

Abstract
Accumulating toxic protein assemblies, including Aβ and tau, and dysfunctional mitochondria are associated with synaptic and neuronal loss in Alzheimer’s disease (AD). Such accumulations are thought to be owing to clearance defects in the autophagy-lysosome pathway. Mitochondrial dysfunction is evident in AD brains and animal models at multiple levels, such as mitochondrial genomic mutations, disrupted bioenergetics, deregulated mitochondrial dynamics and impaired clearance of damaged mitochondria (mitophagy). Slingshot homolog-1 (SSH1) is a phosphatase activated by oxidative stress, high intracellular levels of Ca2+ and Aβ42 oligomers (Aβ42O), known for its function to dephosphorylate/activate cofilin through the N-terminal region. SSH1-mediated cofilin dephosphorylation results in Ab42O-induced severing of F-actin and translocation of cofilin to mitochondria, which promotes mitochondria-mediated apoptosis, synaptic loss and synaptic deficits. On the other hand, SSH1-mediated dephosphorylation/deactivation of the autophagy-cargo receptor p62 (SQSTM1), through its C-terminal region, inhibits p62 autophagy flux. However, the interplay between these two different activities of SSH1 in Aβ42O-induced mitochondrial toxicity remains unclear. In this study, we assessed the role of endogenous SSH1 and different regions of SSH1 in regulating mitochondrial health, mitochondrial respiration, clearance of damaged mitochondria and synaptic integrity in vitro and in vivo. Our results indicate that SSH1 suppresses mitochondrial health and respiration through the cofilin-binding N-terminal region, whereas SSH1 impairs mitophagy through a newly identified ~ 100 residue p62-binding domain in the C-terminal region. These results indicate that both N-terminal and C-terminal regions negatively impact mitochondria by distinct and independent modalities to amplify mitochondrial abnormalities, making SSH1 an excellent target to mitigate AD pathogenesis.
Introduction
Accumulation of toxic protein assemblies, including Aβ and tau, along with dysfunctional mitochondria, is associated with synaptic and neuronal loss in Alzheimer’s disease (AD) (1). Such accumulations are thought to be in large part owing to clearance defects in the autophagy-lysosome pathway (2–5). Mitochondrial dysfunction is evident in AD brains and animal models of AD at multiple levels, such as mitochondrial genomic mutations, disrupted bioenergetics, deregulated mitochondrial dynamics (i.e. fission/fusion) and impaired clearance of damaged mitochondria (mitophagy) (6). At present, however, the mechanistic basis of such mitochondrial dysfunctions in AD remains to be elucidated.
Slingshot homolog-1 (SSH1) is a phosphatase that has dual functions in the cofilin activation pathway and the SQSTM1/p62 inactivation pathway, both of which have crucial implications for mitochondrial homeostasis. At baseline, SSH1 is inactivated by its association with 14–3-3. However, oxidation of 14–3-3 during oxidative stress liberates SSH1 from 14–3-3, which activates SSH1 (7–9). In the SSH1-mediated cofilin activation pathway, SSH1-mediated dephosphorylation of pSer3-cofilin promotes F-actin severing and the translocation of cofilin to mitochondria, the latter resulting in disrupted mitochondrial membrane potential (ΔΨm), swelling of mitochondria and apoptosis (10). Oxidative stress induced by Aβ42 oligomers (Aβ42o), which also rapidly induces SSH1-dependent activation of cofilin, results in dendritic spine shrinkage (11,12), depletion of F-actin-associated synaptic proteins and translocation of cofilin to mitochondria (13). Indeed, isolated mitochondria from AD brains contain a significantly increased level of cofilin in the activated and oxidized form (13), and cofilin activation status is elevated in brains of AD patients (12,14) and APP/PS1 mice (13). In mechanistic support in vivo, APP/PS1 mouse brains contain increased SSH1/cofilin and decreased SSH1/14–3-3 protein complexes, indicative of SSH1-cofilin activation via release of SSH1 from 14–3-3 (13).
SQSTM1/p62 is a major selective autophagy cargo receptor, which binds cargo, including misfolded protein aggregates and damaged mitochondria, and target them for clearance through the autophagy pathway (15). Notably, the binding of p62 to ubiquitinated cargo depends on the phosphorylation of its UBA domain at Ser403 by TBK1, ULK1 or CK2 (15–19). In the SSH1-mediated p62 inactivation pathway, this activating phosphorylation of p62 at pSer403 is removed by SSH1. Hence, SSH1-mediated dephosphorylation of pS403-p62 impedes p62 autophagy flux and the clearance of misfolded tau (20). These activities of SSH1 on p62 and cofilin are modular and separable from each other, as deletion of SSH1 N-terminal cofilin-binding region (SSH1ΔN) retains full activity on p62 inhibition but no activity on cofilin, whereas deletion of the C-terminal p62-binding region (SSH1ΔC) retains full activity on cofilin activation but has no activity on p62 (20). Interestingly, Ser403 phosphorylated p62 is significantly reduced in the brains of AD patients (5), suggesting a failure of p62 to clear cargo, such as misfolded proteins and damaged mitochondria.
While both cofilin and p62 pathways are implicated in AD pathogenesis and mitochondrial homeostasis, how different regions of SSH1 contribute to mitochondrial function and clearance are unknown. In this study, we assessed the role of endogenous SSH1 and different regions of SSH1 in regulating mitochondrial health, synaptic integrity, mitochondrial respiration and clearance of damaged mitochondria in HT22 cells, primary neurons and in vivo. Our results indicate that SSH1 impairs mitochondrial health and synaptic integrity through the cofilin-binding N-terminal region, whereas SSH1 suppresses mitophagy through a newly identified ~ 100 residue core p62-binding domain in the C-terminal region.
Results
SSH1 N-terminal region exacerbates Ab42-induced disruption of mitochondrial membrane potential and synaptic integrity
Mitochondrial membrane potential (ΔΨm) is a key indicator of mitochondrial health and activity, as ΔΨm is maintained by the process of electron transport and oxidative phosphorylation (21). We used the JC-1 dye to visualize ΔΨm, as JC-1 aggregates accumulate and fluoresce red in healthy mitochondria with high ΔΨm. In HT22 mouse neuroblastoma cells co-transfected with control shRNA and GFP, Aβ42 oligomer (Aβ42o) treatment for 24 h significantly decreased ΔΨm as detected by reduced JC-1 red fluorescence (Fig. 1A and B). However, HT22 cells co-transfected with SSH1 shRNA and GFP were significantly resistant to Aβ42-induced disruption of ΔΨm.
Figure 1.

SSH1 N-terminal region exacerbates Aβ42-induced disruption of mitochondrial membrane potential and synaptic integrity. (A) Representative images for HT22 cells co-expressing GFP (green) and Control or SSH1 shRNA with or without 100 nm Aβ treatment for 24 h, showing mitochondrial membrane potential (JC-1. red). (B) Quantification of JC-1 red intensity for A. Data are presented as mean ± SEM. n = 15–20 images/condition from two independent experiments, t-test, *P < 0.05. (C) Representative images for HT22 cells co-expressing GFP (green) with VC, Flag-SSH1ΔN or Flag-SSH1ΔC with or without 100 nm Aβ treatment for 24 h, showing mitochondrial membrane potential (JC-1. red). (D) Quantification of JC-1 red intensity for C. Data are presented as mean ± SEM. n = 15–20 images/condition from three independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, ***P < 0.0005, **P < 0.005. (E) Representative images of DIV18 WT primary hippocampal neurons transduced with Control, Flag-SSH1ΔN or CFP-SSH1ΔC AAV9 with or without 100 nm Aβ treatment for 24 h, showing mitochondrial membrane potential (JC-1. red). (F) Quantification of JC-1 red intensity for E. Data are presented as mean ± SEM. n = 15–20 images/condition from two independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001, **P < 0.005. (G) Representative images of DIV18 WT primary hippocampal neurons transduced with Control, Flag-SSH1ΔN or CFP-SSH1ΔC AAV9 (green) on DIV3, with or without 100 μm Aβ treatment for 24 h stained for synaptophysin (red) and MAP2 (blue). (H) Quantification of synaptophysin intensity for G. Data are presented as mean ± SEM. n = 10–12 images/condition from four independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001, ***P < 0.0005, **P < 0.005.
The N-terminal region of SSH1 binds and activates cofilin, whereas the C-terminal regions of SSH1 binds and inhibits SQSTM1/p62 (20,22). To determine which region of SSH1 compromises mitochondrial health, we took advantage of the SSH1ΔC construct, which is sufficient to activate cofilin, and the SSH1ΔN construct, which is sufficient to inhibit p62 (20,22). Aβ42o treatment to HT22 cells transfected with vector control or SSH1ΔN significantly decreased ΔΨm with no differences between the conditions (Fig. 1C and D). In contrast, Aβ42o treatment to HT22 cells transfected with SSH1ΔC significantly depressed ΔΨm beyond both vector control and SSH1ΔN by ~ 50% (Fig. 1C and D). Essentially identical results were obtained in primary neurons transduced with corresponding control, SSH1ΔN or SSH1ΔC AAVs and treated with Ab42o (Fig. 1E and F).
As mitochondrial health positively regulates synaptic integrity (23), we also assessed synaptophysin immunoreactivity as a presynaptic marker of synaptic integrity. Aβ42o treatment for 24 h significantly reduced synaptophysin intensity by ~ 45% in neuritic processes of mature neurons transduced with control AAV (Fig. 1G and H). Surprisingly, SSH1DN transduced neurons exhibited a ~25% increase in synaptophysin intensity at baseline and were resistant to Aβ42o-induced reduction in synaptophysin (Fig. 1G and H). SSH1DC transduced neurons, in contrast, displayed significantly reduced synaptophysin intensity (by ~ 45%) at baseline, which did not reduce further with Aβ42o treatment. These results collectively indicate that SSH1 N-terminal region, but not C-terminal region, mediates Ab42o-induced disruption of ΔΨm and synaptic integrity in HT22 cells and primary neurons.
SSH1 N-terminal region impairs mitochondrial respiratory capacity under oxidative stress
We next assessed mitochondrial respiration using the Seahorse XF mito stress test in HEK293T cells owing to its high transfection efficiency. HEK293T cells were transfected with vector control, SSH1 shRNA or Flag-SSH1. We treated transfected cells with or without mild oxidative stress (100 μm H2O2) for 1 h prior to Seahorse analysis, as oxidation of 14-3-3 during oxidative stress liberates SSH1 from 14-3-3, which activates SSH1 (7–9). During Seahorse analysis, oligomycin A was added to inhibit ATP synthase (complex V) to derive basal respiration and ATP-linked respiration (Supplementary Material, Fig. S1A). Then the decoupler FCCP was added to derive maximal respiration, followed by rotenone and antimycin A (complex I and III inhibitors) to derive spare respiratory capacity and non-mitochondrial respiration (Supplementary Material, Fig. S1A). Under mild oxidative stress, knockdown of endogenous SSH1 significantly increased basal mitochondrial respiration, maximal respiration, ATP production and non-mitochondrial respiration compared with vector control (Fig. 2A–E), indicating that endogenous SSH1 impairs both mitochondrial and non-mitochondrial respiration. In the absence of H2O2 treatment, SSH1 shRNA increased basal respiration and ATP-linked respiration without reaching statistical significance (Supplementary Material, Fig. S1B–G). In the presence of H2O2, forced expression of Flag-SSH1 did not impact basal respiration or ATP-linked respiration (Fig. 2A, C and E). However, SSH1 overexpression significantly impaired maximal respiration and spare respiratory capacity (Fig. 2A, D and F). In the absence of H2O2, SSH1 overexpression did not significantly impact any measures of mitochondrial respiration (Supplementary Material, Fig. S1B–G).
Figure 2.
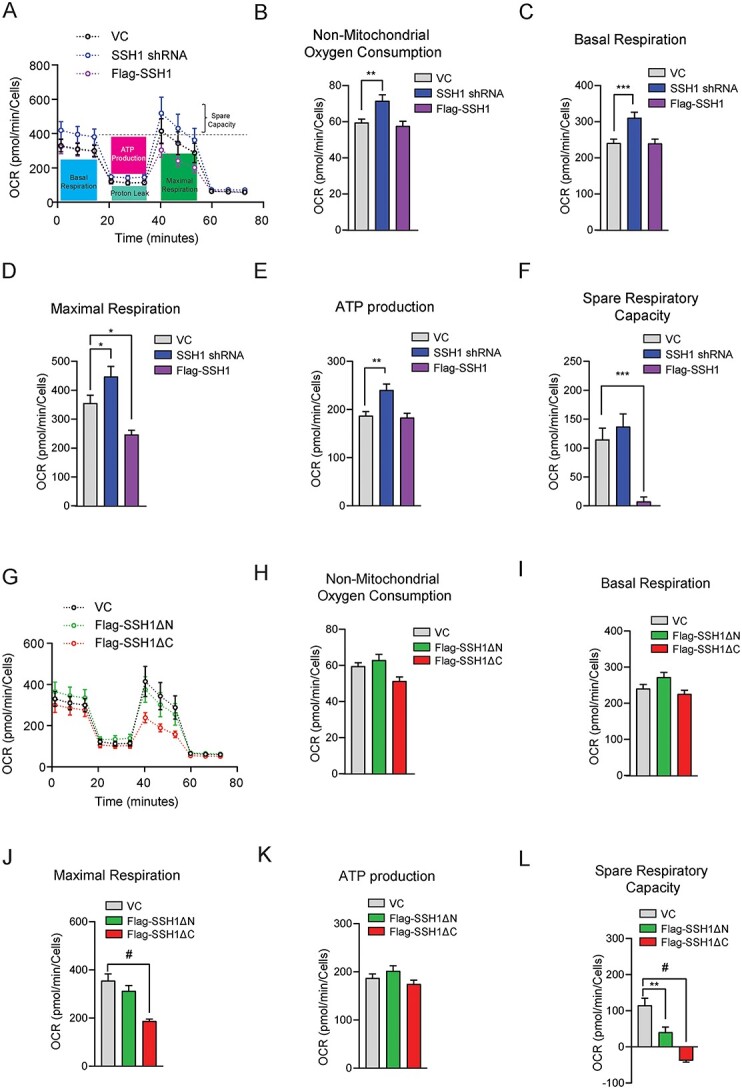
SSH1 N-terminal region impairs mitochondrial respiratory capacity under oxidative stress. (A) Oxygen consumption rate (OCR) measured under basal conditions followed by the sequential addition of oligomycin, FCCP, and rotenone and antimycin A on HEK293t cells expressing Control, SSH1 shRNA or Flag-SSH1 with 100 μm H2O2 treatment for 1 h. (B–F) Calculated measurements from detected OCRs to generate graph A. Data are presented as mean ± SEM. n = 6 independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, ***P < 0.0005, **P < 0.005, *P < 0.05. (B) Non-mitochondrial oxygen consumption. (C) Basal respiration. (D) Maximal respiration. (E) ATP production. (F) Spare respiratory capacity. (G) OCR measured under basal conditions followed by the sequential addition of oligomycin, FCCP, and rotenone and antimycin A on HEK293t cells expressing VC, Flag-SSH1ΔN or Flag-SSH1ΔC with100 μm H2O2 treatment for 1 h. (H–L) Calculated measurements from detected OCRs to generate graph G. Data are presented as mean ± SEM. n = 6 independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001. (H) Non-mitochondrial oxygen consumption. (I) Basal respiration. (J) Maximal respiration. (K) ATP production. (L) Spare respiratory capacity.
Having shown that endogenous and overexpressed SSH1 impairs mitochondrial respiration, we tested the effects of SSH1DN and SSH1DC compared with vector control. In the presence of H2O2, only SSH1DC significantly impaired maximal respiration (Fig. 2G and J), whereas both SSH1DC and SSH1DN significantly decreased spare respiratory capacity (Fig. 2G and L). However, the reduction in spare respiratory capacity by SSH1DN expression was partially owing to increased basal respiration (Fig. 2G and I). In contrast, the negative spare respiratory capacity seen by SSH1DC expression was owing to severely impaired maximal respiration (Fig. 2G and J), indicating that SSH1DC potently limits mitochondrial respiratory capacity under mild oxidative stress. Interestingly, in the absence H2O2, both SSH1DN and SSH1DC significantly increased maximal respiration and spare respiratory capacity (Supplementary Material, Fig. S1H, K and M), while SSH1DN also significantly increased basal respiration and ATP-linked respiration (Supplementary Material, Fig. S1H and J), suggesting a possible gain-of-function or dominant negative activity by the deletion mutants in the absence of oxidative stress. These data overall indicate that under oxidative stress conditions, SSH1 impairs mitochondrial respiratory capacity through its N-terminal region (SSH1DC), results that are in line with ΔΨm and synaptic integrity measures.
Endogenous SSH1 suppresses mitophagy in HT22 cells, primary neurons and in vivo
We previously showed that SSH1 suppresses SQSTM1/p62-mediated autophagy through p62 dephosphorylation at pSer403 (20). To determine whether SSH1 impacts mitophagy, we employed the mitophagy reporter construct mito-QC (mCherry-GFP-Fis1), which targets the fusion protein to mitochondria through the mitochondrial targeting sequence of Fis1. mito-QC fluoresces yellow owing to mCherry-GFP fusion protein expression when present in the neutral pH environment of mitochondria. However, when mitochondria are engulfed by lysosomes during mitophagy, the acidic pH inside lysosomes quenches the GFP but not mCherry signal, thereby providing the mCherry red-only mitophagy signal. To start, we first assessed whether endogenous SSH1 impacts mitophagy in HT22 cells with Ab42o at multiple time points over 48 h. Ab42o treatment increased mCherry red-only positive puncta at 24 h in control shRNA transfected cells, which subsided at 48 h (Fig. 3A and B). SSH1 shRNA transfection increased mCherry red-only puncta at all time points, with 8, 24 and 48 h achieving statistical significance (Fig. 3A and B).
Figure 3.

Endogenous SSH1 suppresses mitophagy in HT22 cells, primary neurons and in vivo. (A) Representative images of HT22 cells co-expressing mito-QC (mCherry-GFP-Fis1) (red and green) and Control or SSH1 shRNA (blue), with 100 nm Aβ treatment for 8, 24 or 48 h. (B) Quantification of red (acidified) mito-QC puncta for A. Data are presented as mean ± SEM. n = 12–15 images/condition from four independent experiments, two-way ANOVA, followed by Sidak’s post hoc, **P < 0.005, *P < 0.05. (C) Representative images of DIV18 mito-QC+/+ (red and green) primary hippocampal neurons transduced with Control or SSH1 shRNA AAV9 (blue) on DIV3, with or without 100 nM Aβ treatment for 4, 8, 24 or 48 h. (D) Quantification of red (acidified) mito-QC for C. Data are presented as mean ± SEM. n = 12–15 images/condition from four independent experiments, two-way ANOVA, followed by Sidak’s post hoc, **P < 0.005. (E) Representative images of brain tissue slices from 7-month APP/PS1;mito-QC+/− injected with Control or SSH1 shRNA in the hippocampus at 5 months, showing mito-QC (red and green) and SSH1 staining (blue). (F) Quantification of red puncta number (acidified mitochondria) for E. Data are presented as mean ± SEM. n = 10–12 images/mouse from four mice/genotype, t-test, #P < 0.0001.
To assess mitophagy in primary neurons, we utilized primary neurons isolated from P0 pups of mito-QC+/+ reporter mice (24). Ab42o treatment similarly increased mCherry red-only mitophagy puncta in control shRNA transduced mito-QC+/+ neurons over 24 h (Fig. 3C and D). SSH1 shRNA transduction increased mCherry red-only puncta at all time points, with 0 h and 4 h achieving statistical significance (Fig. 3C and D). Next, we assessed the role of endogenous SSH1 in mitophagy in vivo in the context of the APP/PS1 AD mouse model. We crossed APP/PS1 mice with mito-QC mice to generate APP/PS1;mito-QC mice. At 5 months of age, we stereotaxically injected AAV9 expressing control shRNA or SSH1 shRNA into the hippocampus of APP/PS1;mito-QC mice and assessed mitophagy at 7 months of age. Indeed, we observed significantly more mCherry red-only puncta in the hippocampus injected with SSH1 shRNA AAV9 compared with control shRNA AAV9 (Fig. 3E and F). As expected, secondary only negative control shows no immunoreactivity (Supplementary Material, Fig. S2A). These data indicate that endogenous SSH1 suppresses mitophagy in a mouse model of Ab pathology.
SSH1 C-terminal but not N-terminal region suppresses mitophagy
We next determined which region of SSH1 suppresses mitophagy in HT22 cells by co-expressing mito-QC with vector control, SSH1DN or SSH1DC and treating cells with Ab42o at different time points over 48 h. SSH1DN expression reduced mCherry only red puncta at all time points, with 24 and 48 h time points being statistically significant (Fig. 4A and B). In contrast, SSH1DC expression significantly increased mCherry red only puncta at all four time points, consistent with the drop in ΔΨm seen with SSH1DC expression (Fig. 4A and B; Fig. 1C and D), which is known to stimulate mitochondrial damage through cofilin (13). To confirm these results in neurons, we expressed control, SSH1DN or SSH1DC by AAV transduction in primary neurons with Ab42o treatment over 48 h. SSH1DN expression significantly decreased mCherry red-only puncta after 24 and 48 h of Ab42o treatment (Fig. 4C and D), similar to that seen in HT22 cells. Likewise, SSH1DC expression increased mCherry red-only puncta at all time points, with the 4 and 8 h time points achieving statistical significance (Fig. 4C and D). These results collectively indicate that endogenous SSH1 inhibits mitophagy and that the C-terminal domain (SSH1DN) mediates such inhibition, whereas the SSH1 N-terminal domain (SSH1DC) can stimulate mitophagy probably by disrupting ΔΨm.
Figure 4.
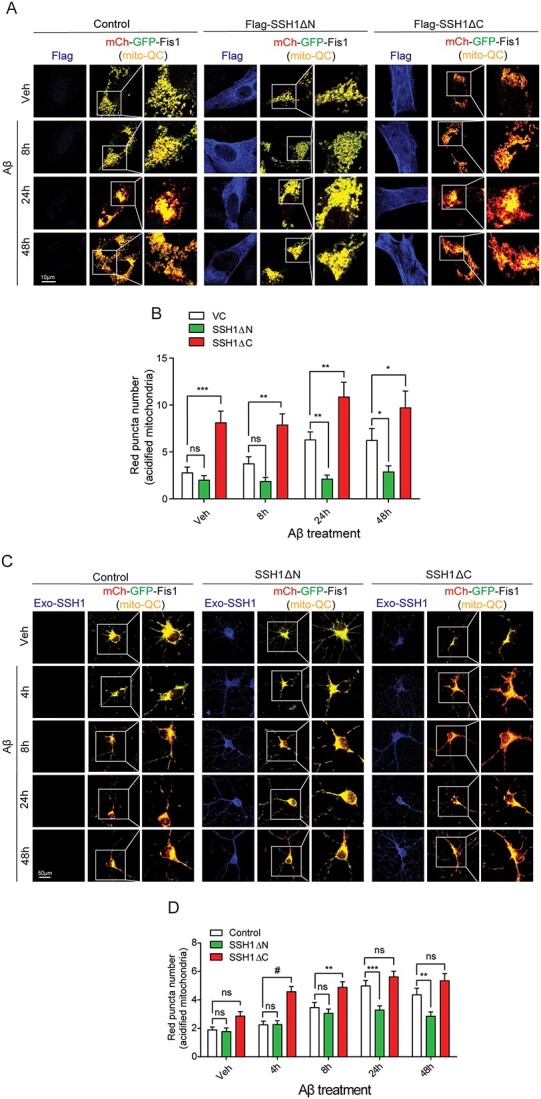
SSH1 C-terminal but not N-terminal region suppresses mitophagy. (A) Representative images of HT22 cells co-expressing mito-QC (mCherry-GFP-Fis1) (red and green) and VC, Flag-SSH1ΔN or Flag-SSH1ΔC (blue), with 100 nm Aβ treatment for 8, 24 or 48 h. (B) Quantification of red (acidified) mito-QC puncta for A. Data are presented as mean ± SEM. n = 12–15 images/condition from three independent experiments, two-way ANOVA, followed by Dunnett’s post hoc, ***P < 0.0005, **P < 0.005, *P < 0.05. (C) Representative images of DIV18 mito-QC+/+ (red and green) primary hippocampal neurons transduced with Control, Flag-SSH1ΔN or CFP-SSH1ΔC AAV9 (blue) on DIV3, with 100 nm Aβ treatment for 4, 8, 24 or 48 h. (D) Quantification of red (acidified) mito-QC puncta for C. Data are presented as mean ± SEM. n = 12–15 images/condition from four independent experiments, two-way ANOVA, followed by Tukey’s post hoc, #P < 0.0001, ***P < 0.0005, **P < 0.005.
SSH1 residues 549–649 constitute the core p62-binding domain
We previously showed that SSH1DN but not SSH1DC suppresses p62-mediated autophagy (20). However, the precise p62-binding site in SSH1DN is unknown. We hypothesized that determining the p62-binding site in SSH1DN would shed light on the mechanism by which SSH1 suppresses mitophagy. To this end, we generated a series of deletion mutants starting with SSH1DN by progressively truncating its C-terminus (Fig. 5A) and confirmed expression (Supplementary Material, Fig. S3A). To determine p62 binding to SSH1 proteins, we employed proximity ligation assay (PLA), which allows the detection of protein–protein interactions in situ (at distances < 40 nm) by utilizing primary antibodies directed at the proteins of interest and secondary antibody probes. As this method detects protein–protein interactions in intact cells, post-lysis artifacts are obviated, and detergent-sensitive interactions are maintained. In HT22 cells co-transfected with GFP-p62 and Flag-SSH1 deletion constructs, we performed PLA with primary antibodies against GFP and Flag followed by secondary antibody probes. Compared with full-length Flag-SSH1, we detected significantly increased binding of GFP-p62 to SSH1DN, SSH1DN-DC849 and SSH1DN-D649, as detected by red PLA puncta, while no PLA puncta were detected in cells transfected with GFP-p62 and vector control (Fig. 5B and C). In contrast, additional C-terminal truncations in SSH1DN-DC549 and SSH1-CAT resulted in a drastic drop in PLA signal comparable to SSH1DC (Fig. 5B and C), the latter having no activity in p62-mediated autophagy (20). The residual PLA puncta observed in SSH1DN-DC549, SSH1-CAT, SSH1DC likely stems from the secondary binding of the catalytic domain to phosphorylated p62, which per se has no phosphatase activity on Ser403 phosphorylated p62 (20). Indeed, we confirmed that the one probe and one primary negative controls show no PLA signals (Supplementary Material, Fig. S3B). Therefore, these results suggest that residues 549–649 constitute the core p62-binding site in SSH1.
Figure 5.
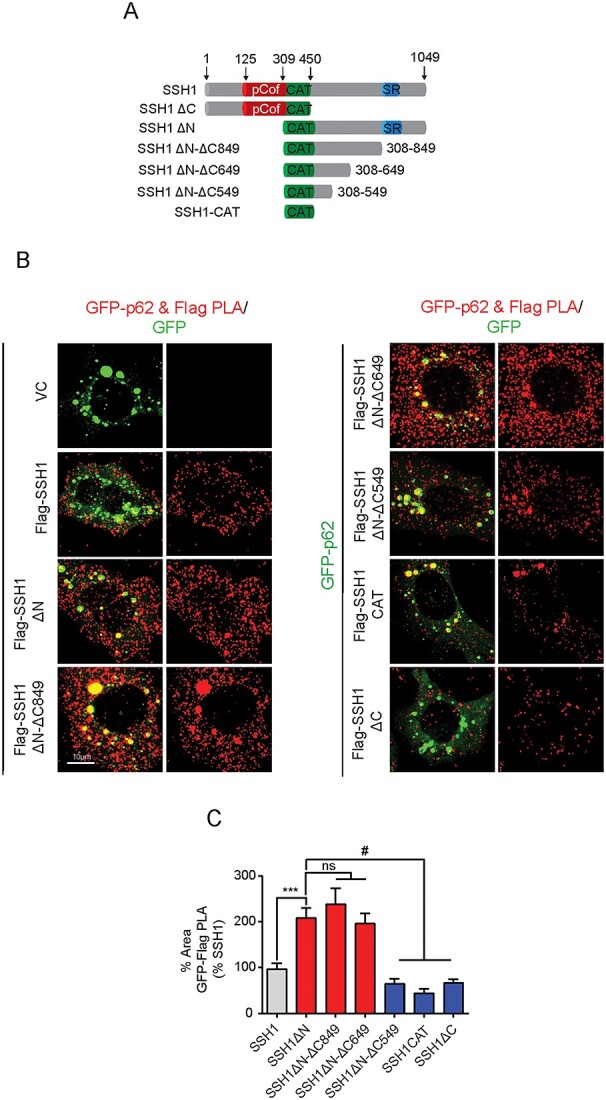
SSH1 residues 549–649 constitute the core p62-binding domain. (A) Schematic for SSH1 full length, SSH1 ΔC, SSH1 ΔN and the truncated forms of SSH1 ΔN generated to determine p62-binding site on SSH1. (B) Representative images of GFP-Flag PLA (red) on HT22 cells expressing the respective Flag tagged SSH1 constructs and GFP-p62 (green). (C) Quantification of the percentage of cell area occupied by Flag-GFP PLA from B. Data are presented as mean ± SEM. n = 10–12 images/condition from six independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001, ***P < 0.0005.
The core p62-binding domain in SSH1 is required to suppress autophagy and mitophagy
Having determined the p62-binding site in SSH1, we carried out functional assays to test the effects of the SSH1 truncation mutants using full-length SSH1 and SSH1DN as positive controls and SSH1DC as the negative control. First, we utilized the mCherry-GFP-p62 construct to assess p62 autophagy flux as we previously showed (20). This construct takes advantage of the quenching sensitivity of GFP and insensitivity of mCherry to acidic pH. Hence, cytosolic mCherry-GFP-p62 fluoresces yellow, whereas lysosomal mCherry-GFP-p62 fluoresces red. We used the mitochondrial decoupler FCCP to induce mitophagy as positive controls. To control for potential differences in Flag-SSH1 expression between constructs, we quantified p62 flux in cells expressing similar levels of Flag-SSH1 variants by staining for Flag. In HT22 cells co-transfected with mCherry-GFP-p62 and vector control, FCCP treatment resulted in a robust and significant increase in the percentage of red-only p62 puncta (Fig. 6A–D), indicative of p62 flux to lysosomes (20). However, co-transfection of mCherry-GFP-p62 with SSH1, SSH1DN, SSH1DN-DC849 or SSH1DN-DC649 strongly suppressed the percentage of red-only p62 puncta even in the presence of FCCP (Fig. 6A–D), indicating these SSH1 constructs potently inhibit p62-mediated autophagy. In contrast, SSH1DN-DC549, SSH1-CAT or SSH1DC failed to suppress red-only p62 puncta (Fig. 6A–D), indicating that the p62-binding domain of SSH1 (residues 549–649) are required to inhibit p62-mediated autophagy. Interestingly, SSH1-CAT and SSH1DC constructs significantly increased the percentage of red-only p62 puncta even in the absence of FCCP treatment (Fig. 6A–C), suggesting that these truncated constructs may exert a dominant negative effect with respect to p62 flux.
Figure 6.

The core p62-binding domain in SSH1 is required to suppress autophagy and mitophagy. (A) Representative images of HT22 cells co-expressing the respective Flag tagged SSH1 constructs (blue) and mCherry-GFP-p62 (red and green), with or without 5 μm FCCP treatment for 4 h. (B, C) Quantification of the percentage of red p62 puncta (acidified) over the total amount of p62 puncta for control condition of A. Data are presented as mean ± SEM. n = 10–12 images/condition from three independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001, ***P < 0.0005, *P < 0.05. (B, D) Quantification of the percentage of red p62 puncta (acidified) over the total amount of p62 puncta for FCCP condition of A. Data are presented as mean ± SEM. n = 10–12 images/condition from three independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001. (E) Representative images of HT22 cells co-expressing mito-QC (mCherry-GFP-Fis1) (red and green) and VC, Flag-SSH1, Flag-SSH1ΔN-ΔC649 or Flag-SSH1ΔN-ΔC549 (blue), with or without 5 μm FCCP treatment for 4 h. (F) Quantification of red (acidified) mito-QC puncta for E. Data are presented as mean ± SEM. n = 8–10 images/condition from four independent experiments, one-way ANOVA, followed by Dunnett’s post hoc, #P < 0.0001.
We next determined if the inhibition of mitophagy by SSH1 requires the core p62-binding domain using the SSH1 truncation constructs and the mitophagy reporter mito-QC (mCherry-GFP-Fis1). As expected, FCCP treatment of HT22 cells transfected with mito-QC and vector control significantly increased mCherry red-only puncta by ~ 3.5-fold (Fig. 6E and F). However, co-transfection of SSH1 or SSH1DN-DC649 with mito-QC nearly completely suppressed FCCP-induced increase in mCherry red-only puncta (Fig. 6E and F). In contrast, SSH1DN-DC549 completely failed to suppress FCCP-induced increase in mCherry red-only puncta, indicating that SSH1 residues 309–649 (SSH1DN-DC649) are sufficient and residues 549–649 (core p62-binding site) are required to suppress mitophagy by SSH1.
Discussion
Mitochondrial dysfunction is one of the earliest pathological features in neurodegenerative diseases, including AD and other tauopathies (25–30). Mitochondrial dysfunction occurs at multiple levels, including impairment in mitochondrial membrane potential, oxidative phosphorylation, mitochondrial dynamics (fission/fusion) and clearance of damaged mitochondria through mitophagy (31–35). In the brains of AD patients, mitochondria are not only excessively damaged but also accumulate owing to the failure of the mitophagy pathway (36). SSH1 is a protein phosphatase implicated in both the impairment of mitochondrial health through cofilin activation (13) and inhibition of autophagy through p62 inactivation (20). These bipartite functions of SSH1 occur through distinct and separable domains (20). In this study, we assessed the role of endogenous SSH1 and different regions of SSH1 in regulating mitochondrial health, mitochondrial respiration, synaptic integrity and clearance of damaged mitochondria in HT22 cells, primary neurons and in vivo. Our findings indicate that while endogenous SSH1 impairs both mitochondrial health and mitophagy, the cofilin-binding N-terminal region selectively compromises mitochondrial health (membrane potential and respiration), whereas the newly identified ~ 100 residue core p62-binding domain in the C-terminal region selectively suppresses mitophagy. Hence, our results validate the dual and separable activities of SSH1 in promoting mitochondrial dysfunction that may impress upon AD pathogenesis.
In light of our current findings, it is notable that cofilin activity is increased in the brains of AD patients and APP/PS1 AD mouse model (12,14). Owing to aberrantly increased cofilin activity, AD patients and animal models also show cofilin-actin rods/aggregates in the brain and exhibit shrinkage of dendritic spines (12,14). Upon oxidative stress and Ca2+ elevation, SSH1 is activated (7), which induces the dephosphorylation and activation of cofilin (22). Likewise, Aβ42 oligomers induce activation and dephosphorylation of cofilin through SSH1, resulting in depletion of F-actin-associated synaptic proteins and translocation of cofilin to mitochondria (13). The translocation of cofilin to mitochondria promotes cytochrome c release and induces apoptosis (13). Indeed, SSH1 reduction prevents Aβ42 oligomer-induced mitochondrial cofilin translocation and apoptosis (13). These studies are consistent with our current observations that endogenous SSH1 impairs mitochondrial membrane potential, mitochondrial respiration and synaptic integrity selectively through the cofilin-binding N-terminal region. Interestingly, the maximal respiration and spare respiratory capacity were impacted the most by full-length SSH1 and SSH1DC, indicating that activated SSH1 renders mitochondria vulnerable to energetic collapse upon oxidative or proteotoxic stress.
In addition to its role in cofilin activation, SSH1 simultaneously inactivates p62 (20), an autophagy cargo receptor mutated in ALS, FTLD and Paget’s disease of bone patients (37–42). Moreover, p62-positive inclusions colocalize with ubiquitin in AD, FTLD-ALS and PD brains (43–46), suggesting a failure of p62 to target ubiquitinated substrates for degradation. The inactivation of p62 by SSH1 is through the dephosphorylation of p62 at Ser403 (20), a phosphorylation event critical for the recognition of ubiquitinated cargo for autophagic clearance (47). Our studies here not only showed that endogenous SSH1 suppresses mitophagy but also that such inhibition occurs through a ~ 100 residue core p62-binding domain in the C-terminal region of SSH1. This region together with the catalytic domain was necessary and sufficient to inhibit both p62 autophagy flux and mitophagy. As SSH1 is activated by oxidative stress (7), it is plausible that the high oxidative stress environment, such as in AD brains, results in the suppression of mitophagy by SSH1. Such mitophagy inhibition through the SSH1 C-terminal region accompanied by impaired mitochondrial health through the SSH1 N-terminal region spells double trouble for mitochondrial dysfunction. Hence, targeting SSH1 could be an effective strategy to improve both mitochondrial health and enhance the clearance of toxic damaged mitochondria in AD and other neurodegenerative conditions.
Materials and Methods
Cells
Mouse hippocampus-derived neuroblastoma cells HT22 were used for immunocytochemistry experiments. Human embryonic kidney cells HEK293T were used for protein extraction experiments (western blotting). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM 1X) (Gibco, 11 965–092) supplemented with 10% fetal bovine serum (Sigma, 12306C), 1% penicillin–streptomycin (P/S) (Gibco, 15140-122) and BM cyclin (Roche, 10799050001). Cells were kept at 37°C with 5% CO2 levels.
Primary neurons
Hippocampal primary neurons were cultured from C57BL6 or mito-QC+/+ P0 mouse pups as previously described (13). Briefly, the hippocampus was dissected in ice-cold HBSS and digested with 0.25% Trypsin–EDTA (1X) (Gibco, 25200-056), then plated on glass coverslips coated with poly-D-lysine (Sigma-Aldrich, 27964-99-4) in neurobasal medium (Invitrogen, 21103049) supplemented with 2% GlutaMAX (Invitrogen, 35050061) and 2% B27 supplement (Invitrogen, 17504044). Cells were kept at 37°C with 5% CO2 levels.
Mice
APP/PS1 mice expressing ‘Swedish’ APP and PS1 ΔE9 mutations were obtained from Jackson Immunoresearch (West Grove, PA, USA) (48); transgenic mito-QC (mCherry-GFP-Fis1) mice were obtained from the Ganley laboratory (University of Dundee, Dundee, Scotland, UK) (24). Animals were bred in the C57BL6 background for at least three generations before interbreeding with each other.
Antibodies
Mouse monoclonal anti-β-Actin (C-4) (Santa Cruz Biotechsc-47778); mouse monoclonal anti-FLAG (M2) (Sigma-Aldrich, F3165); rabbit monoclonal anti-SQSTM1/p62 (D10E10) (Cell Signaling Technologies, 7695); rabbit monoclonal anti-SSH1 (E1K3W) (Cell Signaling Technologies, 13578); rabbit polyclonal anti-SSH1 (ECM Biosciences, SP1711); rabbit monoclonal anti-Synaptophysin (YE269) (abcam, ab32127); chicken polyclonal anti-MAP2 (abcam, ab5392); Alexa Fluor 647 goat anti-mouse (Invitrogen, A21235); Alexa Fluor 647 goat anti-rabbit (Invitrogen, A21245); Alexa Fluor 647 goat anti-chicken (Invitrogen, A32933); Alexa Fluor 405 goat anti-mouse (Invitrogen, A31553); Alexa Fluor 488 goat anti-rabbit (Invitrogen, A11034); Alexa Fluor 594 goat anti-rabbit (Invitrogen, A11037); Dapi (Thermo Scientific, 62248); mouse polyclonal anti-Horseradish Peroxidase (Jackson ImmunoResearch Inc., 223–005-024).
Constructs
pMXs-puro GFP-p62 (Addgene, 38277); mCherry-GFP-Fis1 (mito-QC) was obtained from the Ganley laboratory (24); mCherry-GFP-p62, p3xFlag-SSH1 and p3xFlag-SSH1 ΔN were previously generated by the Kang laboratory (20); SSH1, SSH1ΔC and SSH1ΔN were subcloned into pN3–3XFlag vector using HindIII and SalI sites by PFU-based PCR amplification of SSH1 from EGFP-SSH1 as a template (20). Flag-SSH1 ΔN-ΔC849, Flag-SSH1 ΔN-ΔC649, Flag-SSH1 ΔN-ΔC549 and Flag-SSH1 CAT constructs were generated using the Agilent QuikChange II Site-Directed Mutagenesis Kit from Flag-SSH1 ΔN with additional stop codon TAA. All primers were custom designed as following and ordered from IDT:
Flag-SSH1ΔN-ΔC849-FP:
5′-CAGGGATGGCCCTGCCAGCTAACTGGAGGCCAGCATCCCCG-3′.
Flag-SSH1ΔN-ΔC849-RP:
5′-CGGGGATGCTGGCCTCCAGTTAGCTGGCAGGGCCATCCCTG-3′.
Flag-SSH1ΔN-ΔC649-FP:
5′-GTGAAGCCTTCCTATAAATCCTGTTAAGACTGCATGTACCCTACA.
GCCA-3′.
Flag-SSH1ΔN-ΔC649-RP:
5′-TGGCTGTAGGGTACATGCAGTCTTAACAGGATTTATAGGAAGGCT.
TCAC-3′.
Flag-SSH1ΔN-ΔC549-FP:
5′-GAAGCTGCTCCACCTGCAGAGTAACACAGGCCGGCCAGACAGC-3′.
Flag-SSH1ΔN-ΔC549-RP:
5′-GCTGTCTGGCCGGCCTGTGTTACTCTGCAGGTGGAGCAGCTTC-3′.
Flag-SSH1CAT-FP:
5′-AAGGCATCTTGGATGCAAGCAAATAACGGCACAACAAGCTGTGGCG-3′.
Flag-SSH1CAT-RP:
5′-CGCCACAGCTTGTTGTGCCGTTATTTGCTTGCATCCAAGATGCCTT-3′.
Reagents/chemicals
FCCP (Sigma-Aldrich, C2920), Amyloid-β 1–42 (GenicBio, A-42-T-1), JC-1 Dye (Thermo Fisher, T3168), Duolink In Situ PLA probe Anti-Mouse MINUS (Sigma Aldrich, DUO92004), Duolink In Situ PLA probe Anti-Rabbit PLUS (Sigma Aldrich, DUO92002), Duolink In Situ Detection Reagents Red (Sigma Aldrich, DUO92008), Duolink In Situ Mounting Medium with DAPI (Sigma Aldrich, DUO82040), Duolink In Situ wash buffers, fluorescence (Sigma Aldrich, DUO82049). Amyloid-β 1–42 oligomers were prepared as previously described (49).
DNA transfections and adenoviral transductions
DNA plasmids were transiently transfected using Lipofectamine 2000 (Invitrogen, 11668-019) and reduced serum media Opti-MEM I (Gibco, 31985-070). 4–6 h post transfection, the media was replaced with new complete medium. Cells were grown for 48 h after transfection before being used for experiment. rAAV9 viruses were transduced to primary neurons on DIV3, neurons were cultured until DIV18 before being used for experiments.
Cell lysis and western blotting
Cells were lysed with RIPA buffer (50 mm Tris pH 7.4, 0.1% SDS, 2 mm ethlenediaminetetraacetic acid, 150 mm NaCl, 1% Nonidet P-40) with protease (GeneDEPOT, P3100-010) and phosphatase (GeneDEPOT, P3200-005) inhibitors, then centrifuged for 15 min at 4°C at 17 000 × g, supernatants were equalized in concentrations using Pierce BCA Protein Assay Kit (Thermo Scientific, 23225) and then used for western blotting. Equal amounts of sample were loaded and ran in SDS-PAGE gels and transferred to 0.45 μm nitrocellulose membranes (GE Healthcare, 10600002) for immunoblotting. Blots were blocked with 5% milk in TBST for 1 h at room temperature, then incubated overnight at 4°C with primary antibodies (1:1000 in TBST); after that, blots were washed and incubated with HRP-secondary antibodies for 4 h at room temperature (1:1000 in 5% milk in TBST), then washed and imaged using ECL western blot reagents (Pierce, 34578) and Fuji LAS-4000 imager (LAS-4000, USA).
Immunocytochemistry and immunohistochemistry
Cells were fixed with 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature, then blocked with 3% Normal Goat Serum (NGS) + 0.2% Triton in PBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C (1:100 in blocking solution) and secondary antibodies were incubated for 45 min at room temperature (1:1000 in blocking solution), then cells were mounted on glass slides with Fluoromount-G (Thermo Fisher, 00-4958-02). Mouse brains were extracted after perfusion with PBS and fixed with 4% PFA for 3 days at 4°C before being saturated with 30% sucrose in PBS. Tissues were sliced with 25 μm thickness, washed with 0.2% Triton in TBS and blocked with 3% NGS + 0.2% Triton in PBS for 1 h at room temperature. Tissues were stained with primary antibody overnight at 4°C (1:100 in blocking solution) followed by secondary antibodies at room temperature for 45 min (1:1000 in blocking solution) before being mounted on glass slides with Fluoromount-G (Thermo Fisher, 00-4958-02). Cells and tissues were imaged with Olympus FV10i confocal microscope (Tokyo, Japan) or Keyence BZ-X800 all-in-one fluorescent microscope (Osaka, Japan).
Proximity ligation assay
PLA was performed using commercially available reagents (Duolink, Sigma-Aldrich) following the manufacturer’s manual. Briefly, primary antibodies were applied overnight at 4°C (1:100 in 3% NGS + 0.2% Triton in PBS), probes were diluted in 3% NGS + 0.2% Triton in PBS and incubated at 37°C for 1 h. Ligation reagents were incubated for 30 min at 37°C followed by amplification for 100 min at 37°C. Cells were imaged with Olympus FV10i confocal microscope (Tokyo, Japan).
Mitochondria membrane potential assay (JC-1)
HT22 cells or primary neurons were plated on 35-mm glass bottom dishes coated with poly-D-lysine. Cells were then transiently transfected with DNA plasmids or transduced with AAV9 virus. Approximately 48 h after transfection or 15 days after transduction, cells/neurons were incubated with 2 μg/ml JC-1 dye in PBS for 20 min at 37°C before live cell imaging for ~ 30 min with Olympus FV10i confocal microscope (Tokyo, Japan).
Seahorse mito stress test
Seahorse XF Cell Mito Stress Tests (Seahorse, Agilent Technologies) was performed following the manufacturer’s protocol with the XFe/XF96 instrument. Briefly, HEK293T cells were transiently transfected, and after 24 h, cells were re-plated onto seahorse plates coated with poly-D-lysine and allowed to attach for another 24 h. Sensor cartridges were hydrated with Seahorse XF calibrant overnight at 37°C in the absence of CO2. The day of the assay, media was changed to XF assay medium supplemented with 1 mm pyruvate, 2 mm glutamine and 10 mm glucose; drugs were prepared fresh to the following final concentrations in well during assay: oligomycin A 1.5 μm, FCCP 1 μm, rotenone/antimycin A 0.5 μm. After assay, cells were lysed and protein concentration of each well was measured for normalization. Data analysis was performed using the Seahorse Wave 2.6.1 software.
AAV9 generation and stereotaxic injection
Stereotaxic injection of AAV9 viruses was performed as previously described (20). pTR12.1-MCSW-Flag-SSH1ΔN, pTR12.1-MCSW-ECFP-SSH1ΔC and H1rSC-Ssh1-shRNA constructs were generated by this laboratory (20). Briefly, recombinant AAV9 viruses were generated by co-transfection of serotype vector expressing the gene of interest with pAAV9 and pXX6 in HEK293 cells for 48 h. Cells were then lysed in the presence of 0.5% sodium deoxycholate, 1 mm MgCl2 and 50 U/ml Benzonase by freeze-thawing. Virus was isolated using a gradient of OptiPrep Density Gradient Medium (Sigma Aldrich, D1556) and then purified with Apollo 20 ml High-Performance Centrifugal quantitative concentrators (Orbital Biosciences, 2,015,010). For brain stereotaxic injections, 5-month-old mice were anesthetized with isoflurane and then bilaterally injected with 2 μl of purified rAAV9 (1.3 × 1012 vg/ml) using a 26-gage needle attached to a 10-μl syringe (Hamilton, 80,330) at the coordinates: anteroposterior 2.7 mm, lateral 2.7 mm and vertical 3.0 mm. Mice were sacrificed 2 months post injection.
Quantification and analysis
All experiments were quantified using Fiji ImageJ software and analyzed with Graphpad Prism 8 (Domantics, USA).
Conflict of Interest statement. None declared.
Data Availability
The data underlying this article are available in the article and in its online supplementary material. If additional information is needed please contact the corresponding author.
Funding
National Institutes of Health (R01AG059721 to J.-A.A.W., RF1NS122218, R01AG067741 to D.E.K. and J.-A.A.W.).)
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Sara Cazzaro, Department of Pathology, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA; Department of Molecular Medicine, Byrd Alzheimer’s Center & Research Institute, USF Health Morsani College of Medicine, Tampa, FL 33613, USA.
Xingyu Zhao, Department of Molecular Medicine, Byrd Alzheimer’s Center & Research Institute, USF Health Morsani College of Medicine, Tampa, FL 33613, USA.
Victoria K Zhao, Department of Pathology, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA.
Yenna K Kim, Department of Pathology, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA.
Jung-A A Woo, Department of Pathology, School of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA.
References
- 1. Chakravorty, A., Jetto, C.T. and Manjithaya, R. (2019) Dysfunctional mitochondria and mitophagy as drivers of Alzheimer's disease pathogenesis. Front. Aging Neurosci., 11, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boland, B., Kumar, A., Lee, S., Platt, F.M., Wegiel, J., Yu, W.H. and Nixon, R.A. (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci., 28(27), 6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bordi, M., Berg, M.J., Mohan, P.S., Peterhoff, C.M., Alldred, M.J., Che, S., Ginsberg, S.D. and Nixon, R.A. (2016) Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy, 12(12), 2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng, Q., Luo, Y., Zhang, X.N., Yang, X.F., Hong, X.Y., Sun, D.S., Li, X.C., Hu, Y., Li, X.G., Zhang, J.F. et al. (2020) MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy, 16(4), 641–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanji, K., Miki, Y., Ozaki, T., Maruyama, A., Yoshida, H., Mimura, J., Matsumiya, T., Mori, F., Imaizumi, T., Itoh, K. et al. (2014) Phosphorylation of serine 349 of p62 in Alzheimer's disease brain. Acta Neuropathol Commun, 2, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang, W., Zhao, F., Ma, X., Perry, G. and Zhu, X. (2020) Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol. Neurodegener., 15(1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernstein, B.W. and Bamburg, J.R. (2010) ADF/cofilin: a functional node in cell biology. Trends Cell Biol., 20(4), 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim, J.S., Huang, T.Y. and Bokoch, G.M. (2009) Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell, 20(11), 2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eiseler, T., Döppler, H., Yan, I.K., Kitatani, K., Mizuno, K. and Storz, P. (2009) Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol., 11(5), 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klamt, F., Zdanov, S., Levine, R.L., Pariser, A., Zhang, Y., Zhang, B., Yu, L.R., Veenstra, T.D. and Shacter, E. (2009) Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol., 11(10), 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shankar, G.M., Bloodgood, B.L., Townsend, M., Walsh, D.M., Selkoe, D.J. and Sabatini, B.L. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci., 27(11), 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao, L., Ma, Q.L., Calon, F., Harris-White, M.E., Yang, F., Lim, G.P., Morihara, T., Ubeda, O.J., Ambegaokar, S., Hansen, J.E. et al. (2006) Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat. Neurosci., 9(2), 234–242. [DOI] [PubMed] [Google Scholar]
- 13. Woo, J.A., Zhao, X., Khan, H., Penn, C., Wang, X., Joly-Amado, A., Weeber, E., Morgan, D. and Kang, D.E. (2015) Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1-integrin conformers. Cell Death Differ., 22(6), 921–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim, T., Vidal, G.S., Djurisic, M., William, C.M., Birnbaum, M.E., Garcia, K.C., Hyman, B.T. and Shatz, C.J. (2013) Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science, 341(6152), 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katsuragi, Y., Ichimura, Y. and Komatsu, M. (2015) p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J., 282(24), 4672–4678. [DOI] [PubMed] [Google Scholar]
- 16. Matsumoto, G., Shimogori, T., Hattori, N. and Nukina, N. (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet., 24(15), 4429–4442. [DOI] [PubMed] [Google Scholar]
- 17. Sánchez-Martín, P. and Komatsu, M. (2018) p62/SQSTM1 - steering the cell through health and disease. J. Cell Sci., 131(21). [DOI] [PubMed] [Google Scholar]
- 18. Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M.A., Dupont, N., Ornatowski, W., Jiang, S., Bradfute, S.B. et al. (2012) TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity, 37(2), 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim, J., Lachenmayer, M.L., Wu, S., Liu, W., Kundu, M., Wang, R., Komatsu, M., Oh, Y.J., Zhao, Y. and Yue, Z. (2015) Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet., 11(2), e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang, C., Woo, J.A., Liu, T., Zhao, X., Cazzaro, S., Yan, Y., Matlack, J., Kee, T., LePochat, P. and Kang, D.E. (2021) SSH1 impedes SQSTM1/p62 flux and MAPT/Tau clearance independent of CFL (cofilin) activation. Autophagy, 17(9), 2144–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smiley, S.T., Reers, M., Mottola-Hartshorn, C., Lin, M., Chen, A., Smith, T.W., Steele, G.D., Jr. and Chen, L.B. (1991) Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. U. S. A., 88(9), 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurita, S., Watanabe, Y., Gunji, E., Ohashi, K. and Mizuno, K. (2008) Molecular dissection of the mechanisms of substrate recognition and F-actin-mediated activation of cofilin-phosphatase Slingshot-1. J. Biol. Chem., 283(47), 32542–32552. [DOI] [PubMed] [Google Scholar]
- 23. Cai, Q. and Tammineni, P. (2017) Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J. Alzheimers Dis., 57(4), 1087–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McWilliams, T.G., Prescott, A.R., Allen, G.F., Tamjar, J., Munson, M.J., Thomson, C., Muqit, M.M. and Ganley, I.G. (2016) Mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol., 214(3), 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu, X., Perry, G., Moreira, P.I., Aliev, G., Cash, A.D., Hirai, K. and Smith, M.A. (2006) Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis., 9(2), 147–153. [DOI] [PubMed] [Google Scholar]
- 26. Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R.L., Atwood, C.S., Johnson, A.B., Kress, Y., Vinters, H.V., Tabaton, M. et al. (2001) Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci., 21(9), 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang, X., Su, B., Siedlak, S.L., Moreira, P.I., Fujioka, H., Wang, Y. and Casadesus and G., Zhu, X. (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U. S. A., 105(49), 19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E.K., Jones, P.K., Ghanbari, H., Wataya, T., Shimohama, S. et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol., 60(8), 759–767. [DOI] [PubMed] [Google Scholar]
- 29. Hauptmann, S., Scherping, I., Dröse, S., Brandt, U., Schulz, K.L., Jendrach, M., Leuner, K., Eckert, A. and Müller, W.E. (2009) Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging, 30(10), 1574–1586. [DOI] [PubMed] [Google Scholar]
- 30. Lin, M.T. and Beal, M.F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443(7113), 787–795. [DOI] [PubMed] [Google Scholar]
- 31. Wang, Y., Xu, E., Musich, P.R. and Lin, F. (2019) Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther, 25(7), 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo, C., Sun, L., Chen, X. and Zhang, D. (2013) Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res., 8(21), 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fivenson, E.M., Lautrup, S., Sun, N., Scheibye-Knudsen, M., Stevnsner, T., Nilsen, H., Bohr, V.A. and Fang, E.F. (2017) Mitophagy in neurodegeneration and aging. Neurochem. Int., 109, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frank, S.J. (2006) Dysregulation of mitochondrial fusion and fission: an emerging concept in neurodegeneration. Acta Neuropathol., 111(2), 93–100. [DOI] [PubMed] [Google Scholar]
- 35. Chen, H. and Chan, D.C. (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet., 18(R2), R169–R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pradeepkiran, J.A. and Reddy, P.H. (2020) Defective mitophagy in Alzheimer's disease. Ageing Res. Rev., 64, 101191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., Gabelle, A., Didic, M., De Septenville, A., Millecamps, S. et al. (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol., 70(11), 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Layfield, R. and Hocking, L.J. (2004) SQSTM1 and Paget's disease of bone. Calcif. Tissue Int., 75(5), 347–357. [DOI] [PubMed] [Google Scholar]
- 39. Kwok, C.T., Morris, A. and de Belleroche, J.S. (2014) Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur. J. Hum. Genet., 22(4), 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teyssou, E., Takeda, T., Lebon, V., Boillée, S., Doukouré, B., Bataillon, G., Sazdovitch, V., Cazeneuve, C., Meininger, V., LeGuern, E. et al. (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol., 125(4), 511–522. [DOI] [PubMed] [Google Scholar]
- 41. Hocking, L.J., Lucas, G.J., Daroszewska, A., Cundy, T., Nicholson, G.C., Donath, J., Walsh, J.P., Finlayson, C., Cavey, J.R., Ciani, B. et al. (2004) Novel UBA domain mutations of SQSTM1 in Paget's disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J Bone Miner. Res., 19(7), 1122–1127. [DOI] [PubMed] [Google Scholar]
- 42. Laurin, N., Brown, J.P., Morissette, J. and Raymond, V. (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet., 70(6), 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zatloukal, K., Stumptner, C., Fuchsbichler, A., Heid, H., Schnoelzer, M., Kenner, L., Kleinert, R., Prinz, M., Aguzzi, A. and Denk, H. (2002) p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol., 160(1), 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nagaoka, U., Kim, K., Jana, N.R., Doi, H., Maruyama, M., Mitsui, K., Oyama/ F. and Nukina, N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions . J. Neurochem., 2004. 91(1): p. 57–68. [DOI] [PubMed] [Google Scholar]
- 45. Kuusisto, E., Salminen, A. and Alafuzoff, I. (2002) Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol. Appl. Neurobiol., 28(3), 228–237. [DOI] [PubMed] [Google Scholar]
- 46. Kuusisto, E., Salminen, A. and Alafuzoff, I. (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport, 12(10), 2085–2090. [DOI] [PubMed] [Google Scholar]
- 47. Matsumoto, G., Wada, K., Okuno, M., Kurosawa, M. and Nukina, N. (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell, 44(2), 279–289. [DOI] [PubMed] [Google Scholar]
- 48. Jankowsky, J.L., Fadale, D.J., Anderson, J., Xu, G.M., Gonzales, V., Jenkins, N.A., Copeland, N.G., Lee, M.K., Younkin, L.H., Wagner, S.L. et al. (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet., 13(2), 159–170. [DOI] [PubMed] [Google Scholar]
- 49. Stine, W.B., Jr., Dahlgren, K.N., Krafft, G.A. and LaDu, M.J. (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem., 278(13), 11612–11622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material. If additional information is needed please contact the corresponding author.