

Abstract
Posttranslational spliced peptides (PTSPs) are a unique class of peptides that have been found to be presented by HLA class-I molecules in cancer. Thus far, no consensus has been reached on the proportion of PTSPs in the immunopeptidome, with estimates ranging from 2% to as high as 45% and stirring significant debate. Furthermore, the role of the HLA class-II pathway in PTSP presentation has been studied only in diabetes. Here, we exploit our large-scale cancer peptidomics database and our newly devised pipeline for filtering spliced peptide predictions to identify recurring spliced peptides, both for HLA class-I and class-II complexes. Our results indicate that HLA class-I–spliced peptides account for a low percentage of the immunopeptidome (less than 3.1%) yet are larger in number relative to other types of identified aberrant peptides. Therefore, spliced peptides significantly contribute to the repertoire of presented peptides in cancer cells. In addition, we identified HLA class-II–bound spliced peptides, but to a lower extent (less than 0.5%). The identified spliced peptides include cancer- and immune-associated genes, such as the MITF oncogene, DAPK1 tumor suppressor, and HLA-E, which were validated using synthetic peptides. The potential immunogenicity of the DAPK1- and HLA-E–derived PTSPs was also confirmed. In addition, a reanalysis of our published mouse single-cell clone immunopeptidome dataset showed that most of the spliced peptides were found repeatedly in a large number of the single-cell clones. Establishing a novel search-scheme for the discovery and evaluation of recurring PTSPs among cancer patients may assist in identifying potential novel targets for immunotherapy.
Keywords: HLA class-I, HLA class-II, posttranslational spliced peptides, immunopeptidomics, neoantigens
Graphical Abstract
Highlights
-
•
A novel computational pipeline reveals post-translational spliced peptides (PTSPs).
-
•
PTSPs show recurrence in a large-scale immunopeptidomic database of patients.
-
•
HLA class-I and -II molecules are predicted to bind PTSPs to a different extent.
-
•
Two of seven PTSPs, validated using synthetic peptides, were shown to be immunogenic.
In Brief
Thus far, no consensus has been reached on the extent to which post-translational spliced peptides occur, stirring significant debate. Here, we designed a pipeline for their identification. Key strategies for the filtration of noncanonical peptides are proposed following comparative filtration to WT peptides. We find that only low percentages of the spliced peptides are identified for HLA class-I and II. Spliced peptides are further validated based on synthetic peptides and immune-reactivity assays.
Analyses of the cancer immunopeptidome have predominantly focused on somatic nonsynonymous protein-altering mutations. This is reflected in the types of approaches used, such as whole-genome and whole-exome sequencing, which support mutation analyses at the DNA and RNA levels (1, 2). However, aberrant alterations can occur also posttranslationally (3, 4, 5, 6).
Posttranslational spliced peptides (PTSPs) are generated by the ligation of two splice reactants, in a manner that either follows the same N- to C-terminus orientation of the parental protein (normal cis-spliced peptides) or inverts it (reverse cis-spliced peptides) (7). They can also be formed by the ligation of two splice reactants from different proteins (trans-spliced peptides). So far, no consensus has been reached on the proportion of PTSPs in the immunopeptidome, with estimates ranging from 2% to as high as 45% and stirring significant debate (7, 8, 9, 10, 11, 12, 13). Recently, it has been suggested that trans-splicing occurs as frequently as cis-splicing (14). Others have argued that for trans-splicing to happen, the two source proteins need to be present in the same proteasome at the same time, which is unlikely to happen on a large scale (8, 15). A recently published study has gone as far as to suggest that proteasome-mediated splicing may not occur at all, putting into question the overall existence of proteasome-spliced peptides (12). The authors of this study argued that as a reactant in normal proteolytic reactions, water competes with transpeptidation (a suggested mechanism of peptide splicing) and that high water mobility and abundance in aqueous solutions renders transpeptidation very inefficient and, therefore, unlikely to occur (12). However, these various reasonings are in contrast with in vitro, in-cellular, and in vivo results published since the discovery of proteasome-catalyzed peptides (5, 11, 16, 17, 18, 19). They also do not consider alternative mechanisms that could explain PTSPs, which would refute arguments that PTSPs do not exist due to reasons associated with the proteasome. While experimental study was beyond the scope of the later commentary, a recent study involving in vitro proteasomal digests of a panel of polypeptide substrates in combination with mass spectrometry found that forward and reverse reactions display unique cis-splicing signatures, providing experimental support for proteasome involvement (20).
While a body of research exists regarding HLA class-I–spliced peptides, the only report on an HLA class-II–spliced peptide comes from a study on type 1 diabetes, in which spliced insulin sequences were identified (21). Type 1 diabetes is caused by the T cell–mediated destruction of insulin-producing β cells. The discovery that autoreactive T cells target spliced peptides may explain how immune tolerance is broken in type 1 diabetes (22, 23). The current study presents the first search for HLA class-II PTSPs in cancer and analysis of their extent. As the classical HLA class-II pathway does not involve the proteasome (whereas HLA class-I does), the identification of such spliced peptides would suggest either proteasome splicing via a nonclassical HLA class-II pathway and/or the involvement of proteasome-independent mechanisms for the generation of HLA class-II PTSPs.
Until recently, there was no adequate tool for the study of PTSPs, and computational limitations precluded an accurate assessment of their prevalence. Recent studies that nevertheless did address this topic were not in agreement. According to Liepe et al. (7), the proteasome-generated cis-spliced peptide pool accounts for one-third of the entire HLA class-I immunopeptidome. This estimation was based on generating an in silico posttranslation sequence database. For each protein in the human proteome, all the 9 to 12 N-mer cis-spliced peptides with a maximum intervening sequence length of 25 residues were generated, followed by a database reduction based on finding the precursor masses in the mass spectrometry (MS) data. However, when Mylonas et al. (8) reanalyzed the data, they found that most of the spectra published by (7) could be assigned to WT peptide sequences. This led them to suggest that spliced peptides make up a much smaller portion of the immunopeptidome (2–6%). They further proposed that the large search space of potential PTSPs generated in (7) led to an uncontrollable propagation of false-positives (8).
The lack of specialized tools dedicated to the prediction of posttranslational cis- and trans-spliced peptides led to the development of the Neo-Fusion tool. This tool does not require an in silico posttranslational sequence database or de-novo prediction approaches (24), both of which suffer from a tendency to produce false-positives. Instead, two separate ion database searches (N-terminus ion database search and C-terminus ion database search) are implemented to identify the two WT parents of each PTSP, and then the full spliced sequence is inferred, following mass matching (24).
In this study, we devise a novel pipeline for the identification and evaluation of recurrent cis-spliced peptides. In this pipeline, PTSPs identified by Neo-Fusion serve as a sequence database for a MaxQuant search (in combination with the full WT proteome). The pipeline stringently eliminates possible false-positives based on multiple criteria. Using our large-scale in-lab peptidomics database, we identified 143 recurrently spliced peptide bound to MHC class-I complexes (pMHC-I; from 72 samples and replicates). Interestingly, we also discovered ten recurrently spliced pMHC-II (from 30 samples and replicates). The recurrent PTSPs include multiple cancer and immune-related genes. Implementing this novel approach for searching for recurrent PTSPs shared among patients may assist in identifying important targets for immunotherapy.
Experimental Procedures
HLA Ligandomic Data
The peptidomics samples analyzed include multiple in-lab datasets obtained from various melanoma cell lines 108T, 12T, MDA55, 83T, 14T, 17T, 26T, 32T, 81T, 88T, 8T, A375, MD-Anderson melanoma patients (42RF, 42RS, 27, 51AL, 51B-R, 55A3, 55A7, 55B3, 86A, 86B, 86C, 92B3, 92C3) from (25, 26) and head and neck squamous carcinoma SCC47 cell line. Breast cancer cell line, MCF7 was obtained from (27) and in addition, mouse melanoma B2905 data (three parental tumor replicates and 16 single-cell clones). A data summary is available in supplemental Table S1. The HLA allotyping of the samples used in this study is provided supplemental Table S2.
Patients and Tumor Infiltrating Lymphocytes
Patient-derived melanoma cell lines were collected with informed patient consent under a protocol approved by the NIH Institutional Review Board (IRB) Ethics Committee (03-C-0277A). The protocol for MD-Anderson tumor samples (from patients 42, 51, 55, 86, and 92) was approved by the MD-Anderson IRB (protocol numbers 2012–0846, LAB00–063, and 2004–0069; NCT00338377). Healthy blood donor informed consent was obtained under a protocol approved by the Weizmann IRB Ethics Committee (1218-3). The studies in this work abide by the declaration of Helsinki principles. A disease description, demographical, and clinical features are provided in supplemental Table S3.
Data Processing
Data were processed with the R statistical scripting language (version 3.6.0), Perl (version 5.24.0), and Python (version 2.7.13)
Experimental Design and Statistical Rationale
HLA class-I peptidomics group contains 72 raw files corresponding to 19 samples. HLA class-II group contains 30 raw files corresponding to 11 samples. The selection of this data was based on our in-house HLA-peptidomics dataset, after excluding samples in which no matching RNA-seq was available for the data. As controls, 55 raw files (31 HLA class-I and 24 class-II) from normal skin were used (ten individuals at autopsy) to correspond to the vast majority of samples from skin cancer. In addition, 49 raw files (24 HLA class-I and 25 class-II) from tongue (nine individuals at autopsy) were used, 42 raw files (36 HLA class-I and six class-II) from ovary (four individuals: two from autopsy and two from living donors) derived from (28) and eight raw files (four HLA class-I and four class-II) from our in-house IHW01070 B-cell peptidomics. Between, 3 to 5 technical replicates were available for the normal set, and 1 to 3 replicates for cancer samples. Furthermore, in cancer patients, 2 to 3 metastases data were available for each patient. Large-scale MS immunopeptidomics database was analyzed by MaxQuant (29), and the search engine used was Andromeda, integrated within MaxQuant version 1.5.3.8. The sequence database used was the Uniprot human proteome ID UP000005640 (from June 20, 2021) containing 101,014 proteins and isoforms and mouse proteome ID UP000000589 (downloaded on July 3, 2020) containing 63,738 proteins and isoforms. In addition, in-house source of spliced sequence databases were compiled using Neo-Fusion software (v0.1.3; https://github.com/zrolfs/MetaMorpheus/tree/Neo-Fusion) (24). HLA class-I and class-II spliced databases include 50,550 and 9909 peptide sequences, respectively. No proteases were used to generate the peptides, and nonspecific fragmentation was applied in both Neo-Fusion and MaxQuant. Neo-Fusion was set to search for modifications, in order to reduce false spliced peptide matches to spectra of modified WT peptides. Neo-Fusion uses Global-PTM-Discovery (30) which searched for the following variable modifications in WT peptides: oxidation of M, deamidation on N and Q, ammonia loss, propionamide on C and K, carboxymethylation on C, W, K, dioxidation on W, P, R, K, M, F, Y, C, oxidation on Y, W, P, F, D, E, V, H, K, N, R, C, carbamidomethyl on K, H, D, E, S, T, Y, carboxylation on K, D, E, phospho on S, T, Y, acetyl on S, K, C, T, Acetald+26 on H and K, water loss, H-2 O1 on L, I, P, W, S, oxidation of W to Kynurenine, dehydroalanine from cysteine, didehydro on Y and S, didehydrobutyrine on T and S, formylation on K, S, T, carbamyl on K, R, C, M, methyl on C, H, K, N, Q, R, I, L, D, E, S, T, Ethyl. Spliced peptides (unmodified) were then inputted to MaxQuant, and N-terminal acetylation and methionine oxidation were specifically set as variable modifications (to avoid impractical run durations, of full common posttranslational modification search by MaxQuant). No fixed modifications were set. Precursor mass tolerance was set to the default 15 ppm, and product mass tolerance was set to default 25 ppm in Neo-Fusion. In MaxQuant, precursor mass tolerance was set to the default 12 ppm, and product mass tolerance to default 20 ppm. Known contaminants were excluded in both software runs. Spliced peptides were searched by Neo-Fusion at length of 8 to 14 amino acids (AAs) for HLA class-I similar to (8) and 12 to 25 for HLA class-II, and the resulting spliced peptides were inputted to MaxQuant using the default full range 8 to 25 AAs length. False discovery rate (FDR) 0.01 was used by Neo-Fusion, and MaxQuant was run both with FDR 0.01 and 0.05. The statistical method used to assess false-positives was “randomized” sequence decoys. The detailed tandem mass spectrometry (MS/MS) information of the peptides is made available in supplemental Tables S4 and S5.
HLA-Peptidomics Preparation
The HLA peptides were dried by vacuum centrifugation, resolubilized with 0.1% formic acid and resolved on capillary reversed-phase chromatography on 075 × 300 mm laser-pulled capillaries, self-packed with C18 reversed-phase 3.5 μm beads. Chromatography was performed with the UltiMate 3000 RSLCnano-capillary UHPLC system (Thermo Fisher Scientific), which was coupled by electrospray to tandem mass spectrometry on Q-Exactive-Plus (Thermo Fisher Scientific). The HLA peptides were eluted with a linear gradient over 2 h from 5 to 28% acetonitrile with 0.1% formic acid at a flow rate of 0.15 μl/min. Data was acquired using a data-dependent “top 10” method, fragmenting the peptides by higher-energy collisional dissociation. Full scan MS spectra was acquired at a resolution of 70,000 at 200 m/z with a target value of 3 × 106 ions. Ions were accumulated to an automatic gain control (AGC) target value of 105 with a maximum injection time of generally 100 msec. The peptide match option was set to Preferred. Normalized collision energy was set to 25% and MS/MS resolution was 17,500 at 200 m/z. Fragmented m/z values were dynamically excluded from further selection for 20 s.
Peptide Filtration
The obtained peptides were stringently filtered, by multiple criteria: peptides with MaxQuant scores less than or equal to 60 or PEP larger than 0.1 were discarded. Any peptide not predicted by NetMHCpan (version 4.0) (31) to bind the relevant HLA-alleles (either as strong or weak binders) were removed. Peptides showing MS/MS %fragmentation coverage lower or equal to 60% were filtered out. Alternative ambiguous peptide matching the MS/MS was examined based on the Delta score of MaxQuant. Peptides were discarded in cases where the next best matching MS/MS of MaxQuant score is 20 or less relative to the best matching score. Pseudogenes were checked versus the output provided in (http://www.pseudogene.org/Human/Human90.txt). Furthermore, peptides were compared against long noncoding transcripts obtained from GeneCode v.38 (32). Peptides in which a single AA alteration, results in a WT sequence in the genome, were filtered out as well. Furthermore, iso-baric AA ambiguity was analyzed, by substituting each Leu to Ile (and vice versa) in all possible combinations along the peptide sequence. All peptides produced by a given peptide was then searched by BLAST (using blastp) (33) for 100% identity matches in the WT Uniprot proteome. In case the substituted peptide was identified, the source peptide was discarded. The Leu/Ile analysis was repeated using the RNA-seq as well (RNA-seq Filtration Stages section). Candidate PTSPs in which a single AA exists before/after the splicing junction were filtered out. Peptides detected by Neo-Fusion in normal immmunopeptidomics samples (from skin, tongue, ovary (28), and from our in-house B-cell IHW01070 peptidomics) were filtered out. Peptides matching bacterial WT peptides identified in our tumor samples were filtered out. The identity of the bacteria was determined experimentally in (34) and also the peptides. Further filtrations applied based on RNA-seq and retention time and recurrence are elaborated below.
RNA-seq Filtration Stages
Four RNA-seq analyses were used for filtration: TopHat (version 2.1.1; http://ccb.jhu.edu/software/tophat/index.shtml) and Cufflinks (version 2.2.1; http://cole-trapnell-lab.github.io/cufflinks/) software were used to help identify peptides generated from alternative spliced RNAs (35). TopHat is designed to align RNA-seq reads to a reference genome. Reads that do not align to the genome are split into shorter fragments and are then aligned independently to identify splice-junctions between exons. After mapping the reads to Hg37 human genome, Cufflinks was applied to the alignment file from TopHat to assemble the overlapping “bundles” of aligned reads into transcripts using a probabilistic approach (35). Candidate PTSPs were then searched by BLAST against these transcripts (using tblastn) (33). Lastly, candidate PTSPs aligning to the translated transcript with 100% identity and without any gaps were discarded from the final list.
Second, the transcripts generated by Cufflinks were used also to search for matching peptides in which Leu/Ile replacements can find another match (using tblastn). Third, the RNA-seq was used to detect mutations and short Indels. For that purpose, STAR version 2.5.2b (36) was used to map the reads to the genome in a two-pass mode. scoreDelOpen, scoreDelBase, scoreInsOpen, scoreInsBase were set to zero to allow Indels, and sensitivity of mapping was increased. Samtools Mpileup v.1.3.1 and BCFtools v.1.3.1 (37) were used for variant calling and filtration was done by setting 'DP>10 && QUAL>30'. The VCF file was inputted to Annovar (38) in order to introduce the mutations and short Indels into the WT proteome. Blast was then used to search for the peptides in the modified proteome. Fourth, RPKM (Reads Per Kilobase Million) values were calculated to discard peptides associated with genes that show no expression.
Retention Time Filtration
For true peptides, a linear dependence of retention time on hydrophobicity is expected. This step serves to assess the quality of filtered PTSPs following the filtration steps of the pipeline. Therefore, the SpecL R package was used to calculate the hydrophobicity index using ssrc_2004. For each sample separately, the hydrophobicity index and retention time were modeled using a linear regression model. Absolute-sign residual distribution was used to calculate the interquartile range. Any peptide associated with a residual greater than 1.5 times the interquartile range was considered an outlier. Retention time check serves as a very weak filter at this final stage, and all peptides following this stage are rechecked to assure they remain recurrent.
Patient Recurrence
A PTSP was considered recurrent if it was identified in two patients or more, after a series of filtration steps. For stringency, we excluded from the count, PTSPs identified in different treatments of the same sample or their replicates. Also, identifications in several metastases were counted only once, in case they belonged to the same patient. Furthermore, MaxQuant “match-between-runs” identifications were not used for the recurrence count to avoid false matches between unrelated samples. HLA-restriction (same allele binding) is not set as a requirement in this filtration as the same peptide may bind to different HLA-alleles with different affinities.
Estimation of %PTSPs in Immunopeptidome
The percentage of HLA class-I and class-II PTSPs were calculated by dividing the number of filtered PTSPs by the total number of filtered peptides (PTSPs + WT). This filtration on WT peptides was implemented to assure that the low calculated values obtained were not a result of stringent PTSP filtration implemented in this study. The average of the values obtained for all HLA class-I and class-II MS files represent the final proportion of PTSPs for HLA class-I and class-II correspondingly. Filtration based on normal tissues and RNA-seq was not included in this analysis, as these two filtration steps remove a significant portion of WT peptides, which biases the calculation. Therefore, the estimated low values obtained in this study represent the upper bound values.
Validation Using Synthetic Peptides
Light Synthetic
Light synthetic peptides for spectra validation were ordered from GenScript, as HPLC grade (≥85% purity). These were analyzed using the same LC–MS/MS system and acquisition parameters as indicated above for the endogenous peptides, with the following changes: the gradient was set from 4% to 30% acetonitrile in 20 min, and NCE was set to 27. The data were processed with MaxQuant using the following parameters: all FDRs were set to 1, the individual peptide mass tolerance was set to false. OrgMassSpecR (https://rdrr.io/rforge/OrgMassSpecR/man/SpectrumSimilarity.html) R software package was used for endogenous peptide validation with light synthetic peptides. The synthetic spectra with the best MaxQuant score was selected for each peptide. All endogenous spectra with MaxQuant score greater than 60 were used for head-to-tail comparisons. Default software parameters were used. Normalized dot product and Pearson correlation were correlated. p-value below 0.05 was regarded as significant.
Heavy Synthetic
Ordered from PEPotec as synthetic with one stable isotope-labeled amino acid, at ≥95% purity. The mass spectrometer was operated at a resolution of 70,000 (at m/z = 200) for the MS1 full scan, scanning a mass range from 300 to 1650 m/z with an ion-injection time of 120 ms and an AGC of 3 × 106. Then, each peptide was isolated with an isolation window of 1.7 m/z before ion activation by high-energy collision dissociation (NCE = 27). Targeted PRM MS/MS spectra were acquired at a resolution of 35,000 (at m/z = 200) with an ion-injection time of 100 ms and an AGC of 2 × 105.
PTSP Reactivity Assay in Tumor Infiltrating Lymphocytes
To assess tumor infiltrating lymphocytes (TILs) reactivity towards the spliced peptides, TILs (106 cells) were stimulated for 24 h with 1 μg/ml LLLDKLLSV (DAPK1), WSDSSGGKGGSY (HLA-E), ILDTAGKEEY (NRASQ61K) (>85% purity, GenScript) dissolved in dimethyl sulfoxide or negative control (no peptide). Next, cells were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) to evaluate their viability and labeled with anti-CD8 (clone RPA-T8, Biolegend) and anti-4-1BB (clone 4B4-1, Biolegend). A CytoFLEX LX flow cytometer (Beckman Coulter) was used and the data was analyzed using Kaluza software (https://www.beckman.com/flow-cytometry/software/kaluza; OVO group).
Induction of Healthy Donor’s Reactive T Cells Towards PTSPs
Priming of healthy donors’ T cells with PTSPs was performed as previously described (39) with several modifications. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor’s blood using Ficoll gradient separation (Cytiva). On day 4, monocytes were isolated from PBMCs using CD14-reactive microbeads (Miltenyi Biotec) and cultured for 3 days in CellGro GMP DC medium (CellGenix) supplemented with 1% human serum (Valley Biomedical) and 1% penicillin–streptomycin (DC-T medium) containing 10 ng/ml interleukin (IL)-4 (PeproTec) and 800 IU/ml GM-CSF (PeproTec). On day 1, monocyte-derived–dendritic cells were matured for 16 h with 800 IU/ml GM-CSF, 10 ng/ml IL-4, 10 ng/ml lipopolysaccharide (Sigma-Aldrich), and 5 ng/ml IFNγ (PeproTech) supplemented into the cultures. Autologous T cells were isolated using Pan T Cell MicroBead (Miltenyi Biotec) and cultured overnight in DC-T medium 5 ng/ml IL-7 (PeproTech). On day 0, monocyte-derived–dendritic cells were pulsed for 2 h with one of the following: 10 μg/ml LLLDKLLSV (DAPK1), 10 μg/ml WSDSSGGKGGSY (HLA-E), 10 μg/ml of viral peptides mixture control (Influenza GILGFVFTL, CMV NLVPMVATV, EBV GLCTLVAML), or incubated with ‘no peptide control’. Subsequently, monocyte-derived–dendritic cells were cocultured with isolated T cells in DC-T medium supplemented with 30 ng/ml IL-21 (PeproTech) at a DC:T cell ratio of 1:2. On days 3, 5, and 7, half of the medium was removed and replenished with fresh medium supplemented with 10 ng/ml of both IL-7 and IL-15 (PeproTech). On day 10, 25 IU/ml IL-2 (PeproTech) was also added to the supplemented cytokine cocktail. On day 12, T cells were restimulated with irradiated (35 Gy) human B-LCL 721.221 expressing HLA-A∗01:01 or HLA-A∗02:01 alleles, served as feeder cells. Human B-LCL 721.221 cells were pulsed for 2 h with 10 μg/ml of the abovementioned peptides and cocultured with T cells in DC-T medium at a DC:T cell ratio of 1:2. On days 14 and 17, half of the medium was removed and replenished with fresh medium supplemented with 10 ng/ml of both IL-7 and IL-15 and 25 IU/ml IL-2 or 50 IU/ml IL-2 (PeproTech), respectively. On day 19, T cells were collected and cocultured with pulsed B-LCL 721.221 cells expressing the matching HLA-A allele. For secreted IFN-γ and TNF-α readout, following 2 h of stimulation, Monensin and Brefeldin A were added to the cocultures (1:1000, Biolegend). Upon 6 h of coincubation, cells were stained with Live-dead Fixable Blue Dead Cell Stain Kit (Invitrogen), anti-CD3 (clone HIT3a, Biolegend), anti-CD8 (clone HIT8a, Biolegend), anti-IFN-γ (Clone 4S.B3, Biolegend), and anti-TNF-α (clone MAB11, Biolegend). Following 24 h stimulation, surface expression of 4-1BB was examined. Cells were stained with Live-dead Fixable Blue Dead Cell Stain Kit, CD3 antibodies, CD8 antibodies, and 4-1BB antibodies (Clone 4B4-1, Biolegend). All experiments were evaluated using a CytoFLEX LX flow cytometer (Beckman Coulter) and the data was analyzed using Kaluza software (OVO group).
Defining Peptide-Specific CD8 T Cell Subpopulation via Dextramer Staining
Following the stimulation and expansion of T cells derived from healthy donor PBMCs as mentioned above, 2 × 106 T cells were collected and stained with DAPK1-dextramer, according to the manufacture's protocol (Immudex). Briefly, T cells that were previously exposed to DAPK1-derived peptide or to viral peptide control were stained with Live-dead Fixable Blue Dead Cell Stain Kit (Invitrogen). T cells were next labeled with DAPK1-dextramer followed by staining with anti-CD3 (clone HIT3a, Biolegend) and anti-CD8 (clone HIT8a, Biolegend) and analyzed by flow cytometry (CytoFLEX LX, Beckman Coulter).
RNA-seq and Library Preparation
Total RNA was extracted from melanoma cell lines with Trizol reagent according to the manufacturer’s protocol (Invitrogen). Libraries were prepared with CORALL Total RNA-Seq Library Prep Kit (Lexogen) and sequenced with 200 cycles on the NovaSeq Illumina platform. Samples were next analyzed using an in-house pipeline.
Mouse Single-Cell Clone Immunopeptidomics
The B2905 mouse melanoma data derived from (40) has been run in the same manner as elaborated for the human data. MaxQuant was run against mouse Uniprot data UP000000589. The same filtration process was applied besides filtration based on normal samples, as this data was not available. Second, since the clones belong to the same mouse tumor, both recurrence by MS/MS and by MaxQuant’s “match-between-runs” property were allowed.
Results
Pipeline Design for Robust Prediction of PTSPs
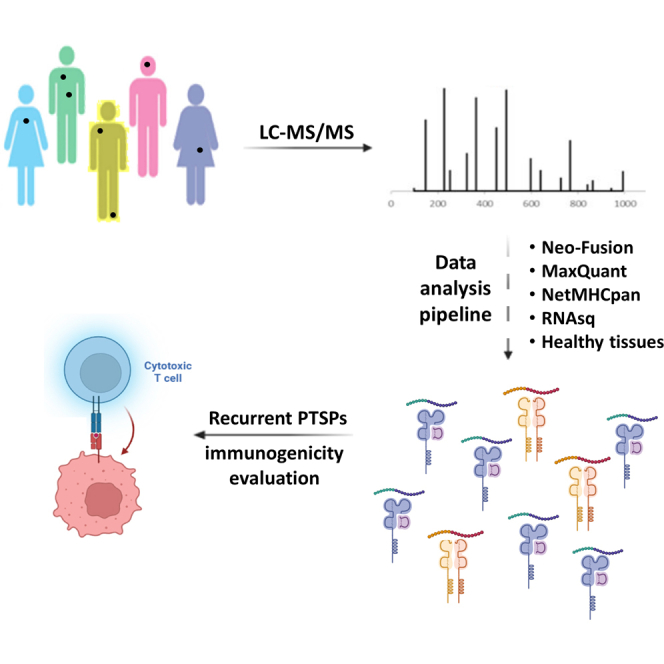
An essential part of any search for PTSPs is the careful filtration of all candidates and the consideration of alternative explanations for their presence in the sample (8). Therefore, our pipeline has been designed to search for as many alternative explanations as possible (both MS-related and biological explanations). The input to the pipeline is MS peptidomics data from human melanoma, breast cancer, and head and neck cancer. This data, together with that of the WT human proteome, served as an input to Neo-Fusion (Fig. 1). As a second prediction check, the candidate PTSPs are then inputted into MaxQuant (29) together with the human proteome. Only PTSPs predicted by both MaxQuant and Neo-Fusion are kept (after checking the associated peptide score and PEP (posterior error probability)). Subsequently, the pipeline determines whether these PTSPs are predicted to bind one of the HLA-alleles associated specifically to their sample (using NetMHCpan). As it is possible that insufficient MS2 peptide fragmentation will result in incorrect peptide-spectra matching, the pipeline selects only peptides with sufficient MS2 %Fragmentation coverage. Furthermore, spectra can be ambiguously associated to more than one peptide sequence with different MaxQuant scores. To avoid this type of peptide ambiguity, the difference between the associated scores (termed Delta score) of the predicted sequence and the top alternative sequence is checked. If the associated scores are close enough, then ambiguity exists, and such PTSPs are removed. Importantly, examining aspects related to peptide MS characteristics alone is insufficient. Therefore, alternative biological explanations are investigated (such as pseudogenes, noncoding, and sequences identified from the microbiome in the tumors (34)). As cancer samples contain both somatic and germline mutations, it is possible that a putative PTSP sequence that originates from a mutation rather than splicing would result with the same sequence. These cases are excluded by substituting all AAs along the PTSP sequence with any alternative possible AA. Similarly, leucine and isoleucine have the same mass and are, therefore, indistinguishable in MS. To address this issue, all sequence permutations are generated for each PTSP by replacing leucine to isoleucine (or vice versa). When a certain permutation matches the WT proteome (or translated transcriptome), the original PTSP is removed. Furthermore, RNA-seq data associated to the patients is utilized by the pipeline to eliminate the possibility of peptides sourced from RNA alternative splicing, mutations, short indels, and nonexpressed genes. To focus only on potential cancer-specific PTSPs, the pipeline further compares the PTSPs to those obtained in normal tissues. An important feature of the pipeline is that it takes all passing PTSPs and checks for recurrence in more than one cancer patient. Peptides passing upstream filtration steps in independent samples are more reliable. Lastly, PTSPs are checked for retention time versus hydrophobicity in order to assure the above filtration steps are sufficient. The number of peptides retained after each filtration step are provided in supplemental Fig. S1.
Fig. 1.

Pipeline design. The full list of peptidomic samples derived from cell lines and patients are inputted to Neo-Fusion (separately for HLA class-I and HLA class-II). Uniprot’s WT proteome-containing proteins and isoforms are used as a sequence database. PTSPs identified by Neo-Fusion are subsequently inputted as a sequence database, together with the Uniprot WT proteome, for a MaxQuant search against the same peptidomics samples. The pipeline stringently eliminates possible false-positives based on multiple criteria. A large-scale in-lab peptidomics database is exploited to identify recurrent PTSPs. PTSP, posttranslational spliced peptide.
Evaluation of Spliced versus Unspliced WT Peptides
To assess the importance of each filtration step of the pipeline, PTSP filtration was compared to that of WT. Steep drops in the filtration process are observed in the NetMHCpan, Delta score, and RNA-seq stages (Fig. 2). The NetMHCpan filtration stage leads to a reduction of 27.1% and 30.8% of peptide spectrum matches (PSMs) for HLA class-I and class-II PTSPs, respectively (in contrast to only 14.1% and 14.6%, respectively, in WT that are filtered out). The Delta score stage leads to a reduction of 31.1% and 25.4% of PSMs for HLA class-I and class-II PTSPs, respectively (in contrast to only 3.6% and 0.4%, respectively, for WT). The RNA-seq stage filters out mostly WT PSMs, as the associated peptides are frequently identified within the transcript sequences generated from the RNA-seq data. However, only a limited fraction of PTSPs is identified in these transcripts (a reduction of 5.1% and 0.7% of HLA class-I and class-II PTSPs versus 43.9% and 37.6% of the corresponding WT PSMs). This indicates that in most cases, the PTSPs identified do not originate from RNA splicing or mutations observed in the RNA-seq data. Importantly, when each filtration step is examined individually (rather than cumulatively in the pipeline), the Leu/Ile replacement filtration yield the greatest fold difference between PTSPs and WT peptides (Table 1). A 31.9-fold and 44.3-fold difference was observed for HLA class-I and class-II, respectively. Therefore, identifying Leu/Ile ambiguity plays an important role in eliminating falsely discovered PTSPs. Altogether, these stark differences emphasize the great importance of checking alternative explanations when attempting to predict PTSPs. Another interesting thing to note is that filtration based on normal samples discards peptides in a biased manner (6.72-fold and 11.13-fold difference between WT and PTSPs, for HLA class-I and class-II, respectively). This may imply that PTSPs identified in cancer cells have a tendency to be cancer-specific. The cumulative pipeline filtration of PSMs associated with PTSPs versus WT peptides indicates that HLA class-I and class-II PSMs stop declining significantly following the Delta score filtration step (Fig. 2). This demonstrates that the final filtration steps mainly remove PSMs that have already been discarded in the previous steps. In addition, the retention time versus hydrophobicity plot does not show greater peptide-dispersal of PTSPs relative to WT peptides (supplemental Fig. S2 is shown prior to the final filtration based on the retention time). Retention time steps serves as a very weak filter at this final stage, and all peptides following this stage remain recurrent. The full lists of HLA class-I and class-II PTSPs prior to the filtration are available in supplemental Tables S6 and S7 and in supplemental Tables S8 and S9 for the WT peptides.
Fig. 2.

Comparison of PTSP versus WT cumulative filtration of PSMs in the pipeline. Both HLA class-I and class-II show a significant decrease in the NetMHCpan and Delta score filtration steps for PTSPs (dark-blue and light-blue lines, respectively). By contrast, WT peptides decrease only mildly (dashed lines). This is due to the significant peptide ambiguity observed only in PTSPs. Furthermore, filtration based on RNA-seq results in a large reduction mainly in WT peptides (consistently in class-I and class-II). This demonstrates that most of the peptides identified in the RNA-seq transcripts are WT (and not, e.g., PTSPs as a result of splice-junctions). HLA class-I and class-II stop declining significantly following the Delta score filtration step. This demonstrates that the final filtration steps remove mainly peptides already removed in previous steps. PSM, peptide spectrum match; PTSP, posttranslational spliced peptide.
Table 1.
The impact of each filtration feature separately (PSM-filtration for PTSP versus WT)
| Feature | HLA class-I |
HLA class-II |
||||
|---|---|---|---|---|---|---|
| %PTSP/%WT | %PTSP filtered | %WT filtered | %PTSP/%WT | %PTSP filtered | %WT filtered | |
| Score (MaxQuant) | 0.50 | 1.77 | 3.54 | 1.13 | 30.86 | 27.42 |
| PEP (MaxQuant) | 0.80 | 13.73 | 17.13 | 1.24 | 20.41 | 16.49 |
| NetMHCpan | 1.86 | 32.09 | 17.22 | 2.08 | 50.38 | 24.22 |
| MS2 %Fragmentation coverage | 0.63 | 1.41 | 2.25 | 1.01 | 14.09 | 13.90 |
| Delta score | 7.45 | 55.64 | 7.46 | 18.10 | 76.85 | 4.25 |
| Pseudogene | 0.01 | 0.07 | 10.99 | 0.00 | 0.00 | 8.89 |
| Noncoding | 0.03 | 0.13 | 3.71 | 0.00 | 0.00 | 2.26 |
| SNP/Mutation | 1.21 | 12.73 | 10.53 | 1.18 | 11.69 | 9.88 |
| Leu/Ile permutations | 31.88 | 14.99 | 0.47 | 44.26 | 14.05 | 0.32 |
| 1-amino acid splicing | - | 12.49 | 0.00 | - | 17.39 | 0.00 |
| RNA-seq | 0.20 | 15.63 | 77.09 | 0.33 | 27.67 | 84.58 |
| Normal tissue | 0.15 | 6.45 | 43.34 | 0.09 | 2.03 | 22.59 |
| Microbiome | - | 0.00 | 0.00 | - | 0.01 | 0.00 |
| Patient recurrence | 0.74 | 19.47 | 26.44 | 0.41 | 21.52 | 52.87 |
| Retention Time outliers | 1.13 | 2.21 | 1.95 | 1.65 | 3.97 | 2.40 |
Spliced Peptides Capture Less Than 3% of the Immunopeptidome Repertoire
To estimate the proportion of PTSPs in the immunopeptidome, we started by applying our pipeline to the HLA class-I and class-II groups separately. We found that, on average, PTSPs account for less than 3.1% and 0.5% of the peptides in HLA class-I and class-II samples, respectively (supplemental Fig. S3). As the proportion of HLA class-II PTSPs has not been published before, the later value cannot be compared. However, the estimated proportion for HLA class-I is in contradiction with the results published in (7), where one-third of the peptides were identified as spliced. They are, however, similar to those of Mylonas et al. (8), who estimated the HLA class-I PTSP proportion to be between 2 and 6%. In addition, our results are consistent with those obtained by a deep learning approach that predicted that PTSPs represent 3% (41).
Splicing Occurs in Different Cancer Types
To check the extent to which splicing is unique to specific cancer types, we compared the filtered peptides, after classifying them by cancer type (breast cancer, head and neck squamous carcinoma, and melanoma). PTSPs were identified in all these cancer types (supplemental Fig. S4).
Splicing Recurs Across Different Patients and Across Metastases of the Same Patient
Recurrent PTSPs, i.e., those identified in mora than one patient, enhance the reliability of the prediction and can serve as important clinical targets. Interestingly, our search led to the identification of 143 and 10 HLA class-I and class-II recurrent PTSPs, respectively (supplemental Tables S6 and S7). Some peptides have been found to recurrently bind to different HLA-alleles. For example, peptide YIAELTQQL is identified recurrently in samples from breast and skin cancer (MCF7 and 92C3, respectively). The peptide is predicted to strongly bind two different HLA-alleles (HLA-A∗02:01 and HLA-C∗05:01) associated with both samples. Notably, HLA-restriction (same HLA allele binding) was not set as a requirement in this study as peptide binding to HLA is promiscuous and does not always follow a one-to-one relation. Nevertheless, we found that 82.8% and 40.0% of HLA class-I and class-II PTSPs, respectively, are recurrent in two or more samples, when considering HLA-restriction, out of the total number of recurrent HLA class-I and class-II PTSPs. It should be noted that HLA class-II PTSPs may show lower recurrence based on allele-restriction due the smaller number of samples available, the lower prediction accuracy for HLA class-II binding, and the larger repertoire of HLA-alleles available.
Splicing Shows Recurrence Across Tumor Single-Cell Clones
To evaluate the clinical value of spliced peptides, it is important to assess the repeated presence of the same spliced peptides not only in different patients but also in different single-cell clones derived from a tumor. We, therefore, reanalyzed our recently published mouse single-cell clone immunopeptidome dataset (40), obtained from the same parental mouse tumor. Eight spliced peptides were identified following peptide filtration in the parental tumor. These spliced peptides were examined in 16 single-cell clones, for which immunopeptidome data was available. As the single-cell clones originate from the same tumor, MaxQuant’s “match-between-runs” property was enabled (allowing MS1-peak identifications based on mass matching in the same retention time window). Spliced peptides were found repeatedly in a large number of single-cell clones (between 5 and 15 single-cell clones when including mass matching and between 4 and 14 clones based on MS/MS hits; supplemental Table S10). For comparison, the search in the peptidomics dataset for somatic point-mutation–derived neoantigens identified in the parental tumor led to the identification of only two peptides. One of these two peptides was identified only once and the other six times in the single-cell clones. Moreover, these somatic point-mutation–derived peptides are not detected in the parental peptidomics itself, possibly due to the dilution (and, consequently, low abundance) of neoantigens associated with private/subclonal somatic mutations in a heterogeneous tumor.
HLA Class-I–Spliced Peptides Are Identified in Cancer and Immune-Associated Genes
Cancer and immune-associated genes were identified in our list of predictions. Interestingly, one of these recurrent PTSPs belongs to IL17RE, a gene encoding for an Interleukin-17 receptor E found to be differentially expressed between responders and nonresponders to anti-PD1 treatment in pretherapy samples (Table 2) (42). Another recurrent PTSP identified is associated to MITF, encoding for the master regulator of melanocyte development and melanoma oncogene (43). In addition, we identified a recurrent PTSP derived from DAPK1, a tumor suppressor gene encoding for the death-associated protein kinase 1 (44, 45). A recurrent PTSP from HLA-E, encoding for the Class-I, E major histocompatibility complex, was identified as well (46, 47). These recurrent peptides were further validated using synthetic peptides.
Table 2.
Human HLA class-I–spliced peptides associated with cancer and immune-related genes that were validated with synthetic peptides
| Spliced peptide (spliced position designated by a hyphen) | Gene | Description | Recurrence (samples; HLA-restricted) | NetMHCpan top HLA (strong/weak binder, rank, score) |
|---|---|---|---|---|
| WSDS-SGGKGGSY | HLA-E | HLA class-I histocompatibility antigen, alpha chain E | 4; 4 | HLA-A∗01:01 (SB 0.055, 0.9481) |
| LLLD-KLLSV | DAPK1 | Death-associated protein kinase 1 (DAP kinase 1) | 4; 2 | HLA-A∗02:01 (SB 0.009, 0.9887) |
| VA-DSSSPEV | PCBP1 | Poly(rC)-binding protein 1 | 12; 5 | HLA-A∗01:01 (WB 1.2066, 0.2203) |
| LASDHHL-YL | TRAPPC10 | Trafficking protein particle complex subunit 10 | 8; 4 | HLA-C∗03:04 (SB 0.020, 0.9274) |
| ATEKEASA-NLY | MCL1 | Induced myeloid leukemia cell differentiation protein | 4; 4 | HLA-A∗01:01 (SB 0.034, 0.9698) |
| DLSDSA-TEV | IL17RE | Interleukin-17 receptor E | 3; 2 | HLA-C∗05:01 (WB 1.742, 0.1255) |
| SFE-GFSPEL | GTF3C3 | General transcription factor 3C polypeptide 3 | 5; 3 | HLA-C∗07:01 (WB 0.980, 0.1435) |
| ES-LPVSGNLIDLY | MITF | Melanoma oncogene; Microphthalmia-associated transcription factor | 2; 2 | HLA-A∗01:01 (SB 0.445, 0.5375) |
| ALWDIETGQQ-SL | GNB2 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | 4; 4 | HLA-A∗02:01 (SB 0.179, 0.7633) |
| LIK-EPVLLL | RPS16 | 40S ribosomal protein S16 | 13; 12 | HLA-A∗02:01 (WB 1.713, 0.1698) |
| VA-FPNEDGSLQK | HEBP1 | Heme-binding protein 1 | 6; 3 | HLA-A∗03:01 (WB 0.985, 0.2563) |
| SSF-QGGGSVTK | LMNA | Prelamin-A/C | 2; 2 | HLA-A∗11:01 (SB 0.104, 0.7651) |
| VSAP-ASGAF | HERPUD1 | Homocysteine-responsive endoplasmic reticulum-resident ubiquitin-like domain member 1 protein | 2; 2 | HLA-B∗15:01 (SB 0.137, 0.7273) |
| EENA-SRNLEY | SHMT1 | Serine hydroxymethyltransferase, cytosolic | 2; 1 | HLA-B∗44:03 (SB 0.082, 0.8619) |
Validation of Spliced Peptides Using Synthetic Peptides
To validate PTSPs, 30 pMHC-I and pMHC-II from human and mouse samples were synthesized as light peptides. Most of the peptides (24 out of 30) yielded significant head-to-tail Pearson correlations of the spectra (p < 0.05; supplemental Table S11). Further manual inspection of the comparisons led to the removal of two additional peptides. Furthermore, WSDSSGGKGGSY PTSP from the HLA-E gene was subjected to isotopically labeled heavy peptide validation (supplemental Fig. S5) and validated in 83T cell line. The head-to-tail plots of all validated peptides are provided in supplemental Fig. S6.
Spliced Peptides can Potentially Elicit an Immune Response
To evaluate the relevance of spliced peptides to the tumor immune response, we examined the immunogenicity of half (7 out of 14) of the HLA class-I–validated PTSPs. We started by assessing 4-1BB expression following peptide stimulation of TILs, as the PTSPs were identified in patient-derived cell lines for which we have autologous TILs. A low though significant increase in 4-1BB expression was detected in TILs stimulated with the LLLDKLLSV peptide derived from the DAPK1 gene and the WSDSSGGKGGSY peptide derived from the HLA-E gene (p = 0.001 and p = 5.7e-5 respectively, χ2-test; supplemental Figs. S7 and S8). A slightly higher 4-1BB upregulation was observed for the positive control peptide, NRAS Q61K, which was identified as a reactive peptide that was recognized by TILs derived from several patients (3.08% of CD8+ T cells versus 2.46% and 2.83% for DAPK1 and HLA-E, respectively) (48). The low signal obtained in 2 of 7 PTSPs may reflect lack of cancer-specificity of PTSPs. In such cases, immunogenicity is expected to be weaker than neoantigens (similar to TAAs) or not exist at all for other PTSPs. It should be noted however that TILs’ reactivity may be restricted to a limited number of tumor peptides due to immune-editing, ineffective priming, or tolerization of these T cells (49). Conversely, T cells derived from healthy donors’ PBMCs represent a diverse repertoire that can recognize a wider range of peptides presented on human tumors (49). Therefore, to further examine the immunogenicity of DAPK1 and HLA-E peptides, we tested their ability to activate CD8 T cells derived from healthy donors. Autologous monocyte-derived dendritic cells isolated from healthy donor PBMCs were pulsed with either DAPK1 or HLA-E peptides or a mixture of viral peptides as a positive control and cocultured with cognate T cells. A flow cytometry analysis of stimulated T cells revealed the presence of reactive T cells against DAPK1- and HLA-E–derived peptides (Fig. 3, supplemental Figs. S9 and S10). An increase was detected in three different activation markers: 4-1BB expression IFN-γ and TNF-α secretion for CD8 T cells stimulated with either the LLLDKLLSV peptide (DAPK1) or the WSDSSGGKGGSY peptide (HLA-E), compared to the control. Moreover, PBMCs educated toward DAPK1 demonstrated peptide-specific CD8 T cells expansion in dextramer staining assay, with 2.57% of the CD8 T cells stained positive for DAPK1 dextramer. CD8 T cells exposed to a control peptide did not exhibit any such positive staining for the DAPK1 dextramer, as expected (Fig. 4). It should be noted that both the LLLDKLLSV and the WSDSSGGKGGSY peptides were found recurrently across four cancer patients. They were further identified in 6 and 16 replicates/treatments for LLLDKLLSV and WSDSSGGKGGSY, respectively. When focusing on the specific ‘DAPK1/HLA-A∗02:01’ and ‘HLA-E/HLA-A∗01:01’ combinations, they recurred in 11.8% and 23.5%, respectively of the melanoma samples analyzed in this study.
Fig. 3.

DAPK1- and HLA-E–derived peptides show potential immunogenicity. T cells derived from healthy donor PBMCs were stimulated and expanded for 24 days. Following coculturing with human B-LCL 721.221 expressing HLA-A∗02:01/HLA-A∗01:01 alleles that were pulsed with 10 μl/ml peptide (right) or no peptide (control; left), T cells were evaluated for activation markers by flow cytometry analysis. IFN-γ, TNF-α secretion, and 4-1BB expression by T cells stimulated with (A) DAPK1 (LLLDKLLSV) or (B) HLA-E (WSDSSGGKGGSY). The images are representative of ≥2 replicates. PBMC, peripheral blood mononuclear cell.
Fig. 4.

HLA-I dextramer staining of DAPK1-specific CD8 T cells. T cells derived from healthy donor PBMCs were stimulated with 721.221 B-LCLs expressing the HLA-A∗02:01 allele pulsed with 10 μg/ml DAPK1 or 10 μg/ml viral peptide (control). Following expansion, T cells were stained with DAPK1 dextramers to evaluate the percentage of DAPK1-specific subpopulations. CD8 T cells were gated as single, live, CD3 positive and CD8 positive cells. The images are representative of two replicates. PBMC, peripheral blood mononuclear cell.
Discussion
Splicing can occur on both the transcriptional and posttranslational level. Posttranslational peptide splicing is a novel peptide-producing mechanism, one that has been suggested to rely on the proteasome and involve the linkage of fragments originally distant in the parental protein (50), although other mechanisms may derive these spliced products (51). Until recently, the lack of dedicated tools suited for peptidomics splicing predictions made it difficult to accurately assess the true prevalence of PTSPs. In this study, we integrated Neo-Fusion with MaxQuant to establish a novel pipeline suited for the stringent filtration of PTSP candidates. This pipeline is based on multiple criteria and considers alternative explanations for the MS/MS matches. This is the first study to rely on a large-scale immunopeptidomic database and search for recurrence in patient-derived cell lines, metastases, and mouse tumor single-cell clones. An identified recurrent PTSP has a greater chance of being a true-positive and would have greater clinical relevance for a wider number of patients and cancer types in case of cancer-specific PTSPs. It should be noted, however, that our study does not attempt to suggest cancer-specificity for our list of predicted PTSPs. This study is the first to validate a PTSP (i.e., a PTSP from HLA-E) not only with a light peptide but also with a heavy peptide, an approach proposed by (12) to be a convincing validation. In addition, this study identifies the immunereactivity of two recurrent PTSPs, one from DAPK1, a tumor suppressor gene, and one from HLA-E, a nonclassical HLA class-I gene.
Although the first spliced peptides were discovered already in 2004, the extent to which they contribute to the immunopeptidome repertoire is still heavily debated (9). At first, they were thought to be very rare curious oddities (encompassing only 0.0002–0.01% of the immunopeptidome) (22). Later, it was suggested that they account for a substantial fraction—30% (7), but a subsequent study estimated that they comprise only 2 to 6% (8). Out of the broad range of reported PTSP frequencies, our calculated percentage is in consensus with the one published by (8), although we utilized an independent computational tool for the detection of PTSPs. Despite the low percentage, the absolute number of spliced peptides is large relative to the very low proportions of neoantigens discovered based on several thousand somatic mutations (1). Therefore, PTSPs widen the pool of presented peptides that can be used to search for immune-reactive targets.
The approach implemented in this study was to filter out the PTSPs in as many ways as possible, so that downstream analyses and conclusions will be based on the most reliable PTSPs. In doing so, we did not only minimize the chances of false-positives but also discarded, to some extent, possible true spliced peptides (e.g., true PTSPs that are not recurrent). However, it is important to note that the low proportion of spliced peptides identified is obtained following attempts to filter out also the WT peptides in a similar degree of stringency. Therefore, the low estimated percentage of PTSPs out of the total filtered peptides is not due to the rigorous approach taken in this study. It should be acknowledged that these values serve as estimations, as omics-data tend to be incomplete and PTSPs may be missed. In addition, the proportion of PTSPs show variability between the different samples analyzed, which can lead to different estimations. Interestingly, the filtrations applied in this study stop declining significantly in the final steps for PTSPs, indicating that most of the peptides were already removed by the previous filtration criteria. By contrast, WT peptides are being filtered almost entirely.
Among many filtering steps applied using our pipeline, the Leu/Ile replacement filtration feature shows the greatest fold difference between PTSPs and WT peptides (14.99% of PTSPs versus only 0.47% of WT peptides; a 31.9-fold difference). This emphasizes the high importance of checking alternative explanations when attempting to predict PTSPs. The importance of this study goes beyond the question what is the true proportion of PTSPs. It suggests to the scientific community key strategies for analyzing and filtering MS data for noncanonical peptides in general. First, we avoid generating a large artificial sequence database for processing MS data, as this naive approach significantly propagates false peptide identifications. Second, we propose a series of filters and identify the most valuable filtration steps, by performing an in-depth comparison to WT peptide filtration. Third, we argue that filtering based on MS characteristics alone followed by validation of the correct sequence using synthetic peptides cannot serve as a proof that a noncanonical peptide is indeed generated by the suggested biological mechanism. Instead, we search for alternative biological explanations such as peptides originating from pseudogenes, noncoding regions, and the microbiome.
Our finding that the normal samples used in this study filter out peptides in a biased manner (6.45% of PTSPSs versus 43.34% of WT peptides in HLA class-I, and 2.03% of PTSPs versus 22.59% of WT peptides in HLA class-II) raises the question whether posttranslational splicing occurs more frequently in cancer cells. Importantly, due to the lack of matching normal samples for our patient data, this type of analysis cannot determine the cancer-specificity (or the lack thereof) of any given peptide in our list.
The vast majority of identified neoantigens based on somatic mutations in immunopeptidomics are derived from patient-specific mutations, deeming such cases of limited clinical importance. Yet, the identification of neoantigens from cancer-associated targets with a recurrent driver mutation, such as the one recently published for NRAS (48) (presented also in our reactivity experiments), would be of importance to the 3% of melanoma patients, having a Q61K clonal mutation and HLA-A∗01:01 allele. However, such neoantigens are rarely identified in immunopeptidomics data and are limited in number. The search for recurrent PTSPs in cancer-associated genes may reveal additional important candidates, whose short sequence could later be incorporated into mRNA vaccines or other treatment strategies. Our finding that splicing occurs repeatedly in single-cell mouse clones derived from the same tumor, combined with the discovery that PTSPs often recur in different tumors/cell lines, suggest that the PTSPs form a valuable pool of potential targets for immunotherapy. It should be noted, however, that a larger number of PTSPs relative to neoantigens does not imply that they are more important immunologically, as some of them may show weaker immunereactivity than neoantigens (or may not be cancer-specific at all). Nevertheless, a larger pool of recurrent PTSPs can enrich the repertoire of presented peptides available for the search for recurrent immune-reactive peptides.
Notably, although the same PTSP may bind more than one specific HLA-allele with different affinities, HLA-restriction can be used to rank top PTSP-candidates. Indeed, we find that most recurrent HLA class-I PTSPs are recurrent also when adding HLA-restriction. Strikingly, our search for HLA-restriction for DAPK1 revealed that the associated PTSP in combination with HLA-A∗02:01 allele recurs in 11.8% of melanoma patients. A similar analysis of the PTSP from HLA-E in combination with the HLA-A∗01:01 allele found it to be relevant for 23.5% of the patients.
Until now, the only examples of spliced CD4+ T-cell epitopes come from the study of the immunology of type 1 diabetes (21, 22, 23). Our study identifies, for the first time, 10 recurrent HLA class-II PTSPs in cancer cells. The identification of a smaller number of HLA class-II PTSPs than HLA class-I PTSPs raises questions concerning the source of the difference between these two types. Future experimental studies are also needed to elucidate the mechanism by which spliced peptides presented to CD4+ T cells by HLA class-II complexes are formed in cancer. There are several described pathways by which endogenous proteins are degraded and gain access to HLA class-II molecules. These include both nonautophagic and autophagic pathways; the first category includes also HLA class-I–like pathways that hijack the HLA class-I machinery (52). In addition, the proteasome may be involved in the autophagic pathway, as proteasomes have been identified as autophagic degradation targets via proteaphagy (53). Other pathways include cancer-induced proteases involved in protein splicing, lysosomal proteases (51), and autophagy (54). The notably lower percentage of PTSPs from HLA class-II compared to HLA class-I may also be explained by a limited number of peptides passing all the pipeline’s filtration steps. Though the sequences of these peptides have been validated, it is possible that true sequences can originate from other mechanisms not considered in this study.
The question whether trans-spliced peptides exist (which is beyond the scope of this study) challenges current bioinformatics tools. Posttranslational sequence databases cannot take into account all the combinatorial possibilities of splices between all the pairs of proteins in the full proteome. It should be noted, however, that Neo-Fusion does not require a posttranslational sequence database. It is, therefore, capable of predicting trans-splicing, similarly to cis-splicing, but with a lower prediction accuracy. Future experimental work is essential in such cases to support the existence of trans-spliced peptides and to determine whether other biological mechanisms can explain trans-splicing.
Data Availability
The mass spectrometry peptidomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (55) partner repository; the dataset identifier is PXD034788.
RNA-seq data have been uploaded to ENA (European Nucleotide Archive) project accession PRJEB53723.
Code for programs are available at: https://github.com/bioinf-dev/Levy_et_al_MCP_2022/blob/main/README.md.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of the article.
Acknowledgments
We would like to thank Moshe Elkabets from Ben-Gurion University, Israel, for sharing the tissue of SCC47.
Funding and additional information
Y. S. is supported by the Israel Science Foundation (grant no. 696/17), European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 770854), MRA (no. 917324), Minerva, Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics, Estate of Alice Schwarz-Gardos, Estate of John Hunter, Knell Family, and Peter and Patricia Gruber Award.
Author contributions
R. L. conceptualization and computational analyses; T. A. R., and N. G. main experiments; Y. S. supervision; R. L., T. A. R., and Y. S. writing–original draft; S. C. S., M. D.-R., O. B. experimental support; T. A. R., N. G, W. P., M. A., M. D. S., Y. L., Y. M., and N. T. experimental validation.
Supplemental Data
Number of spliced peptides retained after each filtration step. The number of spliced peptides that passed each filtration step is indicated for HLA-class-I (blue), and for HLA-class-II (red).
Spliced peptide predictions show a high hydrophobicity versus retention-time correlation for HLA-class-I and -II. A hydrophobicity versus retention-time linear model for spliced (blue circles) and WT (light-purple circles) peptides reveals a similar distribution, in both the HLA-class-I (left panel) and HLA-class-II (right panel) datasets. All the peptides presented (WT and spliced) are those that remained following the pipeline filtration, prior to the final step of retention-time filtration.
Validation of the endogenous HLA-E peptide using an isotopically-labelled peptide. Overlaid extracted ion chromatograms for both endogenous and heavy-peptide spike-in WSDSSGGKGGSY peptides from HLA-E source protein, as identified in a targeted immunopeptidomics of a 83T melanoma cell line. Images produced using Skyline software.
Head-to-tail plots of each peptide validated with synthetic light peptides.
Supplemental Figure S3.

Proportion of Spliced HLA-presented peptides in HLA-class-I and -II. The fraction of HLA-class-I spliced peptides detected (3.1%) is much smaller than that previously detected by Liepe et al. (Science 2016) (30% of HLA-class-I immunopeptidome). Both HLA-class-I and -II spliced peptides are identified, but to different extents.
Supplemental Figure S4.

Peptide splicing occurs across multiple cancer types. Melanoma, breast and head and neck squamous cancer cells all present spliced peptides to the immune system.
Supplemental Figure S7.

TIL reactivity toward spliced peptides. 4-1BB expression was detected in TILs following 24 h stimulation with 1 μg/ml DAPK1 (LLLDKLLSV), HLA-E (WSDSSGGKGGSY), NRAS Q61K (ILDTAGKEEY) or dimethyl sulfoxide (control), using flow cytometry analysis. T cell reactivity was validated by ≥2 independent experiments.
Supplemental Figure S8.

Flow cytometry gating strategy of reactive CD8 T cells derived from TILs.
Supplemental Figure S9.

Flow cytometry gating strategy of reactive CD8 T cells derived from healthy donor PBMCs.
Supplemental Figure S10.

CD8 T cells derived from PBMCs demonstrated potential immunogenicity against a mixture of viral peptides and served as a positive control (A) IFN-γ (B) TNF-α (C) 4-1BB. CD8 T cell reactivity was analyzed by flow cytometry. The images are representative of ≥2 replicates.
References
- 1.Kalaora S., Barnea E., Merhavi-Shoham E., Qutob N., Teer J.K., Shimony N., et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget. 2016;7:5110–5117. doi: 10.18632/oncotarget.6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dao T., Klatt M.G., Korontsvit T., Mun S.S., Guzman S., Mattar M., et al. Impact of tumor heterogeneity and microenvironment in identifying neoantigens in a patient with ovarian cancer. Cancer Immunol. Immunother. 2021;70:1189–1202. doi: 10.1007/s00262-020-02764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faridi P., Woods K., Ostrouska S., Deceneux C., Aranha R., Duscharla D., et al. Spliced peptides and cytokine-driven changes in the immunopeptidome of melanoma. Cancer Immunol. Res. 2020;8:1322–1334. doi: 10.1158/2326-6066.CIR-19-0894. [DOI] [PubMed] [Google Scholar]
- 4.Bartok O., Pataskar A., Nagel R., Laos M., Goldfarb E., Hayoun D., et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature. 2021;590:332–337. doi: 10.1038/s41586-020-03054-1. [DOI] [PubMed] [Google Scholar]
- 5.Vigneron N., Stroobant V., Chapiro J., Ooms A., Degiovanni G., Morel S., et al. An antigenic peptide produced by peptide splicing in the proteasome. Science. 2004;304:587–590. doi: 10.1126/science.1095522. [DOI] [PubMed] [Google Scholar]
- 6.Kacen A., Javitt A., Kramer M.P., Morgenstern D., Tsaban T., Shmueli M.D., et al. Post-translational modifications reshape the antigenic landscape of the MHC I immunopeptidome in tumors. Nat. Biotechnol. 2023;41:239–251. doi: 10.1038/s41587-022-01464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liepe J., Marino F., Sidney J., Jeko A., Bunting D.E., Sette A., et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–358. doi: 10.1126/science.aaf4384. [DOI] [PubMed] [Google Scholar]
- 8.Mylonas R., Beer I., Iseli C., Chong C., Pak H.S., Gfeller D., et al. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell. Proteomics. 2018;17:2347–2357. doi: 10.1074/mcp.RA118.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rolfs Z., Muller M., Shortreed M.R., Smith L.M., Bassani-Sternberg M. Comment on “A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands”. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aaw1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faridi P., Li C., Ramarathinam S.H., Illing P.T., Mifsud N.A., Ayala R., et al. Response to comment on “A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands”. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aaw8457. [DOI] [PubMed] [Google Scholar]
- 11.Mishto M. Commentary: are there indeed spliced peptides in the immunopeptidome? Mol. Cell. Proteomics. 2021;20:100158. doi: 10.1016/j.mcpro.2021.100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Admon A. Are there indeed spliced peptides in the immunopeptidome? Mol. Cell. Proteomics. 2021;20:100099. doi: 10.1016/j.mcpro.2021.100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purcell A.W. Is the immunopeptidome getting darker?: a commentary on the discussion around Mishto et al., 2019. Front. Immunol. 2021;12:720811. doi: 10.3389/fimmu.2021.720811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faridi P., Li C., Ramarathinam S.H., Vivian J.P., Illing P.T., Mifsud N.A., et al. A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aar3947. [DOI] [PubMed] [Google Scholar]
- 15.Berkers C.R., de Jong A., Schuurman K.G., Linnemann C., Meiring H.D., Janssen L., et al. Definition of proteasomal peptide splicing rules for high-efficiency spliced peptide presentation by MHC class I molecules. J. Immunol. 2015;195:4085–4095. doi: 10.4049/jimmunol.1402455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalet A., Robbins P.F., Stroobant V., Vigneron N., Li Y.F., El-Gamil M., et al. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl. Acad. Sci. U. S. A. 2011;108:E323–E331. doi: 10.1073/pnas.1101892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebstein F., Textoris-Taube K., Keller C., Golnik R., Vigneron N., Van den Eynde B.J., et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci. Rep. 2016;6:24032. doi: 10.1038/srep24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warren E.H., Vigneron N.J., Gavin M.A., Coulie P.G., Stroobant V., Dalet A., et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science. 2006;313:1444–1447. doi: 10.1126/science.1130660. [DOI] [PubMed] [Google Scholar]
- 19.Platteel A.C.M., Liepe J., Textoris-Taube K., Keller C., Henklein P., Schalkwijk H.H., et al. Multi-level strategy for identifying proteasome-catalyzed spliced epitopes targeted by CD8(+) T cells during bacterial infection. Cell Rep. 2017;20:1242–1253. doi: 10.1016/j.celrep.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 20.Paes W., Leonov G., Partridge T., Nicastri A., Ternette N., Borrow P. Elucidation of the signatures of proteasome-catalyzed peptide splicing. Front. Immunol. 2020;11:563800. doi: 10.3389/fimmu.2020.563800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mannering S.I., So M., Elso C.M., Kay T.W.H. Shuffling peptides to create T-cell epitopes: does the immune system play cards? Immunol. Cell Biol. 2018;96:34–40. doi: 10.1111/imcb.1015. [DOI] [PubMed] [Google Scholar]
- 22.Delong T., Wiles T.A., Baker R.L., Bradley B., Barbour G., Reisdorph R., et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. 2016;351:711–714. doi: 10.1126/science.aad2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran M.T., Faridi P., Lim J.J., Ting Y.T., Onwukwe G., Bhattacharjee P., et al. T cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8. Nat. Commun. 2021;12:5110. doi: 10.1038/s41467-021-25404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolfs Z., Solntsev S.K., Shortreed M.R., Frey B.L., Smith L.M. Global identification of post-translationally spliced peptides with neo-fusion. J. Proteome Res. 2019;18:349–358. doi: 10.1021/acs.jproteome.8b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reuben A., Spencer C.N., Prieto P.A., Gopalakrishnan V., Reddy S.M., Miller J.P., et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017;2:10. doi: 10.1038/s41525-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalaora S., Wolf Y., Feferman T., Barnea E., Greenstein E., Reshef D., et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 2018;8:1366–1375. doi: 10.1158/2159-8290.CD-17-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komov L., Kadosh D.M., Barnea E., Milner E., Hendler A., Admon A. Cell surface MHC class I expression is limited by the availability of peptide-receptive “empty” molecules rather than by the supply of peptide ligands. Proteomics. 2018;18 doi: 10.1002/pmic.201700248. [DOI] [PubMed] [Google Scholar]
- 28.Marcu A., Bichmann L., Kuchenbecker L., Kowalewski D.J., Freudenmann L.K., Backert L., et al. HLA ligand atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 30.Li Q., Shortreed M.R., Wenger C.D., Frey B.L., Schaffer L.V., Scalf M., et al. Global post-translational modification discovery. J. Proteome Res. 2017;16:1383–1390. doi: 10.1021/acs.jproteome.6b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., et al. Gencode 2021. Nucleic Acids Res. 2021;49:D916–D923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Kalaora S., Nagler A., Nejman D., Alon M., Barbolin C., Barnea E., et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature. 2021;592:138–143. doi: 10.1038/s41586-021-03368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh S., Chan C.K. Analysis of RNA-seq data using TopHat and Cufflinks. Methods Mol. Biol. 2016;1374:339–361. doi: 10.1007/978-1-4939-3167-5_18. [DOI] [PubMed] [Google Scholar]
- 36.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ali M., Foldvari Z., Giannakopoulou E., Boschen M.L., Stronen E., Yang W., et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019;14:1926–1943. doi: 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]
- 40.Wolf Y., Bartok O., Patkar S., Eli G.B., Cohen S., Litchfield K., et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell. 2019;179:219–235.e21. doi: 10.1016/j.cell.2019.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilhelm M., Zolg D.P., Graber M., Gessulat S., Schmidt T., Schnatbaum K., et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 2021;12:3346. doi: 10.1038/s41467-021-23713-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levy C., Khaled M., Fisher D.E. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 44.Wang Q., Weng S., Sun Y., Lin Y., Zhong W., Kwok H.F., et al. High DAPK1 expression promotes tumor metastasis of gastric cancer. Biology (Basel) 2022;11:1488. doi: 10.3390/biology11101488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin R., Melamed S., Yang B., Saxena M., Sheetz M.P., Wolfenson H. Tumor suppressor DAPK1 catalyzes adhesion assembly on rigid but anoikis on soft matrices. Front. Cell Dev. Biol. 2022;10:959521. doi: 10.3389/fcell.2022.959521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salome B., Sfakianos J.P., Ranti D., Daza J., Bieber C., Charap A., et al. NKG2A and HLA-E define an alternative immune checkpoint axis in bladder cancer. Cancer Cell. 2022;40:1027–1043.e9. doi: 10.1016/j.ccell.2022.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llano M., Lee N., Navarro F., Garcia P., Albar J.P., Geraghty D.E., et al. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur. J. Immunol. 1998;28:2854–2863. doi: 10.1002/(SICI)1521-4141(199809)28:09<2854::AID-IMMU2854>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 48.Peri A., Greenstein E., Alon M., Pai J.A., Dingjan T., Reich-Zeliger S., et al. Combined presentation and immunogenicity analysis reveals a recurrent RAS.Q61K neoantigen in melanoma. J. Clin. Invest. 2021;131 doi: 10.1172/JCI129466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stronen E., Toebes M., Kelderman S., van Buuren M.M., Yang W., van Rooij N., et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 50.Michaux A., Larrieu P., Stroobant V., Fonteneau J.F., Jotereau F., Van den Eynde B.J., et al. A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J. Immunol. 2014;192:1962–1971. doi: 10.4049/jimmunol.1302032. [DOI] [PubMed] [Google Scholar]
- 51.Reed B., Crawford F., Hill R.C., Jin N., White J., Krovi S.H., et al. Lysosomal cathepsin creates chimeric epitopes for diabetogenic CD4 T cells via transpeptidation. J. Exp. Med. 2021;218 doi: 10.1084/jem.20192135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leung C.S. Endogenous antigen presentation of MHC class II epitopes through non-autophagic pathways. Front. Immunol. 2015;6:464. doi: 10.3389/fimmu.2015.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beese C.J., Brynjolfsdottir S.H., Frankel L.B. Selective autophagy of the protein homeostasis machinery: ribophagy, proteaphagy and ER-phagy. Front. Cell Dev. Biol. 2019;7:373. doi: 10.3389/fcell.2019.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crotzer V.L., Blum J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009;182:3335–3341. doi: 10.4049/jimmunol.0803458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Number of spliced peptides retained after each filtration step. The number of spliced peptides that passed each filtration step is indicated for HLA-class-I (blue), and for HLA-class-II (red).
Spliced peptide predictions show a high hydrophobicity versus retention-time correlation for HLA-class-I and -II. A hydrophobicity versus retention-time linear model for spliced (blue circles) and WT (light-purple circles) peptides reveals a similar distribution, in both the HLA-class-I (left panel) and HLA-class-II (right panel) datasets. All the peptides presented (WT and spliced) are those that remained following the pipeline filtration, prior to the final step of retention-time filtration.
Validation of the endogenous HLA-E peptide using an isotopically-labelled peptide. Overlaid extracted ion chromatograms for both endogenous and heavy-peptide spike-in WSDSSGGKGGSY peptides from HLA-E source protein, as identified in a targeted immunopeptidomics of a 83T melanoma cell line. Images produced using Skyline software.
Head-to-tail plots of each peptide validated with synthetic light peptides.
Data Availability Statement
The mass spectrometry peptidomics data have been deposited in the ProteomeXchange Consortium via the PRIDE (55) partner repository; the dataset identifier is PXD034788.
RNA-seq data have been uploaded to ENA (European Nucleotide Archive) project accession PRJEB53723.
Code for programs are available at: https://github.com/bioinf-dev/Levy_et_al_MCP_2022/blob/main/README.md.