

Abstract
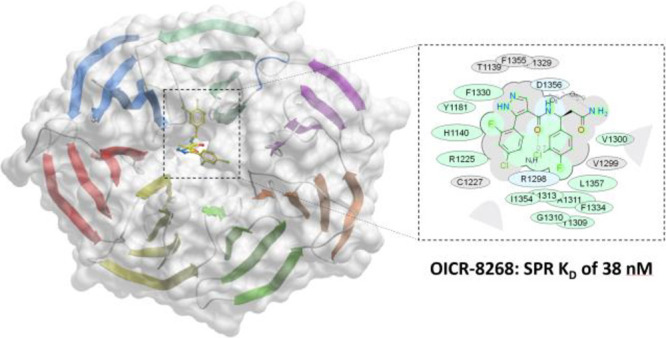
DCAF1 is a substrate receptor of two distinct E3 ligases (CRL4DCAF1 and EDVP), plays a critical physiological role in protein degradation, and is considered a drug target for various cancers. Antagonists of DCAF1 could be used toward the development of therapeutics for cancers and viral treatments. We used the WDR domain of DCAF1 to screen a 114-billion-compound DNA encoded library (DEL) and identified candidate compounds using similarity search and machine learning. This led to the discovery of a compound (Z1391232269) with an SPR KD of 11 μM. Structure-guided hit optimization led to the discovery of OICR-8268 (26e) with an SPR KD of 38 nM and cellular target engagement with EC50 of 10 μM as measured by cellular thermal shift assay (CETSA). OICR-8268 is an excellent tool compound to enable the development of next-generation DCAF1 ligands toward cancer therapeutics, further investigation of DCAF1 functions in cells, and the development of DCAF1-based PROTACs.
Introduction
Targeted protein degradation is an emerging therapeutic modality for the treatment of diseases, especially cancers.1,2 Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules that consist of a chemical entity binding to the target protein linked to another chemical entity binding to an active E3 ubiquitin ligase. A PROTAC molecule brings together the target and E3 ligase into proximity and allows the E3 ubiquitin ligase to ubiquitinate the target protein and mark the target for protein degradation. The ubiquitin proteasome system is one of many mechanisms that regulate protein degradation in the cell. Protein degradation via the ubiquitin-26s proteasome system is a multistep process of covalently linking ubiquitin to substrate proteins. This process involves three classes of enzymes: ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin ligase (E3). There are more than 600 reported E3 ubiquitin ligases, and they can be grouped into different families such as the HECT domain containing, RING-finger containing, and RBR types.3 Current PROTACs are mainly based on utilizing the E3 ligase complexes cereblon (CRBN) and von Hippel–Lindau (VHL). Although there are a few other E3 ligases, e.g., mouse double minute 2 homolog (MDM2), and inhibitors of apoptosis protein (IAPs) used in PROTACs, CRBN-based and VHL-based PROTACs are the most common.4,5 Thus, additional E3 ubiquitin ligases could be explored with ligands designed to avoid limitations and expand the application of PROTACs.
Among the plethora of E3 ubiquitin ligases, the RING-type family is the largest E3 family. The RING-finger E3 family contains a subgroup called the cullin-RING ligases (CRLs).6 CRLs are multi-subunit E3 ligase complexes that contain a cullin (CUL) protein, a RING protein, an adaptor protein, and a substrate receptor protein. There are a few reported adaptor proteins that CRLs utilize and are specific to each CUL such as the DNA damage binding protein 1 (DDB1), Skp1, and Elongin B/Elongin C.7 For example, the CUL4A-DDB1-CRBN (CRL4CRBN) complex has CRBN as the substrate recognition subunit for CUL4A through interaction with DDB1.8 CRL4 also utilizes a class of substrate recognition subunits called the DDB1-CUL4-associated factors (DCAFs).7 DCAFs were first identified as WD40 repeat proteins from proteomic screens using tandem affinity purifications of DDB1 and CUL4A.9−11 Among the DCAFs, DCAF1 is a substrate receptor in the CUL4A-DDB1-DCAF1 (CRL4DCAF1) complex. Initially, DCAF1 was discovered as a target protein hijacked by the human immunodeficiency virus-1 (HIV-1) viral protein Vpr.12 Hence, DCAF1 is also known as Vpr binding protein (VprBP). Vpr binds to DCAF1 to hijack the CRL4DCAF1 E3 ligase complex to induce cell cycle arrest.13 CRL4DCAF1-Vpr also targets host proteins such as uracil DNA glycosylase 2 (UNG2),14 endoribonuclease Dicer,15 and histone deacetylase SIRT716 for ubiquitination leading to protein degradation. The co-crystal structure of DDB1-DCAF1-Vpr-UNG2 (PDB: 5jk7) reveals the detailed interactions on how Vpr mimics DNA binding to UNG2 for subsequent protein degradation.17 Similarly, the Vpx protein of HIV-2/simian immunodeficiency virus hijacks the CRL4DCAF1 complex recruiting SAMHD1, an HIV restriction factor for proteasome-dependent degradation.18,19 These viruses utilize the CRL4DCAF1 complex to modulate the cellular environment to prolong viral replication and pathogenesis.
Since its initial discovery, DCAF1 has been shown to be involved in various normal physiological functions in the cell. These functions include cell cycle regulation/progression, cell division, cellular proliferation, lipid metabolism, and miRNA biogenesis, among others.20 Within these processes, DCAF1 is also a substrate recognition subunit for a different E3 ligase complex called EED-DDB1-VprBP (EDVP) where it plays a role in cytoskeletal organization and cell division.21,22 It has been reported that Vpr hijacks the EDVPDCAF1 complex to disrupt centrosome homeostasis.23 Thus, DCAF1 is unique in which it can service two distinct E3 ubiquitin ligases.20,24 DCAF1 also has an E3 ligase-independent function as it was shown to have kinase activity by phosphorylating histone H2A at Thr120.25 H2AT120 phosphorylation (H2AT120p) by DCAF1 is associated with gene silencing at tumor suppressor genes,25 and overexpression of DCAF1 and H2AT120p has been shown to cause proliferation of colon cancer cells.26
The diverse biological role of DCAF1 makes it an attractive target for drug discovery. We aimed to find small molecule ligands for DCAF1 to help further elucidate its roles in cells and to help enable the development of PROTAC molecules. DNA-encoded library (DEL) technology is a method to identify small molecule binders to a protein target, which has emerged as a popular screening approach in drug discovery. A DEL is a collection of small molecules each covalently linked to a unique DNA sequence that acts as a barcode for identification. The combination of DEL screening, machine learning, and large commercial synthetically accessible enumerate spaces is an alternative to traditional high-throughput screening as it enables the screening of billions of compounds at a fraction of the cost.27
We employed DEL technology to screen the WDR domain of DCAF1 by affinity-mediated selection and used the enrichment data to build machine learning models or conduct similarity searches to predict, optimize, and select commercially available small molecules for further testing. Here, we report the discovery and cellular target engagement of the first nanomolar DCAF1 ligand, OICR-8268, with an SPR KD of 38 nM, and detailed structure-guided hit optimization. This discovery provides a basis for future development of DCAF1-based PROTACs and DCAF1 antagonists. In addition, OICR-8268 could be used as a chemical tool to further investigate DCAF1 functions in cells.
Results and Discussion
Hit Generation from Machine Learning and Similarity Searching of DEL Screening Data
The WDR domain of DCAF1 (1038–1400) with N-terminal 6His-tag (hereafter referred to as DCAF1) was screened at X-Chem using a proprietary 114-billion-compound collection of 59 DNA-Encoded Chemical Libraries, and the data were processed by similarity searching and machine learning algorithms to purchase commercial compounds for assay evaluation as previously reported.27,28 The 2D structure similarity searching workflow comprised searching the large commercially accessible EnamineREAL database (720 M) for 2D structures similar to representative enriched instances and truncates (see the Experimental Section). This led to the acquisition of a collection of 33 commercially available compounds that were evaluated by differential scanning fluorimetry (DSF; Figure S1) and surface plasmon resonance (SPR). From this initial set of 33 compounds, the commercially available compound (Z1391232269) was confirmed as a hit by both DSF and SPR assays. It stabilized DCAF1 with an increase in melting temperature (ΔTm) greater than 2 °C in a concentration-dependent manner, indicating binding (Figure 1A,B). The binding was further confirmed with an SPR KD value of 11.5 ± 4.2 μM (Figure 1C). Isothermal titration calorimetry (ITC) was also used as an additional orthogonal method and confirmed its binding with a KD of 9 μM (Figure S2).
Figure 1.

Confirmation of Z1391232269 binding to the DCAF1 WDR domain by biophysical methods. (A) Z1391232269 stabilized the WDR domain of DCAF1 at 400 μM (green trace) compared with the WDR domain with no compound (black trace), and (B) the effect was concentration dependent. (C) SPR confirmed its binding to DCAF1 with a KD of 11.5 ± 4.2 μM. Experiments were performed in triplicate.
Analysis of the First Crystal Structure of DCAF1 in Complex with the Initial Hit Reveals a Sub-micromolar Enantiomer
Crystallization of DCAF1 in complex with Z1391232269 (3c) further confirmed its binding. However, the analysis of the structure (Figure S3) revealed a clear suboptimal geometry of the bound R-enantiomer (3c). Indeed, the torsion angle of the central amide function was distorted to about 90° to fit the electron density. Molecular modeling and docking experiments were performed to generate reasonable conformations of the ligand that fit the electron density. In parallel, the alternative S-enantiomer (3d) was also considered and was found to provide a better fit for the electron density with a near-optimal trans-conformation amide torsion angle.
Following this molecular modeling study, the pure enantiomers were resynthesized to further confirm our observations. The synthesis of these key analogs was carried out using standard EDCI coupling conditions as shown in Scheme 1. The coupling of the commercially available chiral β-amino amides 1 with the commercially available pyrazole carboxylic acids 2 afforded the target compounds 3 in modest (28–38%) yields.
Scheme 1. Synthesis of Compounds 3a and 3c–d.

Reagents and conditions: (a) EDCI, DIEA, 0 °C (5 min), rt (18 h).
In SPR experiments, a significant difference in binding affinities of the enantiomers was observed (Table 1). These results, in combination with chiral HPLC analysis of 3c and 3d, provided an explanation for the structural observation. Indeed, chiral HPLC showed that compound 3c was contaminated with a minor amount of the other enantiomer 3d. This finding shed light on the discrepancies observed in the X-ray structure of DCAF1 and 3c. Although the initial compound solution contained only about 4% 3d, it was preferentially crystallized with the protein as a result of the ∼27-fold higher binding affinity (Table 1). Finally, small substitutions on both the northern and southern aromatic rings (3d, compared to unsubstituted compound 3b) significantly improved potency.
Table 1. Structure–Activity Relationships of Compounds 3a–d.
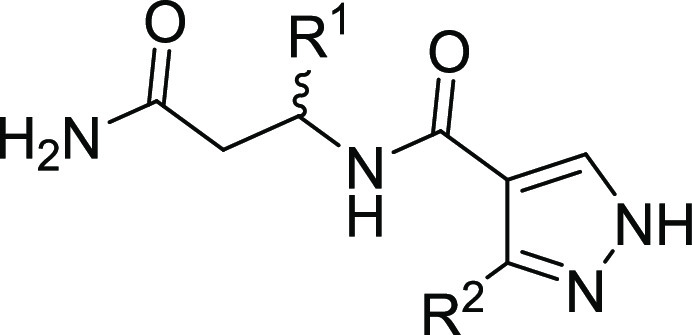
| compound | R1 | R2 | DCAF1 KD (nM) |
|---|---|---|---|
| 3a | (R) Ph | Ph | NBa |
| 3b | (S) Ph | Ph | 16,400; 16,900b |
| 3c | (R) 3-Cl Ph | 2-F Ph | 13,500 ± 200 |
| 3d | (S) 3-Cl Ph | 2-F Ph | 490 ± 90 |
NB = no binding.
This experiment was performed in duplicate.
To confirm our molecular modeling predictions, 3d (S-enantiomer) was synthesized as a pure enantiomer. Our SPR data confirmed that compound 3d (S-enantiomer) has a higher affinity for binding to DCAF1 (Figure 2A) with a KD of 490 ± 90 nM compared to compound 3c (R-enantiomer) with a KD of 13.5 ± 0.2 μM. The observed trend of this set of compounds indicated that S-isomers were more potent DCAF1 binders. Similarly, in the DSF experiments (Figure 2B), 3d showed DCAF1 stabilization at 25 μM, but 3c did not show any significant effect even at 50 μM. As an additional orthogonal confirmation, ITC experiments confirmed binding of compound 3d to DCAF1 with an ITC KD value of 932 ± 150 nM (Figure 2C). The binding interaction between 3d and DCAF1 is enthalpy-driven and exothermic with an enthalpy (ΔH) of −64.1 ± 1.5 kJ/mol and entropy (ΔS) of −99.5 ± 6.0 J/mol·K.
Figure 2.

Biophysical characterization of compounds 3c and 3d. Binding of compound 3c and 3d to DCAF1 WD40 domain was assessed using (A) SPR (KD values of 490 ± 90 nM for 3d and 13.5 ± 0.2 μM for 3c) and (B) DSF (ΔTm of 2.8 ± 0.05 and 3.8 ± 0.08 °C at 25 and 50 μM for 3d, respectively, and no stabilization effect for 3c even at 50 μM). Compound 3d was orthogonally confirmed by (C) ITC with a KD value of 932 ± 150 nM. The chemical structure of 3d is shown. Experiments were performed in triplicate.
Crystal Structure of the Human DCAF1 WDR Domain Bound to 3d
The human DCAF1 WDR domain (residue range 1077 to 1390) was used in crystallization experiments, in which residues 1077 and 1079 were mutated to alanine to promote crystallization. A new DCAF1 co-crystal structure in complex with compound 3d (S-enantiomer), referred to here as DCAF1-3d (PDB ID: 7UFV), was determined to 1.9 Å resolution in P1211 space group symmetry. There are two molecules in the crystal asymmetric unit, and most residues were well resolved, except residues 1315–1325 and 1374–1378 in chain A and residues 1316–1326 and 1373–1379 in chain B. Table 2 summarizes the crystallographic data collection, refinement, and validation statistics.
Table 2. Data Collection and Refinement Statisticsa.
| DCAF1-3d | DCAF1-26e | |
|---|---|---|
| PDB ID | 7UFV | 8F8E |
| Data collection | ||
| wavelength (Å) | 0.97911 | 0.97918 |
| space group | P1211 | P1211 |
| cell dimensions | ||
| a, b, c (Å) | 49.140, 87.875, 73.807 | 48.865, 88.084, 73.562 |
| α, β, γ (°) | 90.00, 98.54, 90.00 | 90.00, 97.56, 90.00 |
| resolution range (Å) | 43.94–1.90 (1.94–1.90) | 44.08–1.55 (1.58–1.55) |
| total reflections | 184,284 | 470,878 |
| unique reflections | 48,379 (3107) | 171,794 (8612) |
| multiplicity | 3.80 (3.80) | 2.7 (2.6) |
| completeness (%) | 99.0 (99.5) | 97.2 (97.4) |
| mean I/sigma(I) | 9.6 (1.5) | 19.7 (1.25) |
| R-merge | 0.062 (0.923) | 0.061 (0.677) |
| R-meas | 0.072 (1.070) | 0.076 (0.836) |
| R-pim | 0.036 (0.534) | 0.044 (0.486) |
| CC1/2 | 0.998 (0.726) | 0.996 (0.658) |
| Refinement | ||
| reflections used in refinement | 46,002 (3420) | 86,348 (6249) |
| reflections used for R-free | 2355 (145) | 1744 (118) |
| R-work | 0.2041 (0.365) | 0.1812 (0.273) |
| R-free | 0.2542 (0.334) | 0.2089 (0.293) |
| number of nonhydrogen atoms | ||
| all atoms | 4696 | 5136 |
| macromolecules | 4580 | 4764 |
| ligands | 55 | 71 |
| solvent | 88 | 330 |
| RMSDs | ||
| bond lengths (Å) | 0.0065 | 0.0072 |
| angles (°) | 1.2412 | 1.4212 |
| average B factors (Å2) | ||
| macromolecule | 47.162 | 25.160 |
| ligands/ions | 42.921 | 24.459 |
| solvent | 42.320 | 32.068 |
| Validation | ||
| Ramachandran plot (%) | ||
| favored | 95.49 | 97.6 |
| allowed | 100 | 100 |
| outliers (%) | 0.0 | 0.0 |
| Clashscore | 3.5 | 1.61 |
| rotamer outliers (%) | 0.0 | 0.78 |
Statistics for the highest-resolution shell are shown in parentheses.
A well-defined electron density map was observed for the entire 3d molecule in both chains of the crystal asymmetric unit (Figure S4), which confirmed the modeled S-enantiomer binding mode (RMSD of 0.5 Å). Thus, we concluded that the original structure of the protein–ligand complex was based on an enantiomerically impure mixture (92% ee (R)) and the S-enantiomer preferentially co-crystallized with the protein because of its higher affinity for DCAF1.
Compound 3d is nestled deep inside the central tunnel of the DCAF1 WDR domain, where it appears to be positioned slightly off center (Figure 3A). It makes extensive hydrogen bonding as well as hydrophobic interactions with residues lining the central tunnel of the DCAF1 WDR domain ring (Figure 3B). Specifically, the central amide group of 3d forms hydrogen bonding interactions with the side chain of Asp1356 and another with the guanidinium group of Arg1298 on the opposite side. The pyrazole ring on the other hand is involved in edge-to-face aromatic interaction with the side chain of Phe1330 and coordinates a nearby water molecule (HOH04). The 3-chlorophenyl group is nestled in a hydrophobic pocket composed of Leu1357, Ile1354, Phe1330, Ala1311, and Val1300 and forms π-stacking interaction with the aromatic ring of Phe1330. In chain A, the terminal amide group of 3d interacts with the imidazole ring of His1140 through hydrogen bonds (Figure 3B).
Figure 3.

Co-crystal structure of DCAF1 WDR domain in complex with compound 3d. (A) The top and side views of the DCAF1 WDR domain shown as cartoon representation in gray bound to 3d shown as yellow sticks. (B) Close-up view of the 3d binding site in chain A of DCAF1-3d. 3d forms a network of interactions with the protein and a water molecule within the central channel of DCAF1 WDR domain ring. The compound is shown as yellow sticks, and the protein residues involved in the interactions are rendered as gray sticks. The hydrogen bonds are shown as black dashes, and an interacting water molecule is shown as a red sphere. (C) An overlay of the DCAF1-3d structure (shown in surface representation in light gray and 3d in yellow sticks indicated by black arrow) onto the DCAF1 WDR domain in complex with lentiviral Vpx (shown in cartoon representation in green) and SAMHD1 (not shown) (PDB ID: 5AJA), showing the compound binding site relative to that of Vpx.
Comparison of the DCAF1-3d structure with that of human DCAF1 in complex with the lentiviral accessory protein Vpx and the Mandrill SAMHD1 (PDB ID: 5AJA) reveals that the compound binds at a site distant from the Vpx peptide binding site and does not overlap with it (Figure 3C). Although compound 3d may not be able to displace the Vpx protein, it could be further optimized by extending it toward the surface of the central pore to disrupt the interaction of DCAF1 with its binding partners.
Methylation of the Pyrazole Core and Amide Modification
With the preferred stereochemistry established, we investigated the impact of N-alkylation of the core pyrazole. With modest yields obtained in EDCI coupling reactions, we explored an alternative coupling protocol using MsCl and DIEA (Scheme 2). This protocol afforded 5b in improved yield; however, when applied to the synthesis of 5a, the result was a complex mixture of products and a lower yield. These reaction conditions did not offer any advantage over the standard EDCI coupling conditions used to synthesize the majority of the compounds described in this manuscript. The synthesis of N-Me amide analog 5c is illustrated in Scheme 3. HATU coupling of methylamine with Boc protected acid 6 afforded 7 in excellent yield. Deprotection of 7 with HCl in 1,4-dioxane afforded the methylated coupling partner 8 also in excellent yield. Variability in the key final coupling reaction of unprotected pyrazole acids 2 with β-amino amides 1 encouraged us to investigate protection of the pyrazole before amide formation. Tosylation of 2a afforded 9 (35% yield), and subsequent HATU coupling (55% yield) and deprotection afforded 5c in 51% yield. Although the yields of the individual reactions of this synthetic scheme were somewhat disappointing, this alternative route could provide access to targets otherwise inaccessible without pyrazole protection. The primary amide in 3d was also dehydrated to the nitrile under standard conditions to afford 5d.
Scheme 2. Synthesis of Compounds 5a–b.

Reagents and conditions: (a) MsCl, DIEA, DCM, 0 °C (10 min), then 1d, 0 °C (30 min).
Scheme 3. Synthesis of Compound 5c.

Reagents and conditions: (a) HATU, DIEA, DMF, MeNH2; (b) HCl in 1,4-dioxane; (c) TsCl, Py; (d) HATU, DIEA, DMF, 8; (e) KOH (aq.), THF.
As shown in Table 3, both possible methylated pyrazoles 5a and 5b exhibited reduced affinity for DCAF1 when compared to 14c. Molecular modeling predicted that this ring decoration (5a) would be detrimental for activity. Indeed, the small hydrophobic methyl group would be surrounded by backbone polar groups from several protein residues (Thr1097, Thr1139, and Phe1355). Also, this methyl group would perturb a water-mediated hydrogen bond network: a key water molecule, which mediated hydrogen bonds between the pyrazole core and backbone from residues Thr1097 and Thr1139, would be displaced. Similarly, the alternative tautomer (5b) was predicted to be even less potent because it would create hydrogen-bond acceptor mismatches between the decorated pyrazole ring and the constrained water molecules. Indeed, both hydrogen bond donor features from solvent molecule were already engaged in polar interactions with the carbonyl backbone group from Thr1097 and Thr1139.
Table 3. Structure–Activity Relationships of Compounds 5a–d.
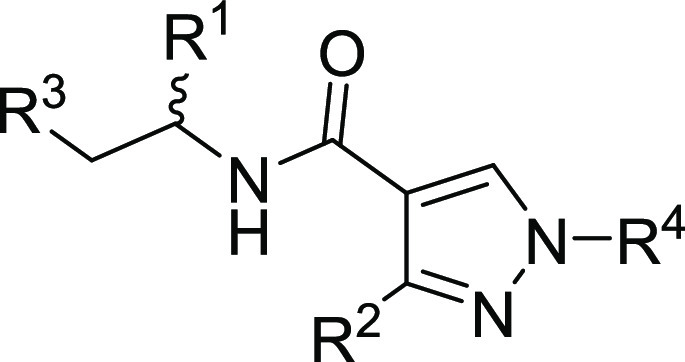

Experiments were performed in duplicate.
Methylation of the primary amide 5c was also detrimental to affinity when compared to 3b. As for the methylated pyrazole case, it was predicted that this modification would perturb a water-mediated hydrogen bond network in the vicinity of Cys1099, His1140, and Asp1356 residues. Finally, the primary amide 3d to nitrile 5d conversion was also detrimental to affinity with about 6-fold loss in potency.
This second set of compounds highlighted that the SAR landscape is sensitive for this series, as exemplified by several activity cliffs with minor structural changes.
Northern Phenyl Ring Optimization
Exploring the substitution of the “northern” phenyl ring was accomplished using EDCI coupling as the final step (Scheme 4). Although yields of products were low to modest (9–45%), the operational simplicity of this reaction between pyrazole acid 2a and β-amino amides 1d–m provided rapid access to the desired compounds 14a–j for testing. Amino amides that were not commercially available 1h–i and 1k–m were prepared in three steps from the corresponding commercial β-amino acids 11 (Scheme 4).
Scheme 4. Synthesis of Compounds 14a–j.

Reagents and conditions: (a) BOC2O, DIEA, DCM; (b) EDCI, NH4Cl, DIEA, DCM; (c) HCl in 1,4-dioxane; (d) EDCI, DIEA, DCM 0 °C (5 min), rt (18 h).
As shown in Table 4 the presence of the northern aromatic ring is required for activity (14a). Analysis of the crystal structure revealed that the binding pocket that accommodates the northern phenyl ring is very deep and hydrophobic. Indeed, this pocket is delimited by V1299, V1300, Y1309, G1310, A1311, F1330, I1354, and L1357 residues. As expected, all favorable modifications concerned the decoration of the aromatic ring with halogen substituents at various positions. Thus, large halogens at the meta-position significantly improved potency (3d, 14c). Indeed, the chlorine atom is located at optimal Van der Waals distances (3.6–4.0 Å) to several hydrophobic sidechains from the pocket. Surprisingly, the meta fluorine substituted analog (14f) displayed no binding, whereas the unsubstituted ring still showed some activity (3b). Similarly, para substitutions were also predicted to have favorable impact on potency. Thus, fluorine (14g) and chlorine (14d) analogs exhibited improved affinity for DCAF1 compared to the unsubstituted northern phenyl ring (3b).
Table 4. Structure–Activity Relationships of Compounds 14a–j.
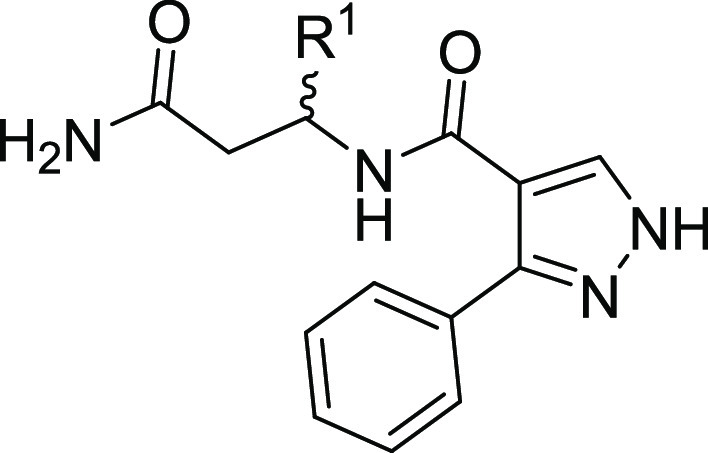
| compound | R1 | DCAF1 KD (nM)a |
|---|---|---|
| 14a | H | NB |
| 3b | (S) Ph | 16,400; 16,900 |
| 14b | (S) 2-Cl Ph | 22,960; 21,940 |
| 14c | (S) 3-Cl Ph | 4220; 4000 |
| 14d | (S) 4-Cl Ph | 6900; 6800 |
| 14e | (S) 2-F Ph | 15,500; 15,800 |
| 14f | (S) 3-F Ph | NB |
| 14g | (S) 4-F Ph | 9600; 9800 |
| 14h | (S) 2-OMe Ph | NB |
| 14i | (S) 3-OMe Ph | NB |
| 14j | (S) 4-OMe Ph | NB |
Experiments were performed in duplicate.
Analysis of the X-ray structure was able to explain why the ortho modifications such as 14b, 14e, and 14h were either suboptimal or even expected to be detrimental for potency (small available space to accommodate chemical moieties and potential intramolecular clashes preventing the observed bioactive conformation). The least sterically demanding ortho fluoro (14e) analog was tolerated but offered no improvement when compared to the unsubstituted (3b) analog. Substitution of the aromatic ring with alkoxy substituents in either the ortho14h, meta14i, or para14j position was also not tolerated.
Southern Phenyl Ring Optimization
A complementary exercise was conducted to determine the optimal substitution for the southern phenyl ring. The effect on substitutions on the biaryl phenyl ring was explored systematically by keeping all other positions constant. The final compounds were purchased if available or synthesized by EDCI coupling of 1b with 2 (Scheme 5).
Scheme 5. Synthesis of Compounds 15a–b, 15f, 15j.

Reagents and conditions: (a) EDCI, DIEA, 0 °C (5 min), rt (18 h).
Similar to the northern phenyl, the presence of the southern aromatic ring is required for activity 15a (Table 5). As for the northern ring, the binding pocket that accommodates the southern phenyl ring is also hydrophobic. Indeed, this cavity is delimited by His1140, Tyr1181, Arg1225, Cys1227, Arg1298, Leu1313, Pro1329, and Phe1330 residues. However, the binding pocket is not deeply buried anymore and contains two polar arginine residues. Again, all favorable modifications concerned the decoration of the aromatic ring with halogen substituents at varied positions. Notably, addition of halogen atoms at the ortho position showed favorable impact on potency (15b and 15e compounds compared to 3b) because we speculate that the ortho substituents can now promote a non-coplanar angle between the two rings. Also, this bioactive conformation exhibited cation-π interactions with a co-planar arginine residue in the vicinity of the southern ring. By contrast, halogenation at the meta position was tolerated but offered no real improvement (15c, 15f), and m-cyano analog 15i displayed ∼1.5-fold loss in potency. Para modifications on the southern ring displayed varied results: whereas both methoxy 15h and fluoro 15g analogs were poor, the very close chloro analog 15d exhibited 5-fold potency gain with the addition of a single atom. This result can be explained by the favorable interaction between the chlorine atom from the ligand and the carbonyl backbone atom from Arg1225 (distance (Cl-O) = 3.3 Å, angle = 167° according to the model and confirmed by the second X-ray structure).29
Table 5. Structure–Activity Relationships of Compounds 15a–j.
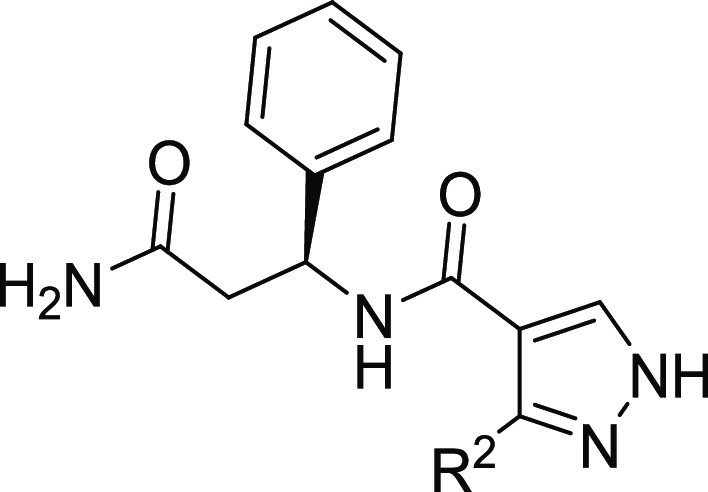
| compound | R2 | DCAF1 KD (nM)a |
|---|---|---|
| 15a | Me | NB |
| 3b | Ph | 16,400; 16,900 |
| 15b | 2-Cl Ph | 4000; 4100 |
| 15c | 3-Cl Ph | 11,100; 11,200 |
| 15d | 4-Cl Ph | 2890; 2890 |
| 15e | 2-F Ph | 3400; 3420 |
| 15f | 3-F Ph | 15,700; 15,700 |
| 15g | 4-F Ph | 13,280; 12,960 |
| 15h | 4-OMe Ph | 35,100; 34,700 |
| 15i | 3-CN Ph | 24,900; 26,500 |
| 15j | 3-Cl, 4-F Ph | 15,900; 15,800 |
Experiments were performed in duplicate.
Northern–Southern Combinations
Combining the findings from the northern and southern SAR studies for finding an optimal compound required an alternate synthetic pathway. Exploring specific and multiple substitutions of both aromatic rings required that we prepare specific enantiomerically enriched β-amino amides 1 and also assemble non-commercially available 3-phenyl pyrazole acids 2. The addition of a Reformatsky reagent to an N-tert-butanesulfinyl imine 17 (Scheme 6) has been reported to proceed in high yield and excellent diastereoselectivity. Subsequent amidation and deprotection afforded the desired intermediates 1 in high enantiomeric excess.
Scheme 6. Synthesis of Intermediates 1n–o.

Reagents and conditions: (a) PPTS, MgSO4, DCM; (b) Zn, CuCl, THF; (c) 7 N NH3 in MeOH; (d) HCl in 1,4-dioxane.
We envisioned that the desired substitution patterns in the southern phenyl ring could be obtained from the palladium catalyzed cross-coupling of the appropriate boronate 22 with the protected bromo-pyrazole ester 21 (Scheme 7). SEM protection of 20 afforded 21 as a mixture of regioisomers (the presumed major regioisomer is drawn) that efficiently underwent Suzuki cross-coupling with commercial boronic acids 22 under microwave irradiation. Saponification of 23 with KOH yielded the pyrazole acid intermediates 24 that coupled smoothly with β-amino amides 1 to afford protected intermediates 25. Treatment of the silyl protected intermediates with TFA afforded the target compounds 26 in high purity but low yield after preparative chromatography.
Scheme 7. Synthesis of Compounds 26a–e.

Reagents and conditions: (a) NaH, SEMCl, THF; (b) PdCl2(PPh3)2, Cs2CO3, 1,4-dioxane, H2O, μw, 110 °C; (c) KOH, THF/MeOH/H2O, 70 °C; (d) HATU, DIEA, DMF; (e) TFA, DCM.
Combining the best substituents on the northern and southern aromatic rings had a dramatic improvement of potency (Table 6). Halogens in both the 3 and the 4 positions of the northern ring (26a, 26b) improved potency ∼2 fold over 3d. Similarly, halogens in the 2 and the 4 position of the southern ring (26c, 26d) provided an additional 1.5- to 5-fold boost in binding affinity for DCAF1. The observed increase in affinity was additive with compound 26e being the most potent compound in this series.
Table 6. Structure–Activity Relationships of Compounds 26a–e.
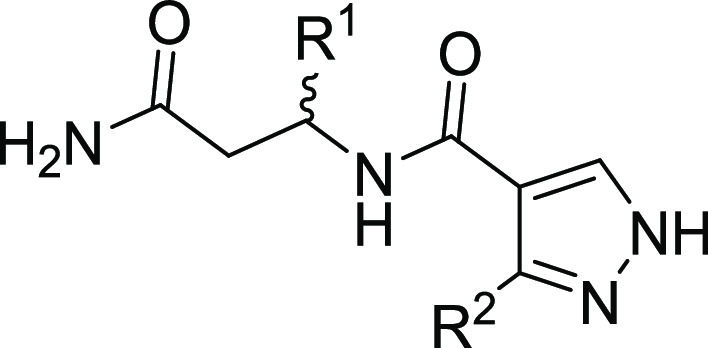
| compound | R1 | R2 | DCAF1 KD (nM) |
|---|---|---|---|
| 26a | (S) 3-Cl, 4-F Ph | 2-F Ph | 170; 180 |
| 26b | (S) 3,4 di-Cl Ph | 2-F Ph | 200; 217 |
| 26c | (S) 3-Cl Ph | 2,4-F Ph | 265; 282 |
| 26d | (S) 3-Cl Ph | 2-F, 4-Cl Ph | 85 ± 10 |
| 26e | (S) 3-Cl, 4-F Ph | 2-F, 4-Cl Ph | 38 ± 1.5 |
Biophysical Characterization and Co-crystal Structure of Compound 26e with DCAF1
Compound 26e showed the highest affinity for binding to the DCAF1 WDR domain with an SPR KD value of 38 ± 1.5 nM (Figure 4A). DCAF1 was also significantly stabilized in the presence of 26e (Figure 4B) in a concentration-dependent manner (Figure 4C) with a ΔTm of 7.4 ± 0.5 °C at 100 μM (Table S1). DCAF1 stabilization of 26e was higher than that for 3d and correlated well with the potency of binding as measured by SPR. Additionally, ITC experiments confirmed 26e binding to DCAF1 with a Kd value of 216 ± 76 nM. The binding interaction was enthalpy-driven and exothermic with a ΔH value of −50.4 ± 1.7 kJ/mol and ΔS value of −41.1 ± 3.9 J/mol·K.
Figure 4.

Biophysical characterization of compound 26e (OICR-8268) with the DCAF1 WDR domain. Binding of compound 26e to the DCAF1 WDR domain was tested by SPR and DSF assays. (A) SPR showed 26e binding to DCAF1 potently with a KD of 38 ± 1.5 nM, and (B) DSF showed that the compound stabilizes DCAF1 in a concentration-dependent manner (C) with increases in the protein’s melting temperature, indicating binding. (D) Orthogonal binding confirmation of 26e by ITC with a KD of 216 ± 76 nM. All experiments were performed in triplicate.
The DCAF1 co-crystal structure in complex with compound 26e, referred to here as DCAF1-26e (PDB ID: 8F8E), was determined to 1.55 Å resolution in P1211 space group symmetry with two molecules in the crystal asymmetric unit. Table 2 summarizes the crystallographic data collection, refinement, and validation statistics. A well-defined electron density map corresponding to 26e was observed in both chains of the crystal asymmetric unit (Figure S5). As expected, 26e binds in the same position deep inside the central pore as 3d, maintaining all the previously observed interactions with the protein and a water molecule (Figure 5). Two additional water molecules coordinate with the compound: one (HOH160) with the N1 atom of the pyrazole ring and the second (HOH292) with the O1 atom of the terminal amide. The added fluoride atom in the 3-chloro-4-fluorophenyl group does not significantly affect the conformation of the protein residues, but it likely contributes to hydrophobic interactions within the hydrophobic pocket involving Leu1357, Ile1354, Phe1330, Ala1311, and Val1300. On the other hand, the added chlorine atom in the 2-fluoro-4-chlorophenyl group contributes to hydrophobic interactions with the protein by packing against the side chains of Arg1225 and Cys1227 (Figure 5). The side chain of Arg1225 adopts a different conformation in the DCAF1-3d structure (Figure 5B). As expected, the binding mode of the ligand (RMSD < 0.5 Å) and the overall binding site conformation have been perfectly conserved through the optimization process. Binding modes of ligands are displayed in Figure 6. Notably, the crystal structure of 26e in complex with DCAF1 protein supported the halogen bond hypothesis, and this special interaction has been automatically detected by the 2D interaction diagram software (LID from Schrodinger).
Figure 5.

Co-crystal structure of DCAF1 WDR domain in complex with 26e. (A) A close-up view of 26e binding site in the central tunnel of the DCAF1 WDR domain. Compound 26e retains the network of interactions observed in DCAF1-3d (Figure 3B) but also makes additional interactions with two extra water molecules, as well as additional hydrophobic interactions with the protein residues. Compound 26e is shown as magenta sticks, and the protein residues involved in the interactions are rendered as cyan sticks. The hydrogen bonds are shown as black dashes, and interacting water molecules are shown as red spheres. (B) An overlay of DCAF1-3d and DCAF1-26e structures showing the location of the additional atoms in the 26e compound and the binding site differences in the two proteins. Compounds and active site residues are rendered as described in Figure 3B and in panel A here.
Figure 6.

2D interaction diagrams of 3d and 26e ligands in complex with DCAF1 protein. The most important interaction classes, including water-mediated hydrogen bonds, are represented. All protein residues within 5.0 Å from ligand are displayed. This picture has been generated using the LID tool from Maestro software (Schrodinger) from the prepared respective X-ray structures (7UFV and 8F8E).
Target Engagement of 26e and 3d
To confirm target engagement in cells, we generated a DCAF1 cellular thermal shift assay (CETSA) using NanoLuc split luciferase technology (Promega), in which we have produced stable H460 cells expressing the DCAF1 WDR domain or DCAF1 full-length tagged with a small fragment of NanoLuc luciferase (HiBit) that, upon complementation with the larger fragment of NanoLuc, will result in luminescence.30 This approach allows the assessment of DCAF1 stability upon compound treatment by monitoring the change in denaturation temperature. Using this approach, 26e significantly (>2 °C) stabilized HiBiT-DCAF1-WDR at 40 μM (Figure 7A, C) and was also dose-dependent (Figure 7B, D). Furthermore, 26e thermally stabilized the WDR domain of DCAF1 to a greater extent than 3d, with an EC50 value of 10.5 μM (Figure 7C,D). However, both compounds failed to thermally stabilize the full-length DCAF1 (HiBiT-DCAF1_FL) at 40 μM (Figure S6). This may simply be the case that binding of 26e to the WDR domain of DCAF1 does not result in significant and detectable stabilization of the full-length protein (169 kDa).
Figure 7.

26e engages the WDR domain of DCAF1 in cells. In a cellular thermal shift assay (CETSA) as described in the materials and methods, the amount of unaggregated flag tagged WDR domain of DCAF1 was quantified. (A) 26e significantly stabilized the Flag-Hb-DCAF1-WD at 40 μM. (B) Titration of 26e showed a dose-dependent pattern of DCAF1-WD stability. Different compound concentrations (40 to 0.2 μM) of 26e were heated at 61 °C for 3.5 min. (C) When compared to the DMSO treatment, 26e thermally stabilized the WDR domain of DCAF1 to a greater extent than 3d, causing a 3.6 and 1.7 °C shift in the aggregation temperature of Flag-Hb-DCAF1_WD at 40 μM. Results shown as average ± SD (n = 3) (D) 26e thermally stabilized Flag-Hb-DCAF1_WD in a dose-dependent manner with an EC50 value of 10.5 μM. Results shown as average ± SD (n = 3). CDC40 was used as a negative control. Note that in C, cells are subjected to a gradient of temperatures (46 to 66 °C). Across this range of temperatures, 3d thermally stabilizes the WDR domain of DCAF1, but the stabilization does not extend beyond 61 °C. In D, the protein is kept at 61 °C, and consistent with our observation in C, 3d was not expected to stabilize the WDR domain at any concentration.
Potential Applications of 26e
The biophysical characterization data showed high affinity binding of 26e to DCAF1. In addition, the co-crystal structures revealed that the compound is buried deep into the central pore of the WD domain. The structural comparison with the Vpx peptide did not show any overlap with the compound, minimizing the possibility of direct antagonism. Most recently, an azetidine acrylamide-based electrophilic PROTAC for DCAF1 was reported.31 This covalent DCAF1-based PROTAC specifically reacts with the cysteine 1113 of DCAF1 leading to degradation of FKBP12 and BRD4 by ectopically expressed, but not endogenous, DCAF1.3126e, however, is a reversible DCAF1 ligand with high affinity and cellular engagement. Therefore, it is an excellent starting tool for development of chemical handles for DCAF1-based PROTACs for degradation of desired targets. In addition, it could be further developed to high affinity DCAF1 antagonists by extending the molecule to the surface of the protein.
Antagonists that disrupt protein–protein interactions between DCAF1 and its interacting partners could be used toward development of therapeutics for cancers when DCAF1 is overexpressed,26 and HIV when DCAF1 is hijacked by the virus.15−17,23 In these cases, the small molecule prevents the binding of Vpr and Vpx to DCAF1, for example, preventing viral hijacking of the CRL4DCAF1 or EDVPDCAF1 complexes. Another potential application of antagonizing CRL4DCAF1 function is in Merlin-deficient tumors. Merlin is a tumor suppressor that negatively regulates CRL4DCAF1-mediated ubiquitination of Lats1/2 in the Hippo pathway.32 In Merlin-deficient tumorigenesis, CRL4DCAF1-mediated ubiquitination is upregulated, and expression of Merlin or depletion of DCAF1 suppresses proliferation of tumor cells.33,34 A DCAF1 antagonist can also function as a tool to discover novel biological functions of DCAF1.
Conclusions
We report the discovery of a novel series of small molecule ligands that target DCAF1. Specifically, 3d and its more potent analog 26e were identified using biophysical assays, showing binding to the DCAF1 WDR domain with Kd of 590 and 38 nM by SPR and Kd of 932 and 278 nM by ITC, respectively. Co-crystallization of 3d in complex with the DCAF1 WDR domain allowed us to employ structure-guided SAR to optimize and develop 26e. Interestingly, we also discovered that the S enantiomers of this chemical series were more potent than the R enantiomers. In addition to the biophysical characterization and co-crystallization of these compounds, we assessed the cellular target engagement and further confirmed that 26e is binding to the DCAF1 WDR domain in cells with EC50 of 10.5 μM. Overall, we discovered a potent and in-cell active small molecule for the DCAF1 WDR domain along with the co-crystal structure of the DCAF1-26e complex that could guide the development of DCAF1 antagonists and DCAF1-based PROTACs as future cancer and viral therapeutics.
Experimental Section
Chemistry
All reagents were purchased from commercial vendors and used without further purification. Volatiles were removed under reduced pressure by rotary evaporation or by using the V-10 solvent evaporator system from Biotage. Very high boiling point (6000 rpm, 0 mbar, 56 °C), mixed volatile (7000 rpm, 30 mbar, 36 °C), and volatile (6000 rpm, 30 mbar, 36 °C) methods were used to evaporate solvents. The yields given refer to chromatographically purified and spectroscopically pure compounds, unless stated otherwise. Compounds were purified using either a Biotage Isolera One system or a Teledyne ISCO CombiFlash Rf system. Normal phase chromatography was performed using Biotage SNAP KP-Sil, Sfär Silica D (part no. FSKO-1107/FSRD-0445), Teledyne ISCO RediSep Rf silica (part no. 69-2203-3), or InnoFlash S series silica (part no. VI-1501) columns. Reverse-phase chromatography was performed using Biotage SNAP KP-C18-HS, Sfär C18 D (part no. FSLO-1118/FSUD-040) or Teledyne ISCO RediSep Rf C18 (part no. 69-2203-4) columns. Preparative chromatography was carried out using either a Waters 2767 injector with the collector attached to PDA UV/vis and SQD mass detectors or a Waters 3767 injector with the collector attached to PDA UV/vis and QDa mass detectors. An XSelect CSH Prep C18 5 μm OBD 19 × 100 mm (part no. 186005421) or Xselect CSH Prep C18 5 μm 10 × 100 mm (part no. 186005415) column was used for purification. Final compounds were dried using the Labconco Benchtop FreeZone Freeze-Dry System (4.5 L model). 1H NMR spectra were recorded on a Bruker Avance-III 500 MHz spectrometer at ambient temperature. 13C NMR spectra were recorded on an Agilent DD2 700 MHz spectrometer at ambient temperature. Residual protons of CDCl3, DMSO-d6, and CD3OD solvents were used as internal references. Spectral data are reported as follows: chemical shift (δ in ppm), multiplicity (br = broad, s = singlet, d = doublet, dd = doublet of doublets, t = triplet, m = multiplet), coupling constants (J in Hz), and proton integration. Compound purity was determined by UV absorbance at 254 nm during tandem liquid chromatography/mass spectrometry (LCMS) using a Waters Acquity separations module. All final compounds had a purity of ≥95% as determined using this method. Low-resolution mass spectrometry was conducted in both positive and negative ion mode using a Waters Acquity SQD mass spectrometer (electrospray ionization source) fitted with a PDA detector. Mobile phase A consisted of 0.1% formic acid in water, whereas mobile phase B consisted of 0.1% formic acid in acetonitrile. Analysis was carried out using an Acquity UPLC HSS T3 (2.1 × 50 mm, 100 Å, 1.8 μm. part no. 186003538) column. The gradient went from 98 to 5% mobile phase A over 1.8 min, maintained at 5% for 0.5 min, and then increased to 98% over 0.2 min for a total run time of 3 min. The flow rate was 0.4 mL/min, and all columns were used with the temperature maintained at 25 °C.
General Procedure A for the Synthesis of Compounds 3a, 3c–d, 14a–j, 15a–b, 15f, and 15j
A suspension of carboxylic acid 2 (0.27 mmol), amine 1 (0.24 mmol), and EDCI (0.27 mmol) in DCM (3 mL) was cooled to 0 °C in an ice bath. DIEA (0.54 mmol) was added, and the reaction was stirred at 0 °C for 5 min before the vial was removed from the ice bath and allowed to gradually warm to room temperature. After stirring at room temperature for 18 h, the reaction mixture was concentrated onto Celite under reduced pressure. Purification by flash chromatography on silica (2.5–10% MeOH in DCM with 0.5% NH4OH), by reverse phase chromatography (5–60% MeCN in water with 0.1% formic acid), or by both if required afforded the products in 9–45% yield.
General Procedure B for the Synthesis of Compounds 5a–b
DIEA (0.22 mmol) and MsCl (0.22 mmol) were added to a stirring solution of carboxylic acid 4 (0.20 mmol) in DCM (2 mL) at 0 °C. After 10 min, an additional portion of DIEA (0.22 mmol) was added, and the solution of activated acid was transferred to a solution of amine 1 (0.20 mmol) in DCM (2 mL) also at 0 °C. After stirring for 30 min, the reaction was quenched with water, and the layers were separated. The aqueous layer was extracted with DCM, and the combined organics were dried and concentrated onto Celite. Purification by flash chromatography on silica (2.5–10% MeOH in DCM with 0.5% NH4OH), by reverse phase chromatography (5–60% MeCN in water with 0.1% formic acid), or by both if required afforded the desired compounds in 5–70% yield.
General Procedure C for the Synthesis of Intermediates 12a–e
DIEA (1.6 mmol) was added to a suspension of β-amino acid 11 (1.4 mmol) and BOC2O (1.6 mmol) in DCM (6 mL) at room temperature. After stirring for 18 h at room temperature, the volatiles were removed under reduced pressure. Purification by reverse phase chromatography (5–60% MeCN in water with 0.1% formic acid) afforded the desired intermediates in 33–78% yield.
General Procedure D for the Synthesis of Intermediates 13a–e
DIEA (2.8 mmol) was added to a stirring suspension of BOC protected β-amino acid 12 (0.57 mmol), NH4Cl (1.7 mmol), and EDCI (0.68 mmol) in DCM (5 mL) at 0 °C. After 5 min, the reaction vial was removed from the ice bath and stirred at room temperature for 18 h. MeOH (0.5 mL) was added, and the reaction mixture was concentrated onto Celite under reduced pressure. Purification by flash chromatography (0.5–9.5% MeOH in DCM with 0.5% NH4OH) afforded the desired intermediates in 19–49% yield.
General Procedure E for the Synthesis of Intermediates 1h–i and 1k–m
BOC protected β-amino amides 13 (0.18 mmol) were dissolved in 1,4-dioxane (2.0 mL) and treated with HCl (4.0 M in 1,4-dioxane) (2.0 mL, 4.0 mmol) at room temperature. After stirring at room temperature for 18 h, the reaction mixture was concentrated to dryness under reduced pressure to afford the desired intermediates in quantitative yield as their HCl salts that could be used without further purification.
General Procedure F for the Synthesis of Intermediates 17a–b
A mixture of benzaldehyde 16 (32 mmol), PPTS (1.6 mmol), (R)-2-methylpropane-2-sulfinamide (32 mmol), and MgSO4 (158 mmol) in DCM (80 mL) was heated to 40 °C for 18 h. The reaction mixture was filtered through a pad of Celite, and the filtrate was concentrated to dryness under reduced pressure. Purification of the residue by flash chromatography on silica (10–20% EtOAc in hexanes) afforded the desired intermediates in 51–62% yield.
General Procedure G for the Synthesis of Intermediates 18a–b
A mixture of zinc powder (177 mmol) and copper(II) chloride (17.7 mmol) in THF (120 mL) was heated at reflux for 30 min under a nitrogen atmosphere. After cooling to room temperature, ethyl bromoacetate (44.2 mmol) in THF (10 mL) was added, and the reaction was heated at 50 °C for 30 min. The reaction was cooled to 0 °C, and intermediate 17 (17.7 mmol) in THF (20 mL) was added. The reaction was stirred for 2 h at 0 °C. The reaction mixture was filtered through a pad of Celite washing with EtOAc. The organic layer was washed with water and brine, dried over sodium sulfate, and concentrated to dryness under reduced pressure. Purification by flash chromatography on silica (40–60% EtOAc in hexanes) afforded the desired intermediates in 28–59% yield.
General Procedure H for the Synthesis of Intermediates 19a–b
A mixture of intermediate 18 (10.0 mmol) in 7 N methanolic ammonia (60 mL) was stirred for 5 h at room temperature. The reaction mixture was concentrated to dryness under reduced pressure. Purification by flash chromatography on silica (1–10% MeOH in DCM) afforded the desired intermediates in 66–93% yield.
General Procedure I for the Synthesis of Intermediates 1n–o
A mixture of intermediate 19 (3.3 mmol) and 4 N HCl in 1,4-dioxane (10 mL) was stirred at room temperature for 2 h. The reaction mixture was concentrated to dryness under reduced pressure. Trituration with Et2O afforded the desired intermediates as powders in 49–64% yield.
General Procedure J for the Synthesis of Intermediates 23a–c
A mixture of intermediate 21 (2.15 mmol), boronic acid 22 (2.58 mmol), Pd(PPh3)2Cl2 (0.215 mmol), and Cs2CO3 (4.72 mmol) in 1,4-dioxane (12 mL) and water (4 mL) under nitrogen was irradiated to 110 °C for 45 min. After cooling to room temperature, the reaction mixture was partitioned between DCM (100 mL) and water (50 mL), and the layers were separated. The aqueous layer was extracted with DCM (50 mL), and the combined organics were dried over sodium sulfate and concentrated to dryness. The residue was purified by flash chromatography on silica gel (0–25% EtOAc in hexanes) to afford the intermediates in 86–95% yield.
General Procedure K for the Synthesis of Intermediates 24a–c
A mixture of intermediate 23 (1.84 mmol) and KOH (5.51 mmol) in THF (6 mL), MeOH (2 mL), and water (6 mL) was heated to 70 °C for 3 h. After cooling to room temperature, the reaction mixture was partitioned between DCM (100 mL) and water (100 mL), and the layers were separated. The aqueous layer was acidified with 1 N HCl and extracted three times with DCM (50 mL). The combined organics were dried over magnesium sulfate and concentrated to dryness to afford the intermediates in 76–81% yield.
General Procedure L for the Synthesis of Intermediates 25a–e
DIEA (0.84 mmol) was added to a solution of intermediate 1 (0.84 mmol) and HATU (0.84 mmol) in DMF (4 mL) at room temperature. Intermediate 24 (0.70 mmol) was added, and the reaction was stirred at room temperature for 2 h and then at 45 °C for 18 h. The reaction mixture was concentrated to dryness under reduced pressure, and the residue was purified by flash chromatography on silica gel (40–100% EtOAc in hexanes) to afford the intermediates in >95% yield.
General Procedure M for the Synthesis of Compounds 26a–e
TFA (0.75 mL) was added to a solution of intermediate 25 (0.70 mmol) in DCM (4 mL) at room temperature. After stirring at room temperature for 2 h, the reaction mixture was neutralized with a 5% solution of NH4OH in MeOH. The reaction mixture was concentrated to dryness under reduced pressure, and the residue was purified by preparative chromatography to afford the desired compounds 7–18% yield.
(S)-3-Amino-3-(2-fluorophenyl)propanamide Hydrochloride (1h)
Quantitative yield; ESI-MS m/z 183.3 [M + H]+.
(S)-3-Amino-3-(3-fluorophenyl)propanamide Hydrochloride (1i)
Quantitative yield; ESI-MS m/z 183.0 [M + H]+.
(S)-3-Amino-3-(2-methoxyphenyl)propanamide Hydrochloride (1k)
Quantitative yield; ESI-MS m/z 195.4 [M + H]+.
(S)-3-Amino-3-(3-methoxyphenyl)propanamide Hydrochloride (1l)
Quantitative yield; ESI-MS m/z 195.3 [M + H]+.
(S)-3-Amino-3-(4-methoxyphenyl)propanamide Hydrochloride (1m)
Quantitative yield; ESI-MS m/z 195.4 [M + H]+.
(S)-3-Amino-3-(3,4-dichlorophenyl)propanamide (1n)
64% yield; ESI-MS m/z 232.8 [M + H]+.
(S)-3-Amino-3-(3-chloro-4-fluorophenyl)propanamide (1o)
49% yield; ESI-MS m/z 217.1 [M + H]+.
(R)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-phenyl-1H-pyrazole-4-carboxamide (3a)
1H NMR (DMSO-d6, 500 MHz) δ 13.39–13.12 (m, 1H), 8.47–8.39 (m, 1H), 8.21–7.92 (m, 1H), 7.73–7.62 (m, 2H), 7.42–7.28 (m, 8H), 7.26–7.20 (m, 1H), 6.83 (br s, 1H), 5.37 (q, J = 7.5 Hz, 1H), 2.59–2.56 (m, 2H); ESI-MS m/z 335.4 [M + H]+; yield 28%; purity, 99.0%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-phenyl-1H-pyrazole-4-carboxamide (3b)
1H NMR (DMSO-d6, 500 MHz) δ 13.41–13.11 (m, 1H), 8.43 (br s, 1H), 8.21–7.92 (m, 1H), 7.73–7.62 (m, 2H), 7.44–7.28 (m, 8H), 7.25–7.20 (m, 1H), 6.83 (br s, 1H), 5.37 (q, J = 7.6 Hz, 1H), 2.59–2.56 (m, 2H); ESI-MS m/z 335.3 [M + H]+; enamine; purity, 98.5%.
(R)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-3-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (3c)
1H NMR (DMSO-d6, 500 MHz) δ 13.45–13.26 (m, 1H), 8.47–8.31 (m, 1H), 8.30–8.01 (m, 1H), 7.50–7.23 (m, 9H), 7.22–7.13 (m, 1H), 6.84 (br s, 1H), 5.31–5.25 (m, 1H), 2.57–2.55 (m, 2H); ESI-MS m/z 387.2 [M + H]+; yield 38%; purity, 97.6%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-3-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (3d)
1H NMR (DMSO-d6, 500 MHz) δ 13.35 (br s, 1H), 8.37 (br s, 1H), 8.31–7.98 (m, 1H), 7.47–7.39 (m, 2H), 7.38–7.31 (m, 3H), 7.30–7.25 (m, 2H), 7.21 (br s, 2H), 6.84 (br s, 1H), 5.28 (q, J = 7.4 Hz, 1H), 2.58–2.55 (m, 2H); ESI-MS m/z 387.3 [M + H]+; yield 24%; purity, 98.1%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-1-methyl-3-phenyl-1H-pyrazole-4-carboxamide (5a)
1H NMR (methanol-d4, 500 MHz) δ 7.91 (d, J = 4.5 Hz, 1H), 7.48–7.44 (m, 2H), 7.28–7.22 (m, 4H), 7.22–7.13 (m, 3H), 5.36–5.30 (m, 1H), 3.84 (d, J = 4.6 Hz, 3H), 2.63–2.56 (m, 2H); ESI-MS m/z 383.3 [M + H]+; yield 5%; purity, 95.1%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-1-methyl-5-phenyl-1H-pyrazole-4-carboxamide (5b)
1H NMR (DMSO-d6, 500 MHz) δ 8.25 (d, J = 8.1 Hz, 1H), 7.99 (s, 1H), 7.48–7.44 (m, 3H), 7.41–7.38 (m, 2H), 7.34–7.29 (m, 3H), 7.28–7.25 (m, 1H), 7.23–7.21 (m, 1H), 6.83 (br s, 1H), 5.24 (q, J = 7.3 Hz, 1H), 3.65 (s, 3H), 2.54–2.52 (m, 2H); ESI-MS m/z 383.3 [M + H]+; yield 70%; purity, 99.8%.
(S)-N-(3-(Methylamino)-3-oxo-1-phenylpropyl)-5-phenyl-1H-pyrazole-4-carboxamide (5c)
To a solution of 10 (0.039 g, 0.077 mmol) in THF (1 mL) was added KOH (1.0 M in water) (1.0 mL, 1.0 mmol). The reaction was heated to 50 °C for 1 h. The reaction was cooled to room temperature and acidified with aqueous HCl (1.0 M in water) (∼1 mL). DCM (10 mL) and water (10 mL) were added, and the layers were separated. The aqueous layer was extracted with DCM (10 mL), and the combined organics were dried over sodium sulfate and concentrated to dryness to afford (S)-N-(3-(methylamino)-3-oxo-1-phenylpropyl)-5-phenyl-1H-pyrazole-4-carboxamide (0.025 g, 51%). 1H NMR (DMSO-d6, 500 MHz) δ 13.41–13.11 (m, 1H), 8.44 (br s, 1H), 8.19–7.90 (m, 1H), 7.78–7.74 (m, 1H), 7.72–7.62 (m, 2H), 7.40 (br s, 1H), 7.37–7.28 (m, 6H), 7.25–7.20 (m, 1H), 5.37 (q, J = 7.5 Hz, 1H), 3.30 (s, 3H), 2.60–2.57 (m, 2H); ESI-MS m/z 349.2 [M + H]+; purity, 95.3%.
(S)-N-(1-(3-Chlorophenyl)-2-cyanoethyl)-3-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (5d)
TFAA (0.31 mL, 2.3 mmol) was added to a solution of 3d (0.025 g, 0.065 mmol) in THF (1 mL) at room temperature. After stirring for 3 h, clean conversion to the nitrile along with TFA protection of the pyrazole was observed by UPLC-MS. The volatiles were removed under reduced pressure, and the residue was dissolved in MeOH (1 mL) and treated with K2CO3 (0.045 g, 0.32 mmol). After stirring for 18 h, the reaction mixture was concentrated onto Celite under reduced pressure and purified by reverse phase chromatography (5–60% MeCN in water with 0.1% formic acid) to afford (S)-N-(1-(3-chlorophenyl)-2-cyanoethyl)-3-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (0.016 g, 67%). 1H NMR (DMSO-d6, 500 MHz) δ 13.34 (b s, 1H), 8.59 (b s, 1H), 8.36–7.95 (b m, 1H), 7.43–7.27 (m, 6H), 7.14 (b s, 2H), 5.26–5.19 (m, 1H), 3.04–2.90 (m, 2H); ESI-MS m/z 369.3 [M + H]+; purity, 95.1%.
tert-Butyl (S)-(3-(methylamino)-3-oxo-1-phenylpropyl)carbamate (7)
DIEA (0.16 mL, 0.94 mmol) was added to a solution of (S)-N-Boc-3-amino-3-phenylpropanoic acid (0.25 g, 0.94 mmol) and HATU (0.36 g, 0.94 mmol) in DMF (3 mL). After stirring for 15 min at room temperature, methylamine (2.0 M in THF) (1.4 mL, 2.8 mmol) was added, and the reaction was stirred at room temperature for 18 h. The reaction mixture was concentrated onto Celite under reduced pressure and purified by flash chromatography on silica gel (0.5–5.0% MeOH in DCM with 0.5% NH4OH) to afford tert-butyl (S)-(3-(methylamino)-3-oxo-1-phenylpropyl)carbamate (0.24 g, 92%). ESI-MS m/z 279.4 [M + H]+.
(S)-3-Amino-N-methyl-3-phenylpropanamide Hydrochloride (8)
A solution of 7 (0.24 g, 0.87 mmol) was dissolved in 1,4-dioxane (5 mL) and treated with HCl (4.0 M in 1,4-dioxane) (0.94 mL, 3.8 mmol) at room temperature. After stirring for 1 h at room temperature, the reaction was concentrated to dryness under reduced pressure. Purification by reverse phase chromatography (0–50% MeCN in water with 0.1% formic acid) afforded (S)-3-amino-N-methyl-3-phenylpropanamide hydrochloride (0.18 g, 96%). ESI-MS m/z 179.3 [M + H]+.
3-Phenyl-1-tosyl-1H-pyrazole-4-carboxylic Acid (9)
A mixture of 2a (0.075 g, 0.40 mmol) and TsCl (0.091 g, 0.48 mmol) in pyridine (2 mL) was allowed to stir at room temperature for 2 h. The reaction was concentrated to dryness and purified by flash chromatography on silica gel (0.5–9.5% MeOH in DCM) to afford 3-phenyl-1-tosyl-1H-pyrazole-4-carboxylic acid (0.048 g, 35%). ESI-MS m/z 343.2 [M + H]+.
(S)-N-(3-(Methylamino)-3-oxo-1-phenylpropyl)-3-phenyl-1-tosyl-1H-pyrazole-4-carboxamide (10)
DIEA (0.03 mL, 0.17 mmol) was added to a solution of 9 (0.048 g, 0.14 mmol) and HATU (0.064 g, 0.17 mmol) in DMF (1 mL) at room temperature. After stirring at room temperature for 15 min, this solution was added to a solution of (S)-3-amino-N-methyl-3-phenylpropanamide hydrochloride (0.036 g, 0.17 mmol) and DIEA (0.03 mL, 0.17 mmol) in DMF (1 mL) also at room temperature. After stirring for 18 h, the reaction mixture was concentrated onto Celite under reduced pressure and purified by flash chromatography on silica gel (0.5–9.5% MeOH in DCM with 0.5% NH4OH) to afford (S)-N-(3-(methylamino)-3-oxo-1-phenylpropyl)-3-phenyl-1-tosyl-1H-pyrazole-4-carboxamide (0.039 g, 55%). ESI-MS m/z 503.3 [M + H]+.
(S)-3-((tert-Butoxycarbonyl)amino)-3-(2-fluorophenyl)propanoic Acid (12a)
67% yield; ESI-MS m/z 282.3 [M – H]−.
(S)-3-((tert-Butoxycarbonyl)amino)-3-(3-fluorophenyl)propanoic Acid (12b)
71% yield; ESI-MS m/z 282.4 [M – H]−.
(S)-3-((tert-Butoxycarbonyl)amino)-3-(2-methoxyphenyl)propanoic Acid (12c)
33% yield; ESI-MS m/z 294.4 [M – H]−.
(S)-3-((tert-Butoxycarbonyl)amino)-3-(3-methoxyphenyl)propanoic Acid (12d)
51% yield; ESI-MS m/z 294.4 [M – H]−.
(S)-3-((tert-Butoxycarbonyl)amino)-3-(4-methoxyphenyl)propanoic Acid (12e)
78% yield; ESI-MS m/z 294.4 [M – H]−.
tert-Butyl (S)-(3-Amino-1-(2-fluorophenyl)-3-oxopropyl)carbamate (13a)
48% yield; ESI-MS m/z 283.4 [M + H]+.
tert-Butyl (S)-(3-Amino-1-(3-fluorophenyl)-3-oxopropyl)carbamate (13b)
19% yield; ESI-MS m/z 283.2 [M + H]+.
tert-Butyl (S)-(3-Amino-1-(2-methoxyphenyl)-3-oxopropyl)carbamate (13c)
42% yield; ESI-MS m/z 295.3 [M + H]+.
tert-Butyl (S)-(3-Amino-1-(3-methoxyphenyl)-3-oxopropyl)carbamate (13d)
48% yield; ESI-MS m/z 295.2 [M + H]+.
tert-Butyl (S)-(3-Amino-1-(4-methoxyphenyl)-3-oxopropyl)carbamate (13e)
49% yield; ESI-MS m/z 295.3 [M + H]+.
N-(3-Amino-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14a)
1H NMR (DMSO-d6, 500 MHz) δ 13.37–13.07 (m, 1H), 8.16–7.86 (m, 2H), 7.80–7.66 (m, 2H), 7.47–7.31 (m, 4H), 6.82 (br s, 1H), 2.31 (t, J = 7.2 Hz, 2H), 1.29–1.24 (m, 2H); ESI-MS m/z 259.4 [M + H]+; yield 45%; purity, 97.7%.
(S)-N-(3-Amino-1-(2-chlorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14b)
1H NMR (DMSO-d6, 500 MHz) δ 13.41–13.05 (m, 1H), 8.42 (br s, 1H), 8.21–7.86 (m, 1H), 7.61 (br s, 2H), 7.41 (br d, J = 6.8 Hz, 1H), 7.36–7.17 (m, 8H), 6.83 (br s, 1H), 5.60–5.53 (m, 1H), 2.52–2.48 (m, 2H); ESI-MS m/z 369.3 [M + H]+; yield 16%; purity, 95.3%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14c)
1H NMR (DMSO-d6, 500 MHz) δ 13.40–13.04 (m, 1H), 8.42 (br d, J = 8.1 Hz, 1H), 8.18–7.82 (m, 1H), 7.58 (br s, 2H), 7.35–7.19 (m, 8H), 6.79 (br s, 1H), 5.27 (q, J = 7.5 Hz, 1H), 2.53–2.50 (m, 2H); ESI-MS m/z 369.3 [M + H]+; yield 12%; purity, 95.9%.
(S)-N-(3-Amino-1-(4-chlorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14d)
1H NMR (DMSO-d6, 500 MHz) δ 13.35–13.05 (m, 1H), 8.40 (br d, J = 7.9 Hz, 1H), 8.16–7.83 (m, 1H), 7.59 (br s, 2H), 7.36–7.22 (m, 8H), 6.77 (br s, 1H), 5.26 (q, J = 7.5 Hz, 1H), 2.52–2.49 (m, 2H); ESI-MS m/z 369.3 [M + H]+; yield 9%; purity, 95.9%.
(S)-N-(3-Amino-1-(2-fluorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14e)
1H NMR (DMSO-d6, 500 MHz) δ 13.36–13.04 (m, 1H), 8.33 (br s, 1H), 8.15–7.84 (m, 1H), 7.65–7.54 (m, 2H), 7.39–7.17 (m, 6H), 7.12–7.03 (m, 2H), 6.78 (br s, 1H), 5.55–5.48 (m, 1H), 2.50–2.46 (m, 2H); ESI-MS m/z 353.5 [M + H]+; yield 10%; purity, 98.0%.
(S)-N-(3-Amino-1-(3-fluorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14f)
1H NMR (DMSO-d6, 500 MHz) δ 13.33–13.07 (m, 1H), 8.39 (br dd, J = 8.7, 11.4 Hz, 1H), 8.15–7.84 (m, 1H), 7.64–7.54 (m, 2H), 7.36–7.21 (m, 5H), 7.14–7.06 (m, 2H), 7.02–6.95 (m, 1H), 6.78 (br s, 1H), 5.30 (q, J = 7.6 Hz, 1H), 2.51 (br t, J = 6.6 Hz, 2H); ESI-MS m/z 353.3 [M + H]+; yield 23%; purity, 95.1%.
(S)-N-(3-Amino-1-(4-fluorophenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14g)
1H NMR (DMSO-d6, 500 MHz) δ 13.36–13.04 (m, 1H), 8.37 (br s, 1H), 8.13–7.83 (m, 1H), 7.65–7.55 (m, 2H), 7.37–7.21 (m, 6H), 7.06 (t, J = 8.9 Hz, 2H), 6.76 (br s, 1H), 5.28 (q, J = 7.6 Hz, 1H), 2.51–2.48 (m, 2H); ESI-MS m/z 353.4 [M + H]+; yield 29%; purity, 98.2%.
(S)-N-(3-Amino-1-(2-methoxyphenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14h)
1H NMR (DMSO-d6, 500 MHz) δ 13.33–13.07 (m, 1H), 8.18–7.84 (m, 2H), 7.65–7.56 (m, 2H), 7.37–7.22 (m, 3H), 7.21–7.08 (m, 3H), 6.89 (d, J = 8.1 Hz, 1H), 6.81 (t, J = 7.3 Hz, 1H), 6.74 (br s, 1H), 5.54–5.48 (m, 1H), 3.71 (s, 3H), 2.48–2.45 (m, 2H); ESI-MS m/z 365.2 [M + H]+; yield 13%; purity, 95.7%.
(S)-N-(3-Amino-1-(3-methoxyphenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14i)
1H NMR (DMSO-d6, 500 MHz) δ 13.33–13.05 (m, 1H), 8.33 (br dd, J = 8.4, 14.6 Hz, 1H), 8.11–7.84 (m, 1H), 7.66–7.56 (m, 2H), 7.36–7.21 (m, 4H), 7.15 (t, J = 7.8 Hz, 1H), 6.88–6.70 (m, 4H), 5.27 (q, J = 7.5 Hz, 1H), 3.66 (s, 3H), 2.50–2.47 (m, 2H); ESI-MS m/z 365.4 [M + H]+; yield 16%; purity, 94.9%.
(S)-N-(3-Amino-1-(4-methoxyphenyl)-3-oxopropyl)-3-phenyl-1H-pyrazole-4-carboxamide (14j)
1H NMR (DMSO-d6, 500 MHz) δ 13.36–13.00 (m, 1H), 8.28 (br d, J = 8.2 Hz, 1H), 8.15–7.81 (m, 1H), 7.60 (br s, 2H), 7.37–7.17 (m, 6H), 6.82–6.77 (m, 2H), 6.72 (br s, 1H), 5.24 (q, J = 7.5 Hz, 1H), 3.65 (s, 3H), 2.49–2.46 (m, 2H); ESI-MS m/z 365.3 [M + H]+; yield 33%; purity, 95.0%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-5-methyl-1H-pyrazole-4-carboxamide (15a)
1H NMR (DMSO-d6, 500 MHz) δ 12.81–12.59 (m, 1H), 8.22–7.81 (m, 2H), 7.32–7.20 (m, 5H), 7.17–7.11 (m, 1H), 6.75 (br s, 1H), 5.29 (q, J = 7.5 Hz, 1H), 2.52 (br d, J = 6.7 Hz, 2H), 2.32–2.21 (m, 3H); ESI-MS m/z 273.4 [M + H]+; yield 28%; purity, 96.9%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(2-chlorophenyl)-1H-pyrazole-4-carboxamide (15b)
1H NMR (DMSO-d6, 500 MHz) δ 13.22 (br s, 1H), 8.16 (br s, 2H), 7.45–7.17 (m, 9H), 7.13 (br d, J = 5.7 Hz, 1H), 6.72 (br s, 1H), 5.19 (q, J = 7.4 Hz, 1H), 2.47 (br d, J = 6.4 Hz, 2H); ESI-MS m/z 369.4 [M + H]+; yield 20%; purity, 97.2%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(3-chlorophenyl)-1H-pyrazole-4-carboxamide (15c)
1H NMR (DMSO-d6, 500 MHz) δ 13.57–13.21 (m, 1H), 8.51 (br d, J = 8.2 Hz, 1H), 8.31–7.77 (m, 2H), 7.67 (br s, 1H), 7.44–7.28 (m, 7H), 7.25–7.20 (m, 1H), 6.83 (br s, 1H), 5.38 (q, J = 7.5 Hz, 1H), 2.59 (br d, J = 7.3 Hz, 2H); ESI-MS m/z 369.3 [M + H]+; Enamine; purity, 97.4%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(4-chlorophenyl)-1H-pyrazole-4-carboxamide (15d)
1H NMR (DMSO-d6, 500 MHz) δ 13.50–13.21 (m, 1H), 8.53–8.46 (m, 1H), 8.28–7.95 (m, 1H), 7.80–7.45 (m, 3H), 7.41–7.29 (m, 6H), 7.26–7.20 (m, 1H), 6.84 (br s, 1H), 5.38 (q, J = 7.5 Hz, 1H), 2.58 (br d, J = 7.2 Hz, 2H); ESI-MS m/z 369.3 [M + H]+; Enamine; purity, 97.9%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (15e)
1H NMR (DMSO-d6, 500 MHz) δ 13.43–13.22 (m, 1H), 8.44–8.00 (m, 2H), 7.43 (br s, 2H), 7.34–7.12 (m, 8H), 6.80 (br s, 1H), 5.30 (q, J = 7.3 Hz, 1H), 2.56 (br d, J = 6.8 Hz, 2H); ESI-MS m/z 353.5 [M + H]+; Enamine; purity, 99.7%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(3-fluorophenyl)-1H-pyrazole-4-carboxamide (15f)
1H NMR (DMSO-d6, 500 MHz) δ 13.52–13.10 (m, 1H), 8.45 (br d, J = 7.7 Hz, 1H), 8.22–7.86 (m, 1H), 7.55–7.43 (m, 2H), 7.32–7.20 (m, 6H), 7.18–7.03 (m, 2H), 6.75 (br s, 1H), 5.30 (q, J = 7.5 Hz, 1H), 2.51 (br dd, J = 3.5, 6.8 Hz, 2H); ESI-MS m/z 353.2 [M + H]+; yield 26%; purity, 95.3%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(4-fluorophenyl)-1H-pyrazole-4-carboxamide (15g)
1H NMR (DMSO-d6, 500 MHz) δ 13.44–13.12 (m, 1H), 8.45 (br d, J = 8.1 Hz, 1H), 8.26–7.92 (m, 1H), 7.75 (br s, 2H), 7.38–7.29 (m, 5H), 7.26–7.11 (m, 3H), 6.84 (br s, 1H), 5.37 (q, J = 7.5 Hz, 1H), 2.60–2.56 (m, 2H); ESI-MS m/z 353.3 [M + H]+; Enamine; purity, 97.5%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(4-methoxyphenyl)-1H-pyrazole-4-carboxamide (15h
1H NMR (DMSO-d6, 500 MHz) δ 13.27–13.03 (m, 1H), 8.41–8.34 (m, 1H), 8.18–7.88 (m, 1H), 7.69–7.59 (m, 2H), 7.38–7.28 (m, 5H), 7.25–7.19 (m, 1H), 7.00–6.81 (m, 3H), 5.36 (q, J = 7.6 Hz, 1H), 3.78 (br d, J = 10.0 Hz, 3H), 2.57 (br d, J = 6.8 Hz, 2H); ESI-MS m/z 365.4 [M + H]+; Enamine; purity, 97.8%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(3-cyanophenyl)-1H-pyrazole-4-carboxamide (15i)
1H NMR (DMSO-d6, 500 MHz) δ 13.61–13.36 (m, 1H), 8.54 (br d, J = 7.8 Hz, 1H), 8.33–7.95 (m, 3H), 7.92–7.74 (m, 1H), 7.67–7.52 (m, 1H), 7.41–7.29 (m, 5H), 7.26–7.20 (m, 1H), 6.83 (br s, 1H), 5.39 (q, J = 7.6 Hz, 1H), 2.59 (br d, J = 7.2 Hz, 2H); ESI-MS m/z 360.5 [M + H]+; Enamine; purity, 95.1%.
(S)-N-(3-Amino-3-oxo-1-phenylpropyl)-3-(3-chloro-4-fluorophenyl)-1H-pyrazole-4-carboxamide (15j)
1H NMR (DMSO-d6, 500 MHz) δ 13.31 (br s, 1H), 8.45 (br d, J = 8.2 Hz, 1H), 8.25–8.02 (m, 1H), 7.91 (br d, J = 6.8 Hz, 1H), 7.83–7.61 (m, 1H), 7.42–7.20 (m, 6H), 7.18–7.13 (m, 1H), 6.76 (br s, 1H), 5.31 (q, J = 7.4 Hz, 1H), 2.51 (br d, J = 6.1 Hz, 2H); ESI-MS m/z 387.3 [M + H]+; yield 26%; purity, 96.0%.
(R,E)-N-(3,4-Dichlorobenzylidene)-2-methylpropane-2-sulfinamide (17a)
62% yield; ESI-MS m/z 278.1 [M + H]+.
(R,E)-N-(3-Chloro-4-fluorobenzylidene)-2-methylpropane-2-sulfinamide (17b)
51% yield; ESI-MS m/z 262.1 [M + H]+.
Ethyl (S)-3-(((R)-tert-Butylsulfinyl)amino)-3-(3,4-dichlorophenyl) Propanoate (18a)
28% yield; ESI-MS m/z 366.2 [M + H]+.
Ethyl (S)-3-(((R)-tert-Butylsulfinyl)amino)-3-(3-chloro-4-fluorophenyl) Propanoate (18b)
59% yield; ESI-MS m/z 350.2 [M + H]+.
(S)-3-(((R)-tert-Butylsulfinyl)amino)-3-(3,4-dichlorophenyl) Propanamide (19a)
66% yield; ESI-MS m/z 337.2 [M + H]+.
(S)-3-(((R)-tert-Butylsulfinyl)amino)-3-(3-chloro-4-fluorophenyl) Propanamide (19b)
93% yield; ESI-MS m/z 321.1 [M + H]+.
Ethyl 5-Bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylate (21)
Ethyl 5-bromo-1H-pyrazole-4-carboxylate (5.0 g, 23 mmol) was added to a stirring suspension of sodium hydride (0.68 g, 28 mmol, 60% dispersion) in THF (40 mL) at 0 °C. After stirring for 45 min, (2-(chloromethoxy)ethyl)trimethylsilane (5.1 mL, 27 mmol) was added dropwise, and the solution was allowed to warm to room temperature. After stirring for 48 h, the reaction mixture was partitioned between DCM (200 mL) and water (100 mL), and the layers were separated. The aqueous layer was extracted with DCM (100 mL), and the combined organics were dried over sodium sulfate and concentrated to dryness. The residue was purified by flash chromatography on silica gel (0–25% EtOAc in hexanes) to afford ethyl 5-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylate (5.3 g, 67%) as a mixture of regioisomers. ESI-MS m/z 349.1 [M + H]+.
Ethyl 5-(2-Fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylate (23a)
95% yield; ESI-MS m/z 365.1 [M + H]+.
Ethyl 5-(2,4-Difluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylate (23b)
95% yield; ESI-MS m/z 383.3 [M + H]+.
Ethyl 5-(4-Chloro-2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylate (23c)
86% yield; ESI-MS m/z 399.1 [M + H]+.
5-(2-Fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylic Acid (24a)
81% yield; ESI-MS m/z 335.2 [M – H]−.
5-(2,4-Difluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylic Acid (24b)
79% yield; ESI-MS m/z 353.1 [M – H]−.
5-(4-Chloro-2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxylic Acid (24c)
76% yield; ESI-MS m/z 369.1 [M – H]−.
(S)-N-(3-Amino-1-(3-chloro-4-fluorophenyl)-3-oxopropyl)-5-(2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxamide (25a)
95% yield; ESI-MS m/z 535.1 [M + H]+.
(S)-N-(3-Amino-1-(3,4-dichlorophenyl)-3-oxopropyl)-5-(2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxamide (25b)
95% yield; ESI-MS m/z 551.3 [M + H]+.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-5-(2,4-difluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxamide (25c)
95% yield; ESI-MS m/z 535.4 [M + H]+.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-5-(4-chloro-2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxamide (25d)
95% yield; ESI-MS m/z 551.2 [M + H]+.
(S)-N-(3-Amino-1-(3-chloro-4-fluorophenyl)-3-oxopropyl)-3-(4-chloro-2-fluorophenyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-4-carboxamide (25e)
95% yield; ESI-MS m/z 569.2 [M + H]+.
(S)-N-(3-Amino-1-(3-chloro-4-fluorophenyl)-3-oxopropyl)-5-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (26a)
1H NMR (DMSO-d6, 500 MHz) δ 13.64–13.04 (m, 1H), 8.38 (br s, 1H), 8.14 (br s, 1H), 7.49 (dd, J = 2.0, 7.2 Hz, 1H), 7.47–7.40 (m, 2H), 7.38–7.28 (m, 3H), 7.26–7.16 (m, 2H), 6.85 (br s, 1H), 5.27 (q, J = 7.5 Hz, 1H), 2.57 (br dd, J = 1.9, 7.3 Hz, 2H); ESI-MS m/z 405.2 [M + H]+; yield 9%; purity, 96.2%.
(S)-N-(3-Amino-1-(3,4-dichlorophenyl)-3-oxopropyl)-5-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (26b)
1H NMR (DMSO-d6, 500 MHz) δ 13.39–13.20 (m, 1H), 8.42–8.26 (m, 1H), 8.24–7.92 (m, 1H), 7.51–7.46 (m, 2H), 7.43–7.26 (m, 3H), 7.24–7.04 (m, 3H), 6.79 (br s, 1H), 5.21–5.15 (m, 1H), 2.49 (br d, J = 6.2 Hz, 2H); ESI-MS m/z 421.2 [M + H]+; yield 8%; purity, 95.1%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-5-(2,4-difluorophenyl)-1H-pyrazole-4-carboxamide (26c)
1H NMR (DMSO-d6, 500 MHz) δ 13.32 (br s, 1H), 8.36–8.18 (m, 1H), 7.40 (br s, 1H), 7.29–7.23 (m, 3H), 7.22–7.18 (m, 2H), 7.05 (br s, 1H), 6.77 (br s, 1H), 5.21 (q, J = 7.4 Hz, 1H), 2.49 (br d, J = 7.3 Hz, 2H); ESI-MS m/z 405.2 [M + H]+; yield 7%; purity, 97.6%.
(S)-N-(3-Amino-1-(3-chlorophenyl)-3-oxopropyl)-5-(4-chloro-2-fluorophenyl)-1H-pyrazole-4-carboxamide (26d)
1H NMR (DMSO-d6, 500 MHz) δ 13.46–13.24 (m, 1H), 8.33 (br s, 1H), 8.27–7.94 (m, 1H), 7.38 (br s, 1H), 7.31–7.17 (m, 6H), 6.77 (br s, 1H), 5.21 (q, J = 7.5 Hz, 1H), 2.49 (br dd, J = 3.2, 7.3 Hz, 2H); ESI-MS m/z 421.2 [M + H]+; yield 8%; purity, 99.1%.
(S)-N-(3-Amino-1-(3-chloro-4-fluorophenyl)-3-oxopropyl)-3-(4-chloro-2-fluorophenyl)-1H-pyrazole-4-carboxamide (26e)
1H NMR (DMSO-d6, 500 MHz) δ 13.54–13.30 (m, 1H), 8.40 (br s, 1H), 8.34–8.01 (m, 1H), 7.52–7.43 (m, 2H), 7.42–7.25 (m, 5H), 6.85 (br s, 1H), 5.27 (q, J = 7.5 Hz, 1H), 2.57 (br dd, J = 2.8, 7.2 Hz, 2H); 13C NMR (DMSO-d6, 176 MHz) δ ppm 171.6, 161.9, 160.0, 155.8, 141.4, 132.9, 130.8, 129.0, 127.8, 124.6, 119.4, 116.9, 116.3, 49.4, 42.0; ESI-MS m/z 439.3 [M + H]+; yield 18%; purity, 98.8%.
DEL Screening and Compound Ordering
The affinity-mediated selections included a pool of 59 DNA-encoded libraries as described in Cuozzo et al.28 Purified protein (3.8 μM) DCAF1 (1038–1400) containing an N-terminal His6-tag was incubated in a solution with DNA-encoded library (40 μM) for1 h in a volume of 60 μL in 1× selection buffer, which consisted of HEPES (20 mM), potassium acetate (134 mM), sodium acetate (8 mM), sodium chloride (4 M), magnesium acetate (0.8 mM), 0.02% Tween20, sheared salmon sperm DNA (1 mg/mL, Invitrogen AM9680), imidazole (5 mM), and TCEP (1 mM) at pH 7.2. An additional selection condition containing the target and 100 μM of the Vpx peptide, as a competitor to possible compounds binding in the peptide binding site, was run in parallel. The Vpx peptide was preincubated with the DCAF1 target in 1× selection buffer for 0.5 h prior to addition of the DNA-encoded library. An additional selection condition containing no target was run in parallel to evaluate matrix binders. For each selection condition (no target, target, or target with competitor), a separate ME200 tip (Phynexus) containing 5 μL of nickel affinity matrix was prewashed three times in 200 μL of fresh 1× selection buffer. The affinity matrix used was cOmplete His-Tag Purification Resin (Sigma 5893682001). Each selection was separately captured with 20 passages over the ME200 tip for a total of 0.5 h. The bound protein/library captured on the ME200tip was washed eight times with 200 μL of, fresh 1× selection buffer. Bound library members were eluted by incubating the ME200tip with 60 μL of 1× fresh selection buffer at 85 °C for 5 min. The solution from the heat elution was incubated with 20 passages over a fresh, prewashed ME200 tip containing 5 μL of nickel affinity matrix to remove any eluted protein. This selection process was run for a second time using the eluate of the first selection in place of the input DNA-encoded library and using no target, fresh target, or fresh target with Vpx peptide. The eluate of the second round of selection was PCR amplified and sequenced as described in Cuozzo et al.
Sequence data were parsed, error-containing sequences were disregarded, amplification duplicates were removed, and building block and chemical scheme encodings were decoded and reported along with associated calculated statistical parameters. Significantly enriched instances from two- and three-cycle libraries were called into groupings based on similar chemical features to obtain 98 families. Up to 10 instances (fully enumerated structure and possible truncates) per family were used to structure similarity search the commercially available EnamineReal space (720 million, downloaded in 2018) applying a 2D Tanimoto ≥0.4 distance cutoff to obtain a representative set of potentially purchasable compounds. Duplicate structures were removed, and up to two identified commercial compounds per family were further evaluated by inspection (chirality check that matches building block chirality and deprioritizing compounds that resembled low count instances) to arrive at 40 EnamineReal diverse compounds, which were requested for synthesis from Enamine (Kyiv, Ukraine). Thirty-three compounds were successfully prepared and were submitted for assay evaluation. The DNA-encoded library selection data were also processed, as described in McCloskey et al., to obtain DCAF1 binding RF and GCNN models, used to score commercial compounds from EnamineReal, MCULE, and enumerated virtual libraries to support hit finding and hit expansion SAR workflows.
Protein Expression and Purification Used in Biophysical Characterization
The DNA fragment encoding human DCAF1 residues 1038–1400 was amplified by PCR and subcloned into pFBOH-MHL (derivative of the pFBOH-LIC vector, SGC). The resulting plasmid was transformed into DH10Bac Competent E. coli (Invitrogen), and recombinant viral DNA bacmid was purified and followed by a recombinant baculovirus expression in Sf9 insect cells. Cells were harvested and lysed. Protein was purified through a pre-equilibrated Hispur Ni-NTA resin (Thermo Scientific) column and by gel filtration on a Superdex200 26/600 using an ÄKTA PURE (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 5% glycerol, and 2 mM 2-mercaptoethanol. The purity of the fractions was confirmed on SDS-PAGE gels, and the pure fractions were pooled, concentrated, and flash frozen. Proteins were stored at −80 °C.
Differential Scanning Fluorimetry Assay
DSF experiments were performed using a Roche LightCycler 480 II in sealed Axygen 384-well plates (PCR-384-LC480). Reactions (20 μL) contained 0.1 mg/mL DCAF1 (1038–1400 aa) in 100 mM Hepes pH 7.5 and 150 mM NaCl, and SYPRO Orange (5000× SYPRO Orange stock in DMSO, Invitrogen, Cat # S6650). Compounds were tested at 200 μM with a final concentration of 1% DMSO. The initial hit compound was serially diluted to a final concentration of 400 to 25 μM with a final concentration of 2% DMSO. Thermal denaturation was monitored from 20 to 95 °C at a rate of 4 °C increase per minute, and data points were collected at 0.5 °C intervals.
Surface Plasmon Resonance
SPR studies were performed using a Biacore T200 (GE Health Sciences Inc.) at 20 °C. Biotinylated DCAF1 (1038–1400 aa) was captured onto two flow cells of a streptavidin-conjugated SA chip (according to manufacturer’s protocol) achieving 4000–7000 response units (RU), whereas another flow cell was left empty for reference subtraction. Biotinylated WDR5 was captured on another flow cell as a negative control protein. Compounds were dissolved in 100% DMSO (20 mM stock) and diluted to working concentrations in 100% DMSO before threefold serial dilutions prepared in buffer with DMSO. For SPR analysis, compounds were diluted in HBS-EP buffer (10 mM Hepes pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% Tween-20), giving a final concentration of 0.5 or 1% DMSO. Kinetic determination experiments were performed using single cycle kinetics with an on time of 60 s, off time of 120 s, and flow rate of 75 μL/min. Kinetic curve fittings and Kd calculations were done with a 1:1 binding model and the Biacore T200 Evaluation software (GE Health Sciences Inc.).
Isothermal Titration Calorimetry
Purified DCAF1 was dialyzed and diluted in HBS-EP buffer. Compounds (0.5 mM) were injected into the sample cell containing 40 μM DCAF1 with 2.5% DMSO. ITC titrations were performed on a Nano ITC from TA Instruments (New Castle, DE) at 25 °C by using 2 μL injections with a total of 24 injections. The final DMSO concentration of the DCAF1 solution was adjusted to 2.5%. Kd values of compound 3d were obtained from three independent experiments. Compound 26e (0.2 mM) was injected into the sample cell containing 20 μM DCAF1 with 2.0% DMSO. ITC titrations were performed on an Affinity ITC from TA Instruments at 25 °C by using 2 μL injections with a total of 24 injections. The final DMSO concentration of the DCAF1 solution was adjusted to 2.0%. Kd values of compound 26e were obtained from three independent experiments. Data were fitted with a one-binding-site model using the Nano Analyze software.
Molecular Modeling
Analysis of X-ray Structure
The analysis of X-ray structures was done using both Pymol and Maestro software from Schrodinger. Low energy conformations for ligands were generated using the LigPrep tool within the Maestro software. The Glide docking tool from Schrodinger was used for the docking simulations with default parameters.
Hit-to-Lead
The SeeSAR software from BioSolveIT was used in the hit-to-lead stage. It relies on the HYDE35 scoring function to evaluate atom-based affinity contributions to the binding energy. This is particularly interesting to predict the impact (favorable, neutral, or detrimental) of a given modification. As a preliminary check, SeeSAR was successfully challenged to explain the main activity cliffs from the known SAR from a 3D point of view. Following this positive control stage, a systematic exploration of the chemical space around the compound series was performed from an in silico point of view. This was achieved by plugging in a dictionary of commonly used organic modifications (functional groups and decorated rings with varied spacer lengths) at varied positions of the compound series (ortho/meta/para-positions of both southern and northern rings, replacement of the side chain amide). The designed libraries were subsequently postprocessed to discard compounds with undesired substructures or with unreasonable physicochemical properties. Besides, proper microspecies and tautomers were also generated using LigPrep. Finally, a single 3D conformer was generated for each kept compound and superimposed onto the experimental binding mode of the reference compound 3d. Finally, the designed compounds were directly evaluated in the SeeSAR software using the template-based docking mode (with compound 3d as the reference ligand). Several parameters were used to prioritize the compounds to be synthesized (HYDE evaluation, visual inspection of predicted binding modes, quality of torsion angles, absence of inter/intra-clashes, and physicochemical properties such as logP).
Crystallization and Structure Determination
DCAF1 WDR Domain Gene Cloning, Protein Expression, and Purification
The WDR domain (residues 1077–1390) of the human DCAF1 gene (UniProtKB Q9Y4B6) was cloned into an in-house insect cell expression vector pFBOH-MHL containing an N-terminal His6-tag followed by a TEV cleavage site. Site-directed mutagenesis was then carried out to change residues 1077 (Phe) and 1079 (Arg) to alanine. The resultant construct was used for protein expression in a baculovirus-Sf9 expression system following an optimized protocol as described by Hutchinson and Seitova.36 The cells were harvested by centrifugation, resuspended in a lysis buffer (50 mM Tris–HCl pH 7.5, 5% glycerol, 0.4 M NaCl, 0.1% NP40, protease inhibitor cocktail, and benzonase endonuclease), and lysed by sonication. The cell lysate was then centrifuged, and the supernatant was filtered prior to purification.
The filtered cell-free extract was preincubated with TALON immobilized cobalt affinity resin equilibrated with the binding buffer containing 50 mM Tris–HCl pH 7.5, 5% glycerol, and 0.4 M NaCl prior to loading onto an open column. The column was washed with binding buffer and then with the same buffer containing 5 and 10 mM imidazole before elution with a buffer containing 250 mM imidazole. The eluted protein was then dialyzed overnight into 50 mM Tris–HCl pH 7.5, 0.4 M NaCl, 5% glycerol, and 10 mM β-mercaptoethanol in the presence of TEV protease to remove the polyhistidine (His6) tag. The following day, the dialyzed protein was passed through the TALON cobalt affinity resin in an open column, and the flow-through (cleaved protein) was collected.
The collected protein sample was further purified by size exclusion chromatography using a HiLoad 26/600 Superdex 200 column (GE Healthcare) on an AKTA Pure chromatography system (GE Healthcare) with a running buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, and 1 mM TCEP. The protein purity was assessed on an SDS-PAGE gel, and the pure peak fractions were then pooled and concentrated using 10 kDa cutoff centrifugal filters (Millipore). The protein concentration was determined using Nanodrop (Thermo Scientific) and calculated using the DCAF1 WDR protein extinction coefficient of 35,410 M–1 cm–1 (calculated from the amino acid sequence using Expasy ProtParam (https://web.expasy.org/protparam/)). The concentrated protein was then flash-frozen in liquid nitrogen before storage at −80 °C.
Protein Crystallization
We first attempted to co-crystallize the longer DCAF1 WDR domain construct (1061–1399 residues) as reported in the deposited APO structure (PDB ID: 4PXW), but crystallization was not reproducible. Also, the N-terminal region outside the WDR ring was not visible in the apo structure, so we considered designing a shorter construct for crystallization trials covering the WDR domain only, which generated robust and well-diffracting crystals. To generate DCAF1-3d and DCAF1-26e co-crystals, the protein (in 20 mM Tris–HCl pH 7.5, 150 mM NaCl, and 1 mM TCEP) at 10 mg/mL (0.2812 mM) was preincubated at room temperature with 4.218 mM (15 times molar excess) 3d and 26e compounds prior to crystallization. Crystallization screenings were performed using a Phenix drop dispensing robot (Art Robbins) with 0.5 μL of protein–compound solution and 0.5 μL of screen solution over a 90 μL reservoir in 96-well vapor diffusion sitting-drop plates. Diamond-shaped crystals were obtained after 72 h at 18 °C in a precipitant solution containing 20% PEG3350 and 0.2 M potassium fluoride for the DCAF1-3d and 20% PEG3350 and 0.2 M di-ammonium citrate for the DCAF1-26e.
Diffraction Data Collection, Structure Determination, and Refinement
Crystals were mounted in a nylon loop and cryoprotected in the reservoir solution supplemented with 10% ethylene glycol and 2 mM compound before flash-freezing in liquid nitrogen. Diffraction data were collected on beamline 24-ID-C at the Advanced Photon Source in Argonne National Laboratory. For the DCAF1-3d, the images were processed using XDS37 and Aimless,38 and the structure was phased by molecular replacement using Phaser39 and the DCAF1 apo crystal structure (PDB ID: 4PXW) as a starting model. On the other hand, the DCAF1-26e images were processed using HKL3000,40 and the structure was phased by molecular replacement using the DCAF1-3d (PDB: 7UFV) as a starting model. Following molecular replacement, the structures were refined using iterative cycles of manual model building in Coot41 and refinement in REFMAC542 within the CCP4 crystallography suite.43 The PDB and CIF files of the 3d and 26e compounds were generated using the Grades global phasing server (http://grade.globalphasing.org/cgi-bin/grade/server.cgi). Model validation was performed using the Molprobity44 server, the structures were analyzed using PoseView45 and UCSF Chimera,46 and the graphical images were rendered using PyMOL.47 The structural data for DCAF1-3d and DCAF1-26e were deposited in the Protein Data Bank (PDB) under the accession codes 7UFV and 8F8E, respectively.
Generation of HiBiT-DCAF1 NCI-H460 Cells
The WD40 domain (1038-1400aa) of DCAF1 with N-terminal 3x-flag and HiBiT tags (Flag-Hb-DCAF1_WD) was synthesized and inserted into the pDONR221 plasmid by GeneArt (Invitrogen). The construct was cloned into a pLV series lentiviral transfer plasmid containing a CMV promoter for expression and blasticidin selectable marker. Lentivirus infections were performed by treating one well of a six-well plate containing 100,000 parental NCI-H460 cells (ATCC) with 20 μL of concentrated virus followed by 2 days of incubation (37 °C, 5% CO2) without removing the virus. The viral medium was replaced with fresh medium containing Blasticidin (InvivoGen; ANT-BL-1) to select and generate NCI-H460 cell lines stably expressing Flag-Hb-DCAF1_WD. The expression of the WD40 domain was confirmed via western blot. NCI-H460 cells stably expressing the full-length DCAF1 (Flag-Hb-DCAF1_FL) were also generated.
HiBit-DCAF1 Cellular Thermal Shift Assay (CETSA)
NCI-H460 cells expressing Flag-Hb-DCAF1_WD or Flag-Hb-DCAF1_FL were seeded at 10,000 cells/50 μL (RPMI+10% FBS) per well of a 96-well black PCR plate. For the thermal shift assay, HP D300e digital dispenser was used to spray DMSO and 40 μM of 26e or 3d, and the plate was mixed using an orbital shaker for 15 s. Cells were incubated at 37 °C and 5% CO2 for 1 h and then heated for 3.5 min at different temperatures using C1000 Touch Thermal Cycler (Bio-Rad; 1851196). For the CETSA dose–response assay, an HP D300e digital dispenser sprayed 12 different compound concentrations (40 to 0.2 μM) of 26e or 3d onto cells that were heated at one fixed 61 °C temperature for 3.5 min. An equal volume of NanoGlo HiBiT Lytic (Promega), containing both LgBiT protein and furimazine substrate, was added to the cells. Together, the HiBiT and the LgBiT protein formed a luminescent nanoluciferase enzyme, which emits light in the presence of the furimazine substrate. The luminesce signal is proportional to the unaggregated HiBiT tagged protein. The plate was mixed on an orbital shaker (500 rpm) for 10 min, and a Cytation 3 (BioTek) plate reader measured the emitted luminescence signal.
Quantitative Protein Expression Using Jess (ProteinSimple) to Detect Unaggregated Protein
One million cells/100 μL (RPMI + 10% FBS) of NCI-H460 cells expressing the HiBiT-tagged protein of interest were seeded per well of a 96-well TC plate. For thermal shift assay, an HP D300e digital dispenser sprayed DMSO or 40 μM of 26e, and the plate was mixed using an orbital shaker for 15 s. Cells were incubated at 37 °C and 5% CO2 for 1 h. Cells were then transferred to 1.5 mL Eppendorf tubes and spun at 500g for 5 min. The medium was aspirated, and cells were resuspended in a 100 μL of PBS with protease inhibitor. Cells were then transferred to PCR strip tubes and heated for 3.5 min at 12 different temperatures (42 to 64 °C with increments of 2 °C) using a Veriti 96-Well Fast Thermal Cycler (Applied Biosystems; 9902). Liquid nitrogen was used to freeze–thaw the cells twice, and then the cell lysate was centrifuged at 20,000g for 20 min at 4 °C. The supernatant with the unaggregated soluble protein was transferred into new 1.5 mL Eppendorf tubes. For the CETSA dose–response assay, the HP D300e digital dispenser sprayed DMSO or 12 different compound concentrations (40 to 0.2 μM) of 26e. Cells were then heated at one fixed 61 °C temperature. The Jess (ProteinSimple; JS-4296), which automates traditional western blot, was used to detect and quantify the N-flag tagged DCAF1 WD40 domain.
Acknowledgments
We would like to thank Yanjun Li, Peter Loppnau, Almagul Seitova, Ashley Hutchinson, Pegah Ghiabi, and Taraneh Hajian for gene cloning, protein expression, and purification; Yelena Arnautova for X-ray structure evaluation and computational modeling; Antoine Dumas for structure enumeration; Surayya Sana for compound logistics; Enamine and MCULE for compound synthesis; and Google Accelerated Science for GCNN modeling. We acknowledge the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Structural Genomics Consortium is a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer, and Takeda. Funding for the Ontario Institute for Cancer Research is provided by the Government of Ontario. Héctor González-Álvarez received a scholarship from the Mexican National Council on Science and Technology (CONACyT).
Glossary
Abbreviations
- BRD4
bromodomain-containing protein 4
- CDC40
cell division cycle 40
- CETSA
cellular thermal shift assay
- CRBN
cereblon
- CRL
cullin-RING ligase
- CUL
cullin
- DCAF
DDB1-Cul4-associated factor
- DDB1
DNA damage binding protein 1
- DEL
DNA-encoded library
- DIEA
N,N-diisopropylethylamine
- DSF
differential scanning fluorimetry
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EDVP
EED-DDB1-VprBP
- FKBP12
12 kDa FK506-binding protein
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- HECT
homologous to the E6-AP carboxyl terminus
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- Lats
large tumor suppressor kinase
- LID
ligand interaction diagram
- PROTACs
proteolysis targeting chimeras
- RBR
ring between ring finger
- RING
really interesting new gene
- SAMHD1
sterile alpha-motif and histidine-aspartate domain-containing protein 1
- SD
standard deviation
- SPR
surface plasmon resonance
- Tm
melting temperature
- UNG2
uracil DNA glycosylase 2
- VHL
von Hippel–Lindau
- Vpr
viral protein R
- Vpx
viral protein X
- WDR
WD repeat
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c02132.
Author Contributions
& A.S.M.L., S.K., and B.W. contributed equally.
The authors declare the following competing financial interest(s): J.P.G, Y.Z., M.A.G., A.D.K. are employees of X-Chem. M.v.R., J.S.D, B.L.S. and J.W.C. are or were (C.J.M.) employees of Relay Therapeutics and were employees of X-Chem at the time of writing. Employees and past employees may hold stocks and shares. All other authors declare no conflict of interest.
Notes
J.P.G, Y.Z., M.A.G., and A.D.K. are employees of X-Chem. M.v.R., J.S.D., B.L.S., and J.W.C. are or were (C.J.M.) employees of Relay Therapeutics and were employees of X-Chem at the time of writing. Employees and past employees may hold stocks and shares.
Notes
Authors will release the atomic coordinates upon article publication.
Supplementary Material
References
- Khan S.; He Y.; Zhang X.; Yuan Y.; Pu S.; Kong Q.; Zheng G.; Zhou D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. 10.1038/s41388-020-1336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalawansha D. A.; Crews C. M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N.; Shabek N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- Zou Y.; Ma D.; Wang Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T.; Ciulli A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discovery 2021, 26, 484–502. 10.1177/2472555220965528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K.; Scott D. C.; Schulman B. A. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr. Opin. Struct. Biol. 2021, 67, 101–109. 10.1016/j.sbi.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Song X.; Wavelet C. M.; Wan Y. Cullin 4-DCAF Proteins in Tumorigenesis. Adv. Exp. Med. Biol. 2020, 1217, 241–259. 10.1007/978-981-15-1025-0_15. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S.; Li T.; Yi X.; MacCoss M. J.; Moon R. T.; Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Jin J.; Arias E. E.; Chen J.; Harper J. W.; Walter J. C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 2006, 23, 709–721. 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- He Y. J.; McCall C. M.; Hu J.; Zeng Y.; Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006, 20, 2949–2954. 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Feng Y.; Narayan O.; Zhao L. J. Cytoplasmic retention of HIV-1 regulatory protein Vpr by protein-protein interaction with a novel human cytoplasmic protein VprBP. Gene 2001, 263, 131–140. 10.1016/S0378-1119(00)00583-7. [DOI] [PubMed] [Google Scholar]
- Tan L.; Ehrlich E.; Yu X. F. DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 2007, 81, 10822–10830. 10.1128/JVI.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J.; Vu T.; Novince Z.; Guerrero-Santoro J.; Rapic-Otrin V.; Gronenborn A. M. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 2010, 285, 37333–37341. 10.1074/jbc.M110.133181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockow L. C.; Sharifi H. J.; Wen X.; Flagg M.; Furuya A. K. M.; Nekorchuk M.; de Noronha C. M. C. The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology 2013, 444, 191–202. 10.1016/j.virol.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Monnie C.; DeLucia M.; Ahn J. HIV-1 Vpr activates host CRL4-DCAF1 E3 ligase to degrade histone deacetylase SIRT7. Virol. J. 2021, 18, 48. 10.1186/s12985-021-01514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zhou X.; Barnes C. O.; DeLucia M.; Cohen A. E.; Gronenborn A. M.; Ahn J.; Calero G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23, 933–940. 10.1038/nsmb.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J.; Hao C.; Yan J.; DeLucia M.; Mehrens J.; Wang C.; Gronenborn A. M.; Skowronski J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 2012, 287, 12550–12558. 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Guo H.; Han X.; Liu X.; Zhou X.; Zhang W.; Yu X. F. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell. Microbiol. 2012, 14, 1745–1756. 10.1111/j.1462-5822.2012.01835.x. [DOI] [PubMed] [Google Scholar]
- Schabla N. M.; Mondal K.; Swanson P. C. DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J. Mol. Cell Biol. 2019, 11, 725–735. 10.1093/jmcb/mjy085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. Y.; Wang X.; Jun S.; Park J. I. Dyrk2-associated EDD-DDB1-VprBP E3 ligase inhibits telomerase by TERT degradation. J. Biol. Chem. 2013, 288, 7252–7262. 10.1074/jbc.M112.416792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain D.; Javadi Esfehani Y.; Das A.; Tsang W. Y. Cep78 controls centrosome homeostasis by inhibiting EDD-DYRK2-DDB1(Vpr)(BP). EMBO Rep. 2017, 18, 632–644. 10.15252/embr.201642377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain D.; Barbosa J. A. F.; Cohen É. A.; Tsang W. Y. HIV-1 Vpr hijacks EDD-DYRK2-DDB1(DCAF1) to disrupt centrosome homeostasis. J. Biol. Chem. 2018, 293, 9448–9460. 10.1074/jbc.RA117.001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T.; Mondal K.; Swanson P. C. VprBP (DCAF1): a promiscuous substrate recognition subunit that incorporates into both RING-family CRL4 and HECT-family EDD/UBR5 E3 ubiquitin ligases. BMC Mol. Biol. 2013, 14, 22. 10.1186/1471-2199-14-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Kim J. M.; Kim J. S.; Choi J.; Lee Y. S.; Neamati N.; Song J. S.; Heo K.; An W. VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription. Mol. Cell 2013, 52, 459–467. 10.1016/j.molcel.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghate N. B.; Kim S.; Spiller E.; Kim S.; Shin Y.; Rhie S. K.; Smbatyan G.; Lenz H. J.; Mumenthaler S. M.; An W. VprBP directs epigenetic gene silencing through histone H2A phosphorylation in colon cancer. Mol. Oncol. 2021, 15, 2801–2817. 10.1002/1878-0261.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey K.; Sigel E. A.; Kearnes S.; Xue L.; Tian X.; Moccia D.; Gikunju D.; Bazzaz S.; Chan B.; Clark M. A.; Cuozzo J. W.; Guié M.-A.; Guilinger J. P.; Huguet C.; Hupp C. D.; Keefe A. D.; Mulhern C. J.; Zhang Y.; Riley P. Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding. J. Med. Chem. 2020, 63, 8857–8866. 10.1021/acs.jmedchem.0c00452. [DOI] [PubMed] [Google Scholar]
- Cuozzo J. W.; Centrella P. A.; Gikunju D.; Habeshian S.; Hupp C. D.; Keefe A. D.; Sigel E. A.; Soutter H. H.; Thomson H. A.; Zhang Y.; Clark M. A. Discovery of a Potent BTK Inhibitor with a Novel Binding Mode by Using Parallel Selections with a DNA-Encoded Chemical Library. ChemBioChem 2017, 18, 864–871. 10.1002/cbic.201600573. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinn M. K.; Machleidt T.; Zimmerman K.; Eggers C. T.; Dixon A. S.; Hurst R.; Hall M. P.; Encell L. P.; Binkowski B. F.; Wood K. V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13, 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- Tao Y.; Remillard D.; Vinogradova E. V.; Yokoyama M.; Banchenko S.; Schwefel D.; Melillo B.; Schreiber S. L.; Zhang X.; Cravatt B. F. Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J. Am. Chem. Soc. 2022, 144, 18688–18699. 10.1021/jacs.2c08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Cooper J.; Zhou L.; Yang C.; Erdjument-Bromage H.; Zagzag D.; Snuderl M.; Ladanyi M.; Hanemann C. O.; Zhou P.; Karajannis M. A.; Giancotti F. G. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; You L.; Cooper J.; Schiavon G.; Pepe-Caprio A.; Zhou L.; Ishii R.; Giovannini M.; Hanemann C. O.; Long S. B.; Erdjument-Bromage H.; Zhou P.; Tempst P.; Giancotti F. G. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 2010, 140, 477–490. 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J.; Li W.; You L.; Schiavon G.; Pepe-Caprio A.; Zhou L.; Ishii R.; Giovannini M.; Hanemann C. O.; Long S. B.; Erdjument-Bromage H.; Zhou P.; Tempst P.; Giancotti F. G. Merlin/NF2 functions upstream of the nuclear E3 ubiquitin ligase CRL4DCAF1 to suppress oncogenic gene expression. Sci. Signaling 2011, 4, pt6. 10.1126/scisignal.2002314. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Lange G.; Hindle S.; Klein R.; Rarey M. A consistent description of HYdrogen bond and DEhydration energies in protein-ligand complexes: methods behind the HYDE scoring function. J. Comput.-Aided Mol. Des. 2013, 27, 15–29. 10.1007/s10822-012-9626-2. [DOI] [PubMed] [Google Scholar]
- Hutchinson A.; Seitova A. Production of Recombinant PRMT Proteins using the Baculovirus Expression Vector System. J. Visualized Exp. 2021, 173, e62510 10.3791/62510. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. III; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stierand K.; Maass P. C.; Rarey M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. 10.1093/bioinformatics/btl150. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.PyMOL; 2002.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.