

Abstract
Aims/hypothesis
Beta cells control glucose homeostasis via regulated production and secretion of insulin. This function arises from a highly specialised gene expression programme that is established during development and then sustained, with limited flexibility, in terminally differentiated cells. Dysregulation of this programme is seen in type 2 diabetes but mechanisms that preserve gene expression or underlie its dysregulation in mature cells are not well resolved. This study investigated whether methylation of histone H3 lysine 4 (H3K4), a marker of gene promoters with unresolved functional importance, is necessary for the maintenance of mature beta cell function.
Methods
Beta cell function, gene expression and chromatin modifications were analysed in conditional Dpy30 knockout mice, in which H3K4 methyltransferase activity is impaired, and in a mouse model of diabetes.
Results
H3K4 methylation maintains expression of genes that are important for insulin biosynthesis and glucose responsiveness. Deficient methylation of H3K4 leads to a less active and more repressed epigenome profile that locally correlates with gene expression deficits but does not globally reduce gene expression. Instead, developmentally regulated genes and genes in weakly active or suppressed states particularly rely on H3K4 methylation. We further show that H3K4 trimethylation (H3K4me3) is reorganised in islets from the Leprdb/db mouse model of diabetes in favour of weakly active and disallowed genes at the expense of terminal beta cell markers with broad H3K4me3 peaks.
Conclusions/interpretation
Sustained methylation of H3K4 is critical for the maintenance of beta cell function. Redistribution of H3K4me3 is linked to gene expression changes that are implicated in diabetes pathology.
Graphical abstract
Supplementary Information
The online version of this article (10.1007/s00125-023-05896-6) contains peer-reviewed but unedited supplementary material.
Keywords: Beta cell, Chromatin, COMPASS, DPY30, H3K4me3, Insulin, Transcription, Type 2 diabetes

Introduction
Tissue-specific transcription programmes rely on precise activation and maintenance of specific genes and stable repression of other genes. Chromatin modifications are central to this system of cellular memory. Beta cells exposed to metabolic stress show widespread changes in gene expression, including activation of developmentally silenced genes and decreased expression of genes that are important for insulin biosynthesis or glucose responsiveness or that enforce cell identity [1, 2]. There is mounting evidence that epigenetic mechanisms maintain mature beta cell function and identity and that the chromatin landscape is reshaped in beta cells exposed to chronic metabolic stress [3–9]. Chromatin-modifying enzymes are therefore promising therapeutic targets. For example, inhibition of histone H3 lysine 4 (H3K4) methyltransferases has been suggested to prevent ectopic gene activation [4] or increase replication [10] in beta cells during type 2 diabetes; however, the consequences of such a strategy are unknown.
Methylation of H3K4 is a highly conserved post-translational modification linked to RNA polymerase II-dependent transcription. H3K4 trimethylation (H3K4me3) is reliably enriched at active promoters, where the level of enrichment correlates with transcriptional output [11]. H3K4 monomethylation (H3K4me1) is enriched at active and primed enhancers, promoters and gene bodies [12]. The extent to which H3K4 methylation regulates transcription is less clear. On the one hand, H3K4 methylation can regulate gene activity by recruitment of transcriptional machinery [13] and chromatin remodellers [14], facilitating enhancer–promoter interactions [15], and by blocking repression [16, 17]. On the other hand, global (i.e. genome-wide) ablation of H3K4 methylation is remarkably well tolerated [18–20]. Evidence in animal models suggests that H3K4 methyltransferases impact the expression of only a fraction of genes and are necessary only for specific roles in development or in response to environmental stress [20–23]. Further, loss of H3K4 methylation is associated with gene upregulation in some circumstances [24, 25]. Rather than being a strict requirement for gene activation, the emerging view is that H3K4 methylation fine-tunes gene expression by providing a context-specific but generally activating signal [26], reducing variability of transcription [27], providing a persistent memory of transcriptional activity [28] and maintaining the potential for future expression [29]. H3K4 methylation also has a non-transcriptional role in DNA damage repair [30]. Thus, while H3K4me3 is predictive of transcription [31], the consequences of its removal are difficult to predict.
Mono-, di- and trimethylation of H3K4 are catalysed by six partially redundant multiprotein complexes distinguished by the central methyltransferase: mixed lineage leukaemia protein-1 to -4 (MLL1, MLL2, MLL3, MLL4) and SET domain containing 1A and 1B (SETD1A and SETD1B). These complexes, called the COMPASS (complex of proteins associated with Set1) family, influence gene activity via distinct catalytic and non-catalytic functions. Recruitment of COMPASS by a variety of factors directs methylation of H3K4 at particular genomic loci [32]. The COMPASS family can also recruit a variety of factors themselves, including cell cycle regulators, tumour suppressors and other chromatin-modifying enzymes [33]. The biological relevance of these interactions is exemplified by reports that cells expressing catalytically inactive mutants of methyltransferase subunits display milder defects to gene expression and cell functions than cells completely lacking the same subunit [18, 34–36]. Non-catalytic subunits of COMPASS vary but always include a core subcomplex of WD repeat-containing protein 5 (WDR5), retinoblastoma-binding protein 5 (RBBP5), absent, small or homeotic 2-like protein (ASH2L) and Dumpy-30 (DPY30) [37]. WDR5, RBBP5 and ASH2L are required for complex stability and enzymatic activity [38]. DPY30 is not required for complex stability or methyltransferase activity but improves coordination of the COMPASS catalytic domain with H3K4 in the context of a complete nucleosome [39]. For this reason, DPY30 is required for methylation of H3K4 genome-wide in vivo [39]. DPY30 and the other core subunits are essential genes [40] but, among 27 tissues tested in the Human Protein Atlas tissue-specific transcriptome project, the median expression of each was lowest in the pancreas [41]. DPY30 is enriched in islets compared with pancreatic exocrine cells in mice [42], indicating the potential relevance of COMPASS activity to pancreatic endocrine functions. Accordingly, we previously demonstrated that Dpy30 fine-tunes cell fate decisions during mouse pancreas development in favour of endocrine lineages [23] and contributes to endocrine cell maturation [43]. By contrast, it is not known whether the continuous activity of H3K4 methyltransferases is necessary in terminally differentiated cells, where lineage-specific transcription programmes have already been established. We therefore sought to define the role of H3K4 methylation in the regulation of gene expression in terminally differentiated beta cells.
Methods
Animals
The following mice strains were used: Dpy30flox/flox [23], Rosa26mTmG (The Jackson Laboratory, USA; #007576 [44]), Pdx1CreERTg (The Jackson Laboratory; #024968 [45]), Ins1Cre (The Jackson Laboratory; #026801 [46]), BKS Leprdb/db and Lepr+/+ (The Jackson Laboratory; #000642). Genotypes used for Pdx1CreER studies were as follows—knockout: Pdx1CreERTg/0;Dpy30flox/flox;Rosa26mTmG/+, wild-type: Pdx1CreER0/0;Dpy30flox/flox;Rosa26mTmG/+, or, if sorting recombined cells was required: Pdx1CreERTg/0;Dpy30+/+;Rosa26mTmG/+. Regardless of genotype, all mice used in Pdx1CreER studies were administered 8 mg tamoxifen (Sigma-Aldrich, USA) by oral gavage three times over 5 days at 8 weeks of age and analysed 45 days later unless otherwise specified. Genotypes used for Ins1Cre studies were as follows—knockout: Ins1Cre/+;Dpy30flox/flox, wild-type: Ins1+/+;Dpy30flox/flox, heterozygous: Ins1Cre/+; Dpy30flox/+. Mice in Ins1Cre studies were analysed at 5 weeks unless otherwise specified. Leprdb/db and Lepr+/+ mice were analysed at 12 weeks. Mice were kept under conventional conditions on a 12 h light/dark cycle with free access to water and food (Teklad 2918, Envigo, UK). Experiments were restricted to male mice of the indicated ages and genotypes; no other exclusion criteria were considered. Mice were not randomised and experimenters were not blinded to genotypes. Experiments were approved by the University of British Columbia Animal Care Committee (certificates A17-0045 and A18-0111).
Unfasted blood glucose was measured from the tail tip between 10:00 and 12:00 using a OneTouch Ultra Mini Glucometer (Johnson & Johnson, USA). For glucose tolerance tests, 2 g/kg body weight of glucose was injected intraperitoneally following a 6 h fast. Blood was sampled from a lateral saphenous vein. Serum was prepared by centrifuging blood samples at 9000 g for 9 min at 4°C. Serum insulin was measured using an ELISA (Alpco, USA).
Islets were isolated using collagenase digestion [43] and dispersed to single cells [47] as described previously. EGFP-positive tdTomato-negative cells were enriched from Pdx1CreERTg/0;Rosa26mTmG mice using a FACSAria II Cell Sorter (BD Biosciences, USA).
Drosophila S2 cells (Life Technologies, USA) were cultured at 25°C in Schneider’s Drosophila medium (Life Technologies) supplemented with 10% (vol/vol) heat-inactivated FBS (Life Technologies).
Co-immunoprecipitation, immunoblotting and immunohistochemistry
Immunoprecipitation from islet cell nuclear lysates, immunoblotting of islet lysates and immunofluorescence analyses of paraffin-embedded pancreas sections were carried out using standard protocols as outlined in the electronic supplementary material (ESM) Methods using the antibodies listed in ESM Table 1.
Chromatin immunoprecipitation–sequencing (ChIP-seq)
For Pdx1CreERTg/0;Dpy30+/+; Rosa26mTmG/+ and Pdx1CreERTg/0;Dpy30flox/flox;Rosa26mTmG/+ mice, 100,000 beta cells were pooled with 50,000 Drosophila S2 cells by FACS. For Leprdb/db and Lepr+/+ mice, 100,000 dispersed islet cells were counted using a hemacytometer and spiked with 50,000 Drosophila S2 cells without sorting. The ULI-NChIP procedure [48] was used to generate ChIP-seq libraries using the antibodies listed in ESM Table 1 in biological duplicate. Immunoprecipitated DNA was prepared for sequencing using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, USA). See ESM Methods for further details.
RNA-seq
Beta cells (70,000–226,000 per mouse, n=3) from Pdx1CreERTg/0;Dpy30+/+;Rosa26mTmG/+ and Pdx1CreERTg/0;Dpy30flox/flox;Rosa26mTmG/+ mice were purified by FACS and spiked with 10% Drosophila S2 cells. mRNA isolation and library preparation were performed as described previously [43]. See ESM Methods for further details.
Single-cell RNA-seq
Islet cells from one Pdx1CreERTg/0;Dpy30+/+;Rosa26mTmG/+ mouse and one Pdx1CreERTg/0;Dpy30flox/flox;Rosa26mTmG/+ mouse were processed through the Chromium Single Cell 3′ protocol using the Chromium Controller with Reagent Kit v3.1 and Dual Index Kit TT Set A (10x Genomics, USA). See ESM Methods for further details.
Pyrosequencing
DNA from 100 islets was bisulphite converted using the EZ DNA Methylation-Direct Kit (Zymo Research, USA). Genomic regions were amplified using the PyroMark PCR Kit with CpG Assay primers Mm_Igf2_04_PM, Mm_Cd81_01_PM or Mm_Cdkn1c_01_PM (Qiagen, Germany). Per cent methylation at amplified CpGs was measured using the PyroMark Q96 MD (Qiagen).
Electron microscopy
Islets were fixed in 2% (wt/vol) glutaraldehyde (Sigma-Aldrich) and then submitted to the Electron Microscopy Facility at McMaster University (Canada). Islets were post-fixed with 1% (wt/vol) osmium tetroxide, dehydrated in ethanol, embedded in Spurr’s resin and sectioned with a Leica UCT ultramicrotome. Sections were stained with uranyl acetate and lead citrate and imaged with a JEM 1200 EX TEMSCAN transmission electron microscope (JEOL, USA). Insulin granules were quantified using ImageJ v1.52a [49].
Islet functional assays
Insulin secretion assays [43] and calcium imaging [50] were performed as described previously. Respiration was measured in dispersed islets using the Seahorse XF24 Extracellular Flux Analyzer (Agilent, USA).
Analysis of publicly available data
Fastq files from GSE50244 [51], GSE50386 [52], GSE107489 [53] and GSE124742 [54] were downloaded from the Sequence Read Archive and analysed as described above. A table of per cent methylation of each cytosine in beta cells from 16- to 20-month-old C57BL6 mice was downloaded from GSE68618 [55] and converted to the GRCm38/mm10 reference genome using the University of California Santa Cruz liftover utility [56].
Statistics
Bar plots show means ± SD with individual data points. In box and whisker plots, the central horizontal line indicates the median, the upper and lower limits of the box indicate the first and third quartiles, and the whiskers span 1.5× the IQR. Correlation was estimated using Spearman’s coefficient. P values were calculated using unpaired Welch’s t tests, Wilcoxon tests, Wald tests, mixed-effect models, ANOVA, Fisher’s exact tests or permutation tests as indicated. The Benjamini–Hochberg correction was applied where indicated. Calculations were performed in R v4.2.1 (http://www.r-project.org/) or Prism v9.5.0 (GraphPad Software, USA).
Results
Reduction of H3K4 methylation in beta cells of adult mice leads to glucose intolerance and hyperglycaemia
To induce synchronised deletion of Dpy30 in mature beta cells, Pdx1CreERTg/0;Dpy30flox/flox;Rosa26mTmG/+ mice were administered tamoxifen at 8 weeks of age (hereafter called Dpy30-KO mice) (Fig. 1a,b). As expected, Dpy30-KO did not impact assembly of the other core subunits of the COMPASS complex or their association with chromatin (Fig. 1c) but did reduce H3K4 methylation (Fig. 1d).
Fig. 1.

Reduction of H3K4 methylation in beta cells of adult mice leads to glucose intolerance and hyperglycaemia. (a) Schematic showing the core COMPASS subunits and a nucleosome methylated on H3K4. (b) Genome-aligned RNA-seq reads at the Dpy30 gene locus in Dpy30-WT and Dpy30-KO beta cells 15 days post tamoxifen. Note that the floxed exon 4 is efficiently deleted in the KO cells. (c) Immunoblots showing the COMPASS subunits RBBP5, ASH2L and WDR5 and the nucleosome protein histone H3, co-immunoprecipitated with WDR5 or an IgG control from Dpy30-WT and Dpy30-KO islet cell nuclei 45 days post tamoxifen administration. Representative immunoblots of three independent co-immunoprecipitations are shown. IP, immunoprecipitation. (d) Immunoblots showing H3K4me3, H3K4me1, histone H3 lysine 27 acetylation (H3K27ac), histone H3 lysine 27 trimethylation (H3K27me3) and total histone H3 in islets from Dpy30-WT and Dpy30-KO mice 45 days post tamoxifen administration. Numbers beneath each band indicate the band intensity normalised to the left-most sample. (e) Mean immunofluorescent intensity of DPY30, H3K4me3 and H3K4me1 in Dpy30-WT and Dpy30-KO beta cell nuclei at the indicated days after tamoxifen administration. Data are normalised to the fluorescence intensity of alpha cell nuclei (n=3). (f) Example immunohistochemical images of Dpy30-KO islets used for measurements in (e) showing H3K4me3 (cyan), insulin (magenta) and glucagon (yellow). Scale bars: 100 μm. (g, h) Scatterplots showing log2(fold change) and –log10(p) of gene expression in Dpy30-KO vs Dpy30-WT beta cells 15 days (g) and 45 days (h) post tamoxifen administration. Genes showing a twofold or greater increase or decrease in expression at p≤0.01 (calculated using Wald tests with Benjamini–Hochberg correction) are coloured red and green, respectively, and enumerated above. The full-length transcript of Dpy30 is blue. (i, j) Blood glucose (i) and serum insulin (j) levels during an IPGTT in Dpy30-WT and Dpy30-KO mice 15, 30 and 45 days after tamoxifen administration. Data are means ± SD (n=8–15). P values were calculated from AUCs using multiple two-tailed t tests with Welch’s and Benjamini–Hochberg corrections. (k, l) Unfasted blood glucose levels (k) and body mass (l) of Dpy30-WT and Dpy30-KO mice up to 60 days after tamoxifen administration. Data are individual measurements with means (n=8; however, tracking was stopped after a blood glucose reading ≥20 mmol/l). *p<0.05, ***p<0.001
H3K4me3 and H3K4me1 were stable for at least 15 days after tamoxifen administration and were then gradually lost during the subsequent 45 days, whereas DPY30 was undetectable by 15 days (Fig. 1e,f). We reasoned that defects arising in Dpy30-KO beta cells by 15 days highlight functions of DPY30 itself. Defects arising later, by 45 days, highlight functions of H3K4 methylation. Remarkably, loss of DPY30 had little effect on gene expression in beta cells, with RNA-seq analysis identifying only three differentially expressed genes (Dpy30, Edn3, C3) 15 days post tamoxifen administration (Fig. 1g, ESM Tables 2 and 3). In contrast, 828 genes were dysregulated at 45 days, the majority of which (634) showed lower expression in Dpy30-KO cells (Fig. 1h, ESM Tables 4 and 5). We conclude that transcriptome remodelling in Dpy30-KO beta cells results from reduction of H3K4 methylation.
Dpy30-KO mice developed impaired glucose tolerance and had reduced serum insulin levels by 45 days post tamoxifen administration (Fig. 1i,j) and then rapidly developed diabetes (Fig. 1k,l), prompting euthanasia by 60 days. As the Pdx1CreER transgene may also drive recombination in delta cells [57] and the hypothalamus [58], we additionally used Ins1Cre mice [46] to verify that Dpy30 deletion specifically in beta cells is sufficient to drive the in vivo phenotype. Deletion of Dpy30 from maturing beta cells using Ins1Cre caused reduction of H3K4 methylation in islets by 5 weeks of age, leading to hyperglycaemia, impaired glucose tolerance and reduced serum insulin levels (ESM Fig. 1).
H3K4 methylation maintains expression of genes involved in insulin production and glucose-stimulated activity
The insulin deficit in Dpy30-KO mice prompted us to examine insulin production and secretion. We first confirmed that H3K4me3 was lost from the Ins1 and Ins2 promoters in Dpy30-KO beta cells (Fig. 2a) 45 days after tamoxifen administration using ChIP-seq. H3K4me1 was also dramatically reduced (Fig. 2a). Despite this, RNA-seq data showed a surprisingly modest reduction in levels of Ins1 and Ins2 transcripts (Fig. 2b,c), indicating that robust expression of Ins1 and Ins2 can occur in the absence of local H3K4me3. However, insulin (but not glucagon) immunofluorescence intensity decreased in Dpy30-KO mice (Fig. 1e and ESM Fig. 2a, b) and expression of several genes involved in insulin maturation and packaging was reduced (Fig. 2d, ESM Fig. 2c–g). This included Nnat (ESM Fig. 2d), encoding neuronatin, part of the signal peptidase complex and knockdown of which redirects preproinsulin for proteasomal degradation [59]. Slc30a8, encoding the zinc transporter ZNT8 required to package insulin into dense core granules, knockout of which impairs proinsulin-to-insulin processing [60], was also downregulated (ESM Fig. 2e). Consistent with impairment of insulin synthesis and/or increased degradation in Dpy30-KO mice, we observed a significant loss of islet insulin content (Fig. 2e). Reduction in insulin granule size and density in individual beta cells from Dpy30-KO mice was confirmed using electron microscopy (Fig. 2f–h). Therefore, Dpy30-KO in beta cells results in a reduction in insulin content.
Fig. 2.

Genes involved in insulin production and glucose-induced activity are regulated by H3K4 methylation. (a) H3K4me3 and H3K4me1 enrichment at the Ins1 and Ins2 gene loci in Dpy30-WT and Dpy30-KO beta cells 45 days after tamoxifen administration. (b, c) Ins1 (b) and Ins2 (c) RNA levels in Dpy30-WT and Dpy30-KO beta cells 45 days after tamoxifen administration. Expression and p values were calculated from RNA-seq data using DESeq2 (Wald tests with Benjamini–Hochberg correction). (d) Heatmap showing expression Z scores in Dpy30-WT and Dpy30-KO beta cells 45 days post tamoxifen administration for selected genes. P values were calculated from RNA-seq data using Wald tests with Benjamini–Hochberg correction (n=3). Significantly downregulated and upregulated genes are shown in green and pink, respectively. (e) Insulin content in Dpy30-WT and Dpy30-KO islets. P values were calculated using two-tailed t tests with Welch’s correction (n=8 WT, n=6 KO). (f) Representative transmission electron micrographs of beta cells from a Dpy30-WT mouse and a Dpy30-KO mouse. Examples of insulin granules and mitochondria are indicated with cyan and yellow arrows, respectively. Scale bars: 2 μm. (g, h) Quantification of median insulin core granule density (g) and size (h). P values were calculated using two-tailed t tests with Welch’s correction (n=3). (i) Enrichment analysis of Gene Ontology: biological process terms in differentially expressed genes 45 days after tamoxifen administration. P values represent EASE scores, modified Fisher’s exact p values, calculated using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v.6.8 [82]. (j) Insulin secretion from Dpy30-WT and Dpy30-KO islets during static in vitro stimulation with glucose and KCl solutions, normalised to islet insulin content. P values were calculated using multiple two-tailed t tests with Welch’s and Benjamini–Hochberg corrections (n=7 WT, n=5 KO). (k) Cytosolic Ca2+ concentration in islets from Dpy30-WT and Dpy30-KO mice during in vitro perifusion of glucose and KCl solutions. P values were calculated for the AUC in each time block by one-way ANOVA between genotypes (n=4 WT, n=3 KO). (l) Oxygen consumption rate in Dpy30-WT and Dpy30-KO dispersed islet cells during treatment with the indicated compounds (n=3 WT, n=4 KO). (m, n) Mitochondrial respiration in Dpy30-WT and Dpy30-KO islet cells in 16.7 mM glucose (m) and their maximal respiration capacity (n), inferred from the data shown in (l). P values were calculated using two-tailed t tests with Welch’s correction (n=3 WT, n=4 KO). (o) Fraction of cytoplasm area occupied by mitochondria in transmission electron micrographs of Dpy30-WT and Dpy30-KO beta cells. P values were calculated using two-tailed t tests with Welch’s correction (n=3). *p<0.05, **p<0.01, ***p<0.001. FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; Glc, glucose; OCR, oxygen consumption rate; RAA, rotenone and antimycin A
In line with their lower insulin content, Dpy30-KO islets secreted less insulin during high glucose or KCl stimulation than Dpy30 wild-type (WT) islets (ESM Fig. 2h). Gene Ontology analysis of downregulated genes suggested the additional impairment of biological processes required for stimulated insulin secretion, namely vesicle fusion, calcium-dependent exocytosis and glucose homeostasis (Fig. 2i, ESM Table 6). We tested whether this gene signature was associated with functional changes. When accounting for their low insulin content, Dpy30-KO islets responded normally to KCl, but glucose-stimulated insulin secretion remained low compared with Dpy30-WT islets (Fig. 2j). Cytosolic calcium influx was also impaired during high glucose but not KCl stimulation (Fig. 2k). Gene Ontology of upregulated genes prioritised energy production processes including oxidation/reduction and the electron transport chain (Fig. 2i, ESM Table 7); accordingly, mitochondrial respiratory capacity (Fig. 2l,n) and area (Fig. 2f,o) were elevated in Dpy30-KO cells. This was not associated with an elevated rate of glucose-stimulated respiration (Fig. 2m), perhaps because expression of the rate-limiting enzyme of glycolysis, Gck, was not altered (Fig. 2d, ESM Fig. 2f). These data suggest that Dpy30-KO impairs insulin secretion owing to a reduction of beta cell insulin content combined with a glucose-specific signalling defect downstream of mitochondrial glucose oxidation, although more work will be required to identify the precise mechanism. Together, these data show that genes with altered expression were enriched for roles in stimulation, secretion and energy production.
Loss of H3K4 methylation is associated with limited gene downregulation in mature beta cells
While H3K4 methylation is predictive of transcription [31], our RNA-seq analysis indicates that only 634 of the 14,677 (4.3%) expressed genes were downregulated by depletion of H3K4 methylation (Fig. 1g, ESM Table 4). This suggests that H3K4 methylation is not required for the expression of most genes in mature beta cells. We therefore explored whether dysregulated genes were distinguished by specific epigenetic features. We first confirmed that H3K4 methylation was depleted at active promoters (Fig. 3a,b) and genome-wide (Fig. 3c,d) by Dpy30-KO. Despite this, the transcriptomes of Dpy30-KO and Dpy30-WT cells were well correlated and the total RNA expression of Dpy30-KO cells was not decreased (Fig. 3e). Therefore, global depletion of H3K4 methylation does not cause a global decrease in gene expression in mature beta cells.
Fig. 3.

Transcription start site (TSS)-associated H3K4 methylation maintains gene expression in mature beta cells. (a, b) Average enrichment profiles of H3K4me3 (a) and H3K4me1 (b) at the TSS of all expressed genes in Dpy30-WT and Dpy30-KO chromatin. (c, d) Scatterplot of H3K4me3 (c) and H3K4me1 (d) enrichment in 10 kb bins spanning the genome in Dpy30-KO vs Dpy30-WT cells. (e) Scatterplot showing gene expression in Dpy30-KO vs Dpy30-WT cells. (f) Examples of genes that retain H3K4me3 (i), lose H3K4me3 but retain H3K4me1 (ii) and lose H3K4me3 and H3K4me1 (iii) from the TSS in Dpy30-KO chromatin. (g) Box and whisker plot showing log2(fold change) in RNA expression for gene groups exemplified in (f) (group i, n=10,045 genes, group ii, n=2302 genes, group iii, n=111 genes). (h) Contour scatterplot showing log2(fold change) in H3K4me1 enrichment (x axis) plotted against log2(fold change) in H3K4me3 enrichment (y axis) in the TSS±1 kb of all expressed genes. The log2(fold change) in RNA expression of the associated genes is shown as a colour gradient. ***p<0.001
To explore further, we segregated genes into three groups based on residual enrichment for H3K4 methylation, determined using model-based analysis of ChIP-seq (MACS2) [61]. Group (i) retained at least some H3K4me3 enrichment at the transcription start site (TSS) (10,045 genes); group (ii) lost H3K4me3 but retained at least some H3K4me1 enrichment (2302 genes); and group (iii) lost H3K4me3 and H3K4me1 (111 genes) (Fig. 3f, ESM Table 8). As a group, genes that retained at least some enrichment for H3K4me3 at the TSS were not downregulated in Dpy30-KO cells (Fig. 3g). Loss of both H3K4me3 and H3K4me1 from the TSS was associated with significant impairment of gene expression, and genes that lost H3K4me3 but retained H3K4me1 showed an intermediate effect (Fig. 3g). To visualise this relationship in a threshold-free manner, we plotted the change in H3K4me3, H3K4me1 and RNA expression for all active TSSs (Fig. 3h). Most genes clustered in the bottom left quadrant, indicating some reduction of both H3K4me3 and H3K4me1 from the TSS. Strikingly, genes that were highly downregulated clustered at the extreme edge, indicating that genes experiencing the greatest relative loss of both H3K4me3 and H3K4me1 were strongly downregulated (Fig. 3h). Thus, while transcriptome remodelling in this model is likely to reflect both direct and indirect effects of reduced methylation, these data show that loss of H3K4me3 from a gene promoter was associated with reduced expression of that gene.
H3K4 methylation enforces expression of weakly active and repressed genes in mature beta cells
We compared the chromatin profile of downregulated and stably expressed genes by measuring histone H3 lysine 27 trimethylation (H3K27me3), a marker of developmentally repressed chromatin, and histone H3 lysine 27 acetylation (H3K27ac), a marker of active chromatin, using ChIP-seq. We also retrieved data for DNA cytosine methylation (DNAme), a modification linked to gene silencing, in C57BL6 mouse beta cells [55], and examined nucleotide content. The downregulated gene set had a weakly active profile even in Dpy30-WT cells: enrichment for H3K4me3 and H3K27ac was low whereas H3K4me1, H3K27me3, G/C content, and DNAme were high in comparison to stably expressed genes (Fig. 4a–l). Accordingly, the downregulated gene set showed lower expression in Dpy30-WT cells than stably expressed genes (Fig. 4m). Interestingly, genes upregulated in Dpy30-KO cells had a similar epigenetic profile to downregulated genes (Fig. 4a–m).
Fig. 4.

Weakly active and developmentally regulated genes are susceptible to downregulation in H3K4me3-deficient beta cells. (a–l) Average enrichment profiles and quantification of H3K4me3 (a, b), H3K4me1 (c, d), H3K27ac (e, f), H3K27me3 (g, h), DNAme (i, j) and G/C nucleotide content (k, l) in Dpy30-WT cells for genes downregulated, stably expressed or upregulated by Dpy30-KO. (m) Box and whisker plot showing RNA expression in Dpy30-WT cells for genes downregulated, stably expressed or upregulated by Dpy30-KO. (n) Running enrichment plot for imprinted genes ([83]), genes up- or downregulated during mouse beta cell maturation (adults vs postnatal day 10, [63]) and mature beta cell transcription factor genes. P values were calculated using Wilcoxon rank-sum (b–m) or permutation (n) tests with Benjamini–Hochberg correction. *p<0.05, **p<0.01, ***p<0.001. NES, normalised enrichment score; ns, not significant; TES, transcription end site
H3K4 methylation deficiency leads to a less active and more repressed epigenome in mature beta cells
During development, active (H3K27ac-positive) and repressed (H3K27me3- or DNAme-positive) chromatin is dynamically regulated; resolution of the genome into active and repressed domains is fundamental to establishment of terminal cell type-specific transcriptomes [29, 62]. We noted that expression of genes that are dynamically regulated during, and important for, beta cell maturation [63] tended to be lower in Dpy30-KO cells (Fig. 4n), suggesting that maintenance of genes in an active state requires continual reinforcement in mature beta cells. Because G/C content tended to be high at downregulated gene loci (Fig. 4k,l) and imprinted genes tended to be downregulated in Dpy30-KO cells (Fig. 4n), we measured DNAme near the TSS of three downregulated genes (Cdkn1c, Igf2 and Cd81) but did not observe a change (ESM Fig. 3), suggesting that reduction of H3K4me3 did not immediately lead to accumulation of DNAme. We explored whether H3K27me3-positive regions expand in H3K4me3-deficient cells. As shown in Fig. 5a, the megabase-scale distribution of H3K27me3-positive compartments was largely unchanged in H3K4me3-deficient beta cells. However, H3K27me3 increased specifically at promoters of downregulated genes (Fig. 5g,h). These data suggest that maintenance of H3K4 trimethylation is necessary to prevent Polycomb-mediated gene repression in mature beta cells.
Fig. 5.

H3K4me3 deficiency leads to a less active and more repressed epigenome in mature beta cells. (a) Genome browser view of H3K4me3, H3K4me1, H3K27ac and H3K27me3 in Dpy30-WT and Dpy30-KO cells showing preservation of the megabase-scale organisation of H3K27me3-positive Polycomb-repressed compartments. (b, c) Scatterplots of H3K27ac (b) and H3K27me3 (c) enrichment in 10 kb bins spanning the genome in Dpy30-KO vs Dpy30-WT cells. (d, e) Average enrichment profiles of H3K27ac (d) and H3K27me3 (e) at the TSS of all expressed genes in Dpy30-WT and Dpy30-KO cells. (f, g) Box and whisker plots showing the log2(fold change) in H3K27ac (f) and H3K27me3 (g) enrichment in the TSS of downregulated, stable and upregulated genes in Dpy30-KO vs Dpy30-WT cells. (h) Genome browser view of a downregulated gene, Rab34, showing loss of H3K4me3, H3K4me1, H3K27ac and RNA and accumulation of H3K27me3 in Dpy30-KO cells. P values were calculated using Wilcoxon rank-sum tests with Benjamini–Hochberg correction. ***p<0.001
In contrast to the locus-specific accumulation of H3K27me3, H3K27ac was diminished throughout the genome in Dpy30-KO cells (Fig. 5b). Like H3K4me, H3K27ac was reduced even at promoters of genes that were stably expressed or upregulated in Dpy30-KO cells (Fig. 5d,f), indicating that the generalised decrease in H3K27ac was not a consequence of gene downregulation. Despite the global reduction, however, H3K27ac was still positively associated with gene expression in Dpy30-KO chromatin: H3K27ac peaks gaining intensity were near upregulated genes, and H3K27ac peaks losing intensity were near downregulated genes, more frequently than expected by chance (ESM Fig. 4). These observations are consistent with a model wherein acetylation of H3K27 is partially a consequence of H3K4 methylation [18] and of transcription [64].
To summarise, Dpy30-KO beta cells showed a global reduction in active histone marks but not of gene expression. Genes that lost an especially large fraction of H3K4 methylation tended to be downregulated; they tended to be developmentally regulated or in a weakly active state in Dpy30-WT beta cells and to accumulate the repressive mark H3K27me3, which was otherwise globally stable.
Reduction in transcriptional consistency in Dpy30-KO beta cells
Transcriptional entropy describes the ‘specialisation’ of a cell’s transcriptome whereby cells with low entropy have a narrow distribution of highly expressed genes and gene expression in cells with high entropy becomes less predictable. In islets, transcriptional entropy decreases during maturation [65] and increases in type 2 diabetes [4]. We explored whether reduction of H3K4 methylation increases the variability or entropy of gene expression. To this end, we processed islets from a Dpy30-WT mouse and a Dpy30-KO mouse for single-cell RNA-seq (ESM Fig. 5a). We obtained transcriptome profiles of 4877 beta cells, which formed two distinct clusters: cluster 1 contained cells from the Dpy30-WT mouse and a small population of beta cells from the Dpy30-KO mouse expressing tdTomato (indicating that they were not Cre-recombined) and cluster 2 contained EGFP-expressing Dpy30-KO beta cells (Fig. 6a,b). To model the divergence of Dpy30-KO beta cells from Dpy30-WT and unrecombined beta cells, we performed pseudotime trajectory analysis using Slingshot [66] (Fig. 6c). Expression of Ins1 and Ins2 showed a narrow distribution in Dpy30-WT and unrecombined beta cells but became more variable as pseudotime progressed in Dpy30-KO cells (Fig. 6d). More generally, overall transcriptional entropy increased in Dpy30-KO cells over pseudotime (Fig. 6e) and tended to be higher than in Dpy30-WT and unrecombined beta cells (Fig. 6f). As lower entropy is associated with greater cell maturity [65], we examined the expression of genes associated with immaturity and dedifferentiation but did not observe induction of these genes in Dpy30-KO cells (see ESM Fig. 5b, c and compare with Fig. 4n). Therefore, Dpy30-KO led to higher transcriptional entropy in mature beta cells but did not cause reversion to a developmentally immature transcriptional state.
Fig. 6.

Reduction in transcriptional consistency in beta cells from Dpy30-KO mice and human donors with type 2 diabetes. (a–c) Uniform manifold approximation and projection (UMAP) visualisation of beta cell transcriptomes coloured according to genotype of the donor mouse (a), expression of tdTomato, EGFP and their overlap (b) and pseudotime (c). (d) Scatterplot showing Ins1 and Ins2 expression in beta cells plotted against pseudotime. (e) Scatterplot showing gene expression entropy scores of beta cells plotted against pseudotime. (f) Box and whisker plot showing transcriptome entropy of beta cells in clusters 1 and 2 (2466 and 2411 cells, respectively). (g) Active transcription factor regulons identified using SCENIC [67, 68], showing the specificity score of each regulon for cluster 1 (purple) and cluster 2 (orange) connected by a line. Regulons are ranked from the most specific for cluster 1 on the left to the most specific for cluster 2 on the right, and some notable regulons are labelled. (h) Box and whisker plot showing the variance/mean ratio of gene expression across beta cells in clusters 1 and 2. (i) Variance/mean ratio of beta cell gene expression in cluster 2 vs cluster 1 (y axis) plotted as a function of H3K4me3 breadth in quantiles (x axis) (622 genes/quantile). Data are presented as means ± SD. The Spearman rank correlation is shown. (j, k) The same analysis as in (h) and (i), respectively, in beta cells from human donors with or without type 2 diabetes, using H3K4me3 ChIP-seq data from Bramswig et al [52] and single-cell RNA-seq (scRNA-seq) data from Camunas-Soler et al [54]. P values were calculated using Wilcoxon rank-sum tests. ***p<0.001
A rise in entropy implies a loss of transcriptional control. To identify transcription factors whose activity is altered following loss of Dpy30 and H3K4 methylation in mature beta cells we used single-cell regulatory network inference and clustering (SCENIC [67, 68]). Unsupervised clustering of beta cells on the basis of their regulon activities (i.e. expression of transcription factors and their putative targets) effectively separated Dpy30-KO beta cells from Dpy30-WT and unrecombined beta cells (ESM Fig. 6a, b), recreating clusters based on gene expression profiles (ESM Fig. 6c) and indicating a shift in the gene regulatory landscape after Dpy30-KO. Of 438 active regulons identified in beta cells, 319 (73%) showed lower activity in Dpy30-KO cells (cluster 2) (Fig. 6g, ESM Table 9). In line with the general downregulation of mature beta cell transcription factor genes (Fig. 4n), Foxa2, Isl1, Mafa, Mnx1, Neurod1, Pax6 and Pdx1 showed lower activity in Dpy30-KO cells cluster 2) (Fig. 6g, ESM Fig. 6d). Activity-regulated factors such as Fos and Junb, and Creb3 factors, which regulate secretory pathways [69], also showed lower activity (Fig. 6g, ESM Fig. 6d). Regulons showing greater activity in the Dpy30-KO cluster 2 included Ctcf and Rad21, which independently regulate chromatin organisation [70], and Mbd2, which binds to a methylated DNA motif [71] (Fig. 6g, ESM Fig. 6d, ESM Table 9). This analysis suggests that coordination between chromatin structure and transcription factor activity is altered in Dpy30-KO cells.
We examined whether transcriptional consistency, that is, the variability of gene expression between cells, was linked to the breadth of the H3K4me3 peak at gene promoters, as broad H3K4me3 peaks may increase transcriptional consistency [27]. We ranked H3K4me3 peaks in Dpy30-WT cells in order of increasing breadth and grouped them into 20 quantiles, each representing 5% of the H3K4me3 peaks (ESM Table 10). We then compared the variance in expression of genes with H3K4me3 peaks in each quantile between beta cell clusters. Transcriptional variability between cells was increased in Dpy30-KO cells (Fig. 6h). Greater H3K4me3 peak breadth was modestly correlated with a greater gain of variability in Dpy30-KO cells (Fig. 6i; Pearson r=0.149, p=1.11 × 10−52). Analysis of H3K4me3 ChIP-seq data from healthy human beta cells [52] and single-cell RNA-seq data from human donors with or without type 2 diabetes [54] suggests that variability of gene expression is also higher in type 2 diabetes (Fig. 6j) and that the gain in variability is weakly correlated with H3K4me3 peak breadth (Fig. 6k; Pearson r=0.071, p=6.04 × 10−14). These data support a minor association between broad H3K4me3 peaks and transcriptional consistency in beta cells, which is nominally linked to the increased transcriptional variability in type 2 diabetes.
H3K4me3 peak breadth stratifies genes dysregulated in type 2 diabetes
We explored the relationship between H3K4me3 peak breadth, gene expression and diabetes further. H3K4me3 forms 18,936 distinct peaks in beta cells. Ranking peaks by breadth revealed a class of exceptionally broad peaks spanning up to 21 kb, including those at essential beta cell transcription factor genes (Fig. 7a, ESM Table 10), consistent with the observation that genes marked by broad peaks are important for maintaining cell identity [27]. For example, Fig. 7b shows the H3K4me3 profiles of a housekeeping gene with a typical H3K4me3 peak (Rplp0), and an expression-matched beta cell transcription factor gene with a broad peak (Nkx6-1). Gene expression was moderately correlated with promoter H3K4me3 peak breadth; however, genes with exceptionally broad peaks did not have exceptionally high levels of RNA expression (ESM Fig. 7a).
Fig. 7.

H3K4me3 peak breadth encodes gene expression changes in a mouse model of type 2 diabetes. (a) H3K4me3 peaks in Dpy30-WT beta cells ranked from narrow to broad. Peaks associated with beta cell transcription factor genes are labelled. (b) Genome browser views showing H3K4me3 at a housekeeping gene (Rplp0) and an expression-matched beta cell transcription factor gene (Nkx6-1). (c) Enrichment p values of genes down- or upregulated in islets from Leprdb/db mice, with genes ranked by H3K4me3 peak breadth (as in [a]) and grouped into 20 quantiles. P values were calculated using one-sided Fisher’s exact tests. (d, e) The same data as in (a, b) for human beta cells. (f) Enrichment p values of genes down- or upregulated in islets from donors with type 2 diabetes, with genes ranked by H3K4me3 peak breadth (as in [d]) and grouped into 20 quantiles. P values were calculated using one-sided Fisher’s exact tests. (g) Immunoblots of H3K4me3 and H3 in islet lysates from Lepr+/+ and Leprdb/db mice. H3-normalised H3K4me3 band densities are listed. (h) Average enrichment profiles of H3K4me3 at the TSS of all expressed genes in Lepr+/+ and Leprdb/db islets. (i) Genome browser views of H3K4me3 in Lepr+/+ and Leprdb/db islets at notable genes that are downregulated (Pdx1, Slc30a8) or induced (Aldh1a3, Ldha) in Leprdb/db islets. (j) Enrichment p values of H3K4me3 peaks showing significant change in peak breadth in Leprdb/db vs Lepr+/+ islets (y axis) in the different gene groups ranked from narrow to broad (x axis). Enrichment p values were calculated using one-sided Fisher’s exact tests. (k) Box and whisker plot showing the log2(fold change) in H3K4me3 peak breadth for genes that are downregulated, stably expressed and upregulated in Leprdb/db islets. P values were calculated using Wilcoxon rank-sum tests with Benjamini–Hochberg correction. ***p<0.001. (l) Stratified rank/rank hypergeometric overlap plot comparing gene expression changes caused by Dpy30-KO (x axis) and by Leprdb/db (y axis) mutations. Colourscale shows the hypergeometric enrichment p value. (m) Same as (c) for genes down- or upregulated in Dpy30-KO cells
To explore the relationship between H3K4me3 peak breadth and diabetes, we used the Leprdb/db mouse model of type 2 diabetes. We retrieved published bulk RNA-seq data from Leprdb/db islets [53] and compared H3K4me3 peak breadth with gene expression changes. Notably, genes that become downregulated in Leprdb/db islets were explicitly enriched in the group of genes with broad H3K4me3 peaks (Fig. 7c). Furthermore, housekeeping genes (GSEA:M11197 [72]), genes involved in endocrine pancreas development (GSEA:M12875) and MODY genes (GSEA:M18312) were enriched in the broadest H3K4me3 peaks (ESM Fig. 7b). In contrast, genes upregulated in Leprdb/db islets were uniquely enriched in the narrowest H3K4me3 peaks (Fig. 7c). As H3K4me3 peak breadth correlates with gene expression (ESM Fig. 7a), we tested whether these observations could be secondary to a relationship wherein highly expressed genes become downregulated and weakly expressed genes become upregulated in Leprdb/db. Ranking genes by RNA expression level instead of H3K4me3 peak breadth proved less effective at stratifying each gene set except for housekeeping genes (ESM Fig. 7b), supporting a primary link with H3K4me3. Therefore, genes that are differentially expressed in diabetic islets are diametrically stratified according to H3K4me3 breadth, with upregulated genes having narrow peaks and downregulated genes having broad peaks in non-diabetic conditions. These observations are replicable in humans using publicly available H3K4me3 ChIP-seq data from beta cells from donors without diabetes [52] and RNA-seq data from islets from donors with or without type 2 diabetes [51] (Fig. 7d–f, ESM Fig. 7c, d, ESM Table 11).
H3K4me3 peak breadth dynamics encode gene expression changes in a mouse model of type 2 diabetes
The positive association between H3K4me3 peak breadth and differential gene expression in diabetes prompted us to examine if H3K4me3 is altered in diabetic islets. Several COMPASS subunit genes were downregulated in Leprdb/db islets (ESM Fig. 8a), although none reached statistical significance in islets from human donors with diabetes (ESM Fig. 8b). Immunoblots and ChIP-seq showed that average H3K4me3 enrichment was not altered in Leprdb/db islets (Fig. 7g,h). However, we observed contraction of H3K4me3 at downregulated genes, for example the transcription factor Pdx1 and zinc transporter Slc30a8, and expansion of H3K4me3 at upregulated genes, for example the disallowed Aldh1a3 and Ldha (Fig. 7i). RNA expression and H3K4me3 peak breadth decreased for mature beta cell transcription factor genes and increased for disallowed genes (ESM Fig. 9a, b). More generally, broad H3K4me3 peaks tended to shrink and narrow H3K4me3 peaks tended to expand in Leprdb/db islets (Fig. 7j). Similarly, genes that were up- or downregulated in Leprdb/db islets showed corresponding expansion or contraction of HK4me3 levels, respectively (Fig. 7k). Therefore, a pattern emerges in Leprdb/db islets wherein weakly active genes tend to gain H3K4me3 and become upregulated, while genes with broad H3K4me3 peaks tend to lose some H3K4me3 and become downregulated.
We examined whether genes that are dysregulated in Leprdb/db islets are regulated by H3K4 methylation, that is, are downregulated in Dpy30-KO cells 45 days after tamoxifen administration. To test the overlap between differential gene expression in the Dpy30-KO and Leprdb/db models, we used a threshold-free rank–rank hypergeometric overlap test [73]. There was significant overlap between genes downregulated in Dpy30-KO mice with genes that were either up- or downregulated in Leprdb/db mice (Fig. 7l). Accordingly, genes downregulated in Dpy30-KO mice were enriched in both the narrow and broad H3K4me3 peak groups (Fig. 7m), thus mirroring the pattern for genes up- or downregulated in Leprdb/db mice (Fig. 7c). Gene Ontology terms for insulin secretion, response to glucose and regulation of transcription were over-represented in genes downregulated in both models (ESM Fig. 9c, ESM Tables 12 and 13). Terms over-represented in genes downregulated in Dpy30-KO but upregulated in Leprdb/db included regulation of cell growth, transport and response to unfolded protein and endoplasmic reticulum stress (ESM Fig. 9d, ESM Table 14, 15), which could indicate that Dpy30-KO mice are not subject to these stresses and/or that methylation of H3K4 is required for induction of stress-responsive genes, as previously suggested in Drosophila [20]. Combined, these results suggest that genes that are dysregulated in Leprdb/db islets show concordant changes in H3K4me3 peak breadth and significantly overlap with genes that are downregulated in Dpy30-KO cells, which perform contextually important biological functions in mature beta cells.
Discussion
In this study we describe a gene regulatory system in mature beta cells with consequences for cell function in health and type 2 diabetes. We find that sustained methylation of H3K4 is essential for the preservation of insulin content and stimulated secretion. Reduction of H3K4 methylation led to a less active and more repressed epigenome. Gene expression became more stochastic, a feature shared with beta cells during type 2 diabetes. We furthermore describe epigenetic features associated with genes that are sensitive to H3K4 demethylation in beta cells in vivo, providing support for a generalisable, but limited, function for H3K4me3 in the regulation of transcription in a terminally differentiated tissue in vivo (Fig. 8).
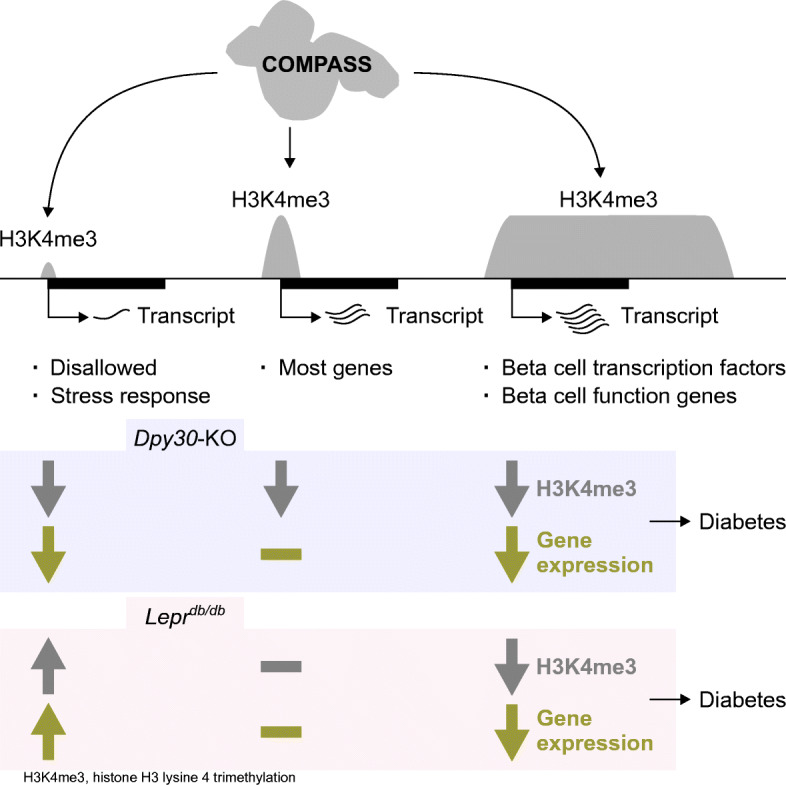
Fig. 8.

Summary of H3K4me3 in mature mouse beta cells. (a) Active gene promoters are enriched for H3K4me3, H3K4me1 and H3K27ac and are depleted of repressive H3K27me3 and DNAme. Reduction of H3K4me3 and H3K4me1 in Dpy30-KO cells was associated with a generalised reduction of H3K27ac and specific gain of H3K27me3 in downregulated gene promoters. (b) A continuum of H3K4me3 peak breadth distinguishes disallowed and lowly expressed genes from critical mature beta cell lineage factors. Global reduction of H3K4me3 in Dpy30-KO cells impaired expression of genes with very narrow or very broad peaks. In Leprdb/db mice, accumulation of H3K4me3 at narrow peaks and loss of H3K4me3 from broad peaks was associated with concordant changes in gene expression. Genes with an intermediate H3K4me3 peak profile were not generally dysregulated in either model
Conditional mutagenesis of Dpy30 revealed a slow turnover of H3K4 methylation in mature beta cells. We observed slower loss of H3K4me1 than H3K4me3, consistent with previously reported relative turnover rates [74], and that loss of methylation was probably driven by enzymatic demethylation and nucleosome eviction rather than dilution due to mitosis or overall H3 degradation [74, 75]. Following an initial delay of between 15 and 30 days after tamoxifen administration, H3K4 methylation was progressively lost from beta cells, leading to impaired glucose tolerance by 45 days, and hyperglycaemia shortly thereafter. The initial delay may result from the action of pre-existing DPY30, while the metabolic dysfunction appearing by 45 days apparently represents a threshold beyond which methylation was too low to sustain normal beta cell functions. Then, unabated decline of H3K4 methylation and/or a feed-forward of beta cell dysfunction led to worsening hyperglycaemia and euthanasia by 60 days. Although reduction of methylation was obvious by 30 days after tamoxifen administration, no defect in glucose tolerance was observed at this time point, indicating that beta cells can maintain normal function when faced with a moderate reduction in methylation levels. This suggests that therapeutic strategies that cause limited demethylation of H3K4 could be tolerated, although long-term effects were not examined here. Further, this may explain why human pathologies caused by heterozygous mutations in COMPASS methyltransferases, such as Wiedemann–Steiner syndrome (caused by mutations in KMT2A) and subgroups of generalised dystonia (KMT2B) and Kabuki syndrome (KMT2D), do not present with glucose intolerance and hyperglycaemia; compensation by other methyltransferases means that it is unlikely that H3K4me3 is reduced to an extent that impairs beta cell function.
DPY30 was depleted in Dpy30-KO cells by 15 days after tamoxifen administration but no phenotype was apparent until H3K4me3 was also depleted, several weeks later. This suggests that DPY30 has little direct impact on gene expression or insulin secretion by beta cells. Still, we cannot exclude the possibility that DPY30 stimulates methylation of stable non-histone substrates, or contributes to non-transcriptional functions, with a subtle or delayed phenotype. For instance, SETD1B-COMPASS has a non-enzymatic role in cytosolic lipid metabolism [36]. Lipid metabolism potentiates glucose-stimulated insulin secretion [76] so, if DPY30 contributes to this role, its ablation conceivably contributed to the impaired glucose-stimulated insulin secretion we observed.
In Dpy30-KO beta cells, changes in RNA expression were linked to local changes in histone methylation, leading to dysregulation of ~5% of genes. This mirrors findings in yeast [25], Drosophila embryos [19] and mouse embryonic stem cells [18] that H3K4 methylation is not required to maintain expression of most genes. H3K4 methylation may, therefore, be important for allocation of transcriptional machinery to particular genes, rather than their absolute activity. In other words, promoter-associated H3K4me3 levels modulate expression of target genes but global H3K4me3 levels should not be assumed to reflect a cell’s transcriptional output. We note, however, that total mature mRNA content, which we measured here, is not a direct measure of transcription, and measurement of nascent RNA may have uncovered more widespread roles in transcription regulation. In either case, our findings raise the important question of why reduction of H3K4me3 was associated with downregulation of a specific group of H3K4me3-marked genes. Part of the answer probably arises from cooperation with specific transcription factors. For example, there is evidence that MAF bZIP transcription factor A (MAFA) and paired box 6 (PAX6) activate target genes by recruiting COMPASS methyltransferases [77, 78] and the activity of both was decreased in Dpy30-KO cells. Additional specificity probably arises from compensation and competition from other epigenetic regulators. H3K27ac, an unambiguous marker of active chromatin, was globally reduced in Dpy30-KO cells and was positively linked to gene up- and downregulation. H3K27me3, a modification that antagonistically drives gene repression, accumulated at the promoters of downregulated genes. Thus, our results are consistent with a cooperative role for H3K4me3 within a larger ‘histone code’ [79]. We focused our analyses on promoters but it should be noted that H3K4 methylation also influences gene expression from distal enhancers [15]. We suspect that consideration of enhancers would further refine the link between gene regulation and H3K4 methylation in beta cells. More broadly, we believe that, provided one has sufficient knowledge of the epigenetic profile of a given gene, the degree to which its expression depends on H3K4 methylation could be accurately predicted.
Global reduction of H3K4 methylation was associated with upregulation of almost 200 genes. This observation is at odds with correlative and mechanistic evidence that H3K4 methylation is an ‘activating’ mark [13, 14, 26] but it is a common observation when COMPASS genes are inactivated (e.g. [20, 24, 25, 43]). Although the mechanism by which this occurs is unknown, it may point to roles for COMPASS/H3K4 methylation in gene repression. In this regard, it is notable that epigenetic features were similar between upregulated and downregulated genes (Fig. 4). It is tempting to speculate that, in the same way that transcription factors can activate or repress target genes from identical DNA motifs, COMPASS/H3K4 methylation can activate or repress genes with common epigenetic features based on the influence of other regulatory inputs. Alternatively, reduction of H3K4 methylation is merely permissive of gene upregulation caused by other factors, such as (1) downregulation of negative transcriptional regulators, (2) metabolic stimuli and (3) reallocation of transcriptional machinery from downregulated genes. Whether H3K4me3 directly suppresses expression in some circumstances will need to be tested by carrying out acute targeted methylation and demethylation at candidate genes.
Peak breadth has emerged as an informative dimension of H3K4me3 enrichment: H3K4me3 peak breadth predicts gene expression levels [31]; breadth dynamics predict gene expression dynamics [80]; and exceptionally broad H3K4me3 peaks are predictive of genes that are critical to the maintenance of a cell’s lineage [27]. From our analyses of peak breadth, we confirmed that markers of beta cells, including Ins1/2 and beta cell transcription factors, possess broad H3K4me3 peaks in mature beta cells. Dpy30-KO led to a genome-wide reduction of H3K4me3 but transcriptional downregulation was largely restricted to genes that had either very narrow or very broad H3K4me3 peaks, indicating that these two classes of promoters rely on H3K4me3 for the maintenance of gene expression. Compared with the effect at narrow peaks, the transcriptional defects for genes with broad H3K4me3 enrichment was modest. For example, Ins1 and Ins2 were downregulated by 15–25% in Dpy30-KO cells despite complete loss of their broad H3K4me3 peaks. As broad peaks colocalise with many other activating chromatin marks [27], the modest effect is likely to result from compensation from other regulatory inputs. In aggregate, our results show that broad H3K4me3 peaks increase expression levels and transcriptional consistency of associated genes but are not required for transcription.
A surprising finding is that weakly active promoters, with little H3K4me3, show the greatest reliance on H3K4 methylation. One explanation is that these promoters are susceptible to silencing as H3K4 methylation opposes repressive H3K27 and DNA methyltransferase activity [16, 17]. In support of this, genes that were downregulated in Dpy30-KO cells showed high initial enrichment of H3K27me3 and further accumulation after reduction of H3K4me3. Weakly active genes are also the most likely to be upregulated after inactivation of the Polycomb system [81] and, in beta cells, inactivation of the Polycomb system leads to activation of previously silenced genes [4]. Therefore, weakly active genes may be the most responsive targets in ongoing competition between the Polycomb and trithorax systems in mature beta cells.
Weakly active genes become more active in Leprdb/db islets, suggesting that the trithorax system is dominant during the development of diabetes. Meanwhile, promoters with broad H3K4me3 peaks showed a unique tendency to shrink and for the associated gene to be downregulated. More work is required to determine what drives H3K4me3 peak dynamics in Leprdb/db islets and to what extent these mechanisms are active in human diabetes. In any case, we favour a model wherein redistribution of H3K4me3 away from promoters of genes with broad H3K4me3 peaks, which enforce beta cell identity or functions, in favour of relatively inactive genes, including stress response and disallowed genes, contributes to transcriptome remodelling and type 2 diabetes.
Supplementary information
(PDF 4873 kb)
(XLSX 8344 kb)
Acknowledgements
The authors thank the staff of the Animal Care Facility at British Columbia Children’s Hospital Research Institute for daily maintenance of the mouse colonies, and W. Gibson (University of British Columbia) for providing Ins1Cre animals. We thank M. Reid at the Electron Microscopy Facility at McMaster University for help with transmission electron microscopy, and L. Xu at the Flow Cytometry Core at British Columbia Children’s Hospital Research Institute for help with FACS.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
BV and BGH conceived of and designed the experiments. BV, NM, DJP, MA, CLM, PS and BGH acquired and analysed the data. BV, NM, DJP, DSL, FCL and BGH interpreted the data. BV drafted the manuscript. All authors read and critically edited the manuscript and approved the final version of the manuscript to be published. BGH is the guarantor of this work.
Abbreviations
- ASH2L
Absent, small or homeotic 2-like protein
- ChIP-seq
Chromatin immunoprecipitation–sequencing
- COMPASS
Complex of proteins associated with Set1
- DNAme
DNA cytosine methylation
- DPY30
Dumpy-30
- Dpy30-KO
Beta cell-specific Dpy30 knockout
- Dpy30-WT
Dpy30 wild-type
- H3K27ac
Histone H3 lysine 27 acetylation
- H3K27me3
Histone H3 lysine 27 trimethylation
- H3K4me1
Histone H3 lysine 4 monomethylation
- H3K4me3
Histone H3 lysine 4 trimethylation
- IHC
Immunohistochemistry
- IP
Immunoprecipitation
- OCR
Oxygen consumption rate
- RBBP5
Retinoblastoma-binding protein 5
- RNA-seq
RNA sequencing
- SCENIC
Single-cell regulatory network inference and clustering
- scRNA-seq
Single-cell RNA-seq
- TSS
Transcription start site
- UMAP
Uniform manifold approximation and projection
- WDR5
WD repeat-containing protein 5
Funding
This work was supported by operating grants from the British Columbia Children’s Hospital Research Institute, the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-04292) and the Canadian Institutes of Health Research (RN310864 – 375894). Fellowship support for BV was provided by the British Columbia Children’s Hospital Research Institute, University of British Columbia and Natural Sciences and Engineering Research Council of Canada.
Data availability
RNA-seq, scRNA-seq and ChIP-seq data generated from Pdx1CreER Dpy30-KO islet cells and ChIP-seq data generated in Leprdb/db islet cells have been deposited in the Gene Expression Omnibus under accession number GSE181951. Requests for further information should be directed to the corresponding author, Brad G. Hoffman (brad.hoffman@ubc.ca).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ebrahimi AG, Hollister-Lock J, Sullivan BA, Tsuchida R, Bonner-Weir S, Weir GC (2020) Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Mol Metab 35:100959. 10.1016/j.molmet.2020.02.002 [DOI] [PMC free article] [PubMed]
- 2.Salinno C, Cota P, Bastidas-Ponce A, Tarquis-Medina M, Lickert H, Bakhti M. β-cell maturation and identity in health and disease. Int J Mol Sci. 2019;20(21):1–20. doi: 10.3390/ijms20215417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim H, Yoon BH, Oh CM, et al. PRMT1 is required for the maintenance of mature β-cell identity. Diabetes. 2020;69(3):355–368. doi: 10.2337/db19-0685. [DOI] [PubMed] [Google Scholar]
- 4.Lu TTH, Heyne S, Dror E, et al. The Polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018;27(6):1294–1308. doi: 10.1016/j.cmet.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20(4):419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dayeh T, Volkov P, Salö S, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10(3):e1004160. doi: 10.1371/journal.pgen.1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG. Methyltransferase set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes. 2009;58(1):185–193. doi: 10.2337/db08-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo T, Kraakman MJ, Damle M, Gill R, Lazar MA, Accili D. Identification of C2CD4A as a human diabetes susceptibility gene with a role in β cell insulin secretion. Proc Natl Acad Sci U S A. 2019;116(40):20033–20042. doi: 10.1073/pnas.1904311116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miguel-Escalada I, Bonàs-guarch S, Cebola I, et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet. 2019;51(7):1137–1148. doi: 10.1038/s41588-019-0457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P, Karakose E, Liu H, et al. Combined Inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metab. 2018;29(3):638–652. doi: 10.1016/j.cmet.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25(1):15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 12.Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 13.Vermeulen M, Mulder KW, Denissov S, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131(1):58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Wysocka J, Swigut T, Xiao H, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442(7098):86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 15.Yan J, Chen SAA, Local A, et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018;28(2):204–220. doi: 10.1038/cr.2018.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitges FW, Prusty AB, Faty M, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42(3):330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 17.Ooi SKT, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorighi KM, Swigut T, Henriques T, et al. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell. 2017;66(4):568–576. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hödl M, Basler K. Transcription in the absence of histone H3.2 and H3K4 methylation. Curr Biol. 2012;22(23):2253–2257. doi: 10.1016/j.cub.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Rickels R, Herz HM, Sze CC, et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet. 2017;49(11):1647–1653. doi: 10.1038/ng.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glaser S, Lubitz S, Loveland KL, et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin. 2009;2(1):1–16. doi: 10.1186/1756-8935-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bledau AS, Schmidt K, Neumann K, et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development. 2014;141(5):1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- 23.Campbell SA, McDonald CL, Krentz NAJ, Lynn FC, Hoffman BG. trxg complex catalytic and non-catalytic activity play distinct roles in pancreas progenitor specification and differentiation. Cell Rep. 2019;28(7):1830–1844. doi: 10.1016/j.celrep.2019.07.035. [DOI] [PubMed] [Google Scholar]
- 24.Andreu-Vieyra CV, Chen R, Agno JE, et al. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8(8):53–54. doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiner A, Chen HV, Liu CL, et al. Systematic dissection of roles for chromatin regulators in a yeast stress response. PLoS Biol. 2012;10(7):17. doi: 10.1371/journal.pbio.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cano-Rodriguez D, Gjaltema RAF, Jilderda LJ, et al. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat Commun. 2016;7:12284. doi: 10.1038/ncomms12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benayoun BA, Pollina EA, Ucar D, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158(3):673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hörmanseder E, Simeone A, Allen GE, et al. H3K4 methylation-dependent memory of somatic cell identity inhibits reprogramming and development of nuclear transfer embryos. Cell Stem Cell. 2017;21(1):135–143. doi: 10.1016/j.stem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar D, Cinghu S, Oldfield AJ, Yang P, Jothi R. Decoding the function of bivalent chromatin in development and cancer. Genome Res. 2021;31(12):2170–2184. doi: 10.1101/gr.275736.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faucher D, Wellinger RJ. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010;6(8):e1001082. doi: 10.1371/journal.pgen.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlić R, Chung HR, Lasserre J, Vlahoviček K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107(7):2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bochyńska A, Lüscher-Firzlaff J, Lüscher B. Modes of interaction of KMT2 histone H3 lysine 4 methyltransferase/COMPASS complexes with chromatin. Cells. 2018;7(3):17. doi: 10.3390/cells7030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugeedha J, Gautam J, Tyagi S. SET1/MLL family of proteins: functions beyond histone methylation. Epigenetics. 2021;16(5):469–487. doi: 10.1080/15592294.2020.1809873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao K, Collings CK, Morgan MA, et al. An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci Adv. 2018;4(1):1–14. doi: 10.1126/sciadv.aap8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sze CC, Cao K, Collings CK, et al. Histone H3K4 methylation-dependent and -independent functions of set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. 2017;31(17):1732–1737. doi: 10.1101/gad.303768.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Collings CK, Zhao Z, et al. A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolism. Genes Dev. 2017;31(20):2056–2066. doi: 10.1101/gad.306092.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ali A, Tyagi S. Diverse roles of WDR5-RbBP5-ASH2L-DPY30 (WRAD) complex in the functions of the SET1 histone methyltransferase family. J Biosci. 2017;42(1):155–159. doi: 10.1007/s12038-017-9666-9. [DOI] [PubMed] [Google Scholar]
- 38.Xue H, Yao T, Cao M, et al. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature. 2019;573:445–449. doi: 10.1038/s41586-019-1528-1. [DOI] [PubMed] [Google Scholar]
- 39.Lee YT, Ayoub A, Park SH, et al. Mechanism for DPY30 and ASH2L intrinsically disordered regions to modulate the MLL/SET1 activity on chromatin. Nat Commun. 2021;12(1):1–13. doi: 10.1038/s41467-021-23268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang T, Birsoy K, Hughes NW et al (2015) Identification and characterization of essential genes in the human genome. Science 350(6264):1096–1101. 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed]
- 41.Fagerberg L, Hallstrom BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tennant BR, Hurley P, Dhillon J, Gill A, Whiting C, Hoffman BG. The TrxG complex mediates cytokine induced de novo enhancer formation in islets. PLoS One. 2015;10(10):e0141470. doi: 10.1371/journal.pone.0141470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell SA, Bégin J, McDonald CL, Vanderkruk B, Stephan TL, Hoffman BG. H3K4 trimethylation is required for postnatal pancreatic endocrine cell functional maturation. Diabetes. 2021;70(11):2568–2579. doi: 10.2337/db20-1214. [DOI] [PubMed] [Google Scholar]
- 44.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 45.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 46.Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkilä E, Ferrer J. Ins1 Cre knock-in mice for beta cell-specific gene recombination. Diabetologia. 2015;58(3):558–565. doi: 10.1007/s00125-014-3468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saunders DC, Brissova M, Phillips N, et al. Ectonucleoside triphosphate diphosphohydrolase-3 antibody targets adult human pancreatic β cells for in vitro and in vivo analysis. Cell Metab. 2018;29(3):745–754. doi: 10.1016/J.CMET.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, Lorincz MC. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat Commun. 2015;6:1–8. doi: 10.1038/ncomms7033. [DOI] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luciani DS, White SA, Widenmaier SB, et al. Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic β-cells. Diabetes. 2013;62(1):170–182. doi: 10.2337/db11-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fadista J, Vikman P, Laakso EO, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A. 2014;111(38):13924–13929. doi: 10.1073/pnas.1402665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bramswig NC, Grompe M, Kaestner KH et al (2013) Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J Clin Invest 123(3):1275–1284. 10.1172/JCI66514DS1 [DOI] [PMC free article] [PubMed]
- 53.Neelankal John A, Ram R, Jiang F-X. RNA-Seq analysis of islets to characterise the dedifferentiation in type 2 diabetes model mice db/db. Endocr Pathol. 2018;29:207–221. doi: 10.1007/s12022-018-9523-x. [DOI] [PubMed] [Google Scholar]
- 54.Camunas-Soler J, Dai XQ, Hang Y, et al. Patch-Seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab. 2020;31(5):1017–1031. doi: 10.1016/j.cmet.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avrahami D, Li C, Zhang J, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015;22(4):619–632. doi: 10.1016/j.cmet.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hinrichs AS, Karolchik D, Baertsch R, et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34(Database issue):590–598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Mckenna LB, Bogue CW, et al. The diabetes gene Hhex maintains δ -cell differentiation and islet function. Genes Dev. 2014;28:829–834. doi: 10.1101/gad.235499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sabatini PV, Speckmann T, Nian C, et al. Neuronal PAS domain protein 4 suppression of oxygen sensing optimizes metabolism during excitation of neuroendocrine cells. Cell Rep. 2018;22(1):163–174. doi: 10.1016/j.celrep.2017.12.033. [DOI] [PubMed] [Google Scholar]
- 59.Millership SJ, Da Silva Xavier G, Choudhury AI et al (2018) Neuronatin regulates pancreatic β cell insulin content and secretion. J Clin Invest 128(8):3369–3381. 10.1172/JCI120115 [DOI] [PMC free article] [PubMed]
- 60.Wijesekara N, Dai FF, Hardy AB, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. 2010;53(8):1656–1668. doi: 10.1007/s00125-010-1733-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schuettengruber B, Bourbon H-M, Di Croce L, Cavalli G. Genome regulation by Polycomb and trithorax: 70 years and counting. Cell. 2017;171(1):34–57. doi: 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30(3):261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin BJE, Brind’Amour J, Kuzmin A, et al. Transcription shapes genome-wide histone acetylation patterns. Nat Commun. 2021;12(210):1–9. doi: 10.1038/s41467-020-20543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kannan S, Farid M, Lin BL, Miyamoto M, Kwon C. Transcriptomic entropy benchmarks stem cell-derived cardiomyocyte maturation against endogenous tissue at single cell level. PLoS Comput Biol. 2021;17(9):1–21. doi: 10.1371/journal.pcbi.1009305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Street K, Risso D, Fletcher RB, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19(477):1–16. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aibar S, González-Blas CB, Moerman T, et al. SCENIC: Single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van de Sande B, Flerin C, Davie K, et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc. 2020;15(7):2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 69.Khan HA, Margulies CE (2019) The role of mammalian Creb3-like transcription factors in response to nutrients. Front Genet 10:1–9. 10.3389/fgene.2019.00591 [DOI] [PMC free article] [PubMed]
- 70.Zuin J, Dixon JR, Van Der Reijden MIJA, et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111(3):996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31(6):1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsiao LL, Dangond F, Yoshida T, et al. A compendium of gene expression in normal human tissues. Physiol Genomics. 2002;2002(7):97–104. doi: 10.1152/physiolgenomics.00040.2001. [DOI] [PubMed] [Google Scholar]
- 73.Cahill KM, Huo Z, Tseng GC, Logan RW, Seney ML. Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci Rep. 2018;8(1):1–11. doi: 10.1038/s41598-018-27903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng Y, Tipton JD, Thomas PM, Kelleher NL, Sweet SMM. Site-specific human histone H3 methylation stability: Fast K4me3 turnover. Proteomics. 2014;14(19):2190–2199. doi: 10.1002/pmic.201400060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chory EJ, Calarco JP, Hathaway NA, et al. Nucleosome turnover regulates histone methylation patterns over the genome. Mol Cell. 2019;73(1):61–72.e3. doi: 10.1016/j.molcel.2018.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Imai Y, Cousins RS, Liu S, Phelps BM, Promes JA. Connecting pancreatic islet lipid metabolism with insulin secretion and the development of type 2 diabetes. Ann N Y Acad Sci. 2020;1461(1):53–72. doi: 10.1111/nyas.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scoville DW, Cyphert HA, Liao L, et al. MLL3 and MLL4 methyltransferases bind to the MafA and MAFB transcription factors to regulate islet β-cell function. Diabetes. 2015;64(11):3772–3783. doi: 10.2337/db15-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun J, Zhao Y, McGreal R et al (2016) Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenetics & Chromatin 9(1):37. 10.1186/s13072-016-0087-z [DOI] [PMC free article] [PubMed]
- 79.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 80.Dai Z, Mentch SJ, Gao X, Nichenametla SN, Locasale JW. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun. 2018;9(1955):1–12. doi: 10.1038/s41467-018-04426-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dobrinić P, Szczurek AT, Klose RJ. PRC1 drives Polycomb-mediated gene repression by controlling transcription initiation and burst frequency. Nat Struct Mol Biol. 2021;28(10):811–824. doi: 10.1038/s41594-021-00661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 83.Brideau CM, Eilertson KE, Hagarman JA, Bustamante CD, Soloway PD. Successful computational prediction of novel imprinted genes from epigenomic features. Mol Cell Biol. 2010;30(13):3357–3370. doi: 10.1128/mcb.01355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 4873 kb)
(XLSX 8344 kb)
Data Availability Statement
RNA-seq, scRNA-seq and ChIP-seq data generated from Pdx1CreER Dpy30-KO islet cells and ChIP-seq data generated in Leprdb/db islet cells have been deposited in the Gene Expression Omnibus under accession number GSE181951. Requests for further information should be directed to the corresponding author, Brad G. Hoffman (brad.hoffman@ubc.ca).