

Abstract
BACKGROUND & AIMS:
Anti-granulocyte macrophage-colony stimulating factor autoantibodies (aGMAbs) are detected in patients with ileal Crohn’s disease (CD). Their induction and mode of action during or before disease are not well understood. We aimed to investigate the underlying mechanisms associated with aGMAb induction, from functional orientation to recognized epitopes, for their impact on intestinal immune homeostasis and use as a predictive biomarker for complicated CD.
METHODS:
We characterized using enzyme-linked immunosorbent assay naturally occurring aGMAbs in longitudinal serum samples from patients archived before the diagnosis of CD (n = 220) as well as from 400 healthy individuals (matched controls) as part of the US Defense Medical Surveillance System. We used biochemical, cellular, and transcriptional analysis to uncover a mechanism that governs the impaired immune balance in CD mucosa after diagnosis.
RESULTS:
Neutralizing aGMAbs were found to be specific for post-translational glycosylation on granulocyte macrophage-colony stimulating factor (GM-CSF), detectable years before diagnosis, and associated with complicated CD at presentation. Glycosylation of GM-CSF was altered in patients with CD, and aGMAb affected myeloid homeostasis and promoted group 1 innate lymphoid cells. Perturbations in immune homeostasis preceded the diagnosis in the serum of patients with CD presenting with aGMAb and were detectable in the noninflamed CD mucosa.
CONCLUSIONS:
Anti-GMAbs predict the diagnosis of complicated CD long before the diagnosis of disease, recognize uniquely glycosylated epitopes, and impair myeloid cell and innate lymphoid cell balance associated with altered intestinal immune homeostasis.
Keywords: Crohn’s Disease, Innate Lymphoid Cells, Macrophages, GM-CSF, Autoantibodies
Graphical Abstract
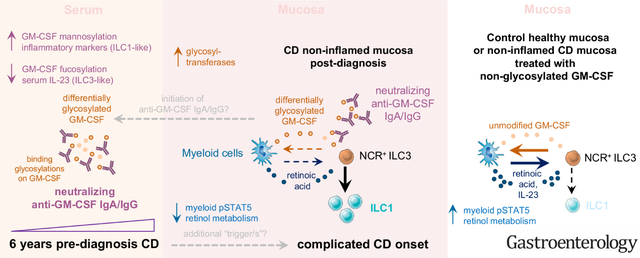
Inflammatory bowel disease (IBD), subclassified into Crohn’s disease (CD) and ulcerative colitis (UC), is an increasingly diagnosed chronic inflammatory pathology of the gastrointestinal tract, affecting 0.3% of the world’s population.1 Although both CD and UC affect the gastrointestinal tract, their causes remain puzzling and are likely multifactorial.2 Genome-wide association studies provide strong support that IBD is a pathology driven by monogenetic and multigenetic variations, but nongenetic and environmental factors are also considered as contributors to the heterogeneity of this disease.3,4 Identifying factors contributing to IBD development that may predict disease onset is, therefore, of high clinical relevance.
A key immunologic characteristic of IBD is the break in intestinal homeostasis, commonly manifested through insufficient barrier integrity, decreased immunologic tolerance, or excessive inflammation. The cytokine granulocyte macrophage-colony stimulating factor (GM-CSF) reportedly plays a dual role in intestinal inflammation and was shown to have both protective and inflammatory properties in CD.5–9 Group 3 innate lymphoid cells (ILC3) produce GM-CSF in the gut, which acts on myeloid immune cells to sustain immune homeostasis.10 Derived from ILC3, GM-CSF promotes antibacterial and immunomodulatory functions in myeloid cells to sustain the transcriptional stability of ILC3 via the metabolite retinoic acid (RA) and the cytokine interleukin (IL) 23.10,11 However, in patients with CD, myeloid cells induce differentiation of ILC3 into inflammatory group 1 ILC (ILC1) via IL12.12–14 ILC3, or GM-CSF deficiency, impairs antimicrobial immunity and mucosal homeostasis aligning with the identification of mutations in GM-CSF signaling in patients with CD.6,10,15 Administration of yeast-produced GM-CSF (sargramostim) improved CD and ameliorated symptoms.5,16 However, larger trials with sargramostim in CD failed to reach statistical significance, possibly due to high placebo group responses and suboptimal study design.17–19
Besides genetic defects in GM-CSF signaling, anti-granulocyte macrophage-colony stimulating factor autoantibodies (aGMAbs) may also contribute to CD in a subset of patients, and they are associated with ileal involvement, disease severity, higher relapse rates, and complications.20–24 Induction of aGMAb is also known to cause another disease, namely pulmonary alveolar proteinosis (PAP), resulting in alveolar macrophage-deficiency and lung pathologies.25 Why patients with PAP do not display intestinal pathologies and wy patients with CD with aGMAb do not show PAP-associated pulmonary symptoms remains unknown. Moreover, both the overproduction and the absence of GM-CSF significantly increase the susceptibility to develop IBD, emphasizing the heterogeneity in CD pathogenesis, prompting us to investigate the biology of aGMAb before diagnosis in CD.5–9,26
Here, we analyzed >1800 sera, collected longitudinally from active component US military personnel over 10 years including subjects who eventually developed CD (n = 220), UC (n = 200), and personnel without CD or UC (healthy donors [HD], n = 200),27 along with controls with established CD or PAP. We analyzed titers, isotype and subclass profiles, epitopes, and functional consequences of aGMAb using enzyme-linked immunosorbent assay, neutralization, and reporter assays, and their effect on immune subsets in noninflamed ileal CD biopsy specimens using RNA sequencing to establish mechanisms of pathology before disease and at its onset that could be exploited for personalized CD therapies.
Methods
Human Specimen
Noninvolved (NI) intestinal resection and involved intestinal resection samples were obtained from patients with CD undergoing ileal resection surgery at the Mount Sinai Medical Center after obtaining informed consent. All protocols were reviewed and approved by the Institutional Review Board (IRB) under IRB 08-1236, IRB HSM 13-00998, NMRC.2012.0007, and NMRC.2014.0019.
Native Polyacrylamide Gel Electrophoresis, Sodium Dodecyl-Sulfate Polyacrylamide Gel Electrophoresis/Western Blot, and Lectin Blot
Recombinant and stripped GM-CSF (sargramostim) (8 μg/well) were separated on 15% resolving native polyacrylamide gels. Gels were stained with Coomassie G Brilliant Blue to confirm stripping. Proteins were then transferred to nitrocellulose membranes and membranes were blocked with 5% nonfat dry milk in Tris-Buffer Saline 0.1% Tween-20 at 4°C. Membranes were then incubated with serum samples (diluted 1:100 in blocking buffer). Bound anti-GM-CSF antibodies were detected using anti-human immunoglobulin (Ig) G alkaline phosphatase at 1:1000 in Tris-Buffer Saline 0.1% Tween-20. The glycoprofile of recombinant forms of GM-CSF was evaluated using lectin blot. Also, 2 μg of purified GM-CSF were separated as previously described and membranes blocked with bovine serum albumine 4%. Phaseolus Vulgaris Leucoagglutinin (L-PHA), Maackia Amurensis Lectin II (MALII), Galanthus Nivalis Lectin (GNA), and Aleuria Aurantia Lectin (AAL) (each 2 μg/mL) were used and bands visualized using the Vectorstain Elite avidin-biotin complex kit and enhanced chemiluminescence reagent. For Western blot analysis, 5 μg of purified GM-CSF separated as described previously and detected using biotinylated GM-CSF antibodies (0.1 μg/mL R&D Systems, Minneapolis, MN). Target bands were visualized as described previously.
Results
CD-Associated aGMAbs are Distinct From Those in PAP and UC
To assess the prevalence and characteristics of aGMAb in IBD, serum IgG titers against sargramostim, a yeast-produced recombinant GM-CSF, were measured in a cohort of patients with active CD (n = 81) or UC (n = 37), HD (n = 43), and patients with PAP (n = 12). Among patients with IBD, 40% of patients with CD and 14% of patients with UC displayed detectable levels of IgG aGMAb (titers >1/100) (Figure 1A). Titers detected in patients with CD but not in patients with UC were significantly higher compared with HD, but lower when compared with patients with PAP (Figure 1A). Only 3 of 53 sera from patients with IBD with aGMAb reacted with other known autoantigens, emphasizing that the vast majority of aGMAb responses were antigen-specific (Supplementary Figure 1A). Sera showing broad nonspecific antigen reactivity were excluded. Even though aGMAb titers differed between patients with PAP and patients with IBD, adjusted titers demonstrated comparable, relative avidity to GM-CSF (Supplementary Figure 1B).
Figure 1.
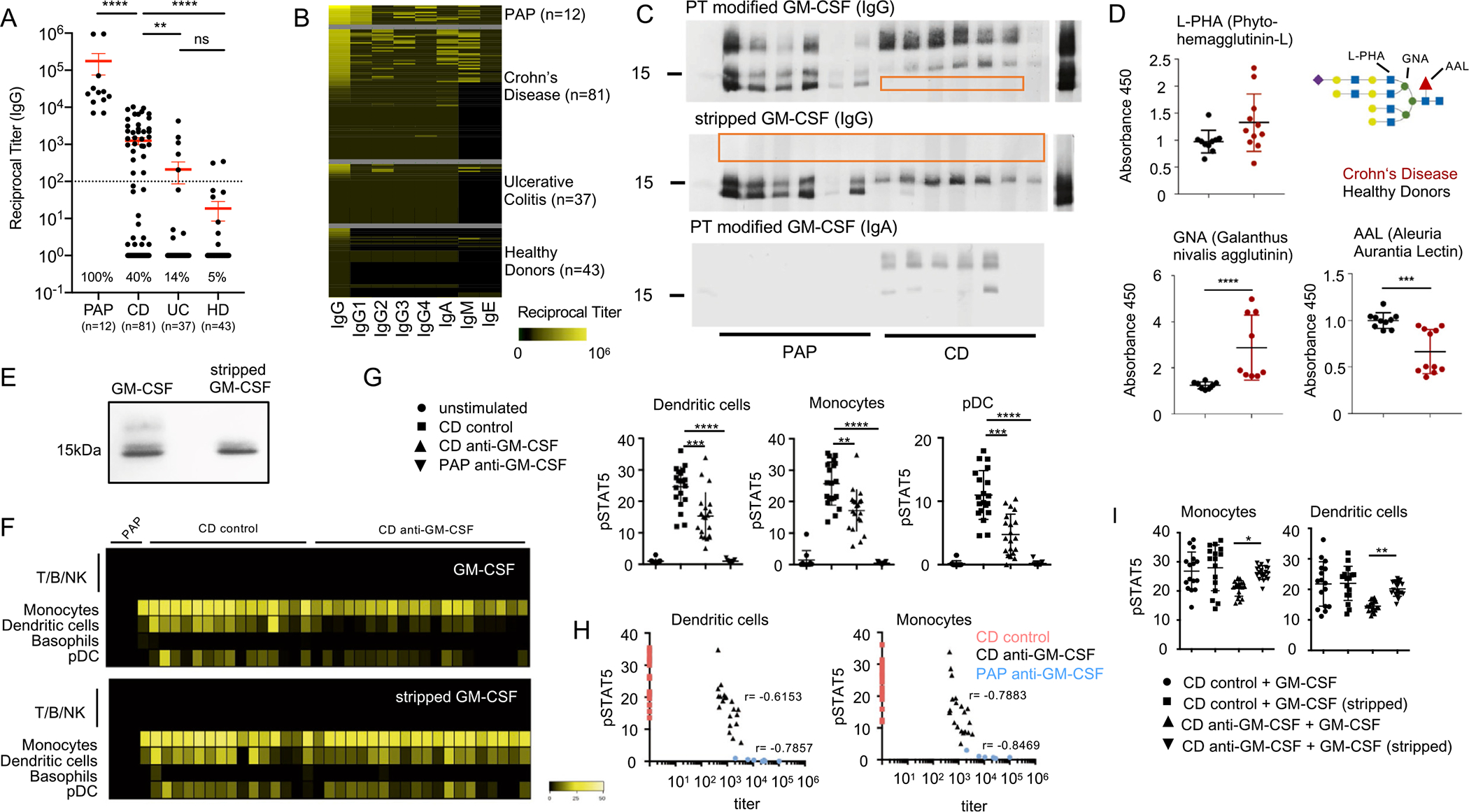
Characterization of aGMAb in patients with CD. (A) Reciprocal titers for total serum IgG aGMAb in sera of HD, patients with PAP, patients with CD, and patients with UC. (B) Isotype profiles of aGMAb in patients with PAP and patients with CD. Horizontal rows represent patients and vertical rows indicate isotypes. (C) Western blots probed with polyclonal sera from patients with CD and PAP show binding of anti-GM-CSF IgG and IgA to glycosylated (post-translationally-modified) and stripped GM-CSF. (D) Levels of GNA, AAL, and L-PHA binding to GM-CSF from HDs (black) and patients with CD (red), normalized for the total levels of GM-CSF in each sample. Schematic representation of N-glycan highlighting lectin recognition. (E) Native polyacrylamide gel electrophoresis of GM-CSF (sargramostim) and stripped GM-CSF stained with Coomassie Brilliant Blue. (F) Heat maps of pSTAT5 signal in peripheral blood mononuclear cells after stimulation with glycosylated (top) or deglycosylated GM-CSF in the presence of PAP serum, aGMAb− CD serum, or aGMAb+ CD serum. Heat maps show signal intensity of anti-pSTAT5 staining in yellow code for individual patients (lanes) within the indicated cell populations identified using mass cytometry (rows). (G) Plots show quantification of pSTAT5 signal in DCs, monocytes, and plasmacytoid DCs either unstimulated or stimulated with GM-CSF preincubated with serum from the indicated patient groups. (H) Loss in pSTAT5 correlates with aGMAb titers. (I) Quantification of pSTAT signal for monocytes and DC shown in (F). One-way analysis of variance (ANOVA) Bonferroni’s multiple comparison test was performed. Mann-Whitney test. *P-value <.05.
We examined the isotypes and IgG subclasses of aGMAb in patients with IBD vs patients with PAP. PAP-associated aGMAbs were enriched in IgG1 and IgG4, isotypes virtually absent in IBD-associated aGMAbs, and were enriched in IgG2 and IgA. IgM and IgG3, but not IgE, were detectable in both PAP and IBD, with higher average IgM aGMAb in patients with CD (Figure 1B). IBD-associated aGMAbs were a specific marker for CD with ileal involvement and complicated disease, confirming previous studies (Supplementary Figure 1C and D and Supplementary Table 1).21–24 Measuring aGMAb thus allows the discrimination of a subgroup of patients with CD among all patients with IBD.
In contrast to previously reported peptide epitopes in PAP, synthetic overlapping 20-mer linear peptides covering GM-CSF failed to react with CD-associated aGMAb (data not shown).25 Suspecting conformational epitopes, binding of aGMAb to denaturated GM-CSF was virtually absent using CD sera (Supplementary Figure 1E). Unlike PAP sera, CD sera did not react with GM-CSF in the absence of post-translational modifications; only PAP sera bound bacterially produced recombinant GM-CSF, whereas CD sera required a eukaryotic GM-CSF product to react (data not shown). Analysis of yeast-produced GM-CSF using gel electrophoresis revealed 3 bands at ~19.5 kDa, ~16.5 kDa, and ~14.5 kDa (Figure 1C). The highest band carried post-translational modifications by glycosylation, lost when enzymatically stripped (Figure 1E). GM-CSF expressed in HEK293 cells lost this band pattern when all glycosylation sites were mutated to alanine (Supplementary Figure 1F). Post-translational modifications of GM-CSF have previously been suggested as ideal antibody-recognition sites.28 We assessed whether sera from patients with CD would react with the different forms of yeast-derived GM-CSF. Whereas PAP sera bound all bands of GM-CSF, CD-associated aGMAb bound larger glycosylated GM-CSF (Figure 1C, Supplementary Figure 1E and F). When sargramostim was enzymatically stripped of glycans, seroreactivity of CD to the 19.5 kDa band was lost but remained to the 16.5 kDa (Figure 1C), whereas PAP sera also recognized the fully deglycosylated 14.5 kDa band of GM-CSF (Figure 1E).25 Similarly to IgG, IgA aGMAb from patients with CD showed high specificity to glycosylation (Figure 1C). We confirmed specificity of CD-associated aGMAb using recombinant human GM-CSF produced in human cells, indicating that reactivity was not exclusive to yeast-specific modifications (data not shown).
To evaluate commonalities in the glycan composition, yeast- and mammalian cell-derived GM-CSF were resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the glycoprofile of GM-CSF assessed via lectin blot (Supplementary Figure 1G). Membranes were probed with the lectins L-PHA, GNA, and AAL, which specifically recognize B1,6GlcNAc branched N-glycans, high-mannose N-glycans, and a1,6/a1,3 fucose, respectively (Supplementary Figure 1I). Mammalian cell-derived GM-CSF displayed a large glycoform reacting positively with L-PHA and AAL implicating the expression of complex branched and fucosylated N-glycans (Supplementary Figure 1I). The 16–17 kDa band of mammalian cell-derived GM-CSF reacted with MALII and GNA, revealing high-mannose N-glycans, and hybrid N-glycans with terminal sialylation (Supplementary Figure 1I). Yeast-derived GM-CSF reacted only with GNA, suggesting high-mannose N-glycans as shared glycol-modification (Supplementary Figure 1I). These results demonstrate that mammalian-derived GM-CSF shares high-mannose N-glycans with yeast-derived GM-CSF and suggest that CD-associated aGMAbs selectively recognize these structures on correctly folded, native GM-CSF. To determine if glycosylations on GM-CSF differed between CD and HD, serum GM-CSF was captured and incubated with lectins. GNA binding was significantly higher on GM-CSF from patients with CD, suggesting an increase in high mannose N-glycans on GM-CSF in CD (Figure 1D). Decreased AAL binding indicated a decrease in the core fucose residues in CD (Figure 1D and Supplementary Figure 2A). The increase in mannose and decrease in the core-fucose structures suggest a CD-specific glycosignature on GM-CSF. The analyzed samples showed comparable levels of serum GM-CSF but no detectable aGMAbs, suggesting that changes in the glycosylation of GM-CSF are no prerequisite for aGMAb formation (Supplementary Figure 2A and B).
Neutralizing Capacity of CD-Associated aGMAb
The enriched abundance of binding of aGMAb to GM-CSF in patients with CD (Supplementary Table 1, Figure 1A and B) supports the hypothesis that aGMAbs have neutralizing capacities20. We next determined whether aGMAbs abrogate granulocyte macrophage-colony stimulating factor receptor (GM-CSFR) signaling in monocytes, dendritic cells (DC), and plasmacytoid DC of healthy controls. Blood leukocytes were stimulated with recombinant human GM-CSF in the presence of PAP-sera (n = 9), CD-sera negative for aGMAb (n = 20), or CD-sera positive for aGMAb (n = 20). Post-stimulation, phosphorylated (p)-STAT5 levels were measured (Supplementary Table 2). Reduced pSTAT5 was observed in the presence of PAP-sera and CD-sera positive for aGMAb (Figure 1F and G, Supplementary Figure 2C). GM-CSF stimulation did not increase pSTAT5 in T/B/natural killer (NK) cells across all groups (Figure 1F). IL3 stimulation of basophils was not affected by aGMAb, suggesting unaltered common beta chain signaling (Supplementary Figure 2D). Decreased pSTAT5 correlated with aGMAb titers, demonstrating neutralizing activity on circulating precursors of intestinal antigen-presenting cells (APCs) (Figure 1F).
Unmodified GM-CSF as a Potential Way to Restore Homeostatic Functions of GM-CSF
Enzymatically stripped and genetically engineered GM-CSF, lacking glycans structures remained biologically active and stimulated pSTAT5 (Supplementary Figure 2E–G). We hypothesize that GM-CSF devoid of glycosylation would render the cytokine capable of escaping neutralization by CD-associated aGMAb. Freshly isolated peripheral blood mononuclear cells stimulated with glycosylated and deglycosylated GM-CSF in the presence or absence of aGMAb were analyzed for pSTAT5. Glycosylated GM-CSF was neutralized by CD-associated aGMAb whereas enzymatically stripped GM-CSF escaped aGMAb recognition (Supplementary Figure 2F and I).
CD-Associated aGMAbs Precede the Onset of Complicated Disease
The presence of neutralizing aGMAbs in patients with CD further suggests that isotypes may contribute to disease pathophysiology and possibly etiology. To address this hypothesis, we tested sera obtained from the Department of Defense Serum Repository, prospectively collected from military service members during their annual routine medical examinations.29 Some of these service members were eventually diagnosed with either UC or CD during the course of their service. Three to four longitudinal serum samples spanning up to 10 years, obtained before diagnosis, at diagnosis, and after diagnosis from 220 CD, 200 UC, and 200 matched individuals remaining healthy (HD), were analyzed in 2 independent runs (Supplementary Table 3). Total IgG and IgA anti-GM-CSF enzyme-linked immunosorbent assays were performed for each time point of collection. Healthy military service members and service members diagnosed with UC had a similar 5%–10% detection rate of anti-GM-CSF IgG, with low mean titers less than the limit of significance (between 1 of 25 and 1 of 50), and nearly no anti-GM-CSF IgA detection (0–1%), without significant change by time point (Figure 2A and B, Supplementary Figure 3A–D). In contrast, IgG and IgA aGMAb were already found 6 years before CD diagnosis in 21% and 7% of samples, with additional patients seroconverting and mean titer increasing from 1 of 190 to 1 of 320 toward the time of diagnosis (Figure 2A–C, Supplementary Figure 3). At the time of diagnosis, IgA aGMAb were exclusively elevated in 12% of CD, whereas IgGs were significantly more frequent (25%) compared with HD and UC (Figure 2A and B, Supplementary Figure 3C). Nearly all patients with CD with detectable aGMAb 6 years before diagnosis maintained or increased their titers until the time of diagnosis (Figure 2C, Supplementary Figure 3E and F). Most (75%) anti-GM-CSF IgA co-occurred with IgG, whereas IgG to GM-CSF was more frequent and was detected in 63% of cases in the absence of IgA.
Figure 2.
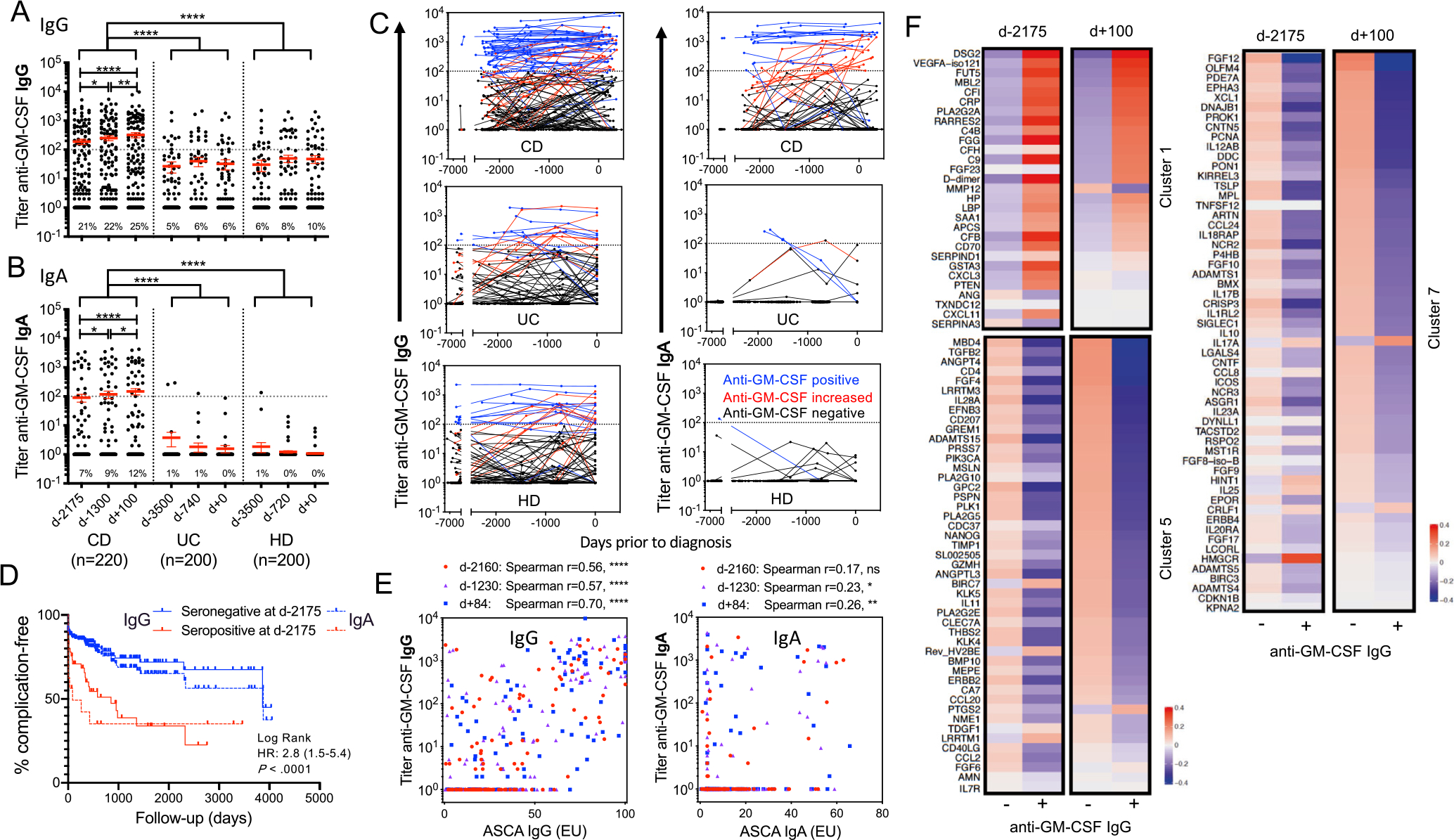
CD-specific aGMAbs precede the onset of disease by years. (A) and (B) show reciprocal titers of aGMAbs (IgG and IgA) in combined serum samples (training and validation cohort) at 2 time points before and 1 time point after diagnosis. (C) Trajectory of aGMAb titers in patients with CD. Blue lines indicate aGMAb+, black lines aGMAb− patients, and red lines indicate sero-converters. (D) Kaplan-Meier analysis with hazard ratio for developing complications after diagnosis in aGMAb+ (red) and aGMAb− patients 6 years before disease. (E) Correlations of aGMAb with anti-saccharomyces cerevisiae antibodies (ASCA) IgG and ASCA IgA antibodies at different time points before diagnosis. (F) Clusters of SOMAmers correlating with aGMAbs before (d-2175) and after (d+100) diagnosis of CD.
Anti-GMAb in CD associated with ileal/ileocolonic involvement and complications within 100 days of diagnosis, with a 2.8 risk hazard ratio of having penetrating and/or stricturing disease or surgery at or soon after clinical diagnosis (Figure 2D, Supplementary Figure 4A–D). Presence of IgA up to 6 years before diagnosis provided a predictor for CD development with >97% specificity and with sensitivity increasing from 15%–21% as time to diagnosis decreased (area under the curve 0.6). The detection of aGMAb did not correlate with date of birth, sex, race, or year of sample acquisition, rendering this biomarker universal across patients (data not shown). Anti-Saccharomyces cerevisiae antibodies, a common serologic marker for IBD, were shown to present before diagnosis using similar cohorts.3,30,31 Anti-GMAb preceded the occurrence of anti-Saccharomyces cerevisiae antibodies IgA and showed no significant correlations 2000 days before diagnosis (Figure 2E, Supplementary Table 4). We next analyzed serologic data from the Somalogics platform to determine if aGMAb correlated with immune-related blood serum markers before diagnosis. Unsupervised clustering by similarity matrix of 1129 circulating analytes in sera collected from 100 patients with CD ~6 years before or around diagnosis revealed 20 distinct clusters with coexpression patterns. Three clusters showed significant mean expression differences depending on the aGMAb status (Figure 2F, Supplementary Figure 4G, Benjamini-adjusted P-value <10%). Markers in cluster 1 were up-regulated in aGMAb+ patients as early as 6 years before diagnosis and remained elevated thereafter. These markers were enriched in inflammation (D-dimer, C-reactive protein), lipid binding protein, opsonization (MBL2, SAA1), plasma cell, the complement cascade (CFB, CFI, CFH, C4B, C9, FGG), and Th1 chemokines (CXCL3 and CXCL11) (Figure 2F). Clusters 5 and 7 contained markers that decreased with aGMAb across both time points. These markers were enriched in cytokines (IL10, IL12AB, IL17B, IL23A), chemokines (CCL2, CCL8, CCL20, CCL24), and fibroblast growth factor signaling implicating an impairment in immune homeostasis. Of note, IL25 and IL17A were exceptions in cluster 7, and were higher with aGMAb. Collectively these findings suggest an altered immune balance in the presence of aGMAb years before the diagnosis of CD, consistent with a shift toward type 1 immunity and the possibility for undetected subclinical inflammation. These results prompted us to next investigate the phenotype and function of GM-CSF–responsive cells in the mucosa of patients with CD.
CD Mucosa Shows Impaired Homeostatic Functions in GM-CSF–Responsive Myeloid Cells
GM-CSF engages the heterodimeric GM-CSFR, composed of the GM-CSF binding alpha chain CD116 (CSF2RA) and the signal transducing common beta chain CD131 (CSF2RB) activating the transcription factor STAT5. To understand impairments in GM-CSFR signaling, we determined CD116 and CD131 expression across most hematopoietic cells in the inflamed (INF) and noninflamed (NI) CD mucosa (Supplementary Table 2B). Although CD16+ or CD14+ monocytes, CD141+ DC, CD1c+ DC, plasmacytoid DC, and neutrophils differed in their abundance, CD116 and CD131 expression and STAT5 phosphorylation remained unchanged between INF and NI CD tissues (Figure 3A–D, Supplementary Figure 5A and B). These findings suggest unperturbed GM-CSFR expression and responsiveness in the INF and NI CD mucosa.
Figure 3.
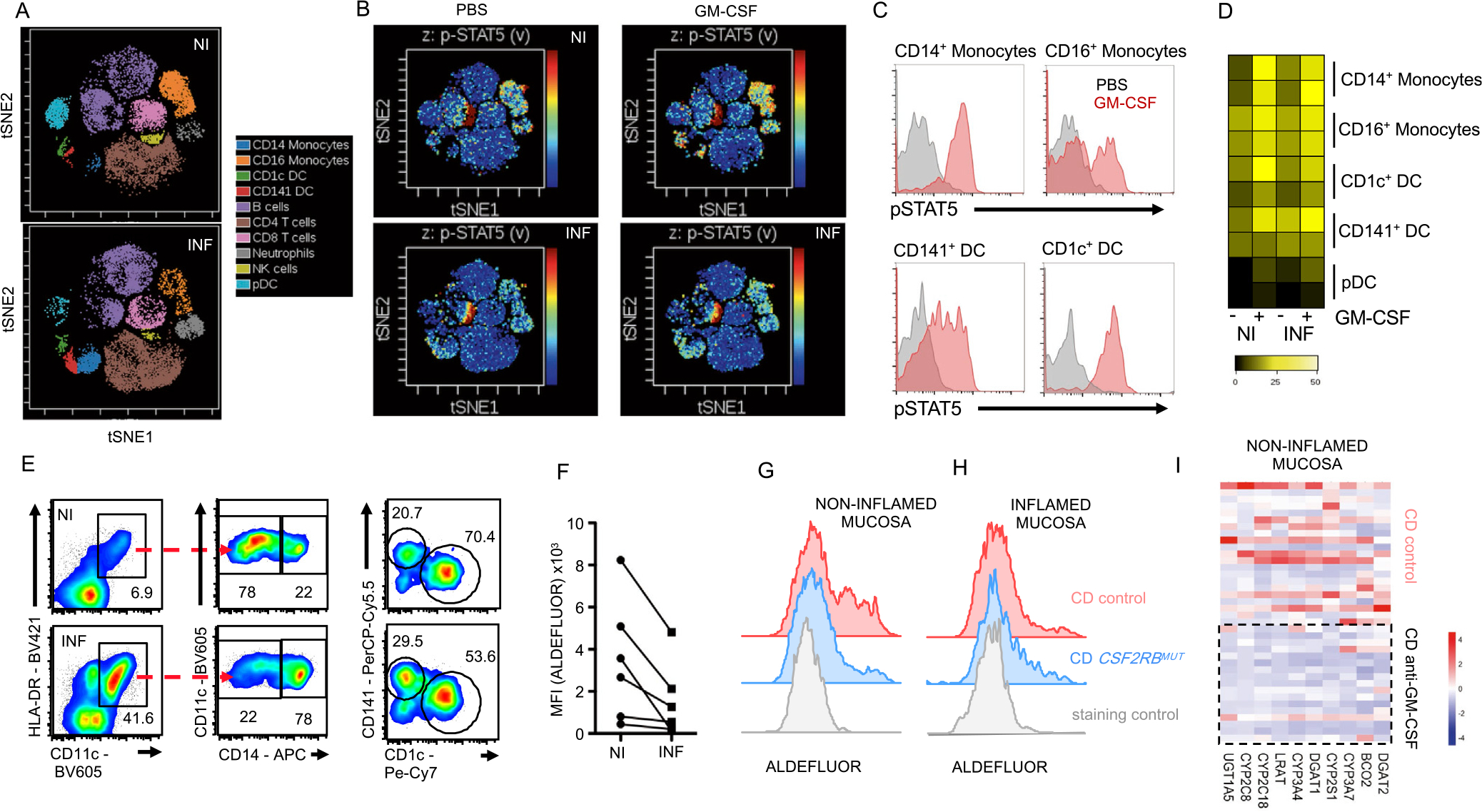
The inflamed CD mucosa shows impaired homeostatic functions in GM-CSF–responsive myeloid cells. Lamina propria leukocytes from NI and INF ileal mucosa were analyzed. (A) Visualized stochastic neighbor embedding analysis shows distribution of leukocyte populations in NI and INF tissues indicating supervised annotation of populations. (B) Stimulation of cells in (A) with GM-CSF, followed by pSTAT5 measurement. Signal intensity is visualized in t-distributed stochastic neighbor embedding plots (blue = low, red = high pSTAT5). (C) Representative histograms show pSTAT5 levels in the indicated myeloid cell populations. (D) Heat map shows pSTAT5 intensity across myeloid populations from (A) and (B). (E) DC subset and macrophage identification by flow cytometry. (F) Mean fluorescence intensity (MFI) of ALDEFLOUR staining in CD45+CD11c+HLA-DR+ cells. ALDEFLUOR staining in CD45+CD11+HLA-DR+ cells in CD control and one CSF2RBMUT carrier from (G) NI and (H) INF tissue biopsies. (I) Heat map shows gene expression for genes associated with retinol metabolism in NI ileal tissues of aGMAb+ or aGMAb− patients with CD.
GM-CSF stimulation controls essential myeloid functions including RA production. We next assessed the production of RA by APCs using ALDEFLUOR staining on HLA-DR+CD11c+ APCs from the INF and NI CD mucosa. APCs including CD14+ macrophages CD141+ DC, and CD1c+ DC showed decreased ALDEFLUOR staining specifically in the INF mucosa (Figure 3E and F, Supplementary Figure 5C and D). Monocytes and precursor DCs continuously infiltrate the intestinal mucosa to differentiate into DCs and macrophages and require GM-CSF for RA production.10,15,32,33 GM-CSF stimulation of blood-derived CD14+ monocytes confirmed a GM-CSF–dependent increase in ALDEFLUOR staining comparable with staining in APCs from the NI CD mucosa (Supplementary Figure 5C–E). APCs isolated from a patient carrying a CSF2RBMUT allele revealed the requirement for GM-CSF in maintaining APC function even in NI tissues (Figure 3G and H). To determine if aGMAb could alter RA-related metabolism in the NI CD mucosa, gene expression of retinol metabolism-associated genes was performed. The expression of DGAT2, BCO2, CYP3A7, CYP2S1, DGAT1, CYP3A4, LRAT, CYP2C18, CYP2C8, and UGT1A5, genes controlling RA metabolism at multiple distinct steps, were decreased in the NI CD mucosa of aGMAb+ patients, but not CD controls (Figure 3I). These findings suggest that GM-CSF contributes to homeostatic RA metabolism in the NI CD mucosa, promoting us to investigate the phenotype and function of GM-CSF–producing cells.
T Cells and ILC3 Contribute to the Pool of GM-CSF in the NI and INF CD Mucosa
Assessment of spontaneously released GM-CSF by CD45+ cells was comparable between INF and NI CD tissues (Figure 4A), confirming unchanged serum GM-CSF levels in CD and HC (Supplementary Figure 2A). A characterization of GM-CSF–producing cells in the NI CD ileal mucosa revealed that 80% were NKp44+CD3− ILC, whereas the remaining 20% were composed of CD3+ T cells or cells lacking both markers (Figure 4B). Interestingly, the number of GM-CSF–producing T cells increased in the INF CD mucosa, whereas the number of GM-CSF+NKp44+ cells decreased (Figure 4B and C). GM-CSF–secreting NKp44+ cells coexpressed CD117, CD127, CD161, CD69, and the transcription factor ROR gamma(γ) t (Figure 4D and E), identifying them as natural cytotoxicity receptor (NCR)+ group 3 ILC (NCR+ILC3).34 Analyzing NCR+ILC3 numbers and cytokine release revealed lower levels of GM-CSF production and GM-CSF producers in the INF mucosa (Figure 4F and G, Supplementary Figure 5F). Despite increased GM-CSF+ T-cell frequencies, a lower per cell output was observed in these cells (Figure 4C, Supplementary Figure 5F). We next determined if NCR+ILC3 increasingly differentiated into ILC1 as reported in CD.12,14 ILC1s were increased in the INF mucosa, releasing higher levels of interferon-γ (Figure 4H).35,36 These findings demonstrate changes in the source and levels of GM-CSF in the INF CD mucosa, accompanied by decreased NCR+ILC3 and increased ILC1 counts (Figure 4F–H). The aberrantly glycosylated GM-CSF observed in CD sera (Figure 1D) inspired the analysis of glycogene-expression in GM-CSF+ leukocytes of the INF and NI CD mucosa. Published single cell RNA-Seq data from patients with CD was analyzed for the expression of glycosyltransferases in T cells, NCR+ILC3s, and ILC1.37 Glycogene signatures differed in T cells and NCR+ILC3s between INF and NI mucosa, implicating the production of differential glycovariants of secreted proteins including GM-CSF. T cells and NCR+ILC3 in the INF mucosa displayed an upregulation of mannose-related (ALG6, ALG8, ALG9), fucosylation-related (FUT2, FUT7), and α2,3-sialylation-related glycogenes, when compared with the NI mucosa, in line with previous reports (Figure 4I and J).38–42 Noteworthy, substrate availability or modulation of glycosyltransferases expression may result in aberrantly glycosylated proteins including GM-CSF. The decreased availability of NCR+ILC3-derived GM-CSF was reported to reduce myeloid RA production in mice, aligning with our findings in patients with CD (Figure 3E–G, Supplementary Figure 5C–F).10,43 Importantly, myeloid-derived RA and IL23 both prevent the accumulation of inflammatory ILC1, suggesting that aGMAbs perturb the equilibrium of tissue-resident ILCs in prediagnostic patients with CD through the modulation of GM-CSF–dependent myeloid RA production.29,30 To determine if the presence of aGMAb coincided with altered ILC3 and ILC1 gene signatures before inflammation, bulk messenger RNA-Seq data from NI ileal CD biopsy specimens (n = 191) and non-IBD controls (n = 121) were reanalyzed for ILC1 and ILC3 signature gene expression. Samples were stratified based on their principal component (PC)-ILC3 and PC-ILC1 signature gene expression profiles in the presence or absence of aGMAbs. ILC3-PC1 showed marginal changes, whereas significant changes in ILC1-PC1 were seen in the presence of aGMAbs (Figure 4K and L). These findings implicate a shift in ILC homeostasis in the NI CD mucosa in the presence of aGMAb, an event that may happen years before diagnosis.
Figure 4.
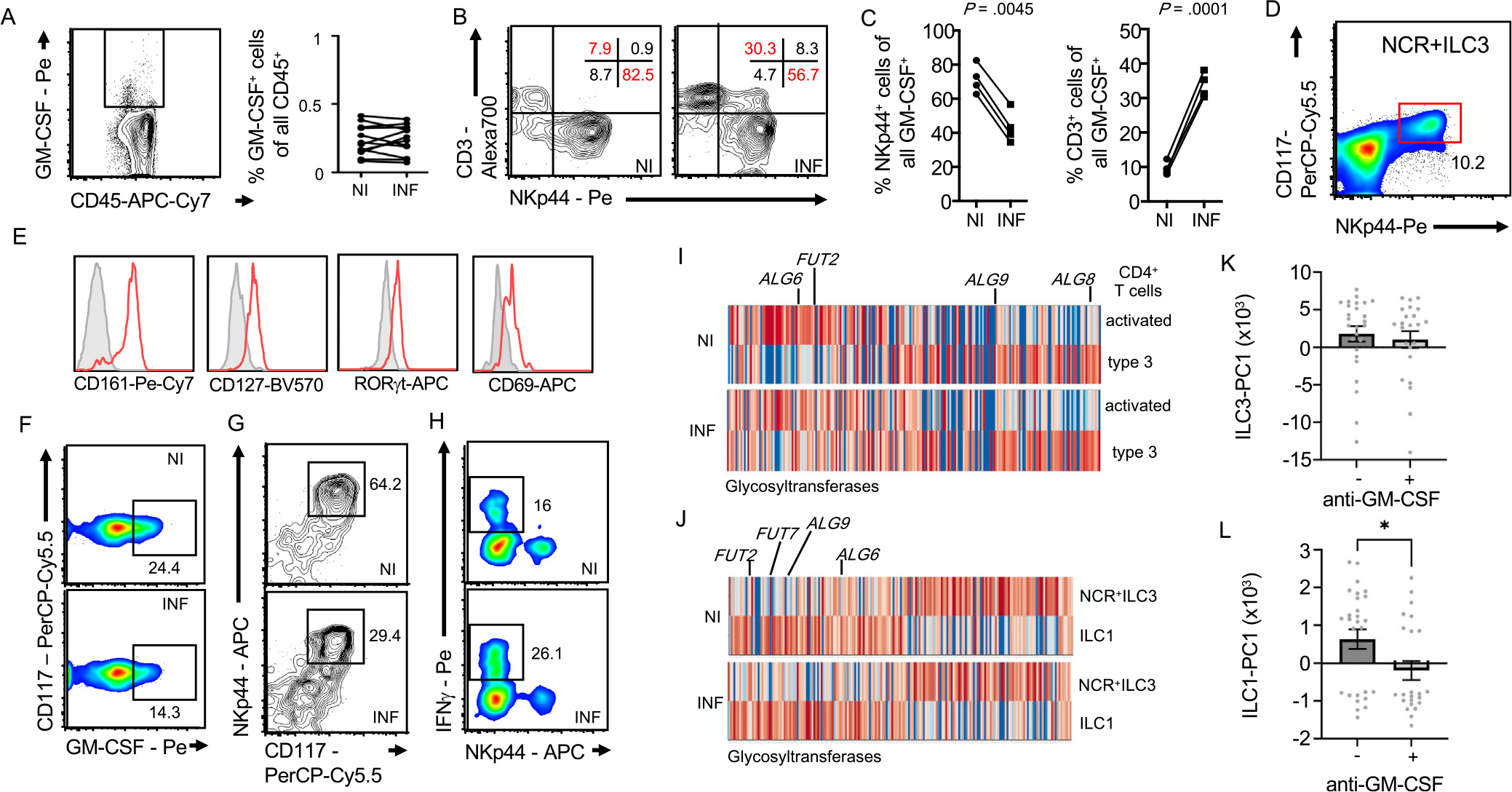
Innate and adaptive sources of intestinal GM-CSF in patients with CD. (A) This shows GM-CSF+CD45+ cells. Adjacent dot plot shows frequency in NI and INF CD mucosa. (B) GM-CSF+CD45+ cells were analyzed for CD3 and NKp44 expression in NI and INF CD mucosa. Numbers in gate represents percentages. (C) Plots show percentages of NKp44+GM-CSF+ and CD3+GM-CSF+ cells. (D) This shows identification of NKp44+ CD117+ cells and (E) expression of CD127, CD161, RORγt, and CD69 on NKp44+ CD117+ cells. (F) GM-CSF production by NCR+ILC3 (CD45+CD3−CD4−CD127+CD161+NKp44+CD117+), (G) NCR+ILC3 frequencies, and (H) interferon-γ-producing ILC1 were quantified. Numbers in gates represent percentages. (I) This image and (J) this image show gene expression analysis of glycosyltransferase genes in the indicated population from INF and NI patients with CD. (K) This image and (L) this image show scatter plots of PC1 values of ILC1-PC and ILC3-PC derived from principal component analysis of signature ILC1 and ILC3 genes against a sub-dataset (195 NI ileal CD and 121 NI ileal non-IBD samples) of the bulk messenger RNAseq samples from the Mount Sinai Crohn’s and Colitis Registry stratified by the presence of aGMAb.
Discussion
Here, we report the presence of aGMAb in the sera of patients with CD years before diagnosis, and propose that these antibodies contribute to the pathophysiology of CD. We demonstrated IgG2- and IgA-skewed aGMAb isotypes in patients with CD, suggesting an origin within the intestinal mucosa. Anti-GM-CSF autoantibodies were associated with ileal disease location, were present up to 6 years before diagnosis in asymptomatic subjects developing CD, and predicted complications at disease presentation (hazard ratio = 2.9 using log rank; P < .001). IgA aGMAbs were an exclusive hallmark of CD, and blocked GM-CSFR signaling by binding to GM-CSF glycovariants. This in turn impaired communication of ILC3 and myeloid cells in the NI CD mucosa at steady-state, resulting in significant alterations in ILC gene signatures in the presence of aGMAb. In support of these findings, retinol metabolism-associated gene expression in the NI mucosa of patients with CD was reduced in the presence of aGMAbs, identifying a subgroup of individuals at high risk of developing complicated CD through an altered intestinal immune balance. Importantly, GM-CSF in patients with CD was aberrantly glycosylated, in line with altered glycosyltransferase expression in GM-CSF–producing ILC3 and T cells, which in turn implicates a possible immunogenicity of post-translational epitopes recognized by aGMAbs. Together, these results support a novel mechanism of disease pathophysiology that may be exploited for developing personalized CD-preventive and therapeutic strategies.
Myeloid cells of the INF CD mucosa displayed markedly reduced GM-CSF–dependent RA production associated with reduced NCR+ILC3 numbers, lower GM-CSF output, and altered glycogene expression. Abrogating GM-CSF–mediated signaling altered retinol metabolism-associated gene expression and increased ILC1 signature gene expression even in NI tissues, suggesting a role for GM-CSF in sustaining homeostasis.13,14,34,36,44 Enrichment in ILC1 signatures in aGMAb+ patients with CD suggests a GM-CSF–dependent myeloid regulation of the ILC3/ILC1 balance. Autoreactive aGMAbs thus promote the accumulation of inflammatory ILC1 by disrupting the NCR+ILC3-myeloid cell circuit. We speculate that CD-associated aGMAbs shift the immune balance during the “prediagnostic” period of CD by altering GM-CSF–dependent homeostasis, a hypothesis supported by our data demonstrating reduced immune-related gene expression even in the NI CD mucosa of aGMAb+ patients. One limitation of our study is that we were unable to determine at what “stage” of the preclinical phase of CD aGMAb occur and whether subclinical inflammation at the histologic or endoscopic level was already present.45 Such subclinical inflammation is indeed commonly observed in patients with CD in clinical remission and has also been described in their first-degree relatives using biomarkers such as calprotectin or endoscopic exploration with video capsule.46,47
Our work further supports the rising interest in exploring the preclinical phase of CD where prevention strategies could be pursued48 and measuring aGMAb adds to the recently identified list of predictive biomarkers, such as microbial antibodies49 and intestinal permeability.50 One specific interest is the association of aGMAb with complicated CD disease at onset, which may one day help to identify relevant candidates for disease prevention at its earliest phase.
The differential glycosylation of GM-CSF and altered glycosyltransferase expression suggest that new (glyco) epitopes on GM-CSF may promote the development of aGMAb, however, such an event will likely be accompanied by genetic predisposition or additional triggering stimulus, given that sera of aGMAb− patients with CD shows aberrant glycosylation too. Engineering of GM-CSF variants with functional activity but capable of circumventing aGMAb may provide a way to re-establish immune homeostasis or delay disease progression. Our data calls for revisiting the use of GM-CSF in clinical trials with modified versions of active GM-CSF not prone to antibody neutralization, with careful preselection of patients based on their aGMAb profiles. Our findings demonstrate an intriguing mechanism for the development of complicated CD and allow the identification of these patients using a predictive serologic biomarker. Collectively, these findings open new roads for the precise diagnosis, classification, and personalized treatment of patients with CD.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Anti-granulocyte macrophage-colony stimulating factor autoantibodies (aGMAb) are enriched in complicated Crohn’s disease (CD) and their epitopes, isotypes, time of appearance, and biological consequence on granulocyte macrophage-colony stimulating factor (GM-CSF) producer and responder cells remain poorly understood.
NEW FINDINGS
Anti-GM-CSF autoantibodies precede CD onset by years, recognize glycosylation on GM-CSF, and are associated with increased group 1 innate lymphoid cells.
LIMITATIONS
Perturbations of immune homeostasis by aGMAbs before disease onset is not demonstrated, but observed in the noninflamed CD mucosa in the presence of aGMAbs.
IMPACT
Anti-GM-CSF autoantibodies predict complicated CD and removal of glycosylation on GM-CSF facilitates escape from autoantibody-mediated neutralization.
Acknowledgments
Our highest gratitude goes to patients donating samples for this study. We acknowledge the service by the Mount Sinai Mass Cytometry Core facility and all surgeons and clinical staff involved in this study.
Funding
A.M. is supported by a Canadian Institute for Health Research (CIHR) Project Grant (388337), a CIHR Team Grant (8833615), a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (RGPIN-2019-04521), and the Helmsley Foundation and is the Tier 2 Canadian Research Chair in Mucosal Immunology through the Tier 2 Canadian Research Chair (CRC)-CIHR program. S.S.P. acknowledges Portuguese funds through the Portuguese Foundation for Science and Technology (FCT) in the framework of the project POCI-01-0145-FEDER-028772 and support by the Broad Medical Research Program at the Crohn’s and Colitis Foundation of America, the International Organization for the study of Inflammatory Bowel Disease, and the Portuguese Group of Study in IBD (GEDII) for funding. S.S.P. also acknowledges the US Department of Defense, US Army Medical Research Acquisition Activity, FY18 Peer Reviewed Medical Research Program Investigator-Initiated Research Award (award number W81XWH1920053). I.A., C.M.A., and J.G. thank FCT for funding (SFRH/BD/128874/2017, 2021.07357.BD and 2020.00088.CEECIND). S.L.T. acknowledges funding through the Dr. Edward KETCHUM Graduate Student Scholarships at the University of Toronto and the Canada Graduate Scholarships – Master’s (CGS-M) award. M.W. is supported by a Undergraduate Research Opportunity Program (UROP) fellowship by the Department of Immunology at the University of Toronto. J.H.C. acknowledges funding from U01 DK062429, U01 DK062422, R01 DK106593, and the Sanford Grossman Charitable Trust. J.F.C and F.P. are supported by the Kenneth Rainin Foundation (grant number 20210021). S.G. and J.F.C. are supported through U01 DK124165 and the Helmsley Foundation. S.G. is additionally supported by grants U24 CA224319 and CA196521. Funding and support of the PREDICTS (PRoteomic Evaluation and Discovery in an IBD Cohort of Tri-service Subjects) study platform was provided through a Cooperative and Research Development Agreement with direct contributions by Janssen Pharmaceuticals, Prometheus Laboratories, and the Naval Medical Research Center (Naval Cooperative Research And Development Agreement [NCRADA] number NMR-11-3920). The mass cytometry instrumentation at the Human Immune Monitoring Center was obtained with support from S10 OD023547 and P01 CA196521.
Abbreviations used in this paper:
- AAL
Aleuria Aurantia Lectin
- aGMAb
anti-granulocyte macrophage-colony stimulating factor autoantibodies
- APC
antigen-presenting cell
- CD
Crohn’s disease
- DC
dendritic cell
- GM-CSF
granulocyte macrophage-colony stimulating factor
- GM-CSFR
granulocyte macrophage-colony stimulating factor receptor
- GNA
Galanthus Nivalis Lectin
- HD
healthy donors
- IBD
inflammatory bowel disease
- Ig
immunoglobulin
- IL
interleukin
- ILC
innate lymphoid cell
- INF
inflamed
- IRB
Institutional Review Board
- L-PHA
Phaseolus Vulgaris Leucoagglutinin
- MALII
Maackia Amurensis Lectin II
- NCR
natural cytotoxicity receptor
- NI
noninflamed
- NK
natural killer
- PAP
pulmonary alveolar proteinosis
- PC
principal component
- RA
retinoic acid
- UC
ulcerative colitis
Footnotes
Conflicts of interest
The University of Toronto and the Mount Sinai Hospital have collectively filed a patent application listing S.G., A.M., J.F.C., M.M., and R.R. as inventors, which is related in part to this publication. S.G. reports past consultancy and/or advisory roles for Merck and OncoMed and research funding from Bristol-Myers Squibb, Genentech, Janssen R&D, Pfizer, Takeda, Boehringer-Ingelheim, and Regeneron. J.F.C. reports receiving research grants from AbbVie, Janssen Pharmaceuticals, and Takeda; receiving payment for lectures from AbbVie, Amgen, Allergan, Inc., Ferring Pharmaceuticals, Shire, and Takeda; receiving consulting fees from AbbVie, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers-Squibb, Celgene Corporation, Eli Lilly, Ferring Pharmaceuticals, Galmed Research, Genentech, Glaxo Smith Kline, Janssen Pharmaceuticals, Kaleido Biosciences, Imedex, Immunic, Iterative Scopes, Merck, Microba, Novartis, PBM Capital, Pfizer, Sanofi, Takeda, TiGenix, Vifor; and holding stock options in Intestinal Biotech Development. C.K.P. is an employee of the US Government. This work was prepared as part of his official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person’s official duties. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the US Government. This is a US Government work. There are no restrictions on its use.
Data Transparency
The data that supports the findings of this study are available from the corresponding authors on reasonable request.
CRediT Authorship Contributions
Arthur Mortha, PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Equal; Funding acquisition: Equal; Investigation: Lead; Methodology: Lead; Project administration: Lead; Validation: Equal; Visualization: Lead; Writing – original draft: Lead; Writing – review & editing: Lead). Romain Remark, PhD (Conceptualization: Supporting; Formal analysis: Equal; Investigation: Supporting; Methodology: Supporting; Visualization: Equal). Diane Marie Del Valle, PhD (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting). Ling-Shiang Chuang, PhD (Data curation: Supporting; Formal analysis: Supporting; Methodology: Supporting; Software: Supporting; Visualization: Supporting). Zhi Chai, PhD (Data curation: Supporting; Formal analysis: Supporting; Software: Supporting; Visualization: Supporting). Inês Alves, PhD (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting; Visualization: Supporting). Catarina Azevedo, BSc (Data curation: Supporting; Formal analysis: Supporting; Visualization: Supporting). Joana Gaifem, PhD (Data curation: Supporting; Formal analysis: Supporting; Visualization: Supporting). Jerome Martin, MD, PhD (Data curation: Supporting; Formal analysis: Supporting; Resources: Supporting; Software: Supporting; Visualization: Supporting). Francesca Petralia, PhD (Formal analysis: Supporting; Visualization: Supporting). Kevin Tuballes, PhD (Formal analysis: Supporting; Validation: Supporting). Vanessa Barcessat, PhD (Formal analysis: Supporting; Validation: Supporting). Siu Ling Tai, MSc (Formal analysis: Supporting; Investigation: Supporting; Validation: Supporting; Visualization: Supporting). Hsin-Hui Huang, MD, PhD (Methodology: Supporting; Software: Supporting; Validation: Supporting). Ilaria Laface, PhD (Formal analysis: Supporting). Yeray Arteaga Jerez, PhD (Investigation: Supporting). Gilles Boschetti, MD, PhD (Investigation: Supporting). Nicole Villaverde, MSc (Investigation: Supporting). Mona D. Wang, B.Sc. (Investigation: Supporting). Ujunwa M. Korie, BSc (Data curation: Supporting; Investigation: Supporting). Joseph Murray, MD, PhD (Resources: Supporting). Rok-Seon Choung, PhD (Resources: Supporting). Takahiro Sato, PhD (Resources: Supporting). Renee M Laird, PhD (Resources: Supporting). Scot Plevy, MD, PhD (Resources: Supporting). Adeeb Rahman, PhD (Formal analysis: Supporting; Methodology: Supporting; Visualization: Supporting). Joana Torres, MD (Data curation: Supporting; Investigation: Supporting; Resources: Supporting). Chad Porter, PhD (Resources: Supporting). Mark S. Riddle, MD, PhD (Resources: Supporting). Ephraim Kenigsberg, PhD (Data curation: Supporting; Formal analysis: Supporting; Methodology: Supporting; Resources: Supporting; Software: Supporting). Salomé S. Pinho, PhD (Conceptualization: Supporting; Formal analysis: Supporting; Investigation: Supporting; Resources: Supporting; Visualization: Supporting; Writing – review & editing: Supporting). Judy H. Cho, MD (Conceptualization: Supporting; Data curation: Supporting; Resources: Equal; Supervision: Supporting). Miriam Merad, MD, PhD (Conceptualization: Supporting; Funding acquisition: Equal; Resources: Supporting; Supervision: Supporting; last authorship: Equal). Jean-Frederic Colombel, MD (Conceptualization: Equal; Data curation: Equal; Funding acquisition: Equal; Methodology: Equal; Resources: Equal; Supervision: Equal; Validation: Lead; Writing – review & editing: Equal; last authorship: Equal). Sacha Gnjatic, PhD (Conceptualization: Equal; Data curation: Equal; Funding acquisition: Equal; Investigation: Equal; Methodology: Supporting; Resources: Equal; Supervision: Equal; Validation: Equal; Visualization: Equal; Writing – original draft: Equal; Writing – review & editing: Equal; last authorship: Equal).
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://dxdoi.org/10.1053/j.gastro.2022.05.029.
References
- 1.Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 2018;390:2769–2778. [DOI] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN, Bernstein CN, Iliopoulos D, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol 2018;15:39–49. [DOI] [PubMed] [Google Scholar]
- 3.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol 2005;19(Suppl A):5A–36A. [DOI] [PubMed] [Google Scholar]
- 4.Verhelst X, Dias AM, Colombel JF, et al. Protein glycosylation as a diagnostic and prognostic marker of chronic inflammatory gastrointestinal and liver diseases. Gastroenterology 2020;158:95–110. [DOI] [PubMed] [Google Scholar]
- 5.Han X, Gilbert S, Groschwitz K, et al. Loss of GM-CSF signalling in non-haematopoietic cells increases NSAID ileal injury. Gut 2010;59:1066–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirata Y, Egea L, Dann SM, et al. GM-CSF-facilitated dendritic cell recruitment and survival govern the intestinal mucosal response to a mouse enteric bacterial pathogen. Cell Host Microbe 2010;7:151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lang RA, Metcalf D, Cuthbertson RA, et al. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell 1987;51:675–686. [DOI] [PubMed] [Google Scholar]
- 8.Sainathan SK, Hanna EM, Gong Q, et al. Granulocyte macrophage colony-stimulating factor ameliorates DSS-induced experimental colitis. Inflamm Bowel Dis 2008;14:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song C, Lee JS, Gilfillan S, et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med 2015;212:1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mortha A, Chudnovskiy A, Hashimoto D, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014;343:1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castro-Dopico T, Fleming A, Dennison TW, et al. GM-CSF calibrates macrophage defense and wound healing programs during intestinal infection and inflammation. Cell Rep 2020;32:107857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonarbourg C, Mortha A, Bui VL, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 2010;33:736–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernink JH, Krabbendam L, Germar K, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 2015;43:146–160. [DOI] [PubMed] [Google Scholar]
- 14.Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 2013;14:221–229. [DOI] [PubMed] [Google Scholar]
- 15.Chuang LS, Villaverde N, Hui KY, et al. A frameshift in CSF2RB predominant among Ashkenazi Jews increases risk for Crohn’s disease and reduces monocyte signaling via GM-CSF. Gastroenterology 2016;151:710–723.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabritz J Granulocyte macrophage colony-stimulating factor and the intestinal innate immune cell homeostasis in Crohn’s disease. Am J Physiol-Gastrointest Liver Physiol 2014;306:G455–G465. [DOI] [PubMed] [Google Scholar]
- 17.Dieckgraefe BK, Korzenik JR. Treatment of active Crohn’s disease with recombinant human granulocyte-macrophage colony-stimulating factor. Lancet 2002;360:1478–1480. [DOI] [PubMed] [Google Scholar]
- 18.Korzenik JR, Dieckgraefe BK, Valentine JF, et al. Sargramostim for active Crohn’s disease. N Engl J Med 2005;352:2193–2201. [DOI] [PubMed] [Google Scholar]
- 19.Roth L, MacDonald JK, McDonald JW, et al. Sargramostim (GM-CSF) for induction of remission in Crohn’s disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis 2012;18:1333–1339. [DOI] [PubMed] [Google Scholar]
- 20.Han X, Uchida K, Jurickova I, et al. Granulocyte-macrophage colony-stimulating factor autoantibodies in murine ileitis and progressive ileal Crohn’s disease. Gastroenterology 2009;136:1261–1271; e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nylund CM, D’Mello S, Kim MO, et al. Granulocyte macrophage-colony-stimulating factor autoantibodies and increased intestinal permeability in Crohn disease. J Pediatr Gastroenterol Nutr 2011;52:542–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gathungu G, Kim MO, Ferguson JP, et al. Granulocyte-macrophage colony-stimulating factor autoantibodies: a marker of aggressive Crohn’s disease. Inflamm Bowel Dis 2013;19:1671–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jurickova I, Collins MH, Chalk C, et al. Paediatric Crohn disease patients with stricturing behaviour exhibit ileal granulocyte-macrophage colony-stimulating factor (GM-CSF) autoantibody production and reduced neutrophil bacterial killing and GM-CSF bioactivity. Clin Exp Immunol 2013;172:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dabritz J, Bonkowski E, Chalk C, et al. Granulocyte macrophage colony-stimulating factor auto-antibodies and disease relapse in inflammatory bowel disease. Am J Gastroenterol 2013;108:1901–1910. [DOI] [PubMed] [Google Scholar]
- 25.Piccoli L, Campo I, Fregni CS, et al. Neutralization and clearance of GM-CSF by autoantibodies in pulmonary alveolar proteinosis. Nat Commun 2015;6:7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griseri T, Arnold IC, Pearson C, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote Interleukin-23 driven chronic colitis. Immunity 2015;43:187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter CK, Riddle MS, Gutierrez RL, et al. Cohort profile of the PRoteomic Evaluation and Discovery in an IBD Cohort of Tri-service Subjects (PREDICTS) study: rationale, organization, design, and baseline characteristics. Contemp Clin Trials Commun 2019;14:100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyajima A, Otsu K, Schreurs J, et al. Expression of murine and human granulocyte-macrophage colony-stimulating factors in S. cerevisiae: mutagenesis of the potential glycosylation sites. EMBO J 1986;5:1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perdue CL, Eick-Cost AA, Rubertone MV. A brief description of the operation of the DoD serum repository. Mil Med 2015;180:10–20. [DOI] [PubMed] [Google Scholar]
- 30.Plevy S, Silverberg MS, Lockton S, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn’s disease, and ulcerative colitis patients. Inflamm Bowel Dis 2013;19:1139–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres J, Petralia F, Sato T, et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 y before diagnosis. Gastroenterology 2020;159:96–104. [DOI] [PubMed] [Google Scholar]
- 32.Bujko A, Atlasy N, Landsverk OJB, et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J Exp Med 2018;215:441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter L, Landsverk OJB, Atlasy N, et al. Transcriptional profiling reveals monocyte-related macrophages phenotypically resembling DC in human intestine. Mucosal Immunol 2018;11:1512–1523. [DOI] [PubMed] [Google Scholar]
- 34.Cella M, Fuchs A, Vermi W, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009;457:722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med 2009;361:2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glatzer T, Killig M, Meisig J, et al. RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 2013;38:1223–1235. [DOI] [PubMed] [Google Scholar]
- 37.Martin JC, Chang C, Boschetti G, et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 2019;178:1493–1508.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dias AM, Pereira MS, Padrao NA, et al. Glycans as critical regulators of gut immunity in homeostasis and disease. Cell Immunol 2018;333:9–18. [DOI] [PubMed] [Google Scholar]
- 39.Pereira MS, Duraes C, Catarino TA, et al. Genetic variants of the MGAT5 gene are functionally implicated in the modulation of T cells glycosylation and plasma IgG glycome composition in ulcerative colitis. Clin Transl Gastroenterol 2020;11:e00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujii H, Shinzaki S, Iijima H, et al. Core fucosylation on T cells, required for activation of T-cell receptor signaling and induction of colitis in mice, is increased in patients with inflammatory bowel disease. Gastroenterology 2016;150:1620–1632. [DOI] [PubMed] [Google Scholar]
- 41.Pereira MS, Alves I, Vicente M, et al. Glycans as key checkpoints of T cell activity and function. Front Immunol 2018;9:2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dias AM, Dourado J, Lago P, et al. Dysregulation of T cell receptor N-glycosylation: a molecular mechanism involved in ulcerative colitis. Hum Mol Genet 2014;23:2416–2427. [DOI] [PubMed] [Google Scholar]
- 43.Samarakoon A, Shim YA, Dosanjh M, et al. CD45 regulates GM-CSF, retinoic acid and T-cell homing in intestinal inflammation. Mucosal Immunol 2016;9:1514–1527. [DOI] [PubMed] [Google Scholar]
- 44.Croxatto D, Micheletti A, Montaldo E, et al. Group 3 innate lymphoid cells regulate neutrophil migration and function in human decidua. Mucosal Immunol 2016;9:1372–1383. [DOI] [PubMed] [Google Scholar]
- 45.Torres J, Ungaro RC, Colombel JF. Is prevention the best way to modify inflammatory bowel disease? How close are we? Gastroenterology 2022;162:1452–1455. [DOI] [PubMed] [Google Scholar]
- 46.Sorrentino D, Avellini C, Geraci M, et al. Tissue studies in screened first-degree relatives reveal a distinct Crohn’s disease phenotype. Inflamm Bowel Dis 2014;20:1049–1056. [DOI] [PubMed] [Google Scholar]
- 47.Thjodleifsson B, Sigthorsson G, Cariglia N, et al. Subclinical intestinal inflammation: an inherited abnormality in Crohn’s disease relatives? Gastroenterology 2003;124:1728–1737. [DOI] [PubMed] [Google Scholar]
- 48.Torres J, Halfvarson J, Rodriguez-Lago I, et al. Results of the Seventh Scientific Workshop of ECCO: precision medicine in IBD–prediction and prevention of inflammatory bowel disease. J Crohns Colitis 2021;15:1443–1454. [DOI] [PubMed] [Google Scholar]
- 49.Choung RS, Princen F, Stockfisch TP, et al. Serologic microbial associated markers can predict Crohn’s disease behaviour years before disease diagnosis. Aliment Pharmacol Ther 2016;43:1300–1310. [DOI] [PubMed] [Google Scholar]
- 50.Turpin W, Espin-Garcia O, Bedrani L, et al. Analysis of genetic association of intestinal permeability in healthy first-degree relatives of patients with Crohn’s disease. Inflamm Bowel Dis 2019;25:1796–1804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.