

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is characterized by a severe fibrotic component that compromises treatment, alters the immune cell profile and contributes to patient mortality. It has been shown that early on in this process, dynamic changes in tissue biomechanics play an integral role in supporting pancreatic cancer development and progression. Despite the acknowledgement of its importance, a granular view of how stromal composition changes during the course of PDAC progression remains largely unknown. To mimic the quasi-mesenchymal phenotype and pronounced desmoplastic response observed clinically, we utilized a genetically engineered mouse model of PDAC that is driven by a KrasG12D mutation and loss of Tgfbr2 expression. Application of compartment resolved proteomics revealed that PDAC progression in this KTC model is associated with dynamic stromal alterations that are indicative of a wound healing program. We identified an early provisional matricellular fibrosis that was accompanied by markers of macrophage activation and infiltration, consistent with the inflammatory phase of wound healing. At 20 weeks a proliferative phenotype was observed with increased fibroblast markers, further collagen deposition and loss of basement membrane and native cell markers.
Keywords: PDAC, solid tumor, extracellular matrix, matrisome, chemical digestion, proteomics, mass spectrometry, fibrosis
Graphical abstract

Introduction
A spectrum of distinct pancreatic malignancies that exhibit distinct histological and molecular features have been observed and described.1 Pancreatic ductal adenocarcinoma (PDAC) is the most common pancreatic neoplasm, accounting for >85% of all pancreatic tumor cases. Additionally, more than 95% of all pancreatic cancers arise from exocrine (i.e. duct where digestive enzymes are produced) elements, while cancers arising from endocrine elements (i.e. neuroendocrine tumors and islet cell tumors) account for <5% of cases. Pancreatic cancer is one of the most lethal malignancies and has a median survival of <6 months and a 5-year survival rate of 7%.2–3
The fibrosis that develops during PDAC progression compromises drug delivery, alters immune cell accessibility and promotes disease aggression and therapy resistance.4–6 Targeting of various stromal components and pathways is considered a promising strategy to biochemically and biophysically enhance therapeutic response. However, none of the efforts have yet led to efficacious and approved therapies in patients. The extracellular matrix (ECM) specifically has been shown to modify almost every hallmark of cancer and is considered an enabling characteristic of solid tumor growth and invasion.7–9 Paradoxically, others have shown that the same desmoplastic stroma that confers drug resistance might also reduce the ability of PDAC cells to invade and metastasize. In mice, PDACs that are depleted of alpha smooth muscle actin positive myofibroblasts at the early or late stages formed more invasive tumors with reduced overall survival.10–11 Taken together, these findings illustrate the complex role of dynamic reciprocity between cellular and stromal compartments to influence PDAC progression. As such, a more detailed understanding of the tumor microenvironment in tumor pathogenesis and therapy resistance is needed to optimize strategies that target stromal components.
Pancreatic tumors share common genetic alterations in a handful of genes including Kras, p53, Smad4, and p16.12 Importantly, these genetic events, combined with accompanying histopathological alterations (i.e. fibrosis), suggest a sequential transformation roadmap of pancreatic cancer from normal pancreatic epithelium to increasing grades of pancreatic intraepithelial neoplasia (PanIN) to, ultimately, invasive PDAC. However, tumors with identical germline mutations can exhibit diverse stromal phenotypes that predict tumor aggressiveness13–15 supporting further study of the tumor microenvironment including the ECM. In support of this, recent studies have demonstrated that the genotype of PDAC tunes epithelial tension to regulate fibrosis and accelerate PDAC progression in mice.15 Pathological fibroblasts (i.e. myofibroblasts) are generally thought to be the precursors to increases in matrix deposition, stiffness and enhanced biomechanical signaling.16–17 Although the biological impact of pancreatic cancer stroma on tumor cells has been investigated for some time18, the molecular mechanisms that underlie the desmoplastic response are not well understood. In part, this is because there is a lack of methods aimed specifically at characterizing covalently cross-linked and highly insoluble extracellular matrices. Here, we investigate a biomechanically driven model of PDAC at early and late timepoints to age matched normal pancreas using compartment resolved proteomics to better understand the stromal remodeling that occurs with disease progression.
Materials and Methods
Reagents
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Sodium chloride was from Acros Organics (part of Thermo Fisher). Microcentrifuge tubes and other consumables were from Axygen Inc. (Union City, CA) and RINO Screw Camp Tubes from Next Advance (Averill Park, NY). Formic acid (FA), and hydroxylamine (NH2OH) hydrochloride were from Fluka (Buchs, Switzerland). Anhydrous potassium carbonate, guanidine hydrochloride, sodium hydroxide, and acetonitrile (LC-MS grade) were from Fisher Scientific (Pittsburgh, PA). Trypsin (sequencing grade, TPCK treated) was from Promega (Madison, WI).
Sample Preparation
All mice were maintained in accordance with University of California Institutional Animal Care and Use Committee guidelines under protocol number AN105326-01D. Three biological replicates of normal pancreas early (5 weeks), normal pancreas late (20 weeks), KTC PDAC early (5 weeks) and KTC PDAC late (20 weeks) were harvested from either normal (C57bl/6) or from a genetically engineered KTC (KrasLSL-G12D/+Tgfbr2flox/+Ptf1a-Cre) mice (n=3 each group), as previously described.15 An equal number of male and female mice were used and matched between the time points. The early tumors visually present as normal pancreas tissue (250mg) and late tumors take over most of the pancreas (300-500 mg). Tissues were flash frozen in liquid nitrogen and milled to a fine powder using a ceramic mortar and pestle. Tissue was dried overnight in a lyophilizer and weighed tissue (approximately 1 mg for each sample) was homogenized in freshly prepared high-salt buffer (50 mM Tris-HCl, 3 M NaCl, 25 mM EDTA, 0.25% w/v CHAPS, pH 7.5) containing a 1x protease inhibitor cocktail (Halt Protease Inhibitor, Thermo Scientific) at a concentration of 10 mg/mL. Homogenization took place in a bead beater (Bullet Blender Storm 24, Next Advance, 1 mm glass beads) for 3 min at 4 °C. Samples were then spun for 20 min 18,000 × g at 4 °C, and the supernatant removed and stored as Fraction 1. A fresh aliquot of high-salt buffer was added to the remaining pellet at 10 mg/mL of the starting weight, vortexed at 4 °C for 15 min, and spun for 15 min (18,000 × g at 4 °C). The supernatant was removed and stored as Fraction 2. This high-salt extraction was repeated once more to generate Fraction 3, after which freshly prepared guanidine extraction buffer (6 M guanidinium chloride adjusted to pH 9.0 with NaOH) was added at 10 mg/mL and vortexed for 1 hour at room temperature. The samples were then spun for 15 min, the supernatant removed, and stored as Fraction 4 (sECM). Fractions 1, 2, & 3 (Cellular) were combined and all fractions were stored at −20 °C until further analysis. Hydroxylamine (NH2OH) Digestion – The remaining pellets from each tissue, representing insoluble ECM proteins, were digested with hydroxylamine as previously described19. Briefly, after chaotrope extraction pellets were treated with freshly prepared hydroxylamine buffer (1 M NH2OH-HCl, 4.5 M guanidine-HCl, 0.2 M K2CO3, pH adjusted to 9.0 with NaOH) at 10 mg/mL of the starting tissue weight. The samples were briefly vortexed, then incubated at 45 °C with vortexing for 16 hours. Following incubation, the samples were spun for 15 min at 18,000 × g, the supernatant removed, and stored as Fraction 5 (iECM) at −80 °C until further proteolytic digestion with trypsin. The final pellet was stored at −80 °C until further analysis.
Trypsin Digestion
For each sample, 100 μL of the Cellular fraction, and 200 μL of the sECM and iECM fractions, respectively, were subsequently subjected to reduction, alkylation, and enzymatic digestion with trypsin. A filter-aided sample preparation (FASP) approach, as well as C18 cleanup, was performed as previously described.20
LC-MS/MS Analysis
Samples were analyzed on an Q Exactive HF mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nanoLC 1000 system through a nanoelectrospray source. The analytical column (100 μm i.d. × 150 mm fused silica capillary packed in house with 2.7 μm 80 Å Cortex C18 resin (Phenomenex; Torrance, CA)). The flow rate was adjusted to 400 nL/min, and peptides were separated over a 120-min linear gradient of 4–28% ACN with 0.1% FA. Data acquisition was performed using the instrument supplied Xcalibur™ (version 2.1) software. The mass spectrometer was operated in positive ion mode. Full MS scans were acquired in the Orbitrap mass analyzer over the 300–2000 m/z range with 60,000 resolution. Automatic gain control (AGC) was set at 1.00E+06 and the fifteen most intense peaks from each full scan were fragmented via HCD with normalized collision energy of 28. MS2 spectra were acquired in the Orbitrap mass analyzer with 15,000 resolution with AGC set at 1.00E+05. All replicates of each tissue were run sequentially and pre-digested yeast alcohol dehydrogenase standard (nanoLCMS Solutions LLC, Rancho Cordova, CA) was run between tissue groups to monitor drift in analytical performance.
Database Searching and Protein Identification
MS/MS spectra were extracted from raw data files and converted into .mgf files using MS Convert (ProteoWizard, Ver. 3.0). Peptide spectral matching was performed with Mascot (Ver. 2.5) against the Uniprot mouse database (release 201701). Mass tolerances were +/− 10 ppm for parent ions, and +/− 0.2 Da for fragment ions. Trypsin specificity was used for cellular and sECM fractions, allowing for 1 missed cleavage. For iECM fraction, C-terminal N and trypsin were used, allowing for 1 missed cleavage. Met oxidation, Pro hydroxylation, protein N-terminal acetylation, and peptide N-terminal pyroglutamic acid formation were set as variable modifications with Cys carbamidomethylation set as a fixed modification. Scaffold (version 4.4.6, Proteome Software, Portland, OR, USA) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Mascot scoring algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least two identified unique peptides. Byonic Preview was used to assess missed cleavage percentages and ragged N- and C-termini.21 All data has been made publicly available on the Proteome Exchange database via PXD007816.
H&E Staining
Paraffin-embedded or Fresh frozen pancreatic tissues were analyzed by H&E, according to the manufacturer’s instructions.
Picrosirius Red Staining and Quantification
Flash frozen formalin fixed paraffin embedded (FFPE) tissues were cryo-sectioned at 5 μm, fixed in 4% neutral buffered formalin and stained using 0.1% picrosirius red (Direct Red 80, Sigma) and counterstained with Weigert’s hematoxylin, as previously described22. Polarized light images were acquired using Olympus IX81 microscope, fitted with an analyzed (U-ANT) and a polarizer (U-POT, Olympus) oriented parallel and orthogonal to each other. Images were quantified using ImageJ as previously described.22
Results
Here we investigate the relationship between tumor progression, fibrosis and ECM composition by utilizing a genetically engineered PDAC mouse model: KTC, KrasLSL-G12D/+Tgfbr2flox/+Ptf1a-Cre.10, 15 In comparison to the common KPC model23, experimental tumors in KTC mice are highly fibrotic and exhibit a pronounced mesenchymal-like phenotype following stromal ablation.10, 15 Clinically, patient PDACs with a quasi-mesenchymal phenotype have more fibrotic tumors that are more aggressive and resistant to treatment than PDACs with a classical, more differentiated histophenotype.24–25
The fibrotic PDAC stroma is used as a readout of stromal activation during disease progression. Considering the rapid and aggressive nature of these mesenchymal tumors in human disease and mouse models, we sought to investigate differences in the matrisome during PDAC progression using compartment resolved proteomics applied to KTC PDACs at early (5 weeks) and late (20 weeks) time points. Based on our near universal observation of an insoluble pellet that remains after chaotrope extraction26 we applied our recently published protocol for chemical digestion using hydroxylamine (NH2OH), which cleaves at asparagine residues.19 The sample preparation protocol yields three distinct fractions: 1) cellular, 2) soluble ECM (sECM), and 3) insoluble ECM (iECM) fractions (Figure 1). We analyzed protein digests using global data-dependent acquisition and were able to identify a total of 3,347 proteins across all tissues (Supplementary Table S1) at a false discovery rate of 0.5%.
Figure 1. Compartment resolved proteomics workflow.
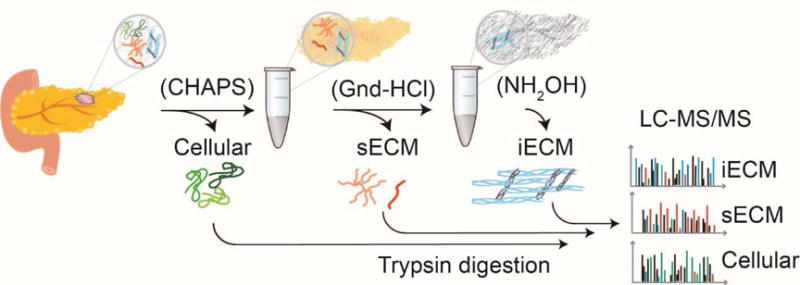
The sample preparation protocol yields three distinct fractions: 1) cellular, 2) soluble ECM (sECM), and 3) insoluble ECM (iECM) fractions. Cellular compartments are isolated using CHAPS detergent (cellular fraction), Gnd-HCl is used to facilitate protein denaturation and the solubilization of an ECM rich fraction (sECM fraction). After this extraction, an insoluble pellet remains that is subjected to chemical digestion with NH2OH to solubilize the remaining insoluble components (iECM fraction). All fractions are digested with trypsin and analyzed separately by quantitative tandem mass spectrometry (LC-MS/MS).
To monitor changes in the abundance of exocrine, endocrine, ductal, and stromal cell populations during tumor progression, pancreatic cell markers were quantified (Supplementary Table S2). A shift in the cell populations between normal pancreas and PDACs was observed in the 5-week time points (Figure 2A). These data show that exocrine cell markers (e.g. Cela2a, Cpa1, Prss2, Ctrb1) are approximately 6-fold higher abundance in normal pancreatic tissue relative to PDAC. These cells are integral to normal pancreatic function as they produce pancreatic enzymes for digestion. The remaining 5% of cells consists of endocrine cells that produce hormones that regulate blood sugar and pancreatic secretions. Although less abundant than exocrine cell population markers, endocrine cell population markers (e.g. Gcg and Ins2) were present in the normal pancreas at 2-fold higher abundance relative to KTC PDACs. Importantly, even in early (5 week) KTC PDACs, a marked decrease in the abundance of exocrine and endocrine markers was observed. As exocrine/endocrine populations decrease and normal cells become transformed, protein markers of tumor cell (i.e. malignant ductal cells) activation increase and begin to release various factors (i.e. TGFβ and Fibronectin) that stimulate ECM protein production and remodeling.27 A 9-fold increase in ductal (i.e. Krt20, Tpm2, and Krt7) and a 2.7-fold increase in stromal (i.e. Gfap, Vim, Mrc1, Cd47, Cd81) cell markers, relative to normal pancreatic tissue, was observed at 5 weeks in the KTC tumors and indicates an expansion of ductal (and stromal) cell populations commensurate with PDAC progression Previous studies which analyzed microdissected PDAC tumors found that the ductal marker Krt7 is specifically overexpressed in the tumor epithelium and is correlated with decreased survival.28 Our analysis also revealed that KTC tumors retain a relatively high abundance of exocrine cell markers early on, however; exocrine cell markers in later stage tumors decreased more than 10-fold (Supplementary Table S2). Multi-variant analysis illustrates that these markers provide sufficient information to discriminate between, normal pancreas, early KTC, and late KTC tumors (Figure 2B).
Figure 2. Characterization of shifts in tissue morphology and cell markers during pancreatic tumorigenesis.
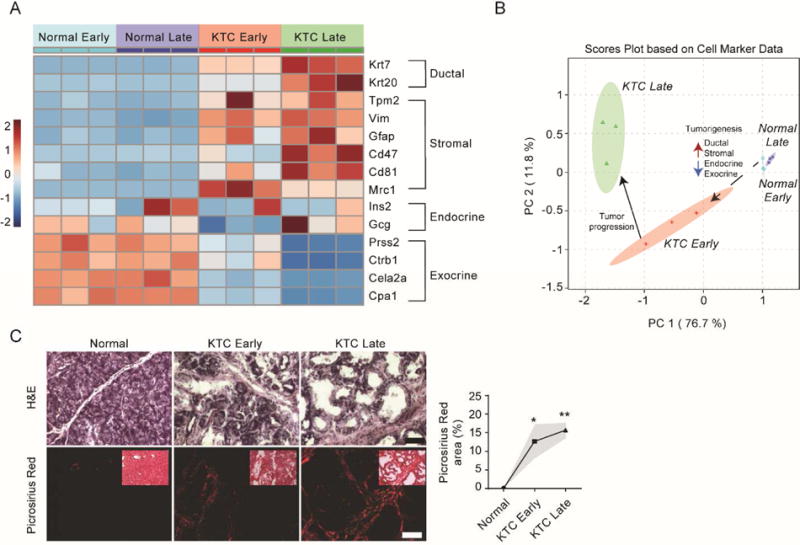
(A) Heatmap of changes in the relative abundance of ductal, stromal, endocrine and exocrine cell population markers in normal early (n=3) and normal late (n=3) pancreas and KTC PDAC early (n=3) and KTC PDAC late (n=3) tumors derived from data dependent global LC-MS/MS data. Early and late time points were taken at 5 weeks and 20 weeks, respectively. (B) Scores plot from principal component analysis (PCA) using cell population markers as defined variables. Red arrows indicated increases in relative abundance and blue arrows indicate decreases in relative abundance. (C) Representative images from histological staining (H&E, picrosirius red with polarized light) of normal pancreas, KTC early and KTC late (left). Quantification of total Picrosirius red area as percentage of total (left). Subsequent statistical analysis was performed using Mann-Whitney test (*P < 0.05; **P < 0.01).
To further investigate the flux from exocrine/endocrine cell populations to more ductal and stromal cell populations, the morphologic changes that form the bases of contemporary PDAC diagnoses were compared. Regions with confirmed malignant transformation were identified on H&E stained sections and revealed stark differences in cellular morphology (Figure 2C). Further, because PDACs are characterized by dynamic stromal remodeling and an influx of cancer-associated stromal cells, we also sought to investigate the relationship between fibrillar collagen and PDAC progression. As expected, evidence of fibrosis was indicated by polarized imaging of picrosirius red stained parallel sections which revealed that fibers progressed in thickness between early and late time points.
Commensurate with increased collagen deposition, several significant changes in the composition of the ECM were observed during disease progression (Supplementary Table S3). First, our results indicated a progressive loss of basement membrane proteins (i.e. Lamb2, Col4a2, and Lamc1) from normal pancreas through KTC PDAC progression (Figure 3A). This is consistent with the observed destruction of the basement membrane as normal cellular organization is lost to facilitate invasion and metastasis in many cancers8. Additionally, a stark increase in matricellular (i.e. Tnc, Thbs1), ECM regulator (i.e. Lox, Tgm2, Plod2), and structural ECM (i.e. Fn1) protein abundance was also observed. This is consistent with previous findings which suggests a strong matricellular-fibrosis phenotype in patients15 and has implications for alterations in cell-matrix interactions known to contribute to tumor invasion and metastasis. Early and late normal pancreatic tissue are distinguished from early and late KTC PDACs based solely on their ECM composition using multivariate analysis (Figure 3C). Furthermore, the variance observed is primarily driven by decreases in basement membrane abundance, and increases in matricellular, collagen, and ECM regulator protein abundance (Figure 3B). These data are consistent with cell marker data which indicated large increases in stromal cell populations.
Figure 3. Comparative matrisome analysis in normal pancreas and during pancreatic ductal adenocarcinoma progression.
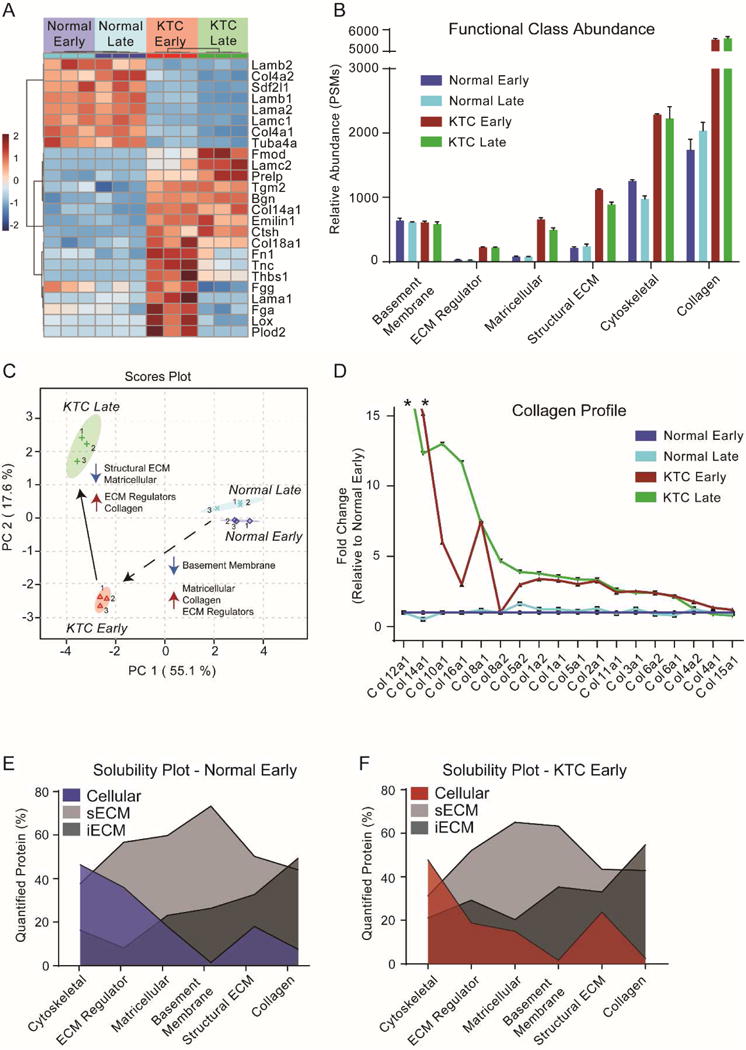
(A) Heatmap of changes in the relative abundance of the top 25 ECM proteins contributing to the separation of groups seen in part C. (B) ECM curated list of proteins from data dependent global LC-MS/MS experiments grouped by their defined function (C) Scores plot from principal component analysis using the ECM curated list of protein from data dependent global LC-MS/MS experiments. (D) Fold change plot of all collagens identified relative to normal early pancreas samples and sorted by largest to smallest fold changes. * above Col12a1 and Col14a1 indicates that these variable were not detected in the normal pancreas matrisome. (E) and (F) Area fill plot based on the relative abundance of the defined protein group across all fractions for normal early pancreas tissue and KTC PDAC early tumors. Values indicate the overall abundance of a protein group that was found in each fraction out of 100%.
KTC PDACs exhibit an approximate 3-fold increase in fibrillar collagen (i.e. Col1a1, Col1a2, Col2a1, Col3a1, and Col5a1/2) after just 5 weeks (Figure 3D, Supplementary Table S3), relative to normal tissue. Notably, fibril-associated collagen with interrupted triple helix (FACIT) collagens were either not detected (Col12a1) or detected at a relatively low abundance (Col14a1) in normal pancreatic tissue, however; their abundance is increased dramatically during tumor progression. Another less abundant FACIT collagen, Col16a1, was identified at very low levels (i.e. one peptide identification) in normal pancreatic tissue but showed significant increases in abundance during tumorigenesis and PDAC progression (Supplementary Table S3). Previous evidence has shown that Col16a1 is extensively overexpressed in glioblastoma and oral squamous cell carcinoma tumorigenesis, compared to normal tissue.29 Significant increases in the abundance of the network collagens VIII and X also occurred during tumor progression. Type VIII and X collagens are known to form networks of polygonal superstructures,30–31 have been shown to be critical to the formation of vessel walls,32 tumor vasculature33 and are associated with vascular injury.34 It is possible this may be a part of the extended would healing program observed in KTC PDACs and the expansion of tumor vasculature. Additionally, previous studies in colorectal cancer have identified Col10a1 as one of the top four genes upregulated in carcinogenic colon tissues, relative to adjacent normal controls.35
To determine if the observed changes in collagen during the course of tumor progression would alter the solubility of the stroma, the percentage of total collagen found in the cellular, sECM, and iECM fractions were compared (Figure 3E & F). This approach revealed changes in the solubility profiles of normal and KTC PDACs (Supplementary Table S4). A progressive decrease in basement membrane solubility was observed from 26.4% to 35.3% present in the iECM fraction of normal and KTC respectively. The solubility of matricellular proteins and collagens remained relatively consistent during tumor progression. However, for individual collagen families we observe some unique trends (Figure 3E & F). In comparing normal early pancreas with normal late pancreas, we observed a 12.9% decrease in the solubility of fibrillar collagen. Interestingly, the same comparison in KTC progression shows a 5.4% decrease in fibrillar collagen solubility in later stage PDACs. These data indicate that more fibrillar collagen is deposited in the stroma in KTC PDACs than in normal pancreatic tissue, but is more soluble (Supplementary Table S4), possibly indicating that in addition to abnormal matrix deposition proper maturation into stable fibers is not occurring.
End products of coagulation (Fga, Fgb, Fgg) increase significantly in abundance in the ECM fractions during the early phase of tumor progression (Figure 2A) and are classic markers of the homeostasis phase of wound healing.36 Coagulation factor XIII A chain (F13a1) which is responsible for crosslinking reactions to stabilize fibrin clots37–38 was detected at very low levels in normal pancreas, however; increased dramatically in the ECM fractions of the KTC model. Interestingly, another transglutaminase, Tgm2, also increased ~5-fold, as well as its known substrate fibronectin. Additionally, Tgm2 decreases substantially in solubility (27% decrease) in PDACs relative to normal pancreas tissue (Supplementary Table S1). This may be explained by the observation that Tgm2 auto-crosslinks to structural ECM substrates such as Fn1.39–40
A significant increase in almost all proteins defined as matricellular41–42 was observed at the early time point in the tumor model. (Figure 4A and Supplementary Table S5). Tenascin-C (Tnc) abundance is highest early in KTC samples and then decreases approximately 5-fold by the KTC 20-week time point. Tnc has been identified as a metastatic niche component involved in colonizing the lung during breast cancer metastasis.43–44 Multivariate analysis revealed significant contributions of small leucine-rich proteoglycans (SLRPs), such as biglycan (Bgn), lumican (Lum), and decorin (Dcn) in the discrimination of sample groups during tumor progression (Figure 4B). The expression of these proteins is related to collagen expression and organization.45
Figure 4. Pancreatic tumorigenesis and progression is characterized by increased matricellular protein abundance.
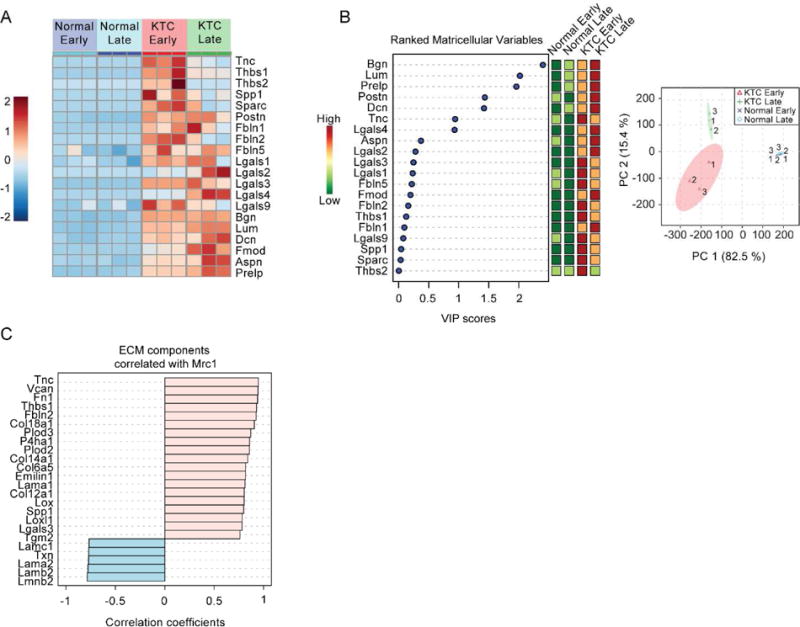
(A) Heatmap of changes in the relative abundance of all matricellular proteins identified by data dependent global LC-MS/MS. (B) Variable of importance (VIP) plot (left) in which matricellular protein are ranked based on their contribution to the separation between groups in principal component analysis scores plot (right). A larger VIP score indicates that the variable is more important in defining the differences between all groups in PCA scores plot. (C) Correlation analysis of ECM components that correlate with Mannose receptor 1 (Mrc1) abundance based on relative abundance measures from data dependent global LC-MS/MS experiments.
To investigate the relationship between macrophage activation and matricellular protein abundance, we performed Pearson correlation analysis (Figure 4C) against a marker of tumor associated macrophages (TAMs); Mannose receptor 1 (Mrc1/Cd206).46 Previous studies have identified Mrc1 as an M2 macrophage subtype termed MRC1-expressing TAMs (MEMs)47 and have established its role as the principal collagen endocytosis receptor in TAMs.48 Our analysis revealed strong correlations with several matricellular proteins, ECM components that regulate ECM post-translational modifications (i.e. Plod2, Lox, Loxl1), ECM deposition (i.e. Fn1, P4ha1), and effect cell adhesion (i.e. Tnc, Fn1, Thbs1) through activation of integrin signaling.49–50
Discussion
PDAC-associated fibrosis creates a tissue-microenvironment that compromises drug delivery, alters immune cell accessibility and promotes tumor aggression and drug resistance. Despite the ECM being recognized as a major contributor to these events and the hallmarks of cancer,8–9 we have a limited understanding of global alterations that occur with disease progression. Here, we applied compartment resolved proteomics to characterize changes in the abundance of cellular populations and ECM composition at early and late time points. We observed formation of a provisional matrix early in tumor progression that exhibited several characteristics of the homeostatic and inflammatory stages of wound healing. Most notably there was increased matricellular protein and fibrillar collagen abundance. The advanced tumors observed at the 20-week time-point exhibited a scar-like fibrosis driven primarily by type I collagen deposition and additional changes, including a decrease in basement membrane proteins and increased abundance of ECM modifying enzymes (i.e. Lox, Plod2). Cellular differentiation markers revealed that KTC PDACs progressively lost exocrine cell populations with a concurrent increase in ductal and stromal cells. These findings also support progression through the first half of the “wound healing” program.
Analysis of the insoluble pellet remaining after chaotrope extraction revealed that over 50% of total fibrillar collagen abundance was found in this often-overlooked fraction. In addition to increased levels of abundant fibrillary collagens we identified FACIT collagens Col12a1, Col14a1, and Col16a1 as an abundant and unique feature of the PDAC stroma. Col12a1 was undetected in normal pancreatic tissue while Col14a1 was detected at a 10-fold lower level than was observed in KTC PDACs. Additionally, our data would suggest that Col16a1 appears to be of particular importance during PDAC tumorigenesis and progression in this model. Of note, FACIT collagens associate with the surface of existing collagen fibrils to create bridges between fibrils.51 Future work to substantiate these findings in human samples should include not only the described collagen levels, but also post-translational modifications associated with fibrosis. Particularly, enzymatic crosslinking that has been shown to promote tumor progression52–53 and limit the reversal of fibrotic diseases.54–55
Perhaps most striking is the change in the matricellular protein phenotype observed in early tumor progression and to a lesser extent at the advanced time point. In general, matricellular protein abundance was very low, or not-detected in normal pancreatic tissue. SLRPs such as biglycan, decorin, fibromodulin and lumican, increased and remained prominent in the tumors. This family of proteins binds collagen fibrils and regulates interfibrillar spacing, in part, through their highly charged hydrophilic glycosaminoglycans.45 In addition, this family of proteins is known to sequester TGF-β into the ECM,56 bind to multiple cell surface receptors,57 and regulate inflammation and innate immunity.58 Ultimately, these ECM components form a permissive or promotional matrix that guide altered fibrillogenesis and cell-ECM contacts that can lead to the development of drug resistance and increased tumor angiogenesis.57 The tumor associated macrophage marker, Mrc1, showed a strong correlation with matricellular protein abundance in KTC PDACs. Of the identified matricellular proteins, tenascin-C was found to be one of the most abundant and had a strongest positive correlation with this marker. It has been shown previously that tenascin-C expression is upregulated in human PDAC and correlates with differentiation.50, 59 Furthermore, there is evidence to suggest that exosome bound tenascin-C is involved in initiating a pre-metastatic niche in the liver.49, 60
In comparison to the widely used KPC model of PDAC that has conditional expression of the tumor-associated p53R172H mutation, the KTC tumors are smaller yet more fibrotic as indicated by imagining modalities and biomechanical measurements.15 The reduction in TGF-β signaling increased actomyosin tension and mechanosignaling in the transformed pancreatic epithelium to induce a stiffer, periductal, matricellular-enriched fibrosis as well. Human PDAC biopsies from patients with shorter median survival supported these findings including the identified signaling pathways converging on YAP1.
Analysis of cellular and stromal compartments revealed a classic wound healing response and formation of a provisional matrix involving fibronectin and fibrinogen. It has been shown that a provisional ECM provides the proper microenvironment for resident and invading cells to proliferate and migrate during normal wound repair process61. It is likely that this process supports the migration of cancer associated fibroblasts and myofibroblasts to the tumor microenvironment to modify the ECM and further the dynamic reciprocity observed in tumor progression.62
Components of the newly synthesized matrix, along with tumor adaptions likely fuel the physiological wound healing response towards a chronic fibrotic state in these tumors. This in turn, perpetuates further interstitial pressure and biochemical queues that advance PDAC progression and impair organ function. Future studies aimed at further temporal resolution of PDAC progression and means to resolve the fibrotic state, or direct the pancreas toward the remodeling stage of wound healing will require a better understanding of tumor microenvironment alterations and cellular responses that result.
Supplementary Material
Supplemental Table S1 – Global LC-MS/MS full results
Supplemental Table S2 – Global LC-MS/MS cell population marker results
Supplemental Table S3 – ECM-specific LC-MS/MS results
Supplemental Table S4 – Fractional solubility analysis from global LC-MS/MS experiments
Supplemental Table S5 – Global LC-MS/MS results for matricellular proteins
Highlights.
First study to characterize changes in the matrisome during PDAC progression
Early and late tumor timepoints revealed a dynamic wound healing stroma
An early provisional matrix was replaced by a more classical fibrotic phenotype
Stromal cells overtook exocrine and endocrine cell populations during PDAC progression
Acknowledgments
The work included in this manuscript includes funding from NIH/CTSA 63007957 and NIH T32 HL007171 (to A.S.B.), and NIH/NCI R33CA183685 (to K.C.H. and V.M.W.). Funding from US DOD Breast Cancer Research Program (BCRP) grant BC122990 (V.M.W), US DOD BCRP grant BC130501 (O.M.), and US NIH T32 grant CA 108462 (M.W.P & O.M.) also supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
The authors declare no conflicts of interest.
Author Information
A.S.B., K.C.H., and V.M.W. were responsible for hypothesis development, conceptual design and final data integrity and interpretation. O.M. and M.W.P. performed H&E and PS staining. All authors contributed to the conceptual design of the manuscript and to data interpretation. A.S.B., K.C.H., and V.M.W. wrote the manuscript and all authors edited and approved the final manuscript submission.
References
- 1.Büchler MW, Uhl W, Malfertheiner P, Sarr M. Diseases of the Pancreas. Diseases of the pancreas. 2004 [Google Scholar]
- 2.Lemke J, Schafer D, Sander S, Henne-Bruns D, Kornmann M. Survival and prognostic factors in pancreatic and ampullary cancer. Anticancer Res. 2014;34(6):3011–20. [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nature communications. 2013:4. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nature Reviews Cancer. 2012;12(3):210–219. doi: 10.1038/nrc3186. [DOI] [PubMed] [Google Scholar]
- 6.Yu M, Tannock IF. Targeting tumor architecture to favor drug penetration: a new weapon to combat chemoresistance in pancreatic cancer? Cancer cell. 2012;21(3):327–329. doi: 10.1016/j.ccr.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. The hallmarks of cancer. cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO reports. 2014;15(12):1243–53. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes & development. 2006;20(10):1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 13.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, Friess H. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nature Reviews Gastroenterology and Hepatology. 2012;9(8):454–467. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 14.Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11(5):497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature medicine. 2016;(22.5):497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinz B. The myofibroblast: paradigm for a mechanically active cell. Journal of biomechanics. 2010;43(1):146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, Ding Q, Thannickal VJ, Zhou Y. Matrix stiffness–induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. American journal of respiratory cell and molecular biology. 2012;47(3):340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer research. 2008;68(3):918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett AS, Wither MJ, Hill RC, Dzieciatkowska M, D’Alessandro A, Reisz JA, Hansen KC. Hydroxylamine Chemical Digestion for Insoluble Extracellular Matrix Characterization. Journal of Proteome Research. 2017 doi: 10.1021/acs.jproteome.7b00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goddard ET, Hill RC, Barrett A, Betts C, Guo Q, Maller O, Borges VF, Hansen KC, Schedin P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. The International Journal of Biochemistry & Cell Biology. 2016;81:223–232. doi: 10.1016/j.biocel.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Current Protocols in Bioinformatics. 2012:13.20. 1–13.20. 14. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang E, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative Biology. 2015 doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 24.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature genetics. 2015;47(10):1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature medicine. 2011;17(4):500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill RC, Calle EA, Dzieciatkowska M, Niklason LE, Hansen KC. Quantification of Extracellular Matrix Proteins from a Rat Lung Scaffold to Provide a Molecular Readout for Tissue Engineering. Molecular & Cellular Proteomics. 2015;14(4):961–973. doi: 10.1074/mcp.M114.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. Journal of Biological Chemistry. 1986;261(9):4337–4345. [PubMed] [Google Scholar]
- 28.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia-The authors reported a combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-gastroenterology. 2008;55(88):2016. [PubMed] [Google Scholar]
- 29.Grässel S, Bauer RJ. Collagen XVI in health and disease. Matrix Biology. 2013;32(2):64–73. doi: 10.1016/j.matbio.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Stephan S, Sherratt MJ, Hodson N, Shuttleworth CA, Kielty CM. Expression and supramolecular assembly of recombinant α1 (VIII) and α2 (VIII) collagen homotrimers. Journal of Biological Chemistry. 2004;279(20):21469–21477. doi: 10.1074/jbc.M305805200. [DOI] [PubMed] [Google Scholar]
- 31.Gordon MK, Hahn RA. Collagens. Cell and tissue research. 2010;339(1):247. doi: 10.1007/s00441-009-0844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plenz GA, Deng MC, Robenek H, Völker W. Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis. 2003;166(1):1–11. doi: 10.1016/s0021-9150(01)00766-3. [DOI] [PubMed] [Google Scholar]
- 33.Chapman KB, Prendes MJ, Sternberg H, Kidd JL, Funk WD, Wagner J, West MD. COL10A1 expression is elevated in diverse solid tumor types and is associated with tumor vasculature. Future oncology. 2012;8(8):1031–1040. doi: 10.2217/fon.12.79. [DOI] [PubMed] [Google Scholar]
- 34.Sibinga NE, Foster LC, Hsieh C-M, Perrella MA, Lee W-S, Endege WO, Sage EH, Lee M-E, Haber E. Collagen VIII is expressed by vascular smooth muscle cells in response to vascular injury. Circulation research. 1997;80(4):532–541. doi: 10.1161/01.res.80.4.532. [DOI] [PubMed] [Google Scholar]
- 35.Yang Q, Feng M, Ma X, Li H, Xie W. Gene expression profile comparison between colorectal cancer and adjacent normal tissues. Oncology letters. 2017;14(5):6071–6078. doi: 10.3892/ol.2017.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson BJ, Longaker MT, Lorenz HP. Scarless fetal wound healing: a basic science review. Plastic and reconstructive surgery. 2010;126(4):1172. doi: 10.1097/PRS.0b013e3181eae781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staindl O. The healing of wounds and scar formation under the influence of a tissue adhesion system with fibrinogen, thrombin, and coagulation factor XIII. European Archives of Oto-Rhino-Laryngology. 1979;222(4):241–245. doi: 10.1007/BF01261169. [DOI] [PubMed] [Google Scholar]
- 38.Clark RA, Lanigan JM, DellaPelle P, Manseau E, Dvorak HF, Colvin RB. Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. Journal of Investigative Dermatology. 1982;79(5):264–269. doi: 10.1111/1523-1747.ep12500075. [DOI] [PubMed] [Google Scholar]
- 39.Cardoso I, Østerlund EC, Stamnaes J, Iversen R, Andersen JT, Jørgensen TJ, Sollid LM. Dissecting the interaction between transglutaminase 2 and fibronectin. Amino acids. 2017;49(3):489–500. doi: 10.1007/s00726-016-2296-y. [DOI] [PubMed] [Google Scholar]
- 40.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. The Journal of cell biology. 2000;148(4):825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bornstein P. Matricellular proteins: an overview. Journal of cell communication and signaling. 2009;3(3–4):163. doi: 10.1007/s12079-009-0069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biology. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, Chilvers ER, Dransfield I, Donnelly SC, Strieter R. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nature medicine. 1999;5(6) doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 44.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature medicine. 2011;17(7):867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalamajski S, Oldberg Å. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biology. 2010;29(4):248–253. doi: 10.1016/j.matbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 47.Scodeller P, Simón-Gracia L, Kopanchuk S, Tobi A, Kilk K, Säälik P, Kurm K, Squadrito ML, Kotamraju VR, Rinken A. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Scientific reports. 2017;7(1):14655. doi: 10.1038/s41598-017-14709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madsen DH, Jürgensen HJ, Siersbæk MS, Kuczek DE, Cloud LG, Liu S, Behrendt N, Grøntved L, Weigert R, Bugge TH. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Reports. 2017;21(13):3662–3671. doi: 10.1016/j.celrep.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juuti A, Nordling S, Louhimo J, Lundin J, Haglund C. Tenascin C expression is upregulated in pancreatic cancer and correlates with differentiation. Journal of clinical pathology. 2004;57(11):1151–1155. doi: 10.1136/jcp.2003.015818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paron I, Berchtold S, Vörös J, Shamarla M, Erkan M, Höfler H, Esposito I. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PloS one. 2011;6(6):e21684. doi: 10.1371/journal.pone.0021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw LM, Olsen BR. FACIT collagens: diverse molecular bridges in extracellular matrices. Trends in biochemical sciences. 1991;16:191–194. doi: 10.1016/0968-0004(91)90074-6. [DOI] [PubMed] [Google Scholar]
- 52.Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73(6):1721–32. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, Mikels-Vigdal A, Smith V, Schuppan D, Popov YV. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66(9):1697–1708. doi: 10.1136/gutjnl-2016-312473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu SB, Ikenaga N, Peng ZW, Sverdlov DY, Greenstein A, Smith V, Schuppan D, Popov Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J. 2016;30(4):1599–609. doi: 10.1096/fj.14-268425. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrand A, Romaris M, Rasmussen L, Heinegård D, Twardzik D, Border W, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor β. Biochemical Journal. 1994;302(2):527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Theocharis AD, Karamanos NK. Proteoglycans remodeling in cancer: Underlying molecular mechanisms. Matrix Biology. 2017 doi: 10.1016/j.matbio.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 58.Frey H, Schroeder N, Manon‐Jensen T, Iozzo RV, Schaefer L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. Febs Journal. 2013;280(10):2165–2179. doi: 10.1111/febs.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Esposito I, Penzel R, Chaib‐Harrireche M, Barcena U, Bergmann F, Riedl S, Kayed H, Giese N, Kleeff J, Friess H. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. The Journal of pathology. 2006;208(5):673–685. doi: 10.1002/path.1935. [DOI] [PubMed] [Google Scholar]
- 60.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, kumar Thakur B, Becker A, Hoshino A, Mark MT, Molina H. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nature cell biology. 2015;17(6):816. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baum CL, Arpey CJ. Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatologic surgery. 2005;31(6):674–686. doi: 10.1111/j.1524-4725.2005.31612. [DOI] [PubMed] [Google Scholar]
- 62.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. The Journal of pathology. 2003;200(4):500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 – Global LC-MS/MS full results
Supplemental Table S2 – Global LC-MS/MS cell population marker results
Supplemental Table S3 – ECM-specific LC-MS/MS results
Supplemental Table S4 – Fractional solubility analysis from global LC-MS/MS experiments
Supplemental Table S5 – Global LC-MS/MS results for matricellular proteins