

SUMMARY
Oncogene-induced senescence is a phenomenon in which aberrant oncogene expression causes non-transformed cells to enter a non-proliferative state. Cells undergoing oncogenic induction display phenotypic heterogeneity, with some cells senescing and others remaining proliferative. The causes of heterogeneity remain unclear. We studied the sources of heterogeneity in the responses of human epithelial cells to oncogenic BRAFV600E expression. We found that a narrow expression range of BRAFV600E generated a wide range of activities of its downstream effector ERK. In population-level and single-cell assays, ERK activity displayed a non-monotonic relationship to proliferation, with intermediate ERK activities leading to maximal proliferation. We profiled gene expression across a range of ERK activities over time and characterized four distinct ERK response classes, which we propose act in concert to generate the ERK-proliferation response. Altogether, our studies map the input-output relationships between ERK activity and proliferation, elucidating how heterogeneity can be generated during oncogene induction.
In brief
Chen et al. show that oncogenic BRAFV600E heterogeneously activates ERK and inhibits proliferation. ERK activity displays a non-monotonic relationship to proliferation, with intermediate levels leading to maximal proliferation. Transcriptional profiling reveals four classes of genes responding to different ranges of ERK levels, leading to a bell-shaped proliferation response.
Graphical Abstract
INTRODUCTION
Activation or aberrant regulation of oncogenes promotes cellular transformation and tumorigenesis, enabling cancer cells to grow and avoid programmed cell death.1 Activating RAS or RAF oncogenic mutations in cancers promotes growth by inducing constitutive MAPK signaling.2,3 However, ectopic expression of oncogenes in non-transformed cells can cause them to undergo stable cell-cycle arrest, a phenomenon known as oncogene-induced senescence (OIS).4,5 OIS, initially reported in primary fibroblasts with HRasG12V expression,5 was later found to be caused by various oncogenes and reported both in vitro and in vivo.4,6 In cultured melanocytes, BRAFV600E expression initially stimulated moderate proliferation (over 3–7 days), which was followed by a progressive decrease in growth rate and eventual cell-cycle arrest.7 Cell-cycle arrest typically involves the p53/p21WAF1 and p16INK4A/RB tumor suppressor genes and their interacting networks, although the roles of these proteins appear to be cell-type and context dependent.8,9 OIS is considered a bona fide tumor-suppressor mechanism, acting alongside cell death programs.
Cell-to-cell heterogeneity is often observed during oncogene induction, with some cells in a culture arresting and others continuing to proliferate. In vivo, malignant and benign tumors harboring the same driver oncogene mutation can co-exist.10,11 However, senescence-associated markers are found only in benign or premalignant lesions and are progressively lost as the lesions become malignant. At a population level, the time between oncogene expression and senescence varies from a few days to several weeks and proceeds asynchronously.5,7 It remains unclear why a subset of cells in a population is better able to tolerate the negative effects of oncogene activation. Contributing factors likely include the type, strength, and duration of the senescence-inducing signal; non-cell-autonomous influences from oncogene expression; and the cell’s susceptibility to (epi)genetic reprogramming.8,12
In this study, we focused on cell-cycle changes induced by BRAFV600E, an oncogenic variant of a MAPK serine/threonine kinase that is commonly found in cutaneous melanomas and is the primary target of current treatments.3,13 Activation of the MAPK cascade is critical, but not sufficient, for initiating melanocytic neoplasia, most likely due to the induction of senescence (Figure 1A). Indeed, BRAFV600E occurs in both benign and malignant skin lesions13,14 and can induce senescence in cell lines.7,15,16 We investigated the relationship between BRAFV600E levels, the activity of its downstream effector kinase ERK,17 and cell proliferation in non-transformed human hTERT-immortalized retinal pigment epithelial (RPE) cells. We showed that a narrow expression range of BRAFV600E protein generated a wide range of ERK activities. We found a non-monotonic relationship between ERK activity level and the proliferation response, which we examined through global transcriptional profiling. This analysis revealed four dynamic categories of cellular responses across different ranges of ERK activities. Our study highlights the various networks of genes that are induced in response to different strengths of ERK signaling and provides important clues as to how individual or combinatorial classes of genes can generate a non-monotonic proliferation response.
Figure 1. BRAFV600E-ERK pathway activation results in a non-monotonic proliferation response.
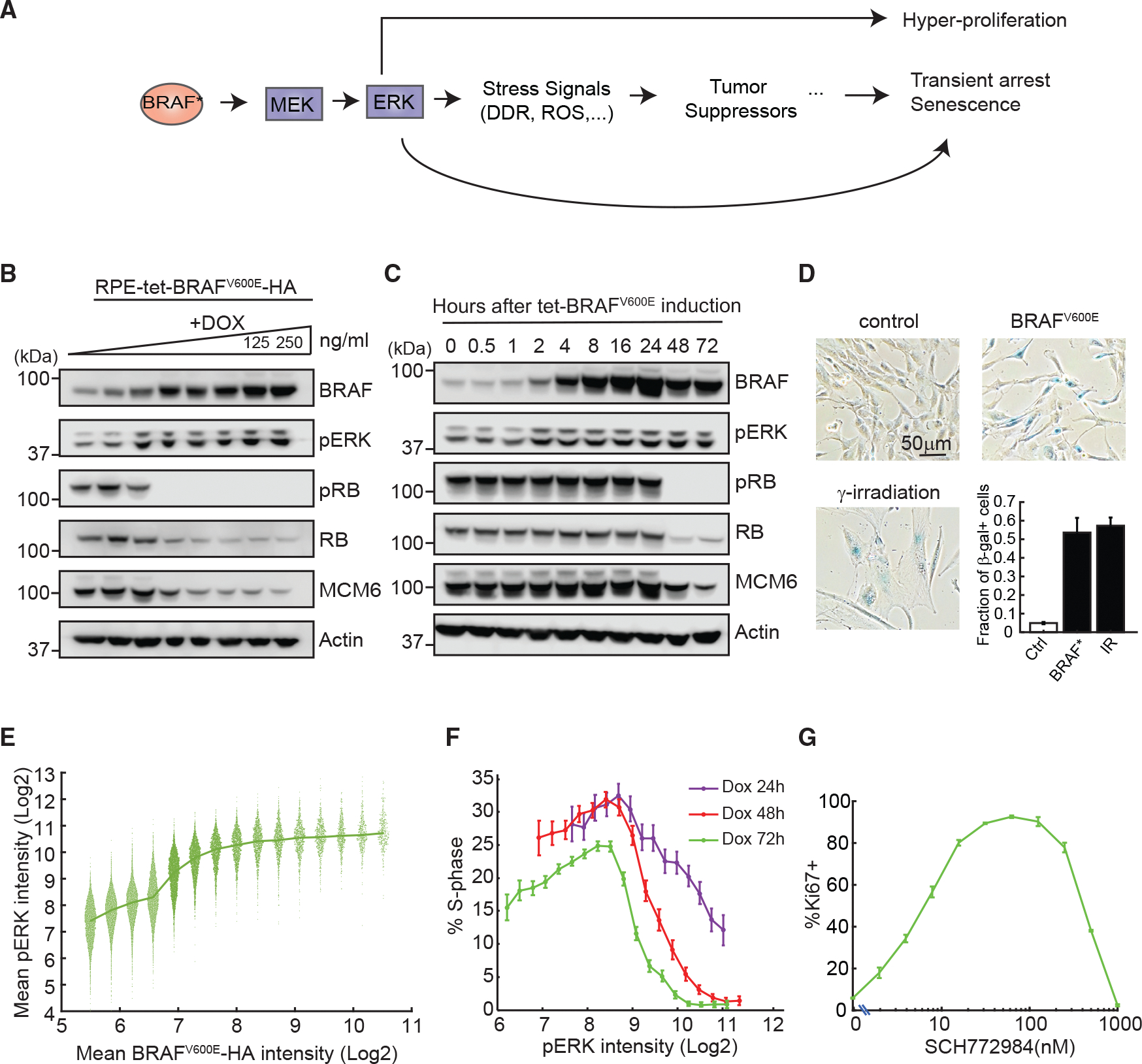
(A) Schematic of pathways activated by oncogenic BRAF (BRAF*). DDR, DNA-damage response; ROS, reactive oxygen species.
(B and C) Western blot analysis of levels of BRAF, active pERK, the proliferation marker pRB, total RB, and MCM6 (B) following BRAFV600E induction by increasing doses of doxycycline (DOX; 0–250 ng/mL, 2-fold dilution from the right) for 72 h or (C) at the indicated time points following BRAFV600E induction with 250 ng/mL DOX. Actin is shown as a loading control.
(D) Representative images of cells assayed for senescence-associated γ-galactosidase (SA-β-gal) activity 7 days after DOX induction of BRAFV600E-HA (BRAF*) at 250 ng/mL or after 10 Gy γ-irradiation. Bottom right: quantification of images. Data represent mean ± SD of three to five replicate wells; n > 560 cells/well for control; n > 180 cells/well for BRAFV600E; n > 50 cells/well for γ-irradiation.
(E) RPE/tet-BRAFV600E cells were treated with DOX as in (B) and immunostained for BRAFV600E-HA and pERK. Data from all DOX doses were pooled together, and single-cell pERK levels were extracted for equally spaced bins of BRAFV600E expression; n > 400 cells/bin. Each dot represents a single cell
(F) RPE/tet-BRAFV600E cells were treated with serial doses of DOX as in (B) for 24, 48, or 72 h before immunostaining. The immunofluorescence data from all DOX doses were pooled, and the percentage of cells in S phase was calculated for equally spaced bins of ERK activity (mean ± 95% bootstrap confidence interval; n > 700 cells per bin).
(G) RPE/tet-BRAFV600E cells were treated with DOX (250 ng/mL) and the indicated doses of ERKi for 72 h and then stained for the proliferation marker Ki67. The percentage of Ki67+ cells at each dose of ERKi is shown (mean ± SD of three replicate wells; n > 5,900 cells/well).
RESULTS
The relationship between ERK activity and proliferation is non-monotonic
To establish a model of OIS, we expressed the oncogenic BRAFV600E variant carrying a C-terminal hemagglutinin (HA) tag in RPE cells under a doxycycline (DOX)-inducible promoter (Figure 1B). Activation of the MAPK cascade by BRAFV600E was assessed by measuring phospho-ERK (pERK) levels using western blotting. At a saturating dose of DOX (250 ng/mL), BRAFV600E levels increased for the first 24 h and then plateaued, while pERK levels plateaued around 2 h. MCM6, a marker of S phase, was largely diminished by 48 h (Figure 1C). In addition, both pRB and total RB also decreased at DOX doses above 15 ng/mL; this reduction was achieved by 48 h at 250 ng/mL DOX. These results align with reports that total RB levels dropped greatly during cell-cycle arrest, likely due to oncogene-mediated protein degradation.18 To determine if BRAFV600E expression induced senescence, cells were stained for β-galactosidase. Cells exposed to γ-irradiation, a well-established inducer of senescence, were used as a positive control. BRAFV600E induction for 7 days resulted in ~55% of cells being positive for β-galactosidase expression, comparable to the level in irradiated cells (Figure 1D). These results suggest that in non-transformed RPE cells, BRAFV600E expression causes cell-cycle exit and promotes senescence. Deactivation of BRAFV600E by washing out DOX (at day 8 post DOX treatment) allowed cells to resume proliferation (Figure S1A), implying that, at least for some cells, exit from the cell cycle requires continuous BRAFV600E expression.
We next investigated the relationships between BRAFV600E expression, ERK activity, and proliferation outcomes. To induce variable BRAFV600E-HA expression levels, RPE/tet-BRAFV600E cells were treated with various doses of DOX for 72 h and then stained for HA, pERK, and incorporation of 5-ethynyl-2′-deoxyuridine (EdU) (a marker for DNA synthesis) (Figures S1B–S1D). Consistent with Figure 1B, proliferation was inhibited by BRAFV600E expression in a DOX-dose-dependent manner, with maximum proliferation occurring in the uninduced condition (Figure S1D). The data were pooled for subsequent analysis of ERK activity, in which we binned BRAFV600E expression levels measured in single cells (irrespective of the DOX dose) and quantified the ERK activity in each bin. This analysis revealed that pERK levels increased with BRAFV600E expression at lower levels but plateaued at higher levels, suggesting that ERK activity saturated at intermediate levels of BRAFV600E expression (Figure 1E). pERK levels varied at any given level of BRAFV600E, demonstrating substantial cell-to-cell variability in the activation of the MAPK cascade (Figure 1E). To determine the relationship between pERK levels and cell-cycle progression, DOX was added to the RPE/tet-BRAFV600E cells at various doses for 24, 48, or 72 h, and the fraction of S-phase cells was quantified by EdU incorporation (Figure 1F). Binning the data on pERK levels (regardless of DOX dose) revealed that the fraction of cycling cells was highest at intermediate pERK levels and decreased at higher and lower pERK levels. This result suggested that a moderate induction of pERK enhanced proliferation; however, beyond a certain level, proliferation was inhibited (Figure 1F). In the absence of BRAFV600E expression, ERK activity levels fell in the lower range (6–9 log2 intensity), and the percentage of proliferating cells associated with these levels ranged from 15% to 25% (Figures S1C and S1E). After BRAFV600E induction, ERK activity levels fell into a higher range (8.5–11.5 log2 intensity), and the associated proliferation frequencies ranged from 25% at the lower end of ERK activity to 1% at the higher end (Figure S1E). Because proliferation was highly sensitive to ERK activity in the range experienced by cells expressing BRAFV600E, it is likely that the heterogeneity in ERK activity during oncogene induction (Figure S1C) led to heterogeneity in proliferation and cell-cycle arrest. To further confirm the non-monotonic relationship between ERK activity and cell proliferation, we overexpressed BRAFV600E at a level sufficient to arrest most cells and treated them with a dose series of ERK inhibitor (ERKi) (SCH772984) to titrate down ERK activity. Consistent with Figure 1F, the fraction of cycling cells (indicated by the Ki67+ fraction) was highest at an intermediate ERKi dose (Figure 1G). By contrast, the fraction of proliferating cells was reduced when cells were treated with higher or lower ERKi doses, suggesting a non-monotonic relationship between proliferation and ERK activity.
Establishment of a cell-cycle reporter that differentiates G1, S, and G2 phases
Our data suggested that cells can make proliferation or arrest decisions in response to ERK activity. The data presented in Figure 1 show the average response of a population of cells. We hypothesized that at the single-cell level, internal cellular states, such as cell-cycle phase or the level or dynamics of oncogenic signaling, may influence the proliferation status. We monitored ERK activity and its relationship to cell-cycle progression using live single-cell imaging. Commonly used live-cell cell-cycle reporters, such as Geminin (1–110), monitor the G1-S transition,19 but growing evidence suggests that G2 plays a pivotal role in proliferation-quiescence decisions.20–22
To distinguish G1, S, and G2 cell-cycle phases, we developed a biosensor based on the PCNA-interacting-protein (PIP)-box motif, which is recognized by Cul4Cdt2 E3 ubiquitin ligase and degraded specifically at S phase (Figure 2A).23 Because ectopic expression of human PIP-box-containing proteins could interfere with normal cell-cycle progression, we developed a sensor based on the PIP-box motif of Drosophila E2F (dE2F).24 The sensor includes the N terminus of dE2F (amino acids 1–187) fused to the red fluorescent protein (FP) mCherry. The resulting mCherry-PIP protein contains PIP boxes functional in humans as well as a naturally occurring nuclear localization signal. RPE cells stably expressing mCherry-PIP, a turquoise FP-tagged nuclear histone marker (H2B-Turq), and a Venus FP-tagged Geminin (1–110) reporter were established. The Geminin (1–110) reporter accumulates in S phase and is degraded in G1 phase,19 while mCherry-PIP exhibited differential degradation/accumulation patterns in the nucleus throughout imaging (Figure 2B and Video S1).
Figure 2. Development and characterization of a live-cell sensor that identifies G1, S, and G2 cell-cycle phases.
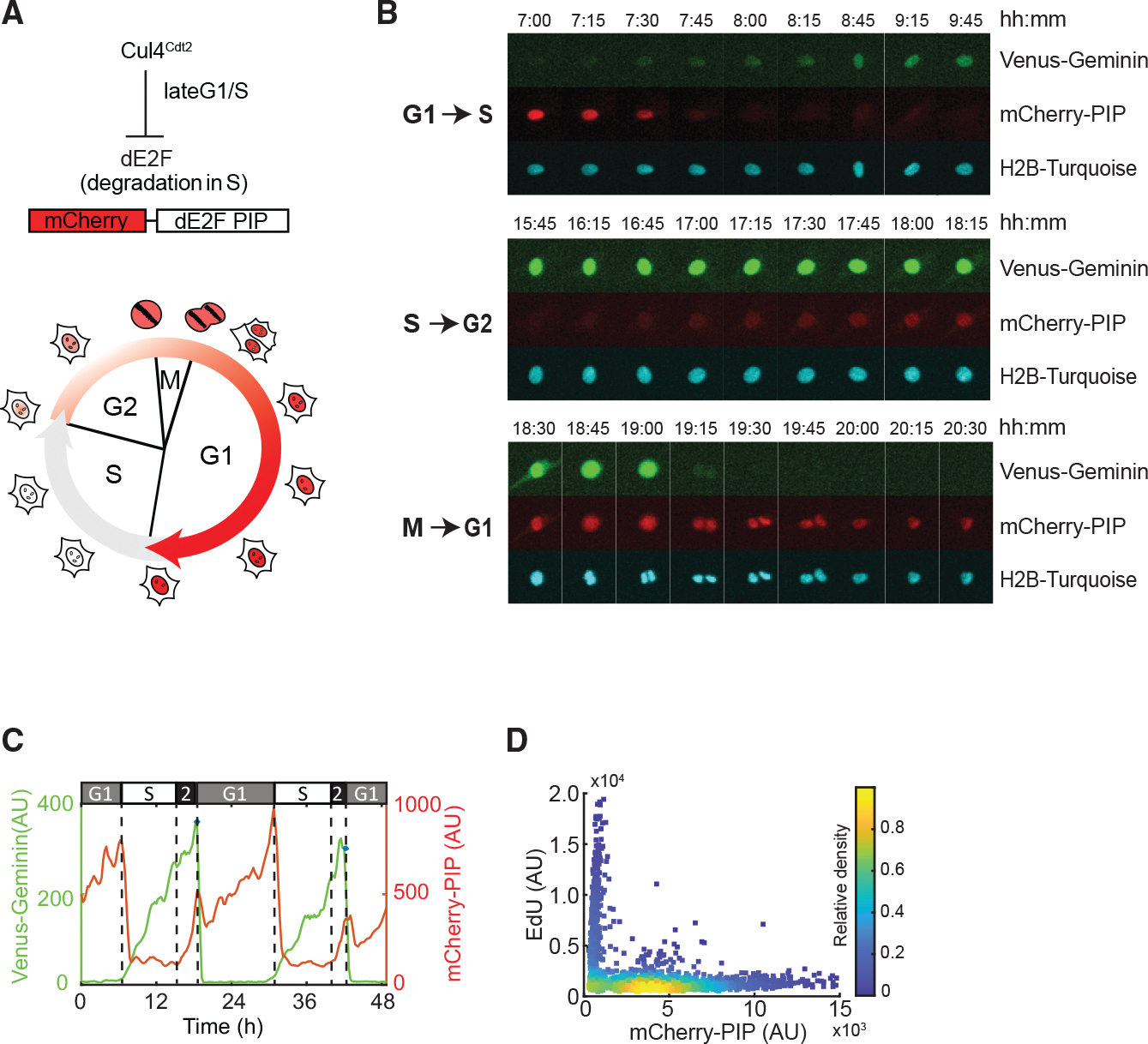
(A) (Top) Schematic of S-phase-specific degradation of Drosophila E2F1 PIP-motif-based biosensor. (Bottom) Cell-cycle progression with nuclear mCherry-dE2F PIP fluorescence changes.
(B) Images of a cycling RPE cell expressing Venus-Geminin (1–110), mCherry-dE2F PIP, and H2B-Turq over 24 h. Image strips of these three markers are shown as three panels corresponding to G1-S (top), S-G2 (middle), and M-G1 (bottom) transitions.
(C) Quantification of mCherry-dE2F PIP (red) and Venus-Geminin (1–110) (green) from (B).
(D) Density scatterplots of mCherry-dE2F PIP intensity versus EdU fluorescence intensity. Each dot represents a single cell; .
Comparing both cell-cycle reporter levels within the same cell, mCherry-PIP rapidly dropped in expression when Venus-Geminin (1–110) began to accumulate at the G1-S transition (Figures 2B and 2C). mCherry-PIP fluorescence rose subsequently, while Venus-Geminin continued to accumulate. Thus, the patterns of Venus-Geminin (1–110) and mCherry-PIP protein accumulation and degradation were consistent with the anticipated properties of the reporter proteins.19,23 By labeling S-phase cells with EdU, we validated that mCherry-PIP levels were lowest in S phase and were detectable only when cells were not in S phase (Figure 2D). In both normally cycling cells (Figure S2A) and cells arrested in G1 or G2 (Figures S2B–S2E), the reporter signal in G1 or G2 scaled with the duration of the phase. Thus, in live-cell experiments, G2 can be identified by the presence of both Venus-Geminin (1–110) and mCherry-PIP (Figure 2C). Cell-cycle phases can also be computationally derived from live-cell data by quantifying only the levels of the mCherry-PIP reporter as follows: G1 corresponds to the period between nuclear division and a rapid drop in mCherry-PIP fluorescence to basal level; S corresponds to the period between this rapid drop and right before resynthesis of mCherry-PIP occurs; and G2 corresponds to the period of mCherry-PIP accumulation prior to the next cell division (Figure 2C; STAR Methods).
Live-imaging traces revealed a bell-shaped relationship between ERK activity and cell-cycle entry in single cells
Given the cell-to-cell heterogeneity in ERK levels and OIS induction, we next established the relationship between ERK activity, cell-cycle phase transitions, and cell fate at a single-cell level. We generated an RPE cell line stably expressing DOX-inducible BRAFV600E, mCherry-PIP, and EKAREN5, a reporter for ERK activity. This line (termed BRAFV600E Dual Reporter cells) allowed us to induce oncogenic BRAFV600E and simultaneously measure ERK activity and cell-cycle progression in the same cells through long-term live imaging. EKAREN525 represents the widely used EKAREV fluorescence resonance energy transfer (FRET)-based ERK activity reporter25,26 engineered to be insensitive to CDK1/cyclin B activity at G2 and M phases. Control experiments confirmed that the EKAREN5 sensor reflected ERK activity in our RPE line and that activation during G2/M phase was strongly reduced relative to EKAREV (Figure S3A).
We imaged asynchronous cultures of BRAFV600E Dual Reporter cells for 24 h to obtain cell-cycle phase information under unperturbed conditions, then added DOX to induce BRAFV600E expression, and imaged live cells for 3 days to monitor ERK activity and cell-cycle progression. We used a semiautomated tracking method to identify individual division events and computationally derived the cell-cycle phases and ERK activities (Figure 3A; see STAR Methods). Note that the PIP signal increased sharply around division (±30 min) due to sudden nuclear shape changes, followed by a brief drop due to partitioning of the reporter proteins to two daughter cells. When BRAFV600E Dual Reporter cells were treated with DOX, ERK activity rapidly increased within 1–4 h (Figure 3B, consistent with data in Figure 1C), while control cells showed basal ERK activity with occasional pulses throughout the imaging period.27 Most DOX-treated cells underwent one or two divisions prior to entering prolonged G1 arrest, whereas cells not treated with DOX continued to proliferate, serving as a control for the effects of long-duration imaging (Figure 3C). A detailed cell-cycle duration analysis revealed that, prior to the prolonged G1 arrest, the G1- and S-phase lengths of the preceding cell cycle remained unaltered, while G2 length (orange blocks in Figure 3B) increased (Figure S3B). The increase in G2 length was ERK dependent, as addition of ERKi shortened the G2 duration (Figure S3C).
Figure 3. Single-cell live-imaging traces revealed a bell-shaped correlation between ERK activity and proliferation.
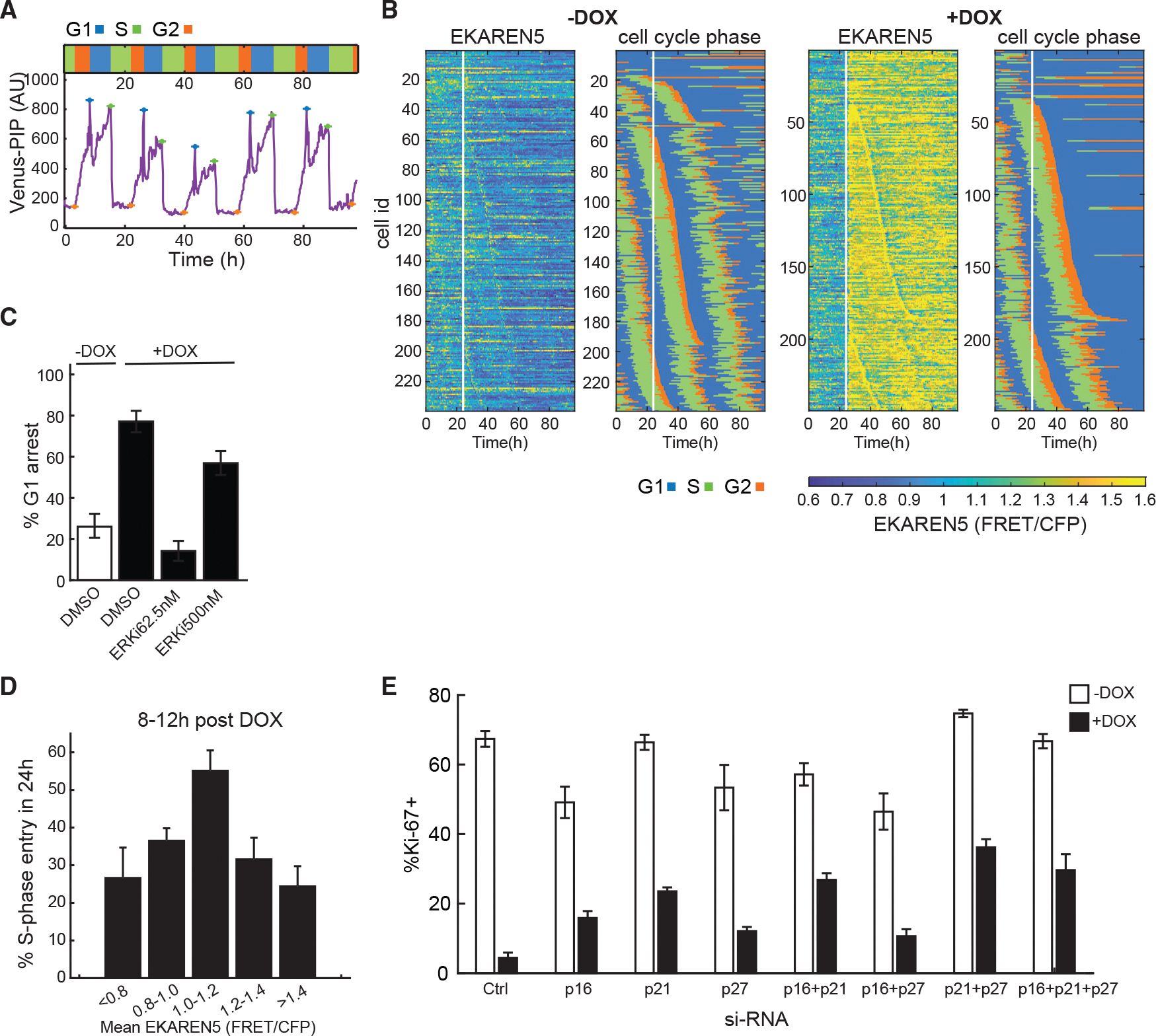
(A) Sample single-cell trace of Venus-dE2F PIP in RPE cells proceeding through the cell cycle. Crosses mark the start of each phase. Relative length of each cell-cycle phase is at the top.
(B) Heatmaps of ERK activity (EKAREN5) and cell-cycle distribution in BRAFV600E Dual Reporter cells treated with or without DOX at 24 h after the start of imaging (white vertical line). Each horizontal line represents a single cell.
(C) Frequency of G1 arrest in BRAFV600E Dual Reporter cells treated with or without DOX as in (B) together with DMSO or ERKi. The percentage of G1-arresting cells was calculated following time-lapse imaging (mean ± 95% confidence interval; n > 200 cells per condition).
(D) Fraction of S-phase entry in response to increasing ERK activity. The BRAFV600E Dual Reporter cells in (B) were treated with or without DOX and 0, 62.5, or 500 nM ERKi 24 h after the start of live imaging. Cells were then imaged for another 72 h. Data from all treatments were pooled and the mean ERK activity between 8 and 12 h post-treatment was calculated. The probability of entering into S phase was quantified within 24 h after the time frame of ERK monitoring (mean ± 95% confidence interval; n > 100 for each ERK activity bin).
(E) Percentage of Ki67-positive RPE/tet-BRAFV600E cells after siRNA-mediated depletion of the indicated CDK inhibitors 2 days after treatment with or without DOX (mean ± SD; replicates).
To map the relationship between ERK activity and cell-cycle progression, cells were treated with ERKi at different doses simultaneous with BRAFV600E induction. While 77% of cells treated with DOX in the absence of ERKi underwent G1 cell-cycle arrest, the addition of 62.5 nM ERKi rescued the arrest (Figures 3C and S3D). However, at 500 nM ERKi, the fraction of cells undergoing G1 arrest increased, consistent with a non-monotonic relationship between ERK levels and proliferation (Figures 3C and S3D). To quantify this relationship in single cells, we pooled single-cell trajectories based on mean ERK activity and then computed the fraction of cells that entered S phase within the following 24 h window. Mean ERK activity was determined between 8 and 12 h post BRAFV600E induction, when ERK activity levels stabilized in cells (Figures 1C and S4A). When the probability of S-phase entry was plotted against mean ERK activity, we again observed a non-monotonic, bell-shaped response (Figure 3D). Cells that entered S phase in Figure 3D completed the cell cycle and divided (Figure S3E). The slight drop in division rate at higher ERK levels was likely due to cells having prolonged G2, such that the division event was not captured during the live-imaging experiment. The non-monotonic relationship between ERK activity and proliferation was also evident at time intervals between 12–16 and 16–24 h after DOX addition (Figure S3F). The high sensitivity of cell-cycle arrest to increases in ERK activity above the optimum value likely explains cell-to-cell heterogeneity in response to BRAFV600E overexpression.
Previous OIS studies have suggested that activation of p16INK4A and p53 provides the two major mechanisms leading to cell-cycle arrest.8,9 To investigate this possibility in RPE/tet-BRAFV600E cells, we used RNAi to acutely knock down p16, p21 (a downstream target of p53), or p27, alone or in various combinations. The CDK inhibitor p27 was included due to its well-documented role in integrating diverse signals that regulate cell-cycle exit.28 The CDK inhibitor knockdowns were verified at the mRNA and protein levels (Figures S3G and S3H) and showed comparable efficiency both individually and in combination. Control and knockdown cells were treated with DOX, and the fraction of cycling cells was measured (Figure 3E). Knockdown of CDK inhibitors individually and in combination had a modest but reproducible effect on BRAFV600E-mediated arrest, but in no case was proliferation fully restored to control levels, consistent with prior observations.29,30 These results imply that additional proteins, beyond those suggested by previous studies, are involved in OIS, prompting us to apply a more systematic approach.
Deep RNA sequencing identifies genes that respond to ERK activity levels
We hypothesized that factors mediating ERK activity-dependent cell-fate decisions must themselves undergo changes in expression or activity in response to varying levels of ERK activity. To systematically identify genes whose expression changes with ERK activity, we performed deep RNA sequencing of RPE/tet-BRAFV600E cells treated with a combination of DOX and ERKi at different concentrations for varying times (including 0—an untreated control—and 1, 2, 4, 8, 16, and 24 h) (Figure 4A). The ERKi concentrations were chosen to sample the full range of proliferation responses (Figure 1G). The time points were selected based on the observation that RPE cells showed a bell-shaped relationship between ERK activity and proliferation (Figure 1F) as early as 24 h after BRAFV600E induction. The early time points allow identification of genes that are more directly responsive to ERK activity changes, while the later time points reveal long-term effects. Of note, the live-imaging experiments using BRAFV600E Dual Reporter cells showed that ERK activity peaked 1–2 h following DOX and ERKi treatment and then slowly decayed, while remaining at distinct levels for different ERKi doses during the subsequent 24 h period of our experiments (Figure S4A). These results suggested fast and stable ERK responses. To evaluate the effects of ERK inhibition on normal cycling cells, cells were treated with different doses of ERKi for 24 h without BRAFV600E induction. The resulting gene expression dataset involved 43 conditions assayed in duplicate. We detected ~13,000–14,000 coding transcripts in each condition (available in GEO: GSE180210) with an average Pearson correlation coefficient of 0.99 for replicates, demonstrating high reliability across the data (Figures 4B and S4B).
Figure 4. Deep RNA sequencing identifies genes with altered expression in response to different ERK activities.
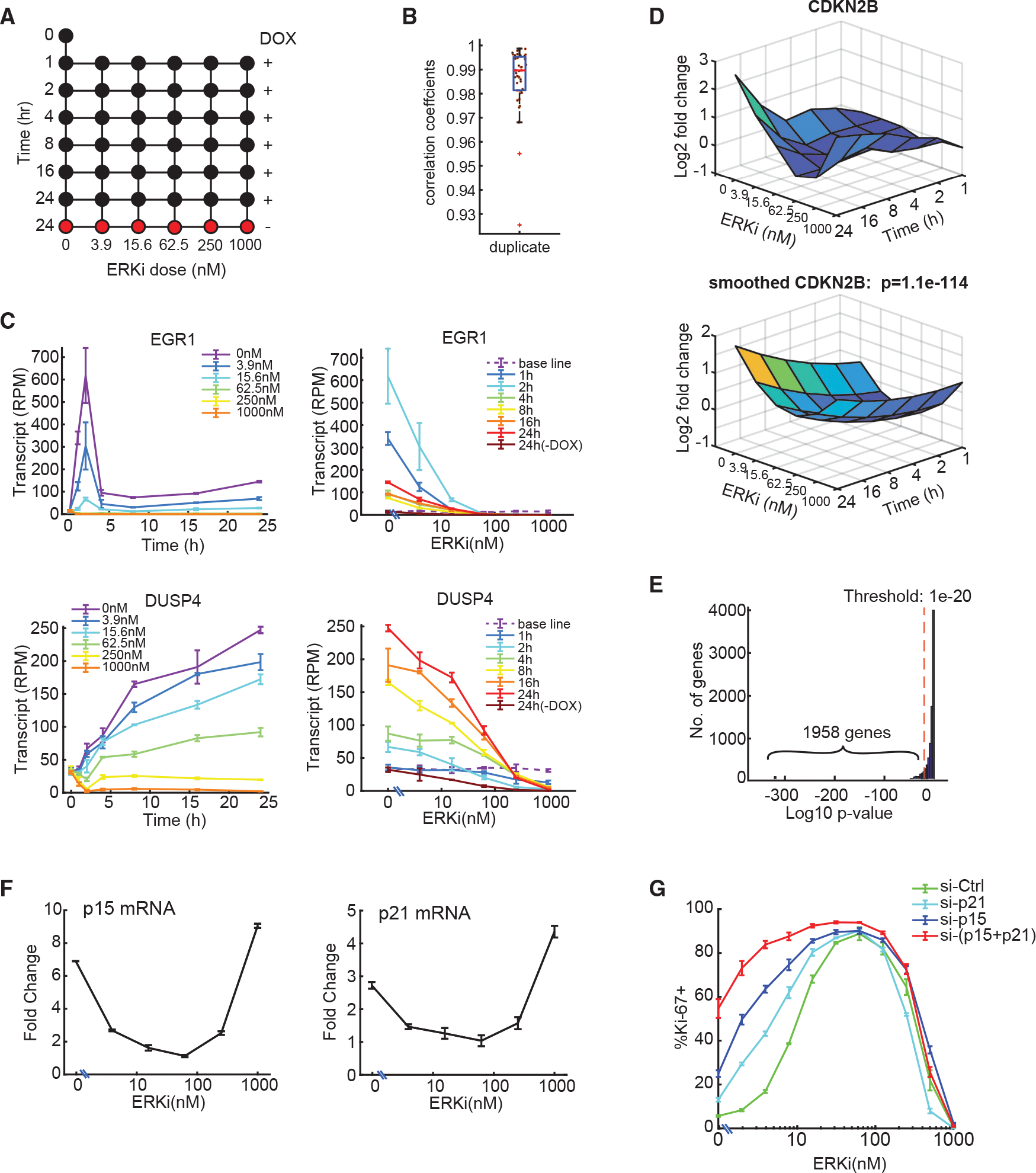
(A) RNA-seq experimental design. RPE/tet-BRAFV600E cells were treated with DOX and the indicated concentrations of ERKi for 1–24 h (43 conditions) in two independent replicates ( for no-treatment control).
(B) Boxplot showing correlation coefficients for RNA-seq replicates. Each dot represents a pair of replicates.
(C) RNA-seq measurements of EGR1 and DUSP4 transcripts as a function of treatment time (left) or ERKi dose (right) (mean ± SD; independent replicates; baseline represents no-treatment control).
(D) (Top) Log2 fold change of CDKN2B transcripts as a function of treatment time and ERKi dose. Values were normalized to untreated control. (Bottom) Smoothed quadratic surface fit for CDKN2B expression. The p value (likelihood ratio test) shows the goodness of fit between the quadratic surface and a flat surface.
(E) Histogram of p values of quadratic surface fits of all genes, as in (D). Genes with were considered differentially expressed.
(F) qPCR measurements of differential p15 and p21 mRNA expression in response to the indicated ERKi doses together with 250 ng/mL DOX for 24 h. Data represent mean ± SD ( replicates). Values were normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT) and reported relative to untreated control at time 0 h in the absence of DOX and ERKi (see also Figure S4F).
(G) RPE/tet-BRAFV600E cells were transfected with the indicated siRNA for 24 h, treated with DOX (250 ng/mL) together with different doses of ERKi for another 48 h, and then stained for the proliferation marker Ki67. The percentage of Ki67+ cells at each ERK dose is shown (mean ± SD of three replicates; n > 3,800 cells/well).
As a first step in validating the approach, we quantified the levels of two genes, EGR1 and DUSP4, whose expression is known to be responsive to ERK activation (Figure 4C).31 EGR1, an immediate-early response gene, exhibited a rapid and dramatic (~50-fold) induction within 2 h of DOX treatment and then decreased rapidly, remaining ~8-fold above its preinduction levels for the duration of the experiment (Figure 4C, left). This time course was consistent with its role as an immediate-early response gene. In contrast, DUSP4 rose steadily by ~6-fold over 24 h, consistent with its role as an early-response gene mediating negative feedback in the MAPK cascade (Figure 4C, left). Both the sequencing analysis (Figure 4C, right) and the targeted qPCR (Figure S4C) showed that induction of EGR1 and DUSP4 decreased in an ERKi dose-dependent manner. These results confirmed that our transcript profiling studies had a large dynamic range and could readily detect different ERK activity-dependent gene expression programs.
While it is common to analyze RNA-sequencing (RNA-seq) data to identify changes in expression associated with a single experimental variable (e.g., time or drug dose as in Figure 4C), given the dynamics of ERK activity, we identified genes differentially expressed as a function of both time and ERKi dose. Comprehensive cross-correlations between different treatment conditions showed that samples collected at different times and/or different ERKi doses could have similar transcriptional programs (high correlation), whereas samples collected at similar times and/or ERKi doses could have low correlation in their transcriptional programs, exemplifying the complexity of our datasets (Figure S4B). Thus, inferring differential expression using traditional approaches posed a substantial challenge, since both dose-response and temporal dynamics need to be accounted for. To address the challenge, we normalized each gene’s expression to the untreated control and used regression with quadratic terms to identify the best-fitting time-dose response for every gene using QR factorization of the Vandermonde regressor matrix32 (Figure 4D). Computing the quadratic surface approximation minimized noise across the landscape of treatments and emphasized time- and dose-dependent trends in the data.
To identify differentially expressed genes, we compared the goodness of fit between the quadratic response surface for each gene and a flat surface. The p values were computed using the standard likelihood ratio test and subjected to multiple-testing correction using Bonferroni-Holm.33 Data for CDKN2B (the p15INK4B CDK inhibitor) are shown in Figure 4D to illustrate the approach: CDNK2B expression was induced steadily over 24 h and exhibited a U-shaped response to ERKi concentration, with a minimum expression at 62.5 nM (Figure 4D, top). Quadratic regression on the data yielded a smoothed surface (Figure 4D, bottom) significantly different from a flat surface (, likelihood ratio test). Using this approach, we identified 1,958 genes that exhibited significant (, likelihood ratio test) differential expression over time and ERKi dose (Figure 4E). Gene ontology (GO) analysis showed that these genes fell into different functional categories, including extracellular matrix signaling, cancer pathways, DNA replication, and cell-cycle control (Figure S4D). We investigated the presence of known BRAFV600E, ERK, and senescence signatures in the 1,958 differentially induced genes using gene set enrichment analysis (GSEA) (Figure S4E), which revealed significant enrichment of the senescence and MAPK signatures examined, validating the effectiveness of our analysis method.
To identify genes likely to have a role in shaping the non-monotonic proliferation response, we searched for cell-cycle regulators whose expression was non-monotonic with respect to ERK activity. This analysis identified p15 and p21 as candidates, both of which showed a U-shaped expression profile with respect to ERK activity. We confirmed their expression profiles by qPCR (Figure 4F) and showed that their mRNA levels increased over time following BRAFV600E induction (Figure S4F). Simultaneous knockdown of p15 and p21 strongly diminished proliferation arrest at high ERK activity (low ERKi doses), without affecting the proliferation at low ERK activity (Figure 4G and validation of the knockdown on Figure S4G). These results show a role for p15 and p21 in mediating high-ERK induced cell-cycle arrest. The lack of proliferation rescue at the lower ERK range suggests that additional players are involved under these conditions.
Characterization and clustering of gene expression in relation to ERK signaling dosage
To identify overall trends in the data, we performed principal-component analysis (PCA) with the transcriptional data from all 43 conditions for the 1,958 differentially expressed genes. The first two principal components (PC1 and PC2) explained 81% of overall variance in the data, with PC1 and PC2 capturing 54.7% and 26.5% of the variance, respectively (Figure 5A). When the weights of PC1 and PC2 were plotted separately for every time point post treatment (Figure 5A), the variance of weights increased in a time-dependent manner, starting at 4 h (top right) and progressively forming a bell shape that reached its full extent at the 24 h time point (bottom right). The bell-shaped curve at 24 h closely resembled the correlation between ERK activity and proliferation response (Figures 1F, 1G, and 3D), suggesting that PC1 corresponded to differences in ERK activity and PC2 to differences in proliferative index.
Figure 5. Gene classification by clustering of gene expression reveals different response types to changing levels of ERK activity and proliferation.
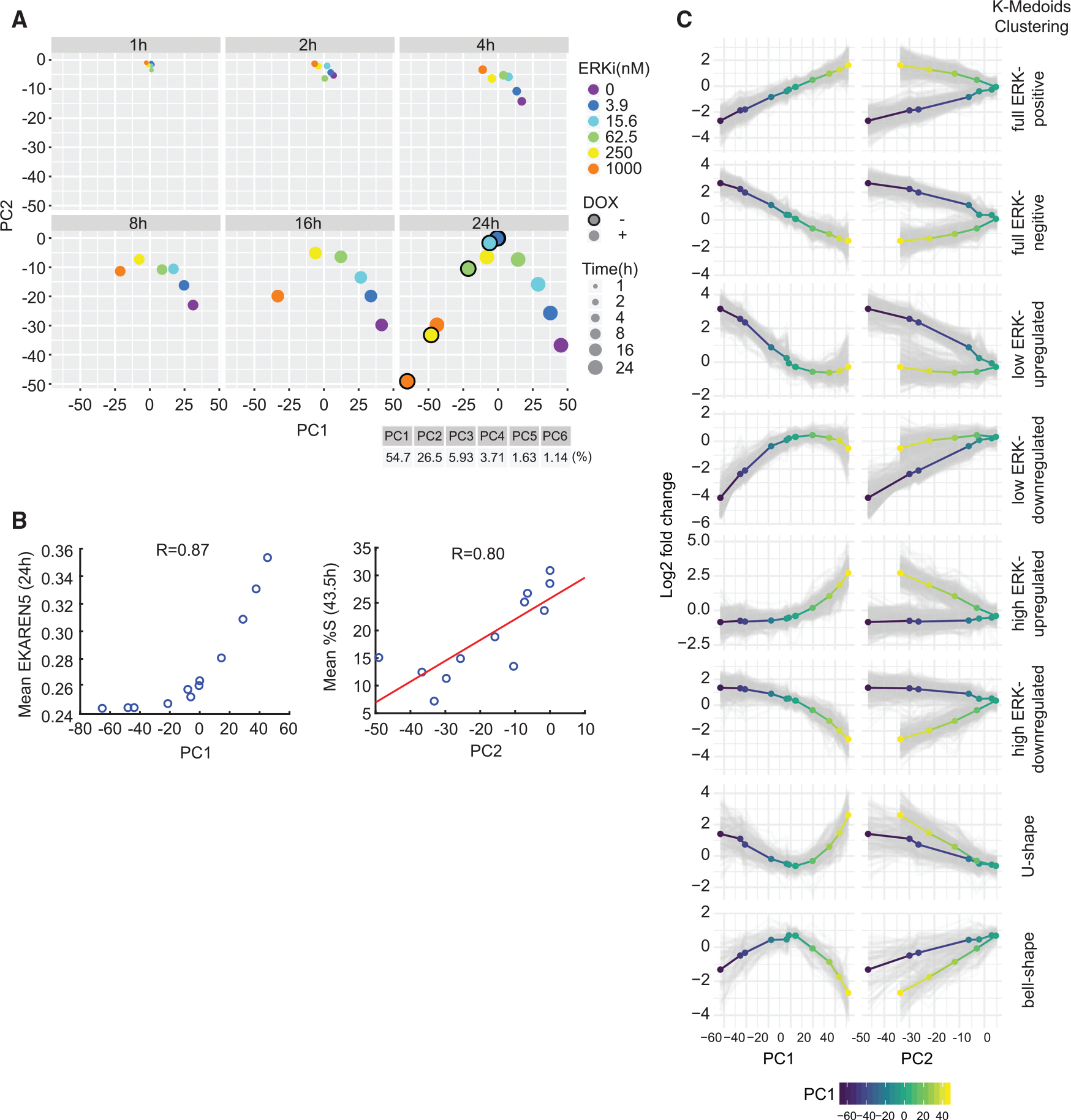
(A) Principal-component analysis (PCA) of log2 fold change in gene expression versus untreated control for each treatment (mean of two replicates) from RNA-seq.
(B) BRAFV600E Dual Reporter cells were treated with ERKi (0, 3.9, 15.6, 62.5, 250, 1,000 nM) with or without DOX and imaged for 43.5 h. PC1 values obtained at 24 h in (A) were plotted against the ERK activity measured at 24 h with corresponding treatments from live-imaging experiments. Similarly, PC2 values at 24 h post-treatment were plotted against the percentage of cells in S phase measured at 43.5 h from live-imaging experiments. We chose 43.5 h to account for the time delay between gene expression and cell-cycle entry.
(C) k-medoids clustering of the 1,958 differentially expressed genes identified from RNA-seq experiments. Log2 fold-change expression data of each differentially expressed gene at 24 h were clustered by k-medoids clustering (). Mean expression levels of each cluster are shown as multi-colored lines (blue-yellow represents low-high PC1 values).
To test this hypothesis, we measured the mean ERK activity and degree of proliferation at each condition assayed by RNA-seq. BRAFV600E Dual Reporter cells were treated with DOX and different doses of ERKi simultaneously (mirroring the experimental conditions in RNA-seq) and subjected to live-cell imaging to monitor mean ERK activity and the fraction of cells in S phase over time. PC1 values (Figure 5A, bottom right) were highly correlated (R = 0.87) with mean ERK activity measured under the same conditions using the EKAREN5 reporter (Figure 5B, left). The fraction of cells in S phase (measured using mCherry-PIP) was highly correlated with the value of PC2 (; Figure 5B, right). Thus, we conclude that the first principal component is a proxy for ERK activity, whereas the second principal component is a proxy for proliferation index. Moreover, we found that the value of PC2 was similar for conditions with similar average ERK activity, regardless of how that activity level was achieved. For example, a PC2 value of approximately −30 was achieved in cells not expressing BRAFV600E and treated with low-dose (250 nM) ERKi, as well as in cells with BRAFV600E induced and treated with high-dose ERKi (1,000 nM) (Figure 5A, 24 h plot, yellow and orange circles). These data also strongly suggest that the two primary drivers of gene expression, over a wide range of conditions, are the ERK activity level and the extent of proliferation.
To investigate the possibility of non-specific ERKi targets among the 1,958 differentially expressed genes, we determined whether the effects of the inhibitor on these genes could be rescued by increasing ERK expression. Of the 1,958 genes, many were differentially expressed in the presence of 250 nM ERKi (compared with untreated cells). However, gene expression was restored to resemble that in the control when cells were treated with 250 nM ERKi plus DOX to induce BRAFV600E and ERK activity (Figure S5A). Furthermore, comparing two conditions with similar ERK activities (ERKi 250 nM − DOX versus ERKi 1,000 nM + DOX; Figure S5B, left) revealed a high correlation in gene expression (; Figure S5B, right), suggesting that the differentially expressed genes were primarily responding to ERK activity. These results together suggested that ERKi effects could be rescued by overexpressing BRAFV600E and that the potential off-target effects of ERKi were likely very minimal.
We expected the expression levels of genes to have a differential pattern of responses to varying levels of ERK signaling (PC1) and proliferation rates (PC2). To further subdivide gene expression programs, we used unsupervised k-medoids clustering based on PC1 and PC2 values at 24 h (Figure 5C). k-medoids is a variant of k-means clustering that is robust to outliers and also allows the use of arbitrary distance metrics. We computed pairwise Euclidean distances for the log2 fold changes (relative to an untreated control) in the set of 1,958 genes and then performed k-medoids clustering. With clusters, differences in relationship between changes in expression and PC1 or PC2 values were evident (Figure 5C). For instance, in the “full ERK-positive” cluster, gene expression increased with increasing PC1 value, suggesting a strong positive correlation. However, in the same set of genes, a single PC2 value could have two different levels of gene expression (from treatments that have either high or low PC1 values), indicating a weak correlation between gene expression and PC2 value. Similar clustering results were obtained with data from all the time points.
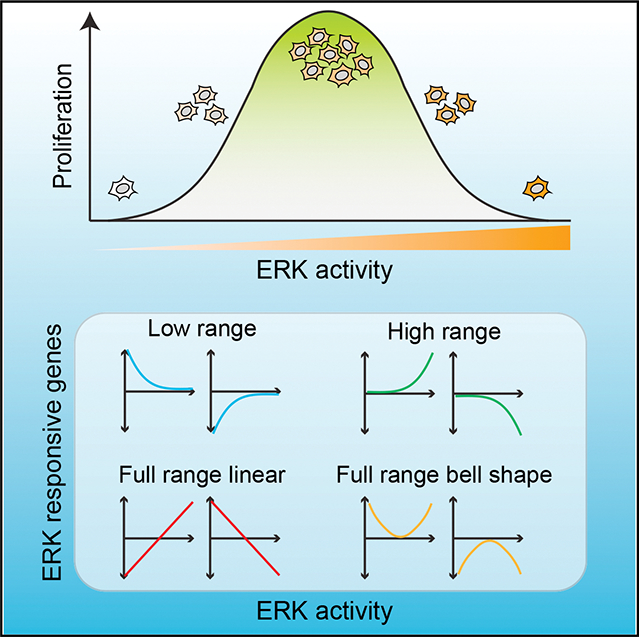
The results of k-medoids clustering yielded clusters that could be grouped by visual inspection into four qualitatively different classes of responses to ERK activity, each having two clusters with opposite trends (Figure 6). Class I included genes whose expression was correlated either positively () or negatively () with ERK activity across its full range, resulting in a linear relationship (“full-range ERK responder”; Figure 6A, red lines). Canonical negative feedback regulators of the MAPK pathway, such as DUSP4/6 and SPRY2, belonged in this group.31 Class II genes were those in which differential gene expression fell with ERK activity in a non-linear, “convex” manner (blue lines). At low ERK activity levels, 248 (12.7%) were significantly upregulated and 404 (20.6%) were downregulated. Class II included genes involved in DNA replication, DNA damage repair, and the G1/S cell-cycle transition (e.g., G1 cyclins) (Figure S6A). Class III was similar to class II with the response window shifted to higher ERK activity ranges (green lines). This class included 281 upregulated genes (14.4%) and 231 (11.8%) downregulated genes. Class III included genes involved in differentiation, migration/motility, cytokine response, and growth factor activity. Class IV (orange lines) comprised genes with a bell-shaped () or U-shaped () response curve, having the greatest differential gene expression at the lowest and highest ERK levels. This class included the CDK inhibitor CDKN2B (p15INK4B). Expression of class IV genes was highly dependent on PC2, as seen by a monophasic response where similar expression levels were obtained for a given PC2, regardless of PC1 value (Figure 5C, bottom). The expression profiles of the 20 most differentially expressed genes in each category are shown in Figure S6B. The classifications of gene expression in Figure 6A exemplify different strategies cells utilize in response to varying levels of ERK activity.
Figure 6. Patterns of ERK-dependent gene expression clustered by ERK activity and time of induction.
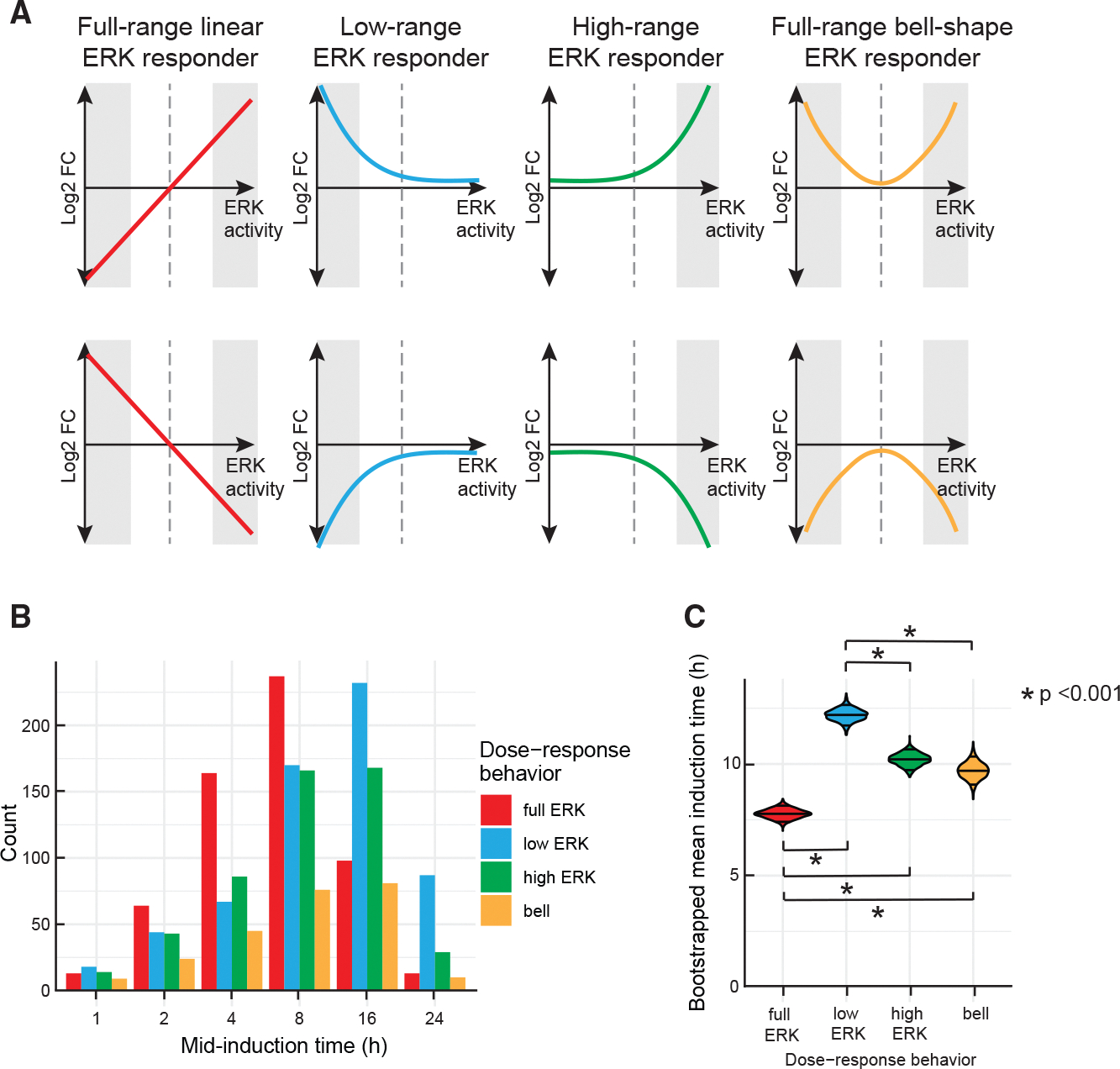
(A) Four distinct classes of gene expression patterns as a function of ERK activity. Full-range ERK responders include genes with expression proportional to ERK activity across the entire range of activity levels. Low- and high-range ERK responders show differential gene expression only at the low and high ranges of ERK activity, respectively, while showing minimal response at the opposite end of the range. Full-range bell-shape ERK responders include genes that have a bell- or a U-shaped response, with greatest differential expression at low and high ERK activities. Shaded areas indicate the ERK range in which the class is most responsive. Dashed line marks the ERK activity for untreated cells used for normalization. For illustration purposes, we substituted ERK activity for PC1, since the two are highly correlated.
(B) Temporal dynamics of each class of genes shown in (A) in response to DOX and ERKi treatment. Genes in each class were grouped according to the earliest time point at which they achieved 50% of the maximal change observed over the course of the experiment (see STAR Methods). The time point of mid-induction is shown on the x axis, and the number of genes falling into each category is shown on the y axis.
(C) Mean induction time of ERK responder classes. The violin plot shows bootstrapped estimates of the means for each cluster. Significant differences in the induction time between ERK-response clusters are highlighted with black bars. The p values are derived from ANOVA followed by Tukey’s honest significant difference test.
We next investigated the timing of gene expression changes. To obtain a simple measure of the timing of gene induction or repression, we considered the time at which each gene first reached half of its maximal differential gene expression value in a weighted average that aggregated data across different ERKi doses (see STAR Methods). Genes were grouped according to the time of this absolute half-maximum change in expression (the “mid-induction time”). This analysis revealed genes responding on rapid (1–2 h), intermediate (4–8 h), and slow (16–24 h) time scales (Figure S6C). For instance, the previously mentioned EGR1 and DUSP4 exhibited rapid and intermediate time scales of induction, respectively.31 In general, genes in any ERK response category from Figure 6 were found in any temporal response category. However, we found that genes in the full-range ERK responder group (class I; red lines in Figure 6A) tended to respond at earlier times in BRAFV600E induction than genes in other groups. Genes in the low-ERK responder group (class II) responded most slowly to BRAFV600E induction (Figure 6C). These results suggest only modest correlations between time- and ERK activity-dependent gene regulation.
We envision that combinations of different temporal and ERK dose-response classes contribute to the overall bell-shaped proliferation response. For instance, low G1 cyclin expression (low ERK downregulated group) combined with high CDK inhibitor expression (U-shaped group) at low ERK activity could trigger cell-cycle arrest. This likely explains the lack of proliferation rescue at low ERK range with co-knockdown of p15 and p21, as G1 cyclin expression was low (Figure 4G). Similarly, upregulation of CDK inhibitors at high ERK activity (U-shaped group) could also trigger cell-cycle arrest (Figure 4G). Even though the proliferation response was stable over time (Figure 1F), the underlying mechanisms could vary over time, as we detected a large variation in the kinetics of ERK-responsive genes. Thus, a combination of ERK activity-responsive genes could function collaboratively to achieve heterogeneous proliferation responses within a population of cells, across time or ERK activity levels.
DISCUSSION
While OIS is an efficient way to halt tumor development, it is not uncommon to see genetically homogeneous cells respond to oncogenes in an asynchronous and heterogeneous manner. In this paper, we showed that cell-to-cell variability in oncogene induction can be traced to differences in ERK activity at a single-cell level. A narrow range of ectopic BRAFV600E expression generated cells with a wide range of ERK activities. Such “noise” is common in intracellular signaling networks,34 commonly arising from transcriptional bursting or unequal partitioning of cytoplasm at cell division,35,36 and generates a log-normal distribution of protein concentrations in a population of cells that can cause dramatic differences in their downstream protein activity and cell fate.37 Using a recently developed single-cell PIP reporter and live-cell imaging, we found that initiation of cell-cycle arrest is preceded by a prolonged G2 phase, but not by a burst of proliferation as previously described.38,39 Single-cell imaging revealed a non-monotonic, bell-shaped relationship between ERK activity and cell proliferation. Our data showed that proliferation is highly sensitive across the entire range of ERK activity. Thus, a population of cells expressing BRAFV600E will continuously exhibit differences in ERK activity and heterogeneous outcomes, with some cells continuing to proliferate and others undergoing OIS. We speculate that cells that can proliferate in the presence of elevated BRAFV600E are likely to give rise to tumors. It is also possible that cells that arrest in the presence of high ERK activity could once again become proliferative in the presence of the BRAF and MEK inhibitors used therapeutically (also see Figure S1A).
Our data support a Goldilocks principle (“just the right amount”) for ERK activity, such that an intermediate amount promotes proliferation, whereas much higher or lower levels prevent it. Accordingly, hyperactivating mutations in ERK are much less common than those in its upstream regulators RAS and RAF,40,41 as ERK mutations are likely to lead to OIS. Oncogenic RAS and RAF mutations may promote complex gene expression programs that either limit ERK activity or bypass the arrest caused by high ERK levels. In line with the Goldilocks principle, hyperactivation of MAPK signaling is deleterious to BRAFV600E melanoma cells.42 Moreover, melanomas with acquired resistance to RAF and MEK inhibitors become drug dependent for their continued proliferation due to elevation of their MAPK signaling.43 In contrast, in normal cells, reducing ERK activity rescued cells from senescence and facilitated cell transformation by oncogenic RAS.18 These results all pointed to a tumor-suppressive role of ERK signaling and support our bell-shaped ERK-proliferation model.
Our study suggests that the intensity of ERK signaling plays a pivotal role in determining the final proliferation outcome. Yet, how the strength of ERK signaling connects to the outputs of this pathway is largely unclear. Induction of BRAFV600E for different amounts of time with different doses of ERKi made it possible to generate cells with a wide range of ERK activities. RNA-seq revealed four distinct classes of ERK-regulated genes, which differed with respect to their relationships between ERK activity and the level of gene expression. These categories responded over the full range (class I) or portions of ranges (classes II–IV) of ERK activity sampled in this study. Previous studies that focused on gene expression at only high or low ERK levels would not have resolved these four classes of ERK dependency. For example, genes that increase with ERK activity in all four classes would have been categorized in a single group when assayed at high ERK activity. The methods used here enable further refinement of specific ERK-dependent signatures by identifying whether the genes are full-range linear ERK responders, low- or high-range ERK responders, or U-shape ERK responders. This analysis suggests that suppression of proliferation at low and high ERK levels likely proceeds via distinct transcriptional programs. Our ERK activity- and time-dependent classifications pave the way for dissecting the relevant pathways at different levels of ERK activity.
Our classification of genes also suggests that a single class of genes or a combination of multiple classes of genes could achieve the bell-shaped proliferation response. For instance, positive cell-cycle regulators in a bell-shaped class, or negative cell-cycle regulators in a U-shaped class could, on their own or in combination, generate a bell-shaped proliferation response. In support of a combinatorial mechanism suppressing proliferation at low ERK activity, we found that G1 cyclins were downregulated and CDK inhibitors were upregulated. It is thus likely that cells take a network approach rather than a single-gene strategy to regulate proliferation across a range of ERK activity levels. It is also important to note that our gene classes are not exhaustive and that additional types of regulation (e.g., protein levels or post-translational modifications) must co-exist for a robust bell-shaped proliferation response. For instance, high ERK activity can prevent cell-cycle progression by inducing degradation of key regulators,18 by modulating senescence-associated secretomes,44 or by engaging homeostasis at tissue levels.12 We envision a broader set of genes, and diverse regulatory mechanisms are needed for cells to engage a robust and coherent bell-shaped proliferation response.
In summary, our studies provide a detailed and comprehensive map of the input-output relationship between ERK activity, proliferation response, and gene expression programs in non-transformed cells. The data provide an explanation for cell-to-cell heterogeneity in OIS induction in a nominally uniform population of proliferating cells. Our data help to explain the bell-shaped relationship between MAPK signaling and proliferation while also revealing substantial complexity in time- and activity-dependent changes in gene expression. Such insights should improve our ability to study OIS in vivo and ultimately develop treatment regimens and therapeutics that exploit OIS to block cancer growth.
Limitations of the study
Our studies began to uncover the sources of heterogeneity in oncogene-induced cell-cycle arrest and suggested that complex interactions of regulatory pathways likely govern the response. However, several unanswered questions remain. This work used a single widely studied human RPE cell line to understand the input-output relationship between ERK activity and proliferation. The cell line was chosen for its sensitive response to BRAFV600E induction and suitability for long-term live-cell imaging. Further studies will be needed to test the generality of our findings in other non-transformed cell types that can recapitulate the early stages of oncogene expression in otherwise healthy cells. Although we found a non-monotonic relationship between proliferation and ERK activity, the duration of G2 phase seemed to scale with ERK activity (Figure S3C). These results could indicate that the duration of G2 phase is not directly proportional to the number of cells that arrest at any given ERK activity level and that the mechanisms regulating arrest duration are separate from those that influence whether a cell arrests in the first place. Our RNA-seq experiments revealed four categories of genes that respond differentially to ERK activity. These results are limited in distinguishing which genes are directly regulated by ERK signaling and which are regulated by the proliferation state of the cell. However, the early time points of sample collection (from 1 to 24 h) increase the likelihood of identifying genes that are direct ERK targets. Our study did not identify a minimal gene expression network responsible for the proliferation response and did not explore mechanisms beyond gene expression changes (e.g., post-translational modifications) that may contribute to this phenomenon. Future work in these areas will be important in understanding responses to oncogene induction and the resulting heterogeneity in cellular outcomes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Galit Lahav (galit@hms.harvard.edu).
Materials availability
Plasmids will be available upon request from the lead contact.
Data and code availability
RNA seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Results of RNA seq analysis used to generate main and supplemental figures in this paper are available at Synapse database (https://www.synapse.org/#!Synapse:syn21411369/files/).
The original code for analysis of RNA seq data is publicly available at GitHub (https://github.com/clemenshug/erk_senescence).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) antibody | Cell Signaling Technology | Cat# 4370, RRID: AB_2315112 |
| Phospho-Rb (Ser807/811) (D20B12) antibody | Cell Signaling Technology | Cat# 8516, RRID: AB_11178658 |
| Rb (4H1) antibody | Cell Signaling Technology | Cat# 9309, RRID: AB_ 823629 |
| p16 (H-156) antibody | Santa Cruz Biotechnology | Cat# sc-759, RRID: AB_632105 |
| p21 antibody | BD Biosciences | Cat# 556430, RRID: AB_396414 |
| Ki-67 (8D5) antibody | Cell Signaling Technology | Cat# 9449, RRID: AB_2797703 |
| p27 Kip1 (D69C12) XP Rabbit monoclonal antibody | Cell Signaling Technology | Cat# 3686, RRID: AB_2077850 |
| Anti-MCM6 antibody [EPR17686] | Abcam | Cat# ab201683, RRID: AB_2924827 |
| Anti-β-Actin monoclonal antibody | Sigma-Aldrich | Cat# 5316, RRID: AB_476743 |
| Anti-Raf-B antibody (F-7) | Santa Cruz Biotechnology | Cat# sc-5284, RRID: AB_626760 |
| Anti-HA High Affinity; Rat monoclonal antibody (clone 3F10) | Roche | Cat# 11867423001, RRID: AB_390918 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7076, RRID: AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID: AB_2099233 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID: AB_2576217 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11077, RRID: AB_2534121 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21235, RRID: AB_2535804 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| CSII-EF1-H2B-mTurquoise (lentiviral) | Spencer et al.21 | N/A |
| CSII-EF1-mVenus-hGeminin (1–110) (lentiviral) | Sakaue-Sawano et al.19 | N/A |
| CSII-EF1-mCherry-dE2F PIP (lentiviral) | This work | N/A |
| CSII-EF1-mVenus-dE2F PIP (lentiviral) | This work | N/A |
| LV-EKAREN5-NLS (lentiviral) | Addgene | Plasmid#167818 |
| LIX402-BRAFV600E-HA-Puro (lentiviral) | This work | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Hoeschst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | Cat# H3570 |
| SCH772984, ERK inhibitor | MedChem Express | Cat# HY-50846 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891-5G |
| SMARTpool: siGENOME Non-targeting siRNA control pools | Horizon Discovery | Cat# D-001206-14-05 |
| SMARTpool: siGENOME Human CDKN1A siRNA | Horizon Discovery | Cat# M-003471-00-0005 |
| SMARTpool: siGENOME Human CDKN2A siRNA | Horizon Discovery | Cat# M-011007-03-0005 |
| SMARTpool: ON-TARGETplus CDKN2B siRNA | Horizon Discovery | Cat# L-003245-00-0005 |
| SMARTpool: siGENOME Human CDKN1B siRNA | Horizon Discovery | Cat# M-003472-00-0005 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668027 |
| Senescence Associated β-Galactosidase staining kit | Cell Signaling Technology | Cat# 9860 |
| Click-iT™ Cell Reaction Buffer Kit | Thermo Fisher Scientific | Cat# C10269 |
| Click-iT™ EdU (5-ethynyl-2’-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Click-iT™ Alexa Fluor® 647 Azide, Triethylammonium Salt | Thermo Fisher Scientific | Cat# A10277 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and processed RNA-seq data | GEO (Gene Expression Omnibus) | GEO: GSE180210 |
| Results of RNA-seq analysis used to generate main and supplemental figures in the manuscript | Synapse database | https://www.synapse.org/#!Synapse:syn21411369/files/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: RPE hTERT | S.J. Elledge Lab (Harvard Medical School) | N/A |
| Human: RPE + tet-BRAFV600E-HA | This work | N/A |
| Human: RPE + H2B-mTurquoise + Venus-dE2F PIP | This work | N/A |
| Human: RPE + tet-BRAFV600E-HA + EKAREN5 + mCherry-dE2F PIP | This work | N/A |
| Human: RPE + EKAREV-NLS | This work | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| CDKN2B PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.25069372.g |
| CDKN1B PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.45564663 |
| CDKN2A PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.40743463.g |
| EGR1 PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.40805543.g |
| DUSP4 PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.18820216 |
| p21 forward qPCR primer: TGTCACTGTCTTGTACCCTTG | Purvis et al.45 | N/A |
| p21 reverse qPCR primer: GGCGTTTGGAGTGGTAGAA | Purvis et al.45 | N/A |
| HPRT forward qPCR primer: GTATTCATTATAGTCAAGGGCATATC | This paper | N/A |
| HPRT reverse qPCR primer: AGATGGTCAAGGTCGCAAG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pPB-CAG-EKAREV-NLS (piggyBac) | Komatsu et al.26 | N/A |
| pCMV-hyPBase | Komatsu et al.26 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Scripts for analysis of RNA sequencing data | This work | https://github.com/clemenshug/erk_senescence |
| Software for automatic segmentation and quantification of immunofluorescence images | Salmeen et al.46 | N/A |
| Software for automatic segmentation, and quantification of fluorescent reporter cells following live imaging | Cappell et al.47 | https://github.com/scappell/Cell_tracking |
| p53 Cinema Single Cell Tracking | Reyes et al.48 | https://github.com/balvahal/p53CinemaManual |
| EllipTrack | Tian et al.49 | https://github.com/tianchengzhe/elliptrack |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human retinal pigment epithelial (RPE) cells immortalized with human telomerase expression (RPE-hTERT, a kind gift from S.J. Elledge, Harvard Medical School) were grown in DMEM/F12 supplemented with 10% fetal bovine serum (FBS), 2 mM L-Glutamine, Antibiotic-Antimycotic (100 U/ml penicillin, 100 μg/ ml streptomycin and 250 ng/ ml Amphotericin B), and 50 μg/ml hygromycin B. RPE cells were treated with 250 ng/ ml DOX to induce BRAFV600E unless otherwise noted.
Cell line construction
To establish RPE/tet-BRAFV600E-HA cell line, C-terminal HA-tagged BRAFV600E construct was made by cloning the full-length BRAFV600E expression cassette (Addgene plasmid # 15269) into a lentiviral HA-containing backbone with tet-inducible promoter (Addgene plasmid # 41394, pLIX402). RPE cells were then infected with lentivirus carrying pLIX402-BRAFV600E-HA and selected with puromycin (2 μg/ ml) to obtain mixed cell clones. Single cell clones were expanded through limited dilution and subsequently screened for HA expression in the presence of doxycycline. To establish dE2F PIP reporter lines, an expression cassette harboring Drosophila E2F1 PIP fragment (comprised of a.a. 1–187) fused to the C-terminus of Venus or mCherry fluorescent protein was cloned into the CSII-EF1 lentiviral vector. RPE cells transduced with lentiviruses carrying H2B-mTurquoise, mCherry-dE2F PIP and Venus-Geminin (1–110) or lentiviruses carrying H2B-mTurquoise and Venus-dE2F PIP were sorted on a BD FACSAria II high speed cell sorter to obtain pure populations expressing the desired fluorescent proteins. To establish RPE/EKAREV-NLS reporter line, RPE cells were co-transfected with pPB-CAG-EKAREV-NLS26 and pCMV-hyPBase transposase vector (A. Bradley, Sanger Institute) and FACS sorted to obtain pure populations. To establish RPE/tet-BRAFV600E-HA + EKAREN5 + mCherry-dE2F PIP dual reporter line, verified RPE/tet-BRAFV600E-HA single cell clone was transduced with EKAREN5 (ERK FRET reporter)25 and mCherry-dE2F PIP lentiviruses and single cell clones harboring both reporters were obtained through single-cell sorting and subsequent expansion.
METHOD DETAILS
Time-lapse microscopy
Cells were plated in poly-D-lysine-coated glass-bottom plates (MatTek Corporation) and switched to phenol-red free culture medium supplemented with 10% FBS prior to live imaging. Cells were imaged using a Nikon Eclipse TE2000 microscope equipped with a chamber for controlled temperature (37%) and CO2 (5%) environment. All live-cell imaging was performed with a 10x Plan Apo objective (Nikon) and a Hamamatsu Orca ER camera using CFP, YFP, mCherry filter sets (Chroma). For EKAREV and EKAREN5 reporter imaging, the FRET signal was collected using customized ECFP/EYFP FRET filter sets with ET436/20x, ET535/30m, and T455lp mounting into the Nikon TE2000/Ti cube.
Cell count assay
RPE cells stably expressing tet-BRAFV600E and EKAREV (served as nuclear marker) were plated in 96-well plates and treated with DOX for 8 days. Cells were then washed 5 times with fresh media to remove DOX (or mock washed) followed by live imaging (45min/frame, 4x objective using GFP filter set) in an IncuCyte Zoom live imager for 8 days. Total cell number per well was derived using IncuCyte analysis software.
siRNA knockdown
Synthetic siRNAs used for this study were from Dharmacon siGenome SMART pool and were used at 13.3 nM with Lipofectamine 2000 reagents (Invitrogen) according to manufacturer’s protocol. The following siRNAs were used: control siRNA (non-targeting #2), siGenome pooled set of four siRNAs for p15, p16, p21 and p27. Cells were treated with DOX and ERKi after 24h of siRNA transfection. The knockdown effects were then measured 2 days post treatments (72h post siRNA transfection). Specific antibodies were used to verify the target knockdown.
Western blot and senescence associated β-galactosidase assay
Cells were harvested in NuPAGE™ LDS Sample Buffer (Invitrogen) for 10 min at 70°C and sonicated to shear genomic DNA. Protein samples were separated by electrophoresis using 4–12% Bis-Tris mini protein gels (Invitrogen) and transferred to Immun-Blot PVDF membranes (Bio-Rad #1620177). Blots were incubated with primary antibodies at 4°C and then with HRP conjugated secondary antibodies for 1 hour at room temperature. HRP was detected using ECL substrates (Thermo Scientific #34076) and Licor Odyssey Fc Imager.
Immunofluorescence
Immunostaining was performed in 96-well plates and all washes were done with the EL406™ Microplate Washer (BioTek). In brief, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 15 min and blocked with Odyssey® blocking buffer (LI-COR) for 1h before applying different antibodies. Primary antibodies were incubated overnight at 4°C. Appropriate Alexa Fluor® conjugated secondary antibodies were then used. For EdU staining, cells were pulsed with 10 μM EdU for 30 min (or as indicated) prior to fixation and processed according to manufacturer’s instructions (Invitrogen #C10340). Cells were imaged with a 10x objective using an Operetta High Content Imaging System (Perkin Elmer, CT) or ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices, CA). 9 sites were imaged in each well for 96-well plates.
Image analysis
Images for the immunostaining experiments were analyzed using MATLAB image analysis programs.46 Briefly, nuclear centroids were identified in images of Hoechst staining after applying a low-pass Gaussian filter and local background subtraction. A nucleus mask was generated for each cell by expansion from the centroid to reach 30% of maximum intensity. The nuclear pERK, BRAFV600E-HA, EdU and pRB mean intensity were measured after local background subtraction. The threshold level used to determine pRB and EdU positive cells was set using a k-means clustering algorithm on a day-to-day experiment basis.
Single-cell tracking and quantification following live-imaging
For population analysis of individual time frames, images were quantified and analyzed using MATLAB scripts.47 To track single-cells following long-term live imaging, cells were tracked semi-automatically using a combined method of EllipTrack49 and p53Cinema Single Cell Tracking Software.48 In brief, EllipTrack segments cells by fitting nuclear contours with ellipses and tracks cells using a machine learning algorithm. The cell tracks were then manually curated using p53Cinema Single Cell Tracking Software that allows for real-time user correction of tracking and annotation of division events. Finally, verified tracks were kept for downstream analysis and signals from each color channel were extracted in the cell nuclei.
Automatic identification of G1/S and S/G2 transitions
Single-cell traces of Venus (or mCherry)-PIP construct intensities were smoothed, normalized to 0–1 range, and partitioned by mitosis (annotated by EllipTrack and p53Cinema). Each trace fragment was then processed in four steps to detect the G1/S and S/G2 transitions. First, seed regions for S phase, defined as time periods whose trace values were smaller than 0.1, were proposed. Only regions longer than 20 frames were kept (10 min/ frame). If no region was proposed but the trace fragment was bounded by two mitoses at least 50 frames apart (indicating that a real cell cycle likely took place), the threshold for the trace values would be relaxed. On the contrary, if multiple regions were proposed (caused by mitosis skipping or human annotation error), each region would be analyzed independently. Second, the S/G2 transition was detected for each seed region. The slopes of the trace values were calculated from the center of the seed region to the end of the trace fragment, and the S/G2 transition was defined as the first time point whose slope was greater than 0.004/ frame (10 min/ frame). To reduce the impact of noise, a second requirement that 3 of 5 of its next five time points should also have slopes greater than 0.004/ frame was imposed. The threshold for the slopes would be gradually relaxed if no such time point was detected. Third, the G1/S transition was detected for each seed region. A reference time point was first proposed following the procedure in the second step, except that 1) the slopes were calculated and examined from the center of the seed region to the beginning of the trace fragment, and 2) the threshold for the slopes was 0.01/ frame. The G1/S transition was then defined as the last time point before the reference whose trace value was a local maximum. Finally, the detection results were manually examined and adjusted to ensure correctness. After cell cycle phases and their transitions were identified, G1 or G2 arrested cells were defined as cells remaining longer than 30 h in G1 or G2 phase at the end of imaging, respectively.
ERK FRET reporter quantification
To quantify ERK activity, CFP, YFP and FRET images were acquired in RPE/ tet-BRAFV600E-HA + EKAREN5 + mCherry-dE2F PIP dual reporter cells. FRET images were taken by CFP excitation and YFP emission. Images were then subjected to flat field correction (to eliminate uneven illumination) and local background subtraction. The FRET signal was calculated on a pixel-by-pixel basis as follows. First, a FRET image was corrected for bleed-through from CFP and YFP channels.
α: bleed-through of CFP into FRET channel upon CFP excitation
β: bleed-through of YFP into FRET channel upon CFP excitation of YFP
The two microscope-specific bleed-through parameters, α (0.53) and β (0.23), were determined using cells transfected with CFP or YFP alone. Then, the corrected FRET image ([FRET]corr) was normalized by the CFP image to obtain the FRET signal ([FRET]corr/[CFP]). ERK activity was calculated from the median value from the nuclear compartment of the FRET signal of each cell.
Total RNA sample preparation and quality control
RPE/tet-BRAFV600E-HA cells were plated in 6-well plates (75,000 per well) and allowed to grow for 72 h till 50% confluency. Cells were then treated with the indicated concentrations of ERK inhibitor (SCH772984) alone for 24 h or in combination with DOX (250 ng/ ml) for variable length of time as indicated by the experimental design. Each condition was performed twice on two different days for a total of two biological replicates. For RNA sequencing experiments, cells were lysed with 600 μl Trizol per well and total RNA was prepared using Direct-zol-96 RNA Kits according to the manufacturer’s protocol. For quantitative PCR experiments, cells were lysed and total RNA extracted using Qiagen RNAeasy Plus mini kits according to manufacturer’s protocol. Sample concentrations were determined by Nanodrop and RNA quality was assessed on a subset of samples by Bioanalyzer (Agilent); all samples scored RINs of > 9.0.
Quantitative PCR (qPCR)
2 μg of total RNA was used to generate complementary DNA (cDNA) using the high-capacity cDNA reverse transcription protocol (Applied Biosystems). q-PCRs were then performed using 1/200 of the total of cDNA (per reaction in a 384-well plate), 300 nM primer, and SYBR Green reagent following the manufacturer’s protocol (Applied Biosystems). Reactions were normalized to HPRT as a loading control. qPCR primers used are listed in key resources table.45
RNA sequencing library preparation
RNA sequencing library preparation was performed with the High Throughput TruSeq Stranded mRNA Library Prep Kit (Illumina) following the manufacturer’s protocol at half reaction volume. Input for each sample consisted of 300–500 ng of RNA and 5 μl of 1:500 diluted ERCC spike-in mix 1 (Ambion). Libraries were amplified for 12 cycles during the final amplification step. Libraries were quantified using the Qubit dsDNA HS assay (Thermo Fisher Scientific). Library size and quality were spot checked for a subset of samples by Bioanalyzer (Agilent). The average size of cDNA fragments in the libraries was 370 base pairs. Libraries were pooled at equimolar concentrations then the pool was quantitated using the KAPA library quantification kit (KAPA Biosystems). Libraries were sequenced single end 114 base pairs using NovaSeq_SP full flow cell (Illumina) at the Bauer Core Facility (Harvard University).
RNA-seq data processing
Reads were processed to counts using the bcbio-Nextgen toolkit (https://github.com/chapmanb/bcbio-nextgen) as follows: (1) Reads were trimmed and clipped for quality control in cutadapt v2.3; (2) Read quality was checked for each sample using FastQC v0.11.8; (3) High-quality reads were then aligned to the human assembly and gene annotation GRCh38.97 using Hisat2 v2.1.050; (4) Gene-level transcript-counts were calculated using HTseq-count v0.9.1. Only data from genes annotated as protein-coding according to annotation from GRCh38.97 were kept for further analysis. Gene expression data (RNA seq) were deposited in the GEO (Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/, accession number: GEO: GSE180210).
Differential expression analysis
Differential expression of genes was analyzed using DESeq251 by fitting a linear mixed effect that expressed the number of reads for each gene using a negative binomial distribution of the form with mean and dispersion . The mean for each gene was modeled by a linear equation taking into account the sample treatments and batch with the untreated sample at time zero as intercept.
Log-fold changes were adjusted using procedures implemented in apeglm,52 which estimates posterior distributions of the coefficients in the linear models that were fitted by DESeq2.
Selecting differentially expressed genes
In this setting, inferring differential expression poses a great challenge, since both dose-response and temporal dynamics need to be accounted for. To account for dependency on both time and dose, we used multiple regression with quadratic terms to describe the time-dose response surface (Log-fold changes) for every gene. As the same time points and doses were measured for all genes, we only computed a single QR-decomposition of the Vandermonde regressor matrix32 and then computed regression using matrix-matrix multiplication. This type of factorization is an effective way to compute least-squares fits for a large number of genes, as computationally expensive factorization only needs to be performed once for a specific set of dose-time combinations. Gene-specific regression then only required computationally cheap matrix-matrix multiplication. To identify differential expression, we compared the goodness of fit between quadratic regression and constant approximation via the mean. p-values were computed using standard likelihood-ratio test and multiple-testing corrected using Bonferroni-Holm.33
Clustering ERK-dependent differences in gene expression
Principal component analysis was performed on the matrix of moderated log2-fold changes of all samples (mean of two replicates) compared to the baseline condition (untreated control). The first principal component was strongly correlated with ERK activity across all samples. In order to find genes that have similar relationships between gene expression and ERK dose, we applied k-medoids clustering53 — a variation of the k-means clustering method that is robust to outliers — on the log2-fold changes at 2 4h. We found that choosing resulted in easily interpretable clusters with different dynamics of ERK signaling responses. The clusters we identified can be grouped into four different response types, each with two variants representing responses with opposite signs.
Time series analysis
In order to group genes into clusters depending on their time of induction or repression, we first normalized the time series log2-fold changes of each gene. First, we computed the range of log2-fold changes for each gene and each ERKi concentration across all time points by subtracting the minimum log2-fold change from the maximum.
Next, we aggregated the time series data across all ERKi doses by computing 33% and 67% quantiles of log2-fold changes at each time point, weighted by the time series range , using the algorithm described in the cited reference.54
with
At each timepoint, the quantile with the highest absolute value was selected as the aggregate log2-fold change , analogous to the procedure used for aggregating expression data across cell lines in the cited reference 55. Finally, genes were grouped into clusters based on when in the time series the absolute aggregated log2-fold change exceeded half of the maximum value across the entire time series. The significance of differences in the induction time between ERK-response clusters were tested using ANOVA followed by Tukey’s honest significant difference test. The distribution of the mean induction time for each ERK-response cluster was estimated using bootstrapping. The induction times of genes from each cluster were resampled 1000 times with replacement and the mean of each sample was computed.
Gene set enrichment analysis
Two variants of gene set enrichment analysis were performed. First, the enrichment of GO-terms in the ERK-response cluster gene sets was assessed using the R Bioconductor package topGO (https://bioconductor.org/packages/release/bioc/html/topGO.html). We considered all GO-terms in the Biological Process (“BP”) and Molecular Function (“MF”) categories. Enrichment was computed using the “weight01” algorithm and Fisher’s exact test. Second, gene set enrichment analysis was performed using gene sets from MSigDB.56 Specifically, gene sets from the Hallmark (H), curated pathways (C2:CP), and ontology (C5) categories, excluding Human Phenotype Ontology (HPO), were considered. We tested for significant enrichment using Fisher’s exact test on the overlap between the 1958 differentially expressed genes and the gene set of interest and p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure (https://mathscinet.ams.org/mathscinet-getitem?mr=1325392). Additionally, we computed the enrichment of a collection of manually curated gene sets related to ERK signaling, containing all gene sets containing “ERK”, “MAPK”, “senescence”, or “melanoma” from MSigDB, as well as the set of differentially expressed genes from a BRAFV600E overexpression experiment57 from GEO (GSE46801).
Replicate similarity
In order to assess the quality of replicates, we computed the Pearson correlation coefficients between the normalized counts of our two replicates, considering the 1000 most differentially expressed genes across all conditions. The genes were ranked by the results of a likelihood-ratio test using DESeq2, comparing the full model described above against a reduced model of the form . The correlation matrix was plotted using the R package ComplexHeatmap.58
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars represent the standard deviation, standard error of the mean, or 95% bootstrap confidence interval as indicated in the legends. Statistical comparisons (p values) were obtained from two-sided t-tests or otherwise as noted. The Pearson’s correlation coefficients (r) were calculated as indicated. Comparison between model fits for the ERK response surfaces was done using standard likelihood-ratio test and multiple-testing corrected using Bonferroni-Holm. Overlap between gene sets and differentially expressed genes was tested using Fisher’s exact test and p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure. Differences in the induction time between ERK-response clusters were tested using ANOVA followed by Tukey’s honest significant difference test.
Supplementary Material
Highlights.
Oncogenic BRAFV600E triggers a heterogeneous ERK activation response
The relationship between ERK activity and proliferation is non-monotonic
Transcriptomics identifies thousands of genes responding to ERK activity over time
ERK controls proliferation via genes that respond to various ranges of its activity
ACKNOWLEDGMENTS
We thank S. Boswell and S. Vallabhaneni for their help; members of the Lahav laboratory for comments, support, and ideas; the Nikon Imaging Center at HMS for support with live-cell imaging; and Harvard Research Computing for use of the O2 High Performance Computing Cluster. This research was supported by NIH grants R35 GM139572 (G.L.) and P50-GM107618 (G.L. and P.K.S.), NCI grant U54-CA225088 (P.K.S.), the Ludwig Center at Harvard (A.J., G.L., and P.K.S.), a Jane Coffin Child Memorial Fund for Medical Research fellowship to J.-Y.C., CONACyT/Fundación México en Harvard (404476) and a Harvard Graduate Merit Fellowship to J.R., and HFSP grant LT000259/2019-L1 to F.F.
Footnotes
DECLARATION OF INTERESTS
P.K.S. is a member of the science advisory board (SAB) or board of directors (BOD) of Glencoe Software, Applied Biomath, and RareCyte and has equity in these companies; P.K.S. is on the SAB of NanoString and a consultant to Montai Health and Merck. In the past 5 years, the Sorger lab has received research funding from Novartis and Merck. P.K.S. declares that none of these relationships are directly or indirectly related to the content of this article.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112252.
REFERENCES
- 1.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hobbs GA, Der CJ, and Rossman KL (2016). RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292. 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holderfield M, Deuker MM, McCormick F, and McMahon M (2014). Targeting RAF kinases for cancer therapy: BRAF mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467. 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collado M, and Serrano M (2010). Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57. 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 6.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, and Chodosh LA (2007). Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 9, 493–505. 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 7.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, Majoor DM, Shay JW, Mooi WJ, and Peeper DS (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724. 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 8.Adams PD (2009). Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol. Cell 36, 2–14. 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 9.Mooi WJ, and Peeper DS (2006). Oncogene-induced cell senescence–halting on the road to cancer. N. Engl. J. Med. 355, 1037–1046. 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 10.Adashek JJ, Kato S, Lippman SM, and Kurzrock R (2020). The paradox of cancer genes in non-malignant conditions: implications for precision medicine. Genome Med. 12, 16. 10.1186/s13073-020-0714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michaloglou C, Vredeveld LCW, Mooi WJ, and Peeper DS (2008). BRAF(E600) in benign and malignant human tumours. Oncogene 27, 877–895. 10.1038/sj.onc.1210704. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Vega R, Chen C-F, Razzak E, Vasudeva P, Krasieva TB, Shiu J, Caldwell MG, Yan H, Lowengrub J, Ganesan AK, and Lander AD (2020). Dynamics of nevus development implicate cell cooperation in the growth arrest of transformed melanocytes. Elife 9, e61026. 10.7554/eLife.61026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 14.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. (2003). High frequency of BRAF mutations in nevi. Nat. Genet. 33, 19–20. 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 15.Vizioli MG, Possik PA, Tarantino E, Meissl K, Borrello MG, Miranda C, Anania MC, Pagliardini S, Seregni E, Pierotti MA, et al. (2011). Evidence of oncogene-induced senescence in thyroid carcinogenesis. Endocr. Relat. Cancer 18, 743–757. 10.1530/ERC-11-0240. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J, Woods D, McMahon M, and Bishop JM (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meloche S, and Pouysségur J (2007). The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239. 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 18.Deschênes-Simard X, Gaumont-Leclerc M-F, Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette FA, Saba-El-Leil MK, Meloche S, et al. (2013). Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 27, 900–915. 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 20.Min M, Rong Y, Tian C, and Spencer SL (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science 368, 1261–1265. 10.1126/science.aay8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spencer SL, Cappell SD, Tsai F-C, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang HW, Chung M, Kudo T, and Meyer T (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. 10.1038/nature23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havens CG, and Walter JC (2009). Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 35, 93–104. 10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibutani ST, de la Cruz AFA, Tran V, Turbyfill WJ, Reis T, Edgar BA, and Duronio RJ (2008). Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev. Cell 15, 890–900. 10.1016/j.devcel.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponsioen B, Post JB, Buissant des Amorie JR, Laskaris D, van Ineveld RL, Kersten S, Bertotti A, Sassi F, Sipieter F, Cappe B, et al. (2021). Quantifying single-cell ERK dynamics in colorectal cancer organoids reveals EGFR as an amplifier of oncogenic MAPK pathway signalling. Nat. Cell Biol. 23, 377–390. 10.1038/s41556-021-00654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, and Matsuda M (2011). Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656. 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerosa L, Chidley C, Fröhlich F, Sanchez G, Lim SK, Muhlich J, Chen J-Y, Vallabhaneni S, Baker GJ, Schapiro D, et al. (2020). Receptor-driven ERK pulses reconfigure MAPK signaling and enable persistence of drug-adapted BRAF-mutant melanoma cells. Cell Syst. 11, 478–494.e9. 10.1016/j.cels.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu IM, Hengst L, and Slingerland JM (2008). The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 8, 253–267. 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 29.Pantoja C, and Serrano M (1999). Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene 18, 4974–4982. 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 30.Lessard F, Igelmann S, Trahan C, Huot G, Saint-Germain E, Mignacca L, Del Toro N, Lopes-Paciencia S, Le Calvé B, Montero M, et al. (2018). Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 20, 789–799. 10.1038/s41556-018-0127-y. [DOI] [PubMed] [Google Scholar]
- 31.Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, et al. (2007). A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 39, 503–512. 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 32.Macon N, and Spitzbart A (1958). Inverses of Vandermonde matrices. Am. Math. Mon. 65, 95–100. 10.2307/2308881. [DOI] [Google Scholar]
- 33.Holm S (1979). A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6, 65–70. [Google Scholar]
- 34.Suderman R, Bachman JA, Smith A, Sorger PK, and Deeds EJ (2017). Fundamental trade-offs between information flow in single cells and cellular populations. Proc. Natl. Acad. Sci. USA 114, 5755–5760. 10.1073/pnas.1615660114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez A, and Golding I (2013). Genetic determinants and cellular constraints in noisy gene expression. Science 342, 1188–1193. 10.1126/science.1242975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huh D, and Paulsson J (2011). Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 43, 95–100. 10.1038/ng.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer SL, Gaudet S, Albeck JG, Burke JM, and Sorger PK (2009). Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432. 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courtois-Cox S, Jones SL, and Cichowski K (2008). Many roads lead to oncogene-induced senescence. Oncogene 27, 2801–2809. 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- 39.Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, and d’Adda di Fagagna F (2014). Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 21, 998–1012. 10.1038/cdd.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deschênes-Simard X, Kottakis F, Meloche S, and Ferbeyre G (2014). ERKs in cancer: friends or foes? Cancer Res. 74, 412–419. 10.1158/0008-5472.CAN-13-2381. [DOI] [PubMed] [Google Scholar]
- 41.Roberts PJ, and Der CJ (2007). Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310. 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 42.Leung GP, Feng T, Sigoillot FD, Geyer FC, Shirley MD, Ruddy DA, Rakiec DP, Freeman AK, Engelman JA, Jaskelioff M, and Stuart DD (2019). Hyperactivation of MAPK signaling is deleterious to RAS/RAF-mutant melanoma. Mol. Cancer Res. 17, 199–211. 10.1158/1541-7786.MCR-18-0327. [DOI] [PubMed] [Google Scholar]
- 43.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, and Stuart DD (2013). Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255. 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Mitri D, and Alimonti A (2016). Non-cell-autonomous regulation of cellular senescence in cancer. Trends Cell Biol. 26, 215–226. 10.1016/j.tcb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, and Lahav G (2012). p53 dynamics control cell fate. Science 336, 1440–1444. 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salmeen A, Park BO, and Meyer T (2010). The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 29, 4473–4484. 10.1038/onc.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cappell SD, Chung M, Jaimovich A, Spencer SL, and Meyer T (2016). Irreversible APC(Cdh1) inactivation underlies the point of No return for cell-cycle entry. Cell 166, 167–180. 10.1016/j.cell.2016.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reyes J, Chen J-Y, Stewart-Ornstein J, Karhohs KW, Mock CS, and Lahav G (2018). Fluctuations in p53 signaling allow escape from cell-cycle arrest. Mol. Cell 71, 581–591.e5. 10.1016/j.molcel.2018.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian C, Yang C, and Spencer SL (2020). EllipTrack: a global-local cell-tracking pipeline for 2D fluorescence time-lapse microscopy. Cell Rep. 32, 107984. 10.1016/j.celrep.2020.107984. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schubert E, and Rousseeuw PJ (2019). Faster k-medoids clustering: improving the PAM, CLARA, and CLARANS algorithms. In Similarity Search and Applications Lecture Notes in Computer Science, Amato G, Gennaro C, Oria V, and Radovanović M, eds. (Springer International Publishing; ), pp. 171–187. 10.1007/978-3-030-32047-8_16. [DOI] [Google Scholar]
- 54.Harrell FE, and Davis CE (1982). A new distribution-free quantile estimator. Biometrika 69, 635–640. 10.1093/biomet/69.3.635. [DOI] [Google Scholar]
- 55.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e17. 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, et al. (2016). MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 30, 321–336. 10.1101/gad.271882.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Results of RNA seq analysis used to generate main and supplemental figures in this paper are available at Synapse database (https://www.synapse.org/#!Synapse:syn21411369/files/).
The original code for analysis of RNA seq data is publicly available at GitHub (https://github.com/clemenshug/erk_senescence).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) antibody | Cell Signaling Technology | Cat# 4370, RRID: AB_2315112 |
| Phospho-Rb (Ser807/811) (D20B12) antibody | Cell Signaling Technology | Cat# 8516, RRID: AB_11178658 |
| Rb (4H1) antibody | Cell Signaling Technology | Cat# 9309, RRID: AB_ 823629 |
| p16 (H-156) antibody | Santa Cruz Biotechnology | Cat# sc-759, RRID: AB_632105 |
| p21 antibody | BD Biosciences | Cat# 556430, RRID: AB_396414 |
| Ki-67 (8D5) antibody | Cell Signaling Technology | Cat# 9449, RRID: AB_2797703 |
| p27 Kip1 (D69C12) XP Rabbit monoclonal antibody | Cell Signaling Technology | Cat# 3686, RRID: AB_2077850 |
| Anti-MCM6 antibody [EPR17686] | Abcam | Cat# ab201683, RRID: AB_2924827 |
| Anti-β-Actin monoclonal antibody | Sigma-Aldrich | Cat# 5316, RRID: AB_476743 |
| Anti-Raf-B antibody (F-7) | Santa Cruz Biotechnology | Cat# sc-5284, RRID: AB_626760 |
| Anti-HA High Affinity; Rat monoclonal antibody (clone 3F10) | Roche | Cat# 11867423001, RRID: AB_390918 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling Technology | Cat# 7076, RRID: AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID: AB_2099233 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID: AB_2576217 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11077, RRID: AB_2534121 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21235, RRID: AB_2535804 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| CSII-EF1-H2B-mTurquoise (lentiviral) | Spencer et al.21 | N/A |
| CSII-EF1-mVenus-hGeminin (1–110) (lentiviral) | Sakaue-Sawano et al.19 | N/A |
| CSII-EF1-mCherry-dE2F PIP (lentiviral) | This work | N/A |
| CSII-EF1-mVenus-dE2F PIP (lentiviral) | This work | N/A |
| LV-EKAREN5-NLS (lentiviral) | Addgene | Plasmid#167818 |
| LIX402-BRAFV600E-HA-Puro (lentiviral) | This work | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Hoeschst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | Cat# H3570 |
| SCH772984, ERK inhibitor | MedChem Express | Cat# HY-50846 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891-5G |
| SMARTpool: siGENOME Non-targeting siRNA control pools | Horizon Discovery | Cat# D-001206-14-05 |
| SMARTpool: siGENOME Human CDKN1A siRNA | Horizon Discovery | Cat# M-003471-00-0005 |
| SMARTpool: siGENOME Human CDKN2A siRNA | Horizon Discovery | Cat# M-011007-03-0005 |
| SMARTpool: ON-TARGETplus CDKN2B siRNA | Horizon Discovery | Cat# L-003245-00-0005 |
| SMARTpool: siGENOME Human CDKN1B siRNA | Horizon Discovery | Cat# M-003472-00-0005 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668027 |
| Senescence Associated β-Galactosidase staining kit | Cell Signaling Technology | Cat# 9860 |
| Click-iT™ Cell Reaction Buffer Kit | Thermo Fisher Scientific | Cat# C10269 |
| Click-iT™ EdU (5-ethynyl-2’-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Click-iT™ Alexa Fluor® 647 Azide, Triethylammonium Salt | Thermo Fisher Scientific | Cat# A10277 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and processed RNA-seq data | GEO (Gene Expression Omnibus) | GEO: GSE180210 |
| Results of RNA-seq analysis used to generate main and supplemental figures in the manuscript | Synapse database | https://www.synapse.org/#!Synapse:syn21411369/files/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: RPE hTERT | S.J. Elledge Lab (Harvard Medical School) | N/A |
| Human: RPE + tet-BRAFV600E-HA | This work | N/A |
| Human: RPE + H2B-mTurquoise + Venus-dE2F PIP | This work | N/A |
| Human: RPE + tet-BRAFV600E-HA + EKAREN5 + mCherry-dE2F PIP | This work | N/A |
| Human: RPE + EKAREV-NLS | This work | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| CDKN2B PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.25069372.g |
| CDKN1B PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.45564663 |
| CDKN2A PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.40743463.g |
| EGR1 PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.40805543.g |
| DUSP4 PrimeTime qPCR primers | Integrated DNA Technologies | Hs.PT.58.18820216 |
| p21 forward qPCR primer: TGTCACTGTCTTGTACCCTTG | Purvis et al.45 | N/A |
| p21 reverse qPCR primer: GGCGTTTGGAGTGGTAGAA | Purvis et al.45 | N/A |
| HPRT forward qPCR primer: GTATTCATTATAGTCAAGGGCATATC | This paper | N/A |
| HPRT reverse qPCR primer: AGATGGTCAAGGTCGCAAG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pPB-CAG-EKAREV-NLS (piggyBac) | Komatsu et al.26 | N/A |
| pCMV-hyPBase | Komatsu et al.26 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Scripts for analysis of RNA sequencing data | This work | https://github.com/clemenshug/erk_senescence |
| Software for automatic segmentation and quantification of immunofluorescence images | Salmeen et al.46 | N/A |
| Software for automatic segmentation, and quantification of fluorescent reporter cells following live imaging | Cappell et al.47 | https://github.com/scappell/Cell_tracking |
| p53 Cinema Single Cell Tracking | Reyes et al.48 | https://github.com/balvahal/p53CinemaManual |
| EllipTrack | Tian et al.49 | https://github.com/tianchengzhe/elliptrack |