

Abstract
Motivation
Biomedical named entity recognition (BioNER) seeks to automatically recognize biomedical entities in natural language text, serving as a necessary foundation for downstream text mining tasks and applications such as information extraction and question answering. Manually labeling training data for the BioNER task is costly, however, due to the significant domain expertise required for accurate annotation. The resulting data scarcity causes current BioNER approaches to be prone to overfitting, to suffer from limited generalizability, and to address a single entity type at a time (e.g. gene or disease).
Results
We therefore propose a novel all-in-one (AIO) scheme that uses external data from existing annotated resources to enhance the accuracy and stability of BioNER models. We further present AIONER, a general-purpose BioNER tool based on cutting-edge deep learning and our AIO schema. We evaluate AIONER on 14 BioNER benchmark tasks and show that AIONER is effective, robust, and compares favorably to other state-of-the-art approaches such as multi-task learning. We further demonstrate the practical utility of AIONER in three independent tasks to recognize entity types not previously seen in training data, as well as the advantages of AIONER over existing methods for processing biomedical text at a large scale (e.g. the entire PubMed data).
Availability and implementation
The source code, trained models and data for AIONER are freely available at https://github.com/ncbi/AIONER.
1 Introduction
Large-scale application of automated natural language processing (NLP) to biomedical text has successfully helped address the information overload resulting from the thousands of articles added to the biomedical literature daily (Sayers et al. 2021). Biomedical NLP is also increasingly used to support quantitative biomedical science, by augmenting manual curation efforts to populate databases via automated extraction (Singhal et al. 2016), or by making inferences directly, through tasks such as literature-based knowledge discovery (Weeber et al. 2001). Biomedical named entity recognition (BioNER), the task of identifying bio-entities such as chemicals and diseases within the text, provides an important foundation for many biomedical NLP applications, and the accuracy of the entities identified by BioNER strongly affects the quality of the downstream applications. However, biomedical entities are with much more complicated naming principles. Compared with NER tasks in the general domain, such as the recognition of the persons or organizations, BioNER is more challenging because biomedical entity names are longer, more complex, and ambiguous (Cariello et al. 2021; Jeong and Kang 2021).
Prior to the deep learning era, conditional random fields (CRF) (Lafferty et al. 2001) with the rich feature sets were the most popular method for BioNER, and consistently performed well on a variety of tasks (Leaman et al. 2013; Leaman et al. 2015; Wei et al. 2015). In recent years, several deep learning-based methods have been widely applied to BioNER tasks with promising results, including bidirectional Long Short-Term Memory with a CRF layer (BiLSTM-CRF) (Lample et al. 2016), Embeddings from Language Models (ELMo) (Peters et al. 2018), and Bidirectional Encoder Representations from Transformers (BERT) (Devlin et al. 2019). Most recently, the BERT pre-trained language model has become among the most popular methods, and several variants trained on biomedical text have been publicly released and widely applied to BioNER tasks [e.g. BlueBERT (Peng et al. 2019), BioBERT (Lee et al. 2020), and PubMedBERT (Gu et al. 2022)].
The success of these machine-learning based methods relies heavily on manually annotated gold-standard data for model training and testing. Hence, significant efforts have been made to develop BioNER corpora for key biomedical entities such as diseases (Doğan et al. 2014) and chemicals (Krallinger et al. 2015). However, unlike the general English domain, manually annotating biomedical text requires domain knowledge and is highly costly. As a result, the current corpora in BioNER are generally limited in size, with a few hundred articles on average, and machine learning models trained on such limited annotations are prone to overfitting. Several recent studies (Galea et al. 2018; Kühnel and Fluck 2022) demonstrate that the accuracy of models trained on individual corpora decreases significantly when applied to independent corpora due to the limited generalizability of entity-related features captured by individual corpora.
Recently, several BioNER methods based on multi-task learning (MTL) (Crichton et al. 2017; Wang et al. 2019; Giorgi and Bader 2020; Zuo and Zhang 2020; Rodriguez et al. 2022) have been proposed, which improve the generalizability of the model by making use of various publicly available datasets. These methods generally share the hidden layers of the deep learning model across the related tasks and have an output layer specific to each task. In particular, MTL can improve the model’s generalizability by leveraging domain-specific information found in the training signals of related tasks (Caruana 1997). However, several studies (Chai et al. 2022; Rodriguez et al. 2022) have found that MTL results are not always stable: MTL can improve performance compared to single-task learning on some datasets, but does not do so universally. Moreover, the MTL-based methods (Wang et al. 2019; Chai et al. 2022) usually require a complex model architecture.
In this work, we propose a novel data-centric perspective to enhance the accuracy and robustness of BioNER models by merging multiple datasets into a single task via task-oriented tagging labels. As a result, our method can achieve better performance more consistently than MTL and is applicable to various machine learning models.
More specifically, we propose AIONER, a new NER tagger that takes full advantage of various existing datasets for recognizing multiple entities simultaneously, despite their inherent differences in scope and quality, through a novel all-in-one (AIO) scheme. Our AIO scheme utilizes a small dataset recently annotated with multiple entity types [e.g. BioRED (Luo et al. 2022a)] as a bridge to integrate multiple datasets annotated with a subset of entity types, thereby recognizing multiple entities at once, resulting in improved accuracy and robustness. Experimental results show that external datasets can help AIONER achieve statistically higher performance compared to that of the model trained only using the original BioRED training data. Vice versa, we demonstrate our AIO scheme can help improve model performance on individual datasets.
We further show that instead of using the entire BioRED training data (500 articles) in AIONER, we can use a minimal set of 10 articles to achieve a competitive result (86.91%) when external datasets are used. Finally, we demonstrate that AIONER can be reused in a versatile manner as a pre-trained model to further improve the performance of other BioNER tasks, even when the entity types are not previously seen in the AIONER training data.
2 Materials and methods
The overall architecture of AIONER for multiple named entity recognition is shown in Fig. 1. We first collected multiple resources for the six target entity types (i.e. gene, disease, chemical, species, variant, and cell line) which were annotated in the BioRED dataset. We then propose an effective all-in-one strategy to merge different resources into a single sequence labeling task. Next, a cutting-edge deep learning model is trained with the merged dataset for this BioNER task. Finally, the trained model is used to recognize the multiple biomedical entities from unseen documents. Further details on each step are provided in the following section.
Figure 1.

Overview of our AIONER pipeline.
2.1 Publicly available BioNER datasets
To develop such a comprehensive BioNER method, we collected multiple resources within the six most popular entities in biomedical literature (i.e. gene, disease, chemical, species, variant, and cell line) as shown in Supplementary Table S1. We defined two criteria to filter the inconsistent datasets: (i) The selected datasets should annotate the interchangeable entities consistently. Some concepts are highly relevant and are usually used interchangeably. For example, a drug is a chemical substance that affects the functioning of living things and is frequently represented by a chemical. Besides, the gene and its products (e.g. RNA and protein) are usually named identically. There are also some other overlapping concepts, like phenotypes to diseases and residues to variants. (ii) The datasets should annotate the concept identifiers of the entities by the same resources (e.g. NCBI Gene for gene/protein and MESH for chemical), which guarantees the definitions of the curated entities are consistent. The narrowed down resources are shown in Table 1.
Table 1.
The BioNER datasets used in our study.a
| Entity type | Dataset | Text size | Entities |
|---|---|---|---|
| All | BioRED(Luo et al. 2022a) | 600 abs | 20 419 |
| Gene | GNormPlus (Wei et al. 2015) | 694 abs | 9986 |
| NLM-Gene (Islamaj et al. 2021,b) | 550 abs | 15 553 | |
| Disease | NCBI Disease (Doğan et al. 2014) | 793 abs | 6892 |
| BC5CDR-Disease (Li et al. 2016) | 1500 abs | 12 850 | |
| Chemical | BC5CDR-Chemical (Li et al. 2016) | 1500 abs | 15 935 |
| NLM-Chem (Islamaj et al. 2021a) | 150 full | 40 467 | |
| Species | Linnaeus (Gerner et al. 2010) | 100 full | 4259 |
| Species-800 (Pafilis et al. 2013) | 800 abs | 3708 | |
| Variant | tmVar3 (Wei et al. 2022) | 500 abs | 1895 |
| Cell line | BioID (Arighi et al. 2017) | 570 full | 5590 |
Abs denotes abstracts; full denotes full-texts. Text genre is scientific article.
However, a few minor inconsistencies remained in the selected corpora after filtering. First, the annotations in the BioID (Arighi et al. 2017) dataset are inconsistent internally. About 30% of the cell line spans with the suffix “cell(s)”. To make it more consistent, we removed all the suffixes before the training and evaluation. Besides, Linnaeus (Gerner et al. 2010) and Species-800 (Pafilis et al. 2013) do not annotate the species relevant clinical terms (e.g. patient). Also, Species-800 excludes the genus name (e.g. Arabidopsis), and the species name which is in the higher level of the taxonomy system (e.g. Fungal), although those are annotated in BioRED. Third, GNormPlus (Wei et al. 2015) and NLM-Gene (Islamaj et al. 2021,b) distinguish gene/protein family (e.g. Dilps) from the specific gene names (e.g. Dilp6), but BioRED did not distinguish the two types of entities. In our implementation, we merged the family names to the gene entity type.
2.2 All-in-one scheme
Like most previous studies, we treated BioNER as a sequence labeling task. Consider a sequence of text , where denotes the length of the text. Each is tagged with a class label , where denotes the tagging scheme set (e.g. BIO scheme). The BIO scheme (Sang and De Meulder 2003), which contains begin tokens (“B”), inside tokens (“I”), and background (outside) tokens (“O”), is the most popular encoding scheme of the BioNER task. Unlike the traditional BIO scheme, we designed and proposed a novel all-in-one (AIO) scheme to accept multiple datasets from different tasks. Specifically, given tasks and considering an input sentence from the task where , we applied an additional pair of tags surrounding the input sentence to indicate the task (e.g. “<Disease></Disease>” to recognize disease entities, and “<ALL></ALL>” to recognize all concept entities). The special tokens indicating the task tags were added to the beginning and end of the input sentence. For the label set , we defined three types of labels, which include “B-EntityType”, “I-EntityType”, and “O-Task”. Note that, the definitions of the “B” and “I” in the AIO scheme are the same as the traditional BIO scheme. However, to avoid entity conflict, we flexibly redesigned the “O” (outside) label since some entities may be curated in some of the datasets but not others. For example, in the scenario of recognizing diseases, the original “O” label is modified to “O-Disease” which can be clearly distinguished from the “O-Chemical” label for the task of recognizing chemical entities. Finally, we defined a total of 19 labels in the label set = {B-Gene, I-Gene, O-Gene, B-Disease, I-Disease, O-Disease, …, B-CellLine, I-CellLine, O- CellLine, O-ALL}.
We merged the datasets listed in Table 1 based on the AIO scheme. Excluding BioRED annotating all entity types, other datasets only focus on partially annotated entity types. If the dataset contains multiple partially annotated entity types, we split it into multiple datasets with a single entity type (e.g. BC5CDR is split into BC5CDR-Disease and BC5CDR-Chemical). Then we merged the datasets with the same entity type into a BioNER task. Figure 2 shows some example sentences annotated based on our tagging scheme. For example, we merged GNormPlus and NLM-Gene datasets for the gene recognition task. All sentences in these two datasets are added “<Gene>” and “</Gene>” tags at the front and end of the sentence. Only the tokens of the gene entity are labeled as “B-Gene” (or “I-Gene”). All other tokens are labeled as “O-Gene”. “B-Gene” represents the status of the first token of the gene span, “I-Gene” represents the tokens of the gene span other than the first one, “O-Gene” represents the background tokens out of the gene spans. Different from other datasets, BioRED is a resource containing all entity types. We added “<ALL></ALL>” tags surrounding the input sentence from BioRED to indicate recognizing all entities of the six entity types. The tokens of the biomedical entity are labeled to B-EntityType (or I-EntityType), and all background tokens are labeled with “O-ALL”. After converting all datasets using the AIO scheme, the integrated data is used to train NER models.
Figure 2.

Some example sentences annotated based on our AIO scheme.
2.3 Deep learning model for BioNER
The BioNER task has been modulated to a sequence labeling task via the AIO scheme. We applied cutting-edge biomedical pre-trained language models (PLMs) [e.g. PubmedBERT (Gu et al. 2022)] for the implementation of our framework. Specifically, given an input sentence consisting of tokens, the model aims to map the token sequence to a corresponding sequence of the label , where . We first used the PLMs to encode the input to a hidden state vector sequence, then computed the network score using a fully connected layer with a ReLU (Glorot et al. 2011) activation. Finally, the CRF output layer is added to optimize the boundary detection of the bio-entities. A score is defined as:
| #(1) |
where is the score matrix of the output from the last fully connected layer and is a transition matrix of the CRF layer. represents the score of the transition from the ith label to the jth label. During the training phase, the objective of the model is to maximize the log-probability of the correct tag sequence: where denotes all possible tag paths. At inference time, we predict the tag path that obtains the maximum score given by:
| #(2) |
| #(3) |
This can be computed using dynamic programming, and the Viterbi algorithm (Viterbi 1967) has been chosen for this inference.
We merged the training and development sets of the datasets for model training and evaluated the models on the official test sets. Since the official test sets of Linnaeus, Species-800, and BioID are not available or released, we randomly split 20% of the dataset as the test set for evaluating the models. We applied the default PLM parameter settings and set main hyper-parameters as follows: learning rate of 5e–6, batch size of 32, and max input length of 256 tokens (split into multiple sentences if the length of a sentence is over the max length). To determine the optimal number of training epochs, we set the patience parameter to 5 with a maximum of 50 epochs. The training process would terminate if there were no significant accuracy improvements in five consecutive epochs. For a detailed account of hyper-parameter settings, please refer to the Supplementary Material.
After training the model, the trained model can be used for recognizing the biomedical entities from the unseen input text. First, the input text was split into sentences and tokenized. Then we inserted the special task tags surrounding the input sentence to indicate the task. Finally, the trained AIONER model can be used to tag the tokens of the sentence to extract the task-specific entities according to the inserted task tags (e.g. “<Disease></Disease>” to only recognize disease entities, and “<ALL></ALL>” to recognize all concept entities). It should be noted that the entity scope and definitions in individual corpora may not align completely with BioRED. Therefore, utilizing the “<ALL> </ALL>” task tags for identifying all concept entities in BioRED and applying individual (IND) task tags for identifying entities in the corresponding individual corpora. Further information can be found in Supplementary Table S3.
3 Results
3.1 Experimental settings
To demonstrate the effectiveness of AIONER, we performed four experiments. First, we examined the performance of AIONER for multiple entity recognition on the BioRED test set. Second, we evaluated the stability and robustness of AIONER by analyzing its overall performance on the test set for each individual dataset. Third, we tested whether AIONER can be applied to support those BioNER tasks with new entity types that are not previously seen during AIONER model training. Finally, we investigated the performance and efficiency of the different variants of the BERT-based pre-trained model in our framework for supporting the processing of PubMed-scale literature data.
In addition, we also implemented the MTL framework with the same deep learning model and training data for comparison. MTL treats each dataset as an individual task, and its model architecture shares the hidden layers across the different tasks where each task has its own task-specific output layers. The final loss is calculated by summarizing the losses of different tasks. In our experiments, we evaluated the model performance using the entity-level micro F1-score (F1), which has been widely applied in BioNER tasks. We further applied the two-sided Wilcoxon signed-rank test to perform statistical significance testing. Note that a few documents exist in both BioRED and some other datasets. To accurately evaluate the performance of different methods, we filtered those overlapped documents in the training set if the documents also exist in the test set.
3.2 Multiple named entity recognition via AIONER on BioRED
We examined the effectiveness of the AIO scheme and the contribution of different datasets for multiple named entity recognition. According to the evaluation in a 2022 study (Luo et al. 2022a), the PubMedBERT-CRF model achieves state-of-the-art (SOTA) performance and compares favorably to other methods such as BiLSTM-CRF and BioBERT-CRF models on the BioRED dataset. Therefore, we use it as the default model in our architecture of AIONER and MTL. We firstly prepared a strong baseline based on the PubMedBERT-CRF model, which trained on the original BioRED training set. Then we merged every external dataset and BioRED training set via our AIO scheme to train the models, respectively. Finally, we integrated all datasets as a union training set and trained the AIONER model [i.e. BioRED+All (AIONER)]. We also implemented two more options to integrate datasets for comparison: (i) BioRED+All (w/o AIONER): the model trained on the naive concatenation of all training datasets. (ii) BioRED+All (MTL): the multi-task learning model trained on all datasets, in which each dataset is treated as an individual task. Table 2 shows the results of evaluating the models on the BioRED test set.
Table 2.
F1 scores for multiple named entity recognition on the BioRED test set.a
| Dataset | Overall | Gene | Disease | Chemical | Species | Variant | CellLine |
|---|---|---|---|---|---|---|---|
| BioRED | 89.34 | 92.35 | 83.47 | 88.55 | 96.98 | 87.34 | 90.53 |
| +NLM-Gene | 89.76 | 92.40 | 84.03 | 90.19 | 97.35 | 85.89 | 86.87 |
| +GNormPlus | 89.95 | 92.74 | 83.57 | 90.05 | 96.82 | 88.98 | 91.67 |
| +NCBI-Disease | 89.55 | 91.68 | 85.19 | 89.46 | 96.52 | 86.01 | 81.72 |
| +BC5CDR-Disease | 89.66 | 91.46 | 85.34 | 89.67 | 96.98 | 84.86 | 90.53 |
| +BC5CDR-Chemical | 89.40 | 91.52 | 84.07 | 89.09 | 96.99 | 88.38 | 87.50 |
| +NLM-Chem | 89.60 | 91.92 | 84.15 | 89.78 | 97.09 | 87.16 | 83.67 |
| +Linnaeus | 89.19 | 91.49 | 84.04 | 88.69 | 96.72 | 88.16 | 86.60 |
| +Species-800 | 89.65 | 92.19 | 83.34 | 90.14 | 97.37 | 88.79 | 80.81 |
| +tmVar3 | 89.01 | 91.08 | 83.77 | 88.09 | 97.08 | 89.21 | 88.66 |
| +BioID | 89.69 | 92.02 | 84.23 | 88.83 | 97.48 | 88.75 | 91.67 |
| +All (w/o AIONER) | 69.96 | 76.85 | 58.86 | 84.82 | 30.57 | 71.77 | 27.12 |
| +All (MTL) | 90.84b | 92.59 | 87.01 | 90.71 | 96.40 | 88.25 | 90.32 |
| +All (AIONER) |
|
|
|
|
|
|
|
The parenthesized numbers are the improvements of AIONER compared to the baseline trained on the BioRED training set only. Bold indicates the best score for each entity type and overall entity.
P < 0.05 (two-sided Wilcoxon signed-rank test compared with baseline). There is no significant difference between MTL and AIONER.
Compared to the baseline, the models trained on both the individual external datasets and BioRED training set can obtain better performance on the corresponding entity type with slightly higher F1-scores in overall performance. The MTL and AIONER models both perform significantly better than the baseline when all datasets are used for training. Particularly, the AIONER model obtained the highest overall score, improving the F1-score from 89.34% to 91.26%. In terms of entity types, disease, and chemical are the most improved (4.60% and 2.43%, respectively). We evaluated the stability and robustness of AIONER and MTL models by comparing the means and standard deviations of their overall F-scores across five runs with different random initial seeds. Our results demonstrate that AIONER achieved a higher mean F-score and lower standard deviation than MTL, indicating its superior stability and robustness. Specifically, the mean F-scores for AIONER and MTL were 91.21 ± 0.15% and 90.64 ± 0.34%, respectively. Compared with the MTL model that shares the same hidden layers but uses independent CRF output layers, our AIONER model merges all datasets with the AIO scheme and output results within a single output layer. Thus, it may be able to better utilize the information from the different datasets. Merging all datasets directly without applying the AIO scheme dropped the F1-score about 20% due to the large number of missing annotations. For example, gene datasets do not annotate diseases or chemicals.
In addition, we tested the performance of the AIONER model trained on partial BioRED training data with all external datasets. We set up seven configurations with different numbers of abstracts (10, 50, 100, 200, 300, 400, and 500 abstracts) as the BioRED training subset. The results are shown in Fig. 3. Here, the baseline is the model trained on the BioRED only. The results indicate that both AIONER and MTL models exhibit similar performances and outperform the baseline across all configurations of the BioRED training subset. Furthermore, it is noteworthy that even with only 10 articles, AIONER performs significantly better than MTL, and it still can achieve a competitive performance compared with the baseline model trained on the entire 500 articles (86.91% versus 89.34%).
Figure 3.
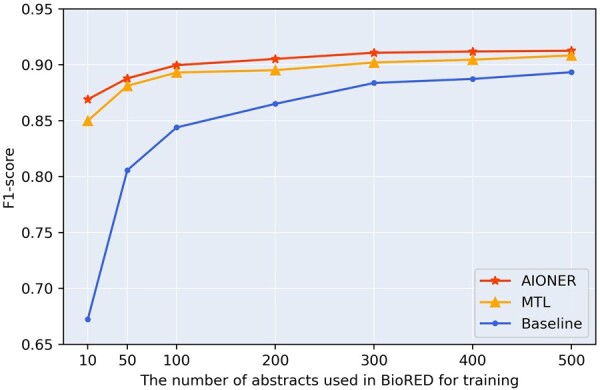
The performance of the models trained on different sizes of the BioRED training data. Baseline: the model trained on the BioRED training set only; MTL: the multi-task learning model trained on the BioRED training set along with external datasets; AIONER: our AIONER model trained on the BioRED training set with external datasets.
3.3 Performance of AIONER on the test sets of individual datasets
The previous experiment demonstrated AIONER successfully utilized external datasets for the BioRED task. In this experiment, we evaluate the performance of the AIONER on those external datasets. We trained the PubMedBERT-CRF model only using the original training data as the baseline 1 (BL1). We provide an analysis of the performance of the model (baseline 2, BL2) on each individual corpus by training it on the combined dataset consisting of the individual corpus and the sub-corpus obtained by selecting the individual entity type from BioRED. The MTL model uses the same training data as AIONER.
As shown in Table 3, we obtained similar results to the previous experiment, both MTL and AIONER methods achieve higher average F1-scores than the baselines across those datasets. However, the performance of MTL is not stable and it performs worse than the baseline 1 on 3 out of 10 BioNER datasets. AIONER brings higher average improvements than MTL across those datasets. It performs better than the baseline 1 on 9 out of 10 datasets and obtains significant advantages over the results of baselines and MTL on three datasets. Furthermore, our experiments revealed that the simple combination of individual corpus and sub-corpus from BioRED by selecting the corresponding entity type (i.e. baseline 2) resulted in an average F1-score lower than that of baseline 1. This suggests that the performance improvement cannot be achieved merely through the combination of the datasets. Notably, we observed a significant drop in the F1-score of baseline 2 on Linnaeus and Species-800 corpora, which could be attributed to the inherent differences in scope and quality between these corpora and BioRED for the species type. This highlights the challenges in leveraging existing datasets by combining them, especially when they differ significantly in scope and quality. In contrast, our proposed AIO schema improved the performance of datasets other than BioRED, demonstrating its high robustness and stability. We also provided a comparison with the state-of-the-art (SOTA) methods in terms of F1-scores for each corpus. AIONER achieved competitive performance compared to the SOTA methods. It is worth noting that a direct comparison with some published SOTA benchmarks for the BioID task may not be applicable due to the revised version of the BioID dataset in our experimental setup.
Table 3.
F1 scores for the single entity recognition on the test sets of individual datasets.a
| Dataset | BL1 | BL2 | MTL | AIO | SOTA |
|---|---|---|---|---|---|
| NLM-gene | 92.09 | 91.88 | 92.34 | 92.51 | 88.10 |
| GNormPlus | 85.09 | 85.92 | 85.62 | 85.98 | 86.70 |
| NCBI-disease | 87.56 | 88.13 | 88.41 | 89.59b | 89.71 |
| BC5CDR-disease | 87.13 | 87.12 | 86.51 | 87.89b | 87.28 |
| BC5CDR-chemical | 93.42 | 92.82 | 93.93b | 92.84 | 93.83 |
| NLM-Chem | 82.40 | 79.23 | 82.95 | 82.51 | 84.79 |
| Linnaeus | 90.36 | 85.19 | 90.14 | 90.63 | 92.70 |
| Species-800 | 78.32 | 76.91 | 78.76 | 79.67 | 76.35 |
| tmVar3 | 89.66 | 89.96 | 90.54 | 90.98 | 91.36 |
| BioID | 89.07 | 88.93 | 88.70 | 91.13b | – |
| Average | 87.51 | 86.61 | 87.79 | 88.37 | – |
BL1: the PubMedBERT-CRF model trained on the original training set. BL2: the PubMedBERT-CRF model trained on a combination of individual corpora and sub-corpora decomposed from BioRED by selecting specific entity types. MTL: the multi-task learning model trained on the BioRED and the external datasets. AIO: the AIONER model trained on the BioRED and the external datasets. SOTA: the published state-of-the-art F1-score of each corpus. Bold denotes the best F1-score (except SOTA) on each dataset.
Denotes statistical significance over Baselines and MTL (two-sided Wilcoxon signed-rank test with a P-value < 0.05). We list the scores of the SOTA models on different datasets as follows: the score of Islamaj et al. (2021,b) on NLM-Gene, the score of Wei et al. (2019) on GNormPlus, the score of Zhang et al. (2021) on Species-800, the scores of Chai et al. (2022) on BC5CDR, the score of Tong et al. (2022) on NLM-Chem, the score of Sung et al. (2022) on Linnaeus, the score of Wei et al. (2022) on tmVar3. It is important to note that we made revisions to the BioID dataset in order to improve its consistency. As a result, certain previously published SOTA benchmarks for the BioID task [such as the F1-score of 74.4% reported in Arighi et al. (2017)] may not be directly comparable to our experimental setup.
3.4 Using AIONER for new entity types
Although our combined training dataset already covers six main entity types in the biomedical domain, there are other biological entities that are outside of our consideration. This experiment investigates whether AIONER can be applied to support those BioNER tasks with new entity types that are not previously seen in our AIONER training data. We conducted experiments with AIONER in two ways. (i) AIONER-Merged: we first merge the training set of the new task with all datasets via our AIO scheme to train a NER model, and then we applied the trained model to the task test set. (ii) AIONER-Pretrained: We utilize the trained AIONER model as a pre-trained model, and then we further fine-tune this model on the new task with its training data. We benchmarked AIONER on three independent datasets with a variety of entity types different from the six which are our primary focus: (i) DMCB_Plant (Cho et al. 2017) contains 3985 mentions of plant names (e.g. “Trichosanthes kirilowii”). (ii) AnEM (Pyysalo and Ananiadou 2014) contains anatomical entities and organism parts between the molecule and the whole organism, with a total of 11 entity types (e.g. multi-tissue structure). (iii) BEAR (Wührl and Klinger 2022) annotates seven groups of biomedical entities (e.g. medical conditions, diagnostics, and environmental factors) on Twitter. More details of these datasets can be found in Supplementary Table S2. We used the PubMedBERT-CRF model trained on the original training set as the baseline method. We also tested the MTL method for comparison. The overall performances of all models on the three individual tasks are shown in Fig. 4.
Figure 4.
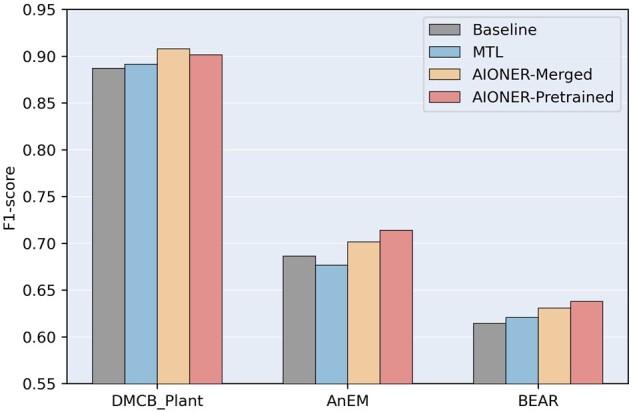
The performance of models on additional BioNER tasks. Baseline: the model trained on the original training set. MTL: the multi-task learning model trained on the new dataset and all previous datasets. AIONER-Merged: the training set of the new task is first merged into all datasets via the AIO scheme, then the data is used to train the NER model. AIONER-Pretrained: the trained AIONER model is used as a pre-trained model, then the model is fine-tuned on the new task. AIONER-Pretrained significantly outperforms the baseline and MTL on AnEM; and it significantly outperforms the baseline on BEAR in a two-sided Wilcoxon signed-rank test with a P-value < 0.05.
Although none of the entity types in these three tasks are covered by BioRED, the dataset used to bridge between the individual datasets, AIONER can still improve the performance of the three tasks consistently. The results in Fig. 4 indicate that the entity information gathered from the AIONER model assists in recognizing other entity types that have not been observed before. Overall, both the AIONER-Merged and -Pretrained models achieve better performance compared to the baseline and the MTL methods. More importantly, the performance gain is consistent for our AIONER method on all three tasks while MTL showed inferior performance on one of the three tasks. Again, this confirms that performance gain with MTL is somewhat task dependent, a limitation that was previously discussed in the literature (Chai et al. 2022; Zhang and Yang 2022).
3.5 Performance of different deep learning model variants
To process large-scale datasets in real-world settings, we further investigated the performance and efficiency of different deep learning models. The BioBERT (Lee et al. 2020) and Bioformer (Fang and Wang 2021) models were additionally evaluated as variants of BERT-based pre-trained language models (PLMs). For options of the output layer, we also tested the Softmax function to classify the label for each token. Table 4 shows the performance of different deep learning model variants on the BioRED test set.
Table 4.
The performance of different deep learning model variants on the BioRED test set.a
| PLM | Output layer | Efficiency |
F1-score |
||
|---|---|---|---|---|---|
| GPU | CPU | Baseline | AIO | ||
| PubMedBERT | CRF | 27s | 116s | 89.34 | 91.26 |
| Softmax | 17s | 110s | 88.98 | 91.00 | |
| BioBERT | CRF | 29s | 120s | 88.66 | 90.29 |
| Softmax | 18s | 113s | 88.33 | 90.06 | |
| Bioformer | CRF | 21s | 43s | 88.65 | 90.28 |
| Softmax | 12s | 40s | 88.35 | 90.19 | |
Baseline: the model trained on the original BioRED training set. AIO: the AIONER model trained on the merged training set. All AIONER models significantly outperform the corresponding baselines in a two-sided Wilcoxon signed-rank test with a P-value < 0.05. Bold indicates the best score in efficiency and F1-score. Note that the numbers of efficiency are the processing time (seconds) on the BioRED test set (100 abstracts). All models were evaluated on the same GPU (Tesla V100-SXM2-32GB) and CPU [Intel(R) Xeon(R) Gold 6226 CPU @ 2.70 GHz, 24 Cores]. The processing times of the BioBERT and PubMedBERT are almost the same, as their model architectures and parameters are similar.
The result shows that our AIONER scheme can be applied in various deep learning models and significantly enhances their performance. In comparison with other PLMs, the PubMedBERT model obtains the highest F1-scores. The lightweight Bioformer is more efficient and achieves a close performance. The efficiency advantage of the Bioformer has also been demonstrated in several recent studies (Fang and Wang 2021; Luo et al. 2022,b). According to the statistical significance analysis, all PubMedBERT models exhibit significant improvements in performance compared to their corresponding BioBERT and Bioformer models. This finding is supported by a two-sided Wilcoxon signed-rank test with a P-value of less than 0.05. On the other hand, there was no significant difference observed between BioBERT and Bioformer models. In addition, the configuration of using CRF as the decoding layer obtains the best performance. The Softmax layer’s performance is slightly lower, but the efficiency is significantly higher on GPU. No significant difference was observed between the performance of the same pre-trained language models with either the CRF or Softmax output layer. In summary, Bioformer-Softmax presents the highest efficiency (2- and 3-times improvement on both GPU and CPU servers respectively) and is very close to the best setting (PubMedBERT-CRF) in performance (about 1% drop in F-score). Moreover, Bioformer-Softmax-AIONER achieves higher performance than PubMedBERT-CRF-Baseline, suggesting it may be a better option for processing the large-scale datasets. Per our previous experience of extracting entities in entire PubMed abstracts (>35 Million) and PMC full texts (>4 Million) in PubTator Central (Wei et al. 2019) by individual entity taggers, the whole process took ∼30 days by using 300 parallel processes via NCBI computer cluster. As an estimation of using Bioformer-Softmax-AIONER instead of the six individual NER tools, the processing time can be reduced to less than 10 days. Thus, the implementation of the AIONER brings significant advantages of entity recognition on large-scale data.
4 Discussion
As mentioned in Section 1, several MTL methods have been explored for BioNER tasks to make full use of various existing resources. These methods share the hidden layers to learn common features, which are generic and invariant to all the tasks. MTL is powerful when all the tasks are related, but it is vulnerable to noisy and outlier tasks, which can significantly degrade performance (Zhang and Yang 2022). Different from MTL, our AIONER scheme successfully integrates multiple resources into a single task via adding task-oriented tagging labels. The learned features are more informative and flexible. The results of our experiments demonstrate our AIONER method achieved performance competitive with the MTL methods for multiple named entity recognition, and it is more stable than the MTL method on multiple BioNER tasks. Moreover, AIONER does not require complex model design and it can be easily implemented with various machine learning models.
The main contribution of the AIONER schema is that it can train on more diverse entity-type corpora. By doing so, the model can learn different synonyms present in diverse texts. We analysed the differences between the results of the AIONER model trained on multiple corpora and the baseline model trained on the BioRED corpus only. Then, we summarized the three main cases where AIONER performs better than the baseline. (i) More precise categorization of entity types: AIONER can better categorize the entity type based on the context. In PMID:15464247, “HCV genotype 1-infected” is incorrectly recognized as a species by the baseline, but AIONER can correctly detect it as a disease. (ii) Better boundary detection: AIONER also presents higher accuracy in detecting the entity boundaries. For example, in the case of “Vogt-Koyanagi-Harada (VKH) syndrome”, the baseline detects the wrong boundaries and wrongly identifies it as two entities, “Vogt-Koyanagi-Harada” and “(VKH) syndrome,” while AIONER correctly recognizes it. (iii) Slightly higher recall: Some entity spans that are not shown in the training set may be missed by the baseline, but AIONER can handle those unseen entities better.
Although AIONER exhibits promising performance for multiple named entity recognition, there are still some errors in the results. We have reviewed the errors of the model with the best performance (BioRED + All via AIONER) on the BioRED test set and sorted the errors based on the percentages as shown in Fig. 5. (i) Incorrect boundary (36.0%): most errors are caused by incorrect boundaries in the extracted mentions. In this error type, the most critical issue is which leading or trailing tokens fall within the mention boundary. For example, “Necrotising fasciitis” (MeSH: D019115) is the disease mention that should be detected, but our method missed the first token “Necrotising” which can help to narrow down the entity more specifically. (ii) Entity type ambiguity (26.6%): This error type contains two major errors. First, the same entity mention may have different entity types in the text. For example, Growth hormone is a protein encoded by the gene, i.e. a member of the somatotropin/prolactin family which plays an important role in growth control and is also a drug (chemical) for the treatment of the growth hormone deficiency. In different context, it can be annotated differently. Second, different entities may be ambiguously named. For example, BMD gene is the corresponding gene of the Becker Muscular Dystrophy (BMD). Both the gene and the disease are named BMD. (iii) Natural language mentions (10.4%): Pre-trained language models are trained on large natural language texts. However, it is still very difficult to accurately identify the entities written in descriptive natural language (e.g. the variant of “phenylalanine to the polar hydrophilic cysteine in exon 6 at codon 482”). Similarly, composite spans—which mention multiple entities—also confuse the models (e.g. D1 or D2 dopamine receptors). Such natural language mentions are rare in the training set and individually unique, making recognition challenging. Other smaller error types include missed entities, which are mostly abbreviations with insufficient definition, entities not in the recognition scope which were wrongly detected, multiple spans of the same entity were detected inconsistently, and others. Our future work will focus on these problems, incorporating linguistic information (e.g. part of speech and syntactic information) and dictionary resources into our method to further enhance the model’s performance.
Figure 5.
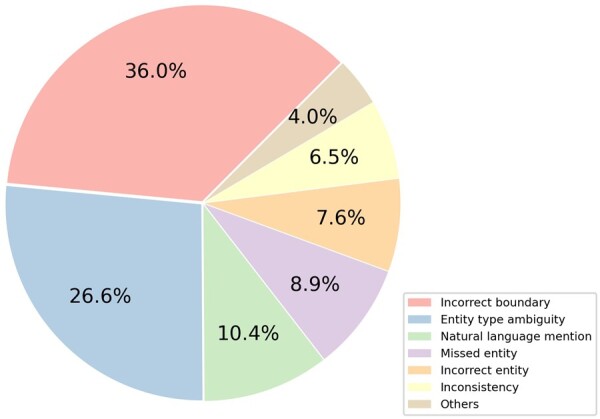
Error analysis of the AIONER results on the BioRED test set.
AIONER is a reliable method for recognizing the entities of different types at once. However, AIONER cannot return multiple entities with overlapping boundaries, such as a nested entity span. For example, growth hormone deficiency contains two mentions, “growth hormone”, and “growth hormone deficiency.” AIONER cannot return both simultaneously.
5 Conclusion
In conclusion, we present an AIONER method to integrate heterogeneous corpora for multiple named entity recognition at once. AIONER can develop a single model for multiple entity types with optimized performance for generalizable usage. This implementation also significantly reduces the effort of the process, especially for large-scale data. We also demonstrate that AIONER can be used to further improve the performance of various BioNER tasks, even when the entity types have never been observed before. These results suggest that AIONER is highly robust and generalizable for BioNER. We released the pre-trained AIONER model for standalone usage to support the BioNER tasks. In the future, we will apply the optimized AIONER model on the entire PubMed (abstracts) and PMC full texts for downstream text mining research (e.g. biomedical relation extraction).
Supplementary Material
Contributor Information
Ling Luo, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States; School of Computer Science and Technology, Dalian University of Technology, Dalian 116024, China.
Chih-Hsuan Wei, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States.
Po-Ting Lai, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States.
Robert Leaman, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States.
Qingyu Chen, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States.
Zhiyong Lu, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), National Institutes of Health (NIH), Bethesda, MD 20894, United States.
Author contributions
Conception and design: L.L., C.-H.W., and Z.L. Data collection: L.L., C.-H.W., and P.-T.L. Analysis and interpretation: L.L., C.-H.W., P.-T.L., R.L., and Q.C. Drafting the manuscript: L.L., C.-H.W., R.L., and Z.L.
Supplementary data
Supplementary data is available at Bioinformatics online.
Conflict of interest
None declared.
Funding
This research was supported by the Intramural Research Program of the National Library of Medicine (NLM), National Institutes of Health; the Fundamental Research Funds for the Central Universities [DUT23RC(3)014 to L.L.]
Data availability
The AIONER data underlying this article are available at https://github.com/ncbi/AIONER/tree/main/data.
References
- Arighi C, Hirschman L, Lemberger T. et al. Bio-ID track overview. In: BioCreative VI Workshop, Bethesda, MD, USA: BioCreative, pp. 28–31, 2017.
- Cariello MC, Lenci A, Mitkov R. A comparison between named entity recognition models in the biomedical domain. In: Proceedings of the Translation and Interpreting Technology Online Conference, Online: INCOMA Ltd, pp. 76–84, 2021.
- Caruana R. Multitask learning. Mach Learn 1997;28:41–75. [Google Scholar]
- Chai Z, Jin H, Shi S. et al. Hierarchical shared transfer learning for biomedical named entity recognition. BMC Bioinformatics 2022;23:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Choi W, Lee H. et al. A method for named entity normalization in biomedical articles: application to diseases and plants. BMC Bioinformatics 2017;18:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton G, Pyysalo S, Chiu B. et al. A neural network multi-task learning approach to biomedical named entity recognition. BMC Bioinformatics 2017;18:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin J Chang MW, Lee Ket al. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In: NAACL-HLT, Minneapolis, Minnesota: Association for Computational Linguistics, pp. 4171–4186, 2019.
- Doğan RI, Leaman R, Lu Z. et al. NCBI disease corpus: a resource for disease name recognition and concept normalization. J Biomed Inform 2014;47:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Wang K. Team Bioformer at BioCreative VII LitCovid track: multic-label topic classification for COVID-19 literature with a compact BERT model. In: Proceedings of the Seventh BioCreative Challenge Evaluation Workshop, Online: BioCreative, pp. 272–274, 2021.
- Galea D, Laponogov I, Veselkov K. et al. Exploiting and assessing multi-source data for supervised biomedical named entity recognition. Bioinformatics 2018;34:2474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner M, Nenadic G, Bergman CM. et al. LINNAEUS: a species name identification system for biomedical literature. BMC Bioinformatics 2010;11:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi JM, Bader GD.. Towards reliable named entity recognition in the biomedical domain. Bioinformatics 2020;36:280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorot X, Bordes A, Bengio Y. Deep sparse rectifier neural networks. In: Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, Fort Lauderdale, FL, USA: PMLR, pp. 315–323, 2011.
- Gu Y, Tinn R, Cheng H. et al. Domain-specific language model pretraining for biomedical natural language processing. ACM Trans Comput Healthcare 2022;3:1–23. [Google Scholar]
- Islamaj R, Leaman R, Kim S. et al. NLM-Chem, a new resource for chemical entity recognition in PubMed full text literature. Sci Data 2021a;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islamaj R, Wei C-H, Cissel D. et al. NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition. J Biomed Inform 2021b;118:103779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Kang J. Regularization for Long Named Entity Recognition. arXiv preprint arXiv:.07249. 2021.
- Krallinger M, Leitner F, Rabal O. et al. The CHEMDNER corpus of chemicals and drugs and its annotation principles. J Cheminform 2015;7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafferty J, McCallum A, Pereira F. Conditional random fields: probabilistic models for segmenting and labeling sequence data. In: Proceedings of the Eighteenth International Conference on Machine Learning, San Francisco, CA, USA: Morgan Kaufmann Publishers Inc., pp. 282–289, 2001.
- Lample G, Ballesteros M, Subramanian S. Neural architectures for named entity recognition. In: NAACL-HLT, San Diego, California, USA: Association for Computational Linguistics, pp. 260–270, 2016.
- Kühnel L, Fluck J.. We are not ready yet: limitations of state-of-the-art disease named entity recognizers. J Biomed Semant 2022;13:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman R, Islamaj Dogan R, Lu Z.. DNorm: disease name normalization with pairwise learning to rank. Bioinformatics 2013;29:2909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman R, Wei C-H, Lu Z.. tmChem: a high performance approach for chemical named entity recognition and normalization. J Cheminform 2015;7:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Yoon W, Kim S. et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020;36:1234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Sun Y, Johnson RJ. et al. BioCreative V CDR task corpus: a resource for chemical disease relation extraction. Database 2016;2016:baw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Lai P-T, Wei C-H. et al. BioRED: a rich biomedical relation extraction dataset. Brief Bioinf 2022a;23:bbac282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Wei C-H, Lai P-T. et al. Assigning species information to corresponding genes by a sequence labeling framework. Database 2022b;2022:baac090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pafilis E, Frankild SP, Fanini L. et al. The SPECIES and ORGANISMS resources for fast and accurate identification of taxonomic names in text. PLoS ONE 2013;8:e65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Yan S, Lu Z. Transfer learning in biomedical natural language processing: an evaluation of BERT and ELMo on ten benchmarking datasets. In: Proceedings of the 18th BioNLP Workshop and Shared Task, Florence, Italy: Association for Computational Linguistics, pp. 58–65, 2019.
- Peters ME, Neumann M, Iyyer M. Deep contextualized word representations. In: NAACL, New Orleans, Louisiana: Association for Computational Linguistics, pp. 2227–2237, 2018.
- Pyysalo S, Ananiadou S.. Anatomical entity mention recognition at literature scale. Bioinformatics 2014;30:868–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez NE, Nguyen M, McInnes BT. et al. Effects of data and entity ablation on multitask learning models for biomedical entity recognition. J Biomed Inf 2022;130:104062. [DOI] [PubMed] [Google Scholar]
- Sang ETK, De Meulder F. Introduction to the CoNLL-2003 shared task: language-independent named entity recognition. In: Proceedings of the Seventh Conference on Natural Language Learning at HLT-NAACL 2003, Edmonton, Canada: Association for Computational Linguistics, pp. 142–147, 2003.
- Sayers EW, Beck J, Bolton EE. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2021;49:D10–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Simmons M, Lu Z. et al. Text mining genotype-phenotype relationships from biomedical literature for database curation and precision medicine. PLoS Comput Biol 2016;12:e1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M, Jeong M, Choi Y. et al. BERN2: an advanced neural biomedical named entity recognition and normalization tool. Bioinformatics 2022;38:4837–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Zhuang F, Zhang H. et al. Improving biomedical named entity recognition by dynamic caching inter-sentence information. Bioinformatics 2022;38:3976–83. [DOI] [PubMed] [Google Scholar]
- Viterbi A. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans Inf Theory 1967;13:260–9. [Google Scholar]
- Wang X, Zhang Y, Ren X. et al. Cross-type biomedical named entity recognition with deep multi-task learning. Bioinformatics 2019;35:1745–52. [DOI] [PubMed] [Google Scholar]
- Weeber M, Klein H, de Jong-van den Berg LT. et al. Using concepts in literature‐based discovery: simulating Swanson's Raynaud–fish oil and migraine–magnesium discoveries. J Am Soc Inf Sci 2001;52:548–57. [Google Scholar]
- Wei C-H, Allot A, Leaman R. et al. PubTator Central: automated concept annotation for biomedical full text articles. Nucleic Acids Res 2019;47:W587–W593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C-H, Allot A, Riehle K. et al. tmVar 3.0: an improved variant concept recognition and normalization tool. Bioinformatics 2022;38:4449–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C-H, Kao H-Y, Lu Z. et al. GNormPlus: an integrative approach for tagging genes, gene families, and protein domains. BioMed Res Int 2015;2015:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wührl A, Klinger R. Recovering patient journeys: a corpus of biomedical entities and relations on Twitter (BEAR). In: Proceedings of the Language Resources and Evaluation Conference, Marseille, France: European Language Resources Association, pp. 4439–4450, 2022.
- Zhang Y, Yang Q.. A survey on multi-task learning. IEEE Trans Knowl Data Eng 2022;34:5586–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Qi P. et al. Biomedical and clinical English model packages for the Stanza Python NLP library. J Am Med Inf Assoc 2021;28:1892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo M, Zhang Y.. Dataset-aware multi-task learning approaches for biomedical named entity recognition. Bioinformatics 2020;36:4331–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The AIONER data underlying this article are available at https://github.com/ncbi/AIONER/tree/main/data.