

Abstract
Checkpoint blockade of the immunoreceptor Programmed Cell Death-1 (PD1) with its ligand-1 (PDL1) by monoclonal antibodies such as pembrolizumab provided compelling clinical results in various cancer types, yet the molecular mechanism by which this drug blocks the PD1/PDL1 interface remains unclear. To address this question, we examined the conformational motion of PD1 associated with the binding of pembrolizumab. Our results revealed that the innate plasticity of both C’D and FG loops is crucial to form a deep binding groove (371 Å3) across several distant epitopes of PD1. This analysis ultimately provided a rational-design to create pembrolizumab H3 loop mimics [RDYRFDMGFD] into β-hairpin scaffolds. As a result, a 20-residue long β-hairpin peptide 1e was identified as a first-in-class potent PD1-inhibitor (EC50 of 0.29 μM; Ki of 41 nM).
Keywords: Programmed Cell Death-1 (PD1), Pembrolizumab, Protein plasticity, Antibody H3 loop mimics, PD1 Inhibitors, β-hairpin peptides
Graphical Abstract
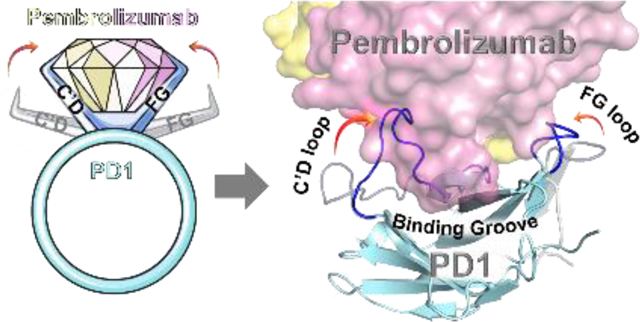
Bent to Bind. Pembrolizumab is the flagship of PD1 blockers used in immuno-oncology. A blueprint of the PD-1•pembrolizumab binding revealed that ~40% of the total interface resides at the rigid CDR-H3 loop periphery through the formation of a dynamic groove on PD1. This was exploited to rationally mimic the pembrolizumab H3-loop into smaller β-hairpin scaffolds as PD1/PDL1 blockers.
Introduction
Immune checkpoint inhibitors have drawn considerable interest in cancer therapy due to their efficacy and their long-lasting clinical responses in various cancer types.[1,2] The immune checkpoint involving PD-1 (i.e. CD279) and its ligand 1 (PDL1, i.e. CD274 or B7-H1) was established as a major co-inhibitory pathway exploited by tumors to evade detection by activated T cells and suppress immune responses.[3–5] PD1 expression in antigen-specific CD8+ cells is indicative of T-cell exhaustion[6], and it was shown that PD1 also promotes differentiation of CD4+ cells into Treg cells further supporting an immune suppressive microenvironment favorable to tumor growth. Monoclonal antibodies (mAbs) blocking this PD1/PDL1 checkpoint have been shown to restore T-cell function and rapidly emerged at the forefront of one of the most significant breakthroughs in cancer (immuno)therapy.[7,8] The anti-PD-1 blocker pembrolizumab[9] is being tested in combination with small molecules in over 750 clinical trials.[10] Despite this success, only a fraction of patients show a durable and complete response to most PD1/PDL1 blockers[11–13], and some immune-related adverse events have been reported.[14,15] While large mAb blockers have demonstrated impressive clinical results[16], only a handful of small-molecule inhibitors of PDL1 have been developed[17–27], and no PD1 inhibitors have been reported to date. For these reasons, the development of effective small-molecule inhibitors of the PD1/PDL1 interaction is needed (Figure 1). To gain insights into the PD1 activity modulation,[28] a comprehensive structural and conformational analysis of several high-resolution PD1•pembrolizumab co-crystal structures is reported herein, thus revealing the dynamic nature of PD1 and the formation of a binding groove between the C’D and FG loops upon contact with pembrolizumab. Our results suggest that the CDR-H3 loop (complementary determining region heavy-chain 3) of pembrolizumab (BSA of 460 Å2, a total of 10 contacts with PD1) can be exploited to design peptide-based PD1 inhibitors (Figure 1). Using a long-loop display technology recently developed by our group[29], several H3 loop mimics [99RDYRFDMGFD108] were synthesized within β-hairpin scaffolds and evaluated for PD1 inhibition in vitro.[30] As a result, two β-hairpin 1e and 2c blockers of the PD1/PDL1 interaction (EC50 < 600 nM) are reported which validates a new promising modality for designing small-molecule PD1 inhibitors.
Figure 1.

Strategy overview for the design of β-hairpin inhibitors targeting the PD1/PDL1 protein-protein interaction inspired by the CDR-H3 loop of pembrolizumab. The corresponding binding groove formed between the C’D and FG loops of PD1 is highlighted in blue with the binding surface to PDL1 in green (overlap in turquoise).
Results and Discussion
The Plasticity of distal C’D and FG Loops Forges a Binding Groove at the Interface between PD1 and Pembrolizumab.
It is becoming increasingly evident that protein plasticity plays a major role in protein–ligand binding, conformational selection and in epitope mapping in comparison to static models.[31–34] Even though the PD1/PDL1 binding was shown to be entropically-driven[35,36], the innate plasticity of PD1 has been largely overlooked until now. Therefore we thought to closely examine the dynamic nature of the PD1/pembrolizumab binding.[37,38] Previous molecular dynamics (MD) simulations by Liu and others suggested that the flexibility of the CC’ loop in apo-PD1 assisted a conformational pre-equilibrium of the receptor before binding PDL1.[39,40] The recent study by Kim and Tang also revealed that PDL2 triggers a major conformational motion of the CC’ and FG loops to create a favourable binding pocket at the surface of PD1.[41] Along these lines, a study by Goa suggested that the PD1FG loop plasticity played a crucial role in binding anti-PD1 mAb blockers.[42] Taken together, these studies support the notion that the apo-PD1 protein possesses a substantial innate plasticity (See Supplementary Video S1), therefore suggesting that pembrolizumab could bind PD1 through a dynamic conformational-fit mechanism.
The lack of complete structural and conformational information about the pembrolizumab•PD1 interface considerably hampered the design of PD1 inhibitors. Indeed, several assumptions in the initial interpretation of the co-crystal structure (PDB code: 5JXE, 2.9 Å resolution) –in which water molecules were missing and the PD1C’D loop backbone was undefined– thwarted the investigation of the main paratopes of pembrolizumab.[43] Thus, we carried out a comparative analysis of these contact interactions with crystal structures of higher atomic resolution between pembrolizumab antigen-binding fragments (Fabs) and PD1 obtained by Nomura[44] (PDB code: 5B8C, 2.1 Å resolution), and by Heo[45] (PDB code: 5GGS, 2.0 Å resolution). As shown in Figure 2A (see also Tables S2-4 and Fig. S1-2, Supporting Information), our structural analysis clearly revealed three distinct epitopes at the PD-1•pembrolizumab interface.[46]
Figure 2.

Overview of distal PD1 epitopes mapping the binding interface with pembrolizumab and their motion upon binding. (A) Ribbon representation of PD1 with patches of contact residues (pink) forming the three epitopes. (B) Conformational motion of the C’D and FG loops creating a binding groove on PD1 around the pembrolizumab H3 loop (yellow). Overlay of 7 selected conformers (light blue) extracted from the conformational motion simulation with the central rigid core of PD1 (grey) showing both extreme open and closed conformations (dark blue). Pro89 and Lys131 on each edge of the receptor are represented to show the Cα–Cα distance closing the groove onto pembrolizumab. see morphing Supplementary Video S2.
In brief, residues forming epitope-1 were mapped along the CC’ hairpin which contains a large hydrophobic core corresponding to the hinge region of the PD1 curvature (Figure 2B). Earlier computational[39] and NMR binding studies[36] as well as a crystallographic structure of the PD1•PDL1 complex (PDB code: 4ZQK)[47,48] also suggested that the CC’ loop is essential to ligand recognition. Given that pembrolizumab (Kd = 27 pM)[49] is an orthosteric inhibitor to PDL1 (Kd = 8.2 μM)[36], contacts generated solely at epitope-1 cannot be responsible for the higher affinity of pembrolizumab as previously speculated.[43] In fact, blockers of protein–protein interaction (PPI) must typically exhibit larger binding surfaces to compete with a native ligand.[50] In contrast, epitope-2 is constituted of a very localized patch of residues within the C’D loop and it was found to display a number of interactions (hydrophobic and H-bonds) across the CDRs H1–3 and CDR-L1,3 (Figure 2A). Those hydrogen bonds mainly involved the backbone amides of the C’D loop with CDR-H2 residues of pembrolizumab through water bridges. Due to the low-resolution crystal diffraction of the original pembrolizumab•PD-1 complex (5JXE), water molecule coordinates could not be determined accurately, leaving several solvent-exposed interactions between the C’D loop and pembrolizumab unexplained (sub-interface I in ref (43); epitope-2 in this work). Finally, epitope-3 is composed of two large patches on the BC and FG loops and the nearby residue V64 from the C strand that generate a large surface of contacts with several CDR-L1,2 and CDR-H3 sections of pembrolizumab. Interestingly, we found that this epitope-3 is highly lipophilic (~10 van der Walls contacts) leading to numerous contacts to the CDR-L2 segment rich in leucine and tyrosine residues (YL53, IL54, YL57, and IL58) in addition to YL34 from the CDR-L1. This analysis is consistent with previous mutagenesis studies which highlighted the important role of the PD1FG loop for ligand binding.[51]
To obtain a more accurate description of the PD1•pembrolizumab interface and refine the interpretation of the “hot spot” residues, a dynamic model was built (Figure 2B, see also Fig. S4-6, Supporting Information). In both unbounded and bounded states (to PDL1), the PD1C’D loop is disordered which is often correlated to an important backbone flexibility. Strikingly, in each pembrolizumab•PD1 cocrystal structures, the C’D loop is visibly structured and engaged in a number of contact interactions (Figure 3A/B). To analyse the conformational changes induced upon binding, the unresolved C’D loop of apo-PD1 (PDB code: 3RRQ) was remodelled using MODELLER[52] through a series of 500 low-energy conformers from which the lowest-energy state was extracted (Fig. S5, Supporting Information). The homology PD1 structure bearing a minimized de novo C’D loop (PDB file 3RRQ**, Supplementary Structure) was then engaged in MD and morphing simulations to assess the dynamic nature of the various loops.[53] As shown in the morphing simulation (Supplementary Videos S2-3), the C’D loop elasticity plays a pivotal role when binding to pembrolizumab. The plasticity was further confirmed by a 500 ns molecular dynamics (MD) simulation of the reconstituted free 3RRQ** and bounded PD1 structures using the ToeLoop algorithm.[54,55] In the calculated MD trajectories, the C’D loop mobility was characterized by “fast” dynamics (Fig. S7, Supporting Information). The overall flexibility of this 14-residue loop as well as the distance between segment termini (dPro83-Arg94 = 16.3 Å) are characteristic of a long Ω-type loop (Fig. S8, Supporting Information).[46,56,57] Our computational model clearly established that the open-closed transition of PD1 is characterized by a significant closure (12.1 Å) of both C’D and FG loops onto the CDR-H3 loop of pembrolizumab (Figure 2B).
Figure 3.

Rational design of pembrolizumab H3 loop mimics. Comparison of binding modes from. (A) Overlay of H3 loops [RH99 – DH108] excised from co-crystal structures (PDB: 5JXE, 5B8C, 5GGS, in magenta) with its unbounded form (PDB: 5DK3, in blue). (B) Close-up view of pembrolizumab CDR-H3 contact interactions at the interface with PD1; Ribbon representation of the binding complex (PDB code: 5GGS) with PD1 in cyan, and the heavy and light chains of pembrolizumab in yellow and pink respectively. The network of H-bonds and hydrophobic contacts from PD1 residues depicted in the same lighter colors. Direct and water mediated H-bonds are displayed with yellow and cyan dashed lines respectively, and one salt bridge with a red dashed line. (C) Summary of peptide H3-mimics 1a-e and 2a-c prepared in this study with variations of the native loop sequence CAR-[RDYRFDMGFD]-YWG and stabilizing strap motifs.
Overall, the two distal C’D and FG loops (epitope-2 and −3 respectively) display an important elasticity across the PD1 receptor to form a groove enveloping the CDR-H3 loop of pembrolizumab (Supplementary Videos S2-3). The dimensions of this binding groove were calculated by POCASA 1.1 using a probe sphere of 6-Å radius (Fig. S3, Supporting Information).[41,58] The total volume of 371 Å3 calculated for this cavity further confirmed the prominence of this deep groove formed at the interface with the CDR-H3 loop.
Rational Design of Pembrolizumab H3 Loop Mimics as PD1 Inhibitors.
The pembrolizumab antibody developed by Merck scientists was reported to be unusually compact and rigid, thus imparting a high degree of preorganization to the entire Fab Fv-region in solution.[9] As shown in Figure 3A, the rigidity and compactness of the H3-loop backbone were further confirmed by aligning the unbounded pembrolizumab structure (in blue, PDB code: 5DK3, 2.3 Å resolution) with other pembrolizumab-Fab structures excised from the corresponding X-ray complexes (PDB codes: 5JXE, 5B8C, and 5GGS). The root mean square deviation (RMSD) of 0.29 ± 0.06 Å over the backbone atoms of these H3 loops [ArgH99 – AspH108] attested of a significant structural preorganization. As shown in Figure 3B, the H3 loop makes contacts not only with residues in epitope-1 (YH101:T76, RH102:N66, and YH101/YH102:Y68/K78), but also with residues in epitope-2 with RH99:S87, the salt bridge RH99+/DH104-: D85-, and residues in epitope-3 YH101/RH102:E136, RH102:A132, and MH105/DH108:K131 alongside of several hydrophobic contacts created around FH103. Strikingly, we found that the H3 loop binding surface accounts for 460 Å2, which represents about 38% of the total antibody buried surface area (BSA estimated to 1,218 Å2).[46] Our calculations support an earlier hypothesis from Iwata and Nomura who postulated that the H3 loop could be exploited to create short pembrolizumab mimics.[44]
Therefore, we hypothesized that the H3 loop could offer a suitable rigidity and spatial distribution of binding “hot spots” dispersed across the PD1 binding groove to serve as a template for the development of PD1 inhibitors. Typically, “hot loops” found at the interface between antibodies proteins can be targeted by small-molecule and other proteomimetic inhibitors.[59,60] Yet designing inhibitors of dynamic PPIs similar to the PD1/PDL1 interface remains challenging.[61,62] To this aim, our strategy was to synthesize β-hairpins that could closely mimic the native fold of the H3 loop found in pembrolizumab (Figure 3C).[29] Given that long loops (N ≥ 10-mer) are inherently challenging to prepare because of the large entropic penalty associated with their nucleation,[63,64] we decided to exploit a technology of stabilizing β-straps developed in our laboratory. To display the 10-residue loop, we crafted the two most stabilizing β-strap motifs RW(VW)•••(WV)WE constituted of a hydrogen bond network, a hydrophobic core and a terminal salt bridge (Table inset, Figure 3C). All peptides 1a-d and 2a-c were synthesized by solid-phase peptide synthesis (SPPS) using a Fmoc-based chemistry.[46] As a control, two analogues 1a and 2a carrying simple unfunctional polyglycine G4KKG(2/4) loops were prepared to ascertain relationships between biological activity, loop sequences, and the overall conformational topology of these hairpins.[64]
Next, we verified the folding of these constructs by CD (circular dichroism) spectroscopy and their relative stability by CD-melts (Figure 4).[65] Facing W/W residues within a β-sheet (edge-to-face indole rings) generate a characteristic and intense exciton couplet on far UV-CD spectra comprised of negative and positive maxima at 214 and 229 ± 1 nm, which is commonly used to probe hairpin folding (Figure 4A).[66] The decrease in amplitude of molar ellipticity [θ(T)]229 upon heating was therefore exploited to measure the hairpin unfolding transitions. Thermal denaturations were performed in an aqueous buffer by heating the peptides from 0 to 95 °C (Figure 4B), and the unfolding transitions were fitted to a two-state model equation (hairpin/coil transition)[67] to obtain accurate melting temperatures (Tm) and the corresponding stability curves (Figure 4C). The best-fitted melting curves were computed to obtain plots of %-folding as a function of temperature for each hairpin and the corresponding thermodynamic parameters (Fig. S11, Table S8, Supporting Information). As shown in Table 1, scaffold 1a built around the shorter strap was found to be fully open (no exciton band at 229 ± 1 nm), while the second scaffold model 2a did fold into a β-hairpin structure characterized by a 87%-folding and a Tm of 52 °C. By judiciously placing P and G residues into the sequence, a synthetic coil model 1b was obtained and exploited to determine the molar ellipticity value corresponding to a fully unfolded random coil (baseline for all other melting curves).[46] As one could expect, the CD spectrum of the pembrolizumab mimic 1c (native sequence) RW-CAR-[RDYRFDMGFD]-YWG-WE did not exhibit the folding exciton at 229 nm, but a distinctive band at 200 ± 1 nm indicative of a coiled structure in solution (Fig. S9, Supporting Information). To improve the short strap structure, the native C3/G18 pair was replaced by a V/V pair in later constructs 1d-e. Excitingly, the intense exciton observed at 229 ± 1 nm in the case of peptides 1d-e and 2b-c (Figure 4A) and the thermal stability curves shown in Figure 4C supported the formation of the first well-folded β-hairpin tertiary structures. For most hairpins, the melting temperatures recorded were superior to physiological temperature which further demonstrated that both short and longer β-straps RW(VW)•••(WV)WE were highly stabilizing. All thermal denaturations were reversible as judged by the quasi-identical recovery of the 229 nm exciton intensity (> 95%) upon cooling the temperature back to 0 °C. Interestingly, the most flexible mimics 1d and 2b harbouring a F10G glycine mutation within the loop were found to be more soluble than the original sequences 1e and 2c and presented higher levels of folding (χF of 91 and 81% versus 55 and 54% respectively).
Figure 4.

Comparison of β-hairpin folding and denaturation properties. (A) Overlay of CD spectra for peptides 1 and 2 recorded at 20 °C in phosphate buffer (15 mM, pH 6.5). (B) Example of temperature dependence on the W/W exciton intensity at 214/229 nm (hairpin 2c) used to plot denaturation melting curves. (C) Thermal stability curves of unfolding free energy for hairpins 1d,e and 2a-c.
Table 1.
Structure−activity relationship of peptide inhibitors against the PD1/PDL1 interaction in comparison to pembrolizumab.
| ID | S (mM)a | Tm (°C) | %-Foldb | ELISAc EC50 (μM) |
|---|---|---|---|---|
| 1a | 5.4 ± 0.2 | n.a. | 0% | 9.69 ± 0.10 |
| 1b | 17.8 ± 0.3 | n.a. | RC | no binding |
| 1c | 0.3 ± 0.1 | n.a. | 0% | 3.70 ± 1.02 |
| 1d | 3.4 ± 0.1 | 59 | 91 ± 1% | 7.05 ± 0.35 |
| 1e | 0.9 ± 0.1 | 37 | 55 ± 1% | 0.29 ± 0.11 |
| 2a | 5.8 ± 0.2 | 52 | 87 ± 1% | 3.50 ± 0.12 |
| 2b | 0.6 ± 0.1 | 47 | 81 ± 1% | 7.08 ± 0.27 |
| 2c | 8.2 ± 0.2 | 25 | 54 ± 1% | 0.58 ± 0.18 |
| Pem | − | − | − | 0.9 10−3 ± 0.2 |
Thermodynamic solubility measured in a phosphate buffer (PB, 50 mM) at pH 7.4 (mean ± s.d., N = 3).
Hairpin folded fractions calculated at 293K from the best-fitted melting curves of thermal denaturation recorded by CD spectroscopy.
EC50 values obtained from the inhibitory dose-response curves of the PD1/PDL1 interaction at 293K. Data are reported as the average of triplicates (N = 3) with the corresponding s.d.
To confirm the tertiary fold of hairpins 1d-e and 2a-c suggested by our CD study, a careful structural assignment and conformational analysis were secured by a combination of 1H, TOCSY, NOESY and HSQC NMR data.[46] The solution structures of these hairpins were confirmed based on the large chemical shifts dispersion of HN and Hα signals, and the significant positive and negative chemical shift deviations (CSDs) observed in both β-strap and loop fragments respectively (see Fig. S12, Supporting Information). The hairpin folds were substantiated by several strong long-range NOESY correlations between backbone protons of the RWVW•••WVWE β-strap motif.[29] Collectively, our NMR data conclusively established that hairpins 1d-e and 2a-c were folded. On the other end, the spectral interpretation of 1a strongly suggested that the peptide was misfolded or existed as unfolded random coil conformers in solution.
As shown in Table 1, the loop-display system developed by our group enabled several analogues of the pembrolizumab H3-loop segment to be crafted into short β-hairpins. Hence, the inhibitory activity of these hairpins was evaluated in a PD1/PDL1 blocking experiment by enzyme-linked immunosorbent assay (ELISA). Two commercially available PDL1 inhibitors (BMS202 and BMS1166) and the full-length pembrolizumab antibody (anti-PD1 blocker) were tested as positive controls in this assay (Fig. S17, Supporting Information). Levels of PD1/PDL1 inhibition measured for both BMS small molecules were consistent with previous reports, with half-maximal effective concentration EC50 values in the submicromolar range of 0.92 ± 0.34 μM and 0.76 ± 0.32 μM respectively, whereas pembrolizumab exhibited a much stronger potency of 0.9 ± 0.2 nM (Table 1).[46] In the control experiments with both model constructs 1a/2a and the synthetic random coil 1b, no or only marginal inhibitory activities were detected (EC50 > 3.0 μM). The lack of activity from both unfunctionalized hairpin scaffold 2a and the related coil 1a suggest that neither the β-straps, nor the polyglycinyl loop strongly bind to PD1. In addition, the unfolded construct 1c (coil conformation established by NMR and CD data) bearing an identical loop sequence to pembrolizumab demonstrated no compelling activity (Table 1, EC50 of 3.70 μM). This result tends to suggest that the native peptide sequence found in 1c is not adequate to achieve a high inhibitory effect without adopting a stable β-hairpin structure. This result is consistent with the idea that rigid molecules may be more effective to bind the highly dynamic PD1 structure (entropic penalty, see rational design section). More surprisingly, whereas both hairpin mimics 1e and 2c blocked the PD1/PDL1 interaction in a dose-dependent manner with respective EC50 values of 0.29 and 0.58 μM, the more soluble and folded hairpins 1d and 2b bearing the F10G mutation revealed no meaningful inhibitory activity (Table 1, EC50 of 7.05 and 7.08 μM respectively). Hairpins 1e and 2c disrupted the PD1/PDL1 interaction at concentrations ~12 to 20-fold lower than their glycine-derived analogues (F10G). This direct comparison supports the notion that the F10 residue within the loop of hairpins 1e and 2c while increasing lipophilicity contributes significantly to the overall pharmacological activity of these inhibitors. Overall, the analysis of this series of peptides has shown that two features seem to affect the potency of inhibition: (1) a H3-loop sequence closely mimicking pembrolizumab and (2) a stable hairpin tertiary fold. These results validated our first design of pembrolizumab CDR-H3 mimics as inhibitors and suggested that hairpin stapling or macrocyclization might provide synthetic manifolds to create superior blockers of the PD1/PDL1 interaction.
Given that PDL1 is highly expressed on the surface of tumor cells and constantly present in large concentrations in its soluble form within the tumor microenvironment[68–70], we sought to evaluate our most potent inhibitor 1e in a more realistic setting through a competitive binding assay against PDL1.[27] The blocking activity profile of hairpin 1e was therefore tested in a competitive binding experiment in which the PD1•PDL1 complex formation was quantified overtime by UV-Vis absorbance (Figure 5).[71] In this assay, PD1 precoated plates were incubated with 1e (two-fold serial dilutions: 3.0 μM to 375 nM) or pembrolizumab at 67 pM, before being treated with an excess concentration of PDL1 (12-fold) to analyse the inhibitor displacement over the course of hundred minutes (Figure 5A). As shown in Figure 5B, even under saturation conditions, the endogenous PDL1 ligand (Kd of 7−8 μM)[36,72] could not outcompete the pembrolizumab blockade. This result verified that pembrolizumab fully blocked the PD1/PDL1 complex (< 5% dissociation) for a prolonged time, thus validating our assay. Then, progress curves generated at varying concentrations of hairpin 1e (with [1e]<<[PD1]<<[PDL1]) were fitted to a competitive binding model of kinetics accounting for a preincubation period.[73] First, the kinetic curves obtained through the PD1•1e disassociation established for the first time, that these blockers actually target PD1. At concentrations close to the IC50 value for hairpin 1e (375 and 750 nM), PDL1 displaced our inhibitor relatively rapidly. In contrast, the kinetic profile of PD1•1e disassociation drastically slowed down for 1e at 1.5 μM as shown by the initial velocities of PD1•PDL1 complex formation. These results were further confirmed by the excellent kinetic profile of 1e at 3.0 μM which exhibited a quasi-complete blockade of the PD1/PDL1 interaction throughout the first 60 minutes of experiment. Such progress curve nearly congruent to a linear regression in the first hour of assay is suggestive of a direct competitive inhibition of PDL1. To fit all the experimental time-course data to a competitive model of PD1-binding kinetics, both kon and koff of 1e were allowed to freely vary assuming the previously reported ka and kd values of PDL1[74] (see Eq. S8, Supporting Information). From this kinetic model, an inhibition constant Ki of 41 nM was obtained, which further demonstrated the high affinity of inhibitor 1e for PD1. In addition, the first-order dissociation rate constant koff of 4.3 10−4 s−1 is very similar to the ones obtained for pembrolizumab and other anti-PD1 antibodies[75], which strongly suggest that 1e forms a stable complex with PD1. Collectively, these results demonstrate that the binding affinity of hairpin 1e to PD1 is significantly superior than PDL1 rendering these β-hairpin peptides as an exciting starting point for future structure−activity guided drug selections.
Figure 5.

Competitive inhibition of the PD1/PDL1 interaction. (A) Illustration of the PDL1-competitive assay setup used to measure the hairpin 1e binding kinetics to PD1. (B) Progress curve analysis of [PD1•PDL1] formation at varying concentrations of inhibitor 1e in comparison to the full-length antibody pembrolizumab.a
a Experimental data points (mean ± s.d.) from two or four replicates (N ≥ 2) with non-linear fitted progress curves (dashed and dotted lines).
Conclusion
In summary, a comprehensive reinterpretation of the PD1•pembrolizumab interface from X-ray crystal structures was carried out to obtain a static blueprint of contact interactions. Our morphing and MD simulations further demonstrated that the two distal FG and C’D loops of PD1 possess enough plasticity to bend the edges of the receptor and form a deep binding groove of 371 Å3 latching onto the pembrolizumab H3 loop through a dynamic induced-fit mechanism (Supplementary Videos S2/S3). The remarkable conformational plasticity of PD1 presented herein is consistent with previous binding studies regarding PDL1[39,40] and PDL2[41], and contributes to understand the overall conformational discrimination mechanism of this receptor.[62] Central to this binding is the rigid H3 loop of pembrolizumab [RH99 – DH108] which was shown to create numerous contacts across a large interface (38% of the total Ab BSA). A series of H3 loop mimics (10 residue-long) were then rationally designed into β-hairpin structures for PD1 inhibition. We found that two hairpins 1e and 2c with a H3-loop display of pembrolizumab block the PD1/PDL1 interaction with EC50 of 0.29 and 0.58 μM respectively, and our results suggested that the central hydrophobic phenylalanine residue (F10) is an important loop pharmacophore. Finally, the most potent blocker 1e was confirmed as a PDL1 competitive inhibitor and a promising “hit” with a Ki of 41 nM. To our knowledge, hairpin 1e is the first non-antibody molecule reported as PD1 inhibitor to date. This study establishes a proof-of-concept that mimicking the pembrolizumab H3 loop is an attractive approach to generating novel peptide-based PD1 inhibitors of medium size. The activity and toxicity associated with such β-hairpin peptides will need to be confirmed on T cells and peripheral blood mononuclear cells (PBMCs) to further develop promising anti-PD1 lead candidates.[76,77]
Experimental Section
Loop Modeling and Morphing Simulation.
Due to the important plasticity of the C’D loop segment in the apo-PD-1 crystal structure (PDB code: 3RRQ), a remodeling and a minimum-energy calculation of this loop was required to determine a model of stable unbounded conformation. A large portion of the C’D loop of PD1 [Pro83 – Arg94] of sequence D85RSQPGQD92 was remodeled to obtain a set of 500 low-energy conformers generated using the loop modeling protocol DOPE-HR from the MODELLER add-on[78] in the UCSF Chimera Software[52] (Supplementary Figure S5A). The lowest-energy loop conformer was selected by comparison of free energy using the zDOPE scoring approach (zDOPE −1.93 au) to obtain an initial apo-PD1 model with a reconstituted C’D loop (PDB: 3RRQ*). Secondly, the missing E61 residue (BC loop) was added with Interactive ROSETTA (KIC protocol > SES loop modeling, De Novo) to generate the complete model of apo-PD-1 (PDB: 3RRQ**) shown in Supplementary Figure S5B. A morphing simulation was achieved to extrapolate the plausible trajectory of PD1 upon binding by using the apo-PD1 (PDB: 3RRQ**) as initial state, and a bounded PD1 structure extracted from the co-crystal with pembrolizumab (PDB: 5GGS) as final state. The new PD1 model (PDB: 3RRQ**) along with the apo-pembrolizumab (PDB: 3DK3) were overlaid with the reported PD1•pembrolizumab-Fab X-ray complex (PDB: 5GGS) (RMSD = 0.41 Å and 0.74 Å respectively, Supplementary Figure S6, STEP 1). Binding trajectories were calculated on each individual chain/object (pembrolizumab heavy-chain, light-chain and PD1 separately) to obtain a conformational simulation from the apo-forms to the bounded states through a motion comprising 60 states (60 refinement cycles, STEP 2). The 3 objects were reassembled together to obtain the full-length simulation with the motion of both pembrolizumab and PD1 and water molecules from the 5GGS crystal were added to the final file to maintain the hydrogen bond and salt bridge contacts involving water (STEP 3). Dynamic polar interactions between the two proteins are displayed in the final state to generate the morphing Supplementary Video S3.
Peptide Synthesis.
Peptides were prepared using Fmoc chemistry by solid-phase peptide synthesis (SPPS) on a Protein Technologies PS-3 peptide synthesizer. A detailed description of the peptide synthesis, cleavage and purity analysis procedures are given in the Supporting Information section: Peptide Synthesis and Purification.
Solubility Measurements.
For thermodynamic solubility measurements, ~1.0 mg of lyophilized peptide was weighed in a vial. Then, a solution of potassium phosphate buffer (50 mM PB, pH 7.4) was added by increments of 50 μL until saturation of the aqueous phase and some solid in suspension can still be observed. The mixture was sonicated (3 minutes) three to four times over the course of the solubilization process. The solution was then heated at 50 °C for 30 minutes and stirred at room temperature overnight. The solution was then centrifuged for 2 minutes to allow any undissolved peptide to separate from the supernatant. The maximum solubility concentration was measured by UV-absorbance based on the combined molar absorptivity of Trp and Tyr residues present in the peptide at 280 nm (ε280 = 5580 M−1.cm−1 per Trp, ε280 = 1280 M−1.cm−1 per Tyr). The maximum solubility concentration S (mM) was accurately calculated from the Beer-Lambert law.
Nuclear Magnetic Resonance (NMR) Data Collection.
NMR samples were prepared by dissolving a freeze-dried peptide (~ 1–2 mg) in a mixture of phosphate buffer (50 mM, pH 6.5) and D2O (9:1, v/v) for 1a,d and 2a; a mixture of phosphate buffer (50 mM, pH 6.5) and DMSO-d6 (7:3, v/v) for 2c; a mixture of water and DMSO-d6 (85:15, v/v) for 2b; and a mixture of water and DMSO-d6 (8:2, v/v) for 1e, using 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) as internal standard for chemical shifts (0 ppm). Samples were prepared in a range of 3–10 mM of peptide for 1H NMR, TOCSY (mixing time 80 ms), NOESY (mixing time 200 ms) and 1H−13C HSQC experiments. A PRESAT experiment was used to suppress the H2O solvent signal, in order to record the initial 1H NMR. All spectra were recorded at 291K (18 °C) on a Varian Mercury500 (500 MHz) spectrometer and processed using the Vnmrj 4.2 software. Signals assignments were obtained on the basis of a set of 1H and TOCSY spectra (for intra-residue connectivities), NOESY spectra (for vicinal and interstrand backbone connectivities), and HSQC spectra (for Hα to Cα connectivities and W side-chains assignments).
Far-UV Circular Dichroism (CD) Spectroscopy.
Peptide solutions were prepared at 20–100 μM concentration range in phosphate buffer (15 mM, pH 6.5), with addition of MeOH (up to 10% v/v) if required to increase solubility. Peptide concentrations were determined accurately by measuring the solution UV-absorbance using a JASCO V-670 spectrophotometer. Before initiating the conformational analysis, the peptides aggregation states were confirmed by several methods.[29] No concentration effects were found on the far-UV CD spectra (< 100 μM). CD spectra were recorded on a JASCO J-810 Spectropolarimeter with a temperature controller module JASCO PFD-425S. In brief, raw CD data were recorded in mdeg from 190 nm to 270 nm every 0.1 nm, and CD spectra of the blank solutions were subtracted for baseline correction. Spectra were smoothed, the baseline was set to 0 between 260 nm to 270 nm, and the ellipticity scale was converted into molar ellipticity (deg.cm2.dmol−1) using SpectraGryph 1.2.[79] To obtain the melting curves of peptides unfolding transitions, variable-temperature CD experiments were recorded at 229 nm (every 0.1 °C) with a temperature ramp speed of 0.75 °C/min from 0 to 95 °C. Likewise, intensities for the corresponding blank solutions were recorded and subtracted to afford the raw experimental melting curves. These melting curve intensities reported in molar ellipticity (deg.cm2.dmol−1) were divided by the number of tryptophan pairs present in the peptide construct ([θ(T)]229/pTrp) in order to normalize spectra. Finally, a protocol has been developed to compute the best-fitted melting curves from the raw data of [θ(T)]229 as function of temperature by using the thermodynamic Gibbs-Helmholtz equation and a nonlinear least square fitting routine programmed in Origin 9.0 (Originlab Corporation, U.S.A.). A detailed description of the %-folding best-fitted curves, and the thermodynamic calculations to obtain the values for ΔGF°, ΔHF°, ΔSF°, and Tm are given in the Supporting Information, section: Characterization of Peptide Constructs by NMR and CD Spectroscopy.
PD1 ImmunoBlocking ELISA.
Peptide inhibitors stock solutions were prepared at ~ 1–5 mM concentration range in DMSO. Assay kits for screening and profiling inhibitors of the PD1/PDL1 interaction were purchased from BPS Bioscience (cat # 72005). Briefly, a 96-well plate was coated with PDL1 at 2.0 μg/mL overnight at 4 °C. Plate was then washed and blocked with blocking buffer for 1 h at room temperature. Inhibitor solutions (0.01–100 μM) and blank (DMSO/PBS buffer solution) were added to designated wells. To initiate the reaction, 0.5 ng/μL of biotin-labeled human PD-1 was then added into positive control wells and inhibitor solution wells. After 2 h incubation at room temperature, plate was washed and blocked again for 10 mins. Streptavidin-HRP (1:1000 dilution) was added and incubate for 1 h at room temperature with slow shaking. Plate was washed and block again for 10 mins. ELISA ECL substrate was then added and the resulting chemoluminescence was immediately read in a luminometer (FLUOstar OPTIMA, BMG Labtech). Luminescence value of the blank is subtracted from all readings according to manufacturer instruction. The half maximal effective concentrations (EC50) values were determined from the fit of experimental normalized luminescence data to the Hill model with RLUmax values representing maximal relative luminescence values from the PD1/PDL1 interaction. Average EC50 values and the corresponding standard deviations were calculated from three independent experiments.
UV/Vis-Colorimetric Competition Assay.
Assay kits were purchased from BPS Bioscience (cat# 72016). Briefly, 96-well plates were coated with PD1 at 2.0 μg/mL overnight at 4 °C. Plates were then washed and blocked with blocking buffer for 1 h at room temperature. The two-fold serial dilutions of peptide competitor 1e (3.0, 1.5, 0.75, and 0.375 μM), and a blank solution were added to designated wells and incubated at room temperature for 60 mins. To initiate the reaction, biotinylated PDL1 in the excess amount of 30 μg/mL was added every 10 mins for 2 h. Plates were then washed and blocked again for 10 mins. Streptavidin-HRP (1:1000 dilution) was added and incubate for 1 h at room temperature with slow shaking. Plates were washed and blocked again for 10 mins. A colorimetric streptavidin-HRP substrate was then added until a blue color is developed in the positive control well, then 100 μl of a HCl solution (1.0 M) was added to each well. The absorbance was measured at 450 nm using a Biorad xMark™ microplate absorbance spectrophotometer. Measured absorbance consistent with the PD1•PDL1 complex formation were plotted as a function of time at the varying concentrations of peptide blocker 1e (Figure 5). Experimental data points at concentrations of 0.375, 0.75, and 1.5 μM for 1e were obtained in duplicates, and in four replicates for 1e at 3.0 μM and pembrolizumab at 67 pM on separate plates. Best-fitted progress curves were computed to the kinetics of a preincubation competitive binding model (Eq. S8, Supporting Information) by a nonlinear least square fitting routine programmed in Origin 9.0. For a detailed description, see Supporting Information section: Competitive Binding Kinetics.
The Supporting Information is available free of charge on the xxx website. Supplementary data to this article presenting the reconstituted apo-PD1 ectodomain (3RRQ**), C’D loop modeling, conformational morphing protocols and videos S1-3 for the binding of PD1 with PDL1 and pembrolizumab, as well as the synthesis and characterization of peptide inhibitors are available online at https//doi…
Supplementary Material
Acknowledgements
We are very grateful for the financial support from the US National Institutes of Health (NIGMS Grant: R15GM116025 to M.Z., and Grant: R21GM132754 to A.D.R., G.Z. and S.P.R.). We thank Mr. Samir Hobloss for the initial study on peptide 1a, as well as Dr. M. Cudic and Dr. D. Du at Florida Atlantic University for the use of their peptide synthesizer. The authors also thank Dr. Giovanni Gonzalez-Gutierrez from the Macromolecular Crystallography Facility at Indiana University Bloomington for the stimulating discussions and sharing his expertise on the reinterpretation of the various crystal structures studied herein and Dr. G. Theodore from Theogen Corp. for proofreading and editing the manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Pardoll DM, Nat. Rev. Cancer 2012, 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sharma P, Allison JP, Cell 2015, 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Boussiotis VA, Engl N. J. Med 2016, 375, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Goodman A, Patel SP, Kurzrock R, Nat. Rev. Clin. Oncol 2017, 14, 203–220. [DOI] [PubMed] [Google Scholar]
- [5].De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P, Front. Immunol 2018, 9, 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen DS, Irving BA, Hodi FS, Clin. Cancer Res, 2012, 18, 6580–6587. [DOI] [PubMed] [Google Scholar]
- [8].Luke JJ, Ott OA, Oncotarget 2015, 6, 3479–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C, Nat. Struct. Mol. Biol 2015, 22, 953–958. [DOI] [PubMed] [Google Scholar]
- [10].Tang J, Yu JX, Hubbard-Lucey VM, Neftelinov ST, Hodge JP, Lin Y, Nat. Rev. Drug Discov 2018, 17, 854–855. [DOI] [PubMed] [Google Scholar]
- [11].Wang X, Teng F, Kong L, Yu J, OncoTargets Ther. 2016, 9, 5023–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].O’Donnell JS, Long GV, Scolyer RA, Cancer Treat. Rev, 2017, 52, 71–81. [DOI] [PubMed] [Google Scholar]
- [13].Nowicki TS, Hu-Lieskovan S, Ribas A, Cancer J. 2018, 24, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Narváez J, Juarez-López P, LLuch J, Narváez JA, Palmero R, García del Muro X, Nolla JM, Domingo-Domenech E, Autoimmun. Rev 2018, 17, 1040–1045. [DOI] [PubMed] [Google Scholar]
- [15].Wang Y, Zhou S, Yang F, Qi X, Wang X, Guan X, Shen C, Duma N, Vera Aguilera J, Chintakuntlawar A, Price KA, Molina JR, Pagliaro LC, Halfdanarson TR, Grothey A, Markovic SN, Nowakowski GS, Ansell SM, Wang ML, JAMA Oncol. 2019, 5, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lepir T, Zaghouani M, Roche SP, Li YY, Suarez M, Irias MJ, Savaraj N, Medicine 2019, 98, e13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miller MM, Mapelli C, Allen MP, Bowshe MS, Boy KM, Gillis EP, Langley DR, Mull E, Poirier MA, Sanghvi N, Sun LQ, Tenney DJ, Yeung KS, Zhu J, Reid PC, Scola PM, (Bristol-Myers Squibb Company), U.S. patent 20140294898 A1, Oct 02, 2014.
- [18].Skalniak L, Zak KM, Guzik K, Magiera K, Musielak B, Pachota M, Szelazek B, Kocik J, Grudnik P, Tomala M, Krzanik S, Pyrc K, Dömling A, Dubin G, Holak TA, Oncotarget 2017, 8, 72167–72187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Magiera-Mularz K, Skalniak L, Zak KM, Musielak B, Rudzinska-Szostak E, Berlicki Ł, Kocik J, Grudnik P, Sala D, Zarganes-Tzitzikas T, Shaabani S, Dömling A, Dubin G, Holak TA, Angew. Chem. Int. Ed 2017, 56, 13732–13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guzik K, Zak KM, Grudnik P, Magiera K, Musielak B, Törner R, Skalniak L, Dömling A, Dubin G, Holak TA, J. Med. Chem, 2017, 60, 5857–5867. [DOI] [PubMed] [Google Scholar]
- [21].Basu S, Yang J, Xu B, Magiera-Mularz K, Skalniak L, Musielak B, Kholodovych V, Holak TA, Hu L, J. Med. Chem 2019, 62, 7250–7263. [DOI] [PubMed] [Google Scholar]
- [22].Konieczny M, Musielak B, Kocik J, Skalniak L, Sala D, Czub M, Magiera-Mularz K, Rodriguez I, Myrcha M, Stec M, Siedlar M, Holak TA, Plewka J, J. Med. Chem 2020, 63, 11271–11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Muszak D, Surmiak E, Plewka J, Magiera-Mularz K, Kocik-Krol J, Musielak B, Sala D, Kitel R, Stec M, Weglar-czyk K, Siedlar M, Dömling A, Skalniak L, Holak TA, J. Med. Chem 2021, 64, 11614–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Butera R, Ważyńska M, Magiera-Mularz K, Plewka J, Musielak B, Surmiak E, Sala D, Kitel R, de Bruyn M, Nijman HW, Elsinga PH, Holak TA, Dömling A, ACS Med. Chem. Lett 2021, 12, 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miao Q, Zhang W, Zhang K, Li H, Zhu J, Jiang S, RSC Adv. 2021, 11, 23270–23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].For a recent overview small-molecule PDL1 inhibitors, see: Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, Kundu JK, Shields J, Agopsowicz KC, Xu L, Tabana Y, Srivastava N, Zhang G, Moon TC, Belovod-skiy A, Hena M, Kandadai AS, Hosseini SN, Hitt M, Walker J, Smylie M, West FG, Siraki AG, Lemieux MJ, Elahi S, Nieman JA, Tyrrell DL, Houghton M, Barakat K, Sci. Rep 2019, 9, 12392. See also references cited herein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yin H, Zhou X, Huang Y-H, King GJ, Collins BM, Gao Y, Craik DJ, Wang CK, J. Am. Chem. Soc 2021, 143, 18536–18547. [DOI] [PubMed] [Google Scholar]
- [28].Sharpe AH, Pauken KE, Nat. Rev. Immunol 2018, 18, 153–167. [DOI] [PubMed] [Google Scholar]
- [29].Richaud AD, Zhao G, Hobloss S, Roche SP, J. Org. Chem 2021, 86, 13535–13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zaghouani M, Zhao G, Richaud AD, Wangpaichitr M, Savaraj N, Roche SP, 2021, DOI 10.26434/chemrxiv.14477730.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Goh CS, Milburn D, Gerstein M, Curr. Opin. Struc. Biol 2004, 14, 104–109. [DOI] [PubMed] [Google Scholar]
- [32].Dobbins SE, Lesk VI, Sternberg MJE, Proc. Nat. Acad. Sci. USA 2008, 105, 10390–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lexa KW, Carlson HA, J. Am. Chem. Soc 2011, 133, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].For a recent review, see: Dishman AF, Volkman BF, ACS Chem. Biol 2018, 13, 1438–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pascolutti R, Sun X, Kao J, Maute RL, Ring AM, Bowman GR, Kruse AC, Structure 2016, 24, 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, Huo J, Yu C, Evans EJ, Leslie AJ, Griffiths M, Stubberfield C, Griffin R, Henry AJ, Jansson A, Ladbury JE, Ikemizu S, Carr MD, Davis SJ, J. Biol. Chem 2013, 288, 11771–11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R, Protein Sci. 2000, 9, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Grünberg R, Nilges M, Leckner J, Structure 2006, 14, 683–693. [DOI] [PubMed] [Google Scholar]
- [39].Liu W, Huang B, Kuang Y, Liu G, Mol. BioSyst 2017, 13, 892–900. [DOI] [PubMed] [Google Scholar]
- [40].Mittal L, Srivastava M, Kumari A, Tonk RK, Awasthi A, Asthana S, J. Chem. Inf. Model 2021, 61, 358–384. [DOI] [PubMed] [Google Scholar]
- [41].Tang S, Kim PS, Proc. Nat. Acad. Sci. USA 2019, 116, 24500–24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen D, Tan S, Zhang H, Wang H, He W, Shi R, Tong Z, Zhu J, Cheng H, Gao S, Chai Y, Qi J, Xiao M, Yan J, Gao GF, iScience 2019, 14, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Na Z, Yeo SP, Bharath SP, Bowler MW, Balıkçı E, Wang CI, Song H, Cell Res. 2017, 27, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Horita S, Nomura Y, Sato Y, Shimamura T, Iwata S, Nomura N, Sci. Rep 2016, 6, 35297–35304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, Lim H, Won Heo T, Park KY, Lee YJ, Ryu SE, Son JY, Lee JU, Heo YS, Nat. Commun 2016, 7, 13354–13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].See Supporting Information for complete experimental details.
- [47].Lin DYW, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN, Proc. Nat. Acad. Sci. USA 2008, 105, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA, Structure 2015, 23, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fessas P, Lee H, Ikemizu S, Janowitz T, Semin. Oncol 2017, 44, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wells JA, McClendon CL, Nature, 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- [51].Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC, Proc. Nat. Acad. Sci. USA 2008, 105, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [53].Shehu A, Kavraki LE, Entropy, 2012, 14, 252–290. [Google Scholar]
- [54].Gu Y, Li DW, Brüschweiler R, Chem J. Theory Comput. 2015, 11, 1308–1314. [DOI] [PubMed] [Google Scholar]
- [55].Gu Y, Li DW, Brüschweiler R, Bioinformatics 2017, 33, 1814–1819. [DOI] [PubMed] [Google Scholar]
- [56].Leszczynski J, Rose G, Science 1986, 234, 849–855. [DOI] [PubMed] [Google Scholar]
- [57].Fetrow JS, FASEB J., 1995, 9, 708–717. [PubMed] [Google Scholar]
- [58].Yu J, Zhou Y, Tanaka I, Yao M, Bioinformatics 2009, 26, 46–52. [DOI] [PubMed] [Google Scholar]
- [59].Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA, Nat. Chem. Biol 2014, 10, 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Horne WS, Grossmann TN, Nat. Chem 2020, 12, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R, Chem. Rev 2016, 116, 6391–6423. [DOI] [PubMed] [Google Scholar]
- [62].Amaral M, Kokh DB, Bomke J, Wegener A, Buchstaller HP, Eggenweiler HM, Matias P, Sirrenberg C, Wade RC, Frech M, Nat. Commun 2017, 8, 2276–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Anderson JM, Kier BL, Shcherbakov AA, Andersen NH, FEBS Lett. 2014, 588, 4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Anderson JM, Shcherbakov AA, Kier BL, Kellock J, Shu I, Byrne AL, Eidenschink LA, Andersen NH, Biopolymers 2017, 107, e22995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].The concentrations of peptides 1a-e and 2a-c were accurately measured in phosphate buffer (pH 6.5) by UV absorbance and the minimum concentrations of aggregation were determined in each case to be above 5.0 mM (see ref 29).
- [66].Anderson JM, Kier BL, Jurban B, Byrne A, Shu I, Eidenschink LA, Shcherbakov AA, Hudson M, Fesinmeyer RM, Andersen NH, Biopolymers 2016, 105, 337–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Greenfield NJ, Nat. Protoc 2006, 1, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, Xia H, Man Q, Zhong W, Antelo LF, Wu B, Xiong X, Liu X, Guan L, Li T, Liu S, Yang R, Lu Y, Dong L, McGettigan S, Somasundaram R, Radhakrishnan R, Mills G, Lu Y, Kim J, Chen YH, Dong H, Zhao Y, Karakousis GC, Mitchell TC, Schuchter LM, Herlyn M, Wherry EJ, Xu X, Guo W, Nature 2018, 560, 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, Besse B, Yanagitani N, Friboulet L, Nishio M, Takeuchi K, Kawamoto H, Fujita N, Katayama R, J. Exp. Med 2019, 216, 982–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Poggio M, Hu T, Pai C-C, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, Blelloch R, Cell 2019, 177, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, Kundu JK, Shields J, Agopsowicz KC, Xu L, Tabana Y, Srivastava N, Zhang G, Moon TC, Belovodskiy A, Hena M, Kandadai AS, Hosseini SN, Hitt M, Walker J, Smylie M, West FG, Siraki AG, Lemieux MJ, Elahi S, Nieman JA, Tyrrell DL, Houghton M, Barakat K, Sci. Rep 2019, 9, 12392–12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Magnez R, Thiroux B, Taront S, Segaoula Z, Quesnel B, Thuru X, Sci. Rep 2017, 7, 17623–17631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hoare SRJ Analyzing Kinetic Binding Data. 2021. Apr 1. In: Markossian S, Grossman A, Brimacombe K, et al. editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences. Available from: https://www.ncbi.nlm.nih.gov/books/NBK569501/ [PubMed] [Google Scholar]
- [74].Tang S, Kim PS, Proc. Natl. Acad. Sci. USA 2019, 116, 24500–24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Brown ME, Bedinger D, Lilov A, Rathanaswami P, Vásquez M, Durand S, Wallace-Moyer I, Zhong L, Nett JH, Burnina I, Caffry I, Lynaugh H, Sinclair M, Sun T, Bukowski J, Xu Y, Abdiche YN, PLoS One 2020, 15, e0229206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bacot SM, Harper TA, Matthews RL, Fennell CJ, Akue A, KuKuruga MA, Lee S, Wang T, Feldman GM, Int. J. Mol. Sci 2020, 21, 9023–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Roberts A, Bentley L, Tang T, Stewart F, Pallini C, Juvvanapudi J, Wallace GR, Cooper AJ, Scott A, Thickett D, Lugg ST, Bancroft H, Hemming B, Ferris C, Langman G, Robinson A, Chapman J, Naidu B, Pinkney T, Taylor GS, Brock K, Stamataki Z, Brady CA, Curnow SJ, Gordon J, Qureshi O, Barnes NM, Sci. Rep 2021, 11, 4030–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Webb B, Sali A, Curr. Protoc. Bioinform 2016, 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Menges F, “Spectragryph - optical spectroscopy software”, Version 1.2.13, 2019, http://www.effemm2.de/spectragryph/ (accessed May 11, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.