

Summary
As COVID-19 evolves from a pandemic to an endemic disease, the already staggering number of people that have been or will be infected with SARS-CoV-2 is only destined to increase, and the majority of humanity will be infected. It is well understood that COVID-19, like many other viral infections, leaves a significant fraction of the infected with prolonged consequences. Continued high number of SARS-CoV-2 infections, viral evolution with escape from post-infection and vaccinal immunity, and reinfections heighten the potential impact of Long COVID. Hence, the impact of COVID-19 on human health will be seen for years to come until more effective vaccines and pharmaceutical treatments become available. To that effect, it is imperative that the mechanisms underlying the clinical manifestations of Long COVID be elucidated. In this article, we provide an in-depth analysis of the evidence on several potential mechanisms of Long COVID and discuss their relevance to its pathogenesis.
Subject areas: Health sciences, Medicine
Graphical abstract
Health sciences; Medicine
Introduction
Since the first reported case of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in Wuhan, China, in December 2019, the rampant COVID-19 pandemic has altered the fate of billions of people around the world.1 The virus has spread to all continents, with over 600 million confirmed cases of COVID-19 and more than 6 million deaths reported worldwide.2 Since only a fraction of acute infections are diagnosed and reported, the overall burden of COVID-19 is considerably underestimated (see below). The genetic evolution of SARS-CoV-2 in the face of immune pressures from natural and vaccinal immunity has perpetuated this burden over time, with the unrelenting emergence of variants of concern and resultant intermittent surges of infections.3 SARS-CoV-2 endemicity appears likely, and future disease transmission will depend on the interplay between virus evolution, host immunity, and the population’s vulnerability and susceptibility to COVID-19.1
Over the past years, the focus has transitioned from public health containment efforts and measures to mitigate mortality risk, via interventions to retard progression to severe disease, to global vaccination efforts to prevent infection or severe disease, and more recently, to our efforts to understand and treat post-acute sequelae of the COVID-19 (also abbreviated PASC or PACS) diseases, often referred to as “Long COVID”, abbreviated here as LC, and used throughout this article. It is estimated that over two-thirds of the global population has been infected with SARS-CoV-2 already, the vast majority of whom were not diagnosed or reported.2,3 A significant proportion of survivors experience a diversity of long-lasting clinical sequelae and have an increased risk for new morbidity and possibly mortality.4,5,6,7,8 Up to 75% of critically ill patients, half of all hospitalized patients, and up to 30% of patients with asymptomatic infection may develop LC,9,10,11,12,13 although some epidemiological studies side with lower estimates.14,15 Regardless of the exact numbers, we are experiencing an unprecedented health crisis from an infection that is likely to become the most significant source of debilitating disease of our lifetime, which can affect nearly every bodily organ in people of any age. There is an urgent need to understand the pathobiology underpinning the development of this major public health threat. The development of diagnostic tests, biomarkers for risk-stratification and prognostication, and therapeutic interventions will depend on a complete and well-characterized understanding of the biological mechanisms responsible for generating the diverse phenotypes of LC.
Several reviews have been published on Long COVID in an attempt to summarize this rapidly advancing field.16,17,18,19 Early reviews were critical for establishing the scientific and clinical landscape of Long COVID but were limited by the lack of scientific data which could incisively discriminate between the competing proposed mechanistic bases of the diverse clinical phenotypes associated with the condition.18 Consequently, many previous reviews sought to consolidate the available scientific literature by working backward using clinical phenotypes and organ-specific deficits as the primary system of categorization. In general, such reviews were less focused on biological mechanisms underpinning Long COVID.16,18,19 Still, others have focused primarily on the cellular and metabolic changes associated with incomplete recovery, which are crucial for formulating mechanistic hypotheses but insufficient to reveal the discrete biological mechanisms contributing to the likely multi-mechanistic pathogenesis of Long COVID.17 Our review builds on the excellent work of our colleagues and provides a focused and comprehensive summary of the biological mechanisms underpinning the development of Long COVID. We further review the latest up-to-date results that support or question the reviewed hypotheses, and thereby contribute a different lens for understanding this complex condition.
We performed a literature search in PubMed and EMBASE for “Post COVID-19 Condition”, “Post-acute sequelae of COVID-19”, “Long COVID”, “Post-COVID”, or “Post-COVID Syndrome” between 1 January 2020 and 1 September 2022. We manually searched references of identified studies and included all studies in the English language. We considered all studies which evaluated a potential biological basis for the persistence, recurrence, or emergence of symptoms following SARS-CoV-2 infection, at least four weeks following acute infection.
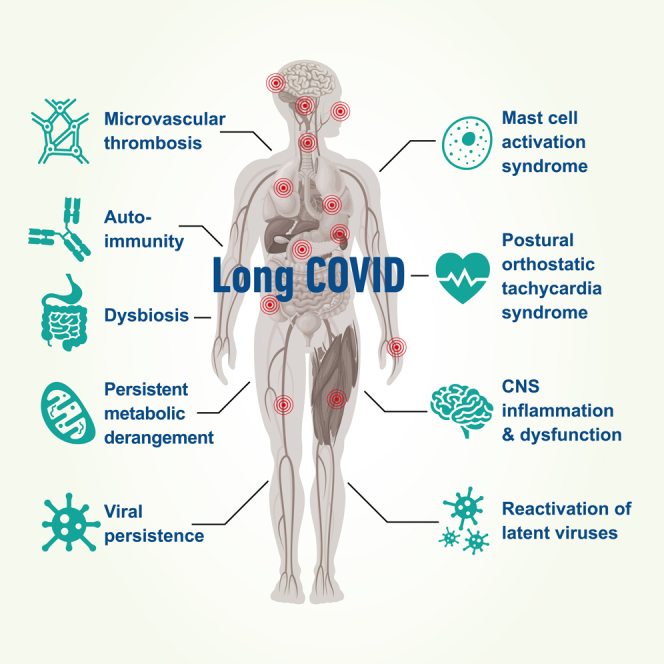
This review identified several potential mechanisms for discussion, supported by published evidence, including viral persistence, SARS-CoV-2 antigen persistence, microclot formation, autoimmunity, reactivation of latent viruses, mast cell activation, persistent systemic inflammation, dysbiosis, persistent central nervous system inflammation, metabolic dysfunction/bioenergetic failure, and autonomic dysfunction (Figure 1).
Figure 1.

The potential biological mechanisms underpinning the development of Long COVID (LC)
Potential contributors to a protracted recovery from COVID-19 are indicated in the blue circles. Potential biological mechanisms underpinning Long COVID are indicated in the red rectangles.
Given that the mechanisms we discuss herewith remain plausible and that none have to date been conclusively demonstrated to be involved in LC pathogenesis, we also list interventional trials that are in progress for each of them. Indeed, in the absence of validated animal models for LC pathogenesis, it is the trials that will be pivotal for distinguishing between the proposed LC mechanisms.
Pathogenic mechanisms of Long COVID
Viral persistence, shedding, and antigen persistence
Many virus infections are controlled and eliminated by the innate immune system, particularly by the induction of interferons, as a first line of host defense, and the subsequent development of the adaptive immune response (virus-specific antibodies and cytotoxic T cells) to the pathogen. Virus replication products, including double-stranded RNA and viral proteins, are recognized by the infected cell as viral pathogen-associated molecular patterns by the cell’s pattern recognition receptors such as Toll-like receptors and RIG-I-like receptors.20 SARS-CoV-2 is very sensitive to interferon, even more so than the original SARS-CoV of the 2003 outbreak and SARS-CoV-2 replication is impaired by a primary, but not a secondary, infection by rhino- or influenza virus.20,21,22 Therefore, SARS-CoV-2 has evolved strategies, involving more than half of its gene products, to evade the induction of the innate immune response.20 Type I and type III interferon responses are associated with better prognosis, while inborn defects in the interferon system or autoantibodies to interferon are associated with more severe symptoms.23,24,25,26
Coronaviruses, including SARS-CoV-2, continuously undergo evolution to manipulate, obstruct, and avoid elimination by the host’s immune response.27 Some authors have suggested viral persistence as a pathogenic mechanism behind both severe acute COVID and LC.15,28,29,30,31,32,33,34,35,36 A study by Truong et al. (2021) demonstrated that immunocompromised children and young adults were highly susceptible to persistent viral infection, prolonged viral shedding, and the accumulation of viral variants. It was proposed that a weak host immune response increased the emergence of SARS-CoV-2 variants to propagate viral persistence through enhanced immune escape.37 Indeed, the evidence on the emergence of the Omicron variant strongly suggests that the major driver of viral evolution is the persistence of SARS-CoV-2 for prolonged periods within an immunocompromised host.38,39,40 Furthermore, certain SARS-CoV-2 antigens have the potential to act as superantigens, activating T cells through non-specific T cell receptor interactions, resulting in immune overstimulation and cytokine hypersecretion, diminished potential for directed viral clearance, and a negative feedback loop resulting in immune exhaustion or inhibition, and unintentional persistence of the virus.41
Two types of persistence can occur for SARS-CoV-2 - persistent replication of infectious virus and persistence of virus macromolecules (RNA and proteins). Either type of viral persistence would be predicted to induce different states of immune system activation compared with acute infection. The current evidence supporting a functional role for prolonged SARS-CoV-2 viral replication in the development of LC is the discovery of viral genomic particles contained within bodily fluids extracted from infected hosts following superficial recovery from the initial acute infection.26,36 Stein et al. reported the detection of SARS-CoV-2 RNA throughout the body, with evidence of viral replication at multiple extra-pulmonary locations during acute infection. In addition, subgenomic RNA (indicative, but not diagnostic, of viral replication) persisted at some sites, especially the CNS, weeks after the initial symptoms.36 Isolation of a replicating virus from tissues in this study has been demonstrated as late as day 13 following symptom onset. However, by definition, these autopsy cases reflect the situation where the host did not control the virus, leading to the host’s death. Moreover, the authors of this study explicitly state that it was not designed to address LC. Therefore, we will likely have to await the results of clinical trials using antivirals in patients with LC to resolve the importance of replicating virus in LC pathogenesis.
Viral RNA has been found in various tissues from patients with acute COVID-19 such as the adrenal glands,42 kidneys,43 intestine,44 lymph nodes,45 spleen,46 and heart.47 To date, the longest detected presence of SARS-CoV-2 RNA was 126 days in feces, 83 days in the upper respiratory tract, 60 days in blood, and 59 days in the lower respiratory tract.31,48,49 In a study of 29 patients with LC, the majority of whom experienced mild or moderate COVID-19, detectable plasma SARS-CoV-2 RNA was demonstrated at ∼8 weeks post-infection.50 Similarly, SARS-CoV-2 RNA was detectable in the stool and urine, albeit at lower rates. Despite the prolonged presence of SARS-CoV-2 RNA in the upper respiratory tract, successful efforts to isolate live replication-competent virus from culture beyond 9 days from symptom onset have not been reported, even in instances where subgenomic viral RNA has been detected.31 However, Xu et al. demonstrated cultivable SARS-CoV-2 in stool and urine samples from three convalescent patients for longer than 4 weeks after infection.51
The presence of SARS-CoV-2 within the gastrointestinal (GI) tract has multiple potential effects on human health. Generally, maintaining appropriate homeostasis in the intestinal microbiome/virome environment plays a significant role in host health [39]. Whether and in what instances the GI tract may be vulnerable to chronic infection with SARS-CoV-2 is poorly understood. There are many reports on successful isolation of infectious SARS-CoV-2 virus from stool samples collected from patients with acute severe COVID-19.52,53,54,55,56 However, it is unclear if infectious virus can be found in stool over the long term.57,58 Notably, a study by Natarajan et al. (2022) observed extended fecal SARS-CoV-2 RNA shedding in participants who had undetectable viral RNA in oropharyngeal swabs.57 Furthermore, these patients reported a host of both GI and systemic symptoms, providing evidence of clinically significant SARS-CoV-2 chronic gut infection. Zollner and co-researchers (2022)30 demonstrated that viral antigens, but not the infectious virions, persisted in the gut mucosa even after recovery from mild acute COVID-19 infection. The persistence of SARS-CoV-2 antigens was determined to occur in 52%–70% of patients up to 7 months after the onset of symptoms. The persistence of the nucleocapsid SARS-CoV-2 antigen was detected adjacent to or within gut-derived CD8+ T cells and epithelial cells. The significance of these observations is unclear, because this persistence was unrelated to the severity of the acute COVID-19 illness or gut inflammation status.30
Through the utilization of conventional immunochemistry techniques, Cheung and co-researchers (2022) detected the nucleocapsid protein of SARS-CoV-2 in the lymph nodes, gall bladder, liver, hemorrhoids, ileum, appendix, and colon of five patients with COVID-19, ranging between 9 and 180 days after testing negative for SARS-CoV-2 by nasal swab RT-PCR.59 SARS-CoV-2 viral nucleic acid was also found in the pulmonary tissue of deceased patients with negative nasopharyngeal swab RT-PCR results.52,59,60,61,62 These findings strongly suggest that negative SARS-CoV-2 RT-PCR results from upper and lower airway samples may not accurately represent complete viral clearance from the host.59 Whether the presence of viral nucleic acid and antigens in the tissues is from persistence of infectious virus is unclear, and in-depth studies will be necessary to determine the mechanism establishment of viral macromolecular persistence and its potential to cause disease long after infection.57 Non-classical monocytes containing S1 protein were found in circulation up to 15 months post-infection, and it was proposed that their senescence and long-term circulation may reflect the acquisition of a proinflammatory phenotype, consistent with the elevations in key inflammatory markers associated with LC.63,64 Finally, an intriguing study by Swank et al. showed the presence of viral antigens in >70% of the 37 patients with LC from a New England cohort using highly sensitive bead-based protein detection, with the whole S protein being most sensitive in detecting LC. Larger confirmatory studies are in progress to validate these findings.64
A separate and relevant, but distinct, scenario is persistent viremia seen in immunocompromised individuals.37,65 In a study by Hagman et al. (2022), an analysis of 121 hospitalized patients with SARS-CoV-2 viremia demonstrated viral clearance at a median of 7 days following hospital admission, corresponding to an average of 15 days after the onset of symptoms.66 Notably, the mortality risk increased with each additional day of viremia.66 Li et al. (2021) discovered upregulated proteomic markers that correlated with SARS-CoV-2 viremia, including upregulation of SARS-CoV-2 entry factors such as angiotensin-converting enzyme-2 (ACE2), cathepsin L, and furin, and elevated markers related to tissue damage affecting the vascular endothelium, gastrointestinal tract, and lungs, and alterations in coagulation pathways.67 Persistent SARS-CoV-2 viremia has been demonstrated for up to 7 weeks after infection in immunocompromised patients, raising the possibility of low-level chronic viremia as a biological mechanism contributing to the genesis of LC.68 However, there is a paucity of data evaluating the burden of LC in immunocompromised people. A study by Basic-Jukic (2022) determined that only 11.5% of renal transplant recipients who survived acute COVID-19 presented with no clinical symptoms or demonstrated no laboratory abnormality during a median follow-up of 64 days. Prolonged symptoms and clinical complications were present in 45.2% of patients, while 71.2% reported one or more laboratory abnormalities.69 The increased risk for developing post-COVID clinical complications may be due to the impaired immunity in transplant recipients, owing to their treatments with immunosuppressants. Common symptoms reported by patients included cognitive impairments (5.7%), dry cough (7.7%), peripheral neuropathy (7.7%), fatigue (11.5%), and dyspnea (19.2%). Patients also had abnormal laboratory findings favoring clot formation, resistance to clot degradation, inflammation, impaired antibody production, and an increased risk of venous thromboembolism.69 Several patients required re-hospitalization for severe complications, including reactivation of herpesvirus and polyoma virus infections such as Epstein-Barr virus (EBV) and cytomegalovirus (CMV), and BK virus, respectively.69 Also, elevated IgM antibodies against EBV or T cell responses against CMV have been seen in LC,70,71 and a recent study suggested that EBV reactivation and pre-existing HIV infection positively correlated with LC, while CMV reactivation negatively correlated with LC.29,72
HIV is an important form of immunodeficiency to consider in relation to SARS-CoV-2. People living with HIV/AIDS (PLWHA) are at a higher risk of developing severe COVID-19 complications because of dysregulated inflammation and a functionally impaired immune system.73 Studies suggest that PLWHA may pose a higher risk of developing COVID-19-related clinical complications, especially in the setting of HIV viremia and immunosuppression.74 In a recent LC study observing PLWHA, the prevalence of LC was 43.6%, with moderate to severe acute COVID-19 being significantly linked to an increased risk for LC. The most common symptoms observed in the participants included fatigue (19.1%) and cough (22.3%). Persistent symptoms ranged between 30 and 109 days following the onset of COVID-19 illness.75 Peluso et al. demonstrated that PLWHA were more likely to develop LC than well-matched HIV-negative participants, had lower SARS-CoV-2-specific CD8+ T cell responses, and mounted SARS-CoV-2-specific CD4+ T cell responses which were functionally impaired by overexpression of the co-inhibitory receptor PD-1.76 Two previous studies revealed the possible beneficial effect of ART on COVID-19 risk in PLWHA.77,78 It was hypothesized that ART might act as pre-exposure prophylaxis against SARS-CoV-2, an idea based on the evidence that several ART drugs exert in vitro inhibitory activity against SARS-CoV-2 viral replication.74 It has subsequently been shown that PLWHA who receive ART benefit from the regimen’s effect on restoring a functional immune system rather than any direct inhibitory effects exerted on the replication capacity of the SARS-CoV-2 virus.79,80,81 Whether and to what extent the aforementioned factors may be further modulated by SARS-CoV-2 within-host evolution and escape in these and other immunocompromised patients remains to be seen.
As shown by several studies, viral persistence and/or virus antigen persistence could potentially have direct and indirect effects on the pathogenesis of LC.26,82,83 Firstly, direct lytic cell death from viral infection would contribute to tissue destruction; secondly, prolonged presence of viral proteins could impact many cellular processes that could affect tissue and organ function.82 Finally, chronic inflammation and cell damage initiated by viral persistence may induce both immune pathogeneses, with an inappropriate and overexuberant immune activation, autoimmune responses, including molecular mimicry.26,84 The smoking gun, in the form of isolation of a replicating SARS-CoV-2, has been lacking so far. Still, there is the possibility of the direct or indirect involvement of viral macromolecules in mechanisms that could perpetrate and/or contribute to LC. However, there is sufficient reason to launch interventional trials to explore whether antivirals can prevent the onset, reduce the incidence, and ameliorate the clinical course of LC. While off-label use of antiviral agents for the treatment of LC has been reported in clinical practice, there are currently ongoing registered clinical trials evaluating antiviral agents for this indication which should shed more light on this issue (NCT05576662, NCT05595369, NCT05668091).
Microclot formation
Recently, Pretorius and team showed that plasma derived from patients with LC contained microclots resistant to fibrinolysis.85 SARS-CoV-2 RNA has been found in platelets, and severe acute infection is linked to the activation and degranulation of platelets.86 The exact mechanism of platelet activation through SARS-CoV-2 viral interactions has not been fully elucidated. However, several mechanisms could explain this phenomenon.85 Firstly, SARS-CoV-2 can interact with platelets and possibly activate them by direct interaction of the viral spike protein with the host platelet receptor. Indeed, SARS-CoV-2 spike protein avidly binds to ACE2 receptors, which are expressed at high levels on platelets. Alternatively, SARS-CoV-2 could bind to platelets when coated with antiviral antibodies capable of binding to the FcγRIIA (CD32a) on platelets.87 The SARS-CoV-2 spike protein binds to CD209 on platelets and also contains an arginine-glycine-aspartic acid amino acid sequence that can develop interactions with integrins. Glycoprotein IIb/IIIa is the primary receptor found on the platelet surface and readily binds multiple ligands that encompass the arginine-glycine-aspartic acid sequence.87
In addition, the envelope protein of SARS-CoV-2 has been shown to interact with Toll-like receptor 2 found on platelets.87 Activated platelets release thrombin (IIa), leading to platelet aggregation and three-dimensional clot formation.88 This event is catalyzed by collagen and von Willebrand factor that is exposed on damaged vessel walls binding to glycoprotein Ib-IX-V, a platelet adhesion receptor located on the surface of platelets. In physiological conditions, there is a balance between procoagulant and anticoagulant systems; this balance is often dysregulated in pathological conditions, which creates the potential for coagulopathy in affected individuals.89
As mentioned previously, these persistent microclots were highly resistant to fibrinolysis and contained high levels of pro-inflammatory molecules, including alpha-2 antiplasmin (α2AP), Von Willebrand factor, fibrinogen, and plasminogen.85 These microclots may release pro-inflammatory cytokines over a prolonged period. Thus, in addition to impeding microcirculatory flow, these microclots have the potential to result in a persistent state of coagulopathy and hyperinflammation.85 On account of the prolonged state of inflammation caused by viral persistence, an increase in dysregulated molecules such as serum amyloid A and α2AP was identified in patient samples from LC and acute COVID-19 patients in comparison to healthy control non-COVID-19 samples.85
Microclots may accumulate and disseminate within the microcirculation, eventually obstructing capillaries and circulatory pathways, leading to hypoperfusion of tissues. Recently, it has been shown that the SARS-CoV-2 spike protein can induce the formation of microclots by binding to soluble fibrinogen molecules causing structural changes to complement 3, prothrombin, β and γ fibrin/fibrinogen, thereby rendering the molecule insoluble.90 Furthermore, the ACE2 and TMPRSS receptors are found universally on platelets and vascular endothelium, to which the virus readily binds to infect host cells.91,92 Microclot formation leading to micro-occlusion may be a central mechanism for the development of LC85,93 and SARS-CoV-2 spike protein could play a vital role in establishing the underlying hypercoagulability.90 Detecting the microclots requires specialized laboratory equipment such as bright-field microscopes or fluorescence microscopy. Hence, when the soluble component of the plasma is analyzed, the inflammatory molecules are not detected, obfuscating their important role in LC.85,90,93
Several trials are underway to evaluate the potential of anticoagulation or plasmapheresis for the treatment of LC (NCT05445674, NCT05543590, ISRCTN10665760).
Mast cell activation
Mast cells (MCs), which are abundant in the interstitial tissue of organs, play a crucial role in the development of diabetes mellitus type 2, rheumatoid arthritis, autoimmune diseases, and hypersensitivity reactions.94 Disordered physiological processes underlying the onset of LC may be in part due to an abnormally increased activation of atypical MCs induced by SARS-CoV-2. Symptoms associated with mast cell activation (MCA) are commonly observed in patients with LC, often mimicking the symptoms and disease severity observed in individuals with idiopathic mast cell activation syndrome.95 It is estimated that up to 17% of the world’s population is predisposed to MCA, which may be triggered by a viral infection such as COVID-19.96 Generally, MCs influence how the innate and adaptive immune system responds to invading viruses, parasites, or bacteria by using their ability to discern products of these invaders.96,97 These cells contain growth factors, cytokines, heparin, and histamine, which they readily release upon MC activation and degranulation to modulate numerous facets of the immune response and to direct the immune response to the location of the antigen. The pattern of the hyperinflammation that occurs during COVID-19 is consistent with an inflammatory response mediated through SARS-CoV-2-mediated MC activation, and such a hyperactivation of MCs has been widely demonstrated in COVID-19 postmortem studies.95,96 Widespread MC degranulation in the airways of SARS-CoV-2-infected mice and nonhuman primates has been indicated by quantitation of MC-specific chymase protease.98 Importantly, pulmonary MCA persisted beyond the acute infection, even after patents have tested negative for SARS-CoV-2 infection by PCR. Persistent MCA is associated with extensive systemic inflammatory pathologies seen in LC, including myocardial infarction, compromised microvasculature, endothelial damage, and intravascular coagulation. In histopathological studies, MCs were observed to organize themselves along the blood vessel walls, placing them in an optimal location to directly exert their effects on the vasculature, where MC-excreted mediators can be disseminated via the circulatory system.94,98 Several studies have hypothesized that persistent hyperinflammation primarily mediated through MCA may be responsible for LC symptoms.95,99,100,101
Interestingly, MCs express coronavirus receptors such as CD26 (dipeptidylpeptidase), a multi-functional type-II transmembrane glycoprotein hypothesized to contribute to SARS-CoV-2-mediated pulmonary inflammation.102,103 This receptor has pleiotropic functions, including cell-cell and cell-matrix interactions, hormone, and metabolic.104 Activated MCs contain and secrete several active mediators, including TNF-α prostaglandins, leukotrienes, reactive nitrogen species, chondroitin sulfates, heparin, lysosomal enzymes, granzyme, serotonin, cathepsin G, serine S1, histamine, carboxypeptidase, chymase, and tryptase. The role of the latter three mediators has been implicated in the pathogenesis of LC.105,106,107,108 Mucosa and connective tissue MC-derived tryptase can trigger eosinophils to secrete IL-6 and -8 for neutrophil recruitment to affected sites. This enzyme may also stimulate inflammatory cytokine release from endothelial cells, such as IL-8 and -1β. Tryptase has also been identified to exert epigenetic effects through its capacity to affect DNA stabilization. Tryptase may contribute to cognitive impairments such as brain fog symptoms due to the induction of vascular leakage, thus causing increased blood-brain barrier permeability.99,106,109 In addition, tryptase may contribute to post-infection fibrosis of the lungs by inciting the movement and division of pulmonary fibroblasts leading to the reorganization and upregulation of fibroblastic collagen synthesis.106 Chymases are members of a large serine protease family whose expression is unique to MCs.110 MC-derived chymase can independently generate angiotensin II from angiotensin I without the catalytic activity of ACE and is often associated with vascular diseases.111,112,113,114,115 Chymases activate transforming growth factor β and matrix metalloproteinases, which contribute to fibrosis of the lungs.
Several clinical trials are registered or underway for the evaluation of antihistamines, leukotriene receptor antagonists, and mast cell stabilizers in the treatment of LC (NCT04695704, EUCTR2021-000605-24-ES, ISRCTN10665760).
Autoimmunity
Extrafollicular B cell expansion
Severe SARS-CoV-2 infection is marked by exaggerated extrafollicular B cell expansion and production of both SARS-CoV-2-specific and autoreactive antibodies.68,116 This is immunophenotypically similar to the picture seen in severe systemic lupus erythematosus (SLE), with a profound lack of follicular T helper cells.68 Extrafollicular B cell expansion in severe COVID-19 is characterized by the induction of broadly SARS-CoV-2-reactive and autoreactive Tbet-driven double-negative 2 (CD27−, IgD−, CD11c+, and CD21−) B cells, again similar to the responses in patients with severe SLE.68,116 Produced autoantibodies shown to clonally originate from these dysregulated naive cells contain few mutations, are immunophenotypically consistent with an extrafollicular origin, and account for clinically relevant autoreactivity. Importantly, during the resolution of severe COVID-19, this autoreactivity subsided and was controlled. However, in a subset of patients with LC, the resolution was incomplete, and autoimmunity against nuclear autoantigens and carbamylated proteins persisted.116
Molecular mimicry by viral antigens
SARS-CoV-2 may elicit autoimmunity due to viral proteins’ mimicry of human molecular chaperones.117 Several studies have shown that in patients with severe COVID-19, SARS-CoV-2 stimulates antibodies which interact with human-derived proteins.118,119,120,121 A process of molecular mimicry may occur when microbial peptides/proteins possess homology to human tissue peptides/proteins leading to an activation of autoreactive T or B cells,122 leading to the onset of transient or chronic autoimmune disorders. Molecular mimicry has also been implicated in heterologous immunity. The latter is described as immunity in which the immune system responds to an unrelated or partially related antigen or pathogen following exposure to another different antigen.123,124 In an in silico study by Nunez-Castilla et al. (2022), molecular mimicry was described as a molecular match of at least five identical sequential amino acids located in the SARS-CoV-2 spike protein and matched human protein sequences.118 In addition to favoring the production of autoantibodies, molecular mimicry by SARS-CoV-2 antigens may result in autoimmunity by stimulating autoreactive T cells which have escaped central and peripheral tolerance mechanisms.125
Autoantibodies and their effects
Autoimmunity has been proposed as a possible pathobiological driver of LC and some, but not all, reports have found autoantibodies in patients with LC.126 At the present, definitive evidence for or against this mechanism is lacking. There are several plausible mechanisms that may contribute toward the onset of autoimmunity in patients with COVID-19. These include SARS-CoV-2-encoded superantigen-mediated T cell activation, viral persistence-mediated chronic activation of the immune system, pathogen-host tissue molecular mimicry, and increased secretion of peripheral and central cytokines. Furthermore, other concurrent microbial or viral infections (including reactivation of persistent herpesviruses) may contribute to autoantibody production under inflammatory conditions, causing diverse autoantibody reactivity.127 Pre-existing autoimmunity is associated with severe COVID-19 disease, especially in individuals with Sjögren syndrome, systemic lupus erythematosus, scleroderma, rheumatoid arthritis, myasthenia gravis, Guillain-Barre syndrome, multiple sclerosis, autoimmune hepatitis, idiopathic pulmonary fibrosis, cardiac sarcoidosis, subacute bacterial endocarditis, Dressler syndrome, and autoimmune myocarditis.128,129,130,131 Autoantibodies produce diverse clinical phenotypes by disrupting and interfering with the body’s biological systems via autoreactive attack against proteins, DNA, and other cellular components.132 However, more recently, multidimensional immune profiling of 99 patients with LC by rapid extracellular antigen profiling, a high-throughput method capable of measuring antibody reactivity to >6000 extracellular and secreted human proteins, failed to identify any stereotypical autoantibodies to distinguish patients with LC from healthy controls.133 However, differential antigen targeting in the LC and control groups revealed shared patterns of autoreactivity in a subset of patients with LC with tinnitus and nausea. Moreover, it remains unclear whether antibodies to intracellular autoantigens and non-protein autoantigens are implicated in the pathogenesis of Long COVID, as these issues were not addressed by the previous study.
Anti-type I interferon (IFN) (IFN-I) autoantibodies play an important role in COVID-19 pathogenesis.128 Pre-existing anti-IFN-I autoantibodies are strongly associated with an increased risk of developing severe COVID-19.128,129,130,131,134 Compared to the general population, an increased anti-IFNα autoantibody prevalence is seen in patients with systemic lupus erythematosus co-infected with COVID-19.130,131 Type I IFN is a central alarm, a warning instruction for the human host to initiate immune responses to fight, neutralize, and eliminate encountered viral infections. Deficiency of type I IFNs is a hallmark of many severe COVID-19 infections.135 Bastard et al. (2020) revealed that 13.7% of patients with severe COVID-19 exhibited anti-IFN-I autoantibodies, mostly neutralizing IFN-α2 and IFN-ω.129,134 In healthy individuals not exposed to SARS-CoV-2, anti-IFN-I autoantibodies were only detected in 0.3% of the study population.134 The anti-IFN autoantibodies neutralize the ability of the corresponding type I IFNs to inhibit SARS-CoV-2 both in vitro and in vivo.128,129 In addition, a B cell autoimmune phenocopy of type I IFN autoimmunity was identified and implicated in severe COVID-19 pneumonia, occurring in 12.5% of men and 2.6% of women.129 While anti-INF antibodies may increase the risk of LC by increasing the risk for severe COVID-19, it is less clear if a direct relationship between anti-IFN antibodies and LC exists. It is plausible that anti-IFN antibodies could delay viral clearance and may potentiate viral persistence. More recently, Peluso and colleagues demonstrated that while anti-IFN antibodies are likely associated with severe COVID-19, they were uncommon among individuals with Long COVID.136
In 2021, Chang and colleagues identified SLE-associated antinuclear antibodies (ANAs) in acute SARS-CoV-2-infected individuals.137 Up to 44% of patients with autoantibodies detected at 2–3 months following initial SARS-CoV-2 infection had shown mature autoantibody profiles, suggesting the possibility that the autoantibodies predated the viral infection71,129 and a small portion of autoantibody-positive patients reported autoimmune symptoms before contracting COVID-19.71 Notably, there was a strong negative correlation between anti-SARS-CoV-2 antibodies and IFN autoantibodies.71 Inhibition of IFN-α2 may potentiate the activity of pro-inflammatory cytokines, thereby provoking the generation of ANAs against self-antigens.71 Chang and coworkers found that patients reporting neurological LC symptoms had relatively higher anti-SARS-CoV-2 nucleocapsid protein IgG concentrations, those with gastrointestinal-related LC symptoms were associated with increased levels of several autoantibodies, and elevated IFN-α2 antibodies were associated with respiratory LC symptoms.137 High levels of anti-Ro/SS-A autoantibodies, as seen in patients with Sjögren syndrome, have also been detected in individuals with severe COVID-19 pneumonia.138 Unfortunately, it could not be determined whether the increased levels of anti-SSA/Ro autoantibodies were the direct cause or consequence of severe COVID-19 pneumonia. However, it was hypothesized that COVID-19 pneumonia may have developed due to an autoimmune response.139 Collectively, the diverse range of autoantibodies seen in patients with LC may provide a potential explanation for the diverse phenotypes seen in LC.71,139,140,141
Arthur et al. (2021) identified the presence of anti-ACE2 autoantibodies in hospitalized and recovered patients with COVID-19.140 The study revealed that individuals with a history of SARS-CoV-2 infection have elevated levels of ACE2 antibodies coupled with reduced activity of soluble plasma ACE2 and suppression of exogenous ACE2 activity. These results are congruent with findings that suggest ACE2 antibodies only develop after SARS-CoV-2 infection and cause a corresponding reduction in ACE2 functional activity, leading to increased angiotensin II via the renin-angiotensin-aldosterone system, stimulating a pro-inflammatory state, which may initiate symptoms related to LC.140 The anti-ACE2 autoantibodies are presumed to facilitate the persistence of inflammation, a central characteristic of autoimmune disease. While SARS-CoV-2 antibodies declined within 5–12 months following the acute infection, LC patients remained ACE2 antibody positive 12 months after the initial acute infection. These findings support the mounting evidence linking LC and autoimmunity.126 It will be of importance to test RAAS inhibitors in ameliorating LC or some of its phenotypes to test this pathogenesis avenue.
Longitudinal timepoint analysis conducted by Su et al. (2022) revealed three significant findings on LC.71 Firstly, LC-associated GI phenotypes correlated with newly expanded cytotoxic CD8+ and CD4+ T cell populations at 2–3 months following the acute infection.71 This includes SARS-CoV-2-specific clonal T cell progenies that become activated during the recovery phase before identification of LC. The authors suggested that this event may be connected to GI viral shedding. LC GI symptoms may involve CMV-specific T cell bystander activation proposing the additional contribution of non-specific T cell activation, which may further spur LC GI symptoms. Secondly, after 2–3 months, patients with LC could be segregated into one of four distinct immune endotypes, providing a potential basis to stratify LC types and pathogenesis. These findings need further longitudinal follow-up and corroboration. Thirdly, these authors found time-evolving connections of measurable LC risk factors from acute infection to convalescence. In the convalescence phase, LC risk factors are relatively independent of each other, suggesting that these risk factors may require independently targeted treatments once convalescence has been reached.71
COVID-19 is associated with diverse forms of coagulopathy and demonstrates a prolific potential for thrombogenesis, and some of them may be autoimmune in nature. A higher-than-expected prevalence of anti-phospholipid antibodies (APLAs) in patients with COVID-19 may partially explain this risk.142,143,144 There are three major types of APLAs, those against beta-2 glycoprotein I (β2GPI), lupus anticoagulant (LAC), and anticardiolipin (aCL).145 These antibodies bind to cell membrane proteins to induce coagulation dysfunction.142 Approximately 47% of severely ill patients with SARS-CoV-2 produced β2GPI or/and aCL autoantibodies.146,147 In addition, patients with severe COVID-19 had significantly higher concentrations of aCL autoantibody than patients with moderate infection.143,148,149 Elevated LAC concentrations are evident among patients with COVID-19 with coagulation disorders.150 The persistence of these antibodies beyond acute infection may result in an increased long-term risk of venous thromboembolism, microcirculatory insufficiency, and pervasive impairment of perfusion in multiple organ systems.151,152,153 Increased prevalence of thrombotic events is often associated with IgG phosphatidylserine/prothrombin (aPS/PT) autoantibodies, frequently observed in individuals with LC.142,154 In 2020, a study by Zuo et al. (2020) reported that 24% of 172 hospitalized SARS-CoV-2-infected patients produced aPS/PT IgG.143 Moreover, a platelet-activating antibody, anti-heparin-PF4 (aPF4), was also found in patients with COVID-19 with severe infection, especially in those who experienced heparin-induced thrombocytopenia. Contrastingly, some patients with SARS-CoV-2 had circulating aPF4 without heparin pre-exposure. These findings support the hypothesis that SARS-CoV-2 can cause coagulation disorders via an autoimmune response.142,155,156,157
Several planned or ongoing clinical trials seek to ameliorate autoimmunity through corticosteroid therapy, intravenous immunoglobulins, human ribonuclease therapy, and plasmapheresis (NCT04944121, NCT05543590, NCT05350774, NCT05445674).
Other potential mechanisms
Pathogenic mechanisms mentioned in the following section are less substantiated and are mentioned for completeness of the discussion of LC pathogenesis.
Inflammation is a common fallout from many of the mechanisms discussed in this paper and should be considered when discussing LC pathogenesis. Acute COVID-19 comes with a plethora of inflammatory changes, the most persistent and distinguishing ones involving IL-6, CRP, and to a lesser extent TNFα and other proinflammatory molecules that correlate with severe COVID-19. However, from the earliest studies onward, it became clear that acute disease markers and phenotypes do not linearly translate into the LC stage, and that was the case with inflammatory markers as well.71 A more recent study by Ruffieux et al. did link inflammatory, immune, and metabolic profiles at the transition from acute COVID-19 to recovery.17 These authors found a remarkable correlation between inflammation, immune, and metabolic abnormalities, clustering around CRP as a convenient, sensitive, and robust biomarker to predict recovery trajectories. As they followed these parameters for up to a year, the authors found persistent disruption of measured markers up to 6 months specifically in the group characterized by high initial inflammation. Moreover, these biological changes, centered around proinflammatory cytokines, correlated well with delayed clinical recovery rates, and were predictive of excess mortality in the most severe group as well.17 However, there were individual patients in that study who exhibited complete biochemical and inflammatory recovery, yet still exhibited LC symptoms. This suggests that inflammation, measured by a simple and reliable clinical test such as CRP, can be a good parameter to predict some, but not all, COVID-19 recovery and LC trajectories. Mechanistic causality, however, remains to be investigated, and that should begin with meta-analysis of large cohorts of patients receiving steroid and non-steroid anti-inflammatory treatments.
SARS-CoV-2 infection-induced reactivation of latent herpesviruses as a possible contributing factor to LC has been studied. In a study of 215 individuals with Long COVID, unexpected increases were observed in antibody responses directed at EBV.133 Also, in a cohort of 280 adults with prior SARS-CoV-2 infection, Peluso et al. observed that LC symptoms, such as fatigue and neurocognitive dysfunction at a median of 4 months following initial diagnosis, were associated with serological evidence of recent EBV reactivation (early antigen-diffuse IgG positivity or high nuclear antigen IgG levels) but not with ongoing EBV viremia. Serological evidence suggesting recent EBV reactivation (early antigen-diffuse IgG positivity) was most strongly associated with fatigue (OR = 2.12). In this study, underlying HIV infection was also associated with neurocognitive LC (OR = 2.5). However, those with serologic evidence of prior CMV infection were less likely to develop neurocognitive LC (OR = 0.52).72 In another study, the authors tested for SARS-CoV-2 RNA in stool and throat washings, and EBV DNA in stool, throat washings, and blood by real-time PCR (EBV) and real-time RT-PCR (SARS-CoV-2) in 30 patients with LC characterized by persistent fatigue, post-exertional malaise, autonomic dysfunction and/or orthostatic intolerance, and who all had had a mild SARS-CoV-2 infection. Twenty age- and sex-matched patients who had fully recovered after the SARS-CoV-2 infection served as controls. SARS-CoV-2 RNA indicating persistent infection was not detected in throat washing or stool. SARS-CoV-2 antibody titers (IgA and IgG) did not differ between the cohorts. EBV real-time PCR was negative in all blood or stool samples. However, EBV DNA was detected in throat washings in 15/30 (50%) of patients with LC, while in only 4/20 (20%) of non-LC patients (p = 0.0411).158 A study of patients with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and healthy donors as controls analyzed whether a mild/asymptomatic SARS-CoV-2 infection imposes latent EBV, HHV6, and HERV-K virus reactivation. At 3–6 months after infection, virus-specific antibodies in saliva were substantially induced, indicating a strong reactivation of all three viruses in both cohorts. In patients with ME/CFS, however, antibody responses were significantly stronger, in particular against EBV-encoded nuclear antigen-1 than in the controls.159
The difficulty in interpreting these findings lies in the fact that many stressors, including infectious diseases, will reactivate persistent infections and that correlation/timing here may reflect an epiphenomenon and not a causal link. Therefore, the role of reactivated EBV infection in the development of LC remains to be fully elucidated.
The metabolic sequelae of SARS-CoV-2 infection are highly complex, involving the dysregulation and dysfunction of multiple organs. Closely related coronavirus diseases, such as the Middle East respiratory syndrome and SARS, have been shown to prompt long-term metabolic modifications in recovered patients causing liver disorders, altered glucose metabolism, and hypertriglyceridemia. The earliest bioenergetic effects of SARS-CoV-2 infection occur through the binding of viral polypeptides to critical host cell mitochondrial proteins, retarding ATP production. However, SARS-CoV-2 produces a pervasive inhibition of the transcription of key mitochondrial genes central to cellular respiration.160 Mitochondrial dysfunction and resultant bioenergetic failure have been demonstrated during acute infection, but establishing impaired cellular respiration in the context of LC requires further research.161,162,163 Interventions seeking to reverse bioenergetic failure, reduce reactive oxygen species production, and restore cellular metabolism are currently under evaluation in several registered clinical trials (NCT05531019, NCT05373043, NCT04604704, NCT05371288, NCT05152849, NCT04960215, EUCTR2020-005961-16-DK).
Recently, evidence has emerged to propose the role of the lung-gut axis in increasing the risk for severe COVID-19.164 Gut microbiota dysbiosis correlates with poor clinical outcomes in mechanically ventilated patients with COVID-19. These findings indicate a strong interrelation between SARS-CoV-2 consequences and host gut microbiota, which may also be associated with an increased risk of LC. The substantial rise in pathogenic bacteria and simultaneous reduction in anti-inflammatory microbiota promotes sustained intestinal inflammation during COVID-19, subsequently leading to prolonged recovery.165 Su and colleagues’ (2022) study followed 155 patients for 14 months after being diagnosed with acute COVID-19 infection; 76.4% were diagnosed with LC at six months and 78.7% at 14 months. In LC-associated dysbiosis, the three most common symptoms were disrupted sleep (35.5%), cognitive difficulties relating to memory (44.5%), and fatigue (50.9%). It was found that the gut microbiota of the patients with LC did not fully recover when compared to the healthy control group (no COVID-19 diagnosis) at one year in patients with LC.166 Metabolic pathways accompanying the generation of proinflammatory molecules were abundantly increased. In contrast, anti-inflammatory pathways were downregulated, which may be one of the main attributes underlying the persistence of LC and associated symptoms.167 Whether and to what extent the microbiome dysregulation may further be potentiated by an infection- or inflammation-mediated increase in gut permeability (’leaky gut”) remains to be evaluated. A clinical trial investigating the role of fecal microbiota transplantation in the treatment of LC is underway (NCT05556733).
Amyloidogenesis is a speculative mechanism for LC.168 Persistent LC symptoms, including cognitive, behavioral, and psychiatric symptoms, resemble those seen in neurodegenerative disorders. Many neurodegenerative diseases such as Parkinson and Alzheimer are distinguished by the abundance of amyloid or amyloid plaque.169 Spike protein amyloid fibrils are known to localize in nerve tissues and to undergo heterologous seeding to promote accumulation and aggregation of endogenous proteins, which may lead to neurodegeneration. In patients with COVID-19, blood clotting is related to extracellular amyloidotic fibrillar assemblages. This was experimentally proven by demonstrating hypercoagulation and impaired fibrinolysis in the plasma from healthy donors when incubated with the viral spike protein. Previous studies have linked amyloidosis to thromboinflammation, activation of surface-activated coagulation system (FXII Kallikrein/Kinin), disruption of the fibrinolytic system, dysregulation of blood coagulation, and cerebral amyloid angiopathy, collectively, suggesting budding associations between COVID-19 phenotypes and S-protein amyloidogenesis. A study conducted by Nystrom and Hammarstrom (2022) found amyloidal sequences in SARS-CoV-2 spike protein and that endoproteolytic cleavage of the protein resulted in amyloid formation.170 Amyloidosis, resulting from the systemic deposition of amyloid proteins, may present as localized or systemic conditions with many phenotypes overlapping with the reported LC clinical phenotypes.170
Some symptoms observed in patients with LC suggest immune or virus-induced autonomic nervous system (ANS) disruption, resulting in temporary or possibly long-term orthostatic intolerance. Many have hypothesized that COVID-19 infection affects the ANS and that the SARS-CoV-2 virus itself may mediate the resultant autonomic dysfunction. Postural orthostatic tachycardia syndrome (POTS) involves many symptoms such as nausea, heart palpitations, blurry vision, headaches, tiredness, cognitive impairments, and lightheadedness. The exact cause underlying the development of POTS in LC remains unknown, but dysautonomia has been commonly implicated. Autonomic disorders like POTS are often associated with abnormal activity of muscarinic receptors and alpha and beta (α/β) adrenoreceptors. Usually, when a healthy individual stands, blood accumulates in the pelvis and limbs, which causes a reduction in venous return to the heart. Cardiac baroreceptors, sensing the volume change, stimulate an increase in sympathetic adrenergic and neural tone modulated by epinephrine and norepinephrine, respectively. Consequently, these events lead to tachycardia and splanchnic vascular bed vasoconstriction for elevated venous cardiac return. During orthostatic intolerance, norepinephrine and epinephrine release induces distinct tachycardia, manifesting as chest pain, shortness of breath, and palpitations, symptoms frequently associated with LC. Extremely high catecholamine levels can cause inconsistent vasodilatation, withdrawal of sympathetic activity, and vagus nerve activation which may compound the symptoms.171,172,173
In a study on the neuropathology in the brain and brainstem of SARS-CoV-2-infected nonhuman primates, Rutkai et al. found neuroinflammation, micro-hemorrhages, signs of brain hypoxia, and a neuropathology that would be consistent with hypoxic-ischemic injury in SARS-CoV-2, including evidence of neuron degeneration and apoptosis.174 This was primarily seen in the cerebellum, brainstem, and basal ganglia. Importantly, this was seen among infected animals that did not develop severe respiratory disease, and therefore could provide insight into neurological symptoms associated with the brain fog seen in LC. Sparse virus antigen or RNA was detected in brain endothelial cells but did not associate with the severity of central nervous system injury. Several interventions aimed at improving cerebral blood flow and oxygenation, reducing neuroinflammation, restoring neurotransmitter activity, and enhancing cognitive performance are under investigation in clinical trials (NCT04997395, NCT05531019, NCT04809974, NCT04604704, NCT05311852, NCT05074888, NCT05507372, NCT05430152, NL9742).
Conclusions and translational perspectives
Post-acute sequelae of viral infections are common, often leading to significant damage and loss of function in a subset of the infected population. COVID-19 and subsequent LC have taken the human population by storm, with SARS-CoV-2 infecting a significant portion of the world population in less than three years since its appearance. As COVID-19 transitions from a pandemic to an endemic disease, there is no doubt that cases of LC will continue to occur for years to come. Thus, there is an urgency to fully understand the cause(s) of LC and post-viral syndromes. From our discussion, and from the heterogeneity of LC presentations, consequently, there probably is no single pathogenic mechanism of LC but rather a complex interplay between virus and host, which may determine the extent and severity of the sequelae in different ways in different patients. Therefore, clinicians will have to arm themselves with knowledge of the multifactorial nature of LC and perform detailed clinical phenotyping while studying diagnostic tools and biomarkers that can guide their understanding of the driving mechanisms of pathology in the different manifestations of the sequelae. Biomedical scientists will, conversely, have to team up with clinicians to simultaneously apply comprehensive, unbiased multidimensional molecular analyses to elucidate the nature of LC and eventually provide the most effective treatment to each patient. While more effective COVID vaccines may become available and protect more individuals from infection, in the short-medium term, we must be focused on understanding the mechanisms underlying the symptoms of Long COVID to inform effective treatment approaches.
In that regard, and in the absence of clear “smoking guns” and “silver bullets”, a special role in understanding LC will belong to pragmatic, small, and deeply studied clinical trials. In other words, as the clinical trials are initiated, there is utmost urgency to make sure that biomedical samples are collected throughout, and that funding be provided for their deep molecular analysis and cross-correlation with clinical endotypes and analyses to understand success, partial success, as well as failure of treatments in different populations of patients with LC. Only then will we be able to reap the most benefit from the trials themselves, and to rapidly and nimbly improve ineffective or partially effective treatments, and to combine best treatments to achieve improved results.
Clinical trials directed at some of the putative biological mechanisms underpinning Long COVID are planned or underway, but there are still many missed opportunities. The roles of vaccination, antiviral therapy, anticoagulation, and immunomodulation in treating Long COVID need urgent evaluation in appropriately designed clinical trials. Other candidate interventions will emerge in parallel from strong basic science research on LC, and we strongly recommend that they be translated through small, flexible, and pragmatic trials in comprehensively phenotyped participants to help many patients suffering from LC, and to simultaneously help illuminate the path ahead.
Acknowledgments
We wish to thank Chris Bohnsack for assistance with the illustration in this manuscript.
Author contributions
R.P. and K.N. conceptualized the manuscript. R.P., J.N., A.V., A.G.D., and L.S. conducted the literature review and produced the original draft. K.N., D.W., and C.B. reviewed the original draft and contributed to the final version. All authors contributed to the synthesis, write-up, and review of the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Estrada L.v., Levasseur J.L., Maxim A., Benavidez G.A., Porter K.M.P. Structural racism, place, and COVID-19: a narrative review describing how we prepare for an endemic COVID-19 future. Health Equity. 2022;6:356–366. doi: 10.1089/HEQ.2021.0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabore J.W., Karamagi H.C., Kipruto H.K., Mungatu J.K., Asamani J.A., Droti B., Titi-ofei R., Seydi A.B.W., Kidane S.N., Balde T., et al. COVID-19 in the 47 countries of the WHO African region: a modelling analysis of past trends and future patterns. Lancet Glob Health. 2022;10:e1099. doi: 10.1016/S2214-109X(22)00233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reese H., Iuliano A.D., Patel N.N., Garg S., Kim L., Silk B.J., Hall A.J., Fry A., Reed C. Estimated incidence of coronavirus disease 2019 (COVID-19) illness and hospitalization—United States, February–September 2020. Clin. Infect. Dis. 2021;72:e1010–e1017. doi: 10.1093/CID/CIAA1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsoularis I., Fonseca-Rodríguez O., Farrington P., Jerndal H., Lundevaller E.H., Sund M., Lindmark K., Fors Connolly A.M. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after covid-19: nationwide self-controlled cases series and matched cohort study. BMJ. 2022;377 doi: 10.1136/BMJ-2021-069590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis A., Wamil M., Alberts J., Oben J., Cuthbertson D.J., Wootton D., Crooks M., Gabbay M., Brady M., Hishmeh L., et al. Original research: multiorgan impairment in low-risk individuals with post-COVID-19 syndrome: a prospective, community-based study. BMJ Open. 2021;11 doi: 10.1136/BMJOPEN-2020-048391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie Y., Xu E., Bowe B., Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022;28:583–590. doi: 10.1038/s41591-022-01689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhaskaran K., Rentsch C.T., Hickman G., Hulme W.J., Schultze A., Curtis H.J., Wing K., Warren-Gash C., Tomlinson L., Bates C.J., et al. Overall and cause-specific hospitalisation and death after COVID-19 hospitalisation in England: a cohort study using linked primary care, secondary care, and death registration data in the OpenSAFELY platform. PLoS Med. 2022;19:e1003871. doi: 10.1371/JOURNAL.PMED.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roca-Fernandez A., Wamil M., Telford A., Carapella V., Borlotti A., Monteiro D., Thomaides-Brears H., Dennis A., Banerjee R., Robson M.D., et al. Cardiac impairment in Long Covid 1-year post SARS-CoV-2 infection. Eur. Heart J. 2022;43 doi: 10.1093/EURHEARTJ/EHAC544. ehac544.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malkova A., Kudryavtsev I., Starshinova A., Kudlay D., Zinchenko Y., Glushkova A., Yablonskiy P., Shoenfeld Y. Post COVID-19 syndrome in patients with asymptomatic/mild form. Pathogens. 2021;10:1408. doi: 10.3390/PATHOGENS10111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wanga V., Chevinsky J.R., Dimitrov L.v., Gerdes M.E., Whitfield G.P., Bonacci R.A., Nji M.A.M., Hernandez-Romieu A.C., Rogers-Brown J.S., McLeod T., et al. Long-term symptoms among adults tested for SARS-CoV-2 — United States, January 2020–April 2021. MMWR Morb. Mortal. Wkly. Rep. 2021;70:1235–1241. doi: 10.15585/MMWR.MM7036A1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groff D., Sun A., Ssentongo A.E., Ba D.M., Parsons N., Poudel G.R., Lekoubou A., Oh J.S., Ericson J.E., Ssentongo P., et al. Short-term and long-term rates of postacute sequelae of SARS-CoV-2 infection: a systematic review. JAMA Netw. Open. 2021;4 doi: 10.1001/JAMANETWORKOPEN.2021.28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang L., Yao Q., Gu X., Wang Q., Ren L., Wang Y., Hu P., Guo L., Liu M., Xu J., et al. 1-year outcomes in hospital survivors with COVID-19: a longitudinal cohort study. Lancet. 2021;398:747–758. doi: 10.1016/S0140-6736(21)01755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayoubkhani D., Khunti K., Nafilyan V., Maddox T., Humberstone B., Diamond I., Banerjee A. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. 2021;372:n693. doi: 10.1136/BMJ.N693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayoubkhani D., Pawelek P., Gaughan C. Technical article: updated estimates of the prevalence of post-acute symptoms among people with coronavirus (COVID-19) in the UK - office for National Statistics. 2021. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/articles/technicalarticleupdatedestimatesoftheprevalenceofpostacutesymptomsamongpeoplewithcoronaviruscovid19intheuk/26april2020to1august2021
- 15.Ballering A.v., van Zon S.K.R., olde Hartman T.C., Rosmalen J.G.M. Persistence of somatic symptoms after COVID-19 in The Netherlands: an observational cohort study. Lancet. 2022;400:452–461. doi: 10.1016/S0140-6736(22)01214-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantovani A., Morrone M.C., Patrono C., Santoro M.G., Schiaffino S., Remuzzi G., Bussolati G., Cappuccinelli P., Fitzgerald G., Bacci M.L., et al. Long Covid: where we stand and challenges ahead. Cell Death Differ. 2022;29:1891. doi: 10.1038/S41418-022-01052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruffieux H., Hanson A.L., Lodge S., Lawler N.G., Whiley L., Gray N., Nolan T.H., Bergamaschi L., Mescia F., Turner L., et al. A patient-centric modeling framework captures recovery from SARS-CoV-2 infection. Nat. Immunol. 2023;24:349. doi: 10.1038/S41590-022-01380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nalbandian A., Sehgal K., Gupta A., Madhavan M.V., McGroder C., Stevens J.S., Cook J.R., Nordvig A.S., Shalev D., Sehrawat T.S., et al. Post-acute COVID-19 syndrome. Nat. Med. 2021;27:601. doi: 10.1038/S41591-021-01283-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis H.E., McCorkell L., Vogel J.M., Topol E.J. Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023;21:133–146. doi: 10.1038/s41579-022-00846-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J.H., Koepke L., Kirchhoff F., Sparrer K.M.J. Interferon antagonists encoded by SARS-CoV-2 at a glance. Med. Microbiol. Immunol. 2022;212:125–131. doi: 10.1007/S00430-022-00734-9/FIGURES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lokugamage K.G., Hage A., de Vries M., Valero-Jimenez A.M., Schindewolf C., Dittmann M., Rajsbaum R., Menachery V.D. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020;94:e01410-20. doi: 10.1128/JVI.01410-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Essaidi-Laziosi M., Alvarez C., Puhach O., Sattonnet-Roche P., Torriani G., Tapparel C., Kaiser L., Eckerle I. Sequential infections with rhinovirus and influenza modulate the replicative capacity of SARS-CoV-2 in the upper respiratory tract. Emerg. Microb. Infect. 2022;11:412–423. doi: 10.1080/22221751.2021.2021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bastard P., Zhang Q., Zhang S.Y., Jouanguy E., Casanova J.L. Type I interferons and SARS-CoV-2: from cells to organisms. Curr. Opin. Immunol. 2022;74:172–182. doi: 10.1016/J.COI.2022.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galani I.E., Rovina N., Lampropoulou V., Triantafyllia V., Manioudaki M., Pavlos E., Koukaki E., Fragkou P.C., Panou V., Rapti V., et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021;22:32–40. doi: 10.1038/S41590-020-00840-X. [DOI] [PubMed] [Google Scholar]
- 25.Galbraith M.D., Kinning K.T., Sullivan K.D., Araya P., Smith K.P., Granrath R.E., Shaw J.R., Baxter R., Jordan K.R., Russell S., et al. Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/PNAS.2116730119. e2116730119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodin P., Casari G., Townsend L., O’Farrelly C., Tancevski I., Löffler-Ragg J., Mogensen T.H., Casanova J.L., Abel L., Aiuti A., et al. Studying severe long COVID to understand post-infectious disorders beyond COVID-19. Nat. Med. 2022;28:879–882. doi: 10.1038/s41591-022-01766-7. [DOI] [PubMed] [Google Scholar]
- 27.Kasuga Y., Zhu B., Jang K.J., Yoo J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021;53:723–736. doi: 10.1038/s12276-021-00602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Proal A.D., VanElzakker M.B. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front. Microbiol. 2021;12:1494. doi: 10.3389/FMICB.2021.698169/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peluso M.J., Deveau T.-M., Munter S.E., Ryder D., Buck A., Beck-Engeser G., Chan F., Lu S., Goldberg S.A., Hoh R., et al. Impact of pre-existing chronic viral infection and reactivation on the development of long COVID. medRxiv. 2022 doi: 10.1101/2022.06.21.22276660. Preprint at. [DOI] [Google Scholar]
- 30.Zollner A., Koch R., Jukic A., Pfister A., Meyer M., Rössler A., Kimpel J., Adolph T.E., Tilg H. Postacute COVID-19 is characterized by gut viral antigen persistence in inflammatory bowel diseases. Gastroenterology. 2022;163:495–506.e8. doi: 10.1053/J.GASTRO.2022.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desimmie B.A., Raru Y.Y., Awadh H.M., He P., Teka S., Willenburg K.S. Insights into SARS-CoV-2 persistence and its relevance. Viruses. 2021;13:1025. doi: 10.3390/V13061025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaspar-Rodríguez A., Padilla-González A., Rivera-Toledo E. Coronavirus persistence in human respiratory tract and cell culture: an overview. Braz. J. Infect. Dis. 2021;25 doi: 10.1016/J.BJID.2021.101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caniego-Casas T., Martínez-García L., Alonso-Riaño M., Pizarro D., Carretero-Barrio I., Martínez-de-Castro N., Ruz-Caracuel I., de Pablo R., Saiz A., Royo R.N., et al. RNA SARS-CoV-2 persistence in the lung of severe COVID-19 patients: a case series of autopsies. Front. Microbiol. 2022;13:1. doi: 10.3389/FMICB.2022.824967/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zahn T., Mhedhbi I., Hein S., Raupach J., Miskey C., Husria Y., Bayanga K., Bartel D., Vieths S., Ivics Z., et al. Persistence of infectious SARS-CoV-2 particles for up to 37 days in patients with mild COVID-19. Allergy. 2021;77:2053–2066. doi: 10.1111/ALL.15138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlis R.H., Green J., Santillana M., Lazer D., Ognyanova K., Simonson M., Baum M.A., Quintana A., Chwe H., Druckman J., et al. Persistence of symptoms up to 10 months following acute COVID-19 illness. medRxiv. 2021 doi: 10.1101/2021.03.07.21253072. Preprint at. [DOI] [Google Scholar]
- 36.Stein S.R., Ramelli S.C., Grazioli A., Chung J.Y., Singh M., Yinda C.K., Winkler C.W., Sun J., Dickey J.M., Ylaya K., et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature. 2022;612:758–763. doi: 10.1038/S41586-022-05542-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Truong T.T., Ryutov A., Pandey U., Yee R., Goldberg L., Bhojwani D., Aguayo-Hiraldo P., Pinsky B.A., Pekosz A., Shen L., et al. Increased viral variants in children and young adults with impaired humoral immunity and persistent SARS-CoV-2 infection: a consecutive case series. EBioMedicine. 2021;67:103355. doi: 10.1016/J.EBIOM.2021.103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maponga T.G., Jeffries M., Tegally H., Sutherland A., Wilkinson E., Lessells R.J., Msomi N., Zyl G. van, Oliveira T. de, Preiser W. Persistent SARS-CoV-2 infection with accumulation of mutations in a patient with poorly controlled HIV infection. Clin. Infect. Dis. 2022 doi: 10.1093/CID/CIAC548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kupferschmidt K. Where did “weird” Omicron come from? Science. 2021;374:1179. doi: 10.1126/SCIENCE.ACX9738. [DOI] [PubMed] [Google Scholar]
- 40.Viana R., Moyo S., Amoako D.G., Tegally H., Scheepers C., Althaus C.L., Anyaneji U.J., Bester P.A., Boni M.F., Chand M., et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature. 2022;603:679–686. doi: 10.1038/S41586-022-04411-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng M.H., Zhang S., Porritt R.A., Rivas M.N., Paschold L., Willscher E., Binder M., Arditi M., Bahar I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA. 2020;117:25254–25262. doi: 10.1073/PNAS.2010722117/-/DCSUPPLEMENTAL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paul T., Ledderose S., Bartsch H., Sun N., Soliman S., Märkl B., Ruf V., Herms J., Stern M., Keppler O.T., et al. Adrenal tropism of SARS-CoV-2 and adrenal findings in a post-mortem case series of patients with severe fatal COVID-19. Nat. Commun. 2022;13:1–12. doi: 10.1038/s41467-022-29145-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z., Hu J., Liu L., Chen R., Wang M., Xiong M., Li Z.Q., Zhao Y., Li H., Guan C., et al. SARS-CoV-2 causes acute kidney injury by directly infecting renal tubules. Front. Cell Dev. Biol. 2021;9:1245. doi: 10.3389/FCELL.2021.664868/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian Q., Fan L., Liu W., Li J., Yue J., Wang M., Ke X., Yin Y., Chen Q., Jiang C. Direct evidence of active SARS-CoV-2 replication in the intestine. Clin. Infect. Dis. 2021;73:361–366. doi: 10.1093/CID/CIAA925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bostancıklıoğlu M. SARS-CoV2 entry and spread in the lymphatic drainage system of the brain. Brain Behav. Immun. 2020;87:122. doi: 10.1016/J.BBI.2020.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang Q., Feng Z., Diao B., Tu C., Qiao Q., Yang H., Zhang Y., Wang G., Wang H., Wang C., et al. SARS-CoV-2 induces lymphocytopenia by promoting inflammation and decimates secondary lymphoid organs. Front. Immunol. 2021;12:1292. doi: 10.3389/FIMMU.2021.661052/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Escher F., Pietsch H., Aleshcheva G., Bock T., Baumeier C., Elsaesser A., Wenzel P., Hamm C., Westenfeld R., Schultheiss M., et al. Detection of viral SARS-CoV-2 genomes and histopathological changes in endomyocardial biopsies. ESC Heart Fail. 2020;7:2440–2447. doi: 10.1002/EHF2.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cevik M., Tate M., Lloyd O., Maraolo A.E., Schafers J., Ho A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. Lancet Microbe. 2021;2:e13–e22. doi: 10.1016/S2666-5247(20)30172-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li N., Wang X., Lv T. Prolonged SARS-CoV-2 RNA shedding: not a rare phenomenon. J. Med. Virol. 2020;92:2286–2287. doi: 10.1002/JMV.25952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tejerina F., Catalan P., Rodriguez-Grande C., Adan J., Rodriguez-Gonzalez C., Muñoz P., Aldamiz T., Diez C., Perez L., Fanciulli C., et al. Post-COVID-19 syndrome. SARS-CoV-2 RNA detection in plasma, stool, and urine in patients with persistent symptoms after COVID-19. BMC Infect. Dis. 2022;22 doi: 10.1186/S12879-022-07153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D., Zhang Z., Jin L., Chu F., Mao Y., Wang H., Liu M., Wang M., Zhang L., Gao G.F., et al. Persistent shedding of viable SARS-CoV in urine and stool of SARS patients during the convalescent phase. Eur. J. Clin. Microbiol. Infect. Dis. 2005;24:165. doi: 10.1007/S10096-005-1299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao F., Sun J., Xu Y., Li F., Huang X., Li H., Zhao J., Huang J., Zhao J. Infectious SARS-CoV-2 in feces of patient with severe COVID-19. Emerg. Infect. Dis. 2020;26:1920. doi: 10.3201/EID2608.200681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Chen C., Zhu S., Shu C., Wang D., Song J., Song Y., Zhen W., Feng Z., Wu G., et al. Isolation of 2019-nCoV from a stool specimen of a laboratory-confirmed case of the coronavirus disease 2019 (COVID-19) China CDC Wkly. 2020;2:123–124. doi: 10.46234/CCDCW2020.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jeong H.W., Kim S.M., Kim H.S., Kim Y.I., Kim J.H., Cho J.Y., Kim S.H., Kang H., Kim S.G., Park S.J., et al. Viable SARS-CoV-2 in various specimens from COVID-19 patients. Clin. Microbiol. Infection. 2020;26:1520–1524. doi: 10.1016/J.CMI.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W., Xu Y., Gao R., Lu R., Han K., Wu G., Tan W. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 2020;323:1843–1844. doi: 10.1001/JAMA.2020.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cerrada-Romero C., Berastegui-Cabrera J., Camacho-Martínez P., Goikoetxea-Aguirre J., Pérez-Palacios P., Santibáñez S., José Blanco-Vidal M., Valiente A., Alba J., Rodríguez-Álvarez R., et al. Excretion and viability of SARS-CoV-2 in feces and its association with the clinical outcome of COVID-19. Sci. Rep. 2022;12:7397. doi: 10.1038/s41598-022-11439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Natarajan A., Zlitni S., Brooks E.F., Vance S.E., Dahlen A., Hedlin H., Park R.M., Han A., Schmidtke D.T., Verma R., et al. Gastrointestinal symptoms and fecal shedding of SARS-CoV-2 RNA suggest prolonged gastrointestinal infection. Med. 2022;3:371. doi: 10.1016/J.MEDJ.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo M., Tao W., Flavell R.A., Zhu S. Potential intestinal infection and faecal-oral transmission of SARS-CoV-2. Nat. Rev. Gastroenterol. Hepatol. 2021;18:269–283. doi: 10.1038/S41575-021-00416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheung C.C.L., Goh D., Lim X., Tien T.Z., Lim J.C.T., Lee J.N., Tan B., Tay Z.E.A., Wan W.Y., Chen E.X., et al. Residual SARS-CoV-2 viral antigens detected in GI and hepatic tissues from five recovered patients with COVID-19. Gut. 2022;71:226–229. doi: 10.1136/gutjnl-2021-324570. [DOI] [PubMed] [Google Scholar]
- 60.Li Y., Hu Y., Yuanyuan Y., Zhang X., Xiaodong Z., Bin L., Wu J., Li J., Wu Y., Xia X., et al. Positive result of Sars-Cov-2 in faeces and sputum from discharged patients with COVID-19 in Yiwu, China. J. Med. Virol. 2020;92:1938–1947. doi: 10.1002/jmv.25905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zuo T., Liu Q., Zhang F., Chung-Yan Lui G., Tso E.Y., Kit Yeoh Y., Chen Z., Shi Boon S., Chan F.K., Chan P.K., et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut. 2021;70:276–284. doi: 10.1136/gutjnl-2020-322294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan B., Liu H.Q., Yang Z.R., Chen Y.X., Liu Z.Y., Zhang K., Wang C., Li W.X., An Y.W., Wang J.C., et al. Recurrence of positive SARS-CoV-2 viral RNA in recovered COVID-19 patients during medical isolation observation. Sci. Rep. 2020;10:1–7. doi: 10.1038/s41598-020-68782-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patterson B.K., Francisco E.B., Yogendra R., Long E., Pise A., Rodrigues H., Hall E., Herrera M., Parikh P., Guevara-Coto J., et al. Persistence of SARS CoV-2 S1 protein in CD16+ monocytes in post-acute sequelae of COVID-19 (PASC) up to 15 Months post-infection. Front. Immunol. 2022;12:746021. doi: 10.3389/FIMMU.2021.746021/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swank Z., Senussi Y., Manickas-Hill Z., Yu X.G., Li J.Z., Alter G., Walt D.R. Persistent circulating severe acute respiratory syndrome coronavirus 2 spike is associated with post-acute coronavirus disease 2019 sequelae. Clin. Infect. Dis. 2023;76:e487–e490. doi: 10.1093/CID/CIAC722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raveendran A.v., Misra A. Post COVID-19 syndrome (“Long COVID”) and diabetes: challenges in diagnosis and management. Diabetes Metabol. Syndr. 2021;15 doi: 10.1016/J.DSX.2021.102235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagman K., Hedenstierna M., Rudling J., Gille-Johnson P., Hammas B., Grabbe M., Jakobsson J., Dillner J., Ursing J. Duration of SARS-CoV-2 viremia and its correlation to mortality and inflammatory parameters in patients hospitalized for COVID-19: a cohort study. Diagn. Microbiol. Infect. Dis. 2022;102 doi: 10.1016/J.DIAGMICROBIO.2021.115595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y., Schneider A.M., Mehta A., Sade-Feldman M., Kays K.R., Gentili M., Charland N.C., Gonye A.L.K., Gushterova I., Khanna H.K., et al. SARS-CoV-2 viremia is associated with distinct proteomic pathways and predicts COVID-19 outcomes. J. Clin. Invest. 2021;131:e148635. doi: 10.1172/JCI148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woodruff M.C., Ramonell R.P., Nguyen D.C., Cashman K.S., Saini A.S., Haddad N.S., Ley A.M., Kyu S., Howell J.C., Ozturk T., et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020;21:1506–1516. doi: 10.1038/S41590-020-00814-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Basic-Jukic N., Juric I., Furic-Cunko V., Katalinic L., Radic J., Bosnjak Z., Jelakovic B., Kastelan Z. Follow-up of renal transplant recipients after acute COVID-19—a prospective cohort single-center study. Immun Inflamm Dis. 2021;9:1563–1572. doi: 10.1002/IID3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gold J.E., Okyay R.A., Licht W.E., Hurley D.J. Investigation of long COVID prevalence and its relationship to Epstein-barr virus reactivation. Pathogens. 2021;10 doi: 10.3390/PATHOGENS10060763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su Y., Yuan D., Chen D.G., Ng R.H., Wang K., Choi J., Li S., Hong S., Zhang R., Xie J., et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell. 2022;185:881–895. doi: 10.1016/J.CELL.2022.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peluso M.J., Deveau T.-M., Munter S.E., Ryder D.M., Buck A.M., Beck-Engeser G., Chan F., Lu S., Goldberg S.A., Hoh R., et al. Chronic viral coinfections differentially affect the likelihood of developing long COVID. J. Clin. Invest. 2023;133:e163669. doi: 10.1172/JCI163669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Danwang C., Noubiap J.J., Robert A., Yombi J.C. Outcomes of patients with HIV and COVID-19 co-infection: a systematic review and meta-analysis. AIDS Res. Ther. 2022;19:1–12. doi: 10.1186/S12981-021-00427-Y/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y., Iwasaki A. Impact of chronic HIV infection on SARS-CoV-2 infection, COVID-19 disease and vaccines. Curr. HIV AIDS Rep. 2022;19:5–16. doi: 10.1007/S11904-021-00590-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pujari S., Gaikwad S., Chitalikar A., Dabhade D., Joshi K., Bele V. Long-coronavirus disease among people living with HIV in western India: an observational study. Immun Inflamm Dis. 2021;9:1037–1043. doi: 10.1002/IID3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peluso M.J., Spinelli M.A., Deveau T.M., Forman C.A., Munter S.E., Mathur S., Tang A.F., Lu S., Goldberg S.A., Arreguin M.I., et al. Postacute sequelae and adaptive immune responses in people with HIV recovering from SARS-COV-2 infection. AIDS. 2022;36:F7–F16. doi: 10.1097/QAD.0000000000003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Altuntas Aydin O., Kumbasar Karaosmanoglu H., Kart Yasar K. HIV/SARS-CoV-2 coinfected patients in Istanbul, Turkey. J. Med. Virol. 2020;92:2288–2290. doi: 10.1002/JMV.25955. [DOI] [PubMed] [Google Scholar]
- 78.del Amo J., Polo R., Moreno S., Díaz A., Martínez E., Arribas J.R., Jarrín I., Hernán M.A. Incidence and severity of COVID-19 in HIV-positive persons receiving antiretroviral therapy : a cohort study. Ann. Intern. Med. 2020;173:536–541. doi: 10.7326/M20-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., et al. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/NEJMOA2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hunt P.W., Martin J.N., Sinclair E., Bredt B., Hagos E., Lampiris H., Deeks S.G. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 2003;187:1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 81.Alrubayyi A., Gea-Mallorquí E., Touizer E., Hameiri-Bowen D., Kopycinski J., Charlton B., Fisher-Pearson N., Muir L., Rosa A., Roustan C., et al. Characterization of humoral and SARS-CoV-2 specific T cell responses in people living with HIV. Nat. Commun. 2021;12:1–16. doi: 10.1038/s41467-021-26137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mehandru S., Merad M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022;23:194–202. doi: 10.1038/s41590-021-01104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Azkur A.K., Akdis M., Azkur D., Sokolowska M., van de Veen W., Brüggen M.C., O’Mahony L., Gao Y., Nadeau K., Akdis C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy. 2020;75:1564–1581. doi: 10.1111/ALL.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jacobs J.J.L. Persistent SARS-2 infections contribute to long COVID-19. Med. Hypotheses. 2021;149 doi: 10.1016/J.MEHY.2021.110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pretorius E., Vlok M., Venter C., Bezuidenhout J.A., Laubscher G.J., Steenkamp J., Kell D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021;20 doi: 10.1186/S12933-021-01359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaid Y., Puhm F., Allaeys I., Naya A., Oudghiri M., Khalki L., Limami Y., Zaid N., Sadki K., ben El Haj R., et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ. Res. 2020;127:1404. doi: 10.1161/CIRCRESAHA.120.317703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cox D. Targeting SARS-CoV-2-platelet interactions in COVID-19 and vaccine-related thrombosis. Front. Pharmacol. 2021;12:708665. doi: 10.3389/FPHAR.2021.708665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koupenova M., Kehrel B.E., Corkrey H.A., Freedman J.E. Thrombosis and platelets: an update. Eur. Heart J. 2017;38:785–791. doi: 10.1093/EURHEARTJ/EHW550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitchell W.B. Reference Module in Biomedical Sciences. 2014. Platelets. [DOI] [Google Scholar]
- 90.Grobbelaar L.M., Venter C., Vlok M., Ngoepe M., Laubscher G.J., Lourens P.J., Steenkamp J., Kell D.B., Pretorius E. SARS-CoV-2 spike protein S1 induces fibrin(ogen) resistant to fibrinolysis: implications for microclot formation in COVID-19. Biosci. Rep. 2021;41 doi: 10.1042/BSR20210611/229418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumar A., Narayan R.K., Kumari C., Faiq M.A., Kulandhasamy M., Kant K., Pareek V. SARS-CoV-2 cell entry receptor ACE2 mediated endothelial dysfunction leads to vascular thrombosis in COVID-19 patients. Med. Hypotheses. 2020;145 doi: 10.1016/J.MEHY.2020.110320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang S., Liu Y., Wang X., Yang L., Li H., Wang Y., Liu M., Zhao X., Xie Y., Yang Y., et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020;13:1–22. doi: 10.1186/S13045-020-00954-7/FIGURES/8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pretorius E., Venter C., Laubscher G.J., Kotze M.J., Oladejo S.O., Watson L.R., Rajaratnam K., Watson B.W., Kell D.B. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Cardiovasc. Diabetol. 2022;21:1–17. doi: 10.1186/S12933-022-01579-5/FIGURES/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Da R., Gorman S., Syed I.U. Connecting the dots in emerging mast cell research: do factors affecting mast cell activation provide a missing link between adverse COVID-19 outcomes and the social determinants of health? Med. Sci. 2022;10:29. doi: 10.3390/MEDSCI10020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weinstock L.B., Brook J.B., Walters A.S., Goris A., Afrin L.B., Molderings G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int. J. Infect. Dis. 2021;112:217. doi: 10.1016/J.IJID.2021.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Afrin L.B., Weinstock L.B., Molderings G.J. Covid-19 hyperinflammation and post-Covid-19 illness may be rooted in mast cell activation syndrome. Int. J. Infect. Dis. 2020;100:327. doi: 10.1016/J.IJID.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morita H., Saito H., Matsumoto K., Nakae S. Regulatory roles of mast cells in immune responses. Semin. Immunopathol. 2016;38:623–629. doi: 10.1007/S00281-016-0566-0. [DOI] [PubMed] [Google Scholar]
- 98.Tan J., Anderson D.E., Rathore A.P.S., O’Neill A., Mantri C.K., Saron W.A.A., Lee C., Cui C.W., Kang A.E.Z., Foo R., et al. Signatures of mast cell activation are associated with severe COVID-19. medRxiv. 2021 doi: 10.1101/2021.05.31.21255594. Preprint at. [DOI] [Google Scholar]
- 99.Theoharides T.C. Potential association of mast cells with coronavirus disease 2019. Ann. Allergy Asthma Immunol. 2021;126:217–218. doi: 10.1016/J.ANAI.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Theoharides T.C., Conti P. Covid-19 and multisystem inflammatory syndrome, or is it mast cell activation syndrome? J. Biol. Regul. Homeost. Agents. 2020;34:1633–1636. doi: 10.23812/20-EDIT3. [DOI] [PubMed] [Google Scholar]
- 101.Kritas S.K., Ronconi G., Caraffa A., Gallenga C.E., Ross R., Conti P. Mast cells contribute to coronavirus-induced inflammation: new anti-inflammatory strategy. J. Biol. Regul. Homeost. Agents. 2020;34:9–14. doi: 10.23812/20-EDITORIAL-KRITAS. [DOI] [PubMed] [Google Scholar]
- 102.Atiakshin D., Buchwalow I., Tiemann M. Mast cells and collagen fibrillogenesis. Histochem. Cell Biol. 2020;154:21–40. doi: 10.1007/S00418-020-01875-9. [DOI] [PubMed] [Google Scholar]
- 103.Raha A.A., Chakraborty S., Henderson J., Mukaetova-Ladinska E., Zaman S., Trowsdale J., Raha-Chowdhury R. Investigation of CD26, a potential SARS-CoV-2 receptor, as a biomarker of age and pathology. Biosci. Rep. 2020;40 doi: 10.1042/BSR20203092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao Q., Zhang W., Li T., Yang G., Zhu W., Chen N., Jin H. Interrelationship between 2019-nCov receptor DPP4 and diabetes mellitus targets based on protein interaction network. Sci. Rep. 2022;12:1–8. doi: 10.1038/s41598-021-03912-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krystel-Whittemore M., Dileepan K.N., Wood J.G. Mast cell: a multi-functional master cell. Front. Immunol. 2016;6:620. doi: 10.3389/FIMMU.2015.00620/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gorman R. da S., Sciences I.S.-M. Connecting the dots in emerging mast cell research: do factors affecting mast cell activation provide a missing link between adverse COVID-19 outcomes and the social determinants of health? Med. Sci. 2022;10:29. doi: 10.3390/medsci10020029. undefined (2022) mdpi.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soria-Castro R., Meneses-Preza Y.G., Rodríguez-López G.M., Romero-Ramírez S., Sosa-Hernández V.A., Cervantes-Díaz R., Pérez-Fragoso A., Torres-Ruíz J.J., Gómez-Martín D., Campillo-Navarro M., et al. Severe COVID-19 is marked by dysregulated serum levels of carboxypeptidase A3 and serotonin. J. Leukoc. Biol. 2021;110:425. doi: 10.1002/JLB.4HI0221-087R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leru P.M. Evaluation and classification of mast cell disorders: a difficult to manage pathology in clinical practice. Cureus. 2022;14:e22177. doi: 10.7759/CUREUS.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karimi N., Morovati S., Chan L., Napoleoni C., Mehrani Y., Bridle B.W., Karimi K. Mast cell tryptase and implications for SARS-CoV-2 pathogenesis. BioMed. 2021;1:136–149. doi: 10.3390/BIOMED1020013. [DOI] [Google Scholar]
- 110.Pejler G. Novel insight into the in vivo function of mast cell chymase: lessons from knockouts and inhibitors. J. Innate Immun. 2020;12:357–372. doi: 10.1159/000506985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miyazaki M., Takai S., Jin D., Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol. Ther. 2006;112:668–676. doi: 10.1016/J.PHARMTHERA.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 112.Ihara M., Urata H., Kinoshita A., Suzumiya J., Sasaguri M., Kikuchi M., Ideishi M., Arakawa K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33:1399–1405. doi: 10.1161/01.HYP.33.6.1399. [DOI] [PubMed] [Google Scholar]
- 113.Zhang H., Wang J., Wang L., Zhan M., Li S., Fang Z., Xu C., Zheng Y., He S. Induction of mast cell accumulation by chymase via an enzymatic activity- and intercellular adhesion molecule-1-dependent mechanism. Br. J. Pharmacol. 2018;175:678. doi: 10.1111/BPH.14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li M., Liu K., Michalicek J., Angus J.A., Hunt J.E., Dell’Italia L.J., Feneley M.P., Graham R.M., Husain A. Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J. Clin. Invest. 2004;114:112–120. doi: 10.1172/JCI20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Company C., Piqueras L., Naim Abu Nabah Y., Escudero P., Blanes J.I., Jose P.J., Morcillo E.J., Sanz M.J. Contributions of ACE and mast cell chymase to endogenous angiotensin II generation and leucocyte recruitment in vivo. Cardiovasc. Res. 2011;92:48–56. doi: 10.1093/CVR/CVR147. [DOI] [PubMed] [Google Scholar]
- 116.Woodruff M.C., Ramonell R.P., Haddad N.S., Anam F.A., Rudolph M.E., Walker T.A., Truong A.D., Dixit A.N., Han J.E., Cabrera-Mora M., et al. Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature. 2022;611:7934. doi: 10.1038/s41586-022-05273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gammazza A.M., Légaré S., Lo Bosco G., Fucarino A., Marino Gammazza A., Giosuè, Bosco L., Angileri F., Conway De Macario E., Macario A.J., et al. Human molecular chaperones share with SARS-CoV-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: possible role of molecular mimicry in COVID-19. Cell Stress Chaperones. 2020;25:737–741. doi: 10.1007/s12192-020-01148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nunez-Castilla J., Stebliankin V., Baral P., Balbin C.A., Sobhan M., Cickovski T., Mondal A.M., Narasimhan G., Chapagain P., Mathee K., et al. Potential autoimmunity resulting from molecular mimicry between SARS-CoV-2 spike and human proteins. Viruses. 2022;14:1415. doi: 10.3390/V14071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kovačić D., Jotanović J., Laković J. The possible role of molecular mimicry in SARS-CoV-2-mediated autoimmunity: an immunobiochemical basis. J. Med. Sci. 2021;90 doi: 10.20883/MEDICAL.E560. [DOI] [Google Scholar]
- 120.Angileri F., Legare S., Marino Gammazza A., Conway de Macario E., JL Macario A., Cappello F. Molecular mimicry may explain multi-organ damage in COVID-19. Autoimmun. Rev. 2020;19 doi: 10.1016/J.AUTREV.2020.102591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kanduc D., Shoenfeld Y. Molecular mimicry between SARS-CoV-2 spike glycoprotein and mammalian proteomes: implications for the vaccine. Immunol. Res. 2020;68:310. doi: 10.1007/S12026-020-09152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Talotta R. Do COVID-19 RNA-based vaccines put at risk of immune-mediated diseases? In reply to “potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2021;224 doi: 10.1016/J.CLIM.2021.108665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Singh S., Yanow S.K., Agrawal B. Editorial: heterologous immunity: implications and applications in vaccines and immunotherapies. Front. Immunol. 2020;11:1408. doi: 10.3389/FIMMU.2020.01408/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Agrawal B. Heterologous immunity: role in natural and vaccine-induced resistance to infections. Front. Immunol. 2019;10:2631. doi: 10.3389/FIMMU.2019.02631/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rojas M., Restrepo-Jiménez P., Monsalve D.M., Pacheco Y., Acosta-Ampudia Y., Ramírez-Santana C., Leung P.S.C., Ansari A.A., Gershwin M.E., Anaya J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018;95:100–123. doi: 10.1016/J.JAUT.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 126.Rojas M., Rodríguez Y., Acosta-Ampudia Y., Monsalve D.M., Zhu C., Li Q.Z., Ramírez-Santana C., Anaya J.M. Autoimmunity is a hallmark of post-COVID syndrome. J. Transl. Med. 2022;20:1–5. doi: 10.1186/S12967-022-03328-4/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ortona E., Malorni W. Long COVID: to investigate immunological mechanisms and sex/gender related aspects as fundamental steps for tailored therapy. Eur. Respir. J. 2022;59:2102245. doi: 10.1183/13993003.02245-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen L.F., Yang C. de, Cheng X.B. Anti-interferon autoantibodies in adult-onset immunodeficiency syndrome and severe COVID-19 infection. Front. Immunol. 2021;12:788368. doi: 10.3389/FIMMU.2021.788368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/SCIENCE.ABD4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gupta S., Nakabo S., Chu J., Hasni S., Kaplan M.J. Association between anti-interferon-alpha autoantibodies and COVID-19 in systemic lupus erythematosus. medRxiv. 2020 doi: 10.1101/2020.10.29.20222000. Preprint at. [DOI] [Google Scholar]
- 131.Beydon M., Nicaise-Roland P., Mageau A., Farkh C., Daugas E., Descamps V., Dieude P., Dossier A., Goulenok T., Farhi F., et al. Autoantibodies against IFNα in patients with systemic lupus erythematosus and susceptibility for infection: a retrospective case-control study. Sci. Rep. 2022;12 doi: 10.1038/S41598-022-15508-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burbelo P.D., Iadarola M.J., Keller J.M., Warner B.M. Autoantibodies targeting intracellular and extracellular proteins in autoimmunity. Front. Immunol. 2021;12:548469. doi: 10.3389/FIMMU.2021.548469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Klein J., Wood J., Jaycox J., Lu P., Dhodapkar R.M., Gehlhausen J.R., Tabachnikova A., Tabacof L., Malik A.A., Kamath K., et al. Distinguishing features of Long COVID identified through immune profiling. medRxiv. 2022 doi: 10.1101/2022.08.09.22278592. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Calabrese L.H., Winthrop K., Strand V., Yazdany J., Walter J.E. Type I interferon, anti-interferon antibodies, and COVID-19. Lancet Rheumatol. 2021;3:e246–e247. doi: 10.1016/S2665-9913(21)00034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718. doi: 10.1126/SCIENCE.ABC6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peluso M.J., Mitchell A., Wang C.Y., Takahashi S., Hoh R., Tai V., Durstenfeld M.S., Hsue P.Y., Kelly J.D., Martin J.N., et al. Low prevalence of interferon-α; autoantibodies in people experiencing long COVID symptoms. J. Infect. Dis. 2022;227:246–250. doi: 10.1093/INFDIS/JIAC372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang S.E., Feng A., Meng W., Apostolidis S.A., Mack E., Artandi M., Barman L., Bennett K., Chakraborty S., Chang I., et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021;12:1–15. doi: 10.1038/s41467-021-25509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang E.Y., Mao T., Klein J., Dai Y., Huck J.D., Jaycox J.R., Liu F., Zhou T., Israelow B., Wong P., et al. Diverse functional autoantibodies in patients with COVID-19. Nature. 2021;595:283–288. doi: 10.1038/S41586-021-03631-Y. [DOI] [PubMed] [Google Scholar]
- 139.Fujii H., Tsuji T., Yuba T., Tanaka S., Suga Y., Matsuyama A., Omura A., Shiotsu S., Takumi C., Ono S., et al. High levels of anti-SSA/Ro antibodies in COVID-19 patients with severe respiratory failure: a case-based review : high levels of anti-SSA/Ro antibodies in COVID-19. Clin. Rheumatol. 2020;39:3171–3175. doi: 10.1007/S10067-020-05359-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arthur J.M., Forrest J.C., Boehme K.W., Kennedy J.L., Owens S., Herzog C., Liu J., Harville T.O. Development of ACE2 autoantibodies after SARS-CoV-2 infection. PLoS One. 2021;16:e0257016. doi: 10.1371/JOURNAL.PONE.0257016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seeble J., Waterboer T., Hippchen T., Simon J., Kirchner M., Lim A., Muller B., Merle U. Persistent symptoms in adult patients 1 Year after coronavirus disease 2019 (COVID-19): a prospective cohort study. Clin. Infect. Dis. 2022;74:1191–1198. doi: 10.1093/CID/CIAB611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dotan A., Muller S., Kanduc D., David P., Halpert G., Shoenfeld Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021;20:102792. doi: 10.1016/J.AUTREV.2021.102792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zuo Y., Estes S.K., Ali R.A., Gandhi A.A., Yalavarthi S., Shi H., Sule G., Gockman K., Madison J.A., Zuo M., et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020;12:eabd3876. doi: 10.1126/SCITRANSLMED.ABD3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tan C.W., Low J.G.H., Wong W.H., Chua Y.Y., Goh S.L., Ng H.J. Critically ill COVID-19 infected patients exhibit increased clot waveform analysis parameters consistent with hypercoagulability. Am. J. Hematol. 2020;95:E156. doi: 10.1002/AJH.25822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Borghi M.O., Beltagy A., Garrafa E., Curreli D., Cecchini G., Bodio C., Grossi C., Blengino S., Tincani A., Franceschini F., et al. Anti-phospholipid antibodies in COVID-19 are different from those detectable in the anti-phospholipid syndrome. Front. Immunol. 2020;11:584241. doi: 10.3389/FIMMU.2020.584241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xiao M., Zhang Y., Zhang S., Qin X., Xia P., Cao W., Jiang W., Chen H., Ding X., Zhao H., et al. Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol. 2020;72:1998–2004. doi: 10.1002/ART.41425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xiao M., Zhang Y., Zhang S., Qin X., Xia P., Cao W., Jiang W., Chen H., Ding X., Zhao H., et al. Brief Report: anti-phospholipid antibodies in critically ill patients with Coronavirus Disease 2019 (COVID-19) Arthritis Rheumatol. 2020;72:1998–2004. doi: 10.1002/ART.41425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bertin D., Brodovitch A., Beziane A., Hug S., Bouamri A., Mege J.L., Heim X., Bardin N. Anti-cardiolipin IgG autoantibodies are an independent risk factor of COVID-19 severity. Arthritis Rheumatol. 2020;72:1953–1955. doi: 10.1002/ART.41409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pavoni V., Gianesello L., Horton A. Antiphospholipid antibodies in critically ill COVID-19 patients with thromboembolism: cause of disease or epiphenomenon? J. Thromb. Thrombolysis. 2021;52:542–552. doi: 10.1007/S11239-021-02470-Y/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Teimury A., Khameneh M.T., Khaledi E.M. Major coagulation disorders and parameters in COVID-19 patients. Eur. J. Med. Res. 2022;27:1–10. doi: 10.1186/S40001-022-00655-6/FIGURES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Angelini D.E., Kaatz S., Rosovsky R.P., Zon R.L., Pillai S., Robertson W.E., Elavalakanar P., Patell R., Khorana A. COVID-19 and venous thromboembolism: a narrative review. Res Pract Thromb Haemost. 2022;6 doi: 10.1002/RTH2.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Giustozzi M., Vedovati M.C., Agnelli G. Venous thromboembolism and COVID-19: mind the gap between clinical epidemiology and patient management. Eur. J. Intern. Med. 2020;82:18. doi: 10.1016/J.EJIM.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang C., Yu C., Jing H., Wu X., Novakovic V.A., Xie R., Shi J. Long COVID: the nature of thrombotic sequelae determines the necessity of early anticoagulation. Front. Cell. Infect. Microbiol. 2022;12:861703. doi: 10.3389/FCIMB.2022.861703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tonello M., Mattia E., Favaro M., Del Ross T., Calligaro A., Salvan E., Hoxha A., Fedrigo M., Ruffatti A. IgG phosphatidylserine/prothrombin antibodies as a risk factor of thrombosis in antiphospholipid antibody carriers. Thromb. Res. 2019;177:157–160. doi: 10.1016/J.THROMRES.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 155.Preti P.S., Russo M., Caneva L., Reduzzi M., Calabretta F., Spataro C., Grimaldi P., De Amici M., Testa G., Iotti G.A., et al. Increased prevalence of heparin induced thrombocytopenia in COVID-19 patients. Thromb. Res. 2021;203:33. doi: 10.1016/J.THROMRES.2021.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu X., Zhang X., Xiao Y., Gao T., Wang G., Wang Z., Zhang Z., Hu Y., Dong Q., Zhao S., et al. Heparin-induced thrombocytopenia is associated with a high risk of mortality in critical COVID-19 patients receiving heparin-involved treatment. medRxiv. 2020 doi: 10.1101/2020.04.23.20076851. Preprint at. [DOI] [Google Scholar]
- 157.Bailly J., Haupt L., Joubert J., Loebenberg P., Jacobson B.F., Louw V.J., Wessels P.F., Opie J.J. Heparin-induced thrombocytopenia: an update for the COVID-19 era. S. Afr. Med. J. 2021;111:841–848. doi: 10.7196/SAMJ.2021.V111I9.15909. [DOI] [PubMed] [Google Scholar]
- 158.Rohrhofer J., Graninger M., Lettenmaier L., Schweighardt J., Gentile S.A., Koidl L., Ret D., Stingl M., Puchhammer-Stöckl E., Untersmayr E. Association between Epstein-barr-virus reactivation and development of long-COVID fatigue. Allergy. 2023;78:297–299. doi: 10.1111/ALL.15471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Apostolou E., Rizwan M., Moustardas P., Sjögren P., Bertilson B.C., Bragée B., Polo O., Rosén A. Saliva antibody-fingerprint of reactivated latent viruses after mild/asymptomatic COVID-19 is unique in patients with myalgic-encephalomyelitis/chronic fatigue syndrome. Front. Immunol. 2022;13:6407. doi: 10.3389/FIMMU.2022.949787/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guarnieri J.W., Dybas J.M., Fazelinia H., Kim M.S., Frere J., Zhang Y., Albrecht Y.S., Murdock D.G., Angelin A., Singh L.N., et al. Targeted down regulation of core mitochondrial genes during SARS-CoV-2 infection. bioRxiv. 2022 doi: 10.1101/2022.02.19.481089. Preprint at. [DOI] [Google Scholar]
- 161.Holmes E., Wist J., Masuda R., Lodge S., Nitschke P., Kimhofer T., Loo R.L., Begum S., Boughton B., Yang R., et al. Incomplete systemic recovery and metabolic phenoreversion in post-acute-phase nonhospitalized COVID-19 patients: implications for assessment of post-acute COVID-19 syndrome. J. Proteome Res. 2021;20:3315–3329. doi: 10.1021/ACS.JPROTEOME.1C00224. [DOI] [PubMed] [Google Scholar]
- 162.Ajaz S., McPhail M.J., Singh K.K., Mujib S., Trovato F.M., Napoli S., Agarwal K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021;320:C57–C65. doi: 10.1152/AJPCELL.00426.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wood E., Hall K.H., Tate W. Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: a possible approach to SARS-CoV-2 “long-haulers”. Chronic Dis Transl Med. 2021;7:14–26. doi: 10.1016/J.CDTM.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Allali I., Bakri Y., Amzazi S., Ghazal H. Gut-lung Axis in COVID-19. Interdiscip Perspect Infect Dis. 2021;2021:6655380. doi: 10.1155/2021/6655380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Giannos P., Prokopidis K. Gut dysbiosis and long COVID-19: feeling gutted. J. Med. Virol. 2022;94:2917. doi: 10.1002/JMV.27684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Su Q., Lau R.I., Liu Q., Chan F.K.L., Ng S.C. Post-acute COVID-19 syndrome and gut dysbiosis linger beyond 1 year after SARS-CoV-2 clearance. Gut. 2022;72:1230–1232. doi: 10.1136/GUTJNL-2022-328319. [DOI] [PubMed] [Google Scholar]
- 167.Haran J.P., Bradley E., Zeamer A.L., Cincotta L., Salive M.C., Dutta P., Mutaawe S., Anya O., Meza-Segura M., Moormann A.M., et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight. 2021;6:e152346. doi: 10.1172/JCI.INSIGHT.152346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jackson C.B., Farzan M., Chen B., Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021;23:3–20. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schweighauser M., Arseni D., Bacioglu M., Huang M., Lövestam S., Shi Y., Yang Y., Zhang W., Kotecha A., Garringer H.J., et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature. 2022;605:310–314. doi: 10.1038/S41586-022-04650-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nyström S., Hammarström P. Amyloidogenesis of SARS-CoV-2 spike protein. J. Am. Chem. Soc. 2022;144:8945–8950. doi: 10.1021/JACS.2C03925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kavi L. Postural tachycardia syndrome and long COVID: an update. Br. J. Gen. Pract. 2022;72:8–9. doi: 10.3399/BJGP22X718037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chadda K.R., Blakey E.E., Huang C.L.H., Jeevaratnam K. Long COVID-19 and postural orthostatic tachycardia SyndromeIs dysautonomia to Be blamed? Front Cardiovasc Med. 2022;9:860198. doi: 10.3389/FCVM.2022.860198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dani M., Dirksen A., Taraborrelli P., Torocastro M., Panagopoulos D., Sutton R., Lim P.B. Autonomic dysfunction in ‘long COVID’: rationale, physiology and management strategies. Clin. Med. 2021;21:e63. doi: 10.7861/CLINMED.2020-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rutkai I., Mayer M.G., Hellmers L.M., Ning B., Huang Z., Monjure C.J., Coyne C., Silvestri R., Golden N., Hensley K., et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat. Commun. 2022;13:1–13. doi: 10.1038/s41467-022-29440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]