

Abstract
Protein–protein interactions (PPI) are involved in all cellular processes and many represent attractive therapeutic targets. However, the frequently rather flat and large interaction areas render the identification of small molecular PPI inhibitors very challenging. As an alternative, peptide interaction motifs derived from a PPI interface can serve as starting points for the development of inhibitors. However, certain proteins remain challenging targets when applying inhibitors with a competitive mode of action. For that reason, peptide‐based ligands with an irreversible binding mode have gained attention in recent years. This review summarizes examples of covalent inhibitors that employ peptidic binders and have been tested in a biological context.
Keywords: bioconjugation, new modalities, peptidomimetics, proteomimetics, structure‐based design
Peptide interaction motifs derived from a protein–protein interaction interface can serve as starting points for the development of inhibitors. However, for certain proteins when applying competitive inhibitors, peptide‐based ligands with an irreversible binding mode may be more active. This review summarizes examples of covalent inhibitors that employ peptidic binders and have been tested in a biological context.
1. INTRODUCTION
Most cellular processes are governed by a complex network of protein–protein interactions (PPI). The sum of these interactions, the interactome, consists of about 650,000 contacts, of which only 14,000 have been studied so far. 1 , 2 Protein–protein interactions (PPI) are determined by the structural characteristics of the involved binding partners. 3 Individual protein stretches can fold into secondary structures like α‐helices, β‐sheets or turns which are crucially involved in the recognition process. Modulators of PPIs are considered to expand the scope of the druggable genome. 4 , 5 , 6 However, addressing PPIs using small molecules has proven challenging due to the rather flat and large interaction interfaces and the absence of traditionally addressed binding pockets or enzymatic cavities. 6 As a consequence, small molecules frequently lack sufficient binding affinity and selectivity. 7 , 8 , 9 As an alternative, peptide binding epitopes derived from a given PPI interface were explored as starting points for the development of inhibitors (Figure 1A). These peptide binding epitopes are defined by their secondary or tertiary structure in their bound state. Stabilizing this bioactive conformation using intramolecular crosslinks and other modifications was found to improve target affinity, cellular uptake and peptide stability (Figure 1A). 10 , 11 , 12 , 13 , 14 , 15
FIGURE 1.
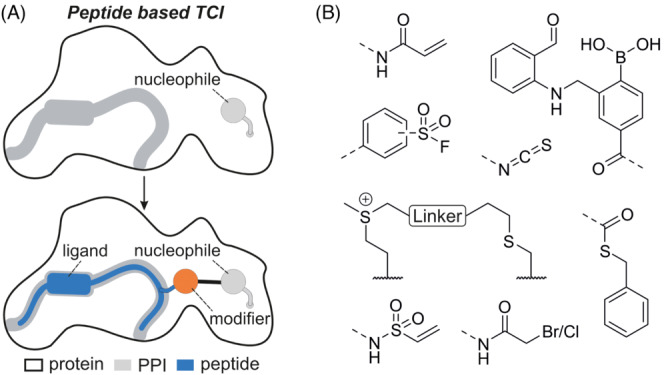
(A) Structure‐based design of a peptide‐based targeted covalent inhibitor. Top: Ligand binding site (grey) in proximity to a nucleophile on the protein surface. Bottom: Introduction of a covalent modifier (orange) in the peptide ligand (blue) results in the desired covalent inhibitor. (B) Chemical structures of selected modifiers discussed in this review.
Nonetheless, the targeting of PPI interfaces remains challenging with classic competitive inhibitors. Alternatively, inhibitors that exhibit an irreversible covalent mode of action have proven efficient in such cases and enabled targeting of traditionally ‘undruggable’ proteins as exemplified by the approval of KRAS(G12C) inhibitor sotorasib. 16 , 17 Prominent examples of approved drugs that trigger the covalent modification of target proteins and bind active sites to inhibit enzymatic activity involve aspirin, penicillin and clavulanic acid. 18 , 19 , 20 In the past, many first‐in‐class covalent drugs were identified serendipitously and their mode of action elucidated thereafter. 21 , 22 Notably, aspirin has been marketed since 1899 and its mechanism was only elucidated in the 1970s. 23 , 24 Many of these mechanism‐based inhibitors mimic a substrate transition state to enable covalent modification of a catalytic amino acid residue. 20 , 25 , 26 , 27 Covalent inhibition offers several advantages: often, even binders with only moderate affinity can show high potency due to prolonged target residence time and extended duration of action. 28 , 29 , 30 This enables the minimization of binding scaffolds, thereby increasing their ‘drug‐likeness’. Furthermore, irreversible inhibition is particularly advantageous when competing with high concentrations of endogenous ligands. 28
In recent years, the clinical success of drugs with a covalent mode of action has led to a renewed interest in irreversible inhibitors. 31 In particular, bifunctional molecules consisting of a binding scaffold decorated with a protein‐reactive functional group (modifier), so‐called targeted covalent inhibitors, attracted attention. 32 , 33 Notably, such inhibitors usually target non‐catalytic nucleophilic residues located in proximity to the binding site, thereby expanding the scope beyond enzyme inhibtion. 34 There is a growing number of examples that use peptide‐based scaffolds as the basis for the design of targeted covalent inhibitors, mainly utilizing cysteine‐ or lysine‐targeting modifiers (Figure 1A). Herein, we summarize peptidic inhibitors, focusing on examples that have shown activity in cell‐based assays.
2. ACRYLAMIDES
Acrylamides are commonly used protein‐targeting electrophiles and have been used to design peptide‐based covalent inhibitors of the E3 ubiquitin ligase seven in absentia homolog (SIAH). 35 SIAH affects HIF‐1α transcription factor levels and regulates key cellular events central to cancer development and progression. The search for an inhibitor of SIAH initially focused on the Drosophila adaptor protein phyllopod due to its inhibition of SIAH‐induced substrate degradation. 36 A phyllopod derived 23‐mer peptide (PHYL) that binds with low micromolar affinity to the substrate‐binding domain of SIAH was developed (Figure 2A) and a crystal structure of the complex was obtained. 37 , 38 In addition, a peptide derived from SIAH‐interacting protein (SIP) was designed showing a similar binding mode as PHYL in complex with SIAH1. 39 The PHYL‐derived sequence was used as starting point for the development of an inhibitor and was initially truncated yielding 13‐mer peptide BI‐107D1 (Figure 2A) without loss of binding affinity. However, efforts to improve the affinity did not result in sufficiently active candidates, and therefore a covalent mode of action was pursued. Different acrylamide‐based modifiers were introduced at position 123 of peptide BI‐107D1 (BI‐107F7, x = 4) to target a solvent‐accessible cysteine adjacent to the binding‐site (SIAH C130, Figure 2A). The electrophiles were incorporated by linking them to the side chain amine of different lysine derivates. Various side chain lengths were tested with ornithine (BI‐107F11, x = 3, Figure 2B) providing the most potent setup. N‐terminal implementation of cell penetrating peptides (CPPs) allowed for biological testing. TAT modified BI‐107F9 and P10 modified BI‐107G3 attenuated SIAH‐mediated degradation of PHD3 with concomitant effects on HIF‐1α levels in cell‐based assays. 35
FIGURE 2.
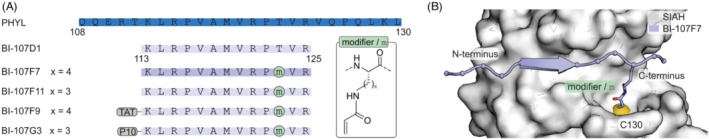
(A) Sequences and modification sites of PHYL‐derived peptides and covalent inhibitors of SIAH. Chemical structure of the tested acrylamide modifiers (x = 3 or 4) is provided. (B) Crystal structure of covalent inhibitor BI‐107F7 bound to SIAH C130 (pdb: 4i7d).
The acrylamide modifier was also used to target anti‐apoptotic B‐cell lymphoma 2 (BCL‐2) protein family member BFL‐1 with the intention of activating apoptosis in BCL‐2 dependent cancer cells. 40 The crystal structure of BFL‐1 in complex with the BH3 helix of the protein NOXA (Figure 3A, top) 41 revealed a BFL‐1 cysteine (C55) in proximity to the binding site, which was selected for covalent targeting. Notably, none of the other BCL‐2 family members contain a cysteine close to the BH3‐binding pocket. Previously, a BID BH3 domain had been stabilized by the introduction of an i, i + 4 staple. 42 , 43 , 44 NOXA BH3 was thus stabilized in an analogous fashion (Figure 3B and C). 42 Based on the crystal structure of NOXA BH3 amino acid, L21 was identified as closest residue to BFL‐1 C55 (d = 3.3 Å, Figure 3A, top) and different acrylamide‐based modifiers N‐terminally introduced (NA‐NOXA‐SAHB A ‐m, Figure 3B and D). The acrylamide bearing D‐nipetoic acid modifier (3) was identified as most promising candidate (Figure 3D). Later, binding of a related i, i + 7 stapled peptide (D‐NA‐NOXA‐SAHB A ‐3) to BFL‐1 was confirmed by a crystal structure (Figure 3A, bottom). 45 In addition, a crystal structure of BFL‐1 bound to the BIM BH3 helix was used as starting point for inhibitor design (Figure 3A and B). 46 Notably, BIM BH3 is a promiscuous binder of BCL‐2 family members. Again, an i, i + 4 stapled helical peptide was used as a scaffold. 43 Based on the crystal structure of BIM BH3 bound to BFL‐1, BIM BH3 W147 was identified as closest to C55 in BFL‐1 (d = 3.6 Å) and thus replaced with different N‐terminal acrylamide modifiers (BIM‐SAHB A , Figure 3A, C and D). Cell‐based testing revealed only limited cellular uptake of NOXA‐derived inhibitors but sufficient uptake of BIM‐derived peptides. BIM‐SAHB A ‐3 showed the expected enhanced apoptotic response in different BFL‐1‐dependent melanoma cells. Also, the covalent inhibitor BIM‐SAHB A ‐3 showed stronger effects than its non‐covalent analog BIM‐SAHB A .
FIGURE 3.
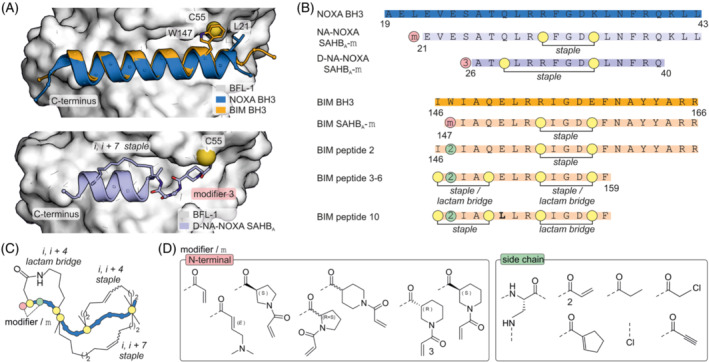
(A) Top: Crystal structure of NOXA BH3 (blue, pdb: 3mqp) and BIM BH3 (orange, pdb: 2vm6) bound to BFL‐1 (pdb: 3mqp). Bottom: Crystal structure of i, i + 7 stapled covalent inhibitor D‐NA‐NOXA SAHB bound to C55 of BFL‐1 (pdb: 5whh). (B) Sequences and modification sites of BFL‐1, BCL‐2A1 and MCL‐1 binding peptides. (C) Scheme of the different employed crosslinks. (D) Chemical structures of tested modifiers.
A similar approach was used to the target oncogenic BCL‐2A1. 47 Again, a stapled BIM BH3‐peptide and acrylamide‐based modifiers were used to target BCL‐2A1 C55 (Figure 3A and B). Here, the covalent mode of action was used to quantify intracellular target engagement and guide ligand optimization. Unlike previously, the modifiers were incorporated as lysine side chain modifications (Figure 3B and D). The resulting BIM peptide 2 was shown to modify BCL‐2A1 in live cells. Data from molecular dynamics (MD) simulations and mutagenesis studies revealed that the C‐terminal region was less important for binding to A1 and MCL‐1 than for other BCL‐2 proteins. 48 Thus, BIM peptide 2 was shortened by seven residues (BIM peptides 3–10, Figure 3B). Moreover, two crosslinks were incorporated, either an i, i + 4 lactam and/or staple (Figure 3B and C), and a variety of different combinations were examined. The BIM variation E151L was found to improve selectivity for MCL‐1/A1 and was thus incorporated into BIM peptide 10 (Figure 3B, bold) which was shown to modify intracellular A1. Interestingly, its non‐covalent derivate bearing a isosteric propionamide instead of the acrylamide modifier also induced apoptosis of human melanoma cells (SKMel‐28), reduced cell viability in a dose‐dependent manner and enhanced cell death by anticancer drug etoposide. 47 , 48
3. CHLOROACETAMIDE
Chloroacetamide is an electrophile that has been widely used to address solvent‐accessible cysteines, 49 , 50 , 51 , 52 and it was also employed to generate covalent BFL‐1 antagonists. 53 Again, a shortened BIM BH3‐derived sequence 54 (Figure 4A) was used as a template. An N‐terminal helix‐inducing cap (Figure 4B) was introduced, which involves a lactam bridge between the N‐terminal backbone amine and the side chain carboxylate of a (homo)glutamic acid at position 4 in the helix. Due to the aforementioned close proximity of BFL‐1 C55 and BIM BH3 W147 (Figure 3A, top), the residue was again chosen for the introduction of the modifier. Chloroacetamide was attached to the side chain of diaminopropionic acid (Dap, Figure 4A and B). The resulting covalent inhibitor 138C5 (Figure 4A) preserved its affinity for BFL‐1 but not for BCL‐xl. Additional alanine substitution by helix inducing aminobutyric acid residues (Aib) in peptides 138C7 and 138C8 (Figure 4A) had no major effect on binding affinity. Notably, inhibitors 138C5 and 138C8 induced apoptosis in SKMEL28 melanoma cell line with high BFL‐1 expression. A crystal structure of peptide 138C7 bound to BFL‐1 confirmed the anticipated binding mode (Figure 4C). 54
FIGURE 4.
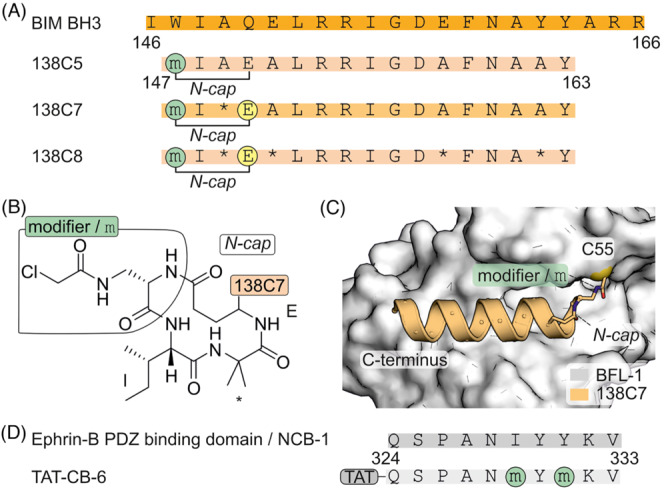
(A) Sequence of BIM BH3 and derived N‐capped peptides with introduced modifications (* α‐aminoisobutyric acid, Aib). (B) Chemical structure of N‐cap and chloroacetamide modifier in 138C5. (C) Crystal structure of 138C7 covalently bound to C55 of BFL‐1 (pdb: 2vm6). (D) Sequence of ephrin‐B PDZ binding domain/NCB1 and derived covalent inhibitor TAT‐CB‐6 with modification sites. TAT refers to a HIV TAT cell penetrating peptide (CPP).
The chloroacetamide modifier was also used to interfere in ephrin‐B signalling, which is induced by stromal derived factor 1 (SDF‐1) and regulates chemotaxis. 55 , 56 Cell–cell contact initiates the interaction of EphB with membrane‐embedded ephrin‐B which recruits PDZ‐RGS3 to its cytosolic C‐terminus. This activates the PDZ‐RGS3 GAP (GTPase‐activating protein) function, which inhibits G‐protein coupled chemoattraction induced by SDF‐1. Inhibition of PDZ‐RGS3 could thus inhibit SDF‐1‐induced chemotaxis. A C‐terminal stretch of ephrin‐B was used as a template for inhibitor design (NCB1, Figure 4D). The chloroacetamide modifier was introduced via diaminopropionic acid (Dap) at different positions of the 10‐mer peptide, aiming at cysteines in the PDZ‐RGS3 binding groove. The two positions showing the highest modification efficiency were combined with the yielding inhibitor CB‐6. Subsequently, the cell‐penetrating TAT sequence 57 , 58 , 59 was attached at the N‐terminus to facilitate cellular uptake. The resulting covalent inhibitor, TAT‐CB‐6 (Figure 4D), was found to modify the target in COS‐7 cells cotransfected with PDZΔRGS3 and efficiently compete with complex formation. Furthermore, TAT‐CB‐6 was observed to prevent chemotaxis of the neuroblastoma SH‐SY5Y cell line towards SDF‐1. 52
To investigate parameters influencing labelling efficiency, chloroacetamide‐modified peptides were used in a model system employing the kinase‐inducible (KIX) domain of human CREB binding protein (CBP). 60 Here, the transactivation domain of mixed lineage leukaemia (MLL) was used as a template for the design of a covalent KIX binder (Figure 5A and B). An NMR structure 61 directed the incorporation of cysteines in the KIX‐domain resulting in six single cysteine variants (Figure 5C, spheres). The modifier was attached to MLL‐inspired peptide L using N‐terminal spacers of different lengths (Figure 5B). The reaction rates of different KIX/L combinations were determined and the highest reactivity was observed for variant KIX(C638) with ligands bearing medium‐sized spacers (number of main chain atoms: y = 9, 13 and 19). It was concluded that accessibility of the targeted cysteine side chain is the crucial factor determining reactivity. Subsequently, covalent ligand Cl‐9 L was modified with a C‐terminal membrane anchor and microinjected into HeLa cells transfected with a fluorescently labelled KIX version. Notably, only the covalent version of the ligand allowed for recruitment of the labelled KIX domain to the HeLa endomembrane system. This is a rare example of directing proteins to a cellular compartment using a synthetic ligand. 60
FIGURE 5.
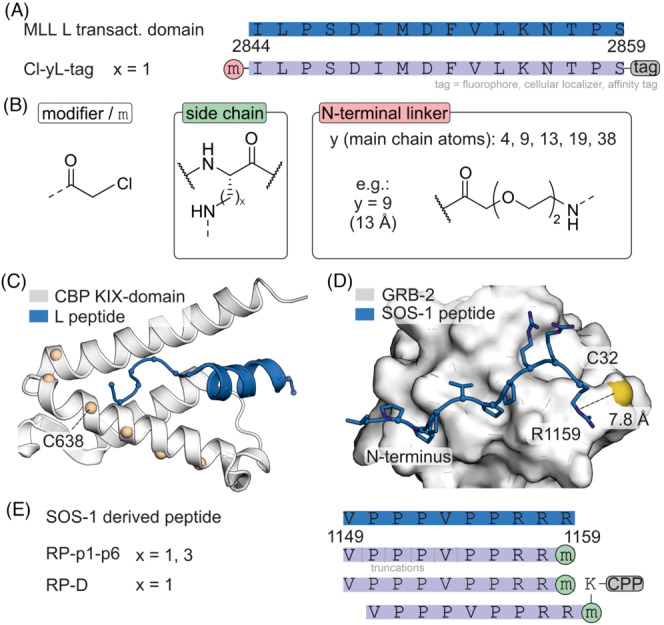
(A) Sequence of MLL‐transactivation domain L and derived covalent binders with modification sites. (B) Chemical structures of modifiers (x = 1 or 3). (C) NMR structure (pdb: 2lxs) of KIX‐domain of CBP (white) and MLL‐transactivation domain (blue) with positions used for the generation of cysteine variants (beige). (D) NMR structure of GRB‐2 bound to a SOS‐1 derived peptide (pdb: 1gbq). (E) Sequence of SOS‐1‐derived peptide and derived monomeric and dimeric covalent inhibitors attached to a cell penetrating peptide (CPP).
A chloroacetamide modifier was also used to address the interaction between growth factor receptor‐bound protein 2 (GRB‐2) and Ras guanine nucleotide exchange factor (SOS‐1), which is essential for Ras signalling propagated activation of cell proliferation in many forms of cancer. 62 Thus, interfering with the GRB‐2/SOS‐1 interaction can affect cancer cell survival. A solution structure of a SOS‐1 derived 10‐mer peptide in complex with the GRB‐2 SH3 domain (Figure 5D) was used as template for the design of a covalent inhibitor utilizing a chloroacetamide modifier. 63 SOS‐1 C‐terminal R1159 is the closest residue to GRB‐2 C32 (d = 7.8 Å) and was selected for the introduction of the modifier using lysine derivatives of varying side chain length (Figure 5B). Covalent inhibitors with lysine derivate Dap showed the highest levels of protein modification (RP‐p1, Figure 5E). N‐terminal peptide truncation led to a loss of covalent labelling (RP‐p2‐p6, Figure 5E). Dimeric versions of RP‐p1 were generated by incorporation of an additional lysine at the C‐terminus that was subsequently linked to another RP‐p1 molecule. Moreover, the sequence was C‐terminally elongated with a CPP (RP‐D, Figure 5E) to facilitate intracellular delivery. Notably, only dimeric RP‐D showed GRB‐2 labelling in COS‐7 cells. Moreover, RP‐D treatment led to a variety of different downstream effects, including inhibition of breast cancer migration, decreased SK‐BR‐3 cell viability and increased apoptosis. 63
4. VINYL SULFONAMIDE
Ras was also directly targeted using vinyl sulfonamide‐bearing SOS‐1‐derived peptides. This approach addresses a particular oncogenic mutation in Ras (G12C). 64 Previously, SOS‐1 F929 and N944 were identified as major contributors to Ras binding. 65 Consequently, this SOS‐1 stretch was used in the design of stabilized SOS‐1 helices (aa 929–944, Figure 6A). An N‐cap replaced a backbone hydrogen bond between the i and i + 4 positions with an isosteric covalent carbon–carbon bond. The introduction of this hydrogen bond surrogate (HBS) resulted in peptide HBS SOS (Figure 6B). This peptide bound SOS‐1 with micromolar affinity and inhibited Ras activation as well as subsequent Erk signalling. 66 To increase proteolytic stability β3‐residues (Figure 6B) were introduced along the non‐interacting face of the helix (α 3 βHBS SOS. ). A covalent mode of action was pursued to increase activity. NMR studies indicated that SOS‐1 L938 is located in proximity to the oncogenic Ras variation C12 and thus chosen as site for modifier incorporation (Figure 6C). The selected electrophiles (acrylamides, vinyl sulfonamides, vinyl sulfones) were attached via the side chain of a lysine (Figure 6D). Vinyl sulfonamide (6) was identified as the most promising electrophile, possessing sufficient reactivity and selectivity. Vinyl sulfones were found to be too reactive, in contrast to acrylamides, which did not show meaningful reactivity. Covalent‐inhibitor α 3 βHBS SOS ‐6 was shown to affect viability of H358 lung cancer cells in a Ras G12C‐dependent manner. This example highlights the importance of modifier screening to identify a group with sufficiently balanced reactivity and selectivity.
FIGURE 6.
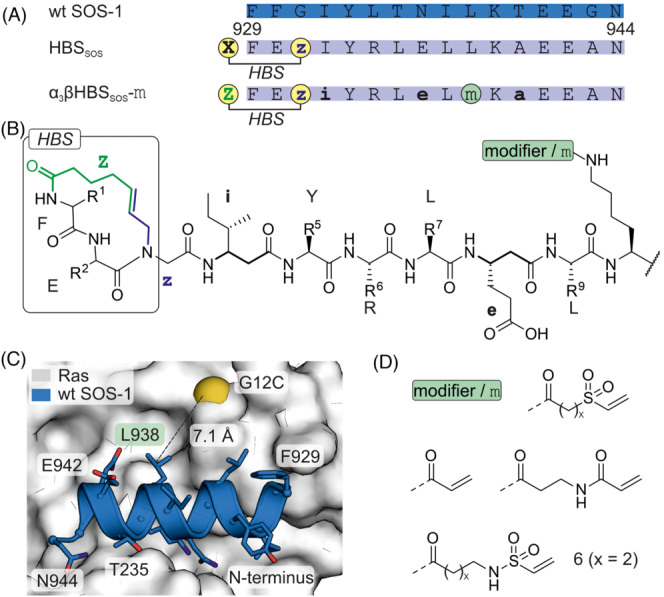
(A) Sequence of wt SOS‐1 and derived inhibitors with modification sites (i: β‐isoleucine, e: β‐glutamic acid, a: β‐alanine, X: 4‐pentenoic acid, Z: 5‐hexenoic acid, z: N‐allylglycine). (B) Chemical structure of the hydrogen bond surrogate (HBS), selected β‐amino acids (bold), position of the modifier m and the HBS are shown. (C) Crystal structure of Ras (white, pdb: 1nvw) in complex with SOS derived peptide (wt SOS, blue, pdb: 1nvw, glycine was varied to cysteine for demonstration purposes). (D) Chemical structure of tested electrophiles (x = 1 or 2).
5. SULFONIUM CROSSLINK
To address the aforementioned protein PDZ‐RGS3 involved in ephrin‐B signalling, another covalent inhibitor targeting a cysteine was reported, 67 , 68 again using the C‐terminal sequence of ephrin‐B as starting point (Figure 7A and B). Notably, an intramolecular sulfonium crosslink was incorporated to stabilize the helical conformation of the peptide ligand and was employed as a cysteine‐directed electrophile (Figure 7C). 69 , 70 The combination of linker (I, Figure 7D) with peptide PD3 (Figure 7A) positioned the reactive sulfonium group in proximity to the target cysteines (C33 and C34, Figure 7B) and consequently resulted in the most promising PDZ‐RGS3 binder (PD3I) that modified PDZΔRGS3 in cells. An analogous strategy was employed to target BFL‐1. 67 , 68 Here, the sulfonium crosslink was incorporated at the N‐terminus of the BIM BH3 α‐helix (Figure 7E) and equipped with different aromatic bridging moieties (Figure 7D, left). The resulting reactive peptides were envisioned to target BFL‐1 C55 in proximity to the binding site (Figure 3B). 69 , 70 Among the tested inhibitors, B4‐MC‐I (Figure 7E) selectively modified BFL‐1 and induced apoptosis in BFL‐1 expressing cell lines.
FIGURE 7.
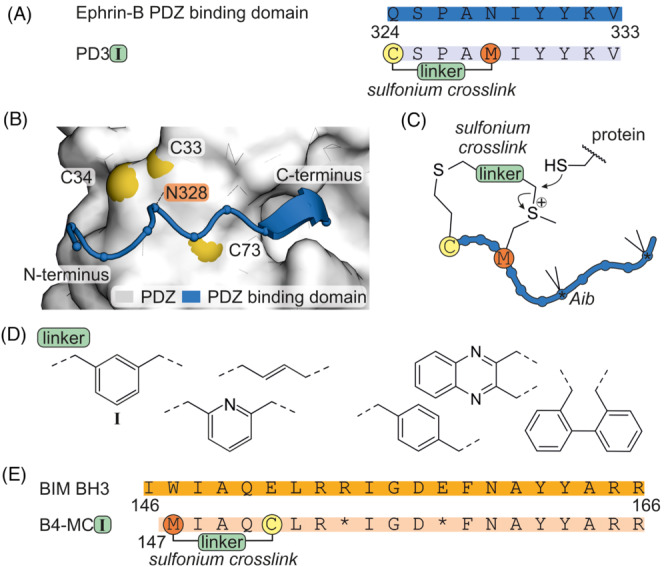
(A) Sequence of PDZ binding domain of ephrin‐B and derived sulfonium crosslinked peptide PD3 with introduced modifications. (B) AlphaFold 71 , 72 prediction of complex between ephrin‐B PDZ binding domain and PDZ. (C) Scheme of sulfonium bridged peptide. (D) Chemical structure of linkers used for crosslinking of ephrin‐B. Only the linkers on the left side were employed in the crosslinking of BIM BH3. (E) Sequence of BIM BH3 and derived sulfonium crosslinked peptide B4‐MCI with location of introduced modifications (*α‐aminoisobutyric acid, Aib).
6. ISOTHIOCYANATE
HIV‐1 infection is initiated by the binding of the gp120/gp41 complex to the target receptor CD4, ultimately leading to virus entry. Gp41 consists of a C‐terminal heptad repeat (CHR, Figure 8A) that folds onto an N‐terminal heptad repeat (NHR) resulting in a six‐helix bundle (6HB). Generation of an inactive bundle by covalent modification of gp41 could thus prevent HIV‐1 infection of host cells. The gp41 crystal structure (Figure 8A) shows a conserved salt bridge between K574 in the NHR and D632 in the CHR that is essential for 6HB formation. To design entry inhibitors, the development of covalent peptidic inhibitors has been pursued. 73 , 74 Notably, one of these examples uses isothiocyanate to target K574 on NHR. 73 For that purpose, CHR‐derived peptide C34 was used as template for inhibitor design and equipped with an isothiocyanate modifier (1) instead of D632 (NC‐C34, Figure 8B). Covalent modification of gp41 NHR K574 thus results in thiourea formation (Figure 8C). To increase the local concentration of the inhibitor at the cell surface, a cholesterol moiety was attached to the C‐terminus of the inhibitor (NC‐C34‐Chol.), which increased the antifusogenic activity in pseudovirus assays.
FIGURE 8.
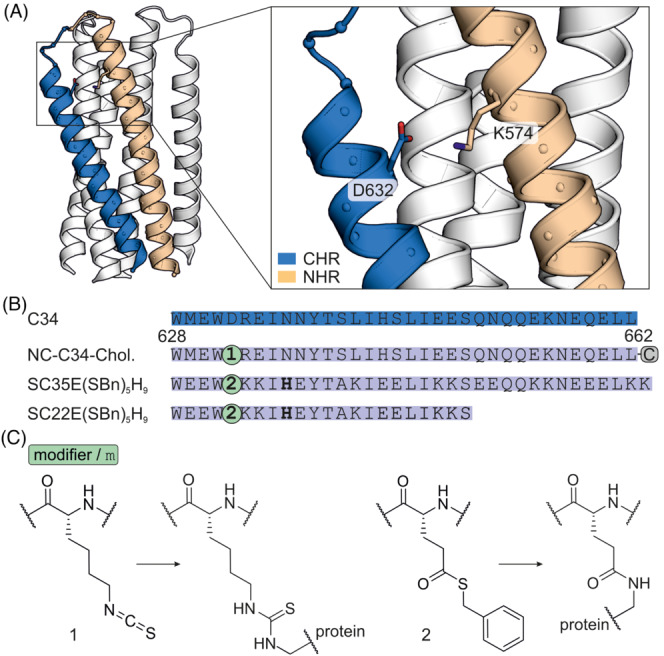
(A) Crystal structure of gp41 helix bundle (pdb: 1szt) with C‐terminal heptad repeat (CHR, blue) and N‐terminal heptad repeat (NHR, beige). 76 (B) Sequence of C34 and derived inhibitors with modifications. SC35E(SBn) 5 H 9 and SC22E(SBn) 5 H 9 have been altered introducing additional salt bridges to stabilize helicity and solubility. The bold histidine residue has been introduced to increase NHR K574 reactivity. C corresponds to a C‐terminally attached cholesterol. (C) Chemical structure of the employed modifiers and their reactivity.
7. THIOESTER
The gp120/gp41 interaction was also addressed using a thioester modifier (2) at position D632 in C34‐derived peptides. 75 The thioester is based on glutamic acid with benzyl mercaptan as leaving group (Figure 8C). Additionally, a His residue was introduced at the i, i + 4 position of the modifier to accelerate the acyl transfer to NHR K574 (SC35E[SBn] 5 H 9 , Figure 8A). Truncation studies revealed that 22‐mer SC22E(SBn) 5 H 9 modified NHR most efficiently in biochemical assays. Notably, in cell‐based assays, these covalent inhibitors showed activities comparable to the non‐reactive analogues, presumably due to very slow reaction kinetics.
8. ARYL SULFONYL FLUORIDE
In various forms of cancer, elevated levels of oncoproteins Mdm2 and Mdm4 prevent the transcriptional activity of p53 by promoting its ubiquitylation and subsequent proteasomal degradation. Inhibition of this interaction thus restores endogenous p53 activity and can reduce tumour growth. Aryl sulfonyl fluorides have been used to design covalent inhibitors targeting lysine and histidine residues in Mdm2 and Mdm4. 77 , 78 For that purpose, the crystal structures of Mdm4 bound to p53 79 (Figure 9A) and of Mdm2 in a complex with stapled peptide SAHp53–8 80 (Figure 9B) were used as starting point. Peptide residue L22 was identified as site closest to lysine and histidine target residues on Mdm2 and Mdm4 (Figure 9A and B) and as such, chosen for modifier incorporation. Aryl sulfonyl fluoride electrophiles with varying phenyl substitution patterns were attached via the Dap side chain (Figure 9D and E). The peptide with a meta‐substituted modifier (four in mSF‐SAH‐4, Figure 9E) showed increased inhibition of p53‐Mdm2/4 interaction in lysates compared to the parental stapled peptide SAHp53–8. Notably, mSF‐SAH‐4 did not affect the viability of p53‐independent Saos‐2 cells, in contrast to the non‐covalent inhibitor SAHp53–8.
FIGURE 9.
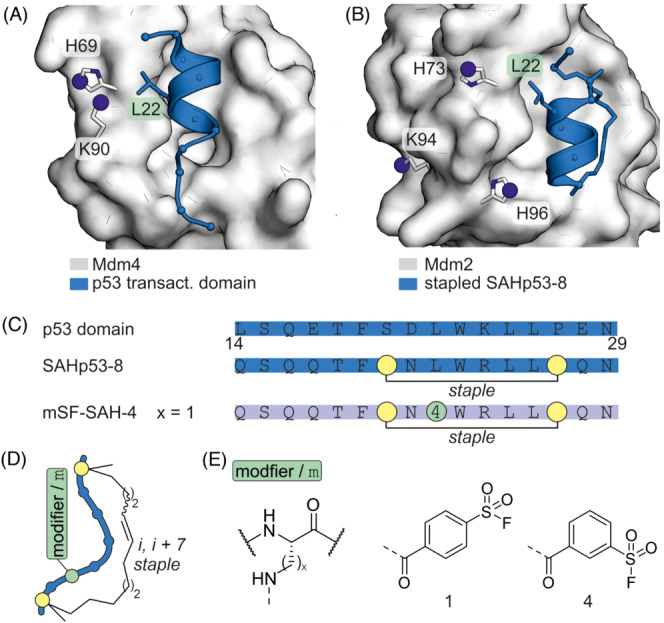
(A) Crystal structure of Mdm4 with p53 transactivation domain (pdb: 3dac). (B) Crystal structure of Mdm2 with SAHp53–8 (pdb: 3v3b). (C) Sequence of p53 (14‐29) and derived inhibitors with modifications. (D) Scheme of i, i + 7 stapled peptide. (E) Chemical structure of tested modifiers (x = 1, 2, 3).
In a series of studies, small peptidic covalent inhibitors of X‐linked inhibitor of apoptosis protein (XIAP) were developed with the intention to trigger programmed cell death. 81 , 82 , 83 , 84 XIAP contains three baculovirus IAP repeats (BIR), with BIR3 inhibiting caspase‐9. Inhibition of the XIAP/caspase 9 interaction results in caspase release and activation. For that purpose, the second mitochondrial activator of caspases (SMAC), which binds XIAP and antagonizes caspase inhibition was used as a scaffold for inhibitor design. The crystal structure of a SMAC‐tetrapeptide (wt) in complex with the XIAP BIR3 domain revealed XIAP K311 as a potential nucleophilic target residue in proximity to the binding site (Figure 10A and B). At position 2, different aryl sulfonyl fluoride‐based modifiers were introduced via the side chain of Dap (Figure 10C) or directly linked to the main chain. Guided by biophysical characterization and computational approaches, the C‐terminus of these SMAC‐derived peptides was replaced by small molecular scaffolds (Figure 10D). The resulting compounds (34, 11, 13) affected cell viability and growth of cancer cells. For instance, compound 34 induced degradation of IAP protein levels and reduced the growth of multiple myeloma‐derived cells. The established design features were later used to address the related protein melanoma‐IAP (ML‐IAP). 84 Ml‐IAP K135 is located in a position equivalent to that of XIAP K311. Introduction of a modifier (4) at position 2 led to an ML‐IAP targeting covalent inhibitor with cellular activity (7, Figure 10B).
FIGURE 10.
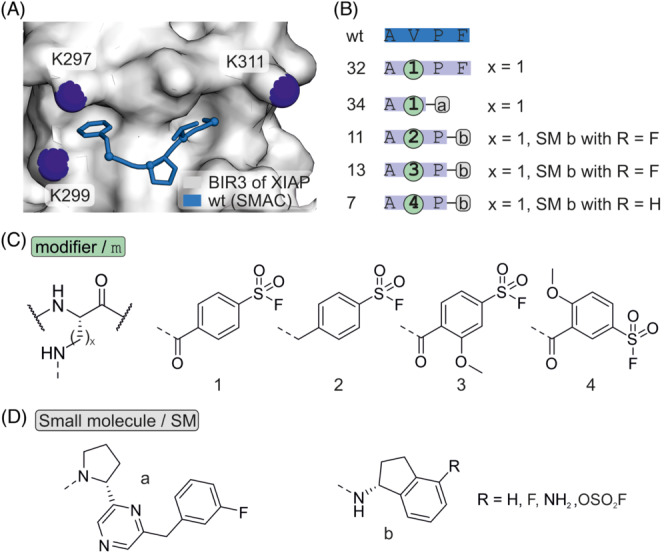
(A) Crystal structure of BIR3 domain of XIAP with SMAC derived peptide AVPF (pdb: 2opz). (B) Sequence of SMAC wt and derived inhibitors with modification sites. (C) Chemical structure of a selection of tested modifiers (x = 1, 2, 3). (D) Chemical structure of N‐terminal modifications.
Aryl sulfonyl fluorides have also been used for the targeting of lysine residues in BCL‐2 family member MCL‐1. 85 The BIM BH3 helix served as the basis for the introduction of a variety of modifications (Figure 11A). Previous structure–activity relationship data and a crystal structure indicated that the Q150E variation increases selectivity for MCL‐1. 86 , 87 The effect of different variations was assessed and peptide 12 (Figure 11B) selected due to its improved affinity and solubility. Notably, shorter peptides were found to retain their binding affinity for MCL‐1 while losing affinity for related BCL‐2 family member BFL‐1. Based on a previous study, 40 , 41 the aryl sulfonyl fluoride modifier was placed at position A147 to provide covalent MCL‐1 inhibitors 15 and 16 (Figure 11C). A crystal structure of inhibitor 15 bound to MCL‐1 was obtained verifying K234 as modification site (Figure 11A). Cell‐based studies with 16 suggested covalent labelling of MCL‐1 and proteasome‐dependent degradation of MCL‐1.
FIGURE 11.
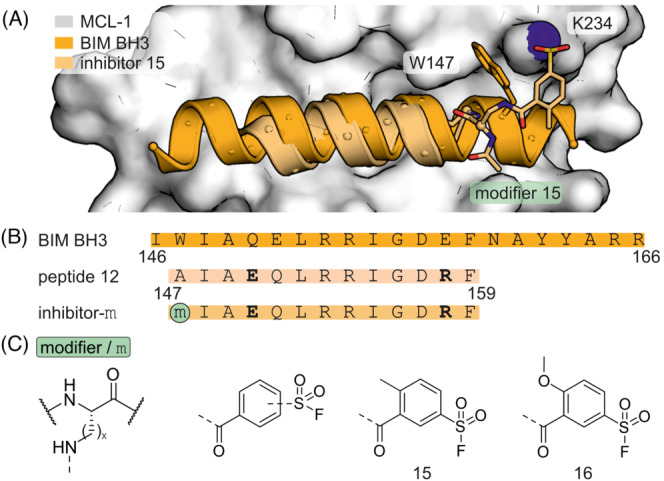
(A) Crystal structure of MCL‐1 (white, pdb: 6vbx) in complex with BIM BH3 peptide (orange, pdb: 2 nl9) or covalent inhibitor 15 (beige, pdb: 6vbx). (B) Sequence of BIM BH3 and derived MCL‐1 inhibitors with modification sites. Additional salt bridges were introduced (bold) to increase helicity. (C) Chemical structure of selected modifiers (x = 1, 2, 3).
9. DIAZOBORINE AND IMINOBORONATE
One way to combine the advantages of reversible and covalent inhibitors is the use of a reversible covalent mode of action. Such inhibitors have a long target residence time whilst not resulting in a permanent covalent modification of the protein. This reduces inhibitor clearance and the risk of immunogenic effects. 22 Diazoborines were used for the design of reversible covalent inhibitors of bacterial Sortase A (SrtA). 88 SrtA is a transpeptidase that attaches surface proteins to the cell wall, and its inhibition has antibacterial effects. In a first step, cyclic SrtA binding peptide W7 was identified and found to exhibit micromolar potency (Figure 12A and B). Modelling of the W7/SrtA complex indicated that the cyclic peptide binds to the protein active site with both termini pointing away from SrtA. Consequently, either N‐ or C‐terminally modified peptides were designed using diazoborine (RMR1) and iminoboronate (APBA) as electrophile (Figure 12C). In both cases, K173 was anticipated as potential modification site on SrtA. The C‐terminal introduction of the RMR1 electrophile resulted in highest affinity inhibitor P5‐RMR1 (Figure 12C). However, the inhibitor P5‐APBA showed more rapid SrtA inhibition, presumably due to the faster reaction kinetics of iminoboronate. Notably, P5‐RMR1 inhibited SrtA transpeptidase activity in Staphylococcus aureus even after extensive washout. Here, P5‐APBA showed a more pronounced loss in activity, most likely due to faster off‐rates which promote washout. Moreover, reversible covalent inhibitors have also been identified in a phage display format. 89 , 90 Here, screening of a cyclic peptide library modified with APBA again led to the discovery of potent SrtA inhibitors (Figure 12D). 89 In the same fashion, a reversible covalent binder of the receptor binding domain (RBD) of the SARS‐CoV‐2 spike protein was identified. Spike interaction with the human ACE2 receptor is well characterized and is an important step in coronavirus infection. A cyclic phage library was constructed using a 9‐mer peptide scaffold which was modified with APBA‐3. This led to the discovery of peptide R1_APBA‐3 which exhibited potent binding to spike RBD in human serum.
FIGURE 12.
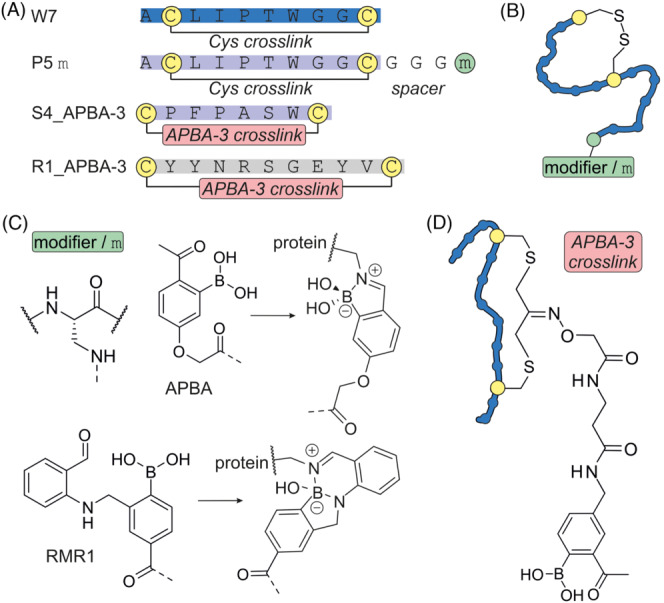
(A) Sequences of reversible covalent inhibitors. (B) Scheme of cyclized W7‐derived inhibitors. (C) Chemical structures of selected reversible modifiers in their bound and unbound state. (D) Scheme of cyclized binders of SrtA and spike RBD including chemical structures of crosslink and modifier APBA‐3.
10. BROMOACETAMIDE AND OTHERS
The bacterial divisome is a complex of membrane (associated) proteins assembling at the mid‐cell plane and orchestrating the organization of cell division with minor interferences leading to cell death. The divisome proteins FtsQ, FtsB and FtsL form a complex which is crucial for bacterial cell division. Inhibition of this interaction is considered an attractive antibiotic target. 91 , 92 , 93 , 94 Based on the crystal structure of a FtsB fragment bound to the periplasmic domain of FtsQ, 91 proteomimetic ligands were designed (Figure 13A). Notably, the stabilization of a tertiary motif 95 , 96 , 97 was necessary to achieve meaningful binding. 98 Here, an essential intramolecular salt bridge between FtsB R72 and E82 was replaced by a covalent crosslink providing 24‐mer cyclic peptide 24f (Figure 13B). 98 An MD‐simulation of the 24f‐FtsQ complex (Figure 13C) suggested 24f T83 as a suitable position to target K239 on FtsQ. Subsequently, a variety of electrophiles were incorporated at position 83 (24 fm, Figure 13D). Screening led to the identification of four modifiers that resulted in the most pronounced FtsQ modification: bromoacetamide (α), acrylamide (β), vinyl sulfonamide (γ) and dichlorotriazine (δ). The size of the 24‐mer peptide ligand was reduced to a 17‐mer, which in combination with bromoacetamide (17fα) showed the strongest growth inhibition of outer membrane permeable Escherichia coli strain lptD4213. Notably, it also attenuated the spread of multi‐drug resistant E. coli 87 in a zebra fish larvae infection model and increased their survival in combination with a potentiator peptide. 98 , 99
FIGURE 13.
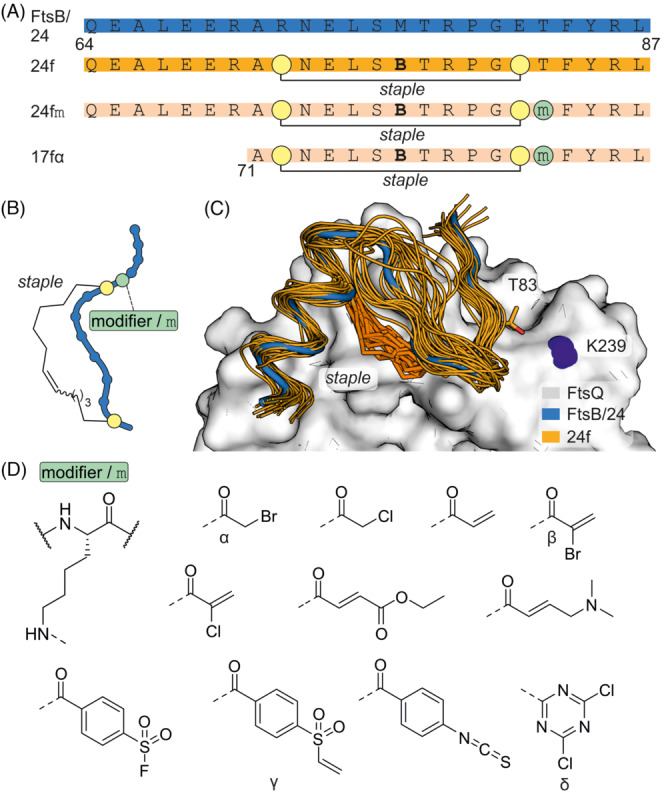
(A) Sequence of FtsB and different FtsB derived peptides (B: Norleucine). (B) Scheme of cyclized FtsB‐derived peptides including chemical structure of hydrocarbon crosslink. (C) MD simulation of 24f bound to FtsQ. (D) Chemical structures of tested modifiers.
11. CONCLUSION
In many case, targeting of PPI interfaces with competitive small molecule inhibitors is not feasible. Peptide‐based inhibitors with an irreversible mode of action have proved useful to address such targets. This involves diverse target proteins that had previously been considered unaddressable and range from cancer‐related signalling hubs to viral receptors. Most of the covalent inhibitors with confirmed bioactivity described herein were designed in a structure‐based process that facilitated the identification of suitable nucleophilic residues on the target protein and thereby guided the incorporation of covalent modifiers. The design process can also be supported by computational tools. 100 Studies that compare the performance of different electrophiles indicate that the identification of a suitable electrophile with an optimal balance between on‐ and off‐target reactivity may not be predictable and therefore requires screening. This is due to the fact that the nature of the peptide ligand and the precise cellular context influence overall reactivity. 48 , 64 , 98 The use of peptide‐based targeted covalent inhibitors is an emerging field and various additional challenges remain to be solved. For example, the prediction of suitable target residues is not straight‐forward as the protein microenvironment influences side chain reactivity. In addition, so far mainly lysine, cysteine and histidine have been addressed calling for an extension of targetable amino acids. Here, inspiration can be taken from small‐molecule covalent inhibitors. 20 Another limitation is related to the intrinsically low cellular uptake of most peptide‐based ligands, which is usually addressed by the reduction of inhibitor size or the attachment of a CPP sequence. However, only a few of these inhibitors have been tested in vivo, and it is not clear if these strategies support sufficient cellular uptake. Therefore, more in vivo studies are required to further explore the applicability of peptide‐based covalent inhibitors. These studies will also reveal which types of electrophiles are most suitable for peptide‐based covalent inhibitors with in vivo activity. Taken together, peptidic covalent inhibitors uniquely combine the enhanced surface‐recognition properties of proteins with an irreversible binding mode, and they have already proven useful to target unaddressable proteins. This class of molecules represents an emerging modality that may have the potential to provide access to therapeutics addressing so far undruggable targets.
ACKNOWLEDGEMENT
F.P. received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant (agreement no. 713669).
Biographies
Felix M. Paulussen received his MSc degree from the Technical University Dortmund in 2018. Since then, he has pursued his PhD in the groups of Tom Grossmann and Joen Luirink at the Vrije Universiteit Amsterdam working on the development of peptide‐inspired antimicrobials with novel mode of action.

Tom N. Grossmann is professor of organic chemistry at the Vrije Universiteit Amsterdam since 2016. He obtained his PhD at the Humboldt‐Universität zu Berlin in the group of Oliver Seitz and, in 2009, joined Gregory L. Verdine's lab at the Harvard University for postdoctoral research. He then started as a group leader at the Chemical Genomics Centre and the Technical University in Dortmund before his group moved to Amsterdam.

Paulussen FM, Grossmann TN. Peptide‐based covalent inhibitors of protein–protein interactions. J Pept Sci. 2023;29(1):e3457. doi: 10.1002/psc.3457
This publication is dedicated to Prof. Ulf Diederichsen.
Funding information H2020 Marie Skłodowska‐Curie Actions, Grant/Award Number: agreement no. 71366
REFERENCES
- 1. Amaral LAN. A truer measure of our ignorance. Proc Natl Acad Sci USA. 2008;105(19):6795‐6796. doi: 10.1073/pnas.0802459105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rolland T, Taşan M, Charloteaux B, et al. A proteome‐scale map of the human interactome network. Cell. 2014;159(5):1212‐1226. doi: 10.1016/j.cell.2014.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Las Rivas J, Fontanillo C. Protein–protein interactions essentials: key concepts to building and analyzing Interactome networks. PLoS Comput Biol. 2010;6(6):e1000807. doi: 10.1371/journal.pcbi.1000807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727‐730. doi: 10.1038/nrd892 [DOI] [PubMed] [Google Scholar]
- 5. Milroy L‐G, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein–protein interactions. Chem Rev. 2014;114(9):4695‐4748. doi: 10.1021/cr400698c [DOI] [PubMed] [Google Scholar]
- 6. Pelay‐Gimeno M, Glas A, Koch O, Grossmann TN. Structure‐based design of inhibitors of protein‐protein interactions: mimicking peptide binding epitopes. Angew Chemie Int Ed. 2015;54(31):8896‐8927. doi: 10.1002/anie.201412070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Villoutreix BO, Kuenemann MA, Poyet JL, et al. Drug‐like protein‐protein interaction modulators: challenges and opportunities for drug discovery and chemical biology. Mol Inform. 2014;33(6‐7):414‐437. doi: 10.1002/minf.201400040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laraia L, McKenzie G, Spring DR, Venkitaraman AR, Huggins DJ. Overcoming chemical, biological, and computational challenges in the development of inhibitors targeting protein‐protein interactions. Chem Biol. 2015;22(6):689‐703. doi: 10.1016/j.chembiol.2015.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bakail M, Ochsenbein F. Targeting protein–protein interactions, a wide open field for drug design. Comptes Rendus Chim. 2016;19(1–2):19‐27. doi: 10.1016/j.crci.2015.12.004 [DOI] [Google Scholar]
- 10. Zhang X‐X, Eden HS, Chen X. Peptides in Cancer nanomedicine: drug carriers, targeting ligands and protease substrates. J Control Release. 2012;159(1):2‐13. doi: 10.1016/j.jconrel.2011.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α‐helix‐mediated protein–protein interactions using designed molecules. Nat Chem. 2013;5(3):161‐173. doi: 10.1038/nchem.1568 [DOI] [PubMed] [Google Scholar]
- 12. Glas A, Bier D, Hahne G, Rademacher C, Ottmann C, Grossmann TN. Constrained peptides with target‐adapted cross‐links as inhibitors of a pathogenic protein‐protein interaction. Angew Chemie ‐ Int Ed. 2014;53(9):2489‐2493. doi: 10.1002/anie.201310082 [DOI] [PubMed] [Google Scholar]
- 13. Horne WS, Grossmann TN. Proteomimetics as protein‐inspired scaffolds with defined tertiary folding patterns. Nat Chem. 2020;12(4):331‐337. doi: 10.1038/s41557-020-0420-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cromm PM, Spiegel J, Grossmann TN. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem Biol. 2015;10(6):1362‐1375. doi: 10.1021/cb501020r [DOI] [PubMed] [Google Scholar]
- 15. Avrutina O, Schmoldt H‐U, Kolmar H, Diederichsen U. Fmoc‐assisted synthesis of a 29‐residue cystine‐knot trypsin inhibitor containing a guaninyl amino acid at the P1‐position. Eur J Org Chem. 2004;2004(23):4931‐4935. doi: 10.1002/ejoc.200400440 [DOI] [Google Scholar]
- 16. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533‐552. doi: 10.1038/s41573-020-0068-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6(1):386 doi: 10.1038/s41392-021-00780-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ray S, Murkin AS. New electrophiles and strategies for mechanism‐based and targeted covalent inhibitor design. Biochemistry. 2019;58(52):5234‐5244. doi: 10.1021/acs.biochem.9b00293 [DOI] [PubMed] [Google Scholar]
- 19. Berdan VY, Klauser PC, Wang L. Covalent peptides and proteins for therapeutics. Bioorg Med Chem. 2021;29:115896. doi: 10.1016/j.bmc.2020.115896 [DOI] [PubMed] [Google Scholar]
- 20. Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022; doi: 10.1038/s41573-022-00542-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee C‐U, Hahne G, Hanske J, et al. Redox modulation of PTEN phosphatase activity by hydrogen peroxide and bisperoxidovanadium complexes. Angew Chem Int Ed. 2015;54(46):13796‐13800. doi: 10.1002/anie.201506338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee C‐U, Grossmann TN. Reversible covalent inhibition of a protein target. Angew Chem Int Ed. 2012;51(35):8699‐8700. doi: 10.1002/anie.201203341 [DOI] [PubMed] [Google Scholar]
- 23. Chandrasekharan NV, Simmons DL. The cyclooxygenases. Genome Biol. 2004;5(9):241. doi: 10.1186/gb-2004-5-9-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin‐like drugs. Nat New Biol. 1971;231(25):232‐235. doi: 10.1038/newbio231232a0 [DOI] [PubMed] [Google Scholar]
- 25. Hatcher JM, Du G, Fontán L, et al. Peptide‐based covalent inhibitors of MALT1 paracaspase. Bioorg Med Chem Lett. 2019;29(11):1336‐1339. doi: 10.1016/j.bmcl.2019.03.046 [DOI] [PubMed] [Google Scholar]
- 26. Pinch BJ, Doctor ZM, Nabet B, et al. Identification of a potent and selective covalent Pin1 inhibitor. Nat Chem Biol. 2020;16(9):979‐987. doi: 10.1038/s41589-020-0550-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu N, Zhang Y, Lei Y, et al. Design and evaluation of a novel peptide–drug conjugate covalently targeting SARS‐CoV‐2 papain‐like protease. J Med Chem. 2022;65(1):876‐884. doi: 10.1021/acs.jmedchem.1c02022 [DOI] [PubMed] [Google Scholar]
- 28. Baillie TA. Targeted covalent inhibitors for drug design. Angew Chem Int Ed. 2016;55(43):13408‐13421. doi: 10.1002/anie.201601091 [DOI] [PubMed] [Google Scholar]
- 29. Lonsdale R, Ward RA. Structure‐based design of targeted covalent inhibitors. Chem Soc Rev. 2018;47(11):3816‐3830. doi: 10.1039/C7CS00220C [DOI] [PubMed] [Google Scholar]
- 30. Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther. 2022;7(1):48. doi: 10.1038/s41392-022-00904-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh J. The ascension of targeted covalent inhibitors. J Med Chem. 2022;65(8):5886‐5901. doi: 10.1021/acs.jmedchem.1c02134 [DOI] [PubMed] [Google Scholar]
- 32. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307‐317. doi: 10.1038/nrd3410 [DOI] [PubMed] [Google Scholar]
- 33. Gehringer M, Laufer SA. Emerging and re‐emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673‐5724. doi: 10.1021/acs.jmedchem.8b01153 [DOI] [PubMed] [Google Scholar]
- 34. Way J. Covalent modification as a strategy to block protein–protein interactions with small‐molecule drugs. Curr Opin Chem Biol. 2000;4(1):40‐46. doi: 10.1016/S1367-5931(99)00049-6 [DOI] [PubMed] [Google Scholar]
- 35. Stebbins JL, Santelli E, Feng Y, et al. Structure‐based design of covalent Siah inhibitors. Chem Biol. 2013;20(8):973‐982. doi: 10.1016/j.chembiol.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Möller A, House CM, Wong CSF, et al. Inhibition of Siah ubiquitin ligase function. Oncogene. 2009;28(2):289‐296. doi: 10.1038/onc.2008.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. House CM, Frew IJ, Huang H‐L, et al. A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA USA. 2003;100(6):3101‐3106. doi: 10.1073/pnas.0534783100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. House CM, Hancock NC, Möller A, et al. Elucidation of the substrate binding site of Siah ubiquitin ligase. Structure. 2006;14(4):695‐701. doi: 10.1016/j.str.2005.12.013 [DOI] [PubMed] [Google Scholar]
- 39. Santelli E, Leone M, Li C, et al. Structural analysis of Siah1‐Siah‐interacting protein interactions and insights into the assembly of an E3 ligase multiprotein complex. J Biol Chem. 2005;280(40):34278‐34287. doi: 10.1074/jbc.M506707200 [DOI] [PubMed] [Google Scholar]
- 40. Huhn AJ, Guerra RM, Harvey EP, Bird GH, Walensky LD. Selective covalent targeting of anti‐apoptotic BFL‐1 by cysteine‐reactive stapled peptide inhibitors. Cell Chem Biol. 2016;23(9):1123‐1134. doi: 10.1016/j.chembiol.2016.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stewart ML, Fire E, Keating AE, Walensky LD. The MCL‐1 BH3 Helix Is an exclusive MCL‐1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6(8):6‐601. doi: 10.1038/nCHeMBIO.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walensky LD, Kung AL, Escher I, et al. Activation of apoptosis in vivo by a hydrocarbon‐stapled BH3 helix. Science (80‐.). 2004;305(5689):1466‐1470. doi: 10.1126/science.1099191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walensky LD, Pitter K, Morash J, et al. A stapled BID BH3 Helix directly binds and activates BAX. Mol Cell. 2006;24(2):199‐210. doi: 10.1016/j.molcel.2006.08.020 [DOI] [PubMed] [Google Scholar]
- 44. Kim Y‐W, Grossmann TN, Verdine GL. Synthesis of all‐hydrocarbon stapled α‐helical peptides by ring‐closing olefin metathesis. Nat Protoc. 2011;6(6):761‐771. doi: 10.1038/nprot.2011.324 [DOI] [PubMed] [Google Scholar]
- 45. Harvey EP, Seo HS, Guerra RM, Bird GH, Dhe‐Paganon S, Walensky LD. Crystal structures of anti‐apoptotic BFL‐1 and its complex with a covalent stapled peptide inhibitor. Structure. 2018;26(1):153‐160.e4. doi: 10.1016/j.str.2017.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herman MD, Nyman T, Welin M, et al. Completing the family portrait of the anti‐apoptotic Bcl‐2 proteins: crystal structure of human Bfl‐1 in complex with Bim. FEBS Lett. 2008;582(25‐26):3590‐3594. doi: 10.1016/j.febslet.2008.09.028 [DOI] [PubMed] [Google Scholar]
- 47. de Araujo AD, Lim J, Good AC, Skerlj RT, Fairlie DP. Electrophilic helical peptides that bond covalently, irreversibly, and selectively in a protein−protein interaction site. ACS Med Chem Lett. 2017;8(1):22‐26. doi: 10.1021/acsmedchemlett.6b00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Araujo AD, Lim J, Wu K‐C, et al. Bicyclic helical peptides as dual inhibitors selective for Bcl2A1 and mcl‐1 proteins. J Med Chem. 2018;61(7):2962‐2972. doi: 10.1021/acs.jmedchem.8b00010 [DOI] [PubMed] [Google Scholar]
- 49. Brauckhoff N, Hahne G, Yeh JTH, Grossmann TN. Protein‐templated peptide ligation. Angew Chem ‐ Int Ed. 2014;53(17):4337‐4340. doi: 10.1002/anie.201400681 [DOI] [PubMed] [Google Scholar]
- 50. Neubacher S, Saya JM, Amore A, Grossmann TN. In situ cyclization of proteins (INCYPRO): cross‐link derivatization modulates protein stability. J Org Chem. 2020;85(3):1476‐1483. doi: 10.1021/acs.joc.9b02490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pelay‐Gimeno M, Bange T, Hennig S, Grossmann TN. In situ cyclization of native proteins: structure‐based design of a bicyclic enzyme. Angew Chem. 2018;130(35):11334‐11340. doi: 10.1002/ange.201804506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blosser SL, Sawyer N, Maksimovic I, Ghosh B, Arora PS. Covalent and noncovalent targeting of the Tcf4/β‐catenin strand interface with β‐hairpin mimics. ACS Chem Biol. 2021;16(8):1518‐1525. doi: 10.1021/acschembio.1c00389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baggio C, Udompholkul P, Gambini L, et al. N‐locking stabilization of covalent helical peptides: application to Bfl‐1 antagonists. Chem Biol Drug Des. 2020;95(4):412‐426. doi: 10.1111/cbdd.13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barile E, Marconi GD, De SK, et al. HBfl‐1/HNOXA interaction studies provide new insights on the role of Bfl‐1 in cancer cell resistance and for the design of novel anticancer agents. ACS Chem Biol. 2017;12(2):444‐455. doi: 10.1021/acschembio.6b00962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu Y, Liu M, Ng TT, et al. PDZ‐reactive peptide activates ephrin‐B reverse signaling and inhibits neuronal chemotaxis. ACS Chem Biol. 2016;11(1):149‐158. doi: 10.1021/acschembio.5b00889 [DOI] [PubMed] [Google Scholar]
- 56. Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin‐B reverse signaling is mediated by a novel PDZ‐RGS protein and selectively inhibits G protein–coupled chemoattraction. Cell. 2001;105(1):69‐79. doi: 10.1016/S0092-8674(01)00297-5 [DOI] [PubMed] [Google Scholar]
- 57. Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans‐activator protein. Cell. 1988;55(6):1179‐1188. doi: 10.1016/0092-8674(88)90262-0 [DOI] [PubMed] [Google Scholar]
- 58. Dietrich L, Rathmer B, Ewan K, et al. Cell permeable stapled peptide inhibitor of Wnt signaling that targets β‐catenin protein‐protein interactions. Cell Chem Biol. 2017;24(8):958‐968.e5. doi: 10.1016/j.chembiol.2017.06.013 [DOI] [PubMed] [Google Scholar]
- 59. Adihou H, Gopalakrishnan R, Förster T, et al. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat Commun. 2020;11:5425. doi: 10.1038/S41467-020-19224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stiller C, Krüger DM, Brauckhoff N, et al. Translocation of an intracellular protein via peptide‐directed ligation. ACS Chem Biol. 2017;12(2):504‐509. doi: 10.1021/acschembio.6b01013 [DOI] [PubMed] [Google Scholar]
- 61. Brüschweiler S, Konrat R, Tollinger M. Allosteric communication in the KIX domain proceeds through dynamic repacking of the hydrophobic core. ACS Chem Biol. 2013;8(7):1600‐1610. doi: 10.1021/cb4002188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Small‐molecule modulation of Ras signaling. Nat Chem Biol. 2014;10(8):613‐622. doi: 10.1038/nchembio.1560 [DOI] [PubMed] [Google Scholar]
- 63. Yu Y, Nie Y, Feng Q, et al. Targeted covalent inhibition of Grb2‐Sos1 interaction through proximity‐induced conjugation in breast cancer cells. Mol Pharm. 2017;14(5):1548‐1557. doi: 10.1021/acs.molpharmaceut.6b00952 [DOI] [PubMed] [Google Scholar]
- 64. Yoo DY, Hauser AD, Joy ST, Bar‐Sagi D, Arora PS. Covalent targeting of Ras G12C by rationally designed peptidomimetics. ACS Chem Biol. 2020;15(6):1604‐1612. doi: 10.1021/acschembio.0c00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Patgiri A, Yadav KK, Arora PS, Bar‐Sagi D. An orthosteric inhibitor of the Ras‐Sos interaction. Nat Chem Biol. 2011;7(9):585‐587. doi: 10.1038/nchembio.612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Douse CH, Maas SJ, Thomas JC, et al. Crystal structures of stapled and hydrogen bond surrogate peptides targeting a fully buried protein‐helix interaction. ACS Chem Biol. 2014;9(10):2204‐2209. doi: 10.1021/cb500271c [DOI] [PubMed] [Google Scholar]
- 67. Shi X, Zhao R, Jiang Y, et al. Reversible stapling of unprotected peptides via chemoselective methionine bis‐alkylation/dealkylation. Chem Sci. 2018;9(12):3227‐3232. doi: 10.1039/C7SC05109C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang D, Yu M, Liu N, et al. A sulfonium tethered peptide ligand rapidly and selectively modifies protein cysteine in vicinity. Chem Sci. 2019;10(19):4966‐4972. doi: 10.1039/c9sc00034h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lin H, Jiang Y, Zhang Q, Hu K, Li Z. An in‐tether sulfilimine chiral center induces helicity in short peptides. Chem Commun. 2016;52(68):10389‐10391. doi: 10.1039/c6cc04508a [DOI] [PubMed] [Google Scholar]
- 70. Liu N, Wang D, Lian C, et al. Selective covalent targeting of anti‐apoptotic BFL‐1 by a sulfonium‐tethered peptide. Chembiochem. 2021;22(2):340‐344. doi: 10.1002/cbic.202000473 [DOI] [PubMed] [Google Scholar]
- 71. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Varadi M, Anyango S, Deshpande M, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50(D1):D439‐D444. doi: 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao L, Tong P, Chen Y‐X, et al. A multi‐functional peptide as an HIV‐1 entry inhibitor based on self‐concentration, recognition, and covalent attachment. Org Biomol Chem. 2012;10(32):6512‐6520. doi: 10.1039/c2ob25853f [DOI] [PubMed] [Google Scholar]
- 74. Jacobs A, Quraishi O, Huang X, et al. A covalent inhibitor targeting an intermediate conformation of the fusogenic subunit of the HIV‐1 envelope complex. J Biol Chem. 2007;282(44):32406‐32413. doi: 10.1074/jbc.M705577200 [DOI] [PubMed] [Google Scholar]
- 75. Bai Y, Xue H, Wang K, et al. Covalent fusion inhibitors targeting HIV‐1 Gp41 deep pocket. Amino Acids. 2013;44(2):701‐713. doi: 10.1007/s00726-012-1394-8 [DOI] [PubMed] [Google Scholar]
- 76. Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV‐1 Gp41. Proc Natl Acad Sci USA. 1997;94(23):12303‐12308. doi: 10.1073/pnas.94.23.12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hoppmann C, Wang L. Proximity‐enabled bioreactivity to generate covalent peptide inhibitors of P53‐Mdm4. Chem Commun. 2016;52(29):5140‐5143. doi: 10.1039/c6cc01226d [DOI] [PubMed] [Google Scholar]
- 78. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the P53 tumor suppressor pathway by a stapled P53 peptide. J Am Chem Soc. 2007;129(9):2456‐2457. doi: 10.1021/ja0693587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Popowicz G, Czarna A, Holak T. Structure of the human Mdmx protein bound to the P53 tumor suppressor transactivation domain. Cell Cycle. 2008;7(15):2441‐2443. doi: 10.4161/cc.6365 [DOI] [PubMed] [Google Scholar]
- 80. Baek S, Kutchukian PS, Verdine GL, et al. Structure of the stapled P53 peptide bound to Mdm2. J Am Chem Soc. 2012;134(1):103‐106. doi: 10.1021/ja2090367 [DOI] [PubMed] [Google Scholar]
- 81. Baggio C, Gambini L, Udompholkul P, et al. Design of potent pan‐IAP and Lys‐covalent XIAP selective inhibitors using a thermodynamics driven approach. J Med Chem. 2018;61(14):6350‐6363. doi: 10.1021/acs.jmedchem.8b00810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gambini L, Baggio C, Udompholkul P, et al. Covalent inhibitors of protein–protein interactions targeting lysine, tyrosine, or histidine residues. J Med Chem. 2019;62(11):5616‐5627. doi: 10.1021/acs.jmedchem.9b00561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gambini L, Udompholkul P, Salem AF, Baggio C, Pellecchia M. Stability and cell permeability of sulfonyl fluorides in the design of Lys‐covalent antagonists of protein‐protein interactions. ChemMedChem. 2020;15(22):2176‐2184. doi: 10.1002/cmdc.202000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Udompholkul P, Baggio C, Gambini L, Alboreggia G, Pellecchia M. Lysine covalent antagonists of melanoma inhibitors of apoptosis protein. J Med Chem. 2021;64(21):16147‐16158. doi: 10.1021/acs.jmedchem.1c01459 [DOI] [PubMed] [Google Scholar]
- 85. Gambini L, Udompholkul P, Baggio C, et al. Design, synthesis, and structural characterization of lysine covalent BH3 peptides targeting mcl‐1. J Med Chem. 2021;64(8):4903‐4912. doi: 10.1021/acs.jmedchem.1c00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hadji A, Schmitt GK, Schnorenberg MR, et al. Preferential targeting of MCL‐1 by a hydrocarbon‐stapled BIM BH3 peptide. Oncotarget. 2019;10(58):6219‐6233. doi: 10.18632/oncotarget.27262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Foight GW, Ryan JA, Gullá SV, Letai A, Keating AE. Designed BH3 peptides with high affinity and specificity for targeting mcl‐1 in cells. ACS Chem Biol. 2014;9(9):1962‐1968. doi: 10.1021/cb500340w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Reja RM, Wang W, Lyu Y, Haeffner F, Gao J. Lysine‐targeting reversible covalent inhibitors with Long residence time. J Am Chem Soc. 2022;144(3):1152‐1157. doi: 10.1021/jacs.1c12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zheng M, Chen F‐J, Li K, Reja RM, Haeffner F, Gao J. Lysine‐targeted reversible covalent ligand discovery for proteins via phage display. J Am Chem Soc. 2022;144(34):15885‐15893. doi: 10.1021/jacs.2c07375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen S, Lovell S, Lee S, Fellner M, Mace PD, Bogyo M. Identification of highly selective covalent inhibitors by phage display. Nat Biotechnol. 2021;39(4):490‐498. doi: 10.1038/s41587-020-0733-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kureisaite‐Ciziene D, Varadajan A, McLaughlin SH, et al. Structural analysis of the interaction between the bacterial cell division proteins FtsQ and FtsB. mBio. 2018;9(5):1‐17. doi: 10.1128/mBio.01346-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Glas M, van den Berg van Saparoea HB, McLaughlin SH, et al. The soluble periplasmic domains of Escherichia Coli cell division proteins FtsQ/FtsB/FtsL form a trimeric complex with submicromolar affinity. J Biol Chem. 2015;290(35):21498‐21509. doi: 10.1074/jbc.M115.654756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. van den Berg van Saparoea HB, Glas M, Vernooij IGWH, Bitter W, den Blaauwen T, Luirink J. Fine‐mapping the contact sites of the Escherichia Coli cell division proteins FtsB and FtsL on the FtsQ protein. J Biol Chem. 2013;288(34):24340‐24350. doi: 10.1074/jbc.M113.485888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. den Blaauwen T, Luirink J. Checks and balances in bacterial cell division. mBio. 2019;10(1):e00149‐19. doi: 10.1128/mBio.00149-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Haim A, Neubacher S, Grossmann TN. Protein macrocyclization for tertiary structure stabilization. Chembiochem. 2021;22(17):2672‐2679. doi: 10.1002/CBIC.202100111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wendt M, Bellavita R, Gerber A, et al. Bicyclic Β‐sheet mimetics that target the transcriptional coactivator Β‐catenin and inhibit Wnt signaling. Angew Chem Int Ed. 2021;60(25):13937‐13944. doi: 10.1002/anie.202102082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kuepper A, McLoughlin NM, Neubacher S, et al. Constrained peptides mimic a viral suppressor of RNA silencing. Nucleic Acids Res. 2021;49(22):12622‐12633. doi: 10.1093/nar/gkab1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Paulussen FM, Schouten GK, Moertl C, et al. Covalent proteomimetic inhibitor of the bacterial FtsQB divisome complex. J Am Chem Soc. 2022;144(33):15303‐15313. doi: 10.1021/jacs.2c06304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schouten GK, Paulussen FM, Kuipers OP, Bitter W, Grossmann TN, van Ulsen P. Stapling of peptides potentiates: the antibiotic treatment of Acinetobacter baumannii in vivo. Antibiotics. 2022;11(2):273. doi: 10.3390/antibiotics11020273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tivon B, Gabizon R, Somsen BA, Cossar PJ, Ottmann C, London N. Covalent flexible peptide docking in Rosetta. Chem Sci. 2021;12(32):10836‐10847. doi. 10.1039/d1sc02322e [DOI] [PMC free article] [PubMed] [Google Scholar]