

Abstract
A complete understanding of a protein folding mechanism requires description of the distribution of microscopic pathways that connect the folded and unfolded states. This distribution can, in principle, be described by computer simulations and theoretical models of protein folding, but is hidden in conventional experiments on large ensembles of molecules because only average properties are measured. A long-term goal of single molecule fluorescence studies is to time-resolve the structural events as individual molecules make transitions between folded and unfolded states. Although such studies are still in their infancy, the work up to now shows great promise and has already produced novel and important information on current issues in protein folding that has been impossible or difficult to obtain from ensemble measurements.
Introduction
A major technological development in recent years has been the capability of investigating protein folding and unfolding at the single molecule level. Two principal methods are currently being used: force-probe techniques and fluorescence. Experiments using atomic force microscopy and laser tweezers have provided important and previously inaccessible information on the mechanical stability and folding of proteins, and the reader is referred to a number of recent reviews on this topic [1-5]. Here we focus on the investigation of single molecule protein folding using the fluorescence method that has produced the most important and interesting results so far, Förster resonance energy transfer (FRET) [6-8], first demonstrated in single molecules by Ha et al. [9] and applied to protein folding in pioneering studies by Hochstrasser, Weiss, and their coworkers [10-12]. Recent applications to protein folding using other single molecule fluorescence techniques and the closely related method of fluorescence correlation spectroscopy can be found elsewhere [13,14].
The exciting prospect of watching individual molecules fold has been the major motivating factor for single molecule FRET studies. Additional motivation comes from computer simulations and analytical models of folding, which are playing an increasingly important role in investigations of protein folding mechanisms. If accurate, everything that one could possibly know about a protein folding mechanism is contained in a complete set of folding trajectories from atomistic molecular dynamics calculations. Such calculations have in fact just recently become possible for microsecond-folding proteins using distributed computing [15]. Moreover, most of what one would want to know about mechanisms is contained in the trajectories of coarse-grained representations of proteins [16]. Although ensemble experiments have provided a wealth of experimental data to test the accuracy of theoretical calculations, the experimental results alone yield relatively little information on actual folding mechanisms.
This point can be made clear by considering the case of a protein exhibiting two-state behavior (Figure 1). No matter what the probe, the same relaxation rates will be observed in kinetic experiments. The measured property at any time, moreover, will be a simple linear combination of the averaged property of the folded and the unfolded state, and the time course will be exponential. The reason for this simple behavior is that there is a separation of timescales; that is, inter-conversion among all of the conformers in each of the two states is rapid compared to the rate of inter-conversion between the two states. Experimental information concerning the mechanism by which the protein proceeds between the folded and unfolded states comes primarily from the systematic analysis of folding kinetics, in particular ϕ values [17]. This approach uses measurements of the relative effect of mutations on rates and equilibria and their interpretation with extra-thermodynamic relations to infer the average structure surrounding a mutated residue in the ensemble of structures that make up the transition state. There is, with few exceptions [18], no structural information at any other point along the reaction coordinate. Such ensemble experiments therefore provide relatively little, although important information concerning mechanism, which for protein folding means describing the sequence of structural events in the microscopic pathways that connect the folded and unfolded states and the distribution of these pathways, as can be obtained from both simulations [15,19] and the solution to the kinetic equations of analytical models [20] (E.R. Henry and WAE, unpublished results).
Figure 1.

Two-state folding dynamics of an individual protein molecule illustrated as a diffusive process on a two-dimensional free energy surface (left) with a corresponding equilibrium folding/unfolding trajectory (white). An intramolecular distance (corresponding to the distance between a donor and an acceptor fluorophore in a single molecule FRET experiment) is plotted as a function of time (right), showing rapid jumps between folded and unfolded state. An ultimate goal of single molecule experiments is to time-resolve these transitions (expanded scale, top right).
In contrast to ensemble studies, the investigation of individual molecules promises direct access to information on microscopic pathways. The ultimate goal of single molecule FRET studies is the time-resolved observation of individual protein folding events, which will allow the acquisition of trajectories of FRET efficiency – and therefore distance – versus time as the molecule transits between the unfolded and folded states. Such data should provide new insights and be much more demanding tests of both simulation and analytical models, thereby speeding progress towards a quantitative and more complete understanding of protein folding mechanisms.
Although single molecule FRET studies are still in their infancy, step by step progress towards the goal of watching single molecules fold has already produced novel and important information which has been either impossible or difficult to obtain from ensemble measurements. Such information includes the ability to count thermodynamic states from the distribution of FRET efficiencies, the separate measurement of properties of sub-populations, such as distance distributions and dynamics of the unfolded state in the presence of an excess of the folded state, and the determination of equilibrium constants and rate coefficients from FRET trajectories by measuring the mean residence times in folded and unfolded states at equilibrium. Examples of each of these will be presented, with brief discussions of their relevance to current issues in protein folding.
Accuracy of distances from single molecule FRET
Unlike most optical methods, FRET provides quantitative information on intermolecular distances1. An immediate question that arises in using the large chromophores that are necessary for single molecule FRET studies is: how accurate is this distance information? This question was first addressed for polypeptides by Schuler et al. [22], who used polyproline as a spacer between donor and acceptor chromophores, as did Stryer and Haugland in their classic study which established FRET as a “spectroscopic ruler” in biophysical research [23]. Schuler et al. measured the FRET efficiency of molecules that produce a burst of photons upon freely diffusing into and out of the illuminated volume of a confocal microscope (Figure 2). They found that the average FRET efficiency at short distances was slightly lower than predicted by Förster theory, but found that it was much higher than predicted for distances longer than R0, assuming that polyproline is a rigid rod in the all-trans conformation. Two possible explanations for the small discrepancy at short distances are the lack of complete orientational averaging during the shortened donor lifetime to result in an orientational factor κ2 < 2/3, and the breakdown of the point dipole approximation of Förster theory, which requires that the inter-chromophore distance be long compared to the size of the chromophores. To explain the much larger discrepancy for longer polyproline spacers, they carried out molecular dynamics simulations using an atomistic representation of polyproline in implicit solvent (i.e. Langevin simulations), which showed that all-trans polyproline is not a rigid rod, and concluded that bending to bring the donor and acceptor closer together is responsible for the higher efficiency. Subsequently, Watkins et al. [24] measured FRET from single immobilized polyproline molecules and discovered a static distribution of conformations, which they attributed to the presence of cis prolines, but made no estimate of their contribution. Doose et al. [25] also concluded that polyproline contains cis isomers from short range electron transfer FCS and ensemble measurements. Best et al. [26] used a combination of NMR spectroscopy to determine the fraction and location of cis-prolines, and single molecule photon trajectories using pulsed, picosecond excitation of freely-diffusing polyproline molecules to obtain accurate FRET efficiencies from subpopulation-specific time-correlated single photon counting histograms, and molecular dynamics calculations that include the dyes and their linkers in explicit solvent to interpret the results. These more realistic and accurate calculations indicate that the all-trans form of polyproline is less flexible than found in the previous simulations, and that the kinks arising from internal cis-prolines in 30% of the molecules and the flexibility of the chromophore linkers appear to be primarily responsible for bringing donor and acceptor closer together to produce the higher mean FRET efficiency. In addition, Best et al. [26] developed a theory for the distribution of donor and acceptor photons in a burst from individual molecules to quantitatively account for the shot noise and the broad distribution of FRET efficiencies [27,28] due to the large number of cis isomers, using the populations determined by NMR and the efficiencies calculated from molecular dynamics simulations.
Figure 2.

Overview of instrumentation and data reduction in confocal single molecule spectroscopy. The scheme on the right illustrates the main components of a 4-channel confocal single molecule instrument, such as the one commercially available [85], that collects fluorescence photons separated by polarization and wavelength and records their individual arrival times. (a) Sample of a trajectory of detected photons recorded from molecules freely diffusing in solution (in this example CspTm in 1.5 M GdmCl [37,38]), where every burst corresponds to an individual molecule traversing the diffraction-limited confocal volume (see upper right of the scheme). (b) 2-dimensional histogram of donor fluorescence lifetime τD versus transfer efficiency E calculated from individual bursts, resulting in subpopulations that can be assigned to the folded and unfolded protein and molecules without active acceptor at E ≈ 0 (shaded in grey). (c) Projection of the histogram onto the E axis. (d) Subpopulation-specific time-correlated single photon counting histograms from donor and acceptor photons from all bursts assigned to unfolded molecules that can be used to extract distance distributions [31,36,38]. (e) Subpopulation-specific donor intensity correlation function, in this case reporting on the nanosecond reconfiguration dynamics of the unfolded protein [58].
The polyproline studies provide an example of the importance of molecular simulations in extracting distance information, and to the extent that the simulations are accurate also suggest that accurate distances can be obtained from single molecule FRET. Additional evidence for the accuracy of distances from single molecule FRET comes from measurements using double-stranded DNA [29,30], which is a perfectly rigid spacer for determining donor-acceptor distances. A more direct test of the accuracy of distance determination in polypeptides was performed by Merchant et al. [31], who studied the distance distribution in the denatured state of protein L. They showed that the fluorescence lifetime decay could be quite well accounted for by the end-to-end distance distribution of a Gaussian chain, assumed to be static during the lifetime of the donor fluorescence (see discussion below on unfolded chain dynamics). This distribution is determined by a single parameter, 〈R2〉, the mean-squared end-to-end distance, which is related to the radius of gyration, Rg, by 〈R2〉 = 6〈Rg2〉. Rg calculated from the FRET-determined 〈R2〉 for protein L fully denatured at 4 M guanidinium chloride (GdmCl) was found to be 2.6 ± 0.1 nm, in remarkably good agreement with the value of 2.60 ± 0.03 nm obtained from SAXS measurements under the same conditions. Kuzmenkina et al. also found good agreement between the radius of gyration calculated from mean transfer efficiencies of single RNase H molecules in 6 M GdmCl (3.8 nm) and previous SAXS results (3.6 nm) [32].
Counting thermodynamic states
The simplest single molecule protein folding experiment is to monitor the photon bursts from freely diffusing molecules (Fig. 2a) at various denaturant concentrations. The limitation of this experiment is that a typical residence time in the confocal volume is only ∼500 μs, but a great advantage is that the experiment is free of artifacts from surface interactions that can be associated with immobilization methods, which will be discussed later. If the folded, unfolded, and partially-folded sub-populations interconvert much more slowly than the residence time, the FRET efficiency distribution will show peaks corresponding to the mean efficiencies of each subpopulation (Fig. 2b,c). Consequently, the number of peaks in the FRET efficiency distribution corresponds to the number of thermodynamic states, or more precisely, allowing for degeneracies, the minimum number of thermodynamic states. Although not yet observed experimentally, there are interesting theoretical studies on how FRET efficiency distributions change under conditions where the rate of interconversion between FRET states is not slow compared to the observation time [33-35].
In all of the free diffusion experiments on proteins that exhibit two-state behavior in ensemble measurements and have sufficiently slow folding and unfolding rates, two peaks are observed in the FRET efficiency distribution at intermediate denaturant concentrations, corresponding to the folded and unfolded populations (Figure 2b,c). The examples so far are chymotrypsin inhibitor 2 (CI2) [12,36], cold shock protein CspTm [31,37,38] acyl-CoA binding protein (ACBP) [36], RNase HI [32,39], protein L [31,40], the B domain of protein A [41], and the immunity protein Im9 [42]. The two-state nature of the kinetics of protein folding was demonstrated for CspTm in a non-equilibrium single-molecule FRET experiment by Lipman et al. [43]. In this work, a microfluidic device was combined with single molecule detection in a continuous flow mixing experiment. The FRET efficiency of individual molecules was determined as the molecules flowed through the confocal volume. Their position in the channel and the velocity of the flow determined the time of the observation following dilution of the chemical denaturant. At the earliest time after mixing (100 ms) a single peak is present, corresponding to the FRET efficiency of unfolded molecules. The amplitude of this peak decreases with time and is associated with a simultaneous increase in a higher efficiency peak corresponding to the folded sub-population (Figure 3). As pointed out above, this is exactly the behavior expected for a two-state system, in which there is simply an exchange of populations with time, with no change in the average property of either population.
Figure 3.
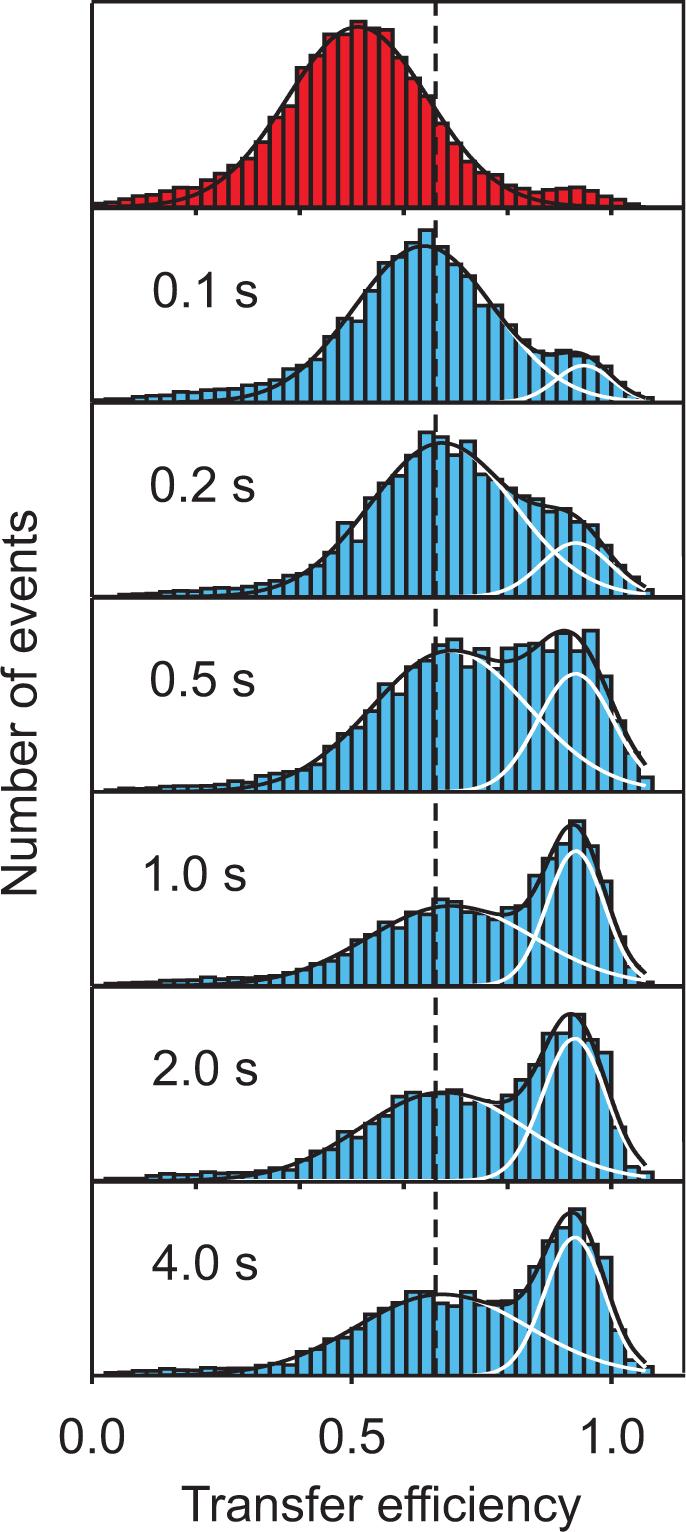
Protein folding kinetics measured with single molecule spectroscopy in a microfluidic mixing device [43]. Starting from CspTm unfolded at high denaturant concentration (top), transfer efficiency histograms are measured at different positions along the channel, corresponding to different times after mixing. The fits to Gaussians having the same peak position and width at all times illustrate the redistribution of populations expected for a two-state system after the initial chain collapse.
An interesting application of FRET efficiency distribution measurements would be to test the so-called “one-state” folding scenario of Muñoz and coworkers, in which there is no free energy barrier separating the folded and unfolded states at all denaturant concentrations [44]. For these proteins the kinetic relaxation rates will be shorter than the residence time and close to the folding speed limit estimated to be ∼(N/100) μs (N is the number of residues) [45]. Only a single peak would be observed, but for proteins folding on that time scale a single peak would also be observed if a barrier were present because of the averaging that results from the multiple folding/unfolding transitions during the ∼500 μs residence time in the confocal volume. However, if it were possible to slow the dynamics, for example by increasing the viscosity without altering the free energy surface, a “one-state” folder would still exhibit a single peak at all denaturant concentrations, but it would continuously shift to lower FRET efficiency as the denaturant concentration is increased. We should point out that it might be more problematic to identify barrierless folding if the barrier exists at high denaturant and disappears as the protein is stabilized by decreasing the denaturant concentration [41], as envisaged in the original “downhill” scenario of Wolynes and Onuchic [46].
Structure of the unfolded subpopulation
A unique aspect of the free diffusion experiments on single molecules is that structural and dynamic properties of the sub-population of unfolded molecules can be separately investigated at equilibrium in the presence of a large excess of folded molecules, where the ensemble averaged properties are dominated by the folded state. A consistent finding in all of the free diffusion experiments is that the overall size of the unfolded protein, as obtained from the FRET efficiency-determined 〈R2〉, increases continuously with increasing denaturant concentration. This behavior, first unequivocally demonstrated for CspTm [37], has been observed in chymotrypsin inhibitor 2 (CI2) [12,36], acyl-CoA binding protein (ACBP) [36], RNase H [32,39], protein L [31,40], the B domain of protein A [41], the immunity protein Im9 [42], and the prion-determining domain of Sup35 [47]. Kuzmenkina et al. [32] were able to fit their data for RNase HI with a model in which the unfolded state is described by a continuum of substates, while Sherman et al. [40] employed an analytical polymer model to describe the change in size as a coil-globule transition and interpreted the results in terms of the solvation properties of the unfolded chain.
The mean FRET efficiencies for unfolded protein L and CspTm are identical at high denaturant concentration [31], consistent with the finding from small X-ray scattering measurements that the size of unfolded proteins depends only on the length of the polypeptide and is independent of sequence [48]. However, they differ at lower denaturant concentration when the folded state is also present, with protein L being more compact, possibly as a result of its more hydrophobic sequence. It would be extremely difficult to reliably detect such differences in equilibrium SAXS experiments where both folded and unfolded molecules contribute to the observed scattering [49]. The single molecule experiments on protein L are, however, at variance with time-resolved SAXS experiments by Plaxco et al. [50] who found no difference in Rg between the initial denatured state at 4 M GdmCl and the denatured state prior to folding at 1M GdmCl. The discrepancy between the ensemble SAXS and single molecule FRET result is yet to be explained [31], particularly as continuous expansion of protein L is observed upon the addition of urea in molecular dynamics simulations in explicit water [31,51].
Interestingly, the Gaussian chain approximation that explains the fluorescence decay discussed above for protein L at GdmCl concentrations where it is fully denatured [31], also applies at lower denaturant concentrations for both the protein L and CspTm unfolded subpopulations. This issue was addressed in more detail for CspTm by Hoffmann et al. [38], who investigated variants of the protein with the dye labels positioned such that different segments of the chain could be probed. A combined analysis of FRET efficiency histograms (Fig. 2c) and subpopulation-specific fluorescence lifetimes (Fig. 2d) gave good agreement with intramolecular distance distributions of a Gaussian chain for all variants, even at low denaturant concentrations, where the chain is compact. This indicates that any residual structure can affect only short segments and is probably highly dynamic. Kinetic synchrotron radiation circular dichroism experiments in fact provided evidence for the presence of some β-structure in the compact unfolded state [38], and kinetic ensemble FRET experiments probing a short segment that forms a β-strand in the folded state indicate the local formation of extended structure in the compact unfolded state of a closely related cold shock protein, CspBc [52]. However, there are also examples for deviations from Gaussian chain behavior. McCarney et al. [53] found slight, but possibly significant, site-specific deviations from Gaussian-chain behavior in single molecule FRET experiments on the FynSH3 domain fully unfolded in GdmCl and trifluoroethanol. Laurence et al. [36] used subpopulation-specific fluorescence lifetime analysis to investigate structural distributions upon collapse of CI2 and ACBP and inferred the presence of transient residual structure in the unfolded state.
These single molecules FRET studies, together with ensemble SAXS and NMR studies on unfolded proteins [54-56], suggest that unfolded proteins behave essentially as homopolymers at the highest denaturant concentration, as evidenced in particular by the finding that Rg depends on chain length and is independent of sequence [48]. But the diversity of observations from single molecule FRET concerning the structure of collapsed unfolded proteins highlight the importance of the amino acid sequence under conditions that favor folding.
Dynamics of the unfolded subpopulation
Dynamics of the unfolded subpopulations can be investigated in multiple time regimes – from the nanosecond time scale of the fluorescence lifetime to the millisecond time scale of the residence time in the confocal volume, and for longer times with immobilized molecules. Dynamics in the millisecond regime have frequently been evaluated from an analysis of the width of the unfolded FRET efficiency peak, and Gopich and Szabo have carried out a comprehensive theoretical analysis of the various contributions to the width in free diffusion experiments [33,35]. As can be seen from their calculations for a Gaussian chain (Figure 4), the width is very sensitive to the relation between the donor-acceptor distance correlation time and the observation time, varying from a very broad distribution in the slow dynamics limit to a δ-function (in the absence of shot noise) in the fast dynamics limit. The analysis of the width for the unfolded subpopulation has been a much discussed subject [37,57] since the first single molecule folding experiments by Hochstrasser, Weiss and their colleagues, in which the width in excess of that produced by shot noise was attributed to slow dynamics of the unfolded molecules [10,12]. Recently, Merchant et al. [31] showed that excess width for the unfolded peaks of protein L and CspTm is not an instrumental, optical, or signal processing artifact, because the donor lifetime is not constant when evaluated from the photons belonging to the high and low efficiency side of the unfolded FRET peaks. The width, moreover, was found to be independent of the observation time, suggesting that the structures responsible for the added dispersion interconvert more slowly than the 1−2 ms bin sizes. However, they could not determine whether the excess width reflects different R0's because of imperfect purification of the two possible permutants, intrinsically slow protein dynamics, or results from slow dynamics of protein-dye interaction.
Figure 4.

Transfer efficiency distribution p(E | t) for a Gaussian chain with different ratios of observation time t and chain reconfiguration time τr, with t/τr = 0 (dark blue), 0.1 (light blue), 1 (green), 5 (yellow), and 10 (red) (assuming R0 = 〈r2〉), illustrating the sensitivity of transfer efficiency distributions to chain dynamics. Gopich and Szabo [33] showed that for a Gaussian chain and t >> τr, τr ≈ 10 t σ2, with , where is the observed variance in E, and is the variance due to noise and other effects not dependent on the interdye distance.
Understanding the dynamics of unfolded polypeptide chains in the fast time regime has taken on special importance, as an increasing number of proteins are discovered to fold on the microsecond time scale. In this regime, the free energy barrier to folding is assumed to be extremely low or even absent, and diffusive chain dynamics become the dominant factor in folding kinetics [45]. The dynamics on this time scale have been studied by Nettles et al. [58], who analyzed the single molecule photon statistics of the fluctuations in intensity of donor and acceptor fluorophores that result from distance fluctuations in the unfolded sub-population of CspTm. The basic idea of their experiment is the following: if, for example, a donor photon is emitted at time τ = 0, the chain ends are likely to be far apart at that instant, corresponding to a low rate of energy transfer. A very short time later, the ends will still be far apart, and the likelihood of emitting another donor photon will still be high. However, at times much greater than the reconfiguration time of the chain, the molecule will have lost the “memory” of its initial configuration, and the probability of donor emission will be determined by the average transfer efficiency. Thus the autocorrelation of the emission intensity is expected to decay approximately on the time scale of chain reconfiguration (Fig. 2e). With information on the unfolded state dimensions available from previous experiments [38], and using a model that describes chain dynamics as a diffusive process on a one-dimensional free energy surface [58-61], the very rapid reconfiguration time could be extracted. This time decreases from about 60 ns to 20 ns between 0 and 6 M GdmCl, after correcting for solvent viscosity. The addition of denaturant thus not only expands the chain, but the reduced transient intramolecular interactions decrease the contribution of internal friction to chain diffusion. Correlation spectroscopy indicates that at times less than ∼100 μs there are no additional long-range dynamics in unfolded CspTm [62]. Moreover, since the dye-labeled protein folds in 12 ms in the absence of denaturant with an exponential time course [37], the requirement that the unfolded state dynamics be fast compared to the folding times indicates that there are no slower unfolded state dynamics.
There have already been three important applications of the results from this study. First, it justifies the approximation by Merchant et al. [31] and Hoffmann et al. [38] that the unfolded CspTm is static during the 1−2 ns donor fluorescence lifetime in their analysis of the lifetime distribution of the unfolded sub-population using various models for the end-to-end distance distribution. Second, the finding of an increasing chain diffusion coefficient with increasing denaturant is directly related to analytical theoretical models in which protein folding kinetics are described by diffusion on a low-dimensional free energy surface [63-65]. Folding simulations using a simplified lattice representation of a protein had shown that the end-to-end diffusion coefficient closely corresponds to the diffusion coefficient for motion along the folding reaction coordinate [63]. This Kramers description of folding kinetics was used by Cellmer et al. [66] to calculate relaxation rates as a function of denaturant for a small ultrafast folding protein – the villin subdomain. The calculations produced a chevron-like plot of relaxation rate versus denaturant concentration, albeit with very small slopes, in contrast to the experimental result of a denaturant-independent relaxation rate. However, introducing the denaturant-dependence for the diffusion coefficient found for CspTm into the calculation compensated for the increasing barrier height to folding, flattening the chevron as observed experimentally.
The third application is the estimation of the free energy barrier height, ΔG≠, separating folded and unfolded states (as opposed to the activation enthalpy which is directly obtained from the temperature dependence of the folding time). Assuming equal diffusion coefficients and curvatures in the denatured well and on the (inverted) barrier top for the free energy versus reaction coordinate, the Kramers equation for the folding time reduces to τf ≈ 2πτr exp(ΔG≠/KBT) [63], where kB is Boltzmann’s constant and T is temperature. With τr = 60 ns as the reconfiguration time obtained by Nettles et al. [58] for the unfolded subpopulation in the absence of denaturant, and τf = 12 ms for the folding time measured for the dye-labeled protein [37], the calculated free energy barrier to folding is 10 kBT [58]. This should be regarded as an upper limit because the diffusion coefficient at the barrier top is expected to be lower as the protein becomes more compact [58,66] (interestingly, the pre-exponential factor, >2πτr, yields an estimate of ≳0.4 μs for the speed limit for this protein, approaching the prediction of ∼0.1N μs = 0.7 μs by Kubelka et al. [45]). The free energy barrier heights have been calculated theoretically for two closely related cold shock proteins that fold with about the same rate (CspB and CspA), using coarse-grained representations in both an Ising-like analytical model [20,67,68] and in Langevin simulations with a bead model representation of the polypeptide [68,69]. In the simulations there is no free energy barrier under folding conditions [69], while the Ising-like models [20,67,68] yield barriers comparable to those estimated from the experiments (e.g., 7−8 kBT, allowing for disordered loops [20]).
Single molecule folding/unfolding trajectories
As pointed out in the introduction to this article, a major goal of single molecule studies is to monitor the actual transitions between the folded and unfolded states. The first requirement for such experiments is to obtain trajectories of proteins undergoing folding and unfolding transitions, as in Figure 1. Because of the short residence time in the confocal volume, experiments up to now have focused on immobilizing the protein. The first experiments by Hochstrasser and coworkers [10,11], however, showed that the technical challenge in attaching the protein to a glass surface is to avoid artifacts resulting from transient sticking to the surface. Rhoades et al. [70,71] addressed this difficulty by encapsulating the protein in lipid vesicles and attaching the vesicles to the surface. They obtained folding/unfolding trajectories of two proteins, adenylate kinase and CspTm. The trajectories for adenylate kinase [70] exhibited broad distributions of step sizes and transition times, indicating a large degree of complexity that was difficult to interpret quantitatively, whereas CspTm [71] showed trajectories for individual molecules close to what would be expected for a two-state protein (Fig. 1): rapid jumps between high and low FRET states, corresponding to the folded and unfolded molecule states, with a distribution of residence times expected from the ensemble unfolding and folding times, respectively. The jumps between states, which correspond to the actual free energy barrier crossing events, could not be resolved, and were estimated to occur in <100 μs. Estimates of both barrier crossing times [64] and of the folding speed limit from experiments [45,58] indicate that jumps between folded and unfolded states should occur in less than a microsecond. It will thus be one of the major challenges in the field to improve the experimental time resolution.
Immobilization of proteins by attaching them to a surface has been revisited by Nienhaus and coworkers. Kuzmenkina et al. [32,39] immobilized RNase H via biotin and streptavidin to a cross-linked polyethylene (“star PEG”) [72] coated surface, and showed that the folding/unfolding equilibrium as a function of denaturant concentration is completely reversible and yields thermodynamic parameters for the surface-immobilized protein in good agreement with ensemble experiments for the unlabeled protein. However, similar to the adenylate kinase studies of Rhoades et al. [70], the fluorescence trajectories were found to be complex, and showed fluctuations over a wide range of FRET efficiencies. Fluctuations with FRET changes >0.05 were interpreted as distance changes and, using a similar procedure as Rhoades et al. [70], were classified according to the initial and final FRET efficiency values as folding transitions, unfolding transitions, transitions among folded states and transitions among unfolded states. From the mean residence times in the folded and unfolded states they obtained rate coefficients (0.01/s) remarkably close to those found in ensemble experiments for the unlabeled protein. Whereas polarization experiments indicated a lack of orientational averaging of the dyes in the folded state, which might explain the transitions among folded state efficiencies, no steady state anisotropy was observed for the unfolded molecules, from which the authors concluded that their slow dynamics (0.4 transition/s) arise from large free energy barriers caused by substantial structure in the denatured state. To explore the unfolded state dynamics on a faster time scale, the donor-acceptor crosscorrelation function was calculated and revealed a 20 μs relaxation.
The interesting result from this study is that reconfiguration of the denatured protein apparently occurs on a wide range of time scales, from microseconds (or less) to seconds, in contrast to the findings from CspTm, in which no unfolded stated dynamics could be identified above the tens of nanoseconds regime (see above) [58,62]. Even though both RNase H [73] and adenylate kinase [74] are known to populate folding intermediates, which may contribute to the complexity observed in single molecule experiments, there is the interesting possibility that the rougher energy landscape revealed in the single molecule trajectories contributes to the slower folding rates of such larger proteins by slowing diffusion along the reaction coordinate.
Concluding Remarks
From the preceding discussion it should be clear that single molecule FRET studies have already contributed to a better understanding of protein folding, particularly on the structure and dynamics of the unfolded state. To reach the goal of time-resolved FRET measurements while an individual molecule folds and unfolds will require overcoming several technical challenges, including improvements in collection efficiency beyond the ∼10% of current instruments, elimination of photo-destruction, and reduction in dark and triplet states of the chromophores, all of which can contribute to increasing the rate of emitted photons towards the limit of the inverse fluorescence lifetime (several nanoseconds). In addition, alternative immobilization methods will most probably be required. A promising technique, that does not require attachment to a surface or bead, has recently been developed by Cohen and Moerner [75], who rapidly vary the direction of an electric field in a microfluidic cell to prevent the molecule from diffusing out of the confocal volume. Imaging single molecules as they flow down a capillary [76] has also been suggested as a way to avoid surface immobilization.
Another important advance would be to measure several distances simultaneously by attaching multiple donor and acceptor chromophores. The feasibility of this approach has been demonstrated for DNA [77-79] and it should also be possible to apply it to proteins, in spite of the more difficult labeling chemistry [80]. The simultaneous time-resolved measurement of even a few distances would place considerable constraints on the evolution of the native fold, and provide critical tests of theoretical models and simulations. The analysis of the correlation among these distances would also directly yield information on the breadth of the microscopic pathway distributions [81]. Finally, there is the possibility that single molecule FRET can be used to investigate protein folding mechanisms in more complex environments. The most obvious application is the influence of cellular factors, ranging from the ribosome and molecular chaperones [82,83] to protein translocation and membrane protein folding, protein unfolding and degradation by proteases, or protein misfolding and aggregation [84]. For most of these processes, the mechanisms of the molecular machinery have been the focus of investigation, but little is known about their influence on protein structure or folding dynamics and mechanism.
Box. Förster resonance energy transfer.

Acknowledgement
Our single molecule FRET studies have profited enormously from the theoretical work of Irina Gopich and Attila Szabo. We would also like to thank them for collaboration and many helpful discussions since we initiated our research in this area and for comments on this manuscript. BS has been supported by the Schweizerische Nationalfonds, the National Center for Competence in Research in Structural Biology, the Human Frontier Science Program, the VolkswagenStiftung, and the Deutsche Forschungsgemeinschaft. WAE is supported by the Intramural Research Program of NIDDK, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1 Box (including figure): Förster [21] showed that the rate of excitation energy transfer, kET, between donor and acceptor chromophores is proportional to the inverse 6th power of the distance separating them, kET = kD(R0/R)6, where kD−1 is the lifetime of the donor in the absence of the acceptor, R is the distance between donor and acceptor and R0 is a proportionality constant that depends on the transition dipole-dipole interaction between donor and acceptor. The probability that an absorbed photon by the donor will lead to excitation energy transfer to the acceptor, called the FRET efficiency, E, is given by kET/( kET + kD), which is determined experimentally either by counting the number of emitted donor and acceptor photons, E = nA/(nA + nD) = 1/(1 + (R / R0)6) or by measuring the donor lifetime in the presence and absence of acceptor: E = 1 – (τDA/τD). The beauty of Förster theory is that R0 can be directly obtained, without any theoretical calculations, from readily measurable spectroscopic quantities - the donor quantum yield and fluorescence spectrum, the acceptor absorption spectrum, and the refractive index of the medium between them. For the additional experimental and theoretical concepts associated with the method, the reader is referred to a number of detailed reviews .
References
- 1.Fisher TE, Oberhauser AF, Carrion-Vazquez M, Marszalek PE, Fernandez JM. The study of protein mechanics with the atomic force microscope. Trends in Biochemical Sciences. 1999;24:379–384. doi: 10.1016/s0968-0004(99)01453-x. [DOI] [PubMed] [Google Scholar]
- 2.Zhuang X, Rief M. Single-molecule folding. Curr Opin Struct Biol. 2003;13:88–97. doi: 10.1016/s0959-440x(03)00011-3. [DOI] [PubMed] [Google Scholar]
- 3.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu Rev Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 4.Forman JR, Clarke J. Mechanical unfolding of proteins: insights into biology, structure and folding. Curr Opin Struct Biol. 2007;17:58–66. doi: 10.1016/j.sbi.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Hummer G, Szabo A. Free energy surfaces from single-molecule force spectroscopy. Accounts Chem Res. 2005;38:504–513. doi: 10.1021/ar040148d. [DOI] [PubMed] [Google Scholar]
- 6.Haran G. Single-molecule fluorescence spectroscopy of biomolecular folding. J Phys-Condens Matter. 2003;15:R1291–R1317. [Google Scholar]
- 7.Schuler B. Single-molecule fluorescence spectroscopy of protein folding. Chemphyschem. 2005;6:1206–1220. doi: 10.1002/cphc.200400609. [DOI] [PubMed] [Google Scholar]
- 8.Michalet X, Weiss S, Jäger M. Single-molecule fluorescence studies of protein folding and conformational dynamics. Chem Rev. 2006;106:1785–1813. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia YW, Talaga DS, Lau WL, Lu HSM, DeGrado WF, Hochstrasser RM. Folding dynamics of single GCN4 peptides by fluorescence resonant energy transfer confocal microscopy. Chem Phys. 1999;247:69–83. [Google Scholar]
- 11.Talaga DS, Lau WL, Roder H, Tang J, Jia Y, DeGrado WF, Hochstrasser RM. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc Natl Acad Sci USA. 2000;97:13021–13026. doi: 10.1073/pnas.97.24.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Single-molecule protein folding: Diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc Natl Acad Sci USA. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuweiler H, Sauer M. Using photoinduced charge transfer reactions to study conformational dynamics of biopolymers at the single-molecule level. Curr Pharm Biotechnol. 2004;5:285–298. doi: 10.2174/1389201043376896. [DOI] [PubMed] [Google Scholar]
- 14.Frieden C, Chattopadhyay K, Elson EL. Adv Protein Chem. Vol. 62. 2002. What fluorescence correlation spectroscopy can tell us about unfolded proteins. pp. 91–109. Advances in Protein Chemistry. [DOI] [PubMed] [Google Scholar]
- 15.Snow CD, Sorin EJ, Rhee YM, Pande VS. How well can simulation predict protein folding kinetics and thermodynamics? Annu Rev Biophys Biomol Struct. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- 16.Levy Y, Onuchic JN. Mechanisms of protein assembly: lessons from minimalist models. Acc Chem Res. 2006;39:135–142. doi: 10.1021/ar040204a. [DOI] [PubMed] [Google Scholar]
- 17.Fersht AR. Structure and Mechanism in Protein Science. W.H. Freeman and Company; New York: 1998. [Google Scholar]
- 18.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 19.Fersht AR, Daggett V. Protein folding and unfolding at atomic resolution. Cell. 2002;108:573–582. doi: 10.1016/s0092-8674(02)00620-7. [DOI] [PubMed] [Google Scholar]
- 20.Henry ER, Eaton WA. Combinatorial modeling of protein folding kinetics: free energy profiles and rates. Chemical Physics. 2004;307:163–185. [Google Scholar]
- 21.Förster T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik. 1948;6:55–75. [Google Scholar]
- 22.Schuler B, Lipman EA, Steinbach PJ, Kumke M, Eaton WA. Polyproline and the “spectroscopic ruler” revisited with single molecule fluorescence. Proc Natl Acad Sci USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watkins LP, Chang HY, Yang H. Quantitative single-molecule conformational distributions: A case study with poly-(L-proline). J Phys Chem A. 2006;110:5191–5203. doi: 10.1021/jp055886d. [DOI] [PubMed] [Google Scholar]
- 25.Doose S, Neuweiler H, Barsch H, Sauer M. Probing polyproline structure and dynamics by photoinduced electron transfer provides evidence for deviations from a regular polyproline type II helix. Proc Natl Acad Sci USA. 2007;104:17400–17405. doi: 10.1073/pnas.0705605104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Best R, Merchant K, Gopich IV, Schuler B, Bax A, Eaton WA. Effect of flexibility and cis residues in single molecule FRET studies of polyproline. Proc Natl Acad Sci USA. in press. A combination of single molecule intensity and subpopulation lifetime measurements, NMR, and molecular dynamics simulations of polyproline, including the dyes and their linkers to the polypeptide, shows that that the kinks arising from internal cis-prolines are primarily responsible for a mean FRET efficiency higher than predicted for a rigid-rod all-trans polyproline. Theoretical analysis also shows that the width in excess of shot noise in the observed efficiency histograms and distributions of donor fluorescence lifetimes are explained by the presence of multiple species with efficiencies consistent with the simulations and populations with NMR.* [Google Scholar]
- 27.Nir E, Michalet X, Hamadani KM, Laurence TA, Neuhauser D, Kovchegov Y, Weiss S. Shot-noise limited single-molecule FRET histograms: Comparison between theory and experiments. J Phys Chem B. 2006;110:22103–22124. doi: 10.1021/jp063483n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antonik M, Felekyan S, Gaiduk A, Seidel CAM. Separating structural heterogeneities from stochastic variations in fluorescence resonance energy transfer distributions via photon distribution analysis. J Phys Chem B. 2006;110:6970–6978. doi: 10.1021/jp057257+. [DOI] [PubMed] [Google Scholar]
- 29.Kapanidis AN, Lee NK, Laurence TA, Doose S, Margeat E, Weiss S. Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proc Natl Acad Sci USA. 2004;101:8936–8941. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. The unfolded subpopulations of protein L and CspTm are compared over a range of denaturant concentrations from FRET efficiencies determined from both intensity and lifetime measurements. The interpretation is guided by Langevin simulations of a simplified representation of the polypeptide, which also suggest that collapse can result either from increased inter-residue attraction or decreased excluded volume.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule FRET study of denaturant induced unfolding of RNase H. J Mol Biol. 2006;357:313–324. doi: 10.1016/j.jmb.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 33.Gopich IV, Szabo A. Single-macromolecule fluorescence resonance energy transfer and free-energy profiles. J Phys Chem B. 2003;107:5058–5063. [Google Scholar]
- 34.Gopich IV, Szabo A. Theory of photon statistics in single-molecule Förster resonance energy transfer. J Chem Phys. 2005;122:1–18. doi: 10.1063/1.1812746. [DOI] [PubMed] [Google Scholar]
- 35.Gopich IV, Szabo A. Single-molecule FRET with diffusion and conformational dynamics. J Phys Chem B. 2007;111:12925–12932. doi: 10.1021/jp075255e. A comprehensive theory is presented for the analysis of single molecule FRET measurements on freely diffusing and immobilized molecules, including the contribution of various processes to the distribution and variance in FRET efficiency.** [DOI] [PubMed] [Google Scholar]
- 36.Laurence TA, Kong XX, Jager M, Weiss S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc Natl Acad Sci USA. 2005;102:17348–17353. doi: 10.1073/pnas.0508584102. This work contains the first use of subpopulation-specific fluorescence lifetime analysis for the investigation of distance distributions in unfolded proteins and infers the presence of transient unfolded state structure in CI2 and ACBP.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann A, Kane A, Nettels D, Hertzog DE, Baumgartel P, Lengefeld J, Reichardt G, Horsley DA, Seckler R, Bakajin O, Schuler B. Mapping protein collapse with single-molecule fluorescence and kinetic synchrotron radiation circular dichroism spectroscopy. Proc Natl Acad Sci USA. 2007;104:105–110. doi: 10.1073/pnas.0604353104. By varying the position of donor and acceptor in the chain, different segments of unfolded CspTm are probed, indicating isotropic collapse and good agreement with Gaussian chain behavior even in the collapsed unfolded state. Synchrotron circular dichroism spectroscopy reveals the formation of some beta-structure concommitant with collapse.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule Forster resonance energy transfer study of protein dynamics under denaturing conditions. Proc Natl Acad Sci USA. 2005;102:15471–15476. doi: 10.1073/pnas.0507728102. Folding/unfolding trajectories are obtained from FRET efficiency measurements on surface-immobilized RNase H, and suggest unfolded state dynamics on time scales from microseconds to seconds.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherman E, Haran G. Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. The mean FRET efficiency and hydrodynamic radius from FCS indicate collapse of the unfolded protein L subpopulation upon decreasing denaturant concentration. The collapse is analyzed in terms of a coil-globule transition to obtain information about the solvation properties of the unfolded state.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang F, Sato S, Sharpe TD, Ying LM, Fersht AR. Distinguishing between cooperative and unimodal downhill protein folding. Proc Natl Acad Sci USA. 2007;104:123–127. doi: 10.1073/pnas.0609717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tezuka-Kawakami T, Gell C, Brockwell DJ, Radford SE, Smith DA. Urea-induced unfolding of the immunity protein Im9 monitored by spFRET. Biophys J. 2006;91:L42–L44. doi: 10.1529/biophysj.106.088344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipman EA, Schuler B, Bakajin O, Eaton WA. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 44.Munoz V. Conformational dynamics and ensembles in protein folding. Annu Rev Biophys Biomol Struct. 2007;36:395–412. doi: 10.1146/annurev.biophys.36.040306.132608. [DOI] [PubMed] [Google Scholar]
- 45.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 47.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. The prion-determining NM domain of the yeast prion protein Sup35 is shown to undergo a continuous expansion rather than a cooperative unfolding transition upon addition of denaturant. Fluorescence intensity fluctuations in the submicrosecond range are assigned to chain dynamics resulting in rapid intramolecular quenching by tyrosine.* [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Millet IS, Townsley LE, Chiti F, Doniach S, Plaxco KW. Equilibrium collapse and the kinetic ‘foldability’ of proteins. Biochemistry. 2002;41:321–325. doi: 10.1021/bi015695a. [DOI] [PubMed] [Google Scholar]
- 50.Plaxco KW, Millett IS, Segel DJ, Doniach S, Baker D. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat Struct Biol. 1999;6:554–556. doi: 10.1038/9329. [DOI] [PubMed] [Google Scholar]
- 51.Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci USA. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magg C, Kubelka J, Holtermann G, Haas E, Schmid FX. Specificity of the initial collapse in the folding of the cold shock protein. J Mol Biol. 2006;360:1067–1080. doi: 10.1016/j.jmb.2006.05.073. [DOI] [PubMed] [Google Scholar]
- 53.McCarney ER, Werner JH, Bernstein SL, Ruczinski I, Makarov DE, Goodwin PM, Plaxco KW. Site-specific dimensions across a highly denatured protein; a single molecule study. J Mol Biol. 2005;352:672–682. doi: 10.1016/j.jmb.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 54.Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem. 2002;62:311–340. doi: 10.1016/s0065-3233(02)62012-1. [DOI] [PubMed] [Google Scholar]
- 55.Millett IS, Doniach S, Plaxco KW. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv Protein Chem. 2002;62:241–262. doi: 10.1016/s0065-3233(02)62009-1. [DOI] [PubMed] [Google Scholar]
- 56.Shi Z, Woody RW, Kallenbach NR. Is polyproline II a major backbone conformation in unfolded proteins? Adv Protein Chem. 2002;62:163–240. doi: 10.1016/s0065-3233(02)62008-x. [DOI] [PubMed] [Google Scholar]
- 57.Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S. Ratiometric single-molecule studies of freely diffusing biomolecules. Annu Rev Phys Chem. 2001;52:233–253. doi: 10.1146/annurev.physchem.52.1.233. [DOI] [PubMed] [Google Scholar]
- 58.Nettels D, Gopich IV, Hoffmann A, Schuler B. Ultrafast dynamics of protein collapse from single-molecule photon statistics. Proc Natl Acad Sci USA. 2007;104:2655–2660. doi: 10.1073/pnas.0611093104. Global chain reconfiguration dynamics on a 50 ns time scale are measured for unfolded CspTm in subpopulation-specific correlation experiments. The dynamics slow down in the more compact denatured state at low denaturant from increased internal friction. This finding may explain the flat chevron observed for an ultrafast-folding protein [66] and raises the possibility of related effects on the folding kinetics of other proteins.** [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang ZS, Makarov DE. Nanosecond dynamics of single polypeptide molecules revealed by photoemission statistics of fluorescence resonance energy transfer: A theoretical study. J Phys Chem B. 2003;107:5617–5622. [Google Scholar]
- 60.Gopich IV, Szabo A. Theory of the statistics of kinetic transitions with application to single-molecule enzyme catalysis. J Chem Phys. 2006;124:154712. doi: 10.1063/1.2180770. [DOI] [PubMed] [Google Scholar]
- 61.Szabo A, Schulten K, Schulten Z. 1st Passage Time Approach to Diffusion Controlled Reactions. J Chem Phys. 1980;72:4350–4357. [Google Scholar]
- 62.Nettels D, Hoffmann A, Schuler B. Unfolded Protein and Peptide Dynamics Investigated with Correlation Spectroscopy from Picoseconds to Seconds. J Phys Chem B. doi: 10.1021/jp076971j. (submitted) [DOI] [PubMed] [Google Scholar]
- 63.Socci ND, Onuchic JN, Wolynes PG. Diffusive dynamics of the reaction coordinate for protein folding funnels. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 64.Best RB, Hummer G. Diffusive model of protein folding dynamics with Kramers turnover in rate. Phys Rev Lett. 2006;96 doi: 10.1103/PhysRevLett.96.228104. [DOI] [PubMed] [Google Scholar]
- 65.Chahine J, Oliveira RJ, Leite VBP, Wang J. Configuration-dependent diffusion can shift the kinetic transition state and barrier height of protein folding. Proc Natl Acad Sci USA. 2007;104:14646–14651. doi: 10.1073/pnas.0606506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cellmer T, Henry ER, Kubelka J, Hofrichter J, Eaton WA. Relaxation rate for an ultrafast folding protein is independent of chemical denaturant concentration. J Am Chem Soc. doi: 10.1021/ja0761939. in press. [DOI] [PubMed] [Google Scholar]
- 67.Muñoz V, Eaton WA. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karanicolas J, Brooks CL. The importance of explicit chain representation in protein folding models: An examination of Ising-like models. Proteins. 2003;53:740–747. doi: 10.1002/prot.10459. [DOI] [PubMed] [Google Scholar]
- 69.Shea JE, Brooks CL. From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu Rev Phys Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- 70.Rhoades E, Gussakovsky E, Haran G. Watching proteins fold one molecule at a time. Proc Natl Acad Sci USA. 2003;100:3197–3202. doi: 10.1073/pnas.2628068100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rhoades E, Cohen M, Schuler B, Haran G. Two-state folding observed in individual protein molecules. J Am Chem Soc. 2004;126:14686–14687. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 72.Groll J, Amirgoulova EV, Ameringer T, Heyes CD, Rocker C, Nienhaus GU, Moller M. Biofunctionalized, ultrathin coatings of cross-linked star-shaped poly(ethylene oxide) allow reversible folding of immobilized proteins. J Am Chem Soc. 2004;126:4234–4239. doi: 10.1021/ja0318028. [DOI] [PubMed] [Google Scholar]
- 73.Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [published erratum appears in Nat. Struct. Biol. 1997 Jun;4(6):505]. [DOI] [PubMed] [Google Scholar]
- 74.Ruan Q, Ruan K, Balny C, Glaser M, Mantulin WW. Protein folding pathways of adenylate kinase from E. coli: hydrostatic pressure and stopped-flow studies. Biochemistry. 2001;40:14706–14714. doi: 10.1021/bi010308i. [DOI] [PubMed] [Google Scholar]
- 75.Cohen AE, Moerner WE. Suppressing Brownian motion of individual biomolecules in solution. Proc Natl Acad Sci USA. 2006;103:4362–4365. doi: 10.1073/pnas.0509976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kinoshita M, Kamagata K, Maeda A, Goto Y, Komatsuzaki T, Takahashi S. Development of a technique for the investigation of folding dynamics of single proteins for extended time periods. Proc Natl Acad Sci USA. 2007;104:10453–10458. doi: 10.1073/pnas.0700267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clamme JP, Deniz AA. Three-color single-molecule fluorescence resonance energy transfer. ChemPhysChem. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 79.Lee NK, Kapanidis AN, Koh HR, Korlann Y, Ho SO, Kim Y, Gassman N, Kim SK, Weiss S. Three-color alternating-laser excitation of single molecules: monitoring multiple interactions and distances. Biophys J. 2007;92:303–312. doi: 10.1529/biophysj.106.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kapanidis AN, Weiss S. Fluorescent probes and bioconjugation chemistries for single- molecule fluorescence analysis of biomolecules. J Chem Phys. 2002;117:10953–10964. [Google Scholar]
- 81.Onuchic JN, Wang J, Wolynes PG. Analyzing single molecule trajectories on complex energy landscapes using replica correlation functions. Chemical Physics. 1999;247:175–184. [Google Scholar]
- 82.Ueno T, Taguchi H, Tadakuma H, Yoshida M, Funatsu T. GroEL mediates protein folding with a two successive timer mechanism. Mol Cell. 2004;14:423–434. doi: 10.1016/s1097-2765(04)00261-8. [DOI] [PubMed] [Google Scholar]
- 83.Yamasaki R, Hoshino M, Wazawa T, Ishii Y, Yanagida T, Kawata Y, Higurashi T, Sakai K, Nagai J, Goto Y. Single molecular observation of the interaction of GroEL with substrate proteins. J Mol Biol. 1999;292:965–972. doi: 10.1006/jmbi.1999.3129. [DOI] [PubMed] [Google Scholar]
- 84.Hillger F, Nettels D, Dorsch S, Schuler B. Detection and analysis of protein aggregation with confocal single molecule fluorescence spectroscopy. J Fluoresc. 2007 doi: 10.1007/s10895-007-0187-z. in press: http://dx.doi.org/10.1007/s10895-10007-10187-z. [DOI] [PubMed]
- 85.Wahl M, Koberling F, Patting M, Rahn H, Erdmann R. Time-resolved confocal fluorescence imaging and spectrocopy system with single molecule sensitivity and sub-micrometer resolution. Curr Pharm Biotechnol. 2004;5:299–308. doi: 10.2174/1389201043376841. [DOI] [PubMed] [Google Scholar]