

Abstract
Osteoarthritis is the most prevalent degenerative joint disease and one of the leading causes of physical impairment in the world's aging population. The human lifespan has significantly increased as a result of scientific and technological advancements. According to estimates, the world's elderly population will increase by 20% by 2050. Aging and age-related changes are discussed in this review in relation to the development of OA. We specifically discussed the cellular and molecular changes that occur in the chondrocytes during aging and how these changes may make synovial joints more susceptible to OA development. These changes include chondrocyte senescence, mitochondrial dysfunction, epigenetic modifications, and decreased growth factor response. The age-associated changes occur not only in the chondrocytes but also in the matrix, subchondral bone, and synovium. This review aims to provide an overview of the interplay between chondrocytes and matrix and how age-related changes affect the normal function of cartilage and contribute to OA development. Understanding the alterations that affect the function of chondrocytes will emerge new possibilities for prospective therapeutic options for the treatment of OA.
Keywords: aging, cartilage, osteoarthritis, oxidative stress, senescence
1. Introduction
Knee Osteoarthritis (OA) is a prevalent chronic degenerative disease that causes severe joint pain and eventually leads to joint dysfunction. Over 500 million people worldwide are affected by OA, and it has a considerable socioeconomic burden on society [1, 2]. Age, genetic predisposition, obesity, inflammation, sports injury, and gender are all risk factors for the development of OA. However, the occurrence of OA increases with advancing age; it is considered the leading risk factor for OA [3]. Even though people of all ages are at risk, the burden of OA falls disproportionately on the elderly. Over 40% of all OA cases occur in those over the age of 65 [4]. Although aging and articular cartilage degeneration in OA are two distinct processes, aging causes changes in the musculoskeletal system that eventually lead to OA development [5, 6]. Several studies have found that as people get older, the frequency and prevalence of synovial joint deterioration increase significantly [7-11]. Moreover, at the age of 50, the occurrence of developing post-traumatic OA following an intraarticular knee fracture increases 3 to 4-fold [12].
The knee joint is the most complex synovial joint composed of multiple structures, including an articular capsule, a subchondral bone, a synovial membrane, and hyaline articular cartilage, which protects the end of bones in a joint. Aging is a multifaceted process that affects the knee joint structure. It leads to changes in the biochemical and biomechanical properties of the knee joint at macroscopic and microscopic levels, affecting its functional performance. These age-related changes include telomere attrition, epigenetic alterations, mitochondrial dysfunction, and, most importantly, changes in the mechanical properties of the cell and changes in the extracellular matrix of the cell [13]. During the process of aging, changes occur at various levels, including cellular changes, changes in the cartilage, osteochondral junction, and also in the synovium, which makes the joint more susceptible to the development of OA [14]. This review provides an overview of the cellular and molecular changes that occur in the synovial joint with age and discusses how age-associated dysregulated cell processes contribute to the development of OA.
2. Effects of aging on chondrocytes and its correlation with OA
2.1. Chondrocyte senescence and aging
Aging contributes to the accumulation of senescent cells and results in age-related tissue dysfunction. Senescence is a complex process where cells undergo metabolic, morphological, and physiological changes in response to various cellular stressors. Cellular senescence is described by stress-induced cell cycle arrest and the generation of pro-inflammatory paracrine chemicals called senescence-associated secretory phenotype (SASP). Articular cartilage deteriorates with age, and chondrocyte senescence is a crucial factor that plays a very significant role in the development and progression of OA [6, 15-17]. The regenerative ability of mesenchymal stem cells is impaired when senescent chondrocytes are injected into the articular cartilage [18]. However, the in vivo study in the mouse OA model shows a decrease in joint degeneration after the local clearance of senescent cells from articular cartilage [19]. These findings reveal that chondrocyte senescence is a critical part of the development of OA and is important for joint degeneration during aging.
Extrinsic and intrinsic senescence are the two types of cellular senescence. The intrinsic senescence, also known as replicative senescence, is caused by telomere shortening. The stress-induced senescence or extrinsic senescence is caused by different stimuli like activation of oncogenes, oxidative stress, or inflammatory cytokines. Chondrocyte’s senescence is more likely due to stress-induced mechanisms than intrinsic ones [20]. In aged cartilage, senescent chondrocytes exhibit upregulation of matrix-degrading enzymes such as matrix metallo-proteinase 3 (MMP-3) and MMP-13, aggrecanases, as well as the accumulation of damaged collagen. During the aging process, increased expression of MMPs, collagenase, and cathepsin K results in cartilage breakdown [21]. Increased production of these matrix-degrading enzymes damages the cartilage, which leads to the progression of OA.
Oxidative stress is a major cause of stress-induced senescence during the process of aging. An increase in the generation of reactive oxygen species (ROS) or a decrease in the number of antioxidants causes oxidative stress [22]. The increased ROS production induces the expression of genes that lead to dedifferentiation or senescence in chondrocytes [20]. Immunohistochemistry analysis of articular cartilage from mouse, human and non-human primates has shown increased nitrotyrosine, an oxidative damage marker in aged and osteoarthritic cartilage [23-25]. Oxidative stress accelerates the telomere shortening process, resulting in chondrocyte senescence and apoptosis of chondrocytes. Another mechanism of induction of chondrocyte senescence by oxidative stress is increased the level of expression of p53 and p21 and activation of p38 mitogen-activated protein kinase (MAPK) and PI3K/Akt signaling pathways which in turn triggers SASP [26]. Brandl and colleagues demonstrated that TRF1, TRF2, XRCC5, and Sirtuin-1 (SIRT1) expression is increased in human chondrocytes during the early passages following acute oxidative stress and decreases in the late passages. This demonstrates that these regulatory proteins deal with oxidative stress and protect DNA from damage in young chondrocytes. Due to reduced amounts of regulatory proteins in aged chondrocytes, they are more sensitive to oxidative stress, which leads to increased DNA damage and senescence [27]. Wnt family proteins are involved in the development of OA. It has been studied that the Wnt/β catenin pathway promotes chondrocyte senescence by increasing the expression of p53 and decreasing the expression of SIRT1 [28].
Apart from growth arrest, senescent cells show other vital features like secretion of proinflammatory cytokines, chemokines, and MMPS, also known as SASP. SASP is an important factor for the immune system to recognize senescent cells. As senescent cells accumulate with aging, it causes chronic inflammation, leading to the development of age-related diseases [29]. In OA cartilage, SASP contributes to the pro-inflammatory state and excessive synthesis of MMPs [30]. Freund and colleagues have studied the various SASP secreted by senescent cells and classified them according to their level of expression, which include high (>4 fold) to intermediate (2-4 fold) to small (<2 fold) [31]. Remarkably, OA tissue and synovial fluid showed all the SASP at high levels. Some of those high fold SASP involve Granulocyte macrophage-colony stimulating factor (GM-CSF), GROα, β, γ, insulin growth factor binding protein 7 (IGFBP-7), Interleukin-1 (IL-1α), IL-6, IL-7, IL-8, monocyte chemoattractant protein-1(MCP-1), MCP-2, macrophage inflammatory protein-1α (MIP1α), MMP-1, MMP-10, and MMP-3. The SASPs which are expressed at intermediates levels are intercellular adhesion molecule 1(ICAM-1), IL-1β, MCP-4, macrophage migration inhibitory factor (MIF), MMP-13, oncostatin M, regulated on activation, normal T cell expressed and secreted (RANTES), and tissue inhibitor of metalloproteinases 2 (TIMP-2). They have also been characterized as potential regulators in OA [30, 32-37]. Intercellular communication can trigger chondrocyte senescence in OA. Senescent chondrocytes secrete extracellular vesicles (EV), which negatively impact the healthy chondrocytes in the cartilage. It has been studied that the normal cells became senescent in human OA cartilage after receiving EV from senescent chondrocytes, and EV production was proportional to the number of senescent cells. This affects the matrix synthesis, which is mediated by the microRNAs derived from EVs [38, 39]. Expression of cellular communication network factor 1 (CCN1) and a gap junction channel protein connexin43 (Cx43) increases in senescent chondrocytes compared to normal chondrocytes [40, 41]. CCN1 stimulates senescence in chondrocytes by activating the p38 pathway and increasing SASP and MMP13 expression. Inhibiting CCN1 reduces inflammaging and maintains cartilage integrity [39, 41].
The reduction in chondrocyte proliferative and anabolic responses to growth factor stimulation is another hallmark of chondrocyte senescence. Stress-induced senescence reduces the ability of chondrocytes to synthesize type II collagen and aggrecan. In human chondrocytes, the increased expression of caveolin-1 protein induced by IL-1β can be inhibited by angiogenic growth factors (AGF) treatment. In the protection against chondrocyte senescence and cartilage aging, AGF may play a critical role [42]. The typical mitogenic response to multiple major growth factors, including TGF, bFGF, PDGF, and IGF-I, has declined with age [3]. This shows that the combined effect of aging, senescence and phenotypic changes in chondrocytes promotes cartilage degeneration, leading to OA progression. Figure 1 depicts how age-related stress impairs chondrocyte function in many ways and contributes to OA development.
Figure 1.
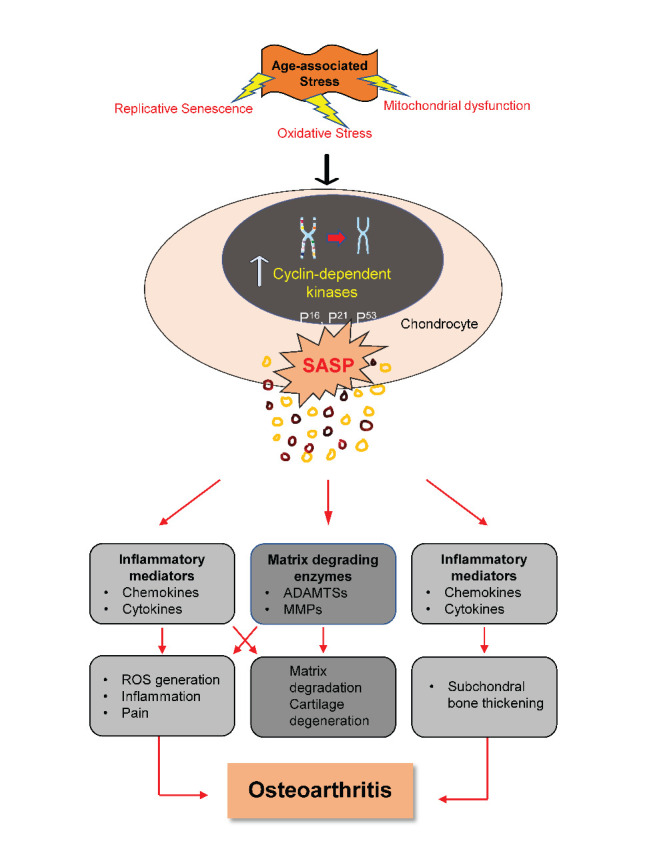
Relationship between age-related changes, senescence, and development of OA. Senescence is induced in chondrocytes by age-related stresses such as mitochondrial dysfunction, oxidative stress, and replicative senescence. These senescent cells express elevated levels of cyclin-dependent kinases such as p53 and secret SASP factors. The SASP factors trigger osteoarthritic changes in the synovial joint. SASP: senescence-associated secretory phenotype; ADAMTSs: A disintegrin and metalloproteinase with thrombospondin motifs; MMPs: Matrix metalloproteinases; IGFB: Insulin-like growth factor bindingproteins TFG- β: Transforming growth factor β; ROS- Reactive oxygen species
2.2. Epigenetic regulation and aging
The expression of an age-dependent mechanism and epigenetics is an important mechanism in the development, aging, and age-related disorders [43]. Cells undergo extensive epigenetic modifications during the aging process. Epigenetics is a mechanism for regulating gene expression without altering the primary DNA sequence. Gene expression is regulated by epigenetic mechanisms such as DNA methylation, histone acetylation, and microRNA (miRNA) [44]. As epigenetic regulations are cell type-specific, disruption of a stable epigenetic state might lead to the development of several diseases, including OA. An increasing number of evidence show that an epigenetic mechanism alters the local transcriptional activity and expression of mRNA in articular cartilage chondrocytes to a significant extent [45, 46]. The epigenetic mechanism is critical for regulating specific cytokines, matrix proteins, and transcription factors gene expression [47].
In chondrocyte differentiation and function, different growth factors like bone morphogenic proteins (BMPs), IGF-1, and TGF-β, play an essential role. BMP-7, also known as osteogenic protein-1 (OP-1), is important to matrix component synthesis. Alteration in the expression level of BMP-7 affects the homeostasis of the matrix. The OP-1 promoter's increasing methylation is one of the factors contributing to the decline in OP-1 mRNA expression with age [48, 49]. An SRY-Box transcription factor 9 (SOX9) transcription factor is involved in the differentiation of chondrocytes and cartilage formation. It has been demonstrated that, at embryonic and newborn stages of mice, SOX9 expression is highest, but it decreases significantly in completely developed joints. This age-dependent SOX9 expression is regulated by DNA methylation and histone methylation in mouse articular cartilage [50]. In mouse cartilage, postnatal inactivation of SOX9 showed a decrease in proteoglycan content but did not show any sign of OA development by the age of 18 months; however, in humans, reduction in SOX9 expression leads to the development of OA [51, 52]. In human chondrocytes, miRNA-145 inhibits the expression of SOX9. The expression of collagen type II alpha one chain (COL2A1) and aggrecan is dramatically reduced when miRNA-145 levels increase, whereas the expression of matrix metalloproteinase is significantly increased. (MMP-13) [53]. Another transcription factor, Nfat1 (NFAT1/NFATc2), is a nuclear factor of activated T cells (NFAT) family member that plays a critical role in maintaining the articular cartilage structure in adult mice. Nfat-deficient mice do not affect skeletal development. Still, it affects the chondrocyte function and exhibits OA-like changes such as increased expression of proinflammatory cytokines, the imbalance between anabolic and catabolic, and loss of cartilage repair abilities [54]. It has been proven that while chondrocyte NFAT1 expression is low at the embryonic stage, it is high in adult chondrocytes in wild-type mice. Histone methylation controls this age-dependent expression of NFAT [55]. Figure 2 depicts the impact of risk factors such as aging in causing significant epigenetic changes, which eventually contribute to the development of OA.
Figure 2.

Changes in epigenetic regulation and the development of OA. Risk factors like age and obesity affect the stable epigenetic state of chondrocytes. DNA methylation and histone methylation changes occurred in the nucleus and miRNA expression in the cytoplasm. This affects the regulation of specific cytokines, matrix proteins, and gene expression and causes their abnormal expression, which affects the chondrocyte functions. These changes will lead to degeneration of cartilage and the pathogenesis of OA.
2.3. Autophagy and aging
Autophagy is a protective mechanism of cells to maintain cellular integrity. It is a process to degrade the cells' damaged and dysfunctional cell organelles, proteins, and other macromolecules. Autophagy enhances the function of the articular chondrocyte. Moreover, autophagy dysfunction is linked with the pathogenesis and increased severity of OA. Autophagy decline or abnormalities are related to accelerated aging-related alterations; however, autophagy stimulation may have anti-aging effects [56-59]. It has been studied that aging cartilage from humans and mice shows the reduced the expression of autophagy proteins like uncoordinated-51-like protein kinase (ULK1), Beclin1, and light chain 3 (LC3) expression [57]. In a mouse model, reduced autophagy activity is linked to the mechanisms of aging-related OA, which affects cell and tissue homeostasis and leads to the development of joint structural defects [60].
Around 30 autophagy-related genes (ATGs) are involved in the process of autophagy. Autophagy is regulated by the mammalian target of rapamycin complex 1 (mTORC-1). It inhibits the ULK1 complex by acting as a negative regulator. Increased mTOR expression is linked to increased chondrocyte apoptosis and decreased expression of key autophagy genes in OA cartilage [61]. In C57Bl/6 OA mice, rapamycin treatment which inhibits mTORC-1 and activates autophagy, retains cartilage cellularity and lowers IL-1β expression in articular cartilage [62]. The damaged or dysfunctional cellular components are enclosed in autophagosomes, a double-membrane vesicle during autophagy. The autophagosome formation is regulated by ATG proteins, including ATG-12, ATG-5, and LC3. During autophagy, LC3-I is converted to LC3-II, an important autophagy marker. SIRT1, an NAD+ dependent histone deacetylase of class III, regulates age-related physiological functions such as stress responses. It has been shown that in human chondrocytes, SIRT1 regulates autophagy via interacting with ATG-7 [63]. SIRT1 expression levels in human chondrocytes decline with age and decreases cartilage integrity with aging. As SIRT1 causes deacetylation of autophagy proteins and increases autophagy, this shows that it is essential to maintain autophagy in chondrocyte and cartilage integrity; therefore, targeting SIRT1 might be important in the prevention of OA [64]. Xu and colleagues have demonstrated that Sirtuin-3 (SIRT3), a deacetylase, activates autophagy by inhibiting PI3K/Akt/mTOR and plays a critical role in protecting against OA pathophysiology [59].
Autophagosome fuses with lysosome where the degradation occurs [57, 65]. Lysosomes play an important role in the degradation of cellular waste and in the process of autophagy. Impaired function of the lysosome is associated with an increase in the chondrocyte apoptosis via release of the Cytochrome c, a proapoptotic protein which induces apoptosis by the Caspase3/7 dependent pathway. Autophagy is continuously activated in cartilage; however, the level of constitutive autophagy in aged mice decreases, and the size and number of autophagosomes are reduced [66]. The autophagy proteins like ATG-5 and LC3 expression levels are reduced in aged mice [67]. While OA cartilage also shows a reduction in levels of autophagy genes like ATG3, ATG12, ULK1, beclin 1, and γ-aminobutyric acid receptor-associated protein-like one and shows an increase in apoptosis [57]. Autophagy has a protective role in cells however, the complete understanding of the mechanism of autophagy and lysosome dysfunction in aged and osteoarthritic cartilage has great therapeutic potential for the treatment of OA because of its ability to target the aging process of chondrocytes.
2.4. Mitochondrial dysfunction and aging
Mitochondria play a critical role in the aging process. Mitochondria are subcellular organelle mainly involved in energy production by oxidative phosphorylation. But the function of mitochondria declines with age. The mitochondrial dysfunction is viewed as a “hallmark of aging.” The mitochondrial dysfunction also promotes the development of age-related diseases like OA [68]. It has been studied that mitochondrial dysfunction results in increased production of ROS, which increases oxidative stress, causes a reduction in ATP production, decreases matrix synthesis ability, and increased apoptosis in osteoarthritic chondrocytes [68, 69]. Mitochondrial dysfunction in aged and OA cartilage activates JNK-MAPK/cFos/AP1 pathway via ROS and triggers the production of matrix-degrading enzymes and the expression of proinflammatory genes [70]. Increased ROS generation activates the MAPK and MAPK/ERK signaling pathways, limiting the formation of extracellular matrix (ECM) components such as collagen II and glycosaminoglycans while promoting type I collagen production [71]. ROS overexpression also damages mitochondrial DNA (mtDNA) and impairs its repair capacity [72].
Superoxide dismutase (SOD) catalyzes the conversion of superoxide to hydrogen superoxide and molecular oxygen, therefore maintaining redox homeostasis. Superoxide dismutase 1, 2, and 3 levels were reduced in osteoarthritic cartilage, but SOD2 knockout mice demonstrated increased cartilage degradation with age [73, 74]. It has been shown that in OA chondrocyte Aurora kinase A (AURKA) binds to SOD2 and causes its lysine-48 (K48) ubiquitination mediated degradation, resulting in mitochondrial dysfunction [75]. Fu et al. have shown that, even though there is an increased expression of superoxide dismutase in mitochondria in aged mice, its specific activity decreases with age due to increased posttranslational lysine acetylation. In cartilage Sirtuin-3 (SIRT3), mitochondrial deacetylase levels decrease with age, driving a decline in the SOD-2 activity. Incubation of aged chondrocyte homogenates with SIRT3 and NAD+ showed an increased level of SOD2 activity [76, 77]. Maintaining SIRT3 levels in aging cartilage could be a potential therapeutic strategy to retain SOD-2 levels in aging cartilage and alleviate age-related mitochondrial dysfunction [76, 78].
2.5. Growth factor response and aging
Various growth factors play a key role in regulating different signaling pathways in articular cartilage. They are important for cell growth, division, and differentiation, thus influencing cartilage development and function. As chondrocytes age, their responsiveness to growth hormones like insulin-like growth factor-1 (IGF-1) and transforming growth factor - β (TGF - β) decreases, which results in impairs chondrocyte ability to repair. IGF-1 is a key factor in articular cartilage that promotes proteoglycan production and is required for cartilage integrity [79]. IGF-1 also promotes chondrocyte proliferation while inhibiting the terminal differentiation of chondrocytes. It has been shown that an increase in IGF binding proteins could result in chondrocytes’ ability to respond to IGF-1. When compared between young and old rats, there is a significant decrease in proteoglycan synthesis [80]. Another study indicates that the age-related reduction in the responsiveness of chondrocytes to IGF-1 could be due to the aged cells’ inability to adequately transduce the signal generated from the IGF-1 receptor [81]. IGF-1 binding to the IGF-1 receptor activates various signaling pathways like Shc/Grb2/Sos/Ras/Raf/MEK/ERK/MAPK pathways. The phosphorylation of insulin receptor substrate 1 (IRS-1) and insulin receptor substrate 2 (IRS-2) occurs when IGF-1 binds to its receptor called IGF-1 receptor, receptor, which further stimulates various signaling pathways, such as the PI3K (phosphoinositide 3-kinase) cascade and ERK (extracellular-signal-regulated kinase). PI3K then leads to the activation of AKT [82]. Activation of Akt signaling by IGF-1 promotes proteoglycan and collagen type II synthesis, while activation of ERK has inhibitory effects [83]. It has been studied that; aged chondrocytes show a decline in proteoglycan synthesis compared to young chondrocytes. The level of Akt phosphorylation is lower in aged chondrocytes than in young chondrocytes [83]. During the development of OA, decreasing levels of IGF-1 are also important in shifting the balance towards catabolic metabolism. Excessive ROS inhibits the IRS-1-PI-3 kinase-Akt signaling pathway. This signaling pathway is important for promoting matrix production. Simultaneously, ROS activates the ERK/MAP kinase and decreases the production of aggrecan, type II collagen, and Sox-9 [84, 85]. Through oxidized low-density lipoprotein (LDL, extracellular ROS may also inhibit the Akt pathway. Stress-induced chondrocyte senescence is triggered by oxidized LDL binding to its receptor, lectin-like ox-LDL receptor 1 (LOX-1), which is associated with a lower Akt phosphorylation than after IGF-1 stimulation [86, 87].
TGF- β is an essential anabolic growth factor in chondrocyte differentiation and plays a vital role in maintaining healthy cartilage. TGF- β acts via two different receptors and generates two different responses. TGF-β signaling via activin receptor-like kinase-5 (ALK5) promotes the synthesis of matrix components like aggrecan and collagen, while signaling via activin receptor-like kinase - 1 (ALK1) induces the expression of MMP-13 [88]. The response of aged chondrocytes and chondrocytes from OA cartilage to TGF-β is reduced, and even the expression of the ratio of receptor ALK1 to ALK5 changes in chondrocytes from aged and OA cartilage. Increased catabolic activity in chondrocytes relative to anabolic activity is the outcome of these alterations in receptor expression, and this imbalance leads to cartilage deterioration [89]. A better understanding of age-associated changes in growth factor response and changes in their receptor interaction would provide an innovative approach for the development of new strategies for OA treatment.
2.6. Chondrocyte metabolism and aging
Articular cartilage is an avascular tissue. Due to a lack of direct oxygen supply, chondrocyte metabolism is constrained by a low degree of anaerobic glycolysis generated by the slow oxygen diffusion and nutrients through the synovial fluid. Under normal oxygen tension, chondrocytes show aerobic glycolysis (Warburg effect) and anaerobic glycolysis [90]. OA cartilage shows a higher rate of anaerobic glycolysis. Glucose transporter (GLUT) 1, a membrane-embedded protein important for glucose transport in chondrocytes, plays a vital role in chondrogenesis [91]. In a hypoxic environment and in response to pro-inflammatory cytokines, GLUT1 expression is increased considerably in chondrocytes [74, 92]. Increased glucose uptake and excessive Advanced Glycation End-products (AGE) are caused by constant, enhanced GLUT1 expression, which can damage cartilage [93]. Pyruvate kinase M2 (PKM2), an isoenzyme of pyruvate kinase, is significantly expressed in OA cartilage chondrocytes; however, PKM2 knockdown reduces COL2A1 and SOX9 expression and promotes chondrocyte death, implying that PKM2 may play a role in the progression of OA [94]. The tricarboxylic acid cycle is the key metabolic pathway in cells, generating more energy than glycolysis. However, due to the increased rate of anaerobic glycolysis in OA chondrocytes, fewer pyruvate molecules are decarboxylated to form acetylated-CoA for the tricarboxylic acid cycle [95, 96].
Cholesterol levels have a critical role in the development of OA. Mice fed with a cholesterol-rich diet and mice without the low-density lipoprotein receptor (LDLr-/-) demonstrated osteophyte development [97]. Intracellular accumulation of cholesterol promotes the severity of OA. Hedgehog (Hh) signaling alters cholesterol accumulation in chondrocytes by regulating cholesterol homeostasis genes. Reduced cholesterol accumulation via HH signaling reduces cartilage degeneration [98].
3. Age-related changes in the ECM composition and characteristics
Articular cartilage (AC) is hyaline cartilage composed of extracellular matrix and chondrocytes. ECM is the complex network of macromolecules, around 95% of the cartilage consist of ECM and chondrocytes. Chondrocytes are responsible for ECM synthesis, and it is composed of collagen, glycosaminoglycans (GAGs), proteoglycans, glycoproteins, minerals, fibrous proteins, and 70 - 80% water. The prime function of ECM is to provide structural support to the tissue and maintain tissue stability [99]. ECM of cartilage directly impacts the tissue's response to mechanical strain, which is critical for maintaining its composition and functionality. The age-related changes increase the susceptibility to cartilage damage, which may lead to OA development. With aging, there is a gradual loss of the cartilage matrix. The loss of chondrocytes, reduced activity of chondrocytes to growth stimuli, or a decrease in water content could all contribute to cartilage thinning [23, 27].
3.1. Collagen II
Collagen type II is predominant in mature articular cartilage. It forms fibrils with proteoglycan and collagen IX and XI. Other collagen types like collagen I, IV, V, VI, IX, and XI are present in less proportion. These collagens support the collagen II fibrils network, and these fibrils provide tensile strength to the cartilage [99]. Numerous collagenases' expression and activity increase with age, resulting in fibrillation and cartilage disintegration, which affect the ECM's stability [100, 101]. Collagen undergoes structural changes with age, resulting in a decrease in the elasticity of the collagen fibers and ultimately altering the collagen's biomechanical properties [99]. With increasing age, the thickness of collagen fiber increases. This is due to non-enzymatic modifications in collagen resulting in the formation of AGE, and it starts to accumulate with increasing age [102]. Accumulation of AGE products makes cartilage stiffer and brittle with advancing age [103]. AGE accumulates in aggrecan, but it accumulates more with collagen, causes excessive crosslinking with collagen, and changes the biomechanical properties of cartilage by increasing the stiffness of the matrix and making cartilage more vulnerable to mechanical failure [99, 103, 104]. Kim and colleagues demonstrated that increasing AGE and the collagen crosslinking enzyme lysyl oxidase increases cartilage stiffness in a surgically induced mouse model [105]. AGEs interact with cell surface receptors called Receptor for Advanced Glycation End-products (RAGE) present on the articular chondrocyte. It has been studied that the levels of RAGE increase during aging and OA. AGE binding activates the RAGE signaling and stimulates the production of MMP-13 [106]. Additionally, AGE accumulations increase inflammation in the cartilage by increasing TNFα as well as the inflammatory mediators like prostaglandin E2 and nitric oxide. It also affects the type II collagen expression while MMPs, a disintegrin, and metalloproteinase with thrombospondin motifs expression are increased (ADAMTS) [107, 108]. It induces apoptosis by activating the NF-кB pathway, leading to further damage to the tissue [104, 109, 110].
3.2. Proteoglycans
Proteoglycans (PG) are highly glycosylated protein cores attached covalently to GAG chains [111]. PGs are highly negatively charged macromolecules that represent 10 to 15% of the wet weight, attract water, and salts in the cartilage, and give hydrophilic properties to cartilage. The ECM includes various proteoglycans that include aggrecan, decorin, biglycan, and fibromodulin [112]. Aggrecan is negatively charged, highly abundant proteoglycans and occurs in the form of aggregates. Aggrecan interacts with GAG and hyaluronan and provides osmotic properties to the cartilage, essential to resist the compressive load [113]. With increasing age, the number and size of proteoglycan aggregate decreases, reducing cartilage water content [114, 115]. An increase in proteolytic activity of aggrecanases leads to the release of aggrecan in the synovial fluid. The concentration of aggrecan fragments "ARGS" in the serum is age-related and increases in OA patients [116, 117]. Proteoglycan plays an important role in the transport of solute and nutrients transport. Age-related changes in cartilage affect the diffusion of nutrients in cartilage [118]. Deposition of calcium-containing crystals, primarily calcium pyrophosphate and basic calcium phosphate (BCP), is a major alteration in aged cartilage. Cartilage calcification due to BCP is associated with chondrocyte hypertrophy and the severity of OA [119, 120]. This cartilage calcification in the synovial joint is primarily due to aging rather than OA, and it serves as a precursor to increased fibrillation and OA[121]. Figure 3 shows the primary changes in ECM that occur with age.
Figure 3.

Changes in ECM structure in healthy and OA cartilage. Under normal physiological conditions, a healthy cartilage structure is maintained by an underlying network of collagen II and aggrecan aggregates with a hyaluronic acid backbone. However, osteoarthritic cartilage exhibits elevated MMPs, and reduced aggrecan molecule size. MMPs: Matrix metalloproteinases
Apart from structural support, ECM is also important for cell to cell signaling and matrix to cell signaling. Matrix to cell signaling plays an important role in the process of mechanotransduction [122]. Integrins are transmembrane proteins that act as a connecting link between ECM and cell cytoskeleton. Integrin enables mechanical signal transmission from ECM to cell and generates the intracellular response. The cytoskeleton acts as a mediator between chondrocytes and ECM interaction and senses the mechanical stimuli [123]. Nofal and Knudson have shown that disruption in the cytoskeletal structure of chondrocytes uncouples it from the extracellular matrix, resulting in altered metabolism and deleterious changes in a matrix structure [124].
4. Conclusion and perspective
The prevalence of OA is increasing with age. Aging affects the musculoskeletal system at a molecular and functional level. The regenerative potential of bone and cartilage is affected by cell renewal and matrix alterations. However, the molecular pathways that link aging and OA are not fully understood. We have summarised current information about the physiological and pathological alterations at the cellular and tissue levels that can be associated with joint aging and contribute to the development of OA.
In this review, we have summarised the role of aging in the development of OA. While aging and OA are closely linked, aging does not always result in the formation of OA. However, age-related alterations in chondrocytes and the ECM increase the joint's susceptibility to OA development. The cumulative effect of risk factors like senescence, altered epigenetics, mitochondrial dysfunction, changes in cell metabolism, and growth factor response affects chondrocyte homeostasis. These changes ultimately affect the homeostasis of ECM and lead to cartilage degradation. All the changes are the prime risk factors for the development of OA.
A deeper understanding of the aging mechanism may enable the creation of novel strategies for delaying these changes and for the development of therapeutics that aid in the slow progression of age-related OA development. Understanding age-related changes in the synovial joint aids in the development of specialized therapies that contribute to disease prevention. Numerous studies have demonstrated that cell-based therapies such as stem cell therapy or autologous chondrocyte implantation may be effective in reducing the severity of OA. Cell-free therapy is another current major therapy that shows promising outcomes in the treatment of OA. Numerous studies have demonstrated that extracellular vesicles are a promising therapy option for OA. Treatment options like use of extracellular vehicles and early detection of OA by targeting biomarkers need to develop to prevent the progression of OA.
Acknowledgments
The figures were made using a smart servier. The study is supported by NHMRC Investigator Fellowship (APP1176298) and the Prince Charles Hospital Research foundation fellowship for Indira Prasadam (RF-01) and ECMR grant from the Centre for Biomedical Technologies (QUT), and Australian Orthopaedic Association grant.
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- [1].Peat G, Thomas MJ (2021). Osteoarthritis year in review 2020: epidemiology & therapy. Osteoarthr Cartil, 29:180-189. [DOI] [PubMed] [Google Scholar]
- [2].Hunter DJ, March L, Chew M (2020). Osteoarthritis in 2020 and beyond: a <em>Lancet</em> Commission. The Lancet, 396:1711-1712. [DOI] [PubMed] [Google Scholar]
- [3].Loeser RF (2009). Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr Cartil, 17:971-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brooks PM (2006). The burden of musculoskeletal disease—a global perspective Clin Rheumatol, 25:778-781. [DOI] [PubMed] [Google Scholar]
- [5].Loeser RF (2010). Age-related changes in the musculoskeletal system and the development of osteoarthritis. Clin Geriatr Med, 26:371-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Loeser RF, Collins JA, Diekman BO (2016). Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol, 12:412-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Postler A, Ramos AL, Goronzy J, Günther K-P, Lange T, Schmitt J, et al. (2018). Prevalence and treatment of hip and knee osteoarthritis in people aged 60 years or older in Germany: an analysis based on health insurance claims data. Clin Interv Aging, 13:2339-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Magnusson K, Kumm J, Turkiewicz A, Englund M (2018). A naturally aging knee, or development of early knee osteoarthritis? Osteoarthr Cartil, 26:1447-1452. [DOI] [PubMed] [Google Scholar]
- [9].Cui A, Li H, Wang D, Zhong J, Chen Y, Lu H (2020). Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinical Medicine, 29-30:100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang Y, Jordan JM (2010). Epidemiology of osteoarthritis. Clin Geriatr Med, 26:355-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Loeser RF (2017). The Role of Aging in the Development of Osteoarthritis. Trans Am Clin Climatol Assoc, 128:44-54. [PMC free article] [PubMed] [Google Scholar]
- [12].Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, et al. (2011). Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res, 29:802-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Phillip JM, Aifuwa I, Walston J, Wirtz D (2015). The Mechanobiology of Aging. Annu Rev Biomed Eng, 17:113-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].He Y, Li Z, Alexander PG, Ocasio-Nieves BD, Yocum L, Lin H, et al. (2020). Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dai S-M, Shan Z-Z, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K, et al. (2006). Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: Possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum, 54:818-831. [DOI] [PubMed] [Google Scholar]
- [16].Vinatier C, Domínguez E, Guicheux J, Caramés B (2018). Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front Physiol, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chin AF, Han J, Gray-Gaillard E, Michel J, Elisseeff JH (2022). Chapter 12 - A framework for addressing senescent cell burden in the osteoarthritic knee: therapeutics, immune signaling, and the local-systemic interface. In Cellular Senescence in Disease. Serrano M, and Muñoz-Espín D, editors: Academic Press. 309-334. [Google Scholar]
- [18].Cao X, Luo P, Huang J, Liang C, He J, Wang Z, et al. (2019). Intraarticular senescent chondrocytes impair the cartilage regeneration capacity of mesenchymal stem cells. Stem Cell Res Ther, 10:86-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shao Y, Zhao C, Pan J, Zeng C, Zhang H, Liu L, et al. (2021). BMP5 silencing inhibits chondrocyte senescence and apoptosis as well as osteoarthritis progression in mice. Aging, 13:9646-9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rim YA, Nam Y, Ju JH (2020). The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int J Mol Sci, 21:2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dejica VM, Mort JS, Laverty S, Antoniou J, Zukor DJ, Tanzer M, et al. (2012). Increased type II collagen cleavage by cathepsin K and collagenase activities with aging and osteoarthritis in human articular cartilage. Arthritis Res Ther, 14:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ramasamy TS, Yee YM, Khan IM (2021). Chondrocyte Aging: The Molecular Determinants and Therapeutic Opportunities. Front Cell Dev Biol, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hui W, Young DA, Rowan AD, Xu X, Cawston TE, Proctor CJ (2016). Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis, 75:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, et al. (2008). Reduction of Age-Associated Pathology in Old Mice by Overexpression of Catalase in Mitochondria. J Gerontol A Biol Sci Med Sci, 63:813-822. [DOI] [PubMed] [Google Scholar]
- [25].Loeser RF, Carlson CS, Carlo MD, Cole A (2002). Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1β and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum, 46:2349-2357. [DOI] [PubMed] [Google Scholar]
- [26].Jeon OH, David N, Campisi J, Elisseeff JH (2018). Senescent cells and osteoarthritis: a painful connection. J Clin Invest, 128:1229-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li Y, Wei X, Zhou J, Wei L (2013). The Age-Related Changes in Cartilage and Osteoarthritis. Biomed Res Int, 2013:916530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li W, Xiong Y, Chen W, Wu L (2020). Wnt/β-catenin signaling may induce senescence of chondrocytes in osteoarthritis. Exp Ther Med, 20:2631-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wei W, Ji S (2018). Cellular senescence: Molecular mechanisms and pathogenicity. J Cell Physiol, 233:9121-9135. [DOI] [PubMed] [Google Scholar]
- [30].Greene MA, Loeser RF (2015). Aging-related inflammation in osteoarthritis. Osteoarthr Cartil, 23:1966-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Freund A, Orjalo AV, Desprez P-Y, Campisi J (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med, 16:238-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Clutterbuck AL, Smith JR, Allaway D, Harris P, Liddell S, Mobasheri A (2011). High throughput proteomic analysis of the secretome in an explant model of articular cartilage inflammation. J Proteomics, 74:704-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Long D, Blake S, Song X-Y, Lark M, Loeser RF (2008). Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res Ther, 10:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. (2012). Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther, 14:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Baker N, Sharpe P, Culley K, Otero M, Bevan D, Newham P, et al. (2012). Dual regulation of metalloproteinase expression in chondrocytes by Wnt-1-inducible signaling pathway protein 3/CCN6. Arthritis Rheum, 64:2289-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Ceuninck F, Dassencourt L, Anract P (2004). The inflammatory side of human chondrocytes unveiled by antibody microarrays. Biochem Biophys Res Commun, 323:960-969. [DOI] [PubMed] [Google Scholar]
- [37].Li WQ, Dehnade F, Zafarullah M (2001). Oncostatin M-Induced Matrix Metalloproteinase and Tissue Inhibitor of Metalloproteinase-3 Genes Expression in Chondrocytes Requires Janus Kinase/STAT Signaling Pathway. J Immunol, 166:3491. [DOI] [PubMed] [Google Scholar]
- [38].Jeon OH, Wilson DR, Clement CC, Rathod S, Cherry C, Powell B, et al. (2019). Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI insight, 4:e125019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xie J, Wang Y, Lu L, Liu L, Yu X, Pei F (2021). Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res Rev, 70:101413. [DOI] [PubMed] [Google Scholar]
- [40].Varela-Eirín M, Varela-Vázquez A, Guitián-Caamaño A, Paíno CL, Mato V, Largo R, et al. (2018). Targeting of chondrocyte plasticity via connexin43 modulation attenuates cellular senescence and fosters a pro-regenerative environment in osteoarthritis. Cell Death Dis, 9:1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Feng M, Peng H, Yao R, Zhang Z, Mao G, Yu H, et al. (2020). Inhibition of cellular communication network factor 1 (CCN1)-driven senescence slows down cartilage inflammaging and osteoarthritis. Bone, 139:115522. [DOI] [PubMed] [Google Scholar]
- [42].Yudoh K, Shi Y, Karasawa R (2009). Angiogenic growth factors inhibit chondrocyte ageing in osteoarthritis: potential involvement of catabolic stress-induced overexpression of caveolin-1 in cellular ageing. Int J Rheum Dis, 12:90-99. [DOI] [PubMed] [Google Scholar]
- [43].Brunet A, Berger SL (2014). Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci, 69 Suppl 1:S17-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Muñoz-Najar U, Sedivy JM (2011). Epigenetic control of aging. Antioxid Redox Signal, 14:241-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reynard LN, Loughlin J (2012). Genetics and epigenetics of osteoarthritis. Maturitas, 71:200-204. [DOI] [PubMed] [Google Scholar]
- [46].Rakyan VK, Down TA, Balding DJ, Beck S (2011). Epigenome-wide association studies for common human diseases. Nat Rev Genet, 12:529-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang M, Wang J (2015). Epigenetic regulation of gene expression in osteoarthritis. Genes Dis, 2:69-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Loeser RF, Im HJ, Richardson B, Lu Q, Chubinskaya S (2009). Methylation of the OP-1 promoter: potential role in the age-related decline in OP-1 expression in cartilage. Osteoarthr Cartil, 17:513-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Merrihew C, Kumar B, Heretis K, Rueger DC, Kuettner KE, Chubinskaya S (2003). Alterations in endogenous osteogenic protein-1 with degeneration of human articular cartilage. J Orthop Res, 21:899-907. [DOI] [PubMed] [Google Scholar]
- [50].Zhang M, Lu Q, Miller AH, Barnthouse NC, Wang J (2016). Dynamic epigenetic mechanisms regulate age-dependent SOX9 expression in mouse articular cartilage. Int J Biochem Cell Biol, 72:125-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee J-S, Im G-I (2011). SOX Trio Decrease in the Articular Cartilage with the Advancement of Osteoarthritis. Connect Tissue Res, 52:496-502. [DOI] [PubMed] [Google Scholar]
- [52].Henry SP, Liang S, Akdemir KC, de Crombrugghe B (2012). The postnatal role of Sox9 in cartilage. J Bone Miner Res, 27:2511-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martinez-Sanchez A, Dudek KA, Murphy CL (2012). Regulation of Human Chondrocyte Function through Direct Inhibition of Cartilage Master Regulator SOX9 by MicroRNA-145 (miRNA-145) *. J Biol Chem, 287:916-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang J, Gardner BM, Lu Q, Rodova M, Woodbury BG, Yost JG, et al. (2009). Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. J Pathol, 219:163-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rodova M, Lu Q, Li Y, Woodbury BG, Crist JD, Gardner BM, et al. (2011). Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. J Bone Miner Res, 26:1974-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chang J, Wang W, Zhang H, Hu Y, Wang M, Yin Z (2013). The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int J Mol Med, 32:1311-1318. [DOI] [PubMed] [Google Scholar]
- [57].Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M (2010). Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum, 62:791-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Rubinsztein David C, Mariño G, Kroemer G (2011). Autophagy and Aging. Cell, 146:682-695. [DOI] [PubMed] [Google Scholar]
- [59].Xu K, He Y, Moqbel SAA, Zhou X, Wu L, Bao J (2021). SIRT3 ameliorates osteoarthritis via regulating chondrocyte autophagy and apoptosis through the PI3K/Akt/mTOR pathway. Int J Biol Macromol, 175:351-360. [DOI] [PubMed] [Google Scholar]
- [60].Caramés B, Olmer M, Kiosses WB, Lotz MK (2015). The Relationship of Autophagy Defects to Cartilage Damage During Joint Aging in a Mouse Model. Arthritis Rheumatol, 67:1568-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li Y-S, Zhang F-J, Zeng C, Luo W, Xiao W-F, Gao S-G, et al. (2016). Autophagy in osteoarthritis. Joint Bone Spine, 83:143-148. [DOI] [PubMed] [Google Scholar]
- [62].Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M (2012). Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis, 71:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liao F-X, Huang F, Ma W-G, Qin K-P, Xu P-F, Wu Y-F, et al. (2019). The New Role of Sirtuin1 in Human Osteoarthritis Chondrocytes by Regulating Autophagy. CARTILAGE, 13:1237S-1248S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sacitharan PK, Bou-Gharios G, Edwards JR (2020). SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov, 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Luo P, Gao F, Niu D, Sun X, Song Q, Guo C, et al. (2019). The Role of Autophagy in Chondrocyte Metabolism and Osteoarthritis: A Comprehensive Research Review. Biomed Res Int, 2019:5171602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ansari MY, Ball HC, Wase SJ, Novak K, Haqqi TM (2021). Lysosomal dysfunction in osteoarthritis and aged cartilage triggers apoptosis in chondrocytes through BAX mediated release of Cytochrome c. Osteoarthr Cartil, 29:100-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pyo J-O, Yoo S-M, Ahn H-H, Nah J, Hong S-H, Kam T-I, et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun, 4:2300-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bolduc JA, Collins JA, Loeser RF (2019). Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med, 132:73-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].López de Figueroa P, Lotz MK, Blanco FJ, Caramés B (2015). Autophagy Activation and Protection From Mitochondrial Dysfunction in Human Chondrocytes. Arthritis Rheumatol, 67:966-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ansari MY, Ahmad N, Voleti S, Wase S, Malik M, Novak K, et al. (2020). Mitochondrial dysfunction in osteoarthritis and aged cartilage triggers inflammatory response and matrix degradation via ros mediated activation of JNK-MAPK/cFos-AP1 axis in chondrocytes. Osteoarthr Cartil, 28:S187. [Google Scholar]
- [71].Rieder B, Weihs AM, Weidinger A, Szwarc D, Nürnberger S, Redl H, et al. (2018). Hydrostatic pressure-generated reactive oxygen species induce osteoarthritic conditions in cartilage pellet cultures. Sci Rep, 8:17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Grishko VI, Ho R, Wilson GL, Pearsall AWI (2009). Diminished mitochondrial DNA integrity and repair capacity in OA & chondrocytes. Osteoarthr Cartil, 17:107-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, et al. (2006). Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum, 54:3533-3544. [DOI] [PubMed] [Google Scholar]
- [74].Zheng L, Zhang Z, Sheng P, Mobasheri A (2021). The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev, 66:101249. [DOI] [PubMed] [Google Scholar]
- [75].Yang C, You D, Huang J, Yang B, Huang X, Ni J (2019). Effects of AURKA-mediated degradation of SOD2 on mitochondrial dysfunction and cartilage homeostasis in osteoarthritis. J Cell Physiol, 234:17727-17738. [DOI] [PubMed] [Google Scholar]
- [76].Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, et al. (2016). Aging Promotes Sirtuin 3-Dependent Cartilage Superoxide Dismutase 2 Acetylation and Osteoarthritis. Arthritis Rheumatol, 68:1887-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Blanco FJ, Rego I, Ruiz-Romero C (2011). The role of mitochondria in osteoarthritis. Nat Rev Rheumatol, 7:161-169. [DOI] [PubMed] [Google Scholar]
- [78].Mao X, Fu P, Wang L, Xiang C (2020). Mitochondria: Potential Targets for Osteoarthritis. Front Med, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lotz M, Loeser RF (2012). Effects of aging on articular cartilage homeostasis. Bone, 51:241-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Martin JA, Ellerbroek SM, Buckwalter JA (1997). Age-related decline in chondrocyte response to insulin-like growth factor-I: The role of growth factor binding proteins. J Orthop Res, 15:491-498. [DOI] [PubMed] [Google Scholar]
- [81].Messai H, Duchossoy Y, Khatib A-M, Panasyuk A, Mitrovic DR (2000). Articular chondrocytes from aging rats respond poorly to insulin-like growth factor-1: an altered signaling pathway. Mech Ageing Dev, 115:21-37. [DOI] [PubMed] [Google Scholar]
- [82].Starkman BG, Cravero JD, Delcarlo M, Loeser RF (2005). IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J, 389:723-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Loeser RF, Gandhi U, Long DL, Yin W, Chubinskaya S (2014). Aging and Oxidative Stress Reduce the Response of Human Articular Chondrocytes to Insulin-like Growth Factor 1 and Osteogenic Protein 1. Arthritis Rheumatol, 66:2201-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yin W, Park J-I, Loeser RF (2009). Oxidative Stress Inhibits Insulin-like Growth Factor-I Induction of Chondrocyte Proteoglycan Synthesis through Differential Regulation of Phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK Signaling Pathways *. J Biol Chem, 284:31972-31981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wei FY, Lee JK, Wei L, Qu F, Zhang JZ (2017). Correlation of insulin-like growth factor 1 and osteoarthritic cartilage degradation: a spontaneous osteoarthritis in guinea-pig. Eur Rev Med Pharmacol Sci, 21:4493-4500. [PMC free article] [PubMed] [Google Scholar]
- [86].Zushi S, Akagi M, Kishimoto H, Teramura T, Sawamura T, Hamanishi C (2009). Induction of bovine articular chondrocyte senescence with oxidized low-density lipoprotein through lectin-like oxidized low-density lipoprotein receptor 1. Arthritis Rheum, 60:3007-3016. [DOI] [PubMed] [Google Scholar]
- [87].Loeser RF (2011). Aging and osteoarthritis. Curr Opin Rheumatol, 23:492-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Blaney Davidson EN, Remst DFG, Vitters EL, van Beuningen HM, Blom AB, Goumans M-J, et al. (2009). Increase in ALK1/ALK5 Ratio as a Cause for Elevated MMP-13 Expression in Osteoarthritis in Humans and Mice. J Immunol, 182:7937. [DOI] [PubMed] [Google Scholar]
- [89].van der Kraan PM, Blaney Davidson EN, van den Berg WB (2010). A role for age-related changes in TGFβ signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther, 12:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sophia Fox AJ, Bedi A, Rodeo SA (2009). The basic science of articular cartilage: structure, composition, and function. Sports health, 1:461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Mobasheri A, Dobson H, Mason SL, Cullingham F, Shakibaei M, Moley JF, et al. (2005). Expression of the GLUT1 and GLUT9 facilitative glucose transporters in embryonic chondroblasts and mature chondrocytes in ovine articular cartilage. Cell Biol Int, 29:249-260. [DOI] [PubMed] [Google Scholar]
- [92].Rasheed Z, Akhtar N, Haqqi TM (2011). Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology, 50:838-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Peansukmanee S, Vaughan-Thomas A, Carter SD, Clegg PD, Taylor S, Redmond C, et al. (2009). Effects of hypoxia on glucose transport in primary equine chondrocytes in vitro and evidence of reduced GLUT1 gene expression in pathologic cartilage in vivo. J Orthop Res, 27:529-535. [DOI] [PubMed] [Google Scholar]
- [94].Yang X, Chen W, Zhao X, Chen L, Li W, Ran J, et al. (2018). Pyruvate Kinase M2 Modulates the Glycolysis of Chondrocyte and Extracellular Matrix in Osteoarthritis. DNA Cell Biol, 37:271-277. [DOI] [PubMed] [Google Scholar]
- [95].Akram M (2014). Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem Biophys, 68:475-478. [DOI] [PubMed] [Google Scholar]
- [96].Maneiro E, Martín MA, de Andres MC, López-Armada MJ, Fernández-Sueiro JL, del Hoyo P, et al. (2003). Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum, 48:700-708. [DOI] [PubMed] [Google Scholar]
- [97].de Munter W, Blom AB, Helsen MM, Walgreen B, van der Kraan PM, Joosten LAB, et al. (2013). Cholesterol accumulation caused by low density lipoprotein receptor deficiency or a cholesterol-rich diet results in ectopic bone formation during experimental osteoarthritis. Arthritis Res Ther, 15:R178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ali SA, Al-Jazrawe M, Ma H, Whetstone H, Poon R, Farr S, et al. (2016). Regulation of Cholesterol Homeostasis by Hedgehog Signaling in Osteoarthritic Cartilage. Arthritis Rheumatol, 68:127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rahmati M, Nalesso G, Mobasheri A, Mozafari M (2017). Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res Rev, 40:20-30. [DOI] [PubMed] [Google Scholar]
- [100].Wu W, Billinghurst RC, Pidoux I, Antoniou J, Zukor D, Tanzer M, et al. (2002). Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum, 46:2087-2094. [DOI] [PubMed] [Google Scholar]
- [101].Brama PAJ, Van Den Boom R, Degroot J, Kiers GH, Van Weeren PR (2004). Collagenase-1 (MMP-1) activity in equine synovial fluid: influence of age, joint pathology, exercise and repeated arthrocentesis. Equine Vet J, 36:34-40. [DOI] [PubMed] [Google Scholar]
- [102].Saudek DM, Kay J (2003). Advanced glycation endproducts and osteoarthritis. Curr Rheumatol Rep, 5:33-40. [DOI] [PubMed] [Google Scholar]
- [103].Verzijl N, DeGroot J, Oldehinkel E, Bank RA, Thorpe SR, Baynes JW, et al. (2000). Age-related accumulation of Maillard reaction products in human articular cartilage collagen. Biochem J, 350 Pt 2:381-387. [PMC free article] [PubMed] [Google Scholar]
- [104].Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im H-J, et al. (2005). Articular chondrocytes express the receptor for advanced glycation end products: Potential role in osteoarthritis. Arthritis Rheum, 52:2376-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kim J-H, Lee G, Won Y, Lee M, Kwak J-S, Chun C-H, et al. (2015). Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc Natl Acad Sci, 112:9424-9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im H-J, et al. (2005). Articular chondrocytes express the receptor for advanced glycation end products: Potential role in osteoarthritis. Arthritis Rheum, 52:2376-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Nah S-S, Choi I-Y, Yoo B, Kim YG, Moon H-B, Lee C-K (2007). Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-α in human osteoarthritic chondrocytes. FEBS Lett, 581:1928-1932. [DOI] [PubMed] [Google Scholar]
- [108].Nah SS, Choi IY, Lee CK, Oh JS, Kim YG, Moon HB, et al. (2008). Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology, 47:425-431. [DOI] [PubMed] [Google Scholar]
- [109].Rasheed Z, Anbazhagan AN, Akhtar N, Ramamurthy S, Voss FR, Haqqi TM (2009). Green tea polyphenol epigallocatechin-3-gallate inhibits advanced glycation end product-induced expression of tumor necrosis factor-α and matrix metalloproteinase-13 in human chondrocytes. Arthritis Res Ther, 11:R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Huang H, Skelly JD, Ayers DC, Song J (2017). Age-dependent Changes in the Articular Cartilage and Subchondral Bone of C57BL/6 Mice after Surgical Destabilization of Medial Meniscus. Sci Rep, 7:42294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Heinegård D, Saxne T (2011). The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol, 7:50-56. [DOI] [PubMed] [Google Scholar]
- [112].Akkiraju H, Nohe A (2015). Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J Dev Biol, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Roughley PJ, Mort JS (2014). The role of aggrecan in normal and osteoarthritic cartilage. J Exp Orthop, 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Leong DJ, Sun HB (2011). Events in Articular Chondrocytes with Aging. Curr Osteoporos Rep, 9:196. [DOI] [PubMed] [Google Scholar]
- [115].Peng Z, Sun H, Bunpetch V, Koh Y, Wen Y, Wu D, et al. (2021). The regulation of cartilage extracellular matrix homeostasis in joint cartilage degeneration and regeneration. Biomaterials, 268:120555. [DOI] [PubMed] [Google Scholar]
- [116].Germaschewski FM, Matheny CJ, Larkin J, Liu F, Thomas LR, Saunders JS, et al. (2014). Quantitation OF ARGS aggrecan fragments in synovial fluid, serum and urine from osteoarthritis patients. Osteoarthr Cartil, 22:690-697. [DOI] [PubMed] [Google Scholar]
- [117].Mitani H, Takahashi I, Onodera K, Bae J-W, Sato T, Takahashi N, et al. (2006). Comparison of age-dependent expression of aggrecan and ADAMTSs in mandibular condylar cartilage, tibial growth plate, and articular cartilage in rats. Histochem Cell Biol, 126:371-380. [DOI] [PubMed] [Google Scholar]
- [118].Mirahmadi F, Koolstra JH, Fazaeli S, Lobbezoo F, van Lenthe GH, Snabel J, et al. (2018). Aging does not change the compressive stiffness of mandibular condylar cartilage in horses. Osteoarthr Cartil, 26:1744-1752. [DOI] [PubMed] [Google Scholar]
- [119].Mobasheri A, Matta C, Zákány R, Musumeci G (2015). Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas, 80:237-244. [DOI] [PubMed] [Google Scholar]
- [120].Jiang Y (2022). Osteoarthritis year in review 2021: biology. Osteoarthr Cartil, 30:207-215. [DOI] [PubMed] [Google Scholar]
- [121].Mitsuyama H, Healey RM, Terkeltaub RA, Coutts RD, Amiel D (2007). Calcification of human articular knee cartilage is primarily an effect of aging rather than osteoarthritis. Osteoarthr Cartil, 15:559-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Birch HL. 2018. Extracellular Matrix and Ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science. Harris JR, and Korolchuk VI, editors. Singapore: Springer Singapore. 169-190. [Google Scholar]
- [123].Duan W, Wei L, Zhang J, Hao Y, Li C, Li H, et al. (2011). Alteration of viscoelastic properties is associated with a change in cytoskeleton components of ageing chondrocytes from rabbit knee articular cartilage. Mol Cell Biomech, 8:253-274. [PubMed] [Google Scholar]
- [124].Nofal GA, Knudson CB (2002). Latrunculin and Cytochalasin Decrease Chondrocyte Matrix Retention. J Histochem Cytochem, 50:1313-1323. [DOI] [PubMed] [Google Scholar]