

SUMMARY
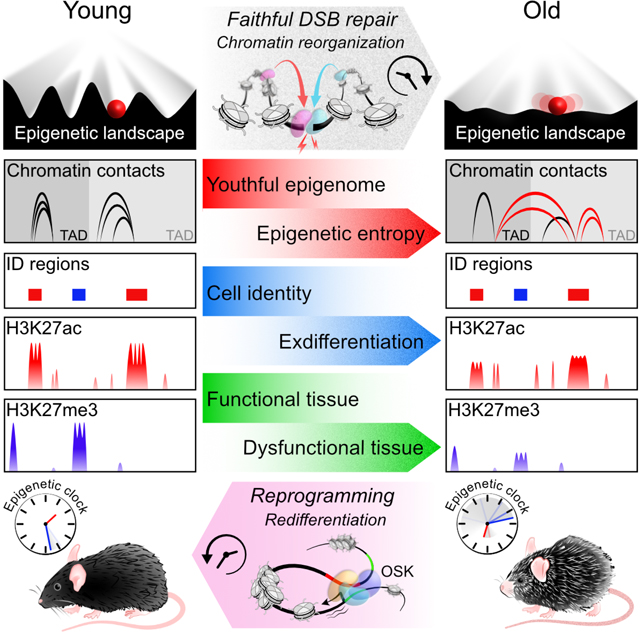
All living things experience an increase in entropy, manifested as a loss of genetic and epigenetic information. In yeast, epigenetic information is lost over time due to the relocalization of chromatin modifying proteins to DNA breaks, causing cells to lose their identity, a hallmark of yeast aging. Using a system called “ICE” (Inducible Changes to the Epigenome), we find the act of faithful DNA repair advances aging at physiological, cognitive, and molecular levels, including erosion of the epigenetic landscape, cellular exdifferentiation, senescence, and advancement of the DNA methylation clock, which can be reversed by OSK-mediated rejuvenation. These data are consistent with the Information Theory of Aging, which states that a loss of epigenetic information is a reversible cause of aging.
Graphical Abstract
INTRODUCTION
Life is based on the complex interplay between the cellular machinery and information stored in the genome and epigenome, which may be thought of as biological hardware and software. Whether aging is caused by a breakdown in the hardware, the software, or both is not yet known. In the 1950s, Szilard and Medawar independently proposed that aging is caused by a loss of genetic information due to DNA damage (Medawar, 1952; Szilard, 1959). The type of DNA damage that is most consistently linked to aging is the double-stranded DNA break (DSB), occurring at a rate of 10 to 50 per cell per day (Tian et al., 2019; Vilenchik and Knudson, 2003).
Recently, however, questions about the primacy of mutations as a driver of aging have been raised. Many types of old cells have a paucity of mutations (De Majo et al., 2021; Kaya et al., 2015), strains of mice or people with higher mutation rates show little to no evidence of premature aging (Narayanan et al., 1997; Robinson et al., 2021), and mammals can be cloned from old somatic cells to produce new individuals with normal lifespans (Burgstaller and Brem, 2017).
During development, cell identity is specified by transcriptional networks and chromatin structures that direct cells into metaphorical valleys in the Waddington landscape (Waddington, 1957). To maintain optimal function, cells must retain their identity by preserving epigenetic information and a state of low Shannon entropy (Hannum et al., 2013; Kane and Sinclair, 2019; Keller, 2009).
A loss of epigenetic information, rather than genetic, as a potential cause of aging emerged from yeast studies in the 1990’s (Kennedy et al., 1997; Sinclair et al., 1997). We and others showed that relocalization of the silent information regulator complex (Sir2/3/4) away from silent mating-type loci to the unstable rDNA is the cause of sterility, a hallmark of yeast aging (Smeal et al., 1996), coincident with alterations in histone occupancy, histone modifications (e.g. H3K56ac and H4K16ac), and gene transcription (Sen et al., 2016). Overexpression of SIR2, histones, or deletion of the histone methyltransferase gene SET2, extends yeast lifespan, indicating that epigenetic changes are not merely a biomarker, but a cause of yeast aging (Dang et al., 2009; Feser et al., 2010; Hu et al., 2014; Kaeberlein et al., 1999; Ryu et al., 2014).
Epigenetic changes linked to aging, including changes in DNA methylation (DNAme) patterns, H3K4me3 and H3K9me3 and H3K27me3 (Benayoun et al., 2015; Pal and Tyler, 2016; Sen et al., 2016) are also seen in multicellular organisms. Examples include lifespan extension in worms deficient in the H3K4 trimethylation complex (Greer et al., 2010; Greer et al., 2011) or in flies overexpressing the Sir2 gene (Jiang et al., 2013; Rogina and Helfand, 2004; Wood et al., 2016), and the relatively stable epigenome of long-lived naked mole rats (Tan et al., 2017). Many epigenetic changes follow a specific pattern, including methylation of specific CpGs (Hannum et al., 2013; Horvath, 2013; Lu et al., 2021; Petkovich et al., 2017; Weidner et al., 2014).
Why the mammalian epigenome changes over time is not yet known. Again, clues have come from yeast. A major driver in yeast is the DSB (Park et al., 1999), the repair of which requires epigenetic regulators Sir2, Hst1, Rpd3, Gcn5, and Esa1 (Martin et al., 1999; McAinsh et al., 1999; Mills et al., 1999; Tamburini and Tyler, 2005). Our Relocalization of Chromatin Modifiers or “RCM” Hypothesis and subsequent Information Theory of Aging propose that aging in eukaryotes is due to the loss of transcriptional networks and epigenetic information over time, driven by a conserved mechanism that evolved to co-regulate responses to cellular damage, such as a DSB or a crush injury (Mills et al., 1999; Oberdoerffer et al., 2008; Oberdoerffer and Sinclair, 2007; Sinclair and LaPlante, 2019).
To test whether epigenetic changes are a cause of mammalian aging, we developed systems to degrade and reset epigenetic information in cells and mice. Our data are consistent with aging in mammals being the equivalent of a software problem, the result of corrupted epigenetic information that can be restored from an existing back-up copy.
RESULTS
A system to induce epigenetic aging
To create DSBs in cells and mice without causing mutations, we employed I-PpoI, an endonuclease from Physarum polycephalum (Berkovich et al., 2007; Dobbin et al., 2013; McCord et al., 2009; Mosammaparast et al., 2013). I-PpoI recognizes the DNA sequence CTCTCTTAA▼GGTAGC (Monnat et al., 1999), which occurs at 20 targets in the mouse genome, 19 of which are non-coding, including the rDNA, and none of which occur in mitochondrial DNA (Berkovich et al., 2007).
The system consists of a fusion of the I-PpoI gene to the C-terminus of a tamoxifen (TAM)-regulated mutant estrogen receptor domain gene (ERT2), a transcriptional loxP-STOP-loxP cassette (Berkovich et al., 2007) (Figure 1A), and a TAM-regulated Cre recombinase gene (Cre-ERT2) upstream of a ubiquitin promoter for whole-body expression (Ruzankina et al., 2007). In the presence of TAM, Cre-ERT2 excises the stop cassette, facilitating transcription of the ERT2-HA-I-PpoI-IRES-GFP cassette that produces nuclear localized ERT2-I-PpoI, which is degraded upon removal of TAM (Figure 1A). C57BL6/J transgenic mice with heterozygous ERT2-I-PpoI and Cre-ERT2 are named “Inducible Changes to the Epigenome” or “ICE” mice. Wildtype (WT), I-PpoI and Cre close relatives served as negative controls (Figure S1A). We reasoned that the 4-complimentary base overhangs that I-PpoI creates would have a far lower rate of mutation than other ways of creating DSBs, such as CRISPR, chemicals, and radiation.
Figure 1. The Inducible Changes to the Epigenome (ICE) system.

(A) The ICE system with a TAM-inducible I-PpoI endonuclease.
(B and C) γH2AX foci in DAPI-stained nuclei of MEFs after TAM (4-OHT, 0.5 μM) treatment. Scale, 10 μm. Two-way ANOVA-Bonferroni.
(D) qPCR analysis of cutting at I-PpoI canonical sites. One-way ANOVA-Bonferroni.
(E) Epigenetic age of 96-hour post-treated ICE cells. All clock DNAme sites (left) and clock DNAme sites post-batch effect correction (right). Two-tailed Student’s t test.
(F and G) Senescence-associated β-galactosidase (SA-β-Gal) staining of post-treated cells. Two-tailed Student’s t test.
(H) mRNA levels of genes known to change during senescence 144 hours post-treatment. Two-tailed Student’s t test.
(I and J) Non-mutated I-PpoI canonical sequences in 96-hour post-treated cells assessed by deep sequencing (>50x).
(K) Mutation frequency of 28S rDNA in 96-hour post-treated cells. Two-tailed Student’s t test.
(L) Experimental design.
(M and N) Immunoprecipitation and quantification of a I-PpoI cut site (Tmem56) in skeletal muscle, liver and kidney during and after TAM treatment (0-,1- and 10-month post-treatment). Two-tailed Student’s t test.
Data are mean (n≥3) ± SD or ± SEM (N). n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001.
Mouse embryonic fibroblasts (MEFs) were isolated from ICE and control embryos. After the addition of TAM, HA-I-PpoI was detected in the nucleus of ICE cells, but not controls (Figure 1B), and the number of serine 139-phosphorylated H2AX (γH2AX) foci, a marker of DSBs, reached a maximum of 4-fold above background after 24-hours, with locus-dependent cutting (Figure 1C and 1D). Compared to etoposide, a topoisomerase II inhibitor, and phleomycin, a free-radical inducer, the number of γH2AX foci, the extent of DNA breakage, and the DNA damage response in ICE MEFs was minimal (Figure S1B–S1D). During and after I-PpoI induction, there was no detectable change in cell cycle profile, apoptosis, or senescence (Figure S1E–S1G). We could not detect changes in mutation frequency at the 28S rDNA (Figure S2A–S2C and 1K), RNA levels (Figure S2D and S2F), or overall translation efficiency (Figure S2K and S2L). Sensitive ligation-mediated PCR (LM-PCR) (Figure S2G) did not detect residual I-PpoI activity at the 96-hours (Figure S2H and S2I).
If the RCM hypothesis is correct, we would expect the post-treated ICE cells to be epigenetically older than untreated cells and show characteristics of cells from older mice. Using reduced representation bisulfite sequencing (RRBS), a weighted sum of 89 age-associated methylation sites, and a refined set of 74 sites (Petkovich et al., 2017), ICE cells were ~1.5-fold older than the Cre control cells (Figure 1E; p=0.0042 and p=0.004, respectively).
One of the most robust and reproducible effects of aging is an increased sensitivity to DNA damaging agents (Li et al., 2016; Mapuskar et al., 2017; Miyoshi et al., 2006). The post-treated ICE cells were significantly more susceptible than Cre controls to DNA damaging agents (Figure S1H and S1I). Another hallmark of aging is reduced Lamin B1, an event that can promote cellular senescence, as indicated by increased SA-β-Gal activity, IL-6, Ccl2, Ccl20, LINE-1 and IAP (Freund et al., 2012; Shah et al., 2013). At later times post-treatment (120–168 hours), ICE cells had lower Lamin B1 levels (Figure S1J and S1K) and increased indicators of cellular senescence (Figure 1F–1H).
Based on extensive whole-genome sequencing, there was no difference in mutation frequency at canonical, non-canonical, or ~100,000 random sites in treated Cre and ICE cells (Wittmayer et al., 1998) (Figure 1I, S1L and S1M), nor was there a difference in mutation frequency between Cre and ICE cells at each I-PpoI recognition site (Figure 1J, 1K and S1N). Thus, in MEFs, faithful DSB repair accelerates aspects of aging, including DNAme age.
The ICE system induces non-mutagenic cuts in vivo
To test our hypothesis in vivo, we performed whole-body I-PpoI expression in 4–6 month-old mice for three weeks (Figure 1L). The extent of STOP cassette removal was similar in muscle (67%), liver (71%), hippocampus (61%) and cortex (72%) (Figure S3A). HA-I-PpoI, γH2AX and eGFP were detectable during TAM treatment but not post-treatment in all tissues tested (Figure S3B–S3E).
To assess the location and extent of I-PpoI cutting, we used a biotinylated oligo with the overhang 5’-TTAA-3’ to capture I-PpoI-cut DNA (Chailleux et al., 2014) (Figure 1M). In skeletal muscle, liver and kidney, the Tmem56 intron and the 28S I-PpoI site were cut during TAM treatment and, as expected, no cutting was detectable 1- and 10-month later (Figure 1N and S3F). At the rDNA in particular, there was no evidence of residual cutting (Figure S2J), changes in copy number (Figure S2O), 28S expression levels (Figure S2P) or protein synthesis (S2M and S2N). There was also no difference in mutation frequency at canonical or non-canonical I-PpoI recognition sites (Wittmayer et al., 1998) or across the genome (Figure S3G–S3I). Thus, the ICE system induces specific DNA breaks with no evidence of mutations or immediate deleterious effects.
The ICE system phenocopies aging in vivo
If the RCM hypothesis is correct, I-PpoI induction should advance DNAme age and accelerate other characteristics of aging (Figure 1L and 2A). As mice age, they undergo characteristic physical and physiological changes, including alopecia, hair greying, kyphosis, decreased body weight, motion in dark phase, and a reduced respiratory exchange ratio (RER) (Ackert-Bicknell et al., 2015; Harkema et al., 2016; Houtkooper et al., 2011; Koks et al., 2016).
Figure 2. ICE mice phenocopy normal aging.

(A) Experimental design.
(B) Images of Cre and ICE mice.
(C-E) Weight and body mass. Two-way ANOVA-Bonferroni (C). One-way ANOVA-Bonferroni (D, left). Two-way ANOVA-Bonferroni (D right and E).
(F) Respiratory Exchange Rate (RER) of 10-month post-treated mice. Repeated measures two-way ANOVA-Bonferroni.
(G) Average motion over 24 hours.
(H) Frailty indices of Cre, ICE, WT 3- and 24-month-old mice. Two-tailed Student’s t test (left) or two-way ANOVA-Bonferroni (right).
(I and J) CT of whole skeleton and micro-CT of trabecular and cortical bones. Kyphosis assessment (I), bone/tissue volume (J, left) and trabecular separation (J, right). Two-tailed Student’s t test.
(K and L) Average damage scores (1+ normal – 4+ global scarring) of glomeruli of 10-month post-treated ICE mice. OC, outer cortex; JM, juxtamedullary glomeruli. Two- tailed Student’s t test.
(M and N) p57 (podocyte) and Periodic acid-Schiff staining, and podocyte density of 10-month post-treated ICE mice. Circles with broken line indicate glomeruli. Scale bar, 50 μm. Two-tailed Student’s t test.
(O and P) Fraction of α-SMA-positive cells in parietal epithelial cells (PEC) along Bowman’s capsule (arrows) of 10-month post-treated ICE mice showing an epithelial to mesenchymal transition (EMT). Circles with broken line indicate glomeruli. Scale bar, 50 μm. Two-tailed Student’s t test.
Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; ***p< 0.001; ****p< 0.0001.
During I-PpoI induction, there were no detectable differences between ICE mice and the negative controls in terms of behavior, activity, or food intake. After one month, however, there were subtle differences in the ICE mice, including alopecia and a loss of pigment on the feet, tail, ears and nose, resembling middle-aged WT mice (Liu et al., 2019; Nishimura et al., 2005) (Figure 2B).
At 10-months post-treatment, the ICE mice exhibited classic features of old age, including reduced body weight and fat mass, independent of food intake (Figure 2B–2E and S4A–S4E), a lower respiratory exchange ratio (RER) during the day (Figure 2F and S4F) and decreased motion in the dark phase (Figure 2G).
The mouse frailty index (FI) is a compendium of health measures including body weight, coat condition, grip strength, mobility, vision, and hearing (Whitehead et al., 2013). At 1-month post-treatment there was no significant difference in FI between ICE mice and controls, but at 10- and 12-months the ICE mice had a higher FI resembling that of 24-month-old WT mice (p=0.0006 and < 0.0001, respectively) (Figure 2H), along with other features of aging, including kyphosis, loss of cortical bone thickness and trabecular bone density in the inner layer (Ferguson et al., 2003; Katzman et al., 2010) (Figure 2I and 2J).
To determine if ICE mice mimicked the histological changes seen during aging, we assessed two tissues, kidney and skin, that undergo well characterized age-related changes, (Roeder et al., 2015; Russell-Goldman and Murphy, 2020). Compared to age-matched controls, the kidneys of 10-month post-treated ICE mice had fewer healthy glomeruli (1+) and podocytes (Figure 2K–N). The glomerular parietal epithelial cells (PECs) of ICE mice experienced a greater epithelial to mesenchymal transition (EMT), another characteristic of aged kidneys (Roeder et al., 2015) (Figure 2O and P). Key features of skin aging were apparent, including subepidermal thinning and hair greying due to a loss of KIT/CD117-positive melanocyte stem cells (Matsumura et al., 2016; Nishimura et al., 2005) (Figure S4J–S4M).
ICE mice phenocopy brain aging
Mammalian aging is associated with a decline in central nervous system function (Figure 3A) (Johnson et al., 2018; Ungvari et al., 2017). During aging, mice move less in the dark phase and have a characteristic loss of coordination. ICE mice moved ~50% less in the dark phase (Figure 3B) with an impaired gait (Figure S4N–S4P).
Figure 3. ICE mice phenocopy brain aging.

(A) Experimental design.
(B) Ambulatory activity of 10-month post-treated mice in light and dark cycles. Two-way ANOVA-Bonferroni.
(C-E) Immediate and contextual freezing in fear conditioning tests in 10-month post-treated mice. One-way ANOVA-Bonferroni (D, left and E, left) or two-tailed Student’s t test (D, right and E, right).
(F and G) Representative images of Barnes maze tests and mean number of pokes at each hole in 10-month post-treated mice. Two-way ANOVA-Bonferroni.
(H-K) Immunofluorescence of the hippocampal CA3 region stained for astrocyte activation (GFAP) and microglia (IBA1) in 10-month post-treated mice. Scale bar, 100 μm. Two-tailed Student’s t test.
Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001; **** p < 0.0001.
Hippocampal function, critical for spatial and memory consolidation, declines with age (Gallagher et al., 2010; Miller and O’Callaghan, 2005; Park and Reuter-Lorenz, 2009) and is often measured by fear-conditioning, which measures short-term memory. On the second day, ~75% of the young mice and ~40% of old mice froze, indicating reduced contextual recall, with a similar difference between Cre and ICE mice (Figure 3C–3E). In the Barnes maze test, a measure of long-term memory, the recall of ICE mice was about half that of Cre controls, similar to that of 24-month-old WT mice (Figure 3F and 3G).
Astrocytes and microglia, mediators of the innate immune response of the central nervous system, become hyper-activated with age (Baruch et al., 2014; Norden and Godbout, 2013). Similar to the hippocampi of aged mice, ICE mice had greater numbers of activated astrocytes (1.6x) and microglia (3.5x) (Figure 3H–3K).
ICE mice phenocopy muscle aging
Exercise endurance, strength, muscle mass, vascularization, and mitochondrial function decrease in skeletal muscle with age (Das et al., 2019; Demontis et al., 2013) (Figure 4A). Molecular hallmarks of muscle aging include reduced ATP, mitochondrial DNA, and alterations in subsarcolemmal and intermyofibrillar morphology (Demontis et al., 2013; Leduc-Gaudet et al., 2015). At 16 months of age, ICE mice had significantly less muscle mass (Figure 4B), reduced endurance (Figure 4C), greater lactate levels post-exercise (Figure S5A), reduced grip strength (Figure S5B) and other molecular hallmarks of muscle aging (Figure 4D–4F and S5C–S5F).
Figure 4. ICE mice phenocopy muscle aging.

(A) Timeline of phenotypic assessments of mice.
(B) Muscle mass of 10-month post-treated mice assessed by MRI. Two-tailed Student’s t test.
(C) Treadmill endurance in WT, 10-month post-treated mice. Two-tailed Student’s t test.
(D) ATP levels in 10-month post-treated muscle. Two-tailed Student’s t test (let) or two-way ANOVA-Bonferroni (right).
(E and F) Mitochondrial morphology and area of 10-month post-treated muscle. Scale bar, 500 nm. Two-tailed Student’s t test.
(G and H) 10-month post-treated gastrocnemius. Laminin (red) and CD31(green), marking extracellular matrix and capillaries, respectively. Two-tailed Student’s t test.
(I) Scatter plot of genes changed (p < 0.01) in muscle from 10-month post-treated ICE and WT 24-month-old mice with significantly changed genes (padj < 0.05) in color.
(J) Heatmaps of the top 200 most significantly altered genes in skeletal muscle.
(K) Epigenetic age of gastrocnemii 1-, 10-, and 14-month post-treatment. Linear regression analysis.
(L and M) Epigenetic age of muscle and blood 10-month post-treatment (Δ age = epigenetic age – chronological age). Two-tailed Student’s t test.
Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001; **** p < 0.0001.
At 10-months post-treatment, the ICE mice had 6-fold fewer COX-positive myofibers (Figure S5G and S5H), about half the capillary to fiber ratio (Figure 4G and 4H), a loss of silencing at repetitive elements and the transcription of retrotransposons, paralleling normal aging (Das et al., 2019; De Cecco et al., 2019; Oberdoerffer et al., 2008; Wenz et al., 2009) (Figure S5I). The ICE mice had thinner left ventricular (LV) posterior walls, implying possible dilated cardiomyopathy (Figure S5K and S5L).
ICE mice undergo accelerated epigenetic aging
Gene expression and DNAme patterns of ICE mice were compared to normal young and old mice. In skeletal muscle, genes that were significantly dysregulated in ICE mice correlated with changes in old mice (Figure 4I, 4J, S5M–S5O and Table S2), including Cdkn1a (Cyclin Dependent Kinase Inhibitor 1A or p21), a mediator of p53-mediated cellular senescence (Beggs et al., 2004; Choudhury et al., 2007; Welle et al., 2004), Myl4 (Myosin light chain 4), a form of myosin upregulated during aging (Lin et al., 2018), Nlrc5 (NLR family CARD domain containing 5), which inhibits NF-κB (Boisvert et al., 2018; Soto et al., 2015; Yoshihama et al., 2016; Zeng et al., 2018), and Mrpl55 (mitochondrial ribosomal protein L55), the methylation of which is associated with longer lifespan (Weidner et al., 2014; Zhang et al., 2017).
Epigenetic clocks serve as a biomarker of biological age in mammals (Hannum et al., 2013; Horvath, 2013; Petkovich et al., 2017; Weidner et al., 2014). We used 61 WT muscle and 28 WT blood samples from male and female mice aged 2 to 30 months to define the training set (Figure S6A and S6C). RRBS identified age-associated CpGs for blood (743) and muscle (2,048) by Elastic Net regression model using CpG sites with >300 reads. The epigenetic ages of muscle and blood samples were calculated as: epigenetic age = inverse. F(b0 + b1CpG1+ ⋯ +bnCpGn), with b as the coefficient from the trained model, b0 as the intercept, and CpGn DNA methylation values. Within the training dataset, epigenetic age was correlated with chronological age (R2 = 0.995 and 0.991 for muscle and blood, respectively) (Figure S6A, S6C and Table S3).
For validation, 18 muscle and 90 blood samples from mice 2–30 months old were used for testing (Figure S6B, S6D and Table S3). Validation sets had R2 = 0.915 and 0.944 for muscle and blood, respectively, demonstrating accurate age estimations using independent datasets. There was no overlap between the clock CpGs and I-PpoI recognition sequences (Figure S6E). Based on the two mouse clocks, the rate of epigenetic aging was ~50% faster in the ICE mice than Cre controls (p < 0.0001), closely paralleling treated ICE fibroblasts (Figure 4K–M).
Faithful DNA repair alters the epigenetic landscape
Aging is associated with specific changes in histone levels and post-translational modifications. Reduced levels of H3K122ac extend the lifespan of budding yeast (Sen et al., 2015) and levels of H3K27ac and H3K56ac decrease in many human immune cell types (Cheung et al., 2018; Dang et al., 2009). Mass spectrometric quantification of 46 different histone modifications detected relatively lower amounts of H3K27ac and H3K56ac and higher amounts of H3K122ac in treated ICE cells (Figure 5A, S6F). Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) with a spiked in Drosophila S2+ cells (Orlando et al., 2014) showed ICE cells had relatively less chromatin-bound H3K27ac and H3K56ac (2% and 5%, respectively) (Figure S6G and Table S4).
Figure 5. Erosion of the epigenetic landscape in ICE cells.

(A) Quantitative mass spectrometry of histone H3 and H4 modifications in 96-hour post-treated ICE cells. %, relative abundance; unmod, unmodified; me, methylation; ac, acetylation.
(B) Genome-wide changes of H3K27ac in 96-hr post-treated cells. Heatmap of ICE/Cre.
(C) Gene Ontology analysis of H3K27ac-increased, H3K56ac-increased, or H3K27me3-decreased peaks ordered by top 20 processes enriched in H3K27ac-increased regions (padj < 0.01). ↑, Cre < ICE peaks, padj < 0.01; ↓, Cre > ICE peaks, padj < 0.01.
(D) TreeFam analysis of gene families with overlapping regions with histone modification changes (padj < 0.01) in ICE cells.
(E) Volcano plot of H3K27ac peaks. All peaks and peaks in Hox genes shown white to yellow and blue to purple, respectively.
(F) ChIP-seq track of histone modifications and mRNA levels across the 120 kb Hoxa locus of post-treated ICE cells. Difference = ICE – Cre
(G) Hi-C contact matrices and HiChIP contact loops in Hoxa. Red, chromatin contacts between Hoxa promoters and other regions. Lower panels, regions with ChIP-seq or RNA-seq peaks. Peak regions, red (Cre<ICE), blue (Cre>ICE) or grey (unchanged).
Based on ChIP-seq and Assay for Transposase Accessible Chromatin using sequencing (ATAC-seq), H3K27ac was enriched in accessible regions, consistent with the known association of H3K27ac with active promoters and enhancers (Heinz et al., 2015; Klemm et al., 2019). In post-treated ICE cells, regions with higher accessibility lost H3K27ac and those with lower accessibility gained it (Figure S6H). Loci with higher H3K27ac peak intensities tended to lose the most signal and vice versa, consistent with a genome-wide erosion of the H3K27ac landscape, with no apparent changes specifically at I-PpoI recognition sites (Figure S6I). Erosion was also seen across H3K27ac peaks genome-wide and in the top or bottom 40% quantile of H3K27ac signals. The ICE:Cre ratio of H3K27ac signals was inversely correlated with basal H3K27ac signals (Figure 5B and S6J), demonstrating that the ICE treatment corrupted epigenetic information and increased Shannon entropy (Hannum et al., 2013).
Erosion of the epigenetic landscape disrupts developmental genes
Consistent with RCM being an ancient stress response that disrupts cell identity (Oberdoerffer et al., 2008), analysis of genes with significant increases in H3K27ac and H3K56ac and decreases in H3K27me3 by Gene Ontology (GO) analysis indicated that cell identity and developmental processes were disrupted in ICE cells. Of the top 20 processes, half of them were involved in development (Figure 5C, Table S5 and S6). Loci that experienced a decrease in H3K27ac were predominately involved in stress responses, nucleobase synthesis, DNA repair, chromatin structure, and metabolism (Table S6).
The intersection of the ChIP-seq datasets was cross-referenced with the TreeFam database, which provides orthology and paralogy predictions of gene families (Li et al., 2006; Ruan et al., 2008) (Figure 5D). At the intersection were two gene families, both comprised of homeobox (Hox) developmental transcription factor genes. In the epigenetically aged ICE cells, all the Hox gene clusters had significant alterations in H3K27ac, H3K56ac, and H3K27me3 peaks, with coincident changes in mRNA levels (Figure 5E and 5F). From Hoxa1 to Hoxa6, levels of H3K27ac and H3K56ac decreased and from Hoxa9 to Hoxa13 they increased, with concomitant changes in H3K27me3 and mRNA levels (Figure 5F and S6K).
To test if the effects of DSB repair on Hoxa were specific to I-PpoI, we isolated MEFs from a mouse strain with an inducible homing endonuclease from budding yeast called I-SceI, which cuts at ~18 cryptic (non-canonical) sites in the mouse genome, far from any I-PpoI sites (Chiarle et al., 2011). Paralleling the effects of I-PpoI on post-recovery gene expression, I-SceI induction altered mRNA levels of genes in the Hoxa cluster. Thus, the effect of DSBs on Hoxa expression apparently does not depend on where the DNA breaks occur (Figure S6L).
Faithful DSB repair alters spatial chromatin contacts
The spatial organization of chromatin, including a variety of long-range DNA interactions, plays a key role in controlling gene expression and maintaining cellular identity (Gorkin et al., 2014; Kundu et al., 2017; Narendra et al., 2015; Schoenfelder et al., 2015; Zirkel et al., 2018). High-resolution spatial chromatin contacts between promoters and enhancers were assessed by Hi-C and HiChIP to assess H3K27ac-accociated chromatin contacts (Mumbach et al., 2016). While topologically associated domains (TADs) remained intact in ICE cells (Figure 5G), H3K27ac-positive posterior Hoxa gene promoters (Hoxa 9–13) formed new contacts with active enhancers in an adjacent TAD, with concomitant increases in mRNA levels consistent with weakened insulation and disordered promoter-enhancer (P-E) communication. As far as we know, this is the first evidence that faithful DNA repair alters multiple layers of epigenetic information, including spatial chromatin contacts, chromatin insulation, and P-E communication.
Epigenetically aged cells lose the ability to maintain cellular identity
The smoothening of the epigenetic landscape, particularly at genes involved in development and differentiation, prompted us to test if the identity of the ICE-treated fibroblasts were compromised. Of the genes with decreased H3K27me3, 4/10 of the top GO processes and 6/10 tissue-specific transcriptional profiles were related to neuronal processes or neuronal tissue types (Figure 6A and 6B). H3K27me3 signals were lower across the promoter regions of the neuronal fate genes Neurod1 and Nefh (Figure 6C). We reasoned that ICE treatment may shift fibroblasts away from their lineage and be primed for differentiating into other cell types, such as neurons. To test this, we reprogrammed the post-treated Cre and ICE cells into neurons by chemical means (Li et al., 2015) and found that Neurod1 and Nefh were 8–15-fold more easily derepressed in the ICE MEFs (Figure 6D and 6E), coincident with increases in neuron count and the neuronal cell marker TUJ1 (Figure 6F and 6G).
Figure 6. Induction of the ICE system disrupts cellular identity.

(A) Gene Ontology analysis of H3K27me3 decreased regions (padj < 0.05). Red, developmental processes. *Neuronal processes.
(B) Mouse tissue types of transcriptional profiles that overlap decreased H3K27me3 regions (padj < 0.05) in ICE cells. Red, neuronal tissues. Numbers indicate rank.
(C) ChIP-seq track of neuronal markers, Neurod1 and Nefh. Difference = ICE – Cre. (D and E) Time-course of mRNA levels of Col1A1 (a fibroblast marker), Neurod1 and Nefh (neuronal markers) during neuronal reprogramming. Two-way ANOVA-Bonferroni. (F and G) Neuronal marker TUJ1 8 d after reprogramming. DNA stained with DAPI. Scale bar, 100 μm. Two-tailed Student’s t test.
(H) Comparison of H3K27ac increased regions (p < 0.01) to epigenome roadmap data from different human tissue types.
(I) Gene Ontology comparison of H3K27ac increased regions in 10-month post-treated ICE mice (16 mo.) (p < 0.01) to RNA-seq data from skeletal muscle from old WT mice (24 mo.) (padj < 0.05).
(J) Aggregation plots of H3K27ac signal in bottom 25% quantile in spleen super-enhancer regions.
(K) H3K27ac ChIP-seq tracks across spleen super-enhancers in the class II major MHC cluster and Nfkbid in 10-month post-treated muscle.
Data are mean (n≥3) ± SD. *p < 0.05; **p < 0.01.
Cellular identity changes in ICE mice
We refer to the loss of cell identity due to damage or aging as “exdifferentiation” and the reversal of it as “redifferentiation” (Cutler, 1982; Sinclair and LaPlante, 2019). To test whether DSBs accelerate exdifferentiation in vivo, we performed H3K27ac ChIP-seq on skeletal muscle of 10-month post-treated ICE mice and compared the datasets to the epigenome roadmap, a consortium of human epigenomic data from different cell types and tissues (Roadmap Epigenomics et al., 2015). Regions with lower H3K27ac (p < 0.01) in Cre vs. ICE showed the strongest enrichment for muscle tissue signatures (p=9.0×10−8) while regions with higher H3K27ac showed an enrichment for immune cell enhancers (p=9.3×10−28) (Figure 6H and S6M). Of the top 20 processes that were increased in ICE muscle, all were elevated in old WT mice, consistent with WT and ICE mice undergoing similar epigenetic changes (Figure 6I).
In mammals, cellular identity is established prenatally and maintained by H3K27ac-enriched super-enhancers (Hnisz et al., 2013; Whyte et al., 2013). Of the regions that gained H3K27ac in ICE cells, there was overlap with super-enhancer regions from immune cells and regulatory regions involved in developmental processes and immune cell activation (Figure 6I and S6N). After ICE treatment, spleen super-enhancers in muscle were derepressed, gaining H3K27ac (Figure 6J), as were the Class II major histocompatibility complex (MHC) cluster and the Nfkbid gene, a regulator of humoral immunity (Figure 6K). These data imply that the muscle of post-treated ICE mice exdifferentiate towards an immune signature, consistent with data from aged mouse tissues (Benayoun et al., 2019). Together with the neuronal priming of ICE cells and increased EMT in the ICE kidney, we conclude that the induction of non-mutagenic DSBs accelerates the DNAme clock and age-related changes to chromatin, gene expression, and cellular identity.
Epigenetic reprogramming restores a youthful epigenome in ICE mice
To further rule out mutations as a cause of the ICE phenotype and gain further insight into epigenetic alterations as a cause of aging, we tested the effect of resetting the epigenome in vitro and in vivo. The cyclic expression of Yamanaka factors Oct4, Sox2, Klf4 and Myc (OSKM) (Takahashi and Yamanaka, 2006) alleviates the symptoms and extends the lifespan of progeroid mice and other aspects of aging (Ocampo et al., 2016; Sarkar et al., 2020). In a parallel study to this one, we found it possible to safely reverse DNAme age and gene expression patterns of old and damaged neurons to cure blindness in mice, a process requiring DNA demethylation (Lu et al., 2020). These findings revealed that cells possess a back-up copy of youthful epigenetic information that can restore cell identity (LaPlante and Sinclair, 2019).
Expression of OSK in treated ICE cells reversed age-associated mRNA changes, including those for Hmgb, Chaf1, Hoxa and canonical histone genes (Figure 7A–7C and S7E), similar to the effect of OSK expression in aged fibroblasts (Figure 7D–7F and S7F), and did not go so far back in age as to begin to erase cell identity or induce the pluripotency gene Nanog (Figure S7A–S7E). Based on four different mouse clocks, the DNAme age of ICE cells was reversed by up to 57% (Figure 7G) (Meer et al., 2018; Petkovich et al., 2017; Stubbs et al., 2017; Thompson et al., 2018). During normal aging, levels of H3K9me3 in kidneys decrease and H3K36me2 in muscle increase (Tvardovskiy et al., 2017). Remarkably, after 5 weeks of inducing AAV-delivered OSK in the whole body of ICE mice, the levels of these aging markers in kidney and muscle were rejuvenated to a point where they resembled negative controls (Figure 7H, 7I and S7H–S7M).
Figure 7. Epigenetic reprogramming restores youthful epigenetic marks.

(A) AAV vectors used for polycistronic OSK expression.
(B) Experimental scheme for AAV-OSK transduction to post-treated ICE cells and mice.
(C) Scatter plot of mRNA changes in ICE (AAV-DJ-rtTA, n=3) and ICE+OSK (AAV-DJ- rtTA and OSK, n=3) fibroblasts from 1-month post-treated ICE mice. Linear regression.
(D) Transgenes in the OSK transgenic mouse.
(E) OSK induction in fibroblasts from young (3 mo., n=8) or old (15 mo., n=3) OSK transgenic mice by Dox treatment.
(F) Scatter plot of mRNA changes by aging or OSK. Linear regression.
(G) Epigenetic age of post-treated ICE (AAV-DJ-tTA, n=8) and ICE+OSK (AAV-DJ-tTA and OSK, n=8) MEFs at 10-day post-AAV transduction. Two-tailed paired Student’s t test.
(H and I) H3K9me3 in Cre (AAV-MYO3-tTA, n=3), ICE (AAV-MYO3-tTA, n=2) and ICE+OSK (AAV-MYO3-tTA and OSK, n=4) kidney at 5-week post-AAV injection. One-way ANOVA-Bonferroni.
(J) Optic nerve head: section used for axon counts (solid blue line). V, retinal blood vessels; MTZ, myelination transition zone; Ax, axon bundles.
(K-N) Density of PPD-stained myelinated optic nerve axons. Scale bar, 10 μm. Two-tailed Student’s t test.
(O) Intravitreal injection of AAV2-OSK and RGC sorting. RGCs isolated by FACS from retinas of young (5 mo., n=5), old (12 mo., n=6) and old mice injected with AAV2-OSK (15 mo., n=4).
(P) Gene Ontology analysis of upregulated genes in RNA-seq data (5 mo. vs 12 mo., padj < 0.01).
(Q) Scatter plot of mRNA changes due to aging or epigenetic reprogramming in age-associated genes (grey) and nervous system development genes (other colors). Data are mean ± SD or ± SEM (L and N). *p < 0.05; **** p < 0.0001.
Old mice develop opaque lenses and lose retinal ganglion cells (RGCs) in the innermost retinal layer (Calkins, 2013; Downs, 2015; Samuel et al., 2011; Wolf et al., 2000) (Figure 7J). In the ICE mice, lens opacity was greater (Figure S7N–S7P) and the number of RGC axons in the myelinated region were fewer than controls (Figure 7K–7N). Remarkably, ectopic expression of OSK in RGCs (Figure 7O) returned mRNA levels to a more youthful pattern (p < 0.0001). Consistent with the in vitro aging forward and reversal data, 7/10 of the top upregulated processes in the RGCs of 12-month-old mice were involved in development (Figure 7P), the majority of which (86% of nervous system developmental genes) were restored by OSK (Figure 7Q).
DISCUSSION
Studies of simple organisms, such as budding yeast, worms and flies, indicate that a loss of epigenomic information, may be an underlying cause of aging in those species (Imai and Kitano, 1998; Jiang et al., 2013; Lu et al., 2014; Mills et al., 1999; Oberdoerffer et al., 2008). Based on yeast work performed in our lab and others, we proposed the RCM and Information Theories of Aging, which state that a loss of epigenetic information driven in part by cellular repair events, are a cause of aging (Oberdoerffer et al., 2008; Sinclair and Guarente, 1997; Sinclair and LaPlante, 2019; Sinclair et al., 1997). In this paper, we show that non-mutagenic DSB repair causes the epigenetic landscape to erode and cells to drift into adjacent Waddington valleys, where they malfunction and cause aging, consistent with the Information Theory of Aging.
There is vigorous debate about the contribution of mutations to aging, relative to other aging hallmarks (Kabacik et al., 2022; Narayanan et al., 1997; Robinson et al., 2021). Separating genomic from epigenomic effects on aging has been challenging because inducing DNA damage, either by chemicals, radiation, or deleting DNA repair genes, causes unnatural and overwhelming DNA damage and disrupts other major cellular processes (Birkisdottir et al., 2021; Zhang et al., 2011).
While it is incontrovertible that certain types of DNA damage mimic aging, including chemotherapy, radiation, smoking, and progeria (Carrero et al., 2016; Hofstatter et al., 2018; Horvath and Levine, 2015; Maccormick, 2006; Nance and Berry, 1992; Salk et al., 1985; White and Vijg, 2016), evidence to the contrary includes the fact that cloned animals or mice engineered to accumulate mutations have normal health and lifespans (Narayanan et al., 1997; Wakayama et al., 2013).
By introducing DSBs without causing mutations, we provide evidence that it is the cell’s reaction to damage and the resulting loss of epigenetic information that drives mammalian aging forward. This would explain why aging proceeds through a predictable series of molecular and physiological changes, even though DNA damage can occur anywhere in the genome. We do not, however, discount the possibility that the effects of genomic and epigenomic information loss on cell senescence and aging are mutually reinforcing. Indeed, we see that advancement of epigenetic age is associated with an increase in DSBs (Figure S1H and S1I).
The fact that the DSB repair efficiency of mammals correlates with lifespan, but not other types of repair, such as NER and BER, indicates that DSB are a special case, perhaps because only severe threats to cell survival disrupt the epigenome sufficiently to cause aging (Brown and Stuart, 2007; Tian et al., 2019). This idea is consistent with our observation that severe nerve damage also causes accelerated aging (Lu et al., 2019).
Chromatin-modifying factors known to relocalize during DSB repair include SIRT1 and SIRT6, HDAC1, and PARP1. Studies of these proteins indicate that repeated triggering of the RCM response disrupts youthful gene expression patterns and liberates retrotransposons, such as those silenced by SIRT1/SIRT6 (De Cecco et al., 2019; Oberdoerffer et al., 2008; Simon et al., 2019). How the DNAme clock is advanced by DSBs is not known, but one possibility is that DSBs cause the relocalization of ten-eleven translocation enzymes (TETs) and DNA methyltransferases (DNMTs) (O’Hagan et al., 2008).
Short-lived species and organoids made from induced pluripotent stem cells (iPSCs) have proven sub-optimal for studying aspects of human aging. The ICE system may help solve this problem by advancing epigenetic age of cells and organisms to resemble old human tissues.
Given that RCM is ultimately lethal to the individual, why did it evolve? We propose that RCM is an example of antagonistic pleiotropy, whereby a process that promotes survival in young individuals does the opposite later in life (Williams, 1957). The benefit is that the relocalization of chromatin factors to sites of repair places cells in survival mode until repair is complete (Kennedy et al., 1997; Mills et al., 1999; Oberdoerffer et al., 2008; Sinclair and LaPlante, 2019; Smeal et al., 1996). Over time, however, repeated activation causes chromatin factors to scatter and long-range DNA interactions to change over time, thereby aging cells and tissues.
RCM may have evolved to allow cells to deal with the abundance of DSBs that occur during rapid DNA replication in microorganisms and then in embryos. Indeed, Wnt and Hox genes direct embryonic development and also facilitate DSB repair (Feltes, 2019; Liu et al., 2007; Rubin et al., 2007; Ye et al., 2007; Zhao et al., 2018). Developmental genes might be more susceptible to DSB-driven information loss because they are poised to turn on rapidly and in response to cellular damage (Brack et al., 2007; Budovskaya et al., 2008; Florian et al., 2013; Salzer et al., 2018).
Even low levels of DSBs caused a surprisingly strong phenotype months later in the ICE mice, which raises the specter that some chemical, enzymatic, and radiological agents that break DNA or otherwise disrupt the epigenome and are regarded as safe because they don’t cause cancer, may promote aging and age-related diseases. Fortunately, it is now apparent that mammals retain a backup copy of youthful epigenetic information that can safely restore the function of old tissues, akin to reinstalling software.
Limitations of the Study
In this study, we did not determine which chromatin factors are relocalized, nor did we study chromatin contacts in vivo. We did not perform epigenomic analyses at the single cell level and induced the ICE system in the whole body but not in a tissue-specific manner, so cannot make conclusions about cell autonomy. We also cannot rule out that some of the effects are due to cutting of the rDNA locus. Indeed, rDNA instability and nucleolar size influence the lifespan of lower organisms (Sinclair et al., 1997; Tiku et al., 2017). That said, we saw no effects on rDNA mutations, rRNA levels, or protein translation, and I-SceI, which does not cut the rDNA, generated a I-PpoI-like gene expression pattern.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David A Sinclair (david_sinclair@hms.harvard.edu).
Materials availability
Reagents and methods in this study will be made available by the lead contact upon request.
Data and code availability
The NGS datasets generated during this study are available through the BioSample database (NCBI) under BioProject ID: PRJNA554729 and PRJNA655981. Details of the analysis are provided in the STAR Methods section. Any additional information is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and treatments
I-PpoISTOP knock-in mouse ES cells were generated. Briefly, an estrogen receptor nuclear translocation domain (ERT2) tagged with HA at N-terminus and I-PpoI were inserted into STOP-eGFP-ROSA26TV plasmid (addgene, plasmid #11739) together followed by IRES and EGFP sequence. HA-ERT2-I-PpoISTOP cassette was integrated at Rosa26 loci and the targeted C57BL6 ES cells were injected into C57BL/6 albino (cBRD/cBRD) blastocysts. After back-crossing I-PpoI STOP/+ chimeric mice with C57BL/6 mice, ICE mice were generated by crossing I-PpoI STOP/+ mice to CreERT2/+ mice harboring a single ERT2 fused to Cre recombinase that is induced whole body (Ruzankina et al., 2007). 4–6 month-old Cre and ICE mice were fed a modified AIN-93G purified rodent diet with 360 mg/kg Tamoxifen citrate for 3 weeks to carry out I-PpoI induction. ERT2 containing three mutations selectively binds to 4-hydroxytamoxifen (4-OHT) but not estradiol. Cre-ERT2 protein is translocated into nucleus by tamoxifen treatment followed by removal of the STOP cassette located at upstream of I-PpoI. In the presence of tamoxifen, Cre- ERT2 and HA-ERT2-I-PpoI localize to the nucleus and induce DNA double strand breaks. Wild-type aged mice were obtained from the NIA aged rodent colonies and acclimatized at least for a month prior to experimentation. Mice were fed LabDiet 5053 diet and all animal care followed the guidelines of Animal Care and Use Committees (IACUCs) at Harvard Medical School.
Cell culture
Mouse Embryonic Fibroblast (MEF) cells were isolated from E13.5 mouse Iittermate embryos at day E13.5 and cultured in low oxygen (3% v/v). After dissecting out the uterus and yolk sac, fetuses were moved in a new dish containing sterile PBS. The liver, heart, head were removed and the remaining part was washed in sterile PBS to remove blood. Fetuses were minced in 0.25% trypsin-EDTA and incubated at 37°C for 30 min. Cells were washed and maintained with MEF growth medium (DMEM containing 20% FBS, 1% pen/strep, 0.1 mM β-mercaptoethanol). For activation of ER (estrogen receptor)-fused Cre in MEFs, 0.5 μM 4-Hydroxytamoxifen (4-OHT) was treated for 24 h and medium was switched to one without 4-OHT to stop I-PpoI-mediated DNA breaks. For activation of GR (glucocorticoid receptor)-fused I-SceI, 200 nM triamcinolone acetonide (TA) were treated in DMEM containing 15% charcoal stripped FBS for 2 d and maintained for 8 d without TA.
Mouse adult fibroblast cells were isolated from ears taken from 3-, 24- and 30-month-old mice. 2 whole ears were washed with 70% EtOH and sterile PBS and minced in DMEM containing 0.14 Wunsch Units/ml Liberase TM and 1% penicillin/streptomycin. After incubation of minced tissues at 37°C for 45 min with shaking, cells were washed with medium twice and plated on collagen coated culture dishes.
All cells were cultured in DMEM containing 20% FBS (Seradigm or Takara Tet-free), 1% penicillin/streptomycin, 0.1 mM β-mercaptoethanol at 37°C, 3% O2 and 5% CO2 unless otherwise specified.
METHOD DETAILS
Western blot analysis
Cell and tissue samples were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA) containing a proteinase inhibitor cocktail (Sigma-Aldrich). An equal amount of lysate was incubated with sample buffer (0.05% Bromophenol blue, 2% sodium dodecyl sulfate, 50 mM Tris-Cl pH 6.8, 5% β-mercaptoethanol) at 95°C for 5 min then separated on an SDS-PAGE gradient gel, transferred to a membrane using transfer buffer (25 mM Tris-HCl pH 8.3, 190 mM glycine 20% methanol), blocked with TBSTM (Tris-buffered saline, 0.1% Tween 20, with 5% skim milk), probed with primary and secondary antibodies and developed using ECL Western Blotting Detection Reagent (Sigma-Aldrich).
Southern blotting
Genomic DNA samples were prepared using EZNATissue DNA Kit (Omega Bio-tek). DNA (3 μg) was run in 0.8% agarose gel, DNA was depurinated in 0.25 N HCl, denatured in 0.4 N NaOH, and washed with 20X SSC. DNA was transferred to a nylon membrane in 0.4 N NaOH using a TurboBlotter (Whatman), washed with 2X SSC, crosslinked by UV then incubated in pre-hybridization solution (6X SSC, 5X Denhardt’s solution, 1X SSD,0.0625 M Tris-HCl pH7 .5, 75 μ/ml salmon sperm DNA) at 65ºC for 3 h with rotation. DNA probes were generated using target-specific PCR with dCTP [α−32P]. Radioactive DNA probes were added to fresh pre-hybridization solution and incubated with the membrane overnight with rotation. The membrane was washed with 2X SSC, 2X SSC containing 1% SDS and 0.1X SSC and exposed to X-ray film at −80°C.
Surveyor assay
I-PpoI target regions were amplified from genomic DNA isolated from either Cre or ICE cells by PCR using flanking primer sets. Hetero- or homo-duplexes were hybridized in thermocycler and hybridized DNA (200 ng) was treated with SURVEYOR nuclease S (Transgenomic) at 42ºC for 60 min. Nuclease reactions were stopped and digestion was analyzed by agarose gel electrophoresis or a Bioanalyzer (Agilent).
Metabolic labeling of MEFs
MEFs were washed twice with pulse-labeling medium (Met- Cys-free DMEM containing 10% dialyzed serum) and incubated in pulse-labeling medium for 1 h to deplete intracellular Methionine. Pulse-labeling medium with 0.2 mCi/ml methionine [35S] was added to cells and incubated for 1h. Cells were lysed and 35S-methionine incorporation was determined by TCA precipitation and scintillation counting.
Quantification of protein synthesis
Protein synthesis was quantified as published (Garlick et al., 1980; Hofmann et al., 2015). L-3H-phenylalanine (1 mCi/mL) was combined with unlabeled phenylalanine (135 mM) to create 100 mCi/ml. After adjusting the solution to pH 7.1 with NaOH, the labeling solution was injected via the lateral tail vein at 1 ml/100 g bodyweight under anesthesia with ketamine (75 mg/kg) and xylazine (10 mg/kg).
Quantification of DSBs
DNA double strand breaks (DSB) generated by I-PpoI were detected as described previously (Chailleux et al., 2014). Briefly, tissue was homogenized in phenol and genomic DNA was purified with chloroform, ethanol and RNase. Genomic DNA carrying I-PpoI specific DSBs was subjected to ligation-mediated purification using biotin-conjugated adaptor nucleotides with 5`-AATT-3` overhangs that bind to the DSB site generated by I-PpoI. Adaptor sequences were as follows: dRbiot-BglII-IPpoI F 5`-CCCTATAGTGAGTCGTATTAGATCTGCGTTAA-3`, dRbiot-BglII-IPpoI R 5`-CGCAGATCTTAATACGACTCACTATAGGG-3`. The biotinylated fragment was digested using EcoRI for 3 h at 37°C followed by purification with streptavidin magnetic beads (Dynabeads M-280 Streptavidin, Invitrogen) in binding buffer (20 mM Tris-HCl pH 8.0, 0.1% SDS, 1% Triton X-100, 2mM EDTA, 150 mM NaCl). After 4 h at 4°C, beads were washed five times with washing buffer (50 mM Tris-HCl pH 8.0, 0.1% SDS, 150 mM NaCl) and once with TE buffer. Cut DNA was eluted by digesting the adaptor with BglII at 37°C overnight. DNA was purified using glycogen, sodium acetate and ethanol. DNA primers were: 5+11 F 5`-ACTTAGAACTGGCGCTGAC −3`, 5+11 R 5`-CTGGCCTGGAACTCAGAAAT-3`, 28S F CCCACTGTCCCTACCTACTATC, 28S R AGCTCAACAGGGTCTTCTTTC.
Indirect Calorimetry
Food consumption, ambulatory activity, oxygen consumption (VO2), carbon dioxide production (VCO2) and respiration exchange ratio (RER) were measured using Columbus Instruments CLAMS. Mice were housed in metabolic cages for 3 d prior to collecting data and body composition was determined by EchoMRI 3-in-1.
MMQPCR
Monochrome multiplex quantitative PCR was performed as described previously (Cawthon, 2009). Briefly, a PCR reaction containing 20 ng of genomic DNA was prepared with SYBR Green system (Applied Biosystems). The PCR program was set up as Step 1: 95°C; 15 min, Step 2; 2 cycles of 94°C for 15 sec and 49°C for 15 sec, Step 3: 32 cycles of 94°C for 15 sec, 62°C for 15 sec, 74°C for 15 sec with signal acquisition for 28S amplification, 84°C for 10 sec, 88°C for 15 sec with signal acquisition for Hbbt1 amplification. Primers are listed in Table 1.
Frailty index assessment
The Frailty Index (FI) was scored as described previously (Whitehead et al., 2013). Briefly 31 health-related deficits were assessed for each mouse. Mice were weighed and body surface temperatures were measured three times with an infrared thermometer (La Crosse Technology), then scored based on the deviation from mean weight and temperature of young mice (Whitehead et al 2014). Twenty-nine other items across the integument, physical/musculoskeletal, oscular/nasal, digestive/urogenital and respiratory systems were scored as 0, 0.5 and 1 based on the severity of the deficit. Total score across the items was divided by the number of items measured to give a frailty index score between 0 and 1.
Lens opacity scoring
Lens opacity scoring was previously described (Wolf et al., 2008). Mice were held without anesthesia and assessed in a dark room using a SL-14 Kowa hand-held slit lamp (Kowa, Tokyo, Japan).
Magnetic Resonance Imaging
Mice were anesthetized with 2% isoflurane gas in oxygen and placed in a 4.7 Tesla Bruker Pharamscan magnetic resonance imager. Rare T1 (TE: 13.4 ms, TR: 900 ms, Rare factor: 4, Matrix: 256 × 256 × 24, Voxel size: 0.215 × 0.156 × 1 mm) and a Rare T2 (TE: 18.26 ms, TR: 2000 ms, Rare factor: 8, Matrix: 256 × 256 × 24, Voxel size: 0.215 × 0.156 × 1 mm) scans of the lower thoracic cavity, abdomen and lower extremities were performed.
Micro CT scanning
Femurs were isolated and placed in 70% ethanol. Micro-CT was performed by using SCANCO Medical μ-CT35 at the core facility at the Harvard School of Dental Medicine (Idelevich et al., 2018).
Quantification of optic nerve axons
To quantify axons, optic nerves were dissected and fixed in Karnovsky’s reagent (50% in phosphate buffer) overnight. Semi-thin cross-sections of the nerve were taken at 1.0 mm posterior to the globe and stained with 1% p-phenylenediamine (PPD) for evaluation by light microscopy. Six to eight non-overlapping photomicrographs were taken at 60x magnification covering the entire area of the optic nerve cross-section. Using ImageJ software, a 100 × 100 μM square was placed on each 60x image and all axons within the square (0.01 mm2) were counted using the threshold and analyze particles function in image J. The average axon counts in 6–8 images was used to calculate the axon density/mm2 of optic nerve. Scorers were blinded to experimental groups.
Immunohistochemistry for mouse skin
Dorsal skin samples were fixed with 4% paraformaldehyde/PBS and kept on ice for 2 h. The fixed skin samples were embedded in OCT (Sakura Finetek) and snap frozen in liquid nitrogen for histology. After washing in PBS, nonspecific staining was blocked with PBS containing 3% skim milk and 0.1% Triton-X for 30 min. Sections were incubated with primary antibodies at 4°C overnight: rat anti-mouse CD117 (BD Pharmingen) and rabbit anti-human KRT5 (COVANCE). Secondary antibodies were conjugated with Alexa Fluor 488 or 594 (Invitrogen). Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) and images were obtained using FV1000 confocal microscope (Olympus). >100 hair follicles per mouse (n=8) were analyzed for the presence of KIT+ melanocytes in the bulge.
Quantification of subepidermal thickness
Site-matched skin tissue was fixed in formalin, embedded in paraffin, and 5 μm sections were cut and stained with hematoxylin and eosin. Representative regions of the subcutaneous layer were measured from the limits of the dermis to the panniculus carnosus (‘subepidermis’) with the assistance of an ocular micrometer. Care was taken to ensure that tissue was embedded perpendicularly and the subdermal thickness determination was not artificially enhanced due to tangential sectioning. Because the subepidermal layer reached maximum thickness in control Cre mice at 17–18 months, this timepoint was selected for comparisons with the ICE mice. A minimum of 10 randomly selected thickness determinations were generated for each tissue section.
Brain immunohistochemistry
For GFAP and Iba1 staining, the tissues were incubated overnight in paraformaldehyde (4% v/v). Fixed brains were embedded in paraffin and 6 μm sections were generated using a manual rotary microtome (Leica). After deparaffinization and re-hydration of tissue slides, an antigen revealing step was performed by using antigen unmasking solution (Vector). Sections were blocked in PBS with 5% BSA and 0.3% Triton-X at 4°C for 1 h and incubated with primary antibodies in PBS with 2% BSA and 0.1% Triton-X at 4°C overnight with Rabbit anti-GFAP antibody (Abcam, ab7260), Rabbit anti-Iba1 antibody (Funakoshi, GTX100042). Secondary antibodies conjugated with Alexa Fluor 488 or 594 (Invitrogen) were used followed by DPAI staining. To localize I-PpoI expression and DNA damage, mice were perfused transcardially and brains were post-fixed overnight with 4% paraformaldehyde/PBS, then cleared by 30% sucrose solution. Brains were embedded in OCT (Sakura Finetek) and 40 μm sections were collected using a cryostat (Leica). Sections were blocked in horse serum/TBS-Triton-X for 30 min at RT, and then incubated with primary antibodies overnight at 4°C with goat anti-GFP (Abcam) and rabbit anti-γ-H2AX (Cell Signaling). Secondary antibodies were conjugated with Alexa Flour 488 and 647 (Jackson ImmunoResearch) and co-stained with DAPI.
ATP and mtDNA measurement
Snap frozen tissue was briefly washed with PBS and 3 ml Tris-HCl TE saturated phenol per 100 mg was added to the tissue followed by homogenizing with a tissue homogenizer (Omni TH, Omni). After centrifugation, cell lysates were added to an equal amount of TE saturated phenol, chloroform and water were added to the same tube. After centrifugation, the supernatant was used for ATP and mtDNA measurement. ATP was measured using an ATP kit (ThermoFisher Scientific) and normalized to tissue weight. Genomic DNA and mtDNA were purified with 2.5-fold ethanol and glycogen. Primers for 18S ribosomal and CytB were used to calculate the ratio of mtDNA to genomic DNA. Primers were: mouse 18S, 5`-TGTGTTAGGGGACTGGTGGACA-3` (forward) and 5`-CATCACCCACTTACCCCCAAAA-3` (reverse), mouse Cytb, 5`-CCCTAGCAATCGTTCACCTC-3` and 5`- TGGGTCTCCTAGTATGTCTGG −3` (reverse).
Contextual fear conditioning test
Contextual fear conditioning was assessed using a TSE system. On day 1, mice were placed into an experimental box (52 cm x 52 cm x 65 cm) and allowed to explore freely for 180 s followed by 0.5 mA electric shock for 1 s. One more 0.5 mA shock for 1 s was given after 30 s and immediate freezing was measured every 10 s by a visual count, after which mice were returned to their home cage. Contextual freezing without a tone was assessed for 180s, 24 hours after the shock, counting freezing every 10 sec.
Barnes maze test
The maze consisted of a circular and white platform (90 cm in diameter) with 20 × 5 cm diameter holes arranged around the edge of the platform, elevated 82 cm above the floor. For visual cues, the platform was surrounded by four pictures with different colors and shapes. A mouse was placed in the center of maze and then, the mouse was guided to a small chamber termed a target hole at adaptation period. After 2 min in the target hole, the mouse was returned to the cage. During the spatial acquisition period, the mouse was allowed to explore the target hole for 3 min. If the mouse entered the target hole or it passed 3 min, the mouse was left for 1 min in the hole. The trial was repeated 3 times/day for 5 d. A probe trial was performed to test long-term memory 7 d later by covering the target hole with a lid. The mouse was allowed to explore the position of target hole for 90 s and the number of pokes in each hole was measured using TopScanLite version 2.
Grip strength test, treadmill test and lactate measurement
To measure muscular strength, a mouse was held by the tail and allowed to grip a mesh grip with the front paws (BIO-G53, BIOSEB) then pulled backward until grip was released. After a 10 min break, the experiment was repeated. Maximum exercise endurance was assessed with a treadmill system (TSE). Mice were trained for 3 d prior to recording the performance to familiarize the mice to the equipment. An electrical stimulation grid was adjusted as 1 mA and slope was set at 15 degrees. The first day of the training, mice walked on the treadmill at 10 m/min speed for 10 min, with a 10 min break, then walked at 10 m/min speed for 10 min. On the second and third day, the initial two steps were the same as first day, then walking was started at 10 m/min and the speed was increased by 1 m/min every minute to a maximum speed of 20 m/min. On day 4, maximum exercise endurance was measured. Six mice were placed on the treadmill and the belt speed was started at 5 m/min for 5 min to allow the mice warm up. The speed was increased by 1 m/min up to 20 m/min. After running for 5 min, the speed was increased from 20 m/min to 21 m/min for 10 min. Mice were then forced to run at 22 m/min until they remained on the electrical stimulation grid for 10 seconds. Details are available upon request. The tail blood at pre-exercising and post-exercising was taken and serum lactate level were measured with a lactate meter (Nova Biomedical).
Ambulatory activity
Animals were maintained in specific-pathogen-free (SPF) facility and single-housed in instrumented individually ventilated cages (IVC) (Digital Smart House, Vium, San Mateo, CA, and Innovive, San Diego, CA) containing corncob bedding with access to Innowheel and Innodome (Innovive, San Diego, CA), Bed-r’Nest (Andersons Lab Bedding, Maumee, OH), and foraging mix (Veggie Relish, LabDiet). Animals had unrestricted access to food (Pico Rodent Diet 5053, Lab Diet, St. Louis, MO) and acidified, sterile water (Innovive, San Diego, CA).
Vium Digital Smart Houses slotted in Vium’s proprietary rack system were outfitted with sensors and a high-definition (HD) camera that enables continuous, 24/7 monitoring of animals and streams data to a secure cloud-based infrastructure. As described elsewhere (Lim et al., 2019; Lim et al., 2017), video is processed using computer vision algorithms to produce a digital history of motion (mm/sec). Motion (mm/s) was averaged across 1 h bins to produce 1 h averages. All 1 h averages from 6am to 7am across 55 d were averaged and repeated for each hour of the day.
Treadmill Gait Analysis
Gait patterns were measured using forced walking on a treadmill (Columbus Instruments; Columbus, OH). A high-speed digital video camera recorded images of the ventral side of the mouse through a transparent treadmill belt reflected off a mirror. Mice for approximately 24 sec at speeds of 13, 19, and 25 cm/s. TreadScan® software (CleverSys, Inc, Reston, VA) identified each individual paw of the mouse in each frame as it walked on the treadmill and measures of stance and swing duration, among other measures, were assessed.
COX and capillary density staining
Freshly isolated quadriceps and gastrocnemius muscles were mounted in OCT (Tissue-Tek), placed in an isopentane bath, and slowly cooled in liquid nitrogen. Transverse sections (20 mm) were sectioned on a cryostat (Leica). Sections were fixed in pre-cooled acetone (−20°C) for 10 min, washed with PBS, then blocked with BlockAid (Invitrogen) for 1 h at RT, and then incubated with CD31 (ab56299, Abcam), Laminin (L9393, Sigma) antibodies diluted in blocking buffer overnight at 4°C. Slides were washed with PBST, then incubated with anti-rat Alexa Fluor 488-conjugated (Life Technologies) and anti-rabbit Alexa Fluor 594-conjugated (Life Technologies) diluted to 1:500 in blocking buffer for 2 h at RT. Slides were washed again with PBST and mounted with Fluoroshield with DAPI mounting medium (Sigma). Images were acquired using a confocal fluorescence microscope (Nikon A1). COX staining was performed according to a protocol (Ross, 2011). Briefly, 20 μm cryostat sections was dried at room temperature for 1 hr and media containing 1X DAB, 100 μM cytochrome c, 2 μg/ml bovine catalase was added to sections and slides were incubated at 37°C for 40 min. Quantification of capillary number and density were performed using ImageJ.
Electron microscopy
Mice at 15 months of age were anesthetized with isoflurane and sacrificed by cervical dislocation or decapitation, in accordance with available ethical permits. Muscle was collected and fixed in electron microscopy fixative (consisting of 3% glutaraldehyde, 2.5% paraformaldehyde, 2 mM calcium chloride, 2% sucrose in 0.1 M cacodylate buffer) and tissue was processed as previously reported (Le Couteur et al., 2001). Two blocks from different parts of the muscle were used and from each section 10 images were taken at 5000X on a Jeol 1210 transmission microscope and photographed using a Gatan US 4000MP digital camera. Mitochondrial network, size and number were quantified blindly using FUJI ImageJ.
Podocyte density p57 and PAS representative images
Podocyte density was quantitated following staining for p57 on formalin fixed, paraffin-embedded, 4 μm kidney sections as previously described (Ohse et al., 2010; Schneider et al., 2017; Zhang et al., 2013). Briefly, Histoclear (National Diagnostics, Atlanta, GA) was used to deparaffinize kidney sections, followed by rehydration using graded 100%, 95%, and 70% ethanol baths. Next, antigen retrieval was performed using 10 mM EDTA pH 6.0. Endogenous peroxidase activity was blocked with 3% v/v H2O2. Non-specific antibody binding was blocked using a 5% non-fat milk in PBS. Rabbit polyclonal p57 antibody (Santa Cruz) was diluted 1:800 in 1% BSA in PBS, applied to the sections, and incubated overnight at 4°C. Rabbit-on-rodent HRP polymer (Biocare Medical, Concord, CA) was applied and incubated at room temperature for 45 min. Diaminobenzidine (DAB) (Sigma-Aldrich, St. Louis, MO) with 0.05% NiCl (Sigma-Aldrich) was used to detect staining. Slides used for podocyte density were not counterstained in order to improve quantitation sensitivity. For representative images, counterstaining was performed with periodic acid–Schiff. Sections were placed in 0.5% periodic acid (Sigma-Aldrich), washed in ddH2O, incubated for 10 min with Schiff’s Reagent (Sigma-Aldrich), washed in 0.5% sodium metabisulfate (Sigma-Aldrich) and incubated with hematoxylin (Sigma-Aldrich). Tissue was dehydrated in 95% and 100% ethanol baths, followed by Histoclear and Histomount (National Diagnostics). Podocyte density was quantitated according to the correction factor method from single histological sections, as previous reported (Venkatareddy et al., 2014). An average of 119 (±14.1) glomeruli for ICE mice and 139 (±16.1) glomeruli for Cre mice were quantified.
Glomerular Injury
Organized matrix accumulation was detected on paraffin-embedded tissue by Jones’ basement membrane stain (Silver Stain) performed by the University of Washington Pathology Research Services Laboratory following standard protocols (Luna, 1968). Silver stained slides were quantitated according to the criteria presented in figure 6G. An average of 156 (±10.8) glomeruli for ICE mice and 187 (±6.36) glomeruli for Cre mice were quantified.
Parietal epithelial cell to mesenchymal transition
Parietal epithelial cells (PECs) were stained for alpha-smooth muscle actin (α-SMA) in order to determine epithelial-mesenchymal transition (EMT) as described above. Non-specific antibody binding was blocked using Background buster (Accurate Chemical & Scientific Corporation, Westbury, NY). Rabbit polyclonal α-SMA antibody (Abcam) was diluted 1:400 in 1% BSA in PBS, applied to the sections, and incubated overnight at 4°C. Detection was performed as described above. Quantification was performed by counting the number of glomeruli with α-SMA staining in PECs as previously described (Schneider et al., 2017). An average of 110 (±6.22) glomeruli for ICE mice and 122 (±19.3) glomeruli for Cre mice were quantified.
5-Ethynyl-2’-deoxyuridine (EdU) staining
% EdU-positive cells was measured using the Click-iT® EdU Flow Cytometry Assay Kits (Invitrogen). Briefly,10 μM EdU was added to the culture medium and incubated for 1h. Cells were trypsinized, washed, fixed and permeabilized for Click-iT reaction. EdU+ cells were analyzed using the BD LSR II flow cytometer.
Microscopy and imaging for kidney
Imaging and quantification were performed on a Leica DMI400B microscope and an EVOS FL Cell Imaging System. ImageJ 1.51 (NIH) was used to measure podocyte density.
Immunocytochemistry
Cells were washed with sterile PBS and fixed with 4% paraformaldehyde. Fixed cells were permeabilized with 0.5% Triton X-100 in PBS, then blocked with 2% PBA (PBS containing 2% bovine serum albumin) overnight. Primary antibodies were incubated in 2% PBA at RT for 1h and cells were washed with PBS 3 times. The secondary antibodies (Alexa Fluor 488 Goat Anti-Mouse IgG or Alexa Fluor 568 Goat Anti-Rabbit IgG) were incubated in 2% PBA at RT for 30 min. After PBS washes, nuclei were stained with antifade mounting medium containing DAPI (Vector Laboratories). Immunofluorescence was examined using Olympus Fluoview FV1000 and FV3000 confocal microscope.
Senescence-associated β-galactosidase (SA-β-Gal) assay
Senescence-associated β-galactosidase assay were performed using Senescence β-galactosidase staining kit (Cell Signaling Technology). At 96-hour post-treatment when ICE cells were not senescent, the standard medium was switched to low serum (1% FBS) medium to preserve senescent cells that appeared later. Cells were fixed at RT for 10 min, washed with PBS and stained in β-galactosidase staining solution containing X-gal (pH 6) at 37ºC for overnight in dry incubator. Stained cells were monitored under bright field microscopy.
Small molecule-driven neuronal reprogramming
Neuronal reprogramming was performed as described in (Li et al., 2015). MEFs were transferred to Matrigel-coated plates. When MEFs were confluent, MEF growth medium was switched to Neurobasal Medium containing 1% N2 and 2% B27 supplements, 1% GlutaMAX (Life technologies), 1% penicillin/streptomycin, 100 ng/ml bFGF (STEM CELL), 20 μM ISX9, 100 μM Forskolin, 0.5 μM I-BET151 (CAYMAN CHEM), 20 μM CHIR99021 (LC Laboratories), 2 μM Fasudil and 1 μM SB203580 (Selleckchem). After 2 d, cells were maintained without Fasudil and SB203580. qPCR to detect neuronal gene activation was performed at day 2 after switching to Neurobasal medium, and TUJ1 immunocytochemistry was performed at day 13.
Quantitative real-time PCR for transcription of repetitive elements
Total RNA was isolated from 30–50 mg of tissue using Trizol reagent (ThermoFisher) according to the manufacturer’s instructions. Prior to the synthesis of cDNA, total RNA was digested with 27.2 Kunitz units of RNase-free DNase (Qiagen) for 45 min at room temperature and further cleaned up on RNeasy columns (Qiagen) (De Cecco et al., 2013). The effectiveness of the digestion was assessed using controls that omitted reverse transcriptase (RT). Digestion with DNase was repeated until the control lacking RT was negative for γ-satellite sequences. RNA integrity was determined using an Agilent Bioanalyzer 2100 and an RNA-nano chip. Total RNA (1 μg) of was transcribed into cDNA in 50 μl reactions using the TaqMan Gold RT-PCR kit (Applied Biosystems) and random hexamers, according to the manufacturer’s protocol. This reaction (1.0 μl) was used in subsequent qPCR reactions, performed using the SYBR Green system (Applied Biosystems) on the ViiA 7 Real Time System (Applied Biosystems), according to the manufacturer’s specifications. Primers were used at a final concentration of 300 nM. Tissue from 6 individual animals was analyzed in triplicate. Statistical analysis was determined using Student’s t-test and SigmaPlot 12.5 (Systat Software).
Design of PCR primers for repetitive elements
All primers used in this study are listed in Table S1. For expression analysis of LINE-1, MusD and pericentromeric γ-satellite sequences (MSAT) we used primers described by Changolkar et al. (Changolkar et al., 2008). Primers for the SINE elements B1 and B2 were designed using the consensus sequence from Repbase (Genetic Information Research Institute, www.girinst.org/repbase/index.html) and Primer-Blast software (www.ncbi.nlm.nih.gov/tools/primer-blast/). Primers against GAPDH and β-actin, used as normalization controls, were designed with Primer-Blast using NCBI reference sequences NC_000072.6 and NM_007393.3, respectively. Primer sequences were analyzed using the UCSC genome browser in silico PCR tool (genome.ucsc.edu/cgi-bin/hgPcr) to determine the number of genomic elements that contribute to the amplification products (De Cecco et al., 2013). All primers were tested with serial dilutions of cDNA to ensure they amplified their target sequences quantitatively.
Mutation frequency of 28S rDNA
Genomic DNA was isolated using E.Z.N.A. Tissue DNA Kit (Omega Bio-tek). 28S rDNA region containing canonical I-PpoI sequence was amplified by PCR using primers that bind to flanking regions of the I-PpoI sequence. Sanger sequencing data of rDNA PCR products were analyzed with the Inference of CRISPR Edits software (Conant et al., 2022) to calculate mutation frequency.
Production and transduction of adeno-associated viruses
Adeno-associated viruses for OSK expression were produced by the Boston Children’s Hospital Viral Core. Fibroblasts were isolated from 1-month post-treated Cre or ICE mice and maintained in DMEM with 15% Tet System Approved FBS (Takara) and 1% Pen/Strep. 24 hours before AAV transduction, 96-hour post-treated MEFs or 1-month post-treated fibroblasts were plated in DMEM containing 1% Tet System Approved FBS. AAV-DJ-TRE-OSK with AAV-CMV-tTA or rtTA at 104 genome copies per cell was transduced, and 2 μg/ml doxycycline was treated from day 7 to day 12 when Tet-On system (rtTA) was used. For AAV transduction in Cre and ICE mice, AAV-MYO3-TRE-OSK with AAV-MYO3-CMV-tTA at 5X1011 genome copies in 100 μl PBS was retro-orbitally injected (El Andari et al., 2022). After 3 weeks, tissues from mice transduced with AAV-MYO3-TRE-GFP and AAV-MYO3-CMV-tTA were imaged in Bio-Rad ChemiDoc MP Imaging System. OSK injected mice were sacrificed after 5 weeks for immunostaining. Intravitreal injection of AAV2 to deliver OSK to RGC was described in detail previously (Lu et al., 2020).
RGC sorting and RNA-seq
Thy1.2+ and Calcein Blue+ RGS were sorted using a BD FACS Aria Cell Sorter with a 130-μm nozzle and sent to Genewiz for ultra-low input RNA sequencing as previously published (Lu et al., 2020).
ChIP-sequencing
MEF ChIP was done following the protocol described in (Yang et al., 2011) with minor modifications. 1/4 number of Drosophila S2R+ cells relative to mouse cells were added as a spiked-in control and combined cells were treated as a single sample during the rest of the procedures. Cells were cross-linked with 1% formaldehyde at RT for 10 min and glycine was added to final concentration 0.125 M for 5 min to quench crosslinking. Fixed cells were washed with PBS and nuclei were isolated using Lysis buffer A (10 mM Tris-HCl pH 7.5, 10 mM KCl, 5 mM MgCl2, 0.5% NP40, protease inhibitor cocktail). Nuclei were resuspended in SDS lysis buffer (50 mM Tris-HCl pH 7.9, 10 mM EDTA, 0.5% SDS, protease inhibitor cocktail). Chromatin was sheared using Covaris E210 Ultrasonicator (duty cycle:5%, intensity:4, cycle/burst:200, time:15–20 min) to generate fragmented chromatin ranging between 200 and 1,000 bp. After centrifugation, sonicated chromatin solution was 5 fold-diluted with ChIP dilution buffer (12.5 mM Tris-HCl pH 7.9, 187.5 mM NaCl, 1.25% Triton X-100, protease inhibitor cocktail). Antibodies and magnetic beads were added to diluted chromatin solutions and immunoprecipitation were performed at 4°C overnight with rotation. Immunocomplexes were washed with Low salt wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl), High salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% NP40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1), and TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Immunocomplexes were eluted in elution buffer (1% SDS, 0.1 M NaHCO3) at RT for 30min with rotation, and RNaseA (final concentration of 0.5 mg/ml at 37°C for 30 min) and proteinase K (final concentration of 0.5 mg/ml at 55°C for 1 h) were treated. Samples were de-crosslinked at 65°C overnight, and ChIP DNA was purified using a ChIP DNA clean and concentrator kit (Zymo).
Muscle ChIP was performed as described previously (Gao et al., 2010). Tissue was chopped into small pieces on the ice, and fix solution (50 mM HEPES pH 7.5, 1 mM EDTA pH 8.0, 0.5 mM EGTA, 100 mM NaCl) and formaldehyde (final 1%) were added to cross-link the tissue sample. After incubation for 15 min at room temperature, glycine was added as 0.125 M final concentration to stop the reaction. The sample was washed using cold PBS three times followed by homogenizing it in cell lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP40). Cell lysate was centrifuged at 12,000 rpm for 5 min and suspended in nuclear lysis buffer (1% SDS, 10 mM EDTA pH 8.0, 50 mM Tris-HCl pH 8.0). Sonication was performed using Covaris E220 Ultrasonicator (duty cycle:5%, intensity:4, cycle/burst:200, time:120 sec). The resulting chromatin was diluted by 10 fold using dilution buffer (1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl pH 8.0). To reduce non-specific binging to beads, the diluted chromatin was mixed with Dynabeads protein A/G for 1 h at 4 °C and the beads were removed prior to incubating chromatin with 2 ug of the appropriate antibodies with Dynabeads protein A/G. After 4 h incubation at 4 °C, beads were washed three times with wash buffer and once with final wash buffer, LiCl Buffer and TE buffer each. Wash buffer contains 1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 0.1% SDS and final wash buffer contains 500 mM NaCl instead of 150 mM NaCl. The composition of LiCl buffer is 0.25 M LiCl, 1% NP40, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris-HCl. ChIP DNA was eluted by incubating at 65°C overnight in elution buffer containing 0.25% SDS, 1 mM EDTA, 10 mM Tris-HCl pH7.5. After treatment of proteinase K and RNase A, DNA was purified using ethanol precipitation and MinElute kit (QIAGEN).
Purified DNA (1–5 ng) was used for ChIP-seq library construction with NEBNext ChIP-Seq Library Prep Master Mix Set. ChIP DNA was end-repaired and added with dA tail using Klenow fragment. Sequencing adaptors were ligated to the dA-tailed libraries, and the libraries raging around 270 bp were selected using AMPure XP beads (Beckman Coulter). Size-selected libraries were enriched by PCR with index primers. The quantity and quality of libraries were respectively monitored by library quantification kit (Kapa Biosystems) and Bioanalyzer (Agilent) for 75bp, paired-end Illumina NextSeq.
Hi-C
Cells were fixed in PBS containing 1% formaldehyde at RT for 15 min, then quenched by adding glycine at final concentration 0.125 M on ice for 10 min. Dovetail Hi-C libraries were prepared in a similar manner as described previously (Lieberman-Aiden et al., 2009). Briefly, for each library, chromatin was fixed in place with formaldehyde in the nucleus and then extracted. Fixed chromatin was digested with DpnII, the 5’ overhangs filled in with biotinylated nucleotides, and then free blunt ends were ligated. After ligation, crosslinks were reversed and the DNA purified from protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. The DNA was then sheared to ~350 bp mean fragment size and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library.
HiChIP
HiChIP assay was performed on 5×106 post-treated Cre and ICE cells. Frozen cells were resuspended in 1X PBS and crosslinked with 3mM DSG and 1% formaldehyde. Washed cells were digested with 0.5 ul MNase in 100 ul of Nuclease digest buffer with MgCl2. Cells were lysed with 1X RIPA and clarified lysate (approximately 1400 ng) was used for ChIP. The Protein A/G bead pulldown, proximity ligation, and libraries were prepared as described in the Dovetail protocol (Dovetail™ HiChIP MNase Kit). Libraries were sequenced on an Illumina HiSeq 4000. Raw fastq files were aligned using BWA mem with the −5SP HiC options with an index containing only the 21 main chromosome from the mouse genome release mm10 (available from the UCSC genome). The aligned paired reads were annotated with pairtools parse (https://github.com/open2c/pairtools) with the following options --min-mapq 40 --walks-policy 5unique --max-inter-align-gap 30 and the --chroms-path file corresponding to the size of the chromosome used for the alignment index. The paired reads were further processed to remove duplicated reads, sorted with unaligned reads removed with the pairtools sort and the pairtools dedup tools with the basic option to produce an alignment file in the bam format as well as the location of the valid pair. The valid pairs were finally converted to the .cool and .mcool format using the cooler cload and cooler zoomify tools (Abdennur and Mirny, 2020) and to the .hic format using the juicer tool (Durand et al., 2016).
Whole-genome sequencing
Genomic DNA was isolated from snap frozen cells or tissues using DNeasy Blood & Tissue Kit. The genomic DNA was fragmented by an ultrasonicator Covaris at 500 bp peak and TruSeq DNA Library Preparation Kit added DNA adaptors to double strand DNA by following the manufacturer’s instructions of Illumina. Deep whole genome sequencing on an Illumina HiSeq X10 platform were performed at BGI.
RRBS and epigenetic (DNA methylation) clock for fibroblasts
RRBS libraries were prepared in two batches. DNA in the first batch was isolated using Quick-DNA Universal kit (Zymo) and in the second batch using E.Z.N.A. Tissue DNA Kit (Omega Bio-tek). 100 μl of 10 mM Tris-HCl buffer was used to elute the samples. Incubation with 2 μl of RNaseA (Life Technologies) was performed for each sample and followed by a purification using Genomic DNA Clean & Concentrator-10 (Zymo). DNA was eluted in 25 μl of TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0). 100ng of each sample, estimated using using a Qubit 2.0 (Life Technologies), was used to prepare RRBS libraries following the previously reported protocol (Petkovich et al., 2017). Libraries included 6–10 samples. The first batch of samples was sequenced on the Illumina HiSeq 2500 platform using 75 bp paired-end sequencing with more than 14 million reads per sample. The second batch was sequenced on the Illumina HiSeq X Ten using 150 paired-end sequencing with more than 32M reads per sample. To compensate for the low complexity of RRBS libraries 10–20% of phiX was spiked in. Raw reads were filtered and mapped as previously described (Meer et al., 2018). More than 3.6 M CpG sites were covered in each sample and 2.7M were covered in all samples. Data were normalized using ComBat from the SVA package in R. Only CpG sites covered in all samples were considered for DNA methylation clocks application. This resulted in 89 out of 90 sites being covered for the blood DNA methylation clock. Increase of the threshold for the CpG sites coverage decreased the number of the clock sites included in the analysis.
For AAV-transduced cells, RRBS was performed by Zymo Research and data were mapped to the mm10 genome with Bismark (Krueger and Andrews, 2011). To construct the methylation matrix, bismark.cov files were produced using the bismark methylation_extractor tool with --no_overlap. For Thompson multi-tissue clock analysis, CpG locations, coefficients, and the formula for calculating age were taken from supplemental information reported in the manuscript (Thompson et al., 2018). Using R (version 4.0.2) bismark.cov files were read in from each file and organized into a methylation matrix by selecting only the CpGs that were present in the clock to be analyzed. The sum of weighted methylation values for each sample for each clock was plugged into the reported formulas for age prediction.
Epigenetic (DNA methylation) clock for blood and muscle
Tissue samples were immediately preserved in DNA/RNA Shield™ (Zymo Research; Cat. No. R1100–50) and genomic DNA were purified using Quick-DNA Plus Kit (Zymo Research; Cat. No. D4068) according to manufacturer’s instructions. Sample library preparation and data analyses were performed by Zymo Research, CA. Briefly, genomic DNA (200 ng) was bisulfite-converted using EZ DNA Methylation-Lightning™ Kit (Zymo Research; Cat. No. D5030). Bisulfite-converted DNA libraries for targeted bisulfite sequencing platform, called SWARM® (Simplified Whole-panel Amplification Reaction Method) were prepared according the to the manufacturer’s instructions, then sequenced on a HiSeq 1500 sequencer at >1,000X coverage. Sequence reads were identified using Illumina basecalling software and aligned to the reference genome using Bismark (Krueger and Andrews, 2011), an aligner optimized for bisulfite sequence data and methylation calling . The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. DNA methylation levels of >500 age-related CpG loci were used for age prediction using epigenetic age algorithms.
In the training of the mouse muscle and blood clocks, first, chronological age was transformed using the following function F. If age <= adult age, F(age) = log (age + 1) – log (adult age + 1), where age is the chronological age; the parameter adult age for mouse was set as 12 weeks, which was (12 * 12)/52 = 2.8 months. If age > adult age, F(age) = (age – adult age) / (adult age + 1). Then the transformed age was regressed to the methylation of CpG sites using the elastic net algorithm. To predict methylation age of a sample, its weighted sum of methylation of CpG sites times their coefficients would be converted/inversed into a predicted age. The age prediction formula inverse.F was the following: If x < 0, predicted age = (1 + adult age) * exp(x) – 1, where x is the weighted methylation sum. The parameter adult age for mouse was set as 12 weeks, which was (12 * 12) / 52 = 2.8 months. If x >= 0, predicted age = (1 + adult age) * x + adult age.
Histone mass spectrometry
Histone extraction and qMS were performed as previously described (Luense et al., 2016). Acid-extracted histones were propionylated, trypsin-digested and stage-tip desalted with C18 mini-disks. Desalted histone peptides were separated by reversed-phase HPLC on a Thermo Scientific™ EASY-nLC 1000 system. Histone peptide quantified as described (Luense et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-seq aligning reads
The techniques described for processing the ChIP-seq and ATAC-seq reads are based on ENCODE/Roadmap guidelines with a few modifications (Gjoneska et al., 2015; Landt et al., 2012; Roadmap Epigenomics et al., 2015). The reads were aligned to the mm10 (GRCm38) genome (Cunningham et al., 2019) using Bowtie 2 (Langmead and Salzberg, 2012). The genome fasta files were first indexed and then aligned using the command: bowtie2 -x /directory/with/reference/genome/rootfilename --fast -U /directoryTree/fastq/SAMPLE.fastq -S /directoryTree/fastq/SAMPLE.sam, where SAMPLE was replaced with a unique sample identifier. Following alignment to the genome, the reads were converted from SAM to BAM format (Li et al., 2009). Low quality reads and reads (q < 20) that did not map to the genome were removed. For visualization and peak calling, the bamToBed command line tool was used to convert the BAM files to a modified BED format, called TAGALIGN, which preserved only the read coordinates (Landt et al., 2012; Quinlan and Hall, 2010).
ChIP-seq spike-in controls
Equal amounts of D. Melanogaster DNA were spiked-into ChIP-seq samples. In addition to aligning to the mouse genome, we aligned reads to the D. Melanogaster dm6 genome (Cunningham et al., 2019). To provide a sense of total ChIP-seq signal strength, the proportion of reads aligning to the dm6 genome were compared to the proportion aligning to the mouse genome (Orlando et al., 2014). To compare Cre and ICE mice, a student’s t-test was used on those proportions.
ChIP-seq visualizing read coverage
For visualizing individual samples, the genomecov tool within BedTools was used to convert from the BED format to a BEDGRAPH format (Quinlan and Hall, 2010). Finally, the BEDGRAPH file was converted to the more efficient BIGWIG format using the UCSC command line tool bedGraphToBigWig (Kent et al., 2010; Speir et al., 2016). The information was uploaded to the NCBI sequence read archive and SRA files representing the raw reads, the BAM file representing the aligned reads, and the BigWig files of read coverage across the genome.
ChIP-seq visualizing signal relative to background
Macs2 bdgcmp command was used to calculate the signal to noise ratio for every position in the genome for each combination of histone modification and experimental condition (Cre and ICE) (Feng et al., 2012). The BEDGRAPH file was converted to the more efficient BIGWIG format using the UCSC command line tool bedGraphToBigWig (Speir et al., 2016).
ChIP-seq peak Calling
For each epigenetic measurement (H3K27ac, H3K56ac, H3K27me3, ATAC-seq) and input samples, the TAGALIGN files were merged using the unix command zcat and sorted using the unix sort command according to the chromosome using the start position. For each histone modification, MACS2 was used to call the peaks relative to the input control: macs2 callpeak -t H3K27ac.tagAlign.gz -f BED -c input.tagAlign.gz -n H3K27ac_signal -g mm -p 1e-2 --nomodel --extsize 73 -B –SPMR (Feng et al., 2012). For ATAC-Seq, no input control was used. We removed peaks with significance (signal relative to noise) of p > 10−5. Peaks that fell into the ENCODE blacklist regions were removed (Landt et al., 2012). The output of the program is a BED file with peak coordinates for the mm10 version of the mouse genome.
ChIP-seq peak annotation
Peaks are annotated based on their mapping to the nearest transcription start site, which was performed using BEDTools closestBed command (Quinlan and Hall, 2010) based on ENSEMBL gene annotations, GRCm38 version 79 (Cunningham et al., 2019).
ChIP-seq counting reads across peaks
For each histone modification and ATAC-seq sample, the reads from each experiment are counted in the called peaks using featureCounts in the subread package (Liao et al., 2013). To perform the counting the peak BED file were converted to SAF format.
ChIP-seq differential peaks between Cre and ICE
The negative binomial model in the DESeq2 R package was used to identify differential peaks between the CRE and ICE mice (Love et al., 2014). For MEF experiments, we used a stringent threshold of adjusted p < 0.01. For muscle ChIP-Seq experiments, very few peaks attained significance levels at that cutoff. Therefore, we restricted our analysis to looking at the group of peaks enriched at p < 0.01. The varianceStabilizingTransformation function in the DESeq2 package was used to normalize the read counts per peak. Sex chromosomes were excluded from analyses due to the inconsistency of the sexes of the MEFs.
ChIP-seq metaplots and heatmaps
Metaplots and heatmaps were produced using deepTools version 3.0.1 (Ramirez et al., 2016). Intermediate matrix files were generated by applying computeMatrix (scale-regions mode) to BIGWIG files over genomic loci in BED format. plotProfile and plotHeatmap functions were applied to the matrix files to generate output data used to graph each metaplot and heatmap.
Gene ontology of ChIP-seq differential peaks
Gene ontology analysis for ChIP-seq were performed using Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al., 2010). Genomic coordinates of differential ChIP-seq regions and all ChIP-seq peaks were used as test regions and background regions, respectively. GO biological processes were ranked by HyperFdrQ and only GO terms made up of at least 5 genes were included. ChIP-seq data were also analyzed using ChIP-Enrich (Welch et al., 2014). GO terms with at least 5 genes were ranked by FDR.
Hi-C analysis
Paired-end reads were aligned with bwa mem (v0.7.17) (Li and Durbin, 2009) using the options -S -P. Interaction were parsed and deduplicated with pairsamtools (v0.0.1). Pairwise interaction frequencies were binned in 50-kb nonoverlapping windows and intra-chromosome interaction frequencies were normalized by dividing each interaction by the average number of interactions observed with that distance. Normalized interaction matrices were binned with smoothMat (Yang et al., 2017). Matrix pearson’s correlations were calculated in R v 3.6.1 (Bunn, 2008, 2010) and used to perform a principal component analysis. The sign of the first eigenvector for each chromosome was adjusted to correlate with GC content, and were smoothed with loess smoothing using 1 megabase windows.
Whole-genome sequencing analysis
Whole-Genome raw sequencing reads from paired-end library was quality‐controlled with FastQC and subsequently mapped to the reference genome GRCm38/mm10 (mm10) using the Burrows–Wheeler Alignment (BWA-MEM, version 0.7.17) (Li et al., 2009). A paired-end mapping strategy with default parameters was utilized. After mapping, the reads were sorted and converted into binary alignment format (BAM) via Sequence Alignment/Map tools (SAMtools, version 1.9). The best practices recommended by the Broad Institute for variant calling were then followed (Van der Auwera et al., 2013). The sorted binary alignments underwent post-processing to remove duplicates via Picard’s MarkDuplicates (v.2.01; http://broadinstitute.github.io/picard) before germline variants were identified using Genome Analysis Toolkit (GATK; v. 3.7) HaplotypeCaller (McKenna et al., 2010).
Muscle RNA-seq analysis
Paired-end reads from gastrocnemius muscle RNA-Seq were mapped to the UCSC mm10 genome build using HISAT2 version 2.1.0 (Kim et al., 2015). The featureCounts function from the Rsubread package (Rsubread 1.32.2) was used to collect read counts for genes. DESeq2 (DESeq2 1.22.2) was applied for differential expression analysis to all genes with rowSums >= 10.
To compare gene expression in gastrocnemius muscles of ICE, Cre, and WT, a table of normalized read counts was exported from a combined DESeq dataset with all replicates and conditions. The 200 genes with the smallest adjusted p-value for differential expression between Cre and ICE were selected and ordered by the log2-fold-change difference between Cre and Ice. The heatmap.2 (gplots 3.0.1) R function was used to produce a plot of Z-score values for each gene.
Supplementary Material
Figure S1. Effect of the ICE system on the cell cycle, senescence and mutations, Related to Figure 1
(A) Breeding scheme to generate ICE mice and controls (WT, Cre and STOP-I-PpoI).
(B and C) DNA damage response induced by I-PpoI (4-OHT, 0.1, 0.5, 1 μM) vs. other damaging agents, etoposide (ETS, 1, 10, 25 μM) and phleomycin (Phleo, 1, 25, 50 μg/ml). Scale bar, 10 μM. One-way ANOVA-Bonferroni.
(D) Western blot of DNA damage response proteins.
(E) Cell cycle profile in 96-hour post-treated cells.
(F) Percentage of SA-β-Gal positive (blue) cells during and after TAM treatment compared to replicative senescent cells. p, passage. One-way ANOVA-Bonferroni.
(G) Cell diameter after recovery from I-PpoI induction vs. irradiated (IR, senescent) cells. One-way ANOVA-Bonferroni.
(H and I) γH2AX and 53BP1 in post-treated ICE cells with and without exposure to the DNA damaging agents (ETS, etoposide; CPT, camptothecin; H2O2, hydrogen peroxide). Scale bar, 10 μm. Two-tailed Student’s t test.
(J and K) Lamin B1 mRNA and protein levels in 144-hour post-treated ICE cells. Lamin A/C and actin are loading controls known not to change during senescence.
(L-N) Percent non-mutated I-PpoI canonical (Teme56), non-canonical recognition and random sequences in 96-hour post-treated ICE cells assessed by deep sequencing (>50x).
Data are mean (n≥3) ± SD. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001; **** p < 0.0001.
Figure S2. No change in 28S rDNA in post-treated ICE cells and muscle, Related to Figure 1 and 2
(A) Southern blot of 28S rDNA in 4-OHT treated and post-treated Cre and ICE cells.
(B) In vitro cutting of I-PpoI targets PCR-amplified from genomic DNA from 96-hour post-treated Cre and ICE cells.
(C) Surveyor nuclease assay of I-PpoI targets in 96-hour post-treated cells. DNA with a point mutation (PM) in the I-PpoI site was the positive control.
(D) 28S rRNA level in 96-hour post-treated ICE cells. Two-tailed Student’s t test.
(E) Bioanalyzer tracks of 28S and 18S rRNA in 96-hour post-treated cells.
(F) 28S:18S rRNA ratio. Two-tailed Student’s t test.
(G) Scheme for ligation-mediated PCR to detect residual rDNA breaks.
(H-J) Ligation-mediated PCR after I-PpoI digestion In vitro (H), in Cre and ICE cells (I) or in 10-month post-treated muscle (J).
(K and L) Protein translation in 96-hour post-treated cells assessed by metabolic 35S-labelling. Two-tailed Student’s t test.
(M and N) Protein translation in 10-month post-treated brain (N) and muscle (O) tissue, assessed by metabolic 35S-labelling. Two-tailed Student’s t test.
(O) 28S rDNA copy number in 10-month post-treated muscle assessed by monochrome multiplex quantitative PCR (MMQPCR). One-way ANOVA-Bonferroni (left) or two-way ANOVA-Bonferroni (right).
(P) 28S rRNA levels in 10-month post-treated muscle. One-way ANOVA-Bonferroni (left) or two-way ANOVA-Bonferroni (right).
Data are mean (n≥3) ± SD or ± SEM (M-P). n.s.: p > 0.05; **** p < 0.0001.
Figure S3. Validation of the ICE mouse, Related to Figure 2
(A) Removal of the transcriptional STOP cassette in major tissues. One-way ANOVA-Bonferroni.
(B) Western blot of tissues probed with anti-HA to detect I-PpoI expression and γH2AX after a 4-week TAM treatment.
(C and D) Hippocampal sections immunostained for GFP (as a proxy for IRES-linked I-PpoI expression) and γH2AX. Scale bar, 200 μm (100X, C) and 50 μm (400X, D).
(E) Western blot of tissues probed with anti-HA to detect I-PpoI expression after 3-weeks of TAM and 1-month post-treatment.
(F) Immunoprecipitation and quantification of a I-PpoI cut site (28S rDNA) in skeletal muscle, liver and kidney during and after tamoxifen treatment (0-,1- and 10-month post-treatment). Two-tailed Student’s t test. Data are mean (n=3–6) ± SEM. * p < 0.05.
(G-I) Percent non-mutated I-PpoI canonical, non-canonical recognition and random sequences in 1-month post-treated ICE muscle assessed by deep sequencing (>50x).
Figure S4. ICE mice phenocopy normal aging, Related to Figure 2 and 3
(A) Food consumption of post-treated mice. Two-tailed Student’s t test.
(B) Body weights of female mice post-treatment. Two-way ANOVA-Bonferroni.
(C) Representative images of 10-month post-treated mice from Dual Energy X-ray Absorptiometry (DEXA).
(D) Body mass of 10-month post-treated mice. Two-tailed Student’s t test.
(E) Subcutaneous fat thickness of 10-month post-treated mice. Two-tailed Student’s t test.
(F) Respiratory Exchange Rate (RER) of female mice. Repeated measures two-way ANOVA.
(G and H) Blood glucose levels of 10-month post-treated mice in the fed or fasted state. Two-tailed Student’s t test.
(I) IGF-1 levels in 10-month post-treated mice. Two-way ANOVA-Bonferroni.
(J and K) H&E staining of subcutaneous fat layers and subepidermal thickness of back skin from old WT, 10-month post-treated Cre and ICE mice. Scale bar, 500 μm. One-way ANOVA-Bonferroni (left) or two-tailed Student’s t test (right).
(L) KIT (CD117), KRT55 and DAPI staining of back skin.
(M) Percent hair follicle bulges without KIT staining. Two-tailed Student’s t test.
(N-P) Gait analysis of 10-month post-treated mice. Two-tailed Student’s t test. Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001.
Figure S5. Progeroid features of ICE mouse muscle, Related to Figure 4
(A) Blood lactate build-up after exercise in WT, 10-month post-treated mice. Two-tailed Student’s t test.
(B) Grip strength measured as maximal “peak force”. Two-tailed Student’s t test.
(C) Mitochondrial DNA copy number. Two-tailed Student’s t test.
(D-F) Mitochondria number (D and E) and mitochondrial perimeter (D and F) of 10-month post-treated muscle determined with electron microscopy. Two-tailed Student’s t test. Scale, 500 nm.
(G and H) Cytochrome oxidase (COX) staining of 10-month post-treated gastrocnemius muscle. Scale bar, 100 μm. Two-tailed Student’s t test.
(I and J) Quantification of RNA from repetitive DNA elements in gastrocnemius muscle
(I) and liver (J) of 10-month post-treated mice. Two-tailed Student’s t test.
(K and L) Echocardiogram of 10-month post-treated mice. LVPWd, Left ventricular posterior wall thickness at end-diastole mm. LVIDd, Left ventricular internal diameter in diastole; LVPWd, Left ventricular posterior wall in diastole; LVEF, Left ventricular ejection fraction. Two-tailed Student’s t test.
(M) Scatter plot of genes altered (p < 0.01) in muscle from 10-month post-treated ICE mice and WT 24-month-old mice. Significantly changed genes (padj < 0.05) are blue, red or green.
(N) Fold change of altered genes (padj < 0.05, Cre vs ICE mice) in WT 24-month-old mice.
(O) Fold change of altered genes (padj < 0.05, 12mo. vs 24mo.) in 10-month post-treated ICE mice.
Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001, ****p< 0.0001.
Figure S6. Altered epigenetic landscapes in ICE cells and muscle, Related to Figure 4–6
(A–D) Muscle and blood training (A and C) and testing sets (B and D) of the clock CpG sites in WT C57BL/6J mice.
(E) Circos plot of genomic locations of I-PpoI cut sites (green), clock GpG sites in CpG islands (red), and all muscle clock sites (blue).
(F) Western blotting for histone modifications in 96-hour post-treated ICE cells. Histone H3 and macroH2A serve as loading and internal controls.
(G) Spike-in normalization of ChIP-seq data.
(H) ATAC signal in loci with altered H3K27ac peaks.
(I) Aggregation plots of H3K27ac signal in H3K27ac changed regions (padj < 0.01).
(J) Aggregation plots of H3K27ac signal in top 40% or bottom 40% quantile.
(K and L) qPCR analysis of Hoxa genes in post-treated cells cut with either I-PpoI (K) or I-SceI (L) homing endonucleases. Two-tailed Student’s t test.
(M) Comparison of H3K27ac decreased regions (p < 0.01) in muscle to epigenome roadmap data from different human tissue types.
(N) Super-enhancers (SEs) in different cell types that overlap with regions with increased H3K27ac signals in 10-month post-treated ICE muscle.
Data are mean (n=3) ± SD. *p < 0.05; **p < 0.01.
Figure S7. Epigenetic reprogramming using adeno-associated virus (AAV)-mediated gene transfer, Related to Figure 7
(A and B) Western blot of Oct4 and Klf4 in 293T cells after AAV-DJ transduction.
(C) Dose-dependent increase in mRNA levels of Oct4, Sox2 and Klf4 in mouse fibroblasts at day 4. Two-tailed Student’s t test.
(D) Immunocytochemistry of Oct4 and Klf4 in mouse fibroblasts at day 4. DNA stained with DAPI. Scale, 50 μm.
(E and F) mRNA levels of Oct4, Sox2 and Klf4 at day 6 of Dox treatment in fibroblasts
(E) or at day 5 of Dox treatment in young and old OSK transgenic fibroblasts (F). One-way ANOVA-Bonferroni.
(G) Epigenetic age of post-treated Cre and ICE MEFs.
(H) Experimental scheme for AAV-OSK injection to Cre and ICE mice.
(I) GFP expression in muscle and kidney at 3-week post-AAV injection.
(J and K) qPCR analysis of Oct4 gene in post-treated Cre and ICE kidney (J) and muscle (K) at 5-week post-AAV injection.
(L and M) Representative images and quantification of H3K36me2 in the ICE muscle at 5-week post-AAV injection. One-way ANOVA-Bonferroni.
(N) Timeline of phenotype assessments.
(O and P) Lens opacity in 10-month post-treated mice. Two-tailed Student’s t test.
Data are mean (n≥3) ± SD or ± SEM (P). *p < 0.05; **p < 0.01; ***p< 0.001; ****p< 0.0001.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3 | Abcam | Cat# ab1791 RRID: AB_302613 |
| Mouse monoclonal anti-H3 | Abcam | Cat# ab10799 RRID: AB_470239 |
| Rabbit polyclonal anti-H3K27ac | Active motif | Cat# 39134 RRID: AB_2722569 |
| Rabbit polyclonal anti-H3K56ac | Millipore | Cat# 07–677-I RRID: AB_390167 |
| Rabbit polyclonal anti-H3K27me3 | Millipore | Cat# 07–449 RRID: AB_310624 |
| Rabbit polyclonal anti-H4K20me3 | Abcam | Cat# ab9053 RRID: AB_306969 |
| Rabbit polyclonal anti-H3K36me2 | Abcam | Cat# ab9049 RRID: AB_1280939 |
| Rabbit polyclonal anti-H3K9me3 | Active Motif | Cat# 39161 RRID: AB_2532132 |
| Rabbit polyclonal anti-γH2AX | Abcam | Cat# ab2893 RRID: AB_303388 |
| Rabbit polyclonal anti-γH2AX | Cell Signaling Technology | Cat# 2577 RRID: AB_2118010 |
| Mouse monoclonal anti-γH2AX | Novus | Cat# NBP1–19255 RRID: AB_1642310 |
| Rabbit polyclonal anti-H2AX | Abcam | Cat# ab11175 RRID: AB_297814 |
| Rabbit polyclonal anti-53BP1 | Novus | Cat# NB100–304 RRID: AB_10003037 |
| Rabbit polyclonal anti-P53p | Cell Signaling Technology | Cat# 9284 RRID: AB_331464 |
| Mouse monoclonal anti-P53 | Cell Signaling Technology | Cat# 2524 RRID: AB_331743 |
| Mouse monoclonal anti-ATMp | Cell Signaling Technology | Cat# 4526 RRID: AB_2062663 |
| Rabbit polyclonal anti-p16 | Santa Cruz Biotechnology | Cat# sc-1207 RRID: AB_632106 |
| Rabbit polyclonal anti-PARP1 | Cell Signaling Technology | Cat# 9542 RRID: AB_2160739 |
| Rat monoclonal anti-CD31 | Abcam | Cat# ab56299 RRID: AB_940884 |
| Rabbit polyclonal anti-Laminin | Sigma-Aldrich | Cat# L9393 RRID: AB_477163 |
| Rabbit polyclonal anti-Iba1 | Wako | Cat# 01919741 RRID: 01919741 |
| Rabbit polyclonal anti-GFAP | Antibodies.com | Cat# A85419 RRID: AB_2752917 |
| Mouse monoclonal anti-Lamin A/C | BD Bioscience | Cat# 612163 RRID: AB_399534 |
| Rabbit polyclonal anti-Lamin B1 | Abcam | Cat# ab16048 RRID: ab16048 |
| Rabbit polyclonal anti-macroH2A1 | Active motif | Cat# 39593 RRID: AB_2793271 |
| Mouse monoclonal anti-GAPDH | Millipore | Cat# MAB374 RRID: AB_2107445 |
| Mouse monoclonal anti-TUJ1 | BioLegend | Cat# 801201 RRID: AB_2313773 |
| Rat monoclonal anti-HA-Peroxidase | Roche | Cat# 12013819001 RRID: AB_390917 |
| Mouse monoclonal anti-Oct3/4 | BD Biosciences | Cat# 611203 RRID: AB_398737 |
| Goat polyclonal anti-Klf4 | R&D systems | Cat# AF3158 RRID: AB_2130245 |
| Alexa Fluor® 488 Goat Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A-11029 RRID: AB_138404 |
| Alexa Fluor® 568 Goat Anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A-11036 RRID: AB_10563566 |
| Alexa Fluor® 568 Donkey Anti- Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A10037 RRID: AB_2534013 |
| Alexa Fluor® 488 Donkey Anti- Goat IgG (H+L) | Thermo Fisher Scientific | Cat# A32814 RRID: AB_2762838 |
| Anti-rabbit IgG | Millipore | Cat# 12–370 RRID: AB_145841 |
| Bacterial and virus strains | ||
| AAV-DJ-rtTA | Lu et al., 2020 | N/A |
| AAV-DJ-tTA | Lu et al., 2020 | N/A |
| AAV-DJ-OSK | Lu et al., 2020 | N/A |
| AAV-MYO3-tTA | This paper | N/A |
| AAV-MYO3-OSK | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| (Z)-4-Hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| Tamoxifen citrate salt | Sigma-Aldrich | Cat# T9262 |
| Triamcinolone acetonide | Sigma-Aldrich | Cat# T6501 |
| Etoposide | CALBIOCHEM | Cat# 341205 |
| Camptothecin | CALBIOCHEM | Cat# 208925 |
| Paraquat | SIGMA | Cat# 36541 |
| Hydrogen peroxide | SIGMA | Cat# 216763 |
| Phleomycin | InvivoGen | Cat# ant-ph-1 |
| dCTP [α−32P] | PerkinElmer | Cat# BLU513H500UC |
| L-[35S]-Methionine | PerkinElmer | Cat# NEG709A500UC |
| Recombinant I-PpoI | Promega | Cat# R7031 |
| Rodent Chow Diet | LabDiet | Cat# 5053 |
| LightCycler 480 SYBR Green I Master | Roche | Cat# 4707516001 |
| Dynabeads® Protein A for Immunoprecipitation | Life Technologies | Cat# 10001D |
| Dynabeads® Protein G for Immunoprecipitation | Life Technologies | Cat# 10003D |
| Dynabeads® M-280 Streptavidin | Life Technologies | Cat# 11205D |
| UltraPure™ Buffer-Saturated Phenol | Thermo Fisher | Cat# 15513039 |
| Phenylalanine, L-[2,3,4,5,6–3H] | PerkinElmer | Cat# NET1122001MC |
| Hematoxylin solution modified acc. to Gill II | Millipore | Cat# 1051750500 |
| Eosin Y-solution 0.5% alcoholic | Millipore | Cat# 1024390500 |
| Fluoroshield mounting medium with DAPI | Sigma-Aldrich | Cat# F6057 |
| bFGF | STEM CELL | Cat# 78003.1 |
| ISX9 | CAYMAN CHEM | Cat# 16165 |
| Forskolin | CAYMAN CHEM | Cat# 11018 |
| CHIR99021 | LC Laboratories | Cat# C-6556 |
| I-BET151 | CAYMAN CHEM | Cat# 11181 |
| Fasudil | Selleckchem | Cat# S1573 |
| SB203580 | Selleckchem | Cat# S1076 |
| Dynabeads® Protein A | Thermo Fisher | Cat# 10001D |
| Dynabeads® Protein G | Thermo Fisher | Cat# 10003D |
| AMPure® XP Beads | BECKMAN COULTER | Cat# A63881 |
| Propidium Iodide | Abcam | Cat# ab14083 |
| Premium Grade Fetal Bovine Serum | Seradigm | Cat# 1500–500 |
| Charcoal stripped FBS | VWR | Cat# 35–072-CV |
| Tet System Approved FBS | TAKARA | Cat# 631106 |
| N-2 Supplement | Thermo Fisher Scientific | Cat# 17502001 |
| B-27™ Supplement | Thermo Fisher Scientific | Cat# 17504044 |
| GlutaMAX™ Supplement | Thermo Fisher Scientific | Cat# 35050061 |
| Neurobasal™ Medium | Thermo Fisher Scientific | Cat# 21103049 |
| 0.1% Gelatin | Millipore | Cat# ES-006-B |
| Formaldehyde | CALBIOCHEM | Cat# 344198 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Adenosine-5’-triphosphate disodium salt hydrate | VWR | Cat# AAJ61125–09 |
| NEBNext® High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541S |
| TRIzol® Reagent | Thermo Fisher Scientific | Cat# 15596–026 |
| VECTASHIELD® PLUS Antifade Mounting Medium | Vector Laboratories | Cat# H-1900–10 |
| cOmplete™ EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693132001 |
| Phosphatase Inhibitor Cocktail 2 | SIGMA | Cat# P5726 |
| Phosphatase Inhibitor Cocktail 3 | SIGMA | Cat# P0044 |
| SuperScript® III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080093 |
| Critical commercial assays | ||
| E.Z.N.A.® Tissue DNA Kit | Omega Bio-tek | Cat# D3396 |
| E.Z.N.A.® Total RNA Kit I | Omega Bio-tek | Cat# R6834 |
| iScript cDNA synthesis kit | Bio-rad | Cat# 1708891 |
| NEBNext® ChIP-Seq Library Prep Master Mix | New England Biolabs | Cat# NEB E6240L |
| NEBNext® Multiplex Oligos for Illumina | New England Biolabs | Cat# NEB E7335S |
| RNeasy MinElute Cleanup Kit | QIAGEN | Cat# 74204 |
| NEXTflex™ Rapid RNA Sequencing Kit | Bioo Scientific | Cat# 5138–01 |
| NEXTflex™ RNA-Seq Barcodes – 12 | Bioo Scientific | Cat# 512912 |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121–1030 |
| Dovetail™ HiChIP MNase Kit | Dovetail Genomics | Cat# 21007 |
| QuantiFluor® dsDNA System | Promega | Cat# E2670 |
| QuantiFluor® RNA System | Promega | Cat# E3310 |
| Library Quantification Kits | Kapa Biosystems | Cat# KK4854 |
| ChIP DNA clean & concentrator | Zymo | Cat# D5201 |
| Surveyor® Mutation Detection Kits | Transgenomic | Cat# 706025 |
| ChIP DNA Clean & Concentrator™ | Zymo Research | Cat# D5201 |
| Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C-10425 |
| Senescence β-Galactosidase Staining | Cell Signaling Technology | Cat# 9860 |
| Deposited data | ||
| Next generation sequencing data (ChIP-seq, ATAC-seq, RNA-seq, WGS and Hi-C data) | BioProject ID: PRJNA554729, PRJNA655981 | N/A |
| Experimental models: Cell lines | ||
| WT mouse embryonic fibroblasts | This paper | N/A |
| I-PpoI mouse embryonic fibroblasts | This paper | N/A |
| Cre mouse embryonic fibroblasts | This paper | N/A |
| ICE mouse embryonic fibroblasts | This paper | N/A |
| Cre mouse adult fibroblasts | This paper | N/A |
| ICE mouse adult fibroblasts | This paper | N/A |
| I-SceI mouse embryonic fibroblasts | This paper | N/A |
| 3 mo. OSK-tg mouse adult fibroblasts | This paper | N/A |
| 15 mo. OSK-tg mouse adult fibroblasts | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 mouse | NIA (USA) | N/A |
| C57BL/6 HA-ERT2-I-PpoI mouse | This paper | N/A |
| C57BL/6 Cre-ERT2 mouse | Ruzankina et al., 2007 | N/A |
| I-SceI mouse | Chiarle et al., 2011 | N/A |
| Oligonucleotides | ||
| PCR primers | This paper | Table S1 |
| Recombinant DNA | ||
| pAAV-CMV-rtTA | Lu et al., 2020 | N/A |
| pAAV-CMV-tTA | Lu et al., 2020 | N/A |
| pAAV-TRE3G-OSK | Lu et al., 2020 | N/A |
| pAAVMYO3 | El Andari et al., 2022 | N/A |
| Software and algorithms | ||
| Cellprofiler | Broad Institute | http://cellprofiler.org |
| Ensembl blat | European Bioinformatics Institute | http://www.ensembl.org/Multi/Tools/Blast?db=core |
| GraphPad Prism | GraphPad Software | https://www.grphpad.com |
| Metacore | GeneGo | https://portal.genego.com |
| Galaxy | Penn State University | https://usegalaxy.org |
| GREAT | Stanford University | http://great.stanford.edu/public/html |
| Enrichr | Mount Sinai | http://amp.pharm.mssm.edu/Enrichr |
| ChIP-enrich | University of Michigan | http://chip-enrich.med.umich.edu/chipMain.jsp |
| Easeq | University of Copenhagen | https://easeq.net |
| Primer-BLAST | NIH | https://www.ncbi.nlm.nih.gov/tools/primer-blast |
Cellular responses to double-stranded DNA breaks erode the epigenetic landscape
This loss of epigenetic information accelerates the hallmarks of aging
These changes are reversible by epigenetic reprogramming
By manipulating the epigenome, aging can be driven forwards and backwards
Aging is marked by the changes in cellular identity and function over time. This process is found to be driven by changes in chromatin factor localization as well as epigenetic regulation during DNA repair. The expression of a subset of Yamanka factors OSK can reverse these changes and modulating aging.
ACKNOWLEDGMENTS
This paper represents the work of many people over a dozen years, some of whom we lost. We dedicate this paper to Michael S. Bonkowski, Norman S. Wolf, and our supporter, Paul F. Glenn. Thanks to all members of the Sinclair laboratory, past and present, and to our financial supporters from the NIA/NIH, The Glenn Foundation for Medical Research, The Dalio Foundation, Susan and Duane Hoff, Edward Schulak, AFAR, and The Aoki Foundation, with advice from Andrea Di Francesco, Phu Huynh, Kristal Kalafut, Erin Wade, Rafael de Cabo, Haeyoung Kim, Peter Adams, and Alex Banks. Thanks to Roberto Chiarle and Frederick Alt for the I-SceI mice. Supported by NIH/NIA (R01AG019719 and R37AG028730 to D.A.S), the Glenn Foundation for Medical Research (D.A.S. and A.J.W.), National Research Foundation of Korea (2012R1A6A3A03040476 to J.-H.Y.), HFSP (LT000680/2014-L to M.H.), JSPS KAKENHI (17K13228, 19K16619, 19H05269 to M.H.), the Uehara Memorial Foundation (to M.H.), NIH T32 (T32AG023480 to D.L.V.), NIA K99/00 (K99AG055683 to J.M.R.), NEI (RO1EY019703 to T.C.J.), NIH (5R01DK056799-10, 5R01DK056799-12, 1R01DK097598-01A1 to S.J.S.), Glenn/AFAR Grants for Junior Faculty (to A.R.P.) and St Vincent de Paul Foundation (to B.R.K.).
Footnotes
DECLARATION OF INTERESTS
D.A.S is a consultant, inventor, board member, and in some cases an investor in Life Biosciences (developing reprogramming medicines), MetroBiotech, InsideTracker, Zymo, EdenRoc Sciences/Cantata/Dovetail, Caudalie, Galilei, Immetas, Animal Biosciences, Tally Health, and more. See https://sinclair.hms.harvard.edu/david-sinclairs-affiliations. E.M.M., M.Bl, M.Bh are employees of Catata Bio/Dovetail. Y.C.C., W.G. and X.Y. are employees of Zymo Research. A.J.W. advises Kate Therapeutics and Frequency Therapeutics and is a co-founder, advisor, and equity holder of Elevian, which sponsors Wagers Lab research. L.S. was an employee of Vium. Y.L., and L.A.R. are equity owners of Life Biosciences. D.L.V. advises Liberty Biosecurity. Other authors declare no competing interests. A patent application was filed on the reprogramming methods.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdennur N., and Mirny LA. (2020). Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics (Oxford, England) 36, 311–316. 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackert-Bicknell CL, Anderson LC, Sheehan S, Hill WG, Chang B, Churchill GA, Chesler EJ, Korstanje R, and Peters LL (2015). Aging Research Using Mouse Models. Current protocols in mouse biology 5, 95–133. 10.1002/9780470942390.mo140195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science (New York, NY) 346, 89–93. 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs ML, Nagarajan R, Taylor-Jones JM, Nolen G, Macnicol M, and Peterson CA (2004). Alterations in the TGFbeta signaling pathway in myogenic progenitors with age. Aging cell 3, 353–361. 10.1111/j.1474-9728.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, and Brunet A. (2015). Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nature reviews Molecular cell biology 16, 593–610. 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, and Brunet A. (2019). Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome research 29, 697–709. 10.1101/gr.240093.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ Jr., and Kastan MB (2007). Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nature cell biology 9, 683–690. 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- Birkisdottir MB, Jaarsma D, Brandt RMC, Barnhoorn S, van Vliet N, Imholz S, van Oostrom CT, Nagarajah B, Portilla Fernandez E, Roks AJM, et al. (2021). Unlike dietary restriction, rapamycin fails to extend lifespan and reduce transcription stress in progeroid DNA repair-deficient mice. Aging cell 20, e13302. 10.1111/acel.13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ (2018). The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell reports 22, 269–285. 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, and Rando TA (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science (New York, NY) 317, 807–810. 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Brown MF, and Stuart JA (2007). Correlation of mitochondrial superoxide dismutase and DNA polymerase beta in mammalian dermal fibroblasts with species maximal lifespan. Mechanisms of ageing and development 128, 696–705. 10.1016/j.mad.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, and Kim SK (2008). An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell 134, 291–303. 10.1016/j.cell.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn AG (2008). A dendrochronology program library in R (dplR). Dendrochronologia 26, 115–124. 10.1016/j.dendro.2008.01.002. [DOI] [Google Scholar]
- Bunn AG (2010). Statistical and visual crossdating in R using the dplR library. Dendrochronologia 28, 251–258. 10.1016/j.dendro.2009.12.001. [DOI] [Google Scholar]
- Burgstaller JP, and Brem G. (2017). Aging of Cloned Animals: A Mini-Review. Gerontology 63, 417–425. 10.1159/000452444. [DOI] [PubMed] [Google Scholar]
- Calkins DJ. (2013). Age-related changes in the visual pathways: blame it on the axon. Investigative ophthalmology & visual science 54, Orsf37–41. 10.1167/iovs.13-12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero D, Soria-Valles C, and López-Otín C. (2016). Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Disease Models & Mechanisms 9, 719–735. 10.1242/dmm.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM (2009). Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic acids research 37, e21. 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailleux C, Aymard F, Caron P, Daburon V, Courilleau C, Canitrot Y, Legube G, and Trouche D. (2014). Quantifying DNA double-strand breaks induced by site-specific endonucleases in living cells by ligation-mediated purification. Nature protocols 9, 517–528. 10.1038/nprot.2014.031. [DOI] [PubMed] [Google Scholar]
- Changolkar LN, Singh G, and Pehrson JR (2008). macroH2A1-dependent silencing of endogenous murine leukemia viruses. Molecular and cellular biology 28, 2059–2065. 10.1128/mcb.01362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, et al. (2018). Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397 e1314. 10.1016/j.cell.2018.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119. 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury AR., Ju Z., Djojosubroto MW., Schienke A., Lechel A., Schaetzlein S., Jiang H., Stepczynska A., Wang C., Buer J., et al. (2007). Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature genetics 39, 99–105. 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- Conant D, Hsiau T, Rossi N, Oki J, Maures T, Waite K, Yang J, Joshi S, Kelso R, Holden K, et al. (2022). Inference of CRISPR Edits from Sanger Trace Data. CRISPR J 5, 123–130. 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. (2019). Ensembl 2019. Nucleic acids research 47, D745–D751. 10.1093/nar/gky1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG (1982). The dysdifferentiative hypothesis of mammalian aging and longevity. The aging brain 20, 1–18. [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, and Berger SL (2009). Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807. 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Huang GX, Bonkowski MS, Longchamp A, Li C, Schultz MB, Kim LJ, Osborne B, Joshi S, Lu Y, et al. (2019). Impairment of an Endothelial NAD(+)−H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 176, 944–945. 10.1016/j.cell.2019.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, and Sedivy JM (2013). Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging cell 12, 247–256. 10.1111/acel.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M., Ito T., Petrashen AP., Elias AE., Skvir NJ., Criscione SW., Caligiana A., Brocculi G., Adney EM., Boeke JD., et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Majo F, Martens L, Hegenbarth JC, Rühle F, Hamczyk MR, Nevado RM, Andrés V, Hilbold E, Bär C, Thum T, et al. (2021). Genomic instability in the naturally and prematurely aged myocardium. Proceedings of the National Academy of Sciences of the United States of America 118. 10.1073/pnas.2022974118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F, Piccirillo R, Goldberg AL, and Perrimon N. (2013). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Disease Models & Mechanisms 6, 1339–1352. 10.1242/dmm.012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al. (2013). SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nature neuroscience 16, 1008–1015. 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JC (2015). Optic nerve head biomechanics in aging and disease. Experimental eye research 133, 19–29. 10.1016/j.exer.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Andari J., Renaud-Gabardos E., Tulalamba W., Weinmann J., Mangin L., Pham QH., Hille S., Bennett A., Attebi E., Bourges E., et al. (2022). Semirational bioengineering of AAV vectors with increased potency and specificity for systemic gene therapy of muscle disorders. Science advances 8, eabn4704. 10.1126/sciadv.abn4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltes BC (2019). Architects meets Repairers: The interplay between homeobox genes and DNA repair. DNA repair 73, 34–48. 10.1016/j.dnarep.2018.10.007. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nature protocols 7, 1728–1740. 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson VL, Ayers RA, Bateman TA, and Simske SJ (2003). Bone development and age-related bone loss in male C57BL/6J mice. Bone 33, 387–398. [DOI] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, and Tyler JK (2010). Elevated histone expression promotes life span extension. Molecular cell 39, 724–735. 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, Eckl C, Andra I, Schiemann M, Oostendorp RA, et al. (2013). A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature 503, 392–396. 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, and Campisi J. (2012). Lamin B1 loss is a senescence-associated biomarker. Molecular biology of the cell 23, 2066–2075. 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, Bakker A, Yassa MA, and Stark CE (2010). Bridging neurocognitive aging and disease modification: targeting functional mechanisms of memory impairment. Current Alzheimer research 7, 197–199. 10.2174/156720510791050867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Wang WY., Mao YW., Graff J., Guan JS., Pan L., Mak G., Kim D., Su SC., and Tsai LH. (2010). A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466, 1105–1109. 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlick PJ, McNurlan MA, and Preedy VR (1980). A rapid and convenient technique for measuring the rate of protein synthesis in tissues by injection of [3H]phenylalanine. The Biochemical journal 192, 719–723. 10.1042/bj1920719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, and Kellis M. (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Leung D, and Ren B. (2014). The 3D genome in transcriptional regulation and pluripotency. Cell stem cell 14, 762–775. 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, and Brunet A. (2010). Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387. 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, and Brunet A. (2011). Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479, 365–371. 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G., Guinney J., Zhao L., Zhang L., Hughes G., Sadda S., Klotzle B., Bibikova M., Fan JB., Gao Y., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell 49, 359–367. 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkema L, Youssef SA, and de Bruin A. (2016). Pathology of Mouse Models of Accelerated Aging. Veterinary pathology 53, 366–389. 10.1177/0300985815625169. [DOI] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology 16, 144–154. 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, et al. (2015). Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488. 10.1016/j.cell.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstatter EW, Horvath S, Dalela D, Gupta P, Chagpar AB, Wali VB, Bossuyt V, Storniolo AM, Hatzis C, Patwardhan G, et al. (2018). Increased epigenetic age in normal breast tissue from luminal breast cancer patients. Clinical epigenetics 10, 112. 10.1186/s13148-018-0534-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. (2013). DNA methylation age of human tissues and cell types. Genome biology 14, R115. 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, and Levine AJ (2015). HIV-1 Infection Accelerates Age According to the Epigenetic Clock. The Journal of infectious diseases 212, 1563–1573. 10.1093/infdis/jiv277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH., Argmann C., Houten SM., Canto C., Jeninga EH., Andreux PA., Thoma C., Doenlen R., Schoonjans K., and Auwerx J. (2011). The metabolic footprint of aging in mice. Scientific reports 1, 134. 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol JH, Chen CC, Li W, and Tyler JK (2014). Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes & development 28, 396–408. 10.1101/gad.233221.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idelevich A, Sato K, Nagano K, Rowe G, Gori F, and Baron R. (2018). Neuronal hypothalamic regulation of body metabolism and bone density is galanin dependent. The Journal of clinical investigation 128, 2626–2641. 10.1172/jci99350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, and Kitano H. (1998). Heterochromatin islands and their dynamic reorganization: a hypothesis for three distinctive features of cellular aging. Experimental gerontology 33, 555–570. 10.1016/s0531-5565(98)00037-0. [DOI] [PubMed] [Google Scholar]
- Jiang N, Du G, Tobias E, Wood JG, Whitaker R, Neretti N, and Helfand SL (2013). Dietary and genetic effects on age-related loss of gene silencing reveal epigenetic plasticity of chromatin repression during aging. Aging 5, 813–824. 10.18632/aging.100614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Wozniak DF, and Imai S. (2018). CA1 Nampt knockdown recapitulates hippocampal cognitive phenotypes in old mice which nicotinamide mononucleotide improves. NPJ aging and mechanisms of disease 4, 10. 10.1038/s41514-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabacik S, Lowe D, Fransen L, Leonard M, Ang S-L, Whiteman C, Corsi S, Cohen H, Felton S, Bali R, et al. (2022). The relationship between epigenetic age and the hallmarks of ageing in human cells. Nature Aging. 10.1038/s43587-022-00220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M., McVey M., and Guarente L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & development 13, 2570–2580. 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane AE, and Sinclair DA (2019). Epigenetic changes during aging and their reprogramming potential. Critical reviews in biochemistry and molecular biology 54, 61–83. 10.1080/10409238.2019.1570075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman WB, Wanek L, Shepherd JA, and Sellmeyer DE (2010). Age-related hyperkyphosis: its causes, consequences, and management. The Journal of orthopaedic and sports physical therapy 40, 352–360. 10.2519/jospt.2010.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A, Lobanov AV, and Gladyshev VN (2015). Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging cell 14, 366–371. 10.1111/acel.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller EF (2009). Knowing As Making, Making As Knowing: The Many Lives of Synthetic Biology. Biological Theory 4, 333–339. 10.1162/BIOT_a_00005. [DOI] [Google Scholar]
- Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, and Guarente L. (1997). Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89, 381–391. 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D. (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics (Oxford, England) 26, 2204–2207. 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL., Shipony Z., and Greenleaf WJ. (2019). Chromatin accessibility and the regulatory epigenome. Nature reviews Genetics 20, 207–220. 10.1038/s41576-018-0089-8. [DOI] [PubMed] [Google Scholar]
- Koks S, Dogan S, Tuna BG, Gonzalez-Navarro H, Potter P, and Vandenbroucke RE (2016). Mouse models of ageing and their relevance to disease. Mechanisms of ageing and development 160, 41–53. 10.1016/j.mad.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics (Oxford, England) 27, 1571–1572. 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, and Kingston RE (2017). Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Molecular cell 65, 432–446.e435. 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research 22, 1813–1831. 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Couteur DG, Cogger VC, Markus AM, Harvey PJ, Yin ZL, Ansselin AD, and McLean AJ (2001). Pseudocapillarization and associated energy limitation in the aged rat liver. Hepatology (Baltimore, Md) 33, 537–543. 10.1053/jhep.2001.22754. [DOI] [PubMed] [Google Scholar]
- Leduc-Gaudet JP., Picard M., St-Jean Pelletier F., Sgarioto N., Auger MJ., Vallee J., Robitaille R., St-Pierre DH., and Gouspillou G. (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. 10.18632/oncotarget.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Coghlan A, Ruan J, Coin LJ, Heriche JK, Osmotherly L, Li R, Liu T, Zhang Z, Bolund L, et al. (2006). TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic acids research 34, D572–580. 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zuo X, Jing J, Ma Y, Wang J, Liu D, Zhu J, Du X, Xiong L, Du Y, et al. (2015). Small-Molecule-Driven Direct Reprogramming of Mouse Fibroblasts into Functional Neurons. Cell stem cell 17, 195–203. 10.1016/j.stem.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang W, Chen Y, Guo W, Zhang J, Tang H, Xu Z, Zhang H, Tao Y, Wang F, et al. (2016). Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell death and differentiation 23, 1765–1777. 10.1038/cdd.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic acids research 41, e108. 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum NL., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie BR., Sabo PJ., Dorschner MO., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, NY) 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Defensor EB, Mechanic JA, Shah PP, Jaime EA, Roberts CR, Hutto DL, and Schaevitz LR (2019). Retrospective Analysis of the Effects of Identification Procedures and Cage Changing by Using Data from Automated, Continuous Monitoring. Journal of the American Association for Laboratory Animal Science : JAALAS 58, 126–141. 10.30802/aalas-jaalas-18-000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Louie B, Ford D, Heath K, Cha P, Betts-Lacroix J, Lum PY, Robertson TL, and Schaevitz L. (2017). Development of the Digital Arthritis Index, a Novel Metric to Measure Disease Parameters in a Rat Model of Rheumatoid Arthritis. Frontiers in pharmacology 8, 818. 10.3389/fphar.2017.00818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin IH, Chang JL, Hua K, Huang WC, Hsu MT, and Chen YF (2018). Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC genetics 19, 55. 10.1186/s12863-018-0660-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, et al. (2007). Augmented Wnt signaling in a mammalian model of accelerated aging. Science (New York, NY) 317, 803–806. 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, Asakawa K, Morinaga H, Mohri Y, De Arcangelis A, et al. (2019). Stem cell competition orchestrates skin homeostasis and ageing. Nature 568, 344–350. 10.1038/s41586-019-1085-7. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT., Fei Z., Haghani A., Robeck TR., Zoller JA., Li CZ., Zhang J., Ablaeva J., Adams DM., Almunia J., et al. (2021). Universal DNA methylation age across mammalian tissues. bioRxiv, 2021.2001.2018.426733. 10.1101/2021.01.18.426733. [DOI] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, et al. (2014). REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454. 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Brommer B, Tian X, Krishnan A, Meer M, Wang C, Vera DL, Zeng Q, Yu D, Bonkowski MS, et al. (2020). Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129. 10.1038/s41586-020-2975-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Krishnan A, Brommer B, Tian X, Meer M, Vera DL, Wang C, Zeng Q, Yu D, Bonkowski MS, et al. (2019). Reversal of ageing- and injury-induced vision loss by Tet-dependent epigenetic reprogramming. bioRxiv, 710210. 10.1101/710210. [DOI] [Google Scholar]
- Luense LJ, Wang X, Schon SB, Weller AH, Lin Shiao E, Bryant JM, Bartolomei MS, Coutifaris C, Garcia BA, and Berger SL (2016). Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenetics & chromatin 9, 24. 10.1186/s13072-016-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna LG (1968). Manual of histologic staining methods of the Armed Forces Institute of Pathology (Third edition. New York: : Blakiston Division, McGraw-Hill, [1968] ©1968). [Google Scholar]
- Maccormick RE (2006). Possible acceleration of aging by adjuvant chemotherapy: a cause of early onset frailty? Medical hypotheses 67, 212–215. 10.1016/j.mehy.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Mapuskar KA., Flippo KH., Schoenfeld JD., Riley DP., Strack S., Hejleh TA., Furqan M., Monga V., Domann FE., Buatti JM., et al. (2017). Mitochondrial Superoxide Increases Age-Associated Susceptibility of Human Dermal Fibroblasts to Radiation and Chemotherapy. Cancer research 77, 5054–5067. 10.1158/0008-5472.can-17-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, and Gasser SM (1999). Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97, 621–633. 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Mohri Y, Binh NT, Morinaga H, Fukuda M, Ito M, Kurata S, Hoeijmakers J, and Nishimura EK (2016). Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science (New York, NY) 351, aad4395. 10.1126/science.aad4395. [DOI] [PubMed] [Google Scholar]
- McAinsh AD, Scott-Drew S, Murray JA, and Jackson SP (1999). DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Current biology : CB 9, 963–966. 10.1016/s0960-9822(99)80424-2. [DOI] [PubMed] [Google Scholar]
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al. (2009). SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 1, 109–121. 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G. (2010). GREAT improves functional interpretation of cis- regulatory regions. Nature biotechnology 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medawar PB. (1952). An unsolved problem of biology (Published for the College; by Lewis HK). [Google Scholar]
- Meer MV, Podolskiy DI, Tyshkovskiy A, and Gladyshev VN (2018). A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7. 10.7554/eLife.40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, and O’Callaghan JP (2005). Aging, stress and the hippocampus. Ageing research reviews 4, 123–140. 10.1016/j.arr.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, and Guarente L. (1999). MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97, 609– 620. 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- Miyoshi N, Oubrahim H, Chock PB, and Stadtman ER (2006). Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proceedings of the National Academy of Sciences of the United States of America 103, 1727–1731. 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnat RJ Jr., Hackmann AF, and Cantrell MA (1999). Generation of highly site- specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochemical and biophysical research communications 255, 88–93. 10.1006/bbrc.1999.0152. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, et al. (2013). The histone demethylase LSD1/KDM1A promotes the DNA damage response. The Journal of cell biology 203, 457–470. 10.1083/jcb.201302092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature methods 13, 919–922. 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MA., and Berry SA. (1992). Cockayne syndrome: Review of 140 cases. American Journal of Medical Genetics 42, 68–84. 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Narayanan L, Fritzell JA, Baker SM, Liskay RM, and Glazer PM (1997). Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proceedings of the National Academy of Sciences of the United States of America 94, 3122–3127. 10.1073/pnas.94.7.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D. (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science (New York, NY) 347, 1017–1021. 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura EK, Granter SR, and Fisher DE (2005). Mechanisms of Hair Graying: Incomplete Melanocyte Stem Cell Maintenance in the Niche. Science (New York, NY) 307, 720–724. 10.1126/science.1099593. [DOI] [PubMed] [Google Scholar]
- Norden DM, and Godbout JP (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathology and applied neurobiology 39, 19–34. 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan HM, Mohammad HP, and Baylin SB (2008). Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS genetics 4, e1000155. 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918. 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P., and Sinclair DA. (2007). The role of nuclear architecture in genomic instability and ageing. Nature reviews Molecular cell biology 8, 692–702. 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, et al. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733.e1712. 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohse T, Vaughan MR, Kopp JB, Krofft RD, Marshall CB, Chang AM, Hudkins KL, Alpers CE, Pippin JW, and Shankland SJ (2010). De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am J Physiol Renal Physiol 298, F702–711. 10.1152/ajprenal.00428.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell reports 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Pal S, and Tyler JK (2016). Epigenetics and aging. Science advances 2, e1600584. 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, and Reuter-Lorenz P. (2009). The adaptive brain: aging and neurocognitive scaffolding. Annual review of psychology 60, 173–196. 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PU., Defossez PA., and Guarente L. (1999). Effects of mutations in DNA repair genes on formation of ribosomal DNA circles and life span in Saccharomyces cerevisiae. Molecular and cellular biology 19, 3848–3856. 10.1128/mcb.19.5.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, and Gladyshev VN (2017). Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell metabolism 25, 954–960 e956. 10.1016/j.cmet.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PS, Coorens THH, Palles C, Mitchell E, Abascal F, Olafsson S, Lee BCH, Lawson ARJ, Lee-Six H, Moore L, et al. (2021). Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nature genetics 53, 1434–1442. 10.1038/s41588-021-00930-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder SS, Stefanska A, Eng DG, Kaverina N, Sunseri MW, McNicholas BA, Rabinovitch P, Engel FB, Daniel C, Amann K, et al. (2015). Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol 309, F164–178. 10.1152/ajprenal.00144.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina ,B, and Helfand ,SL (2004). Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proceedings of the National Academy of Sciences of the United States of America 101, 15998–16003. 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JM. (2011). Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. Journal of visualized experiments : JoVE, e3266. 10.3791/3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Li H, Chen Z, Coghlan A, Coin LJ, Guo Y, Heriche JK, Hu Y, Kristiansen K, Li R, et al. (2008). TreeFam: 2008 Update. Nucleic acids research 36, D735–740. 10.1093/nar/gkm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E, Wu X, Zhu T, Cheung JC, Chen H, Lorincz A, Pandita RK, Sharma GG, Ha HC, Gasson J, et al. (2007). A role for the HOXB7 homeodomain protein in DNA repair. Cancer research 67, 1527–1535. 10.1158/0008-5472.Can-06-4283. [DOI] [PubMed] [Google Scholar]
- Russell-Goldman E, and Murphy GF (2020). The Pathobiology of Skin Aging: New Insights into an Old Dilemma. The American journal of pathology 190, 1356–1369. 10.1016/j.ajpath.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, and Brown EJ (2007). Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell stem cell 1, 113–126. 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HY, Rhie BH, and Ahn SH (2014). Loss of the Set2 histone methyltransferase increases cellular lifespan in yeast cells. Biochemical and biophysical research communications 446, 113–118. 10.1016/j.bbrc.2014.02.061. [DOI] [PubMed] [Google Scholar]
- Salk D., Fujiwara Y., and Martin GM. (1985). Werner’s Syndrome and Human Aging. 10.1007/978-1-4684-7853-2_9. [DOI] [Google Scholar]
- Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, Peixoto FO, Stephan-Otto Attolini C, Prats N, Aguilera M, et al. (2018). Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 175, 1575–1590 e1522. 10.1016/j.cell.2018.10.012. [DOI] [PubMed] [Google Scholar]
- Samuel MA, Zhang Y, Meister M, and Sanes JR (2011). Age-related alterations in neurons of the mouse retina. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 16033–16044. 10.1523/jneurosci.3580-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar TJ, Quarta M, Mukherjee S, Colville A, Paine P, Doan L, Tran CM, Chu CR, Horvath S, Qi LS, et al. (2020). Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nature communications 11, 1545. 10.1038/s41467-020-15174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RR, Eng DG, Kutz JN, Sweetwyne MT, Pippin JW, and Shankland SJ (2017). Compound effects of aging and experimental FSGS on glomerular epithelial cells. Aging 9, 524–546. 10.18632/aging.101176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. (2015). Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature genetics 47, 1179–1186. 10.1038/ng.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Dang W, Donahue G, Dai J, Dorsey J, Cao X, Liu W, Cao K, Perry R, Lee JY, et al. (2015). H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes & development 29, 1362–1376. 10.1101/gad.263707.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P., Shah PP., Nativio R., and Berger SL. (2016). Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839. 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes & development 27, 1787–1799. 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, et al. (2019). LINE1 Derepression in Aged Wild-Type and SIRT6- Deficient Mice Drives Inflammation. Cell metabolism 29, 871–885 e875. 10.1016/j.cmet.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, and Guarente L. (1997). Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 91, 1033–1042. 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, and LaPlante MD (2019). Lifespan: Why We Age—and Why We Don’t Have To (Atria Books; ). [Google Scholar]
- Sinclair DA, Mills K, and Guarente L. (1997). Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science (New York, NY) 277, 1313–1316. 10.1126/science.277.5330.1313. [DOI] [PubMed] [Google Scholar]
- Smeal T, Claus J, Kennedy B, Cole F, and Guarente L. (1996). Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84, 633–642. 10.1016/s0092-8674(00)81038-7. [DOI] [PubMed] [Google Scholar]
- Soto I., Graham LC., Richter HJ., Simeone SN., Radell JE., Grabowska W., Funkhouser WK., Howell MC., and Howell GR. (2015). APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS biology 13, e1002279. 10.1371/journal.pbio.1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic acids research 44, D717–725. 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs TM, Bonder MJ, Stark AK, Krueger F, von Meyenn F, Stegle O, and Reik W. (2017). Multi-tissue DNA methylation age predictor in mouse. Genome biology 18, 68. 10.1186/s13059-017-1203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard L. (1959). ON THE NATURE OF THE AGING PROCESS. Proceedings of the National Academy of Sciences of the United States of America 45, 30–45. 10.1073/pnas.45.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tamburini BA, and Tyler JK (2005). Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Molecular and cellular biology 25, 4903–4913. 10.1128/mcb.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Ke Z, Tombline G, Macoretta N, Hayes K, Tian X, Lv R, Ablaeva J, Gilbert M, Bhanu NV, et al. (2017). Naked Mole Rat Cells Have a Stable Epigenome that Resists iPSC Reprogramming. Stem cell reports 9, 1721–1734. 10.1016/j.stemcr.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MJ., Chwialkowska K., Rubb L., Lusi AJ., Davi RC., Srivastav A., Korstanje R., Churchill GA., Horvath S., and Pellegrini M. (2018). A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854. 10.18632/aging.101590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, Tan R, Simon M, Henderson S, Steffan J, et al. (2019). SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 177, 622–638 e622. 10.1016/j.cell.2019.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V, Jain C, Raz Y, Nakamura S, Heestand B, Liu W, Spath M, Suchiman HED, Muller RU, Slagboom PE, et al. (2017). Small nucleoli are a cellular hallmark of longevity. Nature communications 8, 16083. 10.1038/ncomms16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, and Jensen ON (2017). Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic acids research 45, 9272–9289. 10.1093/nar/gkx696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Tarantini S, Hertelendy P, Valcarcel-Ares MN, Fülöp GA, Logan S, Kiss T, Farkas E, Csiszar A, and Yabluchanskiy A. (2017). Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. GeroScience 39, 33–42. 10.1007/s11357-017-9964-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics 43, 11.10.11–33. 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, and Wiggins RC (2014). Estimating podocyte number and density using a single histologic section. Journal of the American Society of Nephrology : JASN 25, 1118–1129. 10.1681/asn.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilenchik MM., and Knudson AG. (2003). Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences 100, 12871–-12876.. 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH (1957). The strategy of the genes; a discussion of some aspects of theoretical biology (London,: Allen & Unwin; ). [Google Scholar]
- Wakayama S, Kohda T, Obokata H, Tokoro M, Li C, Terashita Y, Mizutani E, Nguyen VT, Kishigami S, Ishino F, et al. (2013). Successful serial recloning in the mouse over multiple generations. Cell stem cell 12, 293–297. 10.1016/j.stem.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, et al. (2014). Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome biology 15, R24. 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RP, Lee C, Imbriano PM, Patil S, Weymouth TE, Smith RA, Scott LJ, and Sartor MA (2014). ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic acids research 42, e105. 10.1093/nar/gku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welle S, Brooks AI, Delehanty JM, Needler N, Bhatt K, Shah B, and Thornton CA (2004). Skeletal muscle gene expression profiles in 20–29 year old and 65–71 year old women. Experimental gerontology 39, 369–377. 10.1016/j.exger.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, and Moraes CT (2009). Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proceedings of the National Academy of Sciences 106, 20405–20410. 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- White RR., and Vijg J. (2016). Do DNA Double-Strand Breaks Drive Aging? Molecular cell 63, 729–738. 10.1016/j.molcel.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead JC, Hildebrand BA, Sun M, Rockwood MR, Rose RA, Rockwood K, and Howlett SE (2013). A Clinical Frailty Index in Aging Mice: Comparisons With Frailty Index Data in Humans. The Journals of Gerontology: Series A 69, 621–632. 10.1093/gerona/glt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC (1957). Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 11, 398–411. 10.2307/2406060. [DOI] [Google Scholar]
- Wittmayer PK, McKenzie JL, and Raines RT (1998). Degenerate DNA recognition by I-PpoI endonuclease. Gene 206, 11–21. 10.1016/s0378-1119(97)00563-5. [DOI] [PubMed] [Google Scholar]
- Wolf N, Pendergrass W, Singh N, Swisshelm K, and Schwartz J. (2008). Radiation cataracts: mechanisms involved in their long delayed occurrence but then rapid progression. Molecular vision 14, 274–285. [PMC free article] [PubMed] [Google Scholar]
- Wolf NS, Li Y, Pendergrass W, Schmeider C, and Turturro A. (2000). Normal mouse and rat strains as models for age-related cataract and the effect of caloric restriction on its development. Experimental eye research 70, 683–692. 10.1006/exer.2000.0835. [DOI] [PubMed] [Google Scholar]
- Wood JG., Jone BC., Jian N., Chang C., Hosier S., Wickremesinghe P., Garcia M., Hartnett DA., Burhenn L., Neretti N., et al. (2016). Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 113, 11277–11282. 10.1073/pnas.1604621113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Song Y, Seol JH, Park JY, Yang YJ, Han JW, Youn HD, and Cho EJ (2011). Myogenic transcriptional activation of MyoD mediated by replication-independent histone deposition. Proceedings of the National Academy of Sciences of the United States of America 108, 85–90. 10.1073/pnas.1009830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang F, Yardimci GG, Song F, Hardison RC, Noble WS, Yue F, and Li Q. (2017). HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome research 27, 1939–1949. 10.1101/gr.220640.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, and Adams PD (2007). Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Molecular cell 27, 183–196. 10.1016/j.molcel.2007.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, Sidiq T, Shipp MA, Lizee GA, and Kobayashi KS (2016). NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proceedings of the National Academy of Sciences of the United States of America 113, 5999–6004. 10.1073/pnas.1602069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Chen X, Ning C, Zhu Q, Yao Y, Zhao Y, and Luan F. (2018). Methylation of the genes ROD1, NLRC5, and HKR1 is associated with aging in Hainan centenarians. BMC medical genomics 11, 7. 10.1186/s12920-018-0334-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Pippin JW., Krofft RD., Naito S., Liu ZH., and Shankland SJ. (2013). Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am J Physiol Renal Physiol 304, F1375–1389. 10.1152/ajprenal.00020.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, Imamichi H, Huang DW, Lempicki RA, Baseler MW, et al. (2011). Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol 186, 4541–4545. 10.4049/jimmunol.1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hapala J, Brenner H, and Wagner W. (2017). Individual CpG sites that are associated with age and life expectancy become hypomethylated upon aging. Clinical epigenetics 9, 9. 10.1186/s13148-017-0315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yi J, Tao L, Huang G, Chu X, Song H, and Chen L. (2018). Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell death & disease 9, 433. 10.1038/s41419-018-0466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkel A, Nikolic M, Sofiadis K, Mallm JP, Brackley CA, Gothe H, Drechsel O, Becker C, Altmuller J, Josipovic N, et al. (2018). HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Molecular cell 70, 730–744.e736. 10.1016/j.molcel.2018.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of the ICE system on the cell cycle, senescence and mutations, Related to Figure 1
(A) Breeding scheme to generate ICE mice and controls (WT, Cre and STOP-I-PpoI).
(B and C) DNA damage response induced by I-PpoI (4-OHT, 0.1, 0.5, 1 μM) vs. other damaging agents, etoposide (ETS, 1, 10, 25 μM) and phleomycin (Phleo, 1, 25, 50 μg/ml). Scale bar, 10 μM. One-way ANOVA-Bonferroni.
(D) Western blot of DNA damage response proteins.
(E) Cell cycle profile in 96-hour post-treated cells.
(F) Percentage of SA-β-Gal positive (blue) cells during and after TAM treatment compared to replicative senescent cells. p, passage. One-way ANOVA-Bonferroni.
(G) Cell diameter after recovery from I-PpoI induction vs. irradiated (IR, senescent) cells. One-way ANOVA-Bonferroni.
(H and I) γH2AX and 53BP1 in post-treated ICE cells with and without exposure to the DNA damaging agents (ETS, etoposide; CPT, camptothecin; H2O2, hydrogen peroxide). Scale bar, 10 μm. Two-tailed Student’s t test.
(J and K) Lamin B1 mRNA and protein levels in 144-hour post-treated ICE cells. Lamin A/C and actin are loading controls known not to change during senescence.
(L-N) Percent non-mutated I-PpoI canonical (Teme56), non-canonical recognition and random sequences in 96-hour post-treated ICE cells assessed by deep sequencing (>50x).
Data are mean (n≥3) ± SD. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001; **** p < 0.0001.
Figure S2. No change in 28S rDNA in post-treated ICE cells and muscle, Related to Figure 1 and 2
(A) Southern blot of 28S rDNA in 4-OHT treated and post-treated Cre and ICE cells.
(B) In vitro cutting of I-PpoI targets PCR-amplified from genomic DNA from 96-hour post-treated Cre and ICE cells.
(C) Surveyor nuclease assay of I-PpoI targets in 96-hour post-treated cells. DNA with a point mutation (PM) in the I-PpoI site was the positive control.
(D) 28S rRNA level in 96-hour post-treated ICE cells. Two-tailed Student’s t test.
(E) Bioanalyzer tracks of 28S and 18S rRNA in 96-hour post-treated cells.
(F) 28S:18S rRNA ratio. Two-tailed Student’s t test.
(G) Scheme for ligation-mediated PCR to detect residual rDNA breaks.
(H-J) Ligation-mediated PCR after I-PpoI digestion In vitro (H), in Cre and ICE cells (I) or in 10-month post-treated muscle (J).
(K and L) Protein translation in 96-hour post-treated cells assessed by metabolic 35S-labelling. Two-tailed Student’s t test.
(M and N) Protein translation in 10-month post-treated brain (N) and muscle (O) tissue, assessed by metabolic 35S-labelling. Two-tailed Student’s t test.
(O) 28S rDNA copy number in 10-month post-treated muscle assessed by monochrome multiplex quantitative PCR (MMQPCR). One-way ANOVA-Bonferroni (left) or two-way ANOVA-Bonferroni (right).
(P) 28S rRNA levels in 10-month post-treated muscle. One-way ANOVA-Bonferroni (left) or two-way ANOVA-Bonferroni (right).
Data are mean (n≥3) ± SD or ± SEM (M-P). n.s.: p > 0.05; **** p < 0.0001.
Figure S3. Validation of the ICE mouse, Related to Figure 2
(A) Removal of the transcriptional STOP cassette in major tissues. One-way ANOVA-Bonferroni.
(B) Western blot of tissues probed with anti-HA to detect I-PpoI expression and γH2AX after a 4-week TAM treatment.
(C and D) Hippocampal sections immunostained for GFP (as a proxy for IRES-linked I-PpoI expression) and γH2AX. Scale bar, 200 μm (100X, C) and 50 μm (400X, D).
(E) Western blot of tissues probed with anti-HA to detect I-PpoI expression after 3-weeks of TAM and 1-month post-treatment.
(F) Immunoprecipitation and quantification of a I-PpoI cut site (28S rDNA) in skeletal muscle, liver and kidney during and after tamoxifen treatment (0-,1- and 10-month post-treatment). Two-tailed Student’s t test. Data are mean (n=3–6) ± SEM. * p < 0.05.
(G-I) Percent non-mutated I-PpoI canonical, non-canonical recognition and random sequences in 1-month post-treated ICE muscle assessed by deep sequencing (>50x).
Figure S4. ICE mice phenocopy normal aging, Related to Figure 2 and 3
(A) Food consumption of post-treated mice. Two-tailed Student’s t test.
(B) Body weights of female mice post-treatment. Two-way ANOVA-Bonferroni.
(C) Representative images of 10-month post-treated mice from Dual Energy X-ray Absorptiometry (DEXA).
(D) Body mass of 10-month post-treated mice. Two-tailed Student’s t test.
(E) Subcutaneous fat thickness of 10-month post-treated mice. Two-tailed Student’s t test.
(F) Respiratory Exchange Rate (RER) of female mice. Repeated measures two-way ANOVA.
(G and H) Blood glucose levels of 10-month post-treated mice in the fed or fasted state. Two-tailed Student’s t test.
(I) IGF-1 levels in 10-month post-treated mice. Two-way ANOVA-Bonferroni.
(J and K) H&E staining of subcutaneous fat layers and subepidermal thickness of back skin from old WT, 10-month post-treated Cre and ICE mice. Scale bar, 500 μm. One-way ANOVA-Bonferroni (left) or two-tailed Student’s t test (right).
(L) KIT (CD117), KRT55 and DAPI staining of back skin.
(M) Percent hair follicle bulges without KIT staining. Two-tailed Student’s t test.
(N-P) Gait analysis of 10-month post-treated mice. Two-tailed Student’s t test. Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001.
Figure S5. Progeroid features of ICE mouse muscle, Related to Figure 4
(A) Blood lactate build-up after exercise in WT, 10-month post-treated mice. Two-tailed Student’s t test.
(B) Grip strength measured as maximal “peak force”. Two-tailed Student’s t test.
(C) Mitochondrial DNA copy number. Two-tailed Student’s t test.
(D-F) Mitochondria number (D and E) and mitochondrial perimeter (D and F) of 10-month post-treated muscle determined with electron microscopy. Two-tailed Student’s t test. Scale, 500 nm.
(G and H) Cytochrome oxidase (COX) staining of 10-month post-treated gastrocnemius muscle. Scale bar, 100 μm. Two-tailed Student’s t test.
(I and J) Quantification of RNA from repetitive DNA elements in gastrocnemius muscle
(I) and liver (J) of 10-month post-treated mice. Two-tailed Student’s t test.
(K and L) Echocardiogram of 10-month post-treated mice. LVPWd, Left ventricular posterior wall thickness at end-diastole mm. LVIDd, Left ventricular internal diameter in diastole; LVPWd, Left ventricular posterior wall in diastole; LVEF, Left ventricular ejection fraction. Two-tailed Student’s t test.
(M) Scatter plot of genes altered (p < 0.01) in muscle from 10-month post-treated ICE mice and WT 24-month-old mice. Significantly changed genes (padj < 0.05) are blue, red or green.
(N) Fold change of altered genes (padj < 0.05, Cre vs ICE mice) in WT 24-month-old mice.
(O) Fold change of altered genes (padj < 0.05, 12mo. vs 24mo.) in 10-month post-treated ICE mice.
Data are mean ± SEM. n.s.: p > 0.05; *p < 0.05; **p < 0.01; ***p< 0.001, ****p< 0.0001.
Figure S6. Altered epigenetic landscapes in ICE cells and muscle, Related to Figure 4–6
(A–D) Muscle and blood training (A and C) and testing sets (B and D) of the clock CpG sites in WT C57BL/6J mice.
(E) Circos plot of genomic locations of I-PpoI cut sites (green), clock GpG sites in CpG islands (red), and all muscle clock sites (blue).
(F) Western blotting for histone modifications in 96-hour post-treated ICE cells. Histone H3 and macroH2A serve as loading and internal controls.
(G) Spike-in normalization of ChIP-seq data.
(H) ATAC signal in loci with altered H3K27ac peaks.
(I) Aggregation plots of H3K27ac signal in H3K27ac changed regions (padj < 0.01).
(J) Aggregation plots of H3K27ac signal in top 40% or bottom 40% quantile.
(K and L) qPCR analysis of Hoxa genes in post-treated cells cut with either I-PpoI (K) or I-SceI (L) homing endonucleases. Two-tailed Student’s t test.
(M) Comparison of H3K27ac decreased regions (p < 0.01) in muscle to epigenome roadmap data from different human tissue types.
(N) Super-enhancers (SEs) in different cell types that overlap with regions with increased H3K27ac signals in 10-month post-treated ICE muscle.
Data are mean (n=3) ± SD. *p < 0.05; **p < 0.01.
Figure S7. Epigenetic reprogramming using adeno-associated virus (AAV)-mediated gene transfer, Related to Figure 7
(A and B) Western blot of Oct4 and Klf4 in 293T cells after AAV-DJ transduction.
(C) Dose-dependent increase in mRNA levels of Oct4, Sox2 and Klf4 in mouse fibroblasts at day 4. Two-tailed Student’s t test.
(D) Immunocytochemistry of Oct4 and Klf4 in mouse fibroblasts at day 4. DNA stained with DAPI. Scale, 50 μm.
(E and F) mRNA levels of Oct4, Sox2 and Klf4 at day 6 of Dox treatment in fibroblasts
(E) or at day 5 of Dox treatment in young and old OSK transgenic fibroblasts (F). One-way ANOVA-Bonferroni.
(G) Epigenetic age of post-treated Cre and ICE MEFs.
(H) Experimental scheme for AAV-OSK injection to Cre and ICE mice.
(I) GFP expression in muscle and kidney at 3-week post-AAV injection.
(J and K) qPCR analysis of Oct4 gene in post-treated Cre and ICE kidney (J) and muscle (K) at 5-week post-AAV injection.
(L and M) Representative images and quantification of H3K36me2 in the ICE muscle at 5-week post-AAV injection. One-way ANOVA-Bonferroni.
(N) Timeline of phenotype assessments.
(O and P) Lens opacity in 10-month post-treated mice. Two-tailed Student’s t test.
Data are mean (n≥3) ± SD or ± SEM (P). *p < 0.05; **p < 0.01; ***p< 0.001; ****p< 0.0001.
Data Availability Statement
The NGS datasets generated during this study are available through the BioSample database (NCBI) under BioProject ID: PRJNA554729 and PRJNA655981. Details of the analysis are provided in the STAR Methods section. Any additional information is available from the lead contact upon request.