

Abstract
Protein kinases have proven to be a very productive class of therapeutic targets, and over 90 inhibitors are currently in clinical use primarily for the treatment of cancer. Repurposing these inhibitors as antimalarials could provide an accelerated path to drug development. In this study, we identified BI-2536, a known potent human polo-like kinase 1 inhibitor, with low nanomolar antiplasmodial activity. Screening of additional PLK1 inhibitors revealed further antiplasmodial candidates despite the lack of an obvious orthologue of PLKs in Plasmodium. A subset of these inhibitors was profiled for their in vitro killing profile, and commonalities between the killing rate and inhibition of nuclear replication were noted. A kinase panel screen identified Pf NEK3 as a shared target of these PLK1 inhibitors; however, phosphoproteome analysis confirmed distinct signaling pathways were disrupted by two structurally distinct inhibitors, suggesting Pf NEK3 may not be the sole target. Genomic analysis of BI-2536-resistant parasites revealed mutations in genes associated with the starvation-induced stress response, suggesting BI-2536 may also inhibit an aminoacyl-tRNA synthetase.
Keywords: malaria, Plasmodium, kinase, PLK, NEK, antiplasmodial, antimalarial
Graphical Abstract
Malaria represents a global threat, with ~241 million cases and 627,000 deaths worldwide in 2020.1 The Plasmodium parasite’s propensity for developing drug resistance has now extended to frontline treatment with artemisinin combination therapy (ACT); ACT resistance has been detected in both Asia and Africa, accelerating the need for new drugs.2,3 The most lethal species, Plasmodium falciparum, displays a complex life cycle from mosquitoes to host, where parasites mature first in the liver before entering into the symptomatic blood stage and finally advancing to the retransmitted gametocyte stage. Protein kinases are heavily involved in the regulation of each of these stages, making them a desirable target for rational drug design. Additionally, the plasmodial kinome has been found to be highly divergent from the human kinome, which offers the potential for high selectivity.4 The repurposing of existent kinase inhibitors offers a viable strategy to target these essential enzymes in Plasmodium while accelerating the drug development timeline. The dissimilarity between human and plasmodial kinomes presents both challenges and opportunities for repurposing; while it hinders efforts to model plasmodial targets in silico based on human orthologues, the pool of available inhibitors has been bolstered by over two decades of kinase inhibitor research, predominantly in the cancer field.5 Considerable progress has been made toward advancing inhibitor scaffold design, target identification, and off-target mitigation strategies, all of which may be leveraged for antimalarial drug development. Large-scale screening efforts of kinase inhibitor libraries have already successfully identified compounds active against both blood and liver-stage parasites, further validating this approach.6
In the following work, we screened a kinase inhibitor library curated by the Harvard Medical School (HMS) Library of Integrated Network-based Cellular Signatures (LINCS) Center (www.lincsproject.org). The LINCS program is a consortium-based project with the goal of illuminating cellular response pathways by observing the impact of pathway perturbations. The resulting data are publicly made available to assist in the development of therapeutics. From these efforts, nine kinase inhibitors with antiplasmodial activity were identified, including the potent and selective polo-like kinase 1 (PLK1) inhibitor BI-2536.7 Polo-like kinases (PLKs) are serine/threonine kinases that regulate various components of cell division in mammals; however, no PLKs have currently been identified in Plasmodium.8,9 Exploration of additional PLK1 inhibitors, comprised of both BI-2536 analogues and structurally distinct chemical scaffolds, confirmed that the activity against the human PLK1 enzyme does not correlate with antiplasmodial activity. Furthermore, we identified the plasmodial NIMA (never in mitosis gene A)-related kinase, Pf NEK3, as a potential target of BI-2536 and other PLK1 inhibitors. Phosphoproteomic analysis of the two most potent structurally distinct Pf NEK3 inhibitors showed little overlap, however, highlighting the complexity of these signaling pathways and the possibility of additional in vitro targets. In BI-2536-resistant parasites, we detected a series of mutations in genes related to the starvation-induced stress response. A hallmark of activated stress response, increased eIF2α phosphorylation, was also observed in BI-2536-treated parasites, corroborating involvement in this pathway and underscoring the likelihood of polypharmacology, potentially with an aminoacyl-tRNA synthetase as a secondary target.
RESULTS AND DISCUSSION
Kinase Library Screening Identifies Antiplasmodial Effects of Polo-Like Kinase Inhibitor BI-2536.
To identify kinase inhibitors with antiplasmodial potential, we performed a fixed concentration screen of 201 compounds from the HMS LINCS library against the multidrug-resistant Plasmodium falciparum strain Dd2. We observed a range of inhibition at 1μM, with the majority of compounds showing <30% inhibition and an overall hit rate of 4.5% (Figure S1). Of the inhibitors tested, nine exhibited >90% inhibition, prompting their EC50 determination (Table S1). Reflecting the diversity within the compound library, these inhibitors were associated with a variety of human targets, including Akt1, PI3K, PI4K, HSP90, and mTOR. The most potent compound, torin2 (Dd2 EC50 =5.30 ± 0.30 nM), has been previously explored as an antimalarial.10 Similarly, the HSP90 inhibitor NVPAUY922,11 the c-Met inhibitors Crizotinib and PHA665752,12 the AKT inhibitor A443654,13 and the PI3K/PI4K inhibitors INK128, PIK-93, and GSK212645814 have all been evaluated in Plasmodium. However, the potent PLK1 inhibitor BI-2536 (hereafter compound 1) displayed a low nanomolar activity in Dd2 (EC50 = 178 ± 12 nM) and had, to our knowledge, not yet been characterized as an antiplasmodial compound. We therefore chose to pursue this compound for further investigation.
Antiplasmodial Activity Has Little Correlation with the Human PLK1 Activity.
Along with other members of the Apicomplexa phylum, Plasmodium parasites appear to lack polo-like kinases.8 To probe the potential relationship between human PLK1 inhibition and antiplasmodial activity of compound 1, we utilized a previously synthesized set of compound 1 analogues with either 7,8-dihydropteridinone (compounds 2–6) or related pyrimido-diazepine (compounds 7–10) core, and various substitution groups were tested for both cellular antiplasmodial activity and human PLK1 enzymatic activity.15 We evaluated the antiplasmodial activity in both the Dd2 strain and the chloroquine-sensitive strain 3D7, shown in Table 1. Interestingly, there was considerable variation in antiplasmodial activity among these compounds regardless of scaffold, with some inhibitors showing no in vitro activity at the highest concentrations tested (5 μM). It is of interest to note that compound 2 while exhibiting good antiplasmodial activity was inactive against PLK1. Presumably, the lack of cytotoxicity could be attributed to the loss of PLK1 activity of this compound.
Table 1.
Antiplasmodial and Kinase Activities of Compound 1 Structural Analogues
| ||||||
|---|---|---|---|---|---|---|
| ID | antiplasmodial activity | cytotoxicity | kinase activity | |||
| Pf Dd2 EC50 (nM) | Pf 3D7 EC50 (nM) | RIa | HepG2 EC50 (nM) | SIb | PLK1 IC50 (nM) | |
| BI-2536 (1) | 178 ± 12 | 135 ± 35 | 1.3 | 3730 ± 520 | 21 | 0.83 |
| compound 2 | 432 ± 47 | 347 ± 52 | 1.2 | >25,000 | > 58 | >10,000 |
| compound 3 | 2330 ± 160 | 1530 ± 250 | 1.5 | 3030 ± 200 | 1.0 | 204 |
| compound 4 | 4190 ± 260 | 3250 ± 550 | 1.3 | 8550 ± 720 | 2.0 | 430 |
| compound 5 | >5000 | nt | - | >25,000 | - | >10,000 |
| compound 6 | >5000 | nt | - | 6200 ± 470 | <1.0 | nt |
| compound 7 | 1490 ± 260 | 1540 ± 350 | 0.97 | 6390 ± 130 | 4.0 | 270 |
| compound 8 | 2420 ± 440 | 2350 ± 650 | 1.0 | 21,800 ± 700 | 9.0 | 6200 |
| compound 9 | >5000 | nt | - | >25,000 | - | 8800 |
| compound 10 | >5000 | nt | - | 22,300 ± 1290 | <4.0 | nt |
Resistance Index (RI) = Dd2 EC50/3D7 EC50.
Selectivity Index (SI) = HepG2 EC50/Dd2 EC50.
nt = not tested.
Having discovered the antiplasmodial activity of human PLK1 inhibitors, we focused our attention on additional PLK1 inhibitors. We compiled a set of seven commercially available PLK1 inhibitors: BI-6727, a close analogue of BI-2536 (compound 11), GSK461364 (compound 12), MLN0905 (compound 13), SBE 13 (compound 14), NMS-P937 (compound 15), ON-01910 (compound 16), and GW843682 (compound 17). While these compounds are all very potent PLK1 inhibitors, displaying IC50 values under 9.0 nM, only two compounds demonstrated nanomolar antimalarial potency—compounds 11 and 12 (Table 2). Additionally, we evaluated a set of six potent PLK1 inhibitor analogues derived from a 2,4-diarylaminopyrimidine scaffold provided by the Deng Laboratory at Xiamen University (compounds 18–23).16 Among these compounds, antiplasmodial potencies varied, with Dd2 EC50 values ranging from 336 nM to 2.78 μM (Table 2). In general, the antiplasmodial activity varied widely among both compound 1 structural analogues and the known PLK1 inhibitors, as reflected in the broad range of EC50 values observed. Of all of the compounds tested, compound 1 displayed the most potent antiplasmodial activity followed by its close analogue compound 11 (Dd2 EC50= 230 ± 14 nM). Compound 2 although being less active against Plasmodium exhibited the highest selectivity (Table 1). Of the inhibitors screened in both the Dd2 and 3D7 parasite lines, no noteworthy difference in activity was noted, with resistance indices (RI) ranging from 0.34 to 1.5. Additionally, evaluation in clinical isolates displaying artemisinin and piperaquine resistance demonstrated that hit compounds such as compound 1 maintain potency (Table S2).
Table 2.
Antiplasmodial and Kinase Activities of PLK1 Inhibitors

| ||||||
|---|---|---|---|---|---|---|
| ID | antiplasmodial activity | cytotoxicity | kinase activity | |||
| Pf Dd2 EC50 (nM) | Pf 3D7 EC50 (nM) | RIa | HepG2 EC50 (nM) | SIb | PLK1 IC50 (nM) | |
| BI-6727 (11) | 230 ± 14 | 284 ± 52 | 0.81 | 1930 ± 460 | 8.0 | 0.87 |
| GSK461364 (12) | 533 ± 78 | 1410 ± 77 | 0.34 | 1910 ± 210 | 4.0 | 2.2 |
| MLN0905 (13) | 1820 ± 260 | nt | - | 3940 ± 110 | 2.0 | 2.0 |
| SBE 13 (14) | 3570 ± 130 | nt | - | >25,000 | >7.0 | 0.20 |
| NMS-P937 (15) | >5000 | nt | - | 6010 ± 79 | <1.0 | 2.0 |
| ON-01910 (16) | >5000 | nt | - | 11,900 ± 42 | <2.0 | 9.0 |
| GW843682 (17) | >5000 | nt | - | >25,000 | - | 2.2 |
| Compound 18 | 336 ± 34 | 315 ± 11 | 1.1 | 1870 ± 190 | 6.0 | 3.3 |
| Compound 19 | 971 ± 58 | 759 ± 96 | 1.3 | 2740 ± 310 | 2.0 | 4.6 |
| Compound 20 | 1990 ± 34 | 1860 ± 330 | 1.1 | 4090 ± 440 | 2.0 | 4.3 |
| Compound 21 | 2020 ± 150 | 2020 ± 340 | 1 | 6840 ± 210 | 3.0 | 29 |
| Compound 22 | 2530 ± 130 | nt | - | 4690 ± 690 | 2.0 | 4.9 |
| Compound 23 | 2780 ± 290 | nt | - | 4300 ± 450 | 2.0 | <0.50 |
To assess the therapeutic potential of these compounds, we also evaluated growth inhibition in the human hepatocarcinoma line, HepG2. We found that these compounds displayed a range of cytotoxicity, with HepG2 EC50 values from 1.87 μM to greater than 25 μM and parasite over human selectivity ranging from 1- to 58-fold (Tables 1 and 2). The most active inhibitor, compound 1, showed a selectivity index (SI) of 21 (HepG2 EC50 = 3730 ± 520 nM), making it the second most selective of the inhibitors tested. Based on the in vitro activity, there appeared to be no positive correlation between PLK1 inhibition and antiplasmodial potency. This finding was validated through a two-tailed regression coefficient t-test, which confirmed no significant correlation (Figure 1A).However, there was a positive correlation (p < 0.0001) between cytotoxicity and PLK1 inhibition among compound 1 structural analogues, but this was not observed among the diverse PLK1 inhibitors (Figure S2). A relationship was not seen between cytotoxicity and inhibition in Dd2 in compound 1 structural analogues (Figure 1B), and while the correlation among PLK1 inhibitors is significant (p = 0.0104), there appears to be an additional source of variation, supporting the separation between antiplasmodial potency and host-cell activity. These findings also suggest that selectivity could theoretically be improved by reducing affinity for PLK1 without sacrificing parasite inhibition.
Figure 1.
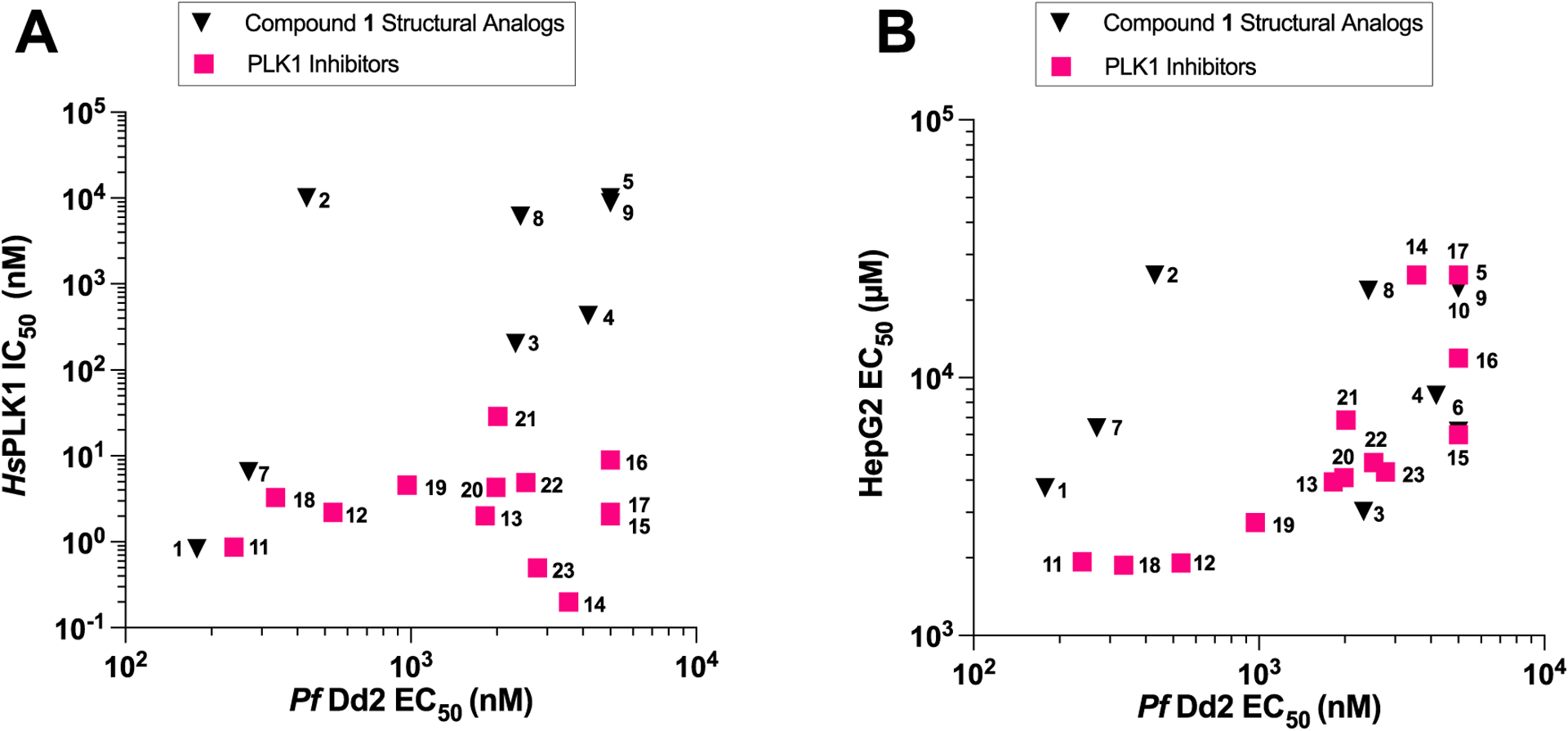
Antiplasmodial potency not correlated with human PLK1 inhibition. (A) Average Dd2 EC50 values are plotted against human PLK1 IC50 values (PLK1 Inhibitors r2 = 3.089 × 10–5, p = 0.9856; compound 1 structural analogues r2 = 0.1578, p = 0.3299). (B) HepG2 EC50 values plotted against Dd2 EC50 values (PLK1 inhibitors: r2 = 0.4635, p = 0.0104; compound 1 structural analogues: r2 = 0.1144, p = 0.3391).
PLK1 Inhibitors Show a Moderate Killing Profile with Early Life Cycle Arrest.
To further characterize the antiplasmodial impact of these PLK1 inhibitors, we assessed their killing profile in Dd2. Included in the assay were compound 1, its close analogue, compound 11, and compound 12, which has a distinct structure. Dd2 cultures were treated with 10 × EC50 of compound or vehicle control for 12, 24, or 48 h before removal. Cultures were then monitored for up to 96 h, or two asexual life cycles. Every 24 h, cultures were evaluated for parasite survival by flow cytometry with SYBR Green I and MitoTracker Deep Red FM staining to monitor the levels of viable parasites over time. Killing profiles were compared to the vehicle, to a slow-acting compound, atovaquone, and to a fast-acting compound, dihydroartemisinin (DHA). As shown in Figure 2A, all three inhibitors had an initial impact to parasitemia after 12 h of exposure. Parasites treated with compound 1 and compound 12 began to recover after 72 h, while compound 11 treated parasites showed a shorter time to recovery, around 48 h. After 24 h of treatment, parasites treated with compound 11 and compound 12 showed marked improvement at 72 and 96 h, respectively, and compound 1 treated parasites remaining more in line with fast-acting DHA, failing to recover within 4 days (Figure 2B). Upon 48 h of compound exposure, all cultures exposed to the PLK1 inhibitors remained under 1% parasitemia for the duration of the assay (Figure 2C). These results suggest that while compound 1 kills more rapidly than compound 11 or compound 12, all three compounds show a moderate killing profile, with 24–48 h of compound incubation required for a full killing effect.
Figure 2.

PLK1 inhibitors display similar killing profile. Dd2 parasites were treated at 1% parasitemia with 10 × EC50 of compound 1, 11, 12, or controls for 12 h (A), 24 h (B), or 48 h (C) prior to compound wash off. Samples were collected every 24 h and treated with nucleic acid stain SYBR Green I and MitoTracker Deep Red FM. Double-positive cells were considered viable. DHA and atovaquone served as fast-acting and slow-acting controls, respectively. The results displayed are the average of three biological replicates ± SEM.
We next sought to determine the stage at which these inhibitors exert their antiplasmodial effects. To accomplish this, synchronized Dd2 parasites were treated at the early ring (6 HPI), late ring (18 HPI), late trophozoite (30 HPI), and schizont (42 HPI) stages with 5 × EC50 of compound 1, 11, or 12. Samples were collected for flow cytometry and Giemsa staining every 12 h until reinvasion and signs of new rings (~54 HPI). When applied during the early ring stage (Figure 3), all three compounds appeared to induce arrest prior to DNA replication based on YOYO-1 staining, although a clear increase in size was noted from ring onward (Figure S3). A similar phenotype was observed when these inhibitors were applied later in the ring stage. However, compound 11 treated parasites showed a delayed increase in the YOYO-1 signal at 54 h, indicating some parasites were able to begin DNA replication. With a 30 HPI addition of compound, parasites treated with all compounds showed signs of entering into schizogony, reflected by an increase in both total DNA content and DNA content per cell at 42 HPI. When applied after schizogony, the effect was much less pronounced, and an increase in DNA content for all compounds was detected, albeit less noticeably for compound 12. Together, this suggests the PLK1 inhibitors have a shared impact on DNA replication when added early in the life cycle.
Figure 3.

Blood stage specificity of PLK1 inhibitors. Synchronized Dd2 parasites were treated at set timepoints and evaluated every 12 h throughout the life cycle until post-reinvasion. Changes in DNA content were assessed in YOYO-1-stained parasites with flow cytometry. For YOYO-1 positive populations, the total DNA content (top) as well as median DNA content per cell (bottom) for compound 1 (A), compound 11 (B), and compound 12 (C) are evaluated. Graphs depict averages of four biological replicates ± SEM. Treatment conditions were each compared to the control with t-test with the Holm–Šídák method for multiple corrections. P-values less than 0.05 are shown.
Having characterized the effect of compound 1 in the plasmodial intraerythrocytic stage, we performed a liver-stage assay to assess compound activity against parasite infection of hepatocytes. HepG2 were pretreated with a serial dilution of compound 1 for 18 h and subsequently infected with luciferase-expressing Plasmodium berghei sporozoites. After 48 h, viable exoerythrocytic parasites, indicative of successful establishment of liver-stage infection, were quantified via bioluminescence. We found that compound 1 displayed nanomolar liver-stage activity, with an EC50 of 367 ± 52 nM (Figure S4), though further investigation would be necessary to confirm that this activity extends to human malaria. Potent activity in both liver and intraerythrocytic stages is a particularly desirable trait in developing antimalarials as these compounds can impart both prophylactic and therapeutic effects.
PLK1 Inhibitors Exhibit Plasmodial Kinase PfNEK3 Inhibition.
Given the similarities between the in vitro profile of the PLK1 inhibitors tested and the lack of apparent correlation with PLK1 activity, we attempted to probe for other possible antiplasmodial protein kinase targets. As Plasmodium lacks polo-like kinases, we began by employing a kinase panel screening assay that utilizes a three-hybrid split-luciferase competitive binding assay17 utilizing a panel of 11 in vitro translated plasmodial kinases. With this assay, we evaluated six of the most potent PLK1 inhibitors identified at a fixed 2.5 μM concentration. We noted that one commonality observed in all compounds was the strong inhibition of Pf NEK3 (PF3D7_1201600), a plasmodial NIMA (never in mitosis gene A)-related kinase (NEK) (Table 3). Compound 1 exhibited the greatest inhibition, with only 4.7% enzyme activity remaining, followed by compound 12 with 9.7%. Of the analogues with the 7,8-dihydropteridinone core, compound 11 retained strong Pf NEK3 inhibition (11.1% activity remaining) yet was less selective than compound 1, while compound 2 displayed the most pan-kinase inhibition, as well as markedly reduced Pf NEK3 inhibition (39.6% activity remaining). The two 2,4-diarylaminopyrimidine compounds 18 and 19 demonstrated pan-kinase inhibition with a similar pattern in addition to notable Pf NEK3 inhibition with just 15.4 and 18.5% activity remaining, respectively. All targets with percent activity remaining lower than 40% were tested for IC50 using the same assay; these values are shown in nanomolars in parentheses in Table 3.
Table 3.
PLK1 Inhibitors Show Pf NEK3 Inhibition In Vitroa
| compound | % activity remaining at 2.5 μM in plasmodial kinases | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ARK1 | ARK3 | CDPK2 | CDPK5 | GSK3 | NEK1 | NEK3 | PK5 | PK6 | PK9 | PKB | |
| 1 | 100.0 | 100.0 | 80.6 | 100.0 | 99.4 | 94.5 | 4.7 (86 ± 11) | 100.0 | 94.6 | 97.2 | 100.0 |
| 2 | 53.4 | 73.5 | 100.0 | 100.0 | 100.0 | 93.3 | 39.6 (1480 ± 150) | 100.0 | 94.8 | 100.0 | 64.2 |
| 11 | 100.0 | 83.6 | 100.0 | 100.0 | 100.0 | 93.8 | 11.1 (130 ± 20) | 100.0 | 67.3 | 100.0 | 49.3 |
| 12 | 99.5 | 96.2 | 100.0 | 100.0 | 100.0 | 100.0 | 9.7 (19 ± 30) | 100.0 | 100.0 | 100.0 | 100.0 |
| 18 | 73.9 | 84.6 | 24.0 (390 ± 50) | 88.1 | 100.0 | 96.4 | 15.4 (630 ± 120) | 100.0 | 78.9 | 78.7 | 12.3 (250 ± 60) |
| 19 | 77.9 | 83.6 | 36.1 (780 ± 90) | 100.0 | 100.0 | 90.1 | 18.5 (1820 ± 210) | 100.0 | 91.0 | 85.7 | 14.8 (790 ± 140) |
Values are % activity remaining.
Values in parentheses are IC50 values in nM ± SD.
Interestingly, while compounds 1 and 12 are touted for their PLK1 inhibitory potency, both compounds have been shown to inhibit multiple human NEKs among their off targets in the KINOMEscan competitive binding assay, with compound 12 displaying more broad activity (NEK1 Kd = 1.8 μM, NEK2 Kd = 260 nM, NEK5 Kd = 1.1 μM, NEK9 Kd = 1.1 μM) than compound 1 (NEK2 Kd = 2.0 μM, NEK3 Kd = 720 nM, NEK7 Kd = 6.1 μM).18 In humans, there are 11 NEK paralogues involved in the cell cycle though P. falciparum contains only four (Pf NEK1–4). Pf NEK1 (PF3D7_1228300) is essential to the asexual blood stage and is expressed in male gametocytes.19 Pf NEK2 (PF3D7_0525900) is implicated in the sexual stage of the parasite, particularly the development of the plasmodial zygote, the ookinete.20 Pf NEK4 (PF3D7_0719200), while not essential for schizogony or gametocytogenesis, is associated with sexual commitment.21 Pf NEK3 had been shown to be expressed late in the asexual blood stage and in gametocytes; however, it is believed to be nonessential for P. falciparum asexual stage development and instead plays a larger role in gametogenesis.22–24 Pf NEK3 is unusual when compared to other plasmodial NEKs and human NEKs. A homology search with protein–protein BLAST (https://blast.ncbi.nlm.nih.gov/) reveals that the kinase domain of Pf NEK3 is most similar to human NEK9, though only at 35% identity. Additionally, the highly conserved glycine-rich loop (GxGxxG) in the ATP-binding site is completely absent in Pf NEK3.22 Pf NEK3 displays a dual kinase function, autophosphorylating serine, threonine, and tyrosine residues, with the tyrosine autophosphorylation playing the largest role in the modulating activity.25 Pf NEK3 has been demonstrated to phosphorylate Pf MAP2 (PF3D7_1113900) in vitro, in addition to myelin basic protein (MBP), a commonly used substrate for experimental kinase assays.22 The exact roles of Pf NEK3 and Pf MAP2 in relation to a broader phosphosignaling cascade is unknown, and inferences based on eukaryotic MAPK pathway provided limited insight as Plasmodium lacks the canonical MAPK signaling module.26 However, the fact that Pf NEK3 does phosphorylate Pf MAP2 highlights the variability in Pf NEK3 functionality and hints at roles aside from mitosis. If the inhibition of Pf NEK3 is to some degree responsible for antiplasmodial activity, these other roles may be more influential, as Pf MAP2 appears not to be essential to the asexual stage.27 Aside from potential therapeutic applications, the discovery of selective inhibitors of Pf NEK3 may also allow chemical interrogation of this atypical kinase and deepen the understanding of the plasmodial kinome.
Along with NEKs, Aurora-related kinases (ARKs) also play an essential role in the regulation of the plasmodial life cycle.29 Furthermore, it has been reported that mutations in either Pf NEK1 or PfARK1 confer resistance to a human Aurora kinase inhibitor, suggesting a relationship between the two kinases.28 Due to the potential biological connection between Pf NEKs and Aurora-related kinases (ARKs), compounds 1 and 12 were also tested against the three known PfARKs.29 PfARK1 and PfARK3 were included in the KinaseSeeker assay, and no significant inhibition was seen (Table 3). Activity against PfARK2, found to be essential to the asexual blood stage, was assessed with a radioactive kinase assay (Figure S5A); however, neither compound displayed inhibitory effects in PfARK2 at the highest concentrations tested (2 μM).24 An ADP-Glo assay confirmed this lack of activity in both PfARK2 and the human Aurora kinase, AurA, up to 5 μM (Figure S5B).
To confirm the split-luciferase assay results, a radioactive kinase assay was performed with purified recombinant Pf NEK3 and MBP as a substrate. When assessed at a concentration up to 2 μM, the analogues with the greatest antiplasmodial potency, compounds 1 and 12, showed a clear dose-dependent response to Pf NEK3 phosphorylation (Figure S6). We then evaluated a subset of compound 1 structural analogues, representing a range of antiplasmodial potencies, at a fixed 1 μM using the same assay (Figure 4A). As shown in Figure 4B, compound 1 and two analogues with the 7,8-dihydropteridinone core, compounds 3 and 4, inhibited Pf NEK3 more strongly than the pan-kinase inhibitor staurosporine. When the percent of Pf NEK3 activity remaining is plotted against the Dd2 EC50, only 6/8 of the analogues show a correlation between enzyme inhibition and in vitro inhibition, and compounds 3 and 4 appear as outliers (Figure 4C). This persisted when the Pf NEK3 IC50 was determined using the same assay, with a significant correlation calculated for 7/8 compounds tested only when values for compound 4 were removed (Table S3). Given these findings, the inhibition of Pf NEK3 is unlikely the sole predictor of in vitro inhibition for these repurposed PLK1 inhibitors. Some of the compounds, such as compound 4, may show a greater proclivity for other targets in a whole cell environment. Furthermore, considering the nonessentiality of Pf NEK3 to the asexual blood stage, there are likely alternate or additional targets.
Figure 4.
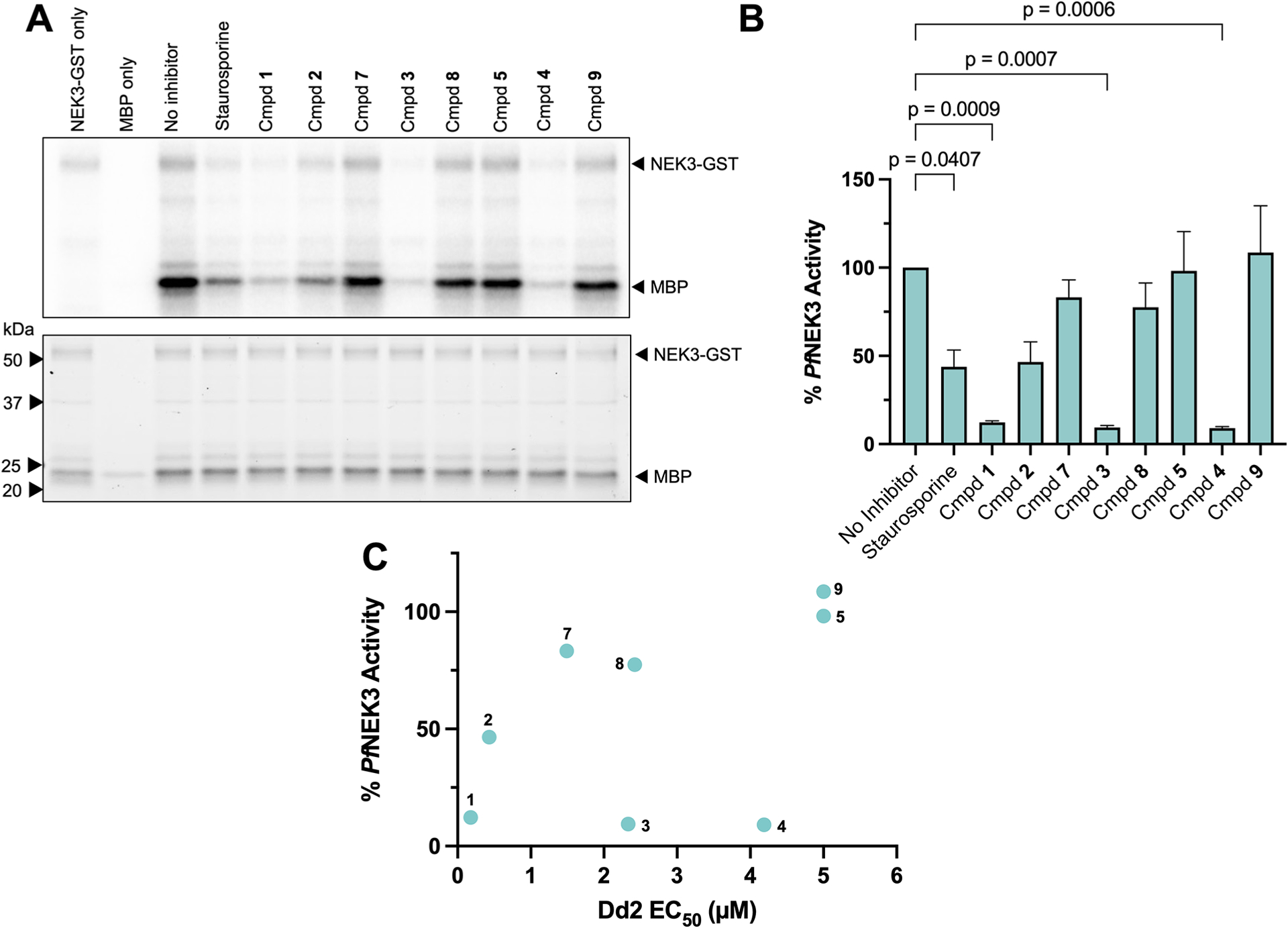
Pf NEK3 inhibition not the sole predictor of antiplasmodial activity. (A) Inhibition of Pf NEK3 following incubation with 1 μM PLK inhibitors. Top: Autoradiogram. Bottom: TCE gel image for protein quantification. A 1 μM pan-kinase inhibitor staurosporine serves as a control. Gel representative of three replicates. (B) Quantification of kinase activity. Autoradiogram band intensity normalized to protein concentration by the TCE gel band. Activity normalized to the no-inhibitor control (100% activity). Data shown represents averages ± SEM of three replicates. Significance of treatments compared to the no-inhibitor control determined by one-way analysis of variance (ANOVA). (C) Relationship between Dd2 EC 50 and Pf NEK3 activity remaining after 1 μM treatment (r2 = 0.2117, p = 0.25).
Compound 1 is known to potently inhibit the human bromodomain-containing protein BRD4 in addition to PLK1.30,31 In fact, along with tubulin, bromodomains are common nonkinase targets of kinase inhibitors.32 In 2016, an open access data set was released by the Structural Genomics Consortium (SGC) that contained the crystal structure of compound 1 cocrystalized with Pf BDP4 (PDB ID: 4PY6) and explored the antiplasmodial effects and binding kinetics.33 They identified the Pf BP4 Kd for compound 1 to be ~100 nM, and the 3D7 EC50 recorded was comparable to our findings (~200 nM).33 The aforementioned crystal structure was later the basis of an in silico docking-based screen of compounds with the bromodomains of Pf BDP1 and Pf GCN5.34 However, when tested in vitro with Dd2 culture, the antiplasmodial effects were modest, with the most potent compound demonstrating an IC50 of 3.2 μM.34 The additional target of a bromodomain alongside Pf NEK3 is intriguing, as bromodomain protein inhibitors are under consideration as emerging antimalarials.35
In Vitro Evolution of Resistance for Target Identification.
In addition to kinase panel screening, we also sought to identify a target through the creation of parasite lines resistant to compound 1. With increased drug pressure applied over ~8 months, resistant parasites were generated in the P.falciparum strains Dd2, 3D7, and Dd2-Polδ, a strain with a DNA polymerase δ mutation that confers hypermutability (Figure 5A). 36 Whole genome sequencing (WGS) was conducted with Dd2 and 3D7 selections. Clonal parasites were obtained from two independent Dd2-Polδ selections, and two clones from each were sequenced: AE9, AG8, BB5, and BE11. Sequencing revealed that, compared to the respective parental strain, resistant parasites from all selections in 3D7 possessed single nucleotide polymorphisms (SNPs) in Pf MDR1 (PF3D7_0523000), an ATP-binding cassette transporter implicated in resistance to multiple antimalarial drugs,37 and amplification of transporter PfABCI3 (PF3D7_0319700), which has also been associated with resistance to numerous antiplasmodial scaffolds (Supporting Files 1 and 2).38 Among resistant Dd2 cultures, SNPs within Pf MDR1 were observed in two of the three selections. In most Dd2-Polδ clones, Pf MDR1 amplification was noted, though BB5 lacked this feature. Interestingly, all four of the Dd2-Polδ clones possessed SNP-induced missense mutations in genes associated with the starvation-induced stress response. In both AE9 and AG8, there were mutations in the methionyl-tRNA synthetase Pf MRSapi (PF3D7_1005000), the autophagy-related E1 activating enzyme PfATG7 (PF3D7_1126100), and an eIF2α kinase, Pf PK4 (PF3D7_0628200). BE11 possessed a mutation in Pf VPS15 (PF3D7_0823000), a kinase that assists in early autophagosome assembly, while BB5 contains a mutation in Pf SYN17 (PF3D7_0210700), a homologue of syntaxin 17 that facilitates fusion of the autophagosome to the lysosome.39 The prevalence of mutations in genes involved in tRNA maturation and aminoacylation and in the starvation response suggested that an additional target of compound 1 may be an aminoacyl-tRNA synthetase (aaRS).
Figure 5.

Compound 1 induces eIF2α-mediated stress response. (A) Parasites with resistance to compound 1 obtained from P. falciparum three strains. (B) Western blot reveals that Dd2 parasites treated with Compound 1 exhibit increased phospho-eIF2α phosphorylation compared to the vehicle control (dimethyl sulfoxide (DMSO)), though not as much as the positive phosphorylation controls, borrelidin and halofuginone. Blot is representative of three biological replicates. (C) Missense mutation is located at the conserved residue in MRSapi (UniProt ID: Q8IJ60). Predicted structure derived from AlphaFold (www.alphafold.ebi.ac.uk).
To determine whether compound 1 does indeed activate the stress response pathway, we investigated increases in eIF2α phosphorylation as an indicator of an activated stress response. The eIF2α subunit plays an important role in post-transcriptional control of protein expression; phosphorylation prohibits the recycling of eIF2α-GDP to eIF2α-GTP, leading to the inability to form the ternary complex, a component of the preinitiation complex, and to a global reduction of translation and a prioritization of translation of stress response-related proteins.40 In vertebrate cells, four kinases are capable of phosphorylating eIF2α, each responding to a different cellular stressor.40 In Plasmodium, eIF2α phosphorylation is conducted by three kinases, PfeIK1 (PF3D7_1444500), PfeIK2 (PF3D7_0107600), and Pf PK4 (PF3D7_0628200), the latter of which is essential to the asexual life cycle.41–43 To evaluate the effect of compound 1 on eIF2α phosphorylation, Dd2 cultures in the primarily late ring stage were treated for 3 h at three concentrations: 10, 1, and 0.1 μM. Two known antiplasmodial compounds served as controls for eIF2α phosphorylation; borrelidin, a threonyl-tRNA synthetase inhibitor,44 and halofuginone, a prolyl-tRNA synthetase inhibitor,45 were used at 200 nM each. Western blot analysis revealed that phosphorylation of eIF2α was notably increased upon compound 1 treatment, though not to the extent caused by treatment with the confirmed aaRS inhibitors, despite the high concentration of compound used (Figure 5B). Since eIF2α phosphorylation can result from activation of disparate stress response pathways, the upstream target of compound 1 eliciting the stress response may be difficult to identify. Since the observed missense mutation in Pf MRSapi alters a conserved residue in one of the enzyme’s two methionyl/leucyl-tRNA synthetase domains (InterPro entry: IPR015413) (Figure 5C), it becomes an intriguing potential target, in addition to Pf NEK3, though further investigation is needed.
PLK1 Inhibitors Compound 1 and Compound 12 Show Divergent Phosphoproteomes.
To explore the signaling pathways disrupted by these inhibitors, we performed a comparative phosphoproteomics evaluation. Asynchronous intraerythrocytic Dd2 cultures were incubated for 3 h with 5 ×EC50 of compound 1, compound 12, or DMSO as a control. The protein lysate was digested with LysC and trypsin and labeled with isobaric tandem mass tags (TMTpro)46 as a 16-plex assay with two other compounds in biological triplicate, as well as a single replicate with a fifth compound; however, these compounds originate from a separate library and will be discussed further in subsequent publications. To quantify proteins and phosphorylation sites, peptides underwent IMAC enrichment to separate phosphorylated and nonphosphorylated peptides before analyzing both peptide populations via quantitative mass spectrometry for comparison to Plasmodium and human databases. The analysis of nonphosphorylated peptides resulted in identification of 3515 unique Plasmodium proteins, all supported by two or more distinct peptides. While a small number of human proteins were observed, all were single-peptide identifications and were thus discarded. The analysis of the phospho-enriched eluate resulted in the identification of 1593 unique phosphorylation sites; 1494 were identified from P. falciparum and 99 from Homo sapiens (Table S4). Differential expression compared to the corresponding control was determined for both proteins and phosphosites. The most differential proteins/phosphosites were ranked by ratio-based paired Z-score, and the top 10% were examined further. The full list of detected proteins with more than one peptide and phosphosites can be found in Supporting Files 3 and 4, and the top differential proteins are detailed in Tables S5 and S6. As seen in Figure 6A and B, when comparing the top differentially expressed phosphosites (|log 2(fc)| > 0.585), there was limited consensus between compounds 1 and 12, though both compounds induce more extensive downregulation of phosphorylation than upregulation (Tables S7 and S8). Despite differences, there were still several downregulated phosphosites shared between the two treatment conditions, including Pf HECT1 site S9833 (PF3D7_0628100), DCP1 site S765 (PF3D7_1032100), Pf PAIP1 site S390 (PF3D7_1107300), and Pf BTP2 (PF3D7_0704100) site S912.
Figure 6.

Differentially expressed phosphosites upon exposure to compound 1 (A) and compound 12 (B) of the top-ranked most downregulated (log 2(fc) < −0.585) motifs per treatment. Dashed lines on volcano plots represent cutoff values of the top 10% of the absolute values of the |Z-score| and |log 2(fc)| > 0.585, and dotted lines represent |log 2(fc)| > 0.322.
To view these differential phosphorylation sites in a network context, we identified those proteins bearing one or more of the 10% most differential phosphorylation sites and extracted their corresponding subnetwork from the STRING database (Figure 7A).47 Pf NEK3 was included as well to visualize its connection to the network. Two clusters appear, one containing Pf HECT1, Pf BTP2, and Pf KIC8, and another containing Pf PAIP1. Pf HECT (PF3D7_0628100), downregulated at site S9833 upon treatment with both compounds, is a HECT family E3 ubiquitin ligase linked to the plasmodial phosphorylation network through direct regulation by Pf PP1 (PF3D7_1414400), a phosphatase involved in erythrocyte development and egress.48 Pf BTP2 (PF3D7_0704100), basal complex transmembrane protein 2, has been implicated in late stages of blood stage schizogony, specifically the formation of the basal complex that mitigates cytokinesis, and its phosphorylation is also downregulated by both compounds at site S912.49 Interestingly, phosphorylation of Pf KIC8 (PF3D7_1014900), a speculated Kelch13 interaction partner,50 was upregulated at site S1500 in the presence of compound 1 but downregulated at site S104 by exposure to compound 12. Pf PAIP1 (PF3D7_1107300), central to the other observed cluster, was also downregulated at site S390 by both compounds. Pf PAIP1 is a putative polyadenylate-binding protein-interacting protein thought to facilitate translation by mediating interaction of translational machinery components, specifically RNA helicase eIF4A and polyadenylate-binding proteins.51 Similarly, phosphorylation of Pf DCP1 (PF3D7_1032100) at site S765 is downregulated by both compounds. Like Pf PAIP1, Pf DCP1 is also involved in translation regulation, as it is a subunit of an m7G 5′decapping enzyme that facilitates mRNA decay.52 Finally, an uncharacterized protein, PF3D7_1036900, was downregulated upon treatment at two sites; treatment with compound 1 led to decreased phosphorylation at site T1037, while compound 12 decreased phosphorylation at both site T1037 and site S805. While the function of this protein is unknown, immunoprecipitation experiments suggest association with the N6-adenosine-methyltransferase MT-A70, which plays a role in post-transcriptional control by influencing mRNA stability.53
Figure 7.
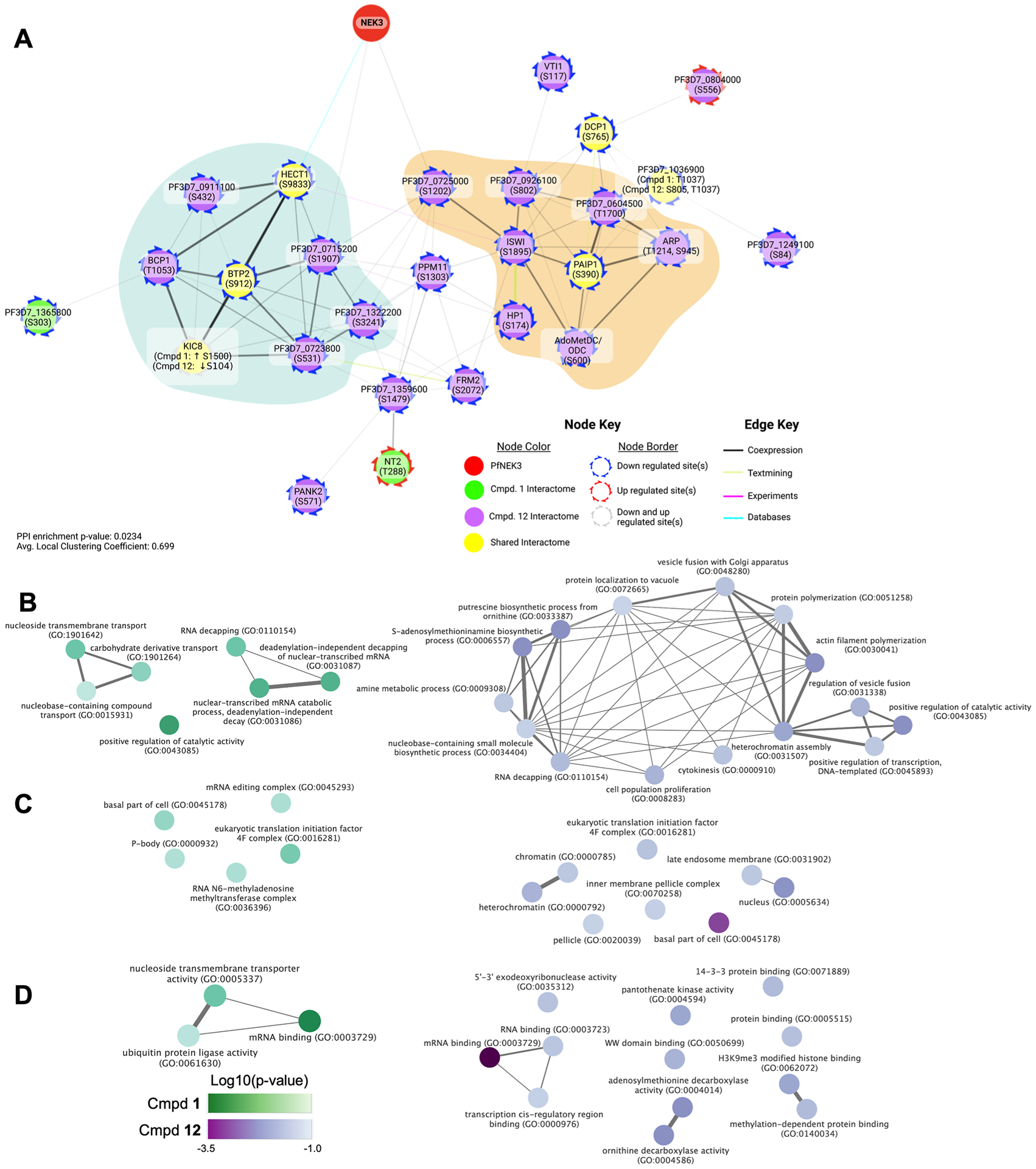
Interactome of compounds 1 and 12 and resulting GO enrichment analysis. (A) STRING network of proteins possessing the top differential (|log 2(fc)| > 0.585) phosphosites with Pf NEK3. Color represents the interactome source (compound 1, compound 12, shared), border color represents the direction of deregulation. Edge color represents strongest contributing source, and edge thickness represents the overall edge score. (B–D) Shared and distinct GO enrichment for compound 1 (green) and compound 12 (purple) for the biological process (B), cellular component (C), and molecular function (D). Color intensity reflects log 10 (enrichment p-value), and edge indicates similarity between terms.
To better assess the similarities and differences between the exposure to compounds 1 and 12, GO enrichment analysis was performed via PlasmoDB (https://plasmodb.org)54 using the gene identifiers of the proteins bearing the top differential phosphosites as the foreground data set and the P. falciparum 3D7 genome as the background. Terms from top differentially phosphorylated proteins were grouped into simplified categories based on commonalities (Figure 7B–D). We found that both treatments shared biological process terms related to RNA decapping, while compound 1 was enriched in transport as well (Figure 7B). Shared cellular component terms were related to the basal component and eukaryotic initiation factor 4F (eIF4F) (Figure 7C), and compound 1 was enriched in terms related to the P-body and RNA N6-methyladenosine methyltransferase complex, both of which are involved in translational regulation. Shared molecular function terms were related to mRNA binding (Figure 7D). Taken together, while there are similarities to be found between the interactomes of these two inhibitors, it is clear that in addition to Pf NEK3, they likely affect disparate signaling pathways. Compound 1 in particular impacts processes related to translational regulation and repression, consistent with downstream effects of the observed eIF2α phosphorylation.
CONCLUSIONS
In this paper, we screened a series of human kinase inhibitors for potential drug repurposing to discover malaria therapeutics. This effort identified compound 1, a potent inhibitor of the human kinase PLK1. As P. falciparum lacks polo-like kinases, we explored compound 1, structural analogues of compound 1 possessing either a 7,8-dihydropteridinone or a pyrimido-diazepine core, and other PLK1 inhibitors for a potential antiplasmodial mechanism. Through these efforts, we found a majority of the PLK1 inhibitors tested displayed inhibition against the plasmodial homologue of NEK3. For some of the inhibitors, the relationship between Pf NEK3 inhibition and antiplasmodial potency was more complex, suggesting additional causative targets. When evaluating the effect of these PLK1 inhibitors in vitro, we found that they exerted their greatest effect early in the asexual life cycle; if applied in ring or early trophozoite stages, they appear to affect DNA replication. Additionally, the compounds displayed a moderate killing profile, in general, requiring 24–48 h for a full killing effect. As a probative look at the in vivo activity of compound 1, we treated mice with 50 mg/kg for 4 days. The treatment was found to be inconclusive, and additional work in needed (Figure S7). However, caution is warranted when using P. berghei models: while the murine-infective strain is a useful tool, there are some genes that have been reported to be essential in P. berghei and dispensable in P. falciparum or vice versa, and NEK3 is one such example. While NEK3 is essential for schizogony in P. berghei, it appears to be refractory in P. falciparum.23,55 This is certainly something to keep in mind when evaluating data from models using P. berghei.
The connection between PLKs and NEKs has been well-established in eukaryotic cells, where mitosis is a coordinated effort utilizing PLKs, Aurora kinase, cyclin-dependent kinases, and NEKs. These mitotic kinases are all involved in the duplication, maturation, and separation of centrosomes through the cell cycle.56 In addition to its function in centrosomal coordination, PLK1 regulates entry from G2 into mitosis, chromosome segregation, cytokinesis, and ciliary assembly.57 Since Plasmodium lacks PLKs, the question arises which kinase (or kinases) executes these functions instead. It has been noted that the paucity of cell-cycle-related kinases in Plasmodium suggests that these kinases perform multiple functions, perhaps also as cell cycle checkpoints.58 Pf NEK3 possesses an N-terminal hydrophobic region, which has been omitted when generating recombinant proteins due to issues it poses in solubility.22 The region is also suspected to contain a localization signal and a transmembrane portion.22,59 Perhaps this region is involved in anchoring to the nuclear membrane, facilitating proximity to the centriolar plaques, the centrosomal equivalent in Plasmodium that likely exerts local control of cell division.60 It is intriguing to consider whether Pf NEK3 is a kinase involved in the localized control of nuclear division in schizogony as part of a complex, perhaps with other cell cycle kinases. In lieu of PLK1, it is possible that Pf NEK3 may fulfill some of the cell cycle duties.
While Pf NEK3 can be inhibited in situ by these PLK1 inhibitors, a full deconvolution of their mechanism of action would require further efforts, and the possibility of action via inhibition of a host kinase cannot be excluded. Targeting multiple kinases can in fact be desirable as it raises the hurdle for resistance and increase efficacy, though promiscuity must be balanced to mitigate off-target effects.61 In addition to kinase targets, certain PLK1 inhibitors like compound 1 may also target aaRSs, triggering the cell to initiate a starvation-induced stress response. Since Plasmodium possesses aaRSs that reside in the apicoplast, a parasite-exclusive organelle, there is a great potential for development of selectivity, and plasmodial aaRSs have been validated as targets for innovative antimalarial compounds with unique mechanisms of action.62,63 In fact, repurposing human kinase inhibitors that target plasmodial aaRSs has been recently explored as a source of novel antimalarials.64 Overall, the results of this study indicate that kinase inhibitors designed to target human PLK1 have potential as antimalarials and as Pf NEK3 probes. Since little of the function of Pf NEK3 has been elucidated, compound 1 and other such PLK1 inhibitors may be used to interrogate the role of Pf NEK3 in the life cycle of Plasmodium, in addition to being a chemical starting point for antiplasmodial medicinal chemistry efforts.
METHODS
Chemistry.
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on a 500 MHz NMR (Bruker-500), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument (Milford, MA). Preparative high-performance liquid chromatography (HPLC) was performed on a Waters Symmetry C18 column (19 mm × 50 mm, 5 μM) using a gradient of 5–95% methanol in water containing 0.05% trifluoacetic acid (TFA) over 8 min (10 min run time) at a flow rate of 30 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by LC-MS analysis. Detailed compound characterization was given in the Supporting Information. Structure graphics were generated by ChemDraw 21.0.0.
Culture Conditions.
P. falciparum Dd2 and 3D7 parasites were cultured with an adapted Trager and Jansen protocol.65 Cultures were maintained at 37 °C in 5% CO2 or in 95% N2/5% CO2 in RPMI 1640 medium (25 mM HEPES, 26 mM sodium bicarbonate, 15 mg/L hypoxanthine, 25 mg/L gentamycin, 2% dextrose), 0.5% AlbuMAX II (Thermo Fisher, Waltham, MA), and L-glutamine as needed. Human A+ erythrocytes were used to maintain 4% hematocrit. HepG2 human hepatocytes were maintained at 37 °C in 5% CO2 in Eagle’s Minimal Essential Medium supplemented with 10% fetal bovine serum and 1% antibiotic–antimycotic solution (Gemini Bio, West Sacramento, CA).
SYBR Green I Proliferation Assay.
This assay was performed as described previously.66 Asynchronous blood stage P. falciparum cultures were plated at 1% parasitemia and 1% hematocrit in a 96-well plate and incubated with serial dilutions of compound for 72 h at 37 °C in 5% CO2. DMSO concentrations were kept at a sublethal percentage (<0.2%). As a negative parasite growth control, 5 μM chloroquine was used. Plates were then frozen at −80 °C, thawed, and wells were given 100 μL of SYBR Green I-containing lysis buffer (20 mM Tris-HCl, 5 mM EDTA, 0.8% Triton X-100, 0.08% saponin, and 0.0001% SYBR Green I) (Thermo Fisher, Waltham, MA). After incubation for 45 min at room temperature protected from light, fluorescence emission was read with a multimode microplate reader (BioTek Synergy Neo2, Winooski, VT) at 485 nm excitation and 530 nm emission. The results were normalized using untreated and chloroquine-treated controls, and EC50 was determined with CDD Vault (www.collaborativedrug.com, Burlingame, CA).
Cytotoxicity Assay.
HepG2 cells were seeded at 2250 cells per well in a flat-bottomed black 384-well plate with a clear bottom and incubated at 37 °C with 5% CO2. After 24 h, a serial dilution of compound was applied, and the plate was incubated further for 48 h. Negative control wells were treated with 5% Triton X-100 for 10 min, and 10 μL of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) reagent (CellTiter 96 AQueous One Solution Reagent, Promega, Madison, WI) was added per well. After 3 h, the plate was read in a microplate reader (BioTek Synergy Neo2, Winooski, VT) at an absorbance of 490 nm. The results were normalized with untreated cells and Triton X-100-treated cells. EC50 was determined with a CDD Vault (Burlingame, CA).
Rate of Killing.
A flow-cytometry-based rate of killing assay was performed as described previously.67 Asynchronous P. falciparum Dd2 cultures were plated at 1% parasitemia and 4% hematocrit into a 24-well plate. Wells were treated with 10 × EC50 of compound for 12, 24, or 48 h before the compound was removed. Every 24 h thereafter for 4 days in total, culture samples were washed in 0.5% bovine serum albumin (BSA) and stained with 1× SYBR Green I (Thermo Fisher, Waltham, MA) and 60 μM MitoTracker Deep Red FM (Thermo Fisher, Waltham, MA) for 20 min at 37 °C. After another wash in 0.5% BSA, samples were resuspended in phosphate-buffered saline (PBS) and analyzed via flow cytometry with a CytoFLEX S (Beckman Coulter, Brea, CA), with 100,000 events recorded per sample. For gating, the following controls were used: uninfected RBCs, untreated culture, DHA-treated culture, and untreated culture without MitoTracker stain. Viable parasite percentage was determined by finding the fraction of SYBR Green/MitoTracker double-positive cells from the total events. The analysis was performed with FlowJo software Version 10 (Ashland, OR).
Stage-Specific Assay.
The stage-specific assay was performed as previously described.68 Asexual stage P. falciparum cultures were first synchronized by a MACS magnetic column (Miltenyi Biotec, Bergisch Gladbach, Germany), followed 3 h later by treatment with 5% sorbitol to select for rings.69,70 Six hours after invasion, parasites were plated onto a 96-well plate at 1% parasitemia and 1% hematocrit, and the compound was added at 5 × EC50 to the 6 HPI wells. Every 12 h, until reinvaded rings (54 HPI), thin blood smear slides for Giemsa stain were taken, and samples were washed and fixed in 4% paraformaldehyde in PBS with 0.04% glutaraldehyde. At the conclusion of the timepoints, all samples were permeabilized with 0.25% Triton X-100 before being treated with 50 μg/mL RNase A and stained with 500 nM YOYO-1 (Thermo Fisher, Waltham, MA). Samples were then washed, and flow cytometric analysis was performed with a CytoFLEX S (Beckman Coulter, Brea, CA), collecting 100,000 events per sample. The analysis was performed with FlowJo software Version 10.
Liver-Stage Activity Assay.
Assessment of activity in the liver stage was done as previously described. 71 Briefly, 3 × 103 of HepG2-A16-CD81 cells were seeded in 1536-well plates (Greiner Bio, Frickenhausen, Germany). Then, 50 nL of the test and control compounds were added in a 12-point 1:3 serial dilution. After 18 h, the cells are infected with 1 × 103 P. berghei sporozoites expressing luciferase. After 48 h post infection, 2 μL of luciferin reagent (BrightGlo, Promega, Madison, WI) was added to each well, and luciferase activity was detected using a PerkinElmer Envision plate reader. IC50 values were determined in CDD Vault (Burlingame, CA) normalized to maximum and minimum inhibition levels for the positive (Atovaquone 0.25 μM) and negative (DMSO) control wells.
ADP-Glo Kinase Assay.
Activities of compounds 1, 11, and 12 were evaluated in recombinant PfARK2-GST and human AurA-GST, which is a kind gift from the laboratory of Dr. Ratna Chakrabarti. Assays were performed in 384-well white round-bottom plates, with a final reaction volume of 10 μL. For both kinases, reactions were comprised of 1× Kinase Buffer (Cell Signaling) and 10 μM ATP (Thermo Fisher, Waltham, MA), with 2 μg of the substrate and 0.4 μg of kinase. For PfARK2, the substrate is dephosphorylated MBP (#31314, ActiveMotif, Carlsbad, CA), and for human AurA, the substrate is cofilin (a gift from the Dr. R. Chakrabarti Laboratory). Inhibitors were added as a serial dilution of 1:3 beginning with 5 μM final concentration, followed by the buffer and enzyme. After 10 min of room-temperature incubation, the ATP and substrate were added, and the reactions were left to incubate for 45 min at 25 °C. Next, 10 μL of ADP-Glo Reagent per well was added to stop the reaction, and the plate was incubated at room temperature for 40 min. Next, 20 μL of Kinase Detection Reagent per well was added, and luminescence was evaluated with a multimode microplate reader (BioTek Synergy Neo2, Winooski, VT). All reactions were performed in duplicate. Per kinase, reactions without inhibitor served as a 100% activity control, and reactions without kinase served as 0% activity controls. The known human AurA inhibitor, VX680 (#HY-10161, MedChemExpress, Monmouth Junction, NJ), was a control for AurA inhibition. Analysis and graphing were generated with GraphPad Prism 9 (San Diego, CA).
Resistance Line Generation and Genomic Analysis.
Resistant parasites were selected with three parental clones: 3D7 clone A10, Dd2 clone B2, and Dd2-pol delta clone H11. For each selection, three independent flasks containing 107 parasites were exposed to BI-2536 for 3–5 days at EC50 concentrations for each parasite clone (288 nM for 3D7, 371 nM for Dd2, and 256 nM for Dd2-pol delta). BI-2536 was removed from the culture media once parasite replication stopped and was started again after parasites resumed to replicate well. The drug off–drug on cycles were continued for 8 months of culturing. During this time, the concentration of BI-2536 was gradually increased (in ~10% steps) until a concentration of 1.8 μM. For each of the different three parental parasite lines, all three flasks yielded compound-resistant parasites. Two flasks originating from 3D7-A10 and three flasks originating from Dd2-B2 were subjected to WGS, while clonal parasites were obtaining from two independent selections with Dd2-pol delta parasites. For Dd2-pol delta parasites, two clones each from two independent selections were sequenced. Genomic DNA (gDNA) was obtained from parasite samples by washing infected RBCs with 0.05% saponin and isolating using the DNeasy Blood and Tissue Kit (Qiagen) following standard protocols. Sequencing libraries were prepared by the UCSD Institute for Genomic Medicine (IGM) Genomics Center using the Nextera XT kit (#FC-131–1024, Illumina, San Diego, CA) and were sequenced on lllumina NovaSeq. 6000 (S4 200, PE100). Sequencing reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB v13.0) and preprocessed following standard GATK version 3.5 protocols.72,73 SNVs and INDELs were called with GATK HaplotypeCaller and filtered to retain high-quality variants as previously described.74 CNVs were detected by the GATK 4 workflow72 adapted for P. falciparum as described in Summers et al.75 Briefly, read counts were collected across the genic regions of the P. falciparum core genome76 and denoised log 2 copy ratios were calculated against a panel of normals constructed from nondrug-selected 3D7 and Dd2 samples. CNVs were retained if at least four sequential genes showed a denoised log 2 copy ratio greater than or equal to 0.5 (copy number increase).
Western Blot Analysis of eIF2α Phosphorylation.
Flasks of P. falciparum Dd2 cultures at 4% hematocrit were sorbitol-synchronized as described above, and cultures were predominantly ring to early trophozoite stages and ~10% parasitemia were treated for 3 h with compound 1 at three concentrations: 10, 1, and 0.1 μM. Borrelidin (SC-200379, Santa Cruz Biotechnology, Dallas, TX) and halofuginone (#55837–20-2, MedChemExpress, Monmouth Junction, NJ) at 200 nM each served as positive controls for eIF2α phosphorylation, and DMSO at 0.1% served as a vehicle control. After incubation, RBCs were lysed with 0.1% saponin and washed three times with Dulbecco’s phosphate buffer (DPBS). Pellets were frozen at −80 °C, thawed on ice, and resuspended in Pierce RIPA buffer (#89900, Thermo Fisher, Waltham, MA) supplemented with 1× Halt Protease & Phosphatase Inhibitor Cocktail (#78443, Thermo Fisher, Waltham, MA). Pellets were incubated on ice for 30 min with periodic vortexing, and lysate cleared with centrifugation at 4 °C at 15 000g for 10 min. After quantification with the Pierce BCA Protein Assay (#23227, Thermo Fisher, Waltham, MA), 30 μg protein per sample was given 1× NuPAGE loading dye (Invitrogen, Waltham, MA) and boiled for 7 min at 80° and loaded onto a 12% Bis–Tris sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel. After electrophoresis, the proteins were blotted onto a poly-(vinylidene fluoride) (PVDF) membrane via the BioRad TransBlot Turbo system and blocked at room temperature for 1 h with 5% milk in tris-buffered saline with 0.1% Tween 20 (TBST). Primary antibody incubations were performed overnight at 4 °C, and secondary antibody incubations were performed at room temperature for 1 h. Between incubations, membranes were washed with TBST 3 × 5 min at room temperature. Rabbit antiphospho-eIF2 (Ser51) primary antibodies (#3597, Cell Signaling) were diluted 1:1000 in 5% BSA in TBST, and anti-rabbit HRP-conjugated secondary antibodies (#7074, Cell Signaling) were diluted 1:20,000 in 5% milk in TBST. Rat anti-BiP primary antibodies (BEI, MRA-1247) were diluted 1:10,000 in 5% milk in TBST and anti-rat HRP-conjugated secondary antibodies (#AP136P, MilliporeSigma, Burlington, MA). Development was performed using the SuperSignal West PLUS Chemiluminescent Substrate (Thermo Fisher, Waltham, MA) and the ChemiDoc MP imaging system (BioRad, Hercules, CA).
Split-Luciferase Assay.
This assay was performed by Luceome Biotechnologies (Tuscon, AZ) as fee per service, using a method previously described.17 The KinaseSeeker Assay offers a panel of 11 P. falciparum kinases, and initially, the compounds are scanned at a fixed concentration of 2.5 μM, and subsequently, an IC50 determination can be made using an eight-point dilution series.
Recombinant Protein Production.
The full coding sequencing (CDS) of Pf NEK3, aside from the first 10 residues on the N-terminus, cloned in a pGEX-4T-3 vector, was a generous gift from Christian Doerig. The PfARK2-GST construct was generated within the Chakrabarti laboratory, and cloning is described in the Supporting Information. E. coli BL21(DE3) competent cells (Agilent, Santa Clara, CA) were transformed via heat shock, and expression was induced with 1 mM IPTG at 20 °C for 16 h. Bacterial lysates were pelleted by centrifugation and resuspended in a HEPES salt buffer (50 mM HEPES, 200 mM NaCl, 0.02% monothiolglycerol, pH 7.5) supplemented with 1× HALT Protease Inhibitor (#78443, Thermo Fisher, Waltham, MA). Lysis was achieved with French press and sonication, and the lysate was clarified with centrifugation and filtration through a 0.2 μm PES membrane. Total protein lysate was run through a GSTrap FF Column (Cytiva, Marlborough, MA) and eluted with the suspension buffer supplemented with 20 mM glutathione. The resulting fractions were desalted with PD-10 Columns (Cytiva, Marlborough, MA) and quantified with a Bradford protein assay in accordance with the manufacturer’s protocol (BioRad, Hercules, CA).
Radioactive Kinase Assays.
For Pf NEK3, individual reactions were prepared in 15 μL, for a final concentration of 1× Kinase Buffer (Cell Signaling), 10 mM MnCl 2, 5 μg of dephosphorylated MBP (#31314, ActiveMotif, Carlsbad, CA), 1.5 μg of purified recombinant Pf NEK3-GST, 10 μM ATP (Thermo Fisher, Waltham, MA), and 0.1 μM γ−32P ATP (3000Ci/mmol 10mCi/ml EasyTide, PerkinElmer, Waltham, MA). For PfARK2, reactions were prepared in 20 μL with 1×Kinase Buffer, 0.5 μg of purified recombinant PfARK2-GST, 5 μg of MBP, 10 μM ATP, and 0.1 μM γ-32 P ATP. Reaction tubes were incubated for 30 min at 30 °C in a water bath before adding 4 μL of 4× NuPAGE loading dye (Invitrogen, Waltham, MA) and boiling for 7 min at 80 °C. The samples were cooled and run on a 12% Bis–Tris SDS-PAGE gel impregnated with 2,2,2-trichloroethanol (TCE). Protein concentration was determined by UV imaging on a ChemiDoc XRS+ Imaging System (BioRad, Hercules, CA). Proteins were blotted onto a PVDF membrane with the BioRad TransBlot Turbo system. Membranes were dried and incubated overnight with a phosphoscreen before imaging with a Personal Molecular Imager (BioRad, Hercules, CA). Densiometric analysis of the TCE gel and membrane was performed with Image Lab 6.1 software (BioRad, Hercules, CA), and correlation plots were generated with GraphPad Prism 9 (San Diego, CA).
Phosphoproteomics Sample Preparation.
Three flasks of asynchronous blood stage P. falciparum Dd2 culture were grown to ~15% parasitemia and 4% hematocrit under standard culture conditions. Per biological replicate, media was aspirated and replaced with fresh culture media, supplemented with either 5 × EC50 or EC90 depending on the slope of the EC50 curve. As a control, culture was treated with an equal volume of DMSO vehicle (0.04% final concentration). After 3 h, parasites were collected as described above through saponin lysis of erythrocytes. Parasite pellets were resuspended in freshly made 8 M urea lysis buffer (8 M urea, 50 mM Tris pH 8. 5, 1% SDS) supplemented with 1× HALT Protease & Phosphatase Inhibitor Cocktail (#78443, Thermo Fisher, Waltham, MA). The parasites were lysed by sonication, and the protein was quantified with the Pierce BCA Protein Assay (#23227, Thermo Fisher, Waltham, MA). The lysate was stored at −80 °C until processing at the Thermo Fisher Center for Multiplexed Proteomics (TCMP) at HMS. At this time, the samples were reduced, alkylated, resuspended, and digested.77 The resulting peptides were quantified and labeled with isobaric tandem mass tags (TMT).46 Following a ratio check, phosphoenrichment was performed with a Pierce High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Fisher, Waltham, MA). The resultant phosphopeptide eluate was analyzed with LC-MS2. The peptide flowthrough was fractionated by basic reverse-phase HPLC and similarly analyzed with LC-MS3. Spectra were searched against a composite P. falciparum and human Uniprot database (https://www.uniprot.org/) using the COMET algorithm.78 Peptide matches were filtered with a 1% false discovery rate (FDR), and peptides with a summed signal-to-noise (SN) threshold of >100 were quantified. For phosphopeptides, a 1% FDR filter and a summed SN threshold of ≥100 were used. Data will be released in the MassIVE repository upon publication.
Phosphoproteomics Analysis.
The analysis was performed with normalized relative abundance values obtained from the TCMP. Only results with more than one peptide were considered, and Z-score from ratio-based paired Z-test (Z = (log 2(r) – 0)/(δ/√n)), where r is the mean of treated/untreated ratios. GraphPad Prism 9 was used to generate the volcano plots (San Diego, CA). Disruptability of proteins was determined through the PhenoPlasm database (http://phenoplasm.org).79 GO-term analysis was performed using gene identifiers of the proteins containing at least one of the 10% most differential phosphorylation sites (|log 2(fc)| > 0.585). Significantly enriched GO terms were obtained using the PlasmoDB Gene Ontology Enrichment tool, which uses the background data set with the P. falciparum 3D7 genome and the Fisher’s Exact test to identify these terms (http://PlasmoDB.org).80 REVIGO81 was then used to remove obsolete or redundant terms and cluster similar terms, and the remaining terms were graphed with Cytoscape version 3.9.82 STRING analysis was performed on all top-ranked differentially expressed phosphosites (highest 10% |Z-score| and |log 2(fc)| > 0.585) with the addition of Pf NEK3 (https://string-db.org/).83 Interactomes for each treatment individually and the shared interactome were considered. The results were transferred into Cytoscape version 3.9,82 and all edges with a minimum interaction of 0.2 were including in groupings. Edge color represented the greatest contribution source for STRING score, and the overall score was indicated by edge thickness. PPI enrichment score was determined based on a more stringent interaction score of 0.4. Legend was created in BioRender (Biorender.com).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Christian Doerig for his generous donation of the Pf NEK3-GST plasmid. The authors also thank the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School (http://tcmp.hms.edu). This work was supported by a grant from NIH AI172066 (to D.C., N.S.G., and E.A.W.). M.R.L. was supported by a Ruth L. Kirschstein Institutional National Research Award from the National Institute for General Medical Sciences, T32 GM008666. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq. 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). The following reagents were obtained through BEI Resources, NIAID, NIH: Plasmodium falciparum, Strain IPC_6261, MRA-1284; Plasmodium falciparum, Strain IPC 5202, MRA-1240; and Plasmodium falciparum, Strain IPC 5188, MRA-1239, all of which were contributed by Didier Ménard.
The authors declare the following competing financial interest(s): Nathanael Gray is a founder, science advisory board member (SAB) and equity holder in Syros, C4, Allorion, Lighthorse, Voronoi, Inception, Matchpoint, CobroVentures, GSK, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield, Springworks, Interline and Sanofi.
ABBREVIATIONS
- P. falciparum
Plasmodium falciparum
- PLK
Polo-like kinase
- NEK
NIMA-related kinase
- EC50
half-maximal effective concentration
- IC50
half-maximal inhibitory concentration
- RI
resistance index
- SI
selectivity index
- HPI
hours post invasion
- ARKs
Aurora-related kinases
- MAPK
mitogen-activated protein kinase
- MPB
myelin basic protein
- SNP
single nucleotide polymorphism
- WGS
whole genome sequencing
- aaRS
aminoacyl-tRNA synthetase
- PDB
Research Collaboratory for Structural Bioinformatics Protein Data Bank
- HPLC
high-pressure liquid chromatography
- IMAC
immobilized metal affinity chromatography
- GO
gene ontology
- BCA assay
bicinchoninic acid assay
- TBST
tris-buffered saline with 0.1% Tween 20
- BSA
bovine serum albumin
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00025.
Supporting figures and tables referred to within the text, chemistry, methods used for supporting figures, and references (PDF)
Genomic sequencing analysis of resistant parasites—copy number variation (Supporting File 1) (XLSX)
Genomic sequencing analysis of resistant parasites—SNP/INDEL (Supporting File 2) (XLSX)
Proteomics analysis of treated parasites (Supporting File 3) (XLSX)
Phosphosite analysis of treated parasites (Supporting File 4) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.3c00025
Contributor Information
Monica J. Bohmer, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States;.
Jinhua Wang, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02215, United States;; Department of Cancer Biolo gy, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States
Eva S. Istvan, Division of Infectious Diseases, Department of Medicine and Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States
Madeline R. Luth, Department of Pediatrics, School of Medicine, University California, San Diego, La Jolla, California 92093, United States
Jennifer E. Collins, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States;
Edward L. Huttlin, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States;
Lushun Wang, Department of Chemical and Systems Biology, ChEM-H, Stanford Cancer Institute, School of Medicine, Stanford University, Stanford, California 94305, United States;.
Nimisha Mittal, Department of Pediatrics, School of Medicine, University California, San Diego, La Jolla, California 92093, United States.
Mingfeng Hao, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02215, United States;; Department of Cancer Biolo gy, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States; Present Address: Affiliations for M.H. and N.P.K. are those in which the research was conducted
Nicholas P. Kwiatkowski, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02215, United States; Department of Cancer Biolo gy, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States; Present Address: Affiliations for M.H. and N.P.K. are those in which the research was conducted
Steven P. Gygi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States;
Ratna Chakrabarti, Division of Cancer Research, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States.
Xianming Deng, School of Life Sciences, Xiamen University, Xiamen, Fujian 361102, China;.
Daniel E. Goldberg, Division of Infectious Diseases, Department of Medicine and Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, Missouri 63110, United States;
Elizabeth A. Winzeler, Department of Pediatrics, School of Medicine, University California, San Diego, La Jolla, California 92093, United States;
Nathanael S. Gray, Department of Chemical and Systems Biology, ChEM-H, Stanford Cancer Institute, School of Medicine, Stanford University, Stanford, California 94305, United States;
Debopam Chakrabarti, Division of Molecular Microbiology, Burnett School of Biomedical Sciences, University of Central Florida, Orlando, Florida 32826, United States;.
REFERENCES
- (1).Organization, W. H. World Health Organization (WHO). World Malaria Report 2021, 2021.
- (2).Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; An SS; Yeung S; Singhasivanon P; Day NP; Lindegardh N; Socheat D; White NJ Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Uwimana A; Legrand E; Stokes BH; Ndikumana JM; Warsame M; Umulisa N; Ngamije D; Munyaneza T; Mazarati JB; Munguti K; Campagne P; Criscuolo A; Ariey F; Murindahabi M; Ringwald P; Fidock DA; Mbituyumuremyi A; Menard D Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med 2020, 26, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Arendse LB; Wyllie S; Chibale K; Gilbert IH Plasmodium Kinases as Potential Drug Targets for Malaria: Challenges and Opportunities. ACS Infect. Dis 2021, 7, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cohen P; Cross D; Janne PA Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discovery 2021, 20, 551–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Derbyshire ER; Zuzarte-Luis V; Magalhaes AD; Kato N; Sanschagrin PC; Wang J; Zhou W; Miduturu CV; Mazitschek R; Sliz P; Mota MM; Gray NS; Clardy J Chemical interrogation of the malaria kinome. ChemBioChem 2014, 15, 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Steegmaier M; Hoffmann M; Baum A; Lenart P; Petronczki M; Krssak M; Gurtler U; Garin-Chesa P; Lieb S; Quant J; Grauert M; Adolf GR; Kraut N; Peters JM; Rettig WJ BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol 2007, 17, 316–322. [DOI] [PubMed] [Google Scholar]
- (8).Miranda-Saavedra D; Gabaldon T; Barton GJ; Langsley G; Doerig C The kinomes of apicomplexan parasites. Microbes Infect. 2012, 14, 796–810. [DOI] [PubMed] [Google Scholar]
- (9).Barr FA; Sillje HH; Nigg EA Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol 2004, 5, 429–440. [DOI] [PubMed] [Google Scholar]
- (10).Hanson KK; Ressurreicao AS; Buchholz K; Prudencio M; Herman-Ornelas JD; Rebelo M; Beatty WL; Wirth DF; Hanscheid T; Moreira R; Marti M; Mota MM Torins are potent antimalarials that block replenishment of Plasmodium liver stage parasitophorous vacuole membrane proteins. Proc. Natl. Acad. Sci. U.S.A 2013, 110, E2838–E2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Murillo-Solano C; Dong C; Sanchez CG; Pizarro JC Identification and characterization of the antiplasmodial activity of Hsp90 inhibitors. Malar. J 2017, 16, No. 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Adderley JD; John von Freyend S; Jackson SA; Bird MJ; Burns AL; Anar B; Metcalf T; Semblat JP; Billker O; Wilson DW; Doerig C Analysis of erythrocyte signalling pathways during Plasmodium falciparum infection identifies targets for host-directed antimalarial intervention. Nat. Commun 2020, 11, No. 4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vaid A; Thomas DC; Sharma P Role of Ca2+/calmodulin-PfPKB signaling pathway in erythrocyte invasion by Plasmodium falciparum. J. Biol. Chem 2008, 283, 5589–5597. [DOI] [PubMed] [Google Scholar]
- (14).Mott BT; Eastman RT; Guha R; Sherlach KS; Siriwardana A; Shinn P; McKnight C; Michael S; Lacerda-Queiroz N; Patel PR; Khine P; Sun H; Kasbekar M; Aghdam N; Fontaine SD; Liu D; Mierzwa T; Mathews-Griner LA; Ferrer M; Renslo AR; Inglese J; Yuan J; Roepe PD; Su XZ; Thomas CJ High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci. Rep 2015, 5, No. 13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Miduturu CV; Deng X; Kwiatkowski N; Yang W; Brault L; Filippakopoulos P; Chung E; Yang Q; Schwaller J; Knapp S; King RW; Lee JD; Herrgard S; Zarrinkar P; Gray NS High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem. Biol 2011, 18, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Deng Z; Chen G; Liu S; Li Y; Zhong J; Zhang B; Li L; Huang H; Wang Z; Xu Q; Deng X Discovery of methyl 3-((2-((1-(dimethylglycyl)-5-methoxyindolin-6-yl)amino)-5-(trifluoromethyl) pyrimidin-4-yl)amino)thiophene-2-carboxylate as a potent and selective polo-like kinase 1 (PLK1) inhibitor for combating hepatocellular carcinoma. Eur. J. Med. Chem 2020, 206, No. 112697. [DOI] [PubMed] [Google Scholar]
- (17).Jester BW; Cox KJ; Gaj A; Shomin CD; Porter JR; Ghosh I A coiled-coil enabled split-luciferase three-hybrid system: applied toward profiling inhibitors of protein kinases. J. Am. Chem. Soc 2010, 132, 11727–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- (19).Dorin-Semblat D; Schmitt S; Semblat JP; Sicard A; Reininger L; Goldring D; Patterson S; Quashie N; Chakrabarti D; Meijer L; Doerig C Plasmodium falciparum NIMA-related kinase Pfnek-1: sex specificity and assessment of essentiality for the erythrocytic asexual cycle. Microbiology 2011, 157, 2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Reininger L; Tewari R; Fennell C; Holland Z; Goldring D; Ranford-Cartwright L; Billker O; Doerig C An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J. Biol. Chem 2009, 284, 20858–20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Reininger L; Garcia M; Tomlins A; Muller S; Doerig C The Plasmodium falciparum, Nima-related kinase Pfnek-4: a marker for asexual parasites committed to sexual differentiation. Malar. J 2012, 11, No. 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lye YM; Chan M; Sim TS Pfnek3: an atypical activator of a MAP kinase in Plasmodium falciparum. FEBS Lett. 2006, 580, 6083–6092. [DOI] [PubMed] [Google Scholar]
- (23).Solyakov L; Halbert J; Alam MM; Semblat JP; Dorin-Semblat D; Reininger L; Bottrill AR; Mistry S; Abdi A; Fennell C; Holland Z; Demarta C; Bouza Y; Sicard A; Nivez MP; Eschenlauer S; Lama T; Thomas DC; Sharma P; Agarwal S; Kern S; Pradel G; Graciotti M; Tobin AB; Doerig C Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun 2011, 2, No. 565. [DOI] [PubMed] [Google Scholar]
- (24).Zhang M; Wang C; Otto TD; Oberstaller J; Liao X; Adapa SR; Udenze K; Bronner IF; Casandra D; Mayho M; Brown J; Li S; Swanson J; Rayner JC; Jiang RHY; Adams JH Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, No. eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Low H; Chua CS; Sim TS Plasmodium falciparum possesses a unique dual-specificity serine/threonine and tyrosine kinase, Pfnek3. Cell. Mol. Life Sci 2012, 69, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ward P; Equinet L; Packer J; Doerig C Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics 2004, 5, No. 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hitz E; Balestra AC; Brochet M; Voss TS PfMAP-2 is essential for male gametogenesis in the malaria parasite Plasmodium falciparum. Sci. Rep 2020, 10, No. 11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Morahan BJ; Abrie C; Al-Hasani K; Batty MB; Corey V; Cowell AN; Niemand J; Winzeler EA; Birkholtz LM; Doerig C; Garcia-Bustos JF Human Aurora kinase inhibitor Hesperadin reveals epistatic interaction between Plasmodium falciparum PfArk1 and PfNek1 kinases. Commun. Biol 2020, 3, No. 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Reininger L; Wilkes JM; Bourgade H; Miranda-Saavedra D; Doerig C An essential Aurora-related kinase transiently associates with spindle pole bodies during Plasmodium falciparum erythrocytic schizogony. Mol. Microbiol 2011, 79, 205–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ember SWJ; Zhu JY; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schonbrunn E Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ciceri P; Muller S; O’Mahony A; Fedorov O; Filippakopoulos P; Hunt JP; Lasater EA; Pallares G; Picaud S; Wells C; Martin S; Wodicka LM; Shah NP; Treiber DK; Knapp S Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Munoz L Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discovery 2017, 16, 424–440. [DOI] [PubMed] [Google Scholar]
- (33).Hui DF; Fonesca M; Josling G; Tallant C; Wernimont A; Fedorov O; Hutchinson A; Loppnau P; Duffy M; Knapp S; Raymond H Plasmodium bromodomain PfBDP4; A Target Enabling Package (Version 5) [Data set]. Zenodo 2016, DOI: 10.5281/zenodo.1209237. [DOI] [Google Scholar]
- (34).Chua MJ; Robaa D; Skinner-Adams TS; Sippl W; Andrews KT Activity of bromodomain protein inhibitors/binders against asexual-stage Plasmodium falciparum parasites. Int. J. Parasitol.: Drugs Drug Resist 2018, 8, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nguyen HHT; Yeoh LM; Chisholm SA; Duffy MF Developments in drug design strategies for bromodomain protein inhibitors to target Plasmodium falciparum parasites. Expert Opin. Drug Discovery 2020, 15, 415–425. [DOI] [PubMed] [Google Scholar]
- (36).Qiu D; Pei JV; Rosling JEO; Li D; Xue Y; Penington JS; Kümpornsin K; Vincent Aw YT; Han Aw JY; Hasemer H; Dennis ASM; Ridgway MC; Papenfuss AT; Lee MCS; van Dooren GG; Kirk K; Lehane AM Generation and character-isation of P. falciparum parasites with a G358S mutation in the PfATP4 Na+ pump and clinically relevant levels of resistance to some PfATP4 inhibitors. bioRxiv 2022, 2022.2001.2011.475938. [Google Scholar]
- (37).Koenderink JB; Kavishe RA; Rijpma SR; Russel FG The ABCs of multidrug resistance in malaria. Trends Parasitol. 2010, 26, 440–446. [DOI] [PubMed] [Google Scholar]
- (38).Murithi JM; Deni I; Pasaje CFA; Okombo J; Bridgford JL; Gnadig NF; Edwards RL; Yeo T; Mok S; Burkhard AY; Coburn-Flynn O; Istvan ES; Sakata-Kato T; Gomez-Lorenzo MG; Cowell AN; Wicht KJ; Le Manach C; Kalantarov GF; Dey S; Duffey M; Laleu B; Lukens AK; Ottilie S; Vanaerschot M; Trakht IN; Gamo FJ; Wirth DF; Goldberg DE; Odom John AR; Chibale K; Winzeler EA; Niles JC; Fidock DA The Plasmodium falciparum ABC transporter ABCI3 confers parasite strain-dependent pleiotropic antimalarial drug resistance. Cell Chem. Biol 2022, 29, 824–839.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Navale R; Atul; Allanki AD; Sijwali PS Characterization of the autophagy marker protein Atg8 reveals atypical features of autophagy in Plasmodium falciparum. PLoS One 2014, 9, No. e113220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Holcik M; Sonenberg N Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol 2005, 6, 318–327. [DOI] [PubMed] [Google Scholar]
- (41).Fennell C; Babbitt S; Russo I; Wilkes J; Ranford-Cartwright L; Goldberg DE; Doerig C PfeIK1, a eukaryotic initiation factor 2alpha kinase of the human malaria parasite Plasmodium falciparum, regulates stress-response to amino-acid starvation. Malar. J 2009, 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang M; Fennell C; Ranford-Cartwright L; Sakthivel R; Gueirard P; Meister S; Caspi A; Doerig C; Nussenzweig RS; Tuteja R; Sullivan WJ Jr.; Roos DS; Fontoura BM; Menard R; Winzeler EA; Nussenzweig V The Plasmodium eukaryotic initiation factor-2α kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J. Exp. Med 2010, 207, 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang M; Mishra S; Sakthivel R; Rojas M; Ranjan R; Sullivan WJ Jr.; Fontoura BM; Menard R; Dever TE; Nussenzweig V PK4, a eukaryotic initiation factor 2alpha(eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 3956–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ishiyama A; Iwatsuki M; Namatame M; Nishihara-Tsukashima A; Sunazuka T; Takahashi Y; Omura S; Otoguro K Borrelidin, a potent antimalarial: stage-specific inhibition profile of synchronized cultures of Plasmodium falciparum. J. Antibiot 2011, 64, 381–384. [DOI] [PubMed] [Google Scholar]
- (45).Keller TL; Zocco D; Sundrud MS; Hendrick M; Edenius M; Yum J; Kim YJ; Lee HK; Cortese JF; Wirth DF; Dignam JD; Rao A; Yeo CY; Mazitschek R; Whitman M Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol 2012, 8, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; Mering CV STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Paul AS; Miliu A; Paulo JA; Goldberg JM; Bonilla AM; Berry L; Seveno M; Braun-Breton C; Kosber AL; Elsworth B; Arriola JSN; Lebrun M; Gygi SP; Lamarque MH; Duraisingh MT Co-option of Plasmodium falciparum PP1 for egress from host erythrocytes. Nat. Commun 2020, 11, No. 3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Rudlaff RM; Kraemer S; Streva VA; Dvorin JD An essential contractile ring protein controls cell division in Plasmodium falciparum. Nat. Commun 2019, 10, No. 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Birnbaum J; Scharf S; Schmidt S; Jonscher E; Hoeijmakers WAM; Flemming S; Toenhake CG; Schmitt M; Sabitzki R; Bergmann B; Frohlke U; Mesen-Ramirez P; Blancke Soares A; Herrmann H; Bartfai R; Spielmann T A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 2020, 367, 51–59. [DOI] [PubMed] [Google Scholar]
- (51).Bennink S; Pradel G The molecular machinery of translational control in malaria parasites. Mol. Microbiol 2019, 112, 1658–1673. [DOI] [PubMed] [Google Scholar]
- (52).Hart KJ; Power BJ; Rios KT; Sebastian A; Lindner SE The Plasmodium NOT1-G paralogue is an essential regulator of sexual stage maturation and parasite transmission. PLoS Biol. 2021, 19, No. e3001434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Baumgarten S; Bryant JM; Sinha A; Reyser T; Preiser PR; Dedon PC; Scherf A Transcriptome-wide dynamics of extensive m(6)A mRNA methylation during Plasmodium falciparum blood-stage development. Nat. Microbiol 2019, 4, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).The Plasmodium Genome Database Collaborative. PlasmoDB: An integrative database of the Plasmodium falciparum genome. Tools for accessing and analyzing finished and unfinished sequence data. Nucleic Acids Res. 2001, 29, 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tewari R; Straschil U; Bateman A; Bohme U; Cherevach I; Gong P; Pain A; Billker O The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 2010, 8, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang G; Jiang Q; Zhang C The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J. Cell Sci 2014, 127, 4111–4122. [DOI] [PubMed] [Google Scholar]
- (57).Zitouni S; Nabais C; Jana SC; Guerrero A; Bettencourt-Dias M Polo-like kinases: structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol 2014, 15, 433–452. [DOI] [PubMed] [Google Scholar]
- (58).Matthews H; Duffy CW; Merrick CJ Checks and balances? DNA replication and the cell cycle in Plasmodium. Parasites Vectors 2018, 11, No. 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Carvalho TG; Doerig C; Reininger L Nima- and Aurora-related kinases of malaria parasites. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1336–1345. [DOI] [PubMed] [Google Scholar]
- (60).Francia ME; Striepen B Cell division in apicomplexan parasites. Nat. Rev. Microbiol 2014, 12, 125–136. [DOI] [PubMed] [Google Scholar]
- (61).Anighoro A; Bajorath J; Rastelli G Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- (62).Khan S Recent advances in the biology and drug targeting of malaria parasite aminoacyl-tRNA synthetases. Malar. J 2016, 15, No. 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Xie SC; Metcalfe RD; Dunn E; Morton CJ; Huang SC; Puhalovich T; Du Y; Wittlin S; Nie S; Luth MR; Ma L; Kim MS; Pasaje CFA; Kumpornsin K; Giannangelo C; Houghton FJ; Churchyard A; Famodimu MT; Barry DC; Gillett DL; Dey S; Kosasih CC; Newman W; Niles JC; Lee MCS; Baum J; Ottilie S; Winzeler EA; Creek DJ; Williamson N; Parker MW; Brand S; Langston SP; Dick LR; Griffin MDW; Gould AE; Tilley L Reaction hijacking of tyrosine tRNA synthetase as a new whole-of-life-cycle antimalarial strategy. Science 2022, 376, 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zhou J; Huang Z; Zheng L; Hei Z; Wang Z; Yu B; Jiang L; Wang J; Fang P Inhibition of Plasmodium falciparum Lysyl-tRNA synthetase via an anaplastic lymphoma kinase inhibitor. Nucleic Acids Res. 2020, 48, 11566–11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Trager W; Jensen JB Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- (66).Dery V; Duah NO; Ayanful-Torgby R; Matrevi SA; Anto F; Quashie NB An improved SYBR Green-1-based fluorescence method for the routine monitoring of Plasmodium falciparum resistance to anti-malarial drugs. Malar. J 2015, 14, No. 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Clements RL; Streva V; Dumoulin P; Huang W; Owens E; Raj DK; Burleigh B; Llinas M; Winzeler EA; Zhang Q; Dvorin JD A Novel Antiparasitic Compound Kills Ring-Stage Plasmodium falciparum and Retains Activity Against Artemisinin-Resistant Parasites. J. Infect. Dis 2020, 221, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wright AE; Collins JE; Roberts B; Roberts JC; Winder PL; Reed JK; Diaz MC; Pomponi SA; Chakrabarti D Antiplasmodial Compounds from Deep-Water Marine Invertebrates. Mar. Drugs 2021, 19, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Mata-Cantero L; Lafuente MJ; Sanz L; Rodriguez MS Magnetic isolation of Plasmodium falciparum schizonts iRBCs to generate a high parasitaemia and synchronized in vitro culture. Malar.J 2014, 13, No. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Lambros C; Vanderberg JP Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979, 65, 418–420. [PubMed] [Google Scholar]
- (71).Antonova-Koch Y; Meister S; Abraham M; Luth MR; Ottilie S; Lukens AK; Sakata-Kato T; Vanaerschot M; Owen E; Jado JC; Maher SP; Calla J; Plouffe D; Zhong Y; Chen K; Chaumeau V; Conway AJ; McNamara CW; Ibanez M; Gagaring K; Serrano FN; Eribez K; Taggard CM; Cheung AL; Lincoln C; Ambachew B; Rouillier M; Siegel D; Nosten F; Kyle DE; Gamo FJ; Zhou Y; Llinas M; Fidock DA; Wirth DF; Burrows J; Campo B; Winzeler EA Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 2018, 362, No. eaat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).McKenna A; Hanna M; Banks E; Sivachenko A; Cibulskis K; Kernytsky A; Garimella K; Altshuler D; Gabriel S; Daly M; DePristo MA The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Van der Auwera GA; Carneiro MO; Hartl C; Poplin R; Del Angel G; Levy-Moonshine A; Jordan T; Shakir K; Roazen D; Thibault J; Banks E; Garimella KV; Altshuler D; Gabriel S; DePristo MA From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinf 2013, 43, 11.101–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Cowell AN; Istvan ES; Lukens AK; Gomez-Lorenzo MG; Vanaerschot M; Sakata-Kato T; Flannery EL; Magistrado P; Owen E; Abraham M; LaMonte G; Painter HJ; Williams RM; Franco V; Linares M; Arriaga I; Bopp S; Corey VC; Gnadig NF; Coburn-Flynn O; Reimer C; Gupta P; Murithi JM; Moura PA; Fuchs O; Sasaki E; Kim SW; Teng CH; Wang LT; Akidil A; Adjalley S; Willis PA; Siegel D; Tanaseichuk O; Zhong Y; Zhou Y; Llinas M; Ottilie S; Gamo FJ; Lee MCS; Goldberg DE; Fidock DA; Wirth DF; Winzeler EA Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 2018, 359, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Summers RL; Pasaje CFA; Pisco JP; Striepen J; Luth MR; Kumpornsin K; Carpenter EF; Munro JT; Lin; Plater A; Punekar AS; Shepherd AM; Shepherd SM; Vanaerschot M; Murithi JM; Rubiano K; Akidil A; Ottilie S; Mittal N; Dilmore AH; Won M; Mandt REK; McGowen K; Owen E; Walpole C; Llinas M; Lee MCS; Winzeler EA; Fidock DA; Gilbert IH; Wirth DF; Niles JC; Baragana B; Lukens AK Chemogenomics identifies acetyl-coenzyme A synthetase as a target for malaria treatment and prevention. Cell Chem. Biol 2022, 29, 191–201.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Miles A; Iqbal Z; Vauterin P; Pearson R; Campino S; Theron M; Gould K; Mead D; Drury E; O’Brien J; Ruano Rubio V; MacInnis B; Mwangi J; Samarakoon U; Ranford-Cartwright L; Ferdig M; Hayton K; Su XZ; Wellems T; Rayner J; McVean G; Kwiatkowski D Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res. 2016, 26, 1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res 2018, 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- (79).Sanderson T; Rayner JC PhenoPlasm: a database of disruption phenotypes for malaria parasite genes. Wellcome Open Res. 2017, 2, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Aurrecoechea C; Brestelli J; Brunk BP; Dommer J; Fischer S; Gajria B; Gao X; Gingle A; Grant G; Harb OS; Heiges M; Innamorato F; Iodice J; Kissinger JC; Kraemer E; Li W; Miller JA; Nayak V; Pennington C; Pinney DF; Roos DS; Ross C; Stoeckert CJ Jr.; Treatman C; Wang H PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Supek F; Bosnjak M; Skunca N; Smuc T REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011, 6, No. e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Szklarczyk D; Gable AL; Nastou KC; Lyon D; Kirsch R; Pyysalo S; Doncheva NT; Legeay M; Fang T; Bork P; Jensen LJ; von Mering C The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.