by in silico predictions (Fig. 2A and B) but as well suggesting an oligomerization potential.
To further refine the mapping of the N- terminal dimerization region we co-expressed Flag-PHF13 with different YFP-PHF13 N-terminal mutants (-ΔNTD, -Δ24–40, and NTD only) and looked for their ability to co-precipitate (Fig. 2D). These experiments confirmed that the NTD and more specifically the N-terminal aggregation motif at aa 24–40, mediate PHF13’s N-terminal dimerization (Fig. 2D) as predicted by TANGO and AlphaFold2 (Fig. 2A and B). A weak residual co-immunoprecipitation of Flag-PHF13 was observed for YFP-ΔNTD and YFP-Δ24–40 proteins (Fig. 2D) consistent with the existence of a second weaker C-terminal dimerization (Fig. 2C). These findings demonstrate that the conserved NTD functions as a homo-dimerizing domain.
The ability of PHF13 to dimerize via its NTD and/or oligomerize via N- and C- terminal self-interactions, is expected to increase its chromatin valence and avidity, potentially explaining why deletion of PHF13’s NTD impairs transcriptional repression in luciferase assays (Fig. 1C–E and Supplementary Fig. S1C–E). To test this possibility, we looked for the ability of recombinant PHF13, PHF13ΔNTD, and PHF13ΔPHD to precipitate H3K4me3 from chromatin nuclear lysates and for the chromatin localization of oligomerization competent (PHF13 and GAL4DD-PHF13ΔNTD) and incompetent (PHF13ΔNTD) proteins (Fig. 2E and F, and Supplementary Fig. S2A). GST-PHF13 efficiently precipitated H3K4me3 an interaction that is lost in the GST-PHF13ΔPHD pulldown and that is absent in the GST-control (Fig. 2E) as previously reported [1]. Furthermore, deletion of PHF13’s NTD, resulted in a reduced precipitation of H3K4me3 (Fig. 2E) in line with PHF13 dimerization/oligomerization increasing its chromatin avidity. Consistently, immunoblotting of fractionated lysates from wild-type PHF13, PHF13ΔNTD, and GAL4DD-PHF13ΔNTD demonstrated that deletion of PHF13’s NTD caused a substantial shift of PHF13 from the chromatin fraction to the nucleoplasmic fraction, which could be recovered by fusion of PHF13ΔNTD to the Gal4 dimerization motif (Fig. 2F). Together, these data support that PHF13’s NTD is a homo-dimerization domain, which increases PHF13 chromatin valence and avidity, explaining in part its importance for gene repression.
PHF13’s PHD domain mediates an indirect C-terminal self-association
In addition to the N-terminal homo-dimerization domain, the results also identified a C-terminal self-interacting region in PHF13, indicating an oligomerization potential. To refine the mapping of the C-terminal self-interactions we co-expressed Flag-PHF13 with YFP-PHF13 C-terminal (Δ7;150–300) mutants (Fig. 3A) devoid of the N-terminal dimerization region and looked for their ability to co-precipitate. Consistent with a C-terminal self-interaction, the C-terminal half of PHF13 (YFP-Δ7;150–300) could co-precipitate Flag-PHF13, albeit less efficiently than full-length YFP-PHF13 (Fig. 3A). Deletion of the PHD domain in the C-terminal Δ7 protein (Δ7ΔPHD) abrogated the C-terminal self-interaction and H3K4me3 interaction (Fig. 3A and Supplementary Fig. S3A), suggesting that the PHD domain mediates PHF13’s C-terminal self-interaction. Surprisingly, however, point mutations in the PHD domain (Δ7M246A and Δ7W255A) which disrupt PHF13 binding to H3K4 methylated histones did not impair the C-terminal interaction with Flag-PHF13 indicating that PHF13’s C-terminal self-association is not simply mediated by histones (Fig. 3A).
Figure 3.

PHF13 oligomerizes via N- and C-terminal dimerization. (A) GFP-Trap:WB from cells co-expressing Flag-PHF13 with YFP, YFP-PHF13, or C-terminal YFP-PHF13 mutant proteins (Δ7;150–300, Δ7-ΔPHD, Δ7-M246A, and Δ-W255A). (B) FACS-FRET analysis of HEK 293 cells co-transfected with YFP and CFP fusion proteins. 10% FRET signal was arbitrarily defined as the minimum threshold and is denoted by a dotted line. (C) Graphical representation and immunodotblot of the size exclusion elution profile (Superose 6 10/300) from GST-cleaved recombinant PHF13 full-length (FL) and deletion proteins (ΔPHD, ΔNTD and Δ24–40). Graphs represent quantified signals obtained by dot blotting of individual fractions and quantification by Image quant.
To better understand PHF13’s N- and C- terminal self-interactions, we next questioned whether they were direct or indirect. To address this, we performed an in vivo FACS-based FRET approach using fluorescently tagged PHF13 and deletion proteins (Fig. 3B, and Supplementary Fig. S3B and C). In FACS-based FRET, YFP-PHF13 was co-expressed with various CFP-PHF13 mutant and full-length proteins and evaluated for the excitation of a FRET signal by FACS, indicating a proximity of 10Å or less and implying a direct interaction (Fig. 3B). The expression levels and nuclear localization of all fused proteins were controlled by immunofluorescence and immunoblot (Supplementary Fig. S3B and C). In addition, a YFP-CFP fusion protein and co-expression of YFP and CFP and of YFP-HP1α and CFP-HP1α served as positive, negative and biological dimer/oligomer FRET controls, respectively (Fig. 3B). Co-expression of CFP-PHF13 with YFP-PHF13 gave an average FRET signal of ∼60% by fluorescent cell sorting, indicating that full length PHF13 can directly self-associate in vivo (Fig. 3B). Notably, this was significantly higher than the FRET signal obtained by co-expression of YFP-HP1α and CFP-HP1α (∼30%), in line with the possibility of PHF13 oligomerization in vivo (Fig. 3B). Consistent with in silico predictions (Fig. 2B) and co-immunoprecipitation experiments (Fig. 2C and D), we were able to map a direct interaction to the first 150 N-terminal amino acids and the NTD (21–70 aa), whereas ΔNTD or Δ24–40 eliminated the FRET signal (Fig. 3B). In contrast to the N-terminal region, CFP-150–300 did not generate a FRET positive signal with YFP-PHF13 (Fig. 3B), indicating that the C-terminal PHD interaction interface in PHF13 is mediated indirectly via other proteins.
To further validate PHF13’s oligomerization potential and to approximate the size of PHF13 homo-oligomers in vitro, we performed size-exclusion column chromatography (Superose6 10/300) of purified recombinant PHF13 and deletion proteins (PHF13ΔPHD, PHF13ΔNTD, and PHF13Δ24–40) after the proteolytic removal of the GST tag (Fig. 3C). Based on calibration standards, monomeric PHF13 (MW = 34 kDa) would be expected to elute around fraction 36 on a Superose 6 column. PHF13 and PHF13-ΔPHD eluted in fractions 22 to 29, with the most predominant peak detected in fraction 25 and only a minor monomeric peak at fraction 36 (Fig. 3C). This indicates that recombinant PHF13 and PHF13-ΔPHD preferentially exists as oligomers between ∼400 and 700 kD in size and is consistent with the PHD domain mediating indirect PHF13–PHF13 interactions. In contrast, deletion of the NTD or residues 24–40 reduced recombinant PHF13 to predominantly monomeric and dimeric fractions (34–38), supporting that the NTD and the 16 residues within mediate direct self-interactions (Fig. 3C), consistent with FACs-FRET findings. Similarly, column chromatography (Superdex 200 5/150) of in vitro transcribed/translated PHF13 or PHF13ΔNTD proteins (Supplementary Fig. S3D) revealed that PHF13 predominantly eluted in higher molecular fractions (∼ 440 kDa) which was shifted to lower molecular weight fractions by deletion of the NTD. Together, these findings support that PHF13 can oligomerize via direct N- and indirect C-terminal interactions.
PHF13 can drive global chromatin reorganization and compaction
The ability of PHF13 to oligomerize creates polyvalent PHF13 chromatin interactions, with alternating DNA- and histone-binding domains. This extended chromatin valence should strengthen PHF13’s chromatin binding avidity and facilitate its ability to span/bridge multiple nucleosomes. Such attributes could potentially explain PHF13’s ability to promote repressive and structural chromatin changes.
In support of this possibility, we found that transient overexpression of PHF13 strikingly coincided with global chromosome condensation visible in phase contrast (PC), differential interference contrast (DIC), and DNA imaging (Fig. 4A–D). We repeated these experiments in U2OS cells stably expressing H2B-GFP and found that likewise H2B-GFP reorganized with the phase contrast compacted structures (Fig. 4B and Supplementary Fig. S4A). Furthermore, 3D confocal analysis revealed a strong spatial correlation between the compacted rod-like structures in DIC and DNA through different z-stack planes (Fig. 4D) confirming chromosome condensation. Notably, the compacted chromosomes did not overlap with H3S10 phosphorylation (Fig. 4E and Supplementary Fig. S4B) indicating interphase chromosome condensation, a phenotype previously reported for condensin and cohesin complexes [66–68].
Interestingly, pre-extraction of soluble nuclear proteins prior to fixation, revealed that PHF13 is stably associated with the compacted chromatin and that under these conditions it forms filaments (Fig. 4F and Supplementary Fig. S4C). Moreover, direct fluorescence of
YFP-PHF13 or CFP-PHF13 likewise revealed that PHF13 is extensively localized to the compacted chromatin (Fig. 4G and H, and Supplementary Fig. S4D). Together these observations demonstrate PHF13’s ability to modulate chromatin architecture, as previously reported [5, 6] and further suggest that PHF13 oligomerization along the chromatin polymer may promote chromatin compaction.
To better understand the level of PHF13 overexpression necessary for this phenotype and the penetrance of this phenotype, we quantified the total fluorescence intensity of PHF13 overexpressing cells and nontransfected cells and the standard deviation (SD) of the bright field channel as a measurement of condensation (Fig. 4I and Supplementary Fig. S4E and F). We found that a SD of the brightfield channel >4 was an accurate indicator of condensation, calling cell death and mitosis in wild-type cells and interphase chromosome condensation in PHF13 overexpressing cells (Supplementary Fig. S4E). Using this threshold, we grouped PHF13 into low (1- to 5-fold) and high (5- to 15-fold) transient expression in comparison to nontransfected cells (Supplementary Fig. S4E and F, and Table 2; “Materials and methods” section). This revealed that the condensation phenotype could be observed starting at ∼2- to 4-fold (or greater) overexpression of PHF13 (Supplementary Fig. S4E) and that the penetrance of the phenotype was ∼25% and 65%, in low and high overexpressing groups, respectively (Fig. 4I). That we could observe this phenotype already at ∼3-fold overexpression is relevant, since PHF13 is overexpressed in different cancers [7, 8, 69] and because PHF13 chromatin levels increase throughout the cell cycle peaking in G2 (Supplementary Fig. S4G), suggesting that PHF13 may influence chromatin architecture under these conditions.
Table 2.
Fluorescent intensity cut offs for determining high and low expression
| Groups | Total sum intensity of PHF13 signal (au) |
|---|---|
| Nontransfected | ≤15 000 (au) |
| Low over-expression | between 15 000 and 75 000 (au) |
| High over-expression | between 75 000 and 225 000 (au) |
| Outliers | All the rest |
PHF13-induced chromatin condensation requires its NTD and PHD domains
Direct immunofluorescence and pre-extraction experiments suggested that PHF13 can oligomerize along the chromatin fiber (Fig. 4F–H), potentially explaining the chromatin compaction phenotype. To demonstrate the importance of PHF13 oligomerization in driving this phenotype, we tested and quantified the impact of N- and C-terminal dimerizing mutant proteins expressed at similar levels (Supplementary Fig. S5A) on PHF13’s ability to condense chromatin (Fig. 5A–H). We noted that condensation initiated in the nucleolus (data not shown) and that nucleolar dissociation coincided with condensation (Supplementary Fig. S5B–D), allowing us to additionally use nucleolar integrity as a second proxy for condensation (Fig. 5A–G).
To this end, we found that deletion of PHF13’s N-terminal domain (ΔNTD) or Δ24-40 abolished PHF13’s ability to compact chromatin (Fig. 5B, C, and H) whereas deletion of the PEST1 (ΔPEST1) domain (aa 50–88) which partially overlaps with the NTD (aa 21–70; Fig. 1A and Supplementary Fig. S1A) but retains the homo-dimerization motif (aa 24–40) still compacted chromatin as efficiently as full-length (Fig. 5D and H, and Supplementary Fig. S5E and F). Similar to ΔNTD and Δ24-40, PHF13ΔPHD failed to compact chromatin into chromosomes (Fig. 5E), however, appeared to compact nucleoli, which was quantifiable by measuring the standard deviation of the bright field channel (Fig. 5H, and Supplementary Fig. S4E and F). This argues that the PHD domain is also important for global chromatin condensation, raising the question of whether this is due to its ability to tether PHF13 to histones or to mediate C-terminal self-interactions. To address this question, we expressed PHF13 PHD domain point mutants (M246A or W255A) which disrupt H3K4me3 binding [1] but not PHF13’s oligomerization potential (Fig. 3A) to see whether oligomerization competent and H3K4me3-binding incompetent PHF13 was able to compact chromatin. Interestingly, in contrast to ΔPHD, PHF13M246A, and PHF13W255A point mutant proteins still compacted chromatin as efficiently as full-length PHF13 (Fig. 5F–H, and Supplementary Fig. S5E and F) indicating that oligomerization and not histone binding was the essential feature for PHF13-induced condensation. However, the PHD point mutant proteins formed chromatin aggregates and not chromosome-like structures (Fig. 5F and G) which are morphologically different from wild-type PHF13 (Fig. 4A–D). This suggests that PHF13’s association with H3K4me3 is not important for condensation per se, but for correct compaction morphology of chromatin into chromosomes and implicates PHF13’s DNA-binding region as an essential chromatin tethering region. Finally, we could rescue the condensation defect of the PHF13ΔNTD protein by fusing it to the exogenous dimerization motif of GAL4DD(50–147aa), confirming the importance of oligomerization, in PHF13 driven higher chromatin order (Fig. 5I). Together, these findings support that the oligomerization mediated by PHF13’s N- and C- terminal domains increase its chromatin binding avidity and allow it to spread along the chromatin fiber which are essential features for PHF13’s ability to promote chromatin architectural changes.
Molecular dynamics simulations recapitulate our in vivo observations
To computationally investigate the folding mechanism driving PHF13-mediated chromatin compaction, we employed a polymer physics model based on the strings and binders switch (SBS) model [52, 70] and the transcription factor (TF) model [51]. In this model, a chromatin filament is represented by a chain composed by  beads, equipped with specific binding sites that can interact with molecules, which populate the surrounding environment (Supplementary Fig. S6A and B). In our simulations, we use polymers with
beads, equipped with specific binding sites that can interact with molecules, which populate the surrounding environment (Supplementary Fig. S6A and B). In our simulations, we use polymers with 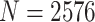 . By imposing a size of 25 kb for each bead, the resulting length equals the size of human chromosome 20 (∼64.4 Mb). The model for the PHF13 protein consists of a small molecule made of three distinct domains, corresponding to the NTD, NLS domain, and the PHD domain (Fig. 6A). PHF13 proteins are at a concentration c (∼0.1–10 pmol/ml), expressed as volume fraction (“Materials and methods” section). PHF13 interacts with chromatin through a strong attractive interaction between the PHD domain and H3K4me3 sites (
. By imposing a size of 25 kb for each bead, the resulting length equals the size of human chromosome 20 (∼64.4 Mb). The model for the PHF13 protein consists of a small molecule made of three distinct domains, corresponding to the NTD, NLS domain, and the PHD domain (Fig. 6A). PHF13 proteins are at a concentration c (∼0.1–10 pmol/ml), expressed as volume fraction (“Materials and methods” section). PHF13 interacts with chromatin through a strong attractive interaction between the PHD domain and H3K4me3 sites (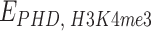 ) and a weaker attractive interaction between PHD domain with H3K4me1/2 sites (
) and a weaker attractive interaction between PHD domain with H3K4me1/2 sites (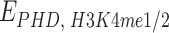 ). Binding sites are regularly located along the polymer chain with density of ∼0.1 (1 site every 10 beads) for H3K4me3 and ∼0.4 for H3K4me1/2 (Supplementary Fig. S6A and B). A nonspecific, weak interaction is also present between NLS (polycationic stretch) and all the polymer beads (
). Binding sites are regularly located along the polymer chain with density of ∼0.1 (1 site every 10 beads) for H3K4me3 and ∼0.4 for H3K4me1/2 (Supplementary Fig. S6A and B). A nonspecific, weak interaction is also present between NLS (polycationic stretch) and all the polymer beads ( ). Finally, PHF13 molecules can attractively interact with themselves through an NTD–NTD interaction (
). Finally, PHF13 molecules can attractively interact with themselves through an NTD–NTD interaction ( ).
).
In addition, we model PHF13’s ability to indirectly oligomerize by adding into the system a second chromatin binding complex, which can interact with PHF13 via its PHD domain and with chromatin, reflecting the indirect C-terminal self-association (Figs 2C and 3A). We model here as putative examples, the chromatin dimers of Cohesin or PRC1, due to our observation that PHF13 can interact with these complexes via its C-terminus (Fig. 6B and C) and their known roles in higher chromatin order [71–76]. Based on approximations from genome-wide datasets (Table 3; “Materials and methods” section), we restrict their binding to H3K4me3 and CTCF or H3K4me3 and H3K27me3 sites, with a density of ∼0.1 and ∼0.2, respectively, and positioned CTCF and H3K27me3 beside H3K4me3 (Supplementary Fig. S6A and B). Finally, we model PHF13 with Cohesin/PRC1 (EPHD, Cohesin/PRC1), allowing PHD domain- Cohesin/PRC1 interactions (Supplementary Fig. S6B). All of the mentioned affinities are taken in the range of 2–8 KBT (Supplementary Fig. S6B), i.e. in the weak biochemical range and consistent with typical affinities between TFs and DNA (Molecular Biology of the Gene: Watson, Baker, Bell, Gann, Levine, Losick, 2013). Overall, the described system includes both protein–chromatin and protein–protein interactions (Fig. 6A), and the resulting equilibrium structure depends on the interplay between these interactions [56, 77].
Table 3.
Published ChIP-seq data sets used to estimate genomic binding for MD simulations
| ChIP | ArrayExpress/GEO dataset | ID ChIP sample | ID control sample | DOI |
|---|---|---|---|---|
| PHF13 | E-MTAB-2636 | ERR689062 | ERR689061 | http://dx.doi.org/10.7554/eLife.10607.001 |
| SMC3 | GSE80049 | GSM2111722, GSM2111723, GSM2111724 | GSM2111696, GSM2111697, GSM2111698 | https://doi.org/10.1038/s41588-017-0015-6 |
| RAD21 | GSE74055 | GSM2099809 | GSM2099810 | https://doi.org/10.1038/cr.2018.1 |
| CTCF | GSE11431 | GSM288351 | GSM288358 | https://doi.org/10.1016/j.cell.2008.04.043 |
| H3K4me3 | GSE120376 | GSM3399477 | GSM3399484 | https://doi.org/10.1186/s13059-019-1860-7 |
| H3K4me1 | GSE120376 | GSM3399476 | GSM3399484 | |
| H3K27me3 | GSE120376 | GSM3399482 | GSM3399484 |
The MD simulations demonstrate that when PHF13 is above the concentration threshold (Supplementary Fig. S6C) that it is able to phase separate chromatin in the absence of cohesin or PRC1, resulting in a collapse globule at the end state equilibrium (Fig. 6E and Supplementary Fig. S6D). Considering that PHF13 can directly interact with DNA and histones [1], its oligomerization capacity (Figs 2 and 3) and its ability to form filaments in pre-extraction IF (Fig. 4F), these observations are consistent with PHF13 driving a linear compaction of chromatin, i.e. compaction of neighboring nucleosomes [78]. Other structural regimes may naturally arise from this model when different affinity ranges are considered. For instance, a significantly higher affinity  could lead to polymer globular structures, irrespective of PHF13 phase separation. More broadly, the interplay between protein phase separation and chromatin interactions adds complexity to the system’s phase diagram, as discussed in recent computational studies [48, 77, 79].
could lead to polymer globular structures, irrespective of PHF13 phase separation. More broadly, the interplay between protein phase separation and chromatin interactions adds complexity to the system’s phase diagram, as discussed in recent computational studies [48, 77, 79].
To check the robustness of the result, we observed that reductions in PHF13 molecule size returned analogous results (Supplementary Fig. S6F, left panel), with stable globular configurations (Supplementary Fig. S6G). Analogously, if two polymers are included in the system, globular structures made of the two polymers are observed (Supplementary Fig. S6F, central panel). Furthermore, by shutting off either the NTD or PHD domain interactions (Fig. 6E), PHF13 loses its ability to phase separate the chromatin fiber, consistent with our in vivo observations (Fig. 5B and E) and that these domains are necessary for global 3D-architectural changes.
Modeling cohesin-only or PRC1-only, induced chromatin loops and drove bridging induced phase separation, resulting in an elongated rod-like phase separated polymer at end state (Fig. 6F and Supplementary Fig. S6E) as has been previously shown for cohesin [75] and similar to polymer models of mitotic compacted chromosomes via condensin-like bridges [80]. Similarly, simulations in which loop extrusion acted solely on the polymer resulted in rod-like configurations (Supplementary Fig. S6F, right panel), though with greater structural variability due to the dynamics of the extruders (Supplementary Fig. S6G, “Materials and methods” section). Finally, modeling PHF13 and cohesin or PRC1 together, resulted in more compacted rod-like chromosome structures at end-state equilibrium (Fig. 6F and G, and Supplementary Fig. S6D), predicting that their cooperative impact promotes chromatin to chromosome-like transitions. In agreement with this if the number of binding sites for Cohesin molecules is varied (Supplementary Fig. S6G), different compaction levels are observed. This suggests that PHF13–cohesin/PHF13–PRC1 can gradually modulate chromatin compaction in a spectrum of structures ranging from globular (few cohesin/PRC1-binding sites) to rod-like (several cohesin/PRC1-binding sites) structures.
Interestingly, when all cohesin/PRC1 and PHF13 interactions were allowed except for PHF13’s PHD interactions with H3K4 methylated histones, phase separated aggregates were still observed (Supplementary Fig. S6H). However, they formed random configurations, similar to what was seen in vivo (Fig. 5F and G, and Supplementary Fig. S5C). These simulations recapitulate our in vivo findings and suggest a two-step process where linear compaction driven by PHF13 and bridging induced compaction mediated by another chromatin factor can cooperate to drive global chromatin compaction (Fig. 6H) resembling chromosomes.
PHF13 can self-associate via its IDRs
Our results indicated that PHF13 can self-associate via an indirect C-terminal interaction localized to the PHD domain that was independent of its histone binding (Fig. 3A). To refine this further, we questioned if the aggregation regions determined by TANGO (Fig. 2A), bookmarking the PHD domain (aa232–238 or aa272–280) were mediating this function. To this end, we co-expressed Flag-PHF13 with full-length YFP-PHF13, YFP-PHF13Δ24–40 or C-terminal YFP-PHF13Δ7 mutants (Δ7;150–300aa) containing deletions of aa232–238 or aa272–280 (Fig. 7A and B), and looked to see how efficiently Flag-PHF13 could co-immunoprecipitate these mutant proteins (Fig. 7B). Deletion of these regions in the full-length protein (YFP-PHF13Δ232-238, YFP-PHF13Δ272-280) or the C-terminal half (YFPΔ7_ Δ232-238, YFPΔ7_ Δ272-280) did not impair the co-precipitation of the Flag- PHF13, nor did they abrogate the residual interaction when combined with Δ24-40 (YFP-PHF13Δ24-40_Δ232-238, YFP-PHF13Δ24-40_Δ272-280), speaking against a role of 232–238 or 272–280 aggregation motifs in mediating PHF13 C-terminal self-interaction (Fig. 7B). Surprisingly however, and in contrast to our speculations, deletion of 272–280 seemed to enhance the interaction between YFP-PHF13 and Flag-PHF13 (Fig. 7B) rather than abrogate it. We repeated these experiments this time expressing full-length YFP-PHF13 with different full-length Flag-PHF13 mutants (Fig. 7C and Supplementary Fig. S7A). Again, we observed that YFP-PHF13 and Flag-PHF13 interact, and that deletion of aa24–40 dramatically reduced this interaction, but that the combined deletion of Δ24-40_Δ272-280 unexpectantly restored PHF13’s self-interaction (Fig. 7C and Supplementary Fig. S7A). These findings indicate that in addition to oligomerization via its ordered domains, that PHF13 is also capable of self-associating independent of its aggregation motifs.
Figure 7.

PHF13 oligomerizes via its IDRs. (A) Overlap of PONDR and TANGO plots depicting deletion (X) of TANGO aggregation regions. (B and C) FlagIP:WB of Flag-PHF13 co-expressed with different YFP-PHF13 full-length and C-terminal (Δ7;150–300) mutant proteins. Flag-IPs were probed by immunoblot for their ability to co-IP the YFP-proteins, detected with GFP and Flag antibodies. (D) IF: U2OS cells expressing YFP-PHF13, YFP-PHF13Δ24-40, YFP-PHF13Δ24-40_Δ272-280 and imaged on a Zeiss Z1 Observer to evaluate protein localization. (E) WB of fractionated lysates (Cytoplasm-Cy, Nucleoplasm-Nu, and Chromatin-Chr) and quantification (% of PHF13/fraction) of cells expressing YFP-PHF13, YFP-PHF13Δ24-40_Δ232-238, and YFP-PHF13Δ24-40_Δ272-280 and detected with GFP, Vinculin, and SMC3 antibodies. (F) Luciferase reporter gene expression in H1299 cells from p53 regulated cyclin G and MDM2 co-transfected with p53 and the indicated pcDNA-4TO expression vectors for 24 h.
To better understand this phenomenon, we evaluated YFPΔ24-40_Δ232-238, YFPΔ24-40_Δ27-280, and FlagΔ24-40_Δ272-280 proteins by immunofluorescence microscopy (Fig. 7D and Supplementary Fig. S7B and C). Interestingly, these proteins did not promote global chromosome condensation and all proteins formed nucleoplasmic condensates, reminiscent of LLPS and suggesting IDR-mediated oligomerization (Fig. 7D, and Supplementary Fig. S7B and C). Consistent with this interpretation, in silico analysis of PHF13’s LLPS potential using the PSPredictor [62], indicated a low potential for PHF13 and PHF13Δ24-40 to form LLPS condensates, whereas it predicted that PHF13 deleted of Δ232-238, Δ272-280, Δ24-40_Δ232-238, and Δ24-40_Δ272-280 has a high propensity to phase separate (Supplementary Fig. S7D), aligning with our in vivo observations (Fig. 7D, and Supplementary Fig. S7B and C). These results suggest that small deletions in PHF13’s ordered aggregation domains alters the localization of PHF13 from chromatin to nucleoplasmic condensates, emphasizing the importance of PHF13’s ordered aggregation domains in regulating its chromatin affinity. Consistently, fractionated immunoblotting confirmed that deletion of PHF13’s ordered aggregation domains (PHF13Δ24-40_Δ232-238 or PHF13Δ24-40_Δ272-280), significantly reduces its chromatin affinity (Fig. 7E and Supplementary Fig. S7E).
For simplicity, from here on in we refer to the LLPS promoting deletions (PHF13Δ24-40_Δ232-238 and PHF13Δ24-40_Δ272-280) as mut1 and mut2, respectively. To further investigate if PHF13 mut1 and mut2 abrogate PHF13 repression similar to PHF13ΔNTD (Fig. 1C–E), we looked at their impact on the cyclin G and MDM2 promoters (Fig. 7F). As expected, mut1 and mut2 abrogated PHF13 repression of cyclin G and MDM2 promoters (Fig. 7F) and modestly activated them in comparison to vector alone, suggesting that the LLPS state modulates PHF13’s transcriptional response.
Together, these findings demonstrate that PHF13’s ordered domains are necessary for its strong polyvalent chromatin association and that PHF13 can self-associate via its IDRs to form LLPS condensates, which have a reduced chromatin affinity and impair PHF13 repression. Moreover, these data imply that PHF13 can phase transition and identify an intrinsic balance between PHF13’s ordered and disordered regions in regulating PHF13 chromatin affinity and transcriptional impact.
PEST domains promote phase separation and regulates PHF13 chromatin association
To demonstrate PHF13’s ability to phase separate using classical in vitro approaches, we tested the ability of recombinant PHF13 to form condensates in the turbidity assay and condensate forming assay in the absence of nucleic acids/chromatin (Fig. 8A and B). As postulated, recombinant PHF13 caused the PEG solution to turn turbid (Fig. 8A) and formed visible condensates by light microscopy at different PEG and salt concentrations (Fig. 8B), in contrast to equimolar amounts of GST (Fig. 8A and B), demonstrating that PHF13 does have the intrinsic ability to phase separate.
Figure 8.

PHF13 phase separates in a PEST2 (IDR3) dependent manner. (A and B) In vitro phase condensate forming assays, demonstrate that recombinant GST-PHF13 can turn a PEG-salt solution turbid (A) and form condensates visible by light microscopy (B) in contrast to equimolar amounts of GST-only (A and B). (C and D) IF: imaging of endogenous PHF13 under wild-type (C and D) or treated conditions (0.5 μg/ml AMD- 4 h or 40 mM LiCl-6 h) (C) showing PHF13’s ability to form polymers or condensates, under different cellular conditions. Cells were imaged on a Leica Stellaris in confocal mode (D) or on a Zeiss Axiophot Phase Fluorescence microscope (C). PHF13 was stained with rat mAb 6F6 (D and F, lower panel) or rabbit polyclonal CR53 (C). (E) WB of fractionated lysates (Cytoplasm-Cy, Nucleoplasm-Nu, and Chromatin-Chr) and quantification (% PHF13/fraction) from cells expressing YFP-PHF13, YFP-PHF13ΔPEST1, and YFP-PHF13ΔPEST2 and detected with GFP, Vinculin, SMC3, and H3 antibodies. (F) Direct and indirect microscopy of YFP-PHF13ΔPEST1, YFP-PHF13ΔPEST2, and 4TO-PHF13 ΔPEST2. (G) Quantification of the penetrance of condensation of 4TO-PHF13, 4TO-PHF13ΔPEST1, 4TO-PHF13ΔPEST2, and PHF13Δ8 (ΔIDR5). Values are derived from three biological replicates.
Moreover, endogenous PHF13 forms condensate-like structures during DNA damage response [5], in response to RNA polymerase I transcriptional inhibition (Fig. 8C and Supplementary Fig. S8A–C), and in response to GSK3β inhibition with LiCl (Fig. 8C and Supplementary Fig. S8A). Whereas PHF13 increases its chromatin association and forms filament-like structures during G2/M phase transitions and during late anaphase/telophase [6] (Fig. 8C and D, Supplementary Figs S4G and S8C). These observations support that endogenous PHF13 forms phase separated condensates and polymers in vivo under physiological/pathological contexts, which likewise coincide with pronounced chromatin architectural and transcriptional changes.
To better understand which IDRs may be involved in PHF13’s LLPS state, we computationally investigated which IDRs in PHF13 are predicted to drive LLPS. To do this, we employed the in silico prediction algorithms PSPredictor [62], ParSe v2 [63] and FuzDrop [64] (Supplementary Fig. S8D–F). PSPredictor indicated that deletion of IDR2 (aa105–135; NLS- and DNA-binding domain) and IDR4 (aa190–230) have a high propensity to phase separate, predicting that they are not driving LLPS (Supplementary Fig. S8D). In contrast, deletion of IDR1 (PEST1), IDR3 (PEST2), and IDR5 all resulted in weak PSP scores of 0.4878, 0.0661, and 0.2494, respectively (Supplementary Fig. S8D), with deletion of IDR3 and IDR5 having a lower propensity to phase separate than wild-type PHF13 (0.4128). Similarly, ParSe2 predicted that PEST1 (IDR1) and PEST2 (IDR3) but not IDR2 are prone to undergo phase separation (Supplementary Fig. S8E) and FuzDrop identified PEST1, PEST2, and IDR5 as droplet promoting regions and identified the majority of aggregation hotspots within these regions (Supplementary Fig. S8F). Collectively, these prediction algorithms converge on IDR1 (PEST1), IDR3 (PEST2), and IDR5 as the relevant IDRs involved in promoting PHF13 LLPS and implicate PEST2 as the strongest catalyst (Supplementary Fig. S8D–F).
Previously, it was shown that deletion of PEST2 (IDR3) enhanced PHF13 chromatin association [6] arguing that PEST2 (IDR3) antagonizes PHF13 chromatin binding, consistent with a putative role in promoting LLPS condensates. Consistently, we found that deleting PEST2 increased PHF13 chromatin affinity in physiological (150 mM) and high salt (400 mM) conditions (Fig. 8E and Supplementary Fig. S8G), whereas deletion of PEST1 (ΔPEST1) increased the amount of PHF13 on chromatin under physiological salt conditions but not high salt conditions (Fig. 8E and Supplementary Fig. S8G). In contrast, deletion of IDR5 (PHF13Δ8; aa 1–285) significantly weakened PHF13’s affinity for chromatin (Supplementary Fig. S8H). Moreover, we found that deletion of IDR1 (ΔPEST1) or IDR3 (ΔPEST2) efficiently promoted global chromatin compaction (Figs 5D and 8F, and Supplementary Fig. S8I) and increased the penetrance of this phenotype (Fig. 8G) whereas deletion of IDR5 (PHF13Δ8), impaired chromatin condensation (Fig. 8G) and showed a similar compacted nucleolar phenotype to PHF13ΔPHD (Fig. 5E and Supplementary Fig. S8J), suggesting that IDR5 is important for PPPS rather than LLPS. These data argue that IDR1 (PEST1) and IDR3 (PEST2) antagonize PHF13 chromatin association consistent with their putative role in PHF13 nucleoplasmic LLPS condensate formation (Supplementary Fig. S8D–F).
PHF13 LLPS and PPPS states differentially modulate gene expression and chromatin accessibility
To further investigate structural and transcriptional impacts of PHF13 LLPS and PPPS states at the genomic level, we performed mRNA and ATAC sequencing on FACs sorted YFP-PHF13 wild-type and mutant expressing cells (Fig. 9). The differentially expressed genes from the RNA-seq analysis and the annotated ATAC-seq peaks can be found in Supplementary Table S1—Annotated differential expressed genes from RNA-seq analysis and Supplementary Table S2—Annotated consensus ATAC-seq peaks. PCA of the ATAC and mRNA-seq data, showed that YFP-PHF13Δ24-40, YFP-mut1 and YFP-mut2 cluster together, and that YFP-PHF13 and YFP-PHF13ΔPEST2 clustered together (Fig. 9A and B), indicating that the transcriptomes and chromatin accessibility of PHF13 LLPS and PPPS states are distinguishable. Consistently, heatmaps of ATAC peaks centered at the TSS, showed that PHF13 and PHF13ΔPEST2 dramatically reduced chromatin accessibility in comparison to the YFP-control, whereas PHF13Δ24-40, mut1 and mut2 had chromatin accessibility levels similar to the YFP-control (Fig. 9C and Supplementary Fig. S9A). This indicates that PHF13 LLPS and PPPS states differentially modulate chromatin structure and accessibility which can be measured and quantified at the genomic (Fig. 9) and optical levels (Figs 4, 5, and 7).
Figure 9.

PHF13 PPPS and LLPS states differentially impact chromatin accessibility and gene expression. (A and B) PCA of the ATAC-seq (A) and RNA-seq expression profiles (B) of U2OS cells expressing various PHF13 variants. (C) Heatmap and line plot representation of ATAC-Seq read densities of all samples centered at TSS within 3 kb window around. The signal represents the read counts normalized to the spike-in. (D) Bar plot showing the number of DEGs when tested against wild-type control sample (left) and tested against to EYFP control sample (right). (E) Heatmap and clustering analysis of RNA-Seq data. Cluster numbers are indicated on the left. (F) Gene ontology enrichment analysis heatmap for biological process of heatmap clusters for top 10 GOs for each cluster. (G) Genome tracks for ATAC-seq and RNA-seq data of two down-regulated genes in WT samples compared to EYFP control (left) and two up-regulated (right). The samples are color-coded as described above. (H). Comparison of z-scores across genes related to Pol-I/nucleoli and Pol II regulation in both ATAC-seq and RNA-seq.
Further comparison of the gene expression profiles revealed that YFP-PHF13 had many differentially expressed genes in comparison to YFP-mut1 (DEGs; 1143) and YFP-mut2 (DEGs; 1212). In contrast, the gene expression profiles of YFP-PHF13 and YFP-PHF13ΔPEST2, were very similar (350 DEGs; Fig. 9D and Supplementary Fig. S9B–D) and of YFP-mut1 and YFP-mut2 were also very similar to each other (S9C), supporting that PPPS and LLPS states are transcriptionally different from each other. Moreover, we additionally noted that YFP-PHF13 and YFP-ΔPEST2 displayed higher fold-changes in gene expression than did YFP-PHF13Δ24–40, YFP-mut1 and YFP-mut2 (Supplementary Fig. S9C and E), suggesting that PPPS has a stronger transcriptional impact than does LLPS.
To better understand the transcriptional impact, we next compared the differentially expressed genes (DEGs) of PHF13 and the mutant proteins to YFP, resulting in five clusters (Fig. 9E and F, and Supplementary Fig. S9E). Clusters 1 and 2 were down regulated and clusters 4 and 5 were up regulated in comparison to YFP. Whereas cluster 3 was differentially up or down regulated between PPPS and LLPS states, respectively, in comparison YFP (Supplementary Fig. S9E). Enrichment analysis revealed that PHF13 positively regulated genes associated with the cell cycle, mitotic cell cycle, chromosome organization and response to virus, and that these processes are differentially regulated between LLPS and PPPS states (Fig. 9F). In addition, PHF13 and mutant proteins positively regulated genes associated with ribonucleoprotein biogenesis and mRNA metabolic processes (Fig. 9F) and negatively regulated genes associated with different developmental and specification pathways, Wnt, Notch and mTor signaling pathways and cell junctions. Interestingly, cluster 3 was enriched in genes associated with cell migration, interleukin-4 (IL4), interleukin-13 (IL13), and Mitogen-activated protein kinase (MAPK) signaling (Fig. 9F), indicating different transcriptional responses for LLPS and PPPS states to immune regulation and cell mobility. Notably, several of these associated functional terms have been previously reported for PHF13, namely cell cycle, mitosis, host viral response, EMT and RNA metabolism [1–4, 6, 7], underscoring the validity of these results.
Finally, we compared the ATAC and RNA-seq data, to understand if accessibility and gene expression are concordant (Fig. 9G and H). To this end we looked at ATAC signals located ±500 bp of the promoters and compared it with gene expression. This analysis showed that PHF13 and PHF13ΔPEST2 down regulated genes showed a concordant decrease in chromatin accessibility (Fig. 9G and H). However, PHF13 and PHF13ΔPEST2 upregulated genes, showed two trends. Either no change in accessibility with increased gene expression, such as the heat shock response genes (Fig. 9G), or decreased accessibility with increased gene expression, such as for RNAPolI related genes (Fig. 9H). Interestingly, when we sorted differentially expressed genes for RNAPolI related or RNAPolII related, we found that while PHF13 and PHF13ΔPEST2 decreased chromatin accessibility for both groups, they concordantly repress RNAPolII related genes whereas they discordantly activate RNAPolI related genes (Fig. 9H). These findings imply a differential impact of PHF13 on RNAPolI and RNA PolII transcriptional regulation and further suggest the possibility that rRNAs may be involved in PHF13 chromatin condensation.
In summary, our results confirm that PHF13 is a potent regulator of chromatin structure, chromatin accessibility and gene expression, and highlight the importance of both ordered and disordered domains in regulating these functions. Finally, our findings identify that polymer-polymer phase separation (PPPS) can orchestrate 3D chromatin architecture and promote gene repression and that phase transitions of chromatin effectors may be a mechanism for regulating dynamic chromatin transitions or states.
Discussion
The discovery that PHF13 can self-associate is highly informative in revealing how PHF13 can perform different chromatin functions. Oligomerization, via its ordered aggregation domains, creates a polyvalent chromatin binding protein with alternating DNA and histone binding domains. Depending on PHF13 valency, this would allow PHF13 to (i) bridge neighboring nucleosomes and/or promoters and enhancers to influence gene expression, (ii) to increase stability and recruitment of other chromatin factors that influence chromatin structure/function, and (iii) to promote global chromatin compaction (Fig. 6H). Furthermore, it seems reasonable that different PHF13 chromatin valences (i.e. monomeric, dimeric, and oligomeric) or different chromatin affinities mediated by different oligomerization mechanisms might contribute to PHF13’s unique chromatin functions, such as gene activation, repression, compaction or repair.
We found that a 3- to 5-fold overexpression of PHF13 was sufficient to drive global chromatin compaction, underlining PHF13’s ability to regulate chromatin structure and function. This phenomenon of global interphase chromatin compaction has only been previously described for a few complexes, namely cohesin [66, 67], condensin [68], and polycomb [31, 81]. Building on their similarities, PRC1 and Cohesin are both reported to phase separate [75, 82], impacting transcription and chromatin architecture [83, 84]. Similar to CBX2 and CBX8 (PRC1 complex members), PHF13 has a highly basic region, a putative RNA-binding domain and a histone binding domain affording it the ability to neutralize DNA’s charge, to interact with nucleic acids, to interact with active/bivalent promoters and enhancers and to compact chromatin. Furthermore, similar to PRC1 Phc1-3 subunits [31, 85], PHF13 is capable of oligomerizing which greatly increases its chromatin valency.
This increased valency, increases PHF13’s binding avidity to the chromatin polymer creating a very stable state consistent with PPPS [28, 34, 86–88]. In support of this understanding, the compacted chromosomes are impermeable to PHF13 antibodies in indirect immunofluorescence but are completely labeled by PHF13 in direct immunofluorescence (Fig. 4A, B, and F–H), suggesting that the chromosomes are phase separated from the nucleoplasm. Secondly, this oligomer-dependent process does not coalesce but rather forms filaments (Fig. 4F and Supplementary Fig. S4C). And lastly chromatin compaction is driven by strong multivalent protein–nucleic acid contacts (Fig. 8C and Supplementary Fig. S8D) [34, 86] instead of weak multi-valent interactions observed in LLPS. Together, our findings implicate PPPS as an underlying biophysical mechanism governing PHF13 condensation and gene repression and opens up the possibility that this may be a general mechanism governing higher chromatin order and function.
Intriguingly, mRNA sequencing indicated that PHF13 promotes RNAPolI transcription, implicating rRNAs in PHF13 condensation and PPPS phenotype. In line with this possibility the rDNA locus was recently shown to be formed by PPPS [89] and rDNAs have been shown by 4C techniques [90, 91] to make inter chromosomal contacts across the genome, which could account for our observations that condensation originates in the nucleolus and spreads to form compacted chromosomes. Moreover, when we inhibit rRNA transcription with AMD, PHF13 forms nucleoplasmic LLPS condensates (Fig. 8C and Supplementary Fig. S8A–C), supporting the idea that rRNAs influence PHF13 chromatin affinity and localization. Notably, PHF13’s IDR5 is a putative RNA binding and nucleolar localizing sequence (Figs 1A and 2A, and Supplementary Fig. S1B). Deletion of IDR5 abrogates PHF13 chromosome condensation, promotes nucleolar compaction and the formation of nucleolar caps similar to PHF13ΔPHD (Figs 5E and 8G, and Supplementary Fig. S8J), and reminiscent of AMD treatment (Supplementary Fig. S8A, upper panel) [92]. This phenotype suggests that PHF13’s PHD domain and IDR5 are important for RNAPolI transcription, and that their deletion inhibits rRNA synthesis, potentially explaining their inability to compact chromatin. It will be important to investigate the PHF13 nucleolar relationship in more detail in the future.
Strikingly, deletion of PHF13’s ordered regions promoted oligomerization via its IDRs (PEST domains) which did coalesce to form condensates, reduced PHF13’s chromatin affinity (Fig. 7F), abrogated PHF13 chromatin structural changes (Fig. 9C) and altered its transcriptional impact (Fig. 9 and Supplementary Fig. S9). This antagonizing biophysical mechanism has been observed previously for PRC1 and other complexes [93–95] underscoring its biological relevance, and suggests that phase transitions may be a general regulation mechanism of chromatin effector proteins. It is tempting to speculate, that post-translation modifications within the ordered regions and IDRs, or allosteric interactions refine PHF13’s chromatin affinity and function. What ligands regulate PHF13’s chromatin interaction and functions will be another important question for the future. Taken together, we uncover the molecular conversation between PHF13’s ordered and disordered regions in regulating PHF13 oligomerization, chromatin valency, and chromatin function.
Limitations of study
In this study, to address the intrinsic features of PHF13 that influence its biological roles in chromatin structure and gene expression, we employed several mapping-based strategies. As such, many experiments required expression of tagged and exogenous proteins to address our questions. Tagging of proteins can alter protein structure and protein interactions. In addition, overexpression of proteins reaches nonphysiological levels and as a result can produce nonphysiological responses. Moreover, the mapping experiments were for the most part not done with recombinant proteins, and therefore the interactions may be indirect. Recombinant in vitro mapping is more informative about direct interactions and mapping of binding domains. Luciferase assays which measure transcriptional impact suffer from two additional limitations: (i) They are analyzed in lysed cells, which disrupts the physiological transcription context potentially inadvertently inducing artifacts in the measurements. (ii) Luciferase plasmids are not chromatinized, which means that it is not suitable for measuring the impact of histone binding domains on transcriptional impact. Lastly, our study does not explore or consider interactions with other chromatin proteins or altered epigenetic landscapes, which may be involved in the observed phenotypes. Therefore, our data do not explore or exclude the possibility that this may occur concomitantly or complimentary to loop extrusion mechanisms. Finally, with regards to our polymer modelling approach, the main limitations are that it was based on a toy model. More realistic models could, e.g. include binding sites located according to the positions of histone modifications to simulate real chromosomes or simulating more chromosomes. We acknowledge these limitations in our study and declare that we made every effort to employ many controls and orthogonal approaches, to reach our conclusions.
Supplementary Material
Acknowledgements
We would like to thank Michael Schindler, Carina Banning, and Kristin Höhne for their technical support with the FACS-FRET assay, M. Schmidt-Zachmann for the NOH61 antibody, Theres Schaub, and Kirsten Reumann for generating some of the plasmids used in this study. We thank the Flow Cytometry Service unit, in particular Melanie Piedavent-Salomon, at the Max Planck Institute for Molecular Genetics for their help with cell sorting. We would like to acknowledge the support of the DFG SPP2191-Molecular Mechanisms of Phase separation for their inclusion in scientific meetings.
Author contributions: Francesca Rossi (Data curation [equal], Formal Analysis [equal], Investigation [equal], Methodology [equal]), Rene Buschow (Data curation [equal], Formal Analysis [equal], Methodology [equal]), Tobias Schubert (Data curation [equal], Formal Analysis [equal], Investigation [equal], Methodology [equal]), Laura V. Glaser (Data curation [equal], Formal Analysis [equal]), Andrea Fontana (Data curation [equal], Formal Analysis [equal]), Julia Mai (Data curation [equal], Formal Analysis [equal]), Hannah Staege (Data curation [equal], Formal Analysis [equal]), Astrid Grimme (Data curation [equal], Methodology [equal], Project administration [equal]), Hans Will (Conceptualization [equal], Resources [equal]), Denes Hnisz (Funding acquisition [equal], Resources [equal], Supervision [equal]), Martin Vingron (Conceptualization [equal], Resources [equal]), Andrea M. Chiariello (Data curation [equal], Formal Analysis [equal], Supervision [equal]), and Sarah Kinkley (Conceptualization [equal], Data curation [equal], Formal Analysis [equal], Funding acquisition [equal], Investigation [equal], Methodology [equal], Project administration [equal], Supervision [equal], Writing—original draft [equal], Writing—review & editing [equal]).
Notes
Present address: Proxygen GmbH, Siemensstsrasse 89, 1210 Vienna, Austria
Present address: Central Institute for Clinical Chemistry and Laboratory Medicine, Klinikum Stuttgart, Kriegsbergstrasse, 60, 70174 Stuttgart, Germany
Contributor Information
Francesca Rossi, Chromatin Structure and Function Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Alexandre P Magalhaes, Multi-level Gene Control Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Rene Buschow, Core Microscopy Facility, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Tobias Schubert, The Department of Virology at the Leibniz Institute for Virology, Martinistrasse 52, Hamburg, 20251, Germany.
Laura Glaser, Chromatin Structure and Function Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Andrea Fontana, Dipartimento di Fisica, Universita’ degli Studi di Napoli Federico II, and INFN Napoli, Complesso Universitario di Monte Sant’Angelo, Naples 80126, Italy.
Julia Mai, Institute of Virology, Medical Center at the University of Freiburg, Hermann-Herder-Str. 11, Freiburg 79104, Germany.
Hannah Staege, The Department of Virology at the Leibniz Institute for Virology, Martinistrasse 52, Hamburg, 20251, Germany.
Astrid Grimme, Chromatin Structure and Function Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Hans Will, The Department of Virology at the Leibniz Institute for Virology, Martinistrasse 52, Hamburg, 20251, Germany.
Denes Hnisz, Multi-level Gene Control Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Martin Vingron, Department of Computational Molecular Biology, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Andrea M Chiariello, Dipartimento di Fisica, Universita’ degli Studi di Napoli Federico II, and INFN Napoli, Complesso Universitario di Monte Sant’Angelo, Naples 80126, Italy.
Sarah Kinkley, Chromatin Structure and Function Group, Max Planck Institute for Molecular Genetics, 63-73 Ihnestrasse, Berlin, 14195, Germany.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
D.H. is a founder and scientific advisor of Nuage Therapeutics.
Funding
This work was supported by the VW-Stiftung 97131, DFG Project 4154/4-1 and the Max Planck Society. A.M.C. acknowledges “Programma per il Finanziamento della Ricerca di Ateneo Linea B” (FRA) 2020, University of Naples Federico II, CINECA ISCRA Grant ID PhaSSep - HP10C8JWU7. SS was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) in the framework of the Research Unit FOR5200 DEEP-DV (443644894) project 08 and under Germany's Excellence Strategy - EXC 2155 - project number 390874280. Funding to pay the Open Access publication charges for this article was provided by the Max Planck Institute for Molecular Genetics.
Data availability
The data underlying this article are available in NCBIs Gene Expression Omnibus (GEO), and can be accessed with GSE289538 and GSE289539.
References
- 1. Chung HR, Xu C, Fuchs A et al. PHF13 is a molecular reader and transcriptional co-regulator of H3K4me2/3. eLife. 2016; 5:e10607. 10.7554/eLife.10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hofmann S, Dehn S, Businger R et al. Dual role of the chromatin-binding factor PHF13 in the pre- and post-integration phases of HIV-1 replication. Open Biol. 2017; 7:2395–413. 10.1098/rsob.170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuderna AK, Reichel A, Tillmanns J et al. Discovery of a novel antiviral effect of the restriction factor SPOC1 against Human Cytomegalovirus. Viruses. 2024; 16:363. 10.3390/v16030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schreiner S, Kinkley S, Burck C et al. SPOC1-mediated antiviral host cell response is antagonized early in human adenovirus type 5 infection. PLoS Pathog. 2013; 9:e1003775. 10.1371/journal.ppat.1003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mund A, Schubert T, Staege H et al. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res. 2012; 40:11363–79. 10.1093/nar/gks868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kinkley S, Staege H, Mohrmann G et al. SPOC1: a novel PHD-containing protein modulating chromatin structure and mitotic chromosome condensation. J Cell Sci. 2009; 122:2946–56. 10.1242/jcs.047365. [DOI] [PubMed] [Google Scholar]
- 7. Sun Y, Li D, Liu H et al. PHF13 epigenetically activates TGFbeta driven epithelial to mesenchymal transition. Cell Death Dis. 2022; 13:487. 10.1038/s41419-022-04940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mohrmann G, Hengstler JG, Hofmann TG et al. SPOC1, a novel PHD-finger protein: association with residual disease and survival in ovarian cancer. Int J Cancer. 2005; 116:547–54. 10.1002/ijc.20912. [DOI] [PubMed] [Google Scholar]
- 9. Bordlein A, Scherthan H, Nelkenbrecher C et al. SPOC1 (PHF13) is required for spermatogonial stem cell differentiation and sustained spermatogenesis. J Cell Sci. 2011; 124:3137–48. 10.1242/jcs.085936. [DOI] [PubMed] [Google Scholar]
- 10. Cermakova K, Hodges HC Interaction modules that impart specificity to disordered protein. Trends Biochem Sci. 2023; 48:477–90. 10.1016/j.tibs.2023.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hnisz D, Shrinivas K, Young RA et al. A phase separation model for transcriptional control. Cell. 2017; 169:13–23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabari BR, Dall’Agnese A, Boija A et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lyons H, Veettil RT, Pradhan P et al. Functional partitioning of transcriptional regulators by patterned charge blocks. Cell. 2023; 186:327–45. 10.1016/j.cell.2022.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gui T, Fleming C, Manzato C et al. Targeted perturbation of signaling-driven condensates. Mol Cell. 2023; 83:4141–57. 10.1016/j.molcel.2023.10.023. [DOI] [PubMed] [Google Scholar]
- 15. Hentze MW, Castello A, Schwarzl T et al. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018; 19:327–41. 10.1038/nrm.2017.130. [DOI] [PubMed] [Google Scholar]
- 16. Malinovska L, Kroschwald S, Alberti S Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim Biophys Acta. 2013; 1834:918–31. 10.1016/j.bbapap.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 17. Brodsky S, Jana T, Mittelman K et al. Intrinsically disordered regions direct transcription factor In vivo binding specificity. Mol Cell. 2020; 79:459–71. 10.1016/j.molcel.2020.05.032. [DOI] [PubMed] [Google Scholar]
- 18. Bhattacharya S, Lange JJ, Levy M et al. The disordered regions of the methyltransferase SETD2 govern its function by regulating its proteolysis and phase separation. J Biol Chem. 2021; 297:101075. 10.1016/j.jbc.2021.101075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alberti S, Gladfelter A, Mittag T Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019; 176:419–34. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lafontaine DLJ, Riback JA, Bascetin R et al. The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. 2021; 22:165–82. 10.1038/s41580-020-0272-6. [DOI] [PubMed] [Google Scholar]
- 21. Faber GP, Nadav-Eliyahu S, Shav-Tal Y Nuclear speckles—a driving force in gene expression. J Cell Sci. 2022; 135:jcs259594. 10.1242/jcs.259594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chappidi N, Quail T, Doll S et al. PARP1–DNA co-condensation drives DNA repair site assembly to prevent disjunction of broken DNA ends. Cell. 2024; 187:945–61. 10.1016/j.cell.2024.01.015. [DOI] [PubMed] [Google Scholar]
- 23. Carruthers LM, Bednar J, Woodcock CL et al. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry. 1998; 37:14776–87. 10.1021/bi981684e. [DOI] [PubMed] [Google Scholar]
- 24. Lu X, Hansen JC Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J Biol Chem. 2004; 279:8701–7. 10.1074/jbc.M311348200. [DOI] [PubMed] [Google Scholar]
- 25. Arya G, Schlick T A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J Phys Chem A. 2009; 113:4045–59. 10.1021/jp810375d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blank TA, Becker PB Electrostatic mechanism of nucleosome spacing. J Mol Biol. 1995; 252:305–13. 10.1006/jmbi.1995.0498. [DOI] [PubMed] [Google Scholar]
- 27. Goytisolo FA, Gerchman SE, Yu X et al. Identification of two DNA-binding sites on the globular domain of histone H5. EMBO J. 1996; 15:3421–9. 10.1002/j.1460-2075.1996.tb00708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korolev N, Allahverdi A, Lyubartsev AP et al. The polyelectrolyte properties of chromatin. Soft Matter. 2012; 8:9322–33. 10.1039/c2sm25662b. [DOI] [Google Scholar]
- 29. Balhorn R The protamine family of sperm nuclear proteins. Genome Biol. 2007; 8:227. 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deckard CE, Sczepanski JT Reversible chromatin condensation by the DNA repair and demethylation factor thymine DNA glycosylase. Nucleic Acids Res. 2021; 49:2450–9. 10.1093/nar/gkab040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grau DJ, Chapman BA, Garlick JD et al. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011; 25:2210–21. 10.1101/gad.17288211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meehan RR, Kao CF, Pennings S HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J. 2003; 22:3164–74. 10.1093/emboj/cdg306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Erdel F Phase transitions in heterochromatin organization. Curr Opin Struct Biol. 2023; 80:102597. 10.1016/j.sbi.2023.102597. [DOI] [PubMed] [Google Scholar]
- 34. Erdel F, Rippe K Formation of chromatin subcompartments by phase separation. Biophys J. 2018; 114:2262–70. 10.1016/j.bpj.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basu S, Mackowiak SD, Niskanen H et al. Unblending of transcriptional condensates in Human repeat expansion disease. Cell. 2020; 181:1062–79. 10.1016/j.cell.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banning C, Votteler J, Hoffmann D et al. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One. 2010; 5:e9344. 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corces MR, Trevino AE, Hamilton EG et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017; 14:959–62. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buenrostro JD, Giresi PG, Zaba LC et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013; 10:1213–8. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dobin A, Davis CA, Schlesinger F et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Love MI, Huber W, Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolberg L, Raudvere U, Kuzmin I et al. g:profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023; 51:W207–12. 10.1093/nar/gkad347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramirez F, Ryan DP, Gruning B et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lopez-Delisle L, Rabbani L, Wolff J et al. pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics. 2021; 37:422–3. 10.1093/bioinformatics/btaa692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Danecek P, Bonfield JK, Liddle J et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021; 10:giab008. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quinlan AR BEDTools: the Swiss-Army tool for genome feature analysis. Curr Protoc Bioinform. 2014; 47:11.12.1–34. 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y, Liu T, Meyer CA et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ancona M, Brackley CA Simulating the chromatin-mediated phase separation of model proteins with multiple domains. Biophys J. 2022; 121:2600–12. 10.1016/j.bpj.2022.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langmead B, Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012; 9:357–9. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kremer K, Grest GS Dynamics of entangled linear polymer melts—a molecular dynamics simulation. J Chem Phys. 1990; 92:5057–86. 10.1063/1.458541. [DOI] [Google Scholar]
- 51. Brackley CA, Taylor S, Papantonis A et al. Nonspecific bridging-induced attraction drives clustering of DNA-binding proteins and genome organization. Proc Natl Acad Sci USA. 2013; 110:E3605–11. 10.1073/pnas.1302950110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiariello AM, Annunziatella C, Bianco S et al. Polymer physics of chromosome large-scale 3D organisation. Sci Rep. 2016; 6:29775. 10.1038/srep29775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Plimpton S Fast parallel algorithms for short-range molecular-dynamics. J Comput Phys. 1995; 117:1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- 54. Rosa A, Everaers R Structure and dynamics of interphase chromosomes. PLoS Comput Biol. 2008; 4:e1000153. 10.1371/journal.pcbi.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tafuri F, Chiariello AM The effect of configurational complexity in hetero-polymers on the coil–globule phase transition. Eur Phys J Plus. 2023; 138:150. [Google Scholar]
- 56. Chiariello AM, Abraham A, Bianco S et al. Multiscale modelling of chromatin 4D organization in SARS-CoV-2 infected cells. Nat Commun. 2024; 15:4014. 10.1038/s41467-024-48370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buckle A, Brackley CA, Boyle S et al. Polymer simulations of heteromorphic chromatin predict the 3D folding of complex genomic loci. Mol Cell. 2018; 72:786–97. 10.1016/j.molcel.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernandez-Escamilla AM, Rousseau F, Schymkowitz J et al. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004; 22:1302–6. 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 59. Obradovic Z, Peng K, Vucetic S et al. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003; 53(Suppl. 6):566–72. 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 60. Mirdita M, Schutze K, Moriwaki Y et al. ColabFold: making protein folding accessible to all. Nat Methods. 2022; 19:679–82. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scott MS, Boisvert FM, McDowall MD et al. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010; 38:7388–99. 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chu X, Sun T, Li Q et al. Prediction of liquid–liquid phase separating proteins using machine learning. BMC Bioinf. 2022; 23:72. 10.1186/s12859-022-04599-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ibrahim AY, Khaodeuanepheng NP, Amarasekara DL et al. Intrinsically disordered regions that drive phase separation form a robustly distinct protein class. J Biol Chem. 2023; 299:102801. 10.1016/j.jbc.2022.102801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hatos A, Tosatto SCE, Vendruscolo M et al. FuzDrop on AlphaFold: visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res. 2022; 50:W337–44. 10.1093/nar/gkac386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hartl B, Zeller T, Blanchette P et al. Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene. 2008; 27:3673–84. 10.1038/sj.onc.1211039. [DOI] [PubMed] [Google Scholar]
- 66. Tedeschi A, Wutz G, Huet S et al. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature. 2013; 501:564–8. 10.1038/nature12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wutz G, Varnai C, Nagasaka K et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017; 36:3573–99. 10.15252/embj.201798004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Houlard M, Cutts EE, Shamim MS et al. MCPH1 inhibits condensin II during interphase by regulating its SMC2-Kleisin interface. eLife. 2021; 10:e73348. 10.7554/eLife.73348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang D, Xiang KF, Xiang C et al. Construction of novel 7 integrin-related gene signatures in thyroid cancer construction of model based on integrin genes. Medicine. 2023; 102:e36412. 10.1097/MD.0000000000036412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Barbieri M, Chotalia M, Fraser J et al. Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci USA. 2012; 109:16173–8. 10.1073/pnas.1204799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eagen KP, Aiden EL, Kornberg RD Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc Natl Acad Sci USA. 2017; 114:8764–9. 10.1073/pnas.1701291114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Loubiere V, Papadopoulos GL, Szabo Q et al. Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci Adv. 2020; 6:eaax4001. 10.1126/sciadv.aax4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Du Z, Zheng H, Kawamura YK et al. Polycomb group proteins regulate chromatin architecture in mouse oocytes and early embryos. Mol Cell. 2020; 77:825–39. 10.1016/j.molcel.2019.11.011. [DOI] [PubMed] [Google Scholar]
- 74. Bsteh D, Moussa HF, Michlits G et al. Loss of cohesin regulator PDS5A reveals repressive role of polycomb loops. Nat Commun. 2023; 14:8160. 10.1038/s41467-023-43869-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ryu JK, Bouchoux C, Liu HW et al. Bridging-induced phase separation induced by cohesin SMC protein complexes. Sci Adv. 2021; 7:eabe5905. 10.1126/sciadv.abe5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Friman ET, Flyamer IM, Marenduzzo D et al. Ultra-long-range interactions between active regulatory elements. Genome Res. 2023; 33:1269–83. 10.1101/gr.277567.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chiariello AM, Corberi F, Salerno M The interplay between phase separation and gene-enhancer communication: a theoretical study. Biophys J. 2020; 119:873–83. 10.1016/j.bpj.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marko JF, Siggia ED Polymer models of meiotic and mitotic chromosomes. Mol Biol Cell. 1997; 8:2217–31. 10.1091/mbc.8.11.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tortora MMC, Brennan LD, Karpen G et al. HP1-driven phase separation recapitulates the thermodynamics and kinetics of heterochromatin condensate formation. Proc Natl Acad Sci USA. 2023; 120:e2211855120. 10.1073/pnas.2211855120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Forte G, Boteva L, Conforto F et al. Bridging condensins mediate compaction of mitotic chromosomes. J Cell Biol. 2024; 223:e202209113. 10.1083/jcb.202209113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Uckelmann M, Davidovich C Chromatin compaction by Polycomb group proteins revisited. Curr Opin Struct Biol. 2024; 86:102806. 10.1016/j.sbi.2024.102806. [DOI] [PubMed] [Google Scholar]
- 82. Plys AJ, Davis CP, Kim J et al. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019; 33:799–813. 10.1101/gad.326488.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Eeftens JM, Kapoor M, Michieletto D et al. Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat Commun. 2021; 12:5888. 10.1038/s41467-021-26147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Conte M, Irani E, Chiariello AM et al. Loop-extrusion and polymer phase-separation can co-exist at the single-molecule level to shape chromatin folding. Nat Commun. 2022; 13:4070. 10.1038/s41467-022-31856-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Niekamp S, Marr SK, Oei TA et al. Modularity of PRC1 composition and chromatin interaction define condensate properties. Mol Cell. 2024; 84:1651–66. 10.1016/j.molcel.2024.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Park J, Kim JJ, Ryu JK Mechanism of phase condensation for chromosome architecture and function. Exp Mol Med. 2024; 56:809–19. 10.1038/s12276-024-01226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hansen JC, Maeshima K, Hendzel MJ The solid and liquid states of chromatin. Epigenetics Chromatin. 2021; 14:50. 10.1186/s13072-021-00424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ng WS, Sielaff H, Zhao ZW Phase separation-mediated chromatin organization and dynamics: from imaging-based quantitative characterizations to functional implications. Int J Mol Sci. 2022; 23:8039. 10.3390/ijms23148039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lawrimore J, Kolbin D, Stanton J et al. The rDNA is biomolecular condensate formed by polymer-polymer phase separation and is sequestered in the nucleolus by transcription and R-loops. Nucleic Acids Res. 2021; 49:4586–98. 10.1093/nar/gkab229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tchurikov NA, Fedoseeva DM, Klushevskaya ES et al. rDNA clusters make contact with genes that are involved in differentiation and cancer and change contacts after heat shock treatment. Cells. 2019; 8:1393. 10.3390/cells8111393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Diesch J, Bywater MJ, Sanij E et al. Changes in long-range rDNA–genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun Biol. 2019; 2:39. 10.1038/s42003-019-0284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shav-Tal Y, Blechman J, Darzacq X et al. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell. 2005; 16:2395–413. 10.1091/mbc.e04-11-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kapur I, Boulier EL, Francis NJ Regulation of polyhomeotic condensates by intrinsically disordered sequences that affect chromatin binding. Epigenomes. 2022; 6:40. 10.3390/epigenomes6040040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li T, Rogers WB, Jacobs WM Interplay between self-assembly and phase separation in a polymer-complex model. Phys Rev E. 2023; 108:064501. 10.1103/PhysRevE.108.064501. [DOI] [PubMed] [Google Scholar]
- 95. Geller M, Cao Y, Simon C et al. Cooperation of a polymerizing SAM domain and an intrinsically disordered region enables full SAMD1 function on chromatin. Nucleic Acids Res. 2025; 53:gkaf259. 10.1093/nar/gkaf259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in NCBIs Gene Expression Omnibus (GEO), and can be accessed with GSE289538 and GSE289539.