

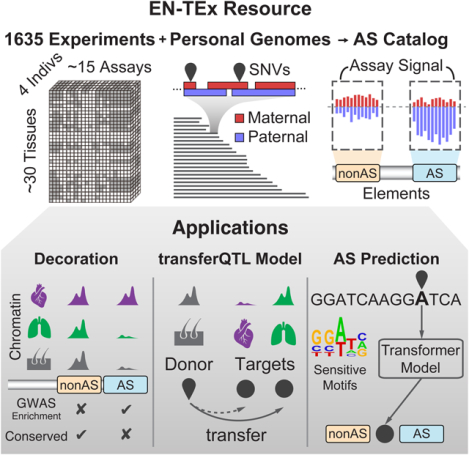
Abstract
Understanding how genetic variants impact molecular phenotypes is a key goal of functional genomics, currently hindered by reliance on a single haploid reference genome. Here, we present the EN-TEx resource of 1635 open-access datasets from four donors (~30 tissues × ~15 assays). The datasets are mapped to matched, diploid genomes, with long-read phasing and structural variants, instantiating a catalog of >1 million allele-specific loci. These loci exhibit coordinated activity along haplotypes and are less conserved than corresponding, non-allele-specific ones. Surprisingly, a deep-learning transformer model can predict the allele-specific activity based only on local nucleotide-sequence context, highlighting the importance of transcription-factor-binding motifs particularly sensitive to variants. Furthermore, combining EN-TEx with existing genome annotations reveals strong associations between allele-specific and GWAS loci. It also enables models for transferring known eQTLs to difficult-to-profile tissues (e.g., from skin to heart). Overall, EN-TEx provides rich data and generalizable models for more accurate personal functional genomics.
In Brief:
Understanding the impact of genetic variants is important to functional genomics. EN-TEx provides epigenomes across tissues, coupled with long-read genome assemblies, to build generalizable models of variant impact.
Graphical Abstract
INTRODUCTION
The Human Genome Project assembled one representative haploid sequence 20 years ago1,2. Since then, many individual genomes have been sequenced3,4. Compared to the reference, an individual’s personal genome typically contains ~4.5 million variants5. The vast majority of these are in non-coding regions and are most often present in the heterozygous state6–8. A goal of functional genomics is to assess the impact of these variants on molecular endophenotypes (e.g., epigenetic activity, RNA expression, or protein levels) and relate these to cell, tissue, and organismal traits, including disease phenotypes9–12.
To this end, researchers have conducted many genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) analyses associating genetic variants with phenotypic traits and changes in gene expression. In particular, the Genotype-Tissue Expression (GTEx) project has performed RNA sequencing (RNA-seq) experiments on >40 human tissues from nearly 1,000 individuals, allowing for the identification of >175K eQTLs13–16. In complementary fashion, the Encyclopedia of DNA Elements (ENCODE) project was initiated to annotate functional regions throughout the human genome17–19. However, these studies have largely been carried out using the generic reference genome, not directly using the variations observed in an individual’s diploid sequence. By using a diploid genome, heterozygous loci can distinguish sequences from each of the two parental chromosomes (haplotypes) that give rise to distinct molecular signals from each (e.g, RNA expression or transcription factor [TF] binding). The imbalance of expression or epigenetic activity between the haplotypes can be accurately measured by taking the reference allele as a baseline, avoiding biological and technical biases. If the imbalance is significant, the heterozygous variant is termed allele-specific (AS). AS variants have been identified in numerous previous studies and are implicated in several diseases18,20–30. Note that only some AS variants are thought to be causal for the observed changes31,32. However, for these loci, the AS experiment provides an ideal way to assess variant impact in a consistent and unbiased fashion33,34.
Here, to better connect personal genomes and functional genomics, we created the EN-TEx resource. This comprises a uniformly processed dataset of ~15 functional genomic assays, consistently collected from four individuals for ~30 tissues, many of them relatively difficult to obtain (e.g. lung). Specifically, it used two representative male and female individuals selected from the GTEx project for which the full battery of ENCODE assays were applied. These assays are coupled with long-read genome assemblies, containing comprehensive sets of structural variants (SVs). Compared to what was previously possible, mapping reads from the assays directly to diploid genomes allows for more precise quantification of differential expression and regulatory-element activity and for directly visualizing the impact on chromatin of single-nucleotide and structural variants (SNVs and SVs, respectively). Moreover, the uniform nature of the dataset makes possible more precise ascertainment of inter-individual vs. inter-tissue differences, and the scale of the resource enables the creation of the largest catalog of non-coding AS variants, an order-of-magnitude beyond what was available previously. We leveraged this catalog to build generalized models of variant impact. In particular, we created a model that predicts the AS imbalance resulting from a SNV just from the extended sequence context around a site (i.e., within a ~250 bp window). It highlights the importance of ~100 key TF motifs we term AS-sensitive. Finally, we can relate the EN-TEx resource to external genome annotations -- eQTLs and regulatory elements already known for the human genome. We built generalized models that transfer eQTLs from a source tissue to a different target one, leveraging the fact that EN-TEx represents a uniform collection of epigenetics data in hard-to-obtain tissues. This is practically quite useful given it is typically much easier to measure eQTLs in blood than other tissues, such as the heart, especially when using large cohorts of individuals. We also show that data from the EN-TEx resource can “decorate” existing regulatory elements, identifying subsets that are much more highly enriched with eQTL and GWAS SNVs than had been previously possible and illuminating broad relationships between conservation, AS activity and tissue-specificity.
RESULTS
Uniform Multi-tissue Data Collection & Diploid Mapping
We sequenced and phased the genomes of four GTEx individuals (identified as 1 through 4) using various complementary sequencing technologies (i.e., short-read Illumina, linked-read 10x Genomics, and long-read PacBio and Oxford Nanopore; STAR Methods “Data Stack” Section). After identifying SNVs, small insertions and deletions (indels), and SVs, we phased the haplotypes of the assembled genomes using linked-reads and proximity ligation sequencing (Hi-C; Data S2)35. This step generated long sequence blocks of phased variants extending across each chromosome, forming diploid personal genomes for each of the four individuals (Figure 1). The paternal/maternal origins of many of the phased blocks were determined by comparing the AS expression levels with known imprinted loci (Figure 1B and Data S2G–H; STAR Methods “Personal Genome” Section).
Figure 1. Uniform Multi-tissue Data Collection, Diploid Mapping and Construction of the AS Catalog.
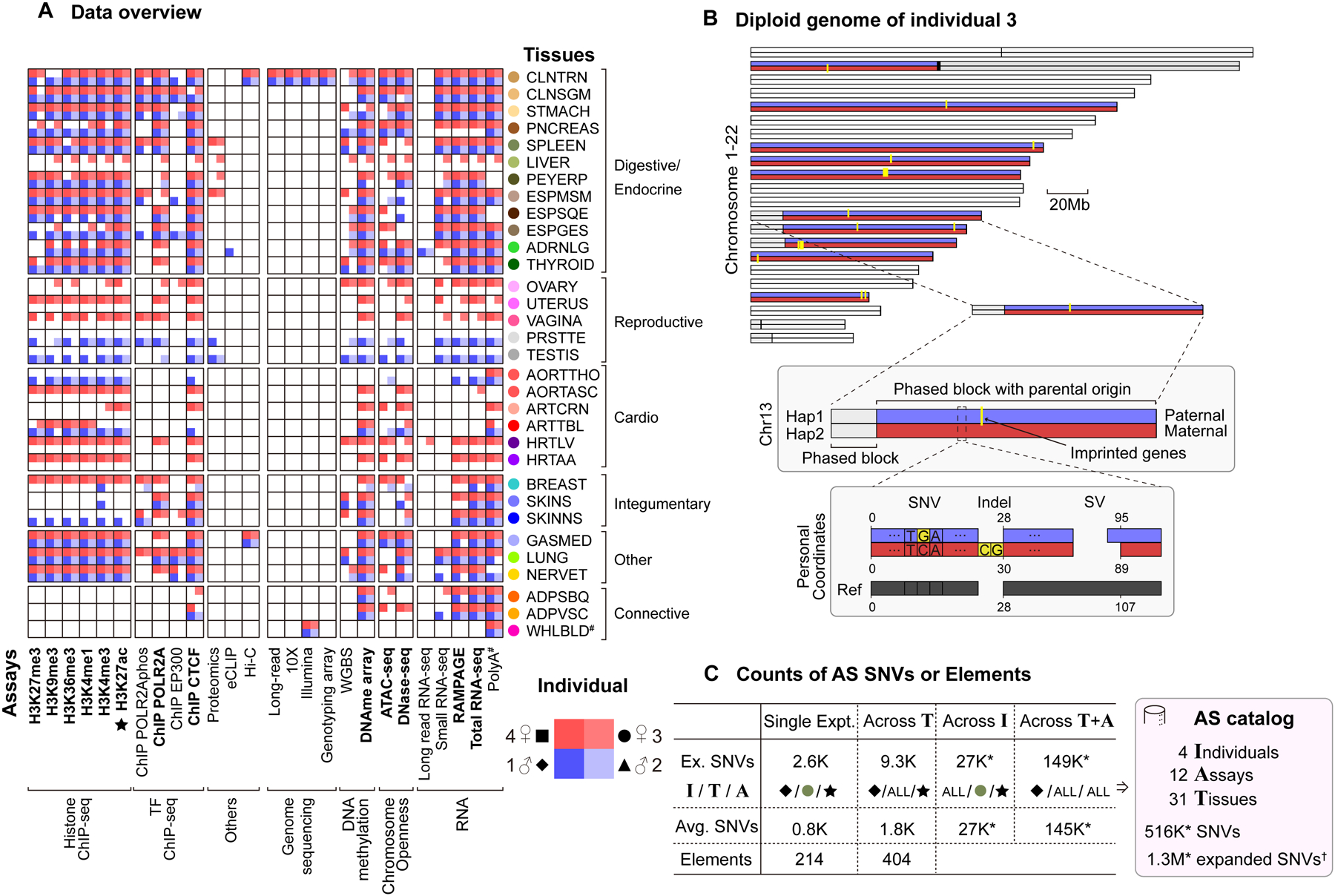
(A) Data matrix. The 13 core assays are indicated in bold; tissue colors from GTEx. (Details in Figure S1A.)
(B) The personal diploid genome of individual 3. The chromosomes are phased with known imprinting events (yellow), allowing the maternal (red) or paternal (blue) origin of many of the phased blocks to be identified. A schematic diagram of a region in chr13 shows the differences between the personal diploid genome and the reference genome, in particular their different coordinate systems and sequences. (Details in Data S2G and STAR Methods “Personal Genome” Section.)
(C) The AS catalog. Key statistics are shown at each level of pooling and averaging. By aggregating across tissues, individuals or assays, we were able to identify a large number of AS SNVs and AS genomic elements, resulting in an AS catalog. “*” indicates the aggregation was done by pooling of reads, instead of the default union method, which significantly increased detection power. Representative numbers in the “Ex. SNVs” row are initially based on a specific H3K27ac experiment in the spleen of individual 1. The I/T/A row shows whether this choice is continued in subsequent columns or whether averaging or pooling is done over “ALL” the individuals, tissues, or assays, respectively. “†” indicates AS SNVs from DNase and WGBS in addition to the 12 RNA/ChIP/ATAC assays. (Details in Figure S3A–D and STAR Methods “AS Catalog” Section.)
We identified ~18K SVs in each of the four individuals (>50 bp in length; Figure S1D and Data S15; STAR Methods “SVs” Section). The SVs tended to be depleted in most functional regions (e.g., genes and enhancers) and to have typical allele-frequency spectra, consistent with previous findings36,37.
In parallel, we carried out 1,635 experiments from ~15 different epigenome, transcriptome, and proteome assays on ~30 tissues obtained from each of four individuals (i.e., 13 core assays -- including ChIP-seq, ATAC-seq, DNase-seq, methylation arrays, short-read RNA-seq -- and several additional ones -- including whole-genome bisulfite sequencing [WGBS], Hi-C, eCLIP, and labeled proteomic mass-spectrometry; Figures 1A and S1A; STAR Methods “Data Stack” Section). This significantly expanded upon the assays available from GTEx using ENCODE technologies (STAR Methods “Sample Selection” Section). The data, analysis and software tools from the project are all open access, with everything being directly available from the EN-TEx portal (entex.encodeproject.org, Details in STAR methods “Portal” Section).
All datasets in the EN-TEx resource were processed using the phased diploid and reference genomes, giving rise to three mappings and three corresponding signal tracks for each assay (maternal and paternal haplotypes and the reference; Figure S1C and Data S2–4). Overall, we found ~2.5% more reads mapped to the personal genomes than the reference (for strict mapping criteria; Figure S1C; STAR Methods “Reference Comparison” Section). This mapping had a measurable effect (>2 fold) on the expression levels of >200 genes across all four individuals. This change is a conservative estimate but still comparable in magnitude to the number of differentially expressed genes often found in comparing between healthy and disease states (Figure S2A and Data S5; STAR Methods “Reference Comparison” Section)38–41. A similar fraction of cCREs, candidate cis-regulatory regulatory elements, exhibited a significant change in activity levels when using the personal compared to the reference genome (specifically, comparing the H3K27ac level on ENCODE cCREs; Figure S2B and Data S5)17.
Because of its uniform data collection and processing, EN-TEx provides an ideal platform to consistently measure inter-individual, inter-tissue, and inter-assay variability (Figure S2C and Data S6). In particular, we can explore all the sources of variation to place each EN-TEx sample in a high-dimensional space. It is readily apparent, as expected, that inter-individual variation is less than inter-tissue variation (e.g. in H3K27ac), which is less than inter-assay variation (e.g. comparing H3K27ac to H3K4me3). Finally, for the specific situation of comparing between tissues, the EN-TEx resource allows us to determine inter-tissue differences with greater accuracy than for equivalent data not matched across individuals (Details in Figure S2D and Data S6N–O).
Large-scale Determination of AS SNVs & Construction of the AS Catalog
We investigated AS behavior on a large scale using EN-TEx. For most assays, we performed these calculations uniformly using a standardized pipeline that dealt with various technical issues such as the reference and ambiguous mapping biases (Data S7 and STAR Methods “AS Calling” Section)18,19,22,42–45. Overall, we ran the pipeline on ~1,000 samples (31 tissues, 12 assays, and four individuals). On average, we detected ~800 AS events at SNVs in each sample, representing about ~4% of the total number of accessible SNVs (Figures 1C and S3A–D). (An accessible SNV is a heterozygous SNV with sequencing depth sufficient to detect AS behavior.) We were also able to group together the AS SNVs within a genomic element together to determine its overall AS status; on average, we found ~200 “AS elements” in each sample.
We had to use a more specialized approach for some of the assays, in particular, WGBS, Hi-C, and mass-spec proteomics. For instance, for Hi-C, we constructed haplotype-resolved contact matrices and then identified haplotype-specific AS interactions (Data S10C). Overall, per sample, we found ~0.5M AS interactions out of a total of ~6.5M Hi-C interactions (Data S10D). We also identified AS peptides exhibiting significant imbalance, corresponding to 696 unique genes (STAR Methods “AS Calling” Section).
After determining the AS SNVs in each sample, we combined them across all tissues, individuals, and assays (Figure 1C or S3C). We used two different combining strategies for the catalog: (i) individually determining AS imbalance (i.e., “calling”) separately on each tissue (or assay) and then taking the union of the calls or (ii) pooling the reads across tissues (or assays) and then jointly calling. We found, in fact, that pooling across tissues dramatically increased our detection power (by ~5X), making it possible to identify ~27K AS SNVs for an assay in an individual (for RNA/ChIP/ATAC assays; Figure 1C and S3A–D; STAR Methods “Aggregation” Section). We then combined the AS SNVs across assays and found ~365K AS SNVs per individual (now including WGBS and DNase). Finally, when we combined these data across all four individuals, we reached a total of about ~1.3M AS SNVs, which constitutes our AS catalog (with 516K coming from RNA/ChIP/ATAC assays; Figure 1C and S3A–D).
The AS catalog has several key aspects. First of all, it is much larger than previous collections of AS chromatin events (STAR Methods “AS Catalog” Section)22,28,46. Moreover, we estimated that the AS SNVs detected in the four EN-TEx individuals cover 76% of common AS SNVs in the European population, suggesting that the catalog includes a majority of the AS events at common SNV loci in Europeans (STAR Methods “AS Catalog”). In addition to the common AS variants detected, some AS sites correspond to rare SNVs: in total, we found that 63K of the 1.3M AS variants were rare (STAR Methods “AS Catalog”). We were also able to cross-reference these rare SNVs with known pathogenic and deleterious variants, including 14 in ClinVar (STAR Methods “AS Catalog” Section)47.
Because of its size, we can leverage the catalog to determine AS SNVs in an entirely new sample with increased sensitivity (Data S12). In addition, using a related strategy, we can develop alternate, “high-power” AS assignments from joint calling across samples (Data S13). A final key aspect of the AS catalog is that most variants are in noncoding regions of the genome and are determined using non-RNA-based assays. In fact, only ~2.5% of the AS variants in the catalog are uniquely detected by RNA-seq, and 95% are only detected by assays other than RNA-seq (Data S8B–C).
Examples of Coordinated AS Activity, involving SNVs & SVs
Using the catalog, we found several examples of coordinated AS activity across different assays. First, we surveyed known imprinted loci, finding that AS activity is fairly consistent across tissues (Data S9B and S2H). A good illustrative example is the classic case of IGF2 and H19. As expected, in several tissues, we observed that H19 is expressed only maternally, and IGF2, only paternally, due to AS CTCF binding at the imprinting control region (Figure S4A)48. Moreover, haplotype-resolved Hi-C showed that, on the maternal haplotype, a cCRE upstream of H19 interacts with this gene but not with IGF2. In contrast, on the paternal haplotype, the same cCRE only interacts with IGF2, suggesting a potential mechanism for the locus.
A second illustrative example shows the coordinated activity over chromosome X. On this chromosome in females, we observed gene expression, active histone marks, POL2R and CTCF binding all skewed toward one haplotype, with repressive marks biased to the other (Figure 2A and Data S14). There are notable exceptions, including genes in pseudoautosomal regions and documented “escaper” genes (e.g., DHRSX and KDM6A, respectively)49. The imbalance in the active chromatin mirrors well what is observed in the RNA-seq. In addition, haplotype-specific Hi-C manifested great differences in AS interactions on chromosome X at some loci (e.g., XACT; Data S14G and STAR Methods “AS Examples” Section). Interestingly, across many tissues, we find a consistent skew in X-chromosome imbalance, in line with recent findings that X-inactivation is completed prior to the specification of the germ layers (Data S14A–F)50.
Figure 2. Examples of Coordinated AS Activity, Involving SNVs and SVs.
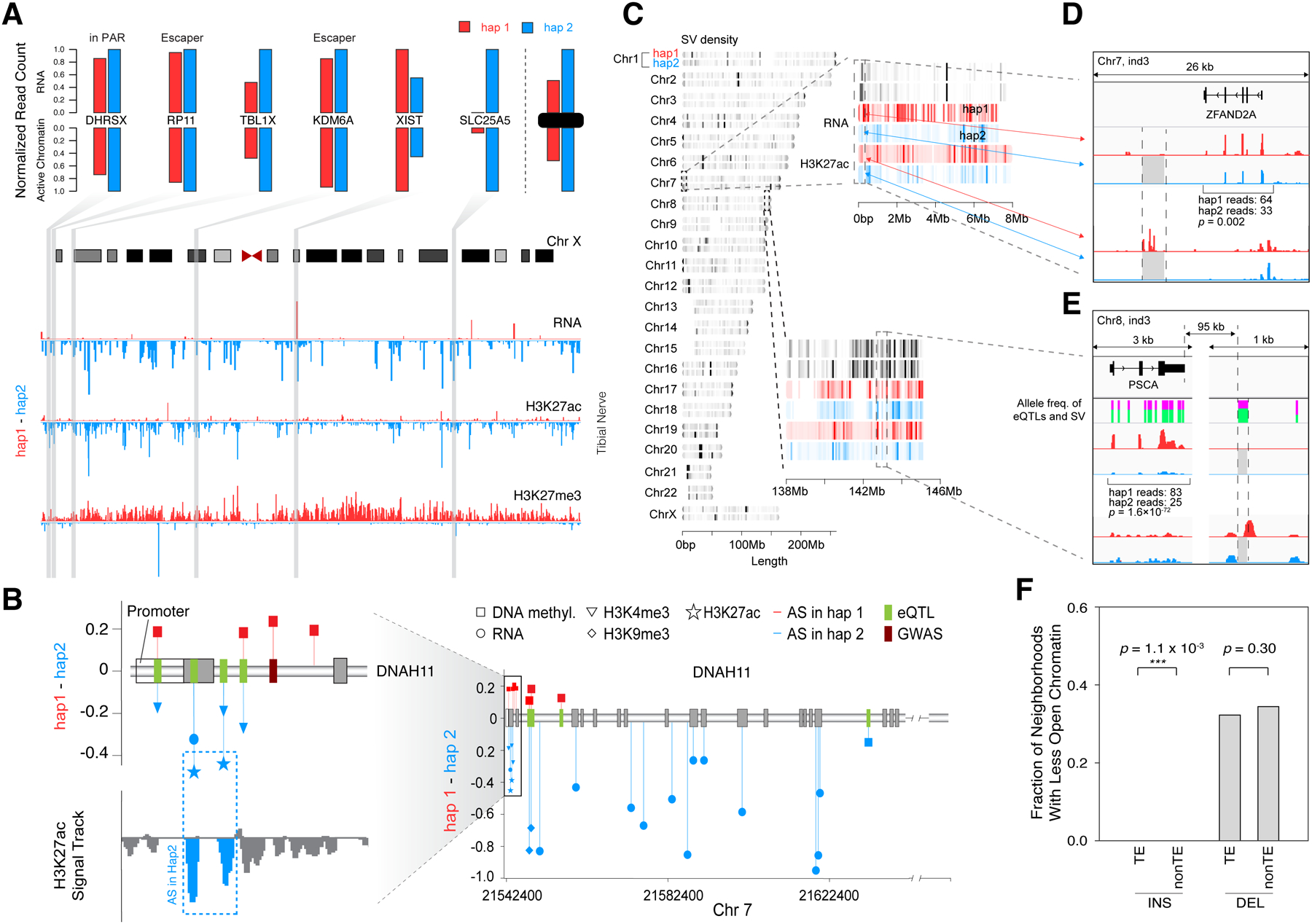
(A) Detecting coordinated AS activity across a chromosome. Signal tracks (bottom) show that for chrX in the tibial nerve of individual 3, hap1 generally has lower expression levels, lower H3K27ac levels, and higher H3K27me3 levels than hap2. The top bar-graphs show the expression and active promoter chromatin of 6 selected genes. (Details in Data S14.)
(B) AS events at a disease-associated locus: the DNAH11 gene. The lollipop diagrams show the degree of AS imbalance for various assays at heterozygous SNPs in individual 1. Those that are GTEx eQTLs and GWAS loci are highlighted. (Details in STAR Methods “AS Examples” Section.)
(C) The chromosomal distribution of SVs on the diploid genome. Colors indicate the density of SVs. Genomic regions of chr7 and chr8 (in individual 3) are enlarged to show the positions of detected SVs and the levels of H3K27ac and RNA expression obtained from transverse colon.
(D) The effect of a 2.6 kb deletion. The deletion in hap2 removed several H3K27ac peaks and reduced ZFAND2A expression in thyroid. (Details in Data S17C–D.)
(E) The effect of a 98-bp deletion. The deletion in hap2 in individual 3 removed a H3K27ac peak in colon downstream of PSCA, potentially contributing to reduced expression. The heights of the green bars indicate the allele frequencies of the deletion and the surrounding GTEx eQTL SNVs, indicating they are potentially in linkage disequilibrium. (Details in Data S17G–H.)
(F) Overall effect of TEs on chromatin. The genomic regions neighboring the TE insertions show reduced chromatin accessibility more often than those of the non-TE insertions. (Details in Data S18 and STAR Methods “SVs” Section.)
A third example that demonstrates coordinated AS activity is DNAH11, a gene associated with ciliary dyskinesia (OMIM #611884; Figure 2B). We observed AS methylation in the promoter regions on the opposite haplotype to the AS expression and H3K4me3 and H3K27ac activity, consistent with transcriptional downregulation.
For SVs as opposed to SNVs, we found many specific examples of variants impacting chromatin and nearby gene expression in an AS fashion. For instance, Figure 2D shows a well-supported example: a heterozygous deletion, overlapping a known SV eQTL, removing an activating region, and a matching decrease in expression of a nearby gene (specifically, an H3K27ac peak near ZFAND2A51). Figure 2E shows a similar example: a heterozygous deletion removing an activating region near PSCA. Here, the deletion is not known to be associated with an eQTL but has a similar allele frequency to nearby eQTL SNVs and thus might represent the causal variant associated with them. On average, we identified ~300 potential SV eQTLs in each individual (STAR Methods “SVs” Section; Figure S4C and Data S17G–J show additional examples and SV-eQTL associations; Data S17N–O shows related examples for homozygous events; Data S17M shows examples of whole-exon deletions).
Figure S4B shows an SV removing a likely repressive region in an intron of PCCB (a H3K9me3 peak). Moreover, this SV is adjacent to several GTEx splicing QTL (sQTL) sites, and long-read RNA-seq indicates that both individuals have different splice isoforms near the SV (Data S17K). Notably, the EN-TEx resource enables direct comparison between SVs and their impact on transcript structure, with both determined by long-read sequencing.
Overall, we found that the SVs were distributed over the diploid genome unevenly with different associations with the chromatin from different haplotypes and that a significant fraction of genes with AS expression were associated with nearby SVs or indels (1.5% for SV deletions and 13.6% for small deletions; Figure 2C–E, Data S16, and Data S17A–B). Furthermore, many of these expression changes were also coupled to chromatin changes, as expected52. In particular, we assessed whether chromatin significantly changes around heterozygous SVs by calculating a “disruption score” (Figure 2F and Data S18A). We found that transposable element (TE) insertions were associated with a reduction in nearby open chromatin (compared with non-TE ones). We observed similar results when comparing the chromatin near SVs between EN-TEx individuals (both heterozygous and homozygous SVs; Data S18B–D). These results agree with findings that cells repress active chromatin to suppress TE expression53–55.
Application 1: Decorating ENCODE Elements with EN-TEx Tissue & AS Information
Up to this point, we have focused on the four EN-TEx individuals; now we turn to leveraging the resource to create generalized knowledge beyond them, broadly applicable in many contexts. We demonstrate 3 applications, focused on predictive modeling of AS behavior and approaches to “decorating” existing genome annotations.
The ENCODE encyclopedia annotations were constructed using a disparate collection of cell lines and tissues; they are also devoid of variant annotations. We can layer the results from EN-TEx onto these annotations, consistently “decorating” them and extending their utility. In particular, we can combine them with the AS catalog, highlighting subsets exhibiting AS activity (Data S19A–B). Next, for each EN-TEx tissue we determined consistently whether each ENCODE element is active, repressed, or bivalent (Figure 3A and Data S19C; STAR Methods “Decoration Process” Section). Overall, 97% of the ~1M cCREs in the ENCODE encyclopedia can be decorated, and we validated our decorations using data from other studies with tissuematched Hi-C (Data S21).
Figure 3. Aspects of Application 1: Decorating ENCODE Elements with EN-TEx Tissue & AS Information.
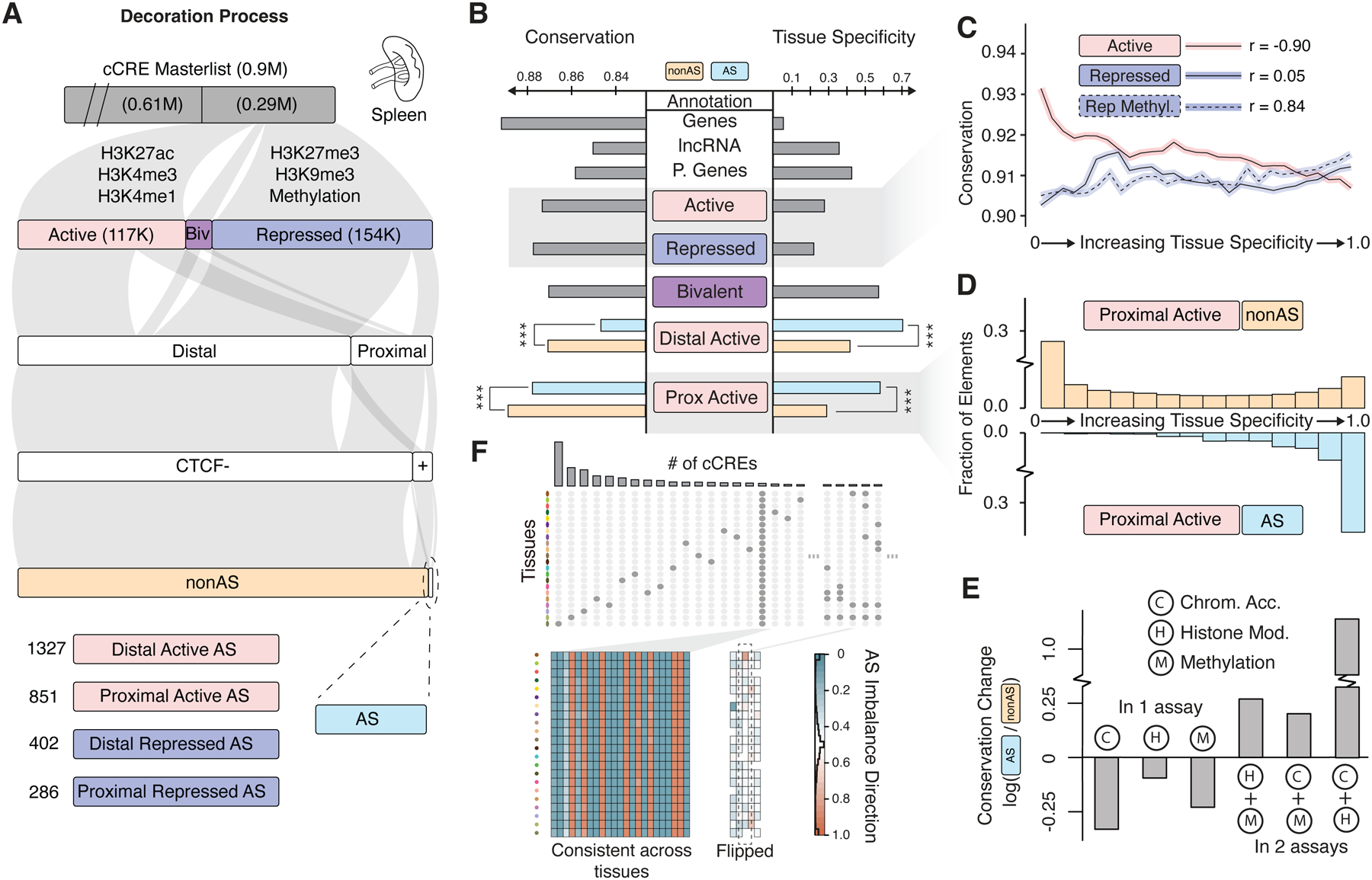
(A) Workflow decorating cCREs with EN-TEx data. The workflow starts with the master list of 0.9M cCREs from ENCODE, which have no tissue-specific information. Representative numbers from spleen are shown along the flowchart. (Details in Figure S5.)
(B) Tissue specificity and conservation of annotations. The tissue specificity of an annotation category is the fraction of the cCREs observed in the category active in only a single tissue. A smaller value indicates that the category members are more ubiquitous. Conservation score is determined by the fraction of rare variants in the genomic regions of an annotation category. Stars indicate statistically significant differences. (Details in in Data S22 and STAR Methods “Tissue Specificity” Section.)
(C) Correlation between tissue specificity and conservation for active and repressed cCREs. Repressed cCREs with methylation show increased significance.
(D) Comparing the tissue distribution of AS and non-AS proximal active cCREs. (Top) Non-AS categories show a “U-shaped” trend, whereas (Bottom) AS categories have an “L-shaped” one. Fraction of Elements is described in the STAR Methods “Tissue Specificity” Section.
(E)AS events occurring in 1 or 2 assays and their relationship to purifying selection. AS events are for chromatin accessibility (Hi-C, DNAse-seq and ATAC-seq), histone modification (H), methylation (M). The change in conservation between an AS category and the corresponding non-AS one is shown as the log ratio of their conservation scores (from B). This ratio is negative for AS events in one assay and positive for AS events in two assays, suggesting that an AS SNV with multiple events is more conserved.
(F)Consistency of AS imbalance across tissues. The heatmap shows the direction of the allelic imbalance across the most ubiquitous AS cCREs (in individual 3). The imbalance direction is consistent across tissues; however, a few tissue-specific cCREs show directional flips. (Details in Data S22G.)
Given our decoration strategy, we used a straightforward approach to measure tissue-specificity, which can be consistently applied to many different types of annotations (STAR Methods “Tissue Specificity” Section). Briefly, the tissue specificity of a given annotation subset (e.g., gene-proximal cCREs) is the fraction of elements active in only one tissue (Figure 3B and Data S22; STAR Methods “Tissue Specificity” Section). As expected, by this measure, only a small percentage of protein-coding genes were tissue-specific (~8% by either RNA-seq or mass spectrometry)56,57; in comparison, pseudogenes, lncRNAs, and active regulatory elements exhibited higher tissue specificity (Figure 3B and 3C). More notably, AS genes and regulatory elements were more tissue-specific than the corresponding non-AS ones (Figures 3B and S5B). Moreover, we observed that unlike many genomic elements that mostly fall into two distinct categories, tissue-specific or ubiquitously active (giving rise to the characteristic “U-shaped” histogram in Figure 3D), AS elements are only tissue-specific for many different assays (an “L-shaped” histogram). (They are also depleted in “housekeeping behavior”; Data S22J and S23D–G). Finally, for the few elements that are AS across all available tissues, we found the haplotype direction of the AS imbalance to be consistent (23 AS cCREs and 20 AS genes; Figure 3F, Data S22G–I, and STAR Methods “Tissue Specificity” Section)58. This finding, plus the fact that we did not observe many loci where the imbalance direction flipped across tissues, supports our joint-calling and aggregation strategy for identifying AS events (Figures 1C and Data S3A–D; STAR Methods “Tissue Specificity” Section).
We examined the relationship between tissue specificity and conservation (Figures 3B and 3C). Notably, we found for active annotations, those with higher tissue specificity had lower purifying selection and for repressed annotations, the opposite trend (Figure 3C). Consistent with previous studies, we found that AS elements are under less purifying selection than non-AS ones (Figure 3B and Data S23D–G)22,28,43,59. Conversely, we detected an increase in purifying selection for loci AS in more than one assay (e.g., methylation and histone modifications), perhaps reflecting their greater functional importance (Figure 3E). In summary, we found that loci demonstrating more activity across tissues, haplotypes, or functional assays showed increased conservation.
Next, we analyzed the relationship between decorated regulatory elements and eQTL and GWAS SNVs. First, we found that AS elements produced significantly better GWAS enrichments for disease traits (compared to an appropriate baseline, Figure 4A–B and Data S25A–B; STAR Methods “Decoration Enrichments” Section). In particular, we found that the subsets of tissue-specific cCREs that were AS showed substantially greater enrichment than those not AS. For example, cCREs that exhibited AS activity in the coronary artery had higher enrichment for cardiovascular-disease GWAS SNVs as compared to non-AS ones60–63. Also, for immune-associated traits, we found that enriched AS cCREs manifest better specificity for their biologically relevant tissue compared to non-AS ones (Figure 4B, showing spleen, and Data S25F).
Figure 4. Aspects of Applications 1 and 2: Relating Decorations and AS SNVs to GWAS & eQTL Loci.
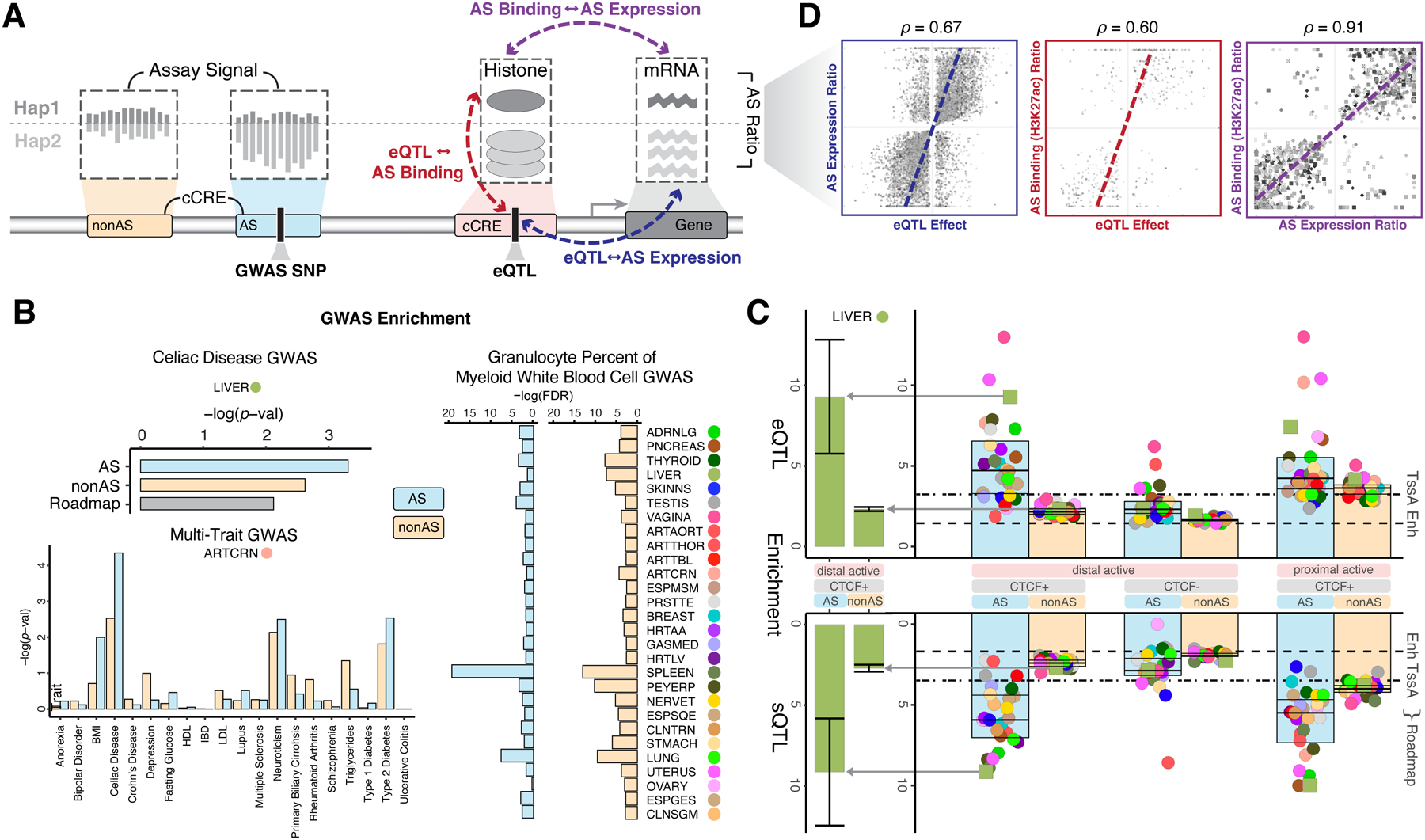
(A) Schematic showing the inter-relationship of AS activity, GWAS SNPs and eQTLs.
(B) Higher GWAS enrichment for AS elements compared to the corresponding non-AS ones. Top left shows one tissue and one trait, compared to the Roadmap Project. Bottom left shows an extension to many traits for one tissue, and right shows many tissues for one trait. (Details in Data S25 and STAR Methods “Decoration Enrichments” Section.)
(C) QTL enrichment for decorated cCREs. Colored dots show the enrichment for each tissue (GTEx colors, Figure 1A and Data S2I). Each bar shows the median enrichment over all tissues for a given annotation subset. As a reference, median enrichment of Roadmap “Enh” and “TssA” annotations are shown as dashed and dotted lines, respectively. The enrichments for the liver are highlighted. Robustness is estimated by resampling genetic variants, providing a range of enrichments shown with whiskers (Details in Data S24 and STAR Methods “Decoration Enrichments” Section.)
(D) Compatibility between AS gene expression, AS binding in the upstream promoter, and eQTL effect. eQTL effect is measured by the beta coefficient, and for AS, the imbalance ratio is plotted. (Details in Figure S5C–D; all correlations are statistically significant.)
Finally, we systematically estimated the enrichment of eQTL and sQTL variants in cCREs active in the matched tissue type (Figure 4C and Data S24A). The enrichment was considerably stronger than previous studies and showed greater magnitude for proximal vs distal cCREs, especially, as expected, for sQTLs (Data S24C)64. As we did for GWAS SNVs, we compared eQTL/sQTL enrichment in AS elements with non-AS ones, finding substantially higher enrichment in AS subsets (Figure 4C). For distal active cCREs, the AS subset showed stronger enrichment across all tissues, with some tissues showing especially large increases (>2X change in enrichment, for cCREs containing CTCF binding sites).
Application 2: Relating AS SNVs to GTEx eQTLs & Modeling eQTLs in Hard-to-obtain Tissues
Another analysis we could do with GTEx eQTLs is to directly relate them to nearby AS activity. First, we analyzed the association of an eQTL with the AS expression of its target gene: as expected, a positive correlation is evident with eQTL effect size, providing an additional confirmation for the eQTL (Figure 4D). Next, we directly relate eQTL effect with the AS imbalance in promotor chromatin at the eQTL SNV (Figure 4D and Data S26A–C). The association here is more direct and provides a way to help prioritize putative causal variants among GTEx eQTLs, in line with previous findings (Data S26B–C; also see Figure S6A and Data S27A for a related, but alternate, approach)65. Finally, to complete the “triad” of comparisons, we interrelated the AS activities in both the promoter and the associated gene (Figure 4D). Here, we found quantitative agreement for the magnitude and direction of the AS imbalance over many different epigenetic and proteomic assays (with an associated list of strongly compatible gene-promoter pairs, STAR Methods “Compatibility” Section; Figure S5D). As expected, we observed negative correlations for repressive marks and DNA methylation and positive correlations for the many different active chromatin modifications (e.g., H3K27ac; Figure S5D and Data S26A–C).
The above correlation between AS activity and eQTLs is an example of how the EN-TEx resource can be integrated with external annotation. This integration can go further: because EN-TEx includes ChIP-seq data from hard-to-obtain tissues (e.g., heart), which is comparatively more difficult to obtain than RNA-seq data, we can use it to extend existing eQTL annotations to additional tissues.
We start with the observation that eQTL SNVs have stronger chromatin signals in the tissues in which they are active than in the tissues in which they are not, suggesting that the chromatin around an SNV may influence its chance of being an eQTL in a particular tissue (Figure S6B and Data S27B). Then, by combining the EN-TEx chromatin data and the GTEx eQTL catalog, we developed a random-forest statistical model that transfers the activity of an eQTL from a given donor tissue to another target tissue by considering the EN-TEx chromatin profile in the target (e.g., from skin to tibial artery; Figures 5A and S6C). Overall, when compared with known GTEx eQTLs, our predictions are highly accurate, independent of which donor or target tissues are employed (0.86 balanced accuracy; Figure 5B and Data S28C–D). Our model tends to transfer stronger GTEx eQTLs to the target (Figure 5C); conversely, it also identifies “likely” eQTLs, not quite reaching the “official” GTEx significance threshold (probably due to sample size) but still achieving greater significance than those not transferred.
Figure 5. Aspects of Application 2: Modeling eQTLs in Hard-to-obtain Tissues.
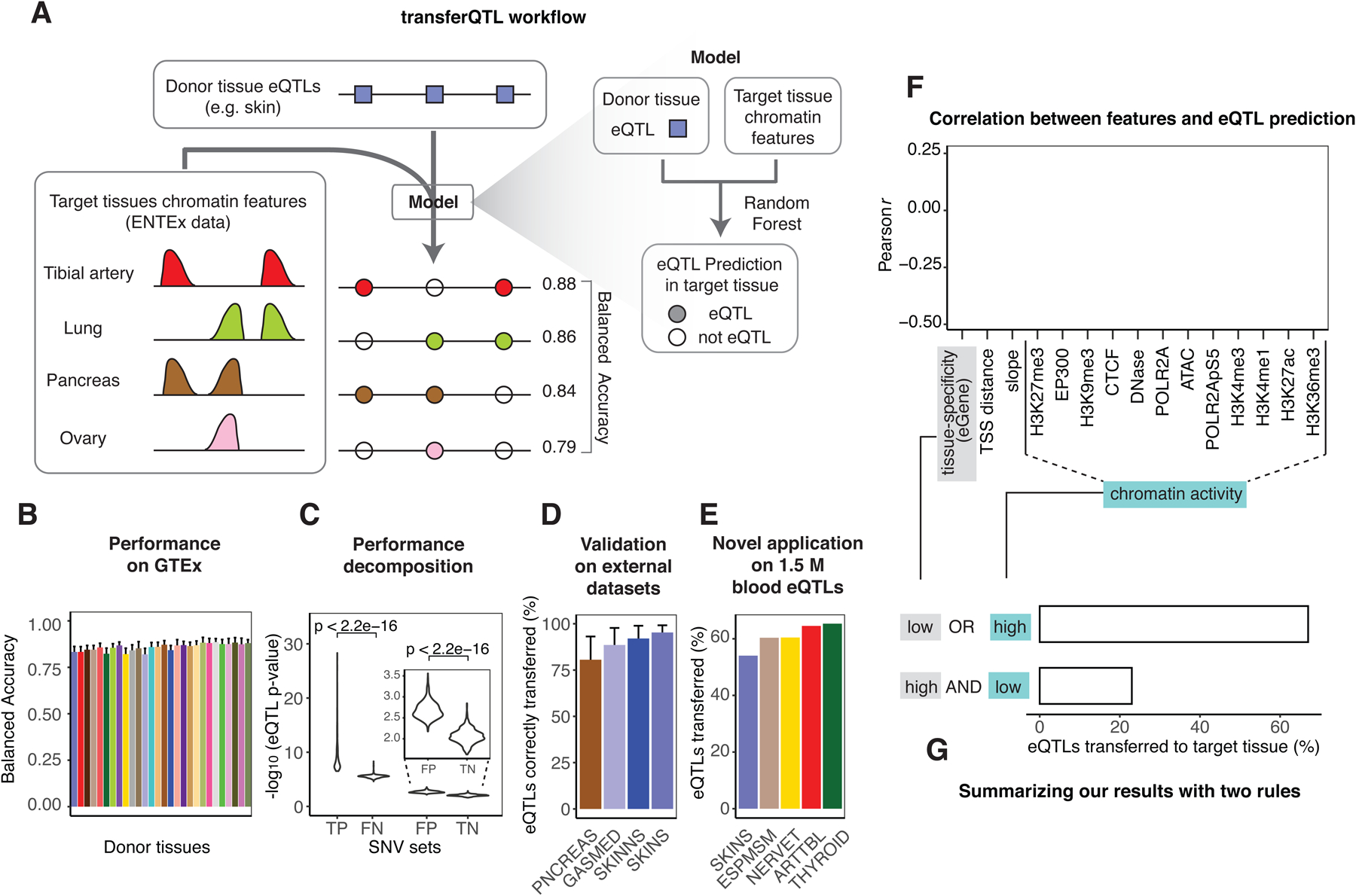
(A) Schema of the transferQTL model. For a catalog of eQTLs active in a source tissue (donor), we transfer them to another tissue (target) by leveraging the chromatin in the target and other features. (Details in Figure S6C.) For several representative target tissues the balanced accuracy is shown for transferring skin eQTLs.
(B) Performance of the model. The X-axis indicates the tissues used as the donors (GTEx coloring), and the Y-axis indicates the average performance (balanced accuracy) across the target tissues. The whiskers indicate variation across targets (standard deviations). (Details in Data S28CD.)
(C) Performance decomposition. For the confusion matrix resulting from applying the model to known GTEx eQTLs, we plotted the distribution of mean p-values on each subset.
(D) External validation. We validated our transferred eQTLs against four eQTLs catalogs other than GTEx: pancreas (PNCREAS), skeletal muscle (GASMED), suprapubic skin (SKINNS), and lower-leg skin (SKINS). The Y axis corresponds to the sensitivity of the prediction (TP / (TP + FN)). (Details in the STAR Methods “transferQTL Model” Section.)
(E) Large-scale application. We applied the model to a set of ~1.5 M eQTLs from blood (as donor). We were able to transfer a large proportion of these to EN-TEx target tissues. The plot shows the five tissues with the largest fractions transferred. (Details in Data S28F–G.)
(F) Importance of the features in the model. We computed the correlation between 15 selected features and the model’s probability of classifying donor-tissue eQTLs as eQTLs in the target tissue. The bar plot shows, for each feature, the strongest correlation observed across all 756 donor-target tissue pairs. (Details in Data S29A.)
(G) Schematic showing how two simple rules help predict eQTLs in a target tissue. To summarize F, we have found that two observations help define transferQTL. As an example, we show the results obtained when transferring eQTLs from testis (donor) to thyroid (target). (Details in STAR Methods “transferQTL Model” Section and Data S29B.)
We further validated our model, trained on GTEx, against other eQTL catalogs66. In particular, it correctly identified >75% of the eQTLs reported in catalogs for pancreas, skeletal muscle, and skin (Figure 5D). Finally, to showcase the value of our approach to enhance existing eQTL catalogs, we applied it to a set of 1.5M blood eQTLs from a large-cohort study; we were able to transfer up to 60% of these, enhancing the GTEx catalog with ~500K new candidate eQTLs per tissue (Figure 5E and Data S28F–G)67. Note the utility of this application: up to now, large-cohort, high-power eQTLs studies so far have been conducted mostly on a few readily available tissues, such as blood or skin67; the uniformly collected EN-TEx chromatin data allow us to leverage these existing annotations to other, more difficult-to-secure tissues.
Finally, we evaluated the relative contribution of the different genomic features to the model (Figure 5F and Data S29A). We found that we could get most of the predictive accuracy from a core model using four histone modifications (H3K36me3, H3K27ac, H3K4me1, H3K27me3, and some non-chromatin features; Figure S6E). Moreover, as expected, we found that SNVs with observable chromatin activity, especially H3K36me3, were more likely to be transferred. We observed the opposite for SNVs associated with genes that are highly tissue-specific or have distant transcription-start sites (Figure 5F and Data S29A). Given this, we can summarize the main features of our model in a simple heuristic: we can likely transfer an eQTL if it has high chromatin activity in the target tissue or its associated gene is not tissue-specific; if neither of these conditions is met, the transfer is probably not possible (Figure 5G and Data S29B).
Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Highlighting “Sensitive” TF Motifs
In our final application, we model the likelihood of a heterozygous variant to cause AS behavior. In particular, the ability of an SNV to disrupt a TF-binding motif suggests a direct relationship to the AS imbalance for a sequence-specific TF. Furthermore, given the importance of TFs in modulating open and closed chromatin, there is also a relationship, though less direct, to AS histone modifications. To study this, we cross-referenced all the AS sites in the EN-TEx ChIP-seq data with the 660 known human TF motifs and then ranked the motifs based on enrichment of AS SNVs (Figure 6A and STAR Methods “Sensitive Motifs” Section)68. Overall, we identified 195 TF motifs that were significantly enriched in AS SNVs and selected further a “top 100” subset (using a logical cutoff, which was robust to tissue selection, Figure S7A and Data S30; STAR Methods “Sensitive Motifs” Section).
Figure 6. Aspects of Application 3: Highlighting “Sensitive” TF Motifs.
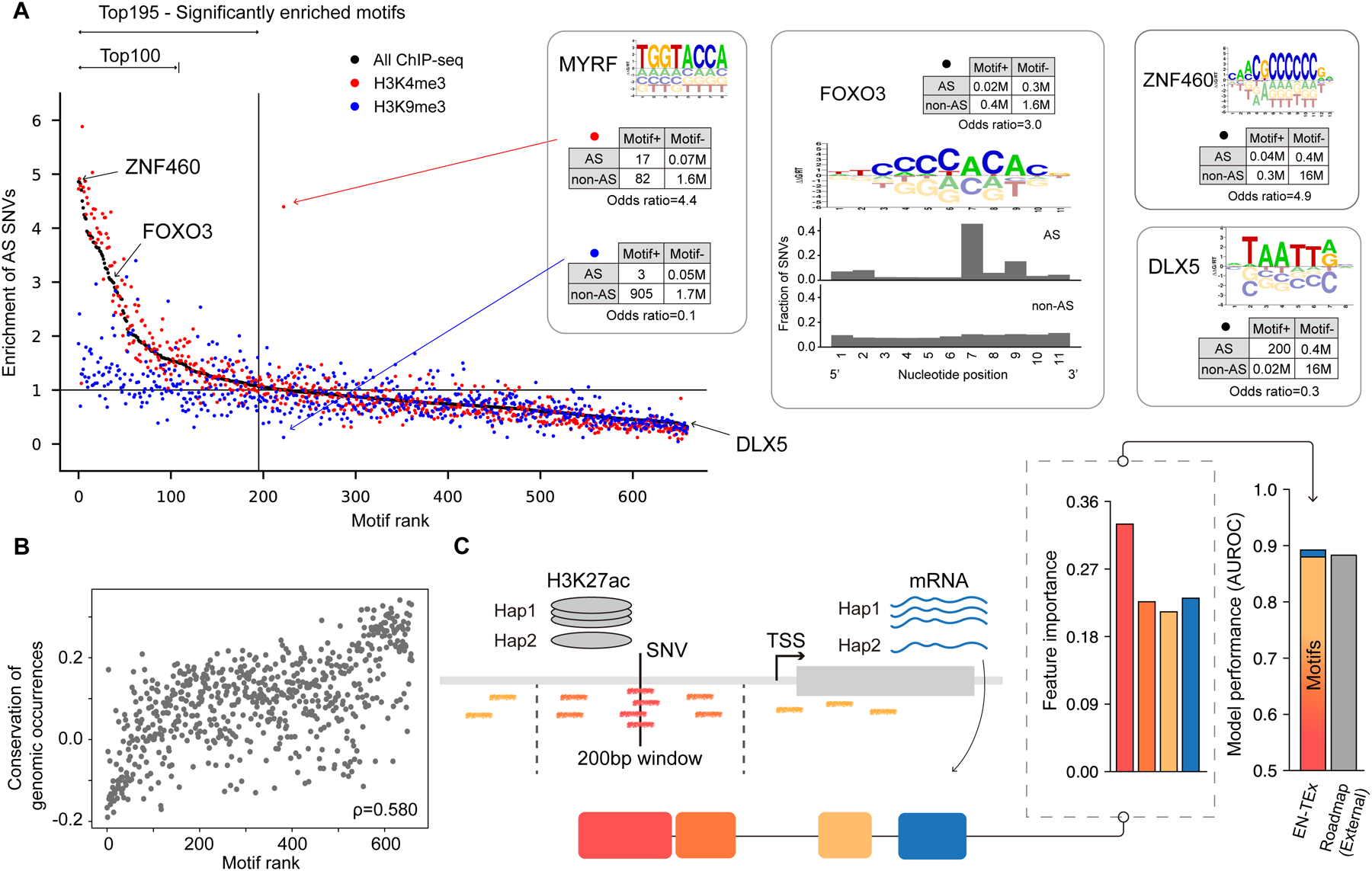
(A) TF Motifs ranked by enrichment of AS SNVs. We calculated the enrichment of AS SNVs for each TF using 2-by-2 contingency tables, with representative ones shown in the figure. For the representative TFs we also show a motif logo (and, for FOXO3, the location of the overlapping AS or non-AS SNVs). In the scatter plot, the dots correspond to TF motifs, which are ranked by AS enrichment. Colors indicate different histone modifications. (Details in Data S30 and STAR Methods “Sensitive Motifs” Section.)
(B) TF motif ranking is correlated with conservation of the motif regions. (Details in STAR Methods “Sensitive Motifs” Section.)
(C) Schematic of a statistical model predicting AS promoter activity. The model predicts whether a promoter exhibits AS H3K27ac activity. Motifs of ranked TFs (colored short lines) were used as features of the model in addition to AS expression ratio. Right-hand-side bar charts show feature weights and the overall performance of the model, in comparison to Roadmap. Model performance is dominated by the motifs, with only marginal improvement from adding AS expression imbalance. (Details in the STAR Methods “AS Promoter” Section.)
These top-ranked motifs represented TF binding sites particularly “sensitive” to mutations and more likely to give rise to AS behavior. They were enriched in C2H2 zinc-finger motifs (e.g. FOXO3 and ZNF460; Figure 6A). In contrast, the bottom-ranked, least-sensitive motifs were more likely to have a homeobox domain (e.g. DLX5). FOXO3, in particular, represents well how AS SNVs affect the zinc-finger motif: the AS SNVs occurred mostly at a single distinct nucleotide positions known to modulate binding, while non-AS SNVs occurred more uniformly (STAR Methods “Sensitive Motifs” Section)69. For many motifs, the enrichment associated with activating and repressive histone marks followed opposite trends (e.g. MYRF). Additionally, we found that the enrichment in AS SNVs anti-correlates with the conservation of the motif regions in the genome but is not correlated at all with a motif’s sequence complexity (i.e., “PWM entropy”; Figure 6B and Data S30A–I; STAR Methods “Sensitive Motifs” Section). The finding that AS-sensitive sites are less conserved dovetails with our earlier finding in Figure 3B that AS elements tend to be less conserved.
We next investigated how variants affecting motifs for AS-sensitive TFs relate to their effect on the expression of the downstream gene. To do this, we built simple statistical models connecting the presence of TF motifs to the AS activities of a gene and its associated promoter (specifically, in terms of AS expression and histone modification; Figure 6C and Data S31B–F). Simple statistics revealed that the promoter-target-gene relationship for AS activity is more nuanced than one might expect (Figure S7B and Data S31E); the complexity potentially results from alternative distal regulation or redundancy of regulatory sites. Nevertheless, we could construct successful models for AS promoter activity (cross-validated AUROCs of 0.81 on the EN-TEx individuals and 0.88 on external validation data; Figure 6C and Data S31A; STAR Methods “AS Promoter” Section). Given that RNA-seq data is much more readily available than ChIP-seq data, the model can be applied in a practical context, e.g., to predict AS promoter activity throughout the 838-individual GTEx cohort, using just RNA-seq data and genotypes (STAR Methods “AS Promoter” Section).
Remarkably, successful models for AS promoter activity needed few features. The most important ones were the number of AS-sensitive TF motifs in the promoter (overlapping or nearby to the central AS SNV), underscoring the importance of variants impacting these motifs. Other relevant features, but of secondary importance, were the occurrence of any TF motif (sensitive or not) distal to the SNV, and the AS expression imbalance of the downstream gene (Figure 6C and Data S31B). Interestingly, features that one might have expected to be important -- including the overall expression level of the gene or the eQTL status of the SNV in the promoter -- were not informative (Data S31D). Related to how the AS-promoter model highlighted the importance of AS-sensitive motifs, we also found an over-representation of TF motifs, particularly AS-sensitive ones, in the decorated subsets of cCREs enriched with eQTLs, discussed earlier (Figure 4C and Data S30J–K). This suggests AS sensitive motifs are key in driving the expression differences between alleles observed in eQTLs.
The impact of SNVs on AS-sensitive TFs implies that we may be able to predict whether an SNV would be associated with AS behavior by whether it overlaps such a motif. To investigate this, we built a simple model to predict whether an SNV would be AS for CTCF binding based on whether it overlapped with a CTCF motif in regulatory regions (STAR Methods “Transformer Model” Section); this “strawman” model had only slight predictive performance (Figure 7B). We then surmised that we could achieve better performance by including sequence context surrounding the CTCF motif. To do this, we built progressively more complex models, culminating in a deep-learning transformer model that took into account the sequence in a 250-bp window around the SNV (using DNABERT70; Figure 7A and STAR Methods “Transformer Model” Section). The transformer model achieved surprisingly good performance (0.69 cross-validated AUROC using EN-TEx samples, for predicting whether or not an accessible SNV for CTCF binding in any tissue would be AS, purely based on the sequence characteristics of the surrounding window; Figure 7B). We were also able to build similar models for POLR2A and various histone marks (Figure S7 and Data S32A). For H3K27ac, we validated our model, trained on EN-TEx, on an external dataset (0.74 AUROC; Figure 7B and Data S32B). Our transformer model predicts whether an SNV would be AS in a tissue-independent fashion. We next tried to enhance it in a tissue-specific fashion, by including additional epigenetic information; this only marginally improved the model, underscoring the overwhelming importance of sequence context in assessing the impact of a variant (Figure 7C).
Figure 7. Aspects of Application 3: Deep-learning Model Predicting AS Activity from Nucleotide Sequence.
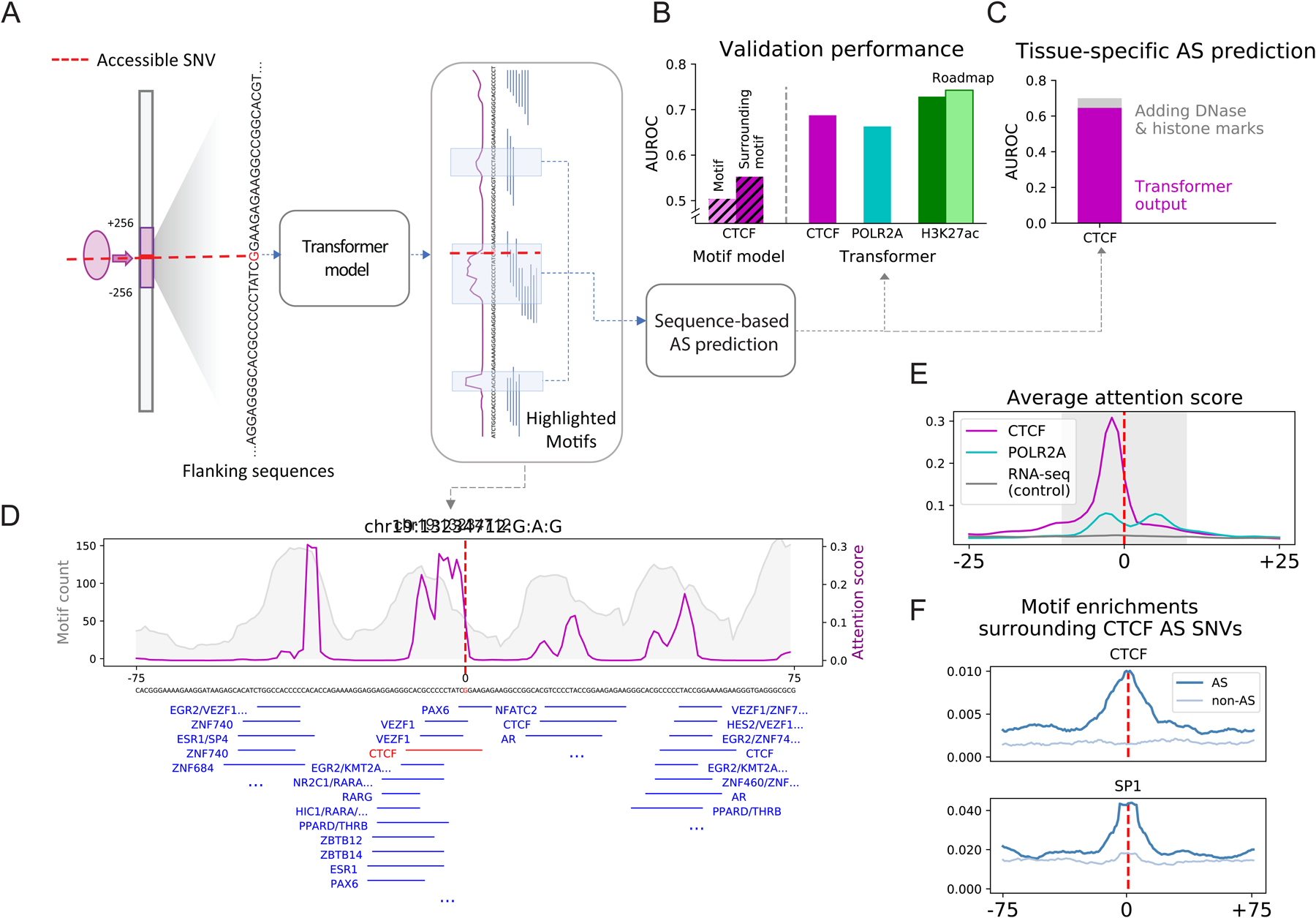
(For all sub-panels, details are in Figure S7, Data S32, and STAR Methods “Transformer Model” Section.)
(A) Schematic of the sequence-based predictive model. A transformer model was trained on the flanking regions (128 bp) of accessible SNVs to predict whether or not they are AS. The attention score (magenta lines) reflects the weights the model attaches to different nucleotide positions in the input sequences.
(B) Average performance of models predicting AS activity. As a reference, the CTCF model was compared to simple logistic regression models with the only information being (1) CTCF-motifs overlapping the SNV or (2) CTCF-motifs in a neighborhood around the SNV. For the H3K27ac model, the prediction was also validated against external data from Roadmap.
(C) Performance of a tissue-specific model for CTCF. Adding epigenomic features only marginally improved the performance over just sequence features.
(D) Attention patterns learned by the model. Those in the flanking regions of a selected CTCF AS SNV (magenta) show strong consistency with motif enrichment (gray). The central peak surrounding the SNV contains a CTCF motif, highlighted in red.
(E) Average attention pattern of sequence-based models for various assays.
(F) Motif enrichment surrounding the AS CTCF SNV agrees with the average attention pattern in E.
To better understand the sequence context that gives rise to AS behavior, we explored one characteristic of transformer models: they direct attention to specific sequence positions, often corresponding to known motifs. An example is shown in Fig 7D; one can see the attention paid to the CTCF motif at the center, and many other locations with known motif clusters are also flagged as important. The attention score from the model averaged over many positions clearly shows that it is more focused on the central SNV than other “control” models. This averaged attention score is ideal for comparing to motif occurrence: as expected, we observed a central enrichment for CTCF, but we also saw an enrichment for other TF motifs, such as SP1 (Figure 7EF and Data S32ACF; also, see Figure S7DE and Data S32ADE for analogous results for additional ChIP-seq datasets). In this way we can partially “re-discover” the key motifs highlighted in Figure 6 by a completely different route (Figure S7B).
DISCUSSION
The main contribution of EN-TEx is the creation of a readily accessible resource of personal epigenomes and the corresponding annotations, decorations, and models. We envision the resource enabling additional analyses outside of the scope of discussion here. Vignettes of methylation data related to aging or the cross-tissue epigenetics of genes associated with COVID-19 provide hints of what is possible (Data S34).
A key aspect of EN-TEx is that it can be easily connected with other human-genome annotation resources, potentially extending them. In particular, by training on the GTEx eQTL catalog, we were able to build a model that can transfer eQTLs from an easily obtained tissue to ones harder to get. With this approach, we leverage the fact that EN-TEx represents a uniform collection of epigenetics data from hard-to-obtain tissues. We also show that EN-TEx can decorate the ENCODE regulatory elements to give a unified view of tissue specificity and conservation and provide subsets of elements that are particularly enriched in GWAS variants. We imagine that in the future, EN-TEx could connect with and extend other genomic resources beyond ENCODE and GTEx, such as the recently initiated IGVF project (IGVF.org).
The second aspect of EN-TEx is that we can leverage the scale of the AS catalog to develop models illuminating the biological impact of variants. These models suggest that the local sequence context around a variant is the dominating factor in determining its impact, with certain TF motifs being particularly sensitive to mutations. That said, it is not just the TF motif right at the SNV position that is relevant, but the surrounding sequence (within a ~250 bp window). This suggests that determining whether a particular site is AS may have to do with other, potentially interacting, TFs binding nearby. For instance, a particular TF-binding site could be stabilized from mutational impact (and AS behavior) by being one of the many DNA-binding sites of a large hetero-oligomeric complex. Alternatively, redundant binding sites for a single factor may act as “backup” against the effects of one mutation71; the concept of “buffering” posits a mechanism for this26.
A final contribution of EN-TEx is demonstrating how the diploid genome is important for future human functional genomics. In particular, we show that diploid genomes provide more accurate quantification of differential expression and regulatory activity, which is essential for disease studies72. Furthermore, the matching of individuals and tissues in EN-TEx allows a precise ascertainment of the relative contribution of inter-tissue and inter-individual variation. We envision that in the near future, with the decreased cost of sequencing, generating a matched personal genome sequence as an accompaniment to each functional genomics experiment will become the norm. Thus, the EN-TEx personalized epigenomics approach for analyzing the impact of genome variation will necessarily become commonplace, potentially providing benefits for precision medicine72.
Limitations of the Study
A key limitation of EN-TEx is that only four individuals were profiled. Due to this, we are not statistically powered to compare the activity of elements between individuals. That said, the EN-TEx approach could be straightforwardly scaled up to larger cohorts. An aspect of this scaling would be the characterization of rare variants. Although the four individuals were considered healthy, their genomes contain many rare variants, including some potentially deleterious. These are not normally accessible to traditional QTL studies, which are mostly targeted to common variants. In contrast, our AS analysis and models can provide information on rare variants, and, in this regard, the EN-TEx resource is particularly informative to precision medicine. Moreover, if the approach piloted by EN-TEx were scaled up to more individuals in the future, it would provide a commensurate amount of information on additional new rare variants. This situation contrasts with common variants, where increasing the cohort size would provide diminishing amounts of additional information (STAR Methods “AS Catalog” Section).
A second limitation of EN-TEx is that due to the many functional assays and tissues used, it was not feasible to do technical replicates for each experiment; only a few tissues and assay combinations were replicated (see STAR Methods “Sample Selection” Section). The absence of replicates limits the utility of the differential expression and comparison of element activity between the personal genome and the reference. This limitation could be addressed in the future by more replicated experiments.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Mark Gerstein (mark@gersteinlab.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data contained in the EN-TEx resource are fully open-consented and accessible without registration as of the date of publication. Raw sequencing data as well as other standard functional genomics data have been deposited at a special page on the ENCODE data center, linked from the EN-TEx portal. Accession numbers are listed in the key resources table or in the supplementary data. Additional ancillary files are available directly on the EN-TEx portal: http://entex.encodeproject.org. The portal is organized into three organized sections: (i) data files, (ii) interactive visualization tools, and (iii) source code. For more details, see “Portal” Section of the STAR Methods.
All original code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
Experimental Model and Subject Details
N/A
Method details
Sections within the STAR Methods are referenced from the main text using the abbreviated section headings. We also indicate the relevant main text sections and figures that each of these STAR Methods sections are related to.
Sample Selection:
Details of the EN-TEx Samples and their Relationship with ENCODE and GTEx (related to “Uniform Multi-tissue Data Collection & Diploid Mapping” in the main text, Figures 1A and S1A)
The EN-TEx project (ENCODE assays applied to GTEx samples) has an intricate relationship with both the ENCODE and GTEx projects. Originally, the four individuals for the EN-TEx project were drawn from the main GTEx cohort. Two males and two females were chosen with a representative age distribution (ENCODE accession numbers: ENCDO845WKR, 37-year-old male; ENCDO451RUA, 54-year-old male; ENCDO793LXB, 53-year-old female; and ENCDO271OUW, 51-year-old female; the corresponding GTEx accession numbers are GTEX-1JKYN, GTEX-1K2DA, GTEX-1LGRB, and GTEX-1LVAN). The other key criterion was that these individuals’ data would be fully open access. This separates them from the consent criteria used for the GTEx cohort. This is non-trivial to obtain and requires a reconsenting process.
The EN-TEx tissues were chosen based on donor availability. The goal was to collect all, or as many as possible, of the exact same tissues collected for the GTEx protocol73. Note that the project specifically targeted organ transplant donors on ventilators, which excluded the collection of brain tissues, but increased the quality of the non-brain tissues due to much shorter collection and ischemic times. As described in74, not all tissues could be collected from all donors, since some were donated for tissue or organ transplant prior to the collection of tissues for research.
A full battery of ENCODE assays were applied to the tissue specimens from each of these four donors. The assays were mostly derived from ENCODE 3 and followed these standards to be consistent with the other ENCODE 3 datasets. However, a few follow-on datasets have been added to the collection, particularly related to histone marks and long-read RNA sequencing, that follow ENCODE 4 rather than ENCODE 3 standards. Based on sample availability, a few tissues were done with technical replicates but most were not. None of the EN-TEx datasets have been described in a publication, including the ENCODE 3 publication in 202075.
As the EN-TEx individuals were drawn from the main GTEx cohort, they were included in the GTEx publication. In that publication, the tissues were subjected to the standard GTEx assays, including short-read DNA sequencing of the blood and short-read RNA sequencing (polyA) also of the blood and a number of other tissues. These standard GTEx assays have data that are under different consent from the ENCODE data. For EN-TEx, in concert with the GTEx project, an Institutional Review Board-approved consent form was written and given to the next-of-kin of each donor. The consent form allows for unrestricted access to data collected as part of the EN-TEx project, including unrestricted use of the primary data and metadata collected from each donor. It was made clear that although no identification of the donor or family constituted part of these data, it is within the realm of possibility that individual identification could be made.
Specific details of the consent document are contained here: https://www.genome.gov/Pages/Research/ENCODE/GTEx_Consent_ENCODE_addendum_10-9-14.pdf. The GTEx consent requires users to undergo a dbGaP registration process to access the associated GTEx data. The GTEx data for these individuals is separately available on the GTEx website (https://gtexportal.org/home/).
For the EN-TEx individuals, there are a wealth of interesting technical comparisons possible between the standards of the GTEx and ENCODE projects, and also between two different versions of short-read DNA sequencing. However, the bulk of the assays and the focus of this paper are on the non-published data derived from the ENCODE assays, which includes the long-read DNA sequencing and all the chromatin and epigenetics assays, which are not part of the standard GTEx assays. As part of the standard GTEx cohort, these individuals fit perfectly into the expression quantitative trait loci (eQTL) calculations done by GTEx and allow us to match the eQTLs to the EN-TEx allele-specific (AS) catalog. Because we have ENCODE assays for all the individuals, we can also perfectly match the ENCODE regulatory elements, particularly the candidate cis-regulatory elements (cCREs). However, the decoration applied to the EN-TEx individuals goes beyond the cCRE annotation described in ENCODE 3, which only included active elements as opposed to repressed or bivalent ones.
The raw data for the ENCODE part of the EN-TEx are housed in the ENCODE data center; the GTEx part is on the GTEx portal. All of this is indicated on the EN-TEx portal. In addition, the EN-TEx portal has a large amount of supplementary analysis and software, all freely available, that are associated with this publication. The EN-TEx assays and analyses were funded by the National Human Genome Research Institute (NHGRI) using ENCODE funds. The relationship of each of the participants in the EN-TEx project to GTEx and ENCODE is described in Document S2 (see Data S36 and S37).
Finally, the Epigenome Roadmap Project data derive from a large set of epigenetic assays consistently applied to many tissues. This project was eventually rolled into the ENCODE project, but did not have consistent standards across projects. Thus, EN-TEx is much like the Epigenome Roadmap Project, but with all the assays being performed consistently with ENCODE. It also includes specific individuals with their personal genome sequence, allowing the impact of variants and inter-individual differences to be precisely ascertained.
Personal Genome:
Construction of the Personal Genome (related to “Uniform Multi-tissue Data Collection & Diploid Mapping” in the main text, Figures 1B and S1BC)
Sequencing of the Personal Genome
Data S2A–F summarizes the technologies used to sequence the whole genomes of the four individuals.
To prepare samples for PacBio sequencing, genomic DNA was isolated as previously described76,77 and evaluated for purity and quantity using UV-Vis (Nanodrop 1000, Thermo Fisher) and fluorometric (Qubit, Thermo Fisher) assays. DNA sizing was checked on the Femto Pulse (Agilent). Samples all exhibited a mode size above 50 kbp (most above 100 kbp) and were considered good candidates for PacBio sequencing. DNA was sheared using Megarupter (Diaganode) to a mode size of ~15 kbp. The sheared material was subjected to SMRTbell library preparation. Fractions were checked via fluorometric quantitation (Qubit) and pulse-field sizing (FEMTO Pulse). For sequencing, isolated gDNA was SMRTbell library prepared using the Express Kit V2 (PacBio) and subjected to size selection on a Blue Pippin instrument (Sage Science) with a 40 kbp size cutoff. Libraries were loaded on a Sequel II using v2.0 binding and v2.0 sequencing kits, no pre-extension, and 24-hour movie times.
For nanopore sequencing, samples were sheared to approximately 60 kbp and size selected by SRE XL (Circulomics). Fragmented DNA was prepared for sequencing with the SQK-LSK110 kit (Oxford Nanopore) following the manufacturer’s instructions. Prepared libraries were sequenced on a PromethION 24 with PROM0002 flow cell for 72 hours. One nuclease flush and reload was performed at 24 hours. Live high accuracy base calling was used.
We generated and analyzed Illumina whole-genome sequencing (WGS) data for each of the four human genome samples. WGS libraries were prepared using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina) in accordance with the manufacturer’s instructions. Briefly, 1 μg of DNA was sheared using a Covaris LE220 sonicator (adaptive focused acoustics). DNA fragments underwent bead-based size selection and were subsequently end-repaired, adenylated, and ligated to Illumina sequencing adapters. Final libraries were evaluated using fluorescent-based assays, including quantitative PCR with the Universal KAPA Library Quantification Kit and Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent 2100). Libraries were sequenced on an Illumina NovaSeq 6000 sequencer using 2 × 150 bp cycles to a minimum depth of 30X.
Variant Calling and Genome Assembly
Personal genomes were assembled from a combination of long-range Hi-C reads, 10x Genomics linked reads, and long reads (PacBio reads and Oxford Nanopore reads were base called with Guppy v4) using the reference-guided assembler CrossStitch (Data S2B)78. This pipeline has been used in several other studies of human and non-human genomes with as many as 100 different genomes at once78–80 for comprehensive single-nucleotide variant (SNV), insertion and deletion (indel), and structural variant (SV) calling. Notably, previous studies have shown that it is possible to accurately identify and phase SVs with variants identified from 10x linked reads and Hi-C data using the approach in CrossStitch to near chromosome-level resolution81.
Specifically, the following preprocessing steps were performed:
-
1
Align all reads (Hi-C, 10X, PacBio) to the human reference (GRCh38).
-
2
Call small variants from the linked reads with Long Ranger (ver. 2.1.2).
-
3
Phase small variants with HapCUT2 (ver. 1.1)35 using HiC and 10X data.
-
4
Call large SVs with Sniffles (ver. 1.0.11)82 using default parameters in samples sequenced with PacBio and using --min_homo_af 0.93 in samples sequenced with Oxford Nanopore. Additionally, in samples sequenced with PacBio, call SVs with pbsv (ver. 2.2.1) and merge the call sets with SURVIVOR (ver. 1.0.6)83, discarding SVs that were only identified by pbsv.
-
5
Filter SVs with low read support (fewer than 10 reads in samples 2 and 3, fewer than 3 reads in sample 1, and fewer than 4 reads in sample 4). Additionally, in samples 2 and 3, filter SVs labeled by Sniffles with the IMPRECISE INFO flag.
Then, the CrossStitch software (commit 53f64af) performed the following steps to obtain a personal genome:
-
6
Refine SVs with Iris (ver. 1.0)80.
-
7
Phase long reads using the phased small variants with which they overlap using an analogous approach to the NanoSV algorithm81.
-
8
Phase large SVs based on the phasing of the reads supporting them (Data S2C).
-
9
Integrate (“splice”) the phased variants into two copies of each human chromosome to produce personal diploid chromosome sequences using vcf2diploid (ver. 1.0)45.
-
10
Assign one sequence of each chromosome to pseudo-haplotype 1 and the other to pseudo-haplotype 2.
Note that each chromosome was phased independently from the other chromosomes, so that pseudo-haplotype 1 of one chromosome may correspond to pseudo-haplotype 2 of another chromosome. Unfortunately, the available data are insufficient to distinguish such cases or assemble full haplotypes genome wide. However, we were able to assign parental origin of the haplotypes for which the AS expression of known imprinted genes was determined (see more in section “Assigning Parental Origin by Imprinted Genes”).
In all four samples, the use of 10x and Hi-C data resulted in chromosome-arm-length phase blocks for all autosomes (Figure 1B and Data S2E). Specifically, the N50 of the phase blocks were 133.65 Mb, 133.68 Mb, 134.99 Mb, and 135.00 Mb for the four individuals, respectively. In addition, in both samples for which long reads were used, more than 90% of the large indels were able to be confidently phased with CrossStitch. For all four individuals, variant call format (VCF) files containing the SNVs and indels are accessible from the ENCODE portal84 (see Data S2D for accession numbers).
We adopted the reference-guided approach over alternative de novo assembly-based approaches because it gave more accurate and comprehensive results for the genome data available. For example, for individual 2 we also applied the leading PacBio-based de novo assembly algorithm FALCON-unzip85 to assemble the genome de novo, but this resulted in a contig N50 of only 7.0 Mbp. Aligning the FALCON-unzip contigs to GRCh38 using MUMmer86/Assemblytics87 identified <13,000 SVs compared with >18,000 for our reference-guided approach, with thousands of variants, especially heterozygous variants, unresolved. De novo assembly of the 10X Genomics linked reads or Illumina paired-end reads was even more limited, with contig N50 values of only 72 kbp and 13 kbp using the 10X Genomics Supernovo88 and Illumina Megahit89 assemblers, respectively. The 10x Genomics de novo assembly was particularly problematic for SV identification, as we observed an enrichment for ~200 bp insertions not observed with other sequencing technologies. In communication with 10X Genomics, we found these to be false positives derived from their assembly algorithm79. However, we and others found the SNV and indel calls to be highly accurate, especially within repetitive elements that could not be mapped using standard short-read paired-end sequencing.
Refining Novel Insertion Sequences with Iris
Iris is an established method for refining the breakpoints and sequences of insertion variants80. This tool has been used in several contexts78,79. Each of the calls, when taken directly from the variant caller, consists of an insertion sequence obtained from the alignment of a single representative read, and Iris improves upon this sequence by integrating all of the reads that support the variant’s presence. The tool gathers the sequences of all of the reads listed in the RNAMES INFO field output by Sniffles, extracts the original insertion sequence with the surrounding context from the reference genome, and uses the gathered reads to polish this sequence with racon (ver. 1.4.0)90. Then, this polished sequence is aligned back to the reference with minimap2 (ver. 2.17)91, and a refined insertion sequence is obtained. If no insertion is found from this alignment, which has a similar length to that of the original variant call, Iris falls back on the original sequence to ensure it does not mask variants in more difficult-to-map regions.
We benchmarked the performance of Iris using data from HG002, a sample sequenced as part of the Genome in a Bottle release. In this individual, we called SVs separately using Oxford Nanopore (ONT) data and PacBio Circular Consensus Sequencing (CCS) data, both sequenced to ~50x coverage using the ngmlr aligner82 and the Sniffles variant caller. Because of the high accuracy of the CCS reads, we used the insertion sequences obtained from these calls as a proxy for the ground truth to evaluate the accuracy of the ONT calls. We compared the CCS and ONT call sets before and after refining the ONT calls with Iris. In each comparison, we evaluated all of the variant calls in the CCS dataset, which had an ONT variant call within 10 kbp in both the refined and unrefined call sets. Among these 14,001 variants, we measured the average sequence similarity between the CCS call and the ONT call, with the similarity of two strings S and T measured as [1 - edit_distance(S, T)] / max[length(S), length(T)]. Using the unrefined calls, the average similarity was 0.854, while the refined calls gave an average similarity of 0.94, demonstrating the ability of Iris to obtain more accurate insertion breakpoints and sequences. Data S2F shows the distribution of sequence similarities before and after refinement.
Assigning Parental Origin by Imprinted Genes
The list of known human imprinted genes was downloaded from the Imprinted Gene Database (geneimprint.com). In total, 216 genes with known parental origin of the expressed allele were used in this analysis. For known imprinted genes that showed AS expression (ASE) in tissues from each individual, the haplotype-specific read counts were combined from these tissues and the potential parental origin of the haplotype blocks was determined based on the direction of the imbalance (haplotype 1 or haplotype 2) and the known expressed allele of the imprinted gene (maternal or paternal allele) (Data S2H). Results were included in the following ancillary files:
File: imprinted_genes_in_ENTEx_ASE.tsv: All known imprinted genes for which allele-specific expression was detected, genome-wide, in the EN-TEx samples.
File: phased_block.tar.gz: Parental origin of each phased block in the four individuals.
The parental origin results of individual 3 are shown in Figure 1B and are available in the file phased_block_ind3.txt within phased_block.tar.gz, where each line is a phased block. The first three columns are genomic coordinates of the phased block. The fourth and fifth columns are the parental origins of haplotype 1 and haplotype 2, respectively. ‘NoInfo’ indicates that there are no imprinted genes in that phased block. ‘Contradict’ indicates that there is at least one AS gene-imprinted gene pair that has a different imbalance direction compared to the other AS gene-imprinted gene pairs, and thus contradictory conclusions are reached for the same phased block. A similar approach can be used for the other EN-TEx individuals (Data S2G). Overall, 97.3% of base pairs in the EN-TEx individuals were assigned to a phased block (on average across the four donors). This corresponds to 98.5% of all heterozygous variants. We were able to determine the parental origin of 45.3%, 43.2%, 36.1%, and 45.3% of the bases in phased blocks for individuals 1–4, respectively.
Data Stack:
Functional Genomics Data in the EN-TEx Resource (related to “Uniform Multi-tissue Data Collection & Diploid Mapping” in the main text, Figures 1A and S1A)
In total, EN-TEx includes more than 25 different biochemical assays performed on multiple (30+) tissues from four individuals (Figure 1A and Figure S1A). The tissues and legend for Figure 1A are detailed in Data S2I. In Figure 1A, we indicate the “core assays” in bold, corresponding to the assays that were performed in EN-TEx across almost all individuals and tissues; these assays include the histone modifications H3K27me3, H3K9me3, H3K36me3, H3K4me1, H3K4me3, and H3K27ac, POL2 and CTCF ChIP-seq, methylation arrays, ATAC-seq, DNase-seq, RAMPAGE, and total RNA-seq. Experiments from GTEx on the four EN-TEx individuals are indicated with asterisks (polyA RNA-seq and whole-blood datasets). Note that EN-TEx encompasses 1,635 total experiments, which includes control experiments and replicates (both of which are not explicitly shown in the data matrix in Figure 1). If we remove replicates and controls, the number of experiments is 1,275.
RNA Sequencing
Multiple RNA-seq experiments were performed in ENCODE Phase III on the 30+ tissue samples sourced from GTEx and included in EN-TEx, including: 1) long RNA-seq, i.e., RNA with a length greater than 200 nt, and total RNA-seq, 2) small RNA-seq, i.e., RNA with a length less than 200 nt, and 3) microRNA-seq, i.e., RNA with a length less than 30 nt. More information about each RNA-seq protocol and data processing pipeline can be found at the ENCODE portal: 1) https://www.encodeproject.org/data-standards/rna-seq/long-rnas/, 2) https://www.encodeproject.org/data-standards/rna-seq/small-rnas/, and 3) https://www.encodeproject.org/microrna/microrna-seq/. RNA-seq data quality was calculated using the number of aligned reads and replicate concordance (as described in the ENCODE pipelines linked above).
RAMPAGE
RNA annotation and mapping of promoters for analysis of gene expression (RAMPAGE) is a biochemical assay that captures 5′-complete cDNA to identify and quantify transcriptional start sites (TSSs) and characterize transcripts. The assay is described in detail at the ENCODE portal: https://www.encodeproject.org/data-standards/rampage/. The ENCODE RAMPAGE data processing pipeline was developed for RAMPAGE libraries containing cDNA sequences longer than 200 nt. The pipeline takes cDNA sequences as input (in FASTQ format) and outputs alignments normalized for both positive and negative strands of the genome. Reproducible peaks between replicates were identified using the irreproducible discovery rate (IDR). The quality of the RAMPAGE data was determined using the read depth and replicate concordance with respect to peaks in the data.
eCLIP
Enhanced crosslinking and immunoprecipitation (eCLIP) is a biochemical assay that identifies RNA-binding protein (RBP) occupancy sites across the transcriptome. The eCLIP experimental protocol is available at the ENCODE portal: https://www.encodeproject.org/documents/842f7424-5396-424a-a1a3-3f18707c3222/@@download/attachment/eCLIP_SOP_v1.P_110915.pdf. Additional assay details are available at https://www.encodeproject.org/eclip/. All eCLIP antibodies were required to undergo primary and secondary characterizations. RBP antibody standards are available at the ENCODE portal: https://www.encodeproject.org/documents/fb70e2e7-8a2d-425b-b2a0-9c39fa296816/@@download/attachment/ENCODE_Approved_Nov_2016_RBP_Antibody_Characterization_Guidelines.pdf. The quality of the eCLIP data was determined using the number of unique RNA fragments, IDR, and fraction of reads in peaks (FRiP).
Histone ChIP-seq
Histone ChIP-seq is a biochemical assay that observes interactions between histone proteins and DNA. This assay selects for a specific histone protein variant or post-translational modification using immunoprecipitation followed by DNA sequencing. The histone ChIP-seq experimental protocol is available at the ENCODE portal: https://www.encodeproject.org/documents/be2a0f12-af38-430c8f2d-57953baab5f5/@@download/attachment/Epigenomics_Alternative_Mag_Bead_ChIP_Protocol_v1.1_exp.pdf. Additional assay details are available at https://www.encodeproject.org/chip-seq/histone/. All commercial histone antibodies were validated by at least two independent experiments. Histone mark antibody standards are available at the ENCODE portal: https://www.encodeproject.org/documents/4bb40778-387a-47c4-ab24-cebe64ead5ae/@@download/attachment/ENCODE_Approved_Oct_2016_Histone_and_Chromatin_associated_Proteins_Antibody_Characterization_Guidelines.pdf. The quality of the histone ChIP-seq data was determined using the read depth, number of uniquely mapping reads over the total number of reads (i.e., non-redundant fraction, NRF), and two PCR bottlenecking coefficients (PBC1 and PBC2).
Transcription Factor (TF) ChIP-seq
ChIP-seq captures DNA and DNA-binding protein (e.g., CTCF, EP300, and Pol II) interactions through immunoprecipitation, pulldown, and DNA sequencing. All ChIP-seq protocols involved in the generation of data included in EN-TEx are available at the ENCODE portal: 1) https://www.encodeproject.org/documents/20ebf60b-4009-4a57-a540-8fd93407eccc/@@download/attachment/Epigenomics_CR_ChIP_Protocol_v1.0.pdf, 2) https://www.encodeproject.org/documents/6ecd8240-a351-479b-9de6-f09ca3702ac3/@@download/attachment/ChIP-seq_Protocol_v011014.pdf, 3) https://www.encodeproject.org/documents/a59e54bc-ec64-4401-8cf6-b60161e1eae9/@@download/attachment/EN-TEx%20ChIP-seq%20Protocol%20-%20Myers%20Lab.pdf, and 4) https://www.encodeproject.org/documents/f2aa60f2-90a6-4e4b-863a-c6831be371a2/@@download/attachment/ChIP-Seq%20Biorupter%20Pico%20TruSeq%20protocol%20for%20Syapse-c5bdc444fe0511e69d6a06346f39f379.pdf. Additional ChIP-seq protocol details are available at https://www.encodeproject.org/chip-seq/transcription_factor/. The quality of the ChIP-seq data was determined using the read depth, NRF, two PCR bottlenecking coefficients (PBC1 and PBC2), replicate concordance (i.e., IDR), and FRiP.
ATAC-seq
ATAC-seq identifies accessible regions of DNA by inserting primers into open chromatin regions via transposase, followed by DNA sequencing. The ATAC-seq experimental protocol is available at the ENCODE portal: https://www.encodeproject.org/documents/404ab3a6-4766-45ca-af80-878a344f07b6/@@download/attachment/ATAC-Seq%20protocol.pdf. Additional details about the ATAC-seq protocol can be found at https://www.encodeproject.org/atac-seq/. The quality of the ATAC-seq data was determined using the number of non-duplicate, non-mitochondrial aligned reads, IDR, NRF, two PCR bottlenecking coefficients (PBC1 and PBC2), number of resulting peaks in the data, DNA fragment length distribution, FRiP, and TSS enrichment.
DNase-seq
DNase-seq is a biochemical method that identifies open regions of chromatin. These regions are identified by performing enzyme digests using endonuclease DNase I, which inserts itself into open regions, followed by DNA sequencing. The DNase-seq experimental protocols are available at the ENCODE portal: https://www.encodeproject.org/documents/c6ceebb6-9a7a-4277-b7be-4a3c1ce1cfc6/@@download/attachment/08112010_nuclei_isolation_human__tissue_V6_3.pdf. Additional protocol information can be found at https://www.encodeproject.org/data-standards/dnase-seq/. The quality of the DNase-seq data was determined using the number of uniquely mapped reads, fraction of mitochondrial reads, and signal portion of tags score.
WGBS
Whole-genome bisulfite sequencing (WGBS) was used to identify DNA methylation. WGBS converts unmethylated cytosine (C) into uracil (U), leaving methylated C unchanged. DNA sequencing followed by read alignment to a genome results in CpG island, CHG, and CHH methylation levels being observed. The WGBS experimental protocol is available at the ENCODE portal: https://www.encodeproject.org/documents/9d9cbba0-5ebe-482b-9fa3-d93a968a7045/@@download/attachment/WGBS_V4_protocol.pdf. Additional WGBS assay details are available at https://www.encodeproject.org/data-standards/wgbs/. The quality of the WGBS data was determined from the genomic read coverage, C-to-T conversion rate, and correlation of CpG methylation levels between replicates.
DNAme Array
DNA methylation profiling by array assay (DNAme) measures CpG island methylation. Similar to WGBS, DNA is treated with bisulfite converting unmethylated C to U. After library amplification and purification, DNA fragments are hybridized to a microarray (Illumina Infinium Methylation EPIC BeadChip) that probes for both methylated and unmethylated states. DNA methylation is then quantified by comparing the signal between the two DNA microarray probes. Illumina Genomestudio (v2011.1) was used to calculate the fraction of methylated reads at each CpG site from the raw microarray output.
Hi-C
High-quality Hi-C data were generated from the four EN-TEx donors using samples collected from the gastrocnemius medialis and transverse colon tissues. The in-situ Hi-C protocol used to produce Hi-C libraries was described previously by Rao et al. (2014)92. A detailed protocol document is provided with each dataset at the ENCODE website: https://www.encodeproject.org/documents/e1ef20c9-7539-40bc-bdbf-a4deab7f72c7/. Approximately 20 mg of tissue was used for each Hi-C experiment, and the MboI restriction enzyme was used for restriction digests. All sequencing was performed on an Illumina 4,000 platform. The data was processed twice, separately utilizing a reference genome or personal genomes constructed for each individual’s tissue.
Hi-C interaction matrices were generated using the Juicer pipeline93, an open-source tool for analyzing large Hi-C libraries. We utilized BWA-MEM94 to align individual reads to the hg38 reference genome, which was obtained from the ENCODE portal. For each paired-end read, the two individual sequences were first separately aligned to the reference genome before being paired based on their read names. Chimeric reads and PCR duplicates were removed prior to the creation of an interaction matrix for each tissue of each individual (Data S3A). Data S3B provides information on the number of reads and number of contacts per sample utilized to create the matrices. Significant intrachromosomal Hi-C interactions were identified with FitHiC2 (ver. 2.0.7)95,96. Preprocessing of EN-TEx Hi-C interaction matrices followed the author’s instructions in the FitHiC2 GitHub repository’s README. Matrices were binned at a resolution of 50 Kb and bin biases were generated using the author’s provided software (HiCKRy.py — with percentOfSparseToRemove set to 0.1). See Data S3E for the number of all vs. significant interactions for each sample.
Determination of the A and B compartments was done using the Juicer pipeline93 at a 1 MB resolution. In detail, the observed/expected interaction matrices were normalized using the Knight-Ruiz matrix-balancing algorithm97. A correlation matrix from these interaction matrices was calculated, with the first eigenvector of the matrix corresponding to A/B compartments. The positive values of the vector indicate genomic regions belonging to the A compartment, while negative values correspond to the B compartment (Data S3C–D).
Topologically associating domains (TADs) were identified using TopDom (ver. 0.9.0)98 from the two tissues of all four donors with a window_size parameter of 3. Before running TopDom, EN-TEx Hi-C libraries were binned at a resolution of 100 Kb and normalized using the Knight-Ruiz matrix-balancing algorithm97 implemented by Juicer93. TopDom’s window_size parameter was optimized for the known enrichment of CTCF motif directionality at TAD boundaries92 and visual consistency/fit with Hi-C interaction matrices. CTCF directionality was identified using paired EN-TEx ChIP-seq peak (narrowPeak format) files and the ‘CTCF_known1’ motif as described by Cameron et al. (2020)99. TAD boundary similarity was calculated by overlapping TAD annotations between individuals/tissues with a buffer of three bins when considering two boundaries to be the same.
The FitHiC2 output and TAD annotations can be found within the ancillary files.
File: fithic2_out.tar.gz: Hi-C genomic data processed by Fit-Hi-C software.
File: TopDomTADcalls.tar.gz: Topologically associating domains of the genome identified by TopDom software.
Proteomics
For 10 mg tissue, 200 μl lysis buffer [50 mM Tris-HCl pH8.5, 50 mM NaCl, 8 M urea, 4% SDS, and Halt protease inhibitor (Thermo)] was added. After the tissue was homogenized by a pestle/mortar, a Dounce homogenizer, or similar device, the sample was heated at 95°C for 10 min, followed by probe sonication until the viscosity was reduced. The sample was then centrifuged at 13,000 rpm for 15 min, and supernatant was collected. RNA was first extracted from samples (see RNA Sequencing).
Protein concentration was measured by the Pierce 660 nm Protein Assay (Thermo). For each sample, 100 μg proteins were taken, and volumes were equalized by 100 mM TEAB to 100 μl, reduced by 20 mM TCEP (Sigma), and then alkylated by 40 mM iodoacetamide (Sigma). Proteins were purified by 20% trichloroacetic acid (TCA) precipitation. A total of 100 mM TEAB was added to the sample, followed by digestion with trypsin (MS grade, Thermo) at 37°C for 18 hours. The peptides were labeled by TMT10plex as per the manufacturer’s instruction, and the labeled samples were pooled and SpeedVac dried. Samples of 300 μg peptides were fractionated on a U3000 HPLC system (Thermo Fisher) using an XBridge BEH C18 column (2.1 mm id × 15 cm, 130 Å, 3.5 μm, Waters) at pH 10, at 200 μl/min on a 30 min linear gradient from 5–35% acetonitrile/NH4OH. The fractions were collected every 30 sec into a 96-well plate, which were concatenated to 35 fractions and dried.
The peptides were resuspended in 0.5% formic acid (FA) and 50% was injected for liquid chromatography with tandem mass spectrometry (MS) analysis on an Orbitrap Fusion Tribrid mass spectrometer coupled with a U3000 RSLCnano UHPLC system (Thermo Fisher). The peptides were loaded onto a PepMap C18 trap (100 μm i.d. × 20 mm, 100 Å, 5 μm) for 10 min at 10 μl/min with 0.1% FA/H2O, and then separated on a PepMap C18 column (75 μm i.d. × 500 mm, 100 Å, 2 μm) at 300 nl/min and a linear gradient of 4–33.6% ACN/0.1% FA in 90 min/cycle at 120 min, or 4–32% ACN/0.1% FA in 150 min or 180 min with a cycle time of 180 min or 210 min for each fraction. For data acquisition, we used the SPS10-MS3 method with the top speed set at 3 s per cycle time. The full MS scans (m/z 380–1,500) were acquired at a 120,000 resolution at m/z 200, and the automatic gain control (AGC) was set at 400,000 with a 50 ms maximum injection time. The most abundant multiply charged ions (z = 2–6, above 5,000 counts) were subjected to MS/MS fragmentation by collision-induced dissociation (35% CE) and detected in an ion trap for peptide identification. The isolation window by quadrupole was set at m/z 1.0, and the AGC at 10,000 with a 35 ms maximum injection time. The dynamic exclusion window was set at ±7 ppm with a duration of 60 s. Following each MS2, the 10-notch MS3 was performed on the top 10 most abundant fragments isolated by synchronous precursor selection. The precursors were fragmented by higher-energy collisional dissociation at 60% CE, and then detected in the Orbitrap at m/z 110–400 at a 50,000 resolution for peptide quantification. The AGC was set at 50,000 with a maximum injection time of 86 ms.
GENCODE v27100 annotation was lifted over from GRCh38 to each EN-TEx donor’s personal genome, to generate eight sets of general feature format annotations. GFFRead utility101 was used to extract the nucleic acid sequence for all protein-coding transcripts. An in-house Python script was then applied to translate each protein-coding transcript into its amino acid sequence. All protein sequences from the eight genomes were combined with the GENCODE v27 reference, redundant sequences were removed, and each unique protein sequence was given a unique accession ID that included the genomes that contain the protein. The final database contained 128,063 unique protein sequences, 82,136 (64%) from the GENCODE reference and 45,927 (36%) unique to the EN-TEx donors. A total of 6,344 protein sequences from GENCODE (8% of the reference proteome) were not matched to any of the alleles in the four individuals. Decoy protein sequences were generated using the DecoyPYrat tool102.
The proteomics results are summarized in this ancillary file.
File: Supp_data_proteomics.xlsx: Proteomics result summary including peptide annotation.
Spectra were processed using ProteomeDiscoverer (ver. 2.4) (Thermo Fisher Scientific) and searched against the personal proteome database using both Mascot (ver. 2.4) (Matrix Science) and SequestHT with target-decoy scoring evaluated using Percolator103. The precursor tolerance was set at 30 ppm and the fragment tolerance was set at 0.5 Da; spectra were matched with fully tryptic peptides with a maximum of two missed cleavages. Fixed modifications included carbamidomethyl [C] and TMT6plex [N-Term]. Variable modifications included TMT6plex [K], oxidation [M], carbamyl [K], methyl [DE], deamidation [NQ], and acetyl [N-term]. The carbamyl and methyl modifications were included due to their high incidence after samples were exposed to high concentrations of urea during the RNA extraction process. Peptide results were initially filtered to a 1% false discovery rate (FDR; 0.01 q-value). The reporter ion quantifier node included a TMT-11-plex quantification method with an integration window tolerance of 15 ppm and integration method based on the most confident centroid peak at the MS3 level. Protein quantification was performed using unique peptides only, with protein groups considered for peptide uniqueness. Peptides were quantified and normalized using tandem mass tags (TMTs) for isobaric labeling. Peptide results from ProteomeDiscoverer were remapped to the protein database and marked as reference, genome, or AS. Gene-level quantification of proteins was conducted by summing normalized unambiguous peptide TMT intensities.
At a 1% FDR, we report 256,512 peptide-to-spectrum matches and 117,934 distinct peptide sequences (0.01 q-value at the peptide level), of which 45,276 were quantified using TMT isobaric labels. Personal peptides were further filtered to unambiguously match one gene and have a posterior error probability below 0.699. These peptides did not map to the reference genome, only matching personal protein sequences. The 4,489 peptides identified were not present in all eight genomes across the four donors, 830 of these peptides were missing in one or more of the donors completely, and 4,334 were only present on a single allele in at least one of the donors. This corresponds to 13% coverage of the possible observable personal peptides across all protein-coding genes in the personal genomes, and a 1% increase in the number of significant distinct peptide sequences quantified (Data S4).
Gene quantification was conducted using only unambiguous peptides summing the peptide isobaric tag intensities. A total of 9,242 genes were quantified, 540 genes had non-reference peptides, 1,333 genes had peptides not present in all eight genomes (personal peptides), 518 genes had peptides absent in at least one donor, and 1,260 genes had peptides specific to a single allele in at least one donor.
For comparison between proteomic and RNA-seq abundances, a paired set of samples and confidently identified genes matching between the proteomic and RNA-seq datasets were extracted. For each dataset, the values were normalized and then scaled to the maximum value across the samples/tissues. A Pearson correlation was then used to test the similarity between the two sets across the samples.
All spectra were also processed via the ICR GENCODE OpenMS novel peptide discovery proteomics pipeline104 against a database containing GENCODE v27 reference proteins and a set of potential novel protein-coding sequences, including many unannotated PhyloCSF conserved regions105. Novel peptide results were filtered according to high-stringency criteria106. This resulted in 291 novel peptides, which were further filtered to remove peptides that could be explained by semi-tryptic cleavage or single amino acid variants. The 27 remaining peptides were assessed, validating eight novel protein models, which have all now been annotated in the GENCODE reference set (Data S4D).
All spectra, results, and supporting files, including the personal proteome database, have been deposited in the PRIDE107 proteomic repository (https://www.ebi.ac.uk/pride/) under project accession number PXD022787.
Reference Comparison:
Comparing Between Personal and Reference Genomes (related to “Uniform Multi-tissue Data Collection & Diploid Mapping” in the main text and Figures S2)
Mapping Functional Genomics Data to the Personal Genomes
We used DNA from transverse colon tissues to construct both haplotype sequences for each individual. Mapping sequences to the derived haplotypes, rather than to the reference genome, resulted in an overall improvement in mapping accuracy across the different assays (RNA-seq, DNA-seq, Hi-C, and ChIP-seq). By applying conventional mapping criteria, we observed an increase in the number of mapped reads of about 0.5–1%. When we applied more stringent filtering criteria to select for high-quality, uniquely mapping sequences, we observed a much larger improvement, reaching an increase of 2–4% across assays over the four individuals (Figure S1C). Data S5A–C summarizes the numbers of reads and percentages for precision mapping across the four individuals for DNA-seq, CHIP-seq, Hi-C, and RNA-seq. Mapping categories include mapping to haplotype 1 (Hap.1), haplotype 2 (Hap.2), union of Hap.1 and Hap.2 (Hap1&Hap2), reference (ref), intersection between Hap.1 and Hap.2 but not ref (Hap1&&Hap2¬Ref), and improvement as a measure of Gain=[(Hap.1 ⋃ Hap.2)-Ref]/Ref.
For all assays, we excluded counting reads that mapped to the X, Y, and M chromosomes for all individuals. In general, to ensure high-quality mapping we selected reads with at most two mismatches and unique mapping. We used raw reads from transverse colon, publicly available at the ENCODE portal, with the exception of DNA-seq. For DNA-seq mapping, we used reads from blood samples that we obtained from GTEx to avoid any bias deriving from the construction of haplotypes using DNA sequences. For DNA-seq and RNA-seq mapping, we used paired-end reads. For RNA-seq, to account for gene splicing, we used *.gtf files with transcript genomic coordinations and STAR Aligner (ver. 2.7). For DNA-seq, Hi-C, and ChIP-seq we used BWA (ver. 0.7.17) and selected reads with at most two mismatches and quality Q>30. For RNA-seq, we used sequences with quality mapping Q=255.
Differential Gene Expression Analysis Between Reference and Personal Genomes
In order to evaluate the impact of personal genomes on gene expression quantification, we performed a differential gene expression (DGE) analysis between gene expression read counts obtained after mapping to reference and personal genomes. Conventional software to perform DGE analysis (such as DESeq2108 or edgeR109) rely on the existence of replicates. Due to the study design, however, the vast majority of RNA-seq experiments in EN-TEx are unreplicated. For this reason, we performed a DGE analysis for each of the four donors, running DESeq2 with default parameters and using RNA-seq experiments for different tissues of the same donor as replicates. For each donor, we identified sets of upregulated and downregulated genes, defined as genes that have significantly higher and lower expression, respectively, when mapped to the reference genome compared to the diploid genome (adjusted p-value [Benjamini–Hochberg] < 0.1 and |log2 FC| > 1; Figure S2A, Data S5D–E).
By taking the union of differentially expressed genes across the four donors, we identified a total of 112 upregulated and 100 downregulated genes. Overall, we observed an enrichment of immune-related genes among our sets of upregulated and downregulated genes (gene ontology term: “MHC class II protein complex assembly,” log10(p-value) = −9.74; gene ontology analysis performed with Metascape110), as well as an enrichment of pseudogenes among downregulated genes (Data S5F). In Data S5G–I, we provide a few examples of either immune-related (HLA-DQA1) or disease-relevant (SMN2, SIK1) genes that show increased expression when mapped to the personal genomes.
We acknowledge that performing the DGE analysis using tissues as replicates is not optimal. However, we argue that it is a conservative approach. To demonstrate this, we performed two additional analyses. First, we identified six RNA-seq experiments with two available technical replicates (from independent sequencing libraries), as well as one tissue (liver for individual 3) with two independent RNA-seq experiments available (biological replicates). For each of these seven experiments, we performed a DGE analysis between the reference and personal genome mappings with the same parameters as the one described above (Data S5J, upper side). We identified 53 upregulated and 59 downregulated genes, including an additional set of 18 and 25 upregulated and downregulated genes, respectively. These genes were not previously reported by the per-donor DGE analysis.
Given that we obtained these results using only two replicates per experiment, we hypothesize that we could potentially identify a larger number of genes differentially expressed between the reference and personal genomes if each tissue had multiple replicates available. Thus, to estimate the reduced discovery power due to the lack of replicated experiments, we generated a personal genome of the cell line GM12878 and performed a DGE analysis using 5 polyA+ RNA-seq experiments available from the ENCODE portal (each experiment with two biological replicates). The list of experiments is provided in Data S5J (lower side). We applied the same pipeline as for the EN-TEx RNA-seq experiments to obtain read counts mapped to both the reference (hg19) and personal assemblies, using GENCODE v19 annotation. We performed DGE analysis running DESeq2 with default parameters as described for the previous two analyses, after specifying batch information per replicate based on the ENCODE experiment identifier (Data S5J, lower side). This analysis identified 46 upregulated and 43 downregulated genes, including an additional set of 31 and 34 upregulated and downregulated genes, respectively.
Overall, these results demonstrate that our reduced discovery power of differentially expressed genes between the reference and personal genomes could be partially due to the lack of multiple replicated experiments available per each tissue. The approach described above, which uses tissues of the same donor as biological replicates, can best measure the impact of personal genomes on genes that are expressed across a wide range of tissues. However, this approach might not be suited for tissue-specific genes, whose changes in expression between the reference and personal genomes in a particular tissue might be masked or underestimated when averaged across all tissues.
The differentially expressed gene lists are available in the following ancillary files. Specifically, novel differentially expressed genes identified in the analysis of experiments with available replicates or in the analysis of GM12878 cells are marked with an asterisk.
File: table.DE.genes.tsv. Union of genes differentially expressed between reference and personal genomes across the four EN-TEx individuals.
File: table.DE.genes.techReps.liver.tsv. Union of genes differentially expressed between reference and personal genomes across seven EN-TEx RNA-seq experiments with available replicates.
File: table.DE.genes.GM12878.tsv. Genes differentially expressed between reference and personal genome in GM12878.
Differential Regulatory Element Activity Between Reference and Personal Genomes
To better characterize the cCRE activity between reference- and diploid-based alignment, we set up a pipeline to accurately estimate the H3K27ac signals of active cCREs from the four EN-TEx individuals by considering their gender information when we performed the ChIP-seq read alignment. Briefly, for the female individuals, we mapped the reads to the autosomal and X chromosomes, while for the male individuals, we mapped the reads to the autosomal and X/Y chromosomes. When considering diploid-based mapping, we performed read alignment to their diploid genomes separately, and calculated the normalized read coverage for each active cCRE, followed by computing the mean value, which was used to represent the activity signals of cCREs under the scenario of diploid-based mapping. Similar to our approach for the differential gene expression analysis, we applied DESeq2108 to identify the active cCREs that show significantly differential activity under reference- versus diploid-based alignment across all the samples (adjusted p-value [Benjamini–Hochberg] < 0.1 and |log2 FC| > 1; Figure S2B, Data S5K–N). See the following ancillary file for the result.
File: differentially_marked_H3K27ac_cCREs.txt: Union of candidate cis-regulatory elements with differential H3K27ac signal between reference and personal genomes across the four EN-TEx individuals.
Variation Analysis:
Analysis of the Variation in Element Activity (related to “Uniform Multi-tissue Data Collection & Diploid Mapping” in the main text and Figures S2CD)
Visualizing the Variation of cCRE Activity with JIVE
To visualize the relationship among the functional genomic data across the tissues, we used a dimension-reduction approach, namely Joint and Individual Variance Explained (JIVE)111. For each functional genomic experiment of histone modifications, we calculated its signals at the cCREs using the UCSC Genome Browser bigWig tools112. For proteomics and RNA-seq experiments, we simply used the normalized protein abundance and RNA abundance of each gene. For each type of assay, we generated a data matrix in which the columns are the tissues from the four individuals and the rows are cCREs or genes, and each element is the signal of the functional genomic activity measured by the assay. For each assay type, we quantile-normalized the signals. For the joint analysis of the different experimental assays, we combined these matrices by column to form a meta-matrix. In each separate data matrix, some columns in each data matrix are not shared by all the assays, and thus these columns are excluded from the metamatrix.
To reduce computation burdens, we removed the rows that have low standard deviation. From this informative meta-matrix, we applied the JIVE algorithm to project the columns into a two-dimensional (2D) space (Figure S2C, Data S6M). As expected, this projection used all the information of the matrix. In addition, from the matrix of each assay, the JIVE algorithm excluded the information that can be explained by the other matrices, and then projected the matrix containing the information unique to the assay into a 2D space (Figure S2C, Data S6M). For example, in the 2D space of RNA-seq, the same tissues from different individuals are well clustered, and the different tissues are well separated. This tendency is weaker for the other assays. Taken together, this observation indicates that RNA-seq likely captures the most unique signatures of different tissues.
Using a Regression-based Approach to Quantify Activity Variation
With a linear regression approach, we used the explained variation of the regression to measure the similarity between two experiments. A larger explained variation of the regression indicated a higher similarity between the two experiments. To elaborate on the variation, we use a concrete example: the H3K27ac signals of cCREs from the spleens of two individuals. In this example, each of these individuals had two technical replicates of the H3K27ac signals measured by ChIP-seq. In each replicate, the signal at a cCRE was the fold change of reads between the immunoprecipitation experiment and the control experiment. For each cCRE, we first calculated the percentage difference of the signals between the two replicates. We focused on the cCREs with differences smaller than a certain cutoff so that the signals of these selected cCREs in one replicate can be largely explained by their counterparts in the other replicate using linear regression (i.e., R2 > 0.95). To compare the two individuals, we used the common set of the selected cCREs with low technical noise. For each of the two individuals, we averaged the signals of the two replicates for the common cCREs. Therefore, we generated two sets of cCREs with H3K27ac signals having little noise, respectively, for the two individuals. Again, using a simple linear regression, we calculated the variance in one of the sets explained by the other. A high value indicates that the two sets of H3K27ac signals are very similar in terms of a linear relationship. As an example, the explained variation between replicates and the explained variation between experiments for different types of histone modifications in spleen is demonstrated in Data S5A–F.
The aforementioned calculation was used for all the available histone modifications and samples (examples shown in Data S6G–H) as well as normalized protein and RNA abundances (Data S6I–L). For each modification, we estimated the variance explained between individuals (i.e., the same tissues of different individuals) and between tissues (i.e., different tissues of the same individual). In addition, we estimated the variance explained between two different histone modifications (i.e., within the same tissue of an individual). For MS, to make the protein abundances of different genes comparable across different tissues, we normalized the protein abundances of each gene across tissues so that the highest and lowest protein abundances were one and zero, respectively. The MS approach we used pooled and labeled multiple samples together to determine protein abundances in a batch, resulting in little technical noise across the samples. To be comparable, we also normalized the RNA-seq data of the samples in the same way.
In general, histone modifications showed high similarity between the same tissue of two individuals; as expected, this number was smaller when comparing different tissues of the same individuals (Data S6G–H). The similarity between different types of functional genomic activities from the same tissue was extremely low (Data S6G–H). For example, H3K27ac between individuals was very similar in spleen and in transverse colon. However, the H2K27ac similarity between the two tissues was substantially reduced (Data S6G–H). In line with this disparity across tissues, the similarity between normalized gene expression and protein abundance also varied substantially across tissues. The lower similarity in prostate is consistent with previous observations113. Full details of the comparison are reported in the following files:
File: Similarity_of_functional_genomic_activities_of_cCREs.xlsx: Similarities between all the available histone modifications.
File: normalized_proteomics_RNA-seq.dat: Normalized proteomics and RNA-seq data of genes.
Prior to the development of the EN-TEx resource, the similarities among assays were usually calculated from unmatched data. For example, a large number of histone modification signals were detected from many different human individuals in Roadmap12. The intrinsic difference between two individuals due to genetic and environmental factors is expected to bias the similarity of two histone modifications. For the histone modifications that are positively correlated to each other, their similarity is expected to be underestimated, whereas for the negatively correlated ones, the similarity may be overestimated. The degree of such bias due to unmatched data has not been investigated for the many types of functional genomic data generated from numerous human samples. With the EN-TEx data, we can finally estimate such bias quantitatively and reliably. For example, we measured both the H3K27ac and H3K4me3 signals from the spleens of two individuals, individual 1 and 2; the average similarity between the two signals from the same individuals was 80% in terms of variance explained, but was reduced to 70% when comparing the two signals from different individuals. We used this approach for all the EN-TEx histone modifications, and thus estimated the influence of unmatched data on the similarity between different types of assays (Data S6N–O). The difference varies with the explained variance. For the two signals with high similarity (i.e., large explained variance), using unmatched data results in about 10% smaller explained variance than using matched data. As expected, this trend was the opposite for two signals with low similarity. In addition, we applied this approach to measure the influence on the similarity between different tissues (Data S6N–O).
AS Calling:
Determining Individual AS Events (related to “Large-scale Determination of AS SNVs & Construction of the AS Catalog” in the main text and Figure S3A)
ASE, AS Binding (ASB), and AS Chromatin Accessibility (ASCA)
ASE, ASB and ASCA were measured with an extended version of the AlleleSeq pipeline, dubbed AlleleSeq2 (see EN-TEx portal for Github with code). Broadly, the pipeline incorporates personal variation, including large SVs, to account for reference bias22,42,45 in a straightforward way. We have included additional filters to mitigate ambiguous mapping biases22,114. In order to account for the overdispersed nature of the functional genomics readcount data, the significance of the allelic imbalance is assessed with the beta-binomial test22 (Data S7A).
For each available replicate of the EN-TEx experiments, functional genomics reads were mapped to both personal haplotypes simultaneously using STAR-2.6.0c115. We required stringent mapping criteria, allowing the maximum number of mismatches to be 3% of the read length. For ChIP-seq, ATAC-seq and DNase-seq datasets, mapping was performed forbidding spliced alignments. Adapters were also removed from the ATAC-seq and DNase-seq reads with cutadapt116. For RNA-seq data, we used GENCODE v24100 annotation converted to personal coordinates. RNA-seq mapping was performed in the two-pass mode to identify and incorporate novel junctions. Read duplicates were identified and removed from all alignments using picard (http://broadinstitute.github.io/picard/). The fraction of assay reads that were preferentially aligned to either haplotype and overlapped heterozygous SNVs (hetSNVs) across all samples ranged from 1.1–7.3%. The allelic imbalance is measured by the fraction of unique reads mapped to each haplotype.
To visualize functional genomics reads on individual haplotypes (Figure 2A), we used SAMtools (ver. 1.9)117 to extract haplotype-specific reads from the BAM files generated by STAR from the last step. If an assay had multiple replicates, we merged all the BAM files. The number of reads mapped to a given region in the personal genome was calculated by bedtools (ver. 2.29.2)118 and stored in bedgraphs, lifted over to the reference genome with UCSC LiftOver119, and converted to bigwigs with bedgraphToBigWig (ver. 2.8)112. Data S7B summarizes the pipeline used to generate the haplotype-specific bigwigs. The bigwigs are displayed with the Integrative Genomics Viewer120. See Data S7C for accession numbers of the data used to generate the signal tracks in Figure 2 and Data S17. A script that generates the haplotype-specific read coverage from the BAM files is provided at https://github.com/gersteinlab/AlleleSeq2. An example of the process is demonstrated in the following files.
File: sample_signal_track.tar.gz: Example output of haplotype-specific signal tracks.
File: AlleleSeq2_workflow_examples.tar.gz: AlleleSeq2 workflow demonstrated using RNA-seq and H3K27ac ChIP-seq experiments from ENC-003 thyroid gland samples.
The number of reads overlapping each hetSNV and carrying the corresponding alleles was calculated after filtering. The filtering included:
Potentially misphased loci;
Reads bearing an incorrect allele;
HetSNVs located in potential copy number variation sites through assessment of the surrounding read depth (+/− 1 Kb);
Non-autosomal chromosomes (for most downstream analyses we used call sets that only include loci from autosomes).
We aggregated read counts from all replicates available for each experiment (sample). We then called AS sites at an FDR of 10% as described previously22,45 (Data S8) by calculating the significance of the imbalance at each heterozygous locus.
We provide the read counts and p-values for all the ASE and ASB sites that are either significantly imbalanced or accessible (SNVs that have at least the minimum number of reads needed to be statistically detectable for allele specificity), which can be found in the following file.
File: hetSNVs_default_AS.tsv: Full list of accessible heterozygous SNV loci with haplotype-specific read counts.
File: hetSNVs_default_AS_DNase.tsv: Full list of accessible heterozygous SNV loci with haplotype-specific read counts from the DNase-seq datasets.
Columns in the hetSNV files are:
| 1) chr | : chromosome |
| 2–3) ref_start, ref_end | : GRCh38 locus positions (0-based, half-open) |
| 4) ref_allele | : reference allele |
| 5–6) hap1_allele/hap2_allele | : haplotype 1/2 allele |
| 7) experiment_accession | : ENCODE experiment ID |
| 8) donor | : EN-TEx individual |
| 9) tissue | : tissue |
| 10) assay | : assay |
| 11–14) cA/cC/cG/cT | : number of reads with A/C/G/T |
| 15) ref_allele_ratio | : number of reads with reference / total number of reads |
| 16) p_betabinom | : p values calculated from the beta-binomial test |
| 17) imbalance significance imbalanced site. | : ‘1’ passes the FDR10% threshold, ‘0’ not a significantly |
Since the EN-TEx samples have had independent genome sequencing completed for more than one tissue (i.e., transverse colon from EN-TEx and blood from GTEx), we can use this information to evaluate the impact of sequencing errors and somatic mutations on our AS call set. We have done this in a limited fashion in Data S35.
Allele-specific Methylation (ASM)
We used WGS variant calls to determine the positions of hetSNVs and identify all homozygous CpG positions in the genome of each donor (Data S9). With such information, and with the fully processed tissue-specific WGBS-aligned reads, an in-house script was then used to identify positions exhibiting significant allelic differences in CpG methylation. Our script counted the number of times a methylated or unmethylated homozygous CpG occurred in the same read as each of the two possible alleles at the hetSNV position for autosomal chromosomes. If the same read overlapped multiple CpGs, they were each considered as independent observations. Reads that overlapped with indels, had a low-quality score (Phred < 20) on the SNP position, or had a base call that did not match either of the two alleles expected in that position based on the WGBS calls were discarded. Due to the nature of bisulfite sequencing data, where cytosines may be observed as thymines during bisulfite conversion, it was not possible to determine which allele the read came from in several cases. In such cases, the read was also discarded. If a low-quality score or an unexpected base call was observed on a CpG position for a particular read, that observation did not contribute to the final counts. The significance of the association between the allele at the hetSNV position and the methylation state of the CpGs in the 300 bp surrounding region was assessed using Fisher’s exact test. The 300 bp windows surrounding the hetSNV position were chosen as the WGBS dataset was composed of paired-end 150 bp reads. The test was only performed for hetSNV positions that showed a minimum of six observations of either a methylated or unmethylated CpG position for both alleles, and the p-values were subsequently corrected with the Benjamini-Hochberg method for FDR control. The difference in the level of methylation between alleles was also computed for each hetSNV. Finally, ASM calls were made by identifying the heterozygous SNP positions with FDR values below a specified threshold (10%), and absolute differences in methylation between alleles above a minimum threshold of 10%. The result can be found in the following file.
File: ENTEx.TissueStacked.phased.final.txt: Assessment of allelic imbalance in CpG methylation.
Explanations of the columns are as follows:
chromosome,start,end: position of the SNV. Coordinates are 0-based in hg38
Allele1[2].[Un]Methylated (int): Number of [un]methylated CpG in the 300 bp region surrounding Allele1[2]
Number.of.good.reads (int): Number of reads used to count methylated and unmethylated CpGs
Is.on.heterozygous.CpG (binary): 0 indicates that the variant is not on CpG; 1 indicates that the variant is on CpG
P.Values (float): p-value of Fisher’s exact test based on Allele1.Methylated, Allele1.Unmethylated, Allele2.Methylated, and Allele2.Unmethylated
FDR (float): Adjusted p-value based on Benjamini-Hochberg method
Methylation.Allele1[2] (float): Fraction of methylated CpG in the 300 bp window of allele1[2]
Methylation.Difference (float): Methylation.Allele1 - Methylation.Allele2
Phasing.Set (string): Phasing set designated by individual VCF
Tissue (string): Tissue from which the sequenced sample originated
Individual (string): EN-TEx individual ID
AS Hi-C Interactions
Each pair of the paired-end reads are aligned separately to both of the parental haplotypes using BWA-MEM94. Sequencing reads are then paired based on their read names. Each paired-end read is then assigned to either one or both of the parental haplotypes as follows: for each paired-end read, a score is assigned to each parental haplotype based on the number of mismatches of the mapping to that haplotype. Paired-end reads are then assigned to either haplotype 1 or haplotype 2 based on their corresponding score. In brief, pairs of reads are assigned to a haplotype if they map exclusively or with a better score to that haplotype. Additionally, pairs of reads that exclusively map to one of the haplotypes are also assigned to that haplotype. After every paired-end read is assigned to a parental haplotype, chimeric reads and PCR duplicates are removed and we generate an interaction matrix for each haplotype of each tissue of each individual (Data S10A–B for the pipeline and Data S10C for the matrices).
For each significant interaction captured by Fit-Hi-C, we found the number of reads that map to haplotype 1 and haplotype 2 using the haplotype-specific interaction matrices. If there was a difference in the number of reads that mapped to one haplotype vs. the other, we then calculated the p-value for the significance of the allelic imbalance using a binomial test. The results are reported in the following file.
File: hic_files.tar.gz: Allele-specific Hi-C interactions.
The above file contains two folders: “ref” and “pgenome”. The “ref” folder contains .hic files for each individual and tissue (each individual and tissue combination is a separate folder, totaling up to eight folders); these files contain information on the genome-wide interaction matrices. The information can be extracted using Juicer tools and the contact matrices can be visualized using Juicebox (Data S10C for an example). The “Pgenome” folder contains two subfolders: “hap1” and “hap2”. Each of these folders contain two .hic files for each chromosome of each individual and tissue. Chr*.hap*.hic files contain the Hi-C data for that chromosome in personal genome coordinates and Chr*.hap*2ref.hic files contain the Hi-C data for that chromosome in a reference genome coordinate (lifted over using personal genome chain files). Data S10D shows the total number of raw AS interactions and significant allelic imbalances per sample (calculated using the binomial test described above).
AS Peptide (ASP) Analysis
The proteomics data were mapped at the gene level and filtered to a set containing one or more ASPs in any donor. These fell into two categories: genes with ASPs for one allele only or those with peptides specific to both alleles. Both groups were considered for ASP ratios. The ASP ratios were calculated for each tissue and donor in which allelic peptides were quantified, based on the ratio of the summed peptide intensities of peptides specific to the two alleles. Individual ASPs were filtered to require a minimum of three distinct peptides unambiguously identifying a gene, an expression level for the tissue of not less than five-fold lower than the highest expressed tissue and an ASP ratio of greater than 0.75. Data S26D summarizes key numbers of genes with allelic peptides. A full list of allelic peptides is included in the Supp_data_proteomics.xlsx file described in the section “Proteomics”.
AS Elements
(related to “Large-scale Determination of AS SNVs & Construction of the AS Catalog” in the main text and Figure S3AB)
Genes and cCREs
We extended our pipeline to measure allelic imbalance at genomic regions and elements of interest. To do so, we aggregated read counts from all hetSNVs within the relevant region and assessed the significance of imbalances between personal haplotypes for individual hetSNVs as described above. We provide a large catalog of genomic elements measured for AS activity (e.g., ASE genes and cCREs) with corresponding haplotype-specific assay read counts and significance scores of the imbalance (Data S11A). Results are summarized in the following files.
File: genes_default_AS.tsv: List of accessible genes with haplotype-specific read counts.
File: cCREs_default_AS.tsv: List of accessible regulatory elements with haplotype-specific read counts.
Columns in these files are similar to those described in the section “ASE, AS Binding (ASB), and AS Chromatin Accessibility (ASCA)” with the following differences:
| 4) region_id | : gene name (GENCODE v24) or cCRE id |
| 5–6) hap1_count/hap2_count | : number of reads mapped to haplotype 1/2 |
Correlation Between AS Genes and Diseases
We compared the set of AS genes to a set of genes associated with certain diseases. The list of disease genes includes those known to be affected by disease-associated mutations and expressed in disease-related tissues121. For each tissue and individual, we noted the genes that were present in both the set of AS genes and the set of disease genes. Many of the correlations were sensible. For example, TSHR, TG, and PAX8, which are associated with hyperthyroidism, showed AS behavior in the thyroid, and TNNT2, LDB3, and SCN5A, associated with cardiomyopathy, showed AS expression in the heart. The list of the overlapping genes and their associated diseases can be found in the following file.
File: Associated_AS_Disease_Genes.xlsx: Allele-specific genes associated with diseases.
Gene Ontology Enrichment Analysis of AS Genes
To determine the characteristics of active AS genes, we performed gene ontology enrichment analysis of protein-coding genes that showed AS activity in different assays (Data S11B). DAVID Bioinformatics Resources 6.8122,123 was used to perform the functional annotation clustering. For ASB, the background list for each assay includes all protein-coding genes with accessible promoters in that assay; for ASE, the background list includes all protein-coding genes with an accessible expression level from RNA-seq. The AS gene list for each assay includes genes showing AS activity in any EN-TEx individual or tissue, and genes were ranked by p-value to be AS genes. For ASE analysis, since DAVID has a 3,000 gene limit, the top 3,000 mostly ASE+ protein-coding genes were selected for the enrichment analysis, and the top 20 enriched terms are shown. We found that protein-coding genes showing AS activity in assays are mostly enriched in phosphoprotein, and their sequences are featured with polymorphisms and variants.
Aggregation:
Aggregating Individual AS Events Across Tissues and Assays (related to “Large-scale Determination of AS SNVs & Construction of the AS Catalog” in the main text, Figures 1C and S3CD)
We use two strategies for aggregating AS events across tissues and assays (see Figure 1C and Figure S3A–D). The first is to simply take the union of AS SNVs from each individual tissue or assay. The second is to pool the reads across different tissues or assays and then re-perform the allelic calculation using the pooled reads as input. This increases the statistical power of the allelic calculation at the expense of distinguishing AS behavior between different tissues or assays. We employ both methods in constructing the EN-TEx AS catalog. In Alleleseq2, we developed an approach to pool reads from alignment files obtained from multiple tissue samples (or assays) to reassess AS imbalance and generate a “pooled” joint call set (see “ASE, ASB, and ASCA” below). We also provide a script that allows for the union of call sets from donors/tissues/assays as part of the AlleleSeq2 repository, creating “union” call sets.
ASE, ASB, and ASCA
We observed a large increase in detection power when we pooled reads for each hetSNV across all tissues in each individual. We calculated the significance of the imbalance at each hetSNV for the pooled call set in the same manner as for individual tissues and called ASE, ASB and ASCA sites at an FDR of 10%22,45. Figure 1C and Figure S3 provide a summary of the AS catalog, including the number of AS hetSNV and AS elements, with different aggregation methods and levels. Results of the aggregation can be found in the following file.
File: hetSNVs_pooled_AS.tsv: List of accessible hetSNVs with haplotype-specific read counts pooled across tissues.
File: hetSNVs_pooled_AS_DNase.tsv: List of accessible hetSNVs with haplotype-specific read counts pooled across tissues from the DNase-seq datasets.
Methylation
We aggregated the counts of methylated and unmethylated homozygous CpG positions surrounding both alleles of each heterozygous SNV across tissues for each individual to assess the cross-tissue association between the allele at the hetSNV position and the methylation state of the homozygous CpGs. The significance of association was computed using Fisher’s exact test; the Benjamini-Hochberg method was used to control the FDR. For aggregated observation, the test was only performed for accessible hetSNV positions that showed a minimum observation of methylated or unmethylated homozygous CpG positions for both alleles. The number of positions n (around 12) was determined by maximizing the sum of p-values. ASMs were called at FDR values under 10% and absolute methylation differences larger than 10%.
We then generated a combined ASM call set that includes cross-tissue counts of methylated and unmethylated homozygous CpG observations surrounding accessible hetSNVs for all four individuals. Identical hetSNVs across individuals were included as separate records of CpG counts. All accessible hetSNVs, their associated gene, distance to gene, and genomic region were annotated based on the refGene database. Alternative allele frequency was annotated based on the Genome Aggregation Database (gnomAD) 3.0 database using ANNOVAR124. cCREs were annotated based on ENCODE. The aggregated result can be found in the following file, with columns similar to those described in the section “Allele-Specific Methylation”.
File: ENTEx.TissueAggregated.final.txt: Assessment of allelic imbalance in CpG methylation with haplotype-specific methylated and unmethylated homozygous CpGs pooled across tissues.
Note that in Figure 2B, because DNA methylation tends to repress gene expression, the polarity (direction of AS imbalance) of the AS DNA methylation in the promoter region is in the opposite direction to that of the AS expression and chromatin active state in the gene body. As expected, the active epigenetic marks H3K4me3 and H3K27ac demonstrate consistent AS imbalances, and most of the AS SNVs associated with DNAH11 are known eQTLs from GTEx. One such SNV (rs11760336) lies within the DNAH11 promoter, likely changing the gene expression directly. In addition, some of the AS SNVs overlap with known GWAS variants as indicated in the figure.
AS Catalog
(related to “Large-scale Determination of AS SNVs & Construction of the AS Catalog” in the main text, Figures 1C and S3BDEF)
Generalizability of the AS catalog
We discovered over one million SNVs that show AS activity in gene expression, DNA methylation, histone modification, and/or TF binding. This catalog should cover a large fraction of AS activity of common SNVs. To estimate the coverage, we started by using the 1,000 Genomes project high-coverage data125 to assign allele frequencies to the EN-TEx SNVs. We found that 76% (i.e., 5,276K) of the common SNVs (EUR MAF > 5%) in 503 European individuals (specifically, individuals of GBR, FIN, IBS, TSI, and CEU) were discovered in EN-TEx, 4,414K of which were heterozygous and unambiguously genotyped in at least one of the four EN-TEx individuals. Among these 4,414K SNVs, 946K (21.4%) show AS activity in at least one assay (whereas 63K of the AS variants are rare, EUR MAF < 1%). If the EN-TEx project was conducted on all 503 European individuals from the 1,000 Genomes project, which contains 6,946K common SNVs with AF < 1, then the number of AS SNVs would be 6,946K * 21.4% = 1,486K (assuming each of the 6,946K SNVs is heterozygous in at least one individual). This number is only a 540K increase from the 946K that are currently in the AS catalog, indicating that our catalog includes a majority of the AS events at common SNV loci in the European population.
While previous studies also compiled AS histone modifications and/or DNA methylation, our catalog is larger. For example, while Onuchic et al.28 reported 125K ASM loci, 36K loci with ASB H3K27ac, and 0.5K loci with ASB H3K27me3 (Table S1 of Onuchic et al.), our catalog includes 469K, 79K, and 96K loci, respectively. Similarly, Chen et al.22 used SNVs discovered by the 1,000 Genomes project (2,504 individuals) to construct a diploid genome for each of the 384 individuals compiled by Geuvadis. They mapped RNA-seq and ChIP-seq to these diploid genomes and identified 63K ASE hetSNVs and 6.1K ASB (ChIP-seq) hetSNVs, the latter of which is a much smaller number than the 361K in our catalog. In terms of AS activity in the regulatory regions, we also found more (28K vs. 11.7K) AS cCREs than a similar study using the Roadmap data46. While we note that the EN-TEx resource does not have more ASE events than GTEx126 or AlleleDB22, we found that ASE is only a small fraction of all the AS events in the genome (Data S8B–C). Most AS events are related to the chromatin states of the regulatory regions.
Calling AS Events in External Datasets Used for Validation
In addition to the EN-TEx AS catalog, we have generated ASE and ASB call sets for the CEPH individual NA12878 and Roadmap12 individuals STL002 and STL003. We used these call sets for external validation of our predictive models.
Personal genome sequences for STL002 and STL003 were constructed using variant calls generated previously28. For NA12878, we used SNVs and indels available from the Illumina Platinum Genomes project127 (2016–1.0) and large deletions generated by the 1,000 Genomes Phase 3 SV Analysis Group128. All datasets with matching assays (and all tissues for STL002 and STL003) that are available from the ENCODE portal were utilized to generate the AS call sets. These personal genome files are available as described below.
File: pgenome_NA12878.tar.gz: Personal genome for NA12878.
File: pgenome_STL-002.tar.gz: Personal genome for STL002.
File: pgenome_STL-003.tar.gz: Personal genome for STL003.
See Data S12 for the number of AS hetSNVs detected in each sample. The files are formatted as described in the section “ASE, AS Binding (ASB), and AS Chromatin Accessibility (ASCA)”. These call sets were used for validation of predictive models described in the sections “Prediction of Promoter AS Activity With a Random Forest Model” and “ASEffect Prediction with the BERT Model”.
High-confidence and High-power Call Sets
We also developed a “high-confidence” call set requiring that at least one read from both alleles was detected in the functional genomics assay, thus accounting for potential false-positive genotype calls. In addition, we generated a “high-power” tissue-specific call set by allowing a more relaxed threshold (FDR 20%) for loci that were detected as significantly imbalanced after read pooling-based joint calling across all tissues (Data S13A). These two call sets can be accessed in the following files.
File: hetSNVs_high-confidence_AS.tsv: List of hetSNVs with high-confidence allelic imbalance calculations.
File: hetSNVs_high-power_AS.tsv: List of hetSNVs with the high-power allelic imbalance calculations.
In addition, we tested two methods for increasing detection power of AS hetSNVs in datasets with low read counts. Both methods impose a less strict test for allele specificity on hetSNVs that have been determined to be AS in prior experiments. This prior knowledge can be taken from other experiments on the same individual, or from experiments on different individuals entirely. The first “high-power” method relaxes the FDR threshold from 10% to 20% for all hetSNVs that have prior evidence of allele specificity. All other hetSNVs are evaluated at the usual 10% FDR threshold. The second method uses a one-sided beta-binomial test, instead of the default two-sided test, to determine whether the direction of imbalance is consistent with prior data. With both high-power methods, new AS hetSNVs are identified that did not meet the threshold using the default calling method.
To validate these high-power methods, we tested them on a deep RNA-seq experiment of the cell line GM12878 (Data S13B–E). We first identified ASE hetSNVs in the dataset using our default calling method. This was our “gold standard” list of 24,685 ASE hetSNVs. Then, we simulated a shallower sequencing experiment by downsampling by a factor of 4. Using the default ASE calling method on the downsampled dataset, we identified 6,928 ASE hetSNVs. Approximately 80% of these hetSNVs were in common with the gold-standard list. We expect that the error rate is a product of the randomness of downsampling. Then, both high-power calling methods were performed on the downsampled dataset.
We generated priors for the high-power methods using the pooled reads across all tissues from the four EN-TEx individuals. If a hetSNV was ASE in at least one individual, it was included in the high-power test. If two or more individuals had ASE of the same hetSNVs, the direction of imbalance should agree (e.g., both favor the reference allele over the alternative allele), otherwise that hetSNV was excluded. We did not take into account the identity of the alternative allele for any hetSNV.
For the relaxed FDR method, 122 new ASE hetSNVs were identified that did not meet the threshold for allele specificity using the default method. For the one-sided method, 275 new ASE hetSNVs were identified. For both, approximately 60% of the new ASE hetSNVs were in common with the “gold standard” list. The validation shows that both methods can be used to identify a modest number of new ASE hetSNVs, at the cost of somewhat reduced specificity.
It should be noted that the results of the high-power methods are dependent on the nature of the prior. Both methods can only be used to evaluate hetSNVs for which there is prior information. Using data from the EN-TEx individuals as a prior for non-EN-TEx cell lines such as GM12878 captures most common hetSNVs but excludes most rare variants. If more of the individual’s hetSNVs are in common with the prior, it is likely that the high-power methods will identify more AS hetSNVs. In the case of the EN-TEx individuals, we circumvent this problem by using AS hetSNVs identified from the all-tissue pooled reads as the prior for high-power analysis of individual tissue datasets. Because both sets come from the same individual, they share both common and rare variants.
Integration with the ClinGen Allele Registry
The variants identified in all four EN-TEx individuals are registered in the ClinGen Allele Registry129, which provides unique variant identifiers for canonical alleles defined at the level of nucleic acid sequences or at the level of proteins. The unique identifier integrates different types of labels and definitions of the same allele across multiple databases including dbSNP130, gnomAD131, ClinVar132, and ExAC133; approximately 24K variants were previously recorded in ClinVar134. A total of 58 variants are classified as ‘pathogenic’ or ‘likely pathogenic’, 14 of which show AS behavior (including ASM) in at least one of the EN-TEx samples. All variants are bulk registered in VCF format using API specified by Allele Registry documentation (http://reg.clinicalgenome.org/doc/AlleleRegistry_1.01.xx_api_v1.pdf). Variants can be queried either programmatically via APIs or via search interface using any type of ID associated with the variant. Metadata for allele(s) are available in machine readable form (JSON) on ClinGen and can be queried in bulk as well.
AS Examples:
Illustrating the Coordination of AS Activity Across Assays (related to “Examples of Coordinated AS Activity, involving SNVs & SVs” in the main text, Figures 2 and S4)
X-chromosome inactivation (XCI) presents an ideal opportunity to showcase coordinated AS activity inferred using the EN-TEx resource while allowing for important biological discoveries. We first categorized the AS activity into three categories: “gene expression”, “active histone mark enrichment”, and “repressive histone mark enrichment”. For gene expression, we used AS RNA-seq data. For the active and repressive histone mark enrichments, we pooled seven (CTCF, EP300, H3K27ac, H3K4me1, H3K4me3, POLR2A, and POLR2AphosphoS5) and two (H3K27me3 and H3K9me3) histone marks for each tissue, respectively. We then analyzed the AS activity of each pooled dataset.
To determine which haplotype is inactivated, we calculated the log2 ratio of activity between the haplotypes as log2(haplotype1/haplotype2). This calculation was performed for all tissues of both female individuals (ENC-003 and ENC-004) as shown in Data S14A. For gene expression, the ratio relates individual gene read counts between haplotypes. For either active or repressive marks, the ratio relates the sum of activity within a +/− 10 Kb region surrounding each gene. We also computed a tissue-level score by calculating the mean log2 ratio across all genes in each tissue (top bar in Data S14A). As shown in Figure 2A and Data S14A, most tissues have the same haplotype inactivated in both individuals. Active histone marks and gene expression showed bias in the same direction, highlighting the coordinated activity across the X chromosome. By contrast, activating and repressive histone marks showed limited (due to data sparsity) bias in opposite directions. These observations were quantified by a cosine similarity analysis in Data S14B.
We then sought to identify genes that escape XCI, denoted as “escaper”, using the EN-TEx resource. Escaper genes (Data S14C, top) were identified as those genes that were expressed in at least eight tissues and showed balanced expression (log2 ratio between −0.4 and 0.4 [inclusive]) in 60% of their expressed tissues. Tissues that showed balanced expression were excluded from this analysis (ENC-003: LIVER and OVARY; ENC-004: ADPSBQ, ADRNLG, ESPMSM, ESPSQE, HRTAA, STMACH, SKINNS, and SKINS). A curated list of escaper genes in both individuals was created (Data S14C, bottom) and their status was validated by a literature review. Data S14D shows three examples of identified escaper genes that show balanced gene expression between both haplotypes despite tissues showing a strong bias towards one haplotype. We also provide a breakdown of haplotype specificity in XCI across different chromatin marks and gene expression in Data S14E–F.
We found an example of AS activity for a less-characterized locus in ENC-003. We detected AS Hi-C interactions in the XACT locus (Data S14G) on the active copy of the X chromosome. We first determined the active copy of the X chromosome by considering the gene expression distribution on both haplotypes and found that haplotype 2 has more gene expression than haplotype 1. We then looked at the differential interaction of the X chromosome by subtracting the Hi-C matrices of the haplotypes. We found that an interaction between the XACT locus and an upstream region is significantly elevated in the active haplotype. We also found that both the XACT locus and the upstream region are bound to CTCF, which might be mediating the interaction. XACT is a long non-coding RNA (lncRNA) found to be active in the active copy of the X chromosome early in cell development. This CTCF-mediated haplotype-specific interaction could play a role in activating the XACT locus established at early stages of cell development. While such observations are interesting, they are provisional on additional supportive data. Our analysis of haplotype-specific Hi-C data revealed an AS skew in Hi-C interactions between another gene, XACT, and its potential distal regulatory element on the active haplotype of the X chromosome (see Data S14G).
SVs:
Illustrating the Impact of Structural Variants (related to “Examples of Coordinated AS Activity, involving SNVs & SVs” in the main text, Figures 2DEF and S4BC)
Analysis of SVs
We focused our analysis on SVs that are larger than or equal to 50 bp, although the VCF files of each individual also contain smaller “SVs.” Note that the SVs identified from Oxford Nanopore data had fewer large insertions than those identified from the PacBio data (Data S15). This likely resulted from differences between the two sequencing technologies79,135.
To analyze the sequence composition of the SVs, we used RepeatMasker (ver. 4.0.7, slow search mode, http://www.repeatmasker.org) to classify the sequences that are inserted, deleted, or inverted. We also estimated the allele frequencies of the SVs. For this purpose, we checked for overlaps in the location between the EN-TEx SVs and those reported by Audano et al. (2019)36. To increase the chance of finding an overlap between these two datasets, we used the confidence intervals (CIs) of an EN-TEx SV’s coordinates as the location of the SV. Specifically, the CIs of the breakpoints were denoted by CI_POS and CI_END in the VCFs of individuals 2 and 3. The SVs of individuals 1 and 4 were called by different tools; therefore, the corresponding VCFs did not have CI_POS and CI_END. Instead, we used +/− 2*STD_quant_start as the Cl of the POS and +/− 2*STD_quant_stop as that of the END. For DELs and INVs, we extended the POS upstream by its CI and the END downstream by its CI. For INSs, we extended the POS upstream and downstream but did not extend the END. When an overlap was found, we further checked whether the two SVs were of the same type (e.g., both are deletions). If the two SVs were not the same type, we considered the two SVs to be different. Through this analysis, we matched SVs in Audano et al. (2019) with 68.3%, 65.9%, 63.4%, and 65.3% of the SVs in the four individuals, respectively. We assigned these EN-TEx SVs an allele frequency in European populations estimated by Audano et al. (2019)36. We performed a similar analysis by using more recent SVs called from long-read DNA sequencing data136 and gnomAD SVs131. A total of 71.4%, 68.9%, 66.5%, and 68.0% of the SVs in the four individuals, respectively, overlap with the former dataset. Because gnomAD annotates SVs differently, we allowed EN-TEx “INS” to match “INS”, “DUP”, “BND”, and “MCNV” in gnomAD, EN-TEx “DEL” to match gnomAD “DEL”, “BND”, and “MCNV”, and EN-TEx “INV” to match gnomAD “INV” and “BND”. In this way, we found a match for 63.4%, 61.4%, 60.3%, and 63.1% of the SVs in the four individuals, respectively.
To understand how SVs distribute in the genome, we generated a null expectation of SV distribution by shuffling the locations of SVs, using a method similar to that used in the 1,000 Genomes SV study128. Specifically, we placed the SVs in random locations on the same chromosome while avoiding gaps in the assembly. We calculated the ratio of the number of unshuffled SVs intersecting a given genomic region over the number of shuffled SVs. We repeated the shuffling 1,000 times.
Associating SVs with eQTLs
We aimed to identify heterozygous SVs that potentially cause AS gene expression and underlie the action of known eQTLs. To do this, we first identified eQTLs73 that are compatible with the ASE of the associated genes. For each ASE gene, we checked if the two alleles at each associated eQTL locus had the expected regulatory effect. The numbers of heterozygous SNPs and indels that were identified as compatible eQTLs in at least one tissue were 219K, 190K, 184K, and 137K in the four individuals, respectively (Data S16A–B). We used the compatible eQTLs associated with a given ASE gene to define a window spanning from –10 Kb of the compatible eQTL on the far 5’ end to +10 Kb of the compatible eQTL on the far 3’ end. For a heterozygous SV that intersects with this window, we determined whether the SV and the compatible eQTLs may locate on the same linkage block by comparing their allele frequency and haplotype. Specifically, for each SV identified in the last step, we identified all compatible eQTLs (with respect to the given ASE gene) that fell within +/− 10 Kb of the SV. Suppose the SV is on haplotype 1, then we calculated the allele frequencies of the alleles of the compatible eQTLs on haplotype 1. Here, we used the allele frequency in the European population reported by the 1,000 Genomes project125 for the alleles at each compatible eQTL. For each individual, about 500 ~ 800 compatible eQTLs carried an alternative allele that could be found in the 1,000 Genomes project. These compatible eQTLs were excluded from the next steps. If at least 30% of the haplotype 1 alleles of the compatible eQTLs within +/− 10 Kb of the SV had similar allele frequencies as the SV’s allele frequency (defined as 80–120% of the SV’s allele frequency), then we considered the SV to be potentially linked to the compatible eQTLs and that it may contribute to the ASE of the given gene. We listed SVs that meet this criteria, the associated ASE gene, and the compatible eQTLs +/− 10 kb from the SVs in the following file.
File: Supp_Data_SVs_associated_with_eQTL.xlsx: List of SV associated with allele-specific expressed genes and eQTLs.
We identified known eQTL-associated SVs (including SV-eQTLs)128,137 in our list of potential eQTL-associated SVs. We considered that an SV was a match if a reported eQTL-associated SV was found within +/−100 bp of this SV and both SVs were associated with the same gene. We searched for matches in tissue-specific and non-tissue-specific ways. For individual 1, our list includes 337 SVs that are associated with eQTLs in at least one tissue, of which 67 match known eQTL-associated SVs. The fractions are 84/317, 70/304, and 46/215 for individuals 2 to 4, respectively. Details of these results are listed in Supp_Data_SVs_associated_with_eQTL.xlsx. For comparison, we also calculated the fraction of known eQTL-associated SVs in our SVs that are close to genes with ASE (Data S16C–E). We pooled genes that have ASE in at least one tissue. Because GTEx eQTLs fall within +/− 1 Mb of the TSS of genes73, we used the same window to look for SVs near the genes with ASE, requiring the SVs to at least partially overlap with the windows. We further required SVs to be heterozygous, clearly phased, and relatively common (i.e., present in Audano et al. (2019)36). We found 4,912 SVs in individual 1 that meet these criteria, of which 596 match known eQTL-associated SVs. This fraction is significantly lower than the observed fraction of 67/337 (p = 3.4e-5, Chi-square test). We observed similar enrichment in the other three individuals (p = 4.2e-12, 3.1e-4, 4.9e-6).
See Data S17 for examples of indels potentially changing the gene expression and examples of splicing variants.
Aggregating the Impact of SVs on Neighboring Chromatin
Our goal is to calculate potential changes in the chromatin state in the neighborhood of SVs. Intuitively, this can be done by comparing the chromatin state near heterozygous SVs between the haplotypes of an individual. We excluded heterozygous SVs within 5 Kb of other SVs in the same individual.
In the remaining heterozygous SVs, we focused on those that have relatively precise breakpoints. Specifically, we kept SVs where the total lengths of the start position’s confidence interval and the end position’s confidence interval are at most 50 bp. To minimize the influence of SVs on mapping sequence reads, we further excluded SVs for which the average mappability of a window +/− 500 bp of the SV is below 0.9. Because EN-TEx requires the length of a ChIP-seq read to be at least 50 bp, we used 50-mer multi-reads Umap mappability138 to filter SVs when calculating potential disruption to chromatin openness (measured by ATAC-seq) and histone modifications (measured by ChIP-seq). We used 100-mer multi-reads Bismap mappability138 to filter SVs when working with WGBS data. We also excluded SVs that fall in blacklist regions that are known to give problematic ChIP-seq reads139. When Umap mappability was used, the numbers of SVs that passed the filters were 3,931, 3,636, 3,006, and 3,258 for the four individuals, respectively. When Bismap mappability was used, the numbers were 4,522, 4,246, 3,497, and 3,777.
For each SV that passed the above filters, we calculated the average chromatin state in the SV’s flanking regions. We defined flanking regions of an SV as the −500 bp ~ −100 bp region and the 100 bp ~ 500 bp region (Data S18A) – the extra 100 bp upstream and downstream of the SV are extra buffer regions that should reduce the influence of SVs on mapping sequencing reads. Because the chromatin state can be tissue specific and individual specific, we treated the SV-sample combinations as independent data points. We summed the allelic ATAC-seq and ChIP-seq reads on hetSNVs that fall into the flanking regions of a given SV in a given sample. For ATAC-seq and each ChIP-seq assay, we excluded SV-sample combinations in which the total reads from both haplotypes were less than 15. This step left about 4.4 K ~ 7.2 K SV-sample combinations (each SV has 2.9 ~ 3.8 samples on average) for the ChIP-seq assay. If the haploid that carries the SV had 70% or less reads (e.g., ATAC-seq reads) than the other haploid, we considered that the SV reduces the given chromatin state (e.g., chromatin openness). For CpG methylation measured by WGBS, we averaged the ASM levels around hetSNVs that fall into the flanking regions of SVs. About 58% of the SV-sample combinations lacked suitable hetSNVs in the SV flanking regions. We similarly required at least 70% reduction in the methylation levels near SVs, but we included all 45.6 K SV-sample combinations (6.2 samples per SV), since the general methylation levels of CpGs were high (in 98% of SV-sample combinations the methylation levels of the SV flanking regions averaged over the two haplotypes were above 50%). For each chromatin state, we reported the fraction of SV-sample combinations where the chromatin state was reduced near the SVs (Data S18B–D).
We also repeated the analysis by comparing one individual that carries the SV with one who does not. In this case, we excluded SVs within 5 Kb of any other SVs in individual 2 or individual 3, and SVs that fall on the sex chromosomes. We similarly filtered out SVs that have imprecise breakpoints and/or low mappability in the neighborhoods. A total of 1,974 SVs in individual 2 and 2,154 in individual 3 passed the filters when Umap mappability was used; the numbers were 2,294 and 2,478 when Bismap mappability was used.
We calculated the average fold-change over control of ATAC-seq and histone ChIP-seq, and the average methylation levels of CpG sites, in the flanking regions of SVs. For ATAC-seq and histone ChIP-seq, we also excluded SV-assay combinations in which the sum of the fold-change over the two individuals is less than 1.0, leaving 43 ~ 93% of the SV-assay combinations to determine reduction in chromatin state. For DNA methylation, we included all SV-assay combinations. To qualify a reduction in chromatin state, the above signal in the individual who carries the given SV must be 70% or lower than in the individual who does not. The right panels of Data S18C show the fraction of SV-assay combinations where a given chromatin state is reduced near the SVs. Again, transposable element-related SVs tended to reduce chromatin openness and H3K27ac levels in the neighboring regions.
Decoration Process:
Layering EN-TEx Information on ENCODE cCREs (related to “Application 1: Decorating ENCODE Elements with EN-TEx Tissue & AS Information” in the main text, Figures 3A and S5A)
Signal Normalization Method
In order to overcome batch effects, matrices of gene expression and histone marks’ values were quantile-normalized across samples (tissues and donors). The choice of the quantile normalization method was made after performing benchmarking of several normalization methods. The methods selected for the benchmarking are among the ones analyzed in a recent publication140: quantile normalization, smooth quantile normalization, upper-quartile normalization, variance stabilization normalization, and local regression normalization (two variants: LoessF and LoessCyc). These normalization techniques are widely applied in other bioinformatics fields, such as microarray and proteomics analyses. The pilot analysis was performed independently for two cell lines, K562 and GM12878, for which different polyA+ RNA-seq evaluation datasets were produced by the Wold, Gingeras, and Graveley labs during ENCODE Phase II. The benchmarking consisted of three steps: i) for each method, we computed the distribution of Pearson’s and Spearman’s correlation coefficients across all genes between each pair of samples; ii) we ranked the methods based on the mean of the distribution of all genes’ variance across samples141, and iii) we calculated the relative log expression distribution (distribution of log2 ratio for a given gene between one particular sample and the median across all samples), which should be close to 0142. Overall, the quantile and smooth quantile normalization techniques performed similarly between each other and better than the other methods. We thus opted for quantile normalization. For each of the histone modifications used in the decoration procedure below, we provide the quantile-normalized fold-change signals of cCREs across all the available tissues and individuals. The data file for each of the histone modifications is a data matrix, in which each row corresponds to a cCRE and each column corresponds to a tissue from an individual. As a result, the element in the matrix is the quantilenormalized signal of the histone modification observed in the cCRE from a tissue. These data are available in the following file.
File: cCRE_histoneSignals_qnorm.tar.gz: Normalized signal matrix of histone modifications in cCREs.
Decoration of Regulatory Annotations
We used the ChIP-seq datasets of both active and repressed marks to decorate (i.e., re-annotate) the cCREs from ENCODE, which are based on a set of high-quality DNase hypersensitive sites75. The ENCODE regulatory elements consist of 0.9 million cCREs averaging ~400 bp. For each type of functional genomic data, we normalized the activity signals of the cCREs from all tissues and focused on the cCREs with relatively strong signals (Data S19A). In the decoration, we considered three active marks (H3K27ac, H3K4me1, and H3K4me3) and three repressed marks (H3K27me3, H3K9me3, and DNA methylation). ChIP-seq datasets were uniformly processed using the ENCODE standard pipeline, including alignment, quality control, and peak calling. With the uniformly processed ChIP-seq datasets, the average epigenomic signals were calculated and normalized for a registry of cCREs from ENCODE (Data S19A). Namely, we first calculated the average fold-change against the control, typically input DNA, for each cCRE. The average fold-change was quantile normalized independently across experiments but jointly between individuals and tissues. Finally, the scores for each experiment were scaled from 1 to 10. For a particular tissue type, we defined a set of cCREs for each epigenomic mark that are considered as “active” (i.e., thresholding the normalized and scaled quantile values of the cCREs). The thresholding value was calculated for each assay by maximizing the similarity – the fraction of shared active cCREs – among the four individuals across tissues. We used the average threshold score across the transverse colon, spleen, and esophagus, since those were the most commonly comprehensive assays across individuals.
For each tissue, we then defined a set of active, repressed, and bivalent cCREs based on their active and repressed epigenomic signals, respectively (Data S19B; Figure S5A as an example from spleen). Briefly, the active cCREs show high activity for only the active marks (i.e., H3K27ac, H3K4me1, and H3K4me3); the repressed cCREs show high activity for only the repressed marks (i.e., H3K27me3, H3K9me3, and DNA methylation); and the bivalent cCREs show high activity for both the active and the repressed marks. Note that repressed and bivalent categories are not included in the current ENCODE encyclopedia. The cCREs were then separated into distal and proximal groups according to their distance to TSSs (proximal as those within 2 Kb of annotated TSSs). We also intersected these cCREs with the CTCF binding sites from the matched tissue type to define CTCF+ and CTCF− cCREs. Finally, the active and repressed cCREs were further annotated using their allelic signature to identify a set of AS and non-AS cCREs, respectively. In the AS decoration, we used the allelic signature from the matched epigenomic marks to define the active/repressed AS and non-AS cCREs. Any active/repressed cCREs intersecting with the AS cCREs were considered to be active/repressed AS cCREs. The active/repressed AS cCREs from different individuals were pooled together to generate a set of active/repressed AS cCREs in the corresponding tissue. We found that the numbers of repressed cCREs are comparable to those of active cCREs in many tissue types, highlighting the necessity of decoration using the repressed markers (Data S19C). Finally, we provided the cCRE decoration results (Data S19D) in all the tissue types in the following files.
File: cCRE_decoration.matrix: Candidate cis-regulatory elements annotated by the EN-TEx resource.
File: active.combined_set.txt.zip: Active candidate cis-regulatory elements of all the human tissues.
File: bivalent.combined_set.txt.zip: Candidate cis-regulatory elements that have both active and repressive signals.
File: repressed.combined_set.txt.zip: Repressed candidate cis-regulatory elements of all the human tissues.
To further explore the association between DNA methylation and other repressive histone modifications (i.e., H3K27me3 and H3K9me3), we partitioned the repressive cCREs from each tissue type into diverse groups, which contain only DNA methylation or only repressive histone modifications or both (Data S20D). We found that on average 53.2% of the repressive cCREs contain DNA methylation (Data S20E), and very few repressive cCREs are supported by multiple repressive epigenomic modifications (i.e., DNA methylation and H3K27me3/H3K9me3) (Data S20D).
File: Repressive_cCRE_DNAmethy_repressiveHM.zip: Binary tables showing whether the repressive cCREs are supported by DNA methylation or repressive histone modifications (H3K27me3 and H3K9me3) in each tissue type.
File: Repressive_cCRE_DNAmethy_repressiveHM_summary.csv: Summary of the binary tables showing the number and percentage of the repressive cCREs with specific patterns of repressive epigenomic modifications (DNA methylation, H3K27me3 and H3K9me3) in each tissue type.
A focused subset of the above just giving the cCREs that are methylated is:
File: cCRE_DNAme_subset.tsv.zip: A set of repressed cCREs with DNA methylation signals.
The first column is the cCRE ID and the second column is the tissues in which the cCRE was found. On average, there are 144K methylated repressed regions per tissue.
In order to further subdivide the active cCREs, we created an annotation set that focuses on regions with high H3K27ac signals. We call this set the “stringent” annotation set. To create this stringent annotation, we intersected the cCRE regions with the top 1% of scored regions as prioritized by the H3K27ac feature from the MatchedFilter program143. This stringent annotation was further used in other analyses and labeled as “stringent” in the main manuscript and figures. A file containing these stringent regions (bed file) can be found in the following file.
File: stringent.regions.MF.hg38.bed: Stringent regions with high MatchedFilter scores.
Completely Repressed Regions
Gene activation and repression can be mediated through the combination of different histone marks. Historically, much effort has been devoted to elucidating how genes are activated; however, emerging evidence suggests that appropriate heterochromatin formation is required for the preservation of genome stability and the cell type-specific silencing of genes144. In the mammalian genome, H3K9me3 and H3K27me3 are well-documented histone marks enriched for “constitutive” and “facultative” heterochromatin, respectively. For genomic regions not containing any active regulatory elements (cCREs), we have identified a set of elements that are marked by either H3K9me3 or H3K27me3 and do not have any active marks (H3K27ac, H3K36me3, H3K4me1, or H3K4me3) or transcriptional activities as fully repressed in the EN-TEx tissues. Regions within the ENCODE4 GRCh38 blacklist (ENCSR636HFF) and GENCODE gene list (GRCh38_v24) were removed. In summary, 45,207 non-overlapping elements of at least 200 bp in size (roughly approximate nucleosome size) are uniquely marked by H3K9me3, spanning 12,655,795 bp (less than 0.4%) of the reference genome, and 24,006 elements by H3K27me3, spanning 7,474,178 bp (less than 0.3%). As shown in Data S20, nearly 75% of these elements are specifically repressed in a certain tissue, and the rest show some degree of tissue specificity. It was previously known that H3K27me3-enriched facultative heterochromatin contains repressed genes in a cell-type-specific manner, whereas H3K9me3-enriched constitutive heterochromatin mainly occurs at the same gene-lacking regions in every cell type145,146. Observations also suggest that large domains of H3K9me2/3 form in a cell-type-specific manner and can influence cell identity by silencing lineage-inappropriate genes and impeding the conversion of terminally differentiated cells into a different cell type, highlighting a role for H3K9me3 in cell-type-specific gene regulation144,147–150. Identified elements can be found in the following file.
File: ENTEx_fully_repressed_regions_independent_of_cCREs.bed: Genomic regions not containing any active regulatory elements and marked only by repressive histone marks, not by active marks or transcriptional activity.
DNA methylation is a major contributor to gene repression and has been reported to interact with H3K9me3 in chromatin repressive pathways151. We further analyzed the methylation rate of CpG sites within these repressed elements. WGBS of CpG sites were available for 11 EN-TEx tissues that also have H3K9me3 and H3K27me3 ChIP-seq data. For the same tissue from different donors, we aggregated the CpG reads by taking the sum of reads from all donors, and considered a CpG site to be methylated (meCpG) when it is covered by at least 5x reads and the ratio of meCpG reads is at least 50%. The overall meCpG rate in each tissue was calculated and used as a control to evaluate the meCpG rate in H3K9me3- and H3K27me3-marked elements. As shown in Data S20A–C, elements uniquely marked by H3K9me3 show significantly (t-test, p-values < 0.05) higher meCpG rates than elements uniquely marked by H3K27me3. Compared with the control, H3K9me3-marked regions seem to be hypermethylated, whereas H3K27me3-marked regions are hypomethylated. This is consistent with the current understanding of constitutive heterochromatin and facultative heterochromatin, of which the former is defined by high levels of DNA methylation and H3K9me3 and the latter displays DNA hypomethylation and high H3K27me3152.
Validating Annotations Using the 3D Genome Organization
Chromosome compartments that are observed from principal component analysis (PCA) on a Hi-C correlation matrix give insight into the activity level of the chromatin. Chromosomes are divided into two distinct compartments, A and B, at a megabase scale153. The A compartment (positive values) corresponds to the active regions on the chromosome and the B compartment (negative values) corresponds to the inactive regions. Chromatin interactions are constrained within the compartment types, e.g., the loci in A compartment interact with the loci in the same compartment. Since A/B compartment assignments are proxies for the activity level of different loci, our tissue-specific regulatory element annotations can be validated by looking at their corresponding compartment in the tissue-level Hi-C data. We showed that our annotated tissue-specific active regulatory elements are dominantly located in the active compartment of the chromosomes of corresponding tissues, with a significantly higher number of regulatory elements per megabase observed in the positive compartment values when layered onto the first principal component of the Hi-C data.
We have assessed where the cCREs are located with respect to the chromatin compartments. To do so, we first binned the genome into 1 MB consecutive bins. We then counted the total number of cCREs in each bin and divided that number by the total number of cCREs in the genome, resulting in the cCRE density per 1 MB. We then plotted this density against the A/B compartment score obtained by the first eigenvector of the correlation matrix calculated from the Hi-C contact matrix. We performed this analysis for the master cCRE list from ENCODE3, tissue-specific active cCRE list derived in this study, more restrictive tissue-specific active cCRE list derived in this study, and tissue-specific repressed cCRE list derived in this study. The scatter plots for two tissues and four individuals are included in Data S21.
Tissue Specificity
(related to “Application 1: Decorating ENCODE Elements with EN-TEx Tissue & AS Information” in the main text, Figures 3BCD and S5B)
There are many methods for determining tissue specificity, most of which are based on continuous positive values154. Here, we chose the simple method of tissue count to determine the tissue specificity of genes/cCREs based on thresholds154. We chose this method because we can consistently apply it across different annotations including cCREs, genes, TSSs, and epigenomic peaks. Most of the other methods that are based on continuous positive values can be only applied on one annotation category (e.g., genes). Briefly, all genes and cCREs (as well as peaks and TSSs) were defined as active or inactive by thresholding their expression/activity level in a particular tissue type. The numbers of tissue types in which these genes/cCREs are active were then summarized. For each gene/cCRE annotation category, we then calculated a tissue-specificity score using the number of genes/cCREs that are active in only one tissue type divided by the total number of genes/cCREs in the category. The uniqueness scores range from 0 to 1, with higher scores indicating stronger tissue specificity.
For the genes, we included three gene types: protein-coding genes (from MS and RNA-seq), lncRNAs, and pseudogenes. To better estimate the expression level of pseudogenes, we applied our previously developed pipeline to quantify the expression level of pseudogenes, which can minimize the effects of multiple mapping bias in RNA-seq data155 (Data S22C). We then applied this pipeline to all three gene types and defined a set of active genes in the tissues by thresholding the FPKM (fragments per kilobase of transcript per million mapped reads) values (FPKM > 1 for protein-coding genes; FPKM > 0.5 for lncRNAs and pseudogenes) (Data S22A). Over 40% and 35% of the detected pseudogenes and lncRNAs, respectively, were actively transcribed in a single tissue, confirming that non-coding RNAs exhibit higher tissue specificity than protein-coding genes156,157. Of the pseudogenes demonstrating tissue specificity, a large fraction showed transcriptional activity only in testis (Data S22B). For the cCREs, we used the decorated annotations in the tissues to calculate the tissue-specificity scores as described above (Figure S5B and Data S22D). We also explored the tissue specificity of regulatory elements and epigenomic peaks (Figure 3B). The epigenetic profiles analyzed, including H3K27ac and DNase-seq, demonstrated tissue specificity, with the exception of DNA methylation, which exhibited ubiquity. An example of the tissue specificity of RAMPAGE data (for TSSs) is shown in Data S22E. The tissue specificity of the genes, cCREs, and epigenomic peaks are shown in the following file.
File: Tissue_Specificity.zip: The tissue specificity of gene expression and functional signals of cis-regulatory elements.
Tissue Specificity of ASB and ASE
Similar to H3K27ac-ASB cCREs (Figure 3), most ASE genes were detected in a single tissue (Data S22F). For the ~20 genes that were detected as ASE across all tissues, the allelic imbalance is in the same direction (Data S22G). We further compared our pan-tissue H3K27ac-ASB and ASE genes with housekeeping genes. Annotation results are shown in Data S22H–I. Furthermore, the non-AS categories show a “U” shape trend between fraction of elements and tissue specificity, indicating that there are many non-AS cCREs either extremely tissue specific or ubiquitous, whereas (bottom) the AS categories demonstrate an “L” shape trend between fraction of elements and tissue specificity, indicating that there are many AS cCREs extremely tissue specific but not ubiquitous. In particular, “fraction” refers to the fraction of elements falling in a given bin of the histogram. In other words, we take the original frequency from the histogram and divide it by the total number of elements for that category.
The Effect of Tissue Specificity on Conservation
The tissue-specificity influence on conservation is shown in Data S23A. Candidates are separated into categories of active, bivalent, and repressed. The number of candidates, rare derived allele frequency (DAF) values, and corresponding total SNP counts (from gnomAD) are given as a function of increasing tissue specificity (shared tissue count). In order to select rare variants, a minor allele frequency (MAF) of 0.05 was used.
Various decorations further subdivide the categories and affect the conservation level, based specifically on whether elements are distal or proximal as well as if they are CTCF bound or not. Conservation is shown for both phastCons (cross-species) and rare DAFs (cross-population) in Data S23B. Furthermore, we check the statistical significance of the difference in conservation and tissue specificity of non-AS and AS active (distal or proximal) cCREs. A proportion test is used in each case and all differences are measured to be p-value<2.2e-16.
We show the conservation across active and repressed cCREs in both ubiquitous and tissue specific cases in Data S23C. We include the results across the 1,000 Genomes, Pan Cancer Analysis Working Group, and gnomAD projects. Additionally, we show an increase in conservation when filtering for high H3K27ac signals (using stringent definitions for active elements with MatchedFilter143), which is supported by all three datasets. The overall conservation calculation is described in the section below.
The Relationship Between Purifying Selection and Regions Exhibiting Allele Specificity
The fraction of causative variants may be estimated by purifying selection. The analysis by the NIH Roadmap Epigenome project of epigenomes from 36 distinct cell and tissue types from 13 donors suggests that the donor genomes harbor on average at least 200 regulatory variants that are under purifying selection and therefore detrimental28.
In order to calculate the purifying selection on AS events, population-scale variants from three cohorts were used. We used two measures of purifying selection and conservation for this analysis. The first is the fraction of rare variants, which is calculated as #rare/(#rare + #common) for variants falling in a given AS region. In order to categorize variants as rare or common, ancestral alleles (i.e., the measurement was a DAF) and a MAF threshold of 0.05 were used. MAF is a commonly used metric for calculating selection in populations22,28,44. When considering the number of variants in each category, we found that across all tissues and individuals, 8,294 out of 128,448 ASB variants were rare. Of the 2,711,078 total non-ASB variants, 274,287 were rare. In total there were 40,123 ASE+ variants, of which 2,961 were rare. Finally, of the 624,210 ASE− variants, 70,370 were rare. The second method we used is phastCons, which measures the cross-species conservation158. All purifying selection analyses (rare DAF and phastCons) were performed for AS/non-AS cCREs, ASB/non-ASB H3K27ac regions, and ASE/non-ASE genes. The results are shown in Data S23D–G. Furthermore, we found that proximal AS events in the promoter were under stronger selection as compared to distal AS events.
Decoration Enrichments:
Relating Encyclopedia Decorations to QTLs and GWAS Loci (related to “Application 1: Decorating ENCODE Elements with EN-TEx Tissue & AS Information” in the main text and Figures 4BC)
We utilized various methods to evaluate the regulatory impact of our cCRE decorations. QTL and GWAS SNPs are important functional genomic variants and are useful for interpreting the function of our decorations. We performed GWAS enrichment analysis using eQTL and GWAS SNPs to assess the disease relevance of our cCRE decorations.
QTL Enrichment Analysis
We estimated the QTL (eQTL and splicing QTL [sQTL]) enrichment in the cCREs by calculating an odds ratio (OR) score using the numbers of real QTL SNPs and control SNPs located in the cCREs compared to those in the baseline regions (Data S24A).
in which a is the number of QTL SNPs in the cCREs; b is the number of control SNPs in the cCREs; c is the number of QTL SNPs in the baseline region; and d is the number of control SNPs in the baseline region.
The eQTL and sQTL SNPs were downloaded from GTEx v8159. The baseline regions are the union of all the functional and putative functional regions in the human genome, including coding regions, untranslated regions, noncoding RNA genes, open chromatin regions, TF binding sites, active and repressed histone peaks from multiple tissue and cell types, and evolutionary conserved regions160. The set of control SNPs was generated with the same number and same MAF distribution as the real QTLs, and this procedure was repeated 30 times to calculate a standard deviation for the SNP enrichment. The results of the QTL enrichment are in the following file.
File: QTL_enrichment.zip: The enrichment of QTL in cis-regulatory elements (cCREs).
We also compared the eQTL/sQTL enrichment in the regulatory elements from EN-TEx with those from Roadmap (Data S24B–C). First, we found that the distal regulatory elements from EN-TEx show stronger enrichment than the enhancer annotation from Roadmap. In addition, the active proximal regulatory elements from EN-TEx show stronger eQTL/sQTL enrichment than the TSS-associated annotations from Roadmap.
GWAS Enrichment Analysis
We downloaded the GWAS tag SNPs from the GWAS Catalog161. We performed several steps of quality control to generate a set of high-quality GWAS tag SNPs by removing some insignificant SNPs (p-values > 5*10–8), low-confidence SNPs, and SNPs from non-European studies. We also removed all SNPs in the human leukocyte antigen locus (for hg38: chr6:29,723,339–33,087,199). Next, we extended the set of tag SNPs by including the SNPs in high linkage disequilibrium (LD scores > 0.6) with the tag SNPs, which can generate more SNPs to increase the statistical power in the enrichment analysis. Some GWAS with very few LD-extended SNPs were removed. This approach resulted in a clean dataset with ~70K unique tag SNPs from 1,140 GWAS covering 717 unique traits. In Figure 4B, we show the LD score regression (LDSC) (left two panels) and GWAS enrichment (right panel) results for the analysis detailed here.
We then applied the hypergeometric test to estimate the enrichment of the GWAS tag SNPs in the cCREs from a particular tissue type (Data S25A).
in which N is the total number of cCREs in the genome; K is the total number of cCREs that carry GWAS tag SNPs; n is the number of cCREs in a particular tissue type; and k is the number of cCREs in a particular tissue type that also carry GWAS tag SNPs. Notably, we extended the cCREs 500 bp on both sides in the calculation (Data S25C). The results of the GWAS tag SNP enrichment are shown in the following file.
File: GWAS_enrichment.zip: GWAS enrichment of cis-regulatory elements (cCREs).
For the active distal cCREs, we identified 141 GWAS that are enriched in at least one tissue type (Data S25D). However, for the active proximal cCREs, we did not find any enriched GWAS in any tissue type. These results are consistent with previous studies showing that the causal GWAS SNPs are enriched in the enhancers instead of the near-gene promoters64,162 and also suggest that the active distal cCREs from our decoration are indeed significantly enriched in enhancers as we observed in the original Roadmap annotations (Data S25E).
Stratified LDSC values were also calculated for each tissue using 1,000 Genomes LD scores and GWAS summary statistics provided by Bulik-Sullivan, et al. This approach regresses chi-square statistics from the GWAS summary statistics with LD scores to estimate partitioned heritability in a disease-specific manner. The p-value indicates enrichment for a particular trait within an annotation.
In Data S25D, we show the p-value enrichment of each tissue with respect to various GWAS traits. Notably, distal active AS regions experienced higher enrichment compared to distal active non-AS regions (Data S25F), and both types of regions experienced higher enrichment compared to the original Roadmap annotations (Data S24C). For LDSC enrichment analysis of distal active elements in coronary artery (Data S25B), we found stronger associations between AS elements with respect to celiac disease, neuroticism, and type II diabetes, which were elucidated in previous clinical studies163–165. These results demonstrate that AS elements can significantly improve GWAS trait enrichment compared with the total set of elements across different traits as well as diverse tissue types, indicating that AS elements are valuable for the interpretation of GWAS data and that they potentially help pinpoint small subsets of regulatory elements driving a trait in specific tissues.
Compatibility:
Analysis of the Compatibility Between Assays (related to “Application 2: Relating AS SNVs to GTEx eQTLs & Modeling eQTLs in Hard-to-obtain Tissues” in the main text, Figures 4AD and S5CD)
Compatible and Incompatible: Single Chromatin Mark vs. Gene Expression
Using the methods described in previous sections, we identified promoters (± 2 Kb from the TSS) with allelic imbalance in the chromatin states measured by H3K27ac, H3K27me3, etc. We determined the compatibility between AS promoter chromatin states and AS gene expression in a straightforward way. The allele with more active promoter chromatin should have higher expression levels, otherwise the promoter and the gene are incompatible. Similarly, alleles with more repressed promoter chromatin are compatible with lower expression levels. We treated histone marks H3K27ac, H3K4me3, and H3K4me1, chromatin openness indicated by ATAC-seq or by DNase-seq, and the binding of EP300, POLR2A, POLR2AP, and CTCF, as marks of active chromatin. Histone marks H3K27me3 and H3K9me3, and CpG methylation were considered as marks of repressed chromatin.
Because AS gene expression and/or AS chromatin state can be tissue specific and/or individual specific, we did not merge compatible (or incompatible) promoter-gene pairs that appeared in multiple samples. Overall, the average number of ASE genes compatible with at least one of the 13 marks was 35 per tissue per individual, while the average number of ASE genes suitable for the compatibility analysis (i.e., the promoters of these genes were accessible for measuring the potential AS chromatin state indicated by any of the 13 marks) was 226 per tissue per individual.
We note that some assays were performed twice for a given tissue of a given individual. For example, the RNA-seq data of individual 3’s liver includes two experiments (ENCSR226KML and ENCSR504QMK), while there is only one H3K27ac ChIP-seq experiment for the same sample. In another example, there are two CTCF ChIP-seq experiments for individual 3’s spleen (ENCSR756URL and ENCSR773JBP), while there is only one RNA-seq experiment for the same sample. In these cases, we combined ASE genes or ASB promoters that were called from either of the duplicated experiments, excluding those where the directions of the allelic imbalance were the opposite in the two experiments. The combined ASE genes and ASB promoters were analyzed for compatibility and the results are listed in the following file.
File: Supp_Data_Compatibility.xlsx: The compatibility of genes with ASE in each tissue and individual.
To test the numbers of compatible vs. incompatible promoter-gene pairs, we identified genes that have ASE in at least one tissue of at least one individual. We shuffled the gene-promoter relation for these genes and calculated the ratio of N_compatible vs. N_incompatible. We repeated this process 1,000 times for each chromatin mark (after excluding replicates where N_incompatible is zero) to calculate a Z-score of the ratios shown in Figure S5D.
Compatibility of AS expression and binding with eQTL effect
We used the GTEx v8 catalog of known tissue-specific eQTLs (GTEx Consortium, 2020) to evaluate compatibility of AS expression and binding in the EN-TEx individuals with eQTL effect. For ASE, we identified all eGene-eQTL pairs where the eGene is AS in matching EN-TEx tissues and the eQTL is a heterozygous variant present in the EN-TEx donors. We used the slope (beta coefficient) of the eQTL and calculated the AS gene ratio of the number of reads mapped to the haplotype with the alternative allele. For ASB, we identify all GTEx eQTLs that are H3K27ac ASB hetSNVs in matching tissues and calculate the AS ratio of reads with the alternative allele. The slope is positively correlated with the AS ratio (see Figure 4A and 4D and more details in Data S26B–C and in the following file).
File: AS_ratios_and_eQTL_effect.tsv: Compatibility between GTEx eQTLs and EN-TEx allele-specific expression and binding.
Compatibility with AS Proteomics
Of the high-stringency ASP set, 114 were overlapped with ASE events calculated from RNA-seq data, 58 showed compatibility, and 56 showed incompatibility (Data S26D). The Z-score 0.26 of the ratio of the compatible to the incompatible (based on ASP/ASE pairs being randomized 1,000 times) was not significant (p<0.05), indicating that the compatibility between the RNA-seq and protein-level allele expression is near random. Although some of this incompatibility is likely due to technical issues, manual examination of the most biased ASPs-overlapping ASEs showed compelling evidence for ASP expression, implicating post-translational regulation (Data S26E)166,167. For some of the incompatible cases, there are clear biological reasons for the difference between the ASP and ASE ratios such as frameshift variants. More results of the compatibility between ASP and ASE are included in the Supp_data_compatibility.xlsx file described in the previous section “Compatible and Incompatible: Single Chromatin Mark vs. Gene Expression”.
Enrichment of ASE Genes Near ASM Promoters
We evaluated the association between the ASM of the promoters and the ASE of the corresponding genes while ignoring the compatibility between the two. We annotated all hetSNVs showing ASM with the closest associated gene based on the refGene database168 using ANNOVAR124. Chi-squared tests were used to determine whether ASE is significantly enriched among genes associated with ASM hetSNVs in promoter-like sequences (PLSs) identified by ENCODE75 with or without AS of TF binding compared to that among genes only associated with ASM hetSNVs in non-cCREs75. Significant enrichment was called at p-values < 0.05 and error in enrichment was estimated based on binomial distribution. Data S26F shows that genes with ASE are more highly enriched near PLSs with ASM and/or TF binding than near non-cCREs with ASM.
transferQTL Model:
Extending eQTL Annotation of Hard-to-obtain Tissues (related to “Application 2: Relating AS SNVs to GTEx eQTLs & Modeling eQTLs in Hard-to-obtain Tissues” in the main text, Figures 5 and S6)
Correlation Between Chromatin Features and eQTL Activity
We overlapped chromatin (histone marks and TFs ChIP-seq, DNase-seq, ATAC-seq) peaks with GTEx catalogs of eQTLs and fine-mapped eQTLs for every EN-TEx sample and observed a higher overlapping proportion in the case of fine-mapped eQTLs (Figure S6A and Data S27A). We obtained fine-mapped eQTLs after intersecting eQTLs with a posterior probability ≥ 0.8 from the three GTEx fine-mapping eQTL catalogs (CAVIAR, CaVEMaN, and DAP-G; see https://gtexportal.org/home/datasets#filesetFilesDiv15).
Next, we identified 1,353,101 SNVs that show tissue-specific eQTL activity: these SNVs are GTEx eQTLs in ≥ 5 EN-TEx tissues and are not GTEx eQTLs in ≥ 5 other EN-TEx tissues. Thus, for every SNV we defined two groups of tissues: i) tissues in which the SNV is an eQTL, and ii) tissues in which the SNV is not an eQTL. In this way, we could compute at which frequency the SNVs are marked by a particular histone modification when they do or do not show eQTL activity. We observed that SNVs are more likely to be marked by a given histone modification in the tissues in which they are eQTLs, compared to the tissues in which they are not eQTLs (Figure S6B and Data S27B). For each histone mark, we excluded lowly marked SNVs, i.e., SNVs overlapping with chromatin peaks in < 10% of all EN-TEx ChIP-seq samples for that particular histone mark.
Predictive Model That Transfers eQTLs From a Donor Tissue to a Target Tissue
In this section, we explain how we trained a machine learning model that can predict the tissue-specific activity of a set of eQTLs. Specifically, one such model takes as input a set of SNV-eQTLs previously identified in a given tissue (i.e., donor tissue) and predicts whether each of these SNVs is also an eQTL in another tissue (i.e., target tissue). Practically, the goal is to transfer eQTLs from a donor tissue to a target tissue (Figure 5A and S6C). Because of this, we called this application “transferQTL”.
For a given donor-target tissue pair, we first retrieved the set of GTEx donor-tissue SNV-eQTLs associated with a single eGene, and randomly partitioned them into training and test sets (containing 70% and 30% of SNV-eQTLs, respectively). Next, we trained a random forest model by providing, for every SNV-eQTL, a number of features related to either the donor or target tissue (see Data S28A). The response class was defined as “yes” if the SNV-eQTL was annotated as GTEx eQTL in the target tissue, and otherwise was defined as “no”. We trained the random forest model using the R package caret169 and by implementing a 5-fold cross-validation schema.
Only chromatin data from EN-TEx assays (histone marks and TFs ChIP-seq, DNase-seq, ATAC-seq) were employed. For this reason, the number of features employed in the model for a given target tissue depends on the type of EN-TEx experiments available for that particular tissue (i.e., if no ATAC-seq experiments were performed for lung tissue, then features “ATAC” and “ATAC_k” would not be employed to predict eQTLs in lung tissue). We downloaded a BED file containing repeated regions annotated in GRCh38 from http://genome.ucsc.edu/cgi-bin/hgTables, after setting “group” = “repeats” and “track” = “Repeatmasker.” We downloaded a BED file containing ENCODE candidate cCREs from https://api.wenglab.org/screen_v13/fdownloads/GRCh38-ccREs.bed.
Because GTEx eQTLs catalogs are available for matched EN-TEx tissues, we considered all possible pairs of donor-target tissues among 28 deeply sampled EN-TEx tissues, leading to a total of 756 (28*27) predictive models. For simplicity, we can consider these as different tissue-specific parameterizations of the general predictive model. We hereafter refer to the 756 donor-target models as “submodels.” Thus, a submodel is defined as the model for a particular donor-target tissue pair. For each target tissue, we have 27 submodels, each using a different donor tissue.
In the case of artery aorta, we combined data from experiments performed on both ascending aorta (individuals 1 and 2) and thoracic aorta (individuals 3 and 4).
The pipeline code to obtain the input matrices and train the predictive models can be found at https://github.com/gersteinlab/transferQTL. The model objects are available in the following ancillary files.
File: R6_RData.objects: All transferQTL sub-models obtained using eQTLs from a given donor tissue.
File: R6_RData.4hm.objects: All transferQTL sub-models obtained using chromatin features from only four histone marks (H3K36me3, H3K27ac, H3K4me1, H3K27me3), in addition to the non-chromatin features.
Model Performance, Validation, and Application
We used several metrics (see Data S28B) to evaluate the performance of each submodel on either the five cross-validation folds (Data S28C) or the test set (Figure 5B and Data S28D). The mean balanced accuracy across donor tissues is 0.86.
We further decomposed the submodels’ performance by considering different sets of SNVs (Figure 5C). Specifically, within a given submodel’s test set, we identified sets of true positives (TPs: SNVs classified as eQTLs in a given target tissue that are also GTEx eQTLs in the same tissue), false negatives (FNs: SNVs not classified as eQTLs but that are GTEx eQTLs in the target tissue), false positives (FPs: SNVs classified as eQTLs but that are not GTEx eQTLs), and true negatives (TNs: SNVs not classified as eQTLs that are not GTEx eQTLs) SNVs. The violin plots in Figure 5C show distributions of GTEx nominal p-values in the corresponding target tissue for these four sets of SNVs (each point of the distribution corresponds to the median p-value of an SNV set in one of the 756 submodels). Of note, the significance of the TP and FP sets is stronger compared to the FN and TN sets. This suggests that our model i) predicts the strongest among all GTEx eQTLs in a target tissue and ii) could help prioritize some of the SNVs with marginally significant p-values discarded by GTEx. The number of “additional likely eQTLs” (i.e., the SNVs classified as eQTLs by our model but that are not present in the GTEx catalog) can be found in Data S28E. We identify an average of ~160K additional “likely” eQTLs per tissue. This list of eQTLs can be found in the following ancillary file.
File: perTissue.likely.eQTLs.tsv: Additional “likely” eQTLs in each target tissue predicted across all transferQTL submodels.
For 4 of the 28 tissues, we focused on additional eQTL catalogs other than GTEx, available from66: i) pancreatic islets eQTLs (van_de_Bunt_2015 dataset) matched to pancreas (PNCREAS); ii) muscle eQTLs (FUSION dataset), matched to skeletal muscle (GASMED); and iii) skin eQTLs (TwinsUK dataset), matched to both suprapubic (SNINNS) and lower-leg (SKINS) skin. Thus, for these four tissues we evaluated the proportion of eQTLs identified by these studies (SNVs with p-value < 10−5) that were also classified as “eQTLs” in the test set of the relevant target tissue (pancreas, muscle, or skin) by our submodels (see Figure 5D). The eQTL catalogs used in this analysis were downloaded from http://ftp.ebi.ac.uk/pub/databases/spot/eQTL/sumstats/ (files “.all.tsv.gz”).
Currently, large-cohort eQTL studies are restricted to tissues such as blood that can be easily obtained from donors (e.g., over 30,000 donors in67), while most other tissues are difficult to profile in a large number of individuals. For this reason, and to showcase the utility of our predictions, we have directly applied our model to a set of >1.5 M blood eQTLs from67. In this way, we could predict which of these blood eQTLs are active in every EN-TEx tissue (Figure 5E and Data S28F–G). We downloaded the eQTL catalog from https://molgenis26.gcc.rug.nl/downloads/eqtlgen/cis-eqtl/ (2019–12-11-cis-eQTLsFDR0.05-ProbeLevel-CohortInfoRemoved-BonferroniAdded.txt.gz). Specifically, we selected 1,547,430 blood eQTLs found to be associated with only one eGene in the original study and lifted them over to the assembly GRCh38 using LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). The metric “GTEx v8 eQTL-eGene regression slope”, which was one of the predictive features employed in the training step (Data S28A), was not available in this second eQTL catalog; thus, for these predictions we instead used the metric log2(Z-score). Since blood tissue is not included in the EN-TEx collection and given that we do not currently have any submodel using blood as donor tissue, we applied each of the 756 submodels trained on GTEx data to this blood eQTL set. As an example, when using artery aorta as donor tissue, we transferred up to 60% of the blood eQTLs to some of the EN-TEx tissues, such as thyroid and tibial artery (Figure 5E and Data S28F–G). These results were computed after excluding those eQTLs contained in the original training sets. The number of additional “candidate” eQTLs transferred from this external blood eQTL catalog to each GTEx tissue is shown in Data S28G. On average, we identified ~500K candidate eQTLs per tissue. Results are available in the following ancillary file.
File: predictions.blood.eQTLs.tar.gz: Predictions of transferQTL submodels using the blood eQTLs from Vosa et al., Nat Genet 2021.
Please note that these “candidate” eQTLs represent a more speculative set of novel eQTLs compared to the average-per-tissue ~160K “likely” eQTLs mentioned above. The main difference is that the 160K “likely” eQTLs are obtained by leveraging donor-tissue eQTL-eGene pairs identified in GTEx tissues, while the ~500K “candidate” eQTLs are obtained by leveraging donor-tissue eQTL-eGene pairs identified in the large-cohort eQTL blood study, many of which are not reported by the GTEx catalog (see67).
Model Interpretation
To facilitate the interpretation of the model, we computed, across the test set of each submodel, the correlation between the level of a particular feature at a donor-tissue eQTL and the probability of classifying the donor-tissue eQTL as an eQTL in the target tissue. In Figure 5F we show, for the first 15 features in Data S28A, the strongest correlation coefficient (i.e., the coefficient with the largest absolute value) obtained across all 756 submodels. This analysis highlights that most chromatin features, with the exception of H3K27me3, are positively correlated with predicting eQTLs in a given tissue, while other features are negatively correlated (e.g., tissue specificity of the eGene and distance from the eGene’s TSS).
Data S29A shows a comprehensive representation of these correlation patterns across all predictive features and submodels. In this case, we discarded 4 of the 39 features (“POLR2A”, “POLR2Aphospho5”, “EP300”, and “POLR2A_k”) since they were not used in a large proportion of the submodels (because ChIP-seq assays for these TFs were performed on a limited number of tissues; for more details on these features see Data S28A). We thus focused on 432 donor-target submodels that did not have missing data for the remaining 35 features. The heatmap in Data S29A shows, for each of these 432 submodels (rows), Pearson’s correlation coefficients between the level of predictive features (columns) at donor-tissue eQTLs in the target tissue and the probability of donor-tissue eQTLs being classified as eQTLs also in the target tissue (clustering method: “Ward.D2”, clustering distance: “manhattan”). The vast majority of the chromatin features show stronger correlations in a specific set of submodels, as highlighted by hierarchical clustering (cluster at the bottom). We found that these submodels use donor tissues with larger GTEx sample sizes (Data S29A, right side). Thus, chromatin features have a stronger impact when transferring eQTLs from donor tissues with larger sample sizes, which tend to detect more eQTLs albeit with lower effects73,159. By contrast, certain features appear to be systematically either negatively (“tissue_specificity”, “tss_distance”, “H3K27me3_k”) or positively (“sum”, “is_proximal”, “H3K36me3”, “H3K36me3_p”) correlated with the SNV’s probability of being an eQTL, independent of the donor tissue. These results suggest that donor-tissue eQTLs with a higher number of chromatin peaks and/or marked by H3K36me3 in the target tissue are more likely to be eQTLs in the target tissue as well. Conversely, donor-tissue eQTLs associated with tissue-specific genes, or that are located far from the eGene’s TSS, are less likely to be transferable from one tissue to another.
Because of this, and with the goal of simplifying the interpretation of these models, we evaluated whether two simple rules could help transfer eQTLs from one tissue to another. In Figure 5G we show that, on one hand, donor-tissue eQTLs either characterized by strong chromatin activity (feature “sum” ≥ 3) in the target tissue, or whose eGene is constitutively expressed across EN-TEx samples (feature “tissue_specificity” < 0.8), tend to be eQTLs also in the target tissue (rule #1: 67% eQTLs, 33% not eQTLs). On the other hand, donor-tissue eQTLs that have low chromatin activity (feature “sum” = 0) in the target tissue and whose eGene shows tissue-specific expression (feature “tissue_specificity” > 5) are less likely to be eQTLs in the target tissue (rule #2: 23% eQTLs, 77% not eQTLs). Figure 5G refers to the specific case employing testis as the donor tissue and thyroid as the target tissue. Data S29B shows that these findings are generalizable across all the 756 donor-target tissue pairs.
Evaluating the Impact of Tissue-specificity on Predicted eQTLs
Many of our predicted eQTLs are fairly ubiquitous, but we also report a considerable fraction of predicted eQTLs that are active only in a small fraction of GTEx tissues (see Data S29C). To demonstrate that our random-forest model (transerQTL) is not simply predicting ubiquitous eQTLs, we built a simple strawman model that transfers eQTLs to a given tissue based on the tissue specificity of the eQTLs. We transferred eQTLs based on different thresholds of tissue specificity, from including very tissue-specific eQTLs (active in at least 10% of GTEx tissues) to transferring only very ubiquitous eQTLs (active in at least 90% of GTEx tissues). In Figure S6D and Data S29D we show that the performance of this simplified model, combined with different thresholds of tissue specificity for eQTL activity, is worse compared to the performance of our transferQTL model.
Sensitive Motifs:
The Relationship Between AS SNPs and TF Motifs (related to “Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Highlighting “Sensitive” TF Motifs” in the main text, Figures 6AB and S7A)
We collected 660 human TF motifs from the Cis-BP database68. Specifically, we required the motifs to be from protein-binding microarray and SELEX-based experiments. Position weight matrices (PWMs) from multiple motifs are combined into a single PWM file for each TF. We used FIMO170 with p<10-e4 to scan the motif occurrence in the human genome. We then intersected the AS SNV file with each motif occurrence file with bedtools and retrieved the contingency table of counts of SNPs depending on whether a SNV was AS and whether a SNV was in a motif. An OR was used as the measurement of AS enrichment and Fisher’s exact test was used for statistical significance (p<0.05). The motifs were then ranked based on the OR. For H3K4me3 ChIP-seq-based or any other assay-based ranking, only the SNVs that were accessible in that assay were used to intersect with the motifs. In addition, we annotated all EN-TEx SNVs with whether they overlap with TF motifs and whether they overlap with cCRE regions. The full result of the motif ranking and SNV annotation can be found in the following files.
File: motif_ranking.tsv: List of motifs with their odds ratio, p-values and rank in overall or in a specific assay.
File: SNPs_motif_cCRE.txt.gz: Relationship among SNVs, motifs, and cis-regulatory elements.
In the SNV annotation file, the first three columns show the coordinates of the SNV. The fourth column is a list of names of TFs whose motifs overlap with this SNV. The sixth column is the ID of the overlapping cCRE regions.
For the conservation score of the motifs, we downloaded the genome-wide phyloP score from the UCSC Genome Browser. For each occurrence of the motif, the conservation score of the region was determined by the mean of each base (from UCSC Kent_tool bigWigAverageOverBed). The conservation of the TF was the mean of the scores from all occurrences of its motifs. The entropy of a motif was calculated by sum(−plog(p)) where p is the relative frequency of each base in each position. The fraction of CG of a motif was calculated as the number of positions where C or G was the most frequent base, divided by the length of the motif. Spearman’s correlation was used for all pairs of the rank and each motif property (Data S30F–G).
To test whether the GC content and motif entropy biased our result, we tried to remove their effect under a linear model. We used the GC content and motif entropy as variables to predict the AS enrichment score, and then re-ranked the TF motifs using the residual of the model only (Data S30H). The formula is:
where OR is the odds ratio and ε is the residual. We solved the linear regression by minimizing the ordinary least squares to get the estimated β1 and β2 as and . The residual with GC content and motif entropy effect corrected is calculated as:
We then re-ranked the TF motifs by εcorrected. We found that the rankings of original top 100 motifs were largely preserved (Data S30H). For example, the Pearson correlation between the original rank and the new residual-corrected rank was 0.64 (p<1.1e-12) for the top 100 but dropped to 0.03 (p<0.416) for the rest of the TF motifs. This result suggests that the top 100 motifs are not strongly affected by the GC content and further justifies using the top 100 for the downstream analysis.
Each motif’s family information was obtained from Cis-BP as well. We noticed that the top-ranked motifs were more likely to be in the C2H2 zinc finger family. A C2H2-ZF domain typically contains 3–4 base-contacting residues, and zinc finger proteins usually contain multiple tandem C2H2-ZF domains. The individual DNA motifs of these tandem domains often overlap with each other and assemble into the full-length motifs we observed in SELEX (e.g., 4-mer and another 4-mer overlapping by one base results in a 7-mer)69. Thus, mutations in the overlapping base in the middle of the motif might be more likely to affect the binding affinity of the TF. Consistent with this reasoning, we observed that AS SNVs occurred more frequently in the “conjunction” base while non-AS SNVs occurred relatively randomly across all positions of the motif (see FOXO3 in Figure 6).
AS Promoter:
Prediction of Promoter AS Activity with a Random Forest Model (related to “Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Highlighting “Sensitive” TF Motifs” in the main text, Figures 6C and S7B)
We trained a random forest model that could predict the ASB state of the gene promoters in an assay and in an individual tissue-specific manner. We call this the “reverse” model as it goes from gene to promoter. The models achieved good performance on both internal EN-TEx and Roadmap STL002/3 data (Data S31A). After testing different combinations of features, models were built using four features (Data S31B) according to Gini impurity-based importance scores. The first feature is the number of the top 100 AS sensitive TF motifs that intersect the SNV. The second feature is those within 100 bp of the SNV that do not intersect. The third feature is the number of the 660 TF motifs distal to the SNV (i.e., >100 bp away). AS bound promoters have significantly more motifs than non-AS bound promoters (also see Data S31D). The fourth feature is the AS imbalance ratio between transcripts from haplotype 1 and haplotype 2. In addition to the feature importance score from the random forest model, we investigated the association of each feature with ASB promoters, indicated by an R2 score (Data S31C). Other features (including gene expression level, eQTL, all 660 non-ranked TF features in the promoter) were tested but proven to not be informative (Data S31D). We applied our model on a large scale to the entire GTEx cohort (>800 people) to predict AS promoters from the available genotypes and ASE data. The GTEx individuals, gene names, assay types, predictions of the associated promoter, and additional results of the model are included in the following file.
File: ASB-predictions-on-GTEx-cohort.tsv: Results of allele-specific binding prediction model on GTEx.
We also constructed a “forward” model to predict ASE from ASB. Specifically, to interrelate AS activities of genes and promoters (Data S31E), a random forest model was trained using assay-based annotations of the promoters. The assay-based ASB of the promoters was informative for the prediction of ASE genes (Data S31F), but we did not have enough validation data for a full evaluation.
Transformer Model:
Prediction of AS Effect with a BERT Model (related to “Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Highlighting “Sensitive” TF Motifs” in the main text, Figures 7 and S7CDE)
Strawman Random Forrest Model for AS Prediction of CTCF Binding Regions
A random forest model was trained with the following features to predict the tissue-specific AS effect labels for CTCF:
the prediction score of the sequence-based transformer model
the tissue-specific epigenomic signals (DNase and histone marks, but excluding CTCF) averaged across the input sequence region
We considered the transformer model score as an indicator of sequential patterns that are genetically related to AS effects, which was then modified with tissue-specific epigenomic features to predict the tissue-specific AS effect.
Training and testing data were split by a 3:1 ratio for each tissue of each individual. For each of the four individuals, we trained a separate model using all the tissue data of the individual. The max depth for the random forest was chosen using a grid search from 2 to 9.
BERT Model
BERT is a natural language model based on the Transformer neural network architecture. This model has been widely applied to natural language processing due to its ability to incorporate long-range contextual information171. Thus, it can also be applied to extract meaningful sequential patterns from genomic sequences, such as to predict AS effects of SNPs.
We extracted the 128 bp sequence upstream and downstream of the SNP in question as the input. The sequences were labeled as positive or negative based on their AS effects. For balancing considerations, the negative set was randomly downsampled to the same size as the positive set. The dataset was then split into training, cross-validation, and testing sets at a 8:1:1 ratio.
We initialized the BERT model with the weights of the pre-trained DNABERT model70. A single-layer classifier was added on top of the output of DNABERT and the model was fine-tuned on the AS datasets. For fine tuning, we selected from a range of hyperparameters (learning rate=1e-5, 5e-5; training epoch = 5, 10, 20). As the pre-trained DNABERT model has different versions with k-mer sizes of 3–6, we report the model with the highest performance.
The model was first trained with only SNPs from donor individual 3 to predict the “pooled” AS SNVs (i.e., SNVs that were active in at least one tissue). For many of the prediction tasks, the model achieved a performance of area under the receiver operating characteristic (AUROC) > 0.7 on the validation set, significantly higher than logistic regression and random forest on sequence embeddings (Data S32; see below for more details). We then tested the model performance on validation sets composed of SNPs exclusive to the other three donors. The validation sets for these three individuals have been randomly downsampled to the same size as the validation set for individual 3. The sampling was repeated ten times and average results are reported. As expected, the performance was lower compared to individual 3. Specifically, the model showed exceptional performance on the prediction for CTCF (AUROC = 0.7936) and generalized well to the other three donors (average AUROC = 0.6876). The model for H3K27ac AS SNVs also showed high validation performance on the test set (AUROC = 0.8001), other individuals (average AUROC = 0.7286), and an external validation set from Roadmap individuals (average AUROC = 0.7426).
For model interpretation, we used the method implemented by70, where the attention scores of the last layer for the first token are averaged over all 12 attention heads, and then regularized by k-mer coverage. As a comparison, we used the dna2vec model released by172 to transform k-mers to continuous-valued vectors, preserving their contextual preference. Using the same training, test, and validation data as above, we represented each input sequence as an average over the embedding of all its k-mers. We then trained a logistic regression classifier based on the average embedding vector. We performed embedding with k-mer sizes of 3–8 and reported the one with the highest performance.
We also implemented a much simpler model based on motif information only (Figure 7). We overlapped the hetSNVs from the same training set as above with identified CTCF motifs from the Cis-BP database. The following features were used to build a random forest classifier:
| Feature | Feature frequency |
|---|---|
| A: SNP overlaps with a CTCF motif | 287/28,891 (positive) 45/28,891 (negative) |
| B: SNP overlaps with conserved positions in a CTCF motif | 161/28,891 (positive) 27/28,891 (negative) |
| C: Presence of a CTCF motif within the 256 bp window | 1489/28,891 (positive) 689/28,891 (negative) |
| D: # of TF motifs within the 256 bp window | Average 56.8 (positive) Average 62.2 (negative) |
A logistic regression model using only features A and B has almost no predictive performance (AUROC=0.504). By adding features C and D, which include some contextual information, the performance increases to AUROC=0.5618, which is still much lower than other models in comparison. This is expected because the frequency of CTCF motifs is quite low and only accounts for a very small portion of the dataset.
Portal: A Central Location for Accessing EN-TEx Data, Analyses & Visualization Tools
The EN-TEx Portal Website
We have a dedicated website (portal) for the EN-TEx resource: entex.encodeproject.org. The portal is organized into three organized sections: (i) data files, (ii) interactive visualization tools, and (iii) source code, as described below.
(i) DATA FILES. The raw and processed EN-TEx data, including the personal genomes, are accessible via a dedicated data-slice page built into the ENCODE data center. In the data-splice page, the EN-TEx assays and data are displayed with a graphic interface for users. The website provides a search function where users can look for the data of particular assays/tissues in which they are interested. In addition, the EN-TEx portal hosts the ancillary analysis files for EN-TEx (e.g., the AS catalog and the cCRE decorations). All data contained in the EN-TEx resource are fully open-consented and accessible without registration as of the date of publication. Accession numbers are listed in the key resources table or the STAR Methods.
(ii) INTERACTIVE VISUALIZATION. The EN-TEx portal provides multiple tools for users to visualize the EN-TEx data in a genomic context. In particular, the genome annotation by EN-TEx data can be visualized using the ENCODE SCREEN Viewer. We also provide Chromosome Painter and the Explorer Tool to visualize the EN-TEx data in a large-scale and high-dimensional fashion. See section “Visualization of the EN-TEx Data” for more information.
(iii) SOURCE CODE. All original code has been deposited on Github and is publicly available as of the publication date. DOIs are listed in the key resources table. Specifically, the EN-TEx portal provides GitHub links to the source codes of the Chromosome Painter, the Explorer Tool, the AlleleSeq2 pipeline, the transferQTL model, and the transformer model for predicting AS activity from sequence (“Application #3”).
Explorer Tool
The EN-TEx Explorer Tool, which can be run in R, installed as an offline executable, or hosted on a website through integration with Amazon Web Services, allows for the interactive exploration of low-dimensional visualizations created by an in-house data analysis pipeline (Data S33A–E). This pipeline performs dimensionality reduction on cCRE signals, genomic data, and proteomic data. Methods include PCA, variational autoencoder, Umap173, potential of heat diffusion for affinity-based transition embedding174, set intersection plots generated by user-specified thresholds, and t-distributed stochastic neighbor embedding175. The pipeline then generates the tool programmatically in R Shiny in one of the three forms above.
The visualizations generally cluster samples from common tissues together. Through extensive precomputation, the tool allows users to interactively adjust analysis parameters, including scaling, normalization, feature subsetting, method-specific hyperparameters, the type of visualization used (ggplot2, plotly 2D, plotly 3D, boxplot, heatmap, UpSetR, Venn diagram), and the appearance of the resulting figures. Users are able to save figures as images, download analysis results as Excel spreadsheets, or bookmark their sessions as short URLs that can be easily shared (Data S33A–E). To install the tool, please consult the Github README. Instructions and documentation regarding tool usage can be found by pressing the “Instructions” button on the tool. The following input files for the explorer tool are available.
File: ENTEx.Explorer.cCRE.Combined.zip: Candidate cis-regulatory elements used in EN-TEx.
File: ENTEx.Explorer.Expression.Combined.zip: Expressed genes analyzed in EN-TEx.
File: ENTEx.Explorer.Expression.Combined.zip: Expressed genes analyzed in EN-TEx. File: ENTEx.Proteomics.cCRE.Combined.zip: Results of mass spectrometry.
Chromosome-Level Data Visualization Tool
Because the EN-TEx data span a wide range of the human genome, it may be useful to visualize the distribution over each chromosome. Accordingly, we present the EN-TEx Chromosome-Level Data Visualization Tool, which generates heatmaps for datasets for all assays, individuals, and tissues present in the EN-TEx data catalog. The data, which were initially in BED format, were preprocessed with in-house Bash and Python scripts and converted to GRCh38 coordinates using LiftOver176 prior to the generation of the plots using the R package chromoMap177. The EN-TEx Chromosome-Level Data Visualization Tool was also used to generate the plots in Figure 2C of the main text.
The EN-TEx Chromosome-Level Data Visualization Tool can be accessed at ENTEx.gersteinlab.org. Users can specify any combination of parameters (individual, assay, ploidy, and color) for a track and subsequently generate interactive plots containing one to four tracks each by pressing the “Submit” button (Data S33F–G). By default, the tool generates heatmaps for the data of each chromosome at a fixed resolution of 2.5 Mb. The user can get information about the data displayed in a specific bin by hovering over the bin with a mouse cursor.
The “Advanced” tab contains tools for custom chromosome and region selections. To view the data in only one chromosome, one can select the chromosome of interest in the “Chromosomes” dropdown menu. To view a subregion of the chromosome, the user can input the region in the format initial_position:final_position in the “Region” text box (e.g., if the user wishes to visualize data between 1 Mb and 2 Mb, the user would input 1000000:2000000). The tool automatically sets the resolution of the data for subregions of the chromosome to the length of the inputted interval divided by a factor of one hundred (e.g., for the 1000000:2000000 interval, the resolution would be 10 Kb). Users can also visualize the data as heatmaps accompanied by either histograms or scatterplots by selecting the desired option in the “Plot Type” dropdown. A series of plots generated with this tool is shown in Data S33H.
Additional Data Exploration with SCREEN
The SCREEN website (https://screen.encodeproject.org/) is a center for the ENCODE cCRE registry and annotation. This website is routinely used by researchers all over the world. The annotations of cCREs using our EN-TEx data are also available at the SCREEN website (Data S33I). The EN-TEx data provide many unique annotations. For example, different from the annotation from other datasets, the EN-TEx data indicate the repressed states of cCREs. In addition, the EN-TEx data specify whether each cCRE is AS in terms of functional genomic signals. Data S33I shows a step-by-step guide from the main webpage of SCREEN to an cCRE with repressed states in multiple human tissues. In line with the repressed states, this cCRE has no CTCF binding and is not AS.
Buffering Hypothesis:
Providing Evidence Connecting AS Elements and Housekeeping Genes (related to the “Discussion” in the main text)
Genetic variants in cCREs can change functional signal and gene expression. For these changes to occur, the variants must escape from buffering effects28. Such effects are strong in important genomic regions. We used allele specificity as a proxy for escaping buffering. Based on our allelic decoration, we evaluated the allele specificity of housekeeping genes expressed in EN-TEx tissues, as shown in Data S22J. For each tissue, expressed protein-coding genes were split into housekeeping genes and non-housekeeping genes according to the Housekeeping and Reference Transcript Atlas (http://www.housekeeping.unicamp.br)58. A two-sided Fisher’s exact test was performed to measure the enrichment of AS housekeeping genes. We found that, compared with non-housekeeping genes, the expression of housekeeping genes show less allele specificity, supporting the buffering hypothesis. We further examined the allele specificity of proximal active (pAct) cCREs in a ± 10 Kb window centered on the TSS (defined by the gene starting site) of each housekeeping and non-housekeeping gene. The cCREs flanking housekeeping genes were significantly (Data S22J, paired-tissue two-sided t-test, p-value < 2.2e-16) longer than the cCREs flanking non-housekeeping genes. To control for this factor, we split genes into 20 bins based on the total length of the flanking cCREs. Within each bin, cCRE length remained similar (paired-tissue two-sided t-test, p-value > 0.05) between housekeeping and non-housekeeping genes. The bins with less than 30 housekeeping or non-housekeeping genes were removed from further analysis. The pAct cCREs flanking housekeeping genes were less likely AS than the ones flanking non-housekeeping genes (two-sided t-test).
The buffering effect is likely due to redundant TFs. To test this, we counted the number of TF motifs that intersect with each CTCF+ and CTCF− cCRE in each tissue. For this calculation, we used the motifs of 206 TFs (CTCF excluded) from Cis-BP68. The total count of all TF motifs was compared between CTCF+ cCREs and CTCF− cCREs using a two-sided t-test. As shown in Data S30K, for both distal and proximal cCREs, CTCF+ cCREs have significantly (p-value < 0.05) more TF motifs than CTCF− cCREs. In addition, we tested whether the genetic variants in large motif clusters tend to be associated with ASB. To this end, we identify the locations of the motifs of 660 TFs in the human genome. For each motif, we counted the number of all motifs within 500 bp of the motif. A larger number suggests that the motif likely has many functionally redundant motifs. According to the proxy of redundancy, we divided all the motifs evenly into two groups: likely redundant or not. The genetic variants were considered ASB if significantly imbalanced reads were observed in any of the tissues of the four individuals with any assays. As a result, we found that the genetic variants in the motifs that are likely redundant tend to not be AS, consistent with the buffering effect.
Quantification and Statistical Analysis
Quantitative and statistical methods are described above within the context of individual analyses in the Method Details section.
Additional Resources
Ancillary files and guidance on raw data of this study can be found on the EN-TEx portal: http://entex.gersteinlab.org/index.html. All files are described in detail in their corresponding sections of STAR Methods. When mentioned, these files are referred to as “File: file_name: short_description.”
Highlights:
EN-TEx includes 1635 datasets mapped to 4 personal genomes, ~30 tissues × ~15 assays
Comprehensive catalog of allele-specific activity, decorating regulatory elements
Model to transfer known eQTLs to difficult-to-profile tissues (e.g., skin→heart)
Transformer model for predicting allelic activity based on local sequence context
Supplementary Material
Figure S1 Additional Details on Uniform Multi-tissue Data Collection and Diploid Mapping, Related to Figure 1, STAR Methods “Data Stack”, “Personal Genome”, “Reference Comparison” and “SVs” Sections.
(A) Data matrix for the EN-TEx resource. Each voxel in the cube stack corresponds to a functional genomic assay for a particular tissue from one of the four individuals. A dark gray voxel indicates that the functional genomic data have been generated. The total experimental counts per assay across the tissues and individuals are shown above the cube stack. The total experimental counts per tissue type are shown on the left. The color and acronym scheme of the tissues is adopted from GTEx (Data S2). We also note that the small RNA-seq column represents the consolidation of both small RNA-seq and targeted micro-RNA-seq assays; the data for both these assays are available from the portal. Due to the hearts from both male donors being selected for organ transplant, we do not have heart derived tissue for both genders. Also the aortic tissue is sublocalized into samples derived from ascending aorta and thoracic aorta. Note further that in GTEx these aortic tissues are combined and considered to be a single tissue.
(B) Constructing personal genomes using the CrossStitch method. This diagram shows an overview of the CrossStitch method with the specific software and data types used. (Details in STAR Methods “Personal Genome” Section.)
(C) Mapping reads to personal and reference genomes. Due to the genetic variants, some reads can be mapped only to the personal genome but not to the reference genome (and to a lesser extent just to the reference genome). Such reads (yellow) are referred to as “personal genome unique,” whereas the reads that can be mapped to both genomes (gray) are referred to as “common.” Compared with the reference genome, using the personal genome results in an average of ~2.5% more mapped reads for the various functional genomic experiments. The percentages range from ~1% for RNA-seq to ~4% for Hi-C data. Note that the numbers for these differences only apply to the high-stringency mapping. (Details in STAR Methods “Reference Comparison” Section.)
(D) Classification and frequency of SVs. The fractions of SVs for each individual are shown and broken down ito insertions (INSs), deletions (DELs), and inversions (INVs). INSs and DELs are further classified based on whether they are related to TEs. (Details in Data S15 showing a breakdown of the SV composition.)
Figure S2 Additional Details on Improvements in Genome Analysis from Uniform Multi-tissue Data Collection and Diploid Genome Mapping, Related to STAR Methods “Reference Comparison” and “Variation Analysis” Sections.
(A) Comparing the quantification of gene expression using personal diploid genomes and the reference haploid genome. The left panel shows the gene expression levels for individual 2 calculated using the reads mapped to the reference genome (x-axis) and to the personal diploid genomes (y-axis). The expression values of the genes are log-transformed, i.e., log10(TPM + 0.001). Differentially expressed genes are color-coded (downregulated: red; upregulated: blue). Genes that are not differentially expressed are shown in gray. The panel on the right is a volcano plot reporting, for every gene, the log2(fold-change) and the −log10(p-value) obtained from the differential-gene expression analysis with DESeq2. The same color schema as in the left panel is applied. Some outlier genes were removed from the volcano plot. The full version of these plots, including all genes and all individuals, is available in Data S5D–E.
(B) Comparing the quantification of regulatory element activity using personal diploid genomes and the reference haploid genome. The left panel shows the H3K27ac signal at cCREs for individual 2 calculated using the reads mapped to the reference genome (x-axis) and to the personal diploid genomes (y-axis). The H3K27ac signals of the cCREs are log-transformed. Differentially marked cCREs are color-coded (downregulated: red; upregulated: blue). cCREs that are not differentially marked are shown in gray. The panel on the right is a volcano plot reporting, for every cCRE, the log2(fold-change) and the −log10(p-value) obtained from the differential analysis performed with DESeq2. The same color schema as in the left panel is applied. Some outlier cCREs were removed from the volcano plot. The full version of these plots, including all cCREs and all individuals, is available in Data S5K–L.
(C) Consistently analyzing functional genomic data across individuals, tissues, and assays. Joint analysis of variation (JIVE, Hellton and Thoresen, 2016) was used to project all of the functional genomic signals from different samples onto two-dimensional planes (one for each assay, e.g., ChIP-seq, ATAC-seq, DNase-seq). For each assay type (e.g., H3K27ac), the samples are represented as dots on the plane. Two dots that are close to each other have similar levels of functional genomic signals. Combining all of the types of assays, we projected the samples onto a single two-dimensional plane. The tissues are colored according to the GTEx convention from Figure 1A and Data S2I. For each tissue, the number of colored dots indicates the number of individuals, up to four. In addition, a linear-regression-based approach was used to measure the difference between two samples in functional genomic signals, (Details in STAR Methods “Variation Analysis” Section.) The variance explained is used to indicate the similarity between two samples. Note that the value of variance explained is equal to the squared correlation coefficient. The difference is measured by one minus the explained variance of the regression. Taking H3K27ac as an example, on average, only 14% of the variation in the functional signals between the spleens of two individuals is unexplained, indicating that the spleen samples have very similar H3K27ac signal patterns. The dissimilarity is also small (15%) between the transverse colons of two individuals. In contrast, the average dissimilarity between these two types of tissues of the same individuals increases to 35%. For H3K27me3, the dissimilarity between different tissues is also high (42%). The dissimilarities between different types of functional signals are even higher. In particular, the dissimilarity between H3K27ac and H3K4me3 in spleen is 51%. (Details in STAR Methods “Variance Analysis” Section and Data S6.)
(D) An example of a more accurate inter-tissue comparison using matched samples. For histone modification signals at cCREs, the spleen and esophagus of the same individual are more similar (i.e., median 0.92 correlation, 84% variance explained [or 16% dissimilarity]) than those of different individuals (0.89 or 79%). This difference indicates the genetic and environmental difference between individuals and its influence on inter-tissue comparisons.
Figure S3 Additional Details on the Construction of the AS Catalog, Related to Figure 1, STAR Methods “AS Calling”, “Aggregation” and “AS Catalog” Sections.
(A) Schematic of calling AS SNVs and AS elements. Examples of the number of reads are shown for each SNV and element. For a functional genomics experiment, if the numbers of reads mapped to the two haplotypes at a heterozygous SNV position are significantly different, the SNV is referred to as an AS SNV. For a given genomic element (e.g., a cCRE) with multiple heterozygous SNVs, we pool the reads of all the SNVs to determine its AS state. Genomic elements with a single AS SNV are identified as AS elements. (Details in STAR Methods “AS Calling” Section.)
(B) Distribution of the number of AS events in different assays. The distribution of H3K27ac AS events in spleen from individual 1 is highlighted as an example here and in panel (D) and further distributions are shown in Data S11A.
(C) Details of union and pooling approaches for read aggregation. We used two approaches to aggregate the information across samples to increase the detection power. In the “union” approach, any SNV observed as AS in any sample is included in our AS catalog - i.e., we call individually on samples and then take the union of the calls. In contrast, in the “pooling” approach, because a given functional genomic assay, e.g., RNA-seq, is usually conducted on many samples, for each SNV, we pool the mapped reads from all the samples together to determine its AS state (see STAR Methods “Aggregation” Section). For instance, to do multitissue pooling, we pool the reads from all tissues and then jointly call AS SNVs on the pooled reads. This approach increases the statistical power of the AS test and thus may be able to identify an SNV as AS, even though the SNV is not AS in each tissue.
(D) Summary of AS catalog. The colored circle on the right shows the aggregation method along that axis. Take the third row as an example. The upper one indicates that the operation is the union across tissues, averaged across assays and individuals, while the bottom one pools reads across tissues, averaged across assays and individuals. Data obtained by merging the tissue-specific AS call sets resulted in ~5.5K unique AS hetSNVs (for either AS binding or expression) and ~1K AS genomic elements (not shown in the figure) for each individual per assay. Pooling the reads from each assay across all tissues dramatically increased the detection power by ~5X fold, making it possible to identify ~27K AS hetSNVs per assay for each individual. Finally, merging across all assays provided a catalog of all loci where AS activity could be assessed in any of the tissues of the four individuals.
(E) Numbers of AS SNVs across assays. The Venn diagram of SNVs shows AS activity in selected assays using the “pooling” approach. (Details in Data S8B.)
(F) Numbers of AS elements across samples. The distribution of the number of genomic elements (genes and cCREs) with AS activity across EN-TEx donors, tissues, and assays is shown. (Details in Data S11A.) Donors and tissues are indicated using the conventions from Figure 1.
Figure S4 Examples of Coordinated AS Activity Involving SNVs and SVs, Related to Figure 2 and STAR Methods “AS Examples” Section.
(A) Detecting AS events at a known imprinted locus: H19/IGF2. Functional genomic signals are measured in the gastrocnemius medialis of individual 2. In agreement with the known imprinting mechanism (top), we detected AS expression of H19 on haplotype 2 (known to be maternally expressed) and AS expression of IGF2 on haplotype 1 (known to be paternally expressed). Consistent with the expression data, we found an AS Hi-C interaction between an upstream cCRE and H19 on haplotype 2 (bold blue arc), and an AS interaction between the same cCRE and IGF2 on haplotype 1 (bold red arc). Other neighboring AS Hi-C interactions (which have fewer read counts or contact irrelevant genes) are also depicted with shaded arcs.
(B) The effect of a 5.2-kb homozygous deletion. This SV removed an H3K9me3 peak in individual 2, potentially resulting in an increase in PCCB expression in the spleen. The sashimi plots, below the signal graphs, show examples of novel splicing isoforms identified by long-read RNA-seq in the adrenal gland of individual 2 and in the heart left ventricle of individual 3 (without the SV). Data S17K shows more details on the splicing isoforms. The differences in splicing between individuals 2 and 3, although supported by only one long read, potentially reflect the SV-disrupting regions important to splicing, as suggested by the known GTEx sQTL sites nearby178. Alternatively, the differences in splicing between the two samples may be due to tissue specificity. In addition, Data S17L provides evidence of novel splicing variants that are potentially associated with an SV in the gene TRDN-AS1.
(C) The effect of a 2.3-kb homozygous deletion. This SV removed an H3K27ac peak downstream of lncRNA RP11–362F19.1 in individual 3, which has lower expression in the spleen sample. In individual 2 without the SV, there are CTCF peaks that point to a potential interaction between the H3K27ac peaks downstream and upstream of the lncRNA. Together, these observations suggest that the loss of the H3K27ac peak due to the SV contributes to the reduction in lncRNA gene expression. (Details in Data S17E–F.)
Figure S5 Additional Details on Applications 1 and 2: Relating Decorations and AS SNVs to GWAS and eQTL Loci, Related to Figures 3, 4, STAR Methods “Decoration Process”, “Tissue Specificity” and “Compatibility” Sections.
(A) Workflow of cCRE decoration. This figure shows the workflow of cCRE decoration and the number of different subgroups of cCREs in the spleen. Note that we define the following abbreviations for the various decorations. dAct: distal active; pAct: proximal active; dBiv: distal bivalent; pBiv: proximal bivalent; dRep: distal repressed; pRep: proximal repressed; CTCF+ and CTCF−: with and without CTCF binding, respectively; and AS and nonAS: with and without allelic signature, respectively. Spleen is used as an example to illustrate the tissue-specific decoration of these cCREs using EN-TEx data. In this tissue, 290k of the cCREs have functional genomic signals and can be categorized as active (117k), repressed (154k), or bivalent (19k). Then, for each category, the cCREs can be consecutively classified according to their genomic locations (proximal or distal), CTCF binding, and allele specificity. (This classification can occur in any order.) As a result, the spleen has 2,866 AS cCREs with various genomic activities and locations. More details on the workflow are included in the STAR Methods “Decoration Process” Section. The numbers of active and repressed cCREs are comparable in each tissue (i.e., on average ~202k active and ~166k repressed cCREs). Only a small subset of the active cCREs exhibit AS activity (~2.5% or 1,750 cCREs, averaged across tissues; see details in Data S19C for all available tissues).
(B) Tissue specificity of different subgroups of cCREs vs. genes and epigenomic peaks. We compared the tissue specificity of protein-coding genes, non-coding genes, different subgroups of decorated cCREs, and various epigenomic peaks. The uniqueness of the activity across tissue types is shown in different colors. Note that the decoration terms are defined in Figure S5A.
(C) Compatibility between AS expression and AS promoter state. As shown in the examples, a high active histone modification signal (e.g., H3K27ac) of a haplotype should correspond to high gene expression of the same haplotype. Note that for a repressive histone modification, this trend is the opposite. For AS genes with promoters accessible to H3K27ac, there is indeed a strong and positive correlation. The exact read counts of H3K27ac and gene expression, respectively, from haplotype 1 and 2 for LAMTOR1 and RPL4P4 are indicated in the examples. In addition, we used the slope (beta coefficient) of the leading eQTL of an AS gene (GTEx Consortium, 2020). The slope is positively correlated with the AS ratio (overall Pearson’s correlation coefficient of 0.6 with p = 0.01). Note that genetic variants with beta coefficients around 0 are unlikely to have sufficient statistical significance to be identified as eQTLs. This also holds for SNVs with read fractions of approximately 0.5. Therefore, there are no data points observed at the center of the plot.
(D) Overall compatibility between AS expression and promoter AS chromatin activity. The compatibility is measured by the fraction of genes for which the AS expression is compatible with promoter AS chromatin state or AS peptide expression (Data S26 and STAR Methods “Compatibility” Section). As a null model, we randomly paired genes with promoters (and peptides) to calculate z-scores. (Details in STAR Methods “Compatibility” Section.) Compatibility between AS expression and AS methylation (meCpG), H3K9me3, and H3K27me3 is weak, potentially because these marks of repressed chromatin also can be associated with genes poised for transcription or genes that are actively transcribed179,180. Note for the bottom entry, compatibility is calculated between ASE and AS peptide expression, which is independent of the other compatibility comparisons between AS promoter activity and ASE.
Figure S6 Additional Details on Application 2: Modeling eQTLs in Hard-to-obtain Tissues, Related to Figure 5 and STAR Methods “transferQTL Model” Section.
(A) Chromatin features can help prioritize causal eQTLs. Barplots showing the percentage of eQTLs overlapping a given feature. The fraction of fine-mapped eQTLs (likely causal, red) overlapping peaks of six histone marks (H3K27ac, H3K4me1, H3K4me3, H3K27me3, H3K9me3, H3K36me3) is higher compared with the total set of GTEx eQTLs (blue) reported in a given tissue. (Details in Data S27A.)
(B) Chromatin-marked loci associated with eQTL activity. We have identified 1,353,101 SNVs that show tissue-specific eQTL activity. These SNVs are GTEx eQTLs in ≥ 5 EN-TEx tissues and are not GTEx eQTLs in ≥ 5 other EN-TEx tissues. Thus, for every SNV we define two groups of tissues: i) tissues in which the SNV is an eQTL (eQTL+, orange) and ii) tissues in which the SNV is not an eQTL (eQTL−, cyan). We observed that SNVs are more likely to be marked by H3K36me3 in the tissues in which they are eQTLs, compared with the tissues in which they are not eQTLs (p-value < 2.2e-16, Wilcoxon paired test). (Details in Data S27B.)
(C) Schema of the predictive model using skin as the donor tissue and tibial artery as the target tissue. We first retrieve GTEx eQTLs associated with one single eGene in sun-exposed skin (n = 967,288). This set of eQTLs is split into training and test sets (70% and 30%, respectively). Next, we train a random forest model in order to predict whether these eQTLs are eQTLs also in the tibial artery. The model uses a number of predictive features described in Data S28A: i) eQTL-gene properties in the skin tissue (blue; features 1–3), ii) chromatin profiles at the eQTL loci in the tibial artery tissue (red; features 4–24), iii) chromatin profiles at the eQTL loci across all EN-TEx tissues (gray; features 25–36), and iv) genomic features of the eQTL loci (white; features 37–39). (Details in Data S28B–G.)
(D) transferQTL outperforms simplified models that transfer eQTLs based only on a tissue-specificity threshold. We built a simple model that transfers eQTLs to a given tissue based on their degree of tissue specificity. We compared the performance of this simple model by using different thresholds of tissue specificity. For instance, 10% corresponds to a model that transfers to a given tissue eQTLs that are active in at least 10% of the GTEx tissues. The performance is compared to our random-forest model trained on multi-omics EN-TEx data (transferQTL). (Details in Data S29C–D.)
(E) The predictive models have high performance (balanced accuracy) even when trained on a reduced number of histone modifications. This modified version of transferQTL utilizes only chromatin features related to the four histone marks that have the strongest predictive power (H3K36me3, H3K27ac, H3K4me1, H3K27me3; also see Figure 5F). The x-axis indicates the tissues used as the donors (GTEx coloring) and the y-axis indicates the average performance (balanced accuracy) across the target tissues that are available. The whiskers indicate variation across targets (standard deviations).
Figure S7 Additional Details on Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Related to Figures 6, 7, STAR Methods “Sensitive Motifs”, “AS Promoter” and “Transformer Model” Sections.
(A) Distribution of the motif rank across tissues. Similar to Figure 6A, we rank the motifs based on the enrichment of H3K4me3 AS SNVs, but using SNVs from each tissue individually. The y-axis shows the tissue-specific rank. The x-axis represents the motifs in the order of the pan-tissue analysis ranking, as in Figure 6A. Each vertical gray line represents the range between the 25% and 75% percentile of the motif rank across all tissues for that motif. The median value is shown in red.
(B) Contingency table for AS genes and AS promoters. The 2-by-2 contingency table shows AS genes vs. non-AS genes (in terms of AS gene expression) and AS promoters vs. non-AS promoters (in terms of H3K27ac activity). The overall odds ratio of the table indicates the association between AS activity in genes and promoters. Tables for other assays are included in Data S31E.
(C) Schematic of the analysis in Figures 6 and 7. In Figure 6, we overlap motif instances with the AS SNPs and discover motifs that are AS sensitive based on enrichment odds ratio. In Figure 7, we use the genomic sequence context surrounding the SNV loci to predict the AS effect using a transformer model, with no motif or ChIP-seq experimental information provided. In the process, the model learns an attention weight over the input sequence. By aggregating the learned attention patterns across the samples, we find that the pattern corresponds to motif enrichment patterns, which in turn agrees with the AS-sensitive motifs in Figure 6. In short, the AS-sensitive motifs are re-discovered by the transformer model.
(D) The average attention scores for H3K4me3 (upper panel) and the enrichment of ZNF460 and FOXO3 surrounding H3K4me3 AS SNPs (lower panel). The two TFs are highly ranked for H3K4me3 in Figure 6. Their enrichment patterns are centered on the SNV position, which agrees with the attention pattern. This result indicates that the transformer model attaches high importance to regions containing these motifs. The attention score was averaged over 2,000 randomly selected test samples. This sub-figure is to be compared with Figure 7E: H3K4me3 appears similar to the CTCF. In Figure 7E, as expected, the attention score peaks around the AS SNV for CTCF binding; by contrast, there are no such strong peaks for POLR2A. This is likely because POLR2A is coordinated with many different regulatory factors instead of particular TFs, and these factors are not necessarily close to POLR2A binding, leading to a less-peaked attention score pattern. RNA-seq is used as a control because the AS SNV for expression is usually present as a tag and is not the causal SNV. In line with the average attention patterns, some TF motifs, including the CTCF motif itself and SP1, show stronger enrichment within the proximity of the AS SNV of CTCF binding as compared to SNVs that are accessible to CTCF binding but do not show AS behavior. This result also agrees with the attention pattern of the model, indicating potentially discriminating motifs.
(E) The maximum difference of the enrichment patterns between AS and non-AS SNPs of top- and bottom-ranked motifs in H3K4me3. The top-ranked motifs show much larger differences between AS and non-AS SNPs than the bottom-ranked ones. Thus, for the top-ranked motifs, there is a much higher peak for the AS SNPs, which is not observed in the non-AS SNPs, while for the bottom-ranked motifs the difference is minimal.
Acknowledgements
Research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under awards: U24HG009446, 1U54HG007004, R01MH101814, 5RM1HG00773509, 3UM1HG009442, R01HG009318, R01MH113005, R01LM012736, U24HG009397, U54HG006991, UM-HG009390, U24HG006620 and U24HG009649. We would also like to acknowledge the administrative support of the NHGRI ENCODE program officers; namely Elise Feingold, Mike Pazin and Daniel Gilchrist. Research was also funded by National Cancer Institute grant U01CA253481 and National Science Foundation grant 1350041 awarded to M.S. We also acknowledge support of the Spanish Ministry of Science and Innovation to the EMBL partnership, to Centro de Excelencia Severo Ochoa and to the CERCA Programme of Generalitat de Catalunya. Also funded in part by the Henry and Emma Meyer Chair in Molecular Genetics to AM. Research also supported by Cold Spring Harbor Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Z.W. co-founded and serves as a scientific advisor for Rgenta Inc. B.E.B. declares outside interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Cell Signaling Technologies, Chroma Medicine and Design Pharmaceuticals. M.G. is on the advisory board for HypaHub, Inc. and Elysium Health.
REFERENCES
- 1.Collins FS, Green ED, Guttmacher AE, Guyer MS, and Institute USNHGR (2003). A vision for the future of genomics research. Nature 422, 835–847. 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. (2001). The sequence of the human genome. Science 291, 1304–1351. 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Stephens ZD, Lee SY, Faghri F, Campbell RH, Zhai C, Efron MJ, Iyer R, Schatz MC, Sinha S, and Robinson GE (2015). Big Data: Astronomical or Genomical? PLoS Biol 13, e1002195. 10.1371/journal.pbio.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. (2022). The complete sequence of a human genome. Science 376, 44–53. 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.French JD, and Edwards SL (2020). The Role of Noncoding Variants in Heritable Disease. Trends Genet 36, 880–891. 10.1016/j.tig.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, and Manolio TA (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106, 9362–9367. 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, and Lupski JR (2015). Non-coding genetic variants in human disease. Hum Mol Genet 24, R102–110. 10.1093/hmg/ddv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Civelek M, and Lusis AJ (2014). Systems genetics approaches to understand complex traits. Nat Rev Genet 15, 34–48. 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight JC (2014). Approaches for establishing the function of regulatory genetic variants involved in disease. Genome Med 6, 92. 10.1186/s13073-014-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning KS, and Cooper TA (2017). The roles of RNA processing in translating genotype to phenotype. Nat Rev Mol Cell Biol 18, 102–114. 10.1038/nrm.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium GT (2013). The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Consortium GT (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working, G., Statistical Methods groups-Analysis Working, G., Enhancing, G.g., Fund, N.I.H.C., Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, et al. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consortium GT (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Consortium EP, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baran Y, Subramaniam M, Biton A, Tukiainen T, Tsang EK, Rivas MA, Pirinen M, Gutierrez-Arcelus M, Smith KS, Kukurba KR, et al. (2015). The landscape of genomic imprinting across diverse adult human tissues. Genome Res 25, 927–936. 10.1101/gr.192278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castel SE, Aguet F, Mohammadi P, Consortium GT, Ardlie KG, and Lappalainen T (2020). A vast resource of allelic expression data spanning human tissues. Genome Biol 21, 234. 10.1186/s13059-020-02122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Rozowsky J, Galeev TR, Harmanci A, Kitchen R, Bedford J, Abyzov A, Kong Y, Regan L, and Gerstein M (2016). A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat Commun 7, 11101. 10.1038/ncomms11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307. 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Do C, Dumont ELP, Salas M, Castano A, Mujahed H, Maldonado L, Singh A, DaSilva-Arnold SC, Bhagat G, Lehman S, et al. (2020). Allele-specific DNA methylation is increased in cancers and its dense mapping in normal plus neoplastic cells increases the yield of disease-associated regulatory SNPs. Genome Biol 21, 153. 10.1186/s13059-020-02059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Dong X, and Li Y (2018). A Genome-Wide Study of Allele-Specific Expression in Colorectal Cancer. Front Genet 9, 570. 10.3389/fgene.2018.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R, and Stamatoyannopoulos JA (2015). Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet 47, 1393–1401. 10.1038/ng.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H, et al. (2015). TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med 372, 341–350. 10.1056/NEJMoa1406829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onuchic V, Lurie E, Carrero I, Pawliczek P, Patel RY, Rozowsky J, Galeev T, Huang Z, Altshuler RC, Zhang Z, et al. (2018). Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 361. 10.1126/science.aar3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pirinen M, Lappalainen T, Zaitlen NA, Consortium GT, Dermitzakis ET, Donnelly P, McCarthy MI, and Rivas MA (2015). Assessing allele-specific expression across multiple tissues from RNA-seq read data. Bioinformatics 31, 2497–2504. 10.1093/bioinformatics/btv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robles-Espinoza CD, Mohammadi P, Bonilla X, and Gutierrez-Arcelus M (2021). Allele-specific expression: applications in cancer and technical considerations. Curr Opin Genet Dev 66, 10–19. 10.1016/j.gde.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castel SE, Levy-Moonshine A, Mohammadi P, Banks E, and Lappalainen T (2015). Tools and best practices for data processing in allelic expression analysis. Genome Biol 16, 195. 10.1186/s13059-015-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White RJ, Mackay E, Wilson SW, and Busch-Nentwich EM (2022). Allele-specific gene expression can underlie altered transcript abundance in zebrafish mutants. Elife 11. 10.7554/eLife.72825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cleary S, and Seoighe C (2021). Perspectives on Allele-Specific Expression. Annu Rev Biomed Data Sci 4, 101–122. 10.1146/annurev-biodatasci-021621-122219. [DOI] [PubMed] [Google Scholar]
- 34.Lupski JR (2022). Biology in balance: human diploid genome integrity, gene dosage, and genomic medicine. Trends Genet 38, 554–571. 10.1016/j.tig.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edge P, Bafna V, and Bansal V (2017). HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27, 801–812. 10.1101/gr.213462.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. (2019). Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675 e619. 10.1016/j.cell.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang Z, Sun W, Zhang M, Xu L, Jia X, Zhang R, and Fu S (2020). Identification of key genes associated with multiple sclerosis based on gene expression data from peripheral blood mononuclear cells. PeerJ 8, e8357. 10.7717/peerj.8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su L, Chen S, Zheng C, Wei H, and Song X (2019). Meta-Analysis of Gene Expression and Identification of Biological Regulatory Mechanisms in Alzheimer’s Disease. Front Neurosci 13, 633. 10.3389/fnins.2019.00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vennou KE, Piovani D, Kontou PI, Bonovas S, and Bagos PG (2020). Multiple outcome meta-analysis of gene-expression data in inflammatory bowel disease. Genomics 112, 1761–1767. 10.1016/j.ygeno.2019.09.019. [DOI] [PubMed] [Google Scholar]
- 41.Zhong M, Wu Y, Ou W, Huang L, and Yang L (2019). Identification of key genes involved in type 2 diabetic islet dysfunction: a bioinformatics study. Biosci Rep 39. 10.1042/BSR20182172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, and Pritchard JK (2009). Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25, 3207–3212. 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. (2012). Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100. 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. (2013). Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342, 1235587. 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. (2011). AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol 7, 522. 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leung D, Jung I, Rajagopal N, Schmitt A, Selvaraj S, Lee AY, Yen CA, Lin S, Lin Y, Qiu Y, et al. (2015). Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 518, 350–354. 10.1038/nature14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison SM, Riggs ER, Maglott DR, Lee JM, Azzariti DR, Niehaus A, Ramos EM, Martin CL, Landrum MJ, and Rehm HL (2016). Using ClinVar as a Resource to Support Variant Interpretation. Curr Protoc Hum Genet 89, 8 16 11–18 16 23. 10.1002/0471142905.hg0816s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Autuoro JM, Pirnie SP, and Carmichael GG (2014). Long noncoding RNAs in imprinting and X chromosome inactivation. Biomolecules 4, 76–100. 10.3390/biom4010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itoh Y, Golden LC, Itoh N, Matsukawa MA, Ren E, Tse V, Arnold AP, and Voskuhl RR (2019). The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J Clin Invest 129, 3852–3863. 10.1172/JCI126250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Werner JM, Ballouz S, Hover J, and Gillis J (2022). Variability of cross-tissue X-chromosome inactivation characterizes timing of human embryonic lineage specification events. Dev Cell 57, 1995–2008 e1995. 10.1016/j.devcel.2022.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, Hadzic T, Damani FN, Ganel L, Consortium GT, et al. (2017). The impact of structural variation on human gene expression. Nat Genet 49, 692–699. 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spielmann M, Lupianez DG, and Mundlos S (2018). Structural variation in the 3D genome. Nat Rev Genet 19, 453–467. 10.1038/s41576-018-0007-0. [DOI] [PubMed] [Google Scholar]
- 53.Goodier JL, and Kazazian HH Jr. (2008). Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35. 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 54.Levin HL, and Moran JV (2011). Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12, 615–627. 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamudio N, and Bourc’his D (2010). Transposable elements in the mammalian germline: a comfortable niche or a deadly trap? Heredity (Edinb) 105, 92–104. 10.1038/hdy.2010.53. [DOI] [PubMed] [Google Scholar]
- 56.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. (2015). Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665. 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang D, Eraslan B, Wieland T, Hallstrom B, Hopf T, Zolg DP, Zecha J, Asplund A, Li LH, Meng C, et al. (2019). A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol 15, e8503. 10.15252/msb.20188503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hounkpe BW, Chenou F, de Lima F, and De Paula EV (2021). HRT Atlas v1.0 database: redefining human and mouse housekeeping genes and candidate reference transcripts by mining massive RNA-seq datasets. Nucleic Acids Res 49, D947–D955. 10.1093/nar/gkaa609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, and Gerstein M (2014). FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15, 480. 10.1186/s13059-014-0480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Einarson TR, Acs A, Ludwig C, and Panton UH (2018). Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol 17, 83. 10.1186/s12933-018-0728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emilsson L, Lebwohl B, Sundstrom J, and Ludvigsson JF (2015). Cardiovascular disease in patients with coeliac disease: A systematic review and meta-analysis. Dig Liver Dis 47, 847–852. 10.1016/j.dld.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 62.Khan SS, Ning H, Wilkins JT, Allen N, Carnethon M, Berry JD, Sweis RN, and Lloyd-Jones DM (2018). Association of Body Mass Index With Lifetime Risk of Cardiovascular Disease and Compression of Morbidity. JAMA Cardiol 3, 280–287. 10.1001/jamacardio.2018.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terracciano A, Lockenhoff CE, Zonderman AB, Ferrucci L, and Costa PT Jr. (2008). Personality predictors of longevity: activity, emotional stability, and conscientiousness. Psychosom Med 70, 621–627. 10.1097/PSY.0b013e31817b9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whalen S, and Pollard KS (2019). Most chromatin interactions are not in linkage disequilibrium. Genome Res 29, 334–343. 10.1101/gr.238022.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown AA, Vinuela A, Delaneau O, Spector TD, Small KS, and Dermitzakis ET (2017). Predicting causal variants affecting expression by using whole-genome sequencing and RNA-seq from multiple human tissues. Nat Genet 49, 1747–1751. 10.1038/ng.3979. [DOI] [PubMed] [Google Scholar]
- 66.Kerimov N, Hayhurst JD, Peikova K, Manning JR, Walter P, Kolberg L, Samovica M, Sakthivel MP, Kuzmin I, Trevanion SJ, et al. (2021). A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat Genet 53, 1290–1299. 10.1038/s41588-021-00924-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S, et al. (2021). Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet 53, 1300–1310. 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Najafabadi HS, Garton M, Weirauch MT, Mnaimneh S, Yang A, Kim PM, and Hughes TR (2017). Non-base-contacting residues enable kaleidoscopic evolution of metazoan C2H2 zinc finger DNA binding. Genome Biol 18, 167. 10.1186/s13059-017-1287-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ji Y, Zhou Z, Liu H, and Davuluri RV (2021). DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 10.1093/bioinformatics/btab083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Payne JL, and Wagner A (2015). Mechanisms of mutational robustness in transcriptional regulation. Front Genet 6, 322. 10.3389/fgene.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coban-Akdemir Z, Song X,. Pehlivan D, Karaca E, Bayram Y, Gambin T, Jhangiani SN, Muzny DM, Lewis RA, et al. , (2022). De novo mutation in ancestral generations evolves haplotypes contributing to disease.
- 73.Consortium GTEx (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, et al. (2015). A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank 13, 311–319. 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Encode Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ardui S, Ameur A, Vermeesch JR, and Hestand MS (2018). Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 46, 2159–2168. 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, Garvin T, Fang H, Gurtowski J, Hutton E, et al. (2018). Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28, 1126–1135. 10.1101/gr.231100.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alonge M, Wang X, Benoit M, Soyk S, Pereira L, Zhang L, Suresh H, Ramakrishnan S, Maumus F, Ciren D, et al. (2020). Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 182, 145–161 e123. 10.1016/j.cell.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aganezov S, Goodwin S, Sherman RM, Sedlazeck FJ, Arun G, Bhatia S, Lee I, Kirsche M, Wappel R, Kramer M, et al. (2020). Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res 30, 1258–1273. 10.1101/gr.260497.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen S, Krusche P, Dolzhenko E, Sherman RM, Petrovski R, Schlesinger F, Kirsche M, Bentley DR, Schatz MC, Sedlazeck FJ, and Eberle MA (2019). Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol 20, 291. 10.1186/s13059-019-1909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cretu Stancu M, van Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, de Ligt J, Pregno G, Giachino D, Mandrile G, Espejo Valle-Inclan J, et al. (2017). Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 8, 1326. 10.1038/s41467-017-01343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, and Schatz MC (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468. 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bahler J, and Sedlazeck FJ (2017). Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 8, 14061. 10.1038/ncomms14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jou J, Gabdank I, Luo Y, Lin K, Sud P, Myers Z, Hilton JA, Kagda MS, Lam B, O’Neill E, et al. (2019). The ENCODE Portal as an Epigenomics Resource. Curr Protoc Bioinformatics 68, e89. 10.1002/cpbi.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054. 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, and Salzberg SL (2004). Versatile and open software for comparing large genomes. Genome Biol 5, R12. 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nattestad M, and Schatz MC (2016). Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023. 10.1093/bioinformatics/btw369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weisenfeld NI, Kumar V, Shah P, Church DM, and Jaffe DB (2017). Direct determination of diploid genome sequences. Genome Res 27, 757–767. 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li D, Liu CM, Luo R, Sadakane K, and Lam TW (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 90.Vaser R, Sovic I, Nagarajan N, and Sikic M (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746. 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaul A, Bhattacharyya S, and Ay F (2020). Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc 15, 991–1012. 10.1038/s41596-019-0273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ay F, Bailey TL, and Noble WS (2014). Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res 24, 999–1011. 10.1101/gr.160374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knight PA, and Ruiz D (2012). A fast algorithm for matrix balancing. IMA Journal of Numerical Analysis 33, 1029–1047. 10.1093/imanum/drs019. [DOI] [Google Scholar]
- 98.Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, and Zhou XJ (2016). TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res 44, e70. 10.1093/nar/gkv1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cameron CJ, Dostie J, and Blanchette M (2020). HIFI: estimating DNA-DNA interaction frequency from Hi-C data at restriction-fragment resolution. Genome Biol 21, 11. 10.1186/s13059-019-1913-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pertea G, and Pertea M (2020). GFF Utilities: GffRead and GffCompare. F1000Res 9. 10.12688/f1000research.23297.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wright JC, and Choudhary JS (2016). DecoyPyrat: Fast Non-redundant Hybrid Decoy Sequence Generation for Large Scale Proteomics. J Proteomics Bioinform 9, 176–180. 10.4172/jpb.1000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spivak M, Weston J, Bottou L, Kall L, and Noble WS (2009). Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J Proteome Res 8, 3737–3745. 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weisser H, Wright JC, Mudge JM, Gutenbrunner P, and Choudhary JS (2016). Flexible Data Analysis Pipeline for High-Confidence Proteogenomics. J Proteome Res 15, 4686–4695. 10.1021/acs.jproteome.6b00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mudge JM, Jungreis I, Hunt T, Gonzalez JM, Wright JC, Kay M, Davidson C, Fitzgerald S, Seal R, Tweedie S, et al. (2019). Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Res 29, 2073–2087. 10.1101/gr.246462.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wright JC, Mudge J, Weisser H, Barzine MP, Gonzalez JM, Brazma A, Choudhary JS, and Harrow J (2016). Improving GENCODE reference gene annotation using a high-stringency proteogenomics workflow. Nat Commun 7, 11778. 10.1038/ncomms11778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hellton KH, and Thoresen M (2016). Integrative clustering of high-dimensional data with joint and individual clusters. Biostatistics 17, 537–548. 10.1093/biostatistics/kxw005. [DOI] [PubMed] [Google Scholar]
- 112.Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kosti I, Jain N, Aran D, Butte AJ, and Sirota M (2016). Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci Rep 6, 24799. 10.1038/srep24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van de Geijn B, McVicker G, Gilad Y, and Pritchard JK (2015). WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 12, 1061–1063. 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 117.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H (2021). Twelve years of SAMtools and BCFtools. Gigascience 10. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuhn RM, Haussler D, and Kent WJ (2013). The UCSC genome browser and associated tools. Brief Bioinform 14, 144–161. 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jiang L, Wang M, Lin S, Jian R, Li X, Chan J, Dong G, Fang H, Robinson AE, GTEx Consortium, and Snyder MP. (2020). A Quantitative Proteome Map of the Human Body. Cell 183, 269–283 e219. 10.1016/j.cell.2020.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 123.Huang da W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Castel SE, Aguet F, Mohammadi P, GTEx Consortium, Ardlie KG, and Lappalainen T (2020). A vast resource of allelic expression data spanning human tissues. Genome Biol 21, 234. 10.1186/s13059-020-02122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eberle MA, Fritzilas E, Krusche P, Kallberg M, Moore BL, Bekritsky MA, Iqbal Z, Chuang HY, Humphray SJ, Halpern AL, et al. (2017). A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 27, 157–164. 10.1101/gr.210500.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pawliczek P, Patel RY, Ashmore LR, Jackson AR, Bizon C, Nelson T, Powell B, Freimuth RR, Strande N, Shah N, et al. (2018). ClinGen Allele Registry links information about genetic variants. Hum Mutat 39, 1690–1701. 10.1002/humu.23637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311. 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ngatchou W, Surdeanu I, Ramadan AS, Essola B, Youatou P, Guimfacq V, Wauty P, and Mols P (2013). Penetrating cardiac injuries in Belgium: 20 years of experience in university hospitals in Brussels. Acta Chir Belg 113, 275–280. 10.1080/00015458.2013.11680927. [DOI] [PubMed] [Google Scholar]
- 135.Kirsche M, Prabhu G, Sherman R, Ni B, Aganezov S, and Schatz MC (2021). Jasmine: Population-scale structural variant comparison and analysis. bioRxiv, 2021.2005.2027.445886. 10.1101/2021.05.27.445886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, Sulovari A, Ebler J, Zhou W, Serra Mari R, et al. (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372. 10.1126/science.abf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, Hadzic T, Damani FN, Ganel L, GTEx Consortium, et al. (2017). The impact of structural variation on human gene expression. Nat Genet 49, 692–699. 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Karimzadeh M, Ernst C, Kundaje A, and Hoffman MM (2018). Umap and Bismap: quantifying genome and methylome mappability. Nucleic Acids Res 46, e120. 10.1093/nar/gky677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Valikangas T, Suomi T, and Elo LL (2018). A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief Bioinform 19, 1–11. 10.1093/bib/bbw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen L, Reeve J, Zhang L, Huang S, Wang X, and Chen J (2018). GMPR: A robust normalization method for zero-inflated count data with application to microbiome sequencing data. PeerJ 6, e4600. 10.7717/peerj.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berghoff BA, Karlsson T, Kallman T, Wagner EGH, and Grabherr MG (2017). RNA-sequence data normalization through in silico prediction of reference genes: the bacterial response to DNA damage as case study. BioData Min 10, 30. 10.1186/s13040-017-0150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sethi A, Gu M, Gumusgoz E, Chan L, Yan KK, Rozowsky J, Barozzi I, Afzal V, Akiyama JA, Plajzer-Frick I, et al. (2020). Supervised enhancer prediction with epigenetic pattern recognition and targeted validation. Nat Methods 17, 807–814. 10.1038/s41592-020-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Becker JS, Nicetto D, and Zaret KS (2016). H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet 32, 29–41. 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gerlitz G (2020). The Emerging Roles of Heterochromatin in Cell Migration. Front Cell Dev Biol 8, 394. 10.3389/fcell.2020.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saksouk N, Simboeck E, and Dejardin J (2015). Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8, 3. 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ninova M, Fejes Toth K, and Aravin AA (2019). The control of gene expression and cell identity by H3K9 trimethylation. Development 146. 10.1242/dev.181180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nicetto D, and Zaret KS (2019). Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr Opin Genet Dev 55, 1–10. 10.1016/j.gde.2019.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Becker JS, McCarthy RL, Sidoli S, Donahue G, Kaeding KE, He Z, Lin S, Garcia BA, and Zaret KS (2017). Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol Cell 68, 1023–1037 e1015. 10.1016/j.molcel.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, Quivy JP, Almouzni G, and Amigorena S (2018). The epigenetic control of stemness in CD8(+) T cell fate commitment. Science 359, 177–186. 10.1126/science.aah6499. [DOI] [PubMed] [Google Scholar]
- 151.Du J, Johnson LM, Jacobsen SE, and Patel DJ (2015). DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16, 519–532. 10.1038/nrm4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Saksouk N, Barth TK, Ziegler-Birling C, Olova N, Nowak A, Rey E, Mateos-Langerak J, Urbach S, Reik W, Torres-Padilla ME, et al. (2014). Redundant mechanisms to form silent chromatin at pericentromeric regions rely on BEND3 and DNA methylation. Mol Cell 56, 580–594. 10.1016/j.molcel.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 153.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kryuchkova-Mostacci N, and Robinson-Rechavi M (2017). A benchmark of gene expression tissue-specificity metrics. Brief Bioinform 18, 205–214. 10.1093/bib/bbw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sisu C, Muir P, Frankish A, Fiddes I, Diekhans M, Thybert D, Odom DT, Flicek P, Keane TM, Hubbard T, et al. (2020). Transcriptional activity and strain-specific history of mouse pseudogenes. Nat Commun 11, 3695. 10.1038/s41467-020-17157-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ransohoff JD, Wei Y, and Khavari PA (2018). The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19, 143–157. 10.1038/nrm.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, and Kaessmann H (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640. 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 158.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050. 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Consortium GTEx (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–1235. 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012. 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yao L, Tak YG, Berman BP, and Farnham PJ (2014). Functional annotation of colon cancer risk SNPs. Nat Commun 5, 5114. 10.1038/ncomms6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gajulapalli RD, and Pattanshetty DJ (2017). Risk of coronary artery disease in celiac disease population. Saudi J Gastroenterol 23, 253–258. 10.4103/sjg.SJG_616_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Almas A, Moller J, Iqbal R, and Forsell Y (2017). Effect of neuroticism on risk of cardiovascular disease in depressed persons - a Swedish population-based cohort study. BMC Cardiovasc Disord 17, 185. 10.1186/s12872-017-0604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Naito R, and Kasai T (2015). Coronary artery disease in type 2 diabetes mellitus: Recent treatment strategies and future perspectives. World J Cardiol 7, 119–124. 10.4330/wjc.v7.i3.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, and Weissman JS (2003). Global analysis of protein expression in yeast. Nature 425, 737–741. 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 167.Greenbaum D, Colangelo C, Williams K, and Gerstein M (2003). Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol 4, 117. 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–745. 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kuhn M (2008). Building Predictive Models in R Using the caret Package. Journal of Statistical Software 28, 1 – 26. 10.18637/jss.v028.i05.27774042 [DOI] [Google Scholar]
- 170.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Devlin J, Chang M-W, Lee K, and Toutanova K (2019). BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding.
- 172.Ng P (2017). dna2vec: Consistent vector representations of variable-length k-mers. ArXiv abs/1701.06279 [Google Scholar]
- 173.McInnes L, and Healy J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv abs/1802.03426 [Google Scholar]
- 174.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen AVD, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol 37, 1482–1492. 10.1038/s41587-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Van der Maaten L, and Hinton G (2008). Visualizing data using t-SNE. Journal of machine learning research 9. [Google Scholar]
- 176.Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. (2006). The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34, D590–598. 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Anand L, and Rodriguez Lopez CM (2020). chromoMap: An R package for Interactive Visualization and Annotation of Chromosomes. bioRxiv, 605600. 10.1101/605600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Garrido-Martin D, Borsari B, Calvo M, Reverter F, and Guigo R (2021). Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat Commun 12, 727. 10.1038/s41467-020-20578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Berger SL (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 180.Suzuki MM, and Bird A (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9, 465–476. 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Additional Details on Uniform Multi-tissue Data Collection and Diploid Mapping, Related to Figure 1, STAR Methods “Data Stack”, “Personal Genome”, “Reference Comparison” and “SVs” Sections.
(A) Data matrix for the EN-TEx resource. Each voxel in the cube stack corresponds to a functional genomic assay for a particular tissue from one of the four individuals. A dark gray voxel indicates that the functional genomic data have been generated. The total experimental counts per assay across the tissues and individuals are shown above the cube stack. The total experimental counts per tissue type are shown on the left. The color and acronym scheme of the tissues is adopted from GTEx (Data S2). We also note that the small RNA-seq column represents the consolidation of both small RNA-seq and targeted micro-RNA-seq assays; the data for both these assays are available from the portal. Due to the hearts from both male donors being selected for organ transplant, we do not have heart derived tissue for both genders. Also the aortic tissue is sublocalized into samples derived from ascending aorta and thoracic aorta. Note further that in GTEx these aortic tissues are combined and considered to be a single tissue.
(B) Constructing personal genomes using the CrossStitch method. This diagram shows an overview of the CrossStitch method with the specific software and data types used. (Details in STAR Methods “Personal Genome” Section.)
(C) Mapping reads to personal and reference genomes. Due to the genetic variants, some reads can be mapped only to the personal genome but not to the reference genome (and to a lesser extent just to the reference genome). Such reads (yellow) are referred to as “personal genome unique,” whereas the reads that can be mapped to both genomes (gray) are referred to as “common.” Compared with the reference genome, using the personal genome results in an average of ~2.5% more mapped reads for the various functional genomic experiments. The percentages range from ~1% for RNA-seq to ~4% for Hi-C data. Note that the numbers for these differences only apply to the high-stringency mapping. (Details in STAR Methods “Reference Comparison” Section.)
(D) Classification and frequency of SVs. The fractions of SVs for each individual are shown and broken down ito insertions (INSs), deletions (DELs), and inversions (INVs). INSs and DELs are further classified based on whether they are related to TEs. (Details in Data S15 showing a breakdown of the SV composition.)
Figure S2 Additional Details on Improvements in Genome Analysis from Uniform Multi-tissue Data Collection and Diploid Genome Mapping, Related to STAR Methods “Reference Comparison” and “Variation Analysis” Sections.
(A) Comparing the quantification of gene expression using personal diploid genomes and the reference haploid genome. The left panel shows the gene expression levels for individual 2 calculated using the reads mapped to the reference genome (x-axis) and to the personal diploid genomes (y-axis). The expression values of the genes are log-transformed, i.e., log10(TPM + 0.001). Differentially expressed genes are color-coded (downregulated: red; upregulated: blue). Genes that are not differentially expressed are shown in gray. The panel on the right is a volcano plot reporting, for every gene, the log2(fold-change) and the −log10(p-value) obtained from the differential-gene expression analysis with DESeq2. The same color schema as in the left panel is applied. Some outlier genes were removed from the volcano plot. The full version of these plots, including all genes and all individuals, is available in Data S5D–E.
(B) Comparing the quantification of regulatory element activity using personal diploid genomes and the reference haploid genome. The left panel shows the H3K27ac signal at cCREs for individual 2 calculated using the reads mapped to the reference genome (x-axis) and to the personal diploid genomes (y-axis). The H3K27ac signals of the cCREs are log-transformed. Differentially marked cCREs are color-coded (downregulated: red; upregulated: blue). cCREs that are not differentially marked are shown in gray. The panel on the right is a volcano plot reporting, for every cCRE, the log2(fold-change) and the −log10(p-value) obtained from the differential analysis performed with DESeq2. The same color schema as in the left panel is applied. Some outlier cCREs were removed from the volcano plot. The full version of these plots, including all cCREs and all individuals, is available in Data S5K–L.
(C) Consistently analyzing functional genomic data across individuals, tissues, and assays. Joint analysis of variation (JIVE, Hellton and Thoresen, 2016) was used to project all of the functional genomic signals from different samples onto two-dimensional planes (one for each assay, e.g., ChIP-seq, ATAC-seq, DNase-seq). For each assay type (e.g., H3K27ac), the samples are represented as dots on the plane. Two dots that are close to each other have similar levels of functional genomic signals. Combining all of the types of assays, we projected the samples onto a single two-dimensional plane. The tissues are colored according to the GTEx convention from Figure 1A and Data S2I. For each tissue, the number of colored dots indicates the number of individuals, up to four. In addition, a linear-regression-based approach was used to measure the difference between two samples in functional genomic signals, (Details in STAR Methods “Variation Analysis” Section.) The variance explained is used to indicate the similarity between two samples. Note that the value of variance explained is equal to the squared correlation coefficient. The difference is measured by one minus the explained variance of the regression. Taking H3K27ac as an example, on average, only 14% of the variation in the functional signals between the spleens of two individuals is unexplained, indicating that the spleen samples have very similar H3K27ac signal patterns. The dissimilarity is also small (15%) between the transverse colons of two individuals. In contrast, the average dissimilarity between these two types of tissues of the same individuals increases to 35%. For H3K27me3, the dissimilarity between different tissues is also high (42%). The dissimilarities between different types of functional signals are even higher. In particular, the dissimilarity between H3K27ac and H3K4me3 in spleen is 51%. (Details in STAR Methods “Variance Analysis” Section and Data S6.)
(D) An example of a more accurate inter-tissue comparison using matched samples. For histone modification signals at cCREs, the spleen and esophagus of the same individual are more similar (i.e., median 0.92 correlation, 84% variance explained [or 16% dissimilarity]) than those of different individuals (0.89 or 79%). This difference indicates the genetic and environmental difference between individuals and its influence on inter-tissue comparisons.
Figure S3 Additional Details on the Construction of the AS Catalog, Related to Figure 1, STAR Methods “AS Calling”, “Aggregation” and “AS Catalog” Sections.
(A) Schematic of calling AS SNVs and AS elements. Examples of the number of reads are shown for each SNV and element. For a functional genomics experiment, if the numbers of reads mapped to the two haplotypes at a heterozygous SNV position are significantly different, the SNV is referred to as an AS SNV. For a given genomic element (e.g., a cCRE) with multiple heterozygous SNVs, we pool the reads of all the SNVs to determine its AS state. Genomic elements with a single AS SNV are identified as AS elements. (Details in STAR Methods “AS Calling” Section.)
(B) Distribution of the number of AS events in different assays. The distribution of H3K27ac AS events in spleen from individual 1 is highlighted as an example here and in panel (D) and further distributions are shown in Data S11A.
(C) Details of union and pooling approaches for read aggregation. We used two approaches to aggregate the information across samples to increase the detection power. In the “union” approach, any SNV observed as AS in any sample is included in our AS catalog - i.e., we call individually on samples and then take the union of the calls. In contrast, in the “pooling” approach, because a given functional genomic assay, e.g., RNA-seq, is usually conducted on many samples, for each SNV, we pool the mapped reads from all the samples together to determine its AS state (see STAR Methods “Aggregation” Section). For instance, to do multitissue pooling, we pool the reads from all tissues and then jointly call AS SNVs on the pooled reads. This approach increases the statistical power of the AS test and thus may be able to identify an SNV as AS, even though the SNV is not AS in each tissue.
(D) Summary of AS catalog. The colored circle on the right shows the aggregation method along that axis. Take the third row as an example. The upper one indicates that the operation is the union across tissues, averaged across assays and individuals, while the bottom one pools reads across tissues, averaged across assays and individuals. Data obtained by merging the tissue-specific AS call sets resulted in ~5.5K unique AS hetSNVs (for either AS binding or expression) and ~1K AS genomic elements (not shown in the figure) for each individual per assay. Pooling the reads from each assay across all tissues dramatically increased the detection power by ~5X fold, making it possible to identify ~27K AS hetSNVs per assay for each individual. Finally, merging across all assays provided a catalog of all loci where AS activity could be assessed in any of the tissues of the four individuals.
(E) Numbers of AS SNVs across assays. The Venn diagram of SNVs shows AS activity in selected assays using the “pooling” approach. (Details in Data S8B.)
(F) Numbers of AS elements across samples. The distribution of the number of genomic elements (genes and cCREs) with AS activity across EN-TEx donors, tissues, and assays is shown. (Details in Data S11A.) Donors and tissues are indicated using the conventions from Figure 1.
Figure S4 Examples of Coordinated AS Activity Involving SNVs and SVs, Related to Figure 2 and STAR Methods “AS Examples” Section.
(A) Detecting AS events at a known imprinted locus: H19/IGF2. Functional genomic signals are measured in the gastrocnemius medialis of individual 2. In agreement with the known imprinting mechanism (top), we detected AS expression of H19 on haplotype 2 (known to be maternally expressed) and AS expression of IGF2 on haplotype 1 (known to be paternally expressed). Consistent with the expression data, we found an AS Hi-C interaction between an upstream cCRE and H19 on haplotype 2 (bold blue arc), and an AS interaction between the same cCRE and IGF2 on haplotype 1 (bold red arc). Other neighboring AS Hi-C interactions (which have fewer read counts or contact irrelevant genes) are also depicted with shaded arcs.
(B) The effect of a 5.2-kb homozygous deletion. This SV removed an H3K9me3 peak in individual 2, potentially resulting in an increase in PCCB expression in the spleen. The sashimi plots, below the signal graphs, show examples of novel splicing isoforms identified by long-read RNA-seq in the adrenal gland of individual 2 and in the heart left ventricle of individual 3 (without the SV). Data S17K shows more details on the splicing isoforms. The differences in splicing between individuals 2 and 3, although supported by only one long read, potentially reflect the SV-disrupting regions important to splicing, as suggested by the known GTEx sQTL sites nearby178. Alternatively, the differences in splicing between the two samples may be due to tissue specificity. In addition, Data S17L provides evidence of novel splicing variants that are potentially associated with an SV in the gene TRDN-AS1.
(C) The effect of a 2.3-kb homozygous deletion. This SV removed an H3K27ac peak downstream of lncRNA RP11–362F19.1 in individual 3, which has lower expression in the spleen sample. In individual 2 without the SV, there are CTCF peaks that point to a potential interaction between the H3K27ac peaks downstream and upstream of the lncRNA. Together, these observations suggest that the loss of the H3K27ac peak due to the SV contributes to the reduction in lncRNA gene expression. (Details in Data S17E–F.)
Figure S5 Additional Details on Applications 1 and 2: Relating Decorations and AS SNVs to GWAS and eQTL Loci, Related to Figures 3, 4, STAR Methods “Decoration Process”, “Tissue Specificity” and “Compatibility” Sections.
(A) Workflow of cCRE decoration. This figure shows the workflow of cCRE decoration and the number of different subgroups of cCREs in the spleen. Note that we define the following abbreviations for the various decorations. dAct: distal active; pAct: proximal active; dBiv: distal bivalent; pBiv: proximal bivalent; dRep: distal repressed; pRep: proximal repressed; CTCF+ and CTCF−: with and without CTCF binding, respectively; and AS and nonAS: with and without allelic signature, respectively. Spleen is used as an example to illustrate the tissue-specific decoration of these cCREs using EN-TEx data. In this tissue, 290k of the cCREs have functional genomic signals and can be categorized as active (117k), repressed (154k), or bivalent (19k). Then, for each category, the cCREs can be consecutively classified according to their genomic locations (proximal or distal), CTCF binding, and allele specificity. (This classification can occur in any order.) As a result, the spleen has 2,866 AS cCREs with various genomic activities and locations. More details on the workflow are included in the STAR Methods “Decoration Process” Section. The numbers of active and repressed cCREs are comparable in each tissue (i.e., on average ~202k active and ~166k repressed cCREs). Only a small subset of the active cCREs exhibit AS activity (~2.5% or 1,750 cCREs, averaged across tissues; see details in Data S19C for all available tissues).
(B) Tissue specificity of different subgroups of cCREs vs. genes and epigenomic peaks. We compared the tissue specificity of protein-coding genes, non-coding genes, different subgroups of decorated cCREs, and various epigenomic peaks. The uniqueness of the activity across tissue types is shown in different colors. Note that the decoration terms are defined in Figure S5A.
(C) Compatibility between AS expression and AS promoter state. As shown in the examples, a high active histone modification signal (e.g., H3K27ac) of a haplotype should correspond to high gene expression of the same haplotype. Note that for a repressive histone modification, this trend is the opposite. For AS genes with promoters accessible to H3K27ac, there is indeed a strong and positive correlation. The exact read counts of H3K27ac and gene expression, respectively, from haplotype 1 and 2 for LAMTOR1 and RPL4P4 are indicated in the examples. In addition, we used the slope (beta coefficient) of the leading eQTL of an AS gene (GTEx Consortium, 2020). The slope is positively correlated with the AS ratio (overall Pearson’s correlation coefficient of 0.6 with p = 0.01). Note that genetic variants with beta coefficients around 0 are unlikely to have sufficient statistical significance to be identified as eQTLs. This also holds for SNVs with read fractions of approximately 0.5. Therefore, there are no data points observed at the center of the plot.
(D) Overall compatibility between AS expression and promoter AS chromatin activity. The compatibility is measured by the fraction of genes for which the AS expression is compatible with promoter AS chromatin state or AS peptide expression (Data S26 and STAR Methods “Compatibility” Section). As a null model, we randomly paired genes with promoters (and peptides) to calculate z-scores. (Details in STAR Methods “Compatibility” Section.) Compatibility between AS expression and AS methylation (meCpG), H3K9me3, and H3K27me3 is weak, potentially because these marks of repressed chromatin also can be associated with genes poised for transcription or genes that are actively transcribed179,180. Note for the bottom entry, compatibility is calculated between ASE and AS peptide expression, which is independent of the other compatibility comparisons between AS promoter activity and ASE.
Figure S6 Additional Details on Application 2: Modeling eQTLs in Hard-to-obtain Tissues, Related to Figure 5 and STAR Methods “transferQTL Model” Section.
(A) Chromatin features can help prioritize causal eQTLs. Barplots showing the percentage of eQTLs overlapping a given feature. The fraction of fine-mapped eQTLs (likely causal, red) overlapping peaks of six histone marks (H3K27ac, H3K4me1, H3K4me3, H3K27me3, H3K9me3, H3K36me3) is higher compared with the total set of GTEx eQTLs (blue) reported in a given tissue. (Details in Data S27A.)
(B) Chromatin-marked loci associated with eQTL activity. We have identified 1,353,101 SNVs that show tissue-specific eQTL activity. These SNVs are GTEx eQTLs in ≥ 5 EN-TEx tissues and are not GTEx eQTLs in ≥ 5 other EN-TEx tissues. Thus, for every SNV we define two groups of tissues: i) tissues in which the SNV is an eQTL (eQTL+, orange) and ii) tissues in which the SNV is not an eQTL (eQTL−, cyan). We observed that SNVs are more likely to be marked by H3K36me3 in the tissues in which they are eQTLs, compared with the tissues in which they are not eQTLs (p-value < 2.2e-16, Wilcoxon paired test). (Details in Data S27B.)
(C) Schema of the predictive model using skin as the donor tissue and tibial artery as the target tissue. We first retrieve GTEx eQTLs associated with one single eGene in sun-exposed skin (n = 967,288). This set of eQTLs is split into training and test sets (70% and 30%, respectively). Next, we train a random forest model in order to predict whether these eQTLs are eQTLs also in the tibial artery. The model uses a number of predictive features described in Data S28A: i) eQTL-gene properties in the skin tissue (blue; features 1–3), ii) chromatin profiles at the eQTL loci in the tibial artery tissue (red; features 4–24), iii) chromatin profiles at the eQTL loci across all EN-TEx tissues (gray; features 25–36), and iv) genomic features of the eQTL loci (white; features 37–39). (Details in Data S28B–G.)
(D) transferQTL outperforms simplified models that transfer eQTLs based only on a tissue-specificity threshold. We built a simple model that transfers eQTLs to a given tissue based on their degree of tissue specificity. We compared the performance of this simple model by using different thresholds of tissue specificity. For instance, 10% corresponds to a model that transfers to a given tissue eQTLs that are active in at least 10% of the GTEx tissues. The performance is compared to our random-forest model trained on multi-omics EN-TEx data (transferQTL). (Details in Data S29C–D.)
(E) The predictive models have high performance (balanced accuracy) even when trained on a reduced number of histone modifications. This modified version of transferQTL utilizes only chromatin features related to the four histone marks that have the strongest predictive power (H3K36me3, H3K27ac, H3K4me1, H3K27me3; also see Figure 5F). The x-axis indicates the tissues used as the donors (GTEx coloring) and the y-axis indicates the average performance (balanced accuracy) across the target tissues that are available. The whiskers indicate variation across targets (standard deviations).
Figure S7 Additional Details on Application 3: Modeling AS Activity from Variant Impact on the Nucleotide Sequence, Related to Figures 6, 7, STAR Methods “Sensitive Motifs”, “AS Promoter” and “Transformer Model” Sections.
(A) Distribution of the motif rank across tissues. Similar to Figure 6A, we rank the motifs based on the enrichment of H3K4me3 AS SNVs, but using SNVs from each tissue individually. The y-axis shows the tissue-specific rank. The x-axis represents the motifs in the order of the pan-tissue analysis ranking, as in Figure 6A. Each vertical gray line represents the range between the 25% and 75% percentile of the motif rank across all tissues for that motif. The median value is shown in red.
(B) Contingency table for AS genes and AS promoters. The 2-by-2 contingency table shows AS genes vs. non-AS genes (in terms of AS gene expression) and AS promoters vs. non-AS promoters (in terms of H3K27ac activity). The overall odds ratio of the table indicates the association between AS activity in genes and promoters. Tables for other assays are included in Data S31E.
(C) Schematic of the analysis in Figures 6 and 7. In Figure 6, we overlap motif instances with the AS SNPs and discover motifs that are AS sensitive based on enrichment odds ratio. In Figure 7, we use the genomic sequence context surrounding the SNV loci to predict the AS effect using a transformer model, with no motif or ChIP-seq experimental information provided. In the process, the model learns an attention weight over the input sequence. By aggregating the learned attention patterns across the samples, we find that the pattern corresponds to motif enrichment patterns, which in turn agrees with the AS-sensitive motifs in Figure 6. In short, the AS-sensitive motifs are re-discovered by the transformer model.
(D) The average attention scores for H3K4me3 (upper panel) and the enrichment of ZNF460 and FOXO3 surrounding H3K4me3 AS SNPs (lower panel). The two TFs are highly ranked for H3K4me3 in Figure 6. Their enrichment patterns are centered on the SNV position, which agrees with the attention pattern. This result indicates that the transformer model attaches high importance to regions containing these motifs. The attention score was averaged over 2,000 randomly selected test samples. This sub-figure is to be compared with Figure 7E: H3K4me3 appears similar to the CTCF. In Figure 7E, as expected, the attention score peaks around the AS SNV for CTCF binding; by contrast, there are no such strong peaks for POLR2A. This is likely because POLR2A is coordinated with many different regulatory factors instead of particular TFs, and these factors are not necessarily close to POLR2A binding, leading to a less-peaked attention score pattern. RNA-seq is used as a control because the AS SNV for expression is usually present as a tag and is not the causal SNV. In line with the average attention patterns, some TF motifs, including the CTCF motif itself and SP1, show stronger enrichment within the proximity of the AS SNV of CTCF binding as compared to SNVs that are accessible to CTCF binding but do not show AS behavior. This result also agrees with the attention pattern of the model, indicating potentially discriminating motifs.
(E) The maximum difference of the enrichment patterns between AS and non-AS SNPs of top- and bottom-ranked motifs in H3K4me3. The top-ranked motifs show much larger differences between AS and non-AS SNPs than the bottom-ranked ones. Thus, for the top-ranked motifs, there is a much higher peak for the AS SNPs, which is not observed in the non-AS SNPs, while for the bottom-ranked motifs the difference is minimal.
Data Availability Statement
All data contained in the EN-TEx resource are fully open-consented and accessible without registration as of the date of publication. Raw sequencing data as well as other standard functional genomics data have been deposited at a special page on the ENCODE data center, linked from the EN-TEx portal. Accession numbers are listed in the key resources table or in the supplementary data. Additional ancillary files are available directly on the EN-TEx portal: http://entex.encodeproject.org. The portal is organized into three organized sections: (i) data files, (ii) interactive visualization tools, and (iii) source code. For more details, see “Portal” Section of the STAR Methods.
All original code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table