

Abstract
The genetic underpinnings and end-stage pathological hallmarks of neurodegenerative diseases are increasingly well defined, but the cellular pathophysiology of disease initiation and propagation remains poorly understood, especially in sporadic forms of these diseases. Altered nucleocytoplasmic transport is emerging as a prominent pathomechanism of multiple neurodegenerative diseases, including amyotrophic lateral sclerosis, Alzheimer disease, frontotemporal dementia and Huntington disease. The nuclear pore complex (NPC) and interactions between its individual nucleoporin components and nuclear transport receptors regulate nucleocytoplasmic transport, as well as genome organization and gene expression. Specific nucleoporin abnormalities have been identified in sporadic and familial forms of neurodegenerative disease, and these alterations are thought to contribute to disrupted nucleocytoplasmic transport. The specific nucleoporins and nucleocytoplasmic transport proteins that have been linked to different neurodegenerative diseases are partially distinct, suggesting that NPC injury contributes to the cellular specificity of neurodegenerative disease and could be an early initiator of the patho-physiological cascades that underlie neurodegenerative disease. This concept is consistent with the fact that rare genetic mutations in some nucleoporins cause cell-type-specific neurological disease. In this Review, we discuss nucleoporin and NPC disruptions and consider their impact on cellular function and the pathophysiology of neurodegenerative disease.
The genetic causes and end-stage pathological hallmarks of neurodegenerative diseases are increasingly well defined. However, the molecular and cellular events that contribute to and initiate disease pathophysiology are still poorly understood. The nuclear pore complex (NPC) — which is made up of many nucleoporin proteins — mediates nucleocytoplasmic transport, genome organization and gene expression1–3. Pathological disruptions to individual nucleoporins or the overall integrity of the NPC are, therefore, likely to have detrimental effects on neuronal function and survival. Consistent with this notion, multiple studies indicate that disruptions of the NPC and the nucleocytoplasmic transport machinery (for example, nuclear transport receptors (NTRs), RAN GTPase-activating protein 1 (RANGAP1) and RAN GTPase energy gradients) are prominent contributors to the pathogenesis of neurodegenerative disease.
Most of these studies have focused on amyotrophic lateral sclerosis (ALS) but some evidence indicates that similar mechanisms exist in Alzheimer disease (AD), frontotemporal dementia (FTD) and Huntington disease (HD)4–11. Genetic interaction studies in flies and yeast suggest that NPC and nucleocytoplasmic transport proteins are potent modifiers of disease phenotypes12,13, and evidence suggests that these proteins can influence the aggregation state of several other proteins associated with neurodegeneration, including TAR DNA-binding protein 43 (TDP43), fused in sarcoma (FUS) and tau6,14–16. Furthermore, mutations in specific nucleoporins cause cell-type-specific neurological diseases17–20, suggesting that defects in the NPC or nucleoporins are sufficient to initiate neurodegeneration.
In this Review, we discuss the latest knowledge about how disruptions in the NPC and nucleocytoplasmic transport contribute to neurogenerative disease pathophysiology. We also highlight the potential of NPC and nucleocytoplasmic transport dysfunction as a therapeutic target in neurodegenerative disease.
Structure and function of the NPC
The mammalian NPC is a multi-protein complex of ~120 MDa that is highly organized and has eightfold rotational symmetry1,21. All NPCs comprise multiple copies of ~30 nucleoporins, although the exact number and stoichiometry of the nucleoporins varies in yeast and mammalian cells22–27. NPCs within a single cell nucleus are not compositionally identical — a heterogeneous distribution of specific nucleoporins has been observed within individual nuclei of neuronal and non-neuronal mammalian cells5,28. The composition of nucleoporins within NPCs also varies between cell types26,27. Despite slight differences in the composition of specific nucleoporins and subcomplexes within yeast and mammalian NPCs, the overall structure and organization are consistent21,23. Nucleoporin nomenclature varies for mammalian and yeast cells; in this Review, we use the mammalian nomenclature.
All NPCs have the same basic structural components that are each made up of various nucleoporins (FIG. 1): the cytoplasmic ring and filaments (NUP88, NUP214 and NUP358), the central channel (NUP54, NUP58, NUP62 and NUP98), the outer ring (ELYS, NUP37, NUP43, NUP85, NUP96, NUP107, NUP133, NUP160, SEC13 and SEH1), the inner ring (NUP35, NUP93, NUP155, NUP188 and NUP205), the transmembrane ring (GP210, NDC1 and POM121), and the nuclear basket (NUP50, NUP153 and TPR). The core NPC scaffold comprises the inner and outer rings, which are anchored within the nuclear envelope via the transmembrane nucleoporins and provide structural support to the cytoplasmic filaments, central channel and nuclear basket. Besides ELYS, which is present only in the nucleoplasmic side of the outer ring subcomplex in mammalian cells, the other nucleoporins are symmetrically localized to the two faces of the NPC21. The nucleoporins that make up the cytoplasmic ring and filaments and the nuclear basket are distributed asymmetrically on the cytoplasmic and nucleoplasmic faces of the NPC, respectively. Thus, the nucleoporins within the scaffold rings on the cytoplasmic and nucleoplasmic faces of the NPC are the same but the nucleoporins that comprise the cytoplasmic filaments and the nuclear basket are distinct (FIG. 1).
Fig. 1 |. Overview of nuclear pore complex structure and subcomplexes.
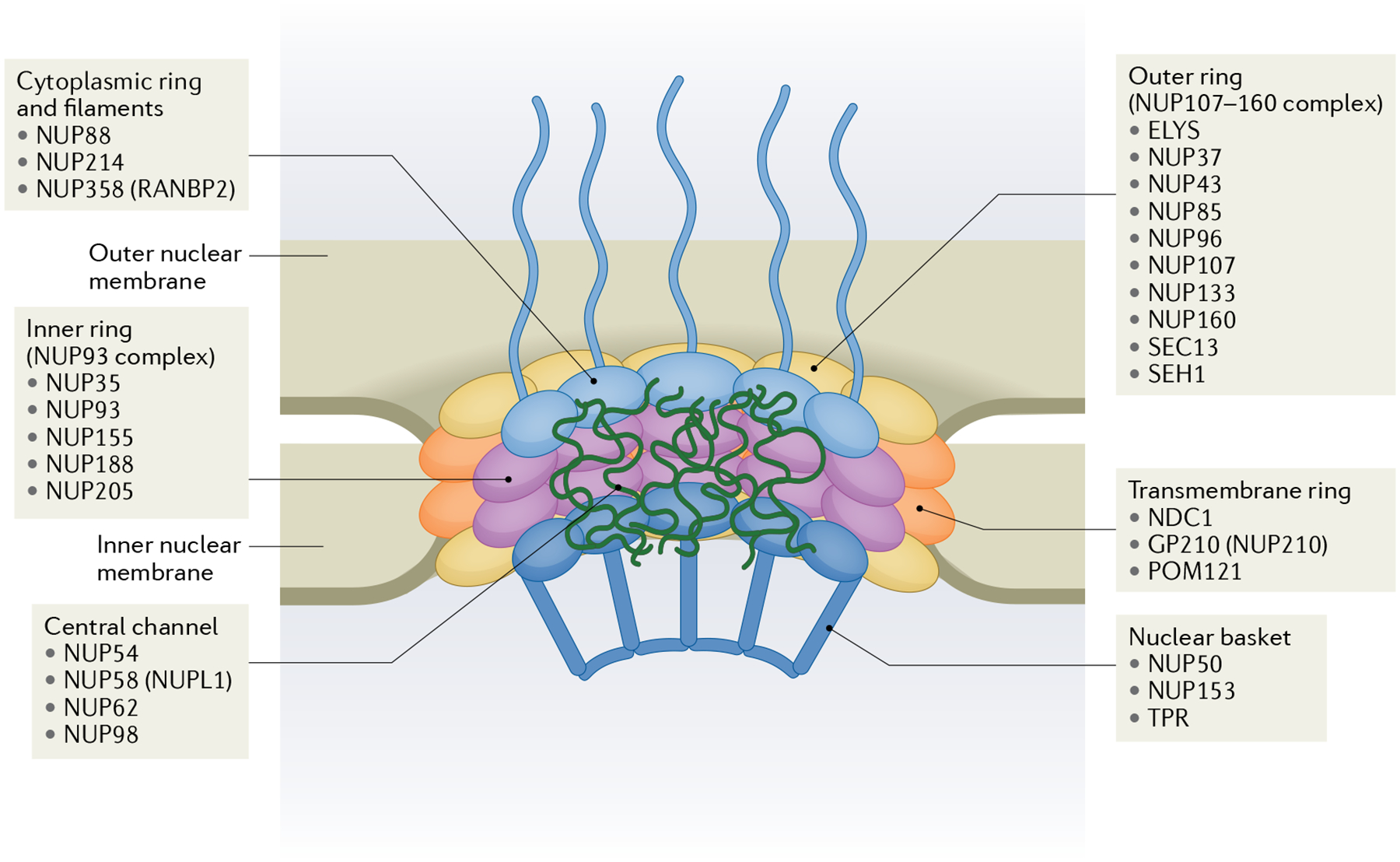
The main subcomplexes of the nuclear pore complex are the cytoplasmic ring and filaments, the outer ring, the inner ring, the transmembrane ring, the central channel and the nuclear basket. Each subcomplex consists of multiple nucleoporins, listed as bullet points for each subcomplex. Adapted from REF.188, Springer Nature Ltd.
The NPC and its nucleoporin constituents collectively regulate nucleocytoplasmic transport of macromolecules, organization of the genome and gene expression. In general, macromolecules of <40–60 kDa can passively diffuse through the NPC. However, the passage of larger molecules relies on active transport facilitated by the binding of the cargo to specific NTRs (karyopherins) via nuclear localization and/or nuclear export sequences29–33. Active nucleocytoplasmic transport is maintained by RAN GTPase and a RAN gradient. In the cytoplasm, RANGAP1 hydrolyses RAN–GTP to RAN–GDP and in the nucleus, RCC1 converts RAN–GDP to RAN–GTP. This process increases the levels of RAN–GTP in the nucleus and of RAN–GDP in the cytoplasm, thereby providing an energy source for active transport34–40.
Approximately one-third of nucleoporins contain FG repeat domains, which form a selective permeability barrier that regulates the transport of macromolecules. These intrinsically disordered domains transiently bind to and interact with NTRs to facilitate active nucleocytoplasmic transport41,42. In the nuclear basket, TPR and NUP153 mediate nuclear mRNA export43–49 and NUP50 plays a role in the dissociation of NTR–cargo complexes upon nuclear protein import50–52 and might also have a role in nuclear protein export53.
In addition to their role in facilitating nucleocytoplasmic transport, the NPC and specific nucleoporins regulate chromatin organization and gene expression. Nucleoporins, including NUP98, NUP50 and NUP153, can interact with chromatin and tether it to specific nuclear landmarks. Furthermore, these nucleoporins have direct and indirect roles in gene expression. They directly regulate gene transcription by interacting with DNA in an RNA polymerase II-dependent manner54–59 and they indirectly influence gene expression by aiding RNA transport, ultimately leading to RNA processing events in the cytoplasm. The functions of the NPC and individual nucleoporins have been reviewed in depth elsewhere1–3,21,41,60–66.
Although the NPC and its functions are common to all cell types, the pathobiology of this macromolecular complex and its constituents seems to differ between cell types and diseases. Most studies of this pathobiology in neurodegenerative disease have focused on ALS. However, increasing evidence suggests that alterations in the NPC and nucleocytoplasmic transport contribute to several neurodegenerative diseases. Differences in the contributions to different diseases might reflect the cell-type-specific compositions of nucleoporins and nucleocytoplasmic transport proteins, although they could also reflect differences in the model systems used to study neurodegenerative disease pathophysiology.
Nucleocytoplasmic transport and the NPC in ALS
Pathogenesis of ALS.
ALS is characterized by progressive degeneration of upper and lower motor neurons, along with degeneration of local interneurons and regional dysfunction of oligodendrocytes and astrocytes, and later activation of microglia67,68. Up to 50% of patients with ALS have mild dementia and a small subset has concomitant FTD69,70.
In up to 10% of affected patients, ALS is inherited, and more than 20 genes can cause familial ALS. The most common causal genetic mutation is a hexanucleotide repeat expansion (HRE) within intron 1 of C9orf72, which accounts for ALS in up to 40% of patients with familial ALS. Mutation of C9orf72 is also the most common cause of familial FTD. Mutations in genes that encode proteins involved in RNA metabolism, including TDP43 and FUS, and cytoskeletal integrity, including profilin 1 (PFN1), cause ALS in a small subset of patients with familial ALS71–75. The remaining 90% of patients with ALS have sporadic disease, although C9orf72 mutations are commonly found among patients with apparent sporadic ALS71,72.
Familial and sporadic ALS have similar clinical manifestations and end-stage pathologies67,76,77. For example, the RNA-binding protein TDP43 is cleared from the nucleus and accumulates and aggregates in the cytoplasm in 97% of all patients with ALS78,79. Whether TDP43 cytoplasmic mislocalization is the result of impaired active and passive nucleocytoplasmic transport or an independent cellular response to neurodegeneration remains unknown, but the fact that the end-stage pathology and clinical manifestations are similar suggests that the cellular and molecular events that initiate the disease are at least partly shared between familial and sporadic ALS.
Nucleocytoplasmic transport alterations.
Two studies published in 2015 were the first to identify defects in nucleocytoplasmic transport as a prominent pathomechanism underlying C9orf72 ALS–FTD10,12. One of these studies, using induced pluripotent stem cell (iPSC)-derived neurons (iPSNs) and Drosophila models of C9orf72 ALS, showed that pathological repeat RNA generated by the C9orf72 HRE disrupts nucleocytoplasmic transport by sequestering RANGAP1, thereby disrupting the RAN GTPase gradient7. Use of a fluorescent nucleocytoplasmic transport reporter and fluorescence recovery after photobleaching (FRAP) demonstrated that nuclear protein import is impaired in iPSNs with the C9orf72 mutation10. In the other study, overexpression of the C9orf72 HRE in Drosophila salivary glands or HeLa cells resulted in retention of poly-A mRNA within the nucleus12. Furthermore, a genetic screen indirectly implicated components of the nuclear transport pathway in C9orf72-mediated retinal degeneration in Drosophila12.
A third study published later in 2015 independently identified a subset of NTRs that are modifiers of C9orf72 dipeptide repeat (DPR) protein toxicity in yeast13. Since 2015, multiple studies have been done using exogenous nucleocytoplasmic transport reporters, permeabilized cell assays and genetic screens to understand how pathological DPR proteins produced by the C9orf72 HRE contribute to disruption of nucleocytoplasmic transport80–87. The precise defects identified in C9orf72 model systems have been reviewed in detail elsewhere88.
All studies indicate dysfunction of elements of the nucleocytoplasmic transport machinery, although many of these studies have yielded conflicting results, making it difficult to interpret exactly how the C9orf72 HRE and DPR proteins disrupt nucleocytoplasmic transport in human neurons. The conflicting results could be due to the technical challenges and limitations of performing these studies in endogenous and relevant cellular model systems. As such, debate remains about the nature of nucleocytoplasmic transport defects in ALS and the mechanisms that could underlie these defects.
In a study published in 2020, experiments in permeabilized HeLa cells and primary mouse cortical neurons showed that arginine (R)-rich DPR proteins impede the function of importin-β in the vicinity of the NPC80. Importin-α–importin-β complexes facilitate nuclear import of many proteins that contain nuclear localization signals (NLSs). Importin-α binds to NLS sequences in target proteins and importin-β binds to importin-α and FG-rich nucleoporins in the NPC to mediate nuclear import of target proteins33, such as TDP43 (REF.89). Thus, disruptions in importin-β function could have detrimental effects on nuclear protein import and might contribute to TDP43 pathology in ALS. Although the study in HeLa cells relied on the artificial expression of short, fluorescently labelled DPR proteins in non-living cells, the result suggests that future studies of DPR protein-mediated NTR disruption could provide novel insights into the mechanistic basis of nucleocytoplasmic transport disruption in C9orf72 ALS–FTD. Furthermore, some evidence suggests that hyperosmotic stress impedes cargo unloading in a similar way to DPR protein overexpression. In non-neuronal mammalian cells, fluorescent nucleocytoplasmic transport reporters accumulated along the nuclear rim upon sorbitol stress and, to a lesser extent, poly(GR) overexpression90, an observation that is similar to poly(GR)-mediated and poly(PR)-mediated defects in importin-α and importin-β cargo unloading in the vicinity of the NPC80. However, the relevance of these artificial cellular stress model systems to true human neurodegenerative disease remains unclear. Studies are needed to ascertain whether physiologically relevant cellular stressors and stress granule assembly functionally affects nucleocytoplasmic transport via similar mechanisms that involve importin-α and importin-β in human neurons.
Though early studies of nucleocytoplasmic transport in ALS focused on C9orf72, nucleocytoplasmic transport is now being investigated in the context of other rare inherited forms of ALS, including those associated with TDP43, FUS and PFN1. The use of fluorescent reporters and static or dynamic FRAP imaging in three independent studies has identified defects in nuclear protein transport in iPSNs or primary mouse cortical neurons that express mutant TDP43 (REF.4), FUS11 or PFN1 (REF.8). Moreover, overexpression of TDP43 variants in primary mouse cortical neurons led to an increase in nuclear levels of poly-A mRNA, suggesting that TDP43 overexpression impairs global protein and mRNA trafficking between the nucleus and cytoplasm. However, the studies of PFN1 and TDP43 were based on artificial overexpression of mutant constructs in primary mouse cortical neurons; whether endogenous levels of mutant PFN1 or TDP43 or cytoplasmic wild-type TDP43 pathology disrupt nucleocytoplasmic transport in human neuronal systems remains to be seen, as extrapolation from non-neuronal and non-mammalian systems to human diseases is difficult, especially when evaluating NPCs, the composition of which varies by cell type26–28. Moreover, these studies did not determine the mechanism by which nucleocytoplasmic transport is impaired in TDP43, FUS, and PFN1 model systems. Studies are needed to ascertain whether nucleocytoplasmic defects result from NPC injury and/or disruptions in NTR function.
NPC disruption.
To date, most studies of nuclear transport in ALS have focused on functional nucleocytoplasmic transport itself, primarily through the use of reporter constructs. However, the NPC tightly governs active and passive nucleocytoplasmic transport. It controls active nucleocytoplasmic transport through interactions between nucleoporins that contain FG repeat domains (FG-Nups) and NTRs and passive nucleocytoplasmic transport by the integrity of the FG-Nup diffusion barrier within the central channel of the NPC, which normally allows macromolecules of >40–60 kDa to freely diffuse through the NPC. Given that FG-Nups are anchored within the NPC via scaffolding nucleoporins, the overall composition and integrity of the NPC is essential for maintaining nucleocytoplasmic transport and other cellular functions1,2,21.
Initial studies in sporadic and C9orf72 ALS–FTD post-mortem human tissue and in mouse models of ALS suggested that mislocalization and cytoplasmic accumulation of the NPC-associated protein RANGAP1 and a subset of nucleoporins, including NUP107, NUP205 and FG-nucleoporins, contribute to deficits in nucleocytoplasmic transport (FIG. 2; TABLE 1). However, these studies provided little direct functional evidence that cytoplasmic nucleoporin pathology is linked to nucleocytoplasmic transport deficiencies10,91–95. Moreover, in a later study, quantification of RANGAP1 pathology in post-mortem human tissue identified no difference in the prevalence of abnormalities between controls and individuals with C9orf72 ALS91, highlighting the necessity of accompanying end-stage pathology studies with robust quantification rather than just crude visual observations when performing histology to identify novel disease pathologies. Nonetheless, histological evidence of protein pathology does not necessarily indicate a functional defect, so the presence or absence of pathology that involves a single protein involved in nucleocytoplasmic transport cannot be used in isolation to draw conclusions about nucleocytoplasmic transport function and its contribution to disease.
Fig. 2 |. Alterations in the nuclear pore complex and nucleocytoplasmic transport in neurodegenerative diseases.
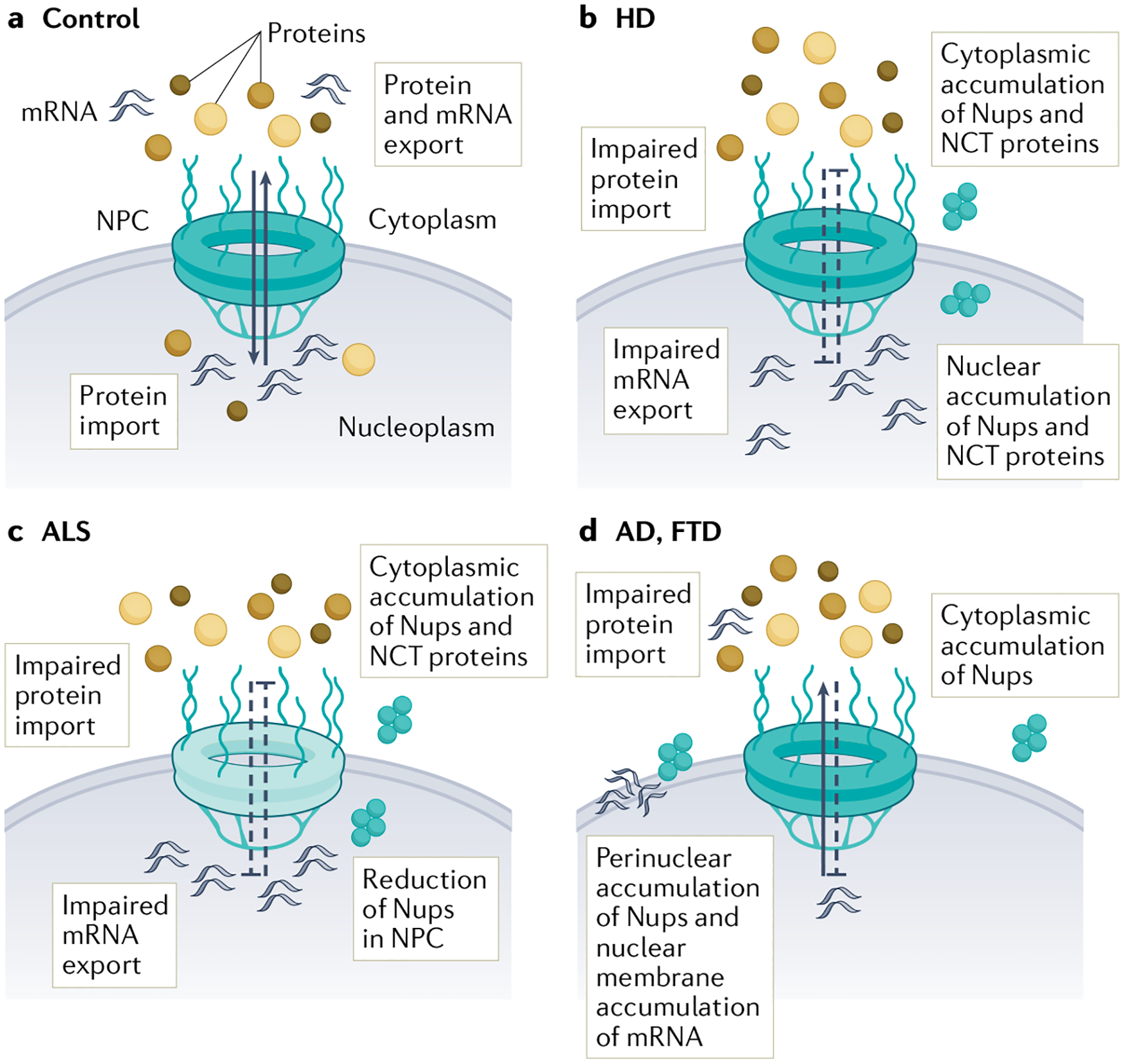
a | Under physiological conditions, mRNA and protein are transported out of and into the nucleus via the nuclear pore complex (NPC). b | In Huntington disease (HD), import and export through the NPC is impaired, leading to impaired protein import and mRNA export. In addition, a subset of nucleoporins (Nups) and nuclear pore-associated proteins with roles in nucleocytoplasmic transport (NCT) are present in nuclear and cytoplasmic aggregates (TABLE 1). c | In amyotrophic lateral sclerosis (ALS), specific Nups (TABLE 1) are reduced from neuronal NPCs, in a manner dependent on CHMP7 and the ESCRT-III complex, with no change in overall NPC number or structure. Consequently, import and export through the NPC is impaired. Moreover, a handful of Nups and nucleocytoplasmic transport proteins (TABLE 1) accumulate in the cytoplasm via unknown mechanisms. d | In Alzheimer disease (AD) and frontotemporal dementia (FTD), cytoplasmic and perinuclear accumulation of a small number of Nups (TABLE 1) occurs. In addition, accumulation of mRNA within nuclear membrane invaginations and impairments in protein import have been described, although the biological implications are unknown and the mechanistic link between all these pathologies remains unclear.
Table 1 |.
Summary of nucleoporin and nuclear transport receptor alterations in neurodegenerative disease models
| Proteins | Model system | Alterations | Biological impact | Refs |
|---|---|---|---|---|
| Amyotrophic lateral sclerosis | ||||
| NUP62 | FUS–ALS iPSN model | Altered nuclear rim staining pattern; possible cytoplasmic mislocalization | Unclear — might affect FUS phase separation | 11 |
| NUP98 | N2A cells that overexpress TDP43 C-terminal fragment | Probable reduction in levels in the nucleus | Unknown | 4 |
| NUP107 | Post-mortem motor cortex tissue from patients with C9orf72 ALS | Cytoplasmic mislocalization | Unknown | 10 |
| NUP205 | Fibroblasts that express mutant TDP43 | Probable reduction in levels in the nucleus | Unknown | 4 |
| NUP205 | Post-mortem frontal and motor cortex tissue from patients with C9orf72 ALS, TDP43 ALS and sporadic ALS | Cytoplasmic mislocalization | Unknown | 4,10 |
| POM121 | AAV GFP–(GR)200 mice | Subtle cytoplasmic mislocalization and/or co-accumulation with (GR)200; no obvious reduction in levels in the nucleus | Unclear in mice | 95 |
| POM121 | Primary spinal neurons that overexpress PFN1 variants | Cytoplasmic mislocalization | Unknown | 8 |
| POM121 | FUS–ALS iPSN model | Altered nuclear rim staining pattern | Unknown | 11 |
| RANGAP1 | Primary spinal neurons that overexpress PFN1 variants; fibroblasts that express mutant C9orf72; post-mortem motor cortex tissue from patients with C9orf72 ALS | Cytoplasmic mislocalization | Unclear — might contribute to deficient nucleocytoplasmic transport | 8,10 |
| RANGAP1 | Primary cortical neurons that overexpress TDP43 variants; C9orf72 ALS iPSN model; post-mortem motor cortex tissue from patients with C9orf72 ALS; AAV (G4C2)149 mice; AAV GFP–(PR)50 mice | Altered nuclear rim staining pattern | Unclear — might contribute to deficient nucleocytoplasmic transport | 4,10,92,94 |
| POM121 and RANGAP1 | AAV GFP–(GA)50 mice | Intranuclear co-accumulation with (GA)50; no obvious reduction in nuclear envelope staining | Unknown | 93 |
| NUP98, importin-α5, KPNA2 | AAV GFP–(GR)200 mice | Cytoplasmic mislocalization and/or co-aggregation with (GR)200 | Unclear — might contribute to deficient nucleocytoplasmic transport | 95 |
| POM121, NUP133, NUP50, NUP153, TPR | iPSN model of sporadic ALS | Reduction in levels in the NPC | Altered cellular distribution of RAN GTPase; impaired nucleocytoplasmic transport; TDP43 dysfunction and mislocalization | 97 |
| POM121, NDC1, GP210, NUP133, NUP107, NUP50, TPR, NUP98 | C9orf72 ALS iPSN model; post-mortem motor cortex and spinal cord tissue from patients with C9orf72 ALS | Reduction in levels in the NPC | Altered cellular distribution of RAN GTPase; impaired nucleocytoplasmic transport; TDP43 dysfunction and mislocalization | 5,97 |
| NUP50, NUP62, NUP93, NUP98, NUP107, NUP214, TPR, GLE1 | Primary cortical neurons that overexpress TDP43 variants | Cytoplasmic mislocalization and/or co-aggregation with TDP43 variants | Unknown | 4 |
| NUP35, NUP43, NUP58, NUP62, NUP88, NUP93, NUP98, NUP107, NUP153, NUP155, NUP160, NUP205, NUP214, NUP358, TPR, Aladin, hCG1, POM121, GP210, XPO5, NXF1, GLE1 | N2A cells that overexpress TDP43 C-terminal fragment | Cytoplasmic mislocalization and/or co-aggregation with TDP43 C-terminal fragment | Unknown | 4 |
| Alzheimer disease and frontotemporal dementia | ||||
| NUP62 | Post-mortem human hippocampal tissue | Perinuclear accumulation | Unknown | 6 |
| NUP98 | Tg4510 mice; post-mortem human hippocampal tissue; iPSNs that express mutant tau | Perinuclear accumulation, possibly within nuclear membrane invaginations | Contributes to altered RAN GTPase localization, which might affect nucleocytoplasmic transport; promotes tau aggregation | 6,123 |
| Huntington disease | ||||
| GLE1 | R6/2 mice; HTTQ175 mice | Intranuclear accumulation | Unknown — might reduce RNA export | 7 |
| NUP62 | R6/2 mice; post-mortem cortical and striatal human tissue; iPSN model of HD | Intranuclear accumulation; possible increase in expression; cytoplasmic mislocalization | Unknown | 9 |
| NUP88 | HTTQ175 mice | Intranuclear accumulation | Unknown | 9 |
| RANGAP1 | R6/2 mice; HTTQ175 mice; post-mortem cortical and striatal human tissue; human neuronal progenitor cells; iPSN model of HD | Intranuclear accumulation; possible increase in expression; abnormal nuclear distribution; abnormal nuclear membrane distribution; cytoplasmic mislocalization | Unknown | 7,9 |
AAV, adeno-associated virus; ALS, amyotrophic lateral sclerosis; FUS, fused in sarcoma; GA, glycine–alanine dipeptide repeat protein; GFP, green fluorescent protein; GP210, transmembrane nucleoporin; GR, glycine–arginine dipeptide repeat protein; HD, Huntington disease; Htt, huntingtin; iPSN, induced pluripotent stem cell-derived neuron; KPNA2, karyopherin-α 2; N2A, neuro2A; NDC1, transmembrane nucleoporin; NPC, nuclear pore complex; NXF1, nuclear RNA export factor 1; PFN1, profilin 1; POM121, transmembrane nucleoporin; RANGAP1, RAN GTPase-activating protein 1; TDP43, TAR DNA-binding protein 43; TPR, translocated promoter region nuclear basket nucleoporin; XPO5, exportin 5.
Subsequent studies in N2A cells, SH-SY5Y cells and mouse primary neurons that overexpressed wild-type or mutant TDP43 or PFN1 have revealed cytoplasmic mislocalization of nucleoporins and NPC-associated proteins4,8,96 (TABLE 1). Intriguingly, overexpression of wild-type TDP43, ALS-associated mutant TDP43 or the carboxy-terminal fragment of TDP43 elicited differential nucleoporin and NPC protein pathology in N2A cells and primary mouse cortical neurons4, suggesting that alterations in the NPC and nucleocytoplasmic transport are TDP43 variant-specific. In addition, in a study of FUS-associated ALS iPSNs, the nuclear rim staining patterns of POM121 and NUP62 were altered11. The decreased nuclear rim immunoreactivity could plausibly be concomitant with cytoplasmic mislocalization of NUP62, although no direct evidence of this was provided11.
Each of these pathology studies evaluated only a small number of nucleoporins and NPC-associated proteins and they fall short of linking cytoplasmic accumulations to defects in the NPC and functional changes in nucleocytoplasmic transport. For example, this cytoplasmic pathology could simply reflect dysregulation of cytoplasmic pools of nucleoporins that has little impact on the composition and function of the NPC. For this reason, none of these studies provides conclusive evidence that cytoplasmic accumulation of nucleoporins and nucleocytoplasmic transport proteins forms the mechanistic basis of dysfunctional nucleocytoplasmic transport or alters expression of nucleoporins within the NPC itself in ALS.
By contrast, injury to the neuronal NPC itself has been demonstrated in a study of a large number of iPSNs that expressed endogenous levels of the C9orf72 HRE5; this injury was characterized by reduced levels of eight specific nucleoporins within the nucleoplasm and NPCs without a change in overall NPC number or distribution5. Importantly, a nearly identical NPC injury has also been observed in iPSNs derived from patients with sporadic ALS97, an observation that is consistent with the notion that familial and sporadic ALS are often clinically and pathologically indistinguishable67. Both of these studies involved the use of super-resolution structured illumination microscopy to accurately and reproducibly resolve individual NPC spots98 and to examine millions of nuclei from a large number of different patient cell lines. Collectively, these two studies illustrate that NPC injury is initiated by reduction of the transmembrane nucleoporin POM121 from the neuronal NPC, which affects overall NPC composition and nucleocytoplasmic transport function, leading to downstream alterations in TDP43 function and localization and reductions in overall neuronal survival5,97.
Most studies of NPC and nucleocytoplasmic transport dysfunction in C9orf72 ALS have centred on the toxicity of DPR proteins rather than repeat RNA88, possibly because early studies in Drosophila indicated that pathological repeat RNA is not toxic99. However, Drosophila lacks the gene that encodes POM121 (REFS1,100,101) so overt toxicity might not have been observed in Drosophila models that expressed only repeat RNA because the POM121 protein is critical for the initiation of NPC injury in human neurons. Consistent with this hypothesis, a study conducted in human iPSCs concluded that pathological repeat RNA is sufficient to induce NPC injury through reduction of POM121 in human iPSNs5. Taken together, studies in C9orf72 ALS model systems suggest that repeat RNA and DPR proteins lead to impairment of nucleocytoplasmic transport via independent mechanisms — repeat RNA initiates an NPC injury cascade, whereas DPR proteins impair nucleocytoplasmic transport, possibly via pathological interactions with NTRs. However, studies are needed to understand the different cellular and molecular events that trigger reductions in POM121 in sporadic ALS that are dependent on C9orf72 repeat RNA in C9orf72 ALS. Moreover, whether C9orf72-mediated deficits in the NPC and in nucleocytoplasmic transport that were initially identified in spinal neurons and post-mortem motor cortex tissue5,10 are also present in neurons that are involved in C9orf72 FTD (for example, frontal cortex neurons) remains unclear.
Although the nature of defects in the NPC in ALS have been characterized, the mechanisms that give rise to these alterations are less well studied. Two biological pathways have been linked to NPC injury and the cytoplasmic accumulation of nucleoporins and NPC-associated proteins: degradation mediated by CHMP7 and the endosomal sorting complex required for transport (ESCRT) III complex97,102, and stress granule assembly90. The ESCRT-III-related protein CHMP7, which recruits the ESCRT-III complex and VPS4 to the nuclear envelope, has a role in NPC turnover in yeast and non-neuronal mammalian cells103–106. Multimodal imaging of iPSNs from patients with familial or sporadic ALS has demonstrated that nuclear accumulation of CHMP7 is sufficient to initiate NPC injury97,102 and could, therefore, be the fundamental disease-initiating pathway in ALS. The studies in iPSNs from patients with sporadic ALS demonstrated that the characteristic loss of nuclear TDP43 function and localization clearly occurs downstream of NPC injury97, suggesting that TDP43 dysfunction and mislocalization is a later biological event in ALS pathophysiology. The mechanisms that underlie pathological nuclear accumulation of CHMP7 in ALS remain unknown, but these studies highlight that the ESCRT-III pathway is a potent mediator of early pathogenic events in ALS; the pathway could have similar importance in related neurodegenerative disorders97,102.
Given that cytoplasmic accumulations of nucleoporins and NPC-associated proteins are prevalent in multiple models of ALS and that these accumulations seem to be independent of NPC injury itself, distinct mechanisms are likely to regulate cytoplasmic nucleoporin pathologies. Consistent with previous work that suggested that nuclear protein export can be abrogated by oxidative stress107, a study of multiple immortalized cell lines has shown that many nucleoporins and nucleocytoplasmic transport proteins can be sequestered in stress granules90. This study also showed that stress granule assembly can affect nucleocytoplasmic transport function90 but it remains unclear whether this effect is the direct result of nucleoporin and nucleocytoplasmic transport protein sequestration in stress granules and whether this phenomenon is more broadly applicable to post-mitotic human neurons and, therefore, human disease.
Studies in iPSNs and post-mortem tissue from patients suggest that only a subset of nucleoporins are reproducibly reduced from neuronal NPCs in ALS5,97. However, these nucleoporins span multiple subcomplexes of the NPC and their functions include maintenance of NPC structure and organization, facilitation of nucleocytoplasmic transport, and regulation of genome organization and gene expression1–3,21, so reduced expression of these specific nucleoporins in the NPC is likely to directly and/or indirectly affect several cellular processes. For example, NUP50 has roles in protein import and transcriptional regulation54, whereas TPR is involved in mRNA export43. Future research should delineate the precise cellular defects induced by loss of specific nucleoporins in human neurons and determine how these disruptions contribute to disease pathogenesis. Moreover, collective nucleocytoplasmic transport impairments downstream of NPC injury could disrupt the import and replenishment of nucleoporins and NPC-associated proteins, leading to end-stage cytoplasmic accumulations in a process that compounds the initial injury to the NPC (FIG. 3).
Fig. 3 |. Pathological progression of changes to the nuclear pore complex and nucleocytoplasmic transport in amyotrophic lateral sclerosis.
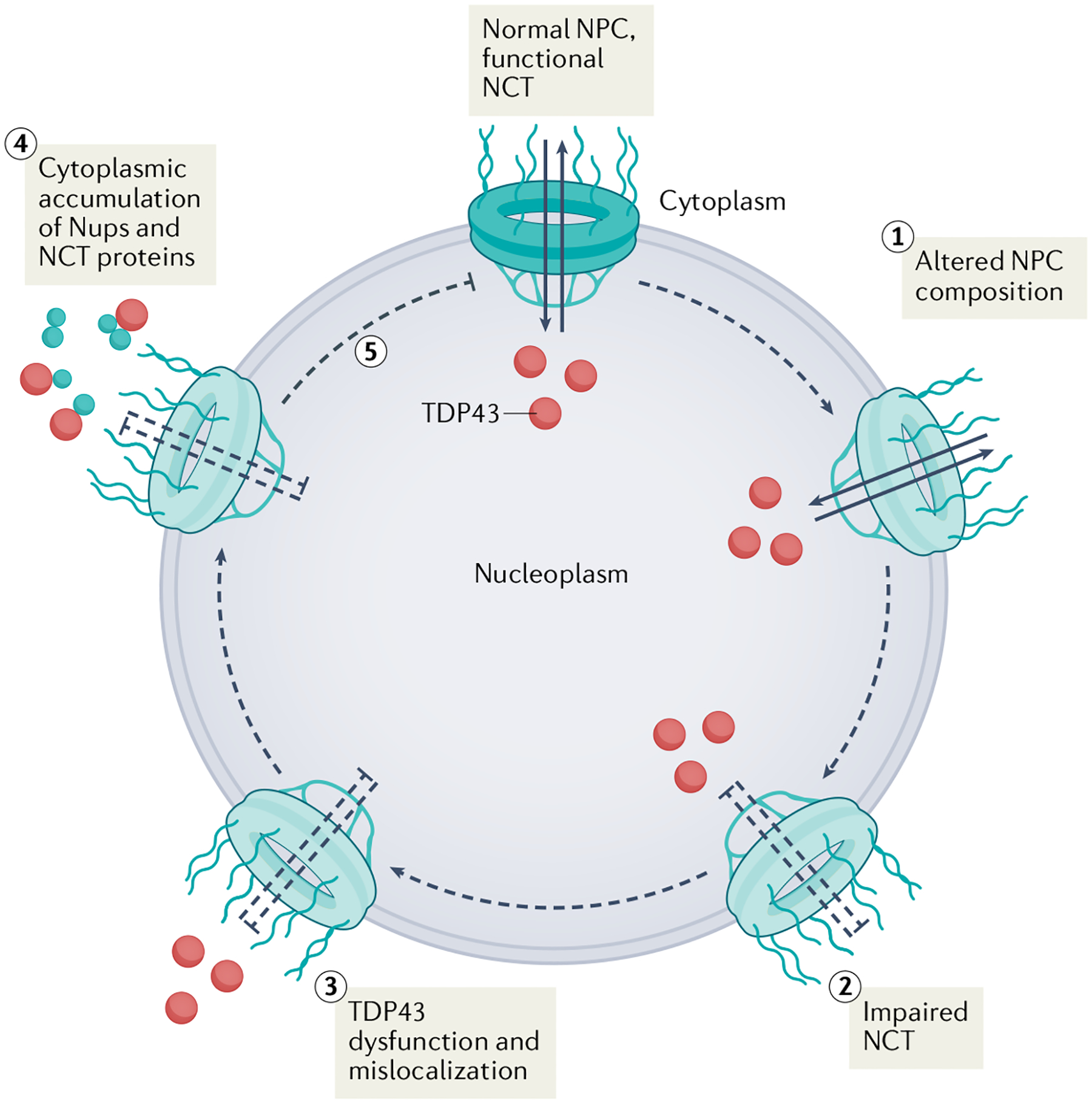
Aberrant nuclear retention of the ESCRT-III-associated protein CHMP7 initiates nuclear pore injury characterized by the reduction in specific nucleoporins (Nups) from the nuclear pore complex (NPC), leading to an altered composition of the NPC (1). Disruption of the NPC in this way alters functional nucleocytoplasmic transport (NCT) (2), which ultimately leads to loss of TAR DNA-binding protein 43 (TDP43) function within the nucleus and translocation of TDP43 from the nucleus to the cytoplasm (3). Impaired NCT might result in cytoplasmic accumulation of TDP43, Nups and nuclear transport receptors in end-stage disease (4). Impaired nuclear import might then inhibit restoration of the NPC composition and integrity (5), thereby exacerbating the process.
Nucleocytoplasmic transport in AD and FTD
AD and FTD — the two most common forms of dementia — are characterized by cognitive decline and memory loss that result from the progressive degeneration of neurons in the hippocampus and, in FTD, the frontal cortex. Mutations in the genes that encode amyloid precursor protein (APP) and presenilin (PSEN) are associated with AD108,109 and mutations in C9orf72 and the genes that encode tau and progranulin are linked to FTD71,72,110–112. The presence of amyloid-β plaques is the hallmark pathology of AD113–116, but cytoplasmic accumulations of TDP43 and tau are present in both AD and FTD113,117–121. The relationship between TDP43 pathology and alterations in the NPC and nucleocytoplasmic transport is discussed above; in this section, we focus on tau-mediated abnormalities in the NPC and nuclear lamina and in nucleocytoplasmic transport.
Although tau inclusions — known as neurofibrillary tangles — within neuronal cell bodies are a common pathological feature of AD and FTD, the mechanisms that lead to this pathology and the cellular consequences are poorly understood. Neurofibrillary tangles occur near the nuclear envelope122, raising the possibility that they interfere with NPC function and/or NPC and nuclear lamina organization. Indeed, one study found that NUP62 and NUP98, but not NUP54 or POM121, were present in perinuclear tau aggregates in post-mortem human brain tissue and in mouse models of FTD-tau6. In addition, NUP98 can promote the aggregation of tau in vitro6 and seems to aggregate in NPCs and the nuclear envelope in AD and FTD6,123. However, the relationship between these phenomena and defects in nucleocytoplasmic transport in cellular models of FTD-tau remain unclear. Intriguingly, in a primary neuron model of FTD-tau, knockdown of NUP98 restored the localization of RAN GTPase6. These findings suggest that sequestration of NUP98 within tau aggregates does not lead to loss of the function of NUP98 in nucleocytoplasmic transport; instead, NUP98-mediated aggregation of tau might affect nucleocytoplasmic transport via currently unknown mechanisms. Therefore, additional work is needed to determine whether cytoplasmic or nuclear pools of NUP98 promote tau aggregation, to understand the contribution of altered NPC permeability and NTR function to disruption of nucleocytoplasmic transport, and to identify whether local or global changes occur in NPC accessibility and function. Moreover, work is needed to understand whether tau-associated alterations in specific nucleoporins translate into a reduction of these proteins in the NPC, as observed in C9orf72 ALS–FTD5, or local aggregation in the vicinity of a subset of NPCs, perhaps owing to altered structural integrity of the nucleus as a result of, for example, abnormal nuclear lamina pathology.
Several studies of post-mortem human brain tissue, iPSNs and Drosophila have demonstrated that nuclear lamina morphology is abnormal in AD and FTD6,123–125. The nuclear lamina interacts with several nuclear envelope proteins, including lamina-associated polypeptide (Lap2) and lamin B receptor and is critical for maintenance of chromatin organization and positioning, thereby facilitating transcription and export of messenger ribonucleoprotein (mRNP) complexes through NPCs126. In iPSNs, nuclear lamina invaginations that are similar to those observed in AD and FTD occur as a result of microtubule depolymerization123, which disrupts the interactions of the linker of the nucleus and cytoskeleton (LINC) complex. This complex normally maintains nuclear integrity and positioning by anchoring the nuclear membranes to cytoplasmic micro tubules, actin and intermediate filaments127. The presence of mutant tau within these invaginations123 raises the possibility that tau mislocalization affects microtubule dynamics and contributes to disruption of nuclear membrane morphology. Moreover, in post-mortem human brain tissue, these nuclear lamina invaginations contain NPCs and LAP2 (REF.125), suggesting that altered nuclear morphology can affect NPC and lamina functions. Indeed, toxic accumulation of mRNA has been observed in nuclear lamina invaginations124, and heterochromatin loss128 occurs in Drosophila models of FTD-tau. In Drosophila, pharmacological or genetic reduction of mRNA export mitigates tau toxicity124.
Taken together, these recent findings suggest that mutations in tau result in its mislocalization to the perinuclear space where NUP98 facilitates its aggregation, leading to impaired microtubule dynamics that deform the nuclear lamina and initiate local disruption of chromatin organization and mRNA export within invaginations. However, additional work is needed to fully define the sequence of pathological events in AD and FTD. Furthermore, future studies are necessary to directly link altered nuclear lamina morphology with impaired nuclear function in AD and FTD.
Nucleocytoplasmic transport in HD
HD is an inherited, autosomal dominant neurodegenerative disorder that is characterized by degeneration of medium spiny neurons within the striatum. Clinically, HD manifests as choreiform movements and dementia. The cause is a CAG trinucleotide repeat expansion in the first exon of the HTT gene, which encodes the huntingtin protein. Expansions of >40 repeats are fully penetrant, and longer expansions are linked to earlier disease onset and greater severity129,130. The repeat expansion leads to production of huntingtin protein that contains poly-glutamine (polyQ) tracts and is consequently aggregate-prone. However, as with the C9orf72 HRE, the HTT CAG repeat can lead to repeat-associated non-AUG (RAN) translation that produces poly-alanine (polyA), poly-serine (polyS), poly-leucine (polyL) and poly-cysteine (polyC) proteins that accumulate in the brain131. However, the molecular mechanisms through which these protein products contribute to neurodegeneration remain largely unknown.
Given that cytoplasmic protein aggregation is known to affect nucleocytoplasmic transport of protein and RNA132, early studies of nucleocytoplasmic transport dysfunction in models of HD have centred on characterization of NPC and nucleocytoplasmic transport pathology that results from accumulation of mutant huntingtin protein. Two studies showed that a subset of nucleoporins and nucleocytoplasmic transport proteins, including NUP62, NUP88, RANGAP1 and GLE1, co-accumulate with mutant huntingtin in mouse models of HD7,9. Furthermore, studies in mouse models of HD showed that the extent of RANGAP1 pathology and huntingtin aggregates are dependent on age and expression level of mutant huntingtin protein7,9. Studies of post-mortem human tissue revealed RANGAP1 and NUP62 alterations, but the pathology was distinct from that in mice7,9. In the mouse models, a few nucleoporins and nucleocytoplasmic transport proteins co-aggregated with mutant huntingtin protein, whereas in end-stage human tissue, mislocalization was more subtle, accumulations of proteins were small but more numerous, and expression of RANGAP1 and NUP62 even seemed to be increased7,9. These findings highlight the complexity of authentic human disease in comparison with defined genetic mouse models. Additional findings that are consistent with the idea that pathology is cell-type-specific and species-specific include abnormal nuclear membrane distribution of RANGAP1 in human neuronal precursor cells in HD7, and severe cytoplasmic mislocalization of RANGAP1 and NUP62 in HD iPSNs9. In a comparison of RANGAP1 and NUP62 pathology in post-mortem tissue from patients with juvenile and adult-onset forms of HD, pathology was more severe in the juvenile form of the disease9, suggesting that a greater extent of this pathology is associated with more severe disease. However, as discussed above, a study of RANGAP1 pathology in post-mortem tissue from patients with C9orf72 ALS91 demonstrated that simple quantification of abnormal-looking cells is not sufficient for accurate quantification of nucleoporin and nucleocytoplasmic transport protein pathology, especially in scenarios where cell-type-specific labels are not employed. Nevertheless, the method of quantification used in this HD study might provide a better estimate of patient-to-patient and mouse-to-mouse variability than studies in ALS, AD, FTD and HD models, in which individual cells or nuclei are counted as biological replicates. For all imaging-based studies across neurodegenerative disease models, in the future it may be best to represent and analyse datasets as superplots133 that depict the full spread of data points across all samples analysed. Another important consideration is that in any nucleoporin or nuclear membrane pathology study in mouse or human tissue, the entire 3D structure of the nucleus should be assessed where possible to avoid mischaracterization on the basis of single z-plane sections. The importance of this approach was demonstrated in C9orf72 ALS: nucleoporin reduction and nuclear lamina pathology were more robustly and accurately characterized in studies in which the 3D structure was analysed than in studies in which single 2D z-plane sections were analysed5,134. This 3D characterization enabled the investigators to conclude that nucleoporin reduction occurs throughout NPCs within the entire nucleus with no change to nuclear lamina structure, at least that is detectable with light microscopy.
Studies of post-mortem tissue from patients with HD and of mouse models have revealed nuclear membrane abnormalities in addition to nucleoporin and nucleocytoplasmic transport protein pathology7. However, whether these abnormalities reflect ageing-associated nuclear membrane folding or pathological nuclear membrane invagination remains unclear. Age-dependent nuclear mRNA retention has been observed in mouse models of HD7 and altered subcellular distribution of RAN GTPase and the S-tdTomato nucleocytoplasmic transport reporter9 has been observed in iPSN models of HD. Together, these observations indicate global defects in nucleocytoplasmic transport. While NUP62, NUP88, RANGAP1 and GLE1 have defined roles in nucleocytoplasmic transport40,135–140, neither study provided any evidence that pathological alterations and/or accumulations of these proteins in HD directly affect nuclear mRNA export or protein import and export.
Quantitative proteomics analysis of non-neuronal cell lines that overexpress proteins involved in neurodegenerative disease suggests that wild-type huntingtin protein associates with NTRs, such as importins β1, 4, 7 and 9, whereas mutant huntingtin protein associates with RANGAP1 and RAE1 (REF.141). However, these studies did not address the functional implications of these associations. Whether sequestration of these nucleoporins and nucleocytoplasmic transport proteins reflects their decreased expression within or association with the NPC itself or reflects accumulation from non-NPC associated pools remains unclear. Therefore, future studies are needed to unravel the functional contribution of nucleoporin pathology to the pathophysiology of HD.
Diseases caused by NPC mutations
Genetic mutations in some nucleoporins and nucleocytoplasmic transport proteins cause cell-type-specific neurological diseases; for example: mutations in NUP62 are implicated in infantile bilateral striatal necrosis, which affects the caudate nucleus and putamen; mutations in NUP214 can cause progressive encephalopathy and cortical atrophy20; and mutations in GLE1 are linked to the fetal motor neuron disease human lethal congenital contracture syndrome 1 (REF.18). Although not addressed in this Review, mutations in some nucleoporins have been linked to other non-neurological, cell-type-specific diseases142,143 (TABLE 2).
Table 2 |.
Summary of selected cell-type-specific or organ-specific diseases resulting from mutations in nucleoporins or NPC-associated proteins
| Disease | Mutated protein | Cell type | Phenotype | Refs |
|---|---|---|---|---|
| Allgrove (AAA) syndrome | Aladin | CNS motor neurons; multiple peripheral cell types | Motor neuron disease, achalasia, adrenal deficiency, alacrima | 177–180 |
| Human lethal congenital contracture syndrome 1 | GLE1 | CNS motor neurons | Infantile motor neuron disease, weakness, death | 18 |
| Lethal arthrogryposis with anterior horn cell disease | ||||
| Striatal nigral degeneration | NUP62 | Striatal neurons | Huntington-like disease | 17 |
| Fetal akinesia | NUP88 | Muscle | Muscle atrophy | 181 |
| Steroid-resistant nephrotic syndrome | NUP93, NUP205, XPO5, NUP107 | Kidney | Renal pathology, diffuse mesangial sclerosis, loss of renal function | 182 |
| Intellectual disability and general developmental delay | NUP107 | CNS and kidney | Simplified cortical gyri, undeveloped frontal lobes, glomerulosclerosis | 183 |
| Atrial fibrillation | NUP155 | Heart | Childhood death | 184 |
| Acute febrile encephalopathy | NUP214 | Purkinje cells, cortex | Febrile-induced ataxia, progressive cortical neurodegeneration | 19,20 |
| Encephalitis and autoimmune myositis | NUP358 | CNS and PNS | Encephalopathy, myopathy often after viral infection | 185 |
| Limb-girdle muscular dystrophy 1F | TNPO3 | Muscle | Selective muscle weakness | 186 |
| Intellectual disability | TPR | Unknown | White matter changes with delayed myelination, gliosis, cerebellar atrophy, microcephaly, ataxia | 187 |
GLE1, mRNA export factor GLE1; TNPO3, transportin 3; TPR, translocated promoter region nuclear basket protein; XPO5, exportin 5.
Although mutations in these nucleoporins and NPC-associated proteins are known to cause system-specific neurological disease, little is known about the functional implications of the mutations. At the RNA level, these proteins are widely expressed in CNS and non-CNS tissues26. However, the composition of nucleoporins and NPC-associated proteins in different cells is not known, especially in the CNS, so variations in the expression and ratio of specific nucleoporins and NPC-associated proteins could contribute to cell-type-specific vulnerability to disease.
GLE1 is an NPC-associated protein with roles in nuclear mRNA export, initiation of translation and termination of translation135,144,145. Disease-associated mutations in GLE1 disrupt its oligomerization, thereby impairing nuclear mRNA export146. NUP62 and NUP214 are FG-repeat-containing nucleoporins that reside, respectively, within the central channel and the cytoplasmic ring and filaments (FIG. 1) to facilitate bidirectional transport. The functional consequences of NUP62 mutations are unknown. However, in fibroblasts, mutations in NUP214 reduce overall NUP214 and NUP88 protein levels without affecting the number and density of NPCs. In addition, in fibroblasts that express mutant NUP214, nuclear protein import and mRNA export are impaired and a high proportion of NPCs have so-called plugs in the central channel. What these plugs are is unknown — one hypothesis is that they are mRNPs that have become trapped during transport through the pore or are transported more slowly, enabling their visualization in NUP214-mutant fibroblasts. These findings suggest that NUP214 mutations affect the passage of large cargo through the NPC19, but future experiments are necessary to understand the precise disruptions to passive and active nucleocytoplasmic transport that result from these mutations. Furthermore, the mechanism by which these functional alterations lead to cell-type-specific pathology remains unclear. Cell-specific expression of these nucleoporins and/or their cellular functions could render specific cell types more susceptible than others to the effects of these rare genetic mutations.
The fact that nucleoporin mutations cause cell-type-specific neurological disease suggests that defects in the NPC and nucleocytoplasmic transport are sufficient to initiate disease. However, few studies have been carried out to test this hypothesis, primarily owing to a lack of mechanistic studies in the context of mutant nucleoporins. In order to determine whether mutations in nucleoporins or NTRs are sufficient to initiate disease cascades in a cell-type-specific manner, studies are needed to comprehensively assess the effects of nucleoporin mutations on NPC composition, nucleo cytoplasmic transport, downstream cellular functions and cellular viability.
Laminopathy in neurodegenerative disease
The nuclear lamina is composed of lamin A/C and lamin B proteins, and provides structural integrity to the nucleus, organizes heterochromatin in the vicinity of the NPC, and is involved in mechanotransduction via interactions with the LINC complex147. Laminopathy has been described in diseases that affect the brain and/or muscle cells. For example, >400 mutations have been identified in LMNA, which encodes lamin A, and these mutations are implicated in a wide range of cellular disorders, including Hutchinson–Guilford progeria syndrome, muscular dystrophy, and Charcot–Marie–Tooth disease148–150. Lamin B1 duplications are associated with adult-onset autosomal dominant leukodystrophy151. Collectively, mutations or duplications in lamin genes alter the integrity of the nuclear lamina, resulting in nuclear membrane invaginations and premature cell ageing147,152,153. However, how nuclear lamina abnormalities contribute to neurodegeneration in these diseases and more broadly is unclear7,8,123,134. Studies that include age-related controls are needed to address whether observed disruptions of the nuclear lamina are related to physiological ageing or pathological processes. For example, one study of iPSNs and C9orf72 ALS–FTD suggested that nuclear lamina invaginations are an age-related phenomenon and not a pathological consequence of the C9orf72 HRE134. Other aspects that need to be investigated are the mechanisms that lead to nuclear lamina folds and/or invaginations and, perhaps most importantly, how these folds and invaginations disrupt NPC composition, nucleocytoplasmic transport, chromatin accessibility and gene expression.
Age-related changes in NPCs
Ageing is a risk factor for neurodegenerative diseases, and age-related disruptions to the NPC and nucleocytoplasmic transport are thought to be exacerbated in neurodegenerative disease4,6,7,9,10,12,13. Few studies have examined age-related declines in NPC integrity and nucleocytoplasmic transport function, but non-dividing human neurons are likely to accumulate age-related damage to NPCs given that they do not undergo frequent rounds of nucleoporin turnover during cell division, as do mitotic cells. Indeed, multiple studies have suggested that, although the overall number of NPCs remains consistent in ageing rat brains, NPC composition changes with age154–156. Specifically, quantitative proteomics analysis has shown that scaffold nucleoporins are extremely long-lived in ageing rat brains, whereas a subset of primarily FG-repeat-containing nucleoporins are exchanged more frequently156. Consistent with this molecular analysis, FRAP analyses in rat kidney cells have revealed that different nucleoporins have different residence times within the NPC, ranging from seconds to days157. Use of electron microscopy has revealed that the overall density of NPCs declines in the ageing rat dentate gyrus but remains fairly stable in hippocampal CA1 neurons158,159, raising the possibility that changes in NPC composition and dynamics with age are cell-type-specific. On the basis of these findings, the slow accumulation of nucleoporin pathology and nucleoporin variants might be expected to eventually lead to dysfunctional nucleocytoplasmic transport and ultimately increase cellular stress in neurodegeneration.
Functionally, NPCs are known to become more ‘leaky’ with age154. However, whether this change results from compositional changes within the NPC or the loss of permeability barrier function and/or integrity remains unclear. Studies in fibroblasts suggest that expression of proteins involved in nucleocytoplasmic transport decreases during ageing160,161 and that nuclear protein import declines161. However, whether these alterations directly affect NPC function or whether age-related declines in nucleocytoplasmic transport result from multiple cellular defects — as is likely to be the case for neurodegenerative disease — is unknown.
Nuclear lamina integrity can also deteriorate with age and in age-related disease162 and could affect NPC functions, such as nucleocytoplasmic transport. Age-associated nuclear envelope invaginations can result in dysfunctional nucleocytoplasmic transport via accumulation of NPCs124,125, but a weakened lamina network also often causes nuclear envelope ruptures, which reduce nuclear–cytoplasmic compartmentalization162,163. Furthermore, nuclear envelope integrity is partly maintained by contact with the cytoskeleton via the LINC complex147 and loss of LINC complexes can facilitate nuclear envelope weakening and lead to ruptures164. Together, these observations raise the possibility that LINC complex and nuclear envelope disruption contribute to age-related neurodegenerative declines in nucleocytoplasmic transport. Indeed, in primary mouse neurons that overexpressed the C9orf72 HRE or mutant PFN1, pharmacologically induced actin depolymerization altered the localization of RANGAP1, FG-repeat-containing nucleoporins and RAN GTPase in primary mouse motor neurons, whereas promotion of actin polymerization partially rescued nucleocytoplasmic transport defects8. Future work is needed to determine the mechanism by which actin dynamics contribute to nucleocytoplasmic transport in human neurons, but these studies suggest that age-related declines in overall nuclear integrity contribute to neurodegenerative disease pathogenesis.
Therapeutic approaches
Where genetic alterations in NPC or nucleocytoplasmic transport proteins contribute to defects in nucleocytoplasmic transport, elimination of the genetic abnormalities by reducing expression of mutant genes would be the most specific therapeutic approach. CRISPR technology enables specific targeting of genetic mutations165,166, and a scenario in which specific mutations in nucleoporins or other proteins that lead to NPC dysfunction are eliminated with CRISPR-based strategies can be envisioned. CRISPR technology has been used to remove pathological C9orf72 repeat expansions in iPSCs, which led to reversal of several C9orf72-associated phenotypes167, highlighting the potential of this new technology. Currently, however, the most common approach to therapeutic targeting of gene mutations is the use of antisense oligonucleotides (ASOs) to eliminate production of mutant protein via RNase H degradation of RNA transcripts168,169. Results from initial preclinical studies in iPSNs with C9orf72 mutations and in mouse models170,171 suggest that targeting the C9orf72 HRE can mitigate molecular hallmarks of disease, including the formation of repeat RNA foci and production of DPR proteins. Subsequent studies in iPSNs with C9orf72 mutations have shown that ASOs that selectively target C9orf72 repeat RNA can alleviate NPC injury and mislocalization of RAN GTPase5,10, highlighting the contribution of NPC injury and nucleocytoplasmic transport dysfunction to disease pathogenesis. An international phase I clinical trial of intrathecal administration of an ASO that specifically targets the G4C2 sense repeat strand of the C9orf72 mutation (BIIB078) was recently completed172. Therapies that modulate the antisense C4G2 repeat strand or the C9ORF72 protein have not yet been explored.
In the absence of a known genetic mutation — as in sporadic ALS — therapies that directly target NPC injury and nucleocytoplasmic dysfunction could prove beneficial in neurodegenerative diseases. Owing to the fact that the involvement of nucleocytoplasmic transport is an emerging concept in our understanding of neurodegenerative disease pathophysiology, few therapeutic studies have been conducted to date. However, in a study published in 2021, use of iPSNs from patients with sporadic or C9orf72 ALS demonstrated that the use of ASOs to reduce expression of an upstream initiator of NPC injury in ALS — CHMP7 — restored NPC composition and function, mitigated downstream deficits in TDP43 function and localization, and improved neuronal survival without overt toxicity97. Functional nucleocytoplasmic transport could also be targeted downstream of NPC injury; for example, the compound KPT-350 inhibits exportin-1 function, thereby preventing nuclear protein export, and variations of this compound have been used to rescue nucleocytoplasmic transport deficits in multiple models of ALS and HD4,8–10,90. Primarily as a result of these preclinical studies, a phase I clinical trial of oral KPT-350 (also known as BIIB100) is being carried out in patients with ALS173. Theoretically, small molecules that increase RAN GTPase activity could also increase nuclear transport capacity in neurodegeneration. Together, these early preclinical studies suggest that direct targeting of NPC injury and nucleocytoplasmic transport dysfunction in the absence of known genetic mutations could be a viable therapeutic strategy for neurodegeneration.
Conclusions
Considerable progress has been made in uncovering pathological changes in the NPC and nucleocytoplasmic transport in neurodegenerative disease but much work is still needed to understand how these alterations affect cellular function and vulnerability and to determine whether NPC and nucleocytoplasmic transport pathology is sufficient to initiate neurodegeneration and/or propagate the cellular injury. Despite the fact that the structure and organization of the NPC is conserved from yeast to flies to mice to humans, its overall composition is highly variable. For example, the transmembrane nucleoporin POM121 is not present in yeast or flies, and rodent POM121 has only ~60% protein sequence homology with human POM121, possibly undermining the interpretation of non-human models in relation to authentic disease. Moreover, expression of POM121 is highly variable across different human cell types26,27. In addition to the fact that genetic mutations in specific nucleoporins can cause cell-type-specific diseases142,143 (TABLE 2), this variability in nucleoporin expression raises the fascinating possibility that NPC heterogeneity contributes to the cell-type specificity of neurodegenerative disease. To address this possibility, studies are needed to understand the composition and stoichiometry of nucleoporins within cell-type-specific NPCs across the CNS.
The mechanisms by which nucleoporin and NTR disruptions contribute to defects in nucleocytoplasmic transport and/or gene expression in neurodegenerative disease remain largely unknown. One interesting insight that has been gained is that a POM121 gene duplication results in production of POM121 and soluble POM121 proteins, which independently mediate nucleocytoplasmic transport and transcription, respectively174. This study raises the intriguing possibility of a two-hit model, whereby alterations in a specific nucleoporin change NPC composition and functionality and could also lead to accumulation of the nucleoporin in the cytoplasm, ultimately leading to a loss of normal function and a gain of toxic cytoplasmic function, as has been proposed for TDP43 (REF.175).
This insight highlights the fact that, in order to understand how NPC injury and nucleocytoplasmic transport dysfunction contribute to disease pathogenesis, nucleoporin and NPC-associated protein pathology in the cytoplasm, the nucleus and the NPC itself must be comprehensively defined. Furthermore, emerging evidence shows that nucleoporins have cytoplasmic functions176 and NTRs have non-transport-related functions14–16, making it critical to understand whether the cytoplasmic, NPC or nucleoplasmic functions of nucleoporins are disrupted in and contribute to disease pathogenesis. Nonetheless, existing evidence demonstrates that alterations to the NPC, nucleoporins and nucleocytoplasmic transport are early and prominent pathogenic events in ALS, AD, FTD and HD, and highlights the potential for therapeutic targeting of these alterations for the treatment of neurodegenerative disease.
Key points.
The nuclear pore complex (NPC), which is made up of multiple nucleoporin proteins, mediates nucleocytoplasmic transport, genome organization and gene expression.
Accumulating evidence suggests that defects in the NPC and nucleocytoplasmic transport contribute to, or possibly initiate, neurodegenerative diseases such as amyotrophic lateral sclerosis, dementia and Huntington disease.
Cell-type-specific NPC composition might underlie differential cellular vulnerability in neurological diseases.
Specific nucleoporin mutations can cause a wide range of neurological disorders, including a Huntington disease-like motor neuron disease.
Therapeutic strategies with antisense oligonucleotides and small molecules to repair NPC defects or restore altered nucleocytoplasmic transport are in development.
Acknowledgements
A.N.C. is supported by funding from NIH grant K99NS123242. J.D.R. is supported by funding from the ALS Association, ALS Finding a Cure, the Chan Zuckerberg Initiative, the Department of Defense, F Prime, NIH grants P01NS099114, P01NS084974, R01NS094239 and R01NS122236TEDCO, the Muscular Dystrophy Association, The Robert Packard Center for ALS Research Answer ALS Program, and the Virginia Gentleman Foundation.
Footnotes
Competing interests
A.N.C. and J.D.R. have submitted a patent application (US Patent Application Serial No. 63/111,882) regarding methods for inhibiting CHMP7 expression in neuronal cells for the treatment of neurodegenerative disorders.
References
- 1.Beck M & Hurt E The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol 18, 73–89 (2017). [DOI] [PubMed] [Google Scholar]; A thorough review of fundamental NPC biology.
- 2.Raices M & D’Angelo MA Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell Biol 13, 687–699 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Raices M & D’Angelo MA Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol 46, 26–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou CC et al. TDP43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci 21, 228–239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coyne AN et al. G(4)C(2) repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron 10.1016/j.neuron.2020.06.027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first study to document a specific NPC defect in neurodegeneration and how this nucleoporin defect can initiate NPC injury.
- 6.Eftekharzadeh B et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99, 925–940.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasset-Rosa F et al. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94, 48–57.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that nucleoporin and NPC-associated protein pathology can be associated with HD.
- 8.Giampetruzzi A et al. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun 10, 3827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grima JC et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron 94, 93–107.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang K et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided the first evidence that nucleocytoplasmic transport dysfunction might underlie C9orf72 ALS–FTD pathogenesis.
- 11.Lin YC et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat. Neurosci 10.1038/s41593-021-00859-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freibaum BD et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jovicic A et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci 18, 1226–1229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofweber M et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Guo L et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshizawa T et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basel-Vanagaite L et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann. Neurol 60, 214–222 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Nousiainen HO et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet 40, 155–157 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fichtman B et al. Pathogenic variants in NUP214 cause “plugged” nuclear pore channels and acute febrile encephalopathy. Am. J. Hum. Genet 10.1016/j.ajhg.2019.05.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shamseldin HE et al. NUP214 deficiency causes severe encephalopathy and microcephaly in humans. Hum. Genet 138, 221–229 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Lin DH & Hoelz A The structure of the nuclear pore complex (an update). Annu. Rev. Biochem 10.1146/annurev-biochem-062917-011901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT & Matunis MJ Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol 158, 915–927 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeGrasse JA et al. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol. Cell Proteom 8, 2119–2130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rout MP et al. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol 148, 635–651 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakhverdyan Z et al. Dissecting the structural dynamics of the nuclear pore complex. Mol. Cell 10.1016/j.molcel.2020.11.032 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ori A et al. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol 9, 648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajoo S, Vallotton P, Onischenko E & Weis K Stoichiometry and compositional plasticity of the yeast nuclear pore complex revealed by quantitative fluorescence microscopy. Proc. Natl Acad. Sci. USA 115, E3969–E3977 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinoshita Y, Kalir T, Dottino P & Kohtz DS Nuclear distributions of NUP62 and NUP214 suggest architectural diversity and spatial patterning among nuclear pore complexes. PLoS ONE 7, e36137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghavami A, van der Giessen E & Onck PR Energetics of transport through the nuclear pore complex. PLoS ONE 11, e0148876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paci G, Zheng T, Caria J, Zilman A & Lemke EA Molecular determinants of large cargo transport into the nucleus. eLife 10.7554/eLife.55963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moroianu J, Blobel G & Radu A Previously identified protein of uncertain function is karyopherin alpha and together with karyopherin beta docks import substrate at nuclear pore complexes. Proc. Natl Acad. Sci. USA 92, 2008–2011 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saitoh H, Cooke CA, Burgess WH, Earnshaw WC & Dasso M Direct and indirect association of the small GTPase ran with nuclear pore proteins and soluble transport factors: studies in Xenopus laevis egg extracts. Mol. Biol. Cell 7, 1319–1334 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moroianu J, Hijikata M, Blobel G & Radu A Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc. Natl Acad. Sci. USA 92, 6532–6536 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melchior F, Guan T, Yokoyama N, Nishimoto T & Gerace L GTP hydrolysis by Ran occurs at the nuclear pore complex in an early step of protein import. J. Cell Biol 131, 571–581 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melchior F, Paschal B, Evans J & Gerace L Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J. Cell Biol 123, 1649–1659 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore MS & Blobel G The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature 365, 661–663 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Moore MS & Blobel G Purification of a Ran-interacting protein that is required for protein import into the nucleus. Proc. Natl Acad. Sci. USA 91, 10212–10216 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moroianu J & Blobel G Protein export from the nucleus requires the GTPase Ran and GTP hydrolysis. Proc. Natl Acad. Sci. USA 92, 4318–4322 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bischoff FR & Ponstingl H Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 354, 80–82 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Klebe C, Bischoff FR, Ponstingl H & Wittinghofer A Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry 34, 639–647 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Knockenhauer KE & Schwartz TU The nuclear pore complex as a flexible and dynamic gate. Cell 164, 1162–1171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Goryaynov A & Yang W The selective permeability barrier in the nuclear pore complex. Nucleus 7, 430–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aksenova V et al. Nucleoporin TPR is an integral component of the TREX-2 mRNA export pathway. Nat. Commun 11, 4577 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee ES et al. TPR is required for the efficient nuclear export of mRNAs and lncRNAs from short and intron-poor genes. Nucleic Acids Res. 48, 11645–11663 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ullman KS, Shah S, Powers MA & Forbes DJ The nucleoporin nup153 plays a critical role in multiple types of nuclear export. Mol. Biol. Cell 10, 649–664 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li P & Noegel AA Inner nuclear envelope protein SUN1 plays a prominent role in mammalian mRNA export. Nucleic Acids Res. 43, 9874–9888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li P et al. The function of the inner nuclear envelope protein SUN1 in mRNA export is regulated by phosphorylation. Sci. Rep 7, 9157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bastos R, Lin A, Enarson M & Burke B Targeting and function in mRNA export of nuclear pore complex protein Nup153. J. Cell Biol 134, 1141–1156 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soop T et al. Nup153 affects entry of messenger and ribosomal ribonucleoproteins into the nuclear basket during export. Mol. Biol. Cell 16, 5610–5620 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makise M et al. The Nup153-Nup50 protein interface and its role in nuclear import. J. Biol. Chem 287, 38515–38522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindsay ME, Plafker K, Smith AE, Clurman BE & Macara IG Npap60/Nup50 is a tri-stable switch that stimulates importin-α:β-mediated nuclear protein import. Cell 110, 349–360 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Matsuura Y & Stewart M Nup50/Npap60 function in nuclear protein import complex disassembly and importin recycling. EMBO J. 24, 3681–3689 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guan T et al. Nup50, a nucleoplasmically oriented nucleoporin with a role in nuclear protein export. Mol. Cell. Biol 20, 5619–5630 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buchwalter AL, Liang Y & Hetzer MW Nup50 is required for cell differentiation and exhibits transcription-dependent dynamics. Mol. Biol. Cell 25, 2472–2484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kadota S et al. Nucleoporin 153 links nuclear pore complex to chromatin architecture by mediating CTCF and cohesin binding. Nat. Commun 11, 2606 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capelson M et al. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140, 372–383 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that nucleoporins can directly influence gene expression.
- 57.Gozalo A et al. Core components of the nuclear pore bind distinct states of chromatin and contribute to polycomb repression. Mol. Cell 77, 67–81.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pascual-Garcia P, Jeong J & Capelson M Nucleoporin Nup98 associates with Trx/MLL and NSL histone-modifying complexes and regulates Hox gene expression. Cell Rep. 9, 433–442 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Vaquerizas JM et al. Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet. 6, e1000846 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capelson M & Hetzer MW The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 10, 697–705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chatel G & Fahrenkrog B Dynamics and diverse functions of nuclear pore complex proteins. Nucleus 3, 162–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D’Angelo MA Nuclear pore complexes as hubs for gene regulation. Nucleus 10.1080/19491034.2017.1395542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dickmanns A, Kehlenbach RH & Fahrenkrog B Nuclear pore complexes and nucleocytoplasmic transport: from structure to function to disease. Int. Rev. Cell Mol. Biol 320, 171–233 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Hampoelz B, Andres-Pons A, Kastritis P & Beck M Structure and assembly of the nuclear pore complex. Annu. Rev. Biophys 10.1146/annurev-biophys-052118-115308 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Ibarra A & Hetzer MW Nuclear pore proteins and the control of genome functions. Genes Dev. 29, 337–349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pascual-Garcia P & Capelson M Nuclear pores in genome architecture and enhancer function. Curr. Opin. Cell Biol 58, 126–133 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masrori P & Van Damme P Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol 27, 1918–1929 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vahsen BF et al. Non-neuronal cells in amyotrophic lateral sclerosis – from pathogenesis to biomarkers. Nat. Rev. Neurol 17, 333–348 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Ringholz GM & Greene SR The relationship between amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Neurol. Neurosci. Rep 6, 387–392 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Wheaton MW et al. Cognitive impairment in familial ALS. Neurology 69, 1411–1417 (2007). [DOI] [PubMed] [Google Scholar]
- 71.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Renton AE et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwiatkowski TJ Jr et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Sreedharan J et al. TDP43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu CH et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andersen PM & Al-Chalabi A Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol 7, 603–615 (2011). [DOI] [PubMed] [Google Scholar]
- 77.Ferrari R, Kapogiannis D, Huey ED & Momeni P FTD and ALS: a tale of two diseases. Curr. Alzheimer Res 8, 273–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ling SC, Polymenidou M & Cleveland DW Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neumann M et al. Ubiquitinated TDP43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Hayes LR, Duan L, Bowen K, Kalab P & Rothstein JD C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 10.7554/eLife.51685 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence that C9orf72 dipeptide repeat proteins can influence NTR function.
- 81.Khosravi B et al. Cytoplasmic poly-GA aggregates impair nuclear import of TDP43 in C9orf72 ALS/FTLD. Hum. Mol. Genet 10.1093/hmg/ddw432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi KY et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl Acad. Sci. USA 114, E1111–E1117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boeynaems S et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep 6, 20877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frottin F, Pérez-Berlanga M, Hartl FU & Hipp MS Multiple pathways of toxicity induced by C9orf72 dipeptide repeat aggregates and G(4)C(2) RNA in a cellular model. eLife 10.7554/eLife.62718 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nanaura H et al. C9orf72-derived arginine-rich poly-dipeptides impede phase modifiers. Nat. Commun 12, 5301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vanneste J et al. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci. Rep 9, 15728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramic M et al. Epigenetic small molecules rescue nucleocytoplasmic transport and DNA damage phenotypes in C9ORF72 ALS/FTD. Brain Sci. 11, 1543 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Semmelink MFW, Steen A & Veenhoff LM Measuring and interpreting nuclear transport in neurodegenerative disease–the example of C9orf72 ALS. Int. J. Mol. Sci 10.3390/ijms22179217 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nishimura AL et al. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133, 1763–1771 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Zhang K et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 10.1016/j.cell.2018.03.025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saberi S et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 135, 459–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chew J et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP43 proteinopathy. Mol. Neurodegener 14, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang YJ et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci 19, 668–677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang YJ et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 10.1126/science.aav2606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook CN et al. C9orf72 poly(GR) aggregation induces TDP43 proteinopathy. Sci. Transl. Med 10.1126/scitranslmed.abb3774 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gasset-Rosa F et al. Cytoplasmic TDP43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP43, and cell death. Neuron 102, 339–357.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Coyne A et al. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP43 dysfunction in sporadic and familial ALS. Sci. Transl. Med 13, 10.1126/scitranslmed.abe1923 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to elucidate a mechanism of NPC injury in sporadic ALS, identify a cause of loss of nuclear TDP43 function in human neurons, and identify a candidate therapeutic strategy to repair NPC injury and subsequent TDP43 dysfunction.
- 98.Schermelleh L et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 320, 1332–1336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mizielinska S et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hallberg E, Wozniak RW & Blobel G An integral membrane protein of the pore membrane domain of the nuclear envelope contains a nucleoporin-like region. J. Cell Biol 122, 513–521 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stavru F, Nautrup-Pedersen G, Cordes VC & Gorlich D Nuclear pore complex assembly and maintenance in POM121-and gp210-deficient cells. J. Cell Biol 173, 477–483 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coyne AN & Rothstein JD The ESCRT-III protein VPS4, but not CHMP4B or CHMP2B, is pathologically increased in familial and sporadic ALS neuronal nuclei. Acta Neuropathol. Commun 9, 127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thaller DJ et al. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. eLife 10.7554/eLife.45284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toyama BH et al. Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J. Cell Biol 10.1083/jcb.201809123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Webster BM, Colombi P, Jager J & Lusk CP Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 159, 388–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified a role for the ESCRT-III pathway in NPC surveillance during cell division.
- 106.Webster BM et al. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 35, 2447–2467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crampton N, Kodiha M, Shrivastava S, Umar R & Stochaj U Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1. Mol. Biol. Cell 20, 5106–5116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schellenberg GD, Bird TD, Wijsman EM, Moore DK & Martin GM The genetics of Alzheimer’s disease. Biomed. Pharmacother 43, 463–468 (1989). [DOI] [PubMed] [Google Scholar]
- 109.Van Broeckhoven C Presenilins and Alzheimer disease. Nat. Genet 11, 230–232 (1995). [DOI] [PubMed] [Google Scholar]
- 110.Wilhelmsen KC, Clark LN, Miller BL & Geschwind DH Tau mutations in frontotemporal dementia. Dement. Geriatr. Cogn. Disord 10, 88–92 (1999). [DOI] [PubMed] [Google Scholar]
- 111.Baker M et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 (2006). [DOI] [PubMed] [Google Scholar]
- 112.Cruts M et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 (2006). [DOI] [PubMed] [Google Scholar]
- 113.DeTure MA & Dickson DW The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Karch CM, Cruchaga C & Goate AM Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Masters CL et al. Alzheimer’s disease. Nat. Rev. Dis. Prim 1, 15056 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Olszewska DA, Lonergan R, Fallon EM & Lynch T Genetics of frontotemporal dementia. Curr. Neurol. Neurosci. Rep 16, 107 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Amador-Ortiz C et al. TDP43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol 61, 435–445 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grundke-Iqbal I et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl Acad. Sci. USA 83, 4913–4917 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Josephs KA et al. Abnormal TDP43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70, 1850–1857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kosik KS, Joachim CL & Selkoe DJ Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl Acad. Sci. USA 83, 4044–4048 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wood JG, Mirra SS, Pollock NJ & Binder LI Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl Acad. Sci. USA 83, 4040–4043 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Metuzals J, Robitaille Y, Houghton S, Gauthier S & Leblanc R Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J. Neurocytol 17, 827–833 (1988). [DOI] [PubMed] [Google Scholar]
- 123.Paonessa F et al. Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 26, 582–593.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence of nuclear lamina invaginations resulting from tau mutations in human neurons.
- 124.Cornelison GL, Levy SA, Jenson T & Frost B Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell 18, e12847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Frost B, Bardai FH & Feany MB Lamin dysfunction mediates neurodegeneration in tauopathies. Curr. Biol 26, 129–136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gruenbaum Y et al. The nuclear lamina and its functions in the nucleus. Int. Rev. Cytol 226, 1–62 (2003). [DOI] [PubMed] [Google Scholar]
- 127.Jahed Z, Soheilypour M, Peyro M & Mofrad MR The LINC and NPC relationship – it’s complicated! J. Cell Sci 129, 3219–3229 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Frost B, Hemberg M, Lewis J & Feany MB Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci 17, 357–366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McColgan P & Tabrizi SJ Huntington’s disease: a clinical review. Eur. J. Neurol 25, 24–34 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Ross CA & Tabrizi SJ Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Bañez-Coronel M et al. RAN translation in Huntington disease. Neuron 88, 667–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Woerner AC et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176 (2016). [DOI] [PubMed] [Google Scholar]; This study provides evidence that cytoplasmic aggregation can impair nucleocytoplasmic transport.
- 133.Lord SJ, Velle KB, Mullins RD & Fritz-Laylin LK SuperPlots: communicating reproducibility and variability in cell biology. J. Cell Biol 10.1083/jcb.202001064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Coyne AN & Rothstein JD Nuclear lamina invaginations are not a pathological feature of C9orf72 ALS/FTD. Acta Neuropathol. Commun 9, 45 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Murphy R & Wente SR An RNA-export mediator with an essential nuclear export signal. Nature 383, 357–360 (1996). [DOI] [PubMed] [Google Scholar]
- 136.Bischoff FR, Klebe C, Kretschmer J, Wittinghofer A & Ponstingl H RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc. Natl Acad. Sci. USA 91, 2587–2591 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bischoff FR, Krebber H, Kempf T, Hermes I & Ponstingl H Human RanGTPase-activating protein RanGAP1 is a homologue of yeast Rna1p involved in mRNA processing and transport. Proc. Natl Acad. Sci. USA 92, 1749–1753 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ben-Efraim I & Gerace L Gradient of increasing affinity of importin β for nucleoporins along the pathway of nuclear import. J. Cell Biol 152, 411–417 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bernad R, van der Velde H, Fornerod M & Pickersgill H Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol. Cell. Biol 24, 2373–2384 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fornerod M et al. The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO J. 16, 807–816 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hosp F et al. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 11, 1134–1146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Juhlen R & Fahrenkrog B Moonlighting nuclear pore proteins: tissue-specific nucleoporin function in health and disease. Histochem. Cell Biol 10.1007/s00418-018-1748-8 (2018). [DOI] [PubMed] [Google Scholar]; A review of nucleoporin mutations in cell-type-specific diseases, including non-neurological diseases.
- 143.Nofrini V, Di Giacomo D & Mecucci C Nucleoporin genes in human diseases. Eur. J. Hum. Genet 24, 1388–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bolger TA & Wente SR Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J. Biol. Chem 286, 39750–39759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weirich CS et al. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat. Cell Biol 8, 668–676 (2006). [DOI] [PubMed] [Google Scholar]
- 146.Folkmann AW et al. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 155, 582–593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stiekema M, van Zandvoort M, Ramaekers FCS & Broers JLV Structural and mechanical aberrations of the nuclear lamina in disease. Cells 10.3390/cells9081884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bonne G et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet 21, 285–288 (1999). [DOI] [PubMed] [Google Scholar]
- 149.De Sandre-Giovannoli A et al. Lamin A truncation in Hutchinson-Gilford progeria. Science 300, 2055 (2003). [DOI] [PubMed] [Google Scholar]
- 150.De Sandre-Giovannoli A et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet 70, 726–736 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Padiath QS et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet 38, 1114–1123 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Kang SM, Yoon MH & Park BJ Laminopathies; mutations on single gene and various human genetic diseases. BMB Rep. 51, 327–337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marcelot A, Worman HJ & Zinn-Justin S Protein structural and mechanistic basis of progeroid laminopathies. FEBS J. 288, 2757–2772 (2021). [DOI] [PubMed] [Google Scholar]
- 154.D’Angelo MA, Raices M, Panowski SH & Hetzer MW Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136, 284–295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examined the contribution of ageing to nucleocytoplasmic transport dysfunction.
- 155.Savas JN, Toyama BH, Xu T, Yates JR III & Hetzer MW Extremely long-lived nuclear pore proteins in the rat brain. Science 335, 942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified a subset of nucleoporins as some of the longest-lived proteins.
- 156.Toyama BH et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rabut G, Doye V & Ellenberg J Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol 6, 1114–1121 (2004). [DOI] [PubMed] [Google Scholar]
- 158.Fifková E, Tonks M & Cullen-Dockstader K Changes in the nuclear pore complexes of the dentate granule cells in aged rats. Exp. Neurol 95, 755–762 (1987). [DOI] [PubMed] [Google Scholar]
- 159.Topple A, Smith G, Fifkova E & Cullen-Dockstader K Nuclear pore complex frequency in CA1 pyramidal cells of the aging rat. Mech. Ageing Dev 51, 33–39 (1990). [DOI] [PubMed] [Google Scholar]
- 160.Mertens J et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pujol G, Soderqvist H & Radu A Age-associated reduction of nuclear protein import in human fibroblasts. Biochem. Biophys. Res. Commun 294, 354–358 (2002). [DOI] [PubMed] [Google Scholar]
- 162.Robijns J, Houthaeve G, Braeckmans K & De Vos WH Loss of nuclear envelope integrity in aging and disease. Int. Rev. Cell Mol. Biol 336, 205–222 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Lusk CP & King MC The nucleus: keeping it together by keeping it apart. Curr. Opin. Cell Biol 44, 44–50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hatch EM & Hetzer MW Nuclear envelope rupture is induced by actin-based nucleus confinement. J. Cell Biol 215, 27–36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Adli M The CRISPR tool kit for genome editing and beyond. Nat. Commun 9, 1911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khadempar S et al. CRISPR-Cas9 in genome editing: Its function and medical applications. J. Cell Physiol 234, 5751–5761 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Ababneh NA et al. Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet 10.1093/hmg/ddaa106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.DeVos SL & Miller TM Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics 10, 486–497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schoch KM & Miller TM Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Donnelly CJ et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lagier-Tourenne C et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl Acad. Sci. USA 110, E4530–E4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03626012 (2022). [DOI] [PubMed]
- 173.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03945279 (2021). [DOI] [PubMed]
- 174.Franks TM et al. Evolution of a transcriptional regulator from a transmembrane nucleoporin. Genes Dev. 30, 1155–1171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee EB, Lee VM & Trojanowski JQ Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci 13, 38–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Agote-Aran A et al. Spatial control of nucleoporin condensation by fragile X-related proteins. EMBO J. 39, e104467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Krull I et al. Two patients with an identical novel mutation in the AAAS gene and similar phenotype of triple A (Allgrove) syndrome. Exp. Clin. Endocrinol. Diabetes 118, 530–536 (2010). [DOI] [PubMed] [Google Scholar]
- 178.Milenkovic T et al. Triple A syndrome: 32 years experience of a single centre (1977–2008). Eur. J. Pediatr 169, 1323–1328 (2010). [DOI] [PubMed] [Google Scholar]
- 179.Vallet AE et al. Neurological features in adult triple-A (Allgrove) syndrome. J. Neurol 259, 39–46 (2012). [DOI] [PubMed] [Google Scholar]
- 180.Cronshaw JM & Matunis MJ The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc. Natl Acad. Sci. USA 100, 5823–5827 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bonnin E et al. Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet. 14, e1007845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Miyake N et al. Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am. J. Hum.Genet 97, 555–566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rosti RO et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J. Med. Genet 54, 399–403 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Zhang X et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135, 1017–1027 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Neilson DE et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am. J. Hum. Genet 84, 44–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Melià MJ et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 136, 1508–1517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Van Bergen NJ et al. Pathogenic variants in nucleoporin TPR (translocated promoter region, nuclear basket protein) cause severe intellectual disability in humans. Hum. Mol. Genet 10.1093/hmg/ddab248 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Strambio-De-Castillia C, Niepel M & Rout M The nuclear pore complex: bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol 11, 490–501 (2010). [DOI] [PubMed] [Google Scholar]