

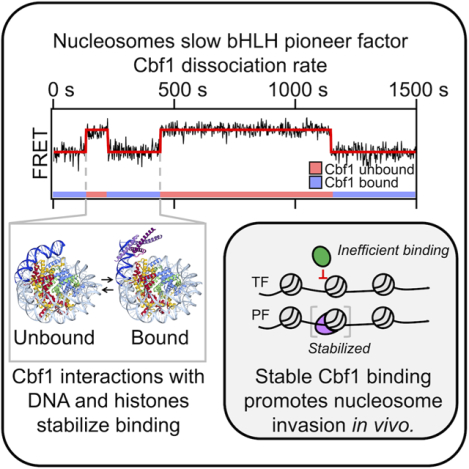
SUMMARY
Nucleosomes drastically limit transcription factor (TF) occupancy, while pioneer transcription factors (PFs) somehow circumvent this nucleosome barrier. In this study, we compare nucleosome binding of two conserved S. cerevisiae basic helix-loop-helix (bHLH) TFs, Cbf1 and Pho4. A Cryo-EM structure of Cbf1 in complex with the nucleosome reveals that the Cbf1 HLH region can electrostatically interact with exposed histone residues within a partially unwrapped nucleosome. Single-molecule fluorescence studies show that the Cbf1 but not the Pho4 HLH region facilitates efficient nucleosome invasion by slowing its dissociation rate relative to DNA through interactions with histones. In vivo studies show that this enhanced binding provided by the Cbf1 HLH region enables nucleosome invasion and ensuing repositioning. These structural, single-molecule, and in vivo studies reveal the mechanistic basis of dissociation rate compensation by pioneer factors and how this translates to facilitating chromatin opening inside cells.
Keywords: gene regulation, chromatin biology, pioneer transcription factors, dissociation rate compensation mechanism, nucleosome depleted regions, single-molecule measurement, cryoelectron microscopy single-particle analysis
Graphical Abstract
eTOC
Donovan, et al. compare structurally similar bHLH TFs: Cbf1, a pioneer TF, and Pho4, a canonical TF. The Cbf1 HLH region interacts with histones facilitating nucleosome targeting and invasion, through the dissociation rate compensation mechanism, and enabling Cbf1’s nucleosome displacing function, while the nucleosome inhibits DNA targeting by Pho4.
INTRODUCTION
Chromatin structure regulates DNA binding protein occupancy genome-wide. For example, nucleosome positioning relative to a transcription factor (TF) binding site can influence TF occupancy by orders of magnitude1–4 and regulate transcription.5–9 However, DNA binding proteins use a myriad of strategies to overcome barriers presented by the nucleosome. Binding upon transient partial nucleosome unwrapping is a primary mechanism TFs use to access nucleosome sites.1,2,10,11 This site exposure mechanism predicts a reduction in TF binding based on DNA target site exposure probability12. Single-molecule studies have revealed that nucleosomes also regulate TF occupancy through influencing TF dissociation. Depending on the TF, nucleosomes can either lead to accelerated or slowed dissociation4,13 and slowed dissociation.14,15
Pioneer factors (PFs) are key TFs that target their sites within nucleosomes as efficiently as naked DNA.16,17 They function to generate nucleosome depleted regions (NDRs)18,19 and as master regulators of cell state.20,21 Some PFs use slowed dissociation from nucleosomes14,15,22 to efficiently targeting their sites within nucleosomes despite limited binding site accessibility. Such “dissociation rate compensation mechanism” may enable, or at least facilitate, PFs to initiate the formation of NDRs and chromatin opening.17,23 However, the mechanisms underlying the nucleosome-dependent slow dissociation, and how essential this PF binding property is for the establishment of NDRs in vivo are unknown.
Cbf1, a budding yeast basic Helix Loop Helix Leucine Zipper (bHLHZ) transcription factor, functions as a pioneer factor and generates NDRs near its binding sites.23 Recent in vitro single molecule assays showed Cbf1 can access transiently exposed binding sites within partially unwrapped nucleosomes stabilizing the unwrapped state without histone eviction. These results agree with structural studies reporting PFs bound to nucleosomes stabilize distorted24 and partially unwrapped DNA.25 Cbf1 binding kinetics to partially unwrapped nucleosomes is reduced by ~100-fold, however the roughly equal (~30 fold) reduction in dissociation rate enables Cbf1 to have similar occupancy within nucleosomes and naked DNA.14 This finding suggests Cbf1 has additional interactions with nucleosomes compared to naked DNA. However, the molecular basis of these nucleosome specific interactions is unknown.
Cbf1 is commonly contrasted with Pho4, another bHLH TF from budding yeast because they target similar E-box motifs.26–29 Interestingly, unlike Cbf1, nucleosomes almost completely block Pho4 binding.30 It is intriguing how two TFs in the same family with similar binding motifs can have such drastic differences in nucleosome binding. Here we report single-molecule fluorescence (SMF), cryo-EM, and live cell studies that investigate key differences between Pho4 and Cbf1 interactions with nucleosomes and how this enables NDR generation. Our findings clarify how PFs are distinct from canonical TFs so that they can efficiently invade and then displace nucleosomes.
RESULTS
Cbf1 pioneer activity is largely determined by the structured bHLHZ domain
Given our previous finding that nucleosomes slow Cbf1 dissociation,14 we sought to localize the region(s) responsible for nucleosome specific interactions. Since Cbf1’s unstructured N-terminus comprises approximately 60% of the protein, we first probed the impact of removing the N-terminus (Cbf1ΔN) on nucleosome binding relative to DNA (Figure 1A, S1A). DNA and nucleosomal templates were prepared with the consensus Cbf1 motif, where nucleosomes contain the binding site 8 bp from the nucleosome edge (Figure S1B).2,10,31 Electrophoresis mobility shift assays (EMSAs) revealed that Cbf1ΔN (residues 209–351) binds DNA and nucleosomes over similar concentration ranges (Figure S1C) as previously observed for full length Cbf1. The overall affinities of Cbf1ΔN were similar to full-length Cbf1.14 These results indicate that, similar to another yeast PF Reb1, the intrinsically disordered N-terminal region of Cbf1 does not significantly contribute to overall binding affinity to DNA and nucleosomes.
Figure 1. The pioneering ability of Cbf1 to efficiently invade a nucleosome is contained within the bHLHZ domain.
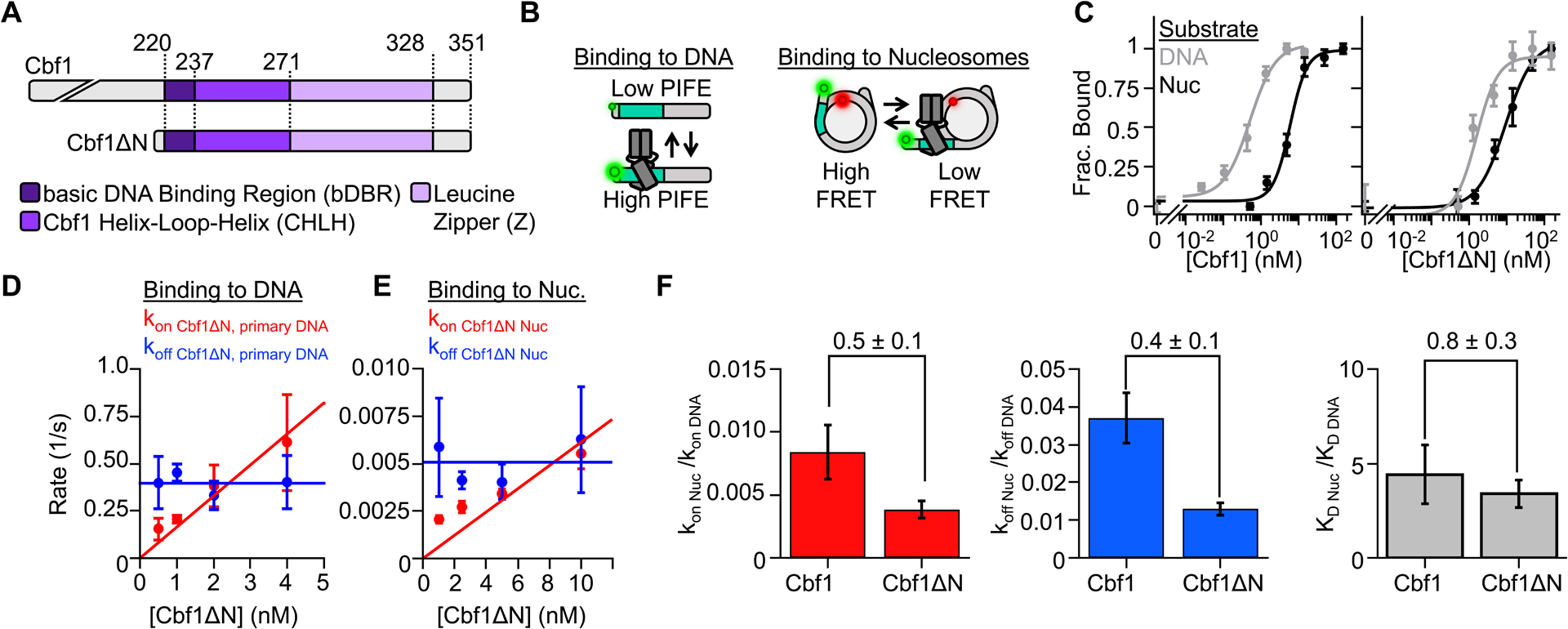
(A) Domain diagrams of Cbf1 and Cbf1ΔN (AA 209–351). (B) Fluorescence approaches to characterize TF binding. TF-DNA interactions are detected by PIFE from Cy3 labeled DNA, and TF-nucleosome interactions are detected with FRET between Cy3-DNA and Cy5-H2A(K119C). (C) Ensemble fluorescence measurements of Cbf1 (left) and Cbf1ΔN (right) binding to DNA (grey) or nucleosomes (black) as determined by PIFE and FRET efficiency, respectively. The PIFE and FRET data were fit to binding isotherms to determine the S1/2 values (Table S1). (D) The primary binding and dissociation rates to and from DNA measured by smPIFE for increasing Cbf1ΔN concentrations. The dissociation rates (blue) are constant with an average rate of koff Cbf1ΔN DNA primary = 0.39 ± 0.02 s−1. The binding rate constant (red) is the slope of a linear fit to the binding rate, kon Cbf1ΔN DNA primary = 0.16 ± 0.01 s−1 nM−1. (E) Cbf1ΔN binding (red) and dissociation (blue) kinetics to and from nucleosomes, measured by smFRET. The dissociation rate (koff Cbf1ΔN Nuc = 0.0051 ± 0.0006 s−1) and the binding rate constant (kon Cbf1ΔN Nuc = 0.0006 ± 0.0001 s−1 nM−1) were determined as described for DNA. (F) Nucleosomes binding rates (red), dissociation rate constants (blue), and binding affinities (gray) relative to DNA for Cbf1 and Cbf1ΔN.
We next measured Cbf1ΔN binding to DNA and nucleosomes using fluorescence-based assays which, in contrast to EMSAs, maintain equilibrium conditions, and are single-molecule amenable. These and all following nucleosome measurements are reported relative to DNA to control for minor experimental variations that change DNA-only interactions. Cbf1ΔN binding to DNA was probed via Protein Induced Fluorescence Enhancement (PIFE) (Figure 1B, left), where a Cy3 fluorophore is positioned adjacent to the Cbf1 binding site. Upon Cbf1 binding, Cy3 fluorescence increases by about 1.5-fold.32–34 We measured nucleosome binding with Förster Resonance Energy Transfer (FRET) efficiency between the Cy3-labeled DNA and Cy5-labeled H2A(K119C) (Figure 1B, right). Cy3-Cy5 FRET efficiency reduces as fully wrapped nucleosome are trapped in partially unwrapped states by TF binding. We quantified the concentration of Cbf1ΔN that binds 50% of DNA molecules or nucleosomes (S1/2).
The nucleosome S1/2 relative to DNA is 3-fold higher (Table S1, S1/2 Cbf1ΔN DNA = 2.4 ± 0.5 nM, S1/2 Cbf1ΔN Nuc = 7.2 ± 1.0 nM, 2 biological replicates), which is similar to full-length Cbf1 (Figure 1C). The gel- and fluorescence-based affinity measurements indicate that the removal of Cbf1’s N-terminus only modestly impacts (a) binding affinity to DNA or nucleosomes and (b) nucleosome targeting relative to DNA.
Since the ratio of the binding and dissociation rates dictates equilibrium affinity, and a defining Cbf1 property is its slow dissociation from nucleosomes, we investigated with SMF experiments if removal of the disordered N-terminal domain influenced the binding and/or dissociation kinetics. The DNA kinetics were determined by single-molecule (sm) PIFE where Cy3-labeled DNA was tethered to the microscope surface through a biotin-streptavidin linkage and excited via Total Internal Reflection Fluorescence (TIRF) microscopy. High and low Cy3 fluorescence reports bound and unbound states of Cbf1ΔN, respectively.4,32 smPIFE data (Figure S2) was acquired at four Cbf1ΔN concentrations and time traces were fit with a 2-state Hidden Markov Model (HMM)35 to determine the corresponding time-scales of binding. Likelihood ratio tests36 indicate the cumulative probabilities are best fit with two characteristic dwell times in both the unbound and bound states (Figure S1E), which determined the binding and dissociation rates to and from DNA (Figure 1D, S1G). We focused on the faster, primary population which comprises 70% of events (Figure S1F). See methods for details.
We measured Cbf1ΔN binding kinetics to nucleosomes through single molecule (sm) FRET,37 where high and low FRET efficiency correspond to the unbound and bound states of Cbf1ΔN, respectively. The time traces (Figure S2) were analyzed as with the smPIFE traces. The likelihood ratio tests indicated a single binding rate constant and dissociation rate, and fits of the cumulative probabilities (Figure S1H) determined the binding and dissociation rates of Cbf1ΔN to and from nucleosomes (Figure 1E).
The Cbf1ΔN dissociation rates from DNA and nucleosomes did not depend on TF concentration and were fit to horizontal lines where the y-intercept represents the dissociation rate (Figure 1D–E blue lines, Table S2–3). The binding rate increased with TF concentration and were fit to a line where the slope represents the binding rate constant (Figure 1D–E red lines Table S2–3). Importantly, both the binding and dissociation rates of Cbf1ΔN to and from nucleosomes relative to DNA were within 2-fold of full length Cbf1 (Figure 1F, Table S3). These results imply that the disordered N-terminal domain has a measurable but modest impact on efficient nucleosome binding, and that the structured bHLHZ domain of Cbf1 is largely responsible for strong nucleosome binding.
The basic helix-loop-helix structural motif can invade nucleosomes as either a pioneer or canonical transcription factor
For comparison with Cbf1, we investigated a second budding yeast bHLH TF, Pho4. Pho4 also recognizes an E-box motif via the basic region of the bHLH domain (Figure 2A)26–28 and, unlike Cbf1, Pho4 occupancy is strongly inhibited by nucleosomes in vivo.30,38 We first investigated the affinity of Pho4 to its consensus motif on DNA with EMSAs. Unless otherwise noted, all experiments matched the Cbf1 or Pho4 consensus target sequence with the corresponding Cbf1 or Pho4 basic region. In this assay, we found that the DNA S1/2 was approximately equal to the total DNA concentration of 0.2 nM (Figure S3A). This implies that Pho4 binds DNA stoichiometrically and that the S1/2 does not accurately reflect the dissociation constant (KD). Instead, the S1/2 is an upper limit of the apparent KD for DNA binding.39 We then investigated the Pho4 binding affinity to DNA with ensemble PIFE, which enables experiments at a lower DNA concentration of 50 pM. We measured a Pho4 binding S1/2 within the uncertainty of this lower DNA concentration (S1/2 Pho4 DNA = 30 ± 20 pM, Figure 2B left), and conclude that the apparent KD of Pho4 binding to naked DNA is below 30 pM, reflecting a high binding affinity.
Figure 2. Nucleosomes restrict Pho4 binding by more than 100-fold.
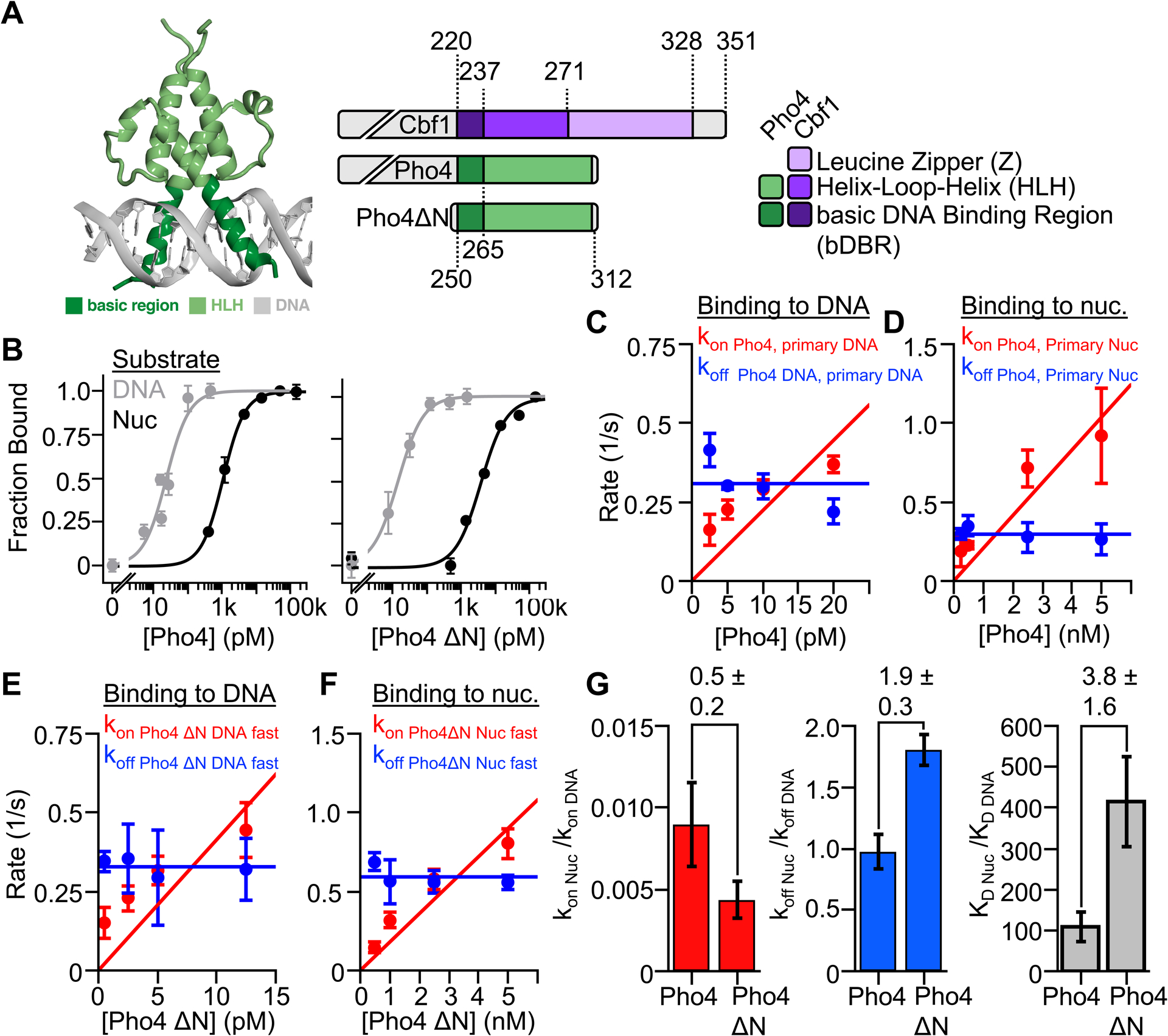
(A) Crystal structure of bHLH TF Pho428 and domain diagrams for Cbf1, Pho4, and Pho4ΔN. (B) Ensemble PIFE (grey) and FRET (black) measurements of full length Pho4 (left) and Pho4ΔN (right) binding their consensus target sequence within DNA and nucleosomes. Binding isotherm fits determine the S1/2 of each titration (Table S1). (C & D) Summary of the smPIFE and smFRET studies of full length Pho4 binding and dissociation kinetics to and from DNA and nucleosomes, respectively (Table S2–S3). (E) Summary of the smPIFE and smFRET studies of Pho4ΔN binding and dissociation kinetics to and from DNA and nucleosome, respectively (Table S2–S3). (G) Summary of the nucleosome binding kinetics (red), dissociation kinetics (blue) and dissociation constant (gray) relative to DNA for both full length Pho4 and Pho4ΔN.
We next characterized Pho4 binding to its consensus sequence within a nucleosome using EMSAs (Figure S3A) and ensemble FRET (Figure 2B left). Here the Pho4-nucleosome affinity measurements occurred over a sufficiently high concentration range that the S1/2 accurately represents the apparent KD. Monitoring FRET while titrating Pho4 reveals it binds via the site exposure mechanism with an S1/2 consistent with our EMSA measurement (S1/2 Pho4 nuc = 1.08 ± 0.04 nM) indicating that nucleosomes impede Pho4 binding by much more than 40-fold (Table S1). We conclude that despite Cbf1 and Pho4 being bHLH TFs that bind nucleosomes via the site exposure model, Cbf1 binds similarly to DNA and nucleosomes, while Pho4 binding is strongly inhibited by nucleosomes.
To understand differences in Cbf1 and Pho4 nucleosome targeting, we considered the possibility that the disordered N-terminal domain of Pho4 inhibits efficient binding. To test this, we purified Pho4ΔN (AA 250–312) and measured DNA and nucleosome binding affinities. We again measured stoichiometric binding to DNA (Figure 2B, right; Figure S3B), which agrees with previous studies.28 Furthermore, Pho4ΔN binds nucleosomes weaker than full length Pho4 (S1/2 Pho4 ΔN Nuc = 3.88 ± 0.93 nM) (Figure 2B right, S3B). These results indicate that, similar to Cbf1, the unstructured N-terminus does not play the dominate role in the differential affinity of Pho4 to nucleosomes and DNA. Instead, the differences in nucleosome binding efficiency between Cbf1 and Pho4 is largely due to differences between their bHLH regions.
Our previous study showed that the canonical TF Gal4 binding within nucleosomes is strongly inhibited because nucleosomes accelerate its dissociation rate.4 We therefore investigated if nucleosomes have the same effect on Pho4 binding. We performed smPIFE and smFRET measurements to determine full-length Pho4 and Pho4ΔN binding and dissociation rates to and from its target site within DNA and nucleosomes (Figures S3C–L). The cumulative probabilities of Pho4 binding and dissociation kinetics to and from DNA and nucleosomes are best fit by two rates, while the cumulative sums of Pho4ΔN kinetics to and from DNA and nucleosomes are best fit by two and one rate, respectively. The concentration dependence of the rates on Pho4 and Pho4ΔN were used to determine the binding rate constants and the dissociation rates (Figure 2C–F). The primary and secondary rates are summarized in Tables S2 and S3. We again focused on primary rates as discussed in the methods.
The nucleosome reduces the binding rate constant of both full-length Pho4 and Pho4ΔN by about 2 orders of magnitude relative to DNA (Figure 2G, Table S3). This is nearly identical to other TFs including Cbf1, Reb1 and LexA.4,14 However, in contrast to Cbf1, the dissociation rate of full-length Pho4 is the same from nucleosomes and DNA, while Pho4ΔN is accelerated off nucleosomes relative to DNA by 2-fold (Figure 2G, Table S3). Overall, these results show that differences between the Cbf1 and Pho4 bHLH domains result in very different interactions with nucleosomes. Cbf1 efficiently invades the nucleosome via the dissociation rate compensation mechanism, while Pho4 occupancy is strongly suppressed by the nucleosome.
The cryo-EM structure of Cbf1 bound to its site within the nucleosome reveals specific interactions between the Cbf1 HLH region and the histone octamer.
We hypothesized the Cbf1’s slowed dissociation from nucleosomes is accomplished through nucleosome-specific interactions. To investigate this hypothesis, we resolved the structure of the Cbf1-nucleosome complex using cryo electron microscopy (cryo-EM) single particle analysis (Figure 3A). We used full-length Cbf1 together with the same consensus Cbf1 motif inserted at the same position within the Widom 601 nucleosome DNA positioning sequence as for our binding experiments. The Cbf1-nucleosome complex was reconstituted, purified by size exclusion chromatography and vitrified without crosslinking. 6339 movies collected on a 300 kV Krios microscope were processed to yield 1.7M particles of the complex. Due to considerable heterogeneity in the Cbf1 region of the map, we employed 3D classification to obtain a more homogenous subset of particles. Reconstructions of subsets containing several hundred thousand particles reached 2.7 Å overall resolution, limited by the Nyquist frequency of the binned images. The volume of the nucleosome core was well-resolved but the region corresponding to Cbf1 displayed considerable flexibility. Multiple rounds of template-guided 3D classification in cryoSPARC, and focused classification without alignment in Relion were performed to obtain a more homogenous subset of particles (Figure S4F). The final subset that was used to build the model consisted of 73k particles and yielded a map with 3.2-Å overall resolution and a local resolution of 4–7 Å in the Cbf1 region (Figure 3C, S4G–J).
Figure 3. Cryo-EM structure of the Cbf1-nucleosome complex reveals specific interactions between the Cbf1 HLH domain and histones.
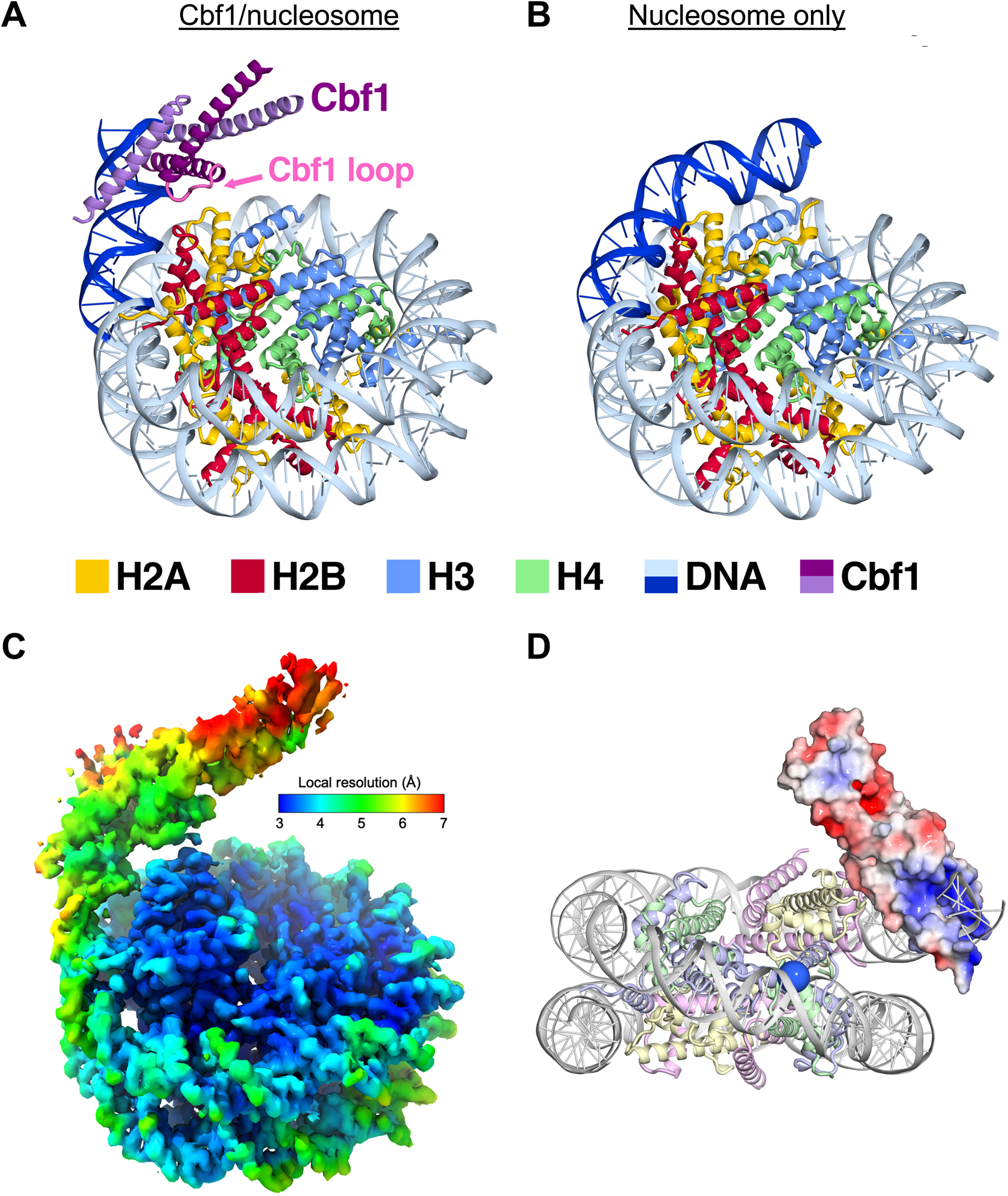
(A) Ribbon representation of Cbf1-nucleosome complex. The Cbf1 dimer is colored in light and dark purple. The unwrapped DNA is in dark blue, while the DNA in contact with the histone octamer is in sky blue. Histone H2A, H2B, H3 and H4 are colored in yellow, red, light blue and light green, respectively. (B) A ribbon representation of the fully wrapped nucleosome.40 The coloring is the same as in panel (A). (C) The 3.2-Å 3D reconstruction used for model building colored by local resolution. (D) Negatively charged patches of Cbf1 (red surfaces) may interact with positively charged histone tails. The APBS-calculated electrostatic86 potential was mapped from −5 to 5 kT/e. The view faces the nucleosome dyad. To indicate where the H3 N-terminal tail extends out, the Cα of the first modeled histone H3 residue, Pro43, is rendered as a blue sphere.
The data supports a model where the homodimer of the bHLHZ domain of Cbf1 (residues 222–288) binds the E-box motif near the nucleosome entry-exit site. In agreement with our FRET studies, our model shows how Cbf1 traps the nucleosome in an unwrapped state with at least 20 bp of DNA peeled off the histone octamer (Figure 3A vs. Figure 3B).40 In addition to unwrapping, the nucleosomal DNA occupied by Cbf1 is angled away from the histone core. In this conformation, the Cbf1 HLH region is positioned to directly interface with the octamer. Extensive 3D classification yielded particle classes where Cbf1 and the unwrapped DNA are positioned at various angles relative to the nucleosome core (Figure S4A). The continuous nature of the structural heterogeneity prevents further improvement of the Cbf1 map. A local resolution of about 4 Å was reached in the vicinity of the interacting histones, but the resolution dropped off to 5 Å and worse in the periphery (Figure 3C).
Despite the dynamic nature of this structure, which makes resolving Cbf1-histone contacts difficult, this model indicates important regions for interaction. The loop of the HLH domain appears to be positioned close to the core histones. We observe map density corresponding to an interaction between Cbf1 E253 and H2A K75 on the H2A histone fold L2 loop. In addition, Cbf1 D247 and R252 can be modeled to interact with H2A K75 and H2B D54, respectively (Figure S4C and S4D). This region in combination with Cbf1-DNA binding likely contribute to a collection of interactions that result in the observed pivoting of Cbf1 relative to the nucleosome (Figure S4A). We also replaced Cbf1 with the known Pho4-DNA structure28 and found Pho4 is positioned similarly. However, the Cbf1 residues that interact with these histone regions are not conserved within Pho4 (Figure S4E), suggesting that Pho4 is unable to interact with the histone octamer.
To investigate the importance of Cbf1-histone interactions we prepared Cbf1ΔN3 Ala Sub, which contains all three Cbf1 residues mentioned above substituted with alanine. We carried out binding titrations with DNA and nucleosomes and find that the relative affinities are decreased by 1.6-fold, suggesting that these interactions modestly contribute to nucleosome specific interactions. In addition, the entire Cbf1 HLH region features many more negatively charged solvent-exposed residues capable of electrostatically interacting with the histone core and/or histone tails (Figure 3D) than Pho4 (Figure S4K). To determine if Cbf1-histone tail interactions contribute to nucleosome specific Cbf1 binding, we prepared nucleosomes with the first 33 residues of H3 removed. We then carried out Cbf1ΔN and Pho4ΔN titrations with H3-tailless nucleosomes, with the latter controlling for the H3 N-terminal tail’s impact on nucleosome unwrapping.41–44 We find that the S1/2 of Cbf1ΔN is reduced by 1.3-fold, while that of Pho4ΔN is reduced by 3.1-fold (Figure S4M). Taken together, we conclude that H3 tail removal only subtly decreases Cbf1ΔN binding affinity because of the opposing effects of increased binding site accessibility and decreased interactions with the H3 tail indicating that Cbf1-H3 N-terminal tail interaction provides about a 3-fold contribution to nucleosome specific interactions.
To provide additional insight into the HLH region of Cbf1, we used Clustal Omega45 to compare Cbf1 to nearly 100 human bHLH TFs and found that Cbf1 has the highest percent identity with USF1, USF2, and USF3 (Figure S5A). While it is not known if these function as PFs, a crosslinking mass spectrometry study showed that the HLH region of USF3 interacts with histone octamer,46 further supporting the conclusion that the Cbf1 HLH region interacts with the histone octamer. We also compared Cbf1 and Pho4 to groups of known human PFs (Ascl1, NGN2, MyoD1)16,47–50 and canonical TFs (Mad1, SRBP2, Myc, Max) (Figure S5B–C).16,51
Most of the homology resides in the helix regions with no homology in the loop region. Interestingly, Cbf1 has higher sequence identity to all 7 of these TFs. Furthermore, Cbf1 is more than 5 percent higher than Pho4 in sequence identity for all 3 PFs, while the percent difference for the canonical TFs was mixed (Figure S5D–E) suggesting that Cbf1 tends to align better to known human PFs than Pho4. Overall, these cryo-EM, binding and sequence alignment studies provide synergistic evidence that Cbf1 interacts with the histone octamer through numerous modest and diffuse electrostatic interactions and provides a mechanism for its slow dissociation from the nucleosome.
The addition of Cbf1 Leucine Zipper to Pho4 does not enhance DNA or nucleosome binding
Our single-molecule measurements indicate that the bHLHZ and bHLH regions of Cbf1 and Pho4, respectively, are largely responsible for binding DNA and nucleosomes. However, much of the Cbf1 leucine zipper is unresolved in our cryo-EM structure. We therefore considered the possibility that the C-terminal leucine zipper extension of Cbf1, which Pho4 does not contain, is important for efficient binding within nucleosomes. To test this hypothesis, we fused Cbf1 residues 270–351 onto Pho4’s C-terminus (Pho4 Zip, Figure S4O). Just like full-length Pho4, we measured Pho4 Zip binding to the Pho4 consensus motif within DNA and nucleosomes. We observe stoichiometric binding of Pho4 Zip to its target site within DNA (Figure S4O), while Pho4 Zip binds nucleosomes with the same affinity as full-length Pho4 (Figure S4O). This indicates that the Cbf1 leucine zipper does not contribute to the increased efficiency of binding within nucleosomes. This result is consistent with the cryo-EM structure where the start of the leucine zipper extends away from the core histones.
The HLH region regulates the dissociation rate from nucleosomes.
Our structure of the Cbf1-nucleosome complex indicates the HLH region interacts directly with the histone octamer. To further investigate this region’s role in controlling binding kinetics, we attempted to eliminate the HLH-nucleosome interaction. To do this, we generated a Cbf1-Pho4 chimera where the Cbf1 HLH region, residues 237–351, were replaced with the corresponding region in Pho4, residues 265–312 (Figure 4A). We refer to this chimera as “Cbf1ΔN PHLH”, where “PHLH” represents the “Pho4 Helix-Loop-Helix” dimerization region. Using ensemble PIFE measurements, we find that Cbf1ΔN PHLH binds stoichiometrically to the Cbf1 consensus motif on naked DNA with a S1/2 of 37 ± 5 pM (Figure S6A–B, Table S1). We then used ensemble FRET measurements to determine that the S1/2 for Cbf1ΔN PHLH binding to nucleosomes is 3.1 ± 0.5 nM (Figure S6C, Table S1), representing an 80 ± 10-fold increase from that of DNA (Table S1). In comparison, Cbf1ΔN binding to nucleosomes relative to DNA (S1/2 Nuc / S1/2 DNA) is only 3.4 ± 0.7-fold higher. These results show that, in contrast to Cbf1ΔN, Cbf1ΔN PHLH chimera behaves more like Pho4ΔN, where it binds tightly to the Cbf1 motif but is unable to efficiently target the nucleosome.
Figure 4. The Cbf1 HLH region facilitates pioneer activity through the dissociation rate compensation mechanism.
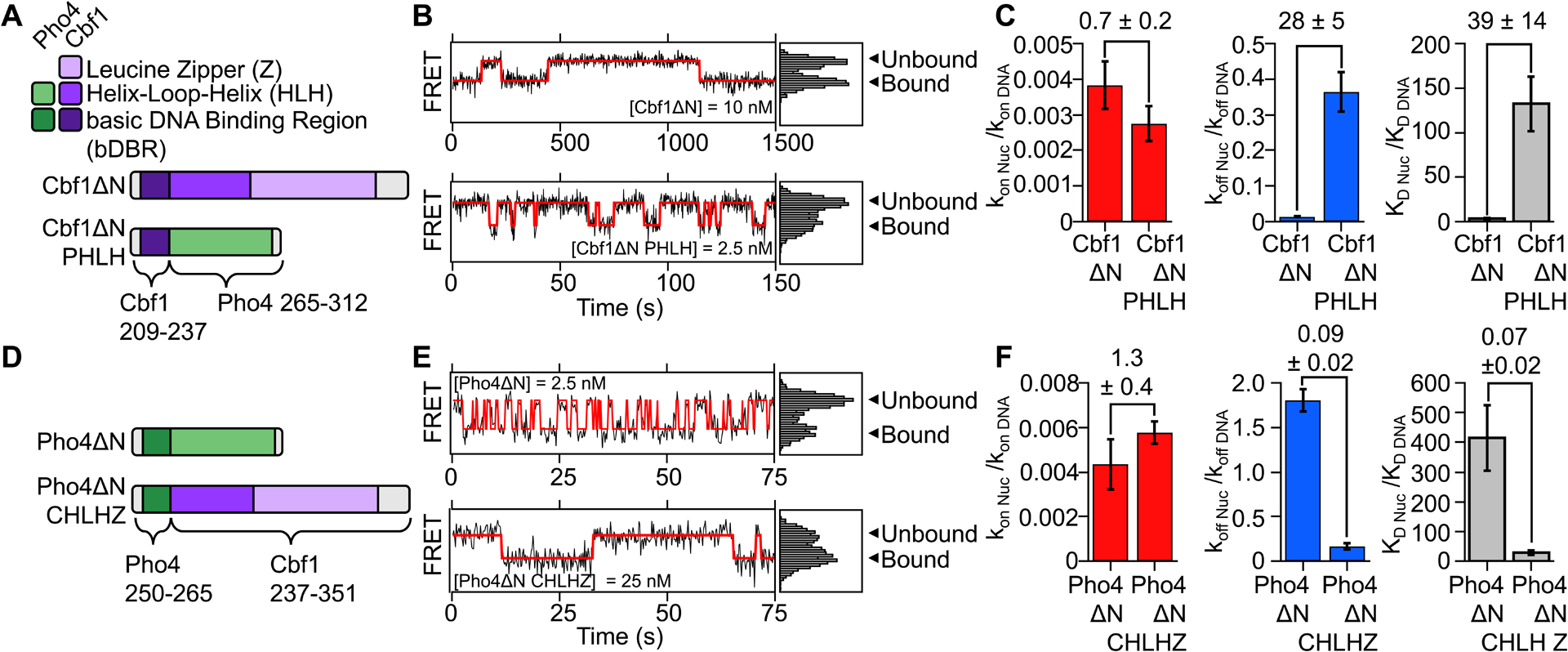
(A) Domain diagram of Cbf1ΔN and the Cbf1ΔN PHLH chimera where Cbf1 residues 237–351 was swapped out for Pho4 residues 265–312. (B) Representative smFRET time traces of Cbf1ΔN (top) and Cbf1ΔN PHLH (bottom) binding to and dissociating from nucleosomes. (C) Comparison of nucleosome binding rates (red), dissociation rates (blue) and dissociation rates relative to DNA for Cbf1ΔN and Cbf1ΔN PHLH. (D) Domain diagram of Pho4ΔN and the Pho4ΔN CHLHZ chimera where Pho4 265–312 was swapped out for Cbf1 residues 237–351. (E) Representative smFRET time traces of Pho4ΔN (top) and Pho4ΔN CHLHZ (bottom) binding to and dissociating from nucleosomes. (F) Comparison of nucleosome binding rates (red), dissociation rates (blue) and dissociation rates relative to DNA for Pho4ΔN and Pho4ΔN CHLHZ.
We then investigated if the low relative affinity of Cbf1ΔN PHLH towards the nucleosome results from the loss of the dissociation rate compensation mechanism. SmPIFE and smFRET experiments were used to determine the Cbf1ΔN PHLH binding rate constant and dissociation rate to and from the Cbf1 consensus motif within DNA (Figure S6D–F, Table S2), and nucleosomes (Figure S6G–H, Table S2), respectively. Visual inspection of time traces shows many binding fluctuations per nucleosome during one video, indicating that Cbf1ΔN PHLH bound dwell times are much shorter to nucleosomes compared Cbf1ΔN (Figure 4B, S2A vs S2D). Quantification of the measured dwell times reveals that nucleosomes reduce the binding rate constant of Cbf1ΔN PHLH by 360-fold (kon nuc / kon DNA = 0.0027 ± 0.0005), similar to the reduction with Cbf1ΔN (Figure 4C, Table S3). However, the Cbf1ΔN PHLH dissociation rate is only 3-fold slower from nucleosomes relative to DNA (koff nuc / koff DNA = 0.36 ± 0.05, Figure 4C, Table S3), while the Cbf1ΔN relative dissociation rate is 80-fold (koff nuc / koff DNA = 0.013 ± 0.002, Figure 4C, Table S3). These results show that replacing the Cbf1 HLH region with the Pho4 HLH region largely abolishes the slow nucleosome dissociation rate relative to DNA and strongly inhibits Cbf1ΔN PHLH binding to nucleosomes.
Our studies of Cbf1ΔN PHLH imply that Cbf1 residues 237–351, in addition to mediating dimerization, are responsible for slowed dissociation from nucleosomes. To further investigate this idea, we prepared another Cbf1-Pho4 chimera, Pho4ΔN CHLHZ (CHLHZ = Cbf1 Helix-Loop-Helix dimerization region), where the Pho4 HLH region (residues 265–312) is replaced with the Cbf1 HLH region (residues 237–351) (Figure 4D). The combination of ensemble PIFE measurements of DNA binding and FRET measurements of nucleosome binding reveal that nucleosomes reduce Pho4ΔN CHLHZ binding by 40-fold (S1/2 Nuc / S1/2 DNA = 40 ± 20). This is a 7-fold reduction in relative nucleosome binding affinity compared to Pho4ΔN (S1/2 Nuc / S1/2 DNA > 270 ± 70) (Figure S6I–K, Table S1), and indicates that addition of the Cbf1 HLH region facilitates nucleosome binding.
To directly probe if Pho4ΔN CHLHZ acquired efficient nucleosome binding via the dissociation rate compensation mechanism, we performed SMF measurements of Pho4ΔN CHLHZ binding and dissociation dynamics to and from DNA (Figure S6L–N, Table S2) and nucleosomes (Figure S6O–P, Table S2). We found that the Pho4ΔN CHLHZ binding rate to its site within the nucleosome is reduced by 170-fold relative to the DNA (kon nuc / kon DNA = 0.0058 ± 0.0005). This is similar to the 250-fold reduced relative binding rate of Pho4ΔN. In contrast, unlike Pho4ΔN, which dissociates 2-fold faster from nucleosomes (koff nuc / koff DNA = 1.8 ± 0.1), the dissociation rate of Pho4ΔN CHLHZ from nucleosomes is slower by 6-fold relative to the dissociation rate from DNA (koff nuc / koff DNA = 0.16 ± 0.04, Figure 4E–F, Table S3). This is more than a factor of 10-fold reduction in the nucleosome dissociation rate relative to DNA between Pho4ΔN CHLHZ and Pho4ΔN. Taken together, these results show that the HLH dimerization region of Cbf1 enables the slow nucleosome dissociation rate of Pho4ΔN CHLHZ resulting in efficient binding within the nucleosome via the dissociation rate compensation mechanism. These combined studies of Cbf1-Pho4 chimeras suggest that the HLH region of Cbf1 provides additional interactions with the nucleosome, which that are absent in Pho4 HLH region, and contribute to efficient targeting of Cbf1 within the nucleosome.
The Cbf1 HLH region imparts nucleosome displacement in vivo.
To address how slowed dissociation from nucleosomes imparted by the Cbf1 HLH region relates to function in live cells, we probed these chimeric TFs’ ability to establish nucleosome depleted regions (NDRs) by generating yeast strains with a few genetic modifications (Table S4, Figure S7A, C). First, we introduced Pho4ΔN, Cbf1ΔN, or chimeras driven by the MET3 promoter so that these factors inducible by methionine depletion. These TFs contain a nuclear localization signal (NLS) and a GFP on their C-terminal to validate protein expression and localization. Note that Pho4ΔN lacks the phosphorylation sites that are required for their cytoplasmic localization in high phosphate media.52 Each factor was nuclear-localized and expressed to similar levels upon methionine depletion (Figure S7A). Second, to eliminate endogenous Cbf1, we replaced the CBF1 promoter with the Gal1 promoter so that the endogenous Cbf1 is depleted in glucose media. Endogenous Pho4 was intact, but all the following measurements were performed in high phosphate conditions so Pho4 was sequestered in the cytoplasm.52 Finally, on a mutated HO promoter assembled into a constitutive nucleosome array (nucleosome -1 to -7),23 we engineered a single Pho4 or Cbf1 binding site 43 bp from the dyad within nucleosome -4. Therefore, accessing these motifs by Pho4 or Cbf1 variants requires direct competition with the nucleosome. Using these strains, we assayed how these mutant TFs influence nucleosome positioning near the engineered motifs using an MNase digestion assay followed by stacking qPCR. We found that Pho4ΔN, Cbf1ΔN, and the chimeras bind to both binding sites specifically within nucleosomes (Figure S7B). So, we investigated all four variants with both binding sites.
Using strains with the Cbf1 motif, we observed a clear shift in nucleosome positioning and the formation of a short NDR upon expression of Cbf1ΔN, indicating that Cbf1ΔN, like the full-length Cbf1, has nucleosome displacement activity in vivo (Figure 5A, top panel). No discernible changes in nucleosome positioning were observed upon induction of Pho4ΔN (Figure 5A, panel 2/4). Remarkably, swapping the HLH region of the two factors reversed their nucleosome invasion activity: Pho4ΔN CHLHZ, but not Cbf1ΔN PHLH, was able to shift nucleosome positioning near the Cbf1 motif (Figure 5A, bottom two panels). These results are highly correlated with our in vitro observations that Pho4ΔN CHLHZ has a slower dissociation rate from nucleosomes than Pho4ΔN, whereas Cbf1ΔN PHLH dissociates faster than Cbf1ΔN.
Figure 5. The Cbf1 HLH region imparts efficient nucleosome targeting and displacement in vivo.
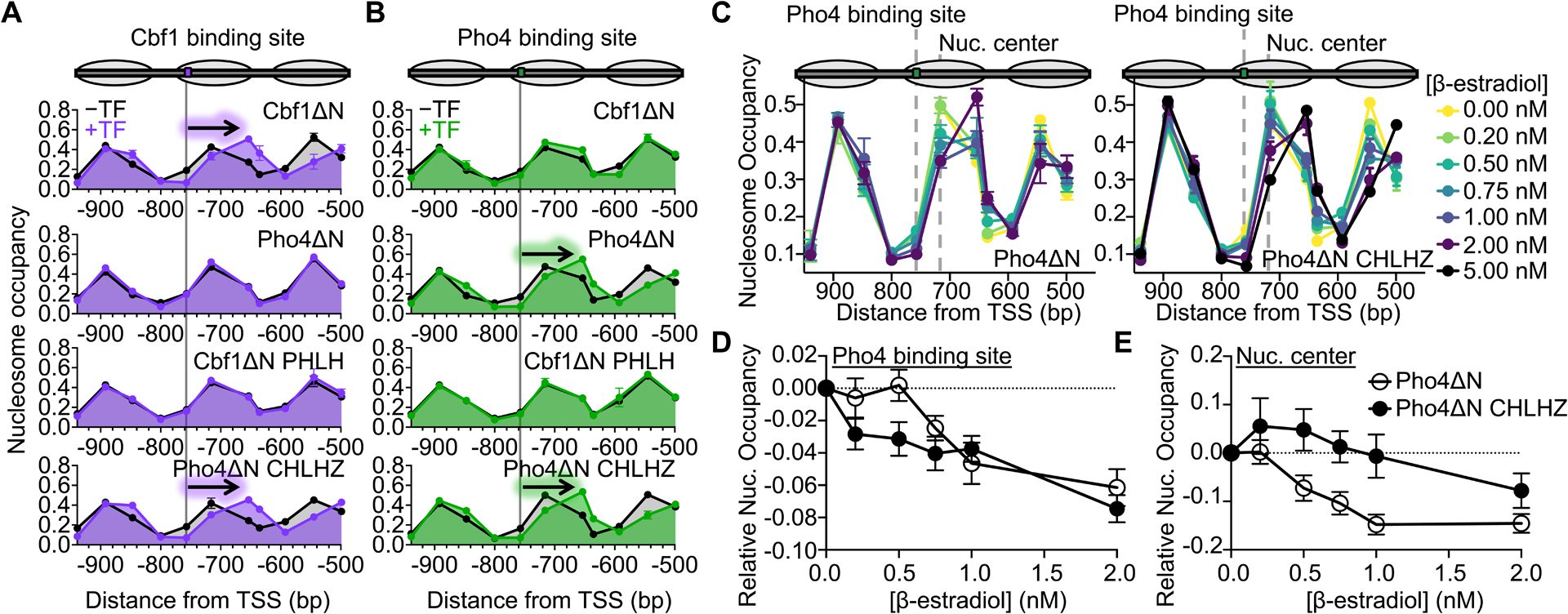
(A) and (B) Nucleosome occupancy within the HO promoter that contains a Cbf1 or Pho4 target site, respectively. The Cbf1 and Pho4 target site positions in the −4 nucleosome are indicated by the gray line, respectively. Nucleosome occupancy was determined in the presence (black) or absence of methionine (purple or green), where methionine depletion induces the expression of either Cbf1ΔN, Pho4ΔN, Cbf1ΔN PHLH, or Pho4ΔN CHLHZ. The arrow indicates which TFs shift the position of the −4 nucleosome. (C) Nucleosome occupancy within the HO promoter with the Pho4 target sequence in the same location as panel (B). β-estradiol titrates the expression level of Pho4ΔN (left) or Pho4ΔN CHLHZ (right), respectively. The grey dotted lines indicate the location of the Pho4 target sequence (−775 bp) and the nucleosome center without TF expression (−716 bp). (D) Relative nucleosome occupancy at the Pho4 binding site (−757 bp from the TSS) upon induction of Pho4 ΔN (white) or Pho4 ΔN CHLHZ (black) with increasing levels of β-estradiol. (E) Relative nucleosome occupancy measured at the center of the −4 nucleosome (−716 bp from the TSS) with increasing levels of β-estradiol.
We also assayed how these proteins influence nucleosome positioning within strains containing the Pho4 motif (Figure 5B, top panel). Not surprisingly, Cbf1ΔN and Cbf1ΔN PHLH, which bind 300-fold weaker to the Pho4 site in vitro (Figure S7B), have no influence on nucleosome positioning. Interestingly, we observed a shift in nucleosome positioning upon expression of Pho4ΔN (Figure 5B, panel 2/4), which is surprising given that Pho4 has been reported to be blocked by nucleosomes.30 However, this is consistent with other very tight DNA binders like the bacterial TF TetR that generates small NDRs.23 Finally, we observed a clear shift in nucleosome positioning when expressing Pho4ΔN CHLHZ (Figure 5B, bottom panel). These combined results indicate two mechanisms for TFs targeting nucleosomal binding sites: (1) through tight DNA binding as in the case of Pho4ΔN and (2) through the dissociation rate compensation mechanism as in the case of TFs possessing the Cbf1 HLH region.
Since highly expressed Pho4ΔN and Pho4ΔN CHLHZ can displace nucleosomes in vivo, we further investigated their functional differences by comparing their nucleosome displacement function at lower concentrations. We constructed strains with either factor under control of a GAL1 promoter activated by Gal4DBD-ER-VP16,53 so the concentration of β-estradiol modulates Pho4ΔN or Pho4ΔN CHLHZ expression level (Figure S7C). We observed concentration-dependent nucleosome displacement activities in both cases, where higher concentrations of these factors decrease nucleosome occupancy near their binding sites and shift the nucleosome away from the binding sites (Figure 5C). For Pho4ΔN, these two events correlate well, and both start at ~ 0.5–1 nM of β-estradiol (Figure 5D–E). For Pho4ΔN CHLHZ, however, 0.2 nM of β-estradiol is sufficient to reduce nucleosome occupancy near its binding site, but the nucleosome shift requires 2 nM of β-estradiol (Figure 5D–E). These observations indicate that Pho4ΔN CHLHZ may bind within the nucleosome through DNA unwrapping at low concentrations, increasing the local DNA accessibility without nucleosome translocation. This is consistent with the more efficient Pho4ΔN CHLHZ-nucleosome binding in vitro. Higher levels of Pho4ΔN CHLHZ traps the nucleosome more in such a partially unwrapped unstable state, which may lead to the recruitment of downstream factors and nucleosome movement. In contrast, Pho4ΔN has shorter dwell times on the nucleosome, and its binding appears more mutually exclusive with the nucleosome.
As a comparison, we also measured the nucleosome occupancy over Cbf1 and Pho4 binding sites in the presence of full-length Cbf1 and Pho4 at high expression level. We used a mutant Pho4 (S100A, S114A, S128A, S152A, and P224A) to keep this factor inside nuclei.54 In comparison with Cbf1ΔN and Pho4ΔN, full-length Cbf1 and Pho4 also reduce the local nucleosome occupancy near their respective binding sites, but they lead to more prominent changes: full-length Cbf1 causes bigger downstream shift of nucleosome −4, while full-length Pho4 significantly reduces the nucleosome −4 occupancy (Figure S6D). We speculate that these larger effects are due to co-activators recruited by the full-length proteins.55
DISCUSSION
Pioneer TFs are the first proteins to bind within silenced chromatin, requiring the ability to target DNA sites within nucleosomes.16,17,56 In contrast, canonical TFs contribute to transcription activation after their DNA target site is released from nucleosomes, which significantly limit binding accessibility.1 In this study, we provide mechanistic insight into how two structurally similar bHLH TFs, Cbf1 and Pho4, can have significantly different pioneering activities (Figure 6A). We find that Cbf1 has comparable affinities towards nucleosome and naked DNA through a dissociation rate compensation mechanism. Neither the N-terminal disordered region, the C-terminal leucine zipper, nor the basic DNA binding region contribute significantly to the slow dissociation of Cbf1 from the nucleosome. Instead, the Cbf1 HLH region enables efficient nucleosome invasion via the dissociation rate compensation mechanism, while the corresponding HLH region of Pho4 does not slow dissociation. Importantly, we show that these HLH regions of Cbf1 and Pho4 can be swapped, resulting in the transfer of the nucleosome dissociation and invasion properties between TFs (Figure 6B). This indicates that the HLH region can function as a key pioneering module.
Figure 6. The Cbf1 and Pho4 HLH regions define their ability to invade nucleosomes in vitro and reposition nucleosomes in vivo.
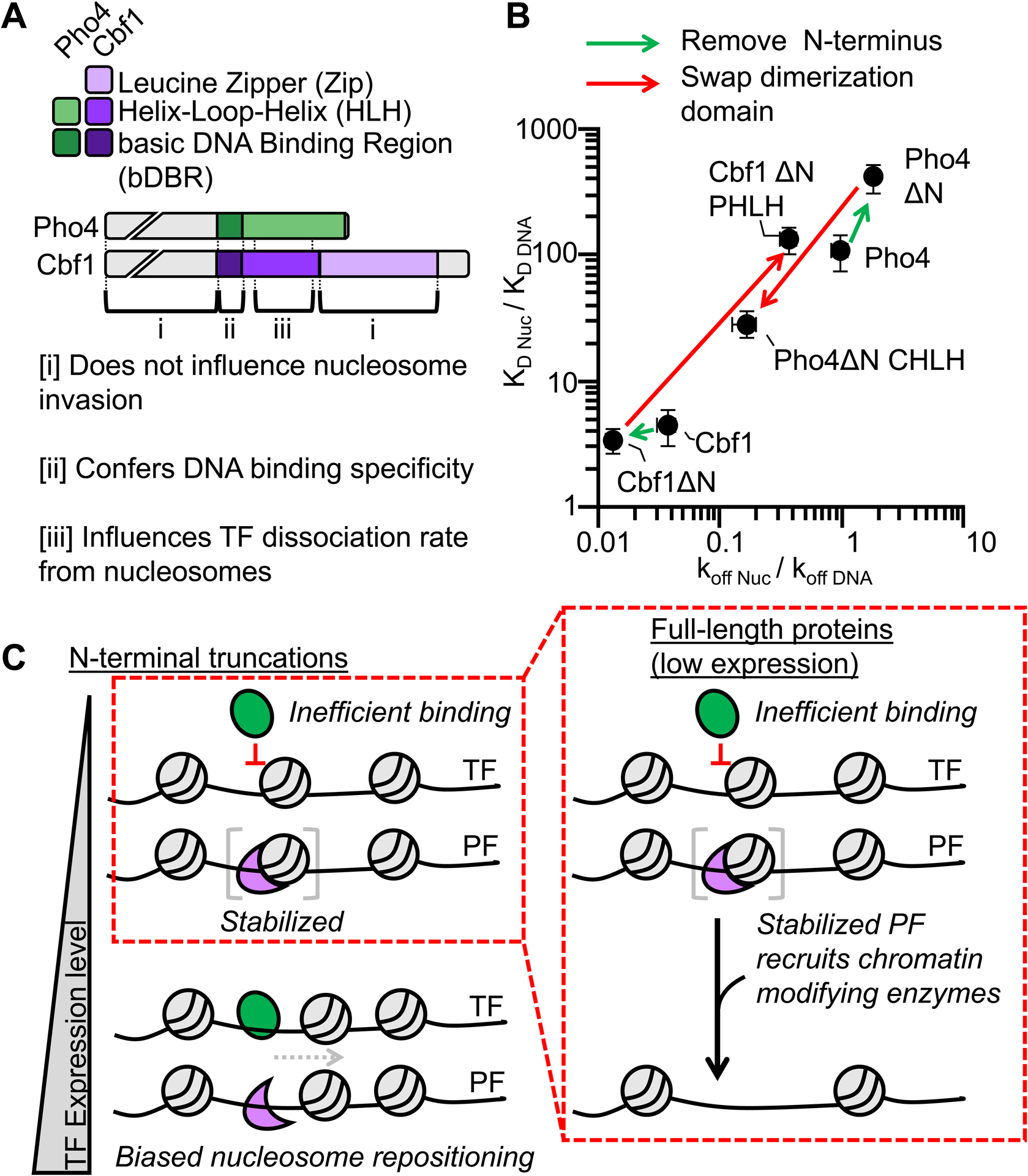
(A) Domain diagrams of full-length Pho4 and Cbf1 with the regions denoted by brackets on how they influence nucleosome invasion. (B) Log-log plot relating the efficiency of nucleosome invasion (KD Nuc/KD DNA, y-axis) to the relative dissociation rate (koff Nuc /koff DNA, x-axis). Each point on this plot represents a different measurement from this study or Donovan et al.14 The arrows between points indicate how specific mutations change relative dissociation rate and the corresponding efficiency of nucleosome invasion. (C) Model of nucleosome invasion and displacement by bHLH PFs that rely on the dissociation rate compensation mechanism and canonical bHLH TFs that are inhibited by nucleosomes.
Our structure of the Cbf1-nucleosome complex provides important molecular insight by confirming that the Cbf1 bHLH domain’s DNA interactions are similar to other bHLH TFs including Pho4, and showing for the first time that the Cbf1 HLH region interacts with histones H2A and H2B. In addition, the acidic region of the Cbf1 HLH region is potentially positioned to interact with the highly basic N-terminal H3 tail, which is disordered and was not resolved by cryo-EM. Cbf1 interactions with histones appear to stabilize Cbf1 binding within the nucleosome resulting in the substantially slower dissociation rate from nucleosomes relative to DNA.
Cbf1-nucleosome structure reveals that a PF can stabilize a partially unwrapped nucleosome state, which potentially provides access to additional DNA binding factors. Similar observations have been made with some human PFs, e.g. Sox2 and Oct4 can disrupt histone-DNA contacts and stabilize partial DNA unwrapping.24,25 However, these PFs increase DNA accessibility via induced DNA distortions upon binding. In contrast, Cbf1 interacts with both DNA and histones, strengthening its stability on nucleosome surface. These additional interactions with the nucleosome provides a molecular picture of the dissociation rate compensation mechanism.14
PFs possess two key properties: (i) they efficiently target sites within nucleosomes14–16,57–59 and (ii) they initiate the formation of NDRs through nucleosome translocation and/or eviction.8,23 However, a quantitative and direct connection between these two PF properties has remained elusive. This work provides a direct connection between these two PF properties by investigating in vivo the Cbf1-Pho4 chimeras we studied in vitro. Our results that Cbf1ΔN and Pho4ΔN CHLHZ, but not Cbf1ΔN PHLH and Pho4ΔN, efficiently reposition nucleosomes away from embedded Cbf1 motif indicate that the Cbf1 HLH region promotes nucleosome displacement activity in vivo. This is highly consistent with the in vitro measurement that the Cbf1 HLH region enables slow dissociation from the nucleosome and directly interacts with the histone octamer. However, our studies also show that Pho4ΔN can displace nucleosomes over a Pho4 motif at high expression level. This is likely due to the high affinity of Pho4ΔN towards its consensus motif. Indeed, depending on the target site affinity and concentration, the canonical TF, HNF4A, has been reported to function as a pioneer factor,60 and even a prokaryotic TF can induce nucleosome displacement at high concentrations.23 Our results combined with these published results indicate that the pioneering ability of a TF depends on multiple factors, and any TF can function as a PF if the overall binding affinity and expression is high enough. Cbf1ΔN and Pho4ΔN appear to use distinct mechanisms to invade nucleosomes: the former may employ an “active” mechanism where Cbf1ΔN directly docks onto nucleosomes and recruits downstream factors to reposition nucleosomes. The latter may use a “passive” mechanism where Pho4ΔN binds transiently exposed nucleosomal DNA and prevents nucleosome formation. Consistent with the “docking” state in the active mechanism, Pho4ΔN CHLHZ unwraps nucleosomal DNA at lower concentrations but does not displace the nucleosome until it is highly expressed. In contrast, Pho4ΔN binding appears more correlated with nucleosome displacement. The detailed molecular pathways for the active vs passive nucleosome invasion require further investigation.
These combined results suggest a model (Figure 6C) where nucleosomes effectively block Pho4 (a canonical TF) binding at low expression levels. However, Cbf1 (a PF), even at low expression levels, binds a nucleosome-embedded motif through the dissociation rate compensation mechanism. The extra interactions of Cbf1 with the histones suppress spontaneous nucleosome repositioning. However, Cbf1’s slow dissociation rate provides time for recruitment of chromatin modifying complexes most likely through the disordered N-terminal region. This results in the large NDR where nucleosomes are significantly shifted (Figure S7D)
These combined results suggest a model (Figure 6C) where nucleosomes effectively block Pho4 (a canonical TF) binding at low expression levels. However, Cbf1 (a PF), even at low expression levels, binds a nucleosome-embedded motif through the dissociation rate compensation mechanism. The extra interactions of Cbf1 with the histones suppress spontaneous nucleosome repositioning. However, Cbf1’s slow dissociation rate may provide time for chromatin modifying complexes to bind and facilitate NDR formation. At higher expression levels, both Cbf1 and Pho4 fully occupy their sites resulting in spontaneous repositioning of the nucleosome. These combined single molecule, cryo-EM structural, and in vivo studies provide significant molecular insight into how a PF interacts with the nucleosome through the dissociation rate compensation mechanism to reposition nucleosomes and initiate transcription activation.
Limitations of the Study
This study has included two Ebox binding site sequences while in vivo a TF targets a range of sequences. In addition, we focused on TF binding within the entry-exit region of the nucleosome. Variation in binding site sequence and the location of the binding site within the nucleosome are anticipated to impact the affinity of both canonical TFs and PFs and potentially their function. Future quantitative studies will be important to resolve the question of how variations in the target site sequence and changes in nucleosome position influence PF specificity and function.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed and will be fulfilled by the Lead Contact, Michael G. Poirier (poirier.18@osu.edu)
Materials Availability
Unique and stable reagents generated in this study are available upon request.
Data and Code Availability
All time traces from single-molecule experiments and unprocessed gel images have been uploaded to Zenodo (10.5281/zenodo.7683126). The time traces can be viewed using vbFRET in matlab.35 The structural model, the cryo-EM map and micrographs of Cbf1-nucleosome complex are available as of the date of publication at PDB 7SSA, EMD-25406 and EMPIAR-10875, respectively.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. cerevisiae were cultured in synthetic complete media + 2% glucose in the presence or absence of methionine. Strain details are in Table S3.
METHODS DETAILS
Preparation of Cbf1, Cbf1ΔN, and Pho4ΔN CHLHZ
Cbf1 cloned into the pET28 expression vector was a gift from S. Diekmann and expresses the Cbf1 coding sequence from the S288C strain of budding yeast. The expression vectors for Cbf1ΔN, and Pho4ΔN CHLHZ were prepared by site directed mutagenesis (Agilent) using the Cbf1 WT expression vector as a template. Expression vectors were transformed into BL21(DE3) cells (Invitrogen). When the OD600 was between 0.4–0.6, cells were induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 4 hours at 37°C. After protein expression, cells were then pelted and stored at −80°C until purification. Cell pellets were thawed in buffer A (50 mM Na2HP04 (pH 7.5), 300 mM NaCl, 5 mM imidazole, 10% glycerol, 1 mM PMSF, 20 ug/mL pepstatin, 20 ug/mL leupeptin), lysed by sonication, clarified by centrifugation (4 C, 23,000 × G, 20 min), and loaded directly onto a 5 mL HisTrap HP Ni-NTA column (Cytiva). The column was first washed with 40 mL buffer A and then 120 mL buffer B (50 mM Na2HP04 (pH 7.5), 300 mM NaCl, 60 mM imidazole, 10% glycerol, 1 mM PMSF, 20 ug/mL pepstatin, 20 ug/mL leupeptin). Protein was then eluted with buffer C (50 mM Na2HP04 (pH 7.5), 300 mM NaCl, 340 mM imidazole, 10% glycerol, 1 mM PMSF, 20 ug/mL pepstatin, 20 ug/mL leupeptin). Pure fractions (as determined by SDS PAGE) were pooled, and imidazole was removed by dialysis into Buffer D (50 mM Na2HP04 (pH 7.5), 300 mM NaCl, 1 mM PMSF, 20 ug/mL pepstatin, 20 ug/mL leupeptin) in a 10 K amicon (Millipore). Concentration was determined by measuring absorbance at 280 nm.14 In all binding assays we report the concentration of protein dimer.
Preparation of Pho4, Pho4ΔN, Pho4 Zip, and Cbf1ΔN PHLH
The Pho4 expression vector was a gift from the R. Kornberg lab.61 Pho4ΔN, Pho4ΔN Zip, and Cbf1ΔN PHLH expression vectors were prepared by site directed mutagenesis (Agilent) and contain a 6x-His Tag. All proteins were expressed in BL21(DE3) (invitrogen) by inducing at OD600 = 0.4–0.6 with 1 mM IPTG for 3 hours at 37°C. Cells were resuspended in 15 mL lysis buffer (20 mM Tris-HCL pH8, 500 mM NaCl, 10 mM imidazole, 0.2% Triton X-100, 5 mM DTT, 1 mM PMSF, 20μg/mL pepstatin, 20 μg/mL leupeptin, 2.1 mM benzamidine hydrochloride) per 600 mL culture and lysed by sonication. Cell debris was removed by centrifugation, loaded onto a 5 mL HisTrap HP Ni-NTA column (Cytiva), washed 150 mL wash buffer (20 mM Tris-HCL pH8, 500 mM NaCl, 5 mM imidazole, 0.02% Triton X-100, 5 mM DTT) followed by 150 mL wash of wash buffer without Triton X-100. Bound proteins were eluted in wash buffer with a gradient from 25–500 mM imidazole. Peak fractions were further purified with a superdex s200 10/300 (GE) size exclusion column equilibrated with storage buffer (40 mM HEPES-NaOH pH 7.4, 200 mM potassium acetate, 1 mM EDTA). Pure fractions, as determined by Coomassie SDS PAGE, were pooled, concentrated, glycerol added to final concentration of 10% v/v, flash-frozen, and stored at −80°C. In all binding assays we report the concentration of protein dimer.
Preparation of DNA molecules
DNA molecules were prepared by PCR using primers (Sigma, Table S5) from a plasmid containing the 601 nucleosome positioning sequence (NPS).62 Fluorescently labeled oligos were labeled at their 5’-ends with Cy3 or, in the case of the reverse primer for smPIFE, internally labeled at an amine modified dT with Cy5. Fluorescently labeled oligos were purified by HPLC with a 218TP C18 column (Grace/Vydac). After amplification, DNA molecules were purified using a MonoQ column (Cytiva).
Preparation of Histone Octamers
Human recombinant histones were purchased from “The Histone Source” which operates at Colorado State University (https://histonesource-colostate.nbsstore.net/). Octamer reconstitution was performed with WT histones with the exception of H3(C110A) and H2A(K119C) which is required for labeling with Cy5 maleimide. The histone octamer was refolded by adding each of the histones together with H2A/H2B in 10% excess of H3/H4 as previously described.63 The histone octamer was labeled with Cy5-maleimide (GE Healthcare) and purified with a Superdex 200 10/300 (Cytiva) size exclusion column as previously described.64
Preparation of nucleosomes
Nucleosomes were reconstituted by salt gradient dialysis as previously described.64 Briefly, Cy3-DNA and Cy5-octamer were mixed at a ratio of 1.25:1 in 0.5x TE (5mM Tris-HCL, 0.5mM EDTA, pH 8.0) + 2M NaCl. Nucleosomes were reconstituted by two rounds of double dialysis into 0.5x TE at 4°C with at least 6 hrs per change.64 Nucleosomes were then loaded onto 5–30% sucrose gradients and purified by centrifugation on an Optima L-90 K ultracentrifuge for 22 hrs (Beckman Coulter) with a SW-41 rotor. Sucrose fractions containing nucleosomes were collected, buffer exchanged into 0.5x TE (pH 8.0). concentrated, and stored on ice.
Electrophoresis mobility shift assays
0.5 nM DNA or nucleosomes were incubated with a specific TF in 10 mM Tris-HCl (pH 8), 130 mM NaCl, 10% glycerol, 0.0075% v/v Tween-20 for at least 5 min and then resolved by electrophoretic mobility shift assay (EMSA) with a 5% native polyacrylamide gel in 0.3x TBE (30mM Tris, 30mM Boric Acid, 6 mM EDTA, pH 8.0). For Pho4 and Pho4ΔN, which bind DNA and nucleosomes very tightly, the experiment was repeated with 0.2 nM DNA and nucleosomes.
Ensemble PIFE measurements
Specific TF binding to Cy3-DNA was determined by protein-induced fluorescence enhancement (PIFE), in which Cy3 fluorescence increases upon protein binding as previously described.14 DNA was incubated for at least 5 min with varying concentrations of TF in 10 mM Tris-HCl (pH 8), 130 mM NaCl, 10% glycerol, and 0.0075% v/v Tween-20. DNA also contained a Cy5 fluorophore to normalize for small pipetting variabilities. Fluorescence spectra were obtained using a Horiba Fluoromax 4 fluorometer and analyzed in MATLAB to (1) remove background autofluorescence from buffer and protein and (2) to determine the relative change in Cy3 fluorescence compared to 0 nM. Background autofluorescence was determined by performing the same fluorescence scan but without DNA (protein + buffer). For each assay, we selected a concentration of DNA that enabled clear visualization of binding. Binding affinity was determined using the concentration of unbound TF in the reaction.
Ensemble FRET measurements
TF binding to Cy3-Cy5 nucleosomes was measured as previously described.2,64 0.2–0.5 nM nucleosomes were incubated for at least 5 min with varying concentrations of TF in T130 buffer (10 mM Tris-HCl (pH 8), 130 mM NaCl, 10% glycerol, and 0.0075% v/v Tween-20). Fluorescence emission spectra were acquired with a Horiba Fluoromax 4 fluorometer31 FRET efficiency was determined using the (Ratio)A method.65
Single-molecule TIRF microscopy
Single-molecule experiments were performed on an Olympus inverted IX73 microscope as previously described.14,66 Cy3 and Cy5 were excited using 532 nm and 638 nm diode lasers (Crystal Lasers), respectively. The excitation beams were focused through a quartz prism (Melles Griot) to establish a TIRF field at the surface of the flow cell. Imaging was performed with a 1.3 N.A. silicone immersion objective (Olympus). Fluorescence emission was directed to the iXon3 EMCCD camera (Andor) with a custom-built emission path containing bandpass filters and dichroic beam splitters (Chroma Tech). Each video was acquired using Micro-Manager software (Open Imaging).67
Flow Cell preparation
Flow cells were functionalized as previously described.14 Briefly, a quartz microscope slide (Alfa Aesar) was patterned with 16 small holes to form 8 flow cells (each flow cell has an inlet and and outlet). #1.5 coverslips (ThermoFisher) and microscope slides were sonicated in toluene, ethanol, and then further cleaned by piranha wash (3:1 mixture of concentrated sulfuric acid to 50% hydrogen peroxide). Slides were washed in water and, once completely dry, incubated in 100 uM mPEG-Si and biotin-PEG-Si (Laysan Bio) overnight in anhydrous toluene. Functionalized quartz slides and coverslips were assembled into microscope flow cells where channels are defined with parafilm. Before each experiment, the flow cell was treated sequentially with 1 mg/ml BSA, 40 ug/ml streptavidin, and biotin-labeled DNA or nucleosomes in T130 + 0.5% Tween-20.
Single-molecule fluorescence measurements of TF binding kinetics
Biotinylated sample molecules (DNA or nucleosomes) were added to the flow cell at room temperature as previously described.14 After a 5 minute incubation, unbound molecules were removed by a wash with T130 buffer. We then added imaging buffer containing the desired TF concentration into the flow cell. The samples were first exposed to 638 nm excitation to determine the location of Cy5-labeled molecules and then to 532 nm excitation for both FRET and PIFE measurements. Acquisition rate is documented in Table S6. Generally, imaging was performed for 2000 frames at 5 Hz. However, for stable binders (i.e. Cbf1ΔN), imaging was slowed to 0.5 Hz. The imaging buffer for FRET experiments contained 10 mM Tris-HCl (pH 8), 130 mM NaCl, 10% glycerol, 0.5% v/v Tween-20, 0.1 mg/ml BSA, 2 mM Trolox, 0.0115% v/v COT (Cyclooctatetraen, Sigma 138924), 0.012% v/v NBA (3-Nitrobenzyl alcohol, Sigma 146056), 450 ug/ml glucose oxidase (Sigma G2133) and 22 ug/ml catalase (Sigma C3155), while the imaging buffer for PIFE experiments contained 10 mM Tris-HCl (pH 8), 130 mM NaCl, 10% glycerol, 0.5% v/v Tween-20, 0.1 mg/ml BSA, 1% v/v BME, 450 ug/ml glucose oxidase and 22 ug/ml catalase.
In each image stack, the imageJ “find maxima” function was applied to the Cy5 channel to determine the coordinates of each molecule. Time traces were generated by measuring the average intensity of a 3×3 pixel area centered on each molecule throughout the entire stack in both channels for FRET measurements and in the cy3 channel for PIFE measurements.14 Traces were selected manually for analysis. For smPIFE, we segregate molecules into three categories: (i) non-fluctuating molecules (typically about 30% of all traces), (ii) molecules that photobleach very quickly or have intensity above an empirically determined fluorescence value (intensity = 600) (approximately 20% of all traces), and (iii) molecules that are included for further analysis (about 40–50% of all traces). Category 3 molecules are then analyzed for interpretable fluctuations. The number of molecules analyzed for each experiment is included in Table S6 for each single-molecule experiment performed herein (typically about 20–50% of traces in category 3). For smFRET, we only analyzed traces that (a) FRET and (b) fluctuate. Again, we end up analyzing about 20–50% of all molecules that meet the initial criteria.
The cumulative probabilities of the DNA smPIFE studies all fit best to two sets of rates. PIFE measurements of TF binding to DNA are sensitive only to site specific binding.4,14 In addition, the separate ratios of the primary rates and secondary rates of each TF with DNA imply very similar KDs and are consistent with the ensemble S1/2 measurements of each TF. So, both the primary and secondary rates are site specific and occur at similar TF concentrations. All smFRET cumulative probabilities, except full length Pho4, fit best to single set of rates. For all TFs, the secondary DNA binding rate constant and dissociation rate are all about 5- to 10-fold slower than the primary rates. So, comparisons of the secondary DNA rates to the nucleosome rates result in a shift in the ratio between nucleosome and DNA kinetic measurements (Figure S5Q–R). However, the comparisons between TF constructs and the conclusions on how the HLH regions of Cbf1 and Pho4 impact relative nucleosome to DNA kinetics are not changed. Therefore, we focused on the primary rates since they occurred the most frequently and thus dominate the binding and dissociation kinetics.
Preparation of nucleosomes for single particle analysis
Nucleosomes containing 149 bp Widom 601 sequence with an added E-box motif (GGTCACGTGACC) positioned 9 bp downstream from the DNA end were prepared essentially as described previously.68 We used the strategy of employing hybrid histone octamers containing yeast H2A and H2B with Xenopus H3 and H469,70 to overcome problems producing large quantities of high-quality nucleosomes containing only yeast histones. This decision is justified by that the fact that yeast histones H3 and H4 are highly similar to Xenopus H3 and H4, and further by our structural observation that Cbf1 appears to contact only histone H2A and H2B proteins.
Expression and purification of Cbf1 for single particle analysis
The coding region of Cbf1(2–351) was cloned into pST50Tr vector,71 and the protein was expressed with a cleavable N-terminal His10 tag in BL21(DE3)pLysS Escherichia coli cells by inducing with 0.5 mM IPTG at 37°C for 3 h. Cells were resuspended in 50 mM sodium phosphate (pH 7), 300 mM NaCl, 1 mM benzamidine, 5 mM β-mercaptoethanol, lysed by freeze-thawing and sonication, and debris was removed by centrifugation. The crude cell extract was loaded on a Talon Co-affinity resin (Clontech), washed with lysis buffer containing 15 mM imidazole, and eluted with 200 mM imidazole. Pooled fractions were digested with tobacco etch virus protease to remove the His10 affinity tag during dialysis into 10 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM PMSF. NaCl concentration was diluted to 50 mM prior to loading on a SourceQ anion exchange column (Cytiva). Sample was eluted in a 50 to 500 mM NaCl gradient. Peak fractions were dialyzed against 10 mM HEPES (pH 7.5), 200 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM PMSF, concentrated and supplemented with 20% v/v glycerol prior to flash freezing in liquid nitrogen and storage at −80°C.
Cbf1-nucleosome complex reconstitution for single particle analysis
Cbf1-nucleosome complexes were reconstituted by mixing Cbf1 with the E-box containing nucleosomes at a 2.5:1 molar ratio in 10 mM HEPES pH 7.5, 75 mM NaCl, 1 mM DTT. The complex was purified over a Superdex 200 Increase 10/300 size exclusion column (Cytiva) in the reconstitution buffer supplemented with 0.1 mM PMSF (see Figure S4L for a representative chromatogram and SDS-PAGE). Peak fractions were diluted and used for cryo-EM grid preparation.
Cryo-EM grid preparation and data collection
3 μl of Cbf1-nucleosome complex at 1 mg/ml was pipetted on a Quantifoil R1.2/1.3 holey carbon, 300 mesh, copper grid (Electron Microscopy Sciences), blotted for 3 s with Grade 595 filter paper (Ted Pella) at 4°C, 100% humidity, and plunge-frozen in liquid ethane at liquid nitrogen temperature using a Vitrobot Mark IV (Thermo Scientific) instrument. Grids were glow discharged for 45 s at 15 mA using an easiGlow device (PELCO) before sample application. The data was collected on a Krios G3i microscope (Thermo Scientific), operated at 300 kV, with a K3 direct electron detector camera (Gatan) using SerialEM at the Pacific Northwest Cryo-EM Center. 6339 movies were collected at a nominal magnification of 18,000x with super-resolution image pixel size of 0.644 Å/px (calibrated nominal pixel size 1.287 Å/px). The total dose per exposure was 50 e−/Å2, and each exposure was fractioned into 51 subframes. Data was recorded in the defocus range of −0.8 to −2.1 μm. The data collection parameters and model statistics are summarized in Table S8.
Single particle data processing
Initial data processing was performed in cryoSPARC v3 (Figure S4F–J).72 Super-resolution movies were motion-corrected with Patch Motion Correction and binned using 2x Fourier cropping. CTF values were estimated with Patch CTF Estimation. Initial model was created by picking particles with Blob Picker, the particles were 2D-classified, nucleosome-like classes were selected for ab initio reconstruction, and the best obtained model was used to generate templates for particle picking with Template Picker. Newly picked particles were again 2D-classified, noise-only classes were discarded, and ab initio reconstructions then generated from the obtained particles. Particles were further filtered through multiple rounds of Heterogeneous Refinement to remove junk and nucleosome-only particles, and to obtain classes of various Cbf1 conformations in relation to the nucleosome core. The 1.9 million particles of the last heterogeneous refinement cycle were subjected to local (per-particle) motion correction.72 To further discriminate between different Cbf1 conformations, particles classes from cryoSPARC’s Heterogeneous Refinement were transferred to Relion 3.1.173,74 for 3D classification without alignment.75 Regularization parameter T was varied in the range of 100–400 and only the volume corresponding to Cbf1 was included in the mask. Several rounds of such classification were performed to obtain a set of conformationally homogenous particles. Final 3D refinement was performed with cryoSPARC’s Homogenous Refinement (Figure S4D). The map was postprocessed using the deep learning algorithm DeepEMhancer (Figure S4D).76
Model building and analysis
Initial models of Cbf1 dimer were generated using HHpred77,78 and MODELLER79 in ChimeraX80 with the Modeller Comparative tool. The best-fitting Cbf1 model and the crystal structure model of the nucleosome (PDB 3LZ0) were used for rigid-body fitting. The Cbf1-nucleosome model was built using Coot81 and UCSF ChimeraX80 with the ISOLDE plugin.82 Real-space refinement was performed with the PHENIX suite83 and validation with MolProbity.84 The model was refined against the DeepEMhanced map. Secondary structure and Ramachandran restraints were employed. Density of Cbf1 residues 1–221 and 293–352 were not visible.
In vivo Nucleosome mapping
For methionine-induced strains, yeast cells were grown in synthetic complete media + 2% glucose+20xMethione (SCD+20xMet) overnight at 30ᐤC until OD660 is 0.15, and then washed by ddH2O for three times. Washed cells were resuspended in SCD-Met medium and incubated for 2hr to fully induce the expression of target proteins. We imaged these cells and quantified the level of GFP-tagged proteins via MATLAB scripts.85 The subsequent nucleosome mapping follows previously described protocol.18 Briefly, cells were collected by centrifugation and then sequentially washed by cold water and 1M sorbitol. 1.5OD unit (OD660 × volume) of pre-washed cells were resuspended in 500ml of spheroplasting buffer (1M sorbitol and 0.5mM β-mercaptoethanol). 30μL of 0.5mg/ml of Zymolyase 100T was added to each sample, and incubated at room temperature for 6.5min. Cells were then centrifuged at 500g for 3min at 4°C and gently washed twice with 1M sorbitol. Each cell pellet was resuspended in 200uL of MNase digestion buffer (0.5mM spermidine, 10mM NaCl, 0.2M Tris-HCl pH7.5, 5mM MgCl2, 1mM CaCl2, 0.075% NP40, 1M sorbitol, and 1mM β-mercaptoethanol), and 5μL of 0.2U/μl MNase was added. Samples were incubated at 37°C for 8min, and then reactions were quenched by adding 20ul of quench buffer (5% SDS and 0.25M EDTA). Quenched samples were incubated at 65°C for 10min. Genomic DNA was extracted with 200μl of Phenol:Chloroform:Isoamyl Alcohol (25:24:1) and precipitated with ethanol. DNA pellets were resuspended in 400ul of TE and digested with 100μg RNase A. DNA was pellet down again with 1ml of ethanol and 6μl of 5M ammonium acetate. Mono-nucleosomal fragments were then gel purified with a Genejet kit and subjected to stacking qPCR (see Table S9 for primers). Nucleosome occupancy was normalized to a well-position nucleosome in the terminator of EXO84.
For β-estradiol-induced strains, cells were grown in SCD+10xMet overnight at 30°C until OD660 is 0.15. β-estradiol was added to cell culture to desired concentrations and incubated for 1.5hr. We analyzed these cells with the same imaging and MNase-qPCR assay as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
For single-molecule datasets, Table S6 reports the total number of molecules identified in each experiment and the fraction of total molecules analyzed. Single-molecule time series were fit to a two-state step function by the Hidden Markov Method using vbFRET.35 From these idealized time series, we determined the cumulative probabilities of the TF bound and unbound states. Each distribution was analyzed using MEMLET to determine the best fit for the data and ultimately to obtain rate constants for the transitions between bound and unbound states.36 Log-likelihood ratio tests were performed to determine whether to fit dwell time distributions to either a single or double exponential distribution. Dwell time distributions were fit to the double-exponential model if 2 out of 3 replicates for all TF concentrations used in this experiment produced a P-value below 0.01 (Table S7).
In the nucleosome mapping assays, we adhered to the following criteria to establish whether a change in nucleosome positioning occurred. We used the characteristic decrease in the nucleosome occupancy at −757 and increase at −654 to define “shift” of nucleosome −4. A shift is detected when the changes in the occupancy at these two positions follow this trend and are both statistically significant (P-value < 0.05; student T test). The changes and P-values are listed in Table S10. Based on these numbers, significant nucleosome shift occurs with Cbf1ΔN and Pho4ΔN CHLHZ over Cbf1 binding site, as well as Pho4ΔN and Pho4ΔN CHLHZ over Pho4 binding site.
Supplementary Material
Key Resource Table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Glucose oxidase | Sigma-Aldrich | G12133 |
| Catalase | Sigma-Aldrich | C3155 |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | 210518 |
| Cy3 NHS Ester | Cytiva | PA13101 |
| Cy5 Maleimide | Cytiva | PA23031 |
| mPEG-silane | Laysan Bio Inc. | MPEG-SIL-2000–1g |
| Biotin-PEG-silane | Laysan Bio Inc. | Biotin-PEG-SIL-3400–1g |
| L-Methionine | Sigma-Aldrich | M9625 |
| β-Estradiol | TOCRIS | 2824 |
| Z1005 Zymolyase | United States Biological | 37340-57-1 |
| nuclease micrococcal from Staphylococcus aureus | Sigma-Aldrich | N3755 |
| Cbf1, Pho4, and chimeras | In house | N/A |
| Histone proteins | In house | N/A |
| TEV protease | In house | N/A |
| Deposited Data | ||
| Cbf1-nucleosome structure | This study | PDB: 7SSA |
| Cbf1-nucleosome, 3.18 Å map | This study | EMDB: EMD-25406 |
| Cbf1-nucleosome, micrographs | This study | EMPIAR: EMPIAR-10875 |
| Single-molecule time traces | This study | DOI: will be added upon acceptance. |
| Critical commercial assays | ||
| HisTrap HP Ni-NTA column | GE healthcare | 17524801 |
| Vydac 218TP C18 reverse phase column | Hichrom | 218TP54 |
| Gen-Pak FAX anion-exchange Column | Waters | WAT015490 |
| Superdex 200 Increase 10/300 GL | Cytiva | 28990944 |
| Talon Superflow metal affinity | Clontech | 635670 |
| Source 15Q anion-exchanger | Cytiva | 17-0947-01 |
| Amicon 10K filters | Millipore | UFC201024 |
| Bacterial and Virus Strains | ||
| E. coli Rosetta (DE3)pLysS cells | Millipore | 70956 |
| E. coli BL21(DE3)pLysS | Invitrogen | C602003 |
| Experimental Models: Organisms/Strains | ||
| yHC54-Cbf1ΔN | This study (Table S4) | N/A |
| yHC54-Cbf1ΔN PHLH | This study (Table S4) | N/A |
| yHC54-Pho4ΔN | This study (Table S4) | N/A |
| yHC54-Pho4ΔN CHLHZ | This study (Table S4) | N/A |
| yHC55-Cbf1ΔN | This study (Table S4) | N/A |
| yHC55-Cbf1ΔN PHLH | This study (Table S4) | N/A |
| yHC55-Pho4ΔN | This study (Table S4) | N/A |
| yHC55-Pho4ΔN CHLHZ | This study (Table S4) | N/A |
| yHC64-Pho4ΔN | This study (Table S4) | N/A |
| yHC64-Pho4ΔN CHLHZ | This study (Table S4) | N/A |
| Oligonucleotides | ||
| Rvs for FRET | Sigma-Aldrich | biotin-CGCATGCTGCAGACGCGTT |
| Rvs for PIFE | Sigma-Aldrich | biotin-GCGGTTAAAAC[\Cy5]CGGGGGACAGCGC |
| Cbf1 FRET fwd | Sigma-Aldrich | Cy3-CTGGAGAGGTCACGTGACCAGGCCGCTC |
| Cbf1 PIFE Fwd | Sigma-Aldrich | Cy3-GGTCACGTGACCTGCCGAGGCCGCTC |
| Pho4 FRET fwd | Sigma-Aldrich | Cy3-CTGGAGAACCCACGTGGGGAGGCCGCTCAATT |
| Pho4 PIFE fwd | Sigma-Aldrich | Cy3-CCCACGTGGGCCGGTGCCGAGGCCGC |
| MNase-qPCR primers | IDT (Table S9) | N/A |
| Recombinant DNA | ||
| Histone expression plasmids | K. Luger, J. Widom | N/A |
| Cbf1 expression vector | S. Diekmann | N/A |
| Pho4 expression vector | R. Kornberg | N/A |
| pHCA/GAL4(1–93).ER.VP16 | D. Picard | Addgene plasmid # 108216 |
| pHC25(SkudMetpr) | This study | N/A |
| pHC30(Gal1pr) | This study | N/A |
| pST50Tr-STRaHSTNyCbf1 | This study | Expresses full-length Cbf1 used for cryoEM |
| pET11a-yH2A | This study | Expresses yeast H2A(2–132) used for cryoEM |
| pET11a-yH2B | This study | Expresses yeast H2B(2–131) used for cryoEM |
| pET3a-xH3 | 61 | Expresses Xenopus H3(1–135) used for cryoEM |
| pET3a-xH4 | 61 | Expresses Xenopus H4(2–102) used for cryoEM |
| pST103–16xNCP601a149c15 | This study | To isolate 149 bp 601 sequence with added E-box used for cryoEM |
| Software and Algorithms | ||
| Matlab | Mathworks | www.mathworks.com |
| vbFRET | 35 | http://vbfret.sourceforge.net/ |
| MEMLET | 36 | https://michaelswoody.github.io/MEMLET/ |
| cryoSPARC v3 | 62 | https://cryosparc.com |
| Relion 3.1.1 | 63,64 | https://github.com/3dem/relion |
| DeepEMhancer | 65 | https://github.com/rsanchezgarc/deepEMhancer |
| HHpred | 66,67 | https://toolkit.tuebingen.mpg.de/tools/hhpred |
| MODELLER | 68 | https://toolkit.tuebingen.mpg.de/tools/modeller |
| Coot | 69 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| UCSF ChimeraX | 70 | https://www.rbvi.ucsf.edu/chimerax/ |
| ISOLDE | 71 | https://isolde.cimr.cam.ac.uk |
| PHENIX | 72 | https://phenix-online.org |
HIGHLIGHTS.
Structurally similar bHLH TFs have divergent nucleosomes interactions
Cbf1 bHLH pioneer factor (PF) targets nucleosomes through HLH-histone interactions
Histone-PF interactions enable the dissociation rate compensation mechanism
HLH-histone interactions facilitate NDR generation in vivo by a bHLH PF
ACKNOWLEDGMENTS
We thank the members of the Poirier, Bai and Tan Labs for helpful discussions. We thank Joseph Cho for preparing cryo-EM grids and Jean-Paul Armache for helping with single particle data processing. This work was supported by NIH grants R01 GM121858 (to LB and MGP), R01 GM131626 and R35 GM139564 (to MGP), R35 GM139654 (to LB), R35 GM127034 (to ST), and T32 GM086252 (to BTD), by Estonian Research Council grant PUTJD906 (to PE) and by NSF grant 1715321 (to MGP). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. We thank PNCC staff Rose Marie Haynes and Theo Humphreys for their expert assistance in cryo-EM data collection.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Polach KJ, and Widom J (1995). Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol 254, 130–149. 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 2.Li G, and Widom J (2004). Nucleosomes facilitate their own invasion. Nat Struct Mol Biol 11, 763–769. 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 3.Adams CC, and Workman JL (1995). Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol 15, 1405–1421. 0270–7306/95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo Y, North JA, Rose SD, and Poirier MG (2014). Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res 42, 3017–3027. 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu Rev Biochem 89, 189–212. 10.1146/annurev-biochem-011520-105250. [DOI] [PubMed] [Google Scholar]
- 6.Donovan BT, Huynh A, Ball DA, Patel HP, Poirier MG, Larson DR, Ferguson ML, and Lenstra TL (2019). Live–cell imaging reveals the interplay between transcription factors, nucleosomes, and bursting. EMBO J 38, e100809. 10.15252/embj.2018100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dadiani M, Van Dijk D, Segal B, Field Y, Ben-Artzi G, Raveh-Sadka T, Levo M, Kaplow I, Weinberger A, and Segal E (2013). Two DNA-encoded strategies for increasing expression with opposing effects on promoter dynamics and transcriptional noise. Genome Res. 10.1101/gr.149096.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai L, Charvin G, Siggia ED, and Cross FR (2010). Nucleosome-Depleted Regions in Cell-Cycle-Regulated Promoters Ensure Reliable Gene Expression in Every Cell Cycle. Dev Cell 18, 544–555. 10.1016/j.devcel.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai L, and Morozov AV (2010). Gene regulation by nucleosome positioning. Trends in Genetics 26, 476–483. 10.1016/j.tig.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Levitus M, Bustamante C, and Widom J (2005). Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol 12, 46–53. 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 11.Tims HS, Gurunathan K, Levitus M, and Widom J (2011). Dynamics of nucleosome invasion by DNA binding proteins. J Mol Biol 411, 430–448. 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.North JA, Shimko JC, Javaid S, Mooney AM, Shoffner MA, Rose SD, Bundschuh R, Fishel R, Ottesen JJ, and Poirier MG (2012). Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res 40, 10215–10227. 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C, and Bundschuh R (2014). Quantitative models for accelerated protein dissociation from nucleosomal DNA. Nucleic Acids Res 42, 1–8. 10.1093/nar/gku719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donovan BT, Chen H, Jipa C, Bai L, and Poirier MG (2019). Dissociation rate compensation mechanism for budding yeast pioneer transcription factors. Elife 8, 1–24. 10.7554/eLife.43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirillo LA, and Zaret KS (1999). An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol Cell 4, 961–969. 10.1016/S1097-2765(00)80225-7. [DOI] [PubMed] [Google Scholar]
- 16.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2014). Pioneer Transcription Factors Target Partial DNA Motifs on Nucleosomes to Initiate Reprogramming. Cell 161, 555–568. 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaret KS, and Carroll JS (2011). Pioneer transcription factors: Establishing competence for gene expression. Genes Dev 25, 2227–2241. 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai L, Ondracka A, and Cross FR (2011). Multiple Sequence-Specific Factors Generate the Nucleosome-Depleted Region on CLN2 Promoter. Mol Cell 42, 465–476. 10.1016/j.molcel.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CS, Friedman JR, Fulmer JT, and Kaestner KH (2005). The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–947. 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 21.Holtzinger A, and Evans T (2006). Erratum: Gata4 regulates the formation of multiple organs (Development vol. 132 (4005–4014)). Development 133, 181. 10.1242/dev.02227. [DOI] [PubMed] [Google Scholar]
- 22.Makowski MM, Gaullier G, and Luger K (2020). Picking a nucleosome lock: Sequence- and structure-specific recognition of the nucleosome. J Biosci 45. 10.1007/s12038-019-9970-7. [DOI] [PubMed] [Google Scholar]
- 23.Yan C, Chen H, and Bai L (2018). Systematic Study of Nucleosome-Displacing Factors in Budding Yeast. Mol Cell, 1–12. 10.1016/j.molcel.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodonova SO, Zhu F, Dienemann C, Taipale J, and Cramer P (2020). Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672. 10.1038/s41586-020-2195-y. [DOI] [PubMed] [Google Scholar]
- 25.Michael AK, Grand RS, Isbel L, Cavadini S, Kozicka Z, Kempf G, Bunker RD, Schenk AD, Graff-Meyer A, Pathare GR, et al. (2020). Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science 368, 1460–1465. 10.1126/science.abb0074. [DOI] [PubMed] [Google Scholar]
- 26.Cave JW, Kremer W, and Wemmer DE (2000). Backbone dynamics of sequence specific recognition and binding by the yeast Pho4 bHLH domain probed by NMR. Protein Sci 9, 2354–2365. 10.1110/ps.9.12.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maerkl SJ, and Quake SR (2007). A systems approach to measuring the binding energy landscapes of transcription factors. Science (1979) 315, 233–237. 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu T, Toumoto A, Ihara K, Shimizu M, Kyogoku Y, Ogawa N, Oshima Y, and Hakoshima T (1997). Crystal structure of PHO4 bHLH domain-DNA complex: Flanking base recognition. EMBO Journal 16, 4689–4697. 10.1093/emboj/16.15.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wieland G, Hemmerich P, Koch M, Stoyan T, Hegemann J, and Diekmann S (2001). Determination of the binding constants of the centromere protein Cbf1 to all 16 centromere DNAs of Saccharomyces cerevisiae. Nucleic Acids Res 29, 1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, and O’Shea EK (2011). Integrated Approaches Reveal Determinants of Genome-wide Binding and Function of the Transcription Factor Pho4. Mol Cell 42, 826–836. 10.1016/j.molcel.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson MD, Brehove M, Luo Y, North J, and Poirier MG (2016). Methods for Investigating DNA Accessibility with Single Nucleosomes 1st ed. (Elsevier Inc.) 10.1016/bs.mie.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 32.Hwang H, and Myong S (2014). Protein induced fluorescence enhancement (PIFE) for probing protein–nucleic acid interactions. Chem. Soc. Rev 43, 1221–1229. 10.1039/C3CS60201J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang H, Kim H, and Myong S (2011). Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc Natl Acad Sci U S A 108, 7414–7418. 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo G, Wang M, Konigsberg WH, and Xie XS (2007). Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc Natl Acad Sci U S A 104, 12610–12615. 10.1073/pnas.0700920104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronson JE, Fei J, Hofman JM, Gonzalez RL, and Wiggins CH (2009). Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys J 97, 3196–3205. 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woody MS, Lewis JH, Greenberg MJ, Goldman YE, and Ostap EM (2016). MEMLET: An Easy-to-Use Tool for Data Fitting and Model Comparison Using Maximum-Likelihood Estimation. Biophys J 111, 273–282. 10.1016/j.bpj.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo Y, North JA, and Poirier MG (2014). Single molecule fluorescence methodologies for investigating transcription factor binding kinetics to nucleosomes and DNA. Methods 70, 108–118. 10.1016/j.ymeth.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossi MJ, Lai WKM, and Pugh BF (2018). Genome-wide determinants of sequence-specific DNA binding of general regulatory factors. Genome Res 28, 497–508. 10.1101/gr.229518.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarmoskaite I, AlSadhan I, Vaidyanathan PP, and Herschlag D (2020). How to measure and evaluate binding affinities. Elife 9. 10.7554/elife.57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasudevan D, Chua EYD, and Davey CA (2010). Crystal Structures of Nucleosome Core Particles Containing the “601” Strong Positioning Sequence. J Mol Biol 403, 1–10. 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 41.Morrison EA, Baweja L, Poirier MG, Wereszczynski J, and Musselman CA (2021). Nucleosome composition regulates the histone H3 tail conformational ensemble and accessibility. Nucleic Acids Res 49, 4750–4767. 10.1093/nar/gkab246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polach KJ, Lowary PT, and Widom J (2000). Effects of core histone tail domains the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J Mol Biol 298, 211–223. 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 43.Anderson JD, Lowary PT, and Widom J (2001). Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol 307, 977–985. 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 44.Nurse NP, Jimenez-Useche I, Smith IT, and Yuan C (2013). Clipping of flexible tails of histones H3 and H4 affects the structure and dynamics of the nucleosome. Biophys J 104, 1081–1088. 10.1016/j.bpj.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fasci D, van Ingen H, Scheltema RA, and Heck AJR (2018). Histone interaction landscapes visualized by crosslinking mass spectrometry in intact cell nuclei. Molecular and Cellular Proteomics 17, 2018–2033. 10.1074/mcp.RA118.000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu ML, Zang T, Zou Y, Chang JC, Gibson JR, Huber KM, and Zhang CL (2013). Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat Commun 4. 10.1038/ncomms3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith DK, Yang J, Liu ML, and Zhang CL (2016). Small Molecules Modulate Chromatin Accessibility to Promote NEUROG2-Mediated Fibroblast-to-Neuron Reprogramming. Stem Cell Reports 7, 955–969. 10.1016/j.stemcr.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu S, Spinner DS, Schmidt MM, Danielsson JA, Wang S, and Schmidt J (2000). Interaction of MyoD family proteins with enhancers of acetylcholine receptor subunit genes in vivo. Journal of Biological Chemistry 275, 41364–41368. 10.1074/jbc.M004172200. [DOI] [PubMed] [Google Scholar]
- 50.Tapscott SJ (2005). The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- 51.Jones S (2004). An overview of the basic helix-loop-helix proteins. [DOI] [PMC free article] [PubMed]
- 52.O’Neill EM, Kaffman A, Jolly ER, and O’Shea EK (1996). Regulation of PHO4 nuclear localization by the PHO80-PH085 cyclin-CDK complex. Science (1979) 271, 209–212. 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- 53.Louvion JF, Havaux-Copf B, and Picard D (1993). Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene 131, 129–134. 10.1016/0378-1119(93)90681-R. [DOI] [PubMed] [Google Scholar]
- 54.Komeili A, and O’Shea EK (1999). Roles of phosphorylation sites in regulating activity of the transcription factor Pho4. Science 284, 977–980. 10.1126/science.284.5416.977. [DOI] [PubMed] [Google Scholar]
- 55.Chen H, Kharerin H, Dhasarathy A, Kladde M, and Bai L (2022). Partitioned usage of chromatin remodelers by nucleosome-displacing factors. Cell Rep 40. 10.1016/j.celrep.2022.111250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwafuchi-doi M, and Zaret KS (2014). Pioneer transcription factors in cell reprogramming. 2679–2692. 10.1101/gad.253443.114.Freely. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sekiya T, Muthurajan UM, Luger K, Tulin AV, and Zaret KS (2009). Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev 23, 804–809. 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, Clark KL, Burley SK, and Zaret KS (1998). Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO Journal 17, 244–254. 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez Garcia M, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural Features of Transcription Factors Associating with Nucleosome Binding. Mol Cell, 1–12. 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansen JL, and Cohen BA (2022). A quantitative metric of pioneer activity reveals that HNF4A has stronger in vivo pioneer activity than FOXA1. Genome Biol 23. 10.1186/s13059-022-02792-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagai S, Davis RE, Mattei PJ, Eagen KP, and Kornberg RD (2017). Chromatin potentiates transcription. Proceedings of the National Academy of Sciences 114, 1536–1541. 10.1073/pnas.1620312114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276, 19–42. 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 63.Luger K, Mäder a W., Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 64.Shimko JC, Howard CJ, Poirier MG, and Ottesen JJ (2013). Preparing Semisynthetic and Fully Synthetic Histones H3 and H4 to Modify the Nucleosome Core. Methods Mol Biol 981, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clegg RM (1992). [18] Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol 211, 353–388. 10.1016/0076-6879(92)11020-J. [DOI] [PubMed] [Google Scholar]
- 66.Roy R, Hohng S, and Ha T (2008). A practical guide to single-molecule FRET. Nat. Methods 5, 507–516. 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsuchida MA, Edelstein AD, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J Biol Methods 1, 10. 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGinty RK, Makde RD, and Tan S (2016). Preparation, Crystallization, and Structure Determination of Chromatin Enzyme/Nucleosome Complexes 1st ed. (Elsevier Inc.) 10.1016/bs.mie.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ranjan A, Mizuguchi G, Fitzgerald PC, Wei D, Wang F, Huang Y, Luk E, Woodcock CL, and Wu C (2013). Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154, 1232. 10.1016/j.cell.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh RK, Fan J, Gioacchini N, Watanabe S, Bilsel O, and Peterson CL (2019). Transient Kinetic Analysis of SWR1C-Catalyzed H2A.Z Deposition Unravels the Impact of Nucleosome Dynamics and the Asymmetry of Histone Exchange. Cell Rep 27, 374–386.e4. 10.1016/j.celrep.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan S, Kern RC, and Selleck W (2005). The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expr Purif 40, 385–395. 10.1016/j.pep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 72.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 73.Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 1–22. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scheres SHW (2016). Processing of Structurally Heterogeneous Cryo-EM Data in RELION 1st ed. (Elsevier Inc.) 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 76.Sanchez-Garcia R, Gomez-Blanco J, Cuervo A, Carazo JM, Sorzano COS, and Vargas J (2021). DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun Biol 4, 1–8. 10.1038/s42003-021-02399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Söding J, Lupas AN, and Alva V (2020). Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr Protoc Bioinformatics 72, 1–30. 10.1002/cpbi.108. [DOI] [PubMed] [Google Scholar]
- 78.Zimmermann L, Stephens A, Nam SZ, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, and Alva V (2018). A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 430, 2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 79.Šali A, and Blundell TL (1993). Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- 80.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Croll TI (2018). ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol 74, 519–530. 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Science 27, 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zou F, and Bai L (2019). Using time-lapse fluorescence microscopy to study gene regulation. Methods 159–160, 138–145. 10.1016/j.ymeth.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Science 27, 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All time traces from single-molecule experiments and unprocessed gel images have been uploaded to Zenodo (10.5281/zenodo.7683126). The time traces can be viewed using vbFRET in matlab.35 The structural model, the cryo-EM map and micrographs of Cbf1-nucleosome complex are available as of the date of publication at PDB 7SSA, EMD-25406 and EMPIAR-10875, respectively.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.