

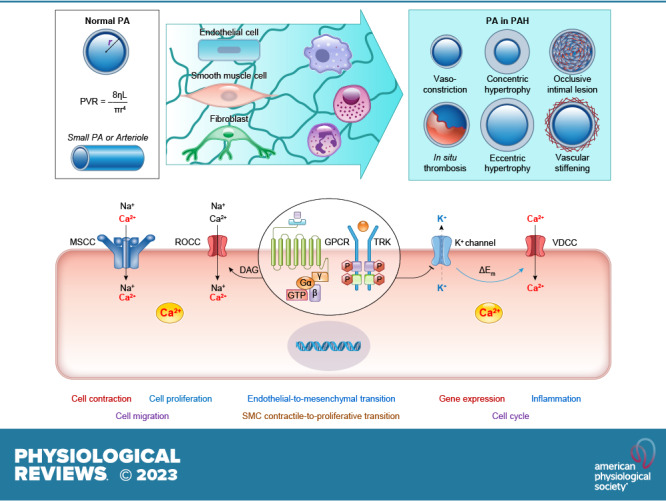
Keywords: Ca2+ signaling, ion channel, membrane receptor, pulmonary arterial hypertension, pulmonary circulation
Abstract
The pulmonary circulation is a low-resistance, low-pressure, and high-compliance system that allows the lungs to receive the entire cardiac output. Pulmonary arterial pressure is a function of cardiac output and pulmonary vascular resistance, and pulmonary vascular resistance is inversely proportional to the fourth power of the intraluminal radius of the pulmonary artery. Therefore, a very small decrease of the pulmonary vascular lumen diameter results in a significant increase in pulmonary vascular resistance and pulmonary arterial pressure. Pulmonary arterial hypertension is a fatal and progressive disease with poor prognosis. Regardless of the initial pathogenic triggers, sustained pulmonary vasoconstriction, concentric vascular remodeling, occlusive intimal lesions, in situ thrombosis, and vascular wall stiffening are the major and direct causes for elevated pulmonary vascular resistance in patients with pulmonary arterial hypertension and other forms of precapillary pulmonary hypertension. In this review, we aim to discuss the basic principles and physiological mechanisms involved in the regulation of lung vascular hemodynamics and pulmonary vascular function, the changes in the pulmonary vasculature that contribute to the increased vascular resistance and arterial pressure, and the pathogenic mechanisms involved in the development and progression of pulmonary hypertension. We focus on reviewing the pathogenic roles of membrane receptors, ion channels, and intracellular Ca2+ signaling in pulmonary vascular smooth muscle cells in the development and progression of pulmonary hypertension.
CLINICAL HIGHLIGHTS.
-
1)
The increased pulmonary arterial pressure (PAP) in patients with pulmonary arterial hypertension (PAH) is caused by elevated pulmonary vascular resistance (PVR). Regardless of the initial genetic and/or environmental trigger, sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, in situ thrombosis, occlusive intimal lesions, and vascular stiffening are the major causes that directly increase PVR and PAP in patients with PAH.
-
2)
The pulmonary artery wall is composed of three layers of cells: a) the intima formed by endothelial cells (ECs), b) the media formed by smooth muscle cells (SMCs), and c) the adventitia formed by fibroblasts (FBs). In addition, the pulmonary artery wall also includes the perivascular progenitor cells and extracellular matrix (ECM). All these cells contribute to the development of pulmonary vasculopathy in PAH.
-
3)
Sustained pulmonary vasoconstriction is caused by pulmonary arterial SMC (PASMC) contraction, whereas concentric vascular remodeling is caused by EC injury and PASMC/FB migration and proliferation. Vascular wall stiffness is increased by increased SMC contractility, SMC/FB migration/proliferation, and ECM remodeling, whereas occlusive vascular lesions contain all types of vascular cells.
-
4)
Membrane receptors, ion channels/transporters, and Ca2+ signaling cascades play a critical role in regulating cell contraction, migration, and proliferation. Activators and inhibitors of these receptors and ion channels are used for the treatment of PAH.
-
5)
This review aims to discuss the basic mechanisms involved in the regulation of normal pulmonary vascular function and structure as well as the pathogenic mechanisms involved in the development and progression of pulmonary vasculopathy based on observations in patients with PAH and animals with experimental pulmonary hypertension.
1. INTRODUCTION: OVERVIEW OF THE PULMONARY CIRCULATION
There are two circulation systems in the human body: the systemic circulation system and the pulmonary circulation system. The pulmonary circulation is a circulatory system that functionally and structurally differs from the systemic circulation. It carries deoxygenated blood, or venous blood, from the right ventricle (RV) through the pulmonary artery (PA) to the lungs. It then returns oxygenated blood, or arterial blood, from the lungs via the pulmonary vein (PV) to the left atria and then the left ventricle (LV). The oxygenated blood is then pumped to the rest of the body by the LV through the systemic arterial system. Given that the predominant function of the lungs is gas exchange, the major function of the pulmonary circulation is to circulate deoxygenated venous blood via the PA to the lung capillaries, where gas exchange occurs and deoxygenated venous blood becomes oxygenated arterial blood. The oxygenated arterial blood is then circulated back to the left heart.
The blood pressure in the two circulatory systems and in the left and right ventricles is significantly different. The mean systemic arterial pressure (mSAP) is ∼100 mmHg, with a left ventricle (LV) systolic pressure at 120 mmHg. The mSAP drops significantly in small arteries and capillaries and then further drops in systemic veins (FIGURE 1A). The blood pressure in the pulmonary circulation system is much lower than that in the systemic circulation system. The mean pulmonary arterial pressure (mPAP) is ∼14 mmHg (14.0 ± 3.3 mmHg) in healthy subjects (1), with a right ventricle (RV) systolic pressure at 25 mmHg (FIGURE 1). Pulmonary hypertension is defined, clinically or hemodynamically, as having an mPAP > 20 mmHg at rest, measured by right heart catheterization (1–3). For example, based on a cohort of patients diagnosed with pulmonary hypertension (PH), the average mPAP reached 50 mmHg (ranging from 25 mmHg to 105 mmHg) in patients with pulmonary arterial hypertension (PAH) and 45 mmHg in patients with chronic thromboembolic pulmonary hypertension (CTEPH) (4). This increase in PAP is caused directly by sustained pulmonary vasoconstriction, concentric pulmonary vascular wall thickening, in situ thrombosis and occlusive vascular lesions, and pulmonary arterial wall stiffening (5–7).
FIGURE 1.
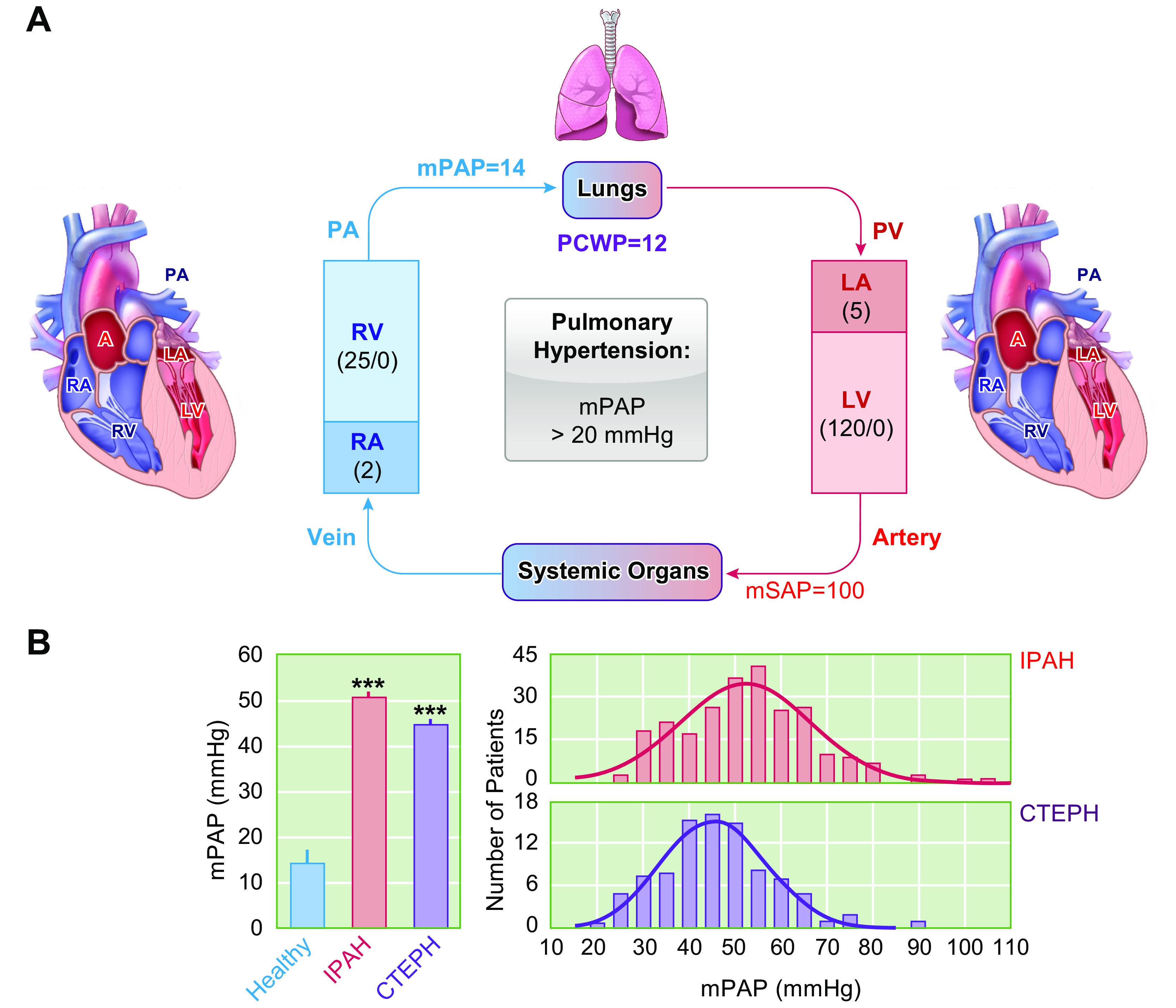
Schematic diagram of the pulmonary circulation and systemic circulation (A) and distribution of mean pulmonary arterial pressure (mPAP) in normal subjects and patients with idiopathic pulmonary arterial hypertension (IPAH) and chronic thromboembolic pulmonary hypertension (CTEPH) (B). A: the venous blood (or deoxygenated blood) is pumped from the right ventricle (RV) to the lungs via the pulmonary artery (PA). After taking in O2 from (and releasing CO2 to) alveoli to the capillary, the reoxygenated blood circulates to the left atria (LA) and is then delivered to systemic organs via the left ventricle (LV) through the systemic circulation. After releasing O2 to (and taking in CO2 from) organs and tissues, the deoxygenated blood circulates to the right atria (RA). The numbers in parentheses indicate the average pressure values in mmHg in healthy human subjects. For example, the mPAP is 14 mmHg, and the mean systemic arterial pressure (mSAP) is 100 mmHg. The pulmonary capillary wedge pressure (PCWP), also called pulmonary arterial wedge pressure (PAWP) or pulmonary artery occlusion pressure (PAOP), is ∼12 mmHg (4–12 mmHg in normal subjects). The RV systolic and diastolic pressure is 25 and 0 mmHg, whereas the LV systolic and diastolic pressure is 120 and 0 mmHg. The RA pressure and LA pressure are 2 and 5 mmHg, respectively. B: average mPAP in healthy control subjects (Healthy) and patients with IPAH and CTEPH (left) and distribution of mPAP in patients with IPAH and CTEPH (right). ***P < 0.001 vs. Healthy control.
The pulmonary and systemic circulatory systems must work together to allow deoxygenated blood to flow from the right ventricle to the lungs and oxygenated blood flow from the left ventricle to the rest of the body, respectively. The stiffening and concentric thickening of the pulmonary arterial wall disturb the pulmonary circulation specifically by increasing the resistance for deoxygenated blood to flow through the pulmonary arteries. These pathophysiological and pathological changes in the pulmonary vascular wall therefore increase PAP, adding more pressure to the heart and weakening the right ventricle over time.
1.1. Pressure Distribution in the Pulmonary Circulation System
In the pulmonary circulation, the pulsatile pressure-flow relation can be expressed in terms of pulmonary vascular impedance, which is the ratio of the amplitude of oscillatory arterial pressure to the oscillatory inflow rate at a given frequency. The distribution of pressure drop, in the systemic and pulmonary circulations, is mainly related to the resistance to blood flow. The pulmonary circulatory system is a low-pressure system compared with the systemic circulation (FIGURE 2). In the systemic circulatory system, the muscular small arteries and arterioles (or resistance arteries) account for 70% of the pressure drop. The systemic capillaries and veins only account for 7% and 19% of the pressure drop, respectively (FIGURE 2, A AND Bb) (8–10). In the pulmonary circulation system, however, the pressure drop is evenly distributed among the pulmonary arteries and veins. Overall, the pressure drop in the pulmonary capillary sheet is greater than that of the systemic circulation (FIGURE 2B). FIGURE 2Ba shows two representative curves depicting the pressure drop from the largest pulmonary artery to the smallest pulmonary arterioles (30 ± 7%), capillaries (17 ± 3), and veins (39 ± 13%). FIGURE 2Bb compares the pressure drops in the pulmonary arteries, capillaries, and veins (left) with the pressure drops in the systemic arteries, capillaries, and veins (right) based on observations by Brody et al. in 1968 (9) and Hakim et al. in 1982 (10). The data on the systemic circulation (i.e., mesenteric and skeletal muscle circulation) are results by Fronek and Zweifach in 1974 (8).
FIGURE 2.
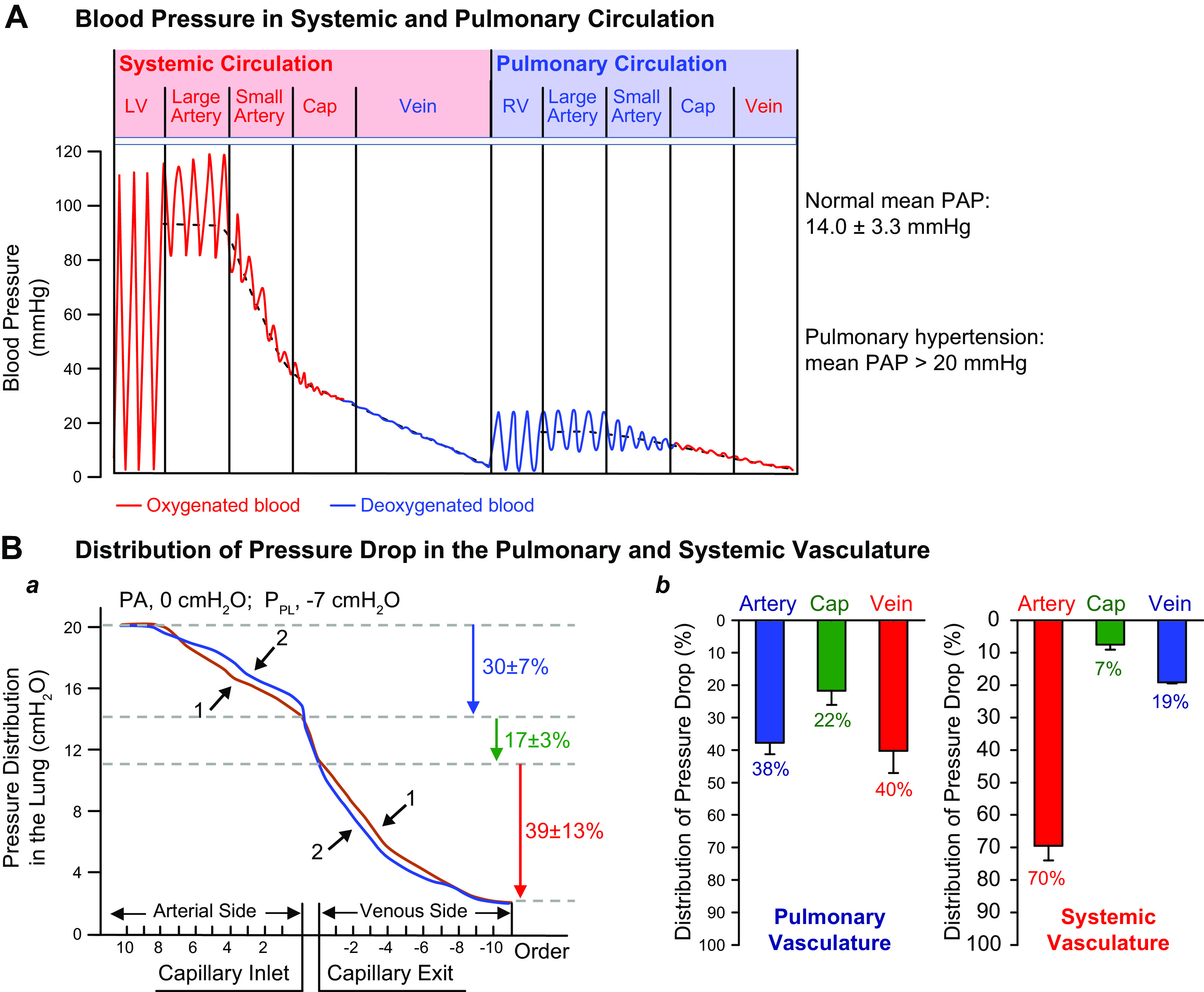
Distribution of pressure changes in the pulmonary circulation. A: blood pressure measured in the systemic circulation system including the left ventricle (LV), large artery, small artery, capillary (Cap), and vein and the pulmonary circulation system including the right ventricle (RV), large and small pulmonary artery, lung capillary, and vein. B: distribution of pressure in the lung (in cmH2O) from the arterial side to the capillary and to the venous side (a). The pressure drop in the pulmonary artery accounts for 30 ± 7% of the total pressure drop, whereas the pressure drop in the capillary and vein accounts for 17 ± 3% and 39 ± 13%, respectively. The pressure drop in the artery, capillary, and vein is compared between the pulmonary (b, left) and systemic (b, right) circulation systems. The bar graphs are based on data from Refs. 8–10.
Based on these data, there are relatively equal amounts of pressure drop in the veins (40% drop) and in the arteries (39% drop) in the pulmonary circulation, implying that both arteries and veins can contribute to an increased pulmonary vascular resistance (PVR) and PAP. Given the observations that the pulmonary artery wall is much thinner than that of the systemic arteries and that vascular smooth muscle cells (SMCs) in pulmonary arteries and veins are similar, it is important to assume that the pulmonary veins are equally effective as the pulmonary arteries in contributing to the increase in PVR and development of PH. The veins exert a remote or downstream control and have a powerful effect on the arteries. They can reduce the resistance of the pulmonary capillaries to blood flow or contribute to an increase in pulmonary arterial pressure. In the future, as much attention should be paid to the pulmonary venous smooth muscle and endothelial cells as to those of the pulmonary arteries in searching for pathogenic triggers of PH. Indeed, the functional and structural changes in pulmonary veins are implicated in the development and progression of chronic hypoxia-induced PH in animal models (11).
In normal lungs, the pressure drop in large pulmonary arteries is similar to that in small arteries, as shown in FIGURE 2B. However, there are still insufficient data to determine the changes in the distribution of pressure drop and vascular resistance in patients with various forms of PH. More studies are needed to define the branching pattern, morphometry, morphology, and the elasticity of pulmonary arterial and venous vessels of diseased human lungs with severe PH.
The systemic and pulmonary circulatory systems differ in how blood pressure is distributed and regulated. In the systemic circulation, the arteries account for the majority of the pressure drop compared with the veins when blood flows from the left ventricle to the right atrium. On the contrary, in the pulmonary circulation arteries and veins contribute equally to the pressure drop when blood flows from the right ventricle to the left atrium. Therefore, a change in the elasticity and/or diameter of either arteries or veins in the pulmonary circulation system can lead to a higher PAP and, potentially, the development of PH. The majority of research done in the field of PH, however, has focused on pathogenic triggers in pulmonary arteries. More attention should be given additionally to the pulmonary venous system, both as a pathogenic contributor and as a potential therapeutic target, as it contributes equally to the regulation of blood pressure in the pulmonary circulation.
1.2. Definition of Pulmonary Hypertension
The lungs are the only organ in the body that receives the entire cardiac output (CO); therefore, maintaining low resistance and low pressure in the pulmonary circulation is crucial to ensure normal blood flow from the right ventricle to the lungs. Pulmonary hypertension (PH) is a condition in which the pulmonary vascular resistance (PVR) and mean PAP (mPAP) are increased because of genetic, epigenetic, and environmental causes. These factors result in the sporadic pulmonary vasculopathy seen in patients with heritable and idiopathic PAH (IPAH) as well as in patients with PH due to other diseases (2, 3, 6, 7, 12, 13).
The updated clinical definition of PH is having an mPAP > 20 mmHg at rest, measured by right heart catheterization (RHC) (1–3, 14). PH is clinically classified into five groups based on pathogenic mechanisms, clinical manifestations, hemodynamic characteristics, and therapeutic approaches by the World Symposium on Pulmonary Hypertension (WSPH) (1–3, 15–18). The new classification of PH has also been recently published as a guideline for clinical management and translational research in the field of pulmonary vascular disease (TABLE 1) (1–3). The hemodynamic definition for each of the five groups of PH is shown in TABLE 2. It is important to note that the hemodynamic definition of PH is determined not only by mPAP but also by pulmonary arterial wedge pressure (PAWP) and PVR (TABLE 2) (1–3, 19). Exercise PH is defined by an mPAP/CO slope > 3 mmHg/L/min between rest and exercise (2, 3, 20). An increase in the mPAP/CO slope defines an abnormal hemodynamic response to exercise; however, it does not allow for differentiation between pre- and postcapillary PH. The PAWP/CO slope with a threshold > 2 mmHg/L/min may best differentiate pre- and postcapillary causes of exercise PH (2, 3, 21, 22).
Table 1.
Revised nomenclature and updated clinical classification of pulmonary hypertension
| Nomenclature | Clinical Classification |
|---|---|
| Group 1 | |
| Pulmonary arterial hypertension (PAH) | 1.1 Idiopathic PAH (IPAH) |
| 1.1.1 Nonresponders at vasoreactivity testing | |
| 1.1.2 Acute responders at vasoreactivity testing | |
| 1.2. Heritable PAH (HPAH) | |
| 1.3 PAH associated with drugs and toxins | |
| 1.4. Associated PAH (APAH) with: | |
| 1.4.1 Connective tissue disease | |
| 1.4.2 HIV infection | |
| 1.4.3 Portal hypertension | |
| 1.4.4 Congenital heart disease | |
| 1.4.5 Schistosomiasis | |
| 1.5 PAH with overt features of venous/capillaries (PVOD/PCH) involvement | |
| 1.6 Persistent PH of the newborn | |
| Group 2 | |
| PH associated with left heart disease | 2.1 Heart failure |
| 2.1.1 with preserved left ventricular ejection fraction (LVEF) | |
| 2.1.2 with reduced or mildly reduced LVEF | |
| 2.2 Valvular heart disease | |
| 2.3 Congenital/acquired cardiovascular conditions leading to postcapillary PH | |
| Group 3 | |
| PH associated with lung diseases and/or hypoxia | 3.1 Obstructive lung disease |
| 3.2 Restrictive lung disease | |
| 3.3 Other lung disease with mixed restrictive/obstructive pattern | |
| 3.4 Hypoventilation syndromes | |
| 3.5 Hypoxia without lung disease (e.g., high altitude) | |
| 3.6 Developmental lung disorders | |
| Group 4 | |
| PH associated with pulmonary artery obstructions | 4.1 Chronic thromboembolic pulmonary hypertension (CTEPH) |
| 4.2 Other pulmonary artery obstructions | |
| Group 5 | |
| PH with unclear and/or multifactorial mechanisms | 5.1 Hematological disorders |
| 5.2 Systemic disorders | |
| 5.3 Metabolic disorders | |
| 5.4 Chronic renal failure with or without hemodialysis | |
| 5.5 Pulmonary tumor thrombotic microangiopathy | |
| 5.6 Fibrosing mediastinitis | |
Table 2.
Hemodynamic definition of pulmonary hypertension
| Definition | Characteristics | Clinical Groups of PH (see TABLE 1) |
|---|---|---|
| Pulmonary hypertension (PH) | mPAP > 20 mmHg | Groups 1–5 |
| Precapillary PH | mPAP > 20 mmHg | Group 1: Pulmonary arterial hypertension (PAH) |
| PAWP ≤ 15 mmHg | Group 3: PH associated with lung diseases and/or hypoxia | |
| PVR > 2 WU | Group 4: PH associated with pulmonary artery obstructions | |
| Group 5: PH with unclear and/or multifactorial mechanisms | ||
| Isolated postcapillary PH (IpcPH) | mPAP > 20 mmHg | Group 2: PH associated with left heart disease |
| PAWP > 15 mmHg | Group 5: PH with unclear and/or multifactorial mechanisms | |
| PVR < 2 WU | ||
| Combined pre- and postcapillary PH (CpcPH) | mPAP > 20 mmHg | Group 2: PH associated with left heart disease |
| PAWP > 15 mmHg | Group 5: PH with unclear and/or multifactorial mechanisms | |
| PVR > 2 WU | ||
| Exercise PH | mPAP/CO slope between rest and exercise > 3 mmHg/L/min |
One of the five forms (or groups) of PH is pulmonary arterial hypertension (PAH). Idiopathic PAH is a subtype of PAH, previously referred to as primary pulmonary hypertension (23–25). It is a rare, progressive, and fatal disease that predominantly affects women (26–28). The high PAP due to increased PVR increases the afterload of the RV and imposes a big strain on the right heart, which eventually causes right heart failure and death if left untreated. Thus, it is also referred to as a right heart failure syndrome. The Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL Registry) is an observational registry of the demographics, disease course, and management of patients with PAH in the United States. Despite the recent progress and advancement of new therapeutic approaches, the 5-yr survival of patients with advanced PAH remains poor (∼52% in men and 62% in women) (29, 30). The RV function estimated by the New York Heart Association Functional Classification (NYHA-FC) is an important predictor of survival. These observations reinforce the importance of continuous monitoring of NYHA-FC on RV function in PAH patients and the continuous development of novel therapies that improve hemodynamics and RV function in PAH patients (31, 32).
2. THE ANATOMY AND PHYSICS OF THE PULMONARY CIRCULATION
The law of physics can be applied to calculate pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) (TABLE 3). PAP is a function of PVR and cardiac output (CO), whereas the whole lung PVR is the sum of the resistance in the pulmonary arteries (PVRA), capillaries (PVRC), and veins (PVRV). In fluid dynamics, the Hagen–Poiseuille equation or the Poiseuille law can be applied to blood vessels in which blood flow (Q) is proportional to the pressure gradient (ΔP) and the fourth power of the intraluminal radius (r4) and inversely proportional to the total length (L) of the pulmonary arteries, capillaries, and veins, as well as the viscosity (η) (33–35). Based on the same law, the PVR is inversely proportional to the fourth power of the intraluminal radius (r4) of pulmonary vessels (TABLE 3). In other words, a very small decrease in the intraluminal diameter of the PA results in a significant increase in PVR and then PAP. It must be noted that the Poiseuille law only applies to laminar flow. Lung blood flow is not always laminar flow because of the 15–17 orders of the human pulmonary arteries and capillaries and the 15–17 orders of the pulmonary veins (36–39) as well as the short branches and multiple junctions of the pulmonary vasculature. Thus, the actual PVR or total PVR is probably greater than the PVR calculated by the Poiseuille equation (33–35).
Table 3.
Formula and equations commonly used to calculate PVR and PAP
| Equations | Definition | Reference(s) |
|---|---|---|
| PAP = PVR × CO | Where PVR is the vascular resistance of the whole lung including the pulmonary arteries (PVRA), capillaries (PVRC), and veins (PVRV) and CO is cardiac output | (33–35) |
| PVR = PVRA + PVRC + PVRV | ||
| PAP = CO × (PVRA + PVRC + PVRV) | ||
| Hagen–Poiseuille equation (or the Poiseuille law) | ||
| Q = ΔP × [(πr4) ÷ (8ηL)] | Where Q is flow; π is the constant of 3.14; ΔP is the pressure difference, r is the inner radius of the cylindrical tube (e.g., blood vessel), η is the viscosity of the fluid (e.g., blood), and L is the length of the tube (e.g., the total the blood vessel tree) | (33–35) |
| PVR = ΔP ÷ Q (in mmHg·min/L) | Where ΔP is the difference between pulmonary arterial pressure (PAP) and left atrial pressure or pulmonary arterial wedge pressure (PAWP), and Q is cardiac output (CO). | (35) |
| PVR = (PAP – PAWP) ÷ CO | ||
| PVR = (8Lη) ÷ (πr4) (in dyn/cm5) | Where L is the total length of the pulmonary vasculature, η is the viscosity of the venous blood through the lung circulation, π is the constant (3.14), and r is the radius of the lumen of pulmonary vessels | (35) |
Ultimately, however, the Poiseuille equation is a useful model in demonstrating the relationship among blood flow, pressure, and vessel diameter. It is thus important to recognize that even the slightest change in intraluminal radius of the pulmonary artery (and vein) can have a major impact on the PVR and PAP. Therefore, any functional (e.g., vasoconstriction) and structural (e.g., vascular wall thickening and intraluminal occlusion) changes in the pulmonary vasculature that decrease the intraluminal radius (or diameter) of a pulmonary vessel can increase the vascular resistance and arterial pressure and the afterload put on the heart (right ventricle).
2.1. Angiogram and Cast of Pulmonary Arteries
There are 15–17 orders of PAs in the human lungs based on early morphological studies (36, 39). The morphometry and structure of the pulmonary vasculature have been well studied with casting, ex vivo angiography, and, more recently, in vivo and ex vivo imaging approaches. These include computerized tomography (CT) scans (40, 41), nuclear magnetic resonance (NMR), and magnetic resonance imaging (MRI) (42–45). Three schemes have been proposed to describe the complex branching structure in the lungs: the Weibel model, the Strahler model, and the diameter-defined Strahler system (36) (FIGURE 3, A–D) (46–48). The Weibel model assumes a symmetrical dichotomic branching structure in which all arteries of the same size (diameter and length) are in parallel, arteries of different sizes are in series, and the largest artery is determined as order 1. The Strahler model system, however, does not require a symmetrical branching pattern, which is believed to be a better physiological representation. In this model, the order 1 branch starts from the smallest arteries, and when two vessels of the same order meet, the order number of the parent vessel increases by 1. If a small artery joins a larger artery, the order number of the larger artery does not change. The Strahler model has been applied to human and cat lungs (49, 50). The diameter-defined Strahler model improves this scheme by adding a new rule stating that when a vessel of order n with diameter Dn meets another vessel of order n, the parent vessel has order n + 1 if its diameter is larger than Dn + (Sn +Sn + 1)/2, where Sn and Sn + 1 are the standard deviations of the diameters of orders n and n + 1 (38, 51). The diameter-defined Strahler model system is potentially the most accurate model of the human lung vasculature, and calculations based on this model best mimic in vivo pulmonary hemodynamics. Using this scheme, Huang et al. (36) identified 15 orders of pulmonary arteries between the main PA and the capillaries and 15 orders of pulmonary veins between the capillaries and the left atrium in healthy human lungs (50). All three models or schemes have been applied to describe the pulmonary vascular networks of humans, cats, dogs, and rats (36, 51–54). However, none of them has been used to describe the pulmonary vasculature in mice.
FIGURE 3.
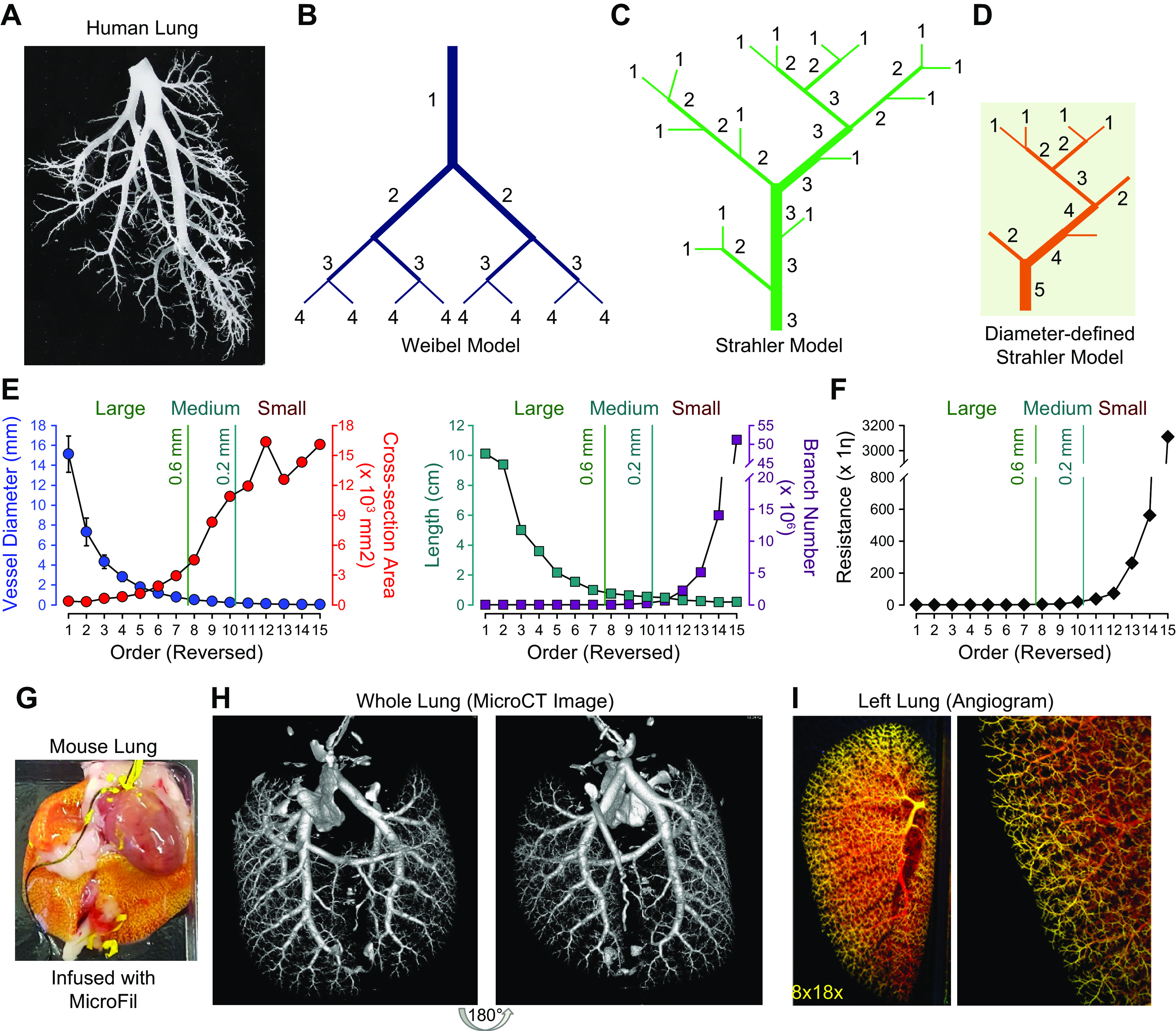
A: typical cast of a small segment of the arterial tree in human lungs shows the complex structure of the vascular tree. B–D: the 3 proposed schemes describing this complex structure are illustrated by the Weibel model (B), the Strahler model (C), and the diameter-defined Strahler system (D). Generation numbers are indicated on each branch. Note that in the Weibel model the largest vessel is designated as a vessel of generation 1. After each bifurcation, the generation number of the offsprings is increased by 1. The exact opposite is true in the Strahler model and the diameter-defined Strahler system, in which the smallest noncapillary blood vessels are defined as order 1. In the Strahler model, when 2 vessels of the same order meet, the order number of the confluent vessel is increased. When 2 vessels of different orders meet, the order number of the confluent vessels remains the same as the larger of the 2. In the diameter-defined Strahler system, when 2 vessels of different order and diameter meet one another, the order number of the confluent vessel is increased only if its diameter is larger than either of the 2 segments by a certain amount. Otherwise, the order number of the confluent segment is not increased. E: lumen diameter (closed circles in blue; left) and length (closed squares in dark cyan; right) of each segment of pulmonary arteries and distribution of total cross-sectional area (closed circles in red; left) and number of branches (closed squares in dark red; right) of all segments of each order of pulmonary arteries in human lungs. Diameter and length of each of the individual pulmonary arterial branches decline exponentially from orders 1 to 15. The number of pulmonary arterial branches increases exponentially from orders 1 to 15. F: distribution of vascular resistance in different orders of pulmonary arteries showing that resistance increases exponentially from order 1 [the largest pulmonary artery (PA)] to order 15 (the smallest PA). Reproduced from Huang et al. (36). The vessels are subjectively classified into large (diameter > 0.6 mm), medium-sized (diameter = 0.6–0.2 mm), and small (diameter < 0.2 mm) based on their arterial diameter. G–I: MICROFIL-filled mouse lung via the right ventricle (RV) (G), high-resolution computerized tomography (CT) scan image of the mouse lung (H), and ex vivo angiogram of mouse lung (I) show peripheral pulmonary vascular branches.η, viscosity.
Based on the diameter-defined Strahler system, the diameter and length of individual PA branches decrease exponentially from order 1 (the main PA) to order 15 (precapillary arteriole), whereas the number of PA branches increases exponentially from order 1 to order 15 (36). Furthermore, the total cross-sectional areas of PAs increase significantly, but not exponentially, from order 1 to order 15 (FIGURE 3E). Human lung histological analyses indicate that 25% of the total cross-sectional area in the pulmonary vasculature stems from large vessels (diameter > 0.6 mm), 44% from medium-sized vessels (diameter between 0.2 and 0.6 mm), and 30% from small vessels (diameter < 0.2 mm) (39, 55).
As discussed above, pulmonary vascular resistance (PVR) is positively proportional to the total length (L) of the PA and inversely proportional to the fourth power of the radius (r). These observations indicate that the total PVR can be significantly altered by changes in the luminal diameters of all large, medium, and small vessels. Specifically, a decrease in the intraluminal diameter of small vessels is the biggest contributor to the increased PVR in patients with PAH and precapillary PH. As shown in FIGURE 3F, the resistance increases exponentially from order 1 (the largest PA) to order 15 (the smallest precapillary arteriole).
The morphology and exact vascular structure (e.g., the number of orders of pulmonary vessels) of lung vessels in experimental animals, such as mice (FIGURE 3, G–I), may be different from humans. However, the basic principles of the Weibel and Strahler models established in human lungs are still applicable to experimental animals. Based on angiogram data, the lung vasculatures of mice, rats, dogs, sheep, and cats all exhibit a branching pattern similar to those described by the Weibel and Strahler models.
The location of the pulmonary vascular abnormality in PH determines its effect on PVR. The cross-sectional area of the pulmonary vascular system enlarges progressively from the central or proximal pulmonary arteries to the capillaries. Therefore, resistance to blood flow in the main PA is much greater than that of the small PAs and arterioles. Concentric wall thickening or obliteration of millions of small PAs and arterioles would be required to equal the effect of occluding one lobar PA. PAH mainly involves vascular lesions in medium-sized and small arteries and precapillary arterioles. Chronic thromboembolic pulmonary hypertension (CTEPH) mainly involves vascular occlusion and remodeling in the central vessels, although concentric vascular remodeling and occlusive lesions are also found in the distal vessels in some CTEPH patients (56, 57).
Overall, the Weibel and Strahler models are useful tools in demonstrating the basic morphometry of pulmonary vessels in humans and experimental animals. Both models mathematically illustrate how the number and size of pulmonary arteries change as they grow more distal throughout the lungs. Through these models, it becomes easier to understand how pathological changes in different parts of the lungs, or different segments of the pulmonary vasculature, affect PVR and to what extent. Idiopathic PAH, for example, is considered a small-vessel disease in which many distal and small arteries and arterioles, rather than a few proximal and large vessels (like in PH associated with pulmonary artery obstruction), become remodeled and obliterated, thereby resulting in a significant increase in PVR and PAP.
3. PHYSIOLOGY AND PATHOPHYSIOLOGY OF THE PULMONARY CIRCULATION
Regardless of the model used to calculate the total resistance in the pulmonary circulation, the decreased intraluminal diameter of pulmonary arteries due to vasoconstriction, concentric vascular wall thickening, and in situ thrombosis, as well as the decreased compliance (or dispensability) of the pulmonary arterial wall, are the major “direct” causes for the elevated PVR in patients with PAH and animals with experimental PH (FIGURE 4A). Pulmonary vascular intimal lesions and fibrosis due to smooth muscle cell migration (58–62), endothelial-to-mesenchymal transition (EndMT) (63–67), and monoclonal endothelial cell proliferation (68–71) all contribute to partial and complete occlusion of pulmonary vessels, especially in patients and animals with severe PH. The development and progression of PH and right heart dysfunction in patients with PAH are generally divided into three stages: 1) the presymptomatic/compensated stage, in which increased PVR and PAP are caused mainly by sustained pulmonary vasoconstriction while CO is maintained normal, 2) the symptomatic or decompensating stage, in which PVR and PAP increase steadily because of pulmonary vascular remodeling while CO starts declining because of compensated RV dysfunction or hypertrophy, and 3) the declining or decompensating stage, in which PVR increases steadily because of severe pulmonary vascular remodeling and occlusive intimal lesions while PAP and CO decline because of decompensated RV failure (FIGURE 4B) (72, 73).
FIGURE 4.
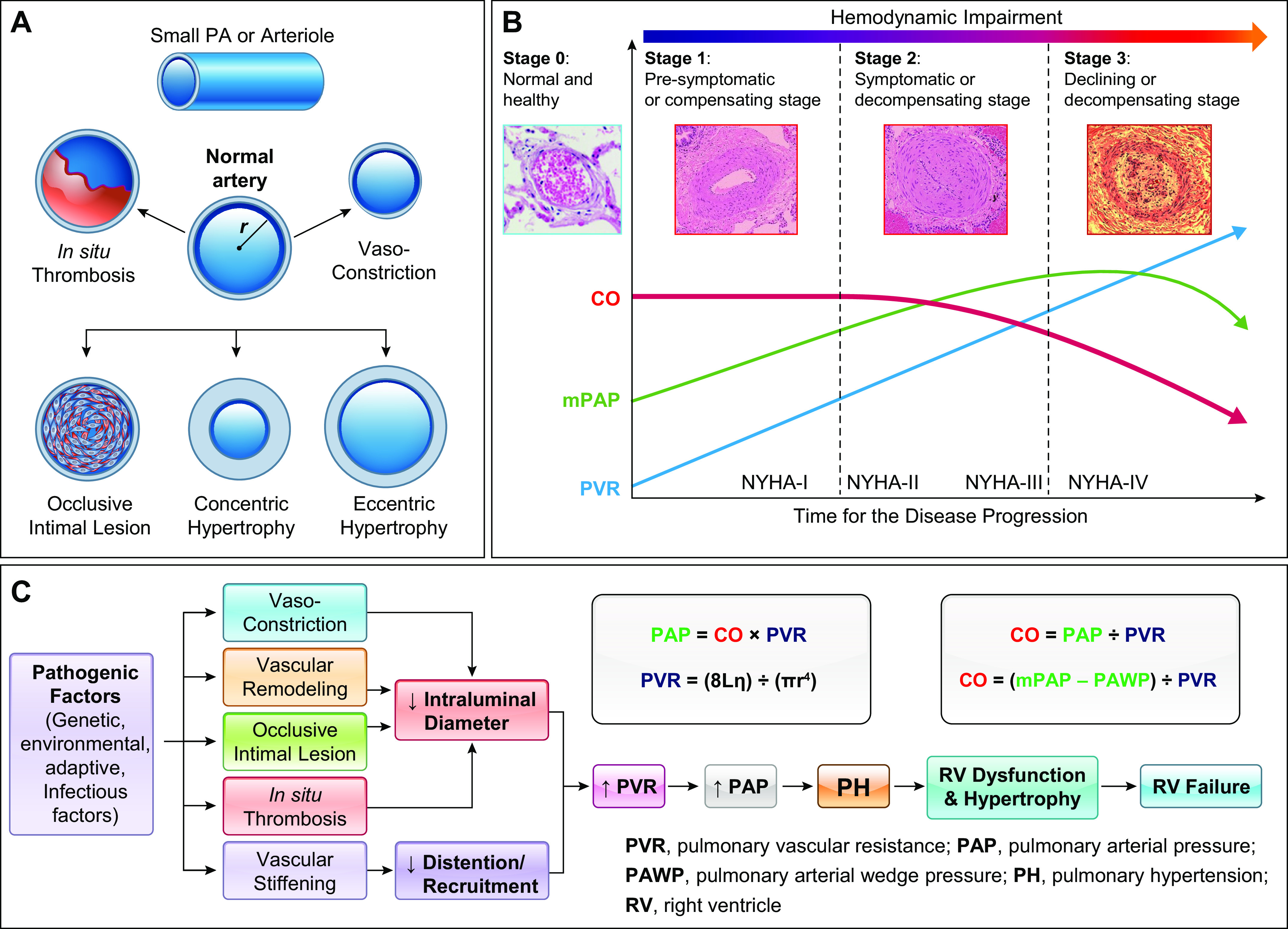
Schematic diagrams showing the patterns of pathological changes in the pulmonary artery (PA) and arteriole (A), the disease progression or the hemodynamic changes and pulmonary vascular remodeling (B), and the proposed mechanisms for increased pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) (C) in patients and animals with pulmonary hypertension. A: intraluminal radius (r) in a small PA is changed or reduced by in situ thrombosis, vasoconstriction, occlusive intimal lesions, and concentric hypertrophy, whereas PA wall stiffness can be increased by eccentric hypertrophy. B: normal cardiac output (CO), mean PAP (mPAP), and PVR are maintained in normal subjects (stage 0) because of a thin PA wall and a relaxed PA. At the early stage (stage 1) of disease initiation, pulmonary vasoconstriction is probably the major cause for increasing PVR and mPAP, whereas CO maintains normal. During the disease progression (stage 2), pulmonary vascular remodeling becomes the major contributor to the increased PVR and mPAP, whereas CO starts declining. At the late stage of the disease, combined pulmonary vascular remodeling and occlusive vascular lesions further increase PVR, whereas mPAP declines because of right ventricular dysfunction and right heart failure. NYHA-I to -IV, New York Heart Association functional classification I–IV. C: flow charts showing the key pathophysiological and pathological changes directly involved in the development and progression of pulmonary hypertension (PH) and right ventricle (RV) failure. The equations for calculating PAP and PVR are also listed to correlate vasoconstriction, concentric vascular remodeling, in situ thrombosis, and vascular wall stiffening to the increased PVR and PAP. L, total length of the pulmonary vasculature; r, intraluminal radius; η, blood viscosity; π, a constant, = 3.14.
Regardless of the initial genetic, epigenetic, environmental, and acquired pathogenic factors, the pathophysiological and pathological bases for the elevated PVR and PAP in PAH are the same (FIGURE 4C). In healthy normal subjects, the RV wall is thin because of its low pressure (RVP, 25/0 mmHg), much thinner than the LV wall. In patients with PAH, elevated PVR due to a reduced intraluminal diameter of the pulmonary vasculature creates a significant burden on the RV, thereby resulting in RV dysfunction and hypertrophy. At the beginning of the disease, we can see compensated RV hypertrophy where CO is maintained in the normal range. When the disease progresses or PVR consistently rises, CO starts declining because of RV dysfunction, ultimately leading to right heart failure.
In this scenario, sustained pulmonary vasoconstriction (74–78), inhibited pulmonary vasodilation (79–81), and/or myogenic tone (82, 83) are the early pathogenic causes for the initiation of the disease. Imbalance of pulmonary vasoconstrictors and vasodilators has been implicated in the development of PH (78, 80). Vasodilators, such as prostacyclin (PGI2) and nitric oxide (NO), were the early drugs developed for treatment of PH (23, 24, 84–86). In addition to the three-stage theory for disease development and progression, variations among patients based on genetic, epigenetic, and environmental influence should be considered for the time, duration, and transition point for the stages. For example, some patients may have extremely high PVR and PAP but have good or normal RV function, whereas others may have mild increases in PVR and PAP, but their RV function deteriorates rapidly.
Although many vasodilators have an antiproliferative effect on vascular smooth muscle cells (SMCs), endothelial cells (ECs), and fibroblasts (FBs), many vasoconstrictors have a mitogenic effect on SMCs and FBs (87–94). In the initial stage (i.e., presymptomatic or compensating stage) of PAH, not only does sustained vasoconstriction contribute to the elevation of PVR and PAP but the contractile-to-proliferative phenotypical transition of PASMCs and the endothelial-to-mesenchymal transition (EndMT) of lung ECs have also begun. Then, in the symptomatic or decompensating stage, increased proliferation and migration of vascular cells [PASMC, EC, FB, myofibroblast (myoFB)], along with accumulated or entrapped inflammatory and progenitor cells (6, 95–99), become the major causes of concentric pulmonary vascular wall thickening and occlusive intimal lesions. These factors ultimately result in the progression of the disease to the declining or decompensating stage. Epoprostenol, a synthetic PGI2, and a variety of PGI2 analogs (e.g., treprostinil, iloprost, beraprost) are clinically used for adult PAH patients with functional classes II–IV (which are defined by how much activity is limited because of PH) (100). Inhaled NO is clinically used for the treatment of pediatric patients, such as neonates with persistent pulmonary hypertension of the newborn and acute hypoxemic respiratory failure (101), and of adult patients with PAH who are considered responders to vasodilators.
Although PAH (or PH) can arise from a number of environmental, genetic, epigenetic, and other initial causes, the clinical manifestations remain consistent across patient groups. There is the initial, presymptomatic stage in which sustained vasoconstriction is the major cause for increased PVR and PAP, the second symptomatic stage in which vascular remodeling becomes the major pathological cause for increased PVR and PAP, and finally the declining stage with severe remodeling and occlusive intimal lesions. Vasodilators and antiproliferative agents have been used as therapeutic agents, with slight variation in the use and timing of these drugs depending on the patient and the specific cause of their PH. However, although these drugs slow down the progression of the disease, they do not reverse or cure it. Future studies could focus on the differences between each variation of the disease depending on its cause and patient population and whether these factors could glean insight into the treatment and potential reversal of a more specific patient group.
3.1. Pulmonary Vasoconstriction Is Dependent on Ca2+
The initial, presymptomatic phase of PAH (and other types of precapillary PH) is characterized by an increase in PVR and PAP through pulmonary vasoconstriction and concentric vascular remodeling. This phenomenon is largely controlled by the regulation of intracellular Ca2+ in pulmonary vascular smooth muscle cells. An increase in the cytosolic free Ca2+ concentration ([Ca2+]cyt) in PASMCs due to Ca2+ release from intracellular stores [e.g., sarcoplasmic reticulum (SR)] and Ca2+ influx through various Ca2+-permeable cation channels is a major trigger for PASMC contraction and thus pulmonary vasoconstriction. Increasing extracellular K+ from 4.7 to 40 mM shifts the equilibrium potential of K+ from −85 mV to −31.3 mV, which results in membrane depolarization, the opening of voltage-dependent Ca2+ channels (VDCCs), Ca2+ influx through VDCCs, and an increase in [Ca2+]cyt (102–108). In isolated PA rings, removal of extracellular Ca2+ (Ca2+ free) from the perfusate abolishes high-K+-mediated pulmonary vasoconstriction, which is mainly due to Ca2+ influx through VDCCs in PASMCs (FIGURE 5, Aa AND Ab, top). In addition to the excitation-contraction coupling through membrane depolarization-mediated opening of VDCCs, agonist-mediated Ca2+ influx through receptor-operated Ca2+ channels (ROCCs) or store-operated Ca2+ channels (SOCCs) is another important mechanism for causing pulmonary vasoconstriction (FIGURE 5, A AND C).
FIGURE 5.
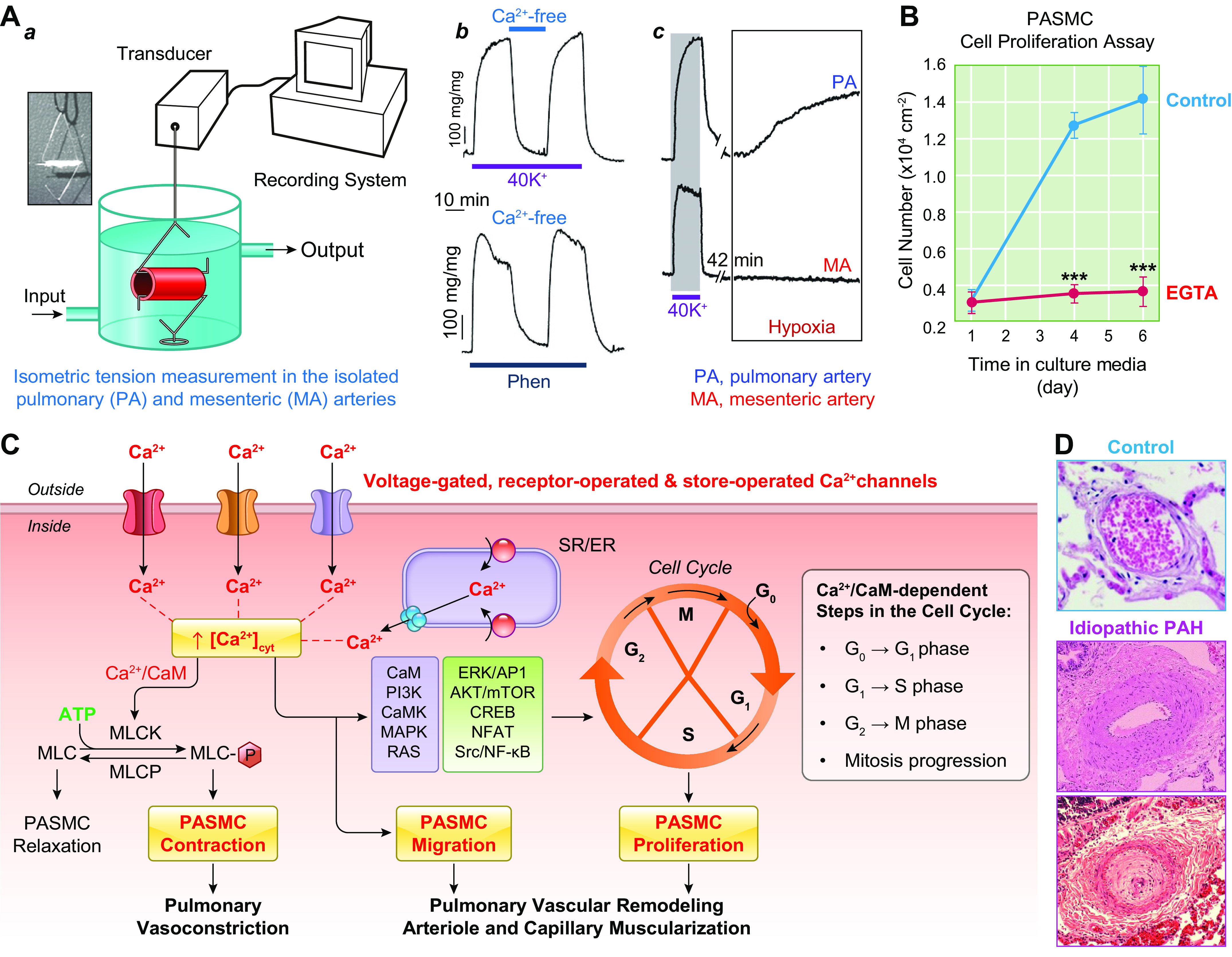
An increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in pulmonary arterial smooth muscle cells (PASMCs) is a trigger for pulmonary vasoconstriction and an important stimulus for cell proliferation. A: pulmonary vasoconstriction, determined by measuring isometric tension in freshly isolated pulmonary artery (PA) (a), is almost abolished by removal of extracellular Ca2+ from superfusate (Ca2+ free) when 40 mM K+-containing solution (40K+) (which induces membrane depolarization and opens voltage-dependent Ca2+ channels) and phenylephrine (Phen; an agonist that activates α-adrenergic receptor and opens receptor-operated Ca2+ channels) (b) are used as stimuli for vasoconstriction. The selective constrictive effect of hypoxia on PA, but not on mesenteric artery (MA), is shown in c. B: PASMC proliferation assay showing increasing cell growth curves of PASMCs in growth media containing serum/growth factors with 1.6 mM Ca2+ (Control) and in media with EGTA (a Ca2+ chelator that decreases free [Ca2+] from 1.6 mM to the nanomolar range). Chelation of extracellular free Ca2+ significantly inhibits serum/growth factor-mediated PASMC proliferation. C: schematic diagram showing the proposed mechanisms for Ca2+-mediated PASMC contraction and proliferation, pulmonary vasoconstriction, and vascular remodeling. A rise in [Ca2+]cyt due to Ca2+ influx through various Ca2+ channels and Ca2+ release from the intracellular Ca2+ stores, the sarcoplasmic (SR) or endoplasmic (ER) reticulum, activates myosin light chain (MLC) kinase (MLCK) by binding to calmodulin (CaM). MLCK-mediated phosphorylation of MLC (MLC-P) is a major step for smooth muscle contraction. The Ca2+-sensitive signaling proteins and transcription factors then propel cells to go through the cell cycle for proliferation. Ca2+/CaM activates at least 4 steps in the cell cycle: transition from G0 to G1, transition of G1 to S, transition of G2 to M, and mitosis itself. D: hematoxylin and eosin (H&E) staining showing cross section of PA in a normal subject (Control, top) and 2 different patients with idiopathic pulmonary arterial hypertension (IPAH). The images show significant adventitial and medial hypertrophy in PA from 1 IPAH patient (middle) and occlusive vascular lesion in another IPAH patient (bottom). MLCP, myosin light chain phosphatase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase.
Activation of membrane receptors like G protein-coupled receptors (GPCRs) and tyrosine kinase receptors (TKRs) on the surface membrane of PASMCs with specific ligands promotes the synthesis of second messengers like diacylglycerol (DAG) and inositol (1,4,5)-trisphosphate (IP3). DAG directly activates ROCCs formed by, for example, transient receptor potential (TRP) channel subunits (109). This activation results in receptor-operated Ca2+ entry triggering PASMC contraction. Furthermore, IP3 activates IP3 receptors (also referred to as Ca2+-release channels) in the membrane of the sarcoplasmic reticulum (SR) or endoplasmic reticulum (ER), leading to Ca2+ mobilization from the SR/ER to the cytosol and an increase in [Ca2+]cyt. Store depletion mediated by agonist-mediated mobilization or release of Ca2+ from intracellular Ca2+ stores (i.e., the SR/ER) results in the opening of SOCCs, formed mainly by STIM/Orai and STIM/Orai/TRP in the plasma membrane (110–116). This then induces store-operated Ca2+ entry (SOCE) and increases [Ca2+]cyt (117).
In addition to membrane depolarization-mediated opening of VDCCs, opening of ROCCs by the adrenergic α-receptor ligand phenylephrine (Phen) also causes rapid and sustained pulmonary vasoconstriction. Removal of extracellular Ca2+ (Ca2+ free) significantly inhibits agonist-mediated pulmonary vasoconstriction, which is mainly due to Ca2+ influx through ROCCs (FIGURE 5Ab, bottom). The agonist (e.g., Phen)-mediated activation of α-receptors also induces Ca2+ release from intracellular stores, such as the SR. Additionally, the store depletion-mediated Ca2+ influx through SOCCs contributes to the Phen-mediated pulmonary vasoconstriction (109, 112, 118–123). The rise in [Ca2+]cyt in PASMCs due to Ca2+ release/mobilization from the SR and Ca2+ influx through various Ca2+ channels enables Ca2+ to bind to and activate calmodulin (CaM) (124). Ca2+/CaM then activates myosin light chain kinase (MLCK), which leads to the phosphorylation of myosin light chain (MLC) and causes PASMC contraction and pulmonary vasoconstriction (FIGURE 5, A AND C) (118, 119). Another example showing Ca2+ dependence of PASMC contraction is hypoxic pulmonary vasoconstriction (HPV) (42, 125, 126). Acute hypoxia causes vasoconstriction in the pulmonary vasculature, whereas hypoxia causes vasodilation in various systemic vessels such as cerebral, coronary, and renal arteries (127). With the same experimental model, hypoxia increases isometric tension in the pulmonary artery (PA) but not in the mesenteric artery isolated from the same animals (rats) (FIGURE 5Ac). Removal of extracellular Ca2+ abolishes or significantly inhibits hypoxia-induced pulmonary vasoconstriction in isolated pulmonary arteries and in isolated perfused/ventilated whole lungs (42, 125, 128).
An increase in [Ca2+]cyt and activation of Ca2+/CaM/CaMK signaling in PASMCs play a major role in PASMC contraction (resulting in pulmonary vasoconstriction), migration, and proliferation (causing pulmonary vascular remodeling). [Ca2+]cyt is controlled, or regulated, by Ca2+ influx through multiple Ca2+-permeable cation channels including VDCCs (activated by membrane depolarization), ROCCs (activated by DAG upon activation of membrane receptors by specific ligands), and SOCCs (activated by the intracellular store depletion due to IP3-mediated active Ca2+ release or SERCA inhibition-associated passive Ca2+ leak or mobilization from the SR/ER to the cytosol). Removal or chelation of extracellular free Ca2+ inhibits agonist-mediated pulmonary vasoconstriction and attenuates PASMC migration and proliferation. Based on a large body of evidence, it becomes clear that Ca2+ and its downstream signaling cascades may play a critical role in the initial, presymptomatic stage of PAH.
3.2. Pulmonary Vascular Remodeling Is Partially Dependent on Ca2+
An increase in [Ca2+]cyt in PASMCs not only is a major trigger for PASMC contraction and pulmonary vasoconstriction but is also an important stimulus for PASMC proliferation and migration leading to pulmonary arterial wall thickening and muscularization of pulmonary arterioles and capillaries. In vitro experiments indicate that chelation of extracellular free Ca2+ with EGTA or BAPTA (129, 130) significantly inhibits proliferation and growth of human and animal PASMCs incubated in culture media containing 10% fetal bovine serum and various growth factors (110, 131) (FIGURE 5B). Ca2+ or Ca2+/CaM regulates cell proliferation via multiple mechanisms and signaling pathways (132–137). Many intracellular signaling cascades associated with cell proliferation (and growth), protein synthesis, and gene expression are regulated by Ca2+/CaM and downstream kinases (38, 133, 134, 138) (FIGURE 5C).
As described above, the major causes for the elevated PVR and PAP in patients with precapillary PH, such as PAH and PH associated with lung diseases and/or hypoxia, are the functional (e.g., sustained vasoconstriction and myogenic tone) (FIGURE 5, A AND C) and structural (e.g., concentric wall thickening, occlusive intimal lesions) (FIGURE 5, B AND D) changes of the pulmonary vasculature that reduce intraluminal diameter or radius of arteries and veins. Given the requirement for Ca2+ in cell growth, proliferation, and migration, the abnormal upregulation and increased activity of Ca2+ channels and/or receptors mediating Ca2+ influx in PASMCs, and other highly proliferative cells like myofibroblasts and fibroblasts, may be an important pathogenic mechanism involved in the development of pulmonary vascular remodeling and increased pulmonary vascular wall stiffness (139).
4. STRUCTURE AND FUNCTION OF PULMONARY VASCULAR ENDOTHELIAL CELLS, SMOOTH MUSCLE CELLS, AND FIBROBLASTS
The pulmonary vascular wall is structurally characterized by three major layers: 1) the intima (or tunica intima) or the endothelium, which contains a single layer of endothelial cells (ECs), 2) the media (or tunica media), which contains mainly smooth muscle cells (SMCs), and 3) the adventitia (or tunica externa), which consists of multiple cells, including fibroblasts (FBs), myofibroblasts (myoFBs), and macrophages (MΦs) and the extracellular matrix (ECM). The intima (or the endothelium) is separated from the media (SMC) by the inner elastic membrane (IEM), and the media is separated from the adventitia by the external elastic membrane (EEM) (FIGURE 6A). The development of PH involves a heterogeneous constellation of genetic, molecular, and humoral abnormalities. The pathogenic factors involved interact in a complicated manner, presenting a final manifestation of vasoconstriction, vascular remodeling, and occlusive lesions in which ECs, SMCs, and FBs all play a role. Despite the relatively thin wall in the pulmonary vasculature, the structure, function, and regulation of the various cell types are complex (39).
FIGURE 6.
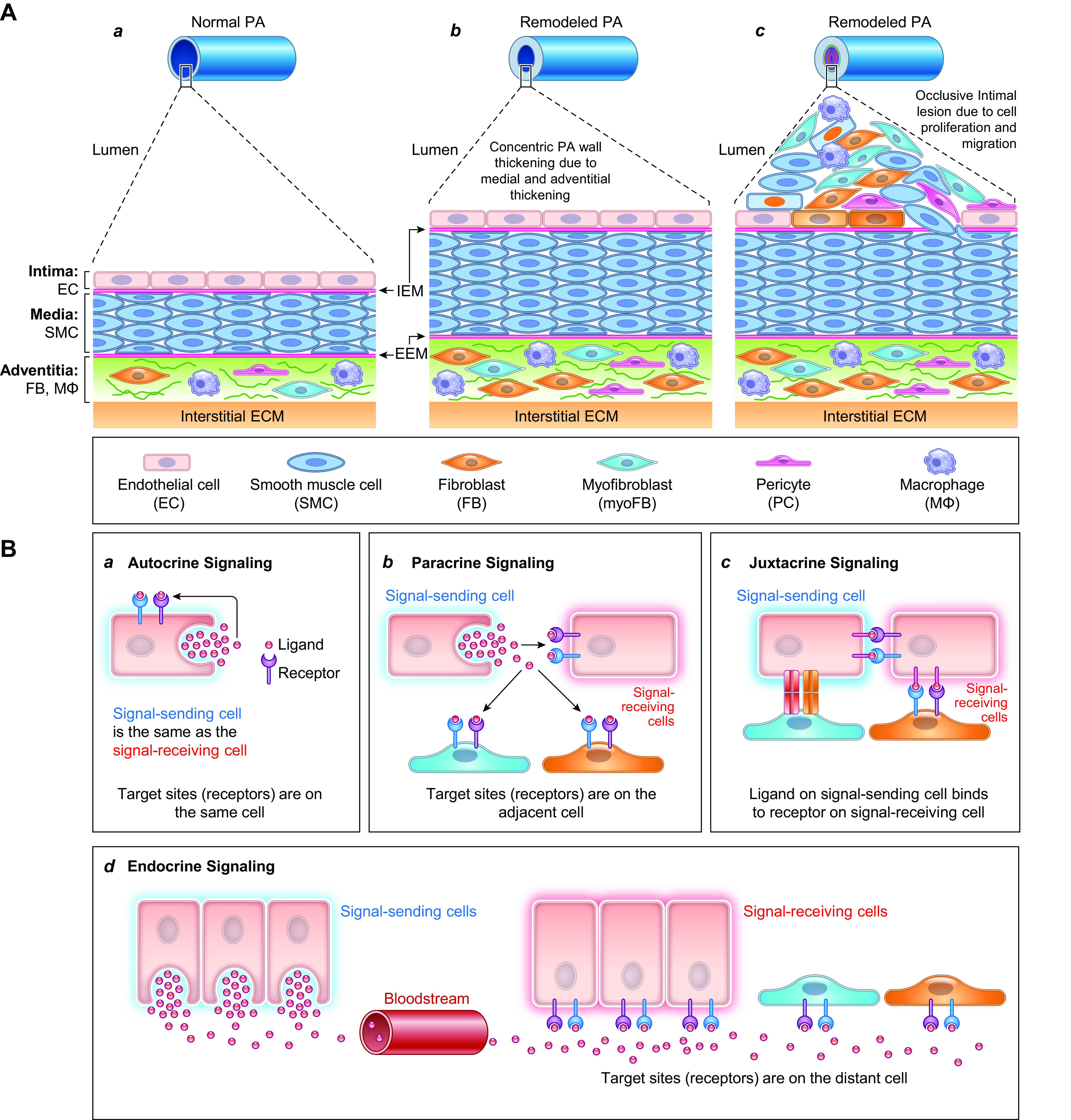
Schematic diagram depicting the progression of pulmonary vascular remodeling caused by changes in adventitial fibroblasts (FBs) and macrophages (MΦs), medial smooth muscle cells (SMCs), and intimal endothelial cells (ECs) (A) and the pathogenic interaction among different cells through the autocrine, paracrine, and juxtacrine mechanisms (B). A: normal pulmonary artery (PA) is thin and composed of the intima or the endothelium (with a monolayer of ECs), the media (mainly contains SMCs and may contain SMC-like pericytes), and the adventitia [mainly contains FBs, MΦs, progenitor cells, and extracellular matrix (ECM)] (a). The intima and media are separated by the internal elastic membrane (IEM), whereas the media is separated by the external elastic membrane (EEM) from the adventitia. Because of the stimulation of pathogenic triggers and self-defects (e.g., somatic mutation, genetic manifestation), increased SMC/FB proliferation results in concentric PA wall thickening (b), whereas EC injury, phenotypical change [e.g., endothelium-to-mesenchymal transition (EndMT)], and SMC migration contribute to the development occlusive intimal lesions (c). All cell types in the PA contribute to the development and progression of the pathological changes that narrow the lumen and increase pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP). B: diagram showing cell interactions through different mechanisms including autocrine (a), paracrine (b), juxtacrine (c), and endocrine (d) signaling. All cells can use the autocrine signaling mechanism for self-regulation or -stimulation. Adjacent cells can regulate each other through paracrine and juxtacrine signaling mechanism in the same layers (e.g., EC↔EC, SMC↔SMC, FB↔FB), whereas EC and SMC (or SMC and FB) can also interact with each other through the myoendothelial junction as well as the internal (and external) elastic lamina.
Pulmonary vasoconstriction results in increases in PVR and PAP in patients and animals with PH. Vasoconstriction is mainly caused by PASMC contraction, which is triggered by a rise in [Ca2+]cyt and the subsequent activation of myosin light chain kinase (MLCK) (FIGURE 5C). However, contractile myofibroblasts and fibroblasts may also contribute to the sustained vasoconstriction and increased myogenic tone in the lung vasculature (140–142). The endothelium, or the tunica intima, is composed of a single layer of ECs in the inner lining of pulmonary vessels. It is one of the major resources for synthesizing and releasing vasodilators and vasoconstrictors, which are referred to as endothelium-derived constricting factors (EDCFs) and endothelium-derived relaxing factors (EDRFs). In the pulmonary circulation, EDRF, including NO and PGI2, is not only an endogenous vasodilator that maintains low resistance and pressure in the lungs but also an exogenous factor or drug used for treatment of pediatric and adult patients with PH (24, 80, 85, 143, 144). EDCF, including thromboxane A2 (TXA2) and endothelin-1 (ET-1), is an endogenous vasoconstrictor that initiates and maintains vasoconstriction causing acute increases in PVR and PAP (145).
In pulmonary arteries from humans and animals, acetylcholine (ACh)-mediated activation of muscarinic receptors in the endothelium (or ECs) causes significant vasodilation due to the release of EDRFs (endothelium-derived relaxing factors) (146–148) and/or EDHFs (endothelium-derived hyperpolarizing factors) (149). The primary EDRF is NO (150), which also causes membrane hyperpolarization by indirectly (via cGMP) or directly (via nitrosylation, oxidation, or nitration of the channel protein) activating various K+ channels in SMCs. The EDRFs also include PGI2 (151). The candidates for EDHFs include K+, NO, and epoxyeicosatrienoic acids (152, 153). Functional removal of the endothelium abolishes ACh-mediated pulmonary vasodilation but does not alter sodium nitroprusside-mediated vasodilation. The endothelium-dependent vasodilation induced by ACh, an activator of endothelial muscarinic receptors, and the endothelium-independent vasodilation induced by sodium nitroprusside, an NO donor, are often used to assess the functional integrity of the endothelium in the pulmonary vasculature. In patients with PAH, reduced EDRF due to downregulated endothelial nitric oxide synthase (eNOS) is implicated in the development of sustained pulmonary vasoconstriction and PH (79, 154). In the human and animal pulmonary vasculature, EDCF- or TXA2/endothelin-mediated pulmonary vasoconstriction plays a significant role in the development of PH and hypoxic pulmonary vasoconstriction (77, 78, 145, 155, 156). Many EDCFs, such as ET-1, also exert a mitogenic effect on PASMCs and FBs, causing vascular remodeling (90, 157). In addition, the endothelium is a major resource for synthesizing and releasing growth factors and mitogenic cytokines, which contribute to pulmonary artery endothelial cell (PAEC) proliferation via an autocrine mechanism as well as to PASMC and FB proliferation via a paracrine mechanism.
Indeed, increased EDCR and decreased EDRF are implicated in the development of PAH (77, 79, 80). In addition, PAEC apoptosis and/or endothelial injury allow circulating growth factors, mitogenic ligands, inflammatory cytokines, and circulating inflammatory cells to penetrate the endothelium and accumulate in the pulmonary vascular wall. This then further stimulates PASMC/FB proliferation and medial/adventitial hypertrophy (FIGURE 6A). In response to the pathogenic cues, a portion of PAECs undergo the endothelial-to-mesenchymal transition (EndMT), which converts slowly proliferative PAECs to highly proliferative myofibroblasts (myoFBs), contributing to the development and progression of intimal thickening and occlusive intimal lesions (64, 67, 158–162). Monoclonal PAEC proliferation is also an important contributor to the obliterative intimal lesions observed in patients with PAH and animals with severe experimental PH (69, 70, 163, 164). Pathological studies on the obliterative intimal lesions (e.g., neointimal and plexiform lesions) show multiple cell types forming the occlusion: ECs, SMCs, (myo)FBs, pericytes, progenitor/stem cells, and inflammatory cells (6, 95–98). These observations indicate that the formation of obliterative intimal lesions at the late stage of some PAH patients is due to not only EC proliferation but also SMC/FB migration and proliferation (58–60, 165), vascular resident or circulating progenitor cell deposition (62, 98, 99, 166–169), and inflammatory cell infiltration and accumulation (6, 97, 170, 171). The various cells and proteins in the pulmonary extracellular matrix (ECM), or interstitial and perivascular ECM, also indirectly play a very important role in determining lung vascular structure and function (172). Among the four direct causes of the elevated PVR (vasoconstriction, concentric vascular wall thickening, occlusive lesions, and increased wall stiffness), all ECs, SMCs, and (myo)FBs contribute to the functional and structural changes observed in patients with PAH and animals with experimental PH (TABLE 4 and FIGURE 6A) (173).
Table 4.
Direct role of pulmonary vascular endothelial cells (intima), smooth muscle cells (media) and fibroblasts (adventitia) in the increased pulmonary vascular resistance in PAH
| Cell Type | Vasoconstriction and Vasodilation | Concentric Vascular PA Wall Thickening | Obliterative Intimal Lesion | Increased PA Stiffness |
|---|---|---|---|---|
| Endothelial cell (EC) | ↑EDCF (TXA2, ET-1) | ↑Mitogenic factors and cytokines | Monoclonal proliferation → intimal and plexiform lesion | EDCF → myogenic tone |
| ↓EDRF (NO, PGI2) | EndMT → myoFB → medial hypertrophy | EndMT → myoFB → occlusive lesion | ||
| ↓EDHF | ↑Thromboembolic factors → embolism | |||
| Interaction with circulating fibrin, plasmin, and thrombin → fibrotic embolism | ||||
| Smooth muscle cell (SMC) | Contraction → vasoconstriction | Proliferation → medial hypertrophy/hyperplasia and muscularization | Migration and proliferation → occlusive lesion | Contraction → myogenic tone |
| Relaxation → vasodilation | Proliferation and migration → medial and intimal/adventitial thickening | |||
| Myofibroblast (myoFB) and fibroblast (FB) | Contraction* | Migration and proliferation → medial hypertrophy/hyperplasia | EC-derived myoFB → intimal lesion | Contraction* |
| Migration and proliferation → occlusive vascular lesion | Proliferation and migration → adventitial hypertrophy/hyperplasia | |||
| Interaction with ECM → collagen deposition | ||||
| Vascular-resident progenitor cells† | Differentiation to SMC and myoFB | ↑Mitogenic factors Differentiation to mesenchymal cells → wall thickening | Differentiation to SMC-like and/or FB → occlusive lesions | ↑Fibrotic factors Differentiation to SMC-like and FB → adventitial hypertrophy |
ECM, extracellular matrix; EDCF, endothelium-derived constricting factor; EDHF, endothelium-derived hyperpolarizing factor; EDRF, endothelium-derived relaxing factor; EndMT, endothelium-to-mesenchymal transition; ET-1, endothelin-1; NO, nitric oxide; PA, pulmonary artery; PAH, pulmonary arterial hypertension; PGI2, prostacyclin; TXA2, thromboxane A2; ↑, increase; ↓, decrease; →, lead to. *Contractile myofibroblasts exist in the lungs and perivascular adventitia of the pulmonary vasculature; it is unclear whether contractile myoFB contributes to pulmonary vasoconstriction and myogenic tone. †Contribution of vascular resident progenitor cells to the normal pulmonary vascular function and pathological pulmonary vascular remodeling is not fully understood yet.
Activity (and/or expression) of the enzymes, such as endothelial nitric oxide synthase (eNOS) and prostacyclin synthase, which are required for synthesis and production of EDRF, EDHF, and EDCF depends on changes of [Ca2+]cyt in PAECs. Whereas an increase in [Ca2+]cyt in PASMCs triggers PASMC contraction and induces pulmonary vasoconstriction, the rise in [Ca2+]cyt in PAECs can result in pulmonary vasodilation due to activation of eNOS. Furthermore, an increase in [Ca2+]cyt in PAECs can 1) upregulate mRNA and protein expression of mitogenic and angiogenic factors in PAECs causing pulmonary vascular remodeling (66, 174) and 2) increase pulmonary vascular endothelial permeability or cause pulmonary vascular barrier dysfunction (by causing PAEC contraction). Lung barrier dysfunction not only enhances infiltration of inflammatory cells (e.g., lymphocytes, neutrophils, macrophages) and circulating progenitor cells but also increases accumulation of circulating growth factors in the vascular wall to further vascular remodeling (164, 175–178).
In muscular arteries in the lungs, including proximal and distal pulmonary arteries, SMC contraction is the foundation for pulmonary vasoconstriction (tonic and phasic vasoconstriction) and generation of myogenic tone. An increase in [Ca2+]cyt in PASMCs, due to Ca2+ influx through Ca2+-permeable cation channels in the plasma membrane and/or Ca2+ release through channels in the SR/ER membrane, is an important trigger for PASMC contraction and, therefore, pulmonary vasoconstriction. SMC contractility is also regulated by increasing Ca2+ sensitivity and by Ca2+-independent mechanisms like RhoA-mediated phosphorylation of contractile proteins (74, 75, 83, 118, 119, 179, 180). SMC contractility in response to chemical and mechanical stimulations is also a major contributor to the increased myogenic tone in the pulmonary vasculature of animals with experimental PH (83, 181, 182). SMC and SMC progenitor migration is also implicated in the development of muscularization of precapillary arterioles and capillaries and in occlusive intimal lesions (55, 58–60, 183, 184). Contractile-to-proliferative phenotype transition of SMCs (61, 185, 186), as well as increased SMC proliferation and decreased SMC apoptosis, contribute to concentric medial hypertrophy (58–60, 165). Some investigators believe that SMC proliferation and SMC transformation into myofibroblasts are responsible for the formation of occlusive intimal or vascular lesions (55, 184, 187, 188).
In the normal PA, FBs are mainly localized in the adventitia and perivascular ECM. FB migration and proliferation is one of the major causes for the perivascular adventitial remodeling that increases PA wall stiffness and decreases PA intraluminal diameter (189, 190). Decreased compliance (or increased stiffness) of the pulmonary vascular wall, due to FB proliferation and extracellular matrix remodeling, is one of the major causes for the decreased distension and recruitment of pulmonary vessels observed in patients with PAH.
The lungs, or more specifically the bronchial and alveolar epithelial cells, are always exposed to inspired air, including 21% O2 and all air pollutants. The environmental changes (e.g., hypobaric and normobaric hypoxia, cold and hot temperature) (191, 192), inhaled air pollutants (e.g., particulate matter, sulfur dioxide, ozone), cigarette smoke, and hyperoxia-mediated oxygen radicals or reactive oxygen species (ROS) can all directly affect airway and alveolar epithelial cells. Epithelial cell-driven or -derived inflammatory cytokines, mitogenic factors, and fibrotic factors can rapidly reach the pulmonary vasculature via the perivascular adventitia. Therefore, it is possible that the initial pathogenic causes for the pathophysiological and pathological changes in the pulmonary vasculature originate from 1) the bronchial and alveolar epithelia, 2) the interstitial ECM, and 3) the perivascular adventitia in patients with PAH and other types of precapillary PH (e.g., PH due to respiratory disease and/or hypoxemia) (99, 193). The lung epithelial-endothelial interaction has been recently demonstrated to be involved in SARS-CoV-2-associated lung vascular disease (194, 195) and mechanosensitive regulation of pulmonary vascular function (196). Furthermore, bronchial and alveolar epithelial cells are the first line of cells to alveolar hypoxia, and the epithelial-vascular interaction is implicated in the development of hypoxic pulmonary vasoconstriction and PH due to respiratory disease and/or hypoxia (197).
Increased FB migration and proliferation, along with potentially enhanced contraction of contractile myoFBs, in the adventitia are major contributors to increased pulmonary vascular wall stiffness and concentric wall thickening (198, 199). The “inward” migration of (myo)FBs from the interstitial and perivascular ECM, and the adventitia to the media and intima, is another source for the development of occlusive vascular lesions in PAH and PH due to respiratory disease and/or hypoxemia. Furthermore, ECM remodeling and stiffening have been proposed as a key early step in pathogenic reprograming and molecular cross talk (through mechanosensitive, metabolic, and posttranscriptional pathways) among FBs, SMCs, and ECs in the development of pulmonary vascular remodeling and occlusive vascular lesions (200).
Cell proliferation, a process that increases the number of cells as a result of cell division, and cell growth, a process that enlarges the volume or size of cells in the absence of cell division, are both implicated in the development and progression of concentric pulmonary wall thickening. Cell growth during the G1 phase in the cell cycle dilutes the cell cycle inhibitor retinoblastoma protein (Rb) to trigger division in human cells (201). Hypertrophy, due to the increased size of cells, and hyperplasia, due to the increased number of cells, are both implicated in pulmonary vascular wall thickening. However, wall hypertrophy is often used to indicate both increased number and size of vascular cells.
Among the cells forming the pulmonary vascular wall (i.e., ECs in the intima, SMCs in the media, and FBs in the adventitia), it has been well documented that all cells contribute to the development of pulmonary vascular remodeling (e.g., concentric PA wall thickening, arteriole and precapillary muscularization, and obliterative intimal lesions) in patients with PAH and animals with severe experimental PH (6, 7, 96). In response to genetic (202, 203) and somatic (204) mutations, as well as localized cues (e.g., EC injury/apoptosis and vascular inflammation) (205, 206), PAECs release EDCFs, mitogenic factors, and inflammatory cytokines to cause PASMC contraction, migration, and proliferation. These factors result in pulmonary vasoconstriction and vascular medial thickening (FIGURE 6A) through autocrine and paracrine signaling mechanisms (FIGURE 6B). SMC migration and proliferation, and their contractile-to-proliferative phenotype transition, ultimately lead to concentric medial hypertrophy. In the adventitia, environmental pollutants leading to inflammation and the recruitment of mitogenic and fibrotic factors can be one of the factors that leads to FB migration and proliferation. An increase in FBs as well as contractile myoFBs in the adventitia and extracellular matrix increases vascular wall stiffness. Ultimately, each cell type in all three layers of the pulmonary vascular wall plays a major role in contributing to the pathogenesis observed in PAH: sustained vasoconstriction, pathogenic remodeling, concentric vascular wall thickening, occlusive lesion, and increased wall stiffness.
4.1. Pathogenic Interaction of Vascular and Perivascular Cells
Regardless of the initial cause of the disease, under pathological conditions all pulmonary vascular wall cells including ECs, SMCs, (myo)FBs, pulmonary vascular resident progenitor cells, and infiltrated (and accumulated) circulating inflammatory cells in the pulmonary vasculature and perivascular tissues are believed to form a “pathogenic network” to enable vasoconstriction, vascular wall remodeling and stiffening, and occlusive lesions in PAH. Cell-to-cell interactions through autocrine, juxtacrine, paracrine, and endocrine mechanisms create a local and/or “self-potentiated” pathogenic cascade to ensure the activation or overactivation of various cells resulting in the pathophysiological and pathological phenotype. TABLE 5 lists some common vasoconstrictive, mitogenic, and inflammatory factors that potentially participate in the local pathogenic interaction among ECs and SMCs via autocrine and paracrine mechanisms.
Table 5.
Selected ligands and receptors implicated in the development of PAH and experimental PH
| Ligands | Receptor (GPCR) | Receptor (TKR) | Effect of Activation on PA | Reference(s) |
|---|---|---|---|---|
| Thromboxane A2 (TXA2) | TP (TBXA2R) | Vasoconstriction, arterial remodeling | (78, 145) | |
| Endothelin-1 (ET-1) | ETA (EDNRA) | Vasoconstriction and arterial remodeling | (77, 145, 207) | |
| Prostacyclin (PGI2) | IP (PTGIR) | Vasodilation, regression of arterial remodeling | (23, 84, 208) | |
| Acetylcholine (ACh) | Muscarinic receptor (M1/M3) | EC-dependent vasodilation, SMC contraction | (209–211) | |
| Bradykinin | Bradykinin Receptor (B1/B2) | EC-dependent vasodilation | (212, 213) | |
| Angiotensin II (ANG II) | ATR1 (or AT1) and ATR2 (or AT2) | Vasoconstriction, vascular remodeling, increased ROS | (214, 215) | |
| ATP* | P2Y | Vasoconstriction, mitogenic effect on SMC | (216–218) | |
| Adenosine | AR or P1 | Vasodilation, increased EC barrier function | (219, 220) | |
| Ca2+, polyamine, neomycin, amyloid-β | CaSR | Arterial remodeling, vasoconstriction, muscularization | (221–226) | |
| Sphingosine 1-phosphate (S1P) | S1PR1–5 | Inflammatory, arterial remodeling, cell migration | (227–231) | |
| Apelin | APJ (APLNR) | Cell proliferation and migration, vascular remodeling, endothelial lesions | (232–235) | |
| Vasoactive intestinal polypeptide (VIP) | VIPR (VPAC1, VPAC2) | Vasodilation via increasing cAMP and PKA | (236–240) | |
| Platelet-derived growth factor (PDGF-AA, -AB, -BB) | PDGRFA, PDGFFB | Arterial remodeling, arteriole muscularization, occlusive intimal lesion | (58, 184, 241–247) | |
| Angiopoietin-1/2 | TIE1, TIE2 | Angiogenesis, vascular remodeling, thromboembolic disease, arteriole muscularization | (248–253) | |
| Nicotinamide phosphoribosyltransferase (NAMPT) | TLR-4 | Arterial remodeling, inflammation, capillary permeability | (254–258) | |
| Vascular endothelial growth factor (VEGF) | VEGFR | Plexiform lesion, disordered angiogenesis | (259) | |
| Epidermal growth factor (EGF) | EGF receptor | Remodeling, arteriole muscularization, occlusive lesions | (260, 261) | |
| Bone morphogenic protein 9 and 10 (BMP9/10) | ACVRL1/ALK1 | Arteriole muscularization, SMC contractile-to-proliferative switch | (262–265) | |
| Midkine | RPTPζ | Thromboembolic vascular remodeling | (261) | |
| Interleukin 6 (IL-6) | IL6R/gp130 | Inflammatory proliferation, vascular remodeling, macrophage recruitment | (266–270) | |
| Interleukin 33 (IL-33) | ST2/IL1RL1 | Vascular fibrosis and remodeling; inflammatory effect | (271–273) | |
| Tumor necrosis factor α (TNF-α) | TNFR-1 | Inflammatory vascular remodeling and RV dysfunction/failure | (274–278) |
CaSR, Ca2+-sensing receptor; EC, endothelial cell; ETA, endothelin receptor A; GPCR, G protein-coupled receptor; IP, prostacyclin receptor; PA, pulmonary artery; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; ROS, reactive oxygen species; RV, right ventricle SMC, smooth muscle cell; TKR, tyrosine kinase receptor; VEGFR, VEGF receptor.*ATP also activates P2X receptor, which is a ligand-gated ion channel.
In addition to “indirect” cell-to-cell interactions via autocrine, paracrine, and endocrine mechanisms, the juxtacrine signaling and adhesion interactions among cells are critical to the development of concentric pulmonary wall thickening and the formation of occlusive intimal lesions. Juxtacrine signaling is a form of cell signaling that occurs in cells that are in direct contact with each other, whereas paracrine signaling is a form of cell signaling between cells that are nearby each other. In paracrine signaling molecules (or ligands) are released into the extracellular space between the signal-sending cell and the signal-receiving cell, whereas in juxtacrine signaling there is physical contact between the signal-sending cell and the signal-receiving cell, so no signaling molecule is required (FIGURE 6B). Under normal conditions, ECs, SMCs, and FBs in the pulmonary arterial wall usually have direct contact with their same cell types. This is because the intimal ECs are separated from medial SMCs by the internal elastic membrane (IEM) and the medial SMCs are separated from the adventitial (myo)FBs by the external elastic membrane (EEM) (39, 279, 280). However, physical contact between two different cells like ECs and SMCs in the smooth muscle-endothelial cell interface [i.e., myoendothelial junctions (MEJs)] (281, 282) enables juxtacrine signaling between ECs and SMCs (281, 283). When the pulmonary vascular endothelium is injured during the early stage of the disease, medial SMCs can form direct contact with injured (or apoptotic) ECs through the juxtacrine signaling mechanism. Furthermore, circulating inflammatory cells infiltrate into the media and adventitia through the injured endothelium and can form direct contact with SMCs and (myo)FBs, causing pathological changes in the vascular wall. Although juxtacrine, or “direct,” interactions between cells of the same type are critical for maintaining homeostasis and coordinating vasoconstriction and vasodilation in a healthy lung, these interactions can be problematic, or pathogenic, in the case of lung injury and vascular barrier dysfunction and can contribute to pulmonary wall thickening and occlusive lesions in PH.
4.2. Juxtacrine Signaling Mechanism: Notch Signaling
Notch signaling is an example of a typical juxtacrine signaling mechanism (284, 285) (FIGURE 7A). In the PA, or the pulmonary vasculature in general, the longitudinal signaling among ECs or SMCs as well as the transverse signaling between ECs and SMCs all play an important role in the development of concentric pulmonary vascular remodeling and occlusive vascular lesions in PAH/PH. Furthermore, circulating cells as signal-sending cells can penetrate the vascular wall and form juxtacrine signaling connections with vascular resident cells such as PASMCs and PAECs to activate Notch signaling and stimulate Notch target gene expression. The Notch ligand-enriched platelets and neutrophils can activate Notch signaling in exposed PASMCs in the vascular area where the endothelial injury takes place. Notch signaling is composed of Notch ligands, Notch receptors, and Notch-responsive nuclear effectors. Notch ligands, including Delta-like (DLL1-4) and Jagged (Jag1-2), are transmembrane proteins characterized by an NH2-terminal Delta, Serrate, and LAG-2 domain (DSL). Notch receptors (Notch1–4) are transmembrane proteins for which the extracellular domains contain 29–36 epidermal growth factor (EGF) repeats, 3 cysteine-rich LIN repeats, and a region that links to the transmembrane and intracellular domain (284). The key cytoplasmic and nuclear transducers of the Notch signaling pathway are the Notch intracellular domain (NICD) and the DNA-binding protein C-promoter binding factor 1 (CBF1) or recombination signal-binding protein for immunoglobulin κJ region (RBPJ or RBP-κJ) (FIGURE 7A). The canonical Notch signaling pathway dictates cell fate and influences cell proliferation, differentiation, and apoptosis (286–288).
FIGURE 7.
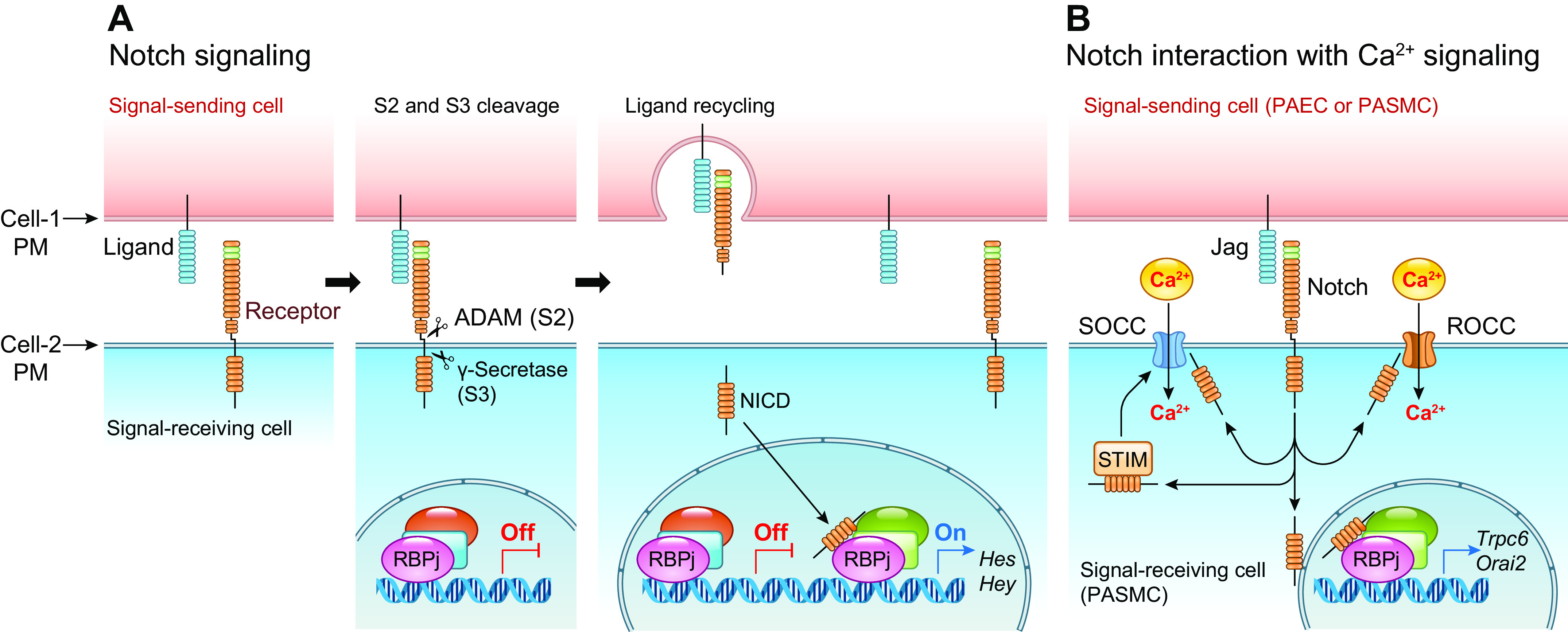
Canonical Notch signaling pathway (A) and Notch interaction with Ca2+ signaling (B). A: Notch ligands, such as Jagged (Jag) 1–2 and Delta-like (DLL) 1–4, in the signal-sending cell [pulmonary arterial smooth muscle cell (PASMC) or endothelial cell (PAEC)] bind to the extracellular domain of the Notch receptor (Notch1–3) in the signal-receiving cell (SMC or EC), inducing a conformational change in Notch that exposes the extracellular ADAM cleavage site for S2 cleavage. A subsequent internal membrane proteolytic cleavage (S3 cleavage) by γ-secretase releases the Notch intracellular domain (NICD) to the cytoplasm. NICD then translocates into the nucleus and interacts with CSL (CBF1, suppressor of hairless, lag-1) or RBPJ and Mastermind (MAM) on the target DNA (the promoter of target genes). In the absence of NICD, CSL/RBPJ recruits corepressors to turn off gene transcription. When NICD binds CSL/RBPJ and MAM, corepressors are replaced by coactivators to turn on gene transcription. A critical group of Notch target genes includes the Hes (hairy/enhancer of split) and Hey (Hes-related repressor Herp, Hesr, Hrt, CHF, gridlock) genes. PM, plasma membrane. B: NICD in the cytoplasm may directly interact with the store-operated (SOCC) and receptor-operated (ROCC) cation channels to increase Ca2+ influx. The cytoplasmic NICD may also interact with STIM protein in the sarcoplasmic (SR) or endoplasmic (ER) reticulum membrane, promote STIM translocation to the plasma-SR/ER membrane junction (or puncta) to recruit Orai channels in the plasma membrane to form SOCC, and ultimately enhance store-operated Ca2+ entry. The canonical Notch signaling pathway may also directly or indirectly stimulate transcription of TRPC6, Orai1/2, and STIM1/2 genes to upregulate ROCC and SOCC.
Canonical Notch signaling is important for regulating the growth, apoptosis, migration, and differentiation of SMCs and is a key mediator of vascular morphogenesis (287, 289, 290). Notch is required for arterial-venous differentiation during embryonic development and regulates arterial specification of vascular SMCs (291, 292). Notch3 is only expressed in the SMCs of arteries, not in veins (293). Notch signaling is involved in vascular development, and Notch3 has been implicated in PAH through enhancing SMC recruitment and proliferation. This process results in the muscularization of precapillary arterioles and capillaries as well as concentric wall thickening of small PAs (65, 206, 221, 248, 294–299). Activation of Notch signaling using the Notch ligand Jag-1 also enhances store-operated Ca2+ entry in human and animal PASMCs (FIGURE 7B), whereas the Jag-1-mediated increase in NICD proceeds the Jag-1-mediated enhancement of Ca2+ influx (294). Ex vivo studies on isolated and perfused/ventilated lungs indicated that Notch signaling is also involved in acute alveolar hypoxia-mediated pulmonary vasoconstriction (299). In addition to the canonical Notch signaling pathway via NICD-mediated transcriptional activation of Hes/Hey genes, it is possible that the functional interaction of cytoplasmic NICD with various ion channels in the plasma membrane and STIM protein in the SR/ER membrane may activate Ca2+ signaling in PASMCs to further enhance cell contraction, migration, and proliferation. The Notch signaling pathway is also directly and indirectly involved in upregulating various Ca2+ channel genes in pulmonary vascular cells.
Overall, the Notch signaling pathway is a prime example of the role that juxtacrine mechanisms play in furthering the pathogenesis of PAH. In the normal lung, it is involved in developing the pulmonary vasculature and differentiating arteries and veins during embryonic development and it regulates the proliferation and differentiation of SMCs. However, in PAH it enhances the recruitment and proliferation of SMCs and upregulates cation channels to result in arteriole muscularization and concentric PA wall thickening. Abnormally increased Notch signaling is also a trigger for EndMT (65, 300) that further enhances occlusive intimal lesion and concentric vascular remodeling in PAH.
4.3. Juxtacrine Signaling Mechanism: Gap Junction
Gap junctions are also a common and important juxtacrine signaling mechanism for intercellular communication (283, 301, 302) (FIGURE 8). Gap junctions are mainly formed by hydrophilic membrane channels that bridge the opposing membrane of neighboring cells. They play an important role in intercellular communication both longitudinally (i.e., between SMCs and/or between ECs) and transversely (i.e., between ECs and SMCs) across the blood vessel axis. The gap junction-associated communication in blood vessels generally includes longitudinal and transverse electrochemical signaling. This coordinates membrane potential changes and intracellular second messenger changes along a segment of a blood vessel and produces more uniform vasoconstriction or vasodilation.
FIGURE 8.
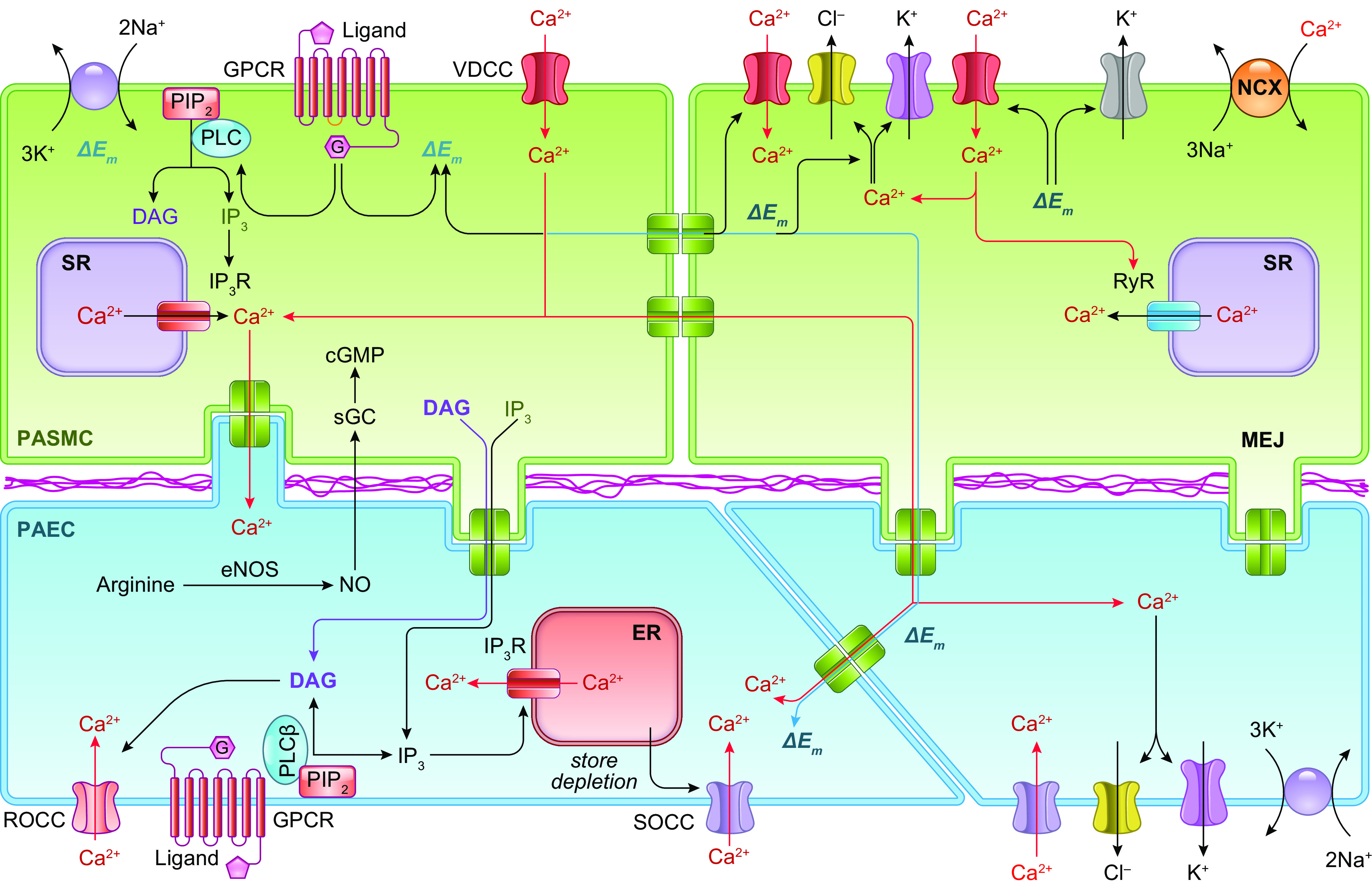
Longitudinal and transverse signaling via gap junction or gap junction channels in pulmonary arterial smooth muscle cells (PASMCs) and endothelial cells (PAECs). Gap junction is formed by a connexon (which consists of 6 connexins) in 1 cell (SMC or EC) that forms a channel with another connexon (also consists of 6 connexins) in an adjacent cell (SMC or EC). Changes in membrane potential and cytosolic free Ca2+ concentration [Ca2+]cyt in 1 cell can be quickly and efficiently communicated to an adjacent cell through the gap junction channel formed by 2 connexons. In addition, the gap junction channels also allow intracellular second messengers, for example, diacylglycerol (DAG), inositol (1,3,5)-trisphosphate (IP3), and cAMP/cGMP to be transferred from 1 cell to another adjacent cell. The changes in membrane potential and [Ca2+]cyt due to Ca2+ influx or release in 1 SMC can be communicated to an adjacent EC through gap junction channels in the myoendothelial junction. Em, membrane potential; ΔEm, change in membrane potential (e.g., membrane depolarization); eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; G, G protein; GPCR, G protein-coupled receptor; IP3R, IP3 receptor; MEJ, myoendothelial junction; NCX, Na+/Ca2+ exchanger; NO, nitric oxide; PIP2, phosphoinositol phosphate 2; PLC, phospholipase C; ROCC, receptor-operated Ca2+ channel; sGC, soluble guanylate cyclase; SOCC, store-operated Ca2+ channel; SR, sarcoplasmic reticulum; VDCC, voltage-dependent Ca2+ channel.
The longitudinal and transverse intercellular communication in SMCs and ECs plays an important role in coordinating vasoconstrictive and vasodilative responses (301–304). When a single cell, or a small number of cells, is stimulated, the response can be propagated to adjacent cells through gap junctions, producing a coordinate effect on a segment of the vessel. For example, a membrane potential change (depolarization or hyperpolarization) from one SMC (or a small group of SMCs) can be rapidly propagated longitudinally to other SMCs. This modulates the activity of VDCCs on the surface membrane and the SR/ER membrane [e.g., ryanodine receptors (RyRs)] to cause Ca2+ influx and/or Ca2+ release, coordinating vasomotor tone (vasoconstriction and vasodilatation) over vessel segments several millimeters in length. Such coordination is critical in minimizing turbulent flow and shear stress in the pulmonary vasculature. Longitudinal transportation of intracellular Ca2+, along with the signaling molecules that induce Ca2+ influx (e.g., diacylglycerol) and Ca2+ release (e.g., IP3), is an important mechanism that causes a uniformly coordinated vasoactive response in resistance arteries and lung capillaries (305, 306). Similar to systemic vascular SMCs and ECs, PASMCs and PAECs are functionally connected by gap junctions (283). This allows a direct transfer of electrical signals (e.g., action potentials, membrane depolarization and hyperpolarization) (307, 308), signaling molecules (i.e., Ca2+, IP3, diacylglycerol, cGMP, cAMP, ATP) (281, 301, 309), microRNAs (miRNAs) (310–314), and intracellular organelles (e.g., mitochondria) (315–317) from cell to cell (FIGURE 8).
In addition to longitudinal signaling, radical or transverse movement of ion current (or potential) or signaling molecules (e.g., Ca2+, diacylglycerol, IP3) between two cell types, such as ECs and SMCs, can occur via gap junctions located at the interface of SMCs and ECs or the myoendothelial junction (MEJ) (281, 282). The direct communication between ECs and SMCs at the myoendothelial junction allows an increase in [Ca2+]cyt, or in intracellular second messengers, in one cell to trigger complementary or opposing signaling events in the other cell type (FIGURE 8). For example, a rise in [Ca2+]cyt in PAECs is a trigger for activating eNOS and inducing NO-mediated pulmonary vasodilation. However, the increased Ca2+ in PAECs can go through the myoendothelial junction to PASMCs, causing pulmonary vasoconstriction. Dysfunction of the myoendothelial junction has been implicated in vascular dysfunction in many cardiopulmonary and metabolic diseases (318). Given the short segment of lung vessels, the coordinated (or disorganized) vascular response through longitudinal and transverse gap junctions is believed to significantly affect pulmonary vascular resistance and pulmonary arterial pressure (304, 308, 319–322).
The gap junction is a juxtacrine mechanism among cells not only to coordinate vascular tone (e.g., synchronized vasoconstriction and vasodilation) but also for “pathogenic” intercellular communication for the formation of segmental arterial wall thickening and occlusive vascular lesions. For example, a rise in [Ca2+]cyt, or an increase in intracellular second messengers and metabolites, in one cell (e.g., an EC or a SMC) can be propagated to other cells to affect the whole segment of the PA. In addition to the electrical signals generated by the changes of cations (e.g., Ca2+, K+, Zn2+) and anions (e.g., Cl−), second messengers (e.g., IP3, diacylglycerol, cAMP, cGMP), metabolites (e.g., ATP), cytoplasmic DNA and RNA (e.g., miRNA, long noncoding RNA), and intracellular organelles (e.g., mitochondria) can also go through gap junctions from one cell to another. The cytoplasmic DNA that causes innate immunity (323, 324) may also be able to go through gap junctions (325) in the pulmonary vasculature to cause vascular lesions.
Gap junctions play an important role in propagating signals across cells, both longitudinally (between the same cell type) and transversely (across different cell types). Ions, signaling molecules, metabolites, and even fragmented intracellular organelles can be transferred through gap junctions so that stimulation of one cell leads to the stimulation of an entire segment of an artery. This juxtacrine mechanism is important in regulating vasomotor tone (dilation and constriction) under normal and physiological conditions. Abnormalities in gap junction channels, however, are implicated in sustained vasoconstriction, vascular inflammation, arteriole muscularization, and vascular barrier dysfunction in the lungs.
4.4. Gap Junctions and Pulmonary Hypertension
A single cell with molecular and cellular defects as a result of, for example, somatic mutations (326) or a single cell (or a small group of cells) with metabolic defects (327–331) can affect adjacent cells by directly transferring electrical signals (321), metabolic products (320, 332), second messengers (283, 301, 321, 333), miRNAs (310, 313, 314), and fragmented or abnormal mitochondria through gap junctions (315–317). These connections can then lead to concentric remodeling and occlusive lesions in the whole segment or branch of the PA. This direct pathogenic intercellular communication among cells may be more efficient than the paracrine mechanism to transmit “cellular defects” or pathogenic triggers from one cell to another, or from a small group of cells to all cells of an arterial segment or branch. At the late stage of the disease, inflammatory cells infiltrated into the pulmonary vascular wall may also form gap junctions with adventitial myoFBs and FBs to further vascular remodeling (FIGURE 9). Circulating and inflammatory cells all highly express Notch ligands like Jag1/2 and DLL1/4. Infiltrated (from airway or alveolar site) or penetrated (from blood) cells in the pulmonary vasculature would also make direct contact with Notch receptors in SMCs and FBs to stimulate pathogenic cell proliferation and migration. It is unknown whether Notch signaling functionally interacts with gap junction channels to enhance or inhibit cell-to-cell communication in PAECs and PASMCs. The Notch-associated enhancement of Ca2+ signaling may indirectly affect cell-to-cell communication via gap junctions.
FIGURE 9.
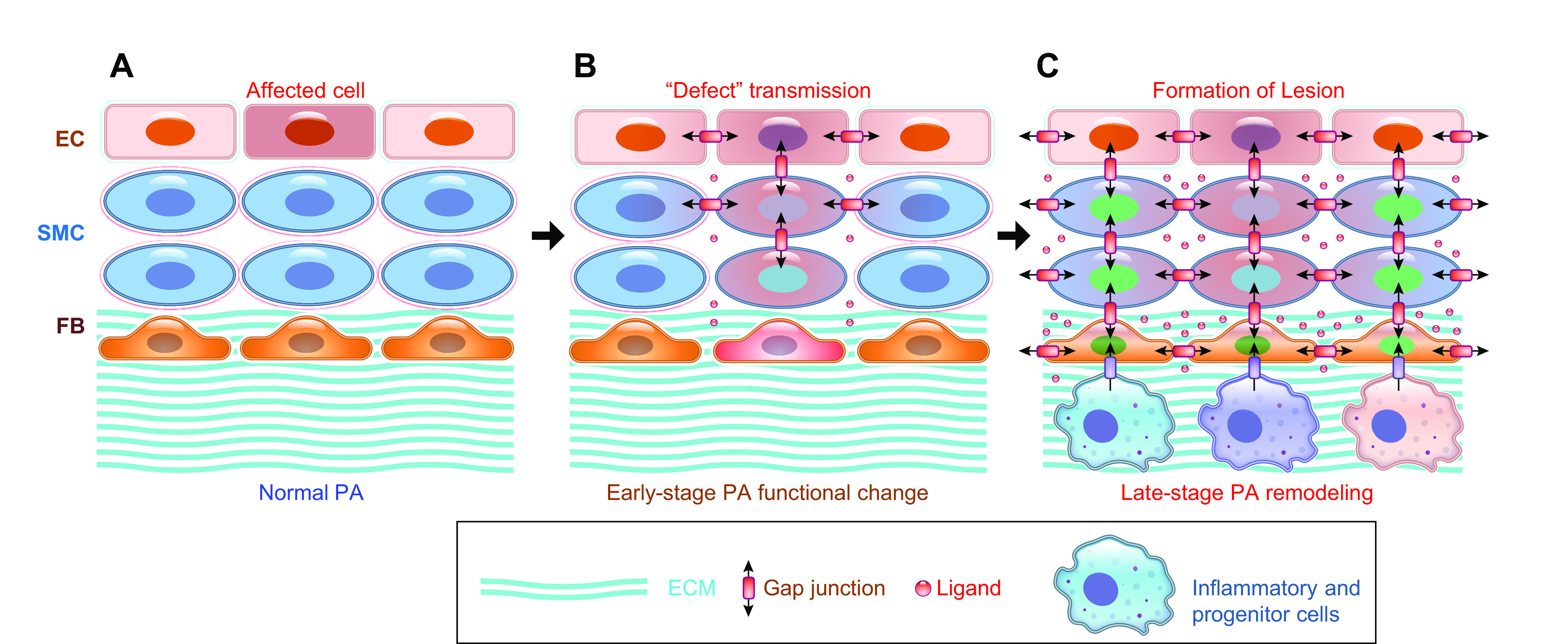
Juxtacrine signaling among pulmonary vascular endothelial cells (ECs), smooth muscle cells (SMCs), and fibroblasts (FBs) may contribute to spread “pathogenic signals.” A: schematic diagram showing the proposed scenario in which a single cell in normal pulmonary artery (PA) may become an affected cell or a “diseased” cell by, for example, somatic mutation and stimulation through localized pathogenic factors. B: the “diseased cell” then spreads the pathogenic signals to adjacent cells through juxtacrine signaling mechanisms to affect other cells in close vicinity. C: the pathogenic communication or interaction among the affected cells eventually forms the local lesion that can further develop and expand and, ultimately, affect the function and structure of the segment of the pulmonary vasculature, especially in the presence of inflammatory cells and pathogenic progenitor cells. ECM, extracellular matrix.
Connexin-40 (Cx40) is a gap junction protein that is predominantly expressed in vascular ECs. The inhibition or knockdown of Cx40 impairs vascular relaxation (334) and inhibits EC proliferation and migration (335, 336). Cx40 expression is decreased in the lungs (337, 338) and PAs (319) of animals with experimental PH and in the PAECs of patients with PAH and mice with experimental PH (308, 339). Systemic Cx40-knockout (KO) mice exhibit a significant increase in blood pressure (308, 340, 341) and a slight increase in right ventricular systolic pressure (RVSP), a surrogate measure of pulmonary arterial systolic pressure (308). In addition, overexpression of the Cx40 negative mutant in ECs showed a significant increase in RVSP with no change in systemic blood pressure. Endothelial overexpression of Cx40 reduces RVSP in chronically hypoxic mice by increasing endothelium-dependent pulmonary vasodilation. Decreased Cx40 is therefore associated with the development of experimental PH. Cx43, however, is increased in PASMCs from rats with hypoxia-induced PH but not from rats with monocrotaline-induced PH (321). Cx43 is upregulated in PAs from mice with hypoxia-induced PH and patients with chronic hypoxic diseases but not in patients with idiopathic PAH (322). In addition, upregulated Cx43 in lung vascular fibroblasts leads to fibroblast proliferation ex vivo (304). There are also reports showing that hypoxia decreases Cx43 expression in PAs (319). Cx37 is another connexin that is significantly increased in PAECs isolated from mice with hypoxia-induced PH (308). Given its potential role in inducing cell apoptosis (342) and cell cycle arrest (343, 344), it is possible that Cx37 is involved in endothelial injury during the development and progression of PH. Despite these reports, the pathogenic and protective effects of various connexins, such as Cx37, Cx40, Cx43, and other isoforms, on the pulmonary vasculature are still in need of further study (308, 319, 321, 322).
Pulmonary vasculopathy in patients with precapillary PH, which includes PAH and PH due to respiratory disease and/or hypoxemia, and in animals with experimental PH is a “local” and specific pulmonary vascular disease. Although pulmonary and systemic vasculature share similar physiological and pathophysiological properties, vascular disease in the pulmonary circulation is not always associated with vascular disease in the systemic circulation. For example, patients with idiopathic PAH often have normal hemodynamics in the systemic circulation, whereas patients with essential hypertension do not have pulmonary hypertension. It is still unknown whether pulmonary vasculopathy initiates from a single cell or a small group of cells in the pulmonary vasculature (like some forms of cancer), but we do know that the initial disorder (e.g., concentric arterial remodeling, increased myogenic tone and wall stiffness) affects all of the vascular branches to increase PVR and PAP. Therefore, it is likely that gap junctions play an important role in furthering the disease to extend throughout vascular branches. Based on the current knowledge, it appears that the decrease of Cx40 and the increase of Cx43 and Cx37 are involved in endothelial cell apoptosis and injury, decreased vessel relaxation, and increased pressure in the pulmonary branches.
4.5. Cell-to-Cell Communication Is the Key in the Development of Pulmonary Vasculopathy
An example of how pathogenic interactions among PAECs and PASMCs lead to the development of sustained pulmonary vasoconstriction, concentric pulmonary vascular wall thickening, and occlusive vascular lesions in PAH is shown in FIGURE 10 (63, 123, 254, 255, 345–357). In this scenario, the increased mitogenic ligands in the lungs bind to membrane receptors, GPCRs, and/or TKRs in PAECs (174, 358). The PKC-mediated activation of eNOS subsequently increases mitochondrial production of ROS (89, 345, 346, 359), an important activator of receptor-operated (ROC) and store-operated (SOC) Ca2+ channels in the plasma membrane (360). The phosphatidylinositol 3-kinase (PI3K)-mediated activation of AKT (or increased phosphorylation of AKT) (361) upregulates hypoxia-inducible factors (HIFs), which then upregulate SNAI1, a trigger for endothelial-to-mesenchymal transition (EndMT) in ECs (63, 362). The Ca2+/CaM- and HIF-mediated production and release of platelet-derived growth factor (PDGF) (174, 184, 241) and nicotinamide phosphoribosyltransferase (NAMPT) (254, 255, 364) trigger the respective receptors in PASMCs through the paracrine mechanism. Extracellular NAMPT, a ligand of Toll-like receptor 4 (TLR4) (256), serves as an autocrine and paracrine stimulator to increase NF-κB in PAECs and PASMCs to activate the inflammatory signaling pathway. The Ca2+/CaM-mediated upregulation of the Notch ligand Jag-1 in PAECs allows ECs to directly activate Notch receptors in PASMCs, through a juxtacrine mechanism. Subsequently, the activated Notch and PI3K/AKT/mammalian target of rapamycin (mTOR) signaling enhances PASMC proliferation and migration. The Notch- and NF-κB-mediated upregulation and activation of ROC and SOC in PASMCs further increase [Ca2+]cyt, causing PASMC contraction (221, 299). Eventually, the increased EC proliferation due partially to HIF/SNAI1- and NF-κB/SNAI1-mediated EndMT, the increased PASMC migration and proliferation due partially to Notch-, PDGF-, and NAMPT-mediated activation of Ca2+ signaling and other signaling pathways responsible for cell proliferation, and the increased PASMC contraction due to enhanced Ca2+ signaling all contribute to the development of pulmonary vascular remodeling and vasoconstriction. Activation of HIF under normoxic conditions and transition of the slow-growing EC phenotype to the highly proliferative myoFB phenotype during HIF-mediated EndMT may play an important role in forming occlusive vascular lesions and/or initiating pathogenic PASMC proliferation and concentric PA wall thickening. The longitudinal and transverse (or radical) signaling among ECs and SMCs in the pulmonary artery through autocrine, paracrine, and juxtacrine mechanisms serves as a critical pathogenic sequence of events for the development and progression of pulmonary vasculopathy in patients with PH.
FIGURE 10.
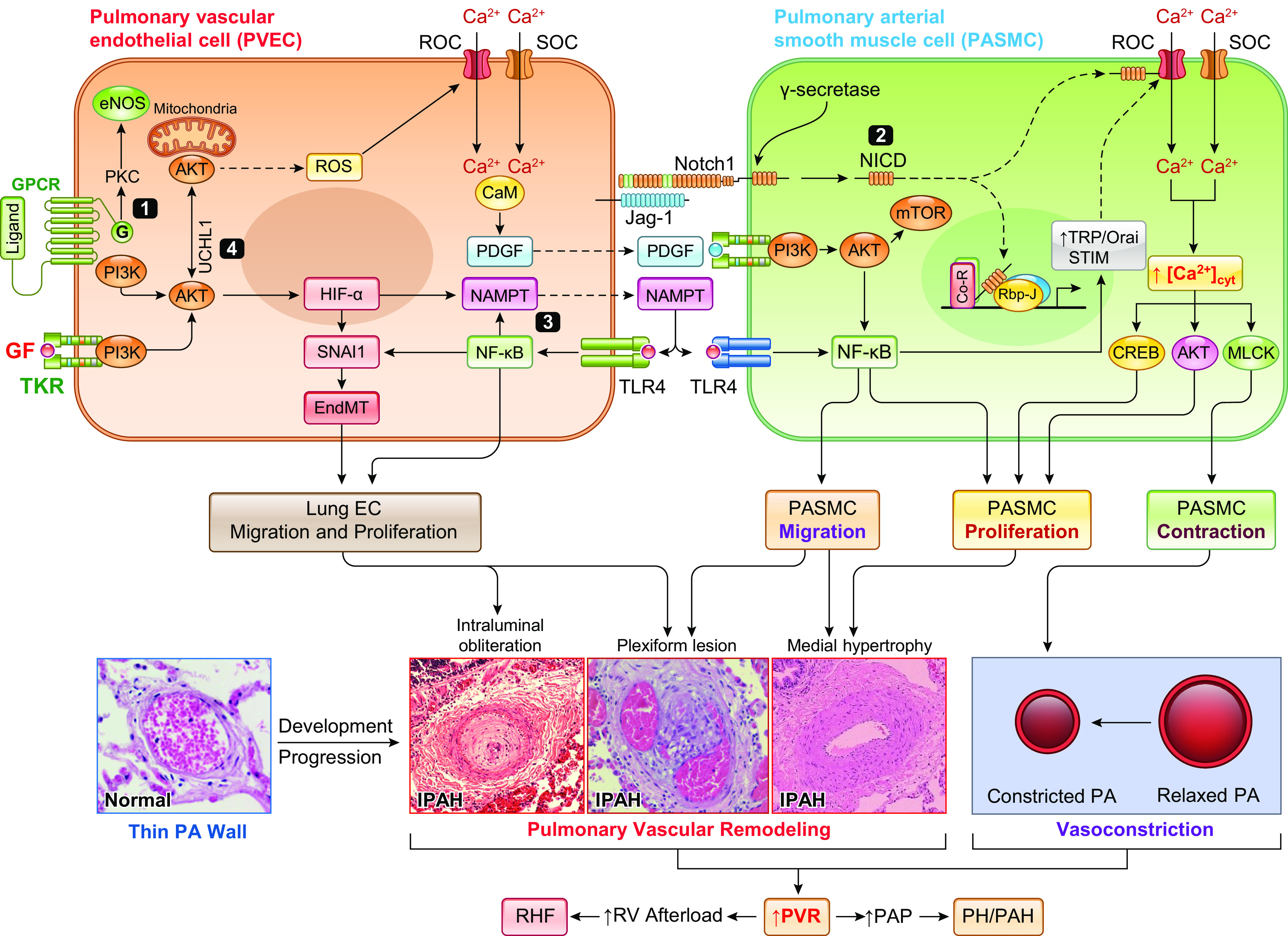
Potential pathogenic role of paracrine and juxtacrine interactions between pulmonary vascular endothelial cells (PAECs) and smooth muscle cells (PASMCs) in the development of pulmonary hypertension (PH). Aberrant phosphorylation of endothelial nitric oxide synthase (eNOS) due to G protein-coupled receptor (GPCR)-mediated activation of protein kinase C (PKC) results in eNOS uncoupling and reactive oxygen species (ROS) generation leading to increased EC proliferation via an eNOS-Akt-mitochondrial hypoxia-inducible factor (HIF) signaling axis (345, 346). The phosphatidylinositol 3-kinase (PI3K)/Akt/HIF/SNAI1 signal axis or the constitutively upregulated HIF-2α due to reduced PHD2 (63, 348, 349) results in endothelium-to-mesenchymal transition (EndMT), upregulates Notch ligand Jag-1, and increases the synthesis and production of platelet-derived growth factor (PDGF) and nicotinamide phosphoribosyltransferase (NAMPT) in PAECs. EndMT converts slow-growing PAECs to highly proliferative myofibroblasts (myoFBs), causing obliterative intimal lesions. Juxtacrine activation of Notch signaling via PAEC-PASMC interaction functionally activates and transcriptionally upregulates receptor-operated Ca2+ channels (ROCs) and store-operated Ca2+ channels (SOCs) to increase cytosolic free Ca2+ concentration ([Ca2+]cyt) in PASMCs, leading to PASMC contraction and pulmonary vasoconstriction and to PASMC proliferation and concentric pulmonary medial hypertrophy. HIF-mediated upregulation of PDGF and NAMPT (254, 255) in PAECs activates their receptors [PDGF receptor (PDGFR) and Toll-like receptor 4 (TLR4), respectively] in PASMCs through a paracrine mechanism, which results in PAMSC migration and proliferation through the PI3K/Akt/mammalian target of rapamycin (mTOR) (350–354) and NF-κB (123, 355) signaling pathways. Increased PASMC migration and proliferation contribute to the development and progression of concentric pulmonary vascular remodeling, arteriole muscularization, and occlusive vascular lesions. Enhanced NAMPT also activates TLR4/NF-κB signaling in PAECs via autocrine mechanism to enhance inflammation-associated PAEC proliferation and migration, whereas UCHL1-mediated regulation of AKT degradation further contributes to enhancing PAEC proliferation by promoting HIF and NF-κB signaling cascades (356, 357). Both NAMPT-NF-κB and Akt1/mTOR signaling also upregulate ROCs and SOCs involved in receptor- and store-operated Ca2+ entry, increase [Ca2+]cyt, and further induce PASMC contraction, migration, and proliferation. Ultimately, concentric PA wall thickening, sustained pulmonary vasoconstriction, and obliterative lung vascular lesions all contribute to increasing pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) in patients with pulmonary arterial hypertension (PAH). IPAH, idiopathic PAH; MCLK, myosin light chain kinase; NICD, Notch intracellular domain; RHF, right heart failure; TKR, tyrosine kinase receptor.
In patients carrying genetic defects [e.g., single-nucleotide polymorphisms (SNPs) or mutations in various genes] associated with heritable and idiopathic PAH, a cell undergoing somatic mutation, metabolic shift, or chromatin remodeling may become a “second hit” to initiate the disease process or function as a pathogenic cell. This initiates a “transmittable” pathogenic signal (e.g., dysfunctional mitochondria, specific composition of miRNAs, abnormal Ca2+ signals, cytoplasmic DNA) to affect other cells through the paracrine and juxtacrine mechanisms. The autocrine and intracrine (365, 366) mechanisms can also be used by the initial pathogenic cell(s) to spontaneously fulfill the transition from a normal cell to a “diseased” cell due to pathogenic stimulators produced as a result of genetic, genomic, and cellular defects.
Based on the current knowledge, this model illustrates what could occur at the molecular level to progress the pathological changes observed in PAH and PH. Mitogenic ligands bind to the membrane receptors of PAECs, which leads to the downstream signaling cascade increasing [Ca2+]cyt and stimulating endothelial-to-mesenchymal transition (EndMT) and endothelial inflammation. The gap junction or juxtacrine interaction among affected PAECs leads to the endothelial dysfunction, a major cause for sustained pulmonary vasoconstriction and vascular remodeling. In summary, gap junction channels like connexins or connexons not only allow intracellular ions (Ca2+, Na+, K+, Cl−) and second messengers (cAMP, cGMP, IP3, DAG, ATP) to transfer quickly from one cell to the other to affect adjacent cells’ function and homeostasis but also allow fragmented intracellular organelles (e.g., mitochondria, ER/SR, nuclear envelope) to travel from an affected cell (or somatically mutated cell) to adjacent normal cells to propagate pathogenic causes to affect the whole tissue segment. In addition to the plasmalemmal membranes, connexins are found to be in the nucleus (as a transcription factor) and on the mitochondrial membrane (as a trigger for apoptosis). The precise (pathogenic or protective) role of each connexin in the disease (e.g., PAH) progression and regression is still unclear, but their involvement creates and promotes an important and active research target for the field.
5. CELLULAR AND MOLECULAR MECHANISMS INVOLVED IN PULMONARY VASCULOPATHY IN PULMONARY ARTERIAL HYPERTENSION
At the cellular level, membrane receptors, ion channels and transporters, intracellular signaling pathways (or signaling proteins), and transcription factors all contribute to the development and progression of PAH/PH by regulating cell contraction (PASMC), migration (FB, PASMC, and EC), proliferation (progenitor cell, FB, PASMC, and EC), apoptosis (EC and SMC), and differentiation (progenitor cell, myoFB, SMC, and EC). A variety of extracellular ligands including vasoactive substances, growth factors, inflammatory and mitogenic cytokines, and metabolites are involved in the regulation of pulmonary vascular function (e.g., vasodilation and vasoconstriction) and structure (e.g., maintaining wall thinness under normal conditions and wall remodeling under pathological conditions). The balance between vasoconstrictive/proproliferative ligands and vasodilative/antiproliferative ligands is one of the early pathogenic changes that result in sustained vasoconstriction and concentric vascular remodeling. The extracellular or intercellular ligands involved in causing pulmonary changes include, but are not limited to, hormones/transmitters (e.g., PE, 5-HT), peptides (e.g., ET-1, ANG II), proteins (e.g., growth factors), ions (e.g., Ca2+, ROS), metabolic products (e.g., ATP, ADP), and extracellular vehicles that can carry more unconventional molecules (e.g., miRNA, ssDNA) to function as extracellular ligands. These ligands stimulate membrane receptors and ion channels in pulmonary vascular cells via autocrine, paracrine, and endocrine mechanisms (TABLE 5) (367, 368).
GPCRs (369) and TKRs [or receptor tyrosine kinases (RTKs)] are two major families of membrane receptors for which activation by mitogenic ligands stimulates pulmonary vascular cell contraction, migration, and proliferation. This then leads to sustained vasoconstriction and concentric vascular remodeling. Using cells isolated from patients with PAH and/or animals with experimental PH, many investigators have studied the potential involvement of specific GPCRs and TKRs in PAH/PH. Through these studies, multiple receptors (and their respective ligands) have been discovered for which the expression and function are enhanced in pulmonary vascular cells (i.e., FBs, SMCs, and ECs) and are involved in the development and progression of PH (TABLE 5).
Different membrane receptors and ion channels use varying downstream signaling cascades to initiate or promote the “pathogenic” signaling pathways for the development and progression of the disease through regulation of genes associated with cell proliferation, apoptosis, and differentiation (FIGURE 11). There are multiple membrane receptors, ion channels and transporters, and intracellular signaling pathways implicated in the development and progression of PAH and experimental PH (TABLE 5). Activation and/or inhibition of these membrane receptors and ion channels are thus important therapeutic strategies for treatment of sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, in situ thrombosis, occlusive vascular lesions, and pulmonary vascular wall stiffening.
FIGURE 11.

Cell signaling. Extracellular ligands (e.g., vasoconstrictive agonists, mitogenic and inflammatory factors) bind to and activate specific receptors [e.g., G protein-coupled receptors (GPCRs) and tyrosine kinase receptors (TKRs) or receptors that are ion channels]. The intracellular domains (e.g., G proteins or kinases) interact with intracellular signaling proteins and produce/activate transcription factors in the cytosol. Then, ligand/receptor-mediated transcription factors (TFs) translocate into the nucleus to bind to the promoters and enhancers of respective genes to stimulate or repress transcription of genes. There are different intracellular signaling cascades that transduce extracellular signals to the nucleus to control and regulate transcription of specific genes. DAG, diacylglycerol; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase.
5.1. Receptor-Operated and Mechanosensitive Ca2+ Signaling
In addition to G protein- and tyrosine kinase-mediated signaling cascades, GPCRs and TKRs also functionally interact with adjacent ion channels and transporters to regulate, for example, Ca2+ signaling in all cell types of the pulmonary vasculature and inflammatory cells. This process is involved in eliciting vasoconstriction via PASMC contraction and developing vascular remodeling (e.g., concentric PA wall thickening, arteriole muscularization, and occlusive vascular lesions) via cell migration and proliferation. One example of a receptor-mediated interaction with ion channels is receptor-operated Ca2+ influx through transient receptor potential (TRP) channels. TRP channels are a family of nonselective cation channels that are regulated by receptors via second messengers like diacylglycerol (DAG) (370). Many membrane receptors (GPCRs and TKRs) implicated in the development of PH are functionally coupled to ion channels like TRP channels. For example, we and others have reported that Ca2+-sensing receptor (CaSR), a GPCR subfamily C member that is activated by extracellular Ca2+ and other agonists (e.g., Mg2+, amino acids, polyamines and antibiotics) (222, 371–373), and TRPC6, a TRP canonical subfamily member of cation channels (370, 374–376), are upregulated in PASMCs from patients with idiopathic PAH and animals with experimental PH (123, 223–226, 377–379). The upregulated CaSR and TRPC6 as well as the enhanced CaSR-associated Ca2+ influx through TRPC6 channels enable the extracellular ligands (e.g., Ca2+, polyamine, neomycin, amyloid-β) of CaSR to become pathogenic stimuli by activating Ca2+ signaling cascades in PASMCs (FIGURE 12). CaSR is a unique GPCR that can be activated directly by a number of factors, many of which have been implicated in PH. These factors include polyvalent cations (e.g., Ca2+, Mg2+, Gd3+), polypeptides (e.g., amyloid-β peptide), polyamines (e.g., spermine, spermidine, putrescine), aminoglycoside antibiotics (e.g., neomycin, gentamicin, streptomycin, kanamycin), and amino acids (e.g., phenylalanine, tyrosine, tryptophan, glutamate) (221, 371, 373). The cations are consistently present in the blood/serum and extracellular fluids. Aminoglycoside antibiotics are often used to treat aerobic gram-negative bacilli (or Enterobacteriaceae) infections and pulmonary tuberculosis (against Staphylococci and Mycobacterium) (380). The blood concentration of amyloid-β is increased in patients with ischemic heart disease and hypertension (381), and the level of spermine is increased in serum from patients with cancer and PAH and in PA tissues from animals with experimental PH (382–384). Additionally, the free amino acid concentration of phenylalanine and tyrosine in blood is 0.97–0.98 mg/dL in normal fasting adults and can be increased to 1.50–2.31 mg/dL after protein/food intake (385). With upregulated and activated CaSR and TRPC6 in PASMCs, all the agonists of CaSR become either pathogenic or priming factors to stimulate vasoconstriction and vascular remodeling (FIGURE 12).
FIGURE 12.

Functional interaction of membrane receptor with ion channels in the plasma membrane via second messengers is important to link extracellular stimuli to the regulation of cell functions (e.g., contraction, migration, proliferation, apoptosis, and differentiation). This schematic diagram depicts the interaction of Ca2+-sensing receptor (CaSR), a unique G protein-coupled receptor (GPCR), with receptor-operated and mechanosensitive cation channels of TRPC6 via the second messenger diacylglycerol (DAG). CaSR can be activated by cations (e.g., Ca2+, Mg2+, Gd3+); amino acids like phenylalanine (Phe), tyrosine (Tyr), tryptophan (Trp), and glutamic acid (Glu); antibiotics like neomycin and streptomycin; and amyloid-β peptide. Activation of CaSR not only stimulates TRPC6 channels to induce extracellular Ca2+-mediated intracellular Ca2+ increase to induce pulmonary arterial smooth muscle cell (PASMC) contraction, migration, and proliferation but also activates the RAS/MAPK signaling pathway to reinforce its mitogenic effect. [Ca2+]cyt, cytosolic free Ca2+ concentration. CSD, caveolin scaffolding domain; MLC, myosin light chain.
Several other forms of TRP channels participating in forming receptor-operated and mechanosensitive Ca2+ channels are implicated in the development of PAH and experimental PH. TRPC1 channels are involved in acute hypoxia-induced pulmonary vasoconstriction and chronic hypoxia-induced PH (386, 387), whereas TRPC1/3/6 and TRPV1/4 are upregulated in PASMCs from patients with PAH and animals with experimental PH (131, 181, 378). TRPC6 and TRPV4 channels in the lungs are also involved in the development of lung edema or barrier function, lung injury, and PH (388, 389). The involvement of multiple TRP channels (including canonical and vanilloid subfamily members) indicates that the initial pathogenic triggers for PAH may transcriptionally upregulate various isoforms of TRP channels to ensure the pathogenic enhancement of Ca2+ signaling, which results in sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling. Indeed, a binding site analysis for the promoter regions of various TRPC and TRPV channel genes shows significant redundancy. In other words, a variety of TRP channel genes share similar binding sites in the promoter regions for many transcription factors.
Upregulated receptor-operated Ca2+ channels (e.g., TRPC6, TRPV1) and membrane receptors (e.g., CaSR) that are functionally coupled to these channels are demonstrated in cell and tissue samples from patients with PAH and animals with various experimental PH. The upregulation of these ion channels and receptors causes extracellular ligands to become pathogenic by enhancing Ca2+ signaling cascades in PASMCs. This demonstrates how external factors in the pulmonary milieu, which are necessary and unproblematic in a healthy subject, can become pathogenic stimuli to further progress vasoconstriction and remodeling. Given the functional role of these channels, it is hypothesized that enhanced Ca2+ influx through these upregulated cation channels is involved in the development and progression of sustained vasoconstriction and concentric vascular remodeling in precapillary PH (i.e., groups 1–3 PH) (1).
5.2. Clustering of Membrane Receptors and Ion Channels in Caveolae
Membrane receptors and ion channels, especially the ones that functionally interact with each other, can be clustered into caveolae to enhance the agonist/receptor/channel/signaling cascade, which then furthers the ligand-initiated effect on cell functions (e.g., contraction, migration, and proliferation) (390–393). In addition, caveolae in PAECs can be used to reduce shear stress-mediated or membrane stretch-mediated activation of mechanosensitive receptors/channels (394). Caveolin-1 (Cav-1), a protein that enables the formation of caveolae, also binds to and inhibits eNOS. In lung endothelial cells, downregulation of Cav-1, due to caveolin-1 mutations for example (395), has been implicated in the development of PAH and experimental PH, whereas Cav-1 deficiency results in experimental PH (396, 397). In PASMCs from remodeled pulmonary vasculature of patients with IPAH and animals with severe experimental PH, the protein expression of Cav-1 and the number of caveolae on the surface membrane are increased in comparison to normal controls (398). It is unclear whether and why mutations or SNPs in Cav-1 gene, identified in PAH patients, can divergently affect Cav-1 expression or caveola formation (see below) in lung vascular endothelial cells and smooth muscle cells.
Caveolae are membrane pits, with a size of 50–100 nm each, that are formed by caveolin/cavin-mediated invagination of the plasma membrane of many cell types, especially in ECs and adipocytes (399). Caveolae have flask-shaped structures and are rich in proteins (e.g., membrane receptors, channels, and transporters) and lipids (e.g., cholesterol, sphingolipids). They play an important functional role in ligand/receptor signal transduction, mechanoprotection and mechanosensation, and the uptake of pathogenic bacteria and viruses (392, 400). Caveolae also have a special type of lipid raft that functionally assembles membrane receptors and ion channels/transporters to enhance the ligand/receptor/signaling cascade. Furthermore, the membrane pits accumulate extracellular ligands and pathogenic factors (e.g., bacteria and viruses and toxins) to enable sustained stimulation or blockade of membrane receptors and ion channels/transporters located within caveolae (390, 391, 393, 398, 401).
In PASMCs isolated from patients with idiopathic PAH, the protein expression of Cav-1 and the number of caveolae are significantly upregulated and increased compared with PASMCs from normal subjects (FIGURE 13A) (398). In lung biopsy tissues obtained from patients with chronic thromboembolic pulmonary hypertension (CTEPH), many caveola structures can be found in lung capillary ECs by electron microscopy (FIGURE 13B) (O. Mathieu-Costello, unpublished observations). The increased number of caveolae due, potentially, to upregulated caveolins may contribute to the pulmonary vascular remodeling in patients with idiopathic PAH and CTEPH. In freshly isolated PA from rats, removal of membrane cholesterol with methyl-β-cyclodextrin (MβCD), which disrupts lipid raft and caveola structure, has little effect on high-K+-mediated pulmonary vasoconstriction but significantly inhibits agonist- or phenylephrine (PE)-mediated pulmonary vasoconstriction (402). The inability of MβCD to affect membrane depolarization-mediated PA contraction due to the opening of VDCCs in PASMCs indicates that VDCCs may distribute evenly in the plasma membrane of PASMCs. Raising extracellular K+ concentration ([K+]) from 4.7 to 40 mM shifts the equilibrium potential for K+ and causes membrane depolarization in the whole cell, subsequently opening VDCCs, increasing [Ca2+]cyt, and inducing PASMC contraction and pulmonary vasoconstriction (or the increase in isometric tension measured in the experiment shown in FIGURE 13C) (402, 403). The time-dependent significant inhibitory effect of MβCD on PE-induced PASMC contraction or pulmonary vasoconstriction indicates that the PE-activated receptor (e.g., α-adrenergic receptor) and its downstream second messengers may physically and functionally colocalize with the receptor-operated Ca2+ channels within caveolae. Disruption of caveolae by removal of membrane cholesterol with MβCD separates the receptor from its functionally coupled downstream effectors (e.g., signaling proteins and ion channels and transporters) and therefore reduces the ligand-mediated vasoconstrictive effect (FIGURE 13C). These experimental data direct us to hypothesize that membrane receptors, ion channels, and membrane transporters are not evenly distributed on the plasma membrane (FIGURE 13D). Instead, they suggest that they are clustered or colocalized together to form a “functional unit” by, for example, the lipid raft or membrane pits or caveolae. This well-organized and selectively formed unit is an important mechanism to enhance signal transduction, but overexpression of the receptors, effectors, and signaling proteins in it may also be pathogenic. The membrane pits can also function as a sac to accumulate and store extracellular ligands (e.g., agonists, growth factors) and, of course, pathogenic factors (e.g., bacteria, virus, heavy metals, ssDNAs) (FIGURE 13E). Alternatively, distribution of membrane receptors and ion channels/transporters in structures like caveolae may also play a critical role in protecting or maintaining normal cell function in tissues and organs under constant shear stress and membrane stretch (e.g., the lungs) (FIGURE 13F). The mechanoprotection effect of caveolae is implicated not only in cardiovascular ECs but also as a general mechanism in other types of cells (400, 404–406).
FIGURE 13.
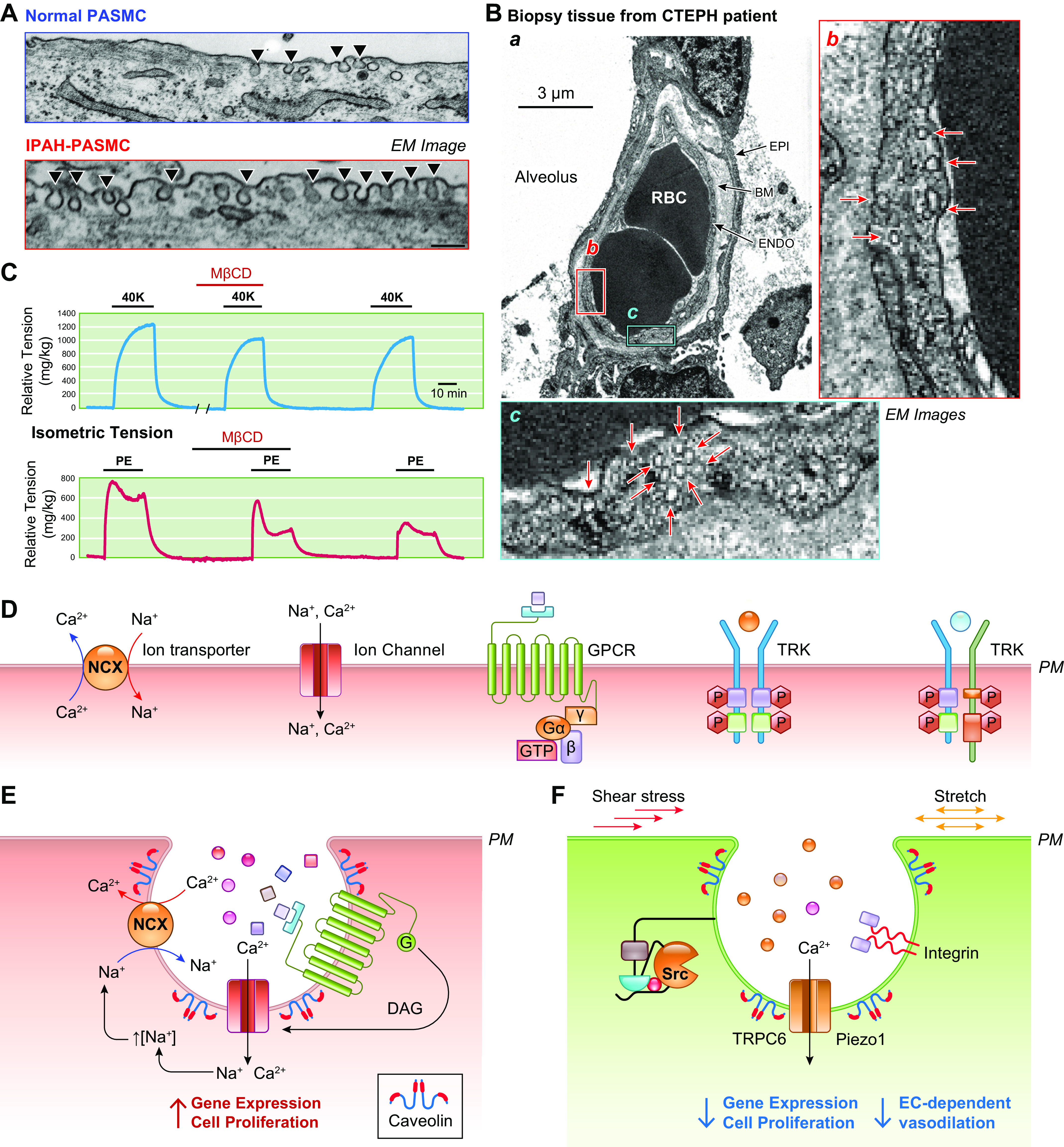
Caveola-mediated clustering of membrane receptors and ion channels in pulmonary arterial smooth muscle cells (PASMCs) is implicated in the development of pulmonary vasoconstriction and remodeling in pulmonary arterial hypertension (PAH) and pulmonary hypertension (PH). A: electron microscopy (EM) images showing caveola (indicated by arrowheads) in the surface membrane of PASMCs from a normal subject (top) and a patient with idiopathic PAH (IPAH) (bottom). The number of caveolae in the plasma membrane of IPAH PASMCs is significantly more than in the membrane of normal control PASMCs; the increased caveola number in IPAH PASMCs is associated with upregulation of caveolin-1/2 (398). B: EM images showing the lung capillary endothelial cell (ENDO) and alveolar epithelial cell (EPI) of the biopsy tissue from a patient with chronic thromboembolic pulmonary hypertension (CTEPH) (a; O. Mathieu-Costello, unpublished observations). Many caveola (indicated by arrows) are found in the different areas of the endothelial cell membrane (b in red box and c in cyan box), as shown in enlarged images (b and c) corresponding to the boxes in the main image. C: isometric tension measured in isolated rat pulmonary artery (PA) challenged with 40 mM K+ (40K)-containing physiological salt solution (PSS) (top) or phenylephrine (PE)-containing PSS (bottom), in the presence or absence of methyl-β-cyclodextrin (MβCD) (402, 403). MβCD, which disrupts caveolae in the plasma membrane by removing cholesterol, had negligible effect on 40K-mediated PA contraction (top) but significantly inhibited PE-mediated PA contraction (bottom). D: schematic diagram depicting membrane receptors, ion channels, and transporters in the plasma membrane (PM) in the absence of caveolae. E: schematic diagram depicting a cluster of G protein-coupled receptor (GPCR), cation channel (e.g., TRPC6), and Na+/Ca2+ exchanger (NCX) in the membrane of caveolae formed by caveolin (Cav) as well as accumulated extracellular ligands in the caveola sac. F: schematic diagram showing mechanosensitive receptor (e.g., Src), integrin, and cation channel (e.g., Piezo1 or TRPC6) in the caveola membrane. Formation of caveolae and relocation of mechanosensitive membrane proteins in caveolae play an important mechanoprotection role in preventing cell membrane from shear stress and/or membrane stretch. DAG, diacylglycerol; NCX, Na+/Ca2+ exchanger. BM, basement membrane; RBC, red blood cell; TRK, tyrosine receptor kinase.
Mutations or SNPs in Cav1 are associated with PAH in patients (395), and genetic deletion of the Cav1 gene in mice results in PH (407). It is, however, still unclear how the Cav-1 mutation and deficiency result in vascular remodeling and obliterative lesions in PAH and experimental PH (391, 393, 395–398). It is possible that the function of caveolae in PASMCs and ECs is very different. For example, an increased number of caveolae and/or an increased release of caveolin in PASMCs clusters receptors with ion channels and downstream signaling proteins to enhance the signal transduction for cell contraction, migration, and proliferation (391, 393). On the other hand, fewer caveolae in PAECs function to make the endothelium more sensitive to mechanical (e.g., shear stress and membrane stretch) stimulation and thus induce EC injury-associated pulmonary vasculopathy. It should be noted that Cav-1 (and other caveolins), although important to form caveolae, may directly bind to membrane proteins and affect their functions. For example, in ECs Cav-1 binds to eNOS and inhibits its activity to reduce the production of NO (392, 408) and stabilizes eNOS expression (409). Cav-1 deficiency-associated activation of eNOS is also implicated in the development of PH because of eNOS uncoupling or eNOS-mediated production of ROS and posttranslational modification of intracellular proteins (397). Cav-1 is critical to retain TRPC1 channels within the regions where STIM1 puncta are localized and enable the interaction of TRPC1 with STIM1 for store-operated Ca2+ entry (393).
As discussed above, the pulmonary vasculopathy may be initiated from one initial pathogenic cell (due to somatic mutations, for example) and then the disease phenotype spreads to other adjacent cells through paracrine and juxtacrine mechanisms in the whole vascular segment. In a single cell, the pathogenic signal may also initiate from a restricted, localized, and selective cluster containing dysfunctional or upregulated receptors, ion channels, and signaling proteins. The localized pathogenic changes in, for example, the “dysfunctional unit” may then spread to the whole cell to convert the cell to a pathogenic cell. It is important to use advanced technology and experimental approaches to find these kinds of pathogenic clusters in single cells.
Based on evidence gathered thus far, it becomes clear that mutations in Cav-1, which is responsible for the formation of caveolae in the lungs, are associated with PAH. An increase in caveolins in PASMCs is seen in patients and animals with PAH, likely because of their ability to enhance “pathogenic” ligand-receptor interactions and the harboring of pathogenic factors such as bacteria and virus, as well as single-stranded RNAs and cell-free DNAs. However, a decrease in caveolins in ECs also contributes to PAH in patients carrying Cav-1 gene mutations, potentially because of the decrease in protection they offer to the EC barrier, leading to endothelial dysfunction and injury. However, the mechanism of the role of Cav-1 deficiency in PAH and PH is still relatively unknown and in need of further study.
5.3. Ion Channels and Ca2+ Signaling in Pulmonary Vasoconstriction and Vascular Remodeling
A rise in [Ca2+]cyt in PASMCs, as discussed above (FIGURE 5C), is a trigger for PASMC contraction (which results in pulmonary vasoconstriction) and migration and proliferation (which result in pulmonary vascular remodeling). In addition, a rise in [Ca2+]cyt, along with activation of the Ca2+ or Ca2+/CaM-sensitive signaling proteins and pathways (e.g., calpains, MAPK, NFAT, AKT, NF-κB, and RhoA), is involved in the activation of inflammatory cells and the migration and proliferation of perivascular progenitor cells and endothelium-derived myofibroblasts. This contributes to the development of concentric pulmonary vascular remodeling and occlusive vascular lesions.
Upon activation of a membrane receptor, cytosolic [Ca2+] is increased by a transient Ca2+ release from intracellular stores, such as the SR/ER, followed by a sustained Ca2+ influx through different Ca2+ channels in the plasma membrane (410). Cytosolic [Ca2+] is decreased by Ca2+ sequestration or uptake into the SR/ER by the SR/ER Ca2+ pump (SERCA, or Ca2+-Mg2+-ATPase) and by Ca2+ extrusion via the plasma membrane Ca2+ pump and Na+/Ca2+ exchanger (NCX) (411). Under resting conditions (when cells are not stimulated by agonists), the extracellular [Ca2+] is 1.4–2.0 mM, the cytosolic [Ca2+] is ∼100 nM, and the membrane potential is at the range of −40 to −85 mV (102, 103). The electrochemical gradient, or the driving force, is thus in great favor of a Ca2+ influx when Ca2+-permeable cation channels in the plasma membrane are opened.
There are at least three major pathways for Ca2+ entry in PASMCs and other cells: 1) receptor-operated Ca2+ entry through receptor-operated Ca2+ channels (ROCCs) upon receptor (e.g., GPCR) activation (FIGURE 14A), 2) store-operated Ca2+ entry (SOCE) due to Ca2+ influx through store-operated Ca2+ channels (SOCCs) when Ca2+ is depleted or reduced from the intracellular stores (e.g., SR and ER) (FIGURE 14B), and 3) voltage-dependent Ca2+ entry (VDCE) due to Ca2+ influx through VDCCs when the cell membrane is depolarized or membrane potential (Em) becomes less negative (membrane depolarization) or becomes more positive (action potential) (412).
FIGURE 14.

Proposed mechanisms of receptor-operated Ca2+ entry (ROCE) and store-operated Ca2+ entry (SOCE). A and B: upon ligand binding to receptor [e.g., G protein-coupled receptor (GPCR)], phospholipase C (PLCβ for GPCR activation) cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to produce diacylglycerol (DAG) and inositol (1,4,5)-trisphosphate (IP3). DAG activates receptor-operated Ca2+ channels (ROCCs) like TRPC6, allowing Ca2+ influx that is often termed as ROCE (A). IP3 activates IP3 receptor (IP3R), a Ca2+ release channel, in the sarcoplasmic reticulum (SR) membrane, allowing Ca2+ release or mobilization to the cytosol. The subsequent store depletion opens store-operated Ca2+ channels (SOCCs) (formed by Orai/STIM and TRPC) in the plasma membrane and induces SOCE (B). Ca2+ influx through ROCC/TRPC6 and/or SOCC/Orai/STIM-TRPC is a major contributor to raising cytosolic free Ca2+ concentration [Ca2+]cyt in pulmonary arterial smooth muscle cells (PASMCs) stimulated by agonists. Na+ influx through ROCC/TRPC6 and/or SOCC/Orai/STIM-TRPC would cause a localized increase in cytosolic [Na+], which triggers Na+/Ca2+ exchanger (NCX) (the reverse mode) to outwardly transport Na+ and inwardly transport Ca2+, which further increases [Ca2+]cyt and eventually results in PASMC contraction, migration, and proliferation. C: changes of the isometric tension in isolated pulmonary arterial (PA) ring before, during, and after application of phenylephrine (PE, an α-adrenergic receptor agonist) in 1.8 mM Ca2+-containing solution or Ca2+-free solution (0Ca) with or without phentolamine (an α-receptor blocker). The data show 3 components of pulmonary vasoconstriction induced by Ca2+ release from intracellular stores [or SR/endoplasmic reticulum (ER)], SOCE, and ROCE. Inset: isolated mouse PA ring. D: single-channel cation currents recorded in the cell-attached membrane patch (inset) of PASMCs before (Control) and during extracellular application of cyclopiazonic acid (CPA), an inhibitor of SERCA that depletes intracellularly stored Ca2+ in the SR/ER and induces SOCE. CPA-mediated store depletion rapidly induces an inward cation current with a slope conductance at the range of 5–15 pS in PASMCs. Popen, steady-state open probability.
One of the most important signaling pathways is through second messengers derived from the membrane lipid phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). Activation of both GPCRs (via phospholipase C-β or PLCβ) and TKRs (via PLC-γ) stimulates PIP2 hydrolysis, which produces two distinct second messengers, diacylglycerol (DAG) and inositol (1,4,5)-trisphosphate (IP3) (413). The second messenger mediating receptor-operated Ca2+ entry is mainly the membrane-attached DAG, which directly activates ROCCs, such as TRPC3/6/7 channels (414), increases Ca2+ influx or ROCE, and raises [Ca2+]cyt. Many ROCCs are nonselective cation channels (e.g., TRP channels), so the DAG-mediated opening of ROCCs/TRPs also allows Na+ to enter the cell. A local or submembrane increase in Na+ concentration ([Na+]) is a major trigger to stimulate NCX (411, 415); the inward transportation of Ca2+ due to NCX is thus another important route for the increased [Ca2+]cyt in cells stimulated by agonists (416–419) (FIGURE 14A). The DAG-mediated activation of protein-serine/threonine kinases (or PKC family protein kinases) also leads to cell proliferation and gene expression through, for example, the MAP kinase pathway and phorbol esters (420). Although DAG remains associated with the plasma membrane, IP3 is a small polar molecule that is released to the cytosol. There it activates IP3 receptors (IP3Rs) in the SR and ER membranes, allowing Ca2+ mobilization to the cytosol (and other intracellular organelles) (421, 422). Activation depletion of intracellularly stored Ca2+ in the SR/ER due to IP3-mediated Ca2+ release or mobilization, along with passive depletion of Ca2+ in the SR/ER due to inhibition of SERCA (by thapsigargin or cyclopiazonic acid), is the major cause for inducing store-operated Ca2+ entry (FIGURE 14B). Low [Ca2+]cyt (∼100 nM) under resting conditions is maintained because of Ca2+ pumps in the plasma membrane (Ca2+ extrusion) and Ca2+ pumps in the SR/ER membrane (Ca2+ sequestration or uptake). Therefore, the [Ca2+] in the SR/ER can be as high as 0.1–1 mM. Activation of IP3R or other Ca2+ release channels, such as ryanodine receptors (RyRs), or the inhibition of SERCA can efficiently result in Ca2+ transportation from the SR/ER to the cytosol based on the chemical gradient and, ultimately, to store depletion and SOCE.
Receptor-operated Ca2+ entry and store-operated Ca2+ entry are both involved in pulmonary vasoconstriction (423–425) and PASMC proliferation (242, 426,427). In isolated PA rings from animals, phenylephrine (PE) caused a transient constriction determined by an increase in isometric tension in the absence of extracellular Ca2+ (0 Ca). This transition contraction was due to IP3-mediated Ca2+ release upon PE-mediated α-adrenergic receptor activation. Blockade of α-receptors with phentolamine in the absence of extracellular Ca2+ further decreases PA contraction initiated by IP3-mediated Ca2+ release, potentially because of a receptor-mediated increase in Ca2+ sensitivity of the contractile apparatus. While α-receptors were blocked by phentolamine, restoration of extracellular Ca2+ (to 1.8 mM) resulted in an increase in tension or PA contraction due apparently to store-operated Ca2+ entry. Then, washout of phentolamine with PE-containing solution further increased the isometric tension due to PA contraction because of receptor-operated Ca2+ entry (FIGURE 14C). These studies indicate that, upon agonist-mediated activation of membrane receptors, IP3-mediated Ca2+ release, store-operated Ca2+ entry, and receptor-operated Ca2+ entry all contribute to the increase in [Ca2+]cyt in PASMCs causing pulmonary vasoconstriction. Furthermore, passive depletion of intracellularly stored Ca2+ in the SR in primary cultured PASMCs is able to increase [Ca2+]cyt via store-operated Ca2+ entry (123, 242, 377) as a result of opening SOCCs (FIGURE 14D) (426, 428).
VDCE through different types of VDCCs, such as the L-type VDCC and the T-type VDCC, is controlled and regulated by changes in membrane potential (Em) (102, 103). Em is generated and regulated by the activity of the electrogenic Na+ pump or Na+-K+-ATPase (429) and various K+ channels in the plasma membrane (411). Decrease of K+ channel activity and downregulation of K+ channel expression are implicated in causing membrane depolarization and opening of VDCCs in PASMCs causing pulmonary vasoconstriction (105). Meanwhile, activation of K+ channels or increase of K+ efflux is involved in membrane repolarization or hyperpolarization causing pulmonary vasodilation (430) (FIGURE 15A). Given the high membrane resistance, a small change of K+ channel activity can have a substantial effect on membrane potential (431). In addition, anion channels like voltage-gated and Ca2+-activated Cl− channels are involved in the regulation of Em in human and animal PASMCs (432–437), whereas changes in Cl− channel activity are implicated in the development of PH (438–442). Since the intracellular concentration of Cl− in SMCs is very high, the opening of Cl− channels at rest (where the resting Em is −40 to −60 mV) would result in inward currents due to Cl− efflux and membrane depolarization in PASMCs.
FIGURE 15.

Proposed mechanisms of voltage-dependent Ca2+ entry. A: membrane potential (Em) is regulated by Na+ pump (or Na+-K+-ATPase) and K+ channels in the plasma membrane. Decreased K+ currents (IK) due to blockade or dysfunction of K+ channels and/or downregulation of K+ channel expression (and inhibited Na+ pump by, for example, ouabain) lead to membrane depolarization that opens voltage-dependent Ca2+ channels (VDCCs), increases Ca2+ influx (or voltage-dependent Ca2+ entry), raises cytosolic free Ca2+ concentration [Ca2+]cyt, and eventually induces pulmonary arterial smooth muscle cell (PASMC) contraction, migration, and proliferation. B: representative records showing whole cell voltage-gated K+ (KV) currents (a), membrane potential (Em, b) and [Ca2+]cyt (c) in PASMCs before (Control), during (4-AP) and after (Recovery) extracellular application of 4-aminopyridine (4-AP), a relatively selective blocker of KV channels. 4-AP-mediated decrease in whole cell KV currents (a) depolarizes the cell and induces Ca2+ action potentials (b) and induces a sustained increase in [Ca2+]cyt due apparently to Ca2+ influx through VDCC (c). C: representative records showing whole cell KV currents (a), elicited by depolarizing the cell from a holding potential of −70 mV to a series of test potentials ranging from −60 to +80 mV (in 20-mV increments) in PASMCs before [normoxia (Nor), Po2 = 155 Torr], during [hypoxia (Hyp)], and after (Recovery, Po2 = 155 Torr) acute superfusion of hypoxic solution (Po2 = 15 Torr). Acute hypoxia-mediated decrease in whole cell KV currents is associated with membrane depolarization (b and c) in PASMCs. Resting Em is measured with the patch-clamp technique in the current-clamp mode (b), whereas the histogram of Em recorded in PASMCs under normoxic (c, top) and hypoxic (c, bottom) conditions is constructed from 29 normoxic cells and 38 hypoxic cells. **P < 0.01 vs. Nor.
There are at least four families of K+ channels functionally expressed in pulmonary vascular cells including PASMCs (443): 1) voltage-gated K+ (KV) channels (444), 2) Ca2+-activated K+ (KCa) channels (445, 446), 3) ATP-sensitive K+ (KATP) channels (447, 448), and 4) two-pore domain K+ (K2P) channels or tandem-pore domain K+ channels (449).
KV channels are a diverse subfamily of K+ channels in vascular smooth muscle cells. A functional KV channel is either a homotetrameric channel composed of four KV channel pore-forming α-subunits (e.g., KCNA5) or a heterotetrametric channel composed of four different KV channel α-subunits (e.g., KCNA1/KCNA2 and 2 KCNA5), whereas the auxiliary KV channel β-subunits alter the KV channels’ biophysical and pharmacological properties (450–452). The KV channel β-subunit confers the sensitivity of KV channels to oxygen, hypoxia, and redox status (451, 453, 454). KV channels are activated by membrane depolarization and play an important role in the regulation of resting membrane potential (e.g., inducing membrane repolarization and hyperpolarization) in PASMCs (105, 455, 456). Extracellular application of 4-aminopyridine (4-AP) significantly and reversibly decreases whole cell KV currents recorded in PASMCs with Ca2+-free and EGTA-containing bath solution and ATP-including pipette solution (which minimizes the contribution of KCa and KATP currents to the whole cell currents) (FIGURE 15Ba). The 4-AP-mediated decrease in KV currents was associated with membrane depolarization, generation of Ca2+-dependent action potentials (FIGURE 15Bb), and an increase in [Ca2+]cyt (FIGURE 15Bc) in PASMCs. Furthermore, acute hypoxia or a change in Po2 from 155 Torr to 15 Torr significantly and reversibly decreases whole cell KV currents in PASMCs and causes membrane depolarization (or a 14-mV change of Em) (FIGURE 15C). It has been demonstrated that acute hypoxia inhibits different K+ channels (e.g., KV, KCa, and K2P channels) and causes membrane depolarization in PASMCs. This subsequently opens VDCCs, promotes Ca2+ influx through L-type VDCCs, increases [Ca2+]cyt, and causes hypoxic pulmonary vasoconstriction (76). In addition, chronic hypoxia downregulates K+ channels and stimulates PASMC proliferation by increasing VDCE through L/T-type VDCCs (457–461) and inhibits PASMC apoptosis by inhibiting apoptotic volume decrease and intracellular caspase activity (93, 462–469).
KCa channels are functionally classified into three subfamilies based on their single-channel conductance: the large-conductance (BK or MaxiK) KCa channel (g = 100–300 pS), the intermediate (IK) KCa channels (g = 25–100 pS), and the small-conductance (SK) KCa channel (g = 2–25 pS) (445, 446, 470). Similar to KV channels, the BK channel is a tetrameric channel that is activated by membrane depolarization and intracellular Ca2+ (445). KCa channels function as a negative-feedback effector in many excitable cells including vascular SMCs in response to membrane depolarization and an increase in [Ca2+]cyt. In PASMCs, the outward K+ currents through large-conductance KCa channels (BKCa or MaxiK channels) are often recorded in cell-attached membrane patches (FIGURE 16A). The slope conductance of BKCa channels in PASMCs is between 200 and 250 pS. The steady-state open probability of the large-conductance BK channels can be significantly and reversibly increased by NO or the NO donor S-nitroso-N-acetylpenicillamine (SNAP) (FIGURE 16B). Similar to KV channels, activity and expression of the large-conductance KCa channels are also associated with PAH and experimental PH. Activation of BK channels inhibits the progression of experimental PH (471–475); however, whether KCa channels are downregulated or upregulated in remodeled PA from animals with experimental PH remains unclear (476–478).
FIGURE 16.

Large-conductance Ca2+-activated K+ (KCa) currents in pulmonary arterial smooth muscle cells (PASMCs). A: representative single-channel K+ currents in cell-attached membrane patch elicited by depolarizing the patch to a series of test potentials in a PASMC bathed in physiological salt solution (left). The current-voltage (I-V) relationship curve (right), which shifts to the right because of the membrane potential (−43 mV), shows a slope conductance (g) of 249 pS. B, top: representative single-channel KCa currents in a cell-attached membrane patch of PASMC before (Control), during (NO), and after (Recovery) extracellular application of the nitric oxide (NO) donor S-nitroso-N-acetylpenicillamine. Bottom: the steady-state open probability (NPopen) of the currents.
K2P channels are a family of 15 members that form the so-called “leak channels” and generate K+ leak currents or “background currents” in excitable and nonexcitable cells. These channels are often regulated by signaling lipids, hypoxia and hyperoxia, and pH (479). Some of the K2P channels are also mechanosensitive and receptor operated (449, 480, 481). For KV channels, each pore-forming α-subunit contains six transmembrane domains (6 TMD) with one pore (P) region and the α-subunits form functional tetramers. K2P channels are characterized by the presence of two pore-forming regions and four transmembrane domains in each channel subunit. It is believed that two subunits of K2P are necessary to form functional dimeric channels. K2P channels are expressed in PASMCs and generate a noninactivating current showing no classic time- and voltage-dependent activity (481, 482). Loss-of-function mutations/SNPs of the KCNK3 gene (483), which encodes the K2P channel TASK-1, have been demonstrated to be partially responsible for the enhanced Ca2+ signaling observed in PASMCs from patients with PAH and animals with experimental PH (443, 484, 485). As a channel contributing to background K+ current, inhibition of the channels initiates membrane depolarization (480, 486, 487), which subsequently opens VDCCs and enhances Ca2+ influx. This increases [Ca2+]cyt in PASMCs (488) and eventually causes pulmonary vasoconstriction and vascular remodeling.
KATP channels are structurally composed of an octameric complex of four pore-forming subunits of inward-rectifier K+ (KIR) channel subunits (e.g., KIR6.1/KCNJ8 and KIR6.2/KCNJ11) and four sulfonylurea receptors (SURs) (447, 448, 489, 490). KATP channels show little or no voltage dependence and have low open probability under basal conditions. KATP channels are inhibited by intracellular ATP with IC50 of 14.5 µM (491). Coexpression of SUR with KIR6.1 (KCNJ8) or KIR6.2 (KCNJ11) produces different KATP channels that are activated by nucleotide diphosphate and inhibited by ATP (492). KIR6.1 (KCNJ8) and SUR2B are expressed in human PASMCs (493). Given the high concentration of intracellular ATP, KATP channels may normally be closed in the pulmonary arteries and therefore cannot be activated by hypoxia that causes pulmonary vasoconstriction (494). Recently, loss-of-function mutations in the ABCC8 gene that encodes sulfonylurea receptor 1 (SUR1) have been associated with PAH (448, 495, 496). These findings will shed further light on the involvement of dysfunctional K+ channels in the development and progression of PAH and experimental PH. Using KATP channel openers to treat PAH and PH due to respiratory diseases and/or hypoxemia could potentially be a good strategy to develop new therapeutic approaches for PAH (443, 448, 497). Indeed, several KATP channel activators (e.g., levosimendan, iptakalim, diazoxide, VU0071063, NN414) show promising therapeutic effects on PAH and experimental PH (498–501).
Among the four different K+ channels, the pore-forming α-subunit of KV channels contains six transmembrane domains (TMDs) with the pore (P) between TMD5 and TMD6 and the voltage sensor in TMD4. Functional KV channels are homotetramers (formed by 4 identical α-subunits) or heterotetramers (formed by 4 different α-subunits) with four regulatory β-subunits. The pore-forming α-subunit of KCa (BKCa) channels has a six-TMD structure similar to the KV channel α-subunit but contains a (TMD0) domain that makes the NH2 terminal outside of the cell. Functional KCa (BKCa) channels are also tetramers with four pore-forming α-subunits and four regulatory β-subunits. K2P channels contain two pore-forming regions (P1 and P2) and four transmembrane domains in each channel subunit. Functional K2P channels are thus dimers. The pore-forming subunits for KATP channels are inward-rectifier channel subunits (e.g., KIR6.x) that contain two TMDs and a pore-forming region. Four KIR6.x subunits and four sulfonylurea receptors (SUR1 or SUR2) are required to form functional octameric KATP channels (FIGURE 17).
FIGURE 17.

Schematic diagram showing structure of K+ channels. Planar membrane topologies of single K+ channel subunits for a voltage-gated (KV) K+ channel (A), a Ca2+-activated (BKCa) K+ channel (B), a 2-pore domain (K2P) K+ channel (C), and a ATP-sensitive K+ (KATP) channel (D), which is composed of inward-rectifier (KIR) K+ channel and sulfonylurea receptor (SUR). The pore-forming loop is indicated (P) and the voltage sensor (+) in the fourth transmembrane domain (TMD4) for KV (A) and BKCa (B) channels, which are homotetramers or heterotetramers with 4 β-subunits (left and center). C: membrane topology of a K2P channel subunit featuring 2 pore regions, P1 and P2, and 4 transmembrane spanning domains and cytoplasmic NH2 and COOH termini. The functional K2P channels are thus dimers. D: the KATP channels are heterooctamers formed by 4 pore-forming KIR subunits (e.g., KIR6.x) and 4 SURs. Right: the channel genes associated with pulmonary arterial hypertension (PAH) and the decreased (↓) whole cell K+ currents through different K+ channels found in PASMCs from PAH patients or related to the mutations/single-nucleotide polymorphisms (SNPs) identified from PAH patients. For BKCa channels, knockout (KO) of KCNMB1, a β-subunit of the large-conductance KCa channel, enhances experimental pulmonary hypertension (PH) in mice.
Downregulated and/or dysfunctional K+ channels (e.g., KV/KCNA5, KCa/KCNMA1-KCNMAB1, K2P/KCNK3, KATP/SUR1 channels) are demonstrated in cell and tissue samples from patients with PAH and animals with experimental PH (104, 485, 488, 500–507). The decreased K+ currents through various K+ channels are not only due to mutations and/or downregulation of the pore-forming α-subunits but also related to the regulatory β-subunits. For example, knockout of KCNMB1, a β-subunit of the BKCa channel (e.g., KCNMA1), enhances the development of experimental PH in mice (506). Activity of K+ channels or intracellular [K+] is not only important for the regulation of membrane potential (102–105) but also pivotal for the regulation of cell apoptosis (462, 467, 508–510) and inflammation or inflammasome formation (511–514). Inhibition of K+ efflux due to reduced number of K+ channel proteins in the plasma membrane and/or reduced open probability and conductance of K+ channels can thus reinforce the antiapoptotic and anti-inflammatory effects on various vascular cells and inflammatory cells contributing to the development of pulmonary vascular remodeling in PAH and other types of precapillary PH (i.e., groups 1–3 PH) (1).
In addition to Ca2+ channels and K+ channels, it has been demonstrated that Na+ channels (e.g., acid-sensing ion channel, ASIC) as well as Na+/H+ (NHE) and Na+/Ca2+ (NCX) exchangers are also involved in the regulation of pulmonary vasoconstriction and vascular remodeling. ASICs are voltage-insensitive cation channels activated by extracellular H+ (515). ASIC1, a nonselective cation channel that conducts both Na+ and Ca2+, is expressed in PASMCs. ASIC1-mediated Na+ influx induces membrane depolarization in PASMCs to trigger the voltage-dependent Ca2+ entry pathway, whereas ASIC1-mediated Ca2+ influx contributes directly to increasing [Ca2+]cyt in PASMCs. Through patch-clamp and in vivo animal experiments, the Jernigan and Resta group (516–519) provides compelling evidence that increased nonselective cation currents through ASIC1 channels and membrane depolarization in PASMCs contribute to the development of experimental PH. Changes in extracellular and intracellular H+ or pH can also contribute to the development and progression of PAH and experimental PH by signaling through the Na+/H+ exchanger expressed in the surface membrane of PASMCs (and other types of pulmonary vascular cells and infiltrated inflammatory cells) (520, 521). Increased NHE1 activity promotes lung vascular cell proliferation and survival, whereas NHE1 inhibition may result in the regression of pulmonary vascular remodeling (522, 523). When intracellular Na+ is increased because of increased Na+ influx through TRP and ASIC channels and inward Na+ transportation through NHE, it also activates the Na+/Ca2+ exchanger in the plasma membrane to induce inward transportation of Ca2+ (411, 415, 524). Upregulated NCX1 in PASMCs is implicated in the increase of [Ca2+]cyt in PASMCs from patients with PAH (417). These observations indicate that Na+ influx through nonselective cation channels upregulated in PASMCs can contribute to sustained pulmonary vasoconstriction (via PASMC contraction) and vascular remodeling (via PASMC proliferation and migration) via membrane depolarization and VDCC-associated Ca2+ influx pathways. The local increase in cytoplasmic [Na+] through TRP and ASIC channels and via Na+ transporters (e.g., NHE1) can also turn on the reverse mode of Na+/Ca2+ exchange to increase [Ca2+]cyt in PASMCs. The NCX-mediated inward Ca2+ transportation is also an important mechanism to enhance the sequestration of Ca2+ into the intracellular Ca2+ stores (i.e., the SR/ER) and enhance agonist- or growth factor-mediated Ca2+ response (306, 411, 419).
The pathogenic roles of ion channels and transporters in PAH and PH due to respiratory diseases and/or hypoxemia have been well established by various laboratories and investigators (443). As discussed above, PAH is a pulmonary vasculopathy that is characterized by sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, and occlusive vascular lesions, which directly contribute to the elevated PVR and PAP in patients with PAH and PH due to respiratory disease and/or hypoxemia. Specifically, the regulation of Ca2+, K+, and Na+ channels and the ionic concentrations, and how they relate to each other, have been extensively shown to play an important role in the pathogenesis of PAH. Increased concentrations of cytoplasmic Ca2+, regulated by ROCCs, SOCCs, and VDCCs, is shown in patients with PAH and animals with experimental PH. Downregulation or mutations in different types of K+ channels (e.g., KCNK3, KCNA5) leads to PH through membrane depolarization and activation of VDCC in PASMCs. Similarly, activation of Na+-permeable cation channels and augmentation of intracellular [Na+] can increase [Ca2+]cyt by causing membrane depolarization and activation of VDCCs and can increase [Ca2+]cyt through the reverse mode of NCX (411, 415, 418, 419), progressing pathogenesis. Expression and function of these ion channels in pulmonary vascular cells are implicated in the regulation of pulmonary vasoconstriction, vascular remodeling, and vascular occlusion. Indeed, genetic mutations or single-nucleotide polymorphisms (SNPs) of various membrane receptors and ion channels have been identified to be associated with PAH and other forms of pulmonary hypertension (525–527).
6. GENETICS AND PATHOPHYSIOLOGICAL MECHANISMS
In 1997, Nichols et al. (528) linked the gene for heritable PAH, previously referred to as familial primary pulmonary hypertension, to chromosome 2q31-32, which led to the discovery of the BMPR2 gene as a predisposing gene for PAH in 2000 (529, 530). Since then, many new genes have been reported to be associated with PAH and other forms of PH based on data obtained from multiple cohorts (TABLE 6). Details on the genetics and genomics of PAH have been reviewed extensively by experts in the field (526, 561–564). Among the genes underlying PAH that are validated from multiple cohorts, many are related to the TGF-β signaling pathway, including genes encoding ligands (e.g., BMP9/10), receptors (e.g., BMPR2, ACVRL1, and ENG), and signaling proteins (e.g., SMAD1, 4, 8, and 9) (TABLE 6). The membrane receptors and ion channels in which mutations and SNPs are associated with PAH include GPCRs (e.g., 5-HT receptor 5HT2B/HTR2B) (565), tyrosine kinase receptors (e.g., BMPR2, ACVRL1, endoglin) (533, 534, 537, 542, 566, 567), K2P channels (e.g., KCNK3) (483–485), KATP channels (e.g., ABCC8/SUR1) (495, 496), KV channels (e.g., KCNA5) (502–505, 540, 557, 568, 569), receptor-operated and mechanosensitive nonselective cation channels (e.g., TRPC6) (123, 549), water channels (e.g., AQP1) (542), and membrane transporters (e.g., 5-HT transporter SLC6A4, polyamine and ATP transporter ATP13A3) (542, 548, 565, 570–572). Furthermore, a couple of genes encoding signaling protein/kinase (e.g., EIF2AK4) (531, 551–553, 562) and transcription factors (e.g., TBX4, Sox17) (203, 531, 532, 557–559) have also been shown to play a role in the development and progression of PAH.
Table 6.
Genes underlying PAH that are validated in multiple cohorts
| Gene | Protein | Cellular and Molecular Function | Reference(s) |
|---|---|---|---|
| Ligand | |||
| GDF2 (BMP9) | Growth differentiation factor 2 or bone morphogenetic protein 9 | Activates TGF-β family receptors (e.g., ACVRL1) and SMAD family transcription factors involved in gene expression and angiogenesis | (264) |
| BMP10 | Bone morphogenetic protein 10 | Activates TGF-β family receptors and SMAD family transcription factors that regulate gene expression and cell proliferation | (264, 531) |
| FBLN2 | Fibulin 2 | An extracellular matrix protein that binds fibronectin and Ca2+. Its function is involved in organ development. | (532) |
| PDGFD | Platelet-derived growth factor D | A growth factor involved in embryonic development as well as cell proliferation, migration, survival, and chemotaxis | (532) |
| Membrane receptor and transporter | |||
| BMPR2 | Bone morphogenetic protein (BMP) receptor type II | A tyrosine kinase receptor activated by BMP family of ligands | (529, 530, 533–536) |
| ACVRL1 | Activin receptor-like kinase 1 | A tyrosine kinase receptor activated by the TGF-β superfamily of ligands | (505, 537–539) |
| ENG | Endoglin | A glycoprotein as a component of the TGF-β receptor complex | (483, 537, 538) |
| Cav1 | Caveolin 1 | A scaffolding protein as the main component of the caveolae plasma membrane | (395, 483) |
| SLC6A4 | Serotonin (5-HT) transporter | Transports extracellular 5-HT (a neurotransmitter and vasoconstrictor) into the cytoplasm | (540, 541) |
| Ion channel and transporter | |||
| KCNK3 (or TASK1) | Two-pore domain K+ channel subfamily K member | A pH-sensitive K+ channel involved in generating background K+ current and regulating membrane potential | (483–485) |
| ABCC8 | Sulfonylurea receptor 1 (SUR1) | Forms ATP-sensitive K+ channels with inward-rectifier K+ channel subunits that are inhibited by intracellular ATP | (448, 490, 495, 496) |
| AQP1 | Aquaporin 1 | Molecular water channel and cGMP-gated nonselective cation channel that allows water, H+, , NO, glycerol, CO2, and O2 to go through according to the electrochemical gradient | (542–547) |
| ATP13A3 | ATPase 13A3 | P-type ATPase family protein that transports cations across membrane | (531, 542, 548) |
| TRPC6 | Transient receptor potential canonical channel 6 | Forms receptor-operated and DAG-activated cation channels for ligand- and mechanical force-mediated Ca2+ influx | (123, 549, 550) |
| Signaling protein and kinase | |||
| EIF2AK4 | Eukaryotic translation initiation factor 2α kinase 4 | Phosphorylates the α-subunit of EIF2 (eukaryotic translation initiation factor-2) and regulates protein expression | (531, 551–553) |
| SMAD1 | SMAD family member 1 | Transduces signals from BMP receptors | (554) |
| SMAD4 | SMAD family member 4 | Transduces signals from TGF-β family receptors | (554) |
| SMAD8 | SMAD family member 8 | Transduces signals from BMP receptors | (555) |
| SMAD9 | SMAD family member 9 | Transduces signals from TGF-β family receptors | (554, 556) |
| Transcription factor | |||
| TBX4 | T-Box transcription factor 4 | Transcriptional regulation of genes involved in the lung development or genesis | (532, 557–559) |
| Sox17 | SRY-box transcription factor 17 | Transcriptional regulation of genes involved in Wnt signaling and embryonic development | (203, 560) |
DAG, diacylglycerol; NO, nitric oxide; PAH, pulmonary arterial hypertension.
How these genes coordinate with each other to cause changes in the pulmonary vasculature in PAH is still unknown. The correlated mutations and SNPs in multiple genes further support the hypothesis that the pathogenic mechanism involved is multifactorial. It can be parallelly and/or sequentially distributed in the different cells (e.g., lung vascular and perivascular cells, infiltrated inflammatory cells, vascular resident and circulating progenitor cells) (573) and have different signaling pathways (e.g., TGF-β/BMP signaling, Ca2+/CaM signaling, PDGF signaling, Notch signaling) (574–577) and varying proteomic and transcriptomic clusters (573, 578–580). The parallel pathogenic mechanism involving multiple genes and signaling pathways may help explain the genetic, molecular, physiological, and clinical heterogeneity of PAH (574). Different patients may acquire the disease through different pathogenic pathways or genetic defects to induce similar functional (e.g., sustained vasoconstriction) and histological (e.g., concentric vascular remodeling and occlusive lesions) alterations leading to the elevated PVR and PAP. The sequential pathogenic mechanism involving ligands, receptors, signaling proteins/kinases, and transcription factors may provide a basic molecular physiological foundation to explain the disease progression and severity in patients with PAH and PH. In the beginning, generic changes in extracellular ligands and extracellular matrix proteins may cause mild changes in the pulmonary vasculature. When the disease progresses, receptors on the surface membranes of affected cells and downstream signaling cascades may undergo significant changes (reversible or irreversible) that enable the cells to change function, structure, interaction network, and, eventually, phenotype. The disease severity is then determined by how many sequential steps are affected or become abnormal and whether the pathogenic changes are reversible. The parallel and sequential mechanisms very likely work together to ensure the development and progression of the lung vasculopathy or vascular lesions in PAH.
There are many studies demonstrating that TGF-β/BMP signaling functionally interacts with other signaling pathways to regulate cell proliferation, migration, apoptosis, and differentiation. For example, the BMP-mediated SMAD signaling in PASMCs is involved in maintaining “normal” expression and function of K+ channels (581), whereas downregulation and/or decreased activity of K+ channels (e.g., KCNK3 and KCNA5) are implicated in causing membrane depolarization, pulmonary vasoconstriction, and vascular remodeling (104, 454, 484, 488, 582, 583). A unique SNP in the promoter region of TRPC6, a receptor-operated and mechanosensitive cation channel, identified in PAH patients results in an extra NF-κB binding site. The SNP is then involved in mediating TNF-α-mediated transcriptional upregulation of the TRPC6 gene in PASMCs. The PAH patients who carry the SNP are more susceptible to inflammation-mediated upregulation of TRPC6, which subsequently enhances Ca2+-associated pulmonary vasoconstriction and vascular remodeling (123, 549). Furthermore, the SNPs or mutations in the KCNK3 gene from PAH patients (483) lead to the decrease of the pH-sensitive KCNK3 channel activity (486), also referred to as TASK-1 (or K2P3.1) channel (487), which subsequently results in membrane depolarization and an increase in [Ca2+]cyt and eventually pulmonary vasoconstriction and vascular remodeling (443, 484, 485). The loss-of-function mutation in KCNK3 (483) also alters inflammatory and metabolic factors synthesized and released from circulating immune cells and eventually changes the susceptibility to inflammation-mediated effect on the pulmonary vasculature (584).
PH is a multifactorial disease further complicated by the fact that it can occur in patients as a result of different initial causes [e.g., chronic obstructive pulmonary disease (COPD), acute/chronic mountain disease, pulmonary embolism, scleroderma, HIV infection]. Therefore, identifying genetic predispositions and mutations specifically for one form of PH is difficult. One good example is that mutations in the BMPRII gene can cause PAH in patients with the heritable form of the disease and that mutations in certain signaling pathways such as TGF-β/BMP, which have downstream effects, lead to changes in ion concentrations, vasoconstriction, and remodeling. More thorough studies are needed to 1) specify the mutation/SNP-associated transcriptional, translational, regulatory, and functional changes of each of the genes that are associated with PAH, 2) investigate the functional cross talk among the proteins for which gene mutations and SNPs are associated with PAH, and 3) examine whether the mutations/SNPs in various predisposing genes of PAH affect the pharmacological properties of the encoded proteins (drug target) or influence the efficacy, kinetics, and effectiveness of varying drugs on the targets. Another important concept is that the predisposing genes of PAH may require wild-type genes or gene products to reinforce the pathogenic effect. It is therefore important to identify the pathogenic partners that initiate and/or enhance the pathogenic effect of the predisposing genes (and proteins) on the pulmonary vasculature.
6.1. Coordinated Role of Ion Channels in PAH and PH
There are three categories of changes of ion channels and transporters that are linked to the development and progression of PAH and PH: 1) SNPs or mutations in certain ion channel (pore-forming subunit) genes and regulatory subunit genes are associated with PAH and other forms of PH; 2) ion channel mRNA and protein expression levels are different in lung tissue and cell (e.g., PASMCs and PAECs) specimens from patients with PAH and animals with experimental PH in comparison to normal controls; and 3) ion channel function (e.g., macroscopic or unitary transmembrane cation and/or anion currents, changes of intracellular free ion concentration due to cation influx and/or anion efflux) is changed in PASMCs/PAECs from PAH patients and PH animals in comparison to normal controls. As shown in TABLE 6, there are several ion channel and transporter genes (e.g., KCNK3/TASK1, ABCC8/SUR1, TRPC6, ATP13A3) for which SNPs or mutations identified in peripheral blood mononuclear cells (PBMCs) are associated with PAH (and PH). The SNPs in the intergenic and promoter regions of an ion channel gene can affect the transcription and expression of the gene, whereas the nonsynonymous SNPs or mutations in the exons of a channel gene can be gain-of-function or loss-of-function SNPs. For example, various gain-of-function SNPs/mutations in KCNK3 are associated with increased K+ currents through KANK3 channels in patients with sleep apnea or developmental delay in sleep apnea (DDSA) (585), whereas loss-of-function SNPs/mutations in KCNK3 are associated with decreased K+ currents through KCNK3 channels in patients with PAH (586).
In addition to these genetically linked channel and transporter genes in PAH, there are multiple ion channels and transporters for which functional and expression changes are observed in lung tissue/cell specimens from patients with PAH and animals with experimental PH (TABLE 7). In vitro studies using lung tissues and PASMCs/PAECs isolated from PAH patients and PH animal models (TABLE 7) and genetic association studies using PBMCs of PAH patients (TABLE 6) are consistent with each other for some genes (e.g., BMPR2 SNPs/mutations in cohort studies of PAH patients and BMPR2 downregulation and BMP/BMPR2 signaling inhibition in lung tissue/cell specimens from PAH patients). However, there are downregulated or upregulated ion channels in lung tissue/cell specimens from patients who do carry loss-of-function or gain-of-function SNPs/mutations in the ion channel genes in comparison to normal subjects. The gap between the in vitro and animal studies and the human genetic/phenotypic studies urges us to conduct more experiments to specify the mechanisms involved in the transcriptional and functional regulation of ion channel and transporter genes in lung vascular cells from PAH patients with or without germline SNPs/mutations associated with the disease. One of the explanations for the gap or difference between human genomic/phenotypic studies and in vitro and animal studies is that the former use PBMCs to analyze the SNP/mutation correlation with human phenotypes whereas the latter use affected tissues/cells (e.g., PASMCs isolated from remodeled pulmonary vascular segments) to define functional and transcriptional changes of the altered genes. Both sets of studies provide important information on the involvement of various ion channels and transporters in the development and progression of pulmonary vascular lesions in PAH.
Table 7.
Selected ion channels that are transcriptionally and functionally changed in lung tissues, PASMCs and PAECs from patients with PAH (in vitro studies) and animals with experimental PH (animal studies)
| Category | Major Functional Effect | References |
|---|---|---|
| Ca2+ channels including VDCC (CaV), ROCC (TRP), SOCC (Orai), and intracellular channels | ||
| ↑CaV1.2 (L-type), ↑CaV3.2 (T-type) | ↑[Ca2+]cyt → ↑SMC contraction → vasoconstriction | (76, 110, 123, 131, 377–379, 387, 587–595) |
| ↑TRPC1/3/6, ↑TRPM6/7, ↑TRPV1/4 | ↑[Ca2+]cyt → cell migration and proliferation → vascular remodeling | |
| ↑Orai1/2-STIM1/2 | ↑Na+ influx → Em depolarization in SMC → ↑VDCC → ↑[Ca2+]cyt | |
| ↑IP3 receptor (IP3R1) | ↑IP3R/RyR → agonist- and Ca2+-induced Ca2+ release → ↑[Ca2+]cyt → ↑ SMC contraction/migration → vasoconstriction and vascular remodeling | |
| ↑Ryanodine receptor (RyR2) | ||
| K+ channels including KV (KCN), KCa (KCNM), and K2P (KCNK) channels | ||
| ↓KCNA1/2/4/5, ↓KCNB1, ↓KCNH2, ↓KCNQ4 | ↓K+ current → Em depolarization → ↑VDCC → ↑[Ca2+]cyt → ↑SMC contraction | (431, 448, 467, 472, 476, 484, 495, 500, 502, 503, 506, 583, 584, 587, 596–600) |
| ↑↓KCNMA1, ↑KCNMB1 | ↑K+ current → Em hyperpolarization and repolarization → ↓VDCC → ↓[Ca2+]cyt → SMC relaxation → vasodilation | |
| ↓KCNK3/KCNK6, ↓KCNK15/KCNK17 | ↑K+ efflux → ↑H2O leak → AVD → apoptosis | |
| ↑K+ efflux → ↓[K+]cyt→ ↑caspase activity → apoptosis | ||
| ↑K+ efflux → ↓[K+]cyt → ↑inflammasome activation and formation → vascular inflammation → vascular remodeling | ||
| Na+ channels including voltage-gated (SCN), acid-sensitive (ASIC), and epithelial (ENaC) Na+ channel | ||
| ↑SCN1A/SCN3A/SCN9A | ↑Na+ influx → Em depolarization in SMC → ↑Ca2+ influx → ↑[Ca2+]cyt → ↑SMC contraction/migration/proliferation → vasoconstriction and vascular remodeling | (517–519, 587, 601, 602) |
| ↑ASIC1/ASIC2/ASIC3 | ↓Na+ influx in epithelial cell → pulmonary edema | |
| ↓ENaC | ||
| Gap junction channels | ||
| ↑Cx37 in EC | ↑Cx37 in EC → ↑lung endothelial injury → vascular remodeling | (283, 304, 308, 309, 319, 321, 322) |
| ↓↑Cx40 in EC | ↓Cx40 in EC → ↓endothelium-dependent relaxation → vasoconstriction | |
| ↑Cx43 in SMC | ↑Cx40 in EC → ↑Em depolarization from EC to SMC → HPV | |
| ↑Cx43 in SMC → ↑SMC proliferation → vascular remodeling | ||
| Cl− channels | ||
| ↑Ca2+-activated Cl− channel (TMEM16A) in SMC | ↑[Ca2+]cyt → ↑Cl− efflux → Em depolarization in SMC → ↑[Ca2+]cyt → SMC contraction/proliferation → vasoconstriction and vascular remodeling | (432, 434–442) |
| ↑CLIC4 → ↑ARF6 → ↓BMPRII → EC proliferation | ||
| ↑Intracellular Cl− channel (CLIC4) in EC and SMC | ↑CLIC4 → ↑ARF6 → ↑NF-kB → EC inflammation | |
| ↑CLIC4 → ↑RhoA/Rac1 → SMC migration/proliferation | ||
| Water channels (aquaporin) | ||
| ↑AQP1 | ↑β-catenin → ↑c-Myc/cyclin D1 → SMC migration/proliferation → vascular remodeling | (542, 545,546) |
↑increase, activate or upregulate; ↓decrease, inhibit, or downregulate. AVD, apoptotic volume decrease; ARF6, ADP ribosylation factor 6; BMPRII, bone morphogenetic protein receptor II; [Ca2+]cyt, cytosolic free Ca2+ concentration; EC, endothelial cell; Em, membrane potential; HPV, hypoxic pulmonary vasoconstriction; IP3, inositol (1,4,5)-trisphosphate; [K+]cyt, cytosolic free K+ concentration; KV, voltage-gated K+ channel; KCa, Ca2+-activated K+ channel; K2P, 2-pore domain K+ channel; PAEC. pulmonary arterial endothelial cell; PAH, pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cell; ROCC, receptor-operated cation channel; SMC, smooth muscle cell; SOCC, store-operated cation channel; VDCC, voltage-dependent Ca2+ channel.
Ion channels and transporters in the plasmalemmal membrane, intracellular organelle membrane, and nuclear envelope in lung vascular cells are involved in maintaining and regulating cell homeostasis under normal or physiological conditions. For example, membrane potential (Em) is mainly set and regulated by the activity of K+ channels and Na+ pump (102, 103, 411). Decreased background or leak K+ currents through KCNK3, an outwardly rectifying pH-sensitive K2P channel, in PAH patients carrying KCNK3 SNPs/mutations (586) would cause or initiate membrane depolarization in PASMCs to open VDCCs, increase [Ca2+]cyt, and thus stimulate PASMC contraction, migration, and proliferation, thereby leading to pulmonary vasoconstriction and vascular remodeling. Downregulated and/or dysfunctional KCNA5, a delayed-rectifier KV channel, in PAH patients with (502, 504, 505, 568) or without (540, 569) germline SNPs/mutations would help maintain the membrane depolarization by inhibiting membrane repolarization and sustain the increase in [Ca2+]cyt in PASMCs. This coordinated or combined effect of KCNK3 and KCNA5 on membrane potential would efficiently cause and maintain a sustained increase in [Ca2+]cyt due to Ca2+ influx through opened VDCCs to reinforce the pathogenic vasoconstriction and vascular remodeling in patients with PAH and PH associated with lung diseases and/or hypoxia. In PAH patients who do not carry KCNK3 SNPs/mutations, the same “pathogenic” mechanism can be used by a different trigger (e.g., a nongenetic trigger). Increased agonists (e.g., ET-1, polyamines) can activate GPCR (some are upregulated in PASMCs from PAH patients) and increase PLC/DAG to inhibit KCNK3 (603, 604) and KCNA5 (605) in PASMCs, causing membrane depolarization, opening of VDCC, and increase in [Ca2+]cyt in PASMCs, which ultimately result in pulmonary vasoconstriction and vascular remodeling.
Functional and transcriptional alterations are observed for many ion channels and transporters in in vitro studies using lung tissue/cell specimens from PAH patients and animal studies using lung tissue/cell specimens from animals with experimental PH (TABLE 7), whereas only a few ion channel and transporter genes for which SNPs/mutations are identified in PBMCs are associated with PAH (TABLE 6). The changes, either an increased/decreased activity or an upregulated/downregulated expression, of the channels in lung tissue/cell specimens from PAH patients and PH animals are not necessarily linked to germline SNPs/mutations in the genes encoding the channels. Ion channel and transporter genes include binding sites for numerous transcription factors and microRNAs in the 5′- and 3′-untranslated regions, respectively. Transcriptional and posttranscriptional regulation of ion channel genes induced by inflammatory (e.g., IL-3/TNF-α-NF-κB) and fibrotic (e.g., TGF-β/BMP-SMAD) factors are well established in lung tissues and pulmonary vascular cells (including ECs, SMCs, and FBs) (6, 7, 42, 443, 578, 606). A transcriptomic profiling study has identified >66 cation channels (including K+ channels, Na+ channels, Ca2+ channels, and nonselective cation channels) that are dysregulated in PAH; the functional and expression changes of mRNAs encoding these channels are similar in patients with idiopathic PAH, associated PAH with connective tissue disease, and congenital heart disease (587). Even in the case with BMPR2 mutations it is, for example, likely that the initial pathogenic cause is the germline SNPs/mutation-associated downregulation of BMPR2 and inhibition of BMPR signaling (and/or activation of TGFβR signaling). Then, the downregulated and/or dysfunctional BMPR2 would, at least in part, use or regulate various ion channels and transporters (by downregulating or upregulating their expression or inhibiting or activating their function) (581, 607, 608) to confer the pathophysiological and pathological effects on the pulmonary vasculature to increase PVR and PAP.
7. THERAPEUTIC TARGETS FOR TREATMENT OF PULMONARY VASCULAR DISEASE
In patients with PAH (group 1 PH) and other forms of precapillary PH (groups 3 and 4), increased PVR is mainly caused by pulmonary vasculopathy characterized by sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, increased pulmonary vascular wall stiffness, in situ thrombosis, and occlusive intimal lesions. An effective therapy for PAH would therefore cause pulmonary vasodilation (or inhibit pulmonary vasoconstriction), exert antiproliferative or proapoptotic effects on highly proliferative vascular cells (e.g., adventitial fibroblasts, medial smooth muscle cells, and intimal endothelial cells), prevent or resolve in situ thromboemboli in small arteries and precapillary arterioles, exert antifibrotic effects to attenuate extracellular matrix stiffness, and reduce pulmonary vascular wall stiffness due to increased myogenic tone and inflammation/cholesterol-associated membrane stiffness (7, 38, 96). Therapeutic strategies for PAH and precapillary PH are thus focused on vasodilative, antiproliferative, anticoagulant, anti-inflammatory, and antifibrotic drugs.
Although the cellular and molecular mechanisms leading to these changes in the pulmonary vasculature are complex, an imbalance of vasoactive mediators, mitogenic and angiogenic factors, and pro- and antiapoptotic proteins plays an important role in PAH development. Relative deficiencies of vasodilators (e.g., NO and prostacyclin) deleteriously accompany an excess of vasoconstrictors (e.g., ET-1 and TXA2). NO released by vascular ECs normally promotes the production of cyclic GMP in PASMCs, resulting in PASMC relaxation and pulmonary vasodilation. PGI2, also released from the vascular endothelium, promotes the synthesis of cAMP, causing PASMC relaxation and pulmonary vasodilation. In addition, NO and PGI2 both have antiproliferative and anticoagulant effects that inhibit concentric pulmonary vascular wall thickening and in situ thrombosis. ET-1 is a potent vasoconstrictor secreted by ECs; it exerts vasoconstrictive and proliferative effects on PASMCs. Other vasoactive mediators such as TXA2 and 5-HT appear to play a role in the development of PAH, but the therapeutic potential of targeting these substances has not been well established clinically.
Sustained vasoconstriction and pulmonary vascular remodeling also result from functional and transcriptional changes in membrane receptors and ion channels on the surface of PASMCs. Several GPCRs and TKRs are implicated in the development and progression of PAH (7, 575). Increased cytosolic [Ca2+] is an important common pathway by which receptor activation and downstream cellular signaling cascades exert their effects on the pulmonary vasculature. A rise in [Ca2+]cyt in PASMCs is a major trigger for PASMC contraction and an important mediator for PASMC proliferation, migration, and vascular remodeling. Furthermore, ion channels, particularly Ca2+-permeable cation channels (e.g., TRPC6 and TRPV1) and K+-permeable channels (e.g., KCNA5 and KCNK3) in the plasma membrane of PASMCs, can directly influence [Ca2+]cyt (38). Downregulation of K+-permeable channels in PASMCs leads to membrane depolarization and opening of VDCCs, enhancing Ca2+ influx, with a consequent increase in [Ca2+]cyt and further sustained vasoconstriction and vascular remodeling (117). Changes in activity and expression of ion channels and transporters, such as VDCCs, ROCCs, SOCCs, and the Na+/Ca2+ exchanger, are implicated in the development of PAH. Thus, all are potential therapeutic targets.
At the cellular and molecular levels, the current therapeutic targets are classified into 1) membrane receptor blockers [e.g., bosentan and ambrisentan to block ETA and imatinib to block PDGF receptor (PDGFR)] and activators (e.g., PGI2/epoprostenol and selexipag to activate IP), 2) ion channel blockers (e.g., nifedipine and verapamil to block VDCC) and activators (e.g., NO to activate K+ channels via cGMP or nitrosylation), and 3) second messenger cGMP modulators (e.g., riociguat to stimulate soluble guanylate cyclase to increase cGMP production and sildenafil to inhibit phosphodiesterase-5 to inhibit cGMP degradation) (TABLE 8). KATP channel (formed by KIR6 and SUR1/2) openers (e.g., levosimendan, iptakalim, diazoxide, VU0071063, NN414) (498–501), selective TRPC6 channel inhibitors (e.g., BI-749327) (645, 646), serotonin transporter/receptor inhibitors (e.g., LY393558) (571, 647–649), allosteric Ca2+-sensing receptor antagonists (e.g., NPS2143 and calhex 231) (223, 225, 650), Notch receptor inhibitors or Notch ligand antibodies (248, 295, 651, 652), vasoactive intestinal peptide receptor (VPAC1 and VPAC2) agonists (VIP) (236, 237), BMP10/TGF-β/ACTRIIA signaling inhibitors (e.g., Sotatercept) (624), and recombinant angiotensin-converting enzyme 2 (ACE2) (214, 653–656) all have recently been tested for treatment of PAH and experimental PH, with promising effect.
Table 8.
Drugs for treatment of PAH
| Drugs | Target | Action Mechanisms | References |
|---|---|---|---|
| Membrane receptor agonist or antagonist | |||
| Epoprostenal/PGI2 (Flolan, Veletri) | ↑ Prostacyclin receptor (IP) | Induces vasodilative, antiproliferative and anticoagulant effect by IP-mediated increase in cAMP and PKA | (24, 32, 84, 609) |
| Treprostinil (Remodulin, Tyvaso) | ↑ IP receptor | Activates IP to induce pulmonary vasodilation and inhibit pulmonary vascular remodeling | (610, 611) |
| Iloprost (Ventavis) | ↑ IP receptor | Activates IP to induce pulmonary vasodilation and attenuate pulmonary vascular remodeling | (612, 613) |
| Selexipag (Uptravi) | ↑ IP receptor | Activates IP to induce pulmonary vasodilation and attenuate pulmonary vascular remodeling | (614, 615) |
| Bosentan (Tracleer) | ↓ Endothelin receptor A (ETA), ↓ endothelin receptor B (ETB) | Blocks both ETA and ETB to inhibit vasoconstriction and vascular remodeling by inhibiting PASMC contraction and proliferation | (90, 156, 243, 605, 616, 617) |
| Ambrisentan (Letairis) | ↓ETA | Selectively blocks ETA to inhibit vasoconstriction and vascular remodeling | (618, 619) |
| Macitentan (Opsumit) | ↓ETA and ↓ETB | Blocks both ETA and ETB to inhibit vasoconstriction and vascular remodeling | (620, 621) |
| Imatinib | Platelet-derived growth factor receptor (PDGFR) | Nonselective blocker of tyrosine kinase receptors to inhibit cell proliferation and concentric pulmonary vascular remodeling | (245, 622, 623) |
| Sotatercept | Activin receptor type 2A (ACTRIIA or ACVR2A) | A recombinant ACTRIIA extracellular domain fused with IgG-Fc domain to neutralize activin proteins and GDF-8 (myostatin) and GDF-11 (BMP11), which then blocks the activin and GDF ligands and indirectly restores or enhances BMP signaling | (624, 625) |
| Ion channel and transporter inhibitor or activator | |||
| Diltiazem | ↓Voltage-dependent Ca2+ channel (VDCC) | Inhibits Ca2+ influx, decreases [Ca2+]cyt in PASMCs and attenuates pulmonary vasoconstriction or induces pulmonary vasodilation | (626, 627) |
| Nifedipine | ↓ L-type VDCC | Inhibits Ca2+ and Na+ influx in cardiomyocytes and vascular smooth muscle cells to inhibit pulmonary vasodilation and induce pulmonary vasodilation | |
| Verapamil | ↓VDCC | Inhibits Ca2+ influx and decreases [Ca2+]cyt in PASMCs to attenuate pulmonary vasoconstriction and induce pulmonary vasodilation | |
| Amlodipine | ↓VDCC | Inhibits Ca2+ influx, decreases [Ca2+]cyt in PASMC, and causes pulmonary vasoconstriction or induces pulmonary vasodilation | |
| Digoxin (Lanoxin) | ↓Na+-K+-ATPase | Exerts positive ionotropic effect to improve RV function and inhibits hypoxia-inducible factor (HIF)-1α to attenuate vasoconstriction and vascular remodeling | (628, 629) |
| Nitric oxide (NO)/cGMP signaling stimulator | |||
| Inhaled NO | ↓ Soluble guanylate cyclase (sGC) | Activates sGC to produce cGMP and induce vasodilation by activating K+ channels and inducing membrane hyperpolarization | (85, 144, 430, 630, 631) |
| Riociguat (Adempas) | ↓ sGC | Stimulates sGC to produce cGMP and induce vasodilation and regression of vascular remodeling | (632–636) |
| Tadalafil (Adcirca) | ↓ Phosphodiesterase 5 (PDE-5) | Inhibits PDE-5 to decrease cGMP degradation and induce cGMP/PKG-mediated vasodilation and regression of vascular remodeling | (637–639) |
| Sildenafil (Revatio) | ↓ PDE-5 | Inhibits PDE-5 to decrease cGMP degradation and induce cGMP/PKG-mediated vasodilation and regression of vascular remodeling | (640, 641) |
| Anticoagulant | |||
| Warfarin (Coumadin) | Vitamin K epoxide reductase | Blocks vitamin K epoxide reductase to inhibit coagulation | (642–644) |
| Other | |||
| Oxygen (O2) | Inhalation of O2 inhibits hypoxic pulmonary vasoconstriction, improves gas exchange,. and increases arterial oxygenation | ||
↑, Increase, activate; ↓, decrease, inhibit. [Ca2+]cyt, cytosolic free Ca2+ concentration; PAH, pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cell; RV, right ventricle.
Owing to the important pathogenic role of membrane receptors, ion channels, and transporters in the development of pulmonary vasoconstriction, vascular remodeling, arteriole muscularization, and occlusive vascular lesions, targeting specific receptors and ion channels associated with the disease pathogenesis is a highly promising strategy to develop novel therapeutic approaches for treatment of PAH and PH.
8. SUMMARY AND SPECULATION
Pulmonary arterial hypertension is a multifactorial pulmonary vasculopathy with complex pathogenic mechanisms. No single genetic defect is the sole trigger for the disease, although multiple genes (e.g., BMPR2, Sox17) are genetically associated with PAH in some patients. Regardless of the initial genetic, genomic, molecular, or environmental cause, sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, in situ thrombosis, occlusive intimal lesion, and vascular wall stiffening are the direct causes for elevating PVR and PAP in patients with PAH and other forms of precapillary PH. All cells in the pulmonary vasculature, i.e., ECs from the intima, SMCs from the media, and (myo)FBs and progenitor cells in the adventitia and ECM, contribute to the pathophysiological and pathological changes in the pulmonary vasculature. The pulmonary vasculopathy in PAH is a self-defect abnormality of the pulmonary vascular cells like cancer or a cancerlike vascular defect. It is initiated by a defected cell(s), likely caused by somatic mutations and disorganized cell reprograming in the pulmonary vasculature, and is facilitated and “regulated” by other cells such as inflammatory and progenitor cells. The initial defect cell becomes a pathogenic cell via an autocrine mechanism, which then spreads the defect to other adjacent cells via paracrine and juxtacrine mechanisms to develop vascular lesions. The initial pathogenic cell(s) can also be alveolar or bronchial epithelial cells that subsequently transmit the disease signals to vascular cells.
Multiple membrane receptors, ion channels, and transporters and intracellular signaling pathways are involved in the pathogenesis and progression of PAH and PH. Partial reprograming of fully differentiated pulmonary vascular wall cells like ECs and SMCs in response to pathogenic cues is an important pathogenic mechanism. Therapeutic strategies should be focused on combination therapy and identifying new drugs that have multiple targets to efficiently inhibit the progression of and/or reverse the established pathophysiological and pathological changes in the pulmonary vasculature and improve right heart function.
GRANTS
This work was supported, in part, by grants from the National Lung, Blood, and Heart Institute of the National Institutes of Health (R35 HL135807, R01 HL146764).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.X.-J.Y. conceived and designed the project; A.B., A.M., and J.X.-J.Y. prepared figures; A.B., A.M., and J.X.-J.Y. drafted, edited and revised manuscript; A.B., A.M., and J.X.-J.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all our colleagues, trainees, and students at the University of Illinois at Chicago, The University of Arizona, and the University of California, San Diego, who have diligently worked with us on defining pathogenic mechanisms of pulmonary hypertension and developing therapeutic interventions for the disease.
REFERENCES
- 1. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53: 1801913, 2019. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RM, Brida M, Carlsen J, Coats AJ, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S; the ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61: 2200879, 2023. doi: 10.1183/13993003.00879-2022. [DOI] [PubMed] [Google Scholar]
- 3. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RM, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 43: 3618–3731, 2022. doi: 10.1093/eurheartj/ehac237. [DOI] [PubMed] [Google Scholar]
- 4. Remillard CY. Characterization of hemodynamics in patients with idiopathic and thromboembolic pulmonary hypertension. Clin Med Insights Circ Respir Pulm Med 2: 59–68, 2008. doi: 10.4137/CCRPM.S696. [DOI] [Google Scholar]
- 5. Hassoun PM. Pulmonary arterial hypertension. N Engl J Med 385: 2361–2376, 2021. doi: 10.1056/NEJMra2000348. [DOI] [PubMed] [Google Scholar]
- 6. Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–S19, 2009. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 7. Morrell NW, Adnot S, Archer SL, Dupuis J, Lloyd Jones P, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fronek K, Zweifach BW. Pre- and postcapillary resistances in cat mesentery. Microvasc Res 7: 351–361, 1974. doi: 10.1016/0026-2862(74)90022-3. [DOI] [PubMed] [Google Scholar]
- 9. Brody JS, Stemmler EJ, DuBois AB. Longitudinal distribution of vascular resistance in the pulmonary arteries, capillaries, and veins. J Clin Invest 47: 783–799, 1968. doi: 10.1172/JCI105773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hakim TS, Michel RP, Chang HK. Partitioning of pulmonary vascular resistance in dogs by arterial and venous occlusion. J Appl Physiol Respir Environ Exerc Physiol 52: 710–715, 1982. doi: 10.1152/jappl.1982.52.3.710. [DOI] [PubMed] [Google Scholar]
- 11. Wu X, Lu W, He M, Chen H, Chen Y, Duan X, Zheng Q, Li Y, Chen J, Liu S, Liao J, Kuang M, Lin Z, Yang K, Wang J. Structural and functional definition of the pulmonary vein system in a chronic hypoxia-induced pulmonary hypertension rat model. Am J Physiol Cell Physiol 318: C555–C569, 2020. doi: 10.1152/ajpcell.00289.2019. [DOI] [PubMed] [Google Scholar]
- 12. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 13. Fishman AP. Primary pulmonary arterial hypertension: a look back. J Am Coll Cardiol 43: S2–S4, 2004. doi: 10.1016/j.jacc.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 14. Rosenkranz S, Preston IR. Right heart catheterisation: best practice and pitfalls in pulmonary hypertension. Eur Respir Rev 24: 642–652, 2015. doi: 10.1183/16000617.0062-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43: S5–S12, 2004. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 16. Humbert M, Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An insider view on the World Symposium on Pulmonary Hypertension. Lancet Respir Med 7: 484–485, 2019. doi: 10.1016/S2213-2600(19)30111-0. [DOI] [PubMed] [Google Scholar]
- 17. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46: 903–975, 2015. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 18. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37: 67–119, 2016. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 19. Wiggers CJ. Pulmonary wedged catheter pressures. Circ Res 1: 371–374, 1953. doi: 10.1161/01.RES.1.5.371. [DOI] [PubMed] [Google Scholar]
- 20. Zeder K, Banfi C, Steinrisser-Allex G, Maron BA, Humbert M, Lewis GD, Berghold A, Olschewski H, Kovacs G. Diagnostic, prognostic and differential-diagnostic relevance of pulmonary haemodynamic parameters during exercise: a systematic review. Eur Respir J 60: 2103181, 2022. doi: 10.1183/13993003.03181-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisman AS, Shah RV, Dhakal BP, Pappagianopoulos PP, Wooster L, Bailey C, Cunningham TF, Hardin KM, Baggish AL, Ho JE, Malhotra R, Lewis GD. Pulmonary capillary wedge pressure patterns during exercise predict exercise capacity and incident heart failure. Circ Heart Fail 11: e004750, 2018. doi: 10.1161/CIRCHEARTFAILURE.117.004750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bentley RF, Barker M, Esfandiari S, Wright SP, Valle FH, Granton JT, Mak S. Normal and abnormal relationships of pulmonary artery to wedge pressure during exercise. J Am Heart Assoc 9: e016339, 2020. doi: 10.1161/JAHA.120.016339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubin LJ, Groves BM, Reeves JT, Frosolono M, Handel F, Cato AE. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation 66: 334–338, 1982. doi: 10.1161/01.CIR.66.2.334. [DOI] [PubMed] [Google Scholar]
- 24. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Clayton LM, Jöbsis MM, Blackburn SD, Shortino D, Crow JW; Primary Pulmonary Hypertension Study Group. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 334: 296–301, 1996. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 25. Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress”. Circulation 102: 2781–2791, 2000. doi: 10.1161/01.CIR.102.22.2781. [DOI] [PubMed] [Google Scholar]
- 26. Mair KM, Johansen AK, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Br J Pharmacol 171: 567–579, 2014. doi: 10.1111/bph.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, Gonin R, Keast S, Badesch D, Barst RJ, Bourge RC, Channick R, Frost A, Gaine S, McGoon M, McLaughlin V, Murali S, Oudiz RJ, Robbins IM, Tapson V, Abenhaim L, Constantine G. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J 152: 521–526, 2006. doi: 10.1016/j.ahj.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 28. Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137: 376–387, 2010. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 29. Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, Benza RL. Five-year outcomes of patients enrolled in the REVEAL Registry. Chest 148: 1043–1054, 2015. doi: 10.1378/chest.15-0300. [DOI] [PubMed] [Google Scholar]
- 30. Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest 141: 363–373, 2012. doi: 10.1378/chest.10-3114. [DOI] [PubMed] [Google Scholar]
- 31. Wang Z, Chesler NC. Pulmonary vascular wall stiffness: n important contributor to the increased right ventricular afterload with pulmonary hypertension. Pulm Circ 1: 212–223, 2011. doi: 10.4103/2045-8932.83453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vanderpool RR, Desai AA, Knapp SM, Simon MA, Abidov A, Yuan JX, Garcia JG, Hansen LM, Knoper SR, Naeije R, Rischard FP. How prostacyclin therapy improves right ventricular function in pulmonary arterial hypertension. Eur Respir J 50: 1700764, 2017. doi: 10.1183/13993003.00764-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Naeije R, Westerhof N. Pulmonary vascular function. In: Textbook of Pulmonary Vascular Disease, edited by Yuan JX, Garcia JG, Hales CA, Rich S, Archer SL, West JB.. New York: Springer, 2011, p. 61–72. [Google Scholar]
- 34. Roldan-Alzate A, Chesler NC. Pulmonary vascular mechanics. In: Textbook of Pulmonary Vascular Disease, edited by Yuan JX, Garcia JG, Hales CA, Rich S, Archer SL, West JB.. New York: Springer, 2011, p. 73–89. [Google Scholar]
- 35. Naeije R. Pulmonary vascular function. In: Pulmonary Circulation Diseases and their Treatment, edited by Peacock AJ, Naeije R, Rubin LJ.. Boca Raton, FL: CRC Press, 2016, p. 11–24. [Google Scholar]
- 36. Huang W, Yen RT, McLaurine M, Bledsoe G. Morphometry of the human pulmonary vasculature. J Appl Physiol (1985) 81: 2123–2133, 1996. doi: 10.1152/jappl.1996.81.5.2123. [DOI] [PubMed] [Google Scholar]
- 37. Makino A, Firth AL, Yuan JX. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr Physiol 1: 1555–1602, 2011. doi: 10.1002/cphy.c100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res 68: 75–103, 2004. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 39. Townsley MI. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol 2: 675–709, 2012. doi: 10.1002/cphy.c100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Razavi H, Dusch MN, Zarafshar SY, Taylor CA, Feinstein JA. A method for quantitative characterization of growth in the 3-D structure of rat pulmonary arteries. Microvasc Res 83: 146–153, 2012. doi: 10.1016/j.mvr.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 41. Knutsen RH, Gober LM, Sukinik JR, Donahue DR, Kronquist EK, Levin MD, McLean SE, Kozel BA. Vascular casting of adult and early postnatal mouse lungs for micro-CT imaging. J Vis Exp 2020: 10.3791/61242, 2020. doi: 10.3791/61242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satoh T, Wang L, Espinosa-Diez C, Wang B, Hahn SA, Noda K, Rochon ER, Dent MR, Levine AR, Baust JJ, Wyman S, Wu YL, Triantafyllou GA, Tang Y, Reynolds M, Shiva S, Hilaire CS, Gomez D, Goncharov DA, Goncharova EA, Chan SY, Straub AC, Lai YC, McTiernan CF, Gladwin MT. Metabolic syndrome mediates ROS-miR-193b-NFYA-dependent down regulation of sGC and contributes to exercise-induced pulmonary hypertension in HFpEF. Circulation 144: 615–637, 2021. doi: 10.1161/CIRCULATIONAHA.121.053889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Priya S, Aggarwal T, Ward C, Bathla G, Jacob M, Gerke A, Hoffman EA, Nagpal P. Radiomics side experiments and DAFIT approach in identifying pulmonary hypertension using Cardiac MRI derived radiomics based machine learning models. Sci Rep 11: 12686, 2021. doi: 10.1038/s41598-021-92155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moher Alsady T, Kaireit TF, Behrendt L, Winther HB, Olsson KM, Wacker F, Hoeper MM, Cebotari S, Vogel-Claussen J. Comparison of dual-energy computer tomography and dynamic contrast-enhanced MRI for evaluating lung perfusion defects in chronic thromboembolic pulmonary hypertension. PLoS One 16: e0251740, 2021. doi: 10.1371/journal.pone.0251740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weibel ER, Gomez DM. Architecture of the human lung. Use of quantitative methods establishes fundamental relations between size and number of lung structures. Science 137: 577–585, 1962. doi: 10.1126/science.137.3530.577. [DOI] [PubMed] [Google Scholar]
- 47. Weibel ER. Morphometry of the Human Lung. New York: Academic, 1963. [Google Scholar]
- 48. Weibel ER. A retrospective of lung morphometry: from 1963 to present. Am J Physiol Lung Cell Mol Physiol 305: L405–L408, 2013. doi: 10.1152/ajplung.00169.2013. [DOI] [PubMed] [Google Scholar]
- 49. Singhal S, Henderson R, Horsfield K, Harding K, Cumming G. Morphometry of the human pulmonary arterial tree. Circ Res 33: 190–197, 1973. doi: 10.1161/01.RES.33.2.190. [DOI] [PubMed] [Google Scholar]
- 50. Yen RT, Zhuang FY, Fung YC, Ho HH, Tremer H, Sobin SS. Morphometry of cat pulmonary venous tree. J Appl Physiol Respir Environ Exerc Physiol 55: 236–242, 1983. doi: 10.1152/jappl.1983.55.1.236. [DOI] [PubMed] [Google Scholar]
- 51. Jiang ZL, Kassab GS, Fung YC. Diameter-defined Strahler system and connectivity matrix of the pulmonary arterial tree. J Appl Physiol (1985) 76: 882–892, 1994. doi: 10.1152/jappl.1994.76.2.882. [DOI] [PubMed] [Google Scholar]
- 52. Burrowes KS, Hunter PJ, Tawhai MH. Anatomically based finite element models of the human pulmonary arterial and venous trees including supernumerary vessels. J Appl Physiol (1985) 99: 731–738, 2005. doi: 10.1152/japplphysiol.01033.2004. [DOI] [PubMed] [Google Scholar]
- 53. Burrowes KS, Hunter PJ, Tawhai MH. Investigation of the relative effects of vascular branching structure and gravity on pulmonary arterial blood flow heterogeneity via an image-based computational model. Acad Radiol 12: 1464–1474, 2005. doi: 10.1016/j.acra.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 54. Burrowes KS, Tawhai MH. Computational predictions of pulmonary blood flow gradients: gravity versus structure. Respir Physiol Neurobiol 154: 515–523, 2006. doi: 10.1016/j.resp.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 55. Yi ES, Kim H, Ahn H, Strother J, Morris T, Masliah E, Hansen LA, Park K, Friedman PJ. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension. A morphometric and immunohistochemical study. Am J Respir Crit Care Med 162: 1577–1586, 2000. doi: 10.1164/ajrccm.162.4.9912131. [DOI] [PubMed] [Google Scholar]
- 56. Dartevelle P, Fadel E, Mussot S, Chapelier A, Hervé P, de Perrot M, Cerrina J, Ladurie FL, Lehouerou D, Humbert M, Sitbon O, Simonneau G. Chronic thromboembolic pulmonary hypertension. Eur Respir J 23: 637–648, 2004. doi: 10.1183/09031936.04.00079704. [DOI] [PubMed] [Google Scholar]
- 57. Dörfmuller P, Günther S, Ghigna MR, Thomas de Montpréville V, Boulate D, Paul JF, Jaïs X, Decante B, Simonneau G, Dartevelle P, Humbert M, Fadel E, Mercier O. Microvascular disease in chronic thromboembolic pulmonary hypertension: a role for pulmonary veins and systemic vasculature. Eur Respir J 44: 1275–1288, 2014. doi: 10.1183/09031936.00169113. [DOI] [PubMed] [Google Scholar]
- 58. Ntokou A, Dave JM, Kauffman AC, Sauler M, Ryu C, Hwa J, Herzog EL, Singh I, Saltzman WM, Greif DM. Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension. JCI Insight, 6: e139067, 2021. doi: 10.1172/jci.insight.139067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 7: 308ra159, 2015. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 23: 1152–1165, 2018. doi: 10.1016/j.celrep.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, Williams CM, Shamsuzzaman S, Mokry M, Henderson CA, Haskins R, Baylis RA, Finn AV, McNamara CA, Zunder ER, Venkata V, Pasterkamp G, Björkegren J, Bekiranov S, Owens GK. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation 142: 2045–2059, 2020. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Firth AL, Yao W, Remillard CV, Ogawa A, Yuan JX. Upregulation of Oct-4 isoforms in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 298: L548–L557, 2010. doi: 10.1152/ajplung.00314.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, Wang Z, Gupta A, Zhou T, Sun X, Dash S, Wang Z, Balistrieri A, Zheng Q, Cordery AG, Desai AA, Rischard F, Khalpey Z, Wang J, Black SM, Garcia JG, Makino A, Yuan JX. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 314: L256–L275, 2018. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sánchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev Dyn 247: 492–508, 2018. doi: 10.1002/dvdy.24589. [DOI] [PubMed] [Google Scholar]
- 65. Chang AC, Fu Y, Garside VC, Niessen K, Chang L, Fuller M, Setiadi A, Smrz J, Kyle A, Minchinton A, Marra M, Hoodless PA, Karsan A. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell 21: 288–300, 2011. doi: 10.1016/j.devcel.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 66. Ranchoux B, Harvey LD, Ayon RJ, Babicheva A, Bonnet S, Chan SY, Yuan JX, Perez VJ. Endothelial dysfunction in pulmonary arterial hypertension: an evolving landscape. Pulm Circ 8: 2045893217752912, 2018. doi: 10.1177/2045893217752912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yun E, Kook Y, Yoo KH, Kim KI, Lee MS, Kim J, Lee A. Endothelial to mesenchymal transition in pulmonary vascular diseases. Biomedicines 8: 639, 2020. doi: 10.3390/biomedicines8120639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62: D4–D12, 2013. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tuder RM, Radisavljevic Z, Shroyer KR, Polak JM, Voelkel NF. Monoclonal endothelial cells in appetite suppressant-associated pulmonary hypertension. Am J Respir Crit Care Med 158: 1999–2001, 1998. doi: 10.1164/ajrccm.158.6.9805002. [DOI] [PubMed] [Google Scholar]
- 70. Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res 88: E2–E11, 2001. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 71. Voelkel NF, Tuder RM. Cellular and molecular mechanisms in the pathogenesis of severe pulmonary hypertension. Eur Respir J 8: 2129–2138, 1995. doi: 10.1183/09031936.95.08122129. [DOI] [PubMed] [Google Scholar]
- 72. Bhatnagar A, Wiesen J, Dweik R, Chaisson NF. Evaluating suspected pulmonary hypertension: a structured approach. Cleve Clin J Med 85: 468–480, 2018. doi: 10.3949/ccjm.85a.17065. [DOI] [PubMed] [Google Scholar]
- 73. Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis 16: 13–18, 2005. doi: 10.1097/00019501-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 74. McMurtry IF, Abe K, Ota H, Fagan KA, Oka M. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol 661: 299–308, 2010. doi: 10.1007/978-1-60761-500-2_19. [DOI] [PubMed] [Google Scholar]
- 75. Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 76. Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013. doi: 10.1152/ajplung.00313.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 328: 1732–1739, 1993. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 78. Yoshimura K, Tod ML, Pier KG, Rubin LJ. Role of venoconstriction in thromboxane-induced pulmonary hypertension and edema in lambs. J Appl Physiol (1985) 66: 929–935, 1989. doi: 10.1152/jappl.1989.66.2.929. [DOI] [PubMed] [Google Scholar]
- 79. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 80. Giaid A. Nitric oxide and endothelin-1 in pulmonary hypertension. Chest 114: 208S–212S, 1998. doi: 10.1378/chest.114.3_Supplement.208S. [DOI] [PubMed] [Google Scholar]
- 81. Turley JE, Nelin LD, Kaplowitz MR, Zhang Y, Fike CD. Exhaled NO is reduced at an early stage of hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 284: L489–L500, 2003. doi: 10.1152/ajplung.00246.2002. [DOI] [PubMed] [Google Scholar]
- 82. Lo CC, Moosavi SM, Bubb KJ. The regulation of pulmonary vascular tone by neuropeptides and the implications for pulmonary hypertension. Front Physiol 9: 1167, 2018. doi: 10.3389/fphys.2018.01167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Broughton BR, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 84. Rubin LJ, Mendoza J, Hood M, McGoon M, Barst R, Williams WB, Diehl JH, Crow J, Long W. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med 112: 485–491, 1990. doi: 10.7326/0003-4819-112-7-485. [DOI] [PubMed] [Google Scholar]
- 85. Roberts JD Jr, Fineman JR, Morin FC 3rd, Shaul PW, Rimar S, Schreiber MD, Polin RA, Zwass MS, Zayek MM, Gross I, Heymann MA, Zapol WM. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med 336: 605–610, 1997. doi: 10.1056/NEJM199702273360902. [DOI] [PubMed] [Google Scholar]
- 86. Kinsella JP, Neish SR, Shaffer E, Abman SH. Low-dose inhalation nitric oxide in persistent pulmonary hypertension of the newborn. Lancet 340: 819–820, 1992. doi: 10.1016/0140-6736(92)92687-B. [DOI] [PubMed] [Google Scholar]
- 87. Kirchengast M, Münter K. Endothelin-1 and endothelin receptor antagonists in cardiovascular remodeling. Proc Soc Exp Biol Med 221: 312–325, 1999. doi: 10.1046/j.1525-1373.1999.d01-88.x. [DOI] [PubMed] [Google Scholar]
- 88. Nagendran J, Sutendra G, Paterson I, Champion HC, Webster L, Chiu B, Haromy A, Rebeyka IM, Ross DB, Michelakis ED. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res 112: 347–354, 2013. doi: 10.1161/CIRCRESAHA.111.300448. [DOI] [PubMed] [Google Scholar]
- 89. Sun X, Kumar S, Sharma S, Aggarwal S, Lu Q, Gross C, Rafikova O, Lee SG, Dasarathy S, Hou Y, Meadows ML, Han W, Su Y, Fineman JR, Black SM. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am J Respir Cell Mol Biol 50: 1084–1095, 2014. doi: 10.1165/rcmb.2013-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Undem C, Rios EJ, Maylor J, Shimoda LA. Endothelin-1 augments Na+/H+ exchange activity in murine pulmonary arterial smooth muscle cells via Rho kinase. PLoS One 7: e46303, 2012. doi: 10.1371/journal.pone.0046303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. J Cell Physiol 214: 434–441, 2008. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- 92. Falcetti E, Hall SM, Phillips PG, Patel J, Morrell NW, Haworth SG, Clapp LH. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 182: 1161–1170, 2010. doi: 10.1164/rccm.201001-0011OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Krick S, Platoshyn O, Sweeney M, McDaniel SS, Zhang S, Rubin LJ, Yuan JX. Nitric oxide induces apoptosis by activating K+ channels in pulmonary vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 282: H184–H193, 2002. doi: 10.1152/ajpheart.2002.282.1.H184. [DOI] [PubMed] [Google Scholar]
- 94. Tsihlis ND, Oustwani CS, Vavra AK, Jiang Q, Keefer LK, Kibbe MR. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem Biophys 60: 89–97, 2011. doi: 10.1007/s12013-011-9179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med 28: 23–42, 2007. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53: 1801887, 2019. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li M, Riddle S, Kumar S, Poczobutt J, McKeon BA, Frid MG, Ostaff M, Reisz JA, Nemkov T, Fini MA, Laux A, Hu CJ, El Kasmi KC, D’Alessandro A, Brown RD, Zhang H, Stenmark KR. Microenvironmental regulation of macrophage transcriptomic and metabolomic profiles in pulmonary hypertension. Front Immunol 12: 640718, 2021. doi: 10.3389/fimmu.2021.640718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hashimoto R, Lanier GM, Dhagia V, Joshi SR, Jordan A, Waddell I, Tuder R, Stenmark KR, Wolin MS, McMurtry IF, Gupte SA. Pluripotent hematopoietic stem cells augment α-adrenergic receptor-mediated contraction of pulmonary artery and contribute to the pathogenesis of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 318: L386–L401, 2020. doi: 10.1152/ajplung.00327.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yeager ME, Frid MG, Stenmark KR. Progenitor cells in pulmonary vascular remodeling. Pulm Circ 1: 3–16, 2011. doi: 10.4103/2045-8932.78095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vazquez ZG, Klinger JR. Guidelines for the treatment of pulmonary arterial hypertension. Lung 198: 581–596, 2020. doi: 10.1007/s00408-020-00375-w. [DOI] [PubMed] [Google Scholar]
- 101. Abman SH. Inhaled nitric oxide for the treatment of pulmonary arterial hypertension. Handb Exp Pharmacol 218: 257–276, 2013. doi: 10.1007/978-3-642-38664-0_11. [DOI] [PubMed] [Google Scholar]
- 102. Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259: C3–C18, 1990. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 103. Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268: C799–C822, 1995. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 104. Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. doi: 10.1161/01.CIR.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 105. Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 77: 370–378, 1995. doi: 10.1161/01.RES.77.2.370. [DOI] [PubMed] [Google Scholar]
- 106. Smirnov SV, Aaronson PI. Ca2+ currents in single myocytes from human mesenteric arteries: evidence for a physiological role of L-type channels. J Physiol 457: 455–475, 1992. doi: 10.1113/jphysiol.1992.sp019387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ko EA, Wan J, Yamamura A, Zimnicka AM, Yamamura H, Yoo HY, Tang H, Smith KA, Sundivakkam PC, Zeifman A, Ayon RJ, Makino A, Yuan JX. Functional characterization of voltage-dependent Ca2+ channels in mouse pulmonary arterial smooth muscle cells: divergent effect of ROS. Am J Physiol Cell Physiol 304: C1042–C1052, 2013. doi: 10.1152/ajpcell.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dospinescu C, Widmer H, Rowe I, Wainwright C, Cruickshank SF. Hypoxia sensitivity of a voltage-gated potassium current in porcine intrapulmonary vein smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 303: L476–L486, 2012. doi: 10.1152/ajplung.00157.2012. [DOI] [PubMed] [Google Scholar]
- 109. Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 95: 645–690, 2015. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Song S, Carr SG, McDermott KM, Rodriguez M, Babicheva A, Balistrieri A, Ayon RJ, Wang J, Makino A, Yuan JX. STIM2 (stromal interaction molecule 2)-mediated increase in resting cytosolic free Ca2+ concentration stimulates PASMC proliferation in pulmonary arterial hypertension. Hypertension 71: 518–529, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437: 902–905, 2005. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443: 226–229, 2006. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456: 116–120, 2008. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA 103: 9357–9362, 2006. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9: 636–645, 2007. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bodnar D, Chung WY, Yang D, Hong JH, Jha A, Muallem S. STIM-TRP pathways and microdomain organization: Ca2+ influx channels: the Orai-STIM1-TRPC complexes. Adv Exp Med Biol 993: 139–157, 2017. doi: 10.1007/978-3-319-57732-6_8. [DOI] [PubMed] [Google Scholar]
- 117. Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562, 2012. doi: 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 119. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 120. Martinsen A, Dessy C, Morel N. Regulation of calcium channels in smooth muscle: new insights into the role of myosin light chain kinase. Channels (Austin) 8: 402–413, 2014. doi: 10.4161/19336950.2014.950537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Firth AL, Remillard CV, Yuan JX. TRP channels in hypertension. Biochim Biophys Acta 1772: 895–906, 2007. doi: 10.1016/j.bbadis.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435–445, 2005. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Saddouk FZ, Ginnan R, Singer HA. Ca2+/calmodulin-dependent protein kinase II in vascular smooth muscle. Adv Pharmacol 78: 171–202, 2017. doi: 10.1016/bs.apha.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 125. Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Contrasting effects of hypoxia on tension in rat pulmonary and mesenteric arteries. Am J Physiol Heart Circ Physiol 259: H281–H289, 1990. doi: 10.1152/ajpheart.1990.259.2.H281. [DOI] [PubMed] [Google Scholar]
- 126. Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Hypoxic and metabolic regulation of voltage-gated K+ channels in rat pulmonary artery smooth muscle cells. Exp Physiol 80: 803–813, 1995. doi: 10.1113/expphysiol.1995.sp003888. [DOI] [PubMed] [Google Scholar]
- 127. Weir EK, Reeve HL, Cornfield DN, Tristani-Firouzi M, Peterson DA, Archer SL. Diversity of response in vascular smooth muscle cells to changes in oxygen tension. Kidney Int 51: 462–466, 1997. doi: 10.1038/ki.1997.62. [DOI] [PubMed] [Google Scholar]
- 128. Jain PP, Hosokawa S, Xiong M, Babicheva A, Zhao T, Rodriguez M, Rahimi S, Pourhashemi K, Balistrieri F, Lai N, Malhotra A, Shyy JY, Valdez-Jasso D, Thistlethwaite PA, Makino A, Yuan JX. Revisiting the mechanism of hypoxic pulmonary vasoconstriction using isolated perfused/ventilated mouse lung. Pulm Circ 10: 2045894020956592, 2020. doi: 10.1177/2045894020956592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 99: 1–26, 2010. doi: 10.1016/B978-0-12-374841-6.00001-3. [DOI] [PubMed] [Google Scholar]
- 130. Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry 19: 2396–2404, 1980. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- 131. Song S, Ayon RJ, Yamamura A, Yamamura H, Dash S, Babicheva A, Tang H, Sun X, Cordery AG, Khalpey Z, Black SM, Desai AA, Rischard F, McDermott KM, Garcia JG, Makino A, Yuan JX. Capsaicin-induced Ca2+ signaling is enhanced via upregulated TRPV1 channels in pulmonary artery smooth muscle cells from patients with idiopathic PAH. Am J Physiol Lung Cell Mol Physiol 312: L309–L325, 2017. doi: 10.1152/ajplung.00357.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Means AR. Calcium, calmodulin and cell cycle regulation. FEBS Lett 347: 1–4, 1994. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- 133. Berridge MJ. Calcium signalling and cell proliferation. Bioessays 17: 491–500, 1995. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 134. Berridge MJ. Smooth muscle cell calcium activation mechanisms. J Physiol 586: 5047–5061, 2008. doi: 10.1113/jphysiol.2008.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 96: 1261–1296, 2016. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 136. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529, 2003. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 137. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11–21, 2000. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 138. Choi J, Husain M. Calmodulin-mediated cell cycle regulation: new mechanisms for old observations. Cell Cycle 5: 2183–2186, 2006. doi: 10.4161/cc.5.19.3265. [DOI] [PubMed] [Google Scholar]
- 139. Shawer H, Norman K, Cheng CW, Foster R, Beech DJ, Bailey MA. ORAI1 Ca2+ channel as a therapeutic target in pathological vascular remodelling. Front Cell Dev Biol 9: 653812, 2021. doi: 10.3389/fcell.2021.653812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ehler E, Babiychuk E, Draeger A. Human foetal lung (IMR-90) cells: myofibroblasts with smooth muscle-like contractile properties. Cell Motil Cytoskeleton 34: 288–298, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 141. Bogatkevich GS, Tourkina E, Abrams CS, Harley RA, Silver RM, Ludwicka-Bradley A. Contractile activity and smooth muscle α-actin organization in thrombin-induced human lung myofibroblasts. Am J Physiol Lung Cell Mol Physiol 285: L334–L343, 2003. doi: 10.1152/ajplung.00417.2002. [DOI] [PubMed] [Google Scholar]
- 142. Huang X, Gai Y, Yang N, Lu B, Samuel CS, Thannickal VJ, Zhou Y. Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am J Pathol 179: 2751–2765, 2011. doi: 10.1016/j.ajpath.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chin KM, Badesch DB, Robbins IM, Tapson VF, Palevsky HI, Kim NH, Kawut SM, Frost A, Benton WW, Lemarie JC, Bodin F, Rubin LJ, McLaughlin V. Two formulations of epoprostenol sodium in the treatment of pulmonary arterial hypertension: EPITOME-1 (epoprostenol for injection in pulmonary arterial hypertension), a phase IV, open-label, randomized study. Am Heart J 167: 218–225.e1, 2014. doi: 10.1016/j.ahj.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 144. Yu B, Ferrari M, Schleifer G, Blaesi AH, Wepler M, Zapol WM, Bloch DB. Development of a portable mini-generator to safely produce nitric oxide for the treatment of infants with pulmonary hypertension. Nitric Oxide 75: 70–76, 2018. doi: 10.1016/j.niox.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Barnard JW, Ward RA, Adkins WK, Taylor AE. Characterization of thromboxane and prostacyclin effects on pulmonary vascular resistance. J Appl Physiol (1985) 72: 1845–1853, 1992. doi: 10.1152/jappl.1992.72.5.1845. [DOI] [PubMed] [Google Scholar]
- 146. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 147. Cremona G, Dinh Xuan AT, Higenbottam TW. Endothelium-derived relaxing factor and the pulmonary circulation. Lung 169: 185–202, 1991. doi: 10.1007/BF02714154. [DOI] [PubMed] [Google Scholar]
- 148. Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res 61: 866–879, 1987. doi: 10.1161/01.RES.61.6.866. [DOI] [PubMed] [Google Scholar]
- 149. Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br J Pharmacol 95: 1165–1174, 1988. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Fleming I, Busse R. NO: the primary EDRF. J Mol Cell Cardiol 31: 5–14, 1999. doi: 10.1006/jmcc.1998.0839. [DOI] [PubMed] [Google Scholar]
- 151. Brandes RP, Schmitz-Winnenthal FH, Félétou M, Gödecke A, Huang PL, Vanhoutte PM, Fleming I, Busse R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA 97: 9747–9752, 2000. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BKCa channels. Circulation 107: 769–776, 2003. doi: 10.1161/01.CIR.0000047278.28407.C2. [DOI] [PubMed] [Google Scholar]
- 153. Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci 23: 374–380, 2002. doi: 10.1016/S0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 154. Varshney R, Ali Q, Wu C, Sun Z. Monocrotaline-induced pulmonary hypertension involves downregulation of antiaging protein klotho and eNOS activity. Hypertension 68: 1255–1263, 2016. doi: 10.1161/HYPERTENSIONAHA.116.08184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wang Y, Coe Y, Toyoda O, Coceani F. Involvement of endothelin-1 in hypoxic pulmonary vasoconstriction in the lamb. J Physiol 482: 421–434, 1995. doi: 10.1113/jphysiol.1995.sp020529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 290: L284–L290, 2006. doi: 10.1152/ajplung.00449.2004. [DOI] [PubMed] [Google Scholar]
- 157. Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 281: L1058–L1067, 2001. doi: 10.1152/ajplung.2001.281.5.L1058. [DOI] [PubMed] [Google Scholar]
- 158. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, Holmes AM. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol 185: 1850–1858, 2015. doi: 10.1016/j.ajpath.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 159. Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131: 1006–1018, 2015. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 160. Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target Slug. Circulation 133: 1783–1794, 2016. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Qiao L, Nishimura T, Shi L, Sessions D, Thrasher A, Trudell JR, Berry GJ, Pearl RG, Kao PN. Endothelial fate mapping in mice with pulmonary hypertension. Circulation 129: 692–703, 2014. doi: 10.1161/CIRCULATIONAHA.113.003734. [DOI] [PubMed] [Google Scholar]
- 162. Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res 90: 1189–1196, 2002. doi: 10.1161/01.RES.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 163. Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 101: 927–934, 1998. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Evans CE, Cober ND, Dai Z, Stewart DJ, Zhao YY. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur Respir J 58: 2003957, 2021. doi: 10.1183/13993003.03957-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Steffes LC, Froistad AA, Andruska A, Boehm M, McGlynn M, Zhang F, Zhang W, Hou D, Tian X, Miquerol L, Nadeau K, Metzger RJ, Spiekerkoetter E, Kumar ME. A Notch3-marked subpopulation of vascular smooth muscle cells is the cell of origin for occlusive pulmonary vascular lesions. Circulation 142: 1545–1561, 2020. doi: 10.1161/CIRCULATIONAHA.120.045750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Dierick F, Solinc J, Bignard J, Soubrier F, Nadaud S. Progenitor/stem cells in vascular remodeling during pulmonary arterial hypertension. Cells 10: 1338, 2021. doi: 10.3390/cells10061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Firth AL, Yao W, Ogawa A, Madani MM, Lin GY, Yuan JX. Multipotent mesenchymal progenitor cells are present in endarterectomized tissues from patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Cell Physiol 298: C1217–C1225, 2010. doi: 10.1152/ajpcell.00416.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Dierick F, Héry T, Hoareau-Coudert B, Mougenot N, Monceau V, Claude C, Crisan M, Besson V, Dorfmüller P, Marodon G, Fadel E, Humbert M, Yaniz-Galende E, Hulot JS, Marazzi G, Sassoon D, Soubrier F, Nadaud S. Resident PW1+ progenitor cells participate in vascular remodeling during pulmonary arterial hypertension. Circ Res 118: 822–833, 2016. doi: 10.1161/CIRCRESAHA.115.307035. [DOI] [PubMed] [Google Scholar]
- 169. Chow K, Fessel JP, Kaoriihida S, Schmidt EP, Gaskill C, Alvarez D, Graham B, Harrison DG, Wagner DH Jr, Nozik-Grayck E, West JD, Klemm DJ, Majka SM. Dysfunctional resident lung mesenchymal stem cells contribute to pulmonary microvascular remodeling. Pulm Circ 3: 31–49, 2013. doi: 10.4103/2045-8932.109912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Frid MG, Thurman JM, Hansen KC, Maron BA, Stenmark KR. Inflammation, immunity, and vascular remodeling in pulmonary hypertension; evidence for complement involvement? Glob Cardiol Sci Pract 2020: e202001, 2020. doi: 10.21542/gcsp.2020.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol 171: 715–727, 2007. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J 50: 1601805, 2017. doi: 10.1183/13993003.01805-2016. [DOI] [PubMed] [Google Scholar]
- 173. Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc 5: 783–791, 2008. doi: 10.1513/pats.200803-027HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wu K, Tang H, Lin R, Carr SG, Wang Z, Babicheva A, Ayon RJ, Jain PP, Xiong M, Rodriguez M, Rahimi S, Balistrieri F, Rahimi S, Valdez-Jasso D, Simonson TS, Desai AA, Garcia JG, Shyy JY, Thistlethwaite PA, Wang J, Makino A, Yuan JX. Endothelial platelet-derived growth factor-mediated activation of smooth muscle platelet-derived growth factor receptors in pulmonary arterial hypertension. Pulm Circ 10: 1–15, 2020. doi: 10.1177/2045894020948470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Humbert M, Montani D, Perros F, Dorfmüller P, Adnot S, Eddahibi S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol 49: 113–118, 2008. doi: 10.1016/j.vph.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 176. Birukova AA, Arce FT, Moldobaeva N, Dudek SM, Garcia JG, Lal R, Birukov KG. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomedicine 5: 30–41, 2009. doi: 10.1016/j.nano.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem 279: 24692–24700, 2004. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 178. Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol Rev 99: 1467–1525, 2019. doi: 10.1152/physrev.00037.2018. [DOI] [PubMed] [Google Scholar]
- 179. Ganitkevich V, Hasse V, Pfitzer G. Ca2+-dependent and Ca2+-independent regulation of smooth muscle contraction. J Muscle Res Cell Motil 23: 47–52, 2002. doi: 10.1023/A:1019956529549. [DOI] [PubMed] [Google Scholar]
- 180. Hori M, Sato K, Miyamoto S, Ozaki H, Karaki H. Different pathways of calcium sensitization activated by receptor agonists and phorbol esters in vascular smooth muscle. Br J Pharmacol 110: 1527–1531, 1993. doi: 10.1111/j.1476-5381.1993.tb13996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yang XR, Lin AH, Hughes JM, Flavahan NA, Cao YN, Liedtke W, Sham JS. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 302: L555–L568, 2012. doi: 10.1152/ajplung.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ducret T, El Arrouchi J, Courtois A, Quignard JF, Marthan R, Savineau JP. Stretch-activated channels in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Cell Calcium 48: 251–259, 2010. doi: 10.1016/j.ceca.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 183. Otsuki S, Sawada H, Yodoya N, Shinohara T, Kato T, Ohashi H, Zhang E, Imanaka-Yoshida K, Shimpo H, Maruyama K, Komada Y, Mitani Y. Potential contribution of phenotypically modulated smooth muscle cells and related inflammation in the development of experimental obstructive pulmonary vasculopathy in rats. PLoS One 10: e0118655, 2015. doi: 10.1371/journal.pone.0118655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yi ES, Lee H, Yin S, Piguet P, Sarosi I, Kaufmann S, Tarpley J, Wang NS, Ulich TR. Platelet-derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am J Pathol 149: 539–548, 1996. [PMC free article] [PubMed] [Google Scholar]
- 185. Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, Cheng P, Kim JB, Pjanic M, Miano JM, Quertermous T. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ Res 126: 517–529, 2020. doi: 10.1161/CIRCRESAHA.119.315968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282: 36112–36120, 2007. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 187. Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol 28: 434–442, 1997. doi: 10.1016/S0046-8177(97)90032-0. [DOI] [PubMed] [Google Scholar]
- 188. Smith P, Heath D, Yacoub M, Madden B, Caslin A, Gosney J. The ultrastructure of plexogenic pulmonary arteriopathy. J Pathol 160: 111–121, 1990. doi: 10.1002/path.1711600204. [DOI] [PubMed] [Google Scholar]
- 189. Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 21: 134–145, 2006. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 190. Stenmark KR, Frid MG, Yeager M, Li M, Riddle S, McKinsey T, El Kasmi KC. Targeting the adventitial microenvironment in pulmonary hypertension: a potential approach to therapy that considers epigenetic change. Pulm Circ 2: 3–14, 2012. doi: 10.4103/2045-8932.94817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Will DH, McMurtry IF, Reeves JT, Grover RF. Cold-induced pulmonary hypertension in cattle. J Appl Physiol Respir Environ Exerc Physiol 45: 469–473, 1978. doi: 10.1152/jappl.1978.45.3.469. [DOI] [PubMed] [Google Scholar]
- 192. Sydykov A, Maripov A, Kushubakova N, Muratali Uulu K, Satybaldyev S, Kulchoroeva C, Kosanovic D, Sarybaev A. An exaggerated rise in pulmonary artery pressure in a high-altitude dweller during the cold season. Int J Environ Res Public Health 18: 3984, 2021. doi: 10.3390/ijerph18083984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 75: 23–47, 2013. doi: 10.1146/annurev-physiol-030212-183802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hui KP, Cheung MC, Lai KL, Ng KC, Ho JC, Peiris M, Nicholls JM, Chan MC. Role of epithelial-endothelial cell interaction in the pathogenesis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Clin Infect Dis 74: 199–209, 2022. doi: 10.1093/cid/ciab406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Niethamer TK, Stabler CT, Leach JP, Zepp JA, Morley MP, Babu A, Zhou S, Morrisey EE. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife 9: e53072, 2020. doi: 10.7554/eLife.53072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lin C, Zheng X, Lin S, Zhang Y, Wu J, Li Y. Mechanotransduction regulates the interplays between alveolar epithelial and vascular endothelial cells in lung. Front Physiol 13: 818394, 2022. doi: 10.3389/fphys.2022.818394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Wang Y, Li X, Niu W, Chen J, Zhang B, Zhang X, Wang Y, Dang S, Li Z. The alveolar epithelial cells are involved in pulmonary vascular remodeling and constriction of hypoxic pulmonary hypertension. Respir Res 22: 134, 2021. doi: 10.1186/s12931-021-01708-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Low RB. Modulation of myofibroblast and smooth-muscle phenotypes in the lung. Curr Top Pathol 93: 19–26, 1999. doi: 10.1186/s12931-021-01708-w. [DOI] [PubMed] [Google Scholar]
- 199. Biasin V, Crnkovic S, Sahu-Osen A, Birnhuber A, El Agha E, Sinn K, Klepetko W, Olschewski A, Bellusci S, Marsh LM, Kwapiszewska G. PDGFRα and αSMA mark two distinct mesenchymal cell populations involved in parenchymal and vascular remodeling in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 318: L684–L697, 2020. doi: 10.1152/ajplung.00128.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Sun W, Chan SY. Pulmonary arterial stiffness: an early and pervasive driver of pulmonary arterial hypertension. Front Med (Lausanne) 5: 204, 2018. doi: 10.1152/ajplung.00128.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zatulovskiy E, Zhang S, Berenson DF, Topacio BR, Skotheim JM. Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science 369: 466–471, 2020. doi: 10.1126/science.aaz6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Karnes JH, Wiener HW, Schwantes-An TH, Natarajan B, Sweatt AJ, Chaturvedi A, Arora A, Batai K, Nair V, Steiner HE, Giles JB, Yu J, Hosseini M, Pauciulo MW, Lutz KA, Coleman AW, Feldman J, Vanderpool R, Tang H, Garcia JG, Yuan JX, Kittles R, de Jesus Perez V, Zamanian RT, Rischard F, Tiwari HK, Nichols WC, Benza RL, Desai AA. Genetic admixture and survival in diverse populations with pulmonary arterial hypertension. Am J Respir Crit Care Med 201: 1407–1415, 2020. doi: 10.1164/rccm.201907-1447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, et al. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med 7: 227–238, 2019. doi: 10.1016/S2213-2600(18)30409-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, Thistlethwaite PA, Tuder RM, Erzurum SC, Geraci MW, Coldren CD. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 182: 1153–1160, 2010. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Alastalo TP, Li M, Perez V, Pham D, Sawada H, Wang JK, Koskenvuo M, Wang L, Freeman BA, Chang HY, Rabinovitch M. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 121: 3735–3746, 2011. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Miyagawa K, Shi M, Chen PI, Hennigs JK, Zhao Z, Wang M, Li CG, Saito T, Taylor S, Sa S, Cao A, Wang L, Snyder MP, Rabinovitch M. Smooth muscle contact drives endothelial regeneration by BMPR2-Notch1-mediated metabolic and epigenetic changes. Circ Res 124: 211–224, 2019. doi: 10.1161/CIRCRESAHA.118.313374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Ross B, D’Orléans-Juste P, Giaid A. Potential role of endothelin-1 in pulmonary fibrosis: from the bench to the clinic. Am J Respir Cell Mol Biol 42: 16–20, 2010. doi: 10.1165/rcmb.2009-0175TR. [DOI] [PubMed] [Google Scholar]
- 208. Wang L, Halliday G, Huot JR, Satoh T, Baust JJ, Fisher A, Cook T, Hu J, Avolio T, Goncharov DA, Bai Y, Vanderpool RR, Considine RV, Bonetto A, Tan J, Bachman TN, Sebastiani A, McTiernan CF, Mora AL, Machado RF, Goncharova EA, Gladwin MT, Lai YC. Treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in pulmonary hypertension associated with heart failure with preserved ejection fraction. Arterioscler Thromb Vasc Biol 40: 1543–1558, 2020. doi: 10.1161/ATVBAHA.119.313883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Vang A, da Silva Gonçalves Bos D, Fernandez-Nicolas A, Zhang P, Morrison AR, Mancini TJ, Clements RT, Polina I, Cypress MW, Jhun BS, Hawrot E, Mende U, O-Uchi J, Choudhary G. α7 Nicotinic acetylcholine receptor mediates right ventricular fibrosis and diastolic dysfunction in pulmonary hypertension. JCI Insight 6: e142945, 2021. doi: 10.1172/jci.insight.142945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Priest RM, Hucks D, Ward JP. Potentiation of cyclic AMP-mediated vasorelaxation by phenylephrine in pulmonary arteries of the rat. Br J Pharmacol 127: 291–299, 1999. doi: 10.1038/sj.bjp.0702525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Norel X, Walch L, Costantino M, Labat C, Gorenne I, Dulmet E, Rossi F, Brink C. M1 and M3 muscarinic receptors in human pulmonary arteries. Br J Pharmacol 119: 149–157, 1996. doi: 10.1111/j.1476-5381.1996.tb15688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Blum-Johnston C, Thorpe RB, Wee C, Opsahl R, Romero M, Murray S, Brunelle A, Blood Q, Wilson R, Blood AB, Zhang L, Longo LD, Pearce WJ, Wilson SM. Long-term hypoxia uncouples Ca2+ and eNOS in bradykinin-mediated pulmonary arterial relaxation. Am J Physiol Regul Integr Comp Physiol 314: R870–R882, 2018. doi: 10.1152/ajpregu.00311.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Song S, Yamamura A, Yamamura H, Ayon RJ, Smith KA, Tang H, Makino A, Yuan JX. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Cell Physiol 307: C373–C383, 2014. doi: 10.1152/ajpcell.00115.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Shen H, Zhang J, Wang C, Jain PP, Xiong M, Shi X, Lei Y, Chen S, Yin Q, Thistlethwaite PA, Wang J, Gong K, Yuan ZY, Yuan JX, Shyy JY. MDM2-mediated ubiquitination of angiotensin-converting enzyme 2 contributes to the development of pulmonary arterial hypertension. Circulation 142: 1190–1204, 2020. doi: 10.1161/CIRCULATIONAHA.120.048191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Fried ND, Morris TM, Whitehead A, Lazartigues E, Yue X, Gardner JD. Angiotensin II type 1 receptor mediates pulmonary hypertension and right ventricular remodeling induced by inhaled nicotine. Am J Physiol Heart Circ Physiol 320: H1526–H1534, 2021. doi: 10.1152/ajpheart.00883.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Song S, Jacobson KN, McDermott KM, Reddy SP, Cress AE, Tang H, Dudek SM, Black SM, Garcia JG, Makino A, Yuan JX. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am J Physiol Cell Physiol 310: C99–C114, 2016. doi: 10.1152/ajpcell.00092.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Cai Z, Tu L, Guignabert C, Merkus D, Zhou Z. Purinergic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc 9: e017404, 2020. doi: 10.1161/JAHA.120.017404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X. Purinergic signaling in pulmonary inflammation. Front Immunol 10: 1633, 2019. doi: 10.3389/fimmu.2019.01633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Verin AD, Batori R, Kovacs-Kasa A, Cherian-Shaw M, Kumar S, Czikora I, Karoor V, Strassheim D, Stenmark KR, Gerasimovskaya EV. Extracellular adenosine enhances pulmonary artery vasa vasorum endothelial cell barrier function via Gi/ELMO1/Rac1/PKA-dependent signaling mechanisms. Am J Physiol Cell Physiol 319: C183–C193, 2020. doi: 10.1152/ajpcell.00505.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Alencar AK, Carvalho FI, Silva AM, Martinez ST, Calasans-Maia JA, Fraga CM, Barreiro EJ, Zapata-Sudo G, Sudo RT. Synergistic interaction between a PDE5 inhibitor (sildenafil) and a new adenosine A2A receptor agonist (LASSBio-1359) improves pulmonary hypertension in rats. PLoS One 13: e0195047, 2018. doi: 10.1371/journal.pone.0195047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Song S, Babicheva A, Zhao T, Ayon RJ, Rodriguez M, Rahimi S, Balistrieri F, Harrington A, Shyy JY, Thistlethwaite PA, Makino A, Yuan JX. Notch enhances Ca2+ entry by activating calcium-sensing receptors and inhibiting voltage-gated K+ channels. Am J Physiol Cell Physiol 318: C954–C968, 2020. doi: 10.1152/ajpcell.00487.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Smith KA, Ayon RJ, Tang H, Makino A, Yuan JX. Calcium-sensing receptor regulates cytosolic [Ca2+] and plays a major role in the development of pulmonary hypertension. Front Physiol 7: 517, 2016. doi: 10.3389/fphys.2016.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca2+-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012. doi: 10.1161/CIRCRESAHA.112.266361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Yamamura A, Nayeem MJ, Al Mamun A, Takahashi R, Hayashi H, Sato M. Platelet-derived growth factor up-regulates Ca2+-sensing receptors in idiopathic pulmonary arterial hypertension. FASEB J 33: 7363–7374, 2019. doi: 10.1096/fj.201802620R. [DOI] [PubMed] [Google Scholar]
- 225. Tang H, Yamamura A, Yamamura H, Song S, Fraidenburg DR, Chen J, Gu Y, Pohl NM, Zhou T, Jiménez-Pérez L, Ayon RJ, Desai AA, Goltzman D, Rischard F, Khalpey Z, Black SM, Garcia JG, Makino A, Yuan JX. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L846–L859, 2016. doi: 10.1152/ajplung.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Peng X, Li HX, Shao HJ, Li GW, Sun J, Xi YH, Li HZ, Wang XY, Wang LN, Bai SZ, Zhang WH, Zhang L, Yang GD, Wu LY, Wang R, Xu CQ. Involvement of calcium-sensing receptors in hypoxia-induced vascular remodeling and pulmonary hypertension by promoting phenotypic modulation of small pulmonary arteries. Mol Cell Biochem 396: 87–98, 2014. doi: 10.1007/s11010-014-2145-9. [DOI] [PubMed] [Google Scholar]
- 227. Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, Sammani S, Zhou G, Raj JU, Garcia JG, Berdyshev E, Yuan JX, Natarajan V, Machado RF. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am J Respir Crit Care Med 190: 1032–1043, 2014. doi: 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Peng X, Sammani S, McVerry BJ, Hassoun PM, Garcia JG. Sphingosine 1-phosphate reduces lung vascular permeability in a murine model of LPS-mediated acute lung injury (Abstract). Am J Respir Crit Care Med 170: A778, 2003. [DOI] [PubMed] [Google Scholar]
- 229. Ji Y, Lisabeth EM, Neubig RR. Transforming growth factor β1 increases expression of contractile genes in human pulmonary arterial smooth muscle cells by potentiating sphingosine-1-phosphate signaling. Mol Pharmacol 100: 53–60, 2021. doi: 10.1124/molpharm.120.000019. [DOI] [PubMed] [Google Scholar]
- 230. Wang J, Yan X, Feng W, Wang Q, Shi W, Chai L, Zhang Q, Chen Y, Liu J, Qu Z, Xie X, Li M. S1P induces proliferation of pulmonary artery smooth muscle cells by promoting YAP-induced Notch3 expression and activation. J Biol Chem 296: 100599, 2021. doi: 10.1016/j.jbc.2021.100599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Ota H, Beutz MA, Ito M, Abe K, Oka M, McMurtry IF. S1P(4) receptor mediates S1P-induced vasoconstriction in normotensive and hypertensive rat lungs. Pulm Circ 1: 399–404, 2011. doi: 10.4103/2045-8932.87309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R, Upton PD, Crosby A, Sawiak SJ, Carpenter TA, Glen RC, Morrell NW, Maguire JJ, Davenport AP. Elabela/Toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135: 1160–1173, 2017. doi: 10.1161/CIRCULATIONAHA.116.023218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Frump AL, Albrecht M, Yakubov B, Breuils-Bonnet S, Nadeau V, Tremblay E, Potus F, Omura J, Cook T, Fisher A, Rodriguez B, Brown RD, Stenmark KR, Rubinstein CD, Krentz K, Tabima DM, Li R, Sun X, Chesler NC, Provencher S, Bonnet S, Lahm T. 17β-Estradiol and estrogen receptor alpha protect right ventricular function in pulmonary hypertension via BMPR2 and apelin. J Clin Invest 131: e129433, 2021. doi: 10.1172/JCI129433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 19: 74–82, 2013. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Andersen CU, Hilberg O, Mellemkjær S, Nielsen‐Kudsk JE, Simonsen U. Apelin and pulmonary hypertension. Pulm Circ 1: 334–346, 2011. doi: 10.4103/2045-8932.87299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Leuchte HH, Baezner C, Baumgartner RA, Bevec D, Bacher G, Neurohr C, Behr J. Inhalation of vasoactive intestinal peptide in pulmonary hypertension. Eur Respir J 32: 1289–1294, 2008. doi: 10.1183/09031936.00050008. [DOI] [PubMed] [Google Scholar]
- 237. Leuchte HH, Prechtl C, Callegari J, Meis T, Haziraj S, Bevec D, Behr J. Augmentation of the effects of vasoactive intestinal peptide aerosol on pulmonary hypertension via coapplication of a neutral endopeptidase 24.11 inhibitor. Am J Physiol Lung Cell Mol Physiol 308: L563–L568, 2015. doi: 10.1152/ajplung.00317.2014. [DOI] [PubMed] [Google Scholar]
- 238. Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, Funk GC, Hamilton G, Novotny C, Burian B, Block LH. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 111: 1339–1346, 2003. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Hamidi SA, Lin RZ, Szema AM, Lyubsky S, Jiang YP, Said SI. VIP and endothelin receptor antagonist: an effective combination against experimental pulmonary arterial hypertension. Respir Res 12: 141, 2011. doi: 10.1186/1465-9921-12-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 115: 1260–1268, 2007. doi: 10.1161/CIRCULATIONAHA.106.681718. [DOI] [PubMed] [Google Scholar]
- 241. Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest 115: 2691–2694, 2005. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 243. Kunichika N, Landsberg JW, Yu Y, Kunichika H, Thistlethwaite PA, Rubin LJ, Yuan JX. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 170: 1101–1107, 2004. doi: 10.1164/rccm.200312-1668OC. [DOI] [PubMed] [Google Scholar]
- 244. Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. doi: 10.1152/ajpcell.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Perros F, Montani D, Dorfmüller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, Hervé P, Emilie D, Eddahibi S, Simonneau G, Souza R, Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 81–88, 2008. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 246. Song S, Zhang M, Yi Z, Zhang H, Shen T, Yu X, Zhang C, Zheng X, Yu L, Ma C, Liu Y, Zhu D. The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell Signal 28: 1489–1501, 2016. doi: 10.1016/j.cellsig.2016.06.022. [DOI] [PubMed] [Google Scholar]
- 247. Ten Freyhaus H, Berghausen EM, Janssen W, Leuchs M, Zierden M, Murmann K, Klinke A, Vantler M, Caglayan E, Kramer T, Baldus S, Schermuly RT, Tallquist MD, Rosenkranz S. Genetic ablation of PDGF-dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol 35: 1236–1245, 2015. doi: 10.1161/ATVBAHA.114.304864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Hobohm L, Kölmel S, Niemann C, Kümpers P, Krieg VJ, Bochenek ML, Lukasz AH, Reiss Y, Plate KH, Liebetrau C, Wiedenroth CB, Guth S, Münzel T, Hasenfuß G, Wenzel P, Mayer E, Konstantinides SV, Schäfer K, Lankeit M. Role of angiopoietin-2 in venous thrombus resolution and chronic thromboembolic disease. Eur Respir J 58: 2004196, 2021. doi: 10.1183/13993003.04196-2020. [DOI] [PubMed] [Google Scholar]
- 250. Richter MJ, Schermuly R, Seeger W, Rao Y, Ghofrani HA, Gall H. Relevance of angiopoietin-2 and soluble P-selectin levels in patients with pulmonary arterial hypertension receiving combination therapy with oral treprostinil: a FREEDOM-C2 biomarker substudy. Pulm Circ 6: 516–523, 2016. doi: 10.1086/688671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Kümpers P, Nickel N, Lukasz A, Golpon H, Westerkamp V, Olsson KM, Jonigk D, Maegel L, Bockmeyer CL, David S, Hoeper MM. Circulating angiopoietins in idiopathic pulmonary arterial hypertension. Eur Heart J 31: 2291–2300, 2010. doi: 10.1093/eurheartj/ehq226. [DOI] [PubMed] [Google Scholar]
- 252. Dewachter L, Adnot S, Fadel E, Humbert M, Maitre B, Barlier-Mur AM, Simonneau G, Hamon M, Naeije R, Eddahibi S. Angiopoietin/Tie2 pathway influences smooth muscle hyperplasia in idiopathic pulmonary hypertension. Am J Respir Crit Care Med 174: 1025–1033, 2006. doi: 10.1164/rccm.200602-304OC. [DOI] [PubMed] [Google Scholar]
- 253. Sullivan CC, Du L, Chu D, Cho AJ, Kido M, Wolf PL, Jamieson SW, Thistlethwaite PA. Induction of pulmonary hypertension by an angiopoietin 1/TIE2/serotonin pathway. Proc Natl Acad Sci USA 100: 12331–12336, 2003. doi: 10.1073/pnas.1933740100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Chen J, Sysol JR, Singla S, Zhao S, Yamamura A, Valdez-Jasso D, Abbasi T, Shioura KM, Sahni S, Reddy V, Sridhar A, Gao H, Torres J, Camp SM, Tang H, Ye SQ, Comhair S, Dweik R, Hassoun P, Yuan JX, Garcia JG, Machado RF. Nicotinamide phosphoribosyltransferase promotes pulmonary vascular remodeling and is a therapeutic target in pulmonary arterial hypertension. Circulation 135: 1532–1546, 2017. doi: 10.1161/CIRCULATIONAHA.116.024557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Sun X, Sun BL, Babicheva A, Vanderpool R, Oita RC, Casanova N, Tang H, Gupta A, Lynn H, Gupta G, Rischard F, Sammani S, Kempf CL, Moreno-Vinasco L, Ahmed M, Camp SM, Wang J, Desai AA, Yuan JX, Garcia JG. Direct extracellular NAMPT involvement in pulmonary hypertension and vascular remodeling. Transcriptional regulation by SOX and HIF-2α. Am J Respir Cell Mol Biol 63: 92–103, 2020. doi: 10.1165/rcmb.2019-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, Moreno-Vinasco L, Mapes B, Zhao J, Gursoy G, Brown ME, Adyshev DM, Siddiqui SS, Quijada H, Sammani S, Letsiou E, Saadat L, Yousef M, Wang T, Liang J, Garcia JG. Unique toll-like receptor 4 activation by NAMPT/PBEF induces NFκB signaling and inflammatory lung injury. Sci Rep 5: 13135, 2015. doi: 10.1038/srep13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Liu P, Li H, Cepeda J, Zhang LQ, Cui X, Garcia JG, Ye SQ. Critical role of PBEF expression in pulmonary cell inflammation and permeability. Cell Biol Int 33: 19–30, 2009. doi: 10.1016/j.cellbi.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Moreno-Vinasco L, Quijada H, Sammani S, Siegler J, Letsiou E, Deaton R, Saadat L, Zaidi RS, Messana J, Gann PH, Machado RF, Ma W, Camp SM, Wang T, Garcia JG. Nicotinamide phosphoribosyltransferase inhibitor is a novel therapeutic candidate in murine models of inflammatory lung injury. Am J Respir Cell Mol Biol 51: 223–228, 2014. doi: 10.1165/rcmb.2012-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 195: 367–374, 2001. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 260. Dahal BK, Cornitescu T, Tretyn A, Pullamsetti SS, Kosanovic D, Dumitrascu R, Ghofrani HA, Weissmann N, Voswinckel R, Banat GA, Seeger W, Grimminger F, Schermuly RT. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med 181: 158–167, 2010. doi: 10.1164/rccm.200811-1682OC. [DOI] [PubMed] [Google Scholar]
- 261. Kinoshita D, Shishido T, Takahashi T, Yokoyama M, Sugai T, Watanabe K, Tamura H, Nishiyama S, Takahashi H, Arimoto T, Miyamoto T, Watanabe T, Kishida S, Kadomatsu K, Abe JI, Takeishi Y, Konta T, Kubota I, Watanabe M. Growth factor midkine aggravates pulmonary arterial hypertension via surface nucleolin. Sci Rep 10: 10345, 2020. doi: 10.1038/s41598-020-67217-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Tu L, Desroches-Castan A, Mallet C, Guyon L, Cumont A, Phan C, Robert F, Thuillet R, Bordenave J, Sekine A, Huertas A, Ritvos O, Savale L, Feige JJ, Humbert M, Bailly S, Guignabert C. Selective BMP-9 inhibition partially protects against experimental pulmonary hypertension. Circ Res 124: 846–855, 2019. doi: 10.1161/CIRCRESAHA.118.313356. [DOI] [PubMed] [Google Scholar]
- 263. Salmon RM, Guo J, Wood JH, Tong Z, Beech JS, Lawera A, Yu M, Grainger DJ, Reckless J, Morrell NW, Li W. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat Commun 11: 1621, 2020. doi: 10.1038/s41467-020-15425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Hodgson J, Swietlik EM, Salmon RM, Hadinnapola C, Nikolic I, Wharton J, et al. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am J Respir Crit Care Med 201: 575–585, 2020. doi: 10.1164/rccm.201906-1141OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Wang L, Rice M, Swist S, Kubin T, Wu F, Wang S, Kraut S, Weissmann N, Böttger T, Wheeler M, Schneider A, Braun T. BMP9 and BMP10 act directly on vascular smooth muscle cells for generation and maintenance of the contractile state. Circulation 143: 1394–1410, 2021. doi: 10.1161/CIRCULATIONAHA.120.047375. [DOI] [PubMed] [Google Scholar]
- 266. Simpson CE, Chen JY, Damico RL, Hassoun PM, Martin LJ, Yang J, Nies M, Griffiths M, Vaidya RD, Brandal S, Pauciulo MW, Lutz KA, Coleman AW, Austin ED, Ivy DD, Nichols WC, Everett AD. Cellular sources of interleukin-6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur Respir J 55: 1901761, 2020. doi: 10.1183/13993003.01761-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244, 2009. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res 10: 6, 2009. doi: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, Fadel E, Savale L, Huertas A, Humbert M, Guignabert C. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest 128: 1956–1970, 2018. doi: 10.1172/JCI96462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest 128: 572S–573S, 2005. doi: 10.1378/chest.128.6_suppl.572S-a. [DOI] [PubMed] [Google Scholar]
- 271. Liu J, Wang W, Wang L, Chen S, Tian B, Huang K, Corrigan CJ, Ying S, Wang W, Wang C. IL-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating HIF-1α and VEGF expression in vascular endothelial cells. EBioMedicine 33: 196–210, 2018. doi: 10.1016/j.ebiom.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Lee JH, Hailey KL, Vitorino SA, Jennings PA, Bigby TD, Breen EC. Cigarette smoke triggers IL-33-associated inflammation in a model of late-stage chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 61: 567–574, 2019. doi: 10.1165/rcmb.2018-0402OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov 7: 827–840, 2008. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, Southwood M, Yang X, Nikolic MZ, Herrera B, Inman GJ, Bradley JR, Rana AA, Upton PD, Morrell NW. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun 8: 14079, 2017. doi: 10.1038/ncomms14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Wang Q, Zuo XR, Wang YY, Xie WP, Wang H, Zhang M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-alpha antagonists via the suppression of TNF-alpha expression and NF-kappaB pathway in rats. Vascul Pharmacol 58: 71–77, 2013. doi: 10.1016/j.vph.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 276. Tang H, Fernandez RA, Yuan JX. miRNA208/Mef2 and TNF-α in right ventricular dysfunction: the transition from hypertrophy to failure. Circ Res 116: 6–8, 2015. doi: 10.1161/CIRCRESAHA.114.305446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Paulin R, Sutendra G, Gurtu V, Dromparis P, Haromy A, Provencher S, Bonnet S, Michelakis ED. A miR-208-Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res 116: 56–69, 2015. doi: 10.1161/CIRCRESAHA.115.303910. [DOI] [PubMed] [Google Scholar]
- 278. Mukai Y, Shibata H, Nakamura T, Yoshioka Y, Abe Y, Nomura T, Taniai M, Ohta T, Ikemizu S, Nakagawa S, Tsunoda S, Kamada H, Yamagata Y, Tsutsumi Y. Structure-function relationship of tumor necrosis factor (TNF) and its receptor interaction based on 3D structural analysis of a fully active TNFR1-selective TNF mutant. J Mol Biol 385: 1221–1229, 2009. doi: 10.1016/j.jmb.2008.11.053. [DOI] [PubMed] [Google Scholar]
- 279. Percival KR, Radi ZA. A modified Verhoeff’s elastin histochemical stain to enable pulmonary arterial hypertension model characterization. Eur J Histochem 60: 2588, 2016. doi: 10.4081/ejh.2016.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Meyrick B, Reid L. Ultrastructural features of the distended pulmonary arteries of the normal rat. Anat Rec 193: 71–97, 1979. doi: 10.1002/ar.1091930106. [DOI] [PubMed] [Google Scholar]
- 281. Isakson BE, Ramos SI, Duling BR. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ Res 100: 246–254, 2007. doi: 10.1161/01.RES.0000257744.23795.93. [DOI] [PubMed] [Google Scholar]
- 282. Schuster A, Oishi H, Bény JL, Stergiopulos N, Meister JJ. Simultaneous arterial calcium dynamics and diameter measurements: application to myoendothelial communication. Am J Physiol Heart Circ Physiol 280: H1088–H1096, 2001. doi: 10.1152/ajpheart.2001.280.3.H1088. [DOI] [PubMed] [Google Scholar]
- 283. Dempsie Y, Martin P, Upton PD. Connexin-mediated regulation of the pulmonary vasculature. Biochem Soc Trans 43: 524–529, 2015. doi: 10.1042/BST20150030. [DOI] [PubMed] [Google Scholar]
- 284. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7: 678–689, 2006. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 285. Thistlethwaite PA, Li X, Zhang X. Notch signaling in pulmonary hypertension. In: Membrane Receptors, Channels and Transporters in Pulmonary Circulation, edited by Yuan JX, Ward JP.. Totowa, NJ: Humana Press, 2010, p. 27. [Google Scholar]
- 286. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science 284: 770–776, 1999. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 287. Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ Res 91: 999–1006, 2002. doi: 10.1161/01.RES.0000044944.99984.25. [DOI] [PubMed] [Google Scholar]
- 288. Miele L, Osborne B. Arbiter of differentiation and death: Notch signaling meets apoptosis. J Cell Physiol 181: 393–409, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 289. Sakata Y, Xiang F, Chen Z, Kiriyama Y, Kamei CN, Simon DI, Chin MT. Transcription factor CHF1/Hey2 regulates neointimal formation in vivo and vascular smooth muscle proliferation and migration in vitro. Arterioscler Thromb Vasc Biol 24: 2069–2074, 2004. doi: 10.1161/01.ATV.0000143936.77094.a4. [DOI] [PubMed] [Google Scholar]
- 290. Morrow D, Sweeney C, Birney YA, Cummins PM, Walls D, Redmond EM, Cahill PA. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ Res 96: 567–575, 2005. doi: 10.1161/01.RES.0000159182.98874.43. [DOI] [PubMed] [Google Scholar]
- 291. Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18: 2730–2735, 2004. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128: 3675–3683, 2001. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- 293. Gridley T. Notch signaling in the vasculature. Curr Top Dev Biol 92: 277–309, 2010. doi: 10.1016/S0070-2153(10)92009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A, Powell FL, Thistlethwaite PA, Yuan JX. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca2+ entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 306: C871–C878, 2014. doi: 10.1152/ajpcell.00221.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Thistlethwaite PA, Li X, Zhang X. Notch signaling in pulmonary hypertension. Adv Exp Med Biol 661: 279–298, 2010. doi: 10.1007/978-1-60761-500-2_18. [DOI] [PubMed] [Google Scholar]
- 296. Babicheva A, Yuan JX. Endothelial Notch1 in pulmonary hypertension. Circ Res 124: 176–179, 2019. doi: 10.1161/CIRCRESAHA.118.314496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Campa VM, Gutiérrez-Lanza R, Cerignoli F, Diaz-Trelles R, Nelson B, Tsuji T, Barcova M, Jiang W, Mercola M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol 183: 129–141, 2008. doi: 10.1083/jcb.200806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Dabral S, Tian X, Kojonazarov B, Savai R, Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, Grimminger F, Seeger W, Savai Pullamsetti S, Schermuly RT. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur Respir J 48: 1137–1149, 2016. doi: 10.1183/13993003.00773-2015. [DOI] [PubMed] [Google Scholar]
- 299. Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev 99: 1281–1324, 2019. doi: 10.1152/physrev.00021.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev 66: 513–569, 2014. doi: 10.1124/pr.112.007351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Lemmey HA, Garland CJ, Dora KA. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr Top Membr 85: 327–355, 2020. doi: 10.1016/bs.ctm.2020.01.004. [DOI] [PubMed] [Google Scholar]
- 303. Heberlein KR, Straub AC, Best AK, Greyson MA, Looft-Wilson RC, Sharma PR, Meher A, Leitinger N, Isakson BE. Plasminogen activator inhibitor-1 regulates myoendothelial junction formation. Circ Res 106: 1092–1102, 2010. doi: 10.1161/CIRCRESAHA.109.215723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. McNair AJ, Wilson KS, Martin PE, Welsh DJ, Dempsie Y. Connexin 43 plays a role in proliferation and migration of pulmonary arterial fibroblasts in response to hypoxia. Pulm Circ 10: 2045894020937134, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Ying X, Minamiya Y, Fu C, Bhattacharya J. Ca2+ waves in lung capillary endothelium. Circ Res 79: 898–908, 1996. doi: 10.1161/01.RES.79.4.898. [DOI] [PubMed] [Google Scholar]
- 306. Miriel VA, Mauban JR, Blaustein MP, Wier WG. Local and cellular Ca2+ transients in smooth muscle of pressurized rat resistance arteries during myogenic and agonist stimulation. J Physiol 518: 815–824, 1999. doi: 10.1111/j.1469-7793.1999.0815p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Behringer EJ, Segal SS. Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+-activated K+ channels. Circ Res 110: 1311–1321, 2012. doi: 10.1161/CIRCRESAHA.111.262592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Si R, Zhang Q, Cabrera JT, Zheng Q, Tsuji-Hosokawa A, Watanabe M, Hosokawa S, Xiong M, Jain PP, Ashton AW, Yuan JX, Wang J, Makino A. Chronic hypoxia decreases endothelial connexin 40, attenuates endothelium-dependent hyperpolarization-mediated relaxation in small distal pulmonary arteries, and leads to pulmonary hypertension. J Am Heart Assoc 9: e018327, 2020. doi: 10.1161/JAHA.120.018327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J, Tabeling C, Barbosa-Sicard E, Chanson M, Kwak BR, Shin HS, Wu S, Isakson BE, Witzenrath M, de Wit C, Fleming I, Kuppe H, Kuebler WM. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest 122: 4218–4230, 2012. doi: 10.1172/JCI59176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Hong X, Sin WC, Harris AL, Naus CC. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 6: 15566–15577, 2015. doi: 10.18632/oncotarget.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Fukuda S, Akiyama M, Niki Y, Kawatsura R, Harada H, Nakahama KI. Inhibitory effects of miRNAs in astrocytes on C6 glioma progression via connexin 43. Mol Cell Biochem 476: 2623–2632, 2021. doi: 10.1007/s11010-021-04118-0. [DOI] [PubMed] [Google Scholar]
- 312. Peng Y, Wang X, Guo Y, Peng F, Zheng N, He B, Ge H, Tao L, Wang Q. Pattern of cell-to-cell transfer of microRNA by gap junction and its effect on the proliferation of glioma cells. Cancer Sci 110: 1947–1958, 2019. doi: 10.1111/cas.14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Lemcke H, David R. Potential mechanisms of microRNA mobility. Traffic 19: 910–917, 2018. doi: 10.1111/tra.12606. [DOI] [PubMed] [Google Scholar]
- 314. Fan X, Teng Y, Ye Z, Zhou Y, Tan WS. The effect of gap junction-mediated transfer of miR-200b on osteogenesis and angiogenesis in a co-culture of MSCs and HUVECs. J Cell Sci 131: jcs216135, 2018. doi: 10.1242/jcs.216135. [DOI] [PubMed] [Google Scholar]
- 315. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, Bhattacharya J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506: 503–506, 2014. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Sinha P, Islam MN, Bhattacharya S, Bhattacharya J. Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr Opin Genet Dev 38: 97–101, 2016. doi: 10.1016/j.gde.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Rogers RS, Bhattacharya J. When cells become organelle donors. Physiology (Bethesda) 28: 414–422, 2013. doi: 10.1152/physiol.00032.2013. [DOI] [PubMed] [Google Scholar]
- 318. Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol 295: C221–C230, 2008. doi: 10.1152/ajpcell.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Htet M, Nally JE, Shaw A, Foote BE, Martin PE, Dempsie Y. Connexin 43 plays a role in pulmonary vascular reactivity in mice. Int J Mol Sci 19: 1891, 2018. doi: 10.3390/ijms19071891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Kizub IV, Lakhkar A, Dhagia V, Joshi SR, Jiang H, Wolin MS, Falck JR, Koduru SR, Errabelli R, Jacobs ER, Schwartzman ML, Gupte SA. Involvement of gap junctions between smooth muscle cells in sustained hypoxic pulmonary vasoconstriction development: a potential role for 15-HETE and 20-HETE. Am J Physiol Lung Cell Mol Physiol 310: L772–L783, 2016. doi: 10.1152/ajplung.00377.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Billaud M, Dahan D, Marthan R, Savineau JP, Guibert C. Role of the gap junctions in the contractile response to agonists in pulmonary artery from two rat models of pulmonary hypertension. Respir Res 12: 30, 2011. doi: 10.1186/1465-9921-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Bouvard C, Genet N, Phan C, Rode B, Thuillet R, Tu L, Robillard P, Campagnac M, Soleti R, Dumas De La Roque E, Delcambre F, Cronier L, Parpaite T, Maurat E, Berger P, Savineau JP, Marthan R, Guignabert C, Freund-Michel V, Guibert C. Connexin-43 is a promising target for pulmonary hypertension due to hypoxaemic lung disease. Eur Respir J 55: 1900169, 2020. doi: 10.1183/13993003.00169-2019. [DOI] [PubMed] [Google Scholar]
- 323. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138: 576–591, 2009. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. O’Neill LA. DNA makes RNA makes innate immunity. Cell 138: 428–430, 2009. doi: 10.1016/j.cell.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 325. Patel SJ, King KR, Casali M, Yarmush ML. DNA-triggered innate immune responses are propagated by gap junction communication. Proc Natl Acad Sci USA 106: 12867–12872, 2009. doi: 10.1073/pnas.0809292106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Frank SA. Somatic mosaicism and disease. Curr Biol 24: R577–R581, 2014. doi: 10.1016/j.cub.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 327. Michelakis ED. Pulmonary arterial hypertension: yesterday, today, tomorrow. Circ Res 115: 109–114, 2014. doi: 10.1161/CIRCRESAHA.115.301132. [DOI] [PubMed] [Google Scholar]
- 328. Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab 20: 827–839, 2014. doi: 10.1016/j.cmet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 329. Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res 115: 148–164, 2014. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 330. Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab 19: 558–573, 2014. doi: 10.1016/j.cmet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 331. Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158: 84–97, 2014. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- 332. Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA, Monyer H, Mammano F. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci USA 105: 18770–18775, 2008. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Shuhaibar LC, Egbert JR, Norris RP, Lampe PD, Nikolaev VO, Thunemann M, Wen L, Feil R, Jaffe LA. Intercellular signaling via cyclic GMP diffusion through gap junctions restarts meiosis in mouse ovarian follicles. Proc Natl Acad Sci USA 112: 5527–5532, 2015. doi: 10.1073/pnas.1423598112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. de Wit C, Roos F, Bolz SS, Kirchhoff S, Krüger O, Willecke K, Pohl U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 86: 649–655, 2000. doi: 10.1161/01.RES.86.6.649. [DOI] [PubMed] [Google Scholar]
- 335. Gärtner C, Ziegelhöffer B, Kostelka M, Stepan H, Mohr FW, Dhein S. Knock-down of endothelial connexins impairs angiogenesis. Pharmacol Res 65: 347–357, 2012. doi: 10.1016/j.phrs.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 336. Alonso F, Domingos-Pereira S, Le Gal L, Derré L, Meda P, Jichlinski P, Nardelli-Haefliger D, Haefliger JA. Targeting endothelial connexin40 inhibits tumor growth by reducing angiogenesis and improving vessel perfusion. Oncotarget 7: 14015–14028, 2016. doi: 10.18632/oncotarget.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Li N, Dai DZ, Dai Y. CPU86017 and its isomers improve hypoxic pulmonary hypertension by attenuating increased ETA receptor expression and extracellular matrix accumulation. Naunyn Schmiedebergs Arch Pharmacol 378: 541–552, 2008. doi: 10.1007/s00210-008-0309-4. [DOI] [PubMed] [Google Scholar]
- 338. Yang L, Yin N, Hu L, Fan H, Yu D, Zhang W, Wang S, Feng Y, Fan C, Cao F, Mo X. Sildenefil increases connexin 40 in smooth muscle cells through activation of BMP pathways in pulmonary arterial hypertension. Int J Clin Exp Pathol 7: 4674–4684, 2014. [PMC free article] [PubMed] [Google Scholar]
- 339. Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, Erzurum SC, Chun HJ. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131: 190–199, 2015. doi: 10.1161/CIRCULATIONAHA.114.013339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Krattinger N, Capponi A, Mazzolai L, Aubert JF, Caille D, Nicod P, Waeber G, Meda P, Haefliger JA. Connexin40 regulates renin production and blood pressure. Kidney Int 72: 814–822, 2007. doi: 10.1038/sj.ki.5002423. [DOI] [PubMed] [Google Scholar]
- 341. de Wit C, Roos F, Bolz SS, Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics 13: 169–177, 2003. doi: 10.1152/physiolgenomics.00169.2002. [DOI] [PubMed] [Google Scholar]
- 342. Seul KH, Kang KY, Lee KS, Kim SH, Beyer EC. Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem Biophys Res Commun 319: 1144–1151, 2004. doi: 10.1016/j.bbrc.2004.05.097. [DOI] [PubMed] [Google Scholar]
- 343. Jacobsen NL, Pontifex TK, Li H, Solan JL, Lampe PD, Sorgen PL, Burt JM. Regulation of Cx37 channel and growth-suppressive properties by phosphorylation. J Cell Sci 130: 3308–3321, 2017. doi: 10.1242/jcs.202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Fang JS, Coon BG, Gillis N, Chen Z, Qiu J, Chittenden TW, Burt JM, Schwartz MA, Hirschi KK. Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat Commun 8: 2149, 2017. doi: 10.1038/s41467-017-01742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Wang H, Sun X, Lu Q, Zemskov EA, Yegambaram M, Wu X, Wang T, Tang H, Black SM. The mitochondrial redistribution of eNOS is involved in lipopolysaccharide induced inflammasome activation during acute lung injury. Redox Biol 41: 101878, 2021. doi: 10.1016/j.redox.2021.101878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Lu Q, Zemskov EA, Sun X, Wang H, Yegambaram M, Wu X, Garcia-Flores A, Song S, Tang H, Kangath A, Cabanillas GZ, Yuan JX, Wang T, Fineman JR, Black SM. Activation of the mechanosensitive Ca2+ channel TRPV4 induces endothelial barrier permeability via the disruption of mitochondrial bioenergetics. Redox Biol 38: 101785, 2021. doi: 10.1016/j.redox.2020.101785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation 133: 2447–2458, 2016. doi: 10.1161/CIRCULATIONAHA.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, Shay S, French JL, West J, Haase VH. The endothelial prolyl-4-hydroxylase domain 2/hypoxia-inducible factor 2 axis regulates pulmonary artery pressure in mice. Mol Cell Biol 36: 1584–1594, 2016. doi: 10.1128/MCB.01055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Babicheva A, Makino A, Yuan JX. mTOR signaling in pulmonary vascular disease: pathogenic role and therapeutic target. Int J Mol Sci 22: 2144, 2021. doi: 10.3390/ijms22042144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. doi: 10.1161/CIRCULATIONAHA.113.004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Goncharova EA, Simon MA, Yuan JX. mTORC1 in pulmonary arterial hypertension. At the crossroads between vasoconstriction and vascular remodeling? Am J Respir Crit Care Med 201: 1177–1179, 2020. doi: 10.1164/rccm.202001-0087ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. doi: 10.1152/ajplung.00242.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Tang H, Wu K, Wang J, Vinjamuri S, Gu Y, Song S, Wang Z, Zhang Q, Balistrieri A, Ayon RJ, Rischard F, Vanderpool R, Chen J, Zhou G, Desai AA, Black SM, Garcia JG, Yuan JX, Makino A. Pathogenic role of mTORC1 and mTORC2 in pulmonary hypertension. JACC Basic Transl Sci 3: 744–762, 2018. doi: 10.1016/j.jacbts.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Farkas D, Alhussaini AA, Kraskauskas D, Kraskauskiene V, Cool CD, Nicolls MR, Natarajan R, Farkas L. Nuclear factor κB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am J Respir Cell Mol Biol 51: 413–425, 2014. doi: 10.1165/rcmb.2013-0355OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Mitra S, Epshtein Y, Sammani S, Quijada H, Chen W, Bandela M, Desai AA, Garcia JG, Jacobson JR. UCHL1, a deubiquitinating enzyme, regulates lung endothelial cell permeability in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 320: L497–L507, 2021. doi: 10.1152/ajplung.00492.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Mitra S, Sammani S, Wang T, Boone DL, Meyer NJ, Dudek SM, Moreno-Vinasco L, Garcia JG, Jacobson JR. Role of growth arrest and DNA damage-inducible alpha in Akt phosphorylation and ubiquitination after mechanical stress-induced vascular injury. Am J Respir Crit Care Med 184: 1030–1040, 2011. doi: 10.1164/rccm.201103-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22: 1276–1312, 2008. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Gross CM, Rafikov R, Kumar S, Aggarwal S, Ham PB 3rd, Meadows ML, Cherian-Shaw M, Kangath A, Sridhar S, Lucas R, Black SM. Endothelial nitric oxide synthase deficient mice are protected from lipopolysaccharide induced acute lung injury. PLoS One 10: e0119918, 2015. doi: 10.1371/journal.pone.0119918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol 3: 1011–1034, 2013. doi: 10.1371/journal.pone.0119918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Sun X, Kellner M, Desai AA, Wang T, Lu Q, Kangath A, Qu N, Klinger C, Fratz S, Yuan JX, Jacobson JR, Garcia JG, Rafikov R, Fineman JR, Black SM. Asymmetric dimethylarginine stimulates Akt1 phosphorylation via heat shock protein 70-facilitated carboxyl-terminal modulator protein degradation in pulmonary arterial endothelial cells. Am J Respir Cell Mol Biol 55: 275–287, 2016. doi: 10.1165/rcmb.2015-0185OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Tang H, Gu Y, Aryon R, Song S, Fernandez RA, Garcia JG, Makino A, Yuan JX. Endothelial-specific deletion of prolyl hydroxylase domain protein 2 (PHD2) results in severe pulmonary hypertension in mice by enhancing the HIF2α signaling pathway (Abstract). Circulation 132: A19126, 2015. doi: 10.1161/circ.132.suppl_3.19126. [DOI] [Google Scholar]
- 364. Adyshev DM, Elangovan VR, Moldobaeva N, Mapes B, Sun X, Garcia JG. Mechanical stress induces pre-B-cell colony-enhancing factor/NAMPT expression via epigenetic regulation by miR-374a and miR-568 in human lung endothelium. Am J Respir Cell Mol Biol 50: 409–418, 2014. doi: 10.1165/rcmb.2013-0292OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Gressner OA. Intracrine signaling mechanisms of activin A and TGF-β. Vitam Horm 85: 59–77, 2011. doi: 10.1016/B978-0-12-385961-7.00004-4. [DOI] [PubMed] [Google Scholar]
- 366. Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KK, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J Clin Invest 121: 4548–4566, 2011. doi: 10.1172/JCI57734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Cullivan S, Murphy CA, Weiss L, Comer SP, Kevane B, McCullagh B, Maguire PB, Ni Ainle F, Gaine SP. Platelets, extracellular vesicles and coagulation in pulmonary arterial hypertension. Pulm Circ 11: 20458940211021036, 2021. doi: 10.1177/20458940211021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Gao Y, Raj JU. Extracellular vesicles as unique signaling messengers: role in lung diseases. Compr Physiol 11: 1351–1369, 2020. doi: 10.1002/cphy.c200006. [DOI] [PubMed] [Google Scholar]
- 369. Calebiro D, Koszegi Z, Lanoiselée Y, Miljus T, O’Brien S. G protein-coupled receptor-G protein interactions: a single-molecule perspective. Physiol Rev 101: 857–906, 2021. doi: 10.1152/physrev.00021.2020. [DOI] [PubMed] [Google Scholar]
- 370. Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 87: 165–217, 2007. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 371. Hendy GN, Canaff L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin Cell Dev Biol 49: 37–43, 2016. doi: 10.1016/j.semcdb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 372. Schepelmann M, Yarova PL, Lopez-Fernandez I, Davies TS, Brennan SC, Edwards PJ, Aggarwal A, Graça J, Rietdorf K, Matchkov V, Fenton RA, Chang W, Krssak M, Stewart A, Broadley KJ, Ward DT, Price SA, Edwards DH, Kemp PJ, Riccardi D. The vascular Ca2+-sensing receptor regulates blood vessel tone and blood pressure. Am J Physiol Cell Physiol 310: C193–C204, 2016. doi: 10.1152/ajpcell.00248.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4: 530–538, 2003. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 374. Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279: 18887–18894, 2004. doi: 10.1074/jbc.M311274200. [DOI] [PubMed] [Google Scholar]
- 375. Azumaya CM, Sierra-Valdez F, Cordero-Morales JF, Nakagawa T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J Biol Chem 293: 10381–10391, 2018. doi: 10.1074/jbc.RA118.003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Bai Y, Yu X, Chen H, Horne D, White R, Wu X, Lee P, Gu Y, Ghimire-Rijal S, Lin DC, Huang X. Structural basis for pharmacological modulation of the TRPC6 channel. Elife 9: e53311, 2020. doi: 10.7554/eLife.53311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 379. Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 380. Ernest JP, Sarathy J, Wang N, Kaya F, Zimmerman MD, Strydom N, Wang H, Xie M, Gengenbacher M, Via LE, Barry CE 3rd, Carter CL, Savic RM, Dartois V. Lesion penetration and activity limit the utility of second-line injectable agents in pulmonary tuberculosis. Antimicrob Agents Chemother 65: e0050621, 2021. doi: 10.1128/AAC.00506-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, Song L, Hanlon D, Tan Hehir CA, Baker D, Blennow K, Hansson O. Plasma β-amyloid in Alzheimer's disease and vascular disease. Sci Rep 6: 26801, 2016. doi: 10.1038/srep26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. He YY, Yan Y, Jiang X, Zhao JH, Wang Z, Wu T, Wang Y, Guo SS, Ye J, Lian TY, Xu XQ, Zhang JL, Sun K, Peng FH, Zhou YP, Mao YM, Zhang X, Chen JW, Zhang SY, Jing ZC. Spermine promotes pulmonary vascular remodelling and its synthase is a therapeutic target for pulmonary arterial hypertension. Eur Respir J 56: 2000522, 2020. doi: 10.1183/13993003.00522-2020. [DOI] [PubMed] [Google Scholar]
- 383. Orlinska U, Olson JW, Gillespie MN. Polyamine content in pulmonary arteries from rats with monocrotaline-induced pulmonary hypertension. Res Commun Chem Pathol Pharmacol 62: 187–194, 1988. [PubMed] [Google Scholar]
- 384. Gillespie MN, Olson JW. Polyamine regulatory pathways as pharmacologic targets in pulmonary arterial hypertension. Adv Exp Med Biol 661: 375–389, 2010. doi: 10.1007/978-1-60761-500-2_24. [DOI] [PubMed] [Google Scholar]
- 385. Stein WH, Moore S. The free amino acids of human blood plasma. J Biol Chem 211: 915–926, 1954. doi: 10.1016/S0021-9258(18)71179-4. [DOI] [PubMed] [Google Scholar]
- 386. Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher-Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A, Weissmann N. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 188: 1451–1459, 2013. doi: 10.1164/rccm.201307-1252OC. [DOI] [PubMed] [Google Scholar]
- 387. Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, Sham JS. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 63: 173–180, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Weber J, Rajan S, Schremmer C, Chao YK, Krasteva-Christ G, Kannler M, Yildirim AÖ, Brosien M, Schredelseker J, Weissmann N, Grimm C, Gudermann T, Dietrich A. TRPV4 channels are essential for alveolar epithelial barrier function as protection from lung edema. JCI Insight 5: e134464, 2020. doi: 10.1172/jci.insight.134464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun 3: 649, 2012. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Adebiyi A, Narayanan D, Jaggar JH. Caveolin-1 assembles type 1 inositol 1,4,5-trisphosphate receptors and canonical transient receptor potential 3 channels into a functional signaling complex in arterial smooth muscle cells. J Biol Chem 286: 4341–4348, 2011. doi: 10.1074/jbc.M110.179747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J Biol Chem 278: 27208–27215, 2003. doi: 10.1074/jbc.M301118200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14: 98–112, 2013. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 393. Pani B, Ong HL, Brazer SC, Liu X, Rauser K, Singh BB, Ambudkar IS. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Natl Acad Sci USA 106: 20087–20092, 2009. doi: 10.1073/pnas.0905002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Del Pozo MA, Lolo FN, Echarri A. Caveolae: Mechanosensing and mechanotransduction devices linking membrane trafficking to mechanoadaptation. Curr Opin Cell Biol 68: 113–123, 2021. doi: 10.1016/j.ceb.2020.10.008. [DOI] [PubMed] [Google Scholar]
- 395. Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, West J, Loyd JE, Chung WK. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 5: 336–343, 2012. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Rathinasabapathy A, Copeland C, Crabtree A, Carrier EJ, Moore C, Shay S, Gladson S, Austin ED, Kenworthy AK, Loyd JE, Hemnes AR, West JD. Expression of a human caveolin-1 mutation in mice drives inflammatory and metabolic defect-associated pulmonary arterial hypertension. Front Med (Lausanne) 7: 540, 2020. doi: 10.3389/fmed.2020.00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 119: 2009–2018, 2009. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J 21: 2970–2979, 2007. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 399. Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341–1379, 2004. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 400. Parton RG, Tillu VA, Collins BM. Caveolae. Curr Biol 28: R402–R405, 2018. doi: 10.1016/j.cub.2017.11.075. [DOI] [PubMed] [Google Scholar]
- 401. Li X, Gu X, Boyce TM, Zheng M, Reagan AM, Qi H, Mandal N, Cohen AW, Callegan MC, Carr DJ, Elliott MH. Caveolin-1 increases proinflammatory chemoattractants and blood-retinal barrier breakdown but decreases leukocyte recruitment in inflammation. Invest Ophthalmol Vis Sci 55: 6224–6234, 2014. doi: 10.1167/iovs.14-14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Schach C, Firth AL, Xu M, Remillard CV, Patel HH, Insel PA, Yuan JX. Regulation of pulmonary vasoconstriction by agonists and caveolae. Exp Lung Res 34: 195–208, 2008. doi: 10.1080/01902140801925471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Schach C, Xu M, Platoshyn O, Keller SH, Yuan JX. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store-operated Ca2+ channels. Am J Physiol Lung Cell Mol Physiol 292: L685–L698, 2007. doi: 10.1152/ajplung.00276.2006. [DOI] [PubMed] [Google Scholar]
- 404. Zhou Y, Ariotti N, Rae J, Liang H, Tillu V, Tee S, Bastiani M, Bademosi AT, Collins BM, Meunier FA, Hancock JF, Parton RG. Caveolin-1 and cavin1 act synergistically to generate a unique lipid environment in caveolae. J Cell Biol 220: e202005138, 2021. doi: 10.1083/jcb.202005138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Lo HP, Hall TE, Parton RG. Mechanoprotection by skeletal muscle caveolae. Bioarchitecture 6: 22–27, 2016. doi: 10.1080/19490992.2015.1131891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Cheng JP, Mendoza-Topaz C, Howard G, Chadwick J, Shvets E, Cowburn AS, Dunmore BJ, Crosby A, Morrell NW, Nichols BJ. Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J Cell Biol 211: 53–61, 2015. doi: 10.1083/jcb.201504042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A 99: 11375–11380, 2002. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Goligorsky MS, Li H, Brodsky S, Chen J. Relationships between caveolae and eNOS: everything in proximity and the proximity of everything. Am J Physiol Renal Physiol 283: F1–F10, 2002. doi: 10.1152/ajprenal.00377.2001. [DOI] [PubMed] [Google Scholar]
- 409. Chen Z, Oliveira SD, Zimnicka AM, Jiang Y, Sharma T, Chen S, Lazarov O, Bonini MG, Haus JM, Minshall RD. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol Cell Biol 29: 1190–1202, 2018. doi: 10.1091/mbc.E17-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3: a004549, 2011. doi: 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am J Physiol Cell Physiol 314: C3–C26, 2018. doi: 10.1152/ajpcell.00196.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol 7: 485–581, 2017. doi: 10.1002/cphy.c160011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Harraz OF, Hill-Eubanks D, Nelson MT. PIP2: a critical regulator of vascular ion channels hiding in plain sight. Proc Natl Acad Sci USA 117: 20378–20389, 2020. doi: 10.1073/pnas.2006737117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 415. Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 416. Zhang J, Wang Y, Chen L, Wier WG, Blaustein MP. Na+/Ca2+ exchanger overexpression in smooth muscle augments cytosolic Ca2+ in femoral arteries of living mice. Am J Physiol Heart Circ Physiol 316: H298–H310, 2019. doi: 10.1152/ajpheart.00185.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C2297–C2305, 2007. doi: 10.1152/ajpcell.00383.2006. [DOI] [PubMed] [Google Scholar]
- 418. Zhang S, Yuan JX, Barrett KE, Dong H. Role of Na+/Ca2+ exchange in regulating cytosolic Ca2+ in cultured human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 288: C245–C252, 2005. doi: 10.1152/ajpcell.00411.2004. [DOI] [PubMed] [Google Scholar]
- 419. Bova S, Goldman WF, Yauan XJ, Blaustein MP. Influence of Na+ gradient on Ca2+ transients and contraction in vascular smooth muscle. Am J Physiol Heart Circ Physiol 259: H409–H423, 1990. doi: 10.1152/ajpheart.1990.259.2.H409. [DOI] [PubMed] [Google Scholar]
- 420. Ringvold HC, Khalil RA. Protein kinase C as regulator of vascular smooth muscle function and potential target in vascular disorders. Adv Pharmacol 78: 203–301, 2017. doi: 10.1016/bs.apha.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Prole DL, Taylor CW. Structure and function of IP3 receptors. Cold Spring Harb Perspect Biol 11: a035063, 2019. doi: 10.1101/cshperspect.a035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Woll KA, Van Petegem F. Calcium-release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol Rev 102: 209–268, 2022. doi: 10.1152/physrev.00033.2020. [DOI] [PubMed] [Google Scholar]
- 423. McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol 280: L870–L880, 2001. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- 424. Xu M, Platoshyn O, Makino A, Dillmann WH, Akassoglou K, Remillard CV, Yuan JX. Characterization of agonist-induced vasoconstriction in mouse pulmonary artery. Am J Physiol Heart Circ Physiol 294: H220–H228, 2008. doi: 10.1152/ajpheart.00968.2007. [DOI] [PubMed] [Google Scholar]
- 425. Xu M, Remillard CV, Sachs BD, Makino A, Platoshyn O, Yao W, Dillmann WH, Akassoglou K, Yuan JX. p75 neurotrophin receptor regulates agonist-induced pulmonary vasoconstriction. Am J Physiol Heart Circ Physiol 295: H1529–H1538, 2008. doi: 10.1152/ajpheart.00115.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280: H746–H755, 2001. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 427. Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283: L144–L155, 2002. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- 428. Golovina VA, Blaustein MP. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca2+ stores in astrocytes. Glia 31: 15–28, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 429. Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem 71: 511–535, 2002. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 430. Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc Natl Acad Sci U S A 93: 10489–10494, 1996. doi: 10.1073/pnas.93.19.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Coppock EA, Martens JR, Tamkun MM. Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am J Physiol Lung Cell Mol Physiol 281: L1–L12, 2001. doi: 10.1152/ajplung.2001.281.1.L1. [DOI] [PubMed] [Google Scholar]
- 432. Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca2+-activated Cl- channels contributes to regulating receptor- and store-operated Ca2+ entry in human pulmonary artery smooth muscle cells. Pulm Circ 1: 269–279, 2011. doi: 10.4103/2045-8932.83447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Yuan XJ. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol Lung Cell Mol Physiol 272: L959–L968, 1997. doi: 10.1152/ajplung.1997.272.5.L959. [DOI] [PubMed] [Google Scholar]
- 434. Ayon RJ, Hawn MB, Aoun J, Wiwchar M, Forrest AS, Cunningham F, Singer CA, Valencik ML, Greenwood IA, Leblanc N. Molecular mechanism of TMEM16A regulation: role of CaMKII and PP1/PP2A. Am J Physiol Cell Physiol 317: C1093–C1106, 2019. doi: 10.1152/ajpcell.00059.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Leblanc N, Forrest AS, Ayon RJ, Wiwchar M, Angermann JE, Pritchard HA, Singer CA, Valencik ML, Britton F, Greenwood IA. Molecular and functional significance of Ca2+-activated Cl- channels in pulmonary arterial smooth muscle. Pulm Circ 5: 244–268, 2015. doi: 10.1086/680189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Wiwchar M, Ayon R, Greenwood IA, Leblanc N. Phosphorylation alters the pharmacology of Ca2+-activated Cl channels in rabbit pulmonary arterial smooth muscle cells. Br J Pharmacol 158: 1356–1365, 2009. doi: 10.1111/j.1476-5381.2009.00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Liang W, Ray JB, He JZ, Backx PH, Ward ME. Regulation of proliferation and membrane potential by chloride currents in rat pulmonary artery smooth muscle cells. Hypertension 54: 286–293, 2009. doi: 10.1161/HYPERTENSIONAHA.109.130138. [DOI] [PubMed] [Google Scholar]
- 438. Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA, Leblanc N. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303: C1229–C1243, 2012. doi: 10.1152/ajpcell.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Sun H, Paudel O, Sham JS. Increased intracellular Cl- concentration in pulmonary arterial myocytes is associated with chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol 321: C297–C307, 2021. doi: 10.1152/ajpcell.00172.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Papp R, Nagaraj C, Zabini D, Nagy BM, Lengyel M, Skofic Maurer D, Sharma N, Egemnazarov B, Kovacs G, Kwapiszewska G, Marsh LM, Hrzenjak A, Höfler G, Didiasova M, Wygrecka M, Sievers LK, Szucs P, Enyedi P, Ghanim B, Klepetko W, Olschewski H, Olschewski A. Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur Respir J 53: 1800965, 2019. doi: 10.1183/13993003.00965-2018. [DOI] [PubMed] [Google Scholar]
- 441. Skofic Maurer D, Zabini D, Nagaraj C, Sharma N, Lengyel M, Nagy BM, Frank S, Klepetko W, Gschwandtner E, Enyedi P, Kwapiszewska G, Olschewski H, Olschewski A. Endothelial dysfunction following enhanced TMEM16A activity in human pulmonary arteries. Cells 9: 1984, 2020. doi: 10.3390/cells9091984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Abdul-Salam VB, Russomanno G, Chien-Nien C, Mahomed AS, Yates LA, Wilkins MR, Zhao L, Gierula M, Dubois O, Schaeper U, Endruschat J, Wojciak-Stothard B. CLIC4/Arf6 pathway. Circ Res 124: 52–65, 2019. doi: 10.1161/CIRCRESAHA.118.313705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Lambert M, Capuano V, Olschewski A, Sabourin J, Nagaraj C, Girerd B, Weatherald J, Humbert M, Antigny F. Ion channels in pulmonary hypertension: a therapeutic interest? Int J Mol Sci 19: 3162, 2018. doi: 10.3390/ijms19103162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev 72: S69–S88, 1992. doi: 10.1152/physrev.1992.72.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- 445. Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, Alvarez O. Molecular determinants of BK channel functional diversity and functioning. Physiol Rev 97: 39–87, 2017. doi: 10.1152/physrev.00001.2016. [DOI] [PubMed] [Google Scholar]
- 446. Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 90: 1437–1459, 2010. doi: 10.1152/physrev.00049.2009. [DOI] [PubMed] [Google Scholar]
- 447. Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 77: 1165–1232, 1997. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 448. McClenaghan C, Woo KV, Nichols CG. Pulmonary hypertension and ATP-sensitive potassium channels. Hypertension 74: 14–22, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605, 2010. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 450. Antz C, Geyer M, Fakler B, Schott MK, Guy HR, Frank R, Ruppersberg JP, Kalbitzer HR. NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature 385: 272–275, 1997. doi: 10.1038/385272a0. [DOI] [PubMed] [Google Scholar]
- 451. Bähring R, Milligan CJ, Vardanyan V, Engeland B, Young BA, Dannenberg J, Waldschutz R, Edwards JP, Wray D, Pongs O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvβ subunits. J Biol Chem 276: 22923–22929, 2001. doi: 10.1074/jbc.M100483200. [DOI] [PubMed] [Google Scholar]
- 452. Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature 369: 289–294, 1994. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 453. Pérez-García MT, López-López JR, González C. Kvβ1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to Kv4.2 but not to Shaker channels. J Gen Physiol 113: 897–907, 1999. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Osipenko ON, Tate RJ, Gurney AM. Potential role for Kv3.1β channels as oxygen sensors. Circ Res 86: 534–540, 2000. doi: 10.1161/01.RES.86.5.534. [DOI] [PubMed] [Google Scholar]
- 455. Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol Heart Circ Physiol 266: H365–H370, 1994. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- 456. Smirnov SV, Aaronson PI. Alteration of the transmembrane K+ gradient during development of delayed rectifier in isolated rat pulmonary arterial cells. J Gen Physiol 104: 241–264, 1994. doi: 10.1085/jgp.104.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Moudgil R, Michelakis ED, Archer SL. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13: 615–632, 2006. doi: 10.1080/10739680600930222. [DOI] [PubMed] [Google Scholar]
- 458. Cribbs LL. T-type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium 40: 221–230, 2006. doi: 10.1016/j.ceca.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 459. Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle-dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res 79: 14–19, 1996. doi: 10.1161/01.RES.79.1.14. [DOI] [PubMed] [Google Scholar]
- 460. Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279: C1540–C1549, 2000. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- 461. Sevilla-Montero J, Labrousse-Arias D, Fernández-Pérez C, Fernández-Blanco L, Barreira B, Mondéjar-Parreño G, Alfaro-Arnedo E, López IP, Pérez-Rial S, Peces-Barba G, Pichel JG, Peinado VI, Cogolludo Á, Calzada MJ. Cigarette smoke directly promotes pulmonary arterial remodeling and Kv7.4 channel dysfunction. Am J Respir Crit Care Med 203: 1290–1305, 2021. doi: 10.1164/rccm.201911-2238OC. [DOI] [PubMed] [Google Scholar]
- 462. Burg ED, Remillard CV, Yuan JX. K+ channels in apoptosis. J Membr Biol 209: 3–20, 2006. doi: 10.1007/s00232-005-0838-4. [DOI] [PubMed] [Google Scholar]
- 463. Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol 153, Suppl: S99–S111, 2008. doi: 10.1038/sj.bjp.0707635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Bortner CD, Cidlowski JA. The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch 448: 313–318, 2004. doi: 10.1007/s00424-004-1266-5. [DOI] [PubMed] [Google Scholar]
- 465. Hughes FM Jr, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem 272: 30567–30576, 1997. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 466. Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Augmented K+ currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol 281: L887–L894, 2001. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- 467. Krick S, Platoshyn O, Sweeney M, Kim H, Yuan JX. Activation of K+ channels induces apoptosis in vascular smooth muscle cells. Am J Physiol Cell Physiol 280: C970–C979, 2001. doi: 10.1152/ajpcell.2001.280.4.C970. [DOI] [PubMed] [Google Scholar]
- 468. Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Cytochrome c activates K+ channels before inducing apoptosis. Am J Physiol Cell Physiol 283: C1298–C1305, 2002. doi: 10.1152/ajpcell.00592.2001. [DOI] [PubMed] [Google Scholar]
- 469. Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol 286: L49–L67, 2004. doi: 10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- 470. Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321–329, 1998. doi: 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 471. Ferraz AP, Seara FA, Baptista EF, Barenco TS, Sottani TB, Souza NS, Domingos AE, Barbosa RA, Takiya CM, Couto MT, Resende GO, Campos de Carvalho AC, Ponte CG, Nascimento JH. BKCa channel activation attenuates the pathophysiological progression of monocrotaline-induced pulmonary arterial hypertension in Wistar rats. Cardiovasc Drugs Ther 35: 719–732, 2021. doi: 10.1007/s10557-020-07115-5. [DOI] [PubMed] [Google Scholar]
- 472. Detweiler ND, Song L, McClenahan SJ, Versluis RJ, Kharade SV, Kurten RC, Rhee SW, Rusch NJ. BK channels in rat and human pulmonary smooth muscle cells are BKα-β1 functional complexes lacking the oxygen-sensitive stress axis regulated exon insert. Pulm Circ 6: 563–575, 2016. doi: 10.1086/688838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Nagaraj C, Tang B, Nagy BM, Papp R, Jain PP, Marsh LM, Meredith AL, Ghanim B, Klepetko W, Kwapiszewska G, Weir EK, Olschewski H, Olschewski A. Docosahexaenoic acid causes rapid pulmonary arterial relaxation via KCa channel-mediated hyperpolarisation in pulmonary hypertension. Eur Respir J 48: 1127–1136, 2016. doi: 10.1183/13993003.01814-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Revermann M, Neofitidou S, Kirschning T, Schloss M, Brandes RP, Hofstetter C. Inhalation of the BKCa-opener NS1619 attenuates right ventricular pressure and improves oxygenation in the rat monocrotaline model of pulmonary hypertension. PLoS One 9: e86636, 2014. doi: 10.1371/journal.pone.0086636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Dumas de la Roque E, Quignard JF, Ducret T, Dahan D, Courtois A, Begueret H, Marthan R, Savineau JP. Beneficial effect of dehydroepiandrosterone on pulmonary hypertension in a rodent model of pulmonary hypertension in infants. Pediatr Res 74: 163–169, 2013. doi: 10.1038/pr.2013.73. [DOI] [PubMed] [Google Scholar]
- 476. Bonnet S, Savineau JP, Barillot W, Dubuis E, Vandier C, Bonnet P. Role of Ca2+-sensitive K+ channels in the remission phase of pulmonary hypertension in chronic obstructive pulmonary diseases. Cardiovasc Res 60: 326–336, 2003. doi: 10.1016/S0008-6363(03)00527-3. [DOI] [PubMed] [Google Scholar]
- 477. Roth M, Rupp M, Hofmann S, Mittal M, Fuchs B, Sommer N, Parajuli N, Quanz K, Schubert D, Dony E, Schermuly RT, Ghofrani HA, Sausbier U, Rutschmann K, Wilhelm S, Seeger W, Ruth P, Grimminger F, Sausbier M, Weissmann N. Heme oxygenase-2 and large-conductance Ca2+-activated K+ channels: lung vascular effects of hypoxia. Am J Respir Crit Care Med 180: 353–364, 2009. doi: 10.1164/rccm.200806-848OC. [DOI] [PubMed] [Google Scholar]
- 478. Peinado VI, París R, Ramírez J, Roca J, Rodriguez-Roisin R, Barberà JA. Expression of BKCa channels in human pulmonary arteries: relationship with remodeling and hypoxic pulmonary vasoconstriction. Vascul Pharmacol 49: 178–184, 2008. doi: 10.1016/j.vph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 479. Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, et al. Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol 51: e1–e62, 2008. doi: 10.1016/j.jacc.2008.02.032. [DOI] [PubMed] [Google Scholar]
- 480. Olschewski A, Veale EL, Nagy BM, Nagaraj C, Kwapiszewska G, Antigny F, Lambert M, Humbert M, Czirják G, Enyedi P, Mathie A. TASK-1 (KCNK3) channels in the lung: from cell biology to clinical implications. Eur Respir J 50: 1700754, 2017. doi: 10.1183/13993003.00754-2017. [DOI] [PubMed] [Google Scholar]
- 481. Patel AJ, Lazdunski M. The 2P-domain K+ channels: role in apoptosis and tumorigenesis. Pflugers Arch 448: 261–273, 2004. doi: 10.1007/s00424-004-1255-8. [DOI] [PubMed] [Google Scholar]
- 482. Gurney AM, Osipenko ON, MacMillan D, Kempsill FE. Potassium channels underlying the resting potential of pulmonary artery smooth muscle cells. Clin Exp Pharmacol Physiol 29: 330–333, 2002. doi: 10.1046/j.1440-1681.2002.03653.x. [DOI] [PubMed] [Google Scholar]
- 483. Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Trégouët DA, Borczuk A, Rosenzweig EB, Girerd B, Montani D, Humbert M, Loyd JE, Kass RS, Chung WK. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 369: 351–361, 2013. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Lambert M, Capuano V, Boet A, Tesson L, Bertero T, Nakhleh MK, Remy S, Anegon I, Pechoux C, Hautefort A, Rucker-Martin C, Manoury B, Domergue V, Mercier O, Girerd B, Montani D, Perros F, Humbert M, Antigny F. Characterization of Kcnk3-mutated rat, a novel model of pulmonary hypertension. Circ Res 125: 678–695, 2019. doi: 10.1161/CIRCRESAHA.119.314793. [DOI] [PubMed] [Google Scholar]
- 485. Lambert M, Mendes-Ferreira P, Ghigna MR, LeRibeuz H, Adão R, Boet A, Capuano V, Rucker-Martin C, Brás-Silva C, Quarck R, Domergue V, Vachiéry JL, Humbert M, Perros F, Montani D, Antigny F. KCNK3 dysfunction exaggerates the development of pulmonary hypertension induced by left ventricular pressure overload. Cardiovasc Res 117: 2474–2488, 2021. doi: 10.1093/cvr/cvab016. [DOI] [PubMed] [Google Scholar]
- 486. Cunningham KP, Holden RG, Escribano-Subias PM, Cogolludo A, Veale EL, Mathie A. Characterization and regulation of wild-type and mutant TASK-1 two pore domain potassium channels indicated in pulmonary arterial hypertension. J Physiol 597: 1087–1101, 2019. doi: 10.1113/JP277275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Inoue M, Matsuoka H, Harada K, Mugishima G, Kameyama M. TASK channels: channelopathies, trafficking, and receptor-mediated inhibition. Pflugers Arch 472: 911–922, 2020. doi: 10.1007/s00424-020-02403-3. [DOI] [PubMed] [Google Scholar]
- 488. Babicheva A, Zhao T, Yuan JX. KCNK3 channel: a new player in the field of pulmonary vascular disease. Circ Res 125: 696–698, 2019. doi: 10.1161/CIRCRESAHA.119.315758. [DOI] [PubMed] [Google Scholar]
- 489. Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP 4th, Boyd AE 3rd, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268: 423–426, 1995. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 490. Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110: 655–664, 1997. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Craig TJ, Ashcroft FM, Proks P. How ATP inhibits the open KATP channel. J Gen Physiol 132: 131–144, 2008. doi: 10.1085/jgp.200709874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Beech DJ, Zhang H, Nakao K, Bolton TB. K channel activation by nucleotide diphosphates and its inhibition by glibenclamide in vascular smooth muscle cells. Br J Pharmacol 110: 573–582, 1993. doi: 10.1111/j.1476-5381.1993.tb13849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of KATP channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am J Respir Cell Mol Biol 26: 135–143, 2002. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- 494. Robertson BE, Kozlowski RZ, Nye PC. Opposing actions of tolbutamide and glibenclamide on hypoxic pulmonary vasoconstriction. Comp Biochem Physiol C Comp Pharmacol Toxicol 102: 459–462, 1992. doi: 10.1016/0742-8413(92)90143-U. [DOI] [PubMed] [Google Scholar]
- 495. Bohnen MS, Ma L, Zhu N, Qi H, McClenaghan C, Gonzaga-Jauregui C, et al. Loss-of-function ABCC8 mutations in pulmonary arterial hypertension. Circ Genom Precis Med 11: e002087, 2018. doi: 10.1161/CIRCGEN.118.002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Lago-Docampo M, Tenorio J, Hernández-González I, Pérez-Olivares C, Escribano-Subías P, Pousada G, Baloira A, Arenas M, Lapunzina P, Valverde D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci Rep 10: 15135, 2020. doi: 10.1038/s41598-020-72089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Huang Y, Hu D, Huang C, Nichols CG. Genetic discovery of ATP-sensitive K+ channels in cardiovascular diseases. Circ Arrhythm Electrophysiol 12: e007322, 2019. doi: 10.1161/CIRCEP.119.007322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Rieg AD, Rossaint R, Verjans E, Maihöfer NA, Uhlig S, Martin C. Levosimendan relaxes pulmonary arteries and veins in precision-cut lung slices—the role of KATP-channels, cAMP and cGMP. PLoS One 8: e66195, 2013. doi: 10.1371/journal.pone.0066195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. He M, Cui T, Cai Q, Wang H, Kong H, Xie W. Iptakalim ameliorates hypoxia-impaired human endothelial colony-forming cells proliferation, migration, and angiogenesis via Akt/eNOS pathways. Pulm Circ 9: 2045894019875417, 2019. doi: 10.1177/2045894019875417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Le Ribeuz H, Masson B, Capuano V, Dutheil M, Gooroochurn H, Boët A, Ghigna MR, De Montpreville V, Girerd B, Lambert M, Mercier O, Chung WK, Humbert M, Montani D, Antigny F. SUR1 as a new therapeutic target for pulmonary arterial hypertension. Am J Respir Cell Mol Biol 66: 539–554, 2022. doi: 10.1165/rcmb.2021-0180OC. [DOI] [PubMed] [Google Scholar]
- 501. Breen E, Yuan JX. Targeting ATP-sensitive K+ channels to treat pulmonary hypertension. Am J Respir Cell Mol Biol 66: 476–478, 2022. doi: 10.1165/rcmb.2021-0549ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Burg ED, Platoshyn O, Tsigelny IF, Lozano-Ruiz B, Rana BK, Yuan JX. Tetramerization domain mutations in KCNA5 affect channel kinetics and cause abnormal trafficking patterns. Am J Physiol Cell Physiol 298: C496–C509, 2010. doi: 10.1152/ajpcell.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O’Connor DT, Yuan JX. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C1837–C1853, 2007. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 504. Wipff J, Dieudé P, Guedj M, Ruiz B, Riemekasten G, Cracowski JL, Matucci-Cerinic M, Melchers I, Humbert M, Hachulla E, Airo P, Diot E, Hunzelmann N, Caramaschi P, Sibilia J, Valentini G, Tiev K, Girerd B, Mouthon L, Riccieri V, Carpentier PH, Distler J, Amoura Z, Tarner I, Degano B, Avouac J, Meyer O, Kahan A, Boileau C, Allanore Y. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum 62: 3093–3100, 2010. doi: 10.1002/art.27607. [DOI] [PubMed] [Google Scholar]
- 505. Pousada G, Baloira A, Vilariño C, Cifrian JM, Valverde D. Novel mutations in BMPR2, ACVRL1 and KCNA5 genes and hemodynamic parameters in patients with pulmonary arterial hypertension. PLoS One 9: e100261, 2014. doi: 10.1371/journal.pone.0100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Barnes EA, Lee L, Barnes SL, Brenner R, Alvira CM, Cornfield DN. β1-subunit of the calcium-sensitive potassium channel modulates the pulmonary vascular smooth muscle cell response to hypoxia. Am J Physiol Lung Cell Mol Physiol 315: L265–L275, 2018. doi: 10.1152/ajplung.00060.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM, Cornfield DN. Hypoxia-inducible factor-1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 302: L352–L359, 2012. doi: 10.1152/ajplung.00302.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 532: 3–16, 2001. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509. Vu CC, Bortner CD, Cidlowski JA. Differential involvement of initiator caspases in apoptotic volume decrease and potassium efflux during Fas- and UV-induced cell death. J Biol Chem 276: 37602–37611, 2001. doi: 10.1074/jbc.M104810200. [DOI] [PubMed] [Google Scholar]
- 510. Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci U S A 97: 9487–9492, 2000. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol 39: 393–406, 2018. doi: 10.1016/j.it.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 513. He Y, Zeng MY, Yang D, Motro B, Nuñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357, 2016. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Xu Z, Chen ZM, Wu X, Zhang L, Cao Y, Zhou P. Distinct molecular mechanisms underlying potassium efflux for NLRP3 inflammasome activation. Front Immunol 11: 609441, 2020. doi: 10.3389/fimmu.2020.609441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Foster VS, Rash LD, King GF, Rank MM. Acid-sensing ion channels: Expression and function in resident and infiltrating immune cells in the central nervous system. Front Cell Neurosci 15: 738043, 2021. doi: 10.3389/fncel.2021.738043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Aaronson PI. Pulmonary hypertension associated with chronic hypoxia: just ASIC-ness? J Physiol 599: 4731–4732, 2021. doi: 10.1113/JP282325. [DOI] [PubMed] [Google Scholar]
- 517. Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. doi: 10.1152/ajpheart.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Jernigan NL, Naik JS, Resta TC. Acid-sensing ion channel 1 contributes to pulmonary arterial smooth muscle cell depolarization following hypoxic pulmonary hypertension. J Physiol 599: 4749–4762, 2021. doi: 10.1113/JP282231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Jernigan NL. Smooth muscle acid-sensing ion channel 1: pathophysiological implication in hypoxic pulmonary hypertension. Exp Physiol 100: 111–120, 2015. doi: 10.1113/expphysiol.2014.081612. [DOI] [PubMed] [Google Scholar]
- 520. Huetsch J, Shimoda LA. Na+/H+ exchange and hypoxic pulmonary hypertension. Pulm Circ 5: 228–243, 2015. doi: 10.1086/680213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Rios EJ, Fallon M, Wang J, Shimoda LA. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 289: L867–L874, 2005. doi: 10.1152/ajplung.00455.2004. [DOI] [PubMed] [Google Scholar]
- 522. Huetsch JC, Jiang H, Larrain C, Shimoda LA. The Na+/H+ exchanger contributes to increased smooth muscle proliferation and migration in a rat model of pulmonary arterial hypertension. Physiol Rep 4: e12729, 2016. doi: 10.14814/phy2.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Walker J, Undem C, Yun X, Lade J, Jiang H, Shimoda LA. Role of Rho kinase and Na+/H+ exchange in hypoxia-induced pulmonary arterial smooth muscle cell proliferation and migration. Physiol Rep 4: e12702, 2016. doi: 10.14814/phy2.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 101: 1030–1038, 2007. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 525. Swietlik EM, Gräf S, Morrell NW. The role of genomics and genetics in pulmonary arterial hypertension. Glob Cardiol Sci Pract 2020: e202013, 2020. doi: 10.21542/gcsp.2020.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 53: 1801899, 2019. doi: 10.1183/13993003.01899-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Welch CL, Chung WK. Channelopathy genes in pulmonary arterial hypertension. Biomolecules 12: 265, 2022. doi: 10.3390/biom12020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND, Siemieniak DR, Wheeler L, Phillips JA 3rd, Newman JH, Conneally PM, Ginsburg D, Loyd JE. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet 15: 277–280, 1997. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- 529. Machado RD, Pauciulo MW, Fretwell N, Veal C, Thomson JR, Vilarino Güell C, Aldred M, Brannon CA, Trembath RC, Nichols WC. A physical and transcript map based upon refinement of the critical interval for PPH1, a gene for familial primary pulmonary hypertension. The International PPH Consortium. Genomics 68: 220–228, 2000. doi: 10.1006/geno.2000.6291. [DOI] [PubMed] [Google Scholar]
- 530. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–744, 2000. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Eyries M, Montani D, Nadaud S, Girerd B, Levy M, Bourdin A, Trésorier R, Chaouat A, Cottin V, Sanfiorenzo C, Prevot G, Reynaud-Gaubert M, Dromer C, Houeijeh A, Nguyen K, Coulet F, Bonnet D, Humbert M, Soubrier F. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J 53: 1801371, 2019. doi: 10.1183/13993003.01371-2018. [DOI] [PubMed] [Google Scholar]
- 532. Zhu N, Swietlik EM, Welch CL, Pauciulo MW, Hagen JJ, Zhou X, Guo Y, Karten J, Pandya D, Tilly T, Lutz KA, Martin JM, Treacy CM, Rosenzweig EB, Krishnan U, Coleman AW, Gonzaga-Jauregui C, Lawrie A, Trembath RC, Wilkins MR; Regeneron Genetics Center, PAH Biobank Enrolling Centers’ Investigators, NIHR BioResource for Translational Research-Rare Diseases, National Cohort Study of Idiopathic and Heritable PAH, Morrell NW, Shen Y, Graf S, Nichols WC, Chung WK. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med 13: 80, 2021. doi: 10.1186/s13073-021-00915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.International PPH Consortium, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81–84, 2000. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 534. Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA 3rd, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 345: 319–324, 2001. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- 535. Wang H, Li W, Zhang W, Sun K, Song X, Gao S, Zhang C, Hui R, Hu H. Novel promoter and exon mutations of the BMPR2 gene in Chinese patients with pulmonary arterial hypertension. Eur J Hum Genet 17: 1063–1069, 2009. doi: 10.1038/ejhg.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA 3rd, Newman J, Williams D, Galiè N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 68: 92–102, 2001. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537. Harrison RE, Flanagan JA, Sankelo M, Abdalla SA, Rowell J, Machado RD, Elliott CG, Robbins IM, Olschewski H, McLaughlin V, Gruenig E, Kermeen F, Halme M, Räisänen-Sokolowski A, Laitinen T, Morrell NW, Trembath RC. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 40: 865–871, 2003. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, Morrell NW, Aldred MA, Trembath RC. Transforming growth factor-β receptor mutations and pulmonary arterial hypertension in childhood. Circulation 111: 435–441, 2005. doi: 10.1161/01.CIR.0000153798.78540.87. [DOI] [PubMed] [Google Scholar]
- 539. Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M, Saji T. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J 72: 127–133, 2008. doi: 10.1253/circj.72.127. [DOI] [PubMed] [Google Scholar]
- 540. Jiao YR, Wang W, Lei PC, Jia HP, Dong J, Gou YQ, Chen CL, Cao J, Wang YF, Zhu YK. 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: a meta-analysis. Gene 680: 34–42, 2019. doi: 10.1016/j.gene.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 541. Willers ED, Newman JH, Loyd JE, Robbins IM, Wheeler LA, Prince MA, Stanton KC, Cogan JA, Runo JR, Byrne D, Humbert M, Simonneau G, Sztrymf B, Morse JA, Knowles JA, Roberts KE, McElroy JJ, Barst RJ, Phillips JA 3rd.. Serotonin transporter polymorphisms in familial and idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 173: 798–802, 2006. doi: 10.1164/rccm.200509-1361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542. Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 9: 1416, 2018. doi: 10.1038/s41467-018-03672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Musa-Aziz R, Chen LM, Pelletier MF, Boron WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci USA 106: 5406–5411, 2009. doi: 10.1073/pnas.0813231106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Boron WF. Sharpey-Schafer lecture: gas channels. Exp Physiol 95: 1107–1130, 2010. doi: 10.1113/expphysiol.2010.055244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Yun X, Philip NM, Jiang H, Smith Z, Huetsch JC, Damarla M, Suresh K, Shimoda LA. Upregulation of aquaporin 1 mediates increased migration and proliferation in pulmonary vascular cells from the rat SU5416/hypoxia model of pulmonary hypertension. Front Physiol 12: 763444, 2021. doi: 10.3389/fphys.2021.763444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Yun X, Jiang H, Lai N, Wang J, Shimoda LA. Aquaporin 1-mediated changes in pulmonary arterial smooth muscle cell migration and proliferation involve β-catenin. Am J Physiol Lung Cell Mol Physiol 313: L889–L898, 2017. doi: 10.1152/ajplung.00247.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Zwiazek JJ, Xu H, Tan X, Navarro-Ródenas A, Morte A. Significance of oxygen transport through aquaporins. Sci Rep 7: 40411, 2017. doi: 10.1038/srep40411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Machado RD, Welch CL, Haimel M, Bleda M, Colglazier E, Coulson JD, Debeljak M, Ekstein J, Fineman JR, Golden WC, Griffin EL, Hadinnapola C, Harris MA, Hirsch Y, Hoover-Fong JE, Nogee L, Romer LH, Vesel S, NIHR Bioresource-Rare Diseases, Gräf S, Morrell NW, Southgate L, Chung WK. Biallelic variants of ATP13A3 cause dose-dependent childhood-onset pulmonary arterial hypertension characterised by extreme morbidity and mortality. J Med Genet 59: 906–911, 2022. doi: 10.1136/jmedgenet-2021-107831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Hamid R, Newman JH. Evidence for inflammatory signaling in idiopathic pulmonary artery hypertension: TRPC6 and nuclear factor-κB. Circulation 119: 2297–2298, 2009. doi: 10.1161/CIRCULATIONAHA.109.855197. [DOI] [PubMed] [Google Scholar]
- 550. Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104: 1399–1409, 2009. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 551. Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, Rosenzweig EB, Bayrak-Toydemir P, Mao R, Cahill BC, Tazelaar HD, Leslie KO, Hemnes AR, Robbins IM, Elliott CG. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 145: 231–236, 2014. doi: 10.1378/chest.13-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Best DH, Sumner KL, Smith BP, Damjanovich-Colmenares K, Nakayama I, Brown LM, Ha Y, Paul E, Morris A, Jama MA, Dodson MW, Bayrak-Toydemir P, Elliott CG. EIF2AK4 mutations in patients diagnosed with pulmonary arterial hypertension. Chest 151: 821–828, 2017. doi: 10.1016/j.chest.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 553. Hadinnapola C, Bleda M, Haimel M, Screaton N, Swift A, Dorfmuller P, et al. Phenotypic characterization of EIF2AK4 mutation carriers in a large cohort of patients diagnosed clinically with pulmonary arterial hypertension. Circulation 136: 2022–2033, 2017. doi: 10.1161/CIRCULATIONAHA.117.028351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JS, Wilkins MR, Ohta-Ogo K, Nakamura K, Girerd B, Coulet F, Soubrier F, Humbert M, Morrell NW, Trembath RC, Machado RD. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 32: 1385–1389, 2011. doi: 10.1002/humu.21605. [DOI] [PubMed] [Google Scholar]
- 555. Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet 46: 331–337, 2009. doi: 10.1136/jmg.2008.062703. [DOI] [PubMed] [Google Scholar]
- 556. Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 49: 403–409, 2013. doi: 10.1165/rcmb.2013-0100OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Hernandez-Gonzalez I, Tenorio-Castano J, Ochoa-Parra N, Gallego N, Pérez-Olivares C, Lago-Docampo M, Palomino Doza J, Valverde D, Lapunzina P, Escribano-Subias P. Novel genetic and molecular pathways in pulmonary arterial hypertension associated with connective tissue disease. Cells 10: 1488, 2021. doi: 10.3390/cells10061488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Kerstjens-Frederikse WS, Bongers EM, Roofthooft MT, Leter EM, Douwes JM, Van Dijk A, Vonk-Noordegraaf A, Dijk-Bos KK, Hoefsloot LH, Hoendermis ES, Gille JJ, Sikkema-Raddatz B, Hofstra RM, Berger RM. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 50: 500–506, 2013. doi: 10.1136/jmedgenet-2012-101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, Hamid R, Nichols WC, Pauciulo MW, Lutz KA, Sawle A, Reid JG, Overton JD, Baras A, Dewey F, Shen Y, Chung WK. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med 11: e001887, 2018. doi: 10.1161/CIRCGEN.117.001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Zhu N, Welch CL, Wang J, Allen PM, Gonzaga-Jauregui C, Ma L, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, Hamid R, Pauciulo MW, Lutz KA, Nichols WC, Reid JG, Overton JD, Baras A, Dewey FE, Shen Y, Chung WK. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med 10: 56, 2018. doi: 10.1186/s13073-018-0566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol 17: 85–95, 2020. doi: 10.1038/s41569-019-0242-x. [DOI] [PubMed] [Google Scholar]
- 562. Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 115: 189–202, 2014. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Garcia-Rivas G, Jerjes-Sánchez C, Rodriguez D, Garcia-Pelaez J, Trevino V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med Genet 18: 82, 2017. doi: 10.1186/s12881-017-0440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Soubrier F, Chung WK, Machado R, Grünig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 62: D13–D21, 2013. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 565. Blanpain C, Le Poul E, Parma J, Knoop C, Detheux M, Parmentier M, Vassart G, Abramowicz MJ. Serotonin 5-HT2B receptor loss of function mutation in a patient with fenfluramine-associated primary pulmonary hypertension. Cardiovasc Res 60: 518–528, 2003. doi: 10.1016/j.cardiores.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 566. Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura C, Phillips JA 3rd, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P, Nichols WC, Elliott G. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol 43: 33S–39S, 2004. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 567. Bochenek ML, Leidinger C, Rosinus NS, Gogiraju R, Guth S, Hobohm L, Jurk K, Mayer E, Münzel T, Lankeit M, Bosmann M, Konstantinides S, Schäfer K. Activated endothelial TGFβ1 signaling promotes venous thrombus nonresolution in mice via endothelin-1: potential role for chronic thromboembolic pulmonary hypertension. Circ Res 126: 162–181, 2020. doi: 10.1161/CIRCRESAHA.119.315259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Wang G, Knight L, Ji R, Lawrence P, Kanaan U, Li L, Das A, Cui B, Zou W, Penny DJ, Fan Y. Early onset severe pulmonary arterial hypertension with ‘two-hit’ digenic mutations in both BMPR2 and KCNA5 genes. Int J Cardiol 177: e167–e169, 2014. doi: 10.1016/j.ijcard.2014.08.124. [DOI] [PubMed] [Google Scholar]
- 569. Bossini-Castillo L, Simeon CP, Beretta L, Broen J, Vonk MC, Callejas JL, Carreira P, Rodríguez-Rodríguez L, García-Portales R, González-Gay MA, Castellvi I, Camps MT, Tolosa C, Vicente-Rabaneda E, Egurbide MV, Schuerwegh AJ, Hesselstrand R, Lunardi C, van Laar JM, Shiels P, Herrick A, Worthington J, Denton C, Radstake TR, Fonseca C, Martin J; Spanish Scleroderma Group. KCNA5 gene is not confirmed as a systemic sclerosis-related pulmonary arterial hypertension genetic susceptibility factor. Arthritis Res Ther 14: R273, 2012. doi: 10.1186/ar4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Gelinas SM, Benson CE, Khan MA, Berger RM, Trembath RC, Machado RD, Southgate L. Whole exome sequence analysis provides novel insights into the genetic framework of childhood-onset pulmonary arterial hypertension. Genes (Basel) 11: 1328, 2020. doi: 10.3390/genes11111328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 108: 1141–1150, 2001. doi: 10.1172/JCI200112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Rabinovitch M. Linking a serotonin transporter polymorphism to vascular smooth muscle proliferation in patients with primary pulmonary hypertension. J Clin Invest 108: 1109–1111, 2001. doi: 10.1172/JCI200114205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Saygin D, Tabib T, Bittar HE, Valenzi E, Sembrat J, Chan SY, Rojas M, Lafyatis R. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm Circ 10: 2045894020908782, 2020. doi: 10.1177/2045894020908782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Oldham WM, Hemnes AR, Aldred MA, Barnard J, Brittain EL, Chan SY, et al. NHLBI-CMREF Workshop Report on Pulmonary Vascular Disease Classification: JACC State-of-the-Art Review. J Am Coll Cardiol 77: 2040–2052, 2021. doi: 10.1016/j.jacc.2021.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 8: 443–455, 2011. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Rowan SC, Keane MP, Gaine S, McLoughlin P. Hypoxic pulmonary hypertension in chronic lung diseases: novel vasoconstrictor pathways. Lancet Respir Med 4: 225–236, 2016. doi: 10.1016/S2213-2600(15)00517-2. [DOI] [PubMed] [Google Scholar]
- 577. Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest 130: 5638–5651, 2020. doi: 10.1172/JCI137558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Hemnes AR, Beck GJ, Newman JH, Abidov A, Aldred MA, Barnard J, Berman Rosenzweig E, Borlaug BA, Chung WK, Comhair SA, Erzurum SC, Frantz RP, Gray MP, Grunig G, Hassoun PM, Hill NS, Horn EM, Hu B, Lempel JK, Maron BA, Mathai SC, Olman MA, Rischard FP, Systrom DM, Tang WHW, Waxman AB, Xiao L, Yuan JX, Leopold JA; PVDOMICS Study Group. PVDOMICS: a multi-center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res 121: 1136–1139, 2017. doi: 10.1161/CIRCRESAHA.117.311737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Romanoski CE, Qi X, Sangam S, Vanderpool RR, Stearman RS, Conklin A, Gonzalez-Garay M, Rischard F, Ayon RJ, Wang J, Simonson T, Babicheva A, Shi Y, Tang H, Makino A, Kanthi Y, Geraci MW, Garcia JG, Yuan JX, Desai AA. Transcriptomic profiles in pulmonary arterial hypertension associate with disease severity and identify novel candidate genes. Pulm Circ 10: 2045894020968531, 2020. doi: 10.1177/2045894020968531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Clair G, Bramer LM, Misra R, McGraw MD, Bhattacharya S, Kitzmiller JA, Feng S, Danna VG, Bandyopadhyay G, Bhotika H, Huyck HL, Deutsch GH, Mariani TJ, Carson JP, Whitsett JA, Pryhuber GS, Adkins JN, Ansong C. Proteomic analysis of human lung development. Am J Respir Crit Care Med 205: 208–218, 2022. doi: 10.1164/rccm.202008-3303OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Fantozzi I, Platoshyn O, Wong AH, Zhang S, Remillard CV, Furtado MR, Petrauskene OV, Yuan JX. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 291: L993–L1004, 2006. doi: 10.1152/ajplung.00191.2005. [DOI] [PubMed] [Google Scholar]
- 582. Pozeg ZI, Michelakis ED, McMurtry MS, Thébaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A, Archer SL. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 107: 2037–2044, 2003. doi: 10.1161/01.CIR.0000062688.76508.B3. [DOI] [PubMed] [Google Scholar]
- 583. Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101: 2319–2330, 1998. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. West JD, Austin ED, Rizzi EM, Yan L, Tanjore H, Crabtree AL, Moore CS, Muthian G, Carrier EJ, Jacobson DA, Hamid R, Kendall PL, Majka S, Rathinasabapathy A. KCNK3 mutation causes altered immune function in pulmonary arterial hypertension patients and mouse models. Int J Mol Sci 22: 5014, 2021. doi: 10.3390/ijms22095014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Sörmann J, Schewe M, Proks P, Jouen-Tachoire T, Rao S, Riel EB, Agre KE, Begtrup A, Dean J, Descartes M, Fischer J, Gardham A, Lahner C, Mark PR, Muppidi S, Pichurin PN, Porrmann J, Schallner J, Smith K, Straub V, Vasudevan P, Willaert R, Carpenter EP, Rödström KE, Hahn MG, Müller T, Baukrowitz T, Hurles ME, Wright CF, Tucker SJ. Gain-of-function mutations in KCNK3 cause a developmental disorder with sleep apnea. Nat Genet 54: 1534–1543, 2022. doi: 10.1038/s41588-022-01185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Bohnen MS, Roman-Campos D, Terrenoire C, Jnani J, Sampson KJ, Chung WK, Kass RS. The impact of heterozygous KCNK3 mutations associated with pulmonary arterial hypertension on channel function and pharmacological recovery. J Am Heart Assoc 6: e006465, 2017. doi: 10.1161/JAHA.117.006465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587. Perez-Vizcaino F, Cogolludo A, Mondejar-Parreño G. Transcriptomic profile of cationic channels in human pulmonary arterial hypertension. Sci Rep 11: 15829, 2021. doi: 10.1038/s41598-021-95196-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Yadav VR, Song T, Mei L, Joseph L, Zheng YM, Wang YX. PLCγ1-PKCε-IP3R1 signaling plays an important role in hypoxia-induced calcium response in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 314: L724–L735, 2018. doi: 10.1152/ajplung.00243.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Suresh K, Servinsky L, Jiang H, Bigham Z, Yun X, Kliment C, Huetsch J, Damarla M, Shimoda LA. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 314: L893–L907, 2018. doi: 10.1152/ajplung.00430.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286: L848–L858, 2004. doi: 10.1152/ajplung.00319.2003. [DOI] [PubMed] [Google Scholar]
- 592. Tanaka S, Yamamoto T, Mikawa M, Nawata J, Fujii S, Nakamura Y, Kato T, Fukuda M, Suetomi T, Uchinoumi H, Oda T, Okuda S, Okamura T, Kobayashi S, Yano M. Stabilization of RyR2 maintains right ventricular function, reduces the development of ventricular arrhythmias, and improves prognosis in pulmonary hypertension. Heart Rhythm 19: 986–997, 2022. doi: 10.1016/j.hrthm.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 593. Castillo-Galán S, Arenas GA, Reyes RV, Krause BJ, Iturriaga R. Stim-activated TRPC-ORAI channels in pulmonary hypertension induced by chronic intermittent hypoxia. Pulm Circ 10: 13–22, 2020. doi: 10.1177/2045894020941484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594. Rode B, Bailey MA, Marthan R, Beech DJ, Guibert C. ORAI channels as potential therapeutic targets in pulmonary hypertension. Physiology (Bethesda) 33: 261–268, 2018. doi: 10.1152/physiol.00016.2018. [DOI] [PubMed] [Google Scholar]
- 595. Masson B, Montani D, Humbert M, Capuano V, Antigny F. Role of store-operated Ca2+ entry in the pulmonary vascular remodeling occurring in pulmonary arterial hypertension. Biomolecules 11: 1781, 2021. doi: 10.3390/biom11121781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Platoshyn O, Brevnova EE, Burg ED, Yu Y, Remillard CV, Yuan JX. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 290: C907–C916, 2006. doi: 10.1152/ajpcell.00028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 280: L801–L812, 2001. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- 598. Platoshyn O, Yu Y, Ko EA, Remillard CV, Yuan JX. Heterogeneity of hypoxia-mediated decrease in IK(V) and increase in [Ca2+]cyt in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 293: L402–L416, 2007. doi: 10.1152/ajplung.00391.2006. [DOI] [PubMed] [Google Scholar]
- 599. Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 351: 726–727, 1998. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- 600. Brevnova EE, Platoshyn O, Zhang S, Yuan JX. Overexpression of human KCNA5 increases IK(V) and enhances apoptosis. Am J Physiol Cell Physiol 287: C715–C722, 2004. doi: 10.1152/ajpcell.00050.2004. [DOI] [PubMed] [Google Scholar]
- 601. Xu N, Ayers L, Pastukh V, Alexeyev M, Stevens T, Tambe DT. Impact of Na+ permeation on collective migration of pulmonary arterial endothelial cells. PLoS One 16: e0250095, 2021. doi: 10.1371/journal.pone.0250095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Breitling S, Ravindran K, Goldenberg NM, Kuebler WM. The pathophysiology of pulmonary hypertension in left heart disease. Am J Physiol Lung Cell Mol Physiol 309: L924–L941, 2015. doi: 10.1152/ajplung.00146.2015. [DOI] [PubMed] [Google Scholar]
- 603. Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, Viana F, Garrison JC, Bayliss DA. Inhibition of a background potassium channel by Gq protein α-subunits. Proc Natl Acad Sci U S A 103: 3422–3427, 2006. doi: 10.1073/pnas.0507710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, Bünemann M, Leitner MG, Oliver D. Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat Commun 5: 5540, 2014. doi: 10.1038/ncomms6540. [DOI] [PubMed] [Google Scholar]
- 605. Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294: L309–L318, 2008. doi: 10.1152/ajplung.00091.2007. [DOI] [PubMed] [Google Scholar]
- 606. Santos-Gomes J, Le Ribeuz H, Brás-Silva C, Antigny F, Adão R. Role of ion channel remodeling in endothelial dysfunction induced by pulmonary arterial hypertension. Biomolecules 12: 484, 2022. doi: 10.3390/biom12040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Hennigs JK, Lüneburg N, Stage A, Schmitz M, Körbelin J, Harbaum L, Matuszcak C, Mienert J, Bokemeyer C, Böger RH, Kiefmann R, Klose H. The P2-receptor-mediated Ca2+ signalosome of the human pulmonary endothelium - implications for pulmonary arterial hypertension. Purinergic Signal 15: 299–311, 2019. doi: 10.1007/s11302-019-09674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608. Fantozzi I, Huang W, Zhang J, Zhang S, Platoshyn O, Remillard CV, Thistlethwaite PA, Yuan JX. Divergent effects of BMP-2 on gene expression in pulmonary artery smooth muscle cells from normal subjects and patients with idiopathic pulmonary arterial hypertension. Exp Lung Res 31: 783–806, 2005. doi: 10.1080/01902140500461026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Boucly A, Savale L, Jaïs X, Bauer F, Bergot E, Bertoletti L, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 204: 842–854, 2021. doi: 10.1164/rccm.202009-3698OC. [DOI] [PubMed] [Google Scholar]
- 610. Benza RL, Seeger W, McLaughlin VV, Channick RN, Voswinckel R, Tapson VF, Robbins IM, Olschewski H, Rubin LJ. Long-term effects of inhaled treprostinil in patients with pulmonary arterial hypertension: the Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension (TRIUMPH) study open-label extension. J Heart Lung Transplant 30: 1327–1333, 2011. doi: 10.1016/j.healun.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 611. McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, Robbins IM, Olschewski H, Rubenfire M, Seeger W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 55: 1915–1922, 2010. doi: 10.1016/j.jacc.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 612. McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, Badesch DB, Barst RJ, Hsu HH, Rubin LJ. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 174: 1257–1263, 2006. doi: 10.1164/rccm.200603-358OC. [DOI] [PubMed] [Google Scholar]
- 613. Fernandes CJ, da Silva TA, Alves JL Jr, Jardim CV, de Souza R. Inhaled iloprost as third add-on therapy in idiopathic pulmonary arterial hypertension. Pulm Circ 11: 2045894020981350, 2021. doi: 10.1177/2045894020981350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R, Rubin LJ, Di Scala L, Tapson V, Adzerikho I, Liu J, Moiseeva O, Zeng X, Simonneau G, McLaughlin VV; GRIPHON Investigators. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 373: 2522–2533, 2015. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 615. Rosenkranz S, Channick R, Chin KM, Jenner B, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, McLaughlin VV, Du Roure C, Rubin LJ, Sitbon O, Tapson V, Lang IM. The impact of comorbidities on selexipag treatment effect in patients with pulmonary arterial hypertension: insights from the GRIPHON study. Eur J Heart Fail 24: 205–214, 2022. doi: 10.1002/ejhf.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Galiè N, Rubin L, Hoeper M, Jansa P, Al-Hiti H, Meyer G, Chiossi E, Kusic-Pajic A, Simonneau G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 371: 2093–2100, 2008. doi: 10.1016/S0140-6736(08)60919-8. [DOI] [PubMed] [Google Scholar]
- 617. Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346: 896–903, 2002. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 618. Oudiz RJ, Galiè N, Olschewski H, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Harrison BC, Despain D, Dufton C, Rubin LJ; ARIES Study Group. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol 54: 1971–1981, 2009. doi: 10.1016/j.jacc.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 619. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, Oudiz RJ, Vonk-Noordegraaf A, White RJ, Blair C, Gillies H, Miller KL, Harris JH, Langley J, Rubin LJ; AMBITION Investigators. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 373: 834–844, 2015. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 620. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369: 809–818, 2013. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 621. Kim KH, Kim HK, Chan SY, Kim YJ, Sohn DW. Hemodynamic and histopathologic benefits of early treatment with Macitentan in a rat model of pulmonary arterial hypertension. Korean Circ J 48: 839–853, 2018. doi: 10.4070/kcj.2017.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622. Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115: 2811–2821, 2005. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med 353: 1412–1413, 2005. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 624. Humbert M, McLaughlin V, Gibbs JS, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, Meyer G, Montani D, Olsson KM, Manimaran S, Barnes J, Linde PG, de Oliveira Pena J, Badesch DB; PULSAR Trial Investigators. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 384: 1204–1215, 2021. doi: 10.1056/NEJMoa2024277. [DOI] [PubMed] [Google Scholar]
- 625. Yung LM, Yang P, Joshi S, Augur ZM, Kim SS, Bocobo GA, Dinter T, Troncone L, Chen PS, McNeil ME, Southwood M, Poli de Frias S, Knopf J, Rosas IO, Sako D, Pearsall RS, Quisel JD, Li G, Kumar R, Yu PB. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci Transl Med 12: eaaz5660, 2020. doi: 10.1126/scitranslmed.aaz5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 327: 76–81, 1992. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 627. Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation 76: 135–141, 1987. doi: 10.1161/01.CIR.76.1.135. [DOI] [PubMed] [Google Scholar]
- 628. Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 109: 1239–1244, 2012. doi: 10.1073/pnas.1120385109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Tsai SH, Huang PH, Hsu YJ, Peng YJ, Lee CH, Wang JC, Chen JW, Lin SJ. Inhibition of hypoxia inducible factor-1α attenuates abdominal aortic aneurysm progression through the down-regulation of matrix metalloproteinases. Sci Rep 6: 28612, 2016. doi: 10.1038/srep28612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630. Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res 81: 34–41, 1997. doi: 10.1161/01.RES.81.1.34. [DOI] [PubMed] [Google Scholar]
- 631. Chester AH, Yacoub MH, Moncada S. Nitric oxide and pulmonary arterial hypertension. Glob Cardiol Sci Pract 2017: 14, 2017. doi: 10.21542/gcsp.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Hoeper MM, Al-Hiti H, Benza RL, Chang SA, Corris PA, Gibbs JS, et al. Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med 9: 573–584, 2021. doi: 10.1016/S2213-2600(20)30532-4. [DOI] [PubMed] [Google Scholar]
- 633. Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ, PATENT-1 Study Group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369: 330–340, 2013. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 634. Rubin LJ, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, Keogh A, Langleben D, Fritsch A, Menezes F, Davie N, Ghofrani HA. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J 45: 1303–1313, 2015. doi: 10.1183/09031936.00090614. [DOI] [PubMed] [Google Scholar]
- 635. Simonneau G, D’Armini AM, Ghofrani HA, Grimminger F, Hoeper MM, Jansa P, Kim NH, Wang C, Wilkins MR, Fritsch A, Davie N, Colorado P, Mayer E. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2). Eur Respir J 45: 1293–1302, 2015. doi: 10.1183/09031936.00087114. [DOI] [PubMed] [Google Scholar]
- 636. Hoeper MM, Gomez Sanchez MA, Humbert M, Pittrow D, Simonneau G, Gall H, et al. Riociguat treatment in patients with pulmonary arterial hypertension: final safety data from the EXPERT registry. Respir Med 177: 106241, 2021. doi: 10.1016/j.rmed.2020.106241. [DOI] [PubMed] [Google Scholar]
- 637. Teymouri Rad R, Dadashzadeh S, Vatanara A, Alavi S, Ghasemian E, Mortazavi SA. Tadalafil nanocomposites as a dry powder formulation for inhalation, a new strategy for pulmonary arterial hypertension treatment. Eur J Pharm Sci 133: 275–286, 2019. doi: 10.1016/j.ejps.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 638. Mamazhakypov A, Weiss A, Zukunft S, Sydykov A, Kojonazarov B, Wilhelm J, Vroom C, Petrovic A, Kosanovic D, Weissmann N, Seeger W, Fleming I, Iglarz M, Grimminger F, Ghofrani HA, Pullamsetti SS, Schermuly RT. Effects of macitentan and tadalafil monotherapy or their combination on the right ventricle and plasma metabolites in pulmonary hypertensive rats. Pulm Circ 10: 2045894020947283, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, Shapiro S, White RJ, Chan M, Beardsworth A, Frumkin L, Barst RJ; Pulmonary Arterial Hypertension and Response To Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119: 2894–2903, 2009. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 640. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G; Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353: 2148–2157, 2005. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 641. Kiss T, Kovacs K, Komocsi A, Tornyos A, Zalan P, Sumegi B, Gallyas F Jr, Kovacs K. Novel mechanisms of sildenafil in pulmonary hypertension involving cytokines/chemokines, MAP kinases and Akt. PLoS One 9: e104890, 2014. doi: 10.1371/journal.pone.0104890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642. Crader MF, Johns T, Arnold JK. Warfarin drug interactions. In: StatPearls. Treasure Island, FL: StatPearls, 2021. [PubMed] [Google Scholar]
- 643. Porres-Aguilar M, Hoeper MM, Rivera-Lebron BN, Heresi GA, Mukherjee D, Tapson VF. Direct oral anticoagulants in chronic thromboembolic pulmonary hypertension. J Thromb Thrombolysis 52: 791–796, 2021. doi: 10.1007/s11239-021-02445-z. [DOI] [PubMed] [Google Scholar]
- 644. Sena S, Bulent M, Derya K, Deniz K, Halil A, Okan E, Bedrettin Y. Real-life data of direct anticoagulant use, bleeding risk and venous thromboembolism recurrence in chronic thromboembolic pulmonary hypertension patients: an observational retrospective study. Pulm Circ 10: 2045894019873545, 2020. doi: 10.1177/2045894019873545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Jain PP, Lai N, Xiong M, Chen J, Babicheva A, Zhao T, Parmisano S, Zhao M, Paquin C, Matti M, Powers R, Balistrieri A, Kim NH, Valdez-Jasso D, Thistlethwaite PA, Shyy JY, Wang J, Garcia JG, Makino A, Yuan JX. TRPC6, a therapeutic target for pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 321: L1161–L1182, 2021. doi: 10.1152/ajplung.00159.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646. Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, Bian H, Zeveleva S, Zhen X, Blair NT, Chong JA, Hessler DP, Bedja D, Zhu G, Muller GK, Ranek MJ, Pantages L, McFarland M, Netherton MR, Berry A, Wong D, Rast G, Qian HS, Weldon SM, Kuo JJ, Sauer A, Sarko C, Moran MM, Kass DA, Pullen SS. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci USA 116: 10156–10161, 2019. doi: 10.1073/pnas.1815354116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647. MacLean MM. The serotonin hypothesis in pulmonary hypertension revisited: targets for novel therapies (2017 Grover Conference Series). Pulm Circ 8: 2045894018759125, 2018. doi: 10.1177/2045894018759125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648. Baranowska-Kuczko M, Kozłowska H, Schlicker E, Göthert M, MacLean MR, Kozłowski M, Kloza M, Sadowska O, Malinowska B. Reduction of the serotonin 5-HT1B and 5-HT2A receptor-mediated contraction of human pulmonary artery by the combined 5-HT1B receptor antagonist and serotonin transporter inhibitor LY393558. Pharmacol Rep 72: 756–762, 2020. doi: 10.1007/s43440-020-00105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649. Morecroft I, Pang L, Baranowska M, Nilsen M, Loughlin L, Dempsie Y, Millet C, MacLean MR. In vivo effects of a combined 5-HT1B receptor/SERT antagonist in experimental pulmonary hypertension. Cardiovasc Res 85: 593–603, 2010. doi: 10.1093/cvr/cvp306. [DOI] [PubMed] [Google Scholar]
- 650. Yamamura A, Ohara N, Tsukamoto K. Inhibition of excessive cell proliferation by calcilytics in idiopathic pulmonary arterial hypertension. PLoS One 10: e0138384, 2015. doi: 10.1371/journal.pone.0138384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651. Morris HE, Neves KB, Montezano AC, MacLean MR, Touyz RM. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin Sci (Lond) 133: 2481–2498, 2019. doi: 10.1042/CS20190835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652. Zhang Y, Hernandez M, Gower J, Winicki N, Morataya X, Alvarez S, Yuan JX, Shyy J, Thistlethwaite PA. JAGGED-NOTCH3 signaling in vascular remodeling in pulmonary arterial hypertension. Sci Transl Med 14: eabl5471, 2022. doi: 10.1126/scitranslmed.abl5471. [DOI] [PubMed] [Google Scholar]
- 653. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 126: 1456–1474, 2020. doi: 10.1161/CIRCRESAHA.120.317015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654. Hemnes AR, Rathinasabapathy A, Austin EA, Brittain EL, Carrier EJ, Chen X, Fessel JP, Fike CD, Fong P, Fortune N, Gerszten RE, Johnson JA, Kaplowitz M, Newman JH, Piana R, Pugh ME, Rice TW, Robbins IM, Wheeler L, Yu C, Loyd JE, West J. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J 51: 1702638, 2018. doi: 10.1183/13993003.02638-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, Zhang Y, Yin Q, Cho Y, Andrade L, Shadel GS, Hepokoski M, Lei T, Wang H, Zhang J, Yuan JX, Malhotra A, Manor U, Wang S, Yuan ZY, Shyy JY. SARS-CoV-2 Spike protein impairs endothelial function via downregulation of ACE 2. Circ Res 128: 1323–1326, 2021. doi: 10.1161/CIRCRESAHA.121.318902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656. Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, Chen L, Shi X, Yin Y, Shang F, Wu Y, Huang HY, Zhang J, Zhang Y, Kang J, Moya EA, Huang HD, Powell FL, Chen Z, Thistlethwaite PA, Yuan ZY, Shyy JY. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am J Respir Crit Care Med 198: 509–520, 2018. doi: 10.1164/rccm.201712-2570OC. [DOI] [PMC free article] [PubMed] [Google Scholar]