

Summary
Cognitive impairment occurs in the vast majority of individuals with Parkinson’s disease (PD), exacting a high toll on patients, their caregivers, and the health care system. In this review, we begin by summarizing the current clinical landscape surrounding cognition in PD. We then discuss how cognitive impairment and dementia may develop in PD, based on the spread of the pathological protein alpha-synuclein (aSyn) from neurons in brainstem regions to those in the cortical regions of the brain responsible for higher cognitive functions, as first proposed in the Braak hypothesis. We appraise the Braak hypothesis from molecular (conformations of aSyn), cell biological (cell-to-cell spread of pathological aSyn), and organ-level (region-to-region spread of aSyn pathology at the whole brain level) viewpoints. Finally, we argue that individual host factors may be the most poorly-understood aspect of this pathological process, accounting for substantial heterogeneity in the pattern and pace of cognitive decline in PD.
Keywords: Parkinson’s disease, dementia, mild cognitive impairment, alpha-synuclein, cognition, Braak hypothesis, dementia, strain, Dementia with Lewy bodies, Lewy body
In brief
Carceles-Cordon et al. discuss molecular and cellular mechanisms of alpha-synuclein spread in the brain and examine their interplay with host factors such as genetic background to provide an overview of the pathogenesis of cognitive heterogeneity in Parkinson’s disease.
Introduction
Parkinson’s disease (PD) and the related disorder dementia with Lewy bodies (DLB) are defined by deposition of misfolded alpha-synuclein (aSyn) into neuropathological inclusions known as Lewy bodies (LBs) and Lewy neurites (LNs) in neurons, and both are considered Lewy body disorders (LBD). Indeed, at autopsy, brain samples from people who bore a clinical diagnosis of PD and those who bore a diagnosis of DLB may be virtually indistinguishable. During life, however, individuals with PD and DLB patients manifest very differently, with DLB patients demonstrating cognitive symptoms and dementia from the onset of their disease, whereas PD patients initially show motor symptoms of bradykinesia, rigidity, tremor, and postural instability.1 In addition, while most people with PD eventually develop cognitive decline and dementia, a minority do not – and even among those PD patients who develop dementia (Parkinson’s disease with dementia, or PDD), the pace varies widely.2–4 Indeed, the time scale over which PDD may develop ranges from months, to years, to decades.
This clinical heterogeneity is a critical factor in disease management. An individual presenting with isolated motor symptoms and newly diagnosed with PD will have different needs from an individual diagnosed with PDD, who may require social or family support to perform activities of daily living. Indeed, cognitive decline in PD has been shown to lead to decreased quality of life for the patient, higher burden for the caregiver, and increased cost of care for the health care system.5–7 Moreover, the number of people affected by PD is growing, projected to exceed 12 million worldwide by 2040.8 Because this increase is largely driven by population aging, and because age is strongly associated with cognitive impairment in PD, the projected increase in PD prevalence will almost certainly come with a substantial increase in PDD.
Here, we first summarize the clinical landscape of cognitive heterogeneity in PD, providing an overview of current treatments for cognitive impairment in PD, as well as experimental approaches in clinical trials. We then discuss how PDD may develop, based on the spread of aSyn pathology from brainstem regions to higher-order cortical regions of the brain, as first proposed in the Braak hypothesis. We appraise the Braak hypothesis from molecular (conformations of aSyn), cell biological (cell-to-cell spread of pathological aSyn), and organ-level (region-to-region spread of aSyn pathology at the whole brain level) viewpoints, pointing out areas that are more vs. less well-supported by existing data. Finally, we suggest that host factors – which we define as characteristics of individuals that influence susceptibility and temporal course of a disease, such as genetic background or environmental exposures – may be the most poorly-understood aspect of cognitive heterogeneity in PD and, as such, represent a particularly valuable area for investigation.
Epidemiology, Presentation, and Assessment of Cognitive Symptoms in Parkinson’s Disease
Between 15-35% of PD patients will meet criteria for mild cognitive impairment (PD-MCI) at the time of diagnosis,9 and several longitudinal studies have demonstrated that approximately 80% of patients will eventually progress to dementia (PDD).10 More recently, changes in cognition have even been reported prior to PD diagnosis11 in population-based studies.12,13 Additionally, prospective studies have demonstrated cognitive worsening in individuals with prodromal or at-risk PD, such as those having impaired olfaction and dopamine transporter (DAT) deficits14 or REM sleep behavior disorder (RBD).15
Traditionally, primary cognitive deficits in PD were attributed to frontal lobe-based changes (manifesting as difficulty with working memory, executive ability and attention).16 However, recent work has found that initial impairments can occur in a range of cognitive domains, including executive, memory, visuospatial, attentional, and even language domains.17–19 Moreover, additional neuropsychiatric symptoms that can either impair cognition or are often comorbid with cognitive impairment in PD include depression, apathy, and psychosis (i.e. hallucinations and delusions). Clinical predictors of cognitive impairment include higher current age, higher age at disease onset, greater disease severity, the postural instability-gait disorder subtype, and presence of features such as RBD, psychosis, depression, and anxiety.20
Advancements in the diagnosis and assessment of cognition in PD include the establishment of diagnostic criteria for both PDD21 and PD-MCI.22 PDD is characterized by cognitive decline over time, cognitive impairment in multiple domains on testing, and significant cognitive functional impairment.21 PD-MCI consists of self- or other-reported cognitive decline, with some cognitive impairment on testing, but no significant cognitive functional impairment (i.e. preserved instrumental activities of daily living). Other improvements in the diagnosis and evaluation of cognitive symptoms in PD are summarized in several recent reviews, which cover recommended cognitive testing,22 global cognitive assessment instruments for use in PD,23 and comparisons of the relative sensitivity of commonly-used cognitive tests in non-demented PD patients.24 Additional recommendations for assessment of cognition in PD can also be found through the National Institute of Neurological Disorders and Stroke Common Data Elements (https://www.commondataelements.ninds.nih.gov).
Dementia with Lewy bodies
A point of controversy related to cognition in PD pertains to the related disorder of DLB, and whether PDD and DLB are categorically, or simply dimensionally, different.7 DLB is characterized by dementia at time of diagnosis and some combination of parkinsonism, psychosis, and RBD. DLB is similar neuropathologically to PDD, particularly in terms of Lewy body pathology, although some have argued for differences between PDD and DLB with respect to (1) the degree of concomitant Alzheimer’s disease (AD) pathology, which may be higher in DLB; (2) biomarker profiles in DLB versus PDD;25–27 and (3) possible genetic variability in SNCA (encoding aSyn, the hallmark protein in the pathological lesions of PD, as described below).28 In practice, a controversial “one-year rule” is used, with dementia that precedes or occurs within one year of onset of parkinsonism being labeled as DLB and all other cases being diagnosed as PDD.1
Management of Cognitive Impairment in PD – Current and Future Prospects
Management options for cognitive impairment in PD consist largely of avoiding medication side effects and treating co-existing conditions that may worsen cognition. For example, medications with anticholinergic properties are sometimes used in PD,29 but they are associated with long-term cognitive decline30 and should be avoided in older individuals with PD. Comorbid vascular diseases, other neuropsychiatric symptoms, obstructive sleep apnea and other sleep disorders,31 and orthostatic hypotension, all associated with worsening of cognitive impairment in PD, should be treated.
To improve cognition in PD, there is evidence that acetylcholinesterase inhibitor (ChEI) treatment is of modest benefit,32,33 with lesser effects reported for memantine.34–36 Currently, no medications have been approved for the symptomatic management of PD-MCI, with negative randomized controlled trials (RCTs) for rasagiline (MAO-B inhibitor),37 atomoxetine (selective noradrenaline reuptake inhibitor),38 and rivastigmine (ChEI).39 Other compounds recently or currently being tested for PD cognitive impairment include SYN120 and itepridine (5-HT6 antagonists), mevidalen (selective positive allosteric modular [PAM] of the D1 receptor),40 blarcamesine (intracellular sigma-1 receptor agonist), IRL752 (a small molecule that selectively enhances norepinephrine, dopamine, and acetylcholine neurotransmission in the cerebral cortex),41 and CST-103 (beta-adrenoreceptor agonist) targeting the locus coeruleus to improve noradrenergic tone. There may also be beneficial effects of physical exercise and cognitive training on cognitive functions in PD.42–44
Taken together, however, current options for treating cognitive impairment and dementia in PD are limited.45 Most therapies aiming to stabilize or reverse the underlying disease process leading to cognitive decline in PD only exist in clinical trials. Examples of these experimental therapies under investigation now include the MAP kinase inhibitor neflamapimod, currently being tested in DLB (NCT04001517); the antibiotic ceftriaxone, purported to reduce glutamatergic hyperactivity and subsequent excitotoxicity; a proprietary plasma fraction (GRF6021) that aims to enhance neurogenesis and reduce neuroinflammation; two N-methyl-D-aspartate (NMDA) receptor PAMs (SAGE-718 and NYX-458); and ambroxol (repurposed expectorant/mucolytic drug, aiming to increase levels of the lysosomal enzyme glucocerebrosidase in order to reduce aSyn aggregation).
Despite the current scarcity of treatment options, however, increasing evidence from prospective studies examining newly diagnosed patients46 and those with prodromal conditions, such as RBD,47 suggest that a therapeutic disease-modifying window exists where progression to significant cognitive impairment could be halted or delayed. Thus, this is an appropriate time to focus research efforts on better understanding the processes leading to cognitive decline in PD, in order to interfere in these processes therapeutically.
Neuropathological substrates for cognitive impairment in PD
The neuropathological hallmark of PD is the neuronal LB, a dense, cytoplasmic inclusion body found at autopsy in individuals who were diagnosed with PD during life. LBs were first described over 100 years ago and are found throughout the PD brain, in areas that are susceptible to neurodegeneration.48 The main protein within LBs is aSyn,49 a normally-occurring protein whose causal role in the development of PD is supported by the fact that both missense mutations in50 and increased copy numbers of51 its coding gene SNCA result in familial PD and DLB. The normal function of aSyn is still unclear, although strong evidence exists for a role in synaptic vesicle function,52,53 where aSyn may exist in equilibrium between cytosolic, unfolded forms and membrane-bound, helical forms.54 In disease contexts, aSyn becomes hyper-phosphorylated, particularly at serine 129,55,56 and aggregates into more insoluble forms.57 In addition, Lewy neurites (LNs), abnormal neuronal processes also containing pathological forms of aSyn, are found in both DLB and PD.
LBs and LNs are found across many brain regions, including areas that exhibit selective vulnerability such as the dopaminergic neurons of the substantia nigra. Moreover, across many neuropathological cohorts, Lewy pathology appears to follow a pattern of regional involvement, leading to the development of a commonly-used staging system by Braak and colleagues.58 In brief, Braak staging proposes that Lewy pathology initially occurs in the dorsal motor nucleus of the vagus (dmX) and the anterior olfactory nucleus (stage I); followed by involvement of the raphe nuclei and the gigantocellular reticular nucleus of the medulla oblongata (stage II). Subsequent stages involve brainstem areas, including the midbrain dopaminergic neurons of the substantia nigra pars compacta (stage III), as well as prosencephalic regions and the anteromedial temporal cortex (stage IV). This rostro-caudal spread culminates in the presence of LBs in the neocortex (stages V and VI).
While this review will concentrate on PD, PDD, and the related condition DLB, we note that aSyn pathology may also occur in other cell types, such as oligodendrocytes. These aSyn glial cytoplasmic inclusions (GCI), found predominantly in oligodendrocytes, characterize multiple system atrophy (MSA), a disorder presenting with parkinsonism and ataxia, to varying degrees.59 Interestingly, MSA patients are less likely to develop cognitive symptoms than PD patients, to the extent that development of dementia was once considered grounds for re-evaluation of an MSA diagnosis,60 despite an overall faster rate of motor progression.
An under-recognized neuropathological feature of PD, and particularly PDD and DLB, is the frequent co-existence of the amyloid-beta-containing plaques and tau-containing neurofibrillary tangles of Alzheimer’s disease (AD).61 While a smaller proportion of PD/PDD patients (~25-30%) have extensive neocortical tangle pathology, and a larger proportion of patients have concomitant amyloid plaque pathology,62,63 approximately half of PD patients demonstrate enough concomitant plaque and tangle pathology to warrant a secondary neuropathological diagnosis of AD.61,64,65 Furthermore, concomitant AD pathology may be even more prominent in DLB than in PD and PDD.26,66
Intriguingly, multiple lines of evidence suggest some degree of synergy between aSyn pathology and AD pathology, and particularly tau pathology. Among human neuropathological studies comparing brain regions in cases with confirmed LBs upon autopsy, the severity of AD pathology appears to correlate with the aSyn pathology burden in both PD and DLB.67,68 Moreover, in vitro studies have long demonstrated that aSyn induces the fibrillization of tau and that co-incubation of tau and aSyn increases the fibrillization of both pathological proteins.69 More recently, Bassil et al. have reported in vivo evidence for synergy between aSyn and tau, demonstrating that co-inoculation of pathological forms of aSyn and tau into mouse brains accelerates the development of tau pathology.70
While neuropathological studies are, by necessity, static “snapshots” of a single time point per individual in a years-long, or even decades-long, process, it is worth considering the inter-individual differences in tempo among patients with LB disorders in both the onset and the progression of cognitive decline. That is, functional neuroanatomy would suggest that brainstem-only-pathology cases would be unlikely to have prominent cognitive deficits, and this is in fact supported by the clinico-pathological literature.71,72 An open question, however, is why some individuals go for years, or decades (or their entire disease course), without development of cognitive decline/cortical pathology, while others develop cognitive deficits/cortical pathology as a presenting symptom.
The Braak hypothesis: A model for the development of cognitive decline in PD?
Current understanding of the pathogenesis of cognitive dysfunction in PD is largely informed by the seminal studies of Braak and colleagues.58,73,74 Braak’s original study consisted of 110 patients with LB inclusions who were stratified into six topographically distinct, progressive stages of PD-related brain lesions,58 wherein dysfunction is posited to be a direct consequence of the neurotoxic effect of misfolded aSyn accumulating in LBs.75 Indeed, aSyn-containing LBs are found in neocortical areas and other brain regions required for higher cognitive functioning,76 and their presence correlates with neuronal cell death and dementia. In the diffuse neocortical stage proposed by Braak (stages V and VI), characterized by widespread aSyn inclusions in the brain, prevalence of dementia has been reported to be nearly universal.58,75
In parallel with this staging system, Braak’s studies proposed that these neuropathological stages are the traces of a disease process whereby pathological species of aSyn spread within the CNS in a rostro-caudal manner, propagating from the lower brain regions to cortical areas where they then perturb normal cognitive processes. Specifically, Braak and colleagues speculated that a neurotropic pathogen could reach the CNS through the nose (anterograde ascension) or the enteric system (retrograde ascension via the enteric plexus and preganglionic fibers of the vagal nerve).77 This pathogen, which was initially speculated to be a viral agent due to other classical examples of rostro-caudal ascension by other viruses,78 would potentially reach the medulla and from there it would spread to the midbrain and above.77 In support of nasal or vagal nerve routes of entry, aSyn inclusions can be found in the enteric nervous system,79–81 as well as in the submandibullary gland,82 and the anterior olfactory bulb83,84 of PD patients. Moreover, clinical symptoms suggesting early involvement of the olfactory system, the vagal nerve, and/or the gut are present in PD. Specifically, hyposmia and dysautonomia may precede parkinsonism in the prodromal stage of PD,85–87 and constipation is highly prevalent among those newly diagnosed with PD, often preceding overt development of parkinsonism.88
Over the last 15 years, no clear evidence of a viral pathogen has emerged. However, evidence suggesting that aSyn pathology might propagate within the brain has steadily accumulated. Early signs came from two independent studies of fetal mesencephalic dopaminergic neuronal grafts in PD patient brains examined by autopsy years after graft implantation. Surprisingly, LB pathology was observed in grafted cells.89,90 Because the grafts came from fetal tissue which would be pathology-naïve at implantation, these studies strongly suggested propagation of aSyn pathology from the surrounding host tissue. Experimental evidence for this possibility emerged shortly thereafter, with the demonstration that neurons can take up and transmit aSyn to other cells,91 and that exogenous preformed fibrils (PFFs) constituted from recombinant aSyn could seed intracytoplasmic aggregation of endogenous aSyn in neuronal culture.92,93 This in cellulo work was followed by a report that a single intracerebral injection of aSyn PFFs in the dorsal striatum of both wild-type and aSyn-transgenic mice could recapitulate the in vivo spread of aSyn pathology to many areas of the brain, in a time-dependent manner, with subsequent neurodegeneration and development of motor and cognitive deficits.94,95 These key findings have subsequently been replicated widely.96–100 Moreover, studies applying this PFF-injection model to non-human primates have confirmed aSyn pathological spread far from the injection site, accompanied by neurodegeneration in those brain regions affected by the pathological spread.101,102 More recently, aSyn pathological spread from an initial site of injection has been demonstrated for human brain-derived pathological material,103 which may be more potent than PFFs generated from recombinant aSyn,104 on a variety of mouse model backgrounds.97,105 In addition, injection of aSyn PFFs into the stomach and small intestine has been reported to result in the subsequent development of brain pathology in wild-type mice and rats,106,107 although in other studies using similar models the development of brain lesions has been transient108 or restricted to the dmX.109
The stereotypical patterns of Lewy pathology demonstrated by Braak in human brain tissue,58 together with evidence for cell-to-cell transmission of aSyn in cellular and mouse models, in the absence of evidence for an exogenous viral agent, has led to the current prevailing view: that misfolded aSyn itself acts as the neurotropic pathogen responsible for spreading pathology across the CNS in a prion-like manner.110–112 Specifically, pathologically-misfolded aSyn might serve as the template for subsequent misfolding of aSyn in a pathology-naïve cell in order to convert aSyn in the recipient cell into a pathological form. In support of this view is the dependence of aSyn spread models on the existence of endogenous non-pathological aSyn in the host, because injection of PFFs in alpha-synuclein knockout mice causes no pathology.94
Evidence for cell-to-cell transmission of alpha-synuclein as the mechanism underlying cognitive decline in PD
For a prion-like version of the Braak hypothesis to account for cognitive decline in PD, several conditions must be met. First, pathological forms of aSyn should be able to transfer from affected cells to pathology-naïve cells. Second, pathological aSyn from the affected cells should then cause templated misfolding of endogenous aSyn in the naive cells into pathological forms. Third, pathological aSyn should cause neurodegeneration and cognitive decline. Fourth, at the human brain level, there should be an orderly progression of pathological involvement of rostral-to-caudal brain regions. We take each of these conditions in turn and examine existing evidence (Figure 1).
Figure 1. Appraising the Braak hypothesis as the mechanistic basis for cognitive decline in Parkinson’s Disease.
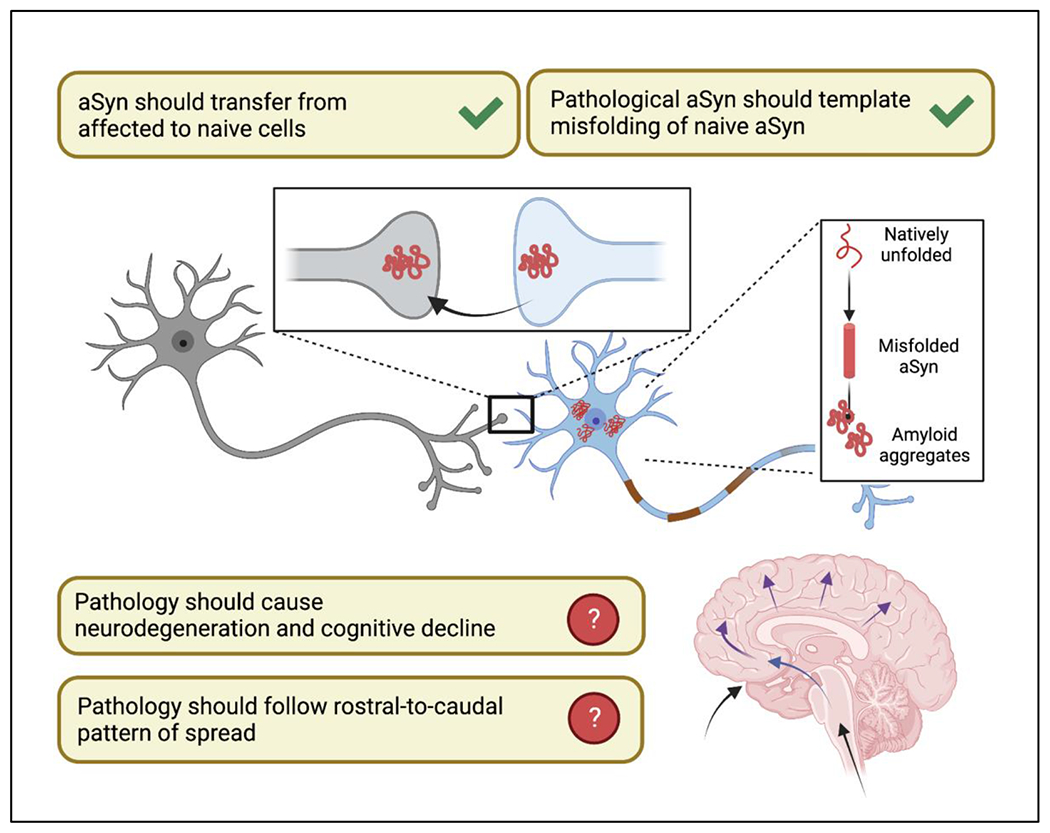
The literature supports cell-to-cell transfer of aSyn from affected to pathology-naïve cells and the subsequent ability of pathologic aSyn to cause templated misfolding of endogenous aSyn in a prion-like manner. However, disconnects emerge with respect to whether aSyn pathology always forms the basis for neuronal dysfunction and cognitive impairment, and whether aSyn pathology consistently follows a rostro-to-caudal spread pattern in the human brain.
Pathological forms of aSyn should be able to transfer from affected to pathology-naïve cells
For pathological aSyn to be able to transfer from affected to pathology-naïve cells, it must exist in an available space, and mechanisms must exist for uptake into recipient cells, including neurons that will ultimately degenerate. Despite lacking a secretory signaling peptide sequence and localizing to the cytosol, both monomeric and aggregated forms of aSyn are found in the extracellular space,113,114 and aSyn is found in the CSF, plasma, and serum of both healthy individuals and PD patients.115,116 Non-classical secretory pathways, including both passive and active mechanisms, are thought to be involved in the release of aSyn from cells into the extracellular space.117 A fraction of the secreted aSyn fraction has been linked to exosomes, extracellular vesicles originating from the endosomal compartment,118,119 which have been reported to induce Lewy pathology in vivo.120 Specifically, exosomes harvested through ultracentrifugation of DLB brain tissue induced intracellular and dendritic aSyn aggregation in neurons and astrocytes of WT mice.120 Moreover, exosomal aSyn is increased in PD patients when compared to controls and has been proposed as a biomarker of PD severity and a blood-based diagnostic test.121–123
Neuronal uptake of amyloid aggregates of aSyn occurs at the somatodendritic compartment as well as at the axon and presynaptic terminals through the endosomal pathway.124–126 Several cell surface proteins have been proposed as receptors that facilitate internalization of aSyn into the cell.127 For example, lymphocyte activation gene 3 (LAG3) reportedly binds to PFFs and mediates internalization of aSyn,128 although these findings have been questioned.129 Recent evidence also suggests that cellular prion protein (PrPC) mediates uptake and toxicity of aSyn130 and that physical interaction between aSyn fibrils and PrPC can mediate cognitive dysfunction through alteration of hippocampal long-term potentiation.131 Once internalized, aSyn can be transported in both anterograde and retrograde directions along neuronal processes;125,132,133 however, it is still unclear how fibrils escape from endocytic vesicles and trigger intracellular pathology.
Recently, we have demonstrated that the PD risk-associated gene GPNMB, which encodes glycoprotein non-metastatic melanoma protein B (GPNMB), may be involved in the cell-to-cell transfer of pathological aSyn.134 Specifically, we have shown that decreasing GPNMB levels in human iPSC-derived cortical neurons results in greatly reduced uptake of exogenously-added aSyn PFFs, with subsequent reduction of pathology (i.e. insoluble, hyperphosphorylated aSyn aggregates) in the recipient cells. Of interest, GPNMB is a transmembrane protein expressed on the cell surface which can be cleaved to generate a soluble form, and addition of GPNMB is sufficient to increase uptake of aSyn PFFs in cell types that normally do not express GPNMB highly. GPNMB and aSyn co-localize and co-immunoprecipitate, suggesting that the interaction between these proteins may be responsible for these cellular effects. Finally, higher GPNMB levels associate with PD risk-associated genotypes in human CSF and brain, and higher GPNMB levels are found in PD patient plasma (compared to neurologically-normal controls), with the highest levels in the most severely affected patients.
Pathological aSyn from affected cells should cause templated misfolding of naive aSyn into pathological forms
Evidence for templated misfolding of aSyn has been reported at in vitro, in cellulo, and in vivo levels. In the in vitro context, protein misfolding cyclic amplification (PMCA) and real-time quaking-induced conversion (RT-QuIC) are two diagnostic assays developed in recent years that take advantage of the self-templating capacity of misfolded proteins.135–137 In aSyn PMCA and RT-QuIC assays, adapted from similar assays developed in the prion field, samples derived from patient biofluids that contain aSyn “seeds” (presumably possessing a particular conformation) are first incubated with unfolded recombinant aSyn protein substrates. These “seeds” serve as templates for the folding of substrate aSyn into a form that can bind amyloid dyes. Through multiple rounds of templating, followed by mechanical fragmentation of the resulting amyloid fibrils, an exponential amplification is achieved. The kinetics of this amplification can then be quantified using amyloid-binding dyes. The finding that both of these assays can distinguish PD from controls with high sensitivity and specificity supports the idea that CSF from PD patients contains “strains” (i.e. specific conformations) of aSyn that can be propagated.138–140 We note, in particular, that the aSyn-PMCA assay developed by Shahnawaz, Soto, and colleagues has been reported to diagnose PD patients with >88% sensitivity and >96% specificity based on their CSF samples.139 Moreover, CSF PMCA assays detecting aSyn oligomers have been reported, in conjunction with enzyme-linked immunoassay measures of CSF neurofilament light chain (NFL), to discriminate between early MSA and LBD.141
Distinct aSyn strains, defined as forms of aSyn with differential seeding capacity and pathogenic behavior, can be generated in cells. For example, Guo, V. Lee, and colleagues discovered two synthetic aSyn PFF strains with differential ability to recruit tau into pathological inclusions.142 In parallel, another group also characterized two different morphologies of aSyn pathology, fibrils and ribbons,143 reporting that when inoculated into rats, each type of aSyn conformation produced a distinct histopathological and behavioral phenotype.144 In addition, aSyn GCI derived from human MSA brain appear to be conformationally and behaviorally distinct from aSyn strains found in LBs in PD.103,145 Indeed, GCI-derived aSyn demonstrated approximately 1000-fold more seeding capacity than LB-derived aSyn in cultured oligodendrocytes, in cultured neurons, and in WT mouse brain. More recently, Yang and colleagues reported, using cryo-electron microscopy, that the ultrastructure of aSyn filaments derived from MSA patients differs markedly from aSyn filaments derived from PD, PDD, and DLB patients.146 Thus, ultrastructural studies, work in cell and rodent models, and human biomarker studies all support the existence of distinct “strains” of aSyn (Figure 2). We note that while aSyn does not exhibit the infectivity of prion proteins, the concept of “strains,” borrowed from the prion literature, is relevant here. Specifically, the prion field has recognized for many years that the same protein – PrPc – underlies different clinical manifestations in humans, with clinical syndromes resulting from the biological behavior of the misfolded prion protein, referred to as PrPsc (reviewed in Aguzzi et al.,147 and Sigurdson et al.148). Not all conformations of PrPsc are the same, though, and these distinct conformations manifest as distinct, yet reproducible, syndromes in affected individuals, resulting in the “strain” concept. Thus, the accumulating evidence for distinct conformations of aSyn, with different biological properties, supports the hypothesis that aSyn conformations may dictate “strains” of aSyn, which in turn contribute to the cognitive heterogeneity seen in the LBD.
Figure 2. Evidence for distinct aSyn “strains” in the pathogenesis of synucleinopathies.
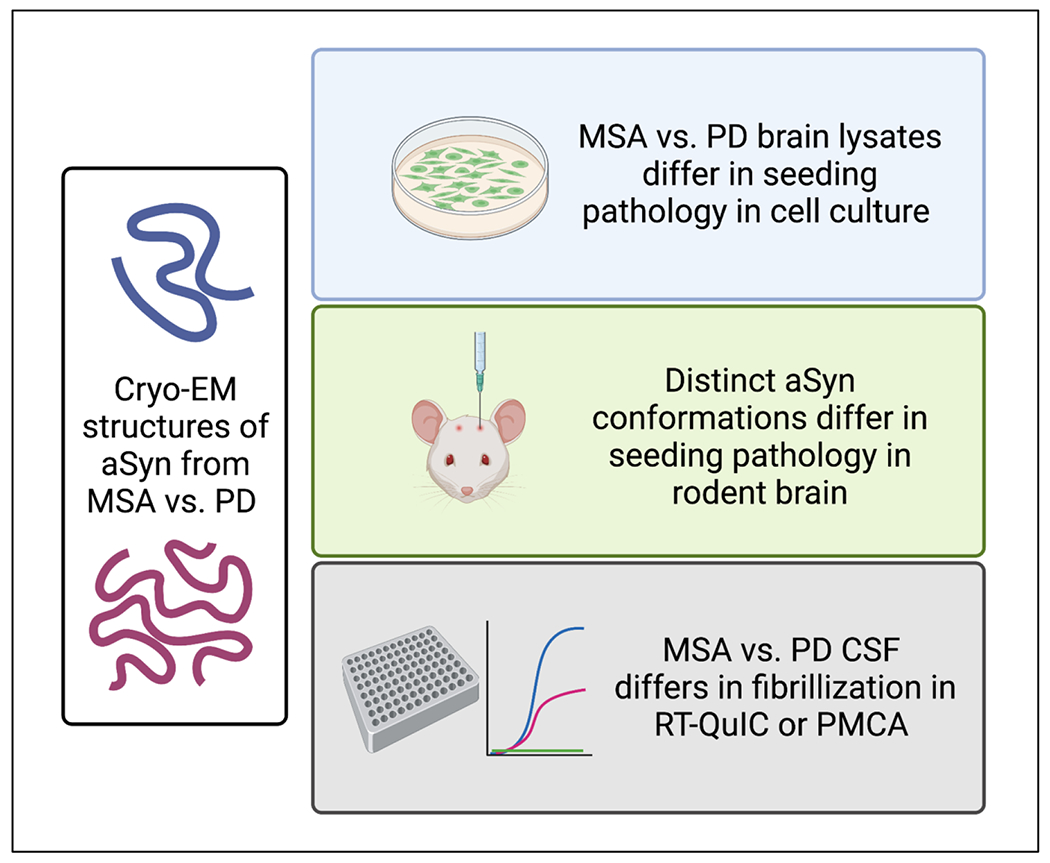
Cryo-EM studies support the existence of distinct conformations of aSyn in MSA vs. PD, both characterized by the presence of aSyn pathology. Studies of distinct conformations of aSyn, or aSyn species isolated from MSA vs. PD brain, demonstrate differential seeding and spreading properties in cell and animal models. CSF samples from individuals with MSA vs. PD exhibit distinct fibrillization properties in in vitro amplification assays.
Dysfunction of clearance mechanisms within the cell likely contributes to both cell-to-cell transmission of pathological forms of aSyn and opportunity for pathological forms of aSyn to template the misfolding of native aSyn. In support of this point, antibody-mediated clearance of aSyn reduces extracellular levels and decreases intercellular transmission in a transgenic mouse PD model;149 whereas inhibition of clearance mechanisms results in the opposite effect.150 Notably, dysfunction of autophagosome-lysosome pathways responsible for clearance of aggregates has been demonstrated in PD, contributing to worsening of aSyn aggregation,151–156 and lysosomal pathway functions are enriched among PD risk genes.157–159 In this context, we highlight the work of Mazzuli, Krainc, and colleagues, demonstrating that dysfunction of the lysosomal enzyme glucocerebrosidase (encoded by the PD-associated risk gene GBA) results in increased aSyn aggregate formation in neurons.160 These aggregates in turn accumulate in lysosomes, causing greater impairment in glucocerebrosidase function in a “feed-forward” loop. Recently, the same group has demonstrated that directly boosting wild-type glucocerebrosidase activity may reduce pathogenic phenotypes in patient-derived dopaminergic neurons.161
“Pathological aSyn” should cause neurodegeneration and cognitive decline
While experimental evidence considered so far has been largely supportive of aSyn as a prion-like protein, disconnects do emerge when we consider the evidence for this prion-like activity as a mechanism for development of cognitive decline and dementia. For example, the connection between cortical aSyn pathology and cognitive decline and dementia is not straightforward.162 Incidental LBs (iLBs) are found in approximately 8-18% of neurologically normal individuals on post-mortem examination, including in higher-order cognitive areas.163,164 The accumulation of iLBs may correlate with age: widespread aSyn iLBs without cognitive impairment can often be observed among centenarians.165 Notably, in one of the largest iLB pathology cohorts reported to date, 55% of subjects who would have been classified as Braak stages V and VI (diffuse neocortical involvement with predicted dementia) lacked signs of dementia or extrapyramidal symptoms.166 According to Braak’s original cohorts, cognitive decline would be expected with an increasing prevalence from stage III (36% with cognition affected) to stage VI (100% with cognition affected).75
At the same time, a number of disease-focused neuropathological studies have found that most PD cases are compatible with the Braak staging system,167–170 and Braak staging has proven particularly useful in early-onset and prolonged-duration PD.167,171 Importantly, when only subjects with a clinical diagnosis of dementia were considered, correlation with a high Braak stage was excellent.166 These seemingly-contradictory results may be reconciled if LBs do not necessarily result in cell dysfunction and death, as evidenced by the presence of iLBs in non-diseased subjects. Rather, LBs could be required, but not sufficient, for cell death,172 accounting for their high prevalence in autopsy series of PD and DLB cases. Alternately, some have argued that mature LBs could play a protective role in the brain by accumulating increasing quantities of aSyn oligomers in vulnerable individuals and thus preventing their neurotoxic effects.173
Cortical pathology in the human brain should follow a rostral-to-caudal pattern of spread
While the most prevalent pattern of LB pathology appears to follow Braak staging, it is worth noting that many cases do not follow this pattern. That is, collective evidence from multiple studies suggests that approximately 7-29% of LBD confirmed diagnoses do not show LBs in brainstem structures,174–177 arguing that rostro-caudal spread of aSyn is not the only route of PD progression. Moreover, correlation between the severity of LBs in lower brainstem regions and severity of LB pathology in the neocortex is poor,177 suggesting that factors other than abundance of LB pathology in lower-order brain regions govern which individual cases go on to develop cortical pathology. Indeed, Surmeier, Obeso, and Halliday have argued that the pattern of aSyn pathology found in human studies, connectome-mapping studies, and the Braak hypothesis cannot be fully reconciled, also suggesting the existence of other factors that “gate” or regulate the pace of pathological aSyn spread in the brain.178 In contrast, in mouse models in which aSyn fibrils have been introduced by a single intrastriatal injection, neuronal connectivity, together with aSyn expression, explains a substantial proportion of the resultant aSyn pathology observed throughout the brain at subsequent time points.98,100 Taken together, these data suggest that variability in humans, in the pace and pattern of aSyn pathological spread to those areas of the brain responsible for cognition, is far greater than in model systems. Moreover, neither the Braak hypothesis, nor neuroanatomical connectivity, can adequately account for this observed variability.
Individuals “host” differences: Impact on mechanisms of cognitive decline in PD
If LB are required, but not sufficient, for degeneration of cortical neurons and cognitive decline, how might we then explain the heterogeneous cognitive outcomes observed in subjects with abundant aSyn pathology? Moreover, if neither the Braak hypothesis nor current knowledge of the human brain connectome can fully explain whether aSyn pathology spreads from the brainstem to the limbic and neocortical areas in a given individual, what, then, might account for this variability? We argue here that “host” factors – genetic background, exposures and other modulators of host state during life – are the “black box” that has been understudied from a mechanistic standpoint.
Evidence that host genetic background impacts cognitive presentation in DLB, PD, and PDD comes from two major sources: clinicogenetic studies linking candidate genetic variants to cognitive trajectories in PD, and genome-wide association studies (GWAS) aimed at finding variants that associate with PDD or DLB. APOE genotype has been widely linked to cognition in LBD, with carriers of the APOE4 allele more likely to develop cognitive decline and dementia, in studies from our group and others.179–182 Similarly, GBA mutations have been widely reported to associate with poorer cognition in PD patients.183–187 Finally, since SNCA was first linked to familial PD, carriers of SNCA mutations (and multiplications) have been noted to have prominent cognitive symptoms,28,188 with common variants at the SNCA locus also associating with cognitive outcomes in PD. More recently, GWAS have been performed to discover, in an unbiased manner, common genetic variants that associate with cognitive phenotype in PD. These GWAS have confirmed associations for GBA and APOE, and nominated other loci near RIMS2, TMEM108, and WWOX as novel variants associating with cognition in PD.189 Additionally, GWAS performed in DLB have also confirmed associations between GBA, APOE, and SNCA and this cognitive-symptom-first form of LBD, while nominating loci near BIN1 and TMEM175 as DLB risk loci.190
Notably, genotypes at the BIN1 locus, first associated with risk of AD by GWAS, have also been shown by our group to associate with the presence of AD neuropathological change (ADNC) at autopsy in LBD cases,191 again underlining the importance of AD pathology in LBD patients. Indeed, we have argued that the high prevalence of concomitant AD pathology in LBD patients at autopsy – with only 43/208 (20.7%) of LBD patients in our study demonstrating no ADNC at autopsy, and 79/208 (37.98%) demonstrating intermediate to high levels of ADNC191 – raises the question of whether PD patients might represent the ideal pre-AD group in which to target mechanisms leading to AD pathology therapeutically.61 A role for AD pathology in determining the cognitive course of PD is also supported by biofluid biomarker studies (beta-amyloid and tau biomarker levels associated with AD are enriched in PD patients with cognitive deficits)192,193 and imaging studies (an AD-like structural MRI pattern is enriched in PD patients with cognitive deficits, who also have higher levels of beta-amyloid positivity by PET).194–197
Host exposures have been linked to the development of PD for decades, with consistent epidemiological evidence for exposure to pesticides (particularly paraquat), the solvent trichloroethylene (TCE), and mild-to-moderate head injury contributing to increased risk for PD.198,199 Moreover, animals exposed to paraquat,200 TCE.201,202 or traumatic brain injury (TBI)203 have been reported to develop varying degrees of neurotoxicity or neurodegeneration. In addition, caffeine and tobacco use inversely associate with risk for PD in many epidemiological studies.204 Whether these host exposures also increase risk for cognitive decline in PD is less well-studied, but it is worth noting that traumatic brain injury (TBI) has also been associated with subsequent development of dementia.205 Mechanistically, the disruption of the blood brain barrier,206 resultant inflammatory cascade,207 and microglial activation208 that may occur with TBI may certainly modulate the aSyn spreading events discussed earlier in this review. In addition to these environmental exposures, alterations in host state such as altered glucose metabolism209 or inflammation210 may impact both the development of LBD and cognitive decline in the LBD. Indeed, we recently found, in a screen of 940 plasma proteins, that modestly elevated C-reactive protein (CRP) levels, often indicative of mild inflammation,211 associated with slower rates of cognitive decline in PD.212 Whether glucose metabolism and systemic inflammatory state relate mechanistically to the aSyn aggregation and spreading events discussed in this review is not currently clear.
Until recently, most work on the impact of host genetics and exposures on cognitive decline in PD has been correlative. In the last few years, however, detailed mechanistic studies have begun to link specific host factors to cellular events in the pathogenesis of cognitive decline in PD. In 2020, two separate groups demonstrated in mouse models that APOE genetic background modulated development of aSyn pathology through pathways independent of amyloid-beta. Davis, Holtzman, and coauthors found that APOE4 mice demonstrated accelerated spread of aSyn pathology following intrastriatal injection of aSyn PFFs, compared to APOE2 or APOE3 mice.213 Zhao, Bu, and coauthors delivered SNCA via adenop-associated virus, finding that APOE4 mice showed increased aSyn pathology and more motor and cognitive deficits than APOE2 or APOE3 mice.214 These two studies provide a mechanistic counterpart for the observation that human APOE4 carriers with PD consistently show accelerated cognitive decline.179–182 In an example involving a different host gene, Wie, Ren and colleagues reported that missense mutations in TMEM175, linked by GWAS to risk for PD and for DLB as noted above, impair the function of the encoded lysosomal potassium channel.215 This organelle-level impairment in turn leads to vulnerability to damage in neurons, accelerated spreading of aSyn pathology in neurons and mice, and accelerated rates of cognitive and motor decline in PD patients.216 Importantly, the TMEM175 lysosomal channel defect may be targetable, since treating cells with a downstream pathway activator restored channel function. In a final example already mentioned in this review, we have shown that PD risk genotypes at the GWAS-nominated GPNMB locus associate with higher levels of GPNMB expression. GPNMB expression, in turn, is both necessary and sufficient for cellular internalization of fibrillar aSyn, and subsequent development of aSyn pathology in cortical neurons.134
These investigations in disease models amenable to manipulation, together with the clinicogenetic candidate gene studies and the GWAS cited above, provide strong support for the concept that host factors mechanistically impact the development of cognitive decline in LBD. Moreover, we underline three aspects of the evidence to date. First, multiple different approaches have converged on the same loci (GBA, APOE, SNCA, BIN1, TMEM175), increasing confidence that these host factors reliably influence risk for cognitive decline, across many cohorts and research strategies. Second, early mechanistic studies of APOE, TMEM175 and GPNMB demonstrate that biological pathways involving these genes may be amenable to therapeutic targeting, suggesting that investigating host genetics in disease-tailored, mechanistic experiments could help the field develop strategies for slowing or stopping cognitive decline in the LBD. Finally, for therapies aimed at GBA, APOE, TMEM175, GPNMB – or, for that matter, individuals who have been exposed to pesticides or TBI – a tailored approach matching the individual (who has a particular genetic or exposure profile) to the appropriate intervention is likely to be most effective.
Concluding Remarks
Cognitive impairment in PD is common, existing management options are limited, and the burden PD-related cognitive impairment and PDD impose on patients, caregivers, and health care systems are projected to grow at a breathtaking pace. As a result, we urgently need to understand underlying mechanisms and whether these mechanisms can be therapeutically targeted. Twenty years after its initial formulation, a refined version of the “Braak hypothesis” of pathological aSyn spread from brainstem to limbic and neocortical regions continues to inform. Specifically, evidence that pathological forms of aSyn may template naïve forms of aSyn in a prion-like manner, that pathological aSyn may exist as different “strains” with distinct conformations and behaviors, and that pathological aSyn may spread from cell-to-cell within the brain, eventually reaching the limbic and cortical regions responsible for many cognitive functions, is strong. At the same time, the frequency with which LBs are found in cognitively-unimpaired individuals, and the overall patterns of LB pathology in human autopsy studies, highlight areas that are poorly explained by an aSyn-focused, “Braak hypothesis” view. Specifically, we do not understand why some individuals with cortical aSyn pathology have PDD, while others do not show obvious cognitive symptoms. Similarly, we do not understand why some individuals with abundant brainstem aSyn pathology develop cortical aSyn pathology, while others do not (or why other individuals with abundant cortical aSyn pathology appear to have “bypassed” the brainstem entirely).
We argue that “host” factors – individual genetic characteristics and exposures or other modulators of host state during life – are the likely “black box” where answers to these questions may be found (Figure 3). We provide evidence from recent mechanistic work on APOE, TMEM175, and GPNMB, all genes in which common variants are known to associate with PD or with cognition in PD. Most encouraging, these recent developments suggest that pathways implicated by these “host” factors could be therapeutically exploited, offering more targets for intervention.
Figure 3. Factors influencing heterogeneity of cognitive outcomes in the Lewy body diseases.
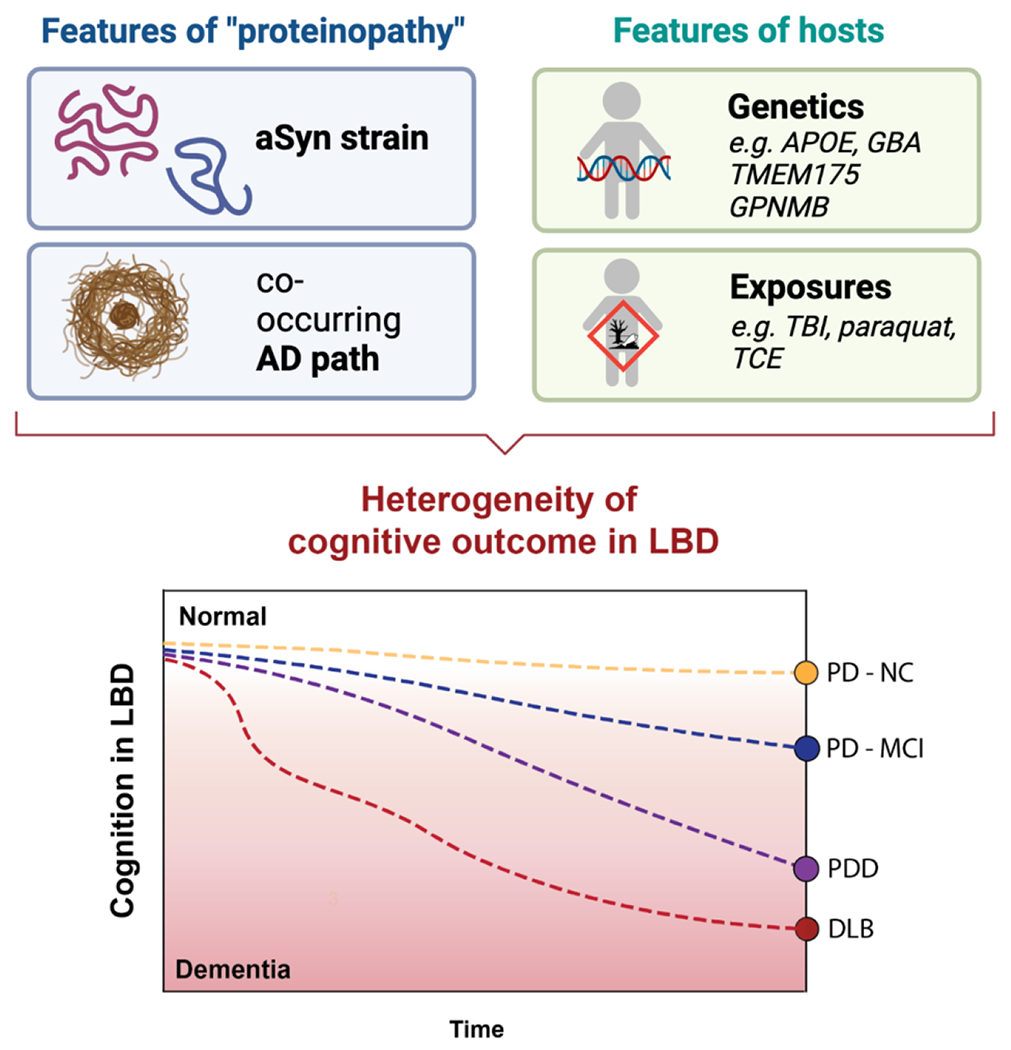
Inherent pathologic features of “proteinopathy” (as exemplified by aSyn strain, or amount of co-existing AD pathology) are modulated by individual host features (genetic background and environmental exposures throughout life), resulting in varying cognitive presentations. PD-NC = Parkinson’s disease with normal cognition, PD-MCI = Parkinson’s disease with mild cognitive impairment, PDD = Parkinson’s disease with dementia, DLB = dementia with Lewy bodies.
Acknowledgments
This work was supported by NIH grants RO1 NS082265 (ACP), RO1 NS115139 (ACP), U19 AG062418 (DW and ACP). ACP is also supported by the Chan Zuckerberg Initiative Neurodegeneration Challenge Network, the American Heart Association/Allen Institute Brain Health Initiative, and the Parker Family Chair. Figures in this review were created with BioRender.com.
Declaration of Interests
ACP is listed as an inventor on US patent application #20190328906 entitled “Therapy for Frontotemporal Dementia” from the Children’s Hospital of Philadelphia and the Trustees of the University of Pennsylvania and has received royalties from this patent. ACP is listed as an inventor on US provisional patent application No. 63/319,623 entitled “Methods for Treating Parkinson’s Disease” from the Trustees of the University of Pennsylvania. DW received honoraria in the past year for consultancy from Acadia Pharmaceuticals, Alkahest, Aptinyx, Cerevel Therapeutics, CHDI Foundation, Clintrex LLC (Otsuka), EcoR1 Capital, Eisai, Ferring, Gray Matter Technologies, Great Lake Neurotechnologies, Intra-Cellular Therapies, Janssen, Merck, Sage, Scion and Signant Health. He has received license fee payments from the University of Pennsylvania for the QUIP and QUIP-RS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, et al. (2017). Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100. 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reid WGJ, Hely MA, Morris JGL, Loy C, and Halliday GM (2011). Dementia in Parkinson’s disease: a 20-year neuropsychological study (Sydney Multicentre Study). Journal of Neurology, Neurosurgery & Psychiatry 82, 1033. 10.1136/jnnp.2010.232678. [DOI] [PubMed] [Google Scholar]
- 3.Williams-Gray CH, Foltynie T, Brayne CEG, Robbins TW, and Barker RA (2007). Evolution of cognitive dysfunction in an incident Parkinson’s disease cohort. Brain 130, 1787–1798. 10.1093/brain/awml11. [DOI] [PubMed] [Google Scholar]
- 4.Pigott K, Rick J, Xie SX, Hurtig H, Chen-Plotkin A, Duda JE, Morley JF, Chahine LM, Dahodwala N, Akhtar RS, et al. (2015). Longitudinal study of normal cognition in Parkinson disease. Neurology 85, 1276–1282. 10.1212/wnl.0000000000002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudlicka A, Clare L, and Hindle JV (2014). Quality of life, health status and caregiver burden in Parkinson’s disease: relationship to executive functioning. International Journal of Geriatric Psychiatry 29, 68–76. 10.1002/gps.3970. [DOI] [PubMed] [Google Scholar]
- 6.Vossius C, Larsen JP, Janvin C, and Aarsland D (2011). The economic impact of cognitive impairment in Parkinson’s disease. Movement Disorders 26, 1541–1544. 10.1002/mds.23661. [DOI] [PubMed] [Google Scholar]
- 7.Aarsland D, Ballard CG, and Halliday G (2004). Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol 17, 137–145. 10.1177/089198870426747b. [DOI] [PubMed] [Google Scholar]
- 8.Dorsey ER, Sherer T, Okun MS, and Bloem BR (2018). The Emerging Evidence of the Parkinson Pandemic. Journal of Parkinson’s Disease 8, S3–S8. 10.3233/JPD-181474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foltynie T, Brayne C, Robbins T, and Barker R (2004). The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 127, 550–560. [DOI] [PubMed] [Google Scholar]
- 10.Aarsland D, Andersen K, Larsen J, Lolk A, and Kragh-Sørensen P (2003). Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Archives of Neurology 60, 387–392. [DOI] [PubMed] [Google Scholar]
- 11.Fengler S, Liepelt-Scarfone I, Brockman K, Schaeffer E, Berg D, and Kalbe E (2017). Cognitive changes in prodromal Parkinson’s disease: a review. Movement Disorders 32, 1655–1666. [DOI] [PubMed] [Google Scholar]
- 12.Darweesh SKL, Wolters F, Postuma R, Stricker BH, Hofman A, Koudstaal PJ, Ikram MK, and Ikram MA (2017). Association between poor cognitive functioning and risk of incident parkinsonism: The Rotterdam Study. JAMA Neurol 74, 1431–1438. 10.1001/jamaneurol.2016.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darweesh SK, Verlinden VJ, Stricker BH, Hofman A, Koudstaal PJ, and Ikram MA (2017). Trajectories of prediagnostic functioning in Parkinson’s disease. Brain 140, 429–441. 10.1093/brain/aww291. [DOI] [PubMed] [Google Scholar]
- 14.Chahine LM, Weintraub D, Hawkins KA, Siderowf A, Eberly S, Oakes D, Seibyl J, Stem MB, Marek K, Jennings D, and PARS (2016). Cognition in individuals at risk for Parkinson’s: Parkinson associated risk syndrome (PARS) study findings. Mov Disord 31, 86–94. 10.1002/mds.26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fantini ML, Farini E, Ortelli P, Zucconi M, Manconi M, Cappa S, and Ferini-Strambi L (2011). Longitudinal study of cognitive function in idiopathic REM sleep behavior disorder. Sleep 34, 619–625. [PMC free article] [PubMed] [Google Scholar]
- 16.Kehagia A, Barker R, and Robbins T (2010). Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in Parkinson’s disease. Lancet Neurol 9, 1200–1213. [DOI] [PubMed] [Google Scholar]
- 17.Cronin-Golomb A, and Braun A (1997). Visuospatial dysfunction and problem solving in Parkinson’s disease. Neuropsychology 11, 44–52. [DOI] [PubMed] [Google Scholar]
- 18.Lewis S, Cools R, Robbins T, Dove A, Barker R, and Owen A (2003). Using executive heterogeneity to explore the nature of working memory deficits in Parkinson’s disease. Neuropsychologia 41, 645–654. [DOI] [PubMed] [Google Scholar]
- 19.Aarsland D, Bronnick K, Williams-Gray C, Weintraub D, Marder K, Kulisevsky J, and Burn D (2010). Mild cognitive impairment in Parkinson’s disease: A multicenter pooled analysis. Neurology 75, 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Liu FT, Hou XH, Li JQ, Cao XP, Tan L, Wang J, and Yu JT (2021). Predictors of cognitive impairment in Parkinson’s disease: a systematic review and meta-analysis of prospective cohort studies. J Neurol 268, 2713–2722. 10.1007/s00415-020-09757-9. [DOI] [PubMed] [Google Scholar]
- 21.Emre M, Aarsland D, Brown R, Burn D, Duyckaerts C, Mizuno Y, Broe G, and al., e. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement Disorders 22, 1689–1707. [DOI] [PubMed] [Google Scholar]
- 22.Litvan I, Goldman J, Troster A, Schmand B, Weintraub D, Petersen R, Mollenhauer B, Adler C, Marder K, Williams-Gray C, et al. (2012). Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force Guidelines. Movement Disorders 27, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skorvanek M, Goldman JG, Jahanshahi M, Marras C, Rektorova I, Schmand B, van Duijn E, Goetz CG, Weintraub D, Stebbins GT, et al. (2018). Global scales for cognitive screening in Parkinson’s disease: Critique and recommendations. Mov Disord 33, 208–218. 10.1002/mds.27233. [DOI] [PubMed] [Google Scholar]
- 24.Hoogland J, van Wanrooij LL, Boel JA, Goldman JG, Stebbins GT, Dalrymple-Alford JC, Marras C, Adler CH, Junque C, Pedersen KF, et al. (2018). Detecting mild cognitive deficits in Parkinson’s disease: comparison of neuropsychological tests. Mov Disord 33, 1750–1759. 10.1002/mds.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuang D, Leverenz J, Lopez O, Hamilton R, Bennett D, Schneider J, Buchman A, Larson E, Crane P, Kaye J, et al. (2013). APOE E4 increases risk for dementia in pure synulceinopathies. JAMA Neurology 70, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jellinger KA, and Korczyn AD (2018). Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 16, 34. 10.1186/sl2916-018-1016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, et al. (2017). Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16, 55–65. 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guella I, Evans DM, Szu-Tu C, Nosova E, Bortnick SF, Group SCS, Goldman JG, Dalrymple-Alford JC, Geurtsen GJ, Litvan I, et al. (2016). α-synuclein genetic variability: A biomarker for dementia in Parkinson disease. Annals of Neurology 79, 991–999. 10.1002/ana.24664. [DOI] [PubMed] [Google Scholar]
- 29.Crispo JA, Willis AW, Thibault DP, Fortin Y, Hays HD, McNair DS, Bjerre LM, Kohen DE, Perez-Lloret S, Mattison DR, and Krewski D (2016). Associations between anticholinergic burden and adverse health outcomes in Parkinson disease. PLoS One 11, e0150621. 10.1371/joumal.pone.0150621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrt U, Broich K, Larsen J, Ballard C, and Aarsland D (2010). Use of drugs with anticholinergic effect and impact on cognition in Parkinson’s disease: a cohort study. Journal of Neurology, Neurosurgery and Psychiatry 81, 160–165. [DOI] [PubMed] [Google Scholar]
- 31.Maggi G, Trojano L, Barone P, and Santangelo G (2021). Sleep disorders and cognitive dysfunctions in Parkinson’s disease: a meta-analytic study. Neuropsychol Rev 31, 643–682. 10.1007/sll065-020-09473-l. [DOI] [PubMed] [Google Scholar]
- 32.Rolinski M, Fox C, Maidment I, and McShane R (2012). Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database of Systematic Reviews Mar 14;3:CD006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emre M, Aarsland D, Albanese A, Byrne E, Deuschl G, De Deyn P, Durif F, Kulisevsky J, van Laar T, Lees A, et al. (2004). Rivastigmine for dementia associated with Parkinson’s disease. New England Journal of Medicine 351, 2509–2518. [DOI] [PubMed] [Google Scholar]
- 34.Emre M, Tsolaki M, Bonuccelli U, Destee A, Tolosa E, Kutzelnigg A, Ceballos-Baumann A, and al., e. (2010). Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomized, double-blind, placebo-controlled trial. Lancet Neurol 9, 969–977. [DOI] [PubMed] [Google Scholar]
- 35.Leroi I, Overshott R, Byrne EJ, Daniel E, and Burns A (2009). Randomized controlled trial of memantine in dementia associated with Parkinson’s disease. Mov Disord 24, 1217–1221. 10.1002/mds.22495. [DOI] [PubMed] [Google Scholar]
- 36.Aarsland D, Ballard C, Walker Z, Bostrom F, Altman D, Kossakowski K, Leroi I, and al., e. (2009). Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 8, 613–618. [DOI] [PubMed] [Google Scholar]
- 37.Weintraub D, Hauser RA, Elm JJ, Pagan F, Davis MD, Choudhry A, and Investigators M (2016). Rasagiline for mild cognitive impairment in Parkinson’s disease: A placebo-controlled trial. Mov Disord 31, 709–714. 10.1002/mds.26617. [DOI] [PubMed] [Google Scholar]
- 38.Hinson V, Delambo A, Elm J, and Turner T (2017). A randomized clinical trial of atomoxetine for mild cognitive impairment in Parkinson’s disease. Movement Disorders Clinical Practice 4, 416–423. 10.1002/mdc3.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mamikonyan E, Xie SX, Melvin E, and Weintraub D (2015). Rivastigmine for mild cognitive impairment in Parkinson disease: a placebo–controlled study. Movement Disorders 30, 912–918. [DOI] [PubMed] [Google Scholar]
- 40.Biglan K, Munsie L, Svensson KA, Ardayfio P, Pugh M, Sims J, and Brys M (2022). Safety and efficacy of mevidalen in Lewy body dementia: a phase 2, randomized, placebo-controlled trial. Mov Disord 37, 513–524. 10.1002/mds.28879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svenningsson P, Odin P, Dizdar N, Johansson A, Grigoriou S, Tsitsi P, Wictorin K, Bergquist F, Nyholm D, Rinne J, et al. (2020). A phase 2a trial investigating the safety and tolerability of the novel cortical enhancer IRL752 in Parkinson’s disease dementia. Mov Disord 35, 1046–1054. 10.1002/mds.28020. [DOI] [PubMed] [Google Scholar]
- 42.Goldman JG, Vernaleo BA, Camicioli R, Dahodwala N, Dobkin RD, Ellis T, Galvin JE, Marras C, Edwards J, Fields J, et al. (2018). Cognitive impairment in Parkinson’s disease: a report from a multidisciplinary symposium on unmet needs and future directions to maintain cognitive health. NPJ Parkinsons Dis 4, 19. 10.1038/s41531-018-0055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dauwan M, Begemann MJH, Slot MIE, Lee EHM, Scheltens P, and Sommer IEC (2021). Physical exercise improves quality of life, depressive symptoms, and cognition across chronic brain disorders: a transdiagnostic systematic review and meta-analysis of randomized controlled trials. J Neurol 268, 1222–1246. 10.1007/s00415-019-09493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orgeta V, McDonald KR, Poliakoff E, Hindle JV, Clare L, and Leroi I (2020). Cognitive training interventions for dementia and mild cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2, CD011961. 10.1002/14651858.CD011961.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weintraub D, Aarsland D, Biundo R, Dobkin R, Goldman J, and Lewis Simon (2022). Management of psychiatric and cognitive complications in Parkinson’s disease. BMJ 379, e068718. 10.1136/bmj-2021-068718. [DOI] [PubMed] [Google Scholar]
- 46.Schrag A, Siddiqui UF, Anastasiou Z, Weintraub D, and Schott JM (2017). Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: a cohort study. Lancet Neurol 16, 66–75. 10.1016/S1474-4422(16)30328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Postuma RB, Iranzo A, Hu M, Hogl B, Boeve BF, Manni R, Oertel WH, Arnulf I, Ferini-Strambi L, Puligheddu M, et al. (2019). Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain 142, 744–759. 10.1093/brain/awz030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, and Litvan I (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. The Lancet Neurology 8, 1150–1157. 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 49.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, and Goedert M (1997). α-Synuclein in Lewy bodies. Nature 388, 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 50.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. (1997). Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 276, 2045–2047. 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 51.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. (2003). α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 302, 841–841. 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 52.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, and Edwards RH (2010). Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 65, 66–79. 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, and Südhof TC (2010). α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 329, 1663–1667. 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burré J, Sharma M, and Südhof TC (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proceedings of the National Academy of Sciences 111, E4274–E4283. 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, and Iwatsubo T (2002). α-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology 4, 160–164. 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 56.Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, et al. (2006). Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. Journal of Biological Chemistry 281, 29739–29752. 10.1074/jbc.m600933200. [DOI] [PubMed] [Google Scholar]
- 57.Zhou J, Broe M, Huang Y, Anderson JP, Gai W-P, Milward EA, Porritt M, Howells D, Hughes AJ, Wang X, and Halliday GM (2011). Changes in the solubility and phosphorylation of α-synuclein over the course of Parkinson’s disease. Acta Neuropathologica 121, 695–704. 10.1007/s00401-011-0815-l. [DOI] [PubMed] [Google Scholar]
- 58.Braak H, Del Tredici K, Rub U, de Vos AR, and Janster Steur NE (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- 59.Krismer F, and Wenning GK (2017). Multiple system atrophy: insights into a rare and debilitating movement disorder. Nature Reviews Neurology 13, 232–243. 10.1038/nrneurol.2017.26. [DOI] [PubMed] [Google Scholar]
- 60.Stankovic I, Krismer F, Jesic A, Antonini A, Benke T, Brown RG, Burn DJ, Holton JL, Kaufmann H, Kostic VS, et al. (2014). Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group. Movement Disorders 29, 857–867. 10.1002/mds.25880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tropea TF, and Chen-Plotkin A (2021). Are Parkinson’s Disease Patients the Ideal Preclinical Population for Alzheimer’s Disease Therapeutics? Journal of Personalized Medicine 11, 834. 10.3390/jpml1090834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kotzbauer PT, Cairns NJ, Campbell MC, Willis AW, Racette BA, Tabbal SD, and Perlmutter JS (2012). Pathologic Accumulation of α-Synuclein and Aβ in Parkinson Disease Patients With Dementia. Archives of Neurology 69, 1326–1331. 10.1001/archneurol.2012.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller RL, Dhavale DD, O’Shea JY, Andruska KM, Liu J, Franklin EE, Buddhala C, Loftin SK, Cirrito JR, Perrin RJ, et al. (2022). Quantifying regional α -synuclein, amyloid β, and tau accumulation in lewy body dementia. Annals of Clinical and Translational Neurology 9, 106–121. 10.1002/acn3.51482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith C, Malek N, Grosset K, Cullen B, Gentleman S, and Grosset DG (2019). Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. Journal of Neurology, Neurosurgery & Psychiatry, jnnp-2019-32111. 10.1136/jnnp-2019-321111. [DOI] [PubMed] [Google Scholar]
- 65.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, et al. (2018). Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141, 2181–2193. 10.1093/brain/awy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jellinger KA (2022). Are there morphological differences between Parkinson’s disease-dementia and dementia with Lewy bodies? Parkinsonism & Related Disorders 100, 24–32. 10.1016/j.parkreldis.2022.05.024. [DOI] [PubMed] [Google Scholar]
- 67.Colom-Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Martí MJ, Clarimon J, and Lleó A (2013). Confluence of α-Synuclein, Tau, and β-Amyloid Pathologies in Dementia With Lewy Bodies. Journal of Neuropathology & Experimental Neurology 72, 1203–1212. 10.1097/NEN.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 68.Coughlin DG, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, McMillan CT, Wolk DA, Akhtar RS, Hurtig H, et al. (2018). Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Annals of Neurology. 10.1002/ana.25392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, and Lee VMY (2003). Initiation and Synergistic Fibrillization of Tau and Alpha-Synuclein. Science 300, 636–640. 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 70.Bassil F, Meymand ES, Brown HJ, Xu H, Cox TO, Pattabhiraman S, Maghames CM, Wu Q, Zhang B, Trojanowski JQ, and Lee VMY (2021). α-Synuclein modulates tau spreading in mouse brains. Journal of Experimental Medicine 218. 10.1084/jem.20192193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ryman SG, Yutsis M, Tian L, Henderson VW, Montine TJ, Salmon DP, Galasko D, and Poston KL (2021). Cognition at Each Stage of Lewy Body Disease with Co-occurring Alzheimer’s Disease Pathology. Journal of Alzheimer’s Disease 80, 1243–1256. 10.3233/JAD-201187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, and Bennett DA (2012). Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135, 3005–3014. 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braak H, Ghebremedhin E, Rüb U, Bratzke H, and Del Tredici K (2004). Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research 318, 121–134. 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 74.Hawkes CH, Del Tredici K, and Braak H (2010). A timeline for Parkinson’s disease. Parkinsonim & Related Disorders 16, 79–84. 10.1016/j.parkreldis.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 75.Braak H, Rub U, Jansen Steur ENH, Del Tredici K, and De Vos RAI (2005). Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64, 1404–1410. 10.1212/01.wnl.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 76.Hurtig H, Trojanowski JQ, Galvin J, Ewbank D, Schmidt M, Lee VMY, Clark C, Glosser G, Stem M, Gollomp S, and Arnold S (2000). Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921. 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 77.Hawkes CH, Del Tredici K, and Braak H (2007). Parkinson’s disease: a dual-hit hypothesis. Neuropathology and Applied Neurobiology 33, 599–614. 10.Ill1/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blessing WW, Li Y, and Wesselingh S (1991). Transneuronal transport of herpes simplex virus from the cervical vagus to brain neurons with axonal inputs to central vagal sensory nuclei in the rat. Neuroscience 42, 261–274. 10.1016/0306-4522(91)90163-i. [DOI] [PubMed] [Google Scholar]
- 79.Wakabayashi K, Takahashi H, Ohama E, and Ikuta F (1990). Parkinson’s disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathologica 79, 581–583. 10.1007/bf00294234. [DOI] [PubMed] [Google Scholar]
- 80.Wakabayashi K, Takahashi H, Takeda S, Ohama E, and Ikuta F (1988). Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathologica 76, 217–221. 10.1007/bf00687767. [DOI] [PubMed] [Google Scholar]
- 81.Braak H, de Vos AR, Bohl J, and Del Tredici K (2006). Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neuroscience Letters 396, 67–72. 10.1016/j.neulet.2005.ll.012. [DOI] [PubMed] [Google Scholar]
- 82.Del Tredici K, Hawkes CH, Ghebremedhin E, and Braak H (2010). Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathologica 119, 703–713. 10.1007/s00401-010-0665-2. [DOI] [PubMed] [Google Scholar]
- 83.Stevenson TJ, Murray HC, Turner C, Faull RLM, Dieriks BV, and Curtis MA (2020). α-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson’s disease olfactory bulb. Scientific Reports 10. 10.1038/s41598-020-63412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pearce RKB, Hawkes CH, and Daniel SE (1995). The anterior olfactory nucleus in Parkinson’s disease. Movement Disorders 10, 283–287. 10.1002/mds.870100309. [DOI] [PubMed] [Google Scholar]
- 85.Schapira HVA, and Tolosa E (2010). Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nature Reviews Neurology 6, 309–317. 10.1038/nmeurol.2010.52. [DOI] [PubMed] [Google Scholar]
- 86.Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, et al. (2015). MDS research criteria for prodromal Parkinson’s disease. Movement Disorders 30, 1600–1611. 10.1002/mds.26431. [DOI] [PubMed] [Google Scholar]
- 87.Heinzel S, Berg D, Gasser T, Chen H, Yao C, and Postuma RB (2019). Update of the MDS research criteria for prodromal Parkinson’s disease. Movement Disorders 34, 1464–1470. 10.1002/mds.27802. [DOI] [PubMed] [Google Scholar]
- 88.Adams-Carr KL, Bestwick JP, Shribman S, Lees A, Schrag A, and Noyce AJ (2016). Constipation preceding Parkinson’s disease: a systematic review and meta-analysis. Journal of Neurology, Neurosurgery & Psychiatry 87, 710–716. 10.1136/jnnp-2015-311680. [DOI] [PubMed] [Google Scholar]
- 89.Kordower JH, Chu Y, Hauser RA, Freeman TB, and Olanow CW (2008). Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nature Medicine 14, 504–506. 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 90.Li J-Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nature Medicine 14, 501–503. 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 91.Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, and Lee S-J (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proceedings of the National Academy of Sciences 106, 13010–13015. 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, and Lee VMY (2009). Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences 106, 20051–20056. 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Volpicelli-Daley Laura A., Luk Kelvin C., Patel Tapan P., Tanik Selcuk A., Riddle Dawn M., Stieber A, Meaney David F., Trojanowski John Q., and Lee Virginia M.Y. (2011). Exogenous α-Synuclein fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 72, 57–71. 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, and Lee VMY (2012). Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 338, 949–953. 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, and Lee VMY (2012). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. Journal of Experimental Medicine 209, 975–986. 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mougenot A, Nicot S, Bencskik A, Morignat E, Verchere J, Lakhdar L, Lagastelois S, and Baron T (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiology of Aging 33, 2225–2228. 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 97.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, and Hasegawa M (2013). Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138. 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henderson MX, Comblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ, Sandler RM, Bassett DS, Trojanowski JQ, and Lee VMY (2019). Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nature Neuroscience 22, 1248–1257. 10.1038/s41593-019-0457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, et al. (2015). Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiology of Disease 82, 185–199. 10.1016/j.nbd.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mezias C, Rey N, Brundin P, and Raj A (2020). Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiology of Disease 134. 10.1016/j.nbd.2019.104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga S-I, and Hasegawa M (2017). Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathologica Communications 5. 10.1186/s40478-017-0413-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Recasens A, and Dehay B (2014). Alpha-synuclein spreading in Parkinson’s disease, frontiers in Neuroanatomy 8. 10.3389/fhana.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi ML, Luk KC, et al. (2018). Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 557, 558–563. 10.1038/s41586-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marotta NP, Ara J, Uemura N, Lougee MG, Meymand ES, Zhang B, Petersson EJ, Trojanowski JQ, and Lee VMY (2021). Alpha-synuclein from patient Lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta Neuropathologica Communications 9. 10.1186/s40478-021-01288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut P-O, Blesa J, Parent A, Perier C, et al. (2014). Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Annals of Neurology 75, 351–362. 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 106.Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, et al. (2019). Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 103, 627–641.e627. 10.1016/j.neuron.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, Tsung-Pin P, Riess O, Nyengaard JR, Tamgüney G, et al. (2019). Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathologica 138, 535–550. 10.1007/s00401-019-02040-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manfredsson FP, Luk KC, Benskey MJ, Gezer A, Garcia J, Kuhn NC, Sandoval IM, Patterson JR, O’Mara A, Yonkers R, and Kordower JH (2018). Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiology of Disease 112, 106–118. 10.1016/j.nbd.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, and Takahashi R (2018). Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Molecular Neurodegeneration 13. 10.1186/s13024-018-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brettschneider J, Tredici KD, Lee VMY, and Trojanowski JQ (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nature Reviews Neuroscience 16, 109–120. 10.1038/nm3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peng C, Trojanowski JQ, and Lee VMY (2020). Protein transmission in neurodegenerative disease. Nature Reviews Neurology 16, 199–212. 10.1038/s41582-020-0333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Olanow CW, and Prusiner SB (2009). Is Parkinson’s disease a prion disorder? Proceedings of the National Academy of Sciences 106, 12571–12572. 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee H-J, Baek SM, Ho D-H, Suk J-E, Cho E-D, and Lee S-J (2011). Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Experimental and Molecular Medicine 43, 216. 10.3858/emm.2011.43.4.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jang A, Lee H-J, Suk J-E, Jung J-W, Kim K-P, and Lee S-J (2010). Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. Journal of Neurochemistry. 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 115.Atik A, Stewart T, and Zhang J (2016). Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathology 26, 410–418. 10.llll/bpa.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.El-Agnaf OMA, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DMA, Ikeda S-I, et al. (2003). α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. The FASEB Journal 77, 1–16. 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- 117.Brás IC, and Outeiro TF (2021). Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 10, 375. 10.3390/cellsl0020375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, and McLean PJ (2011). Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. The FASEB Journal 25, 326–336. 10.1096/ij.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJA, and Cooper JM (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiology of Disease 42, 360–367. 10.1016/j.nbd.2011.01.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ngolab J, Trinh I, Rockenstein E, Mante M, Florio J, Trejo M, Masliah D, Adame A, Masliah E, and Rissman RA (2017). Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathologica Communications 5. 10.1186/s40478-017-0445-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, Li Y, Aro P, Dator R, He C, et al. (2014). Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathologica 128, 639–650. 10.1007/s00401-014-1314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gao L, Tang H, Nie K, Wang L, Zhao J, Gan R, Huang J, Zhu R, Feng S, Duan Z, et al. (2015). Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. International Journal of Neuroscience 125, 645–654. 10.3109/00207454.2014.961454. [DOI] [PubMed] [Google Scholar]
- 123.Kluge A, Bunk J, Schaeffer E, Drobny A, Xiang W, Knacke H, Bub S, Lückstädt W, Arnold P, Lucius R, et al. (2022). Detection of neuron-derived pathological α-synuclein in blood. Brain 145, 3058–3071. 10.1093/brain/awacll5. [DOI] [PubMed] [Google Scholar]
- 124.Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, and Lee S-J (2008). Assembly- dependent endocytosis and clearance of extracellular a-synuclein. The International Journal of Biochemistry & Cell Biology 40, 1835–1849. 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 125.Brahic M, Bousset L, Bieri G, Melki R, and Gitler AD (2016). Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathologica 131, 539–548. 10.1007/s00401-016-1538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, and Brahic M (2012). Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Annals of Neurology 72, 517–524. 10.1002/ana.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Domingues R, Sant’Anna R, da Fonseca ACC, Robbs BK, Foguel D, and Outeiro TF (2022). Extracellular alpha-synuclein: Sensors, receptors, and responses. Neurobiology of Disease 168, 105696. 10.1016/j.nbd.2022.105696. [DOI] [PubMed] [Google Scholar]
- 128.Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin J-H, et al. (2016). Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374. 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Emmenegger M, De Cecco E, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, Tantardini E, De Rossi P, Bacioglu M, Langston RG, et al. (2021). LAG3 is not expressed in human and murine neurons and does not modulate α-synucleinopathies. EMBO Molecular Medicine 13. 10.15252/emmm.202114745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aulić S, Masperone L, Narkiewicz J, Isopi E, Bistaffa E, Ambrosetti E, Pastore B, De Cecco E, Scaini D, Zago P, et al. (2017). α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Scientific Reports 7. 10.1038/s41598-017-10236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ferreira DG, Temido-Ferreira M, Vicente Miranda H, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, et al. (2017). α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nature Neuroscience 20, 1569–1579. 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- 132.Xie YX, Naseri NN, Fels J, Kharel P, Na Y, Lane D, Burré J, and Sharma M (2022). Lysosomal exocytosis releases pathogenic α-synuclein species from neurons in synucleinopathy models. Nature Communications 13, 4918. 10.1038/s41467-022-32625-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, Pierce S, Burmeister AR, Madaj Z, Steiner JA, et al. (2019). Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Molecular Neurodegeneration 14, 34. 10.1186/sl3024-019-0335-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Diaz-Ortiz ME, Seo Y, Posavi M, Carceles Cordon M, Clark E, Jain N, Charan R, Gallagher MD, Unger TL, Amari N, et al. (2022). GPNMB confers risk for Parkinson’s disease through interaction with a-synuclein. Science 377, eabk0637. 10.1126/science.abk0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, et al. (2011). Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nature Medicine 17, 175–178. 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- 136.Saborio GP, Permanne B, and Soto C (2001). Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813. 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 137.Soto C, Saborio GP, and Anderes L (2002). Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends in Neurosciences 25, 390–394. 10.1016/s0166-2236(02)02195-l. [DOI] [PubMed] [Google Scholar]
- 138.Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Annals of Clinical and Translational Neurology 3, 812–818. 10.1002/acn3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, and Soto C (2017). Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurology 74, 163. 10.1001/jamaneurol.2016.4547. [DOI] [PubMed] [Google Scholar]
- 140.Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orrù CD, Antelmi E, Mammana A, Hughson AG, Calandra-Buonaura G, et al. (2020). Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathologica 140, 49–62. 10.1007/s00401-020-02160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Boeve BF, Sletten DM, Gehrking TL, Gehrking JA, Olson AD, Savica R, et al. (2020). Alpha-Synuclein Oligomers and Neurofilament Light Chain in Spinal Fluid Differentiate Multiple System Atrophy from Lewy Body Synucleinopathies. Annals of Neurology 88, 503–512. 10.1002/ana.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo Jing L., Covell Dustin J., Daniels Joshua P., Iba M, Stieber A, Zhang B, Riddle Dawn M., Kwong Linda K., Xu Y, Trojanowski John Q., and Lee Virginia M.Y. (2013). Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 154, 103–117. 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, and Melki R (2013). Structural and functional characterization of two alpha-synuclein strains. Nature Communications 4. 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peelaerts W, Bousset L, Van Der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van Den Haute C, Melki R, and Baekelandt V (2015). α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 145.Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song E-S, Cairns NJ, Kotzbauer PT, and Diamond MI (2019). Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics. Journal of Biological Chemistry 294, 1045–1058. 10.1074/jbc.RA118.004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang Y, Shi Y, Schweighauser M, Zhang X, Kotecha A, Murzin AG, Garringer HJ, Cullinane PW, Saito Y, Foroud T, et al. (2022). Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 610, 791–795. 10.1038/s41586-022-05319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aguzzi A, Heikenwalder M, and Polymenidou M (2007). Insights into prion strains and neurotoxicity. Nature Reviews Molecular Cell Biology 8, 552–561. 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 148.Sigurdson CJ, Bartz JC, and Glatzel M (2019). Cellular and Molecular Mechanisms of Prion Disease. Annual Review of Pathology: Mechanisms of Disease 14, 497–516. 10.1146/annurev-pathmechdis-012418-013109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, and Lee SJ (2012). Antibody-Aided Clearance of Extracellular -Synuclein Prevents Cell-to-Cell Aggregate Transmission. Journal of Neuroscience 32, 13454–13469. 10.1523/jneurosci.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee H-J, Cho E-D, Lee KW, Kim J-H, Cho S-G, and Lee S-J (2013). Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Experimental & Molecular Medicine 45, e22–e22. 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, and Engelender S (2011). α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proceedings of the National Academy of Sciences 108, 18666–18671. 10.1073/pnas.1105725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rubinsztein DC (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 153.Winslow AR, Chen C-W, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. (2010). α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. Journal of Cell Biology 190, 1023–1037. 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, and Jensen PH (2004). Proteasomal Inhibition by α-Synuclein Filaments and Oligomers. Journal of Biological Chemistry 279, 12924–12934. 10.1074/jbc.m306390200. [DOI] [PubMed] [Google Scholar]
- 155.Malkus KA, and Ischiropoulos H (2012). Regional deficiencies in chaperone-mediated autophagy underlie α-synuclein aggregation and neurodegeneration. Neurobiology of Disease 46, 732–744. 10.1016/j.nbd.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, and Sulzer D (2004). Impaired Degradation of Mutant a-Synuclein by Chaperone-Mediated Autophagy. Science 305, 1292–1295. 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 157.Robak LA, Jansen IE, Van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P, Shulman JM, Nalls MA, Plagnol V, et al. (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. 10.1093/brain/awx285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, Gana-Weisz M, Raymond D, Rozenkrantz L, Deik A, et al. (2013). The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80, 1606–1610. 10.1212/wnl.0b013e31828fl80e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Foo J-N, Liany H, Bei J-X, Yu X-Q, Liu J, Au W-L, Prakash KM, Tan LC, and Tan E-K (2013). A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiology of Aging 34, 2890.e2813–2890.e2815. 10.1016/j.neurobiolaging.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 160.Mazzulli Joseph R., Xu Y-H, Sun Y, Knight Adam L., McLean Pamela J., Caldwell Guy A., Sidransky E, Grabowski Gregory A., and Krainc D (2011). Gaucher Disease Glucocerebrosidase and ;l;-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 146, 37–52. 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Burbulla LF, Jeon S, Zheng J, Song P, Silverman RB, and Krainc D (2019). A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Science Translational Medicine 11, eaau6870. 10.1126/scitranslmed.aau6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Parkkinen L, Kauppinen T, Pirttilä T, Autere JM, and Alafuzoff I (2005). α-Synuclein pathology does not predict extrapyramidal symptoms or dementia. Annals of Neurology 57, 82–91. 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- 163.Dickson DW, Fujishiro H, Delledonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, and Ahlskog JE (2008). Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathologica 115, 437–444. 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 164.Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, et al. (2003). Neuropathology of Cognitively Normal Elderly. Journal of Neuropathology & Experimental Neurology 62, 1087–1095. 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 165.Ding Z-T, Wang Y, Jiang Y-P, Hashizume Y, Yoshida M, Mimuro M, Inagaki T, and Iwase T (2006). Characteristics of alpha-synucleinopathy in centenarians. Acta Neuropathologica 111, 450–458. 10.1007/s00401-005-0015-y. [DOI] [PubMed] [Google Scholar]
- 166.Parkkinen L, Pirttilä T, and Alafuzoff I (2008). Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathologica 115, 399–407. 10.1007/s00401-008-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Halliday G, Hely M, Reid W, and Morris J (2008). The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathologica 115, 409–415. 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
- 168.Jellinger KA (2009). Formation and development of Lewy pathology: a critical update. Journal of Neurology 256, 270–279. 10.1007/s00415-009-5243-y. [DOI] [PubMed] [Google Scholar]
- 169.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, et al. (2009). Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathologica 117, 613–634. 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mikolaenko I, Pletnikova O, Kawas CH, O’Brien R, Resnick SM, Crain B, and Troncoso JC (2005). Alpha-Synuclein Lesions in Normal Aging, Parkinson Disease, and Alzheimer Disease: Evidence from the Baltimore Longitudinal Study of Aging (BLSA). Journal of Neuropathology & Experimental Neurology 64, 156–162. 10.1093/jnen/64.2.156. [DOI] [PubMed] [Google Scholar]
- 171.Halliday GM, and McCann H (2010). The progression of pathology in Parkinson’s disease. Annals of the New York Academy of Sciences 1184, 188–195. 10.1111/j.1749-6632.2009.05118.x. [DOI] [PubMed] [Google Scholar]
- 172.Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, and Duda JE (2012). Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 79, 2307–2314. 10.1212/wnl.0b013e318278fe32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chartier S, and Duyckaerts C (2018). Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell and Tissue Research 373, 149–160. 10.1007/s00441-018-2854-6. [DOI] [PubMed] [Google Scholar]
- 174.Jellinger KA (2019). Is Braak staging valid for all types of Parkinson’s disease? Journal of Neural Transmission 126, 423–431. 10.1007/s00702-018-1898-9. [DOI] [PubMed] [Google Scholar]
- 175.Jellinger KA, and Attems J (2008). The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathology and Applied Neurobiology 34, 466–467. 10.llll/j.1365-2990.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- 176.Zaccai J, Brayne C, McKeith I, Matthews F, and Ince PG (2008). Patterns and stages of α-synucleinopathy. Neurology 70, 1042. 10.1212/01.wnl.0000306697.48738.b6. [DOI] [PubMed] [Google Scholar]
- 177.Kingsbury AE, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, Maeir H, Poewe W, and Lees AJ (2010). Brain stem pathology in Parkinson’s disease: An evaluation of the Braak staging model. Movement Disorders 25, 2508–2515. 10.1002/mds.23305. [DOI] [PubMed] [Google Scholar]
- 178.Surmeier DJ, Obeso JA, and Halliday GM (2017). Selective neuronal vulnerability in Parkinson disease. Nature Reviews Neuroscience 18, 101–113. 10.1038/nm.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Williams-Gray CH, Goris A, Saiki M, Foltynie T, Compston DAS, Sawcer SJ, and Barker RA (2009). Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s Disease. Journal of Neurology 256, 493–498. 10.1007/s00415-009-0119-8. [DOI] [PubMed] [Google Scholar]
- 180.Morley JF, Xie SX, Hurtig HI, Stern MB, Colcher A, Horn S, Dahodwala N, Duda JE, Weintraub D, Chen-Plotkin AS, et al. (2012). Genetic influences on cognitive decline in Parkinson’s disease. Movement Disorders 27, 512–518. 10.1002/mds.24946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, Van Deerlin VM, Ritz B, Rausch R, Rhodes SL, Factor SA, et al. (2014). APOE, MAPT, and SNCA Genes and Cognitive Performance in Parkinson Disease. JAMA Neurology 71, 1405. 10.1001/jamaneurol.2014.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tropea TF, Xie SX, Rick J, Chahine LM, Dahodwala N, Doshi J, Davatzikos C, Shaw LM, Van Deerlin V, Trojanowski JQ, et al. (2018). APOE , thought disorder, and SPARE-AD predict cognitive decline in established Parkinson’s disease. Movement Disorders 33, 289–297. 10.1002/mds.27204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Alcalay RN, Caccappolo E, Mejia-Santana H, Tang MX, Rosado L, Orbe Reilly M, Ruiz D, Ross B, Verbitsky M, Kisselev S, et al. (2012). Cognitive performance of GBA mutation carriers with early-onset PD. Neurology 78, 1434. 10.1212/WNL.0b013e318253d54b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Seto-Salviaó N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, Paisán-Ruiz C, Campolongo A, Antón-Aguirre S, Martín I, et al. (2012). Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Movement Disorders 27, 393–399. 10.1002/mds.24045. [DOI] [PubMed] [Google Scholar]
- 185.Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, Duran R, Mencacci NE, Sawcer SJ, and Barker RA (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136, 392–399. 10.1093/brain/aws318. [DOI] [PubMed] [Google Scholar]
- 186.Chahine LM, Qiang J, Ashbridge E, Minger J, Yearout D, Horn S, Colcher A, Hurtig HI, Lee VM-Y, Van Deerlin VM, et al. (2013). Clinical and Biochemical Differences in Patients Having Parkinson Disease With vs Without GBA Mutations. JAMA Neurology 70, 852–858. 10.1001/jamaneurol.2013.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, Ritz B, Rausch R, Factor SA, Wood-Siverio C, et al. (2016). GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Movement Disorders 31, 95–102. 10.1002/mds.26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, and Langston JW (2004). Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Annals ofNeurology 55, 174–179. 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 189.Liu G, Peng J, Liao Z, Locascio JJ, Corvol J-C, Zhu F, Dong X, Maple-Grødem J, Campbell MC, Elbaz A, et al. (2021). Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nature Genetics 53, 787–793. 10.1038/s41588-021-00847-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E, Walton RL, Ahmed S, Viollet C, Ding J, et al. (2021). Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nature Genetics 53, 294–303. 10.1038/s41588-021-00785-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dai DL, Tropea TF, Robinson JL, Suh E, Hurtig H, Weintraub D, Van Deerlin V, Lee EB, Trojanowski JQ, and Chen-Plotkin AS (2020). ADNC-RS, a clinical-genetic risk score, predicts Alzheimer’s pathology in autopsy-confirmed Parkinson’s disease and Dementia with Lewy bodies. Acta Neuropathologica 140, 449–461. 10.1007/s00401-020-02199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Berlyand Y, Weintraub D, Xie SX, Mellis IA, Doshi J, Rick J, McBride J, Davatzikos C, Shaw LM, Hurtig H, et al. (2016). An Alzheimer’s Disease-Derived Biomarker Signature Identifies Parkinson’s Disease Patients with Dementia. PLOS ONE 11, e0147319. 10.1371/joumal.pone.0147319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mollenhauer B, Rochester L, Chen-Plotkin A, and Brooks D (2014). What can biomarkers tell us about cognition in Parkinson’s disease? Movement Disorders 29, 622–633. 10.1002/mds.25846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Charissé D, Erus G, Pomponio R, Gorges M, Schmidt N, Schneider C, Liepelt-Scarfone I, Riedel O, Reetz K, Schulz JB, et al. (2022). Brain age and Alzheimer’s-like atrophy are domain-specific predictors of cognitive impairment in Parkinson’s disease. Neurobiology of Aging 109, 31–42. 10.1016/j.neurobiolaging.2021.08.020. [DOI] [PubMed] [Google Scholar]
- 195.Weintraub D, Dietz N, Duda JE, Wolk DA, Doshi J, Xie SX, Davatzikos C, Clark CM, and Siderowf A (2012). Alzheimer’s disease pattern of brain atrophy predicts cognitive decline in Parkinson’s disease. Brain 135, 170–180. 10.1093/brain/awr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Petrou M, Dwamena BA, Foerster BR, Maceachern MP, Bohnen NI, Müller ML, Albin RL, and Frey KA (2015). Amyloid deposition in Parkinson’s disease and cognitive impairment: A systematic review. Movement Disorders 30, 928–935. 10.1002/mds.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shirvan J, Clement N, Ye R, Katz S, Schultz A, Johnson KA, Gomez-Isla T, Frosch M, Growdon JH, and Gomperts SN (2019). Neuropathologic correlates of amyloid and dopamine transporter imaging in Lewy body disease. Neurology 93, e476–e484. 10.1212/wnL0000000000007855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tanner CM, Goldman SM, Ross GW, and Grate SJ (2014). The disease intersection of susceptibility and exposure: Chemical exposures and neurodegenerative disease risk. Alzheimer’s & Dementia 10, S213–S225. 10.1016/jjalz.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 199.Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, and Yaffe K (2018). Mild TBI and risk of Parkinson disease. A Chronic Effects of Neurotrauma Consortium Study 90, e1771–e1779. 10.1212/wnl.0000000000005522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tieu K (2011). A Guide to Neurotoxic Animal Models of Parkinson’s Disease. Cold Spring Harbor Perspectives in Medicine 1, a009316–a009316. 10.1101/cshperspect.a009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu M, Shin E-J, Dang D-K, Jin C-H, Lee PH, Jeong JH, Park S-J, Kim Y-S, Xing B, Xin T, et al. (2018). Trichloroethylene and Parkinson’s Disease: Risk Assessment. Molecular Neurobiology 55, 6201–6214. 10.1007/sl2035-017-0830-x. [DOI] [PubMed] [Google Scholar]
- 202.Keane PC, Hanson PS, Patterson L, Blain PG, Hepplewhite P, Khundakar AA, Judge SJ, Kahle PJ, LeBeau FEN, and Morris CM (2019). Trichloroethylene and its metabolite TaClo lead to degeneration of substantia nigra dopaminergic neurones: Effects in wild type and human A30P mutant α-synuclein mice. Neuroscience Letters 711, 134437. 10.1016/j.neulet.2019.134437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.de Oliveira PA, Ben J, Matheus FC, Schwarzbold ML, Moreira ELG, Rial D, Walz R, and Prediger RD (2017). Moderate traumatic brain injury increases the vulnerability to neurotoxicity induced by systemic administration of 6-hydroxydopamine in mice. Brain Research 1663, 78–86. 10.1016/j.brainres.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 204.Simon DK, Tanner CM, and Brundin P (2020). Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clinics in Geriatric Medicine 36, 1–12. 10.1016/j.cger.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Crane PK, Gibbons LE, Dams-O’Connor K, Trittschuh E, Leverenz JB, Keene CD, Sonnen J, Montine TJ, Bennett DA, Leurgans S, et al. (2016). Association of Traumatic Brain Injury With Late-Life Neurodegenerative Conditions and Neuropathologic Findings. JAMA Neurology 73, 1062. 10.1001/jamaneurol.2016.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Saw MM, Chamberlain J, Barr M, Morgan MPG, Burnett JR, and Ho KM (2014). Differential Disruption of Blood–Brain Barrier in Severe Traumatic Brain Injury. Neurocritical Care 20, 209–216. 10.1007/sl2028-013-9933-z. [DOI] [PubMed] [Google Scholar]
- 207.Morganti-Kossmann MC, Satgunaseelan L, Bye N, and Kossmann T (2007). Modulation of immune response by head injury. Injury 38, 1392–1400. 10.1016/jinjury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 208.Zhang C, and Chen S (2022). Role of TREM2 in the Development of Neurodegenerative Diseases After Traumatic Brain Injury. Molecular Neurobiology, 10.1007/sl2035-022-03094-w. [DOI] [PubMed] [Google Scholar]
- 209.Chohan H, Senkevich K, Patel RK, Bestwick JP, Jacobs BM, Bandres Ciga S, Gan-Or Z, and Noyce AJ (2021). Type 2 Diabetes as a Determinant of Parkinson’s Disease Risk and Progression. Movement Disorders 36, 1420–1429. 10.1002/mds.28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Amin J, Erskine D, Donaghy PC, Surendranathan A, Swann P, Kunicki AP, Boche D, Holmes C, McKeith IG, O’Brien JT, et al. (2022). Inflammation in dementia with Lewy bodies. Neurobiology of Disease 168, 105698. 10.1016/j.nbd.2022.105698. [DOI] [PubMed] [Google Scholar]
- 211.Custodero C, Mankowski RT, Lee SA, Chen Z, Wu S, Manini TM, Hincapie Echeverri J, Sabbà C, Beavers DP, Cauley JA, et al. (2018). Evidence-based nutritional and pharmacological interventions targeting chronic low-grade inflammation in middle-age and older adults: A systematic review and meta-analysis. Ageing Research Reviews 46, 42–59. 10.1016/j.arr.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Shen J, Amari N, Zack R, Skrinak RT, Unger TL, Posavi M, Tropea TF, Xie SX, Van Deerlin VM, Dewey RB, et al. (2022). Plasma MIA , CRP, and Albumin Predict Cognitive Decline in Parkinson’s Disease. Annals of Neurology. 10.1002/ana.26410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Davis AA, Inman CE, Wargel ZM, Dube U, Freeberg BM, Galluppi A, Haines JN, Dhavale DD, Miller R, Choudhury FA, et al. (2020). APOE genotype regulates pathology and disease progression in synucleinopathy. Science Translational Medicine 12, eaay3069. 10.1126/scitranslmed.aay3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, Meneses AD, Li F, Shue F, Zheng J, et al. (2020). APOE4 exacerbates a-synuclein pathology and related toxicity independent of amyloid. Science Translational Medicine 12, eaay1809. 10.1126/scitranslmed.aayl809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Cang C, Aranda K, Seo Y-J, Gasnier B, and Ren D (2015). TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function. Cell 162, 1101–1112. 10.1016/j.cell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 216.Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H, Liang Y, Cang C, Aranda K, Lohmann J, et al. (2021). A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 591, 431–437. 10.1038/s41586-021-03185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]