

Abstract
Astrocytes provide key neuronal support, and their phenotypic transformation is implicated in neurodegenerative diseases. Metabolically, astrocytes possess low mitochondrial oxidative phosphorylation (OxPhos) activity, but its pathophysiological role in neurodegeneration remains unclear. Here, we show that the brain critically depends on astrocytic OxPhos to degrade fatty acids (FAs) and maintain lipid homeostasis. Aberrant astrocytic OxPhos induces lipid droplet (LD) accumulation followed by neurodegeneration that recapitulates key features of Alzheimer’s disease (AD) including synaptic loss, neuroinflammation, demyelination, and cognitive impairment. Mechanistically, when FA load overwhelms astrocytic OxPhos capacity, elevated acetyl-CoA levels induce astrocyte reactivity by enhancing STAT3 acetylation and activation. Intercellularly, lipid-laden reactive astrocytes stimulate neuronal FA oxidation and oxidative stress, activate microglia via IL-3 signaling, and inhibit the biosynthesis of FAs and phospholipids required for myelin replenishment. Along with LD accumulation and impaired FA degradation manifested in an AD mouse model, we reveal a lipid-centric, AD-resembling mechanism by which astrocytic mitochondrial dysfunction progressively induces neuroinflammation and neurodegeneration.
INTRODUCTION
Mitochondria are the major cellular sources of ATP via the oxidative metabolism of glucose or other fuels. A bioenergetic deficit, encompassing the decline in mitochondrial bioenergetic function and glucose hypometabolism, is associated with brain aging and emerges in early stages of neurodegenerative diseases such as Alzheimer’s disease (AD)1. However, brain metabolic profile is highly diverse with different cell types manifesting differential fuel preference and susceptibility to bioenergetic deficit2. Across cell types, mitochondrial dysfunction in neurodegeneration has been best documented in neurons3, and more recently in microglia4,5, yet the pathological role of mitochondria in astrocyte, the most abundant cell type in the brain, remains to be defined.
Astrocytes regulate essential processes including synaptic development and transmission, blood flow, and inflammatory response by interacting with all cell types in the brain6,7. Upon brain injury or pathological insults, astrocytes undergo a transformation from homeostatic state towards reactive state (reactive astrocyte), which is not only a prominent hallmark, but also a mechanistic contributor, of AD pathogenesis and progression8.
The energy demand of astrocytes is predominantly met by glycolysis that generates lactate9. Accordingly, compared to their neuronal counterparts, astrocytic mitochondria are less abundant with less efficient electron transport chain (ETC) and lower oxidative phosphorylation (OxPhos) capacity10. Mitochondrial OxPhos has been found dispensable for the bioenergetics, proliferation, and survival of astrocytes11. Nevertheless, astrocytic mitochondria express all tricarboxylic acid (TCA) cycle enzymes and maintain a modest level of OxPhos activity12, which indicates their metabolic role other than glucose oxidation and ATP production.
The brain is a highly lipidated organ, and fatty acids (FAs) are the essential building blocks for nearly all lipid classes. Thus, the fine regulation of FAs is vital for the homeostasis and functionality of all brain lipids. In metabolic organs, mitochondria are the main cellular site for the enzymatic and oxidative degradation of FAs via β-oxidation (FAO, generating acetyl-CoA), TCA cycle, and ultimately OxPhos. In the brain, cell type-specific analysis has revealed that the enrichment of FAO enzymes is a key feature that distinguishes astrocytic mitochondrial proteome from that of neuronal mitochondria13, which indicates a unique role of astrocytic mitochondria in FA metabolism. However, the exact role of astrocytic mitochondria in regulating brain lipid homeostasis is unknown.
Clinically, abundant evidence has demonstrated disrupted lipid homeostasis –including the accumulation of lipid droplets (LDs)– in early stages of AD14. A variety of lipid metabolism genes have also been identified as top risk factors of the disease15. Nevertheless, how lipid dyshomeostasis and LD accumulation emerge in the degenerating brain and how they modify disease progression, remain elusive. We reported previously that APOE-ε4 (APOE4), the greatest genetic risk factor for AD, induces a metabolic shift in astrocytes towards diminished FA catabolism16, which indicates a role of astrocytic FA degradation in the initiation of lipid accumulation, and potentially, in promoting neurodegeneration and AD.
Here, by using a mouse model of astrocyte specific OxPhos deficit, we report that astrocytic OxPhos is indispensable in maintaining brain lipid homeostasis by degrading FAs. Loss of astrocytic OxPhos induces: 1) pathological, structural, functional, and behavioral hallmarks that resemble human AD, and 2) metabolic and transcriptional signatures that highly overlap with that of an AD mouse model. We reveal that deficient astrocytic FA degradation triggers lipid accumulation and reactive astrogliosis, which subsequently suppress the biosynthesis of lipids required for oligodendrocyte-mediated myelin turnover. Moreover, we identify cell non-autonomous mechanisms by which lipid-laden reactive astrocytes promote synaptic dysfunction and neuroinflammation by triggering neuronal metabolic reprogramming and microglial activation, respectively.
RESULTS
Astrocytic OxPhos Deficit Causes Cognitive Impairment
Tfam (transcription factor A, mitochondrial) is a key regulator of mitochondrial DNA (mtDNA) that encodes essential subunits of OxPhos, and its deletion results in mtDNA depletion and OxPhos deficiency17. To model astrocyte-specific deficit in mitochondrial OxPhos, we crossed mice with loxP-flanked Tfam (Tfamflox/flox)18 with the GFAP-Cre 77.6 mice19 to generate the GFAP-Cre:Tfamflox/flox mice (hereinafter referred to as TfamAKO, Extended Data Fig. 1a). Their Cre-negative littermates were used as controls (Tfamflox/flox; hereinafter referred to as WT). Consistent with the original report that GFAP-Cre mice do not exhibit Cre activity during embryonic development19, Tfam expression was reduced in the hippocampus and cortex of TfamAKO mice at 3- and 6-month-of-age, but not in those at 1.5-month-of-age (Fig. 1a; Extended Data Fig. 1b). In forebrain cells isolated by magnetic-activated cell sorting, we confirmed that the depletion of Tfam mRNA was specific to astrocytes, but not neurons or microglia (Extended Data Fig. 1c). Upon Tfam depletion, mtDNA copy numbers and mtDNA-encoded transcripts of complexes I, III, IV and V were significantly reduced in 6-month-old mouse brains (Fig. 1b,c). Functionally, astrocytes isolated from TfamAKO brains showed reduced basal and maximal respiration but unaffected proton leak-linked respiration (Extended Data Fig. 1d,e), confirming the OxPhos deficit.
Fig. 1. Astrocyte-specific Tfam deletion induces cognitive impairment and neurodegeneration.

(a) Protein levels of Tfam in the hippocampus and cortex of 6-month-old WT and TfamAKO mice. (b) mtDNA copy numbers in the hippocampus of 6-month-old mice. (c) Levels of mtDNA-encoded transcripts of complexes I, III, IV and V in the cortex of 6-month-old mice. (d) Discrimination index of 6-month-old mice by the novel object recognition (NOR) test. (e) Representative 5-minute tracks and the time spent in the center area (marked by the orange squares) from the open field test of 6-month-old mice. (f) Recorded fEPSP slopes of hippocampal slices from 6-month-old mice normalized to the pre-tetanus baseline (left) and LTP quantified using %fEPSP for the last 5 min of the response to TBS stimulation (right). n = 10–15 slices from 4 mice per group. (g and h) Representative images and quantifications (positive area %) of 6-month-old mouse hippocampus sections stained for MAP-2 (g) and PSD95 (h). (i) Western blots showing protein expression of MAP-2, PSD95, and SNAP25 in 6-month-old mouse hippocampi (quantified in Extended Data Fig. 2a). (j and k) Heatmap (j) and PCA plot (k) showing the expression of top 500 variant genes in the hippocampus of 6-month WT and TfamAKO mice. (l) Top 10 GO Biological Processes enriched in 6-month WT hippocampus compared to that of TfamAKO mice. n = 5 (a, k) or 4 (g and h) mice; n = 6 (WT) or 5 (AKO) mice (b); n = 9 (WT) or 6 (AKO) mice (c); n = 13 (WT) or 9 (AKO) mice (d, e); n = 10 (WT) or 15 (AKO) slices from 4 mice (f). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 100 μm (g and h).
Having established TfamAKO mouse as a model for astrocytic OxPhos deficit, we characterized the cognitive function of these mice. Strikingly, TfamAKO mice developed severe cognitive impairment at 6-month-of-age with unchanged brain weight (Extended Data Fig. 1f). Compared to WT mice, they exhibited deficits in recognition memory and exploratory behavior, as revealed by novel object recognition (NOR) test and open field test, respectively (Fig. 1d,e; Extended Data Fig. 1g). No cognitive deficit was observed in 1.5-month TfamAKO mice (Extended Data Fig. 1h–j). TfamAKO did not affect motor function at 6-month (Extended Data Fig. 1k). Of note, Tfam expression was not affected in the cerebellum or the brainstem (Extended Data Fig. 1l), which could be relevant to the lower astrocyte-neuron ratio in the cerebellum20 and the lower GFAP positive rate in the brainstem21. The lack of significant impact on motor function could also indicate differential susceptibility to astrocytic OxPhos deficit across brain regions, which warrants further investigations.
In parallel to the cognitive impairment, astrocytic OxPhos deficit resulted in a decline in hippocampal long-term potentiation (LTP), the principal mechanism underlying long-term memory and learning. Compared to WT controls, hippocampal slices of 6-month TfamAKO mice failed to sustain synaptic activity following high frequency stimulation (Fig. 1f). TfamAKO-induced cognitive and synaptic deficits were accompanied by compromised dendrite complexity as indicated by MAP-2 immunostaining (Fig. 1g), reduced synaptic density as indicated by the immunostaining of post-synaptic marker PSD-95 (Fig. 1h) and the levels of PSD-95 and pre-synaptic protein SNAP25 in the hippocampus (Fig. 1i; Extended Data Fig. 2a). These synaptic parameters were undistinguishable between 3-month-old WT and TfamAKO mice (Extended Data Fig. 2b,c). Further, hippocampal transcriptomic analysis confirmed synaptic deficits in TfamAKO brains. Heatmap of the top differentially expressed genes (DEGs) and principal component analysis (PCA) revealed a dramatic distinction between the transcriptomes of 6-month WT and TfamAKO hippocampus (Fig. 1j,k), with 3,438 gene upregulated and 3,016 genes downregulated in the TfamAKO group (FDR-corrected p < 0.05). Key synaptic genes were downregulated in the TfamAKO group (Extended Data Fig. 2d), and the top 10 Gene Ontology (GO) Biological Processes underrepresented in TfamAKO hippocampi were all related to synaptic function or neurotransmission (Fig. 1l). We further ruled out the possibility that TfamAKO-induced cognitive decline at 6-month was caused by impaired neurogenesis or neuronal loss. Immunostaining of TfamAKO brains for mature neurons (NeuN) and neuronal progenitor cells (doublecortin; DCX) showed no differences in the counts of NeuN+, DCX+, or NeuN+DCX+ cells from WT brains (Extended Data Fig. 2e,f). Likewise, bromodeoxyuridine (BrdU) tracing did not detect a difference in the counts of BrdU+NeuN+ cells (newly generated mature neurons) in the dentate gyrus of 6-month-old WT versus TfamAKO mice (Extended Data Fig. 2g), confirming unaffected adult neurogenesis. Further, TUNEL assay did not detect apoptosis in any cell type in TfamAKO brains (Extended Data Fig. 2h).
Together, these data suggest that a functional mitochondrial OxPhos machinery in astrocytes is required for brain function, and its loss induces neurodegeneration characterized by memory impairment and disrupted synaptic transmission that recapitulate clinical and pathological features of AD.
Astrocytic OxPhos Maintains Brain Lipid Homeostasis
We next sought to explore the metabolic changes underlying the TfamAKO-initiated neurodegeneration. As astrocytic OxPhos is dispensable for brain ATP production11 and the loss of Tfam did not affect their proliferation in vivo or survival in vitro (Extended Data Fig. 3a), we explored for other mitochondrial pathways responsible for TfamAKO-induced phenotype. 1,140 genes encoding the mitochondrial proteome from the transcriptomes of WT and TfamAKO hippocampi were subjected to GeneSet Enrichment Analysis (GSEA) of defined mitochondrial pathways (MitoCarta 3.022). Besides the suppression of OxPhos and mitochondrial complexes I, III, IV and V (Fig. 2a,b; Extended Data Fig. 3b–f), lipid metabolism and FAO were the only pathways that were significantly altered in the TfamAKO brains (Fig. 2a,c). At transcript level, genes involved in both mitochondrial and peroxisomal β-oxidation were significantly upregulated (Fig. 2d). Moreover, the protein levels of PPARα, a key FAO regulator primarily expressed by astrocytes in the brain, was upregulated in TfamAKO hippocampi (Fig. 2e; Extended Data Fig. 3g). These data strongly support a unique role of astrocytic mitochondria in regulating brain FA metabolism.
Fig. 2. Aberrant astrocytic OxPhos disrupts brain lipid homeostasis.

(a) GSEA identified significantly enriched (enrichment score > 0) or underrepresented (enrichment score < 0) mitochondrial pathways in 6-month TfamAKO mouse hippocampi relative to WT. (b and c) GSEA plots showing enrichment profiles of OXPHOS genes (b) and FAO genes (c). (d) Heatmap showing DEGs (FDR-corrected p < 0.05) related to mitochondrial and peroxisomal β-oxidation in 6-month mouse hippocampi. (e) Western blots showing increased levels of PPARα in 6-month TfamAKO hippocampi (quantified in Extended Data Fig. 3g). (f and g) TLC assay showing altered levels of major lipid classes in 6-month mouse hippocampi (f), which are quantified by normalizing to protein concentrations (g). Std., standard mix. (h) PCA plot of hippocampal lipidomic profile of 6-month WT and TfamAKO mice. (i-o) Heatmaps of the levels of different classes of lipid species that are significantly changed in 6-month TfamAKO vs. WT cortices, including ceramide (i), cardiolipin (j), PS (k), PI (l), PC (m), PE (n), and DAG (o). n = 3 mice (g); n = 10 (WT) or 9 (AKO) mice (h). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons.
Since the complete degradation of FA requires OxPhos to accept electrons from reducing equivalents (NADH and FADH2) generated from FAO and the TCA cycle, we speculated that the upregulated FAO pathway in the TfamAKO brain could be an adaptive response to the defective astrocytic OxPhos and indicative of lipid accumulation. Indeed, the levels of lipid classes including free FAs (FFA), triacylglycerol (TAG), monoacylglycerol (MAG), free cholesterol, and cholesteryl esters (CEs) were increased in the hippocampus and cortex of 6-month TfamAKO mice by thin layer chromatography (TLC) and/or quantitative fluorometric assays (Fig. 2f,g; Extended Data Fig. 3h–n). Notably, an accumulation of TAG, but not FFA, was observed in 3-month TfamAKO brains (Extended Data Fig. 3o,p).
To determine the changes in polar lipids in TfamAKO brains, a targeted lipidomic panel was applied with cortices from 6-month-old mice. TfamAKO led to a substantially distinctive lipid profile (Fig. 2h), with 101 of the 153 detected species differentially expressed (Extended Data Fig. 3q). Specifically, most ceramide and cardiolipin species were increased in TfamAKO brains (Fig. 2i,j), whereas phosphatidylserine (PS) and phosphatidylinositol (PI) species were predominantly decreased (Fig. 2k,l). The levels of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) species showed mixed changes with the majority of PC and lysoPC species increased in TfamAKO cortices (Fig. 2m,n). In contrast to the elevation in TAG, the levels of diacylglycerol (DAG), especially those with saturated or monounsaturated FA chains were decreased in TfamAKO brains (Fig. 2o). Together, our lipid profiling suggests that loss of OxPhos in astrocytes dramatically perturbs brain lipid homeostasis, characterized by accumulations of FFAs and neutral lipids (TAG and CE) and an extensive shift in the composition of phospholipids.
TfamAKO Induces LD Accumulation and Reactive Astrogliosis
LDs are organelles that store and sequester excess FFAs in TAGs and CEs. In peripheral organs, LD formation serves as a protective mechanism against FFA-induced lipotoxicity23. In agreement with the elevated TAG and CE levels, brain immunostaining identified a progressive accumulation of perilinpin-2 (Plin2, LD surface marker)-positive LDs in the hippocampus and cortex of TfamAKO mice; these LDs initially emerged at 3-month and peaked at 6-month (Fig. 3a,b; Extended Data Fig. 4a–d). Importantly, these LDs were exclusively localized to astrocytes, but not neurons or microglia (Fig. 3c; Extended Data Fig. 4e). Moreover, reactive astrogliosis was prominent in LD-accumulating regions, as suggested by increased GFAP+ area and morphological features of reactive astrocytes (Fig. 3c,d; Extended Data Fig. 4f). This was accompanied by upregulation of reactive astrocyte marker genes in the hippocampus (Fig. 3e) and elevated Tnfa and Il6 mRNA in acutely isolated astrocytes (Fig. 3f). Temporally, reactive astrocytes first appeared in TfamAKO brains at 3-month (Extended Data Fig. 4c,d), which coincided with Tfam depletion and LD accumulation but was prior to any synaptic deficit. The reactive phenotype of TfamAKO astrocytes was further confirmed in vitro, where they exhibited higher GFAP intensity (Fig. 3g; Extended Data Fig. 4g). Notably, astrocyte phenotype upon TfamAKO resembles that seen in AD patients and animal models, in which astrocyte reactivity, but not proliferation, is increased24.
Fig. 3. Loss of astrocytic FA degradation leads to LD accumulation and reactive astrogliosis.

(a) Representative images of 6-month hippocampal or cortical staining for Plin2 and LD. (b) Quantification of LD+ area in hippocampal sections. (c) Representative images of hippocampal and cortical sections of 6-month mice. (d) Quantification of GFAP+ area in hippocampal sections. (e) Heatmap of reactive astrocyte DEGs in 6-month hippocampi. (f) Tnfa and Il6 mRNA levels in acute 6-month astrocytes. (g) GFAP intensity of cultured 6-month astrocytes. (h) Relative OCR of 6-month acute hippocampal slices on oleate-BSA. (i) Etomoxir-induced OCR decreases of 6-month acute hippocampal slices. (j) Etomoxir-induced OCR decreases of cultured 6-month astrocytes. (k) FA-induced basal and maximal OCRs in 6-month astrocytes presented as differences in the presence and absence of exogenous FA. (l) LD volume in cultured 6-month astrocytes. (m and n) Quantification and representative images of BODIPY-C12 localized to mitochondria in 6-month astrocytes. (o) 13C enrichment of TCA metabolites in astrocytes incubated with U-13C-oleate. (p) Ratio of mitochondrial fission to fusion products in 6-month astrocytes. (q) Representative images and quantification of LDs in 6-month WT astrocytes treated with oleate-BSA. (r) Representative images and quantification of GFAP intensity of WT astrocytes after oleate-BSA treatment. (s) Levels of p-STAT3Tyr705 in oleate-BSA-treated WT astrocytes. (t) Medium IL-6, TNFα, and PGE2 levels of oleate-BSA-treated WT astrocytes. (u and v) Representative ECAR curves and quantification of acute hippocampal slices before and after FA addition under glycemic or aglycemic conditions. n = 4 (b, d, p, s, t), 7 (f), 3 (g, r), 5 (l, m), 6 (o-WT), 7 (o-AKO), 3 (q-WT), or 4 (q-AKO) mice; n = 13 (h), 28 (i-WT), 22 (i-AKO) slices; n = 8 (j) or 11 (k) independent samples; n = 14 (WT-glucose), 13 (AKO-glucose), 14 (WT-sucrose), 15 (AKO-sucrose) slices (u,v). Bars and plots are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons except for the comparisons before and after FA injections in v, where two-sided paired t-test was used. Scale bars, 500 μm (r); 100 μm (a and c); 25 μm (q); 20 μm (n).
We next investigated whether LD accumulation in TfamAKO brains is triggered by compromised astrocytic FA degradation. Acute hippocampal slices from 6-month TfamAKO mice showed a reduced oxygen consumption rate (OCR) on exogenous FAs (Fig. 3h). In addition, blocking carnitine palmitoyltransferase I (CPT1)-mediated long-chain FA transport into the mitochondria using etomoxir induced a lesser OCR drop in TfamAKO brain slices (Fig. 3i), suggesting lower endogenous FA catabolism. In vitro analysis also confirmed that TfamAKO astrocytes exhibited decreased endogenous FA catabolism (etomoxir-sensitive OCR), decreased FA-induced OCR, and increased intracellular LD volume (Fig. 3j–l; Extended Data Fig. 4h). Moreover, by tracing a fluorescent FA analog (BODIPY-C12), we found strong colocalization of the probe with mitochondria in TfamAKO but not WT astrocytes, suggesting arrested mitochondrial FA degradation upon Tfam deletion (Fig. 3m,n). Pulse-chase of BODIPY-C12 by TLC confirmed that TfamAKO astrocytes accumulated more BODIPY-C12 as free FAs and esterified FAs (Extended Data Fig. 4i,j). Further, an isotope tracing assay suggested significantly reduced carbon flux from U-13C-oleate into TCA cycle metabolites citrate, isocitrate, and oxaloacetate in TfamAKO astrocytes (Fig. 3o). Consistent with the notion that fused mitochondria are more capable of performing FAO25, the mitochondria of TfamAKO astrocytes are more fragmented than that of WT astrocytes (Fig. 3p). In addition, multiple acylcarnitines (C6, C8, C10, C16, C18), intermediates generated during FA transport into mitochondria before entering FAO, accumulated in TfamAKO brains (Extended Data Fig. 4k,l). Overall, these data strongly suggest that compromised FA degradation due to astrocytic OxPhos deficit is responsible for the LD accumulation and aberrant lipid profile in TfamAKO brains.
FA Overload Promotes Astrocyte Reactivity
The temporospatial concurrence of lipid accumulation and reactive astrogliosis in TfamAKO brains indicates a mechanistic link between FA metabolism and astrocyte reactivity. To test this, we incubated WT astrocytes with exogenous oleate. Oleate not only promoted LD accumulation (Fig. 3q), but also substantially increased GFAP and S100β intensities indicative of higher reactivity (Fig. 3r; Extended Data Fig. 4m). Moreover, STAT3, a canonical mediator of astrocyte reactivity26, was activated by oleate (Fig. 3s, p-STAT3Tyr705), which was accompanied by increased secretion of inflammation mediators including IL-6, TNFα and prostaglandin E2 (PGE2) but unchanged ROS levels (Fig. 3t; Extended Data Fig. 4n).
The high metabolic dependence of astrocytes on glycolysis over OxPhos is augmented when they become reactive27. In parallel with reactive astrogliosis, the hippocampus of TfamAKO mice showed a higher glycolysis rate ex vivo (Fig. 3u,v, before FA injection), which was accompanied by higher levels of lactate and upregulation of key glycolytic enzymes including hexokinase 2 (HK2) and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) at both mRNA- and protein levels (Extended Data Fig. 4o–q). Increased glycolysis was confirmed in astrocytes: HK2 and GLUT1 (astrocytic glucose transporter) expression and lactate levels were increased in acutely isolated and cultured TfamAKO astrocytes, respectively (Extended Data Fig. 4r,s). Further, while oleate treatment only modestly elevated glycolysis in WT brain slices, the response was drastically enhanced for TfamAKO slices (Fig. 3u,v). This suggests that astrocytic OxPhos protects the brain from FA overload-induced metabolic reprogramming towards glycolysis that is coupled with astrocyte reactivity. Together, our data suggest that FA metabolic stress, caused by excess supply or impaired degradation, is sufficient to induce astrocyte reactivity and shift the metabolic flux further towards glycolysis, and that astrocytes with defective OxPhos are much more susceptible to such a phenotypic shift.
STAT3 Acetylation Mediates FA-Induced Astrocyte Reactivity
We next sought to determine the mechanism by which OxPhos deficit and FA overload induce astrocyte reactivity. We first tested whether elevated LD levels are involved in the process. Intriguingly, while the inhibition of adipose triglyceride lipase (ATGL, enzyme that breaks down TAG in LDs) substantially increased LD volume in astrocytes, it had no effect on GFAP intensity or p-STAT3 levels (Extended Data Fig. 5a–c). Given that increased LDs are not sufficient to trigger astrocyte reactivity, we speculated that elevated cytosolic FFAs may play a role. We thus blocked long-chain FA transport into the mitochondria with etomoxir (CPT1i). Surprisingly, while etomoxir exaggerated oleate-induced LD accumulation as expected (Fig. 4a), it abrogated the increase in p-STAT3 and the release of IL-6, TNFα, and PGE2 (Fig. 4b,c). This suggests that the entry of FAs into the mitochondria are required for FA-induced reactivity, and that the metabolites generated by FAO could be involved.
Fig. 4. Acetylation of STAT3 mediates FA- and TfamKO-induced astrocyte reactivity.

(a) Representative images and qualification of LD volumes in WT astrocytes treated with BSA (vehicle) or 150 μM oleate-BSA for 24 h with or without pretreatment of 10 μM etomoxir (CPT1i). (b) Representative western blots and quantification of p-STAT3Tyr705 in BSA- or oleate-BSA-treated astrocytes with or without CPT1i pretreatment. (c) IL-6, TNFα, and PGE2 levels in culture medium of BSA- or oleate-BSA-treated astrocytes with or without CPT1i pretreatment. (d) Representative western blots and quantification of acetyl-STAT3Lys685 in BSA- or oleate-BSA-treated astrocytes with or without CPT1i pretreatment. (e) Representative western blots and quantification of acetyl-STAT3Lys685 and p-STAT3Tyr705 in astrocytes treated with BSA or 150 μM oleate-BSA for 24 h with or without pretreatment of 50 μM BMS-303141(ACLYi) or 50 μM S3I-201 (STAT3i). (f) IL-6, TNFα, and PGE2 levels in culture medium of BSA- or oleate-BSA-treated astrocytes with or without ACLYi or STAT3i pretreatment. (g) Western blots and quantifications of acetyl-STAT3Lys685 and p-STAT3Tyr705 levels in the hippocampus of 6-month WT and TfamAKO mice. (h) Acetyl-CoA levels in the cortex of 6-month WT and TfamAKO mice. (i) Schematic representation of the mechanism by which Tfam deletion or exogenous FA induce astrocyte reactivity; key components and inhibitors involved are shown (image created with BioRender.com). n = 4 (a, e, g) or 5 (b and d) mice; n = 6 (IL6 and TNFα) or 3 (PGE2) mice (c); n = 8 (IL6 and TNFα) or 6 (PGE2) mice (f); n = 6 (WT) or 5 (AKO) mice (h). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test (g and h) and one-way ANOVA with post-hoc Tukey test (a-f) was used. Scale bar, 15 μm (a).
Because mitochondrial FAO produces acetyl-CoA and the acetylation of STAT3 is known to promote its phosphorylation and activation28, we reasoned that increased acetyl-CoA may increase upon FA overload and promote STAT3 activation. Consistent with this hypothesis, the levels of acetylated STAT3 (acetyl-STAT3Lys685) were increased by oleate, and more importantly, the effect was abolished by CPT1i pretreatment (Fig. 4d). Mitochondrial acetyl-CoA is exported by first being converted to citrate, which then exits the organelle and regenerate acetyl-CoA by ATP-citrate lyase (ACLY) in the cytosol or the nucleus, before being used for Lysine acetylation29. Pretreatments with either ACLY inhibitor BMS-303141 (ACLYi) or STAT3 inhibitor S3I-201 (STAT3i; inhibits STAT3 DNA binding) nullified oleate-induced STAT3 acetylation and phosphorylation (Fig. 4e) as well as the release of IL-6, TNFα and PGE2 (Fig. 4f). These data demonstrate that STAT3 acetylation is required for FA overload-induced astrocyte reactivity.
We next investigated whether the mechanism is involved in TfamAKO-induced reactive astrogliosis in vivo. Consistent with our in vitro findings, the levels of acetyl-STAT3 and p-STAT3 were significantly higher in the hippocampus of TfamAKO mice (Fig. 4g), which was accompanied by elevated acetyl-CoA (Fig. 4h) and PGE2 (Extended Data Fig. 5d) levels. We confirmed that increased acetyl-STAT3 levels in TfamAKO hippocampus was primarily contributed by astrocytes as suggested by the predominant localization of STAT3 to astrocytes (Extended Data Fig. 5e). Collectively, these results show that in astrocytes with either FA overload or OxPhos deficit, elevated acetyl-CoA levels –caused by an imbalance between FA load and acetyl-CoA metabolism in the mitochondria– induces astrocyte reactivity by promoting STAT3 acetylation (Fig. 4i).
Disrupted FAO Diminishes Astrocytic Support to Neurons
We next determined how astrocytes with OxPhos deficit affect neuronal function and elicit synaptic dysfunction. Primary neurons and astrocytes were cocultured in shared medium without physical contact (Transwell system) to allow subsequent separation for cellular analyses. We found that astrocytes with defective OxPhos negatively affect neurite outgrowth and neuronal metabolic function. Compared to WT astrocytes, TfamAKO astrocytes exhibited a substantially diminished capability to promote neurite outgrowth in terms of both length (Fig. 5a) and volume (Extended Data Fig. 6a). Glucose metabolism in neurons, including both glycolysis and mitochondrial oxidation, was suppressed when they were cocultured with TfamAKO astrocytes (Fig. 5b,c; Extended Data Fig. 6b,c). When WT astrocytes were overloaded with exogenous FA, they also showed diminished neuronal metabolic support (Extended Data Fig. 6h,i). Further, reduced synaptic and metabolic support by TfamAKO astrocytes was accompanied by their reduced capacity to eliminate and accept neuronal lipids, as suggested by a lesser reduction of LDs in neurons (Fig. 5d) and a lesser increase of LDs in astrocytes after coculture (Extended Data Fig. 6d). These data suggest that a functional FA catabolism machinery is required for astrocyte to provide critical neuronal support and highlight the essential role of astrocyte as the major recipient and degrader of excess neuronal lipids.
Fig. 5. Impaired astrocytic FA degradation diminishes neurotrophic support and triggers neuronal FAO and oxidative stress.

(a) Representative images of WT hippocampal neurons cultured alone (on coverslips in 6-well plates) or cocultured with WT or TfamAKO astrocytes (in 6-well inserts) for 7 days and immunostained for MAP-2 with neurite length quantified. (b) Mito Stress Test of WT neurons (in 24-well Seahorse plate) cultured with WT or TfamAKO astrocytes (in 24-well insert) for 7 days with quantifications of maximal respiration (after FCCP injection) shown. (c) Lactate levels in WT neurons (in 6-well plates) cultured with WT or TfamAKO astrocytes (in 6-well inserts) for 7 days. (d) Representative images and qualification of LD volumes in WT neurons cultured alone or with WT or TfamAKO astrocytes for 7 days. (e) mRNA levels of Ppara in neurons acutely isolated from 6-month mouse brains. (f) mRNA levels of genes involved in FA transport and FAO in neurons acutely isolated from 6-month mouse brains. (g) mRNA levels of Cpt2, Acadvl, and Ppara in WT neurons cultured with WT or TfamAKO astrocytes for 7 days. (h) Relative ROS levels (CellROX intensity) in WT neurons (in 6-well plates) cultured with WT or TfamAKO astrocytes (in 6-well inserts) for 7 days. (i and j) Representative images of hippocampal sections of 6-month WT and TfamAKO mice stained for NeuN and 4-HNE (i) or NeuN and 8-oxo-2’-deoxyguanosine (8-OHdG) (j). n = 3 (no-astrocyte) or 5 (+WT and +AKO) independent samples (a,d); n = 6 (WT) or 5 (AKO) independent samples (b); n =3 (c), 5 (g), or 6 (h) independent samples; n = 4 (WT) or 3 (AKO) mice (e); n = 6 mice (f). Bar graphs are presented as mean ± SEM. Two-sided unpaired Student’s t-test (b,c,e-h) or one-way ANOVA with post-hoc Tukey test (a,d) was used. Scale bars, 100 μm (i, j); 50 μm (a); 20 μm (d).
Glia-generated lactate can be transferred to neurons for FA synthesis, which can be transported back to glia for storage in LDs30. To determine if this mechanism contributes to the higher LD volume in TfamAKO astrocytes, we pretreated WT neurons with an inhibitor of lactate update (monocarboxylate transporter inhibitor AR-C155858; MCTi) or an inhibitor of FA synthesis (ACC inhibitor ND630; ACCi) before coculturing them with WT or TfamKO astrocytes (Extended Data Fig. 6e). Interestingly, while either MCTi or ACCi pretreatment of neurons reduced LD volume in cocultured astrocytes relative to astrocytes cocultured with vehicle treated neurons, such reductions were not different between WT and TfamAKO astrocytes (Extended Data Fig. 6f). In addition, there was no difference in the expression of key FA synthesis genes (Acaca and Fasn) in neurons cocultured with WT or TfamAKO astrocytes (Extended Data Fig. 6g). Together, these data support the notion that lactate-derived neuronal lipids contribute to astrocytic LDs, but the higher LD volume in TfamAKO astrocytes is unlikely a result of increased neuronal FA synthesis from lactate and the subsequent transport to astrocytes.
Impaired Lipid Clearance Induces Neuronal Oxidative Stress
Neurons maintain low FA degrading capacity and low FAO gene expression than astrocytes13. The impaired lipid-clearing capacity of TfamAKO astrocytes prompted us to ask whether the reduction in neuronal glucose metabolism represents an adaptive response to elevated intraneuronal lipid burden. Strikingly, we discovered a global upregulation of key FA degradation genes in neurons acutely isolated from 6-month TfamAKO brains, including the upstream regulator Ppara and genes controlling mitochondrial FA import and FAO (Fig. 5e,f). Conversely, these genes were significantly lower in acutely isolated TfamAKO astrocytes (Extended Data Fig. 6j), which suggests that the upregulation in FAO genes in TfamAKO hippocampus (Fig. 2c,d) was primarily contributed by neurons. To determine whether FAO activation in these neurons was a direct effect of impaired FA degradation in astrocytes, we cultured WT neurons with TfamAKO or WT astrocytes. Consistent with the in vivo results, neurons cultured with TfamAKO astrocytes had higher expression of FAO genes (Ppara, Cpt2, Acadvl) (Fig. 5g) and PPARα protein (Extended Data Fig. 6k) than those cultured with WT astrocytes.
The low FAO capacity of neuron is related to 1) its low antioxidative capacity and 2) the higher reactive oxygen species (ROS) production by FAO than glucose oxidation31. We thus reasoned that the activation of neuronal FAO may provoke oxidative stress, another early pathological feature of AD. Indeed, neurons cultured with TfamAKO astrocytes produced more ROS than those cultured with WT astrocytes (Fig. 5h). Evidence in TfamAKO brains also supported the link between FAO and ROS production in neurons, where oxidative stress makers including 4-hydroxynonenal (4-HNE, for lipid peroxidation) and 8-oxo-2’-deoxyguanosine (8-OHdG, for DNA oxidative damage) were prominent in neurons (NeuN+), but not astrocytes (GFAP+), in the hippocampus of 6-month TfamAKO mice (Fig. 5i,j; Extended Data Fig. 6l,m). ROS levels were lower in both cultured and acutely isolated TfamAKO astrocytes (6-month) (Extended Data Fig. 6n,o). Temporally, neither 4-HNE or 8-OHdG signal was detected in 3-month TfamAKO brains (Extended Data Fig. 6p,q), suggesting that neuronal oxidative stress occurs after brain lipid accumulation. Together, these data demonstrate that loss of astrocytic OxPhos induces an adaptive activation of FAO and oxidative stress in neurons, which could underlie the compromised synaptic function.
Astrocytic OxPhos Deficit Triggers Neuroinflammation
Neuroinflammation and microglial activation are common features of neurodegenerative diseases including AD32. Consistent with the upregulation of microglial activation genes and immune response genes and the enrichment of immune response pathways in TfamAKO hippocampi (Fig. 6a; Extended Data Fig. 7a), IBA-1 expression was substantially higher in the hippocampus and cortex of 6-month TfamAKO mice (Fig. 6b,c; Extended Data Fig. 7b). Further analyses by immunostaining and flow cytometry confirmed that TfamAKO increased the number of activated microglia (CD74+ and MHCII+CD11b+) (Fig. 6d,e). Temporally, microglial activation initially appeared in 3-month TfamAKO brains (Extended Data Fig. 7c,d), prior to any synaptic impairment.
Fig. 6. Astrocytes with OxPhos deficit promote microglial activation and neuroinflammation via IL-3.

(a) Heatmap showing DEGs (FDR-corrected p < 0.05) related to microglial activation and immune response in 6-month mouse hippocampi. (b) Representative images of hippocampal or cortical sections of 6-month mice stained for IBA-1. (c) IBA-1+ area % in hippocampal sections of 1.5-, 3-, and 6-month WT and TfamAKO mice. (d) MHCII-positive rate in microglia acutely isolated from 6-month mouse brains with anti-CD11b microbeads. (e) Representative images of hippocampal or cortical sections of 6-month mice stained for CD74. (f) TNFα and IL-1β levels in the cortex of 6-month mice. (g) Western blots of NFκB in 6-month mouse hippocampi (quantified in Extended Data Fig. 7e). (h and i) mRNA level of Tnfa, Il1b, Trem2, and Apoe in microglia acutely isolated from 6-month mouse brains. (j) IBA-1 intensity (normalized to cell count) in WT primary microglia (on coverslips in 6-well plates) cocultured with WT or TfamAKO astrocytes (in 6-well inserts) for 24 h. (k) Representative images of hippocampal sections of 6-month mice stained for IL-3 and GFAP. (l) IL-3+ area in the hippocampus of 3- and 6-month mice. (m) Il3ra mRNA levels in microglia acutely isolated from 6-month mouse brains. (n) Il3ra mRNA levels in WT microglia (in 24-well plates) cultured with WT or TfamAKO astrocytes (in 24-well inserts) for 24 h. (o) IBA-1 intensity (normalized to cell count) in WT microglia (on coverslips in 6-well plates) pretreated with vehicle or IL-3Rα neutralizing antibody before being cocultured with TfamAKO astrocytes (in 6-well inserts) for 24 h. n = 4 (c, h), 5 (j), 3 (d, l, n, o), or 8 (m) mice or independent samples; n = 6 (WT) or 5 (AKO) mice (f); n = 7 (Trem2-WT), 8 (Trem2-AKO), or 4 (Apoe) mice (i). Bar graphs and dot plots are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 100 μm (b, e, and k).
Concomitant with strong microglial activation, TfamAKO brains showed increased mRNA and protein levels of proinflammatory cytokines including TNFα and IL-1β, and the upstream regulator NFκB (Fig. 6f,g; Extended Data Fig. 7e,f). Analysis of acutely isolated microglia confirmed increased Tnfa and Il1b expression by these cells (Fig. 6h). Interestingly, Trem2 and Apoe, both being AD risk genes and upregulated in the microglia of human AD brains, were elevated in microglia isolated from TfamAKO mice (Fig. 6i). Using the Transwell coculture system, we found that primary WT microglia cocultured with TfamAKO astrocytes had higher activation rate than those cultured with WT astrocytes (Fig. 6j; Extended Data Fig. 7g–i), suggesting that microglia can be directly activated by OxPhos-deficient astrocytes.
IL-3 Mediates TfamAKO Astrocyte-Induced Microglia Activation
We next determined the mechanism by which TfamAKO astrocytes activate microglia. Interleukin-3 (IL-3) was recently revealed as a pivotal mediator of astrocyte-derived signal triggering microglial response via IL-3Rα33. In the hippocampus and cortex of TfamAKO mice, Il3 expression was strongly stimulated specifically in reactive astrocytes at 6-month, and to a lesser extent, at 3-month (Fig. 6k,l; Extended Data Fig. 7j–l). Correspondingly, Il3ra expression was dramatically elevated in microglia acutely isolated from TfamAKO mice and in primary microglia cultured with TfamAKO astrocytes, when compared to acute microglia from WT mice and microglia cultured with WT astrocytes, respectively (Fig. 6m,n). Moreover, IL-3Rα blockade by a neutralizing antibody abrogated microglial activation induced by TfamAKO astrocytes (Fig. 6o; Extended Data Fig. 7m). These results reveal that IL-3 is upregulated and released by OxPhos deficit-induced reactive astrocytes and subsequently promotes microglial activation by binding to microglial IL-3Rα.
Astrocytic OxPhos Deficit Compromises White Matter Integrity
Brain is the second most lipidated organ with the white matter areas being highly enriched with lipids (up to 80% of the dry weight). It is thus not surprising that myelinated axonal tracts within white matter are particularly vulnerable to perturbed lipid metabolism, and that white matter loss and demyelination are established features of AD brains34. As lipid homeostasis is severely disrupted in TfamAKO brains, we investigated its impact on myelin integrity. Magnetic resonance imaging (MRI) and diffusion weighted analysis suggested significantly decreased indices of white matter microstructural integrity, including fractional anisotropy, mean diffusivity, axial diffusivity, and radial diffusivity in TfamAKO brains (Fig. 7a; Extended Data Fig. 8a), but no volumetric difference was detected (Extended Data Fig. 8b). Connectometry analysis further revealed lower quantitative anisotropy in TfamAKO brains (0.0313 ± 0.0047) relative to WT brains (0.0456 ± 0.0027) across all fibers (Fig. 7b), suggesting reduced myelin compactness and restricted connectivity. Consistent with MRI outcomes, the density and thickness of white matter tracts were substantially reduced in TfamAKO versus WT brains, with the most affected corpus callosum exhibiting a ~40% reduction in fiber tract area (Fig. 7c; Extended Data Fig. 8c). Demyelination in the TfamAKO brain was further supported by a global decline in the expression of myelin-basic protein (MBP) (Fig. 7d; Extended Data Fig. 8d). None of these myelin changes was observed in 3-month TfamAKO mice (Extended Data Fig. 8e,f), thus ruling out the possibility of a developmental defect in myelination.
Fig. 7. Loss of astrocytic FA degradation suppresses lipid synthesis and induces demyelination.
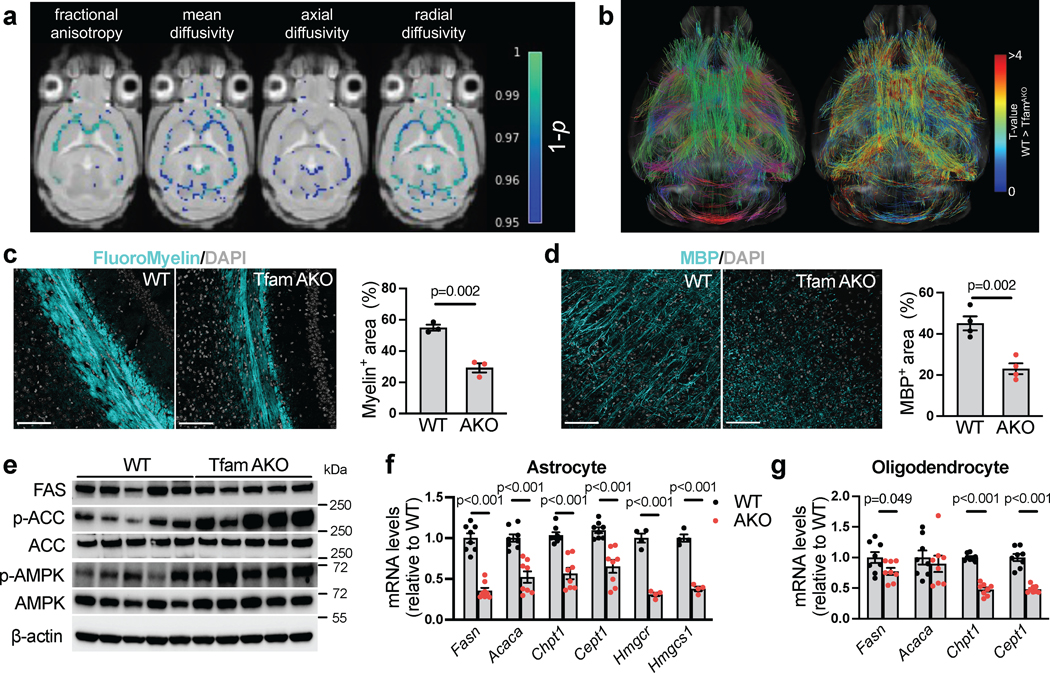
(a) Voxel-wise analyses of fractional anisotropy and mean-, axial-, and radial diffusivity. Colors reflect voxels with statistically significant TfamAKO < WT family-wise error corrected 1-p values. (b) Connectometry analysis comparing TfamAKO to WT brains using quantitative anisotropy. Fiber color indicates principal fiber direction (left) and local T-value (right). (c) Representative images and quantification of the corpus callosum area of brain sections of 6-month mice stained for FluoroMyelin. (d) Representative images and quantification of cortical area of 6-month mouse brains stained for MBP. (e) Western blots showing the levels of FAS, p-ACC, ACC, p-AMPK and AMPK in 6-month mouse hippocampi (quantified in Extended Data Fig. 8l, m, p). (f) mRNA levels of genes involved in FA synthesis (Fasn and Acaca), phospholipid synthesis (Chpt1 and Cept1), and cholesterol synthesis (Hmgcr and Hmgcs1) in astrocytes acutely isolated from 6-month mouse brains. (g) mRNA levels of genes involved in FA synthesis (Fasn and Acaca) and phospholipid synthesis (Chpt1 and Cept1) in oligodendrocytes acutely isolated from 6-month mouse brains. n = 3 (c), 4 (d), or 8 (g) mice; n = 8 (Fasn, Acaca, Chpt1 and Cept1) or 4 (Hmgcr and Hmgcs1) mice (f). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 100 μm (c and d).
Oligodendrocytes are the myelinating cells in the brain, and impairments in their differentiation (from oligodendrocyte progenitor cells) and survival are implicated in demyelination diseases. Surprisingly, there was no significant differences in the count of matured oligodendrocytes (CC1+Olig2+) in the hippocampus or white matter regions between WT and TfamAKO mice (Extended Data Fig. 8g,h), and we found no evidence of oligodendrocyte death or myelin debris in the corpus callosum of either strain (Extended Data Fig. 8i,j), Furthermore, a set of positive regulators of myelination were upregulated, whereas multiple negative regulators of myelination were downregulated, in TfamAKO brains (Extended Data Fig. 8k). These data suggest that the global demyelination in TfamAKO brains is unlikely to be caused by deficits in oligodendrocyte differentiation or survival, or a suppressed myelinating machinery. Instead, the myelination machinery is adaptively activated upon TfamAKO, as seen in an AD mouse model35.
FA- and Phospholipid Synthesis Is Inhibited in TfamAKO Brain
The lipid contents of myelin, in particular phospholipids, have a much higher turnover rate than oligodendrocytes themselves36. We reasoned that TfamAKO-induced disruption to lipid homeostasis may affect lipid synthesis and hence myelin turnover. As speculated, a decreased expression of FA synthase (FAS) and an increased inhibitory phosphorylation of acetyl-CoA carboxylase (p-ACCSer79) suggested suppressed de novo FA synthesis in 6-month TfamAKO brains (Fig. 7e; Extended Data Fig. 8l,m), which, like the demyelination, was not seen at 3-month (Extended Data Fig. 8n,o). Accordingly, AMP-activated protein kinase (AMPK), which suppresses FA synthesis by phosphorylating ACC at Ser79, was activated (p-AMPKThr172) in TfamAKO brains (Fig. 7e; Extended Data Fig. 8p).
Both astrocytic and oligodendrocyte lipid synthesis is required for myelination37,38. Consistent with tissue-levels changes, mRNA levels of key FA synthesis genes were decreased in astrocytes (Fasn and Acaca) and oligodendrocytes (Fasn) acutely isolated from TfamAKO brains (Fig. 7f,g). In addition, key genes catalyzing phospholipid synthesis from FA (Chpt1 and Cept1) were downregulated in both astrocytes and oligodendrocytes of TfamAKO brains (Fig. 7f,g), whereas genes controlling cholesterol synthesis (Hmgcr and Hmgcs1) were decreased in astrocytes only (Fig. 7f). Combined, these findings suggest that impaired FA degradation and lipid accumulation adaptively suppress FA-, cholesterol-, and phospholipid synthesis, which could lead to demyelination by limiting the lipids available to oligodendrocytes for myelin replenishment.
Impaired Astrocytic FAO and LD Accumulation in AD Mice
Given the strong AD-like phenotype of TfamAKO mice and the substantial activation of AD-related genes, disease associated astrocyte (DAA) genes and disease associated microglia (DAM) genes in the hippocampus of TfamAKO mice (Fig. 8a; Extended Data Fig. 9a,b), we explored whether a deficit in FA degradation could be involved in AD. In a mouse model of familial AD carrying five mutations to APP and PSEN1 (5xFAD), LDs were detected in the subiculum, but not other brain regions at 6-month (Fig. 8b), and Plin2 protein was also upregulated in the cortex (Fig. 8c). These lipid markers were not observed in 5xFAD mice at 4-month (Extended Data Fig. 9c and unshown negative LD detection). Of note, subiculum is also where Aβ deposition first appears in 5xFAD mice39. Importantly, a subset of LDs in 5xFAD brains were localized to astrocytes (GFAP+), but none of the detected LDs were localized to neurons (NeuN+) or microglia (IBA-1+) (Fig. 8b; Extended Data Fig. 9d,e). Moreover, compared to WT brains or LD-negative areas, LD-containing areas in 5xFAD brains showed substantially higher GFAP immunoreactivity (Fig. 8b), corroborating the link between lipid accumulation and astrocyte reactivity as observed in the TfamAKO mice.
Fig. 8. Impaired FA degradation, LD accumulation, and TfamAKO-induced transcriptional signatures are resembled in a mouse model of AD.

(a) Heatmap showing disease associated astrocyte (DAA)-related DEGs in 6-month WT and TfamAKO mouse hippocampi. (b) Representative images showing subiculum of 6-month WT and 5xFAD brain sections stained for LD and GFAP. (c) Western blots and quantification of Plin2 expression in 6-month WT and 5xFAD mouse cortices. (d) Relative OCR of hippocampal slices from 6-month mice on oleate-BSA. (e) Etomoxir-induced decreases in OCR of acute hippocampal slices from 6-month mice. (f) Quantification of LDs in cultured astrocytes from 4-month mouse brains. (g) FA-induced basal and maximal OCRs in cultured astrocytes from 4-month brains presented as OCR differences in the presence or absence of exogenous FA. (h) Etomoxir-induced decreases in OCR in cultured astrocytes from 4-month WT or 5xFAD mice. (i) Ppara mRNA levels in cultured astrocytes from 4-month WT or 5xFAD brains. (j and k) Quantifications (j) and representative images (k) of BODIPY-C12 localized to mitochondria in primary astrocytes from 4-month WT or 5xFAD mice. (l) Left: Venn diagram of unique and shared DEG counts in 6-month TfamAKO mouse hippocampi (relative to 6-month WT) and in 12-month 5xFAD mouse hippocampi (relative to 12-month WT) with upregulated DEG counts in red and downregulated DEG counts in blue; right: percentage of DEGs that are co-upregulated, co-downregulated, or showing different directions across the two models. (m) Correlation of overlapped DEGs described in panel j plotted by their signed log2(fold change) across the two models. (n) Hierarchical networks of GO BP terms (p < 0.05) enriched in the shared DEG across models using REVIGO with Resnik measurement and 0.5 distance. n = 4 (c, f), 5 (j), or 6 (i) mice; n = 11 slices (d); n = 23 (WT) or 25 (5xFAD) slices (e); n = 7 (g) or 8 (h) independent samples. Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 50 μm (b); 20 μm (k).
To investigate whether FA degradation is impaired in 5xFAD mouse brain, FA-induced respiration was measured with acute hippocampal slices. Compared to WT slices, 6-month 5xFAD slices showed lower respiration on exogenous FA and lower etomoxir-sensitive respiration (Fig. 8d,e). Interestingly, cultured astrocytes from 4-month-old 5xFAD mice already accumulated substantially more LDs and exhibited reduced FA catabolism than WT astrocytes without altering mitochondrial dynamics (Fig. 8f–h; Extended Data Fig. 9f,g). In parallel with impaired FA degradation, 4-month 5xFAD astrocytes also had lower expression of PPARα (Fig. 8i). Importantly, by tracing BODIPY-C12, we found markedly higher mitochondrial localization of the fluorescent FA in astrocytes from 4- or 6-month 5xFAD mice than those from age-matched WT mice, confirming a deficit in mitochondrial FA degradation (Fig. 8j,k; Extended Data Fig. 9h,i) that resembles TfamAKO astrocytes. These data collectively reveal that signs of lipid accumulation in 5xFAD brains are spatially coupled with Aβ pathology while temporally preceded by astrocytic mitochondrial dysfunction and astrocyte reactivity.
TfamAKO and 5xFAD Hippocampi Share Transcriptomic Signatures
To systematically assess to what extent TfamAKO-induced, AD-like features resemble that of 5xFAD, we compared their hippocampal transcriptomes for overlapped genes, pathways, and mechanisms by leveraging a 5xFAD RNA-sequencing dataset40. By comparing transcriptomic signatures of these two models at their advanced stages (6-month for TfamAKO and 12-month for 5xFAD, each compared to their respective age-matched controls), 1,127 overlapping DEGs were identified (Fig. 8l, hypergeometric test p<0.001). 94% of these common DEGs changed in the same direction (853 upregulated and 206 downregulated), and the fold changes of all common DEGs highly correlated between the two models (r = 0.64; p < 0.00001; Fig. 8m). Pathway enrichment analysis of these shared DEGs followed by removing redundant GO terms41,42 identified lipid metabolism, inflammatory response, cytokine production, and synaptic function as common mechanisms perturbed by both TfamAKO and 5xFAD transgenes (Fig. 8n). Together, outcomes of these transcriptomic level comparisons corroborate the phenotypic similarities between these models and underscore a mechanistic role of astrocytic mitochondria in AD-associated lipid dysregulation and neurodegeneration.
DISCUSSION
While the brain depends on glucose as their primary fuel source, lipids are structurally and functionally vital for brain function. As excess FFAs –the essential building blocks of lipids– are associated with lipotoxicity, their levels need to be finely regulated. Although peroxisomes metabolize very-long-chain FAs, the complete oxidation of the resultant medium-chain FAs and the more abundant long-chain FAs are performed by the mitochondria. Our findings suggest that in the brain, astrocytic OxPhos is required for the degradation of FAs and thereby the homeostasis of all lipid classes. Upon astrocytic OxPhos deficit, brain lipid profile is severely disrupted, and a profound program of pathological mechanisms are activated, including synaptic loss and dysfunction, reactive astrogliosis, microglial activation, oxidative stress, and demyelination, which collectively lead to neuroinflammation, neurodegeneration, and cognitive impairment.
Astrocytes are highly glycolytic, and OxPhos has been found largely dispensable for the proliferation and viability of these cells. Our study reveals that while astrocytic mitochondria are functionally less active (lower OxPhos activity) and bioenergetically less significant (less ATP production) than their neuronal counterparts, a modest level of OxPhos activity is indispensable for the clearance of lipid waste and the protection of the brain against lipotoxicity and degeneration. These findings are consistent with the inhibition of pyruvate dehydrogenase (PDH; generates acetyl-CoA from glucose-derived pyruvate) in astrocytes43 and highlight the metabolic specialty of astrocytic mitochondria in performing FAO. Notably, the effect of Tfam deletion on cellular metabolic function is tissue specific, which could be relevant to the distinct mitochondria abundance and basal OxPhos activity across cell types. In contrast to our findings in astrocytes, Tfam deletion in adipocytes reduces fat content while increases FA degradation and ROS production by elevating mitochondrial uncoupling44.
Astrocyte mtDNA depletion has been shown to induce reactive astrogliosis45, but the mechanism was unclear. Due to the suppression of PDH, mitochondrial metabolic profile in astrocytes is more affected by FA load than glucose levels. Indeed, our data show that FA overload is sufficient to trigger astrocyte reactivity, and astrocytes with OxPhos deficit are more susceptible to FA-induced metabolic stress and phenotypic transformation. Mechanistically, when FA load overwhelms astrocytic OxPhos capacity, acetyl-CoA is accumulated and exported from mitochondria and then promotes the acetylation and activation of STAT3, which ultimately leads to astrocyte reactivity and the release of proinflammatory factors. From a cell autonomous perspective, we reveal a mechanism of astrocyte reactivity and neuroinflammation mediated by lipid metabolic stress, which can be triggered by increased FA load and/or disrupted metabolism of acetyl-CoA (i.e., TCA and OxPhos).
Besides FA degradation, another mechanism adopted by the cell to mitigate lipotoxicity is the sequestration of FAs in LDs. Increased brain LD levels have been reported in aging and AD brains46,47. However, it remains to be determined as to the origin of these LDs and whether they serve as a protective mechanism or rather a driver of pathologies. Our study identified disrupted astrocytic FA degradation as a potential mechanism of LD production in degenerating brains. Further, our findings provide two aspects of new evidence supporting astrocytic LD formation as a first-line protective mechanism to avert brain lipotoxicity rather than an inducer of reactivity and neuroinflammation: 1) the accumulation of LD and TAG in TfamAKO brains (at 3-month) precedes the elevation of FFAs (at 6-month when intracellular LDs are at their capacity), and 2) increasing intracellular LDs –either by blocking TAG breakdown or by inhibiting FA entry into mitochondria– has no effect on astrocyte reactivity.
As a metabolic-inflammatory hub in the brain, astrocytes communicate with other cell types by releasing neurotrophic factors, cytokines, ROS, lipids, and miRNAs48,49. In the present study, we reveal cell non-autonomous mechanisms activated by astrocytic OxPhos deficit and lipid accumulation, which jointly promote neurodegeneration.
Elevated intraneuronal FFAs lead to impaired metabolic function and neurite outgrowth, and astrocyte-mediated lipid clearance is essential for neuronal health16. Here we mechanistically connect these events and discover that a deficit in astrocytic clearance of neuronal lipids maladaptively triggers neuronal FAO, which, due to the limited antioxidant capacity of neurons, is associated with oxidative stress and accumulation of peroxidized lipids. In contrast, oxidative stress is absent in the Tfam-deleted astrocytes, which is consistent with their greater antioxidative capacity and lower ROS production by the diminished FAO and collapsed ETC. These results strongly support the notion that suppressed FAO in neurons protects them from oxidative damage, whereas increased neuronal FAO is an important source of oxidative stress in the brain31.
As the resident innate immune cells in the brain, microglia are highly implicated in AD. While previous findings primarily focus on microglia-induced neurotoxic responses in disease-state astrocytes50, we show that astrocyte-initiated metabolic abnormalities could activate microglia, in part via the IL-3/IL-3Rα pathway. Notably, microglial activation and increased IL-3 expression in TfamAKO brains occur before FFA accumulation and synaptic and myelin abnormalities, suggesting that microglial activation, as an early event, is triggered by reactive astrocytes rather than a consequence of altered lipid signaling.
Saturated FAs released from reactive astrocytes induce oligodendrocyte death in vitro51. Interestingly, oligodendrocyte differentiation and survival are not affected by TfamAKO-induced reactive astrogliosis and FFA accumulation in vivo. Instead, upon OxPhos deficit and lipid accumulation, AMPK, which can be directly or indirectly activated by FFAs52,53, is activated and inhibits FA synthesis to mitigate the elevation of FFAs and lipotoxicity. Since phospholipids in myelin have a high turnover rate, insufficient supply of newly synthesized FAs and phospholipids by astrocytes and oligodendrocytes could compromise the structural and functional integrity of myelin. Given that the synthesis and secretion of lipids by astrocytes are also critical for synaptogenesis and synaptic plasticity54, suppressed lipid synthesis could also contribute to the synaptic loss in TfamAKO brains.
Brain lipid dyshomeostasis is an early and persistent hallmark of AD, and a variety of AD risk factors are involved in lipid trafficking and metabolism14. However, despite abundant correlational evidence, the mechanisms by which disrupted lipid metabolism is triggered and how it subsequently contributes to AD pathologies, remain elusive. The present study provides evidence from two directions that links disrupted FA degradation with AD-related neurodegenerative mechanisms.
First, the phenotypic and transcriptional signatures of the TfamAKO mice resemble key AD features in the absence of amyloid or tau pathologies. Metabolically, astrocyte OxPhos deficit results in a shift towards glycolysis, which agrees with the findings in postmortem AD brains that proteins involved in glucose metabolism and glial activation are enriched55. Beyond the accumulation of LDs, multiple lipid species show similar changes in TfamAKO mice as in human AD brains. For example, the levels of ceramide, CEs, and selected glycerolipids are higher in AD versus control brains (reviewed in14). Additionally, oxidative stress, manifested as lipid peroxidation, is also an early pathological marker of AD56.
In parallel, findings from the 5xFAD mice suggest that disrupted astrocytic FA degradation emerges early in the brain shortly after amyloid aggregation and is followed by LD appearance in astrocytes. Spatially, the subiculum, where LDs first appear in 5xFAD brain, is also where Aβ deposition is initially detected. The deficit in FA degradation by 5xFAD astrocytes also agrees with previous findings in astrocytes derived from iPSCs of familial AD patients carrying PSEN1 mutations57. These lines of evidence indicates that amyloidosis may play a role in initiating astrocytic metabolic deficits, which warrants further investigation. Intriguingly, we previously reported a negative effect of APOE4 on neuron-to-astrocyte FA transport and astrocytic FA degradation16. Based on these findings, we propose lipid metabolism as a central mechanism implicated in AD where the amyloid cascade58, the mitochondrial cascade59 and the ApoE cascade60 converge at astrocytic FA catabolism. Broadly, altered lipid metabolism and lipid accumulation also pertain to Parkinson’s disease61, amyotrophic lateral sclerosis62, Huntington’s disease63, and multiple sclerosis64. Since mitochondrial dysfunction is also implicated in these diseases, it is tempting to determine whether defects in astrocytic FAO and OxPhos are involved in, and can be therapeutically targeted for, these lipid-involving neurodegenerative disorders. Specifically, our findings suggest that strategies that sustain or restore an equilibrium between FA load and mitochondrial catabolic capacity hold therapeutic promises to contain the detrimental reactive astrogliosis.
METHODS
Animals
All animal procedures were approved by the University of Arizona Institutional Animal Care and use Committee (IACUC) and in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Mice were housed in a temperature and humidity-controlled room with a 12-hour light and 12-hour dark cycle with ad libitum access to water and standard laboratory diet. B6.Cg-Tg(Gfap-cre)77.6Mvs/2J mice (Jackson Lab, 024098) were bred with B6.Cg-Tfamtm1.1Ncdl/J mice (Jackson Lab, 026123) to generate GFAP-Cre:Tfamflox/flox (TfamAKO) mice. TfamAKO mice were born at the expected Mendelian ratio. 1.5-, 3-, and 6-month-old, male and female mice were used for experiments. The 5xFAD mouse strain on a congenic C57BL/6J background (B6.Cg-Tg(APPSwFlLon, PSEN1*M146L*L286V)6799Vas/Mmjax) was obtained from the Jackson Lab (008730). Hemizygous, 4- and 6-month-old, male and female transgenic mice and their non-transgenic littermates were used.
Novel Object Recognition Test
Mice were transferred to the behavior analysis room 1 week prior to testing for habituation to the environment. Habituation session was performed on Day 1 when mice were placed into the box measuring 30 cm x 30 cm (L x W) without objects for 5 min. On the training day (Day 2), mice were placed in the box for 5 min with two identical objects. On the testing day (Day 3), one of the training objects was replaced with a novel object and mice were allowed to explore the two objects for 5 min. Mice were scored by interacting with an object when its nose was in contact with the object or was directed toward the object within 2 cm. Discrimination index of novel object was calculated as the ratio of the interaction time with novel object to total interaction time with both objects.
Open Field Test
Open field test was conducted using a 30 cm × 30 cm x 40 cm (L x W x H) box. The area within 7.5 cm from the wall was considered as peripheral, the remaining area was considered as the central area. Each mouse was allowed to move freely and explore the environment for 5 min with its behavior videotaped and its trace recorded. The time spent in the central area and the ratio between ambulatory distance in central area and total ambulatory distance was analyzed with AnyMaze 7.0.
Locomotion Test
Locomotor functions of mice were measured using the Catwalk XT system (Noldus, Netherlands). Mice were allowed 1 h of acclimation to the system before being placed in the corridor of the Catwalk apparatus and allowed an interrupted crossing of the recording field of the runway repeatedly at a 60% variation threshold until three consecutive uninterrupted runs were recorded. Run duration longer than 5 seconds and shorter than 0.5 seconds were automatically excluded. After footprint classification in the CatWalk XT software, gait analysis was performed for step cycle, speed, and swing.
Isolation and culture of primary adult astrocytes
Primary adult astrocytes were isolated from mouse brains as described previously16. Fresh brain tissues (excluding cerebellum and brainstem) were processed with the gentleMACS Dissociator with Heaters. After dissociation, cell debris and red blood cell were removed. After 2 weeks of culture in DMEM/F12 with 10% FBS and 0.2% penicillin/streptomycin, astrocytes were shaken on an orbital shaker to remove microglia and oligodendrocytes. The purity of isolated astrocytes was > 95% as determined by GFAP positive rate. For treatment studies, astrocytes seeded in 6-well plate (1×106/well) or 12-well plate (300,000/well) were pretreated with 10 μM etomoxir, 50 μM S3I-201, 50 μM BMS-303141, or 10 μM atglistatin for 24 h and were then incubated with 150 μM oleate-BSA or BSA (vehicle) for another 24 h before sample collection.
Isolation and culture of primary embryonic hippocampal neurons
Primary embryonic hippocampal neurons were prepared as described16. Hippocampi of E17 fetuses were dissociated followed by repeated passages through fire-polished constricted Pasteur pipettes. Experiments were performed after 7 days of culture. The purity of neurons was > 94% while GFAP-positive cells are < 1% as determined by NeuN and GFAP immunostaining.
Isolation and culture of primary microglia
Primary microglia were isolated from the cerebral cortices and hippocampi of postnatal day 4–7 mice. Dissociated cells were filtered with 40 μm cell strainer and grown in DMEM/F12 with 10% FBS and 0.2% penicillin/streptomycin as mixed glial culture. After 8–10 days, microglia were collected from the mixed glia by shaking and tapping the flask and were confirmed with >95% purity.
Neuron-astrocyte coculture
As described previously16, primary astrocytes were seeded in poly-D-lysine coated 6-well or 24-well 0.4 μm cell culture inserts at a density of 1×106 or 50,000 cells/well, respectively, for 24 h. Primary neurons were seeded in poly-D-lysine coated 6-well plates or 24-well Seahorse plates at a density of 1×106 or 50,000 cells/well respectively. Inserts with astrocytes were then placed into the Seahorse plate with neuronal cultures in neuronal medium. Co-cultures were maintained for 7 days, after which neurons were subject to Mito Stress Test. For neuronal LD and MAP-2 immunostaining experiment, astrocytes were seeded in 6-well 0.4 μm cell culture inserts for 24 h and primary neurons were seeded in 22 mm glass coverslips placed in 6-well plate and cultured for 12 h. The inserts were placed into the 6-well plate with neuronal culture coverslips. After 7 days, the coverslips with neurons were immunostained for imaging analyses. For MCTi (AR-C155858) and ACCi (ND-630) treatment experiments, primary neurons were seeded in 6-well plates at a density of 1×106/well. Primary astrocytes were seeded in 22 mm glass coverslips at a density of 20,000 cells/coverslip. After pretreatment with MCTi (200 nM) or ACCi (20 nM) for 12 h, coverslips with astrocyte cultures (with paraffin dots underneath to support the coverslips) were placed into the 6-well plates with neuron cultures. After 24 h of coculture, coverslips with astrocytes were immunostained for LDs.
Microglia-astrocyte coculture
Primary astrocytes were seeded in 6-well or 24-well 0.4 μm inserts at a density of 1×106 or 200,000 cells/well for 48 h. Primary microglia were plated in coverslip (for imaging analyses) or 24-well plate (for RT-qPCR analyses) at a density of 30,000 cells/coverslip or 200,000 cells/well, respectively. Microglia were allowed to adhere overnight, and astrocyte inserts were then placed into the plates with microglia for 24 h or 48 h.
Acute Cell isolation
Acute isolation of major brain cell types, including neuron, astrocytes, microglia, and oligodendrocytes were conducted using Adult Brain Dissociation Kit (Miltenyi Biotech) and specific magnetic microbeads as described previously16,49. Different cell types were isolated with astrocyte-specific anti-ACSA-2 microbeads, microglia-specific anti-CD11b microbeads, oligodendrocyte-specific anti-O4 microbeads, and the Adult Neuron Isolation Kit.
Flow cytometry of astrocytic and microglial markers
Brain cell suspensions were stained with GLAST (ACSA-1)-APC antibody for analysis of astrocytes. For microglia analysis, dissociated cells were incubated with anti-CD11b microbeads, and isolated microglia were stained with anti-MHCII-PE-Vio770. Flow cytometry was conducted using MACSQuant Analyzer 10 Flow Cytometer and data were analyzed by FlowLogic 8.6. Gating strategies are exemplified in Supplementary Figure 1.
Cell Mito Stress Test
Seahorse XF Mito Stress Test was performed as described16. Primary neurons or astrocytes were cultured in Seahorse 24- or 96-well plates. Baseline OCR (oxygen consumption rate) and ECAR (extracellular acidification rate) were measured prior to sequential injection of oligomycin A at 4 μM, FCCP at 1 μM, and rotenone / antimycin A at 1 μM / 1 μM. OCR and ECAR were normalized to protein concentrations. Basal respiration was calculated as the differences between OCRs before oligomycin injection and after rotenone/antimycin A. Maximal respiration was calculated as the difference between OCRs after FCCP injection and after rotenone/antimycin A injection. Experiments from cells isolated from at least three animals were performed in these assays. For each animal or cell batch, there were 6–8 wells/group. Data were analyzed with Seahorse Wave 2.4.2.
Mitochondrial fatty acid metabolic assay
Mitochondrial FA metabolic test was conducted using the Seahorse XF FA Oxidation Test Kit. Astrocytes were incubated for 12 h in substrate-limited medium (DMEM base medium supplemented with 0.5 mM glucose, 1.0 mM GlutaMAX, 0.5 mM carnitine, and 1% FBS, pH 7.4). On the assay day, the medium was replaced with FAO assay buffer and incubated at 37 °C in a CO2-free incubator for 1 h. BSA or palmitate-BSA were added to each well prior to the initiation of XF assay. To determine CPT1-mediated FA catabolism, astrocytes were incubated for 1 h in unbuffered DMEM (DMEM base medium supplemented with 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, 2 mM glutamine; pH 7.4) before Seahorse XF assay using etomoxir (10 μM) in port A, followed by sequential injections of oligomycin, FCCP, and rotenone/antimycin A.
Ex vivo brain metabolic assessment
Metabolic assessment of acute brain slices was performed as described65. 250 μm coronal acute brain sections were prepared using a vibratome. Slices containing hippocampal regions were transferred to a biopsy chamber and 1-mm stainless steel biopsy punches were used to excise punches. Punches were then ejected into the XF Islet Capture Microplate. Baseline OCR and ECAR were measured prior to the injection of oleate-BSA (150 μM), followed by sequential injections of oligomycin (20 μg/mL), FCCP (10 μM), and antimycin A (20 μM). To determine CPT1-mediated FA catabolism ex vivo, slice punches were incubated in CO2-free incubator for 1 h. After adding 150 μM oleate-BSA, plate was then analyzed by Seahorse XFe24 Analyzer. Baseline OCRs were recorded prior to the injection of etomoxir (200 μM) in Port A, followed by sequential injections of oligomycin, FCCP, and antimycin A.
Long-term potentiation (LTP)
LTP recordings with hippocampal slices were performed as previously described66. Horizontal 350-μm-thick brain slices were prepared using a vibratome and transferred to a recovery chamber with oxygenated aCSF. After 1.5 h of recovery, each brain slice was placed in MED64 (Alpha MED Sciences) probe. The field excitatory postsynaptic potentials (fEPSPs) in CA1 region were recorded by stimulating the CA3 region of the hippocampus. Baseline responses were first recorded until the variation was <5% for at least 10 min. LTP was then induced by Theta Burst Stimulation (four pulses with 200 μs width and 10000 Hz frequency) which was given at the stimulation intensity that was adjusted to elicit 30% of the maximal response. The amplitudes of fEPSPs were normalized and presented as a percentage of the average values of the last 5 min of baseline period.
Immunoblotting and quantification
Proteins were extract from brain homogenates or cultured cells with T-Per or M-Per reagents, respectively. Equal amounts of proteins were loaded and separated on a 4%−15% Criterion Precast Midi Protein Gel (Bio-Rad) and then transferred to a 0.22 mm pore size PVDF membrane. After blocking with 5% non-fat milk in TBST and washing for 3 times, the membrane was immunoblotted with the primary antibody overnight at 4 °C and with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (Vector Laboratories). The bands were visualized by Pierce SuperSignal Substrate or Femto Maximum Sensitivity Substrate, and the images were captured by ChemiDoc Imaging System. Band intensities were quantified using Image Lab 6.0.1.
Immunostaining
For brain immunostaining, mice were deeply anesthetized with isoflurane and intracardially perfused with PBS for 5 min and subsequently with 4 % PFA in phosphate-buffered saline for 5 min. Fixed brains were transferred into 30% sucrose in PBS overnight at 4 °C. Brain embedded in O.C.T. Tissue Tek (Sakura) were cut into 10 μm sections using a cryostat. Sections were rinsed with PBS and blocked with 5% normal goat serum and 0.2% Triton X-100 in PBS for 1 h. For cultured cells on coverslips, they were fixed with 4% PFA for 15 min and rinsed and incubated in blocking buffer for 1 h. Sections or coverslips were then incubated with primary antibody overnight and with fluorescent secondary antibodies for 1 h. After 3 times of washing, sections or coverslips were co-stained with other primary antibodies or mounted with mounting medium containing DAPI. Images were then captured with Zeiss Laser-Scanning Microscopy (LSM) 880 with Airyscan detector or Cytation 5 Cell Imaging Multi-Mode Reader. Fluorescent intensities were analyzed by BioTek Gen5.
BrdU incorporation assay
5-month mice were injected with BrdU (100 mg/kg) on day 1, 7, 14, 21, and 27 and tissues were collected on day 28. After blocking, frozen sections were co-stained for BrdU and NeuN before image capturing. BrdU+NeuN+ cells in the dentate gyrus were counted.
Morphological analysis of astrocytes
Sholl analysis was used to assess the complexity of astrocyte. Single astrocyte images were transformed to 8-bit binary images, which were analyzed by the Image J Sholl analysis plugin. Intersects were defined as points where the astrocytic processes crossed a concentric ring. Approximately 30–40 cells from 3 mice per group were analyzed and data were present as sum of intersects, ramification index, and ending radius.
Primary and secondary antibodies for Immunoblotting and immunostaining
Immunoblotting or immunostaining was performed with: 4-Hydroxynonenal Monoclonal (12F7) antibody (Invitrogen, MA527570, 1:50), acetyl-CoA carboxylase antibody (Cell Signaling Technology, 3662, 1:1000), AMPKα (D63G4) antibody (Cell Signaling Technology, 5832, 1:1000), 8-OHdG monoclonal antibody (N45.1) (JaICA, MOG-100P, 10 μg/mL), APC (Ab-7) antibody (Milipore, OP80, 1:50), CD74 antibody (Novus Biologicals, NBP229465, 1:50), dMBP antibody (Millipore, AB5864, 1:2000), Glial fibrillary acidic protein antibody (Millipore, MAB360, 1:500), NeuN antibody (Sigma, MAB377, 1:100), Olig2 antibody (Millipore, MABN50, 1:300), PPAR alpha (phospho S12) antibody (Abcam, ab3484, 1:1000), Tfam (mtTFA) antibody (Abcam, ab131607, 1:1000), β-Actin (8H10D10) antibody (Cell Signaling Technology, 3700, 1:1000), cPLA2 antibody (Cell Signaling Technology, 2832, 1:1000), doublecortin antibody (Cell Signaling Technology, 4604s, 1:1000), fatty acid synthase (C20G5) antibody (Cell Signaling Technology, 3180, 1:1000), GLAST (ACSA-1) antibody (Miltenyi Biotec, 130–123-641, 1:50), Hexokinase II (C64G5) antibody (Cell Signaling Technology, 2867, 1:1000), Iba1/AIF-1 (E4O4W) antibody (Cell Signaling Technology, 17198, 1:100), MHC Class II Antibody (Miltenyi Biotec, 130–112-232, 1:50), MAP2 antibody (Sigma, M4403, 1:1000), Myelin Basic Protein (D8X4Q) antibody (Cell Signaling Technology, 78896, 1:50), NF-κB antibody (Cell Signaling Technology, 8242, 1:1000), perilipin-2/ADFP antibody (Novus Biologicals, NB110–40877, 1:200), PFKFB3 (D7H4Q) antibody (Cell Signaling Technology, 13123s, 1:1000), Phospho-AMPKα (Thr172) (40H9) antibody (Cell Signaling Technology, 2535, 1:1000), Phospho-CoA Carboxylase (Ser79) antibody (Cell Signaling Technology, 3661, 1:1000), PSD95 (D74D3) antibody (Cell Signaling Technology, 3409, 1:1000), IL-3 antibody (Biolegend, 503902. 1:5), SNAP25 (D7B4) antibody (Cell Signaling Technology, 5308, 1:1000), Acetyl-STAT3 (Lys685) antibody (Cell Signaling Technology, 2523, 1:500), Phospho-STAT3 (Tyr705) antibody (Cell Signaling Technology, 9145, 1:1000), STAT3 antibody (Cell Signaling Technology, 12640, 1:1000), S100β antibody (Abcam, ab215989, 1:100), Horse anti-mouse secondary antibody (Vector Laboratories, PI-2000, 1:5000), Goat anti-rabbit secondary antibody (Vector Laboratories, PI-1000, 1:5000), Alexa Fluor 488 goat anti-mouse secondary antibody (Invitrogen, A11001, 1:500), Alexa Fluor 488 goat anti-rabbit secondary antibody (Invitrogen, A11008, 1:500), Alexa Fluor 555 goat anti-mouse secondary antibody (Invitrogen, A21422, 1:500), Alexa Fluor 555 goat anti-rabbit secondary antibody (Invitrogen, A21429, 1:500).
Cellular oxidative stress
Cells were incubated with 5 μM CellROX Green Reagent for 30 min and lysed in 400 μL M-Per buffer and centrifuged for 10 min at 4 °C. Fluorescence intensity of the supernatants was measured using Cytation 5 at Ex/Em = 485/520 nm and normalized to protein concentrations.
Lipid droplets staining and quantification
Lipid droplets in cells or brain sections were detected with HCS LipidTOX Red Neutral Lipid Stain or Lipid Droplet Screen-Certified Kit. For cells, Z stack images were captured to generate three-dimensional volumetric data. The number and volume of intracellular lipid droplets were quantified using the surface module of Imaris 9.8.0. For brain sections, they were mounted with the probe before image capturing. Quantification of positive areas was performed with Imaris 9.8.0.
TUNEL staining
TUNEL staining was performed using TUNEL Assay Apoptosis Detection Kit . Brain sections permeabilized with Triton X-100 were incubated with equilibration buffer, followed by incubation with TUNEL Reaction Buffer at 37 °C. Sections were then co-stained with primary antibodies of interest or mounted with mounting medium with DAPI for imaging.
Myelin staining
Myelin staining was performed using the FluoroMyelin Green fluorescent stain. Brain section were incubated with 1 x FluoroMyelin stain for 20 min and washed with PBS for 3 × 10 min before image capturing.
Fluorescent fatty acid tracing
Coverslips with primary astrocytes were incubated with 20 nM BODIPY FL C12 probe, followed by incubation with 100 nM MitoTracker Green FM. After 3 × 5 min washes with PBS, astrocytes were fixed in 4% PFA for imaging. The quantification of MitoTracker+ area and Red C12 overlay with MitoTracker were performed with Imaris 9.8.0.
IL-6 and TNFα levels
IL-6 and TNFα levels in cell culture medium were determined by V-PLEX Proinflammatory Panel 1 Mouse Kit (MSD). Plates were analyzed with MSD instrument.
Lactate levels
Lactate levels in brain tissue homogenates were measured by the Lactate Assay Kit (Sigma). After removing lactate dehydrogenase, the lysate was incubated with reaction mix for 30 min in the dark and the fluorescence was determined at Ex/Em = 535/587 nm and normalized to protein concentrations.
Total FFA levels
Total FFA levels in the cortex were determined using Free Fatty Acid Assay Kit (Abcam) with Ex/Em = 535/587 nm and normalized to protein concentrations.
Cholesterol levels
Total cholesterol, cholesteryl ester, and free cholesterol levels were measured by the Cholesterol Quantitation Kit (Sigma). Cholesterol levels were fluorescently measured at Ex/Em = 535/587 nm with or without cholesterol esterase and adjusted to protein concentrations. Cholesterol ester levels were calculated as the difference of total and free cholesterol.
Total triacylglycerol (TAG) levels
Total TAG levels were determined using Triglyceride Colorimetric Assay Kit (Cayman) by measuring 540 nm absorbance and adjusted to protein concentrations.
Acetyl-CoA levels
Acetyl-CoA levels in the cortex were determined using the Acetyl-CoA Assay Kit (Sigma) with Ex/Em = 535/587 and adjusted to protein concentrations.
Prostaglandin E2 levels
PGE2 levels in cell culture medium or brain tissues were measured by the Prostaglandin E2 ELISA Kit (Cayman Chemical). Culture medium or tissue homogenates were incubated with PGE2 AChE Tracer and PGE2 antibody and then with Ellman’s reagent. 410 nm absorbance was measured and normalized to protein concentrations.
Cell viability Assay
The viability of cultured astrocytes was assessed with the Cell Counting Kit-8 by measuring absorbance at 450 nm.
Thin Layer Chromatography (TLC)
Brain homogenates were extracted with lipid extraction solvent [chloroform: heptane: methanol (4:3:2 v/v)]. After centrifugation at 2,000 g for 5 min, the organic phase was collected and dried with N2 gas. Dried extracted lipids were redissolved in chloroform: methanol (2:1 v/v). TLC was performed on Silica gel 60 plates (Merck Millipore). Extracted lipids and the lipid standard mix [equal weights (2 μg/μL) of phospholipid (PL), monoacylglycerol (MAG), cholesterol, diacylglycerol (DAG), FFA, and TAG] were developed in heptane: diethyl ether: acetic acid (55:45:1 v/v). After separation, TLC plates were dried and soaked in copper sulfate-phosphoric acid solution [10% CuSO4·5H2O (w/v) and 8% H3PO4 (v/v) in water]. For visualization of cholesterol and other lipid classes, plates were heated for 10 min at 60 °C and an additional 5 min at 150 °C. Images of TLC plates were captured and band intensities were quantified by qTLC (www.qtlc.app).
Fluorescent BODIPY-C12 pulse-chase assay
Assay was performed as described25 with minor modifications. Astrocytes were incubated with 2 μM BODIPY-C12 for 16 h and then in substrate-limited medium for 24 h. Astrocytes were trypsinized and centrifuged, and the pellets were homogenized in 300 μL chloroform. After centrifuge at 15,000 x g for 5 min, the lower organic phase was collected for TLC separation of lipids and fluorescent lipids were visualized and quantified.
Metabolite and lipid extraction
For aqueous metabolites, pulverized cortices were mixed with 8:2 (v:v) methanol:H2O and incubated on dry ice for 30 min. The mixture was homogenized at 4 °C and centrifuged at 18,400 x g for 5 min. Soluble extracts were collected and dried using the Speedvac (Eppendorf Vacufuge) at 30 °C.
For lipid extraction, cortices were mixed with MeOH:PBS (1:3, v:v, 4 °C) and internal standard solution (100 uM PC (17:0/17:0) and 100 uM PG (17:0/17:0) in MeOH) . After adding 3 mL MTBE (Methyl tert-butyl ether) into each sample, the tube was vortexed for 30 s and incubated at −20 °C for 30 min and then sonicated in ice bath for 10 min. After centrifugation (840 x g, 10 min), 2 mL upper MTBE layer was collected and dried. Samples were reconstituted in 100 μL 1:1 CHCl3:MeOH. 80 μL of each sample was transferred to a LC-MS vial, and the remaining 20 μL was pooled every 10 study samples to create a quality control (QC) sample to ensure consistent LC-MS/MS outputs.
LC-MS/MS analysis of acylcarnitines
LC-MS/MS analysis of acylcarnitines was performed on an Agilent 1290 LC-6490 QQQ-MS system. 5 μL sample was injected for analysis using positive ionization mode. Chromatographic separation was performed using a Waters XSelect HSS T3 column with flow rate of 0.3 mL/min. The mobile phase was composed of solvent A (100% H2O with 0.1% formic acid) and B (100% ACN with 0.1% formic acid). After the initial 0.5 min isocratic elution with 100% A, the percentage of A gradually decreased to 5% at t=10 min and maintained at 5% for 5 min (t=15 min). The extracted MRM peaks were integrated using Agilent Masshunter software.
LC-MS/MS targeted lipidomics
LC-MS/MS targeted lipidomics were performed as described previously67. Each sample was run for both positive and negative ion mode with injection volume of 4 μL and 6 μL, respectively. Both modes used reverse phase chromatography with a Waters XSelect HSS T3 column. Flow rate was set at 0.3 mL/min. The mobile phase was composed of solvent A (10 mM ammonium acetate in 60% H2O/40% CAN) and B (10 mM ammonium acetate in 90% IPA/10% CAN). After isocratic elution with 50% B for 3 min, the percentage of B was gradually increased to 100% in 12 min. Following 10 min of continued 100% B, at 25 min time point, the percent of B was gradually decreased to 50% to prepare for the next sample. MS experiment parameters were integrated using Agilent’s MassHunter Quantitative Analysis.
GC-MS-based metabolic flux analysis
Metabolic flux analysis was performed as described68 with minor modifications. Astrocytes were incubated with 150 μM U-13C18-labled oleate-BSA in substrate-limited medium for 24 h. Cells were collected with 1.2 mL 8:2 (v:v) methanol:H2O and centrifuged 18,400 x g at 4 °C for 10 min, and the supernatant were dried using SpeedVac. Dried samples were incubated with a solution of 40 μL of 20 mg/mL O-methylhydroxylamine hydrochloride in pyridine at 60 °C for 90 min. Next, 70 μL of N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) was added followed by 30 min incubation at 60 °C. An Agilent 8860 GC-5977 MSD system was used for spectral acquisition. Chromatographic separation was obtained by a Zorbax DB5-MS + 10 m Duragard Capillary Column. The temperature of the column was held at 60 °C for 1 min before being increased at a rate of 10 °C / min until 325 °C. The column was then kept at this temperature for 10 min. Mass spectral signals were recorded in full scan mode utilizing electron ionization (EI, 70 eV), and the mass range was 50–600 Da. Agilent MassHunter software was applied to process the raw GC-MS data. Mass isotopomer distributions (MIDs) were calculated by integrating metabolite ion fragments, and the IsoCor software was used for the correction of natural abundance of isotopes and the calculation of enrichment values.
mtDNA copy number
Total cortical DNA was isolated with PureLink Genomic DNA Mini Kit and analyzed by qPCR. mtDNA/nDNA ratios were calculated as the relative ratios of mt-nd1 (mtDNA) to Hk2 (nDNA) using primers: mt-nd1 forward: 5′-CTAGCAGAAACAAACCGGGC-3′; mt-nd1 reverse: 5′-CCGGCTGCGTATTCTACGTT-3′; Hk2 forward: 5′-GCCAGCCTCTCCTGATTTTAGTGT-3′; and Hk2 reverse: 5′-GGGAACACAAAAGACCTCTTCTGG-3′.
RNA extraction and RT-qPCR
Total RNA was isolated from acutely isolated cells, cultured cells, or brain tissues using PureLink RNA Mini Kit. Extracted RNAs were reverse transcribed to cDNA using the SuperScript IV VILO Master Mix with ezDNase Enzyme kit. TaqMan probes (Mm00607939_s1 for Actb, Mm00434273_m1 for Il3ra, and Mm00447485_m1 for Tfam) were used for qPCR using a QuantStudio 6 Real-Time PCR System. mRNA levels of other genes were assessed by SYBR Green real-time PCR assays, with the primer sequences listed in Supplementary Table 1. Relative gene expression to the reference group was calculated by the comparative Ct (ΔΔCt) method, with Ct denoting threshold cycle and ΔCt being calculated as the difference in average Ct of the target genes and Actb.
RNA Sequencing
RNA sequencing was performed using NovaSeq 6000 (3M reads per sample). Transcripts were mapped to mouse cDNA (ensembl release 95) using Salmon. Tximport V1.15.0 was used to generate a counts table and DESeq2 V1.26.0 was used to calculate normalized read counts. Fold changes were determined by dividing the average normalized read counts of one group over the control group. P-values were corrected using the Benjamini-Hochberg False Discovery Rate (FDR), and genes with adjusted p-values < 0.05 were identified as differentially expressed genes (DEGs).
Principal component analysis (PCA)
Principal components were calculated based on VST transformed expression of top 500 variant genes across all samples in RNA-Sequencing. The PCA plot was prepared using R package: factoextra. To show sample clustering, heatmap of the same top variant genes as in PCA analysis was presented using R:pheatmap package.
Gene set enrichment analysis (GSEA)
The ranked gene list was used to identify pathways that were significantly enriched or underrepresented using R:clusterprofile package.
Gene ontology (GO) enrichment and REVIGO analyses
Overlap analysis of DEG lists and shared pathways between TfamAKO and 5xFAD models was performed using hypergeometric test. GO term enrichment analysis was performed using Enrichr with DEGs shared between 6-month-old TfamAKO (versus 6-month-old WT) and 12-month 5xFAD (versus 12-month-old WT) hippocampal RNAseq datasets. GO-BP enrichment was further refined to exclude redundant and similar GO-BP terms using Python package GOATOOLS. Significant GO terms were visualized based on their semantic similarity by REVIGO41, using Resnik as the clustering algorithm and 0.5 as allowed similarity measure.
Magnetic resonance imaging
Imaging data acquisition
Magnetic resonance imaging (MRI) data were acquired for mouse on a Bruker Biospec 70/20 7.0T scanner. High resolution structural images were acquired with T2-weighted rapid acquisition with relaxation enhancement sequence (TE: 30 ms; TR: 1800 ms; flip angle 180°; RARE factor: 8; number of averages: 2; FOV: 2.4 × 1.44 × 0.96; acquisition matrix 320 × 192 × 120; reconstructed voxel size 75 μm; total acquisition time: 3 h 4 min). Diffusion weighted imaging included separate 2D EPI diffusion weighted sequences (TE: 45 ms; TR: 1000 ms; flip angle: 90°; FOV: 2.56 × 1.28 × 1.28; acquisition matrix 128 × 64 × 64; slice thickness 12.8 μm; reconstructed voxel size: 200 μm, 8 b = 0 images) for b values = 1000 s/mm2, 2000 s/mm2, and 3000 s/mm2. Data were converted to NIFTI format using brkraw (v. 0.3.7).
Anatomical pre-processing and volumetric analyses
Volumetric structural analyses were conducted using ANTs 2.3.5. Preprocessing steps included denoising, warping of an initial mask using the DSURQE template brain and mask. Final image intensities were truncated and rescaled to the image range of the DSURQE template image. Using these minimally preprocessed images, a study-specific template was created. Relative log-Jacobian maps were used for deformation-based volumetric analyses. Using the relative Jacobians controls for differences in brain size and therefore was not included as a covariate. Voxelwise analyses were conducted using a two-sample t-test in FSL’s randomize69 on these images. Groupwise differences were assessed across atlas regions of interest using a total brain volume adjusted ANCOVA with R:rstatix. For both deformation-based and atlas-based analyses, results are reported for FDR-corrected p < 0.05.
Diffusion analyses
Initial preprocessing the diffusion weighted data including gradient checking, denoising, Gibbs unringing, motion and eddy current correction, and bias correction using a combination of DIPY and MRtrix70. A study-specific DWI template was generated from the mean b0 images of the pre-processed DWI data and the DSURQE template mask was inverse warped to the individual brain images. Two complementary analytic pipelines were run on these pre-processed diffusion weighted images.
Tract-based spatial statistics (TBSS)
Using the pre-processed diffusion weighted images, diffusion tensor and kurtosis scalars were estimated using RESTORE71. Resulting maps included fractional anisotropy and kurtosis fractional anisotropy maps as well as scalar maps for axial, radial, and mean diffusivity, and kurtosis. Scalar maps were then warped to the study-specific DWI template space for analysis. These scalar maps were analyzed using a modified version of TBSS implemented using FSL72. The individual fractional anisotropy maps were projected onto the skeleton, which was also used for each of the other scalar maps. For the TBSS data, all scalars were analyzed using two-sample t-tests performed using FSL’s randomise (maximum permutations = 70).
Connectometry analyses
The pre-processed diffusion weighted images were analyzed using DSI Studio in a Singularity image. Prior to analyses, the data were linearly upsampled and regridded to the same voxel resolution as the anatomical T2-weighted images. The diffusion data were reconstructed in the MNI space (CIVM mouse template) using q-space diffeomorphic reconstruction to obtain the spin distribution function. A diffusion sampling length ratio of 0.6 was used. Diffusion MRI connectometry was used to derive the correlational tractography that has quantitative anisotropy (QA) correlated with group membership. A nonparametric Spearman correlation was used to derive the correlation. A T-score threshold of 2.5 was assigned and tracked using a deterministic fiber tracking algorithm to obtain correlational tractography. The tracks were filtered by topology-informed pruning73. A length threshold of 20 voxel distance was used to select tracks. To estimate the FDR, a total of 4000 randomized permutations were applied to the group label to obtain the null distribution of the track length. Results are reported at FDR-corrected p < 0.05.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.0 except for RNAseq and MRI analyses. Statistical significance was determined by two-tailed student t-test for pairwise comparisons and one-way ANOVA with post-hoc Tukey test for multiple comparisons. Data were presented as means ± SEM. The exact number of biological replicates for each assay and the p-values for each comparison are provided in figure legends. Primary cell cultures or co-cultures of the same group were randomly assigned to different experimental conditions.
Extended Data
Extended Data Fig. 1. Astrocytic Tfam deletion-induced neurodegeneration in 6- but not 1.5-month-old mice.

(a) Schematic diagram of the generating of astrocyte specific Tfam knockout allele. (b) Western blots and quantification showing protein levels of Tfam in 3-month and 1.5-month WT and TfamAKO mouse hippocampi. (c) Tfam mRNA levels in neurons, astrocytes, and microglia acutely isolated from 6-month WT and TfamAKO mouse brains. (d and e) Representative mitochondrial stress test performed with primary astrocytes isolated from 6-month WT or TfamAKO mouse brains with sequential injections of mitochondrial inhibitors including oligomycin A, FCCP and rotenone + antimycin A (d); basal, maximal, and uncoupling-linked respiration were shown in e. (f) Brain weight of 6-month WT and TfamAKO mice. (g) The ratio between distance travelled in the center area and total distance travelled for 6-month mice. (h) Discrimination index of 1.5-month WT and TfamAKO mice in NOR test. (i and j) Representative tracks (i) and time spent in the center area (marked by orange squares) and the ratio between distance travelled in the center and total distance travelled (j) for 1.5-month WT and TfamAKO mice. (k) Speed, LF swing, and LF step cycle of 6-month WT and TfamAKO mice in CatWalk tests. (l) Tfam mRNA levels in the cerebellum and brainstem of 6-month mice. n = 5 (3-mon) or 4 (1.5-mon) mice (b); n = 5 (astrocyte and neuron) or 6 (microglia) mice (c); n = 14 (WT) or 16 (AKO) wells (d,e); n = 5 mice (f, j, l); n = 13 (WT) or 9 (AKO) mice (g); n = 5 (WT) or 4 (AKO) mice (h); n = 14 (WT) or 17 (AKO) mice (k). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons.
Extended Data Fig. 2. TfamAKO has no effect on neurogenesis or neuronal death, and synaptic deficits occur after 3-month-of-age.

(a) Densitometric analyses of MAP-2, PSD95, and SNAP25 protein expression in the hippocampus of 6-month mice (Fig. 1i). (b) Representative images and quantification of MAP-2-positive area in hippocampal sections of 3-month mice. (c) Western blots and quantifications showing protein expression of MAP-2 and PSD-95 in 3-month mouse hippocampi. (d) Heatmap showing DEGs (FDR-corrected p < 0.05) related to synaptic function from RNAseq data of 6-month mouse hippocampi. (e) Representative images showing the hippocampal area of brain sections of 6-month mice co-stained for NeuN and Doublecortin. (f) Representative images and quantification for NeuN+ areas of the hippocampus and cortex of 6-month mouse brains. (g) Representative images and quantification of BrdU+NeuN+ cells in dentate gyrus of 6-month mouse brains. n = 5 (a, c), 4 (b, f), or 3 (g) mice. Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 100 μm (b, e, f, g, h).
Extended Data Fig. 3. Lipid dyshomeostasis in TfamAKO mouse brains.

(a) Glast-1 positive rate of isolated brain cells by flow cytometry (left) and cell viability of primary astrocytes from 6-month WT and TfamAKO mice. (b-f) Heatmaps of DEGs (FDR-corrected p < 0.05) of mitochondrial complexes I (b), II (c), III (d), IV (e) and V (f) that are encoded by either mtDNA or nuclear DNA (nDNA) in 6-month mouse hippocampi. (g) Densitometric analyses of PPARα protein in 6-month WT and TfamAKO hippocampus (Fig. 2e). (h and i) TLC assay showing altered levels of major classes of lipid species in 6-month mouse cortices (h), which are quantified by normalizing to protein concentrations (i). Std., standard mix. (j and k) Total TAG (j) and FFA (k) levels in 6-month mouse cortices measured by fluorometric assays. (l-n) Cortical levels of total cholesterol, cholesteryl ester, and free cholesterol of 6-month mice. (o and p) Total TAG (o) and FFA (p) levels in 3-month mouse cortices measured by fluorometric assays. (q) Heatmap of all 153 lipid species detected by the targeted lipidomic panel in 6-month mouse cortices. n = 3 (a-left, i), 5 (g), or 7 (l-n) mice; n = 11 (WT) or 14 (AKO) independent samples (a-right); n = 5 (WT) or 4 (AKO) mice (j); n = 5 (WT) or 6 (AKO) mice (k); n = 7 (WT) or 4 (AKO) mice (o); n = 6 (WT) or 4 (AKO) mice (p). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all analyses.
Extended Data Fig. 4. Lipid accumulation and metabolic shift in astrocytic FA degradation-deficient brains.

(a) Western blots and quantification showing Plin2 expression in 6-month WT and TfamAKO mouse hippocampi. (b) Representative Montage images of Plin2 staining of coronal brain sections of 6-month mice. (c) Representative images of hippocampal sections of 3-month mice stained for LD and GFAP. (d) Representative images of hippocampal sections of 1.5-month mice stained for LD and GFAP. (e) Representative images of hippocampal sections of 6-month mice stained for LD and IBA-1 (left) or LD and NeuN (right). (f) Sholl analysis, including ending radius, sum of intersects, and ramification index, of astrocytes (GFAP stained) in the hippocampus of 6-month mouse brains. (g) Representative GFAP staining images of cultured astrocytes from 6-month WT or TfamAKO mice. (h) Representative images of LD staining of 6-month primary astrocytes. (i and j) Quantification (i) and fluorescent TLC image (j) of esterified and free BODIPY-C12 from pulse-chase assay with 6-month primary astrocytes. Std., BODIPY-C12 standard. (k) PCA plot of cortical acylcarnitine profiles of 6-month WT and TfamAKO mice. (l) Heatmap of significantly changed acylcarnitines in 6-month WT vs. TfamAKO cortices. (m) Representative images and quantification of S100β immunostaining of vehicle- or oleate-treated 6-month WT astrocytes. (n) Relative CellROX intensity in oleate treated WT astrocytes. (o) Lactate levels in 6-month mouse cortices. (p) Heatmap of DEGs involved in glycolysis in 6-month mouse hippocampi. (q) Western blots and quantification of HK2 and PFKFB3 expression in 6-month mouse hippocampi. (r) mRNA levels of Hk2 and Glut1 in astrocytes acutely isolated from 6-month mice. (s) Lactate levels in cultured astrocytes from 6-month WT and TfamAKO mice. n = 5 (a, m, o, q), 3 (f, i), or 4 (s) mice; n = 12 independent samples (n); n = 7 (Hk2), 4 (Glut1-WT), or 3 (Glut1-AKO) mice (r). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 1000 μm (b), 500 μm (g, m); 100 μm (d, e); 20 μm (c); 15 μm (h).
Extended Data Fig. 5. LD accumulation is insufficient to induce astrocyte reactivity.

(a) Representative images and qualification of LD volumes in 6-month WT astrocytes treated with 10 μM atglistatin for 24 h. (b) Representative GFAP staining images and intensity (normalized to cell counts) of 6-month WT astrocytes treated with 10 μM atglistatin for 24 h. (c) Representative western blots and quantification of p-STAT3Tyr705 in 6-month WT astrocytes treated with 10 μM atglistatin for 24 h. (d) PGE2 levels in the cortex of 6-month WT and TfamAKO mice. (e) Representative images of hippocampal section of 6-month TfamAKO mouse showing that STAT3 is predominantly localized to GFAP+ astrocytes. n = 4 mice (a, b, c); n = 6 (WT) or 5 (AKO) mice (d). Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 1000 μm (b); 100 μm (e); 15 μm (a).
Extended Data Fig. 6. Loss of OxPhos diminishes astrocytic support to neurons and induces neuronal oxidative stress.

(a) Neurite volume of WT neurons cultured alone or with 6-month astrocytes. (b) Basal OCR of WT neurons cultured with 6-month astrocytes. (c) ECAR of WT neurons cocultured with 6-month astrocytes. (d) Increases in LD volume for WT or TfamAKO astrocytes cocultured with WT neurons relative to these astrocytes cultured alone. (e) Design for data in (f). WT neurons pretreated with vehicle, MCTi (AR-C155858) or ACCi (ND630) were cultured with WT or TfamAKO astrocytes. Astrocytes were stained for LD (image created with BioRender.com). (f) Reductions in LD volume in WT or TfamAKO astrocytes induced by neuronal MCTi (left) or neuronal ACCi (right). Presented values were calculated as: LD volume in astrocytes cocultured with vehicle-pretreated neurons – LD volume in genotype-matched astrocytes cocultured with ACCi- or MCTi-pretreated neurons. (g) mRNA levels of Acaca and Fasn in WT neurons cocultured with 6-month astrocytes. (h and i) OCR of WT neurons cocultured with 6-month oleate-BSA-treated WT astrocytes. (j) mRNA levels of genes involved in FA transport and β-oxidation in acute 6-month astrocytes. (k) Protein levels of PPARα in WT neurons cultured with 6-month astrocytes. (l and m) Representative images of hippocampal sections of 6-month mice co-stained for 4-HNE and GFAP or 8-OHdG and GFAP. (n) Relative CellROX intensity in cultured 6-month astrocytes. (o) CellROX positive rate of Glast1+ astrocytes in 6-month brains by flow cytometry analysis. (p and q) Representative images of hippocampal sections of 3-month mice stained for 4-HNE (p) or 8-OHdG (q). n = 3 (no-astrocyte) or 5 (+WT and +AKO) independent samples (a); n = 6 (WT) or 5 (AKO) independent samples (b, c); n = 4 (d, f), 5 (g), 6 (j), 3 (k, o), or 4 (n) independent samples; n = 5 (+WT) or 4 (+Oleate) independent samples (h, i). Bars are presented as mean ± SEM. Two-sided unpaired t-test was used except for a, where one-way ANOVA with post-hoc Tukey test was used. Scale bars, 100 μm (m, p, q); 25 μm (l).
Extended Data Fig. 7. Microglial activation and neuroinflammation are induced by astrocytes with OxPhos deficit via IL-3 signaling.
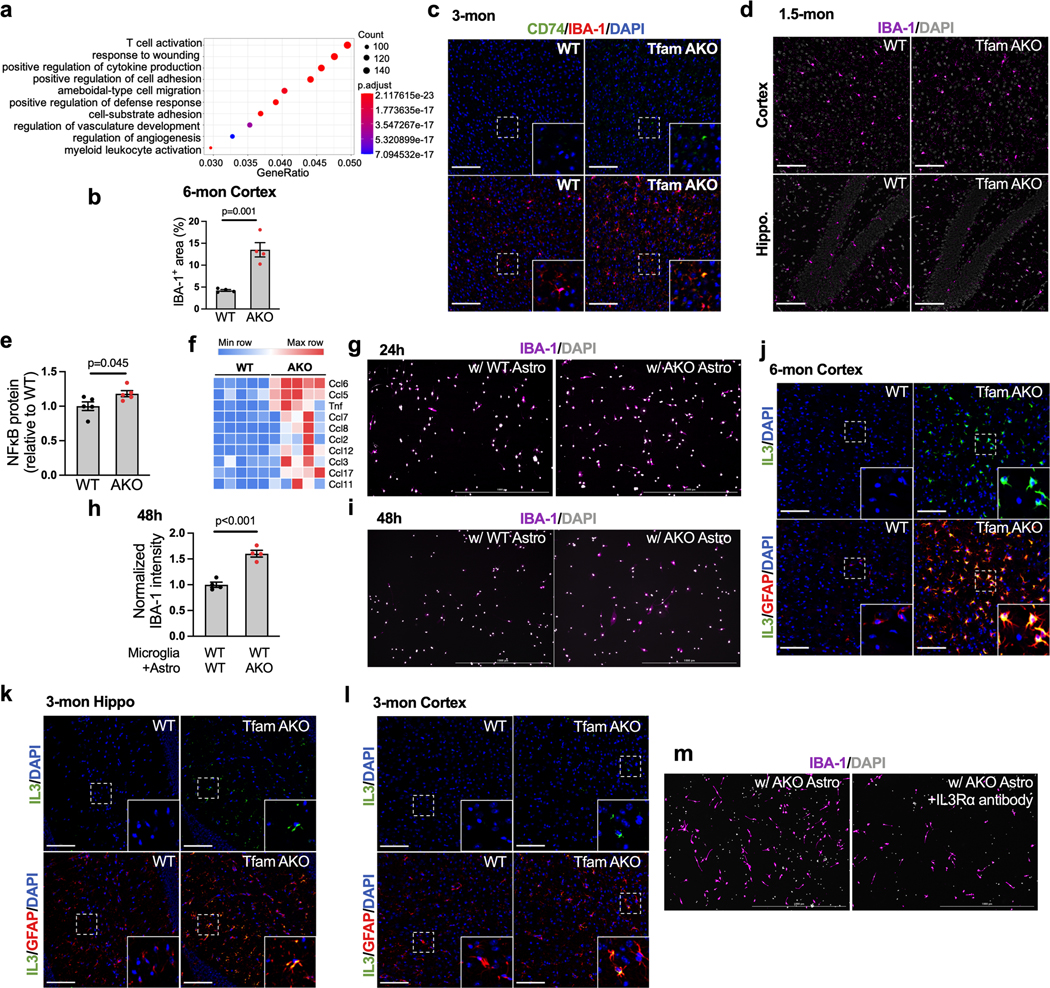
(a) Top 10 GO Biological Processes enriched in 6-month TfamAKO hippocampi compared to WT mice. (b) IBA-1 positive area in cortical sections of 6-month WT and TfamAKO mouse brains. (c) Representative images of hippocampal sections of 3-month WT and TfamAKO mice stained for CD-74 and IBA-1. (d) Representative images of hippocampal and cortical sections of 1.5-month mice stained for IBA-1. (e) Densitometric analysis of NFκB protein levels in 6-month mouse hippocampi (Fig. 6g). (f) Heatmap showing DEGs (FDR-corrected p < 0.05) encoding cytokines in 6-month mouse hippocampi. (g) Representative images of IBA-1 staining of WT primary microglia (on coverslips in 6-well plates) cocultured with WT or TfamAKO astrocytes (in 6-well inserts) for 24 h. (h and i) Quantification (h) and representative images (i) of IBA-1 intensity (normalized to cell count) in WT primary microglia cultured with WT or TfamAKO astrocytes for 48 h. (j) Representative images of cortical sections of 6-month WT and TfamAKO mice stained for IL-3 and GFAP. (k and l) Representative images of hippocampal (k) and cortical (l) sections of 3-month WT and TfamAKO mice stained for IL-3 and GFAP. (m) Representative images of WT primary microglia (on coverslips in 6-well plates) pretreated with vehicle or IL-3Rα neutralizing antibody, cocultured with TfamAKO astrocytes (in 6-well inserts) for 24 h and then stained for IBA-1. n = 4 (b, h) or 5 (e) mice or independent samples. Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 1000 μm (g, i, m); 100 μm (c, d, j, k, l).
Extended Data Fig. 8. Loss of myelin integrity and suppressed lipid synthesis in TfamAKO brains.

(a) Voxel-wise analyses of fractional anisotropy, mean-, axial-, and radial diffusivity of coronal slices on the study specific template. Blue voxels identify statistically significant TfamAKO < WT voxels (family-wise error corrected p < 0.05) for each index. (b) Total brain volume of 6-month WT and TfamAKO mice. (c and d) Representative Montage images of 6-month WT and TfamAKO mouse coronal sections stained for FluoroMyelin and MBP. (e) Representative images and quantification of FluoroMyelin staining of the corpus callosum area of 3-month brain sections. (f) Representative images and quantification of 3-month cortical sections stained for MBP. (g and h) Representative images of the hippocampus (g) or white matter (h) areas of 6-month brain sections stained for CC1 and Olig2. (i and j) Representative images of corpus callosum (CC) of 6-month brain sections stained for TUNEL and Olig2 and dMBP. (k) Heatmap showing DEGs (FDR-corrected p < 0.05) that are positive (Mag, Myrf, Sox10, Nkx6–2, Ckap5) and negative (Omg, Chrm1, and Id4) regulators of myelination in 6-month mouse hippocampi. (l and m) Densitometric analysis of FAS and p-ACC/ACC levels in 6-month mouse hippocampi (Fig. 7e). (n and o) Western blots and quantification showing protein expression of FAS, p-ACC, and ACC in 3-month mouse hippocampi. (p) Densitometric analysis of p-AMPK/AMPK ratio in 6-month mouse hippocampi (Fig. 7e). n = 4 (b, f), 3 (e), or 5 (l, m, o, p) mice. Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 1000 μm (c and d); 100 μm (e, f, g, h, i, j).
Extended Data Fig. 9. Impaired FA degradation, LD accumulation, and TfamAKO-induced transcriptional signatures are resembled in a mouse model of AD.

(a and b) Heatmaps showing AD-related (a) and DAM (b) DEGs (FDR-corrected p < 0.05) in 6-month mouse hippocampi. (c) Western blot and quantification showing protein levels of Plin2 in 4-month WT and 5xFAD mouse cortices. (d and e) Representative images showing the subiculum area of 6-month WT or 5xFAD mouse brain sections stained for LD and IBA-1 (d) or LD and NeuN (e) suggest no localization of LD to neuron or microglia. (f) Representative images of LDs in primary astrocytes isolated from 4-month WT or 5xFAD mouse brains (quantified in Fig. 8f). (g) The ratio of fission to fusion products in cultured astrocytes from 4-month WT or 5xFAD mice by subtype analysis of mitochondria reticulum images. (h and i) Quantification and representative images of BODIPY-C12 localized to mitochondria (MitoTracker+) in primary astrocytes from 6-month WT or 5xFAD mice. n = 4 (c, g) or 3 (h) mice. Bar graphs are presented as mean ± SEM. Two-sided unpaired t-test was used for all comparisons. Scale bars, 100 μm (d, e); 20 μm (f, i).
Supplementary Material
ACKNOWLEDGEMENTS
This work has been supported by the National Institute on Aging (NIA) grants RF1AG068175 to FY, P01AG026572 (Project 1 and Analytic Core to FY), University of Arizona Center for Innovation in Brain Science Startup Fund to FY, Arizona Alzheimer’s Consortium Pilot Project grants to FY, and the Packer-Wenz research endowment to FY. HG is supported by R21AG072561. We thank Avnish Bhattrai and the University of Arizona Translational Bioimaging Resource for their assistance in small animal MRI scans. The 5xFAD mouse transcriptomics data were obtained from the AD Knowledge Portal (https://adknowledgeportal.synapse.org/), and these data were generated by IU/JAX/UCI MODEL-AD Center with funding from NIA (U54 AG054345-01, U54 AG054349 and P30 AG0380770).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
DATA AVAILABILITY
The RNAseq data of the TfamAKO mice have been deposited in Gene Expression Omnibus (GEO) repository under accession number GSE203234 and can be accessed at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203234. Source data are provided with this paper.
REFERENCES
- 1.Cunnane SC et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 19, 609–633, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qi G, Mi Y. & Yin F. Cellular Specificity and Inter-cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration. Frontiers in physiology 10, 1531, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang W, Zhao F, Ma X, Perry G. & Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener 15, 30, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baik SH et al. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab 30, 493–507 e496, (2019). [DOI] [PubMed] [Google Scholar]
- 5.March-Diaz R. et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nature Aging 1, 385–399, (2021). [DOI] [PubMed] [Google Scholar]
- 6.Sofroniew MV & Vinters HV Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linnerbauer M, Wheeler MA & Quintana FJ Astrocyte Crosstalk in CNS Inflammation. Neuron 108, 608–622, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escartin C. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24, 312–325, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almeida A, Almeida J, Bolanos JP & Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A 98, 15294–15299, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong-Riley MT Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12, 94–101, (1989). [DOI] [PubMed] [Google Scholar]
- 11.Bonvento G. & Bolanos JP Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab 33, 1546–1564, (2021). [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Fabuel I. et al. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci U S A 113, 13063–13068, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fecher C. et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci, (2019). [DOI] [PubMed] [Google Scholar]
- 14.Yin F. Lipid metabolism and Alzheimer’s disease: clinical evidence, mechanistic link and therapeutic promise. FEBS J, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karch CM & Goate AM Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi G. et al. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep 34, 108572, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustafsson CM, Falkenberg M. & Larsson NG Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem 85, 133–160, (2016). [DOI] [PubMed] [Google Scholar]
- 18.Hamanaka RB et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal 6, ra8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregorian C. et al. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci 29, 1874–1886, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endo F. et al. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science 378, eadc9020, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taft JR, Vertes RP & Perry G. Distribution of GFAP+ astrocytes in adult and neonatal rat brain. International journal of neuroscience 115, 1333–1343, (2005). [DOI] [PubMed] [Google Scholar]
- 22.Rath S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olzmann JA & Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20, 137–155, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano-Pozo A, Gomez-Isla T, Growdon JH, Frosch MP & Hyman BT A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 182, 2332–2344, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rambold AS, Cohen S. & Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32, 678–692, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L. & Escartin C. The complex STATes of astrocyte reactivity: How are they controlled by the JAK-STAT3 pathway? Neuroscience 330, 205–218, (2016). [DOI] [PubMed] [Google Scholar]
- 27.Allaman I. et al. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30, 3326–3338, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan ZL, Guan YJ, Chatterjee D. & Chin YE Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273, (2005). [DOI] [PubMed] [Google Scholar]
- 29.Wellen KE et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M. & Bellen HJ The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737 e716, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonfeld P. & Reiser G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem Int 109, 68–77, (2017). [DOI] [PubMed] [Google Scholar]
- 32.Heneka MT et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAlpine CS et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595, 701–706, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor ANW et al. Tract-specific white matter hyperintensities disrupt neural network function in Alzheimer’s disease. Alzheimers Dement 13, 225–235, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JF et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron 109, 2292–2307 e2295, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeung MS et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 159, 766–774, (2014). [DOI] [PubMed] [Google Scholar]
- 37.Camargo N. et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 15, e1002605, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dimas P. et al. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oakley H. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forner S. et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci Data 8, 270, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Supek F, Bosnjak M, Skunca N. & Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Miguel Z. et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 600, 494–499, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halim ND et al. Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58, 1168–1176, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vernochet C. et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 16, 765–776, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ignatenko O. et al. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun 9, 70, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marschallinger J. et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23, 194–208, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conte M. et al. Expression pattern of perilipins in human brain during aging and in Alzheimer’s disease. Neuropathol. Appl. Neurobiol, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sofroniew MV Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 41, 758–770, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vicente-Gutierrez C. et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat Metab 1, 201–211, (2019). [DOI] [PubMed] [Google Scholar]
- 50.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guttenplan KA et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinkosky SL et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK beta1 isoforms. Nat Metab 2, 873–881, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia D. & Shaw RJ AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Molecular cell 66, 789–800, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Deijk AF et al. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 65, 670–682, (2017). [DOI] [PubMed] [Google Scholar]
- 55.Johnson ECB et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature medicine 26, 769–780, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams TI, Lynn BC, Markesbery WR & Lovell MA Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol Aging 27, 1094–1099, (2006). [DOI] [PubMed] [Google Scholar]
- 57.Konttinen H. et al. PPARbeta/delta-agonist GW0742 ameliorates dysfunction in fatty acid oxidation in PSEN1DeltaE9 astrocytes. Glia 67, 146–159, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karran E, Mercken M. & De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712, (2011). [DOI] [PubMed] [Google Scholar]
- 59.Swerdlow RH, Burns JM & Khan SM The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 1842, 1219–1231, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martens YA et al. ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fanning S. et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Molecular cell 73, 1001–1014 e1008, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee H. et al. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat Neurosci 24, 1673–1685, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Polyzos AA et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab 29, 1258–1273 e1211, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong Y. et al. Oxidized phosphatidylcholines found in multiple sclerosis lesions mediate neurodegeneration and are neutralized by microglia. Nat Neurosci 24, 489–503, (2021). [DOI] [PubMed] [Google Scholar]
- 65.Qi G, Mi Y. & Yin F. Characterizing brain metabolic function ex vivo with acute mouse slice punches. STAR Protoc 2, 100559, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin F. et al. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol Aging 36, 2282–2295, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eghlimi R, Shi X, Hrovat J, Xi B. & Gu H. Triple Negative Breast Cancer Detection Using LC-MS/MS Lipidomic Profiling. J. Proteome Res 19, 2367–2378, (2020). [DOI] [PubMed] [Google Scholar]
- 68.Shi X. et al. Comprehensive Isotopic Targeted Mass Spectrometry: Reliable Metabolic Flux Analysis with Broad Coverage. Anal. Chem 92, 11728–11738, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winkler AM, Ridgway GR, Webster MA, Smith SM & Nichols TE Permutation inference for the general linear model. Neuroimage 92, 381–397, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tournier JD et al. MRtrix3: A fast, flexible and open software framework for medical image processing and visualisation. Neuroimage 202, 116137, (2019). [DOI] [PubMed] [Google Scholar]
- 71.Chang LC, Jones DK & Pierpaoli C. RESTORE: robust estimation of tensors by outlier rejection. Magn Reson Med 53, 1088–1095, (2005). [DOI] [PubMed] [Google Scholar]
- 72.Smith SM et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23, S208–S219, (2004). [DOI] [PubMed] [Google Scholar]
- 73.Yeh F-C et al. Automatic removal of false connections in diffusion MRI tractography using topology-informed pruning (TIP). Neurotherapeutics 16, 52–58, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data of the TfamAKO mice have been deposited in Gene Expression Omnibus (GEO) repository under accession number GSE203234 and can be accessed at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203234. Source data are provided with this paper.