

ABSTRACT
The etiology of colorectal cancer (CRC) is influenced by bacterial communities that colonize the gastrointestinal tract. These microorganisms derive essential nutrients from indigestible dietary or host-derived compounds and activate molecular signaling pathways necessary for normal tissue and immune function. Associative and mechanistic studies have identified bacterial species whose presence may increase CRC risk, including notable examples such as Fusobacterium nucleatum, Enterotoxigenic Bacteroides fragilis, and pks+ E. coli. In recent years this work has expanded in scope to include aspects of host mutational status, intra-tumoral microbial heterogeneity, transient infection, and the cumulative influence of multiple carcinogenic bacteria after sequential or co-colonization. In this review, we will provide an updated overview of how host-bacteria interactions influence CRC development, how this knowledge may be utilized to diagnose or prevent CRC, and how the gut microbiome influences CRC treatment efficacy.
KEYWORDS: Colorectal cancer, microbiome, genotoxin, inflammation, probiotics, immunotherapy
Introduction
The human colon is colonized by a symbiotic community of microbes containing an estimated 1014 bacteria, similar to the total number of mammalian cells.1 Under healthy conditions, these commensal bacteria promote intestinal homeostasis by facilitating digestion, metabolic outputs, immune tolerance, and epithelial maturation or function. The advent of next-generation sequencing has allowed researchers to catalog how microbial community composition and function change during the progression of various gastrointestinal diseases, including colorectal cancer (CRC). Broadly, these studies have shown that there are specific bacterial taxa that are associated with protection against or promotion of CRC.2–4 For example, some bacteria potentially introduce cancer-initiating mutations by producing microbial genotoxins5 while others produce metabolites that interfere with core metabolic processes in cancer cells.6 Moreover, the bacterial species present within the intestinal tract alter local and systemic cellular or metabolic profiles to influence treatment efficacy.7 The wide impact of microbiota on tumorigenesis has resulted in its incorporation in the hallmarks of cancer.8 Such beneficial or deleterious host-microbe interactions are influenced by a complex metabolic environment, other microbial community members, host mutational status, immune landscapes, and single-cell heterogeneity. In this review, we will summarize the current evidence for several proposed carcinogenic bacteria in CRC, with specific consideration for how these cancer-promoting strains operate in various tumor developmental and mutational contexts. Then we will consider how polymorphic communities or species may find application in the clinic as tools for CRC diagnosis, prevention, and treatment modulation.
Colorectal cancer etiology
An overwhelming majority of CRC cases (~80-85%) are not driven by hereditary mutation, a phenomenon termed sporadic CRC, suggesting that environmental determinants such as lifestyle, diet and microbial community trigger tumorigenesis. Sporadic CRC most commonly occurs after the acquisition of mutations in the adenomatous polyposis coli (APC) gene triggers the cascade of events leading to CRC.9 APC acts as a negative regulator of WNT/β-Catenin, a proliferative signaling pathway whose upregulation is associated with cancer development. Subsequent mutations in other tumor suppressor genes, such as tumor protein 53 (TP53) and Kirsten rat sarcoma viral oncogene homolog (KRAS), further promote malignant transformation.10 In approximately 15% of CRC patients, a heritable mutation or epigenetic silencing of mismatch repair genes (mismatch repair deficiency, MMRd) results in a disproportionate number of mutations in repetitive DNA satellites called microsatellite instability (MSI-high), that may increase the likelihood of CRC driver mutations.11 Alternatively, chronic inflammation can drive dysplastic transformation giving rise to colitis-associated cancer (CAC) that has been recently reviewed elsewhere.12 While the same driver mutations are implicated in CAC, the frequency and timing of these mutations is different, with mutations in TP53 typically preceding APC.12 Regardless of etiology, most CRC cells are characterized by the activation of pro-survival (e.g. nuclear factor-κB; NF-κB), proliferative (e.g. WNT/β-Catenin), or immunogenic (e.g. signal transducer and activator of transcription 3; STAT3) pathways.13–15 Most studies aimed at determining how bacteria may promote CRC utilize susceptible animal models generated to recapitulate these changes. These can include mice with germline mutations in Apc (ApcMin/+) or the addition of exogenous mutagens (azoxymethane; AOM) coupled with additional mutations in immunosuppressive genes (e.g. interleukin-10; Il-10) or the addition of inflammatory chemicals (dextran-sodium sulfate; DSS) to mimic CAC. While this discussion provides an overview of the models commonly discussed in this review, this description is by no means exhaustive. For a more detailed overview, readers should refer to prior reviews.16,17
Proposed carcinogenic bacteria
It is now well established that intestinal bacteria influence the intestinal homeostasis, which has led to the theory that these microbes may act as an environmental trigger for CRC. Bacteria may promote CRC through diverse mechanisms, and we have likely only characterized a small portion of bacteria that might modulate cancer risk. The identification of novel carcinogenic bacteria may come from a better understanding of the consequences of pathogenic infection, or perhaps more intriguingly, from the discovery of novel cancer-promoting activity of commensal microbes. For example, Cao et al.18 recently identified 18 commensal strains from patients with inflammatory bowel disease that produce DNA damaging small molecules. One strain investigated in depth, Morganella morganii, produces a novel genotoxic indolimine through a previously uncharacterized biosynthetic pathway, and promoted tumor formation in a chemically induced model of colitis associated cancer.18 Another layer of complexity arises when considering the cumulative bacterial exposure of an individual over a lifetime, during which microbial community composition and activity may fluctuate. When considering this scenario, an individual’s risk of developing CRC may be influenced by a series of successive microbial “hits”. Such a concept is supported by differential bacterial colonization and metabolic outputs across CRC staging.19 While each exposure may not cause cancer by itself, the cumulative effect of multiple encounters with cancer-promoting bacteria may outweigh the sum of its parts. The processes by which these microbes are thought to promote CRC risk are highly diverse, encompassing changes in genomic integrity, oncogenic signaling, cellular migration, inflammatory states, and epigenetic changes, among others (Figure 1). In this section, we will review these mechanisms in detail, highlighting key proposed carcinogenic species that may promote CRC development.
Figure 1.

Carcinogenic mechanisms of intestinal bacteria. a) pks+ Escherichia coli produce a genotoxin known as colibactin, that induces interstrand crosslinks in host cells resulting in a defined mutational signature detected in colorectal cancer (CRC) genomes. The right panel shows several microbial genotoxins with nonspecific DNA degrading activity, including the cytolethal distending toxin (CDT) found in the human enteric pathogen Campylobacter jejuni, UshA in the murine bacteria Citrobacter rodentium, and indolimines isolated from a commenstal strain of Morganella morganii obtained from patients with inflammatory bowel disease. b) Enterotoxigenic bacteroides fragilis (ETBF) secretes a toxin known as the bacteroides fragilis toxin (BFT), that degrades E-cadherin promoting nuclear translocation of β-Catenin and the activation of proliferative signaling pathways to promote tumor formation. Fusobacterium nucleatum produces a membrane-bound Fusobacterium adhesin A (FadA) protein that binds to E-cadherin to upregulate expression of the Annexin A1/β-Catenin complex to activate proliferative signaling pathways. AvrA is a virulence factor produced by Salmonella spp. promoting epithelial adherence and persistent colonization in the gastrointestinal tract, while simultaneously activating AKT serine/threonine kinase 1 (AKT)-mediated β-Catenin phosphorylation, facilitating nuclear translocation and the activation of signaling pathways to promote proliferation and cell survival. Peptostreptococcus anaerobius are selectively enriched in CRC tissue, facilitated in part by the binding of an outer membrane protein putative cell wall binding repeat 2 (PCWBR2) to α2/β1 integrins overexpressed in cancer cells. This interaction promotes phosphoinositide 3-kinase (P13K) and AKT phosphorylation to promote cell proliferation. c) A superoxide producing strain (OG1RF) of the human pathogen Enterococcus faecalis causes chromosomal instability after infection in cell lines and intestinal ligation models, resulting from the production of reactive oxygen species (ROS). Alternatively, OG1RF infected macrophages elicit similar effects in human cell lines. On the right, infection with ETBF upregulates expression of a spermine oxidase that generates ROS and DNA damage in colonic epithelial cells. d) F. nucleatum produces short-chain fatty acids (SCFA) that bind to Free Fatty Acid Receptor 2 (Ffar2) receptors on T helper 17 (Th17) macrophages or an undetermined intermediate dendritic cell to stimulate interleukin 17 (IL-17) production. The human pathogens Enterotoxigenic Bacteroides fragilis (ETBF) and Clostridioides difficile (C. difficile) secrete toxins (the Bacteroides fragilis toxin, BFT, and Clostridioides difficile toxin B, TcdB, respectively) that promote signal transducer and activator of transcription 3 (STAT3) phosphorylation and the recruitment of IL-17 producing Th17 cells. In both cases, these microbes promote IL-17 mediated inflammation that contributes to neoplastic transformation. e) ETBF infection promotes hypermethylation in ApcMin/+BRAFV600ELgr5Cre mice, resulting in the formation of proximal tumors and activation of IFNγ gene signatures. F. nucleatum infection downregulates methyltransferase 3 (METTL3) expression in a Yes1 associated transcriptional regulator (YAP)-dependent manner to inhibit m6A RNA methylation, altering mRNA translation to promote cancer metastasis.
Pks+ Escherichia coli
Certain strains of Escherichia coli (E. coli) carry a 54-kb biosynthetic gene cluster (BGC) found primarily in the B2-phylogroup called polyketide synthase (“pks” or “clb”) that encodes for a secondary metabolite named colibactin that causes DNA damage in mammalian cells.20,21 While E. coli commonly exists as a commensal constituent of the human microbiome,22 epidemiological evidence suggests that pks+ E. coli are more prevalent in stool or tissue samples CRC patients than those with inflammatory bowel disease (IBD) or healthy controls.23–27 Administration of pks+ E. coli in pre-clinical models of colitis-associated cancer25,28,29 promotes tumor formation with a concomitant increase in a phosphorylated histone variant associated with DNA damage (γH2AX) attenuated by the deletion of pks genes, suggesting that these bacteria promote tumor development mediated by the genotoxic effects of colibactin. Because somatic mutations may result from DNA damage, genotoxic agents are commonly implicated as sources of oncogenic transformation, and a case can be made for pks+ E. coli in this process. Chronic exposure of human organoids to pks+ E. coli generates a unique transcriptional signature characterized by single-base substitutions (SBS) or insertion-deletions (ID) within AT-rich DNA motifs, and similar signatures are identified in approximately 5% of metastatic tumors of predominately CRC-derived primary sites.5 This mutagenic activity is consistent with colibactin’s proposed structure characterized by two cyclopropane warheads that form interstrand DNA crosslinks (ICLs) by adenosine addition,30,31 linked by an unstable α-aminoketone susceptible to nucleophilic cleavage.32,33 Under mild conditions, an estimated >98% of the molecule is lost to 1,2-diketone degradation during isolation.34 This rapid degradation may explain why the genotoxic effects of pks+ E. coli require cell-to-cell contact21 and can be exacerbated by the addition of a mucolytic agent to cell culture35 or co-inoculation of ApcMin/+ mice with biofilm-initiating enterotoxigenic Bacteroides fragilis (ETBF) that facilitate deeper mucosal invasion.23,36
While spatial pks+ E. coli distribution in the gut may influence the carcinogenic effects of colibactin, environmental regulation of pks genes and thus colibactin biosynthesis likely also plays a role.37 Genetic studies indicate that all genes involved in colibactin production are required for pks-associated genotoxicity,21 and multiple studies have shown that pks gene expression is influenced by inflammation,28,29,38 iron availability,39–41 microbial metabolites,42 oxygen availability43 and dietary oligosaccharides.44 These findings suggest that modifiable risk factors, such as diet, may influence the likelihood of individuals colonized by pks+ E. coli developing CRC. This hypothesis is supported by a recent study quantifying cancer risk associated with a western diet in patients with positive pks gene detection in formaldehyde-fixed paraffin-embedded samples gathered from 1175 patient records with dietary history.45 While a Western diet (i.e. high red meat and low dietary fiber consumption) only weakly correlated with CRC in a large cohort of male and female health professionals (Health Professionals Follow-up Study/Nurses Health Study, n = 134,775), the Western diet hazard ratio was significantly increased in patients with high microbial clbB expression.45 Associations such as this may explain why not all individuals colonized by pks+ E. coli develop CRC, despite the implication of colibactin as a carcinogenic metabolite.
Related to these findings, it remains unclear if colibactin’s role in CRC etiology is primarily related to tumor initiation, progression, or both. Several lines of evidence suggest that pks+ E. coli promote tumor-initiating mutagenesis. First, the tumorigenic phenotype observed in pre-clinical models is primarily characterized by an increase in tumor number rather than an increase in tumor size.25 Additionally, in CRC cases harboring the pks+ E. coli mutational signature, the highest proportion of mutations matching the pks motif (AAWWTT) occur within the APC gene (5.3%),5 the most commonly mutated gene in CRC patients. Finally, the evidence suggests that these mutational signatures are acquired during childhood46 and that pks+ E. coli may be acquired as early as the first month of life via mother-to-infant transmission.47,48 On the other hand, the prevalence of pks-associated mutational signatures is much lower (~5%) than the observed number of CRC patients colonized by pks+ E. coli (~55-60%), suggesting these bacteria may promote tumor progression through a non-mutagenic mechanism. For example, pks+ E. coli infection promotes a pro-tumorigenic microenvironment by generating a senescence-associated secretory phenotype in bystander cells,49,50 and reduces the number of cytotoxic T cells within the invasive margins of CRC tumor biopsies.51 Finally, while some studies have shown an increased prevalence of pks+ E. coli in normal tissue and early adenomas, a recent study found that pks genes were enriched only in stage IV tumors in a French cohort.52 Collectively, these studies implicate pks+ E. coli in CRC, but more work is required to fully understand the mechanistic role of colibactin itself in the multifaceted aspects of tumor initiation and cancer progression.
Enterotoxigenic bacteroides fragilis
Enterotoxigenic Bacteroides fragilis (ETBF) strains produce a ~ 20 kDa toxin termed the B. fragilis toxin (bft), a zinc metalloprotease associated with intestinal disease and persistent colitis in humans.53–55 Moreover, several epidemiological studies have demonstrated a higher incidence of ETBF infection in CRC patients,23,52,56,57 suggesting bft may play a causative role in CRC. Pre-clinical studies utilizing the ApcMin/+ mouse model have demonstrated that colonization with ETBF promotes colitis and the formation of distal colon tumors.55,58,59 In this model, tumor formation is inextricably linked to the immune system, dependent on the recruitment of T helper 17 (Th17) and CXC motif chemokine receptor 2+ (CXCR2+) myeloid derived suppressor cells driven by the activation of epithelial NF-κB/STAT3/interleukin 17 (IL-17) signaling cascades.55,58,59 In this context ETBF-associated tumorigenesis can be abrogated by IL-17 blockade or gene knockout,55,58 regulatory T cell (Treg) depletion,59 or epithelial STAT3 deletion.55 Additionally, bft intoxication directly activates transcriptional pathways associated with proliferation and stemness typical of multiple cancers. After binding to intestinal epithelial cells, bft facilitates E-cadherin degradation to activate WNT/β-Catenin signaling pathways to directly increase cell proliferation.60 Collectively, this data suggests that ETBF-mediated tumors are the result of multi-faceted pro-neoplastic changes to the epithelial microenvironment and cell-intrinsic processes.
DNA hypermethylation a hallmark of CpG methylator phenotype (CIMP) CRC cases.61 Recent evidence suggests that ETBF infection can alter epigenetic profiles in CRC cells, and that these changes may promote cancer cell growth. In murine organoids, ETBF infection upregulates expression of the JmjC-domain containing histone demethylase 2B (JMJD2B) that decreases Histone H3 Lysine 9 (H3K9me3) methylation levels in the Nanog homeobox promoter, a core regulator of embryonic stem cell proliferation.62 Consequently, these changes promote the preservation of a stem-like phenotype in organoids and promote tumor growth in HCT 116 xenografts.62 Although btf does not have direct genotoxic activity, several lines of evidence suggest that the epigenetic changes induced by ETBF infection may also promote the accumulation of DNA damage. In HT29 cells, 24-hour treatment with purified bft generates specific “bft-open” chromatin regions with enhanced gene expression.63 In a mouse model of CIMP (ApcMin/+BRAFV600ELgr5Cre; BLM) ETBF colonization promoted tumor formation in the mid-proximal colon mimicking proximal tumors observed in CRC patients with BRAFV600E mutation, in contrast to the distal tumors canonically observed in ApcMin/+-ETBF mice.64 Moreover, ETBF colonization enhanced hypermethylation in BLM-ETBF tumors relative to distal ApcMin/+-ETBF tumors, with concomitant increase in interferon gamma (IFNγ)/STAT3/NF-κB signaling pathways.64 Notably, both BLM and ApcMin/+ mice develop few spontaneous tumors, suggesting that ETBF can act as a microenvironmental trigger for CRC in multiple mutational contexts. Interestingly, in these cases hypermethylation was associated with a higher amount of DNA damage; a slightly higher single nucleotide variant (SNV) rate in bft-open chromatin,63 or a significantly higher number of γH2AX foci in BLM-ETBF tissue relative to ApcMin/+-ETBF tissue.64 These findings suggest ETBF infection may alter chromatin accessibility and indirectly facilitate DNA damage in CIMP CRCs. The genotoxic mechanism for such observations remains unclear, but they may be attributed to the generation of reactive oxygen species (ROS) mediated by bft. Purified bft upregulates the polyamine catabolic enzyme spermine oxidase to increase ROS production and subsequent γH2AX phosphorylation in HT29 cells, while administration of a polyamine catabolism inhibitor reduces ETBF-induced tumorigenesis in the ApcMin/+ mouse model.65 Similarly, ETBF-induced tumors from a mismatch repair deficient mouse combining mutations in Apc and MutS homolog 2 (ApcMin/+Msh2fl/flVillinCre) exhibit a higher mutational burden but no distinct signature, suggesting that ETBF colonization increases mutations attributed to deficient DNA-repair randomly throughout the genome, and consequently may increase the likelihood of a second truncating Apc mutation.66 These findings collectively reinforce the idea that ETBF colonization may generate epigenetic or genomic changes that, in specific contexts, may influence CRC etiology.
Given the high prevalence of ETBF in CRC cases, it is reasonable to consider their application as a CRC biomarker. The efficacy of such an approach is influenced by multiple factors, including the sampling method, reliability of detection, and the stage at which these bacteria can be detected. In most studies using tissue biopsies and assessing ETBF prevalence by quantitative real-time PCR (qPCR), ETBF abundance is positively correlated with tumor stage56,57,62,67 or equally enriched across tumor stage.68 However, in a recent study directly comparing the prognostic value of ETBF to four other CRC-associated bacteria in a French cohort, ETBF was the only species differentially enriched in fresh fecal samples from patients with early adenomas assessed by qPCR.52 These findings suggest ETBF detection in fresh stool may facilitate early detection of individuals with an increased risk of developing invasive disease or identify a subset of patients with early adenomas more efficiently than invasive colonoscopy-based methods.
Fusobacterium nucleatum
Fusobacterium nucleatum (F. nucleatum) is commonly identified as a potential microbial carcinogen, a designation attributed to its frequent enrichment in CRC tumor tissue69–71 and a plethora of studies demonstrating that F. nucleatum colonization promotes tumor growth in ApcMin/+ mice.19,72–74 The mechanisms underlying F. nucleatum-associated tumorigenesis have been reviewed thoroughly elsewhere,71 and include: the production of virulence factors such as Fusobacterium adhesin A (FadA) and fibroblast activation protein 2 (Fap2) that facilitate adhesion or colonization,73,75 activation of β-Catenin signaling pathways in cancer cells that promote tumor proliferation,76,77 the generation of immunosuppressive microenvironments that restrict anti-tumor immunity,72,78 and the promotion of colitis-associated cancer via an IL-17 or myeloid dependent mechanism.73,74 In addition to promoting the proliferation of primary cancer cells, several studies suggest colonization of CRC tissue by F. nucleatum also promotes metastasis.79–83 Enhanced metastatic capabilities may be attributed to exosomes secreted by infected CRC cells containing microRNAs (miRNAs) or cytokines that simultaneously reduce macrophage tumor infiltration and enhanced proliferative signaling cascades, and the production of these molecules correlates with F. nucleatum abundance in CRC patients.79,80 Furthermore, F. nucleatum infection may promote migratory phenotypes by altering epigenetic profiles via non-coding RNA81 or methyltransferase activity.83 However, an open and important question is whether F. nucleatum acts as an initiator of genomic or environmental remodeling to promote carcinogenesis,84 or if these bacteria are selectively enriched in neoplastic lesions and subsequently enhance malignant processes while supplanting tumor-initiating species.85 Epidemiological evidence suggests F. nucleatum are preferentially enriched in late-stage CRC tissue, an observation that may support their role as a tumor-potentiating bacterium, after initiation by another hereditary or environmental event. For example, Kostic et al.72 show that relative F. nucleatum abundance is significantly higher in stool samples from CRC patients with carcinoma relative to adenoma as assessed by qPCR analysis. Multiple studies have found a similar significant positive correlation between F. nucleatum abundance and CRC stage.19,80 Consistent with this hypothesis, F. nucleatum does not stably colonize the intestinal tract after oral administration in SPF mice, requiring daily gavage at a relatively high level (108–109 colony forming units, CFU72,86,87), but readily colonizes tumor tissue in an orthotopic CRC model after intravenous injection.75,88 This model of colonization is consistent with the theory that CRC-colonizing F. nucleatum strains originate in the oral cavity and translocate to intestinal tumor tissue after damage to the oral-intestinal barrier.71,89 Moreover, F. nucleatum-positive patient-derived xenografts (PDX) maintained F. nucleatum positivity for 29 weeks and eight sequential murine passages,90 highlighting the persistence of this bacteria in CRC tumors. In this study, F. nucleatum maintained a PDX growth-enhancing effect demonstrated by a reduction in tumor growth after metronidazole treatment.90 In contrast, colonization of germ-free mice with a mixture of six fadA+, fap2− or + F. nucleatum CRC clinical isolates did not promote tumor formation in ApcMin/+ mice despite persistent F. nucleatum colonization quantified by fecal CFU.91 In a similar study no tumorigenic effect was observed after colonization of germ-free ApcMin/+ mice with five unique CRC-derived F. nucleatum isolates after weekly gavage, despite successful colonization by four of the five strains.92 Collectively, this evidence supports a paradigm in which malignant transformation of colonic epithelial cells facilitates higher levels of F. nucleatum colonization.
Beyond these tumor-promoting effects, F. nucleatum colonization may influence clinical decision-making via its association with specific CRC subtypes or prognoses. Multiple studies have demonstrated that F. nucleatum abundance is enriched in premalignant lesions from MSI-high patients, CIMP patients, and patients presenting with sessile serrated adenomas.93–96 Moreover, F. nucleatum abundance is predictive of shorter survival in CRC patients.96 In such cases, simple clinical interventions aimed at reducing F. nucleatum abundance or inhibiting the production of virulence factors may be warranted. One practical approach may be the addition of an aspirin regimen, as aspirin (1–2.5 mM) inhibits F. nucleatum FadA/Fap2 expression, and the addition of a physiologically relevant dose of aspirin (200 ppm) to mouse chow inhibited F. nucleatum-induced tumorigenesis after daily gavage in ApcMin/+ mice.97 These findings suggest that F. nucleatum may be a consistent biomarker for late-stage disease or premalignant lesions in a subset of CRC patients, and that therapeutic interventions aimed at reducing F. nucleatum abundance may improve patient outcomes.
Enteric pathogens
The advent of large-scale metagenomic studies has ushered in a new age of microbiome research and resulted in the identification of candidate oncogenic bacteria associated with CRC. These approaches necessitate the species be present at the time of (or shortly preceding) diagnosis, and thus may inadvertently miss biologically relevant species that transiently promote cancer initiation at an earlier time point. Furthermore, such techniques may not detect significant changes in low-abundance microbiota. As a result, many bacterial species implicated as a risk factor for CRC are commensal species that persistently colonize the intestinal tract, while the effects of recurrent or transient infection with intestinal pathogens on CRC risk remains unclear. Pathogen infection may cause intestinal inflammation. Given that inflammation is a well-established CRC risk factor it is possible that such infections increase an individual’s risk for developing CRC. A summary of proposed CRC-associated enteric pathogens and their proposed carcinogenic mechanisms is given in Table 1.
Table 1.
Enteric pathogens implicated in CRC.
| Bacterium | Virulence Factors | Experimental/Epidemiological Evidence | Proposed Mechanism | Reference |
|---|---|---|---|---|
| Salmonella spp. | AvrA FliC |
Increased incidence ratio and FliC-reactive antibodies in Dutch cohorts. Increased CRC risk after non-Enteritis or Typhimurium infections. | AvrA mediated colitis, activation of WNT/β-Catenin and STAT3 signaling pathways. | 98,99,100,101,102,103 |
| Pathogenic Escherichia coli spp. |
Cif Cnf Cdt |
EPEC detected in 55.9% of CRC patient biopsies, compared to 20.6% in healthy patient biopsies. Cif (7.9%), Cnf (36.7%), Cdt (8.2%) of CRC biopsies. | Activation of inflammatory signaling cascades, Cancer cell detachment and survival | 26, 27, 104,105,106,107,108,109,110,111,112,113 |
| Clostridium difficile | TcdB | Experimental validation of a carcinogenic strain from biofilm-positive fecal slurries | Increased WNT/β-Catenin signaling, pro-carcinogenic IL-17 mediated immune response | 114 |
| Klebsiella pneumoniae | Clb? | Experimental validation of tumorigenesis after infection in colitis-associated cancer models; mechanism unclear | Carcinogenic activity modulated by microbiome status | 117 |
| Citrobacter rodentium | UshA | Experimental validation, identification of UshA-induced mutational signature in CRC cases | DNA damage and mutagenesis after transient infection | 118 |
| Campylobacter jejuni | Cdt | Experimental validation of tumorigenesis after infection with a CRC clinical isolate | DNA damage | 121 |
Abbreviations: colorectal cancer, (CRC); avirulence A, AvrA; flagellar structural protein, FliC; cycle inhibiting factor, Cif; cytotoxic necrotizing factor 1, Cnf1; cytolethal distending toxin, Cdt; Clostridium difficile Toxin B, TcdB; colibactin, Clb; UDP-sugar hydrolase, UshA; signal transducer and activator of transcription 3, STAT3; interleukin 17, IL-17.
The most thorough epidemiological evidence in this context comes from several studies assessing CRC occurrence in large cohorts of patients in the Netherlands after Salmonella infection. In one example, Mughini-Gras et al.98 cross-referenced population-based registries to investigate a cohort of 14,264 Salmonella cases for CRC incidence, finding an increased standardized incidence ratio (1.54) in early-onset CRC occurring primarily in the ascending/transverse colon. Furthermore, an updated analysis identified an increased risk of CRC more than 1-year post-infection with non-Enteritis or Typhimurium Salmonella serovars,99 and the detection of a Salmonella flagellar structural protein (FliC) antibodies is higher in Dutch and American CRC patients relative to healthy individuals.100 Salmonella colonization may also lead to chronic infection and inflammation that can exacerbate disease in this context. For example, the virulence factor avirulence A (AvrA) is necessary for chronic Salmonella colonization and resultant colitis.101 In a standard model utilizing the carcinogen AOM and DSS (AOM/DSS) to induce colitis-associated cancer, colonization with avrA+ Salmonella Typhimurium for 25–45 weeks simultaneously activates WNT/β-Catenin and STAT3 to promote tumor development.102,103 Similarly, multiple epidemiological studies have shown an increase in mucosal-invasive E. coli in patients with inflammatory bowel disease and colon cancer.104–106 Pathogenic E. coli strains can produce a variety of toxins (e.g. cycle inhibiting factor, Cif; cytotoxic necrotizing factor 1, Cnf1) that modulate the cell-cycle and are often found in CRC patients.26,27 However, unlike pks, the direct tumorigenic potential of these toxins has not been demonstrated in pre-clinical models. In contrast to the direct mutagenic activity attributed to pks+ E. coli, infection with pathogenic E. coli strains may contribute to tumor progression indirectly by promoting inflammation,107 senescence-associated secretory phenotypes,108–111 or cancer cell detachment and survival.112,113 Other enteric pathogens have been shown to promote CRC through similar inflammatory mechanisms. For example, a recent study by Drewes et al.114 isolated a strain of Clostridioides difficile (C. difficile) from biofilm-bearing CRC tissue that induced colonic tumorigenesis in germ-free and SPF ApcMin/+ mice in a toxin-B dependent manner. C. difficile induced tumors occurred with concomitant activation of myeloid and IL-17 producing lymphoid cells, reminiscent of those observed after ETBF infection in the same model.55,58,59 Importantly, tumorigenesis was dependent upon consistent colonization for 10 weeks, as vancomycin administration via intraperitoneal injection beginning 1-week post-infection successfully inhibited microadenoma formation.114
The clinical relevance of these findings and epidemiological associations are difficult to parse because enteric pathogens are not typically identified in metagenomic studies from CRC patients, and symptomatic infections are ideally temporary after patients receive treatment. Thus, another clinically relevant question is whether transient infections by enteric pathogens might increase the risk of cancer development later in life. Temporally distinct associations such as these can be investigated by the detection of antibodies against specific pathogens in CRC patients100 but are limited by antigen specificity. Another approach can be to use tumor mutational signatures, in which exposure to a specific bacterium leaves a permanent genetic fingerprint in transformed host cells, most often as the result of exposure to microbial genotoxins. Such an approach has been described earlier using pks+ E. coli, but the pks gene island is distributed among other Enterobacteriaceae as well. For example, the opportunistic pathogen Klebsiella pneumoniae (K. pneumoniae) can also carry pks genes as part of a mobile genetic element also encoding yersiniabactin, and these genes are observed more frequently in hypervirulent strains.115,116 Moreover, these bacteria frequently colonize the gut during early life when the colibactin-associated mutational signature is thought to be acquired in colonic epithelial cells.46 Accordingly, a neonatal K. pneumoniae isolate promoted tumor formation in ApcMin/+;IL-10−/− mice, although this phenotype was still observed in pks-deficient K. pneumoniae.117 Thus, it is unclear whether the mechanism underlying K. pneumoniae-driven tumorigenesis is related to colibactin production in this model, but it is logical to theorize that other colibactin-producing microbes may elicit similar mutational signatures as pks+ E. coli. Liu et al118 recently demonstrated that transient infection with an attaching/effacing pathogen (Citrobacter rodentium, C. rodentium) producing a novel genotoxin previously characterized as a UDP-sugar hydrolase (UshA), generates mutational signatures that can be identified by whole-exome sequencing in resultant tumors. In this context, mice completely clear ushA+ C. rodentium infection 28-days post-colonization and ushA+ C. rodentium exposed mice develop a higher number of tumors approximately 4 weeks after clearance.118 These tumors have an increased proportion of single-nucleotide substitutions attributed to the COSMIC mutational signature SBS26 canonically attributed to mismatch-repair deficiency (MMRd), but not the related MMRd signature SBS15.119,120 Collectively, these findings suggest that exposure to microbial genotoxins may generate mutations that result in tumor formation long after bacterial clearance, and that the pattern of mutations in these cases may provide insight into transient carcinogenic events in the past. However, these applications may be limited, as in this case the observed mutations overlap previously defined signatures with etiologies proposed in high confidence (i.e. mismatch repair deficiency). In another case, some strains of the pathogenic bacterium Campylobacter jejuni (C. jejuni) produce a genotoxin known as the cytolethal distending toxin (cdt) that promotes colorectal cancer via its DNA-damaging activity.121 This toxin’s activity is mediated by its active subunit CdtB, which exhibits DNase-I-like activity,122 an enzyme that readily digests DNA with limited sequence specificity primarily dictated by chromatin accessibility.123,124 Whether CdtB exposure promotes a specific mutational signature that can be used to estimate its role in CRC initiation remains to be seen. Given the compound’s homology to DNase-I, it may be hypothesized that DNA damage after infection with CdtB-producing bacteria will result in an increase in overall mutational burden without a distinct identifiable signature. In cases like this, the methods of identifying a causative link between transient pathogenic infection and CRC risk remain difficult and require careful investigation.
Enterococcus faecalis
Enterococcus faecalis (E. faecalis) is among the most prevalent commensal enterococci found in human stool and is enriched in fecal125 and formalin-fixed paraffin-embedded (FFPE)67 samples from CRC patients. However, whether E. faecalis colonization modules intestinal tumor development is debated126 and recent evidence suggests strain-specific differences may alternatively elicit pro- or anti-tumorigenic effects in normal intestinal epithelial and tumor cells.127
E. faecalis’ carcinogenic activity is primarily attributed to its ability to directly damage host DNA via superoxide production, or through a macrophage-induced bystander effect. Infection with the superoxide-producing strain E. Faecalis OG1RF increased chromosomal instability in a hybrid hamster cell line harboring a human version of chromosome 11.128 Similar effects were observed after treating these cells with RAW264.7 macrophages that had been infected with OG1RF.128 These findings were later replicated in studies showing more anaphase bridges and aneuploidy in a CRC cell line HCT 116 after direct culture with E. faecalis or infected macrophages.129 Moreover, γH2AX foci were observed in colonic epithelial cells in a 6 h intestinal ligation model after direct mucosal exposure of E. faecalis.129 In both studies,128,129 these effects were abrogated by the addition of a superoxide dismutase, suggesting that resultant DNA damage and chromosomal instability resulted at least in-part from reactive-oxygen species. Interestingly, while testing various free radical scavenging molecules, Wang & Huycke128 noticed that an inactive control metabolite, γ-CEHC, also inhibited chromosomal instability in these cells without reducing superoxide concentrations in bacterial cultures. Instead, this molecule’s protective function was attributed to its inhibition of cyclooxygenase-2 (COX-2) signaling, a key inflammatory mediator in active macrophages upregulated during E. faecalis-associated colitis.130 Consistent with these studies, Wang et al.131 later showed that OG1RF-polarized macrophages induced heritable mutations in a normal colonic epithelial cell line (YAMC), and that allografts of these cells developed into poorly differentiated tumors in immunodeficient mice. These effects are observed with a concomitant increased expression of intestinal stem cell markers (e.g. doublecortin like kinase 1, Dclk1) and the activation of WNT/β-Catenin signaling pathways.130–132 In addition to activating proliferative signaling pathways, epidemiological evidence has linked E. faecalis colonization to distinct genomic and transcriptomic profiles in patients. These associations are primarily characterized by increased detoxification enzymes in pre-cancerous lesions (e.g. glutathione S-transferase alpha 4, GSTA4133) or increased inflammatory signaling pathways in E. faecalis-colonized CRC cases.134
Most of these studies utilize a single strain of superoxide producing Enterococcus faecalis, OG1RF, while strain diversity in the human gut microbiome may be very high. Several recent studies have focused on identifying strain-specific virulence factors that may elicit different phenotypes in colonic epithelial cells. For example, a collagenolytic rodent-derived E. faecalis strain (E2) promotes anastomotic tumor formation 21 days after surgical resection and delivery of bacteria via enema in mice fed a Western diet.135 These tumors can be attenuated by antibiotic depletion or inhibition of microbial collagenase activity by adding a phosphate carrier compound (Pi-PEG) to the drinking water.135 Consistent with these findings, Williamson et al.136 found that the human-derived strain E. faecalis V583 promotes invasion and migration in a human colon cancer cell line dependent on its collagenolytic activity of a secreted gelatinase (gelE). This collagenolytic activity may also facilitate the translocation of immunogenic bacterial metabolites facilitating tumor progression at distant sites. Such a mechanism was recently described for V583 in a murine model of hepatocellular carcinoma.137 In this model, administration of V583 producing a related gelatinase (GelA) promoted liver tumor formation dependent upon myeloid differentiation primary response 88 (Myd88)-dependent innate immune signaling and increased gelA-dependent gut permeability.137 Moreover, liver dysfunction and unbalanced gut bile-acid concentrations may selectively promote E. faecalis expansion in the intestine.137 In contrast, some E. faecalis strains have proposed probiotic function primarily attributed to the bacterium’s lactic acid fermenting activity. At least one study found a decrease in culturable E. faecalis strains from CRC patients relative to healthy donors using a combination of culture-based matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) identification and culture-independent qPCR analysis.127 Pre-fermented media from three E. faecalis strains isolated from healthy donors inhibited the growth of human CRC cell lines in vitro, while no effect was observed in two CRC-derived strains.127 Moreover, these authors found a decrease in E. faecalis prevalence in a small cohort of CRC patients relative to healthy subjects using a combination of culturomics/MALDI-TOF identification and real-time PCR analysis.127 If lactic-acid production facilitates anti-tumorigenic activity in this species, it is reasonable to theorize that this probiotic function requires metabolically active E. faecalis colonization in the intestine. This hypothesis is consistent with a study demonstrating that a heat-killed probiotic E. faecalis strain (EC-12) did not significantly reduce colonic tumor development in an ApcMin/+ mouse model, although a slight reduction in polyp β-Catenin staining was reported.138 Collectively, this evidence suggests that the pro- or anti-tumorigenic consequence of E. faecalis colonization in the gut may be dictated by strain-specificity, the presence/absence of specific virulence factors, and metabolic activities associated with live bacteria.
Parvimonas micra & Peptostreptococcus spp.
Parvimonas micra (P. micra) and Peptostreptococcus spp. are gram-positive anaerobic cocci that commonly exist as commensal members of the intestinal or oral microbiome. Both genera are enriched in CRC patients particularly in late-stage tumors,19,52,94,139 and incorporation of P. micra and Peptostreptococcus stomatis (P. stomatis) into a four-species biomarker panel facilitated noninvasive CRC diagnosis with high accuracy.140 Furthermore, both genera occupy a similar ecological niche and associate with related molecular CRC subtypes. For example, P. micra and P. stomatis were enriched in CRC patients from consensus molecular subtype 1 (CMS1) cases characterized by CpG-island hypermethylation, microsatellite instability, and serrated adenoma presentation.141 MSI-high tumors have high intra-tumoral microbial diversity that increases with tumor stage, with Parvimonas and Peptostreptococcus being among the most highly variable colonizers.94 In this case and others142,143 these bacteria are frequently found in mucosal biopsies from CRC tumors relative to adjacent normal tissue or healthy controls. Collectively, these findings suggest that these strains may share similar niche-specific colonization of tumor tissue in the gut, and this hypothesis is borne out by several studies using CRC cell lines and ApcMin/+ mouse models. For example, P. micra and Peptostreptococcus anaerobius (P. anaerobius) adhere to the CRC cell-line HT-29 after co-culture and promote proliferative signaling markers (e.g. β-Catenin, PCNA; proliferating cell nuclear antigen) although a precise mechanism for these observations is not clear.139,143 A P. micra strain isolated from a CRC patient increased tumor formation in ApcMin/+ mice with a concomitant upregulation of proliferative signaling pathways, but this phenotype required strain delivery by oral gavage at 108 CFU every 3 days for 10 weeks.143 Germ-free ApcMin/+ mice administered a single dose of P. micra did not form tumors but did generate a higher level of the proliferative markers Ki67 and PCNA in epithelial nuclei.143 Similarly, P. anaerobius increased the number of colonic tumors in ApcMin/+ mice after a daily gavage of 108 CFU for 10 weeks144 or high-grade dysplasia after a single gavage at the same dose after antibiotic treatment in mice administered AOM.139 Preferential adherence to CRC cells may facilitate these phenotypes, as P. anaerobius express a surface membrane protein, putative cell wall binding repeat 2 (PCWBR2), that binds integrin α2/β1 overexpressed in CRC cells to active phosphoinositide 3-kinase-AKT serine/threonine kinase 1 (p13k-AKT) proliferative signaling pathways.144 Consistent with this hypothesis, intraperitoneal administration of a peptide that binds integrin α2/β1 attenuated P. anaerobius-induced tumor formation.144 As in animal models of F. nucleatum, the requirement for frequent gavage of these strains and the lack of tumorigenicity in germ-free models suggests that these microbes may preferentially colonize cancer tissue rather than initiate tumor formation. Epidemiological associations with CIMP/MSI-high phenotypes suggests that higher mutational burden may somehow facilitate host-microbe interactions in these genera.
Intestinal bacteria in CRC diagnosis, prevention, and treatment
The goal of microbiome research in the context of CRC is to integrate this knowledge to improve clinical outcomes. Currently, most studies focus on leveraging bacterial communities for one of three applications: noninvasive diagnostic methods, the prevention of CRC by restoring microbial dysbiosis or providing probiotic supplementation, or the modulation of gut microbes to boost therapeutic responses. Although progress has been made in each of these areas, major barriers remain before such techniques are routinely utilized in the clinic. In this section, we will discuss these advances as well as some of the barriers that must be addressed to realize the potential of microbiome-based medicine.
Multi-omics models for CRC diagnosis
CRC diagnosis primarily focuses on two strategies: the early identification of asymptomatic patients via routine colonoscopy, and the identification of symptomatic patients via clinical presentation.145 Given that CRC is often associated with the enrichment or depletion of specific bacterial genera or species, it is possible that a diagnostic set of microbes, microbially derived metabolites, or bacterial gene signatures may provide clinical information without the need for invasive procedures.146 Fecal metagenomics has shown a CRC-associated microbial signature primarily characterized by the increased relative abundance of several core species, namely: F. nucleatum, P. stomatis, P. micra, Solobacterium moorei, and Bacteroides fragilis.147,148 Recent work has focused on developing a set of universal CRC biomarker species that may be used in diverse cohorts with unique ethnicities or geographical origins, a difficult task given the non-uniformity of the microbiome in healthy individuals or cancer patients. Notably, Yu et al.148 present a panel of four microbial genes from these bacteria identified in Chinese patients with high diagnostic accuracy (73%) in validation cohorts from Denmark, France, and Australia. These results are consistent with a later study that utilized a machine-learning method to identify globally conserved bacterial CRC biomarkers from metagenomic sequencing data using three independent cohorts in the United States, France, and China.149 These studies suggest that fecal metagenomic screening may be a viable noninvasive screening method or could be used in conjunction with standard screening methods (such as a fecal occult blood test) to improve accuracy. However, these approaches have important limitations. For one, even the most effective models operate with a predictive capacity between approximately 73–85%. Related to this, the diagnostic accuracy of these models typically inversely correlates with the cancer stage,147,148 limiting the efficacy of using microbial biomarkers to identify patients before advanced disease. Finally, these approaches utilized fecal samples that are not routinely collected from patients. Poore et al.150 provide a proof-of-concept study that addresses some of these limitations by filtering microbial reads from metagenomic sequencing of CRC patient blood samples, providing more accessible material for microbial diagnostics. After in silico contaminant removal and training, machine-learning models were able to predict CRC cases with high accuracy using circulating microbial DNA from blood or plasma, in some cases discriminating between stage I/II cancers before circulating tumor DNA.150 Moreover, combining microbial biomarkers with genomic and metabolomic signatures may enhance screening or clinical decision-making. A recent study151 showed that patients with no disease, adenomas, or CRC could be differentiated with high accuracy from a set of discriminating microbial, amino acid, or proteomics features in patient feces. This multi-omics approach identified correlations between CRC-enriched microbes and biologically relevant metabolites, and outperformed traditional fecal immunochemical tests currently used in the clinic.151 Continued advancements in these techniques may facilitate the routine inclusion of microbial signatures in the clinic as part of a holistic diagnostic approach and allow for more accurate noninvasive cancer diagnosis or staging (Figure 2).
Figure 2.
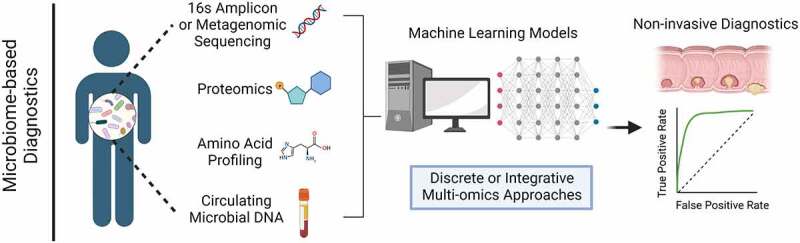
Multi-omics microbiome-based diagnostic methods. Microbiome-based diagnostic models typically utilize three primary technologies derived from patient fecal samples: 1) 16s amplicon or metagenomic sequencing to determine microbial taxa, 2) Liquid chromatography-mass spectrometry (LC-MS) metabolomics/proteomics to identify microbially derived metabolites, and 3) High-performance liquid chromatography (HP-LC) profiling of amino stool amino acid profiles. A fourth and more recently proposed approach amplifies circulating microbial DNA from patient blood or plasma samples, followed by stringent computational filtering. In all methods, marker selection is conducted using machine learning models to identify discriminating markers correlating with tumor stage, and accuracy can be improved by integrating one or more of these datasets.
A major limitation of these approaches is that they all require the application of next-generation sequencing or proteomic analyses and complex machine-learning algorithms (Figure 2), techniques that require expensive dedicated equipment. Moreover, using these techniques in the clinic will require employing expert personnel, particularly with respect to data analysis and the application of predictive machine-learning models. Incorporating these methods in routine clinical diagnostics would require a large economic investment and is unlikely to become commonplace until significant technological advancements reduce the cost of these approaches.
Probiotics and prebiotics
The healthy gut microbiome is typically characterized by the presence of beneficial species that degrade complex polysaccharides derived from indigestible dietary fibers to produce lactic acid (lactic acid bacteria, LAB) and other metabolites that promote intestinal homeostasis.152 CRC-associated dysbiosis is characterized both by enrichment of CRC-associated bacteria and a depletion of probiotic LAB153 (e.g. Bifidobacterium, Lactobacillus, Streptococcus). While there are many associative and mechanistic studies implicating carcinogenic bacteria in CRC initiation or progression, there are fewer studies investigating the preventive potential of probiotic microbes. In the context of CRC tumor prevention, Lactobacillus is perhaps the most thoroughly studied genus. For example, daily administration of Lactobacillus acidophilus (L. acidophilus) probiotics to the drinking water of AOM-treated BALB/c mice inhibits tumor development.154 These findings are consistent with another study showing that oral gavage of L. acidophilus lysates every other day reduced tumor burden in an AOM/DSS BALB/c mouse model.155 These effects may be attributed to the production of exopolysaccharides and other metabolites identified in related Lactobacillus spp. that promote anti-tumor immunity or directly induce cancer cell apoptosis.156,157 Similar anti-tumorigenic effects have been observed after colonization by other LAB such as Bifidobacterium longum (B. longum)158 and Streptococcus thermophilus (S. thermophilus)159 (Figure 3).
Figure 3.
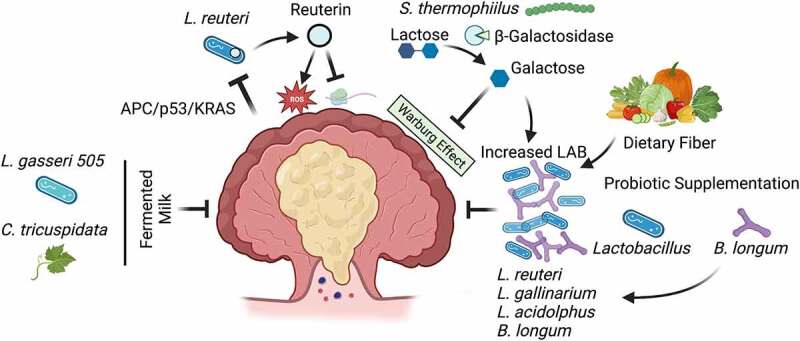
Lactic acid bacteria inhibit colorectal cancer tumorigenesis. Colorectal cancer (CRC) is characterized by a reduction in lactic acid bacteria (LAB). In a murine model of adenomatous polyposis coli/tumor protein 53/Kirsten rat sarcoma viral oncogene homolog (APC/p53/KRAS) mutant CRC, host metabolites directly inhibit Lactobacillus reuteri (L. reuteri) growth and the production of the microbial metabolite reuterin, that inhibits protein translation and generates cytotoxic reactive oxygen species in CRC cells to restrict cell growth. Streptococcus thermophilus (S. thermophilus) secretes the enzyme β-Galactosidase that produces galactose, that inhibits oxidative phosphorylation and Warburg metabolism in CRC cells. Various LAB inhibit tumor growth through a variety of indeterminate mechanisms, and the CRC-associated depletion of these species can be offset by increased abundance of galactose, dietary fibers, or the presence of other probiotic species. Finally, LAB may ferment dietary components such as Cudrania triscuspidata (C. tricuspidata) to produce antioxidants that restrict tumor growth.
While the specific factors resulting in LAB depletion in CRC patients are unclear, the current evidence suggests that the relative abundance of these bacteria is influenced by the tumor microenvironment, the presence of cross-feeding microbes, and the patient’s diet. Using a tamoxifen-inducible model with sequential mutations in key CRC driver genes (APC/p53/KRAS), Bell et al.6 show that homocysteine-degrading metabolites enriched in stool samples from triple mutant tumors inhibit Lactobacillus reuteri (L. reuteri) growth. Reuterin derived from these bacteria inhibited ribosomal biogenesis and induced cytotoxic ROS in multiple CRC cell lines, but not the normal colonic epithelial cell line NCM460 or HeLa cells.6 Moreover, reuterin supplementation in APC/p53/KRAS-mutant mice increased tumor-associated ROS and dysplastic transformation6 (Figure 3). The depletion of probiotic strains like L. reuteri in pre-clinical models of CRC can be offset by the presence of other probiotic strains. For example, oral administration of a high dose of B. longum (four 14-day cycles with 7 days between, 1.5 × 1010 CFU) inhibited tumor initiation in rats treated with AOM/DSS and consequently ameliorated Lactobacillus depletion observed in AOM/DSS rats not gavaged with B. longum.158 Similarly, daily gavage with Lactobacillus gallinarium (L. gallinarium) reduced tumor number and size in both ApcMin/+ and AOM/DSS mouse models, with a concomitant increase in other probiotic species like L. reuteri.160 While these findings suggest that colonization with LAB promotes the proliferation or persistence of other probiotic species, the underlying mechanism remains unclear (Figure 3). LAB may promote the outgrowth of other probiotic strains simply by reducing the relative abundance of non-LAB species with an advantage in competition for limited resources. Alternatively, metabolic interactions between these microbes may favor the entire group. Another probiotic strain, S. thermophilus, suppresses CRC tumor growth in ApcMin/+ mice by producing the enzyme β-Galactosidase that inhibits Warburg metabolism, a mode of aerobic glycolosis used for respiration in cancer cells.159 β-Galactosidase metabolizes a specific glucose moiety of lactose to produce galactose, and the addition of S. thermophilus or galactose to ApcMin/+ mice increased the relative abundance of other probiotic species from the genera Lactobacillus and Bifidobacterium159 (Figure 3). These findings suggest that, in this context, S. thermophilus promotes the expansion of probiotic bacteria through a metabolic interaction. Furthermore, the ApcMin/+ model used in this study developed relatively few colonic tumors (~1-2), suggesting competition within the tumor environment would not lead to pronounced changes in bacterial abundance.
Because nearly all probiotic strains discussed here ferment dietary fiber as a primary nutrient source, it is reasonable to consider dietary interventions aimed at enriching the abundance of these bacteria as a potential means of CRC prevention. One set of studies has used such an approach to demonstrate the efficacy of combining dietary modification with the anti-tumorigenic properties of Lactobaillus gasseri 505 (L. gasseri 505) in pre-clinical models. L. gasseri 505 was identified from a screen of neonatal isolates capable of fermenting milk supplemented with leaf extract from Cudrania tricuspidata (C. tricuspidata), a traditional herbal remedy in Asia, to produce lactic acid and a variety of antioxidant compounds.161,162 Accordingly, treating mice with C. tricuspidata leaf extract in milk fermented with L. gasseri 505 reduced tumor number and inflammatory gene expression in an AOM/DSS mouse model, while causing an expansion of Bifidobacterium spp.163 Several early-stage clinical trials have been conducted to assess how short-term dietary fiber interventions influence the gut microbiome in humans. In one study utilizing a cohort of undergraduate students, individuals were directed to increase fiber intake from ~21 g/day to 46.4 g/day for two weeks.164 Shannon diversity decreased after dietary intervention, but the relative abundance of Bifidobacterium increased, and Bifidobacterium abundance positively correlated with Lactobacillus.164 A second study utilized a random cross-over design in which the same individuals were given varying concentrations of inulin, arabinoxylan, or mixed fibers.165 In this study, a similar decrease in Shannon diversity correlating with the abundance of fiber added to the diet was observed, and the relative abundance of Bifidobacterium and biochemical pathways involved in fructose metabolism were observed after inulin administration.165 Collectively, these findings suggest that even short-term dietary interventions may promote the outgrowth of beneficial probiotic species in humans and that these bacteria exhibit anti-tumorigenic activity in pre-clinical models. Whether such an approach can reduce CRC risk in humans remains to be seen. Of note, and contrary to typical association between healthy status and high microbiota diversity, both studies reported a reduction in gut microbe biodiversity after short-term increased fiber intake, highlighting the unpredictable nature of dietary interventions in diverse human populations. A more precise clinical approach may be to directly administer bacterial metabolites responsible for these prophylactic effects, possibly from fermented supplements, as future CRC-preventive therapies.
The influence of gut bacteria on CRC treatment
Adjuvant chemotherapy (5-fluorouracil, 5-FU; oxaliplatin) is the recommended treatment option for advanced CRC cases that cannot be completely resolved by surgical excision.166 Medication use has a profound influence on gut bacterial community structure and metabolism,167 suggesting that the intestinal microbiome may modulate the efficacy of CRC therapies. One early study from Iida et al.168 demonstrated that antibiotic treatment significantly reduced the efficacy of oxaliplatin, a platinum-based chemotherapeutic, in a pre-clinical model using MC38 xenografts. Interestingly, antibiotic treatment had no effect on the proportion of platinum-bound DNA but rather reduced ROS generation by tumor-infiltrating CD11b+Gr-1hi neutrophils and F4/80+Gr-1int macrophage-like cells168 (Figure 4a). This finding suggests that commensal microbes in the normal gut biota regulate therapeutic efficacy by maintaining immune homeostasis. Alternatively, the outgrowth or persistence of deleterious species may inhibit these processes. Epidemiological evidence suggests that the persistence of F. nucleatum in patients undergoing neoadjuvant chemoradiotherapy is associated with a reduced rate of relapse-free survival (Hazard ratio = 7.5) over 140 months, with a concomitant reduction in tumor-infiltrating CD3+ and CD8 + T cells.169 Consistent with these findings, F. nucleatum is enriched in patients that exhibit a poor clinical response to 5-FU treatment, and F. nucleatum infection in HCT 116 and HT29 cell lines inhibits 5-FU mediated apoptosis via a toll-like receptor 4 (TLR4)/baculoviral IAP repeat containing 3 (BIRC3)-dependent mechanism in CRC xenograft models.170,171 Collectively, these data show that intestinal dysbiosis may influence platinum- or nucleoside-based chemotherapeutics (Figure 4a). The current evidence suggests that these effects are largely driven by disruption of immune homeostasis resultant from altered gut microbiota, although other mechanisms still need to be explored in more detail. For example, intravenous injection of E. coli Nissle 1917 confers gemcitabine resistance in MC26 xenografts dependent on expression of a long isoform of the enzyme cytidine deaminase (CDDL)172 (Figure 4a). In contrast, CDDL-deficient E. coli did not confer resistance to oxaliplatin.172 In this case, the reduced therapeutic efficacy was attributed to bacterial metabolism of the drug itself, as incubated CDDL+ strains with gemcitabine in minimal media led to nearly complete depletion of detectable drug within 4 hours.172 Whether 5-FU therapeutic efficacy is influenced by bacteria-mediated drug depletion in CRC patients remained to be established. Thus, the gut microbiome may influence chemotherapeutics by direct biotransformation of the active compound or by indirectly inhibiting the drug’s cytotoxic mode-of-action.
Figure 4.
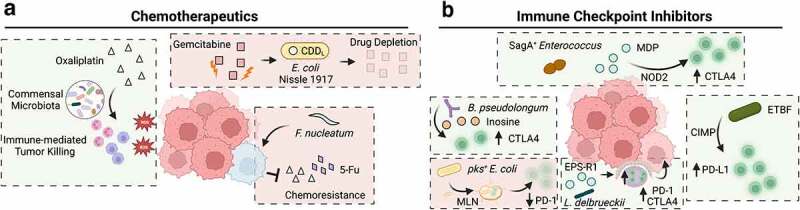
The impact of intestinal bacteria on colorectal cancer treatment. a) In mice harboring a commensal microbiota, neutrophils and macrophages invade tumors and produce reactive oxygen species (ROS), enhancing the tumor-killing effect of oxaliplatin. In mice administered antibiotics, the number of infiltrating immune cells and ROS-mediated cytotoxicity is reduced. In the presence of commensal bacteria (such as Escherichia coli Nissle 1917) harboring the long form of a cytidine deaminse (CDDL), gemcitabine levels are reduced after degradation by this bacterial enzyme, resulting in a reduced therapeutic response. Similarly, infection with Fusobacterium nucleatum confers resistance to oxaliplatin and 5-Fu by downregulating miRNAs that suppress autophagy and survival signaling. b) Several bacterial species have been linked to enhanced immunotherapy response in murine models of CRC or MC-38 xenografts. The species Bifidobacterium longum produces a metabolite inosine, which activates tumor-infiltrating T cells and exacerbates tumor killing after anti-CTLA4 treatment. Enterococcus spp. harboring the secreted antigen A (sagA) gene generate high levels of muramyl dipeptide (MDP) that activates nucleotide-binding oligomerization domain-containing protein 2 (NOD2) signaling pathways in colonic epithelial cells that drives immune recruitment and synergizes with anti-CTLA4 treatment. In a murine model of CpG island methylator phenotype (CIMP) CRC, infection with Enterotoxigenic Bacteroides fragilis promotes the recruitment of interferon gamma (IFNγ)-producing CD8+ T cells to enhance anti-PD-1 efficacy. Lactobacillus delbrueckii subsp. bulgaricus exopolysaccharides (EPS-R1) promote the activation of CCR6+CD8+ T cells in intestinal Peyer’s patches, as well as the number of IFNγ producing CD8+ tumor infiltrating cells, promoting the efficacy of anti-PD-1 and anti-CTLA4 treatment. Conversely, pks+ E. coli can migrate to mesenteric lymph nodes (MLN) and reduce systemic levels of CD3+ and CD8+ T cells, as well as the number of these cells observed in invasive tumor margins and reduces anti-PD-1 treatment efficacy.
Immune checkpoint inhibitors (ICI) rely on the host immune system to mediate their anti-tumoral effect. Given that gut microbes influence local and systemic immune responses, many recent studies have investigated how intestinal bacteria modulate ICI therapies. For example, fecal microbiota transplant (FMT) from responders has been shown to sensitize ICI non-responsive tumor xenografts in pre-clinical models173–176 and human patients.177,178 Establishing a consistent mechanism for these observations has proven difficult, with responder-enriched taxa (most commonly: Akkermansia spp., Bifidobacterium spp., Roseburia spp., or Faecalibacterium spp.) varying across cohorts and cancer type.179–181 How microbial communities influence ICI response in CRC patients is less clear. CRC patients typically exhibit a poor response to ICI, with the exception being MSI-high tumors with higher mutational burdens, and thus a higher variable antigen presentation more likely to engage anti-tumor immune responses.182 MSI-high CRC can result from germline mutations in mismatch-repair associated genes or hypermethylation of the MLH1 promoter; accordingly, MSI-high CRC is often associated with CIMP subtypes. Thus, an interesting question is whether gut microbes boost ICI efficacy in this context. DeStefano Shields et al. show that in a model of CIMP colon cancer (BLM, previously discussed), ETBF colonization increases the number of tumor-infiltrating IFNγ-producing CD8+ T cells relative to ApcMin/+-ETBF mice, and that anti-programmed death-ligand 1 (anti-PD-L1) is effective in reducing tumor burden in these animals64 (Figure 4b). To determine if microbiota composition can boost ICI efficacy in microsatellite stable (MSS) tumors, Mager et al.183 monitored treatment response and microbiome composition in an AOM/DSS model of colitis-associated cancer along with a heterotopic MC38 xenograft model. The authors found that Bifidobacterium pseudolongum (B. pseudolongum) mono-colonization enhanced intratumoral CD8+ T-cell activation while significantly enhancing the effects of ICI therapy183 (Figure 4b). While this bacterium only induced modest tumor growth inhibition in the absence of ICI, anti-cyototxic T-lymphocyte associated protein 4 (anti-CTLA4) treatment increased intratumoral B. pseudolongum abundance and systemic inosine levels, a B. pseudolongum metabolite that activated TH1 differentiation factor expression.183 Moreover, ICI treatment enriched the intratumoral abundance of these species in multiple mutational contexts, including: Msh2LoxP/LoxP;Villin-Cre and Apc;2lox14/+Kras;LSL-G12D/+Fabpl-Cre derived tumors.183 Several other studies have identified bacteria associated with ICI efficacy in murine xenograft models, most often utilizing the MSS cell line MC38. For example, Griffin et al.184 demonstrated that expression of a peptidoglycan hydrolase (secreted antigen A, SagA) in Enterococcus spp. facilitates the release of muramyl dipeptides that activate NOD2 signaling pathways and the expression of proinflammatory NF-κB genes. These changes promote tumor-infiltration by CD45+ and CD8+ cells, while enhancing the efficacy of anti-CTLA4 treatment in MC38 xenografts184 (Figure 4b). In some cases, the metabolite responsible for synergizing with immune checkpoint inhibitors can be isolated and administered as a bacteria-free adjuvant. For example, a recent study showed that exopolysaccharides derived from L. delbrueckii subsp. bulgaricus can activate CD8+ T cells in Peyer’s patches that preferentially infiltrate CCL20 positive tumors and promote the therapeutic efficacy of anti-CTLA4 or anti-programmed cell death protein 1 (anti-PD-1) treatment185(Figure 4b). In contrast, pks+ E. coli have been shown to translocate to the mesenteric lymph nodes where they lower the abundance of cytotoxic T cells, which in turn reduces immune cell invasion in tumor tissue margins and inhibits anti-PD-1 treatment in MC38 tumor-bearing mice51 (Figure 4b). This important area of research may begin to elucidate several important aspects of CRC treatment. Specifically, these studies suggest that a patient’s treatment response can be modulated by the presence or absence of specific bacterial species or microbial metabolites.
Discussion
Systematic efforts to characterize the human microbiome began over a decade ago and were closely followed by an innumerable number of studies cataloging associations between changes in microbial community structure and human diseases such as CRC. These approaches have identified several bacterial species that are more frequently found in CRC patients, many of which have demonstrable carcinogenic effects when administered to susceptible murine models of CRC or colitis-associated cancer. These effects may be attributed to direct host-microbe interactions that activate proliferative73,144 or immunomodulatory55,58,59 signaling pathways. Alternatively, bacteria may produce toxins or metabolites that similarly activate oncogenic gene expression, induce epigenetic changes,63,83 or directly generate mutations in cancer-driver genes through their genotoxic activity.5,186 It is becoming clearer that the oncogenic potential of such microbes is influenced by the complex milieu of host- and microbe-derived factors occurring in the microenvironment. For example, colibactin-associated mutations may pre-dispose individuals to cancer development but require successive microbial “hits” to fully realize a malignant consequence. Such a mechanism may explain the increased risk of CRC in patients colonized with pks+ bacteria that adhere to a high-fat Western diet, or the co-occurrence of pks+ E. coli and ETBF in CRC patients with hereditary APC mutations and the observation that increased tumorigenesis after ETBF + pks+ E. coli dual association in ApcMin/+ mice can be abrogated by the deletion of either bft or pks genes, respectively.23 The recent identification of multiple genotoxins encoded by commensal bacteria isolated from IBD patients18 suggests that there may be a plethora of similar interactions that remain to be discovered. Thus, developing an accurate assessment of the role for bacterial drivers of CRC will likely require incorporating proteomic, genomic, and transcriptomic data capable of holistically anatomizing complex microbial ecosystems. Several technological advances may facilitate the acquisition of knowledge that can more directly answer these questions. Using spatial bacterial transcriptomics and transcriptional recording techniques, researchers can begin probing the effects of heterogenous microbial communities on host gene expression at the single-cell level187 or how transient changes in the intestinal environment may influence microbial gene expression and alter carcinogenic potential.188 Moreover, the advent of novel in situ metabolomics may allow scientists to directly study the interaction of microbial metabolites with immune cells or pre-cancerous lesions.189 This complexity is compounded by cancer progression itself, during which tumor heterogeneity is influenced by a unique “intra-tumoral microbiome”. In such cases, intracellular bacteria modulate gene expression187 and antigen presentation190 in ways that may enhance malignant transformation. Microbial ecosystems in the gut also contain extensive fungal and viral communities that can promote CRC development.191–195 While significant advances have been made, cancer microbiome research is a nascent field. The hope of cancer microbiome research is that unraveling these complexities may lead to a better understanding of the factors influencing an individual patient’s disease progression, and thus lead to better risk-assessment or treatment.
How can one leverage this knowledge to improve CRC outcomes? One possibility is to “edit” the intestinal bacteriome by the targeted removal of carcinogenic bacterial species using bacteriophages. Pre-clinical studies suggest phage-targeting of pks+ E. coli can reduce their oncogenic effects in vivo.196 Such an approach may be feasible in humans using species-specific phage cocktails, as demonstrated by early-phase clinical trials targeting K. pneumoniae in a small cohort of healthy patients.197 Alternatively, the biochemical characterization of microbial metabolic pathways involved in promoting CRC may allow for the development of small molecule inhibitors directly inhibiting these pathways.198,199 On the other hand, the addition of bacteria with preventative or treatment-enhancing effects may be administered to counteract cancer progression and boost treatment efficacy. A more complete mechanistic understanding of these effects may allow for the engineering of microbes that produce specific antigens or metabolites that activate anti-tumor immunity or apoptotic pathways in cancer cells.6,200,201 In cases where microbial metabolism interferes with therapy by reducing efficacy or exacerbating dose-limiting side effects, synthetic inhibitors may be used to interrupt these biosynthetic pathways.202 In theory microbial colonization may be bypassed altogether, and cell-free reactive microbial metabolites administered as treatment or therapeutic adjuvants.185 Collectively, the microbiome holds great promise as a source of individual variability that may help explain diverse patient outcomes. Future clinical approaches may utilize this knowledge to understand the root cause of tumor development in CRC on a patient-by-patient basis, or to help inform therapeutic approaches.
Acknowledgments
C. Jobin acknowledges support from NIH R01DK073338, R01CA215553 and University of Florida Department of Medicine Gatorade Fund. Figures were created using BioRender.
Funding Statement
This work was supported by NIH NCI under R01CA215553; NIH NIDDK under DK073338.
Data Availability Statement
No new data were generated or analyzed in support of this research.
Disclosure Statement
The authors report they have no competing interests to declare.
Author Contributions
Writing – Original Draft: MWD; Writing – Review & Editing: MWD, CJ; Supervision: CJ; Funding Acquisition: CJ.
References
- 1.Sender R, Fuchs S, Milo R.. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tilg H, Adolph TE, Gerner RR, Moschen AR. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33:954–27. doi: 10.1016/j.ccell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in colorectal cancer: how to get from meta-omics to mechanism? Trends Microbiol. 2020;28:401–423. doi: 10.1016/j.tim.2020.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. Coli Nature. 2020;580:269–273. doi: 10.1038/s41586-020-2080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell HN, Rebernick RJ, Goyert J, Singhal R, Kuljanin M, Kerk SA, Huang W, Das NK, Andren A, Solanki S, et al. Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell. 2022;40(185–200.e6):185–200.e6. doi: 10.1016/j.ccell.2021.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33:570–580. doi: 10.1016/j.ccell.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D. Hallmarks of cancer: new Dimensions. Cancer Discov. 2022;12:31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 9.Fodde R. The APC gene in colorectal cancer. Eur J Cancer. 2002;38:867–871. doi: 10.1016/S0959-8049(02)00040-0. [DOI] [PubMed] [Google Scholar]
- 10.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–47. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 11.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–162. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(715–730.e3):715–730.e3. doi: 10.1053/j.gastro.2021.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Constitutive OM. NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res. 2009;15:2248–2258. doi: 10.1158/1078-0432.CCR-08-1383. [DOI] [PubMed] [Google Scholar]
- 14.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng H, Shao J, Williams CS, Pereira MA, Taketo MM, Oshima M, Reynolds AB, Washington MK, DuBois RN, Beauchamp RD. Nuclear translocation of beta-catenin in hereditary and carcinogen-induced intestinal adenomas. Carcinogenesis. 1998;19:543–549. doi: 10.1093/carcin/19.4.543. [DOI] [PubMed] [Google Scholar]
- 16.Bürtin F, Mullins CS, Linnebacher M. Mouse models of colorectal cancer: past, present and future perspectives. World J Gastroenterol. 2020;26:1394–1426. doi: 10.3748/wjg.v26.i13.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIntyre RE, Buczacki SJA, Arends MJ, Adams DJ. Mouse models of colorectal cancer as preclinical models. Bioessays. 2015;37:909–920. doi: 10.1002/bies.201500032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Y, Oh J, Xue M, Huh WJ, Wang J, Gonzalez-Hernandez JA, Rice TA, Martin AL, Song D, Crawford JM, et al. Commensal microbiota from patients with inflammatory bowel disease produce genotoxic metabolites. Science. 2022;378:eabm3233. doi: 10.1126/science.abm3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25:968–976. doi: 10.1038/s41591-019-0458-7. [DOI] [PubMed] [Google Scholar]
- 20.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, Oswald E, Nougayrède J-P. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. MBio. 2018;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.J-P N, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 22.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 23.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359:592–597. doi: 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubinsky V, Dotan I, Gophna U. Carriage of colibactin-producing bacteria and colorectal cancer risk. Trends Microbiol. 2020;28:874–876. doi: 10.1016/j.tim.2020.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R, Battista JR. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE. 2013;8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, Déchelotte P, Bonnet R, Pezet D, Darfeuille-Michaud A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859–867. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Gharaibeh RZ, Newsome RC, Jobin C. Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat Cancer. 2020;1:723–734. doi: 10.1038/s43018-020-0078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue M, Kim CS, Healy AR, Wernke KM, Wang Z, Frischling MC, Shine EE, Wang W, Herzon SB, Crawford JM. Structure elucidation of colibactin and its DNA cross-links. Science. 2019;365(6457). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dziubańska-Kusibab PJ, Berger H, Battistini F, Bouwman BAM, Iftekhar A, Katainen R, Cajuso T, Crosetto N, Orozco M, Aaltonen LA, et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat Med. 2020;26:1063–1069. doi: 10.1038/s41591-020-0908-2. [DOI] [PubMed] [Google Scholar]
- 32.Healy AR, Wernke KM, Kim CS, Lees NR, Crawford JM, Herzon SB. Synthesis and reactivity of precolibactin 886. Nat Chem. 2019;11:890–898. doi: 10.1038/s41557-019-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wernke KM, Xue M, Tirla A, Kim CS, Crawford JM, Herzon SB. Structure and bioactivity of colibactin. Bioorg Med Chem Lett. 2020;30:127280. doi: 10.1016/j.bmcl.2020.127280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou T, Hirayama Y, Tsunematsu Y, Suzuki N, Tanaka S, Uchiyama N, Goda Y, Yoshikawa Y, Iwashita Y, Sato M, et al. Isolation of new colibactin metabolites from wild-type escherichia coli and in situ trapping of a mature colibactin derivative. J Am Chem Soc. 2021;143:5526–5533. doi: 10.1021/jacs.1c01495. [DOI] [PubMed] [Google Scholar]
- 35.Reuter C, Alzheimer M, Walles H, Oelschlaeger TA. An adherent mucus layer attenuates the genotoxic effect of colibactin. Cell Microbiol. 2018;35(20). [DOI] [PubMed] [Google Scholar]
- 36.Tomkovich S, Jobin C. Microbial networking in cancer: when two toxins collide. Br J Cancer. 2018;118:1407–1409. doi: 10.1038/s41416-018-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dougherty MW, Jobin C. Shining a light on colibactin biology. Toxins (Basel). 2021;13. doi: 10.3390/toxins14010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu W, Miyata N, Winter MG, Arenales A, Hughes ER, Spiga L, Kim J, Sifuentes-Dominguez L, Starokadomskyy P, Gopal P, et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J Exp Med. 2019;216:2378–2393. doi: 10.1084/jem.20181939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tronnet S, Garcie C, Rehm N, Dobrindt U, Oswald E, Martin P, Bäumler AJ. Iron homeostasis regulates the genotoxicity of escherichia coli that produces colibactin. Infect Immun. 2016;84:3358–3368. doi: 10.1128/IAI.00659-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tronnet S, Garcie C, Brachmann AO, Piel J, Oswald E, Martin P. High iron supply inhibits the synthesis of the genotoxin colibactin by pathogenic Escherichia coli through a non-canonical Fur/RyhB-mediated pathway. Pathog Dis. 2017;75(5). [DOI] [PubMed] [Google Scholar]
- 41.Wallenstein A, Rehm N, Brinkmann M, Selle M, Bossuet-Greif N, Sauer D, Bunk B, Spröer C, Wami HT, Homburg S, et al. ClbR Is the Key Transcriptional Activator of Colibactin Gene Expression in Escherichia coli. mSphere. 2020;5(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chagneau CV, Garcie C, Bossuet-Greif N, Tronnet S, Brachmann AO, Piel J, J-P N, Martin P, Oswald E. The polyamine spermidine modulates the production of the bacterial genotoxin colibactin. mSphere. 2019;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bossuet-Greif N, Guyonnet C, Chagneau C, Tang-Fichaux M, Penary M, Branchu P, Oswald E, Nougayrede J-P. Oxygen inhibits colibactin production by Escherichia coli. BioRxiv. 2022. [Google Scholar]
- 44.Oliero M, Calvé A, Fragoso G, Cuisiniere T, Hajjar R, Dobrindt U, Santos MM. Oligosaccharides increase the genotoxic effect of colibactin produced by pks+ Escherichia coli strains. BMC Cancer. 2021;21:172. doi: 10.1186/s12885-021-07876-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arima K, Zhong R, Ugai T, Zhao M, Haruki K, Akimoto N, Lau MC, Okadome K, Mehta RS, Väyrynen JP, et al. Western-style diet, pks island-carrying escherichia coli, and colorectal cancer: analyses from two large prospective cohort studies. Gastroenterology. 2022;163:862–874. doi: 10.1053/j.gastro.2022.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, Georgakopoulos N, Torrente F, Noorani A, Goddard M, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–537. doi: 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- 47.Secher T, Brehin C, Oswald E. Early settlers: which E. coli strains do you not want at birth?. Am J Physiol Gastrointest Liver Physiol. 2016;311:G123–9. doi: 10.1152/ajpgi.00091.2016. [DOI] [PubMed] [Google Scholar]
- 48.Tsunematsu Y, Hosomi K, Kunisawa J, Sato M, Shibuya N, Saito E, Murakami H, Yoshikawa Y, Iwashita Y, Miyoshi N, et al. Mother-to-infant transmission of the carcinogenic colibactin-producing bacteria. BMC Microbiol. 2021;21:235. doi: 10.1186/s12866-021-02292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, Sauvanet P, Darcha C, Déchelotte P, Bonnet M, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932–1942. doi: 10.1136/gutjnl-2013-305257. [DOI] [PubMed] [Google Scholar]
- 50.Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5:675–680. doi: 10.4161/19490976.2014.969989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopès A, Billard E, Casse AH, Villéger R, Veziant J, Roche G, Carrier G, Sauvanet P, Briat A, Pagès F, et al. Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int J Cancer. 2020;146:3147–3159. doi: 10.1002/ijc.32920. [DOI] [PubMed] [Google Scholar]
- 52.Périchon B, Lichtl-Häfele J, Bergsten E, Delage V, Trieu-Cuot P, Sansonetti P, Sobhani I, Dramsi S. Detection of streptococcus gallolyticus and four other crc-associated bacteria in patient stools reveals a potential “driver” role for enterotoxigenic bacteroides fragilis. Front Cell Infect Microbiol. 2022;12:794391. doi: 10.3389/fcimb.2022.794391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prindiville TP, Sheikh RA, Cohen SH, Tang YJ, Cantrell MC, Silva J. Bacteroides fragilis enterotoxin gene sequences in patients with inflammatory bowel disease. Emerging Infect Dis. 2000;6:171–174. doi: 10.3201/eid0602.000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franco AA, Mundy LM, Trucksis M, Wu S, Kaper JB, Sears CL. Cloning and characterization of the Bacteroides fragilis metalloprotease toxin gene. Infect Immun. 1997;65:1007–1013. doi: 10.1128/iai.65.3.1007-1013.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, Dejea CM, Fathi P, Chen J, Finard BB, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(203–214.e5). doi: 10.1016/j.chom.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Viljoen KS, Dakshinamurthy A, Goldberg P, Blackburn JM, McDowell A. Quantitative profiling of colorectal cancer-associated bacteria reveals associations between fusobacterium spp., enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS ONE. 2015;10:e0119462. doi: 10.1371/journal.pone.0119462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60:208–215. doi: 10.1093/cid/ciu787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Regulatory T-cell HF. Response to Enterotoxigenic Bacteroides fragilis Colonization Triggers IL17-Dependent Colon Carcinogenesis. Cancer Discov. 2015;5:1098–1109. doi: 10.1158/2159-8290.CD-15-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124:392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 61.Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015;149(1204–1225.e12):1204–1225.e12. doi: 10.1053/j.gastro.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu -Q-Q, Li C-M, Fu L-N, Wang H-L, Tan J, Wang Y-Q, Sun D-F, Gao Q-Y, Chen Y-X, Fang J-Y. Enterotoxigenic Bacteroides fragilis induces the stemness in colorectal cancer via upregulating histone demethylase JMJD2B. Gut Microbes. 2020;12:1788900. doi: 10.1080/19490976.2020.1788900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen J, Hao S, Sears CL, Timp W. Epigenetic Changes Induced by Bacteroides fragilis Toxin. Infect Immun. 2019;87(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeStefano Shields CE, White JR, Chung L, Wenzel A, Hicks JL, Tam AJ, Chan JL, Dejea CM, Fan H, Michel J, et al. Bacterial-driven inflammation and mutant braf expression combine to promote murine colon tumorigenesis that is sensitive to immune checkpoint therapy. Cancer Discov. 2021;11:1792–1807. doi: 10.1158/2159-8290.CD-20-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, Hacker-Prietz A, Rabizadeh S, Woster PM, Sears CL, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011;108:15354–15359. doi: 10.1073/pnas.1010203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen J, Rosendahl Huber A, Pleguezuelos-Manzano C, Puschhof J, Wu S, Wu X, Boot C, Saftien A, O’Hagan HM, Wang H, et al. Colon tumors in enterotoxigenic bacteroides fragilis (ETBF)-colonized mice do not display a unique mutational signature but instead possess host-dependent alterations in the APC Gene. Microbiol Spectr. 2022;10:e0105522. doi: 10.1128/spectrum.01055-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khodaverdi N, Zeighami H, Jalilvand A, Haghi F, Hesami N. High frequency of enterotoxigenic Bacteroides fragilis and Enterococcus faecalis in the paraffin-embedded tissues of Iranian colorectal cancer patients. BMC Cancer. 2021;21:1353. doi: 10.1186/s12885-021-09110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zamani S, Taslimi R, Sarabi A, Jasemi S, Sechi LA, Feizabadi MM. Enterotoxigenic Bacteroides fragilis: a Possible Etiological Candidate for Bacterially-Induced Colorectal Precancerous and Cancerous Lesions. Front Cell Infect Microbiol. 2019;9:449. doi: 10.3389/fcimb.2019.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang N, Fang J-Y. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2022;31(2):159–172. doi: 10.1016/j.tim.2022.08.010. [DOI] [PubMed] [Google Scholar]
- 72.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brennan CA, Clay SL, Lavoie SL, Bae S, Lang JK, Fonseca-Pereira D, Rosinski KG, Ou N, Glickman JN, Garrett WS. Fusobacterium nucleatum drives a pro-inflammatory intestinal microenvironment through metabolite receptor-dependent modulation of IL-17 expression. Gut Microbes. 2021;13:1987780. doi: 10.1080/19490976.2021.1987780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abed J, JEM E, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan KA, et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. 2016;20:215–225. doi: 10.1016/j.chom.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, Peng Y, Yu J, Chen T, Wu Y, Shi L, Li Q, Wu J, Fu X. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget. 2017;8:31802–31814. doi: 10.18632/oncotarget.15992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, Dalerba P, Wang TC, Han YW. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019;20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015;1:653–661. doi: 10.1001/jamaoncol.2015.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo S, Chen J, Chen F, Zeng Q, Liu W-L ZG. Exosomes derived from Fusobacterium nucleatum-infected colorectal cancer cells facilitate tumour metastasis by selectively carrying miR-1246/92b-3p/27a-3p and CXCL16. Gut. 2020. doi: 10.1136/gutjnl-2020-321187. [DOI] [PubMed] [Google Scholar]
- 80.Xu C, Fan L, Lin Y, Shen W, Qi Y, Zhang Y, Chen Z, Wang L, Long Y, Hou T, et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes. 2021;13:1980347. doi: 10.1080/19490976.2021.1980347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen S, Su T, Zhang Y, Lee A, He J, Ge Q, Wang L, Si J, Zhuo W, Wang L. Fusobacterium nucleatum promotes colorectal cancer metastasis by modulating KRT7-AS/KRT7. Gut Microbes. 2020;11:511–525. doi: 10.1080/19490976.2019.1695494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Y, Chen Y, Zhang J, Cao P, Su W, Deng Y, Zhan N, Fu X, Huang Y, Dong W. Fusobacterium nucleatum promotes metastasis in colorectal cancer by activating autophagy signaling via the upregulation of CARD3 expression. Theranostics. 2020;10:323–339. doi: 10.7150/thno.38870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen S, Zhang L, Li M, Zhang Y, Sun M, Wang L, Lin J, Cui Y, Chen Q, Jin C, et al. Fusobacterium nucleatum reduces METTL3-mediated m6A modification and contributes to colorectal cancer metastasis. Nat Commun. 2022;13:1248. doi: 10.1038/s41467-022-28913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis. 2011;203:306–311. doi: 10.1093/jinfdis/jiq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10:575–582. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 86.Guo P, Tian Z, Kong X, Yang L, Shan X, Dong B, Ding X, Jing X, Jiang C, Jiang N, Guo P, Tian Z, Kong X, Yang L, Shan X, Dong B, Ding X, Jing X, Jiang C, Jiang N, et al. FadA promotes DNA damage and progression of Fusobacterium nucleatum-induced colorectal cancer through up-regulation of chk2. J Exp Clin Cancer Res. 2020;39:202. 10.1186/s13046-020-01677-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu Y-N, Yu T-C, Zhao H-J, Sun -T-T, Chen H-M, Chen H-Y, An H-F, Weng Y-R, Yu J, Li M, et al. Berberine may rescue Fusobacterium nucleatum-induced colorectal tumorigenesis by modulating the tumor microenvironment. Oncotarget. 2015;6:32013–32026. doi: 10.18632/oncotarget.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, JEM E, Klutstein M, Tayeb S, Almogy G, Atlan KA, et al. Colon cancer-associated fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front Cell Infect Microbiol. 2020;10:400. doi: 10.3389/fcimb.2020.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Komiya Y, Shimomura Y, Higurashi T, Sugi Y, Arimoto J, Umezawa S, Uchiyama S, Matsumoto M, Nakajima A. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut. 2019;68:1335–1337. doi: 10.1136/gutjnl-2018-316661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358:1443–1448. doi: 10.1126/science.aal5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomkovich S, Yang Y, Winglee K, Gauthier J, Mühlbauer M, Sun X, Mohamadzadeh M, Liu X, Martin P, Wang GP, et al. Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Res. 2017;77:2620–2632. doi: 10.1158/0008-5472.CAN-16-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Queen J, JC D, JR W, Stevens C, Udayasuryan B, Ttd N, Wu S, Ding H, Fan H, McMann M, et al. Comparative Analysis of Colon Cancer-Derived Fusobacterium nucleatum Subspecies: inflammation and Colon Tumorigenesis in Murine Models. MBio. 2022;13:e0299121. doi: 10.1128/mbio.02991-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kong C, Liang L, Liu G, Du L, Yang Y, Liu J, Shi D, Li X, Ma Y. Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer. Gut. 2022;gutjnl-2022–327156. doi: 10.1136/gutjnl-2022-327156. [DOI] [PubMed] [Google Scholar]
- 94.Liu W, Zhang X, Xu H, Li S, Lau -HC-H, Chen Q, Zhang B, Zhao L, Chen H, Sung JJ-Y, et al. Microbial community heterogeneity within colorectal neoplasia and its correlation with colorectal carcinogenesis. Gastroenterology. 2021;160:2395–2408. doi: 10.1053/j.gastro.2021.02.020. [DOI] [PubMed] [Google Scholar]
- 95.Ito M, Kanno S, Nosho K, Sukawa Y, Mitsuhashi K, Kurihara H, Igarashi H, Takahashi T, Tachibana M, Takahashi H, et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer. 2015;137:1258–1268. doi: 10.1002/ijc.29488. [DOI] [PubMed] [Google Scholar]
- 96.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–1980. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brennan CA, Nakatsu G, Gallini Comeau CA, Drew DA, Glickman JN, Schoen RE, Chan AT, Garrett WS. Aspirin modulation of the colorectal cancer-associated microbe fusobacterium nucleatum. MBio. 2021;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mughini-Gras L, Schaapveld M, Kramers J, Mooij S, Neefjes-Borst EA. Pelt W van, Neefjes J. Increased colon cancer risk after severe Salmonella infection. PLoS ONE. 2018;13:e0189721. doi: 10.1371/journal.pone.0189721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Duijster JW, Hansen JV, Franz E, Neefjes JJC, Frisch M, Mughini-Gras L, Ethelberg S. Association between Salmonella infection and colon cancer: a nationwide registry-based cohort study. Epidemiol Infect. 2021;149:e56. doi: 10.1017/S0950268821000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kato I, Boleij A, Kortman GAM, Roelofs R, Djuric Z, Severson RK, Tjalsma H. Partial associations of dietary iron, smoking and intestinal bacteria with colorectal cancer risk. Nutr Cancer. 2013;65:169–177. doi: 10.1080/01635581.2013.748922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu R, Bosland M, Xia Y, Zhang Y-G, Kato I, Sun J. Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget. 2017;8:55104–55115. doi: 10.18632/oncotarget.19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu R, Wu S, Zhang Y-G, Xia Y, Zhou Z, Kato I, Dong H, Bissonnette M, Sun J. Salmonella protein avra activates the STAT3 signaling pathway in colon cancer. Neoplasia. 2016;18:307–316. doi: 10.1016/j.neo.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu R, Wu S, Zhang YG, Xia Y, Liu X, Zheng Y, Chen H, Schaefer KL, Zhou Z, Bissonnette M, et al. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis. 2014;3:e105. doi: 10.1038/oncsis.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 105.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115:281–286. doi: 10.1016/S0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 106.Magdy A, Elhadidy M, Abd Ellatif ME, El Nakeeb A, Abdallah E, Thabet W, Youssef M, Khafagy W, Morshed M, Farid M. Enteropathogenic Escherichia coli (EPEC): does it have a role in colorectal tumourigenesis? A Prospective Cohort Study. Int J Surg. 2015;18:169–173. doi: 10.1016/j.ijsu.2015.04.077. [DOI] [PubMed] [Google Scholar]
- 107.Roxas JL, Koutsouris A, Viswanathan VK. Enteropathogenic Escherichia coli-induced epidermal growth factor receptor activation contributes to physiological alterations in intestinal epithelial cells. Infect Immun. 2007;75:2316–2324. doi: 10.1128/IAI.01690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taieb F, J-P N, Watrin C, Samba-Louaka A, Oswald E. Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. Cell Microbiol. 2006;8:1910–1921. doi: 10.1111/j.1462-5822.2006.00757.x. [DOI] [PubMed] [Google Scholar]
- 109.Samba-Louaka A, Nougayrède J-P, Watrin C, Oswald E, Taieb F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect Immun. 2009;77:5471–5477. doi: 10.1128/IAI.00860-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nougayrède J-P, Taieb F, De Rycke J, Oswald E. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005;13:103–110. doi: 10.1016/j.tim.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Z, Aung KM, Uhlin BE, Wai SN. Reversible senescence of human colon cancer cells after blockage of mitosis/cytokinesis caused by the CNF1 cyclomodulin from Escherichia coli. Sci Rep. 2018;8:17780. doi: 10.1038/s41598-018-36036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Choi HJ, Kim J, Do KH, Park SH, Moon Y. Enteropathogenic Escherichia coli-induced macrophage inhibitory cytokine 1 mediates cancer cell survival: an in vitro implication of infection-linked tumor dissemination. Oncogene. 2013;32:4960–4969. doi: 10.1038/onc.2012.508. [DOI] [PubMed] [Google Scholar]
- 113.Fabbri A, Travaglione S, Rosadi F, Ballan G, Maroccia Z, Giambenedetti M, Guidotti M, Ødum N, Krejsgaard T, Fiorentini C. The Escherichia coli protein toxin cytotoxic necrotizing factor 1 induces epithelial mesenchymal transition. Cell Microbiol. 2020;22:e13138. doi: 10.1111/cmi.13138. [DOI] [PubMed] [Google Scholar]
- 114.Drewes JL, Chen J, Markham NO, Knippel RJ, Domingue JC, Tam AJ, Chan JL, Kim L, McMann M, Stevens C, et al. Human colon cancer-derived clostridioides difficile strains drive colonic tumorigenesis in mice. Cancer Discov. 2022;12:1873–1885. doi: 10.1158/2159-8290.CD-21-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lam MMC, Wick RR, Wyres KL, Gorrie CL, Judd LM, Jenney AWJ, Brisse S, Holt KE. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. 2018;4(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Strakova N, Korena K, Karpiskova R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development - A systematic review. Toxicon. 2021;197:126–135. doi: 10.1016/j.toxicon.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 117.Pope JL, Yang Y, Newsome RC, Sun W, Sun X, Ukhanova M, Neu J, Issa J-P, Mai V, Jobin C. Microbial Colonization Coordinates the Pathogenesis of a Klebsiella pneumoniae Infant Isolate. Sci Rep. 2019;9:3380. doi: 10.1038/s41598-019-39887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu Y, Fu K, Wier EM, Lei Y, Hodgson A, Xu D, Xia X, Zheng D, Ding H, Sears CL, Liu Y, Fu K, EM Wier, Lei Y, Hodgson A, Xu D, Xia X, Zheng D, Ding H, CL Sears, et al. Bacterial genotoxin accelerates transient infection-driven murine colon tumorigenesis. Cancer Discov. 2022;12:236–249. 10.1158/2159-8290.CD-21-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Meier B, Volkova NV, Hong Y, Schofield P, Campbell PJ, Gerstung M, Gartner A. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018;28:666–675. doi: 10.1101/gr.226845.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, A-L B-D, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, Pons B, Mirey G, Vignard J, Hendrixson DR, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68:289–300. doi: 10.1136/gutjnl-2018-317200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37:952–963. doi: 10.1046/j.1365-2958.2000.02070.x. [DOI] [PubMed] [Google Scholar]
- 123.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green R, Meltzer PS, Wolfsberg TG, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–509. doi: 10.1038/nmeth888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Koohy H, Down TA, Hubbard TJ, Mariño-Ramírez L. Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS ONE. 2013;8:e69853. doi: 10.1371/journal.pone.0069853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Balamurugan R, Rajendiran E, George S, Samuel GV, Ramakrishna BS. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol. 2008;23:1298–1303. doi: 10.1111/j.1440-1746.2008.05490.x. [DOI] [PubMed] [Google Scholar]
- 126.de Almeida CV, Taddei A, Amedei A. The controversial role of Enterococcus faecalis in colorectal cancer. Therap Adv Gastroenterol. 2018;11:1756284818783606. doi: 10.1177/1756284818783606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.De Almeida CV, Lulli M, Di Pilato V, Schiavone N, Russo E, Nannini G, Baldi S, Borrelli R, Bartolucci G, Menicatti M, et al. Differential responses of colorectal cancer cell lines to enterococcus faecalis’ strains isolated from healthy donors and colorectal cancer patients. J Clin Med. 2019;9:8. doi: 10.3390/jcm9010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology. 2007;132:551–561. doi: 10.1053/j.gastro.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 129.Wang X, Allen TD, May RJ, Lightfoot S, Houchen CW, Huycke MM. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 2008;68:9909–9917. doi: 10.1158/0008-5472.CAN-08-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM. Colon Macrophages Polarized by Commensal Bacteria Cause Colitis and Cancer through the Bystander Effect. Transl Oncol. 2013;6:596–606. doi: 10.1593/tlo.13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut. 2015;64:459–468. doi: 10.1136/gutjnl-2014-307213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang X, Yang Y, Huycke MM. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget. 2017;8:102176–102190. doi: 10.18632/oncotarget.22250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y, Huycke MM, Herman TS, Glutathione WX. S-transferase alpha 4 induction by activator protein 1 in colorectal cancer. Oncogene. 2016;35:5795–5806. doi: 10.1038/onc.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lennard KS, Goosen RW, Blackburn JM, Goel A. Bacterially-associated transcriptional remodelling in a distinct genomic subtype of colorectal cancer provides a plausible molecular basis for disease development. PLoS ONE. 2016;11:e0166282. doi: 10.1371/journal.pone.0166282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gaines S, van Praagh JB, Williamson AJ, Jacobson RA, Hyoju S, Zaborin A, Mao J, Koo HY, Alpert L, Bissonnette M, et al. Western diet promotes intestinal colonization by collagenolytic microbes and promotes tumor formation after colorectal surgery. Gastroenterology. 2020;158(958–970.e2):958–970.e2. doi: 10.1053/j.gastro.2019.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Williamson AJ, Jacobson R, van Praagh JB, Gaines S, Koo HY, Lee B, Chan W-C, Weichselbaum R, Alverdy JC, Zaborina O, et al. Enterococcus faecalis promotes a migratory and invasive phenotype in colon cancer cells. Neoplasia. 2022;27:100787. doi: 10.1016/j.neo.2022.100787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Iida N, Mizukoshi E, Yamashita T, Yutani M, Seishima J, Wang Z, Arai K, Okada H, Yamashita T, Sakai Y, et al. Chronic liver disease enables gut Enterococcus faecalis colonization to promote liver carcinogenesis. Nat Cancer. 2021;2:1039–1054. doi: 10.1038/s43018-021-00251-3. [DOI] [PubMed] [Google Scholar]
- 138.Miyamoto S, Komiya M, Fujii G, Hamoya T, Nakanishi R, Fujimoto K, Tamura S, Kurokawa Y, Takahashi M, Ijichi T, et al. Preventive Effects of Heat-Killed Enterococcus faecalis Strain EC-12 on Mouse Intestinal Tumor Development. Int J Mol Sci. 2017;19:18. doi: 10.3390/ijms19010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, Chan AWH, Chan FKL, Sung JJY, Yu J. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology. 2017;152(1419–1433.e5):1419–1433.e5. doi: 10.1053/j.gastro.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 140.Osman MA, Neoh H-M, N-S AM, Chin S-F, Mazlan L, Raja Ali RA, Zakaria AD, Ngiu CS, Ang MY, Jamal R. Parvimonas micra, Peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci Rep. 2021;11:2925. doi: 10.1038/s41598-021-82465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Purcell RV, Visnovska M, Biggs PJ, Schmeier S, Frizelle FA. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep. 2017;7:11590. doi: 10.1038/s41598-017-11237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, Ng SC, Tsoi H, Dong Y, Zhang N, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhao L, Zhang X, Zhou Y, Fu K, Lau -HC-H, Chun TW-Y, Cheung AH-K, Coker OO, Wei H, Wu W-K-K, et al. Parvimonas micra promotes colorectal tumorigenesis and is associated with prognosis of colorectal cancer patients. Oncogene. 2022;41:4200–4210. doi: 10.1038/s41388-022-02395-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, Coker OO, Chan AWH, Chan FKL, Sung JJY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol. 2019;4:2319–2330. doi: 10.1038/s41564-019-0541-3. [DOI] [PubMed] [Google Scholar]
- 145.Vega P, Valentín F, Cubiella J. Colorectal cancer diagnosis: pitfalls and opportunities. World J Gastrointest Oncol. 2015;7:422–433. doi: 10.4251/wjgo.v7.i12.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yuan B, Ma B, Yu J, Meng Q, Du T, Li H, Zhu Y, Sun Z, Ma S, Song C. Fecal Bacteria as Non-Invasive Biomarkers for Colorectal Adenocarcinoma. Front Oncol. 2021;11:664321. doi: 10.3389/fonc.2021.664321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, Tang L, Zhao H, Stenvang J, Li Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66:70–78. doi: 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 149.Avuthu N, Guda C, Claesen J. Meta-analysis of altered gut microbiota reveals microbial and metabolic biomarkers for colorectal cancer. Microbiol Spectr. 2022;10:e0001322. doi: 10.1128/spectrum.00013-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bosch S, Acharjee A, Quraishi MN, Bijnsdorp IV, Rojas P, Bakkali A, Jansen EE, Stokkers P, Kuijvenhoven J, Pham TV, et al. Integration of stool microbiota, proteome and amino acid profiles to discriminate patients with adenomas and colorectal cancer. Gut Microbes. 2022;14:2139979. doi: 10.1080/19490976.2022.2139979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pessione E. Lactic acid bacteria contribution to gut microbiota complexity: lights and shadows. Front Cell Infect Microbiol. 2012;2:86. doi: 10.3389/fcimb.2012.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kvakova M, Kamlarova A, Stofilova J, Benetinova V, Bertkova I. Probiotics and postbiotics in colorectal cancer: prevention and complementary therapy. World J Gastroenterol. 2022;28:3370–3382. doi: 10.3748/wjg.v28.i27.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Agah S, Alizadeh AM, Mosavi M, Ranji P, Khavari-Daneshvar H, Ghasemian F, Bahmani S, Tavassoli A. More Protection of Lactobacillus acidophilus Than Bifidobacterium bifidum Probiotics on Azoxymethane-Induced Mouse Colon Cancer. Probiotics Antimicrob Proteins. 2019;11:857–864. doi: 10.1007/s12602-018-9425-8. [DOI] [PubMed] [Google Scholar]
- 155.Zhuo Q, Yu B, Zhou J, Zhang J, Zhang R, Xie J, Wang Q, Zhao S. Lysates of Lactobacillus acidophilus combined with CTLA-4-blocking antibodies enhance antitumor immunity in a mouse colon cancer model. Sci Rep. 2019;9:20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Khalil MA, Sonbol FI, Al-Madboly LA, Aboshady TA, Alqurashi AS, Ali SS. Exploring the therapeutic potentials of exopolysaccharides derived from lactic acid bacteria and bifidobacteria: antioxidant, antitumor, and periodontal regeneration. Front Microbiol. 2022;13:803688. doi: 10.3389/fmicb.2022.803688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Konishi H, Fujiya M, Tanaka H, Ueno N, Moriichi K, Sasajima J, Ikuta K, Akutsu H, Tanabe H, Kohgo Y. Probiotic-derived ferrichrome inhibits colon cancer progression via JNK-mediated apoptosis. Nat Commun. 2016;7:12365. doi: 10.1038/ncomms12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bi Z, Cui E, Yao Y, Chang X, Wang X, Zhang Y, Xu G-X, Zhuang H, Hua Z-C. Recombinant Bifidobacterium longum carrying endostatin protein alleviates dextran sodium sulfate-induced colitis and colon cancer in rats. Front Microbiol. 2022;13:927277. doi: 10.3389/fmicb.2022.927277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li Q, Hu W, Liu W-X, Zhao L-Y, Huang D, Liu X-D, Chan H, Zhang Y, Zeng J-D, Coker OO, et al. Streptococcus thermophilus Inhibits Colorectal Tumorigenesis Through Secreting β-Galactosidase. Gastroenterology. 2021;160(1179–1193.e14):1179-1193. [DOI] [PubMed] [Google Scholar]
- 160.Sugimura N, Li Q, Chu ESH, Lau HCH, Fong W, Liu W, Liang C, Nakatsu G, Su ACY, Coker OO, et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut. 2021;71:2011–2021. doi: 10.1136/gutjnl-2020-323951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Oh NS, Lee JY, Oh S, Joung JY, Kim SG, Shin YK, Lee K-W, Kim SH, Kim Y. Improved functionality of fermented milk is mediated by the synbiotic interaction between Cudrania tricuspidata leaf extract and Lactobacillus gasseri strains. Appl Microbiol Biotechnol. 2016;100:5919–5932. doi: 10.1007/s00253-016-7414-y. [DOI] [PubMed] [Google Scholar]
- 162.Oh NS, Lee JY, Kim Y. The growth kinetics and metabolic and antioxidant activities of the functional synbiotic combination of Lactobacillus gasseri 505 and Cudrania tricuspidata leaf extract. Appl Microbiol Biotechnol. 2016;100:10095–10106. doi: 10.1007/s00253-016-7863-3. [DOI] [PubMed] [Google Scholar]
- 163.NS O, JY L, Y-T K, SH K, Lee J-H. Cancer-protective effect of a synbiotic combination between Lactobacillus gasseri 505 and a Cudrania tricuspidata leaf extract on colitis-associated colorectal cancer. Gut Microbes. 2020;12:1785803. doi: 10.1080/19490976.2020.1785803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Oliver A, Chase AB, Weihe C, Orchanian SB, Riedel SF, Hendrickson CL, Lay M, Sewall JM, Martiny JBH, High-Fiber WK. Whole-Food Dietary Intervention Alters the Human Gut Microbiome but Not Fecal Short-Chain Fatty Acids. mSystems. 2021;6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lancaster SM, Lee-McMullen B, Abbott CW, Quijada JV, Hornburg D, Park H, Perelman D, Peterson DJ, Tang M, Robinson A, et al. Global, distinctive, and personal changes in molecular and microbial profiles by specific fibers in humans. Cell Host Microbe. 2022;30(848–862.e7):848–862.e7. doi: 10.1016/j.chom.2022.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stintzing S. Management of colorectal cancer. F1000Prime Rep. 2014;6:108. doi: 10.12703/P6-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nagata N, Nishijima S, Miyoshi-Akiyama T, Kojima Y, Kimura M, Aoki R, Ohsugi M, Ueki K, Miki K, Iwata E, et al. Population-level metagenomics uncovers distinct effects of multiple medications on the human gut microbiome. Gastroenterology. 2022;163:1038–1052. doi: 10.1053/j.gastro.2022.06.070. [DOI] [PubMed] [Google Scholar]
- 168.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Serna G, Ruiz-Pace F, Hernando J, Alonso L, Fasani R, Landolfi S, Comas R, Jimenez J, Elez E, Bullman S, et al. Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann Oncol. 2020;31:1366–1375. doi: 10.1016/j.annonc.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170:548–563.e16. doi: 10.1016/j.cell.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang S, Yang Y, Weng W, Guo B, Cai G, Ma Y, Cai S. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J Exp Clin Cancer Res. 2019;38:14. doi: 10.1186/s13046-018-0985-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–1160. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Newsome RC, Gharaibeh RZ, Pierce CM, da Silva WV, Paul S, Hogue SR, Yu Q, Antonia S, Conejo-Garcia JR, Robinson LA, et al. Interaction of bacterial genera associated with therapeutic response to immune checkpoint PD-1 blockade in a United States cohort. Genome Med. 2022;14:35. doi: 10.1186/s13073-022-01037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 175.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, Luke JJ, Gajewski TF. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602–609. doi: 10.1126/science.abb5920. [DOI] [PubMed] [Google Scholar]
- 178.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, Davar D, AK Dzutsev, JA McCulloch, RR Rodrigues, J-M Chauvin, RM Morrison, RN Deblasio, Menna C, Ding Q, Pagliano O, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595–602. 10.1126/science.abf3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jobin C. Precision medicine using microbiota. Science. 2018;359:32–34. doi: 10.1126/science.aar2946. [DOI] [PubMed] [Google Scholar]
- 180.Lee KA, Thomas AM, Bolte LA, Björk JR, de Ruijter LK, Armanini F, Asnicar F, Blanco-Miguez A, Board R, Calbet-Llopart N, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med. 2022;28:535–544. doi: 10.1038/s41591-022-01695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Derosa L, Routy B, Thomas AM, Iebba V, Zalcman G, Friard S, Mazieres J, Audigier-Valette C, Moro-Sibilot D, Goldwasser F, et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat Med. 2022;28:315–324. doi: 10.1038/s41591-021-01655-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369:1481–1489. doi: 10.1126/science.abc3421. [DOI] [PubMed] [Google Scholar]
- 184.Griffin ME, Espinosa J, Becker JL, Luo J-D, Carroll TS, Jha JK, Fanger GR, Hang HC. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science. 2021;373:1040–1046. doi: 10.1126/science.abc9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kawanabe-Matsuda H, Takeda K, Nakamura M, Makino S, Karasaki T, Kakimi K, Nishimukai M, Ohno T, Omi J, Kano K, et al. Dietary lactobacillus-derived exopolysaccharide enhances immune-checkpoint blockade therapy. Cancer Discov. 2022;12:1336–1355. doi: 10.1158/2159-8290.CD-21-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iftekhar A, Berger H, Bouznad N, Heuberger J, Boccellato F, Dobrindt U, Hermeking H, Sigal M, Meyer TF. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat Commun. 2021;12:1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Galeano Niño JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, Futran N, Houlton J, Sather C, Sicinska E, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. 2022;611:810–817. doi: 10.1038/s41586-022-05435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Schmidt F, Zimmermann J, Tanna T, Farouni R, Conway T, Macpherson AJ, Platt RJ. Noninvasive assessment of gut function using transcriptional recording sentinel cells. Science. 2022;376:eabm6038. doi: 10.1126/science.abm6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Smith AB, Jenior ML, Keenan O, Hart JL, Specker J, Abbas A, Rangel PC, Di C, Green J, Bustin KA, et al. Enterococci enhance Clostridioides difficile pathogenesis. Nature. 2022;611:780–786. doi: 10.1038/s41586-022-05438-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, Ketelaars SLC, Cheng K, Vervier K, Shental N, et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature. 2021;592:138–143. doi: 10.1038/s41586-021-03368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhu Y, Shi T, Lu X, Xu Z, Qu J, Zhang Z, Shi G, Shen S, Hou Y, Chen Y, et al. Fungal-induced glycolysis in macrophages promotes colon cancer by enhancing innate lymphoid cell secretion of IL-22. EMBO J. 2021;40:e105320. doi: 10.15252/embj.2020105320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stern J, Miller G, Li X, Saxena D. Virome and bacteriome: two sides of the same coin. Curr Opin Virol. 2019;37:37–43. doi: 10.1016/j.coviro.2019.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lin Y, Lau HCH, Liu Y, Kang X, Wang Y, Ting NLN, Kwong TNY, Han J, Liu W, Liu C, Lin Y, HC-H Lau, Liu Y, Kang X, Wang Y, NL-N Ting, TN-Y Kwong, Han J, Liu W, Liu C, et al. Altered mycobiota signatures and enriched pathogenic aspergillus rambellii are associated with colorectal cancer based on multicohort fecal metagenomic analyses. Gastroenterology. 2022;163:908–921. 10.1053/j.gastro.2022.06.038. [DOI] [PubMed] [Google Scholar]
- 194.Nakatsu G, Zhou H, Wu WKK, Wong SH, Coker OO, Dai Z, Li X, Szeto C-H, Sugimura N, Lam -TY-T, et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology. 2018;155(529–541.e5):529–541.e5. doi: 10.1053/j.gastro.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 195.Coker OO, Nakatsu G, Dai RZ, Wu WKK, Wong SH, Ng SC, Chan FKL, Sung JJY, Yu J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut. 2019;68:654–662. doi: 10.1136/gutjnl-2018-317178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, Ajami NJ, Wong MC, Ghazaryan A, Valentine JF, et al. Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell Host Microbe. 2019;25(285–299.e8):285–299.e8. doi: 10.1016/j.chom.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Federici S, Kredo-Russo S, Valdés-Mas R, Kviatcovsky D, Weinstock E, Matiuhin Y, Silberberg Y, Atarashi K, Furuichi M, Oka A, et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell. 2022;185(2879–2898.e24):2879–2898.e24. doi: 10.1016/j.cell.2022.07.003. [DOI] [PubMed] [Google Scholar]
- 198.Volpe MR, Velilla JA, Daniel-Ivad M, Yao JJ, Stornetta A, Villalta PW, Huang H-C, Bachovchin DA, Balbo S, Gaudet R, et al. A small molecule inhibitor prevents gut bacterial genotoxin production. Nat Chem Biol. 2023;19:159–167. doi: 10.1038/s41589-022-01147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Velilla JA, Volpe MR, Kenney GE, Walsh RM, Balskus EP, Gaudet R. Structural basis of colibactin activation by the ClbP peptidase. Nat Chem Biol. 2022;19(2):151-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yue Y, Xu J, Li Y, Cheng K, Feng Q, Ma X, Ma N, Zhang T, Wang X, Zhao X, et al. Antigen-bearing outer membrane vesicles as tumour vaccines produced in situ by ingested genetically engineered bacteria. Nat Biomed Eng. 2022;6:898–909. doi: 10.1038/s41551-022-00886-2. [DOI] [PubMed] [Google Scholar]
- 201.Wang W, Xu H, Ye Q, Tao F, Wheeldon I, Yuan A, Hu Y, Wu J. Systemic immune responses to irradiated tumours via the transport of antigens to the tumour periphery by injected flagellate bacteria. Nat Biomed Eng. 2022;6:44–53. doi: 10.1038/s41551-021-00834-6. [DOI] [PubMed] [Google Scholar]
- 202.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L-A, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.