

Abstract
Background
Circulating transforming growth factor-β (TGF-β)-specific T cells that recognize TGF-β-expressing immune regulatory cells have been described in patients with cancer. TGF-β-derived peptide vaccination modulates the tumor microenvironment and has shown clinical effects in animal models of pancreatic cancer (PC). TGF-β-expressing regulatory cells are especially elevated in PC and may prevent the clinical response to immune checkpoint inhibitors (ICIs). Thus, in the present study we investigated the significance of TGF-β-specific T-cell immunity in patients with PC treated with ICI combined with radiotherapy in a randomized phase 2 study (CheckPAC).
Methods
Immune responses to a TGF-β-derived epitope entitled TGF-β-15 as well as epitopes from Clostridium tetani (tetanus) and influenza were measured in peripheral blood mononuclear cells (PBMCs) with interferon-ɣ enzyme-linked immunospot assays. PBMCs were isolated before and after treatment. Correlations between immune response data and clinical data were evaluated with parametric and non-parametric statistical methods. Survival was analyzed with univariate and multivariate Cox-regression. TGF-β-15 specific T cells were isolated and expanded and examined for recognition of autologous regulatory immune cells by flow cytometry.
Results
PBMCs from 32 patients were analyzed for immune responses to the TGF-β-derived epitope entitled TGF-β-15. Patients with a strong TGF-β-specific immune response at treatment initiation had longer progression-free and overall survival, compared with patients with a weak or no TGF-β-specific immune response. This remained significant in multivariate analysis. Patients with weak and strong TGF-β-specific responses displayed similar responses towards viral antigens. Furthermore, we show that TGF-β-specific T cells from a clinical responder specifically reacted to and lysed autologous, regulatory immune cells. Finally, mimicking a TGF-β-15 vaccination, we showed that repeated stimulations with the TGF-β-15 epitope in vitro enhanced the immune response to TGF-β-15.
Conclusion
A strong TGF-β-15 specific immune response was associated with clinical benefit and improved survival after ICI/radiotherapy for patients with PC. Importantly, the lack of TGF-β-specific T cells in some patients was not caused by a general immune dysfunction. TGF-β-specific T cells recognized regulatory immune cells and could be introduced in vitro in patients without spontaneous responses. Taken together, our data suggest that combining TGF-β-based vaccination with ICI/radiotherapy will be beneficial for patients with PC.
Keywords: Immunotherapy, T-Lymphocytes, Tumor Microenvironment, Antigens, Vaccination
WHAT IS ALREADY KNOWN ON THIS TOPIC
Transforming growth factor-β (TGF-β) is a key molecule involved in immunosuppression and fibrosis, which is elevated in the tumor microenvironment of pancreatic cancer (PC). Circulating TGF-β-specific T cells have been described in patients with cancer. TGF-β-derived peptide vaccination can control tumor growth in a murine model of PC.
WHAT THIS STUDY ADDS
Here we show that patients with PC with clinical benefit from treatment with immune checkpoint inhibitors (ICIs) and radiotherapy harbored TGF-β-specific T cells in the periphery at treatment initiation. These TGF-β-specific T cells recognized regulatory immune cells and could be introduced in vitro in non-responding patients.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These findings suggest that combining TGF-β-based vaccination with ICI/radiotherapy will be beneficial for patients with PC.
Background
The discovery of the immune checkpoints, cytotoxic T-lymphocytes-associated protein 4 and programmed cell death protein-1, and the subsequent introduction of immune checkpoint inhibitors (ICIs) have revolutionized the treatment and prognosis of several cancers.1 However, not all cancers are sensitive to ICI. In particular pancreatic cancer (PC) remains highly refractory to ICI. Several trials that investigated the ICI in patients with advanced PC failed to demonstrate clinically relevant efficacy.2–4 Recently, a phase II trial in patients with refractory metastatic PC (CheckPAC) investigated nivolumab with or without ipilimumab combined with stereotactic body radiotherapy (SBRT).5 A clinical benefit rate of 37.2% was observed in the combination group, however only 14% of patients achieved a partial response. The low clinical response to ICI in patients with PC suggests that other immunosuppressive mechanisms in PC should be targeted to improve the clinical response. One potential target is the cytokine, transforming growth factor-β (TGF-β).
TGF-β is a pleiotropic cytokine important in organogenesis and embryogenesis. It displays both profibrotic and immunosuppressive properties.6 7 In PC, transformed cells secrete TGF-β.8 9 In addition, regulatory immune cells, such as regulatory T cells (Tregs),10 11 tumor associated macrophages (TAMs), myeloid derived suppressor cells (MDSCs) and neutrophils infiltrate the tumor microenvironment (TME) in PC.12–14 All these cell types express TGF-β7 and high TGF-β levels in PC tissue are associated with reduced overall survival (OS).15 Tumor-derived TGF-β converts naïve T cells (Tn) to Tregs, and the level of tumor infiltrating Tregs in PC is inversely correlated with OS.10 16 Another important regulatory immune cell in PC is the cancer-associated fibroblast (CAF). These cells secrete collagen into the TME, which results in the fibrotic/desmoplastic stroma characteristic of PC.17 18 CAFs are attracted to and activated by inflammatory cytokines, such as TGF-β, in the TME.19 CAFs display several tumor-promoting properties, including the secretion of immunosuppressive cytokines, including TGF-β.17 Thus, several cells in the PC TME increase local immune suppression by expressing TGF-β.
Previously, we have demonstrated that naturally occurring T cells specifically targeted immunosuppressive proteins, such as indoleamine 2,3-dioxygenase (IDO), programmed death ligand 1 (PD-L1)/L2, and arginase-I/II.20–25 The function of these pro-inflammatory, auto-reactive T cells is opposite to regulatory cells and they are therefore termed anti-regulatory T cells (anti-Tregs).26 27 Anti-Tregs are a natural part of the immune system; they kill immunosuppressive cells to curtail local immune suppression. Thus, anti-Treg levels must delicately balance regulatory immune cells to maintain immune homeostasis.26 However, in the TME, immune homeostasis is tipped in favor of immune suppression, which hinders immune-mediated tumor-cell killing. Several lines of evidence have shown that the immune microenvironment in cancer can be modulated by anti-Tregs. First, we showed that increasing anti-Treg levels in in vitro cultures increased the amplitude of virus- and tumor-specific T-cell responses.28 29 Second, vaccination with epitopes derived from anti-regulatory proteins induced clinical responses in several in vivo models of cancer.25 30–32 Third, and most importantly, vaccinations with an IDO-derived peptide showed clinical potential in patients with stage III–IV non-small cell lung cancer.33 Additionally, a vaccine consisting of PD-L1 and IDO epitopes induced clinical responses in an unprecedent 80% of patients with metastatic melanoma, when administered in combination with nivolumab.34 The likely explanation is that, by inducing the production of T cells specific to immunosuppressive mechanisms, these anti-Tregs reacted to the regulatory immune cells in the TME, and thus, curtailed immune suppression.
Recently, we described the existence of TGF-β-specific anti-Tregs. Hence, we demonstrated that healthy individuals and patients with cancer harbored naturally occurring T cells that were specific for TGF-β-derived epitopes.35 Furthermore, we showed that T cells specific for TGF-β could recognize and react to TGF-β-expressing cancer cell lines in a TGF-β-dependent manner.35 36 In addition, in a murine model of PC, we described that TGF-β-derived peptide vaccination controlled tumor growth by reducing fibrosis and by generating a pro-inflammatory TME.37
Given the fact that in PC, TGF-β is highly involved in local immune suppression, we explored in the present study whether patients with PC treated with SBRT and ICI might harbor TGF-β-specific T cells and we evaluated the impact these cells had on the disease course. We found that patients with PC harbored TGF-β-specific T cells and that the TGF-β-specific immune response measured before treatment with ICI combined with SBRT was independently associated with improved progression-free survival (PFS) and OS. Furthermore, we showed that the lack of TGF-β-specific T cells in some patients was not caused by a general immune dysfunction.
Materials and methods
Patients and donors
Buffy coats from healthy donors were attained anonymously from the blood bank at Rigshospitalet, Copenhagen, Denmark. The usage of anonymized biological material does not require approval from an ethics committee according to Danish Law. Material was examined from the Clinical Study Protocol CheckPAC. All patients provided informed consent, in agreement with the Helsinki declaration. Patient selection for the in vitro analyses was performed blinded. After initial findings showed responses in patients with partial response (PR) and stable disease (SD), the remaining unexamined PR and SD patients were included in our analysis.
Peptide
The sequence for the used peptide TGF-β-15 was: REAVPEPVLLSRAELRLLRL. The peptide was acquired at a high purity (>90%) from Schafer (Copenhagen, Denmark) and was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM.
In vitro cultures and enzyme-linked immunospot assay
Peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved, as previously described.38 We analyzed PBMCs that had received prior in vitro stimulation with the TGF-β-15 epitope, as previously described.35 After incubation in 9–10 days, the cells were counted using a Countess II Automated Cell Counter (Thermo Fisher) and the occurrence of T cells specific to TGF-β-15 was assessed with an interferon-ɣ (IFN-ɣ) enzyme-linked immunospot (ELISpot) assay. Cells were plated out in triplicates with a concentration of 200,000 cells per well and stimulated with peptide for a final concentration of 5 µM in the well. PVDF membrane plates (Merck, Germany) coated with primary IFN-ɣ specific antibody (Mabtech, Sweden) were used for the ELISpot assays. After incubation overnight the cells were poured off, and following manufacturer’s protocol (Mabtech, Sweden) the wells were coated with secondary antibody, streptavidin-alkaline phosphatase (ALP), and enzyme substrate with washing of wells with phosphate buffered saline before and between each step. The plates were counted using the ImmunoSpot S6 Ultimate Analyzer (CTL Analyzers, Shaker Heights, Ohio, USA) when dry. The normalized mean spots were defined as the mean number of spots in peptide-stimulated wells subtracted by the mean number of spots in negative control wells.
Repeated stimulations assays
In the repeated stimulations assays the in vitro cultures were stimulated with 2 µL of 10 mM TGF-β-15 day 0 and 120 U/mL of interleukin (IL)-2 (Novartis, Switzerland) at day 1. This was repeated every 7 days one time per day or two times per day, and after the final stimulation cells were incubated for 9–10 days after which they were assayed in ELISpot as described above.
Establishment of TGF-β expressing target cells and target recognition assay
Monocytes were isolated using the MACS Miltenyi CD14 MicroBead Kit using the manufacturers protocol. After isolation of the monocytes, cells were plated at a density of 106/mL in Ross Park Memorial Institute Medium (RPMI)+10% fetal bovine serum supplemented with 1000 U/mL granulocyte macrophage colony-stimulating factor (GM-CSF) and 5 ng/mL TGF-β1. After 5 days of culturing, the cells were transfected with TGF-β-1 siRNA using methods described earlier35 and restimulated with cytokines. Immunoblotting of cell lysates from stimulated monocytes from a healthy donor showed effective knockdown of TGF-β at 48 hours after transfection, and in our earlier studies knock down was still evident at 72 hours after transfection.36 Hence, we chose to assay the target cells at 72 hours post siRNA transfection. The cells were assayed as target cells in an intracellular cytokine staining with autologous TGF-β-15 specific T-cell clones as effector cells with an effector:target ratio of 4:1 or 20:1. The intracellular cytokine staining (ICS) was performed as previously described35 with samples acquired on an FACSCanto II flow cytometer (BD Biosciences), whereas we used an expanded panel for detection of cytotoxic markers in one experiment (online supplemental table 1). These samples were acquired on a NovoCyte Quanteon flow cytometer (Agilent).
jitc-2022-006432supp008.pdf (210.2KB, pdf)
Establishment of TGF-β-15 specific T-cell clones
PBMC from patient 15 were thawed and stimulated with TGF-β-15 peptide and IL-2 (120 U/mL) for 14 days. Next, cells were restimulated and antigen specific cells were isolated using the MACS Miltenyi IFN-γ capture kit using the manufacturers protocol. Isolated cells were resuspended in x-vivo supplemented with 5% human serum containing irradiated feeder cells (105/100 µL) in addition to phytohemagglutinin (PHA) and 120 U/mL IL-2. Cells were plated in 100 µL medium and restimulated every third day with IL-2 for a total concentration of 120 U/mL. When cultures started to expand, they were cultures in larger volumes of medium with 120 U/mL IL-2 and then expanded using our rapid expansion protocol as previously described.35
Analysis of expression of TGF-β by myeloid cells
We did not have enough myeloid cells from the patient to analyze the expression of TGF-β by these cells. However, healthy donor monocytes were isolated, stimulated and transfected using exactly the same methods as for the siRNA mediated silencing of patient monocytes. Expression of TGF-β was analyzed by western blotting using a TGF-β specific antibody (CST3711) on cells lysed at 48 hours post transfection using previously published methods.35
Statistics
Statistical analysis of paired observations was done using two-tailed paired t-test and when sample size was small, non-parametric Wilcoxon matched pairs signed-rank test was used. For unpaired observations two-tailed non-parametric Mann-Whitney test was used. These tests were done using the GraphPad Prism V.9.
Survival analysis was performed with the statistical software R using the survival package for analysis. All variables that were found to be associated with OS and PFS in the original trial5 were tested for association with these in our cohort of patients tested for immune responses. This univariate analysis was performed using the log-rank analysis. All statistically significant parameters were included in a Cox proportional hazards model to analyze the independent association of immune responses to OS and PFS. Kaplan-Meier curves were made using GraphPad Prism V.9. Analysis of the ELISpot data also used the distribution-free resampling (DFR) method and the more conservative DFR2x method.39 Statistical analysis for specific recognition of TGF-β-expressing targets by T cells were performed with a paired t-test.
Results
Patient characteristics
We examined T-cell responses in patients with metastatic PC enrolled in the CheckPAC trial and showed spontaneous immune responses to a previously published TGF-β-15 epitope.35 36 CheckPAC was a phase II, single-center trial in patients with refractory metastatic PC conducted at Copenhagen University Hospital, Herlev, Denmark. The clinical outcome was reported previously.5 The trial included 84 patients that received ICI combined with SBRT. The trial included two arms: arm A received single agent nivolumab, and arm B received nivolumab combined with ipilimumab. Among the 84 patients included, we analyzed immune responses to the TGF-β-15 epitope in 32 patients. The patient characteristics are displayed in online supplemental table 2. Of the 32 patients analyzed, 7 (22%) achieved a PR, 12 (37%) had SD, and 13 (41%) displayed progressive disease (PD). The achievement of either PR or SD was defined as clinical benefit from treatment.5 PD was defined as no clinical benefit from therapy. Within a median follow-up time of 219 days (range 65–1511), two patients remained alive (6%).
jitc-2022-006432supp009.pdf (160.2KB, pdf)
Patients with clinical benefit harbored T cells specific to TGF-β
We chose to analyze TGF-β-15-specific responses, because our previous study showed that both CD4+ and CD8+ T cells specific to this epitope recognized and killed TGF-β-expressing cells in a TGF-β-dependent manner.35 36 We found, that T cells isolated both at baseline and after four series of treatment displayed responses to TGF-β-15 (figure 1A, B). Patients with clinical benefit had significantly higher baseline immune-response amplitudes than patients with PD, whereas we found no difference in response amplitude after treatment (8 weeks) (figure 1C). These data suggested that a TGF-β-specific immune response present before initiating treatment could influence the clinical response to ICI/SRBT. Of note, we found that patients with clinical benefit displayed a significant drop in the TGF-β-specific response amplitude 8 weeks after treatment start (figure 2A, B). Furthermore, patients with a durable clinical benefit to therapy, defined as a PR or SD lasting longer than 6 months, displayed a significantly lower TGF-β-specific response 8 weeks after treatment, compared with patients without a durable clinical benefit, (online supplemental figure 1A, B).
Figure 1.

TGF-β-15 specific responses in patients with pancreatic cancer (PC) PBMC. (A) PBMC from patients with PC were stimulated once in vitro with TGF-β-15 peptide and interleukin-2, and incubated for 14 days before plating in interferon-γ enzyme-linked immunospot assays at a concentration of 2×105 cells/well with overnight incubation. The experiments were performed in triplicates with negative control wells left unstimulated. The responses were characterized in 32 samples isolated at baseline (left) and in 31 samples isolated after four series of treatment (right). (B) Representative pictures of a response at baseline (top) and a response after four series of treatment (bottom). (C) The normalized counts from A were used to compare the amplitude of response in patients with clinical benefit and response in patients with progressive disease. Normalization of the data was performed by subtracting the mean spot count of the control wells from the mean spot count of peptide stimulated wells. Error bar depicts SEM. Statistics made using Mann-Whitney test. PBMC, peripheral blood mononuclear cell; TGF-β, transforming growth factor-β.
Figure 2.

The amplitude of TGF-β-15 specific immune responses fluctuates over time. (A) Normalized counts at baseline were compared with counts after four series of treatment for patients with clinical benefit (left) and patients with progressive disease (right). Statistics made using Wilcoxon matched-pairs signed-rank test. (B) Representative pictures of baseline response and response after four series in a patient with clinical benefit (top), and of a response in a patient with progressive disease (bottom). (C) Normalized TGF-β-15 specific immune responses analyzed over time in three patients with clinical benefit. Normalization of spot counts performed as described previously. TGF-β, transforming growth factor-β.
jitc-2022-006432supp001.pdf (809.4KB, pdf)
Next, we analyzed serial PBMC samples from three patients with a follow-up time longer than 2 years. We analyzed TGF-β-15-specific immune responses to assess temporal variations in the response. Interestingly, we found that the response amplitude fluctuated over time (figure 2C).
T-cell responses specific to TGF-β-15 were independently associated with improved survival
Our data strongly suggested that an intact TGF-β-specific immune response could impact the response to therapy. Therefore, we investigated whether the baseline TGF-β-15-specific immune response was associated with survival in our patient cohort. We stratified patients based on whether they showed responses above or below the median normalized TGF-β-15-specific immune response of 51 spots per 2×105 cells. Interestingly, we found that patients with a response amplitude above the median had significantly longer OS than those with responses below the median (univariate Cox-regression, HR: 0.171, p=2.54×10–4; figure 3A and table 1). Next, we investigated whether the TGF-β-15-specific immune response was independently associated with OS. We performed univariate survival analysis on clinical parameters that could be associated with survival, with a special focus on parameters that were included in the survival analysis in the original CheckPAC trial report.5 Strangely, a univariate Cox-regression analysis showed that <2 metastases was associated with a shorter OS (HR=2.64, p=0.03), but no other parameters showed statistical significance (table 1). To further examine this, a multivariate analysis that included the metastases parameter and the TGF-β-15-specific immune response showed that only the TGF-β-15-specific immune response was independently associated with OS (HR: 0.18) for patients with a response above the median amplitude (p=8×10–4; table 1). Of note, the univariate analysis showed borderline significance for reduced survival among patients who received single-agent nivolumab (HR: 2.04, p=0.055). Hence, we included this parameter in another multivariate analysis, together with metastases and the TGF-β-15-specific immune response. In the latter model, importantly neither the metastases variable nor the treatment variable was associated with OS; however, a TGF-β-15-specific response above the median remained associated with improved survival (HR: 0.19, p=0.0032; data not shown). The same analyses were performed for PFS. In the univariate analysis, the TGF-β-15-specific response was associated with a prolonged PFS (HR: 0.227, p=0.0015; figure 3B and table 2). Other significant parameters from the univariate analysis were: nivolumab treatment (HR: 2.86, p=0.007), male sex (HR: 0.406, p=0.029), and >36 g/L albumin in peripheral blood (HR: 4.68, p=0.038). When these significant parameters were included in the multivariate analysis, together with the TGF-β-15-specific immune response, only the TGF-β-15 response above the median remained independently associated with PFS (HR: 0.322, p=0.023; table 2). Interestingly, the patients with a TGF-β-15 response above the 75% percentile had the highest OS and PFS of examined samples (online supplemental figure 2A, B). These results added impetus to the notion that the level of TGF-β-15-specific T cells in patients with PC was important in achieving a clinical response to ICI/SBRT treatment.
Figure 3.

Strong TGF-β-15 specific responses before treatment initiation predict superior survival. (A) Kaplan-Meier curve displaying overall survival in patients with a response above or below the median response amplitude to TGF-β-15. (B) Kaplan-Meier curve displaying progression-free survival in patients with a response above or below the median response amplitude to TGF-β-15. Time-to-event analyses were performed using the log-rank test. TGF-β, transforming growth factor-β.
Table 1.
Univariate Cox-regression on overall survival
| Covariate | HR | Lower 95% CI | Upper 95% CI | P value |
| Treatment arm | ||||
| SBRT+nivolumab+ipilimumab | 1 | |||
| SBRT+nivolumab | 2.04 | 0.986 | 4.214 | 0.055 |
| Age, years | ||||
| >66.5 years | 1 | |||
| ≤66.5 years | 1.12 | 0.545 | 2.314 | 0.754 |
| Sex | ||||
| Female | 1 | |||
| Male | 0.608 | 0.287 | 1.287 | 0.193 |
| WHO performance status | ||||
| 0 | 1 | |||
| 1 | 1.297 | 0.620 | 2.715 | 0.49 |
| Weight loss >5% | ||||
| No | 1 | |||
| Yes | 1.437 | 0.693 | 2.977 | 0.33 |
| Number of metastatic sites | ||||
| ≥2 | 1 | |||
| <2 | 2.644 | 1.103 | 6.338 | 0.0292 |
| Whipple procedure | ||||
| No | 1 | |||
| Yes | 0.856 | 0.363 | 2.02 | 0.722 |
| Biliary stent | ||||
| No | 1 | |||
| Yes | 0.857 | 0.391 | 1.88 | 0.7 |
| Median CA19-9, kU/L | ||||
| ≤median | 1 | |||
| >median | 1.577 | 0.731 | 3.4 | 0.246 |
| NLR | ||||
| ≥5 | 1 | |||
| < 5 | 1.66 | 0.748 | 3.69 | 0.213 |
| Bilirubin, 25 µmol/L | ||||
| >25 | NA | NA | NA | NA |
| ≤25 | NA | NA | NA | NA |
| Albumin, 36 g/L | ||||
| ≤36 | 1 | |||
| >36 | 3.295 | 0.774 | 14.03 | 0.107 |
| CRP, 10 mg/L | ||||
| ≤10 | 1 | |||
| >10 | 0.920 | 0.445 | 1.902 | 0.823 |
| mGPS | ||||
| 0 | 1 | |||
| ≥1 | 0.920 | 0.445 | 1.902 | 0.823 |
| Prior lines of therapy | ||||
| <2 | 1 | |||
| ≥2 | 1.52 | 0.724 | 3.191 | 0.269 |
| Best optimal response to prior last therapy | ||||
| Clinical benefit | 1 | |||
| No benefit | 1.41 | 0.674 | 2.94 | 0.363 |
| ELISpot response to TGF-β-15 at baseline | ||||
| ≤median | 1 | |||
| >median | 0.171 | 0.066 | 0.44 | 0.000254 |
| Multivariate Cox-regression on overall survival | ||||
| Covariate | HR | Lower 95% CI | Upper 95% CI | P value |
| Number of metastatic sites | ||||
| ≥2 | 1 | |||
| <2 | 1.7 | 0.71 | 4.44 | 0.22 |
| ELISpot response to TGF-β-15 at baseline | ||||
| ≤median | 1 | |||
| >median | 0.18 | 0.071 | 0.5 | 0.0008 |
CRP, C-reactive protein; ELISpot, enzyme-linked immunospot assay; mGPS, modified Glasgow Prognostic Score; NLR, neutrophil-to-lymphocyte ratio; SBRT, stereotactic body radiotherapy; TGF-β, transforming growth factor-β.
Table 2.
Univariate Cox-regression on progression-free survival
| Covariate | HR | Lower 95% CI | Upper 95% CI | P value |
| Treatment arm | ||||
| SBRT+nivolumab+ipilimumab | 1 | |||
| SBRT+nivolumab | 2.86 | 1.34 | 6.10 | 0.007 |
| Age, years | ||||
| >66.5 years | 1 | |||
| ≤66.5 years | 1.123 | 0.545 | 2.314 | 0.754 |
| Sex | ||||
| Female | 1 | |||
| Male | 0.406 | 0.1815 | 0.911 | 0.029 |
| WHO performance status | ||||
| 0 | 1 | |||
| 1 | 1.3 | 0.62 | 2.73 | 0.486 |
| Weight loss >5% | ||||
| No | 1 | |||
| Yes | 1.316 | 0.6323 | 2.737 | 0.463 |
| Number of metastatic sites | ||||
| ≥2 | 1 | |||
| < 2 | 2.336 | 0.972 | 5.612 | 0.0579 |
| Whipple procedure | ||||
| No | 1 | |||
| Yes | 0.681 | 0.29 | 1.60 | 0.377 |
| Biliary stent | ||||
| No | 1 | |||
| Yes | 1.169 | 0.548 | 2.50 | 0.69 |
| Median CA19-9, kU/L | ||||
| ≤median | 1 | |||
| >median | 1.329 | 0.635 | 2.80 | 0.452 |
| NLR | ||||
| ≥5 | 1 | |||
| < 5 | 1.957 | 0.878 | 4.36 | 0.1 |
| Bilirubin, 25 µmol/L | ||||
| >25 | NA | NA | NA | NA |
| ≤25 | NA | NA | NA | NA |
| Albumin, 36 g/L | ||||
| ≤36 | 1 | |||
| >36 | 4.68 | 1.084 | 20.18 | 0.038 |
| CRP, 10 mg/L | ||||
| ≤10 | 1 | |||
| >10 | 0.8 | 0.387 | 1.66 | 0.547 |
| mGPS | ||||
| 0 | 1 | |||
| ≥1 | 0.8 | 0.387 | 1.66 | 0.547 |
| Prior lines of therapy | ||||
| <2 | 1 | |||
| ≥2 | 1 | 0.489 | 2.05 | 0.996 |
| Best optimal response to prior last therapy | ||||
| Clinical benefit | 1 | |||
| No benefit | 1 | 0.493 | 2.08 | 0.972 |
| ELISpot response to TGF-β-15 at baseline | ||||
| ≤median | 1 | |||
| >median | 0.227 | 0.091 | 0.566 | 0.0015 |
| Multivariate Cox-regression on progression-free survival | ||||
| Covariate | HR | Lower 95% CI | Upper 95% CI | P value |
| Treatment arm | ||||
| SBRT+nivolumab+ipilimumab | 1 | |||
| SBRT+nivolumab | 1.94 | 0.832 | 4.53 | 0.125 |
| Sex | ||||
| Female | 1 | |||
| Male | 0.446 | 0.192 | 1.034 | 0.061 |
| Albumin, 36 g/mL | ||||
| ≤36 | 1 | |||
| >36 | 2.60 | 0.55 | 12.31 | 0.229 |
| ELISpot response to TGF-β-15 at baseline | ||||
| ≤median | 1 | |||
| >median | 0.322 | 0.121 | 0.857 | 0.023 |
CRP, C-reactive protein; ELISpot, enzyme-linked immunospot assay; mGPS, modified Glasgow Prognostic Score; NLR, Neutrophil-to-lymphocyte ratio; SBRT, stereotactic body radiotherapy; TGF-β, transforming growth factor-β.
jitc-2022-006432supp002.pdf (888.6KB, pdf)
Association between TGF-β-15-specific response and survival was not due to general immune dysfunction in non-responding patients
After these intriguing results, we investigated whether patients with poor survival might harbor a dysfunctional immune system and that the low response amplitude observed in these patients was caused by general T-cell dysfunction. We analyzed the PBMC response to two broadly immunogenic epitopes—one derived from Clostridium tetani40 (tetanus-long) and the other derived from influenza virus (C18 A2 influenza). The latter was a nonamer epitope restricted to human leukocyte antigen (HLA)-A2; consequently, only samples with HLA-A2+ were analyzed for responses against the influenza epitope. We analyzed 23 patients samples for responses against tetanus and 16 for responses against influenza. The normalized median response were 124 spots for the tetanus epitope and 114 spots for the influenza epitope with 2×105 plated cells (data not shown). Samples from patients with strong and weak TGF-β-15 specific immune responses had similar response amplitudes to both tetanus and influenza epitopes (figure 4). Additionally, we showed that the TGF-β-15-response amplitude was not correlated with neither the tetanus response (r2=0.05) nor the influenza response (r2=0.15) (online supplemental figure 3A, B). Perhaps more importantly, the response amplitude to neither the tetanus nor the influenza epitope was not associated with either OS or PFS (figure 5A–D).
Figure 4.

The amplitude of TGF-β-15 specific responses is not coupled to the amplitude of responses to tetanus peptide. (A) Peripheral blood mononuclear cell were tested for response to the tetanus epitope ‘tetanus-long’ using in vitro interferon-ɣ enzyme-linked immunospot assays. The amplitude of the normalized tetanus-long specific response was compared between patients with a response above or below the median TGF-β-15 specific immune response. Responses against tetanus-long were not analyzed in baseline samples in five patients from the group with a response above the median and in one patient from the group with a response below the median, but in samples acquired within 2 weeks of baseline. (B) Representative pictures of a patient with an absent TGF-β-specific immune response (top) but an intact tetanus specific immune response (bottom). (C) Same analysis as in A with a short influenza virus derived epitope ‘C18 A2 Flu’ instead of tetanus derived epitope. (D) Representative pictures of a patient with an absent TGF-β-specific immune response (top) but an intact influenza virus specific immune response (bottom). Error bars depict SEM. Statistics made using Mann-Whitney test. TGF-β, transforming growth factor-β.
Figure 5.

Clostridium tetani and influenza specific responses are not associated with survival. (A) Kaplan-Meier curve displaying overall survival in patients with a response above or below the median response amplitude to tetanus-long. (B) Kaplan-Meier curve displaying progression-free survival in patients with a response above or below the median response amplitude to tetanus-long. (C) Kaplan-Meier curve displaying overall survival in patients with a response above or below the median response amplitude to C18 A2 influenza. (D) Kaplan-Meier curve displaying progression-free survival in patients with a response above or below the median response amplitude to C18 A2 influenza. Time-to-event analyses were performed using the log-rank test.
jitc-2022-006432supp003.pdf (592KB, pdf)
TGF-β-15 specific T cells recognize autologous regulatory myeloid cells in a TGF-β-dependent manner
According to our data, the TGF-β-15 specific immune response is independently associated with both OS and PFS. We have previously demonstrated that TGF-β-15 specific CD4+ and CD8+ T cells recognize TGF-β-expressing myeloid cancer cells.35 36 Hence, the TGF-β-15 specific T cells identified above are able to recognize and kill TGF-β-expressing immunoregulatory cells in the TME thereby lowering the levels of immune suppression. To show this, we isolated monocytes from the best clinical responding patient and stimulated those with TGF-β1 and GM-CSF cytokines to generate regulatory myeloid cells that expressed TGF-β. Four days later, cells were transfected with TGF-β siRNA as described in materials and methods and after 72 hours of incubation the myeloid cells were used as target cells in an ICS assay. Western blot analysis of myeloid cells from a healthy donor stimulated and transfected using exactly the same methods showed effective knock down of TGF-β 48 hours post transfection (figure 6A). Hence, TGF-β siRNA transfected regulatory myeloid cells from the patient were used as target cells at this time point using two different autologous CD4+ TGF-β-15 specific T-cell clones as effector cells. Experiments were performed in three independent experiments with two different TGF-β-15 specific T-cell clones as effector cells with a 4:1 effector:target ratio in two experiments and a 20:1 ratio in one experiment. The T-cell clones were significantly more activated on stimulation with mock transfected regulatory myeloid cells compared with TGF-β siRNA transfected myeloid cells (figure 6B, C and online supplemental figure 4A, B). The TGF-β-15 specific T-cell clones killed mock transfected regulatory myeloid cells whereas TGF-β silencing rescued regulatory myeloid cells from T-cell mediated lysis (figure 6D, E and online supplemental figure 5A, B). In this experiment we used an expanded antibody panel to analyze the expression of additional activation markers by the T-cell clones (online supplemental table 1). We found that expression of the cytotoxic markers CD107a and granulysin was increased by the T-cell clone (clone 27) on stimulation with mock transfected regulatory myeloid cells (figure 6F). Even though the experiments were performed at different time points of effector T-cell expansion and at different effector:target ratios we demonstrated a statistically significant lower number of cells in the monocyte gate in mock transfected cells compared with siRNA transfected monocytes (p=0.0184) (online supplemental figure 6A). In addition, TGF-β-15 specific T-cell clones produced significantly higher levels of tumor necrosis factor (TNF)-α on stimulation with mock transfected target cells compared with siRNA transfected target cells (p=0.040) (online supplemental figure 6B). Stimulation of TGF-β-15 specific T-cell clones with mock transfected target cells resulted in higher levels of IFN-γ expressing and IFN-γ/TNF-α double positive cells, however this difference was not statistically significant, probably due to low sample size (6)online supplemental figure 6C, D). Importantly, the TGF-β1 cytokine used for stimulation is not included in the TGF-β-15 epitope which resides in the latency associated peptide (LAP) part of TGF-β.35 Hence, recognition of TGF-β stimulated target cells is not due to processing and presentation of TGF-β cytokine but instead due to increased expression of endogenous TGF-β.
Figure 6.

T cells specific for TGF-β-15 from a patient with pancreatic cancer with complete response to immune checkpoint inhibitor and stereotactic body radiotherapy recognize and kill autologous regulatory myeloid cells in a TGF-β-dependent manner. (A) Western blot analysis of TGF-β expression by regulatory myeloid cells that had been either mock transfected or transfected with TGF-β siRNA. Monocytes were stimulated for 4 days with GM-CSF and TGF-β1, then transfected and restimulated with GM-CSF and TGF-β1 for 2 days before harvesting and lysing of cells. (B) Two TGF-β-15 specific T-cell clones were stimulated with siRNA or mock transfected autologous regulatory myeloid cells in an intracellular cytokine staining (ICS) assay. (C) Contour plot of results shown in B. (D) The amount of regulatory myeloid cells detected in the forward-side scatter plot during the ICS experiment was quantified. (E) Contour plot of results shown in D. (F) Expression of the cytotoxicity markers CD107a and granulysin by TGF-β-15 specific T-cell clones on stimulation with autologous regulatory myeloid cells. GM-SCF, granulocyte-macrophage colony stimulating-factor; IFN, interferon-γ; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor-α.
jitc-2022-006432supp004.pdf (1.4MB, pdf)
jitc-2022-006432supp005.pdf (1.4MB, pdf)
jitc-2022-006432supp006.pdf (810.4KB, pdf)
Repeated antigen stimulations with a TGF-β-15 peptide led to T-cell responses in PBMCs that did not display a response after one in vitro stimulation
The results provided above strongly suggests that a measurable TGF-β-15-specific immune response is important in the clinical response to ICI/SBRT treatment in patients with PC and that the mechanism beyond the beneficial effect of TGF-β-15 specific T cells is their ability to kill immunosuppressive TGF-β expressing cells. Thus, inducing a TGF-β-15 response with therapeutic peptide vaccines might be beneficial, when combined with ICI/SBRT. Not all patients and healthy donors displayed an immune response to TGF-β-15 after one in vitro stimulation. We investigated whether a response could be induced by repeated in vitro stimulations with the TGF-β-15 epitope as this mimics the repeated antigen stimulations performed during a therapeutic vaccination schedule. We chose to work with PBMC samples from earlier experiments that showed weak or absent responses to TGF-β-15 after one in vitro stimulation. T-cell responses were analyzed after one in vitro stimulation and after one or two additional in vitro stimulations. We showed that repeated antigen stimulations increased the TGF-β-15-specific immune response in PBMCs from both patients with PC (figure 7A) and healthy individuals (figure 7B, C). Of tested patients with PC, 27% had a DFR2x response after one stimulation which increased to 66% after repeated stimulations (online supplemental figure 7). Within the tested healthy donors, 9% had a DFR2x response after one stimulation which increased to 71% after repeated stimulations (online supplemental figure 7). These data showed that repeated antigen stimulations with the TGF-β-15 peptide increased the numbers of TGF-β-specific T cells in PBMCs.
Figure 7.
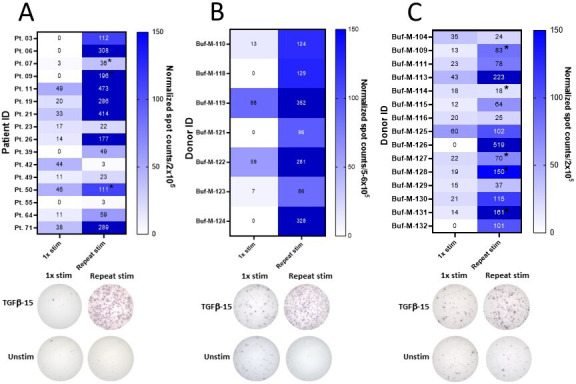
Increased amplitude of the TGF-β-15 specific immune response through repeated in vitro stimulations. (A) PBMC from 16 patients with pancreatic cancer with a weak TGF-β-15 response were cultured in vitro and tested for response to TGF-β-15 after one in vitro stimulation with TGF-β-15 peptide or after three in vitro stimulations with TGF-β-15 peptide with normalized responses shown in a heatmap (top) and representative responses after one stimulation and three stimulations (bottom). Some cultures only received two in vitro stimulations denoted by a black star next to spot count. (B) The ability of repeated stimulations to enhance the TGF-β-15 specific immune response was analyzed in PBMC from seven healthy donors as described in A using 5–6×105 cells/well with normalized results (top) and representative responses (bottom). (C) The ability of repeated stimulations to enhance the TGF-β-15 specific immune response was analyzed in PBMC from 15 healthy donors as described in A using 2×105 cells/well with normalized results (top) and representative responses (bottom). PBMC, peripheral blood mononuclear cell; TGF-β, transforming growth factor-β.
jitc-2022-006432supp007.pdf (611.4KB, pdf)
Discussion
In this study, we scrutinized spontaneous T-cell responses specific to a TGF-β-derived epitope in patients with PC treated with ICI and SBRT. Recently, we showed that both patients with cancer and healthy individuals harbor T cells specific to TGF-β-derived epitopes, and that TGF-β-specific T cells recognized and killed TGF-β-expressing target cells.35 36 An increased expression of TGF-β is associated with inferior survival in PC,15 and TGF-β is heavily involved in local immune suppression in PC. Thus, the identification of TGF-β-specific T-cell responses in patients with PC is interesting as the specific T cells could possibly recognize and kill TGF-β-expressing regulatory cells in the tumor. Consequently, we investigated relationships between the TGF-β-15-specific immune response and clinical outcomes. Interestingly, compared with patients who did not show clinical benefit to treatment, patients with clinical benefit had a significantly stronger TGF-β-15-specific immune response before treatment initiation and a significantly stronger decline in the TGF-β-15-specific T-cell response after initiation of treatment. The latter phenomenon could be explained by migration of TGF-β-specific T cells to the tumor after initiating ICI therapy. These results were consistent with the striking observation that patients with a TGF-β-15-specific immune response above the median had significantly longer PFS and OS, compared with patients with a response below the median.
We also investigated whether the identified difference in OS between patients with strong and weak TGF-β-15-specific immune responses might be attributed to differences in the general immune status of the patients. We compared the spontaneous immune responses in these two groups against two highly immunogenic epitopes derived from common pathogens—influenza virus and C. tetani.40 We found no difference between groups in the response amplitude to neither influenza nor tetanus. Additionally, there was no correlation between the response amplitude to TGF-β-15 and the response to tetanus or influenza epitopes. Interestingly, patients with a strong pathogen specific immune response did not show neither improved OS nor improved PFS after ICI treatment. This implied that the strength of the TGF-β-15-specific immune response did not depend on the general immune constitution; instead it reflected the number of anti-regulatory TGF-β-specific T cells.
These findings could imply that TGF-β-specific anti-Tregs play a role in the response to ICI/SBRT in PC, and we speculated that the TGF-β-15-specific T cells identified were able to recognize TGF-β expressing regulatory cells. Hence, we isolated TGF-β-15-specific T-cell clones from a patient without any signs of disease at 2038 days of follow-up and showed that these cells indeed recognized regulatory myeloid cells in a TGF-β dependent manner. TGF-β-15-specific T-cells both released pro-inflammatory cytokines and cytotoxic mediators on activation and were able to directly kill TGF-β-expressing myeloid cells. This strongly suggests that T cells specific to TGF-β exert their effect by the recognition of TGF-β-expressing cells thereby lowering the local immune suppression in the tumor. Conversion of monocytes into regulatory myeloid cells was performed in vitro through stimulation with TGF-β1 cytokine. Importantly, the TGF-β-15 epitope is not found in the TGF-β1 sequence, but instead in the LAP part of the sequence. Thus, the increased recognition of in vitro generated regulatory cells is not explained by processing and presentation of the TGF-β1 cytokine but can only be by increased endogenous expression of TGF-β. Given the many effects of TGF-β in PC, it is worthwhile to consider the potential immunomodulating and tumor-suppressive effects that TGF-β-specific T cells could have in patients. In PC, approximately 95% of patients harbor an activating mutation in the KRAS-gene.41 These mutations induce the production of TGF-β in transformed cells.8 9 Hence, TGF-β-specific T cells would act directly on the transformed cells. However, several other prevalent immunosuppressive cells would also be targeted.
PC is characterized by a highly desmoplastic stroma and CAFs are the main culprits of this feature.17 18 CAFs are thought to originate from both bone marrow-derived mesenchymal stem cells and pancreatic stellate cells.42 Through secretion of TGF-β, PC-derived cell lines can activate CAF, which increases the deposition of extracellular matrix proteins and enhances fibrosis.19 43 Furthermore, activated CAFs also secrete TGF-β, which acts in an autocrine fashion43 thus resulting in additional secretion of TGF-β and notably intratumoral TGF-β expression was shown to correlate with tissue fibrosis.43 Another study demonstrated that patients with PC with a high level of fibrosis in combination with the CAF-marker alpha smooth muscle actin (aSMA), showed inferior survival.44 This finding underscored the importance of CAFs in PC. The immunomodulatory effects of CAFs have been studied extensively, and CAF populations are similar among different cancers.45 Of note, one group of CAFs is characterized by increased TGF-β signaling—so called myofibroblastic CAFs (myCAFs).46 Interestingly, a subset of myCAFs was associated with a lack of response to ICI in several cancers.45 47
The composition of immune cells in PC is heterogeneous12 but generally, most are of myeloid origin.12–14 These myeloid cells include TAMs, MDSCs, and neutrophils, which all express TGF-β and modulate the TME. Neutrophils secrete high amounts of TGF-β in the PC TME, which attracts and activates CAFs.48 Additionally, local TGF-β converts myeloid cells into M2 macrophages, TAMs, and MDSCs, which are elevated in PC,11 and they negatively impact both PFS and OS in patients.12 44 49 These myeloid cells express TGF-β50 and, apart from their own immunosuppressive properties, they also mediate the conversion of Tn into Tregs.51 52 Hence, the emergence of both MDSCs and Tregs in the TME of PC depends on TGF-β. Moreover, the level of CD8+ T cells in the TME is inversely correlated with the levels of myeloid cells and Tregs.11 12 52 Importantly, we showed that TGF-β expressing regulatory myeloid cells are targets of TGF-β-15 specific T cells. This suggests that TGF-β-specific T cells may alter the composition of immune cells in the PC TME into a more pro-inflammatory phenotype. Consequently, TGF-β is an attractive target for enhancing the effect of cancer immune therapy in PC, and several methods for inhibiting TGF-β signaling are undergoing preclinical and clinical testing for different cancers.53
We recently described an alternative way to target a TGF-β-rich TME. We showed the antitumor activity of TGF-β-derived peptide vaccination in a murine tumor model of PC.37 We described that TGF-β-derived peptide vaccination therapy indeed targets immunosuppression in the TME by differentiating the cellular composition towards a more pro-inflammatory phenotype. Our findings highlight TGF-β-derived peptide vaccination as a novel immunotherapeutic approach. Based on these data and the results of this study, we suggest that therapeutic cancer vaccines of TGF-β-derived epitopes may be an effective future treatment modality in combination with SBRT and ICI. Moreover, repeated peptide vaccinations could enhance the TGF-β-specific immune response in patients. The rationale for repeated vaccinations is to increase the numbers of TGF-β-specific T cells that migrate to the TME; these cells will attack TGF-β-expressing cells and release Th1 cytokines thus, TGF-β signaling will be reduced, and the TME will be converted to an immunopermissive environment that favors the killing of transformed cells by tumor-specific T cells. In the present study, we mimicked therapeutic vaccinations by performing repeated antigen stimulations in PBMCs that displayed a weak/absent response to TGF-β-15. We found that repeated stimulations induced a strong TGF-β-15-specific immune response in almost all cultures and showed that specific T cells recognized regulatory TGF-β expressing myeloid cells. This finding supports the notion that repeated vaccinations with TGF-β-derived peptides can induce a TGF-β-specific immune response. Moreover, these findings suggested that TGF-β-15-specific T cells were not terminally exhausted or absent in samples that did not respond.
We previously described spontaneous T-cell responses to several other immunosuppressive proteins.21 26 54 Earlier clinical trials of vaccinations against immunosuppressive targets, such as IDO and PD-L1, have demonstrated potency in patients with cancer in combination with ICI.34 Accordingly, we are initiating a phase I clinical trial to test the safety and efficacy of vaccinations with TGF-β-derived peptides, in combination with radiotherapy and ICI, in patients with metastatic PC resistant to chemotherapy (EudraCT 2022-002734-13).
Conclusion
PBMC samples from patients with PC treated with ICI and SBRT displayed immune responses to the TGF-β-15 epitope. Patients with clinical benefit from treatment had stronger TGF-β-15-specific T-cell responses than patients with PD. Furthermore, a strong TGF-β-15-specific T-cell response before treatment initiation was independently associated with prolonged PFS and OS. We also showed that low-level TGF-β-15-specific responses observed in some patients were not due to a general dysfunction of the immune system, as these patients retained a normal T-cell response to common pathogen-derived epitopes. Furthermore, we showed that TGF-β-15-specific T cells recognized and killed autologous regulatory myeloid cells in a TGF-β dependent manner, and that immune responses could be induced/enhanced by repeated antigen stimulations. Consequently, we believe that administering therapeutic cancer vaccinations to deliver repeated TGF-β-15-antigen stimulations in patients with PC will induce a specific T-cell response that is likely to lead to a clinical response.
Acknowledgments
We thank Merete Jonassen and Anette Højgaard Andersen for excellent technical assistance.
Footnotes
Contributors: REJM performed experiments, interpreted the data and wrote the manuscript. MOH planned the experiments, performed experiments, interpreted the data and wrote the manuscript. TLL performed experiments. JPH provided resources. GLW performed research. ÖM provided resources. IMS provided resources. JJ provided resources and edited the manuscript. DLN provided resources. IMC provided project administration, contributed vital material and resources and edited the manuscript. MHA conceived the project and the experiments, interpreted the data, wrote the manuscript and is guarantor for this work.
Funding: This work was supported by University of Copenhagen [-], Herlev Hospital [-], the Novo Nordisk Foundation, Vissing Foundation and the Independent Research Fund Denmark. The funders had no role in the study design, collection of data, data analysis, decision to publish or manuscript preparation. This study was in addition supported by the BRIDGE – Translational Excellence Programme (bridge.ku.dk) at the Faculty of Health and Medical Sciences, University of Copenhagen, funded by the Novo Nordisk Foundation. Grant agreement no. NNF20SA0064340.
Competing interests: MHA has made an invention based on the use of TGF-β for vaccinations. The rights of the invention have been transferred to Copenhagen University Hospital Herlev, according to the Danish Law of Public Inventions at Public Research Institutions. The capital region has licensed the rights to the company IO Biotech ApS. The patent application was filed by IO Biotech ApS. MHA is founder, advisor and shareholder in IO Biotech. IMS is co-founder, advisor and shareholder in IO Biotech. The additional authors declare no competing financial interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by Clinical Study Protocol CheckPAC Version 5, 2021 03 12. Approval by Danish Medicines Authority (Lægemiddelstyrelsen) Eudract nr. 2016-001883-12, The Regional Ethics Committee for Capital Region Denmark ID H-16031247 and Videnscenter for Dataanmeldelser j-nr. P-2021-216. Participants provided informed consent to participate in the study before taking part.
References
- 1.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun 2020;11:3801. 10.1038/s41467-020-17670-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Reilly EM, Oh D-Y, Dhani N, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol 2019;5:1431–8. 10.1001/jamaoncol.2019.1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33:828–33. 10.1097/CJI.0b013e3181eec14c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen IM, Johansen JS, Theile S, et al. Randomized phase II study of nivolumab with or without ipilimumab combined with stereotactic body radiotherapy for refractory metastatic pancreatic cancer (checkpac). J Clin Oncol 2022;40:3180–9. 10.1200/JCO.21.02511 [DOI] [PubMed] [Google Scholar]
- 6.Massagué J. Tgfβ in cancer. Cell 2008;134:215–30. 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019;50:924–40. 10.1016/j.immuni.2019.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zdanov S, Mandapathil M, Abu Eid R, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res 2016;4:354–65. 10.1158/2326-6066.CIR-15-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng H, Fan K, Luo G, et al. KrasG12D mutation contributes to regulatory T cell conversion through activation of the MEK/ERK pathway in pancreatic cancer. Cancer Lett 2019;446:103–11. 10.1016/j.canlet.2019.01.013 [DOI] [PubMed] [Google Scholar]
- 10.Carstens JL, Correa de Sampaio P, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 2017;8:15095. 10.1038/ncomms15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518–27. 10.1158/0008-5472.CAN-07-0175 [DOI] [PubMed] [Google Scholar]
- 12.Steele NG, Carpenter ES, Kemp SB, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer 2020;1:1097–112. 10.1038/s43018-020-00121-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Väyrynen SA, Zhang J, Yuan C, et al. Composition, spatial characteristics, and prognostic significance of myeloid cell infiltration in pancreatic cancer. Clin Cancer Res 2021;27:1069–81. 10.1158/1078-0432.CCR-20-3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elyada E, Bolisetty M, Laise P, et al. Cross-Species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 2019;9:1102–23. 10.1158/2159-8290.CD-19-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friess H, Yamanaka Y, Büchler M, et al. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993;105:1846–56. 10.1016/0016-5085(93)91084-u [DOI] [PubMed] [Google Scholar]
- 16.Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013;108:914–23. 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16:582–98. 10.1038/nrc.2016.73 [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi H, Enomoto A, Woods SL, et al. Cancer-Associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol 2019;16:282–95. 10.1038/s41575-019-0115-0 [DOI] [PubMed] [Google Scholar]
- 19.Löhr M, Schmidt C, Ringel J, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res 2001;61:550–5. [PubMed] [Google Scholar]
- 20.Sørensen RB, Køllgaard T, Andersen RS, et al. Spontaneous cytotoxic T-cell reactivity against indoleamine 2,3-dioxygenase-2. Cancer Res 2011;71:2038–44. 10.1158/0008-5472.CAN-10-3403 [DOI] [PubMed] [Google Scholar]
- 21.Munir S, Andersen GH, Met Ö, et al. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res 2013;73:1764–76. 10.1158/0008-5472.CAN-12-3507 [DOI] [PubMed] [Google Scholar]
- 22.Ahmad SM, Martinenaite E, Holmström M, et al. The inhibitory checkpoint, PD-L2, is a target for effector T cells: novel possibilities for immune therapy. Oncoimmunology 2018;7:e1390641. 10.1080/2162402X.2017.1390641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinenaite E, Munir Ahmad S, Hansen M, et al. CCL22-specific T cells: modulating the immunosuppressive tumor microenvironment. Oncoimmunology 2016;5:e1238541. 10.1080/2162402X.2016.1238541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinenaite E, Mortensen REJ, Hansen M, et al. Frequent adaptive immune responses against arginase-1. Oncoimmunology 2018;7:e1404215. 10.1080/2162402X.2017.1404215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weis-Banke SE, Hübbe ML, Holmström MO, et al. The metabolic enzyme arginase-2 is a potential target for novel immune modulatory vaccines. Oncoimmunology 2020;9:1771142. 10.1080/2162402X.2020.1771142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen MH. Anti-regulatory T cells. Semin Immunopathol 2017;39:317–26. 10.1007/s00281-016-0593-x [DOI] [PubMed] [Google Scholar]
- 27.Ødum N. Anti-regulatory T cells are natural regulatory effector T cells. Cell Stress 2019;3:310–1. 10.15698/cst2019.10.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad SM, Larsen SK, Svane IM, et al. Harnessing PD-L1-specific cytotoxic T cells for anti-leukemia immunotherapy to defeat mechanisms of immune escape mediated by the PD-1 pathway. Leukemia 2014;28:236–8. 10.1038/leu.2013.261 [DOI] [PubMed] [Google Scholar]
- 29.Munir Ahmad S, Martinenaite E, Hansen M, et al. Pd-L1 peptide co-stimulation increases immunogenicity of a dendritic cell-based cancer vaccine. Oncoimmunology 2016;5:e1202391. 10.1080/2162402X.2016.1202391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bendtsen SK, Perez-Penco M, Hübbe ML, et al. Peptide vaccination activating galectin-3-specific T cells offers a novel means to target galectin-3-expressing cells in the tumor microenvironment. Oncoimmunology 2022;11:2026020. 10.1080/2162402X.2022.2026020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aaboe Jørgensen M, Ugel S, Linder Hübbe M, et al. Arginase 1-based immune modulatory vaccines induce anticancer immunity and synergize with anti-PD-1 checkpoint blockade. Cancer Immunol Res 2021;9:1316–26. 10.1158/2326-6066.CIR-21-0280 [DOI] [PubMed] [Google Scholar]
- 32.Nandre R, Verma V, Gaur P, et al. Ido vaccine ablates immune-suppressive myeloid populations and enhances antitumor effects independent of tumor cell IDO status. Cancer Immunol Res 2022;10:571–80. 10.1158/2326-6066.CIR-21-0457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iversen TZ, Engell-Noerregaard L, Ellebaek E, et al. Long-Lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res 2014;20:221–32. 10.1158/1078-0432.CCR-13-1560 [DOI] [PubMed] [Google Scholar]
- 34.Kjeldsen JW, Lorentzen CL, Martinenaite E, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med 2021;27:2212–23. 10.1038/s41591-021-01544-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmström MO, Mortensen REJ, Pavlidis AM, et al. Cytotoxic T cells isolated from healthy donors and cancer patients kill TGFβ-expressing cancer cells in a TGFβ-dependent manner. Cell Mol Immunol 2021;18:415–26. 10.1038/s41423-020-00593-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mortensen REJ, Holmström MO, Andersen MH. Characterization of TGFβ-specific CD4+T cells through the modulation of TGFβ expression in malignant myeloid cells. Cell Mol Immunol 2021;18:2575–7. 10.1038/s41423-021-00770-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Penco M, Weis-Banke SE, Schina A, et al. TGFβ-derived immune modulatory vaccine: targeting the immunosuppressive and fibrotic tumor microenvironment in a murine model of pancreatic cancer. J Immunother Cancer 2022;10:e005491. 10.1136/jitc-2022-005491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmström MO, Andersen MH. Healthy donors harbor memory T cell responses to ras neo-antigens. Cancers (Basel) 2020;12:1–16. 10.3390/cancers12103045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moodie Z, Price L, Gouttefangeas C, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother 2010;59:1489–501. 10.1007/s00262-010-0875-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slingluff CL, Petroni GR, Chianese-Bullock KA, et al. Trial to evaluate the immunogenicity and safety of a melanoma helper peptide vaccine plus incomplete Freund’s adjuvant, cyclophosphamide, and PolyICLC (mel63). J Immunother Cancer 2021;9:e000934. 10.1136/jitc-2020-000934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. 10.1158/0008-5472.CAN-11-2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moir JAG, Mann J, White SA. The role of pancreatic stellate cells in pancreatic cancer. Surg Oncol 2015;24:232–8. 10.1016/j.suronc.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 43.Principe DR, DeCant B, Mascariñas E, et al. Tgfβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res 2016;76:2525–39. 10.1158/0008-5472.CAN-15-1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadozai H, Acharjee A, Eppenberger-Castori S, et al. Distinct stromal and immune features collectively contribute to long-term survival in pancreatic cancer. Front Immunol 2021;12:643529. 10.3389/fimmu.2021.643529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kieffer Y, Hocine HR, Gentric G, et al. Single-Cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 2020;10:1330–51. 10.1158/2159-8290.CD-19-1384 [DOI] [PubMed] [Google Scholar]
- 46.Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–96. 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dominguez CX, Müller S, Keerthivasan S, et al. Single-Cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov 2020;10:232–53. 10.1158/2159-8290.CD-19-0644 [DOI] [PubMed] [Google Scholar]
- 48.Aoyagi Y, Oda T, Kinoshita T, et al. Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br J Cancer 2004;91:1316–26. 10.1038/sj.bjc.6602141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsujikawa T, Kumar S, Borkar RN, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep 2017;19:203–17. 10.1016/j.celrep.2017.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu L, Fu X, Chen X, et al. M2 macrophages induce EMT through the TGF-β/smad2 signaling pathway. Cell Biol Int 2017;41:960–8. 10.1002/cbin.10788 [DOI] [PubMed] [Google Scholar]
- 51.Huang B, Pan P-Y, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006;66:1123–31. 10.1158/0008-5472.CAN-05-1299 [DOI] [PubMed] [Google Scholar]
- 52.Siret C, Collignon A, Silvy F, et al. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory T cells in pancreatic ductal adenocarcinoma. Front Immunol 2019;10:3070.:3070. 10.3389/fimmu.2019.03070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciardiello D, Elez E, Tabernero J, et al. Clinical development of therapies targeting TGFβ: current knowledge and future perspectives. Ann Oncol 2020;31:1336–49. 10.1016/j.annonc.2020.07.009 [DOI] [PubMed] [Google Scholar]
- 54.Munir S, Larsen SK, Iversen TZ, et al. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS One 2012;7:e34568. 10.1371/journal.pone.0034568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-006432supp008.pdf (210.2KB, pdf)
jitc-2022-006432supp009.pdf (160.2KB, pdf)
jitc-2022-006432supp001.pdf (809.4KB, pdf)
jitc-2022-006432supp002.pdf (888.6KB, pdf)
jitc-2022-006432supp003.pdf (592KB, pdf)
jitc-2022-006432supp004.pdf (1.4MB, pdf)
jitc-2022-006432supp005.pdf (1.4MB, pdf)
jitc-2022-006432supp006.pdf (810.4KB, pdf)
jitc-2022-006432supp007.pdf (611.4KB, pdf)
Data Availability Statement
Data are available upon reasonable request.