

Abstract
Objective
Primary Sjögren's syndrome (SS) is the second most frequent systemic autoimmune disease, affecting 0.1% of the general population. To characterize the molecular and clinical variabilities among patients with primary SS, we integrated transcriptomic, proteomic, cellular, and genetic data with clinical phenotypes in a cohort of 351 patients with primary SS.
Methods
We analyzed blood transcriptomes and genotypes of 351 patients with primary SS who were participants in a multicenter prospective clinical cohort. We replicated the transcriptome analysis in 3 independent cohorts (n = 462 patients). We determined circulating interferon‐α (IFNα) and IFNγ protein concentrations using digital single molecular arrays (Simoa).
Results
Transcriptome analysis of the prospective cohort showed a strong IFN gene signature in more than half of the patients; this finding was replicated in the 3 independent cohorts. Because gene expression analysis did not discriminate between type I IFN and type II IFN, we used Simoa to demonstrate that the IFN transcriptomic signature was driven by circulating IFNα and not by IFNγ protein levels. IFNα protein levels, detectable in 75% of patients, were significantly associated with clinical and immunologic features of primary SS disease activity at enrollment and with increased frequency of systemic complications over the 5‐year follow‐up. Genetic analysis revealed a significant association between IFNα protein levels, a major histocompatibility (MHC) class II haplotype, and anti‐SSA antibody. Additional cellular analysis revealed that an MHC class II HLA–DQ locus acts through up‐regulation of HLA class II molecules on conventional dendritic cells.
Conclusion
We identified the predominance of IFNα as a driver of primary SS variability, with IFNα demonstrating an association with HLA gene polymorphisms.
INTRODUCTION
Primary Sjögren's syndrome (SS) is a systemic autoimmune disease affecting 0.1% of the general population (1) that mainly targets the exocrine system, such as the salivary and lachrymal glands. The clinical presentation of primary SS is highly heterogeneous. Fatigue, dryness, and pain are hallmarks of the disease, but one‐third to one‐half of patients develop systemic complications (notably, articular involvement, lung involvement, peripheral neuropathy, vasculitis), and 5–10% develop mucosa‐associated lymphoid tissue–type lymphoma (2). No clinical biomarker is currently available to identify the patients with primary SS at risk of systemic complications.
To date, no specific immunomodulatory drug has demonstrated efficacy for primary SS. Disappointing results from randomized clinical trials (3, 4, 5) can be attributed to our current lack of understanding of the pathogenesis and molecular basis of this disease and to the clinical and biologic heterogeneity of the patients.
In this study, we aimed to 1) identify molecular endotypes of the disease associated with clinical phenotypes and serum biomarkers that might provide therapeutic guidance in a precision medicine approach, 2) identify major physiologic correlates of the molecular endotypes, and 3) determine possible genetic associations with the molecular endotypes.
PATIENTS AND METHODS
Description of cohorts of patients with primary SS
The Assessment of Systemic Signs and Evolution in Sjögren's Syndrome (ASSESS) cohort is a multicenter prospective French clinical cohort (6) (Supplementary Figure 1A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). ASSESS enrolled 395 patients (see Appendix A for a list of the study investigators) (see Supplementary Methods, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265).
We performed molecular stratification of patients from the ASSESS cohort, based on transcriptomics. To replicate our findings, we repeated our transcriptome analysis in an independent cohort of patients with primary SS and in 2 public data sets. The independent cohort enrolled 141 consecutive patients with primary SS who were referred for specialist consultation at the Department of Rheumatology, Haukeland University Hospital, Bergen, Norway. The 2 public data sets included 190 patients with primary SS from Oklahoma (7) and 131 patients with primary SS from the UK (8), with both cohorts included in Gene Expression Omnibus (GEO) (a database at the National Center for Biotechnology Information; accession no. GSE51092 and accession no. GSE66795, respectively).
To analyze HLA–DR expression in blood cells among population subsets, we reanalyzed data from a mass cytometry study of blood cells from 49 patients with primary SS performed in the Paris‐Sud University Hospital (9).
Unsupervised transcriptome analysis
Results of the transcriptome analysis were stratified using a robust consensus clustering algorithm based on the PhenoGraph method (10) (Supplementary Methods). The number of clusters was determined using 2 criteria. First, we aimed our analysis on clusters with a high total number of cluster‐associated markers, i.e., those genes whose high expression would be specific to one of the clusters. Second, for robustness, we aimed to examine a smaller set of larger clusters. Without attempting to formally combine these 2 criteria (in a necessarily ad hoc manner), we selected solutions directly from plots representing the different possible tradeoffs between the 2 criteria (Supplementary Figures 1F and 2B, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). Cluster‐associated marker genes were identified using the Limma R package (11). For the detection of significant enrichment of biologic pathways, we performed gene set enrichment analysis (12) against “hallmark” gene sets that are available in the Molecular Signatures Database (13).
For further analysis, we computed an interferon (IFN) score that represented the aggregate expression of 5 key IFN genes (IFI44, IFI44L, IFIT1, IFIT3, MxA) (14) that were standardized (see Supplementary Methods).
IFNα and IFNγ quantification in the ASSESS cohort
Simoa assays were developed using a Quanterix Homebrew Simoa assay kit in accordance with the manufacturer's instructions (15, 16) (see Supplementary Methods).
Statistical analysis of clinical data
All statistical analyses were performed using the R statistical software package (version 3.5.0). Because of the non‐Gaussian distribution of several continuous variables (even if log‐transformed), we used the Kruskal‐Wallis rank test to detect the significant differences of continuous clinical variables across clusters. We then applied the Bonferroni method for adjustment of P values involving multiple comparisons. Application of different linear model analyses is explained in the Supplementary Methods.
HLA imputation and fine mapping
The patient data from the ASSESS cohort had been previously genotyped using ImmunoChip (17) (see Supplementary Methods for imputation methods) (18, 19, 20, 21).
The imputed single‐nucleotide polymorphisms (SNPs) and classic HLA alleles were tested for associations with IFNα concentrations using linear regression and SSA status with logistic regression, with both analyses corrected for the first 10 principal components. We tested multiallelic amino acid positions for associations using the multiple degree of freedom omnibus test, which included the same covariates as used for the regression analyses.
For performance of HLA fine mapping, we only included samples of European descent (N = 246), as determined by principal components analysis, with EigenStrat (22) and HapMap3 (from the International HapMap Consortium, 2010) used as references.
Manhattan plots (23) and the online LocusZoom tool (24) were used to determine the results of the association tests on the SNPs related to their location within the genome.
We calculated the posterior probabilities and frequency of HLA–DR/DQ haplotypes using the R package Haplo.Stats, as the phases of these HLA alleles cannot be resolved from genotyping data. To analyze the associations between haplotypes and circulating IFN, we used the Haplo.glm method within Haplo.Stats and HLA–DRB1*11:01;DQA1*01:02;DQB1*06:02 as the baseline.
RESULTS
Patient stratification using unsupervised transcriptome analysis of 4 primary SS cohorts
We analyzed whole blood transcriptome and genotype results in patients with primary SS from the multicenter prospective French clinical cohort (6) (Supplementary Figure 1A [https://onlinelibrary.wiley.com/doi/10.1002/art.42265]). Implementation of strict quality control over clinical, serologic, genetic, and transcriptome data resulted in a high‐quality database of 351 patients with primary SS. Clinical descriptions of the cohort and quality controls of the data are shown in Supplementary Figures 1B–D.
The use of a clustering approach for transcriptome data allowed us to identify 4 patient clusters of different sizes (comprising a total of 63 patients, 110 patients, 91 patients, and 87 patients per cluster), which we named clusters 1, 2, 3, and 4, respectively (Figure 1A, as well as Supplementary Figures 1E and 1F). Data projection with low‐dimensional embedding validated the consistency of our approach (Supplementary Figures 1G–I).
Figure 1.
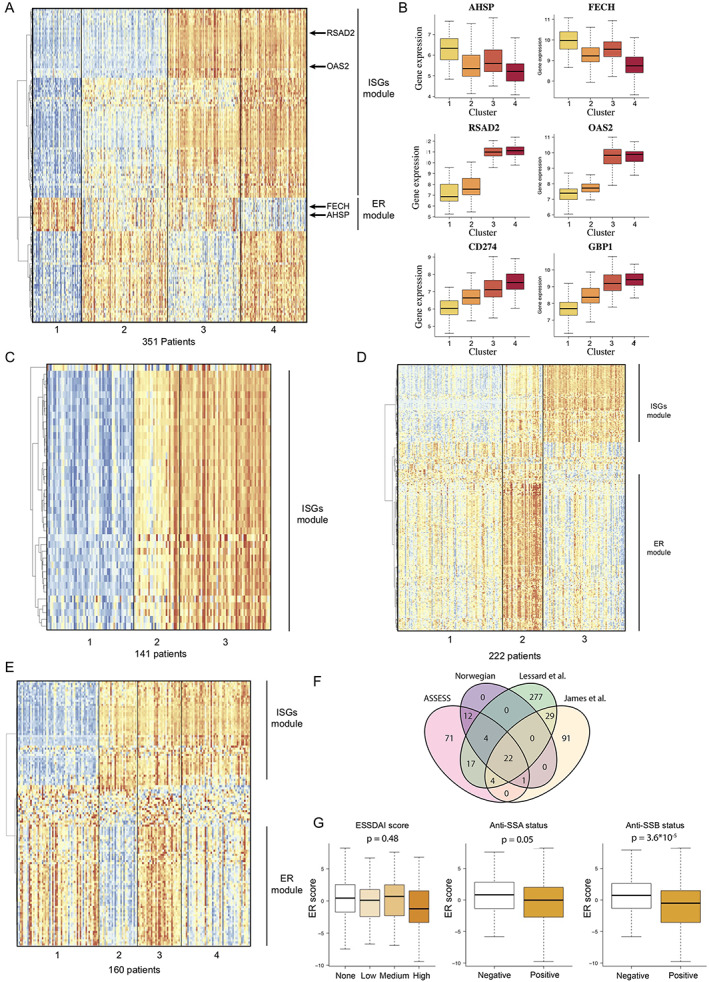
Unsupervised transcriptomic analysis enables robust stratification of patients with primary Sjögren's syndrome (SS) in 4 different cohorts. A, Expression of marker genes across the 4 clusters of patients from the Assessment of Systemic Signs and Evolution in Sjögren's Syndrome (ASSESS) cohort, normalized by row, and annotation of the identified gene modules based on hierarchical clustering. B, Expression of 6 genes identified as marker genes across the patients from the ASSESS cohort. Values are shown as box plots, where the line inside the box represents the median, the box represents the interquartile range, and the whiskers represent the 10th and 90th percentiles. C, Expression of marker genes across the 3 clusters of patients identified in the Norwegian cohort and annotation of the identified gene modules based on hierarchical clustering. D, Expression of marker genes across the 4 clusters of patients identified in the cohort from Lessard et al (7) and annotation of the identified gene modules based on hierarchical clustering. E, Expression of marker genes across the 4 clusters of patients identified in the cohort from James et al (8) and annotation of the identified gene modules based on hierarchical clustering. F, Intersection between the sets of marker genes identified in the 4 different primary SS cohorts. G, Association between erythroid (ER) transcriptomic score and EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI) score, anti‐SSA status, and anti‐SSB status. ISG = interferon‐stimulated gene.
To understand the biology underlying each of these 4 clusters, we considered the genes that were significantly differentially expressed among clusters of patients with primary SS as cluster‐associated marker genes (Supplementary Table 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). We identified 131 gene markers differentially expressed between the 4 clusters (Supplementary Methods). Among these genes, IFN‐stimulated genes (ISGs) were strongly enriched in cluster 3 and cluster 4, with 45 genes for the IFNγ response signature (P = 2.90 × 10−72) and 38 genes for the IFNα response signature (P = 1.49 × 10−71) in the hallmark gene sets. ISGs such as RSAD2 and OAS2 were overexpressed specifically in clusters 3 and 4 (i.e., in 52% of patients) compared with presence in clusters 1 and 2 (Figure 1B). Because hierarchical clustering revealed a strong correlation between ISG expression, those genes were grouped in a gene module that we named the ISG module.
We also detected a significant enrichment of genes related to heme metabolism (8 genes, P = 1.53 × 10−6), which included AHSP and FECH, 2 hemoglobin‐related genes that were overexpressed in clusters 3 and 1 but not in clusters 2 and 4 (Figure 1B). In further analysis using the Human Tissue Compendium database (25), we observed that these genes were specifically expressed by erythroid and erythroid progenitor cells but not by immune cells (Supplementary Figure 1J). These genes were highly coexpressed and clustered together in a specific gene module that we named the erythroid module (Figure 1A).
Molecular stratification strategy replicates in 3 independent primary SS cohorts
To probe the robustness of our findings, we repeated our analysis in an independent cohort of patients with primary SS and in 2 public transcriptome data sets of patients with primary SS (Supplementary Table 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265).
For the independent cohort (141 consecutive patients with primary SS referred for specialist consultation in Norway), the whole blood transcriptome data were generated with the same microarray and hybridization techniques that were used for the ASSESS cohort, with data analysis performed in an identical manner. Our analysis revealed 3 clusters with 39 differentially expressed cluster‐associated markers (Supplementary Table 3, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). We found that ISGs were strongly enriched among these markers, with 22 genes (P = 1.22 × 10−48) shown for the IFNα signature and 29 genes (P = 1.30 × 10−59) shown for the IFNγ signature in the hallmark gene sets (Figure 1C, as well as Supplementary Figures 2A and 2B, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). Consistency of the clustering was successfully verified with low‐dimensional embedding (Supplementary Figures 2D and 2E). In this cohort, we did not detect any significant enrichment in genes linked to erythroid cell and heme metabolism.
For the 2 public transcriptome data sets, which included 190 patients (7) and 131 patients (8) with primary SS, the whole blood transcriptome analysis was performed using different microarray technologies (HumanWG‐6 version 3.0 Illumina BeadChip kit and HumanHT‐12 version 4 Illumina BeadChip kit, respectively). Our analysis of the 2 cohorts revealed clusters identified as clusters 3 and 4, as defined by differential expression of 353 and 147 genes, respectively (Figures 1D and 1E, as well as Supplementary Figures 2B and 2C and Supplementary Tables 4 and 5, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). In both of the public cohorts, an IFN gene signature was identified through gene set enrichment analysis, with 27 and 31 genes (P = 8.77 × 10−47 and P = 2.55 × 10−53), respectively, belonging to the IFNα predicted signature and 46 and 35 genes (P = 5.17 × 10−57 and P = 1.13 × 10−52), respectively, belonging to the IFNγ predicted signature, according to the hallmark database. In addition, we detected an erythroid signature in both data sets (38 and 30 genes [P = 4.9 × 10−57 and P = 6.23 × 10−53], respectively) (Figures 1D and 1E).
We then studied the overlap between the different markers identified in the ASSESS cohort and the 3 other primary SS cohorts (Figure 1F). Of the 22 genes that were validated as marker genes across all 4 cohorts, nearly all were ISGs, with 20 genes belonging to the IFNγ predicted signature (P = 2.56 × 10−44) and 17 genes belonging to the IFNα predicted signature (P = 4.84 × 10−41), thus strongly supporting the critical role of IFN signaling in primary SS.
Lastly, we investigated the potential role of the erythroid gene module. We therefore computed an erythroid expression score for each patient of the ASSESS cohort and looked for associations with clinical and biologic parameters. We did not observe a significant association with the EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI, the international score of systemic disease activity) (26) (P = .48) but observed associations with anti‐SSA (P = .045) and anti‐SSB status (P = 3.63 × 10−5) (Figure 1G).
Thus, our results demonstrated that the stratification of patients with primary SS by whole blood transcriptome data and through our analytic pipeline was highly reproducible for determination of ISG signatures across different independent cohorts and microarray technologies.
Association of IFNα but not IFNγ with the transcriptome signature and with disease activity
IFNα and IFNγ, antiviral cytokines that trigger similar transcriptional changes in immune cells, are challenging to discriminate using gene expression data. To assess whether the transcriptional changes underlying the molecular stratification were regulated by IFNα and/or by IFNγ, we measured baseline circulating IFNα and IFNγ protein concentrations. Detectable concentrations of IFNα were observed in 277 (74.9%) of the 370 patients assessed, and detectable concentrations of IFNγ were observed in 364 (96.8%) of the 376 patients assessed. Significant differences in IFNα concentrations between clusters were observed, with IFNα levels in patients from clusters 1 and 2 close to the lower limit of detection (0.6 fg/ml) and with IFNα levels in patients from clusters 3 and 4 detected at high concentrations (median 60 fg/ml) (Figure 2A). Similarly, IFNγ concentrations significantly differed across clusters, with cluster 3 having the highest levels (median IFNγ concentration of 192, 430, 567, and 475 fg/ml in clusters 1, 2, 3 and 4, respectively) (Figure 2A).
Figure 2.
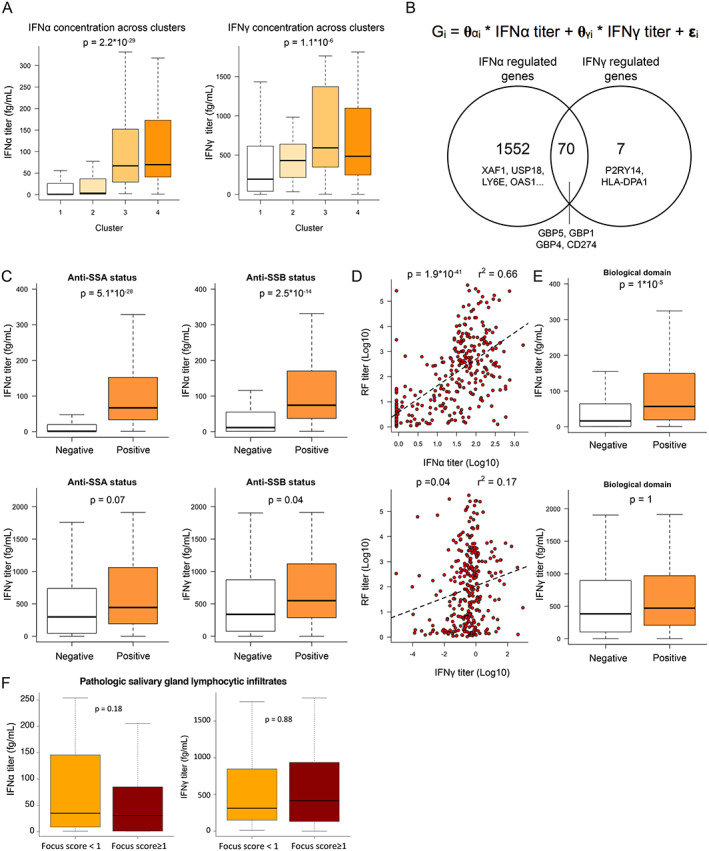
Quantification of interferon‐α (IFNα) and IFNγ protein serum concentrations by digital enzyme‐linked immunosorbent assay reveals the pivotal role of IFNα in patients with primary Sjögren's syndrome (SS). A, IFNα (left) and IFNγ (right) serum titers across clusters. B, Description of the linear model used to describe gene expression (top) and Venn diagram showing the number of genes transcriptionally controlled by IFNα, IFNγ, or both (bottom). C, IFNα (top) and IFNγ (bottom) concentrations based on anti‐SSA (left) and anti‐SSB (right) status. D, Correlations between IFNα and rheumatoid factor (RF) concentrations (top) and between IFNγ and RF concentrations (bottom). Dashed lines are based on the linear regression between the 2 variables. E, IFNα (top) and IFNγ (bottom) concentrations based on presence versus absence of an active biologic domain according to components of the EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI). F, Concentrations of IFNα (left) and IFNγ (right) according to focus score of inflammatory infiltrates in the salivary glands of patients with primary SS. For box plots, the line inside the box represents the median, the box represents the interquartile range, and the whiskers extend to the most extreme data point that is no more than 1.5 times the interquartile range from the box. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42265/abstract.
When we applied linear function modeling to the whole transcriptome data of ASSESS patients, 95% of the IFN‐inducible genes (1,552 genes) were specifically correlated with serum IFNα concentration, whereas only 7 genes were specifically correlated with serum IFNγ concentration. We also found that 70 genes, including CD274 and GBP1/4/5, were correlated with both IFNα and IFNγ concentrations (Figure 2B). Among the 82 cluster‐associated markers of the ASSESS cohort, 14 were solely correlated with serum IFNα concentration, 38 were correlated with serum concentrations of both IFNα and IFNγ, and none of the markers were correlated solely with serum IFNγ concentrations.
We observed a strong association between IFNα concentration and antibody status, including anti‐SSA status (P = 5.09 × 10−28), anti‐SSB serum positivity (P = 2.45 × 10−14), and increased serum concentrations of rheumatoid factor (RF) (r2 = 0.662, P = 1.19 × 10−41) (Figures 2C and 2D). This association with the autoantibodies was considerably weaker when we examined IFNγ, especially for RF (r2 = 0.172) (Figures 2C and 2D). Significant associations between IFNα concentrations and the B cell activation markers (B2M, BAFF), immunoglobulin free light chains, and CCL19 were observed; however, for IFNγ, associations with these markers were weaker and barely significant (Supplementary Figures 3A and 3B, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265).
The low r2 values indicated that not much variance can be explained by single predictors, such as blood biomarkers, suggesting more complex relationships between IFNα and blood serum markers. Significant differences were observed in IFNα concentrations but not in IFNγ concentrations between patients with or without an active biologic domain of the ESSDAI (the domain is considered active when complement components are low, gamma globulin or IgG levels are high, and/or a cryoglobulinemia is detected) (Figure 2E). We found that, within the biologic domain, IgG and gamma globulins were the main drivers of the correlation to IFNα, showing a positive correlation to the IFNα concentration in the blood (for correlation with IgG, Spearman's r = 0.5, P ≤ 0.01; for correlation with total gamma globulins, Spearman's r = 0.47, P ≤ 0.01); however, C3 and C4 showed only small, negative correlations (for correlation with C3, Spearman's r = –0.16, P = 0.003; for correlation with C4, Spearman's r = –0.27, P = 0.02), with no difference visible for the patients having or not having cryoglobulins (P = 0.5) (see Supplementary Figures 3B–E). Together, our results suggested a dominant role of IFNα, compared with IFNγ, for inducing B cell activation and systemic activity of the disease.
We also analyzed IFNα and IFNγ concentrations in patient salivary gland lymphocytic infiltrates but could not detect differences in blood IFNα or IFNγ concentrations between patients with focus score of ≥1 and those with a focus score of <1 according to the results of a minor salivary gland biopsy done any time prior to enrollment (Figure 2F).
We then analyzed associations between IFNα and IFNγ concentrations and clinical involvement at enrollment and during follow‐up. At enrollment, systemic complications were more frequent in patients with detectable IFNα serum concentrations. The mean ESSDAI at enrollment was higher (mean score 4 [range 0–31] versus mean score 2 [range 1–18], P = 0.0004) in patients with detectable IFNα serum concentrations. The proportions of patients with active disease on the ESSDAI, according to the cutaneous domain (22.8% versus 0%, P = 0.028), hematologic domain (24.6% versus 8.1%, P = 0.0038), and biologic domain (28.5% versus 11.4%, P < 0.0001) of the ESSDAI were also higher in patients with detectable IFNα serum concentrations.
We next analyzed the course of systemic complications that occurred in these patients prospectively over 5 years according to baseline IFNα serum concentrations. The ESSDAI values repeated across times were therefore modeled using a beta mixed regression analysis. During the 5‐year prospective follow‐up, patients with baseline detectable IFNα developed significantly more frequent systemic complications (odds ratio [OR] 1.54 [95% confidence interval 1.14–2.13]), with a similar, but nonsignificant, trend for anti‐SSA positivity and type I IFN gene score at enrollment (OR 1.24 [95% confidence interval 0.92–1.69] and OR 0.97 [95% confidence interval 0.95–0.99], respectively).
No association was observed between 1) blood IFNα concentration and patient symptoms, as assessed according to the EULAR Sjögren's Syndrome Patient Reported Index (ESSPRI) (27), 2) blood IFNγ concentrations and systemic clinical complications (according to the ESSDAI), and 3) IFNγ concentrations and patient symptoms (according to the ESSPRI) at enrollment and during follow‐up.
Genetic determinant for IFNα serum concentration by genetic analysis
We also investigated any indications of genetic contributions to stratification of patients with primary SS. When we investigated any statistical associations between circulating levels of IFNα protein and 102,744 SNPs among 307 patients from the ASSESS cohort who were previously genotyped, we observed a quantitative trait locus in the major histocompatibility complex (MHC) locus (top SNP was rs9273012, P = 4.64 × 10−9) and a suggestive association with the KIF3A locus in chromosome 5 (top SNP was rs7732667, P = 3.37 × 10−5) (Figure 3A). A detailed analysis of the MHC locus revealed that the SNP with the strongest association with circulating levels of IFNα, rs9273012, was located in the HLA–DQA1 gene, a member of the HLA class II gene family (Figures 3B and 3C). A conditioning analysis on rs9273012 (i.e., including this SNP as a covariate in the regression model) revealed no further independent associations (Supplementary Figure 4D, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42265). Interestingly, more than 13% of the variance observed in IFNα concentration was solely explained by this SNP.
Figure 3.
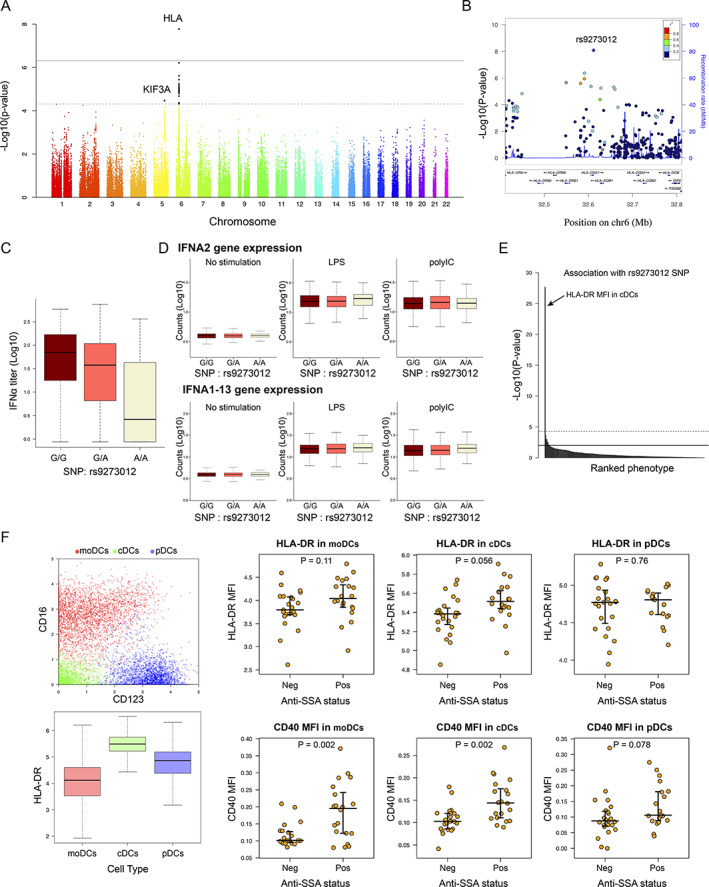
Genome‐wide association study reveals a genetic determinant in patient stratification and interferon‐α (IFNα) blood concentration in patients with primary Sjögren's syndrome. A, Genome‐wide association between single‐nucleotide polymorphisms (SNPs) and IFNα concentration. Dashed line corresponds to the threshold for a suggested genome‐wide association, and solid line corresponds to a significant genome‐wide association. B, LocusZoom plot of the HLA region. C, IFNα concentration according to the rs9273012 SNP status. D, IFNα gene expression in whole blood from 1,000 healthy donors from the Milieu Intérieur cohort, under conditions of no stimulation versus stimulation with lipopolysaccharide (LPS) versus stimulation with poly(I‐C). E, P values indicating possible statistical significance of associations between the rs9273012 SNP and the 166 immunophenotypes measured in the Milieu Intérieur study from Patin et al (30). The top horizontal line represents the threshold after Bonferroni correction for multiple testing at P = 0.05. F, Mass cytometry analysis of data from Mingueneau et al (9). Panels show the 3 dendritic cell (DC) populations defined using unsupervised analysis (top left) and HLA–DR expression in the 3 DC populations (bottom left), as well as mean fluorescence intensity (MFI) results for HLA–DR (top right) and CD40 (bottom right) in the 3 DC populations based on anti‐SSA status. For box plots, the line inside the box represents the median, the box represents the interquartile range, and the whiskers extend to the most extreme data point that is no more than 1.5 times the interquartile range from the box. moDCs = monocyte‐derived DCs; cDCs = conventional DCs; pDCs = plasmacytoid DCs; Neg = negative; Pos = positive. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42265/abstract.
To obtain a more detailed view of the MHC class II locus, we performed a detailed fine mapping of the MHC region using the SNP2HLA v1.0 software and the Type 1 Diabetes Genetics Consortium reference panel (21) (Supplementary Figure 4A) among the 291 patients from the European HapMap Consortium samples with clustering data. Our analysis revealed a significant and specific association between IFNα concentrations and the HLA–DQA1*05:01 allele, an allele previously identified as strongly associated with primary SS (7) and also as part of a larger HLA–DR/DQ haplotype. The DRB1*03:01;DQA1*05:01;DQB1*02:01 haplotype was the most frequent in our cohort (Supplementary Figure 4D). Furthermore, the DRB1*03:01;DQA1*05:01;DQB1*02:01 haplotype was the only one significantly associated with IFNα levels (data not shown).
To investigate the possibility that this HLA haplotype could directly regulate IFNα gene expression, we looked for possible associations between IFNα gene expression in whole blood of the ASSESS patients and rs9273012 status. No significant difference in IFNα gene expression was observed in the presence of the rs9273012 polymorphism (Supplementary Figure 4B). Because IFNα gene expression is highly transient in nature, we utilized the Milieu Intérieur cohort of healthy donors (28) to examine possible genetic associations between the rs9273012 polymorphism and induced IFN gene expression following whole blood stimulation with the poly(I‐C) (synthetic analog of double‐stranded RNA recognized by the Toll‐like receptor 3) (29) (see Appendix A for a list of the study investigators). No associations were observed between the different alleles and induced gene expression of IFNA2 as measured by Nanostring array analysis (Figure 3D).
We also took advantage of extensive cellular phenotypes previously described in the same healthy donors and observed that the rs9273012 G allele was significantly and specifically associated with higher protein expression levels of HLA–DR in conventional dendritic cells (cDCs) (30) (P = 2.14 × 10−28) (Figure 3E). Because a mix of 166 distinct immunophenotypes (for details, see Supplementary Table S3 in ref. 31) had been analyzed, the second‐lowest P value of HLA–DR in conventional dendritic cell subset 3 cells (cDC3s) was below the global significance threshold. HLA–DR expression by cDCs in primary SS was then investigated by reanalysis of data from a mass cytometry study of primary SS blood cells (9). Unsupervised analysis of these data revealed 3 cellular clusters corresponding to DCs: monocyte‐derived DCs (CD16 + CD123−), cDCs (CD16 − CD123−), and plasmacytoid DCs (CD16 − CD123+). Both the monocyte‐derived DCs and the cDCs exhibited higher expression of CD40 in anti‐SSA–positive patients compared with anti‐SSA–negative patients (P = 2.01 × 10−3 and P = 2.01 × 10−3, respectively); however, only cDCs exhibited higher expression of HLA–DR in anti‐SSA–positive patients compared with anti‐SSA–negative patients (P = 0.056) (Figure 3F). Thus, we observed that the rs9273012 polymorphism was associated with increased HLA class II expression in cDCs from healthy controls and that HLA class II expression was increased in cDCs from patients with anti‐SSA autoantibodies.
In our investigation of the relationship between HLA gene polymorphisms and anti‐SSA status in the ASSESS cohort, we observed a consistent signal in the HLA–DQA1 locus (Supplementary Figure 4C). The SNP that had the strongest association with IFNα levels, rs9273012, also had the strongest association with anti‐SSA autoantibody positivity (P = 4.31 × 10−12).
DISCUSSION
Our unsupervised gene expression analytic pipeline, newly applied to blood transcriptome data from 813 patients with primary SS, identified a consistent stratification with clusters associated with IFN and erythroid signatures across different cohorts and microarray technologies. Combining this approach with digital enzyme‐linked immunosorbent assay in a well‐characterized cohort revealed the key role of circulating IFNα protein, as opposed to IFNγ, through its association with clinical and immunologic phenotypes, highlighting its relevance as a therapeutic target. Furthermore, our analysis revealed a significant association between a specific HLA class II gene polymorphism, anti‐SSA antibody, and circulating IFNα. The use of well‐defined healthy donor data from the Milieu Intérieur cohort and the confirmation in anti‐SSA–positive patients with primary SS strongly suggested that this HLA gene polymorphism affects HLA expression on cDCs, thus likely leading to increased autoantigen presentation, autoantibody secretion, and immune complex formation, which can subsequently trigger IFNα secretion.
Limitations of our study are mostly related to the observational design, its focus on peripheral blood only, and the absence of longitudinal biologic assessments.
A strength of our study is the innovative use of an unsupervised clustering method, which has been only previously reported for single cell analysis, to analyze the transcriptome data across different primary SS cohorts. Most previous transcriptomic analyses in primary SS compared limited population samples (31, 32, 33) with healthy controls and used bioinformatics prediction, resulting in the description of an IFN signature that did not discriminate between contributions of IFNα and contributions of IFNγ to primary SS. The new bioinformatic pipeline in our study, which involved a much larger data set, confirmed the presence of an IFN module but also identified an erythroid module that was previously reported in patients with systemic lupus erythematosus (34). Further analysis may shed light on the potential pathogenic mechanisms associated with the action of this erythroid module.
Both type I and type II IFNs are relevant pathogenic suspects in primary SS, based on genetic predisposition to the disease (involving IFN regulatory factor 5 in the IFNα pathway and interleukin‐12A and STAT4 in the IFNγ pathway) and the pathogenic cell populations involved (plasmacytoid DCs, the major IFNα‐producing cells, and natural killer and CD8 T cells, which secrete IFNγ) (32, 35, 36). A deeper understanding of the respective contributions of IFNα and IFNγ is therefore crucial for selective therapeutic targeting.
Of note, most of the genes induced by IFNα are also induced by IFNγ, making such a “signature” actually a broader marker of both IFNα and IFNγ activity (37). To our knowledge, our study is the first to measure circulating IFN proteins in a large prospective cohort of patients with primary SS concomitantly with their transcriptomic signature. The quantification of both IFN proteins in the circulation at attomolar concentrations allowed us to determine that 95% of the IFN‐inducible genes are correlated with serum IFNα but not with IFNγ. A recent multiomic profiling study in a cross‐sectional cohort of patients with primary SS also showed a correlation between serum IFNα concentration and type I IFN signature (38) but did not assess serum IFNγ concentration. Of note, our study, which focused on peripheral blood, did not exclude a possible role of IFNβ (another type I IFN), IFNγ (type II IFN), or type III IFNs in salivary glands (39, 40, 41).
Our multiomic study also allowed us to analyze the relationship between IFN protein concentrations and patient genotypes, whole blood transcriptome results, and clinical phenotypes. A highly significant association between HLA class II gene polymorphisms and circulating IFNα protein concentrations was identified. Specifically, in primary SS, we demonstrated an association between an HLA allele, HLA–DQA1*05:01, and both anti‐SSA antibody and blood IFNα concentrations. The associations between HLA class II polymorphisms and autoantibodies and between autoantibodies and the IFN signature have long been known (42, 43, 44), as well as the associations between anti‐SSA antibodies, cutaneous involvement, and hematologic and biologic domains (45, 46). SNPs associated with HLA class II genes were also associated with the IFN signature and autoantibodies in a recent multiomic study of patients with primary SS (38). Our present results add mechanistic explanations underlying this association. In healthy donors, HLA expression and this specific polymorphism were not associated with IFNα induction upon stimulation, indicating that they do not directly influence IFNα secretion. However, in the same healthy donors, this HLA allele was associated with HLA–DR protein up‐regulation in conventional DCs, but not in plasmacytoid DCs. In addition, HLA–DR was up‐regulated in cDCs from patients with primary SS who were anti‐SSA positive compared with patients with primary SS who were anti‐SSA negative. This suggests that HLA–DQA1*05:01, as part of the HLA DRB1*03:01;DQA1*05:01;DQB1*02:01/DQB1*03:01 haplotype, promotes HLA class II molecule expression at the cDC surface, and thus SSA antigen presentation by cDCs, resulting in anti‐SSA secretion and immune complex formation, which in turn increases IFNα secretion.
In primary SS, and perhaps in other autoimmune diseases, HLA might therefore predispose to IFNα secretion indirectly, by favoring classic presentation by cDCs of SSA peptides to T cells, leading to anti‐SSA antibodies and immune complexes stimulating IFNα secretion. This analysis on DCs and the expression of HLA–DR on B cells, which are also pivotal antigen‐presenting cells, deserves further investigation.
Our results also revealed the potential of circulating IFNα as a biomarker in primary SS. Previous studies have suggested the use of quantitative IFNα and IFNγ signatures as biomarkers on the basis of messenger RNA expression by quantitative polymerase chain reaction of IFN‐inducible genes; however, these studies were mainly cross‐sectional and had limited sample sizes (39, 41, 47). The strength of our present study was the direct quantification of IFNα and IFNγ protein using a highly sensitive method in a cohort prospectively observed for 5 years. The fact that the proportion of patients with detectable circulating IFNα was higher than the proportion of patients with detectable IFN signature suggests that the detection of circulating IFNα protein is a more sensitive measure of IFNα activity than is a transcriptomic signature. In agreement with previous studies of IFN signatures (8, 38, 39), fatigue, pain, and dryness were not associated with either circulating IFNα or circulating IFNγ levels. However, in contrast to IFNγ, we found that circulating IFNα was highly significantly associated with autoantibodies and markers of B cell activation. In addition, in contrast to IFNγ, circulating IFNα was significantly associated with systemic complications at enrollment. Baseline detectable IFNα was also associated with more frequent systemic complications during the 5‐year prospective follow‐up. Further studies that assess IFNα longitudinally at different time points are necessary to confirm the potential predictive role of IFNα.
Although hydroxychloroquine treatment has been shown to decrease the strength of IFNα signatures (14), we observed that circulating IFNα levels were not significantly different among 113 of 352 patients in our analyses who were prescribed hydroxychloroquine at enrollment (see Supplementary Figure 3F). This might be related to nonadherence of some patients to hydroxychloroquine (blood levels of hydroxychloroquine were not assessed).
In conclusion, the strong transcriptomic stratification of patients with primary SS in our analysis was clinically relevant, supported by our observation that it was driven by IFNα rather than by IFNγ, and was associated with HLA gene polymorphisms. Beyond the specific implications for primary SS, this gene analysis approach may also be useful to move to precision therapy in other complex autoimmune diseases.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Gottenberg had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Trutschel, Bost, Mariette, Duffy, Schwikowski, Gottenberg.
Acquisition of data
Trutschel, Bost, Mariette, Bondet, Llibre, Posseme, Charbit, Thorball, Jonsson, Lessard, Felten, Ng, Chatenoud, Dumortier, Sibilia, Fellay, Brokstad, Appel, Tarn, Murci, Mingueneau, Meyer, Duffy, Schwikowski, Gottenberg.
Analysis and interpretation of data
Trutschel, Bost, Mariette, Bondet, Llibre, Posseme, Charbit, Thorball, Jonsson, Lessard, Felten, Ng, Chatenoud, Dumortier, Sibilia, Fellay, Brokstad, Appel, Tarn, Murci, Mingueneau, Meyer, Duffy, Schwikowski, Gottenberg.
Supporting information
Disclosure Form
Supplementary Methods
Appendix A
Figure S1
A) Clinical parameters of the ASSESS cohort with patients having primary SS. “A family history AI” corresponds to patients who have a autoimmune diseases among 1st degree relatives.
B) Probe expression distribution across samples from the ASSESS cohort (after the normalization).
C) Principal Component Analysis (PCA) of the samples from the ASSESS cohort using all genes as input.
D) Global strategy used to define variable genes. Red dots correspond to genes considered as variable.
E) Co‐occurrence matrix of the ASSESS cohort. Color indicates the frequency of co‐occurrence of a pair of patients among the clusterings with varying K parameter.
F) Combinations of number of clusters and number of marker genes for different sets of clusters obtained from hierarchical clustering. The arrow indicates the selected trade‐off between small number of clusters and large number of cluster‐associated markers.
G) UMAP plot with samples labeled by their corresponding consensus Phenograph cluster.
H) UMAP plot with samples labeled by their corresponding anti‐SSA status.
I) UMAP plot with samples labeled by their corresponding originating clinical center.
J) Gene expression of genes from the ER module across tissues according to the Tissue Human Compendium database.
Figure S2
A) and James et al [8] cohorts (from left to right).
B) Number of marker genes based on the number of clusters on a log scale in the Norwegian, Lessard et al [7] and James et al [8] cohorts (from left to right). Arrows indicate selected trade‐off between small number of clusters and large number of cluster‐associated markers.
C) Principal Component Analysis (PCA) of the samples from the Norwegian cohort using all genes as input.
D) UMAP plot with samples from the Norwegian cohort labeled based on their corresponding consensus Phenograph cluster.
E) UMAP plot with samples from the Norwegian cohort labeled based on their corresponding anti‐SSA status.
Figure S3
A) Correlation between B2M or BAFF (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
B) Correlation between Ig free chain or CCL19 (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
C) Correlation between IgG or total Gamma (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
D) Correlation between C3 or C4 (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
E) IFNα (left panel) and IFNγ (right panel) serum titers across patients with or without presence of Cryoglobulinemia (Kruskal‐Wallis ranked sum test to test differences in IFN concentrations at baseline between patients with Cryoglobulinemia and patient without).
F) IFNα (left panel) and IFNγ (right panel) serum titers across patients with or without actual HCQ treatment (Kruskal‐Wallis ranked sum test to test differences in IFN concentrations at baseline between patients with actual HCQ treatment and patient without).
Figure S4
A) Fine mapping analysis of the HLA region. Solid line corresponds to the threshold for a significant association.
B) Mean IFNα gene expression level according to the rs9273012 SNP status in the ASSESS cohort.
C) Genome‐wide association of the SNPs with anti‐SSA status. The dashed line corresponds to the threshold for a genome‐wide suggestive association, and the plain corresponds to a significant association.
D) Results on calculated HLA‐DR‐DQ haplotypes.
Table S1 Description of the 5 cohorts included in the analysis: ASSESS, Norwegian, Lessard et al., James et al., Mingueneau et al.)
Table S2 Marker genes in ASSESS cohort for patient cluster analysis (differentially expressed between the 4 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by an F‐test and the maximal difference between cluster mean expressions.
Table S3 Marker genes in Norwegian cohort for patient cluster analysis (differentially expressed between the 3 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.
Table S4 Marker genes in Lessard et al. cohort for patient cluster (differentially expressed between the 3 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.
Table S5 Marker genes in James et al. cohort for patient cluster (differentially expressed between the 4 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.
Supported by the Innovative Medicines Initiative 2 Joint Undertaking (JU) (grant 806975). The JU receives support from the European Union's Horizon 2020 research and innovation program and the European Union and the European Federation of Pharmaceutical Industries and Associations. This work was also supported by the National Institutes of Health (National Institute of Arthritis and Musculoskeletal Skin Disease grant R01‐AR‐065953). The Assessment of Systemic Signs and Evolution in Sjögren's Syndrome (ASSESS) national multicenter prospective cohort was formed in 2006 with a French Ministry of Health grant (Programme Hospitalier de Recherche Clinique 2005 P060228). The ASSESS cohort is promoted by the French Society of Rheumatology and receives research grants from the French Society of Rheumatology. Dr. Gottenberg's work was supported by Bristol Myers Squibb for transcriptomic analysis of the ASSESS and Norwegian cohorts and by Geneviève Garnier (Association Française du Syndrome de Gougerot‐Sjögren et des syndromes secs). Drs. Trutschel's and Schwikowski's work was supported by Geneviève Garnier (Association Française du Syndrome de Gougerot‐Sjögren et des syndromes secs).
The present article reflects only the authors’ view, and the contents are the sole responsibility of the authors; the JU is not responsible for any use that may be made of the information it contains, and the contents do not necessarily reflect the official views of the National Institutes of Health.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42265&file=art42265‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Benno Schwikowski, Email: benno@pasteur.fr.
Jacques Eric Gottenberg, Email: jacques-eric.gottenberg@chru-strasbourg.fr.
The Milieu Intérieur Consortium, ASSESS study investigators, and NECESSITY Consortium:
Emmanuelle Dernis, Valerie Devauchelle‐Pensec, Philippe Dieude, Jean‐Jacques Dubost, Anne‐Laure Fauchais, Vincent Goeb, Eric Hachulla, Pierre Yves Hatron, Claire Larroche, Véronique Le Guern, Jacques Morel, Aleth Perdriger, Carinne Salliot, Stephanie Rist, Alain Saraux, Jean Sibilia, Olivier Vittecoq, Gaétane Nocturne, Philippe Ravaud, Raphaèle Seror, Laurent Abel, Andres Alcover, Hugues Aschard, Kalla Astrom, Philippe Bousso, Pierre Bruhns, Ana Cumano, Caroline Demangel, Ludovic Deriano, James Di Santo, Françoise Dromer, Gérard Eberl, Jost Enninga, Jacques Fellay, Odile Gelpi, Ivo GompertsBoneca, Milena Hasan, Serge Hercberg, Olivier Lantz, Claude Leclerc, Hugo Mouquet, Sandra Pellegrini, Stanislas Pol, Antonio Rausell, Lars Rogge, Anavaj Sakuntabhai, Olivier Schwartz, Benno Schwikowski, Spencer Shorte, Vassili Soumelis, Frédéric Tangy, Eric Tartour, Antoine Toubert, Mathilde Touvier, Marie‐Noëlle Ungeheuer, Matthew L. Albert, Darragh Duffy, and Lluis Quintana‐Murci
REFERENCES
- 1. Mariette X, Criswell LA. Primary Sjögren's syndrome. N Engl J Med 2018;378:931–9. [DOI] [PubMed] [Google Scholar]
- 2. Nocturne G, Pontarini E, Bombardieri M, et al. Lymphomas complicating primary Sjögren's syndrome: from autoimmunity to lymphoma. Rheumatology (Oxford) 2019;60:3513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gottenberg JE, Ravaud P, Puéchal X, et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjögren syndrome: the JOQUER randomized clinical trial. JAMA 2014;312:249–58. [DOI] [PubMed] [Google Scholar]
- 4. Devauchelle‐Pensec V, Mariette X, Jousse‐Joulin S, et al. Treatment of primary Sjögren syndrome with rituximab. Ann Intern Med 2018;160:233–42. [DOI] [PubMed] [Google Scholar]
- 5. Bowman SJ, Everett CC, O'Dwyer JL, et al. Randomized controlled trial of rituximab and cost‐effectiveness analysis in treating fatigue and oral dryness in primary Sjögren's syndrome. Arthritis Rheumatol 2017;69:1440–50. [DOI] [PubMed] [Google Scholar]
- 6. Gottenberg JE, Seror R, Miceli‐Richard C, et al. Serum levels of beta2‐microglobulin and free light chains of immunoglobulins are associated with systemic disease activity in primary Sjögren's syndrome. Data at enrollment in the prospective ASSESS cohort. PLoS One 2013;8:e59868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lessard CJ, Li H, Adrianto I, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren's syndrome. Nat Genet 2013;45:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. James K, Al‐Ali S, Tarn J, et al. A transcriptional signature of fatigue derived from patients with primary Sjogren's syndrome. PLoS One 2015;10:e0143970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mingueneau M, Boudaoud S, Haskett S, et al. Cytometry by time‐of‐flight immunophenotyping identifies a blood Sjogren's signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol 2016;137:1809–21. [DOI] [PubMed] [Google Scholar]
- 10. Levine JH, Simonds EF, Bendall SC, et al. Data‐driven phenotypic dissection of AML reveals progenitor‐like cells that correlate with prognosis. Cell 2015;162:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The Molecular Signatures Database hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bodewes IL, Al‐Ali S, van Helden‐Meeuwsen CG, et al. Systemic interferon type I and type II signatures in primary Sjögren's syndrome reveal differences in biological disease activity. Rheumatology (Oxford) 2018;57:921–30. [DOI] [PubMed] [Google Scholar]
- 15. Rodero MP, Decalf J, Bondet V, et al. Detection of interferon α protein reveals differential levels and cellular sources in disease. J Exp Med 2017;214:1547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer S, Woodward M, Hertel C, et al. AIRE‐deficient patients harbor unique high‐affinity disease‐ameliorating autoantibodies. Cell 2016;166:582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther 2011;13:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durbin R. Efficient haplotype matching and storage using the positional Burrows‐Wheeler transform (PBWT). Bioinformatics 2014;30:1266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loh PR, Danecek P, Palamara PF, et al. Reference‐based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016;48:1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia X, Han B, Onengut‐Gumuscu S, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PloS One 2013;8:e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- 23. Gibson G. Hints of hidden heritability in GWAS. Nat Genet 2010;42:558–60. [DOI] [PubMed] [Google Scholar]
- 24. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics 2010;26:2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein‐encoding transcriptomes. Proc Natl Acad Sci U S A 2004;101:6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seror R, Ravaud P, Bowman SJ, et al, on behalf of the EULAR Sjögren's Task Force. EULAR Sjögren's Syndrome Disease Activity Index: development of a consensus systemic disease activity index for primary Sjögren's syndrome. Ann Rheum Dis 2010;69:1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seror R, Ravaud P, Mariette X, et al, on behalf of the EULAR Sjögren's Task Force. EULAR Sjögren's Syndrome Patient Reported Index (ESSPRI): development of a consensus patient index for primary Sjögren's syndrome. Ann Rheum Dis 2011;70:968–72. [DOI] [PubMed] [Google Scholar]
- 28. Thomas S, Rouilly V, Patin E, et al. The Milieu Intérieur study: an integrative approach for study of human immunological variance. Clin Immunol 2015;157:277–93. [DOI] [PubMed] [Google Scholar]
- 29. Urrutia A, Duffy D, Rouilly V, et al. Standardized whole‐blood transcriptional profiling enables the deconvolution of complex induced immune responses. Cell Rep 2016;16:2777–91. [DOI] [PubMed] [Google Scholar]
- 30. Patin E, Hasan M, Bergstedt J, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol 2018;19:302–14. [DOI] [PubMed] [Google Scholar]
- 31. Emamian ES, Leon JM, Lessard CJ, et al. Peripheral blood gene expression profiling in Sjögren's syndrome. Genes Immun 2009;10:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gottenberg JE, Cagnard N, Lucchesi C, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren's syndrome. Proc Natl Acad Sci U S A 2006;103:2770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hjelmervik TO, Petersen K, Jonassen I, et al. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren's syndrome patients from healthy control subjects. Arthritis Rheum 2005;52:1534–44. [DOI] [PubMed] [Google Scholar]
- 34. Banchereau R, Hong S, Cantarel B, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016;165:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rusakiewicz S, Nocturne G, Lazure T, et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren's syndrome. Sci Transl Med 2013;5:195ra96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tasaki S, Suzuki K, Nishikawa A, et al. Multiomic disease signatures converge to cytotoxic CD8 T cells in primary Sjogren's syndrome. Ann Rheum Dis 2017;76:1458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hall JC, Casciola‐Rosen L, Berger AE, et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc Natl Acad Sci U S A 2012;109:17609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soret P, Le Dantec C, Desvaux E, et al. A new molecular classification to drive precision treatment strategies in primary Sjögren's syndrome. Nat Commun 2021;12:3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bodewes IL, Huijser E, van Helden‐Meeuwsen CG, et al. TBK1: a key regulator and potential treatment target for interferon positive Sjogren's syndrome, systemic lupus erythematosus and systemic sclerosis. J Autoimmun 2018;91:97–102. [DOI] [PubMed] [Google Scholar]
- 40. Hall JC, Baer AN, Shah AA, et al. Molecular subsetting of interferon pathways in Sjögren's syndrome. Arthritis Rheumatol;67:2437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nezos A, Gravani F, Tassidou A, et al. Type I and II interferon signatures in Sjogren's syndrome pathogenesis: contributions in distinct clinical phenotypes and Sjogren's related lymphomagenesis. J Autoimmun 2015;63:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gottenberg JE, Busson M, Loiseau P, et al. In primary Sjogren's syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003;48:2240–5. [DOI] [PubMed] [Google Scholar]
- 43. Harley JB, Reichlin M, Arnett FC, et al. Gene interaction at HLA‐DQ enhances autoantibody production in primary Sjogren's syndrome. Science 1986;232:1145–7. [DOI] [PubMed] [Google Scholar]
- 44. Lovgren T, Eloranta ML, Bave U, et al. Induction of interferon‐α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum 2004;50:1861–72. [DOI] [PubMed] [Google Scholar]
- 45. Brito‐Zerón P, Acar‐Denizli N, Ng WF, et al. How immunological profile drives clinical phenotype of primary Sjögren's syndrome at diagnosis: analysis of 10,500 patients (Sjögren Big Data Project). Clin Exp Rheumatol 2018;36 Suppl:102–12. [PubMed] [Google Scholar]
- 46. Kyriakidis NC, Kapsogeorgou EK, Tzioufas AG. A comprehensive review of autoantibodies in primary Sjögren's syndrome: clinical phenotypes and regulatory mechanisms. J Autoimmun 2014;51:67–74. [DOI] [PubMed] [Google Scholar]
- 47. Brkic Z, Maria NI, van Helden‐Meeuwsen CG, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjögren's syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis 2013;72:728–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Supplementary Methods
Appendix A
Figure S1
A) Clinical parameters of the ASSESS cohort with patients having primary SS. “A family history AI” corresponds to patients who have a autoimmune diseases among 1st degree relatives.
B) Probe expression distribution across samples from the ASSESS cohort (after the normalization).
C) Principal Component Analysis (PCA) of the samples from the ASSESS cohort using all genes as input.
D) Global strategy used to define variable genes. Red dots correspond to genes considered as variable.
E) Co‐occurrence matrix of the ASSESS cohort. Color indicates the frequency of co‐occurrence of a pair of patients among the clusterings with varying K parameter.
F) Combinations of number of clusters and number of marker genes for different sets of clusters obtained from hierarchical clustering. The arrow indicates the selected trade‐off between small number of clusters and large number of cluster‐associated markers.
G) UMAP plot with samples labeled by their corresponding consensus Phenograph cluster.
H) UMAP plot with samples labeled by their corresponding anti‐SSA status.
I) UMAP plot with samples labeled by their corresponding originating clinical center.
J) Gene expression of genes from the ER module across tissues according to the Tissue Human Compendium database.
Figure S2
A) and James et al [8] cohorts (from left to right).
B) Number of marker genes based on the number of clusters on a log scale in the Norwegian, Lessard et al [7] and James et al [8] cohorts (from left to right). Arrows indicate selected trade‐off between small number of clusters and large number of cluster‐associated markers.
C) Principal Component Analysis (PCA) of the samples from the Norwegian cohort using all genes as input.
D) UMAP plot with samples from the Norwegian cohort labeled based on their corresponding consensus Phenograph cluster.
E) UMAP plot with samples from the Norwegian cohort labeled based on their corresponding anti‐SSA status.
Figure S3
A) Correlation between B2M or BAFF (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
B) Correlation between Ig free chain or CCL19 (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
C) Correlation between IgG or total Gamma (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
D) Correlation between C3 or C4 (left and right panel) and IFNα (top) or IFNγ (bottom). The dashed line, R^2, p‐value corresponds to the linear regression between the two variables and t‐test on regression coefficient.
E) IFNα (left panel) and IFNγ (right panel) serum titers across patients with or without presence of Cryoglobulinemia (Kruskal‐Wallis ranked sum test to test differences in IFN concentrations at baseline between patients with Cryoglobulinemia and patient without).
F) IFNα (left panel) and IFNγ (right panel) serum titers across patients with or without actual HCQ treatment (Kruskal‐Wallis ranked sum test to test differences in IFN concentrations at baseline between patients with actual HCQ treatment and patient without).
Figure S4
A) Fine mapping analysis of the HLA region. Solid line corresponds to the threshold for a significant association.
B) Mean IFNα gene expression level according to the rs9273012 SNP status in the ASSESS cohort.
C) Genome‐wide association of the SNPs with anti‐SSA status. The dashed line corresponds to the threshold for a genome‐wide suggestive association, and the plain corresponds to a significant association.
D) Results on calculated HLA‐DR‐DQ haplotypes.
Table S1 Description of the 5 cohorts included in the analysis: ASSESS, Norwegian, Lessard et al., James et al., Mingueneau et al.)
Table S2 Marker genes in ASSESS cohort for patient cluster analysis (differentially expressed between the 4 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by an F‐test and the maximal difference between cluster mean expressions.
Table S3 Marker genes in Norwegian cohort for patient cluster analysis (differentially expressed between the 3 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.
Table S4 Marker genes in Lessard et al. cohort for patient cluster (differentially expressed between the 3 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.
Table S5 Marker genes in James et al. cohort for patient cluster (differentially expressed between the 4 clusters). Given are the gene ID, the mean expression value in each cluster, ‐10log of the BH‐corrected p‐value determined by F‐test and the maximal difference between cluster mean expressions.