

Abstract
Epidemiologic studies detected an inverse relationship between HDL-C levels and atherosclerotic cardiovascular disease (ASCVD) identifying HDL-C as a major risk factor for ASCVD and suggesting atheroprotective functions of HDL. However, the role of HDL-C as a mediator of risk for ASCVD has been called into question by the failure of HDL-C raising drugs to reduce cardiovascular events in clinical trials. Progress in understanding the heterogeneous nature of HDL particles in terms of their protein, lipid and small RNA composition has contributed to the realization that HDL-C levels do not necessarily reflect HDL function. The most examined atheroprotective function of HDL is reverse cholesterol transport (RCT), whereby HDL removes cholesterol from plaque macrophage foam cells and delivers it to the liver for processing and excretion into bile. Indeed, in several studies HDL have shown inverse associations between HDL cholesterol efflux capacity (CEC) and ASCVD in humans. Inflammation plays a key role in the pathogenesis of atherosclerosis and vulnerable plaque formation, and a fundamental function of HDL is suppression of inflammatory signaling in macrophages and other cells. Oxidation is also a critical process to ASCVD in promoting atherogenic oxidative modifications of LDL and cellular inflammation. HDL and its proteins including apolipoprotein AI (apoAI) and paraoxonase 1 (PON1) prevent cellular oxidative stress and LDL modifications. Importantly, HDL in humans with ASCVD is oxidatively modified rendering HDL dysfunctional and proinflammatory. Modification of HDL with reactive carbonyl species, such as malondialdehyde (MDA) and isolevuglandins (IsoLG), dramatically impairs the antiatherogenic functions of HDL. Importantly, treatment of murine models of atherosclerosis with scavengers of reactive dicarbonyls improves HDL function and reduces systemic inflammation, atherosclerosis development, and features of plaque instability. Here, we discuss the HDL anti-atherogenic functions in relation to oxidative modifications and the potential of reactive dicarbonyl scavengers as a therapeutic approach for ASCVD.
Keywords: Cholesterol efflux, High-density lipoprotein (HDL), Malondialdehyde (MDA), Macrophage, Myeloperoxidase (MPO), 2-hydroxybenzylamine, 5’-O-Pentyl-pyridoxamine (PPM), atherosclerosis
Introduction
In contrast to the direct association of serum LDL-C levels with risk of coronary heart disease, the discovery of the inverse relationship of serum HDL-C levels with the incidence of coronary heart disease in The Framingham Heart Study, resulted in the description of HDL as a protective factor against coronary heart disease.1 This association was found to be independent of other risk factors, including LDL-C, in several early prospective studies.2 These observations identified HDL-C levels as a risk factor and a potential target for therapy of atherosclerotic cardiovascular disease and led to decades of research describing multiple atheroprotective functions of HDL. However, the role of HDL-C as a mediator of risk for ASCVD has been called into question by the failure of clinical trials of HDL-C raising drugs, including niacin, fibric acid derivatives, and several CETP inhibitors, to demonstrate a benefit in reducing cardiovascular events.3 In addition, Mendelian Randomization studies failed to show an association of genetic polymorphisms (SNPs) that impact levels of HDL-C on risk for myocardial infarction and CAD.4, 5 Furthermore, studies with large cohorts found that the number of HDL particles is a better marker for ASCVD risk than HDL-C levels.3 Interestingly, mounting evidence shows that very high levels of HDL-C can be associated with increased risk of ASCVD.6 In this regard, HDL is a collection of subparticles that differ in size, charge, lipid subspecies, and apolipoprotein/protein compositions, all of which can impact HDL functions. Progress in understanding the composition of HDL subpopulations in the areas of proteomics and lipidomics has revealed the complex heterogeneous nature of HDL particles in terms of their protein and lipid composition as well as oxidative modifications.3 Furthermore, HDL carries microRNAs and small noncoding RNAs that may impact its function.7, 8 Together these findings suggest that HDL-C levels do not necessarily reflect the HDL particle number or more importantly, HDL function.3 Consequently, there has been growing interest in HDL’s antiatherogenic functions, such as reverse cholesterol transport (RCT), and its anti-inflammatory and antioxidant properties, as markers of risk and potential therapeutic targets.9 Several HDL based therapies, including recombinant HDL infusions, ApoA-I mimetic peptides, ApoA-I transcriptional supraregulator BET inhibitor RVX-208 (apabetalone), and antagonism of miR-33 are in various stages of development and have been recently reviewed.3 In this review, we will focus on advances in our understanding of three of HDL’s antiatherogenic functions: RCT, anti-inflammatory and anti-oxidant functions of HDL. Furthermore, we discuss recent evidence showing that modification of HDL with reactive dicarbonyl species, such as malondialdehyde (MDA) and isolevuglandins (IsoLG), dramatically impairs all three of these antiatherogenic functions of HDL, and that prevention of these modifications with dicarbonyl scavengers holds promise as a treatment to improve HDL function and reduce the development of atherosclerosis.
The deleterious effects of oxidative modifications of HDL on atherosclerosis have been demonstrated in mouse models showing that administration of oxidized HDL or apoAI promotes inflammatory plaque development, whereas native HDL/apoAI promote lesion regression and anti-inflammatory remodeling.10, 11 Oxidative modification of HDL occurs upon exposure to reactive oxygen species (ROS) and peroxidized lipids. Enzymes that generate ROS and have been implicated in atherosclerosis include myeloperoxidase (MPO), xanthine oxidase, and NADPH oxidases. Indeed, the majority of apoAI in human atherosclerotic plaques is oxidized and associated with MPO.12 ROS generated by these enzymes include hypochlorous acid (HOCl), hypobromous acid (HOBr), hypothiocyanous acid (HOSCN), isocyanate (CNO-), nitrogen dioxide radical (●NO2), superoxide (O2●-), hydroxyl radical (OH●) and peroxynitrite (ONOO−).13 These ROS act on the amino acids of HDL proteins to generate oxidized amino acid species including cysteine (Cys) disulfide bonds (Cys-Cys crosslinks), oxidized Met (oxMet), oxidized tryptophan (oxTrp), modified and tyrosine, including 3-chlorotyrosine (3-Cl-Tyr), nitrotyrosine (NO2-Tyr), tyrosine dimers (Tyr-Tyr), and N-chloroamines. CNO- acts on lysine (Lys) to produce carbamylated-Lys (Cbl-Lys). ROS (especially ●NO2 and ONOO− ) also act on the polyunsaturated fatty acid (PUFA) tail of a phospholipid (PL) or cholesteryl ester (CE) to extract hydrogen, generating a lipid radical that then reacts with molecular oxygen to form a lipid peroxyl radical. If this lipid peroxyl radical can then extract a hydrogen from an adjacent PUFA, the lipid radical reaction rapidly propagates generating both a lipid hydroperoxide and another lipid radical at each step. Lipid hydroperoxides undergo secondary reactions to form reactive lipid dicarbonyls species, including MDA, IsoLG, 4-oxo-nonenal (ONE), and methylglyoxal (MGO), and also reactive lipid monocarbonyl species such as acrolein (ACR) and 4-hydroxy-nonenal (HNE) (Figure 1). Lipid peroxidation also generates oxidized phospholipids, where the lipid dicarbonyls form esterified to phospholipids, including esterified IsoLGs14 and structural analogs of ONE such as 9-keto-12-oxo-10-dodecenoic acid (KODA-PC).15 Various amino acids can then be adducted by these reactive lipid carbonyls to form stable modifications.13 MDA and IsoLG preferentially modify Lys, but Lys can also be modified by HNE, ONE, KODA-PC, and MGO. Cys are readily modified by ACR, HNE, and ONE. Histidines (His) are modified by HNE, ONE, KODA-PC and ACR and arginines (Arg) are modified by HNE, ONE, and MGO. Detailed evidence for contributions of various reactive lipid carbonyls to reducing individual HDL functions will be given in later sections.
Figure 1. Lipid carbonyls react with amino acid residues to form adducts and crosslinks.
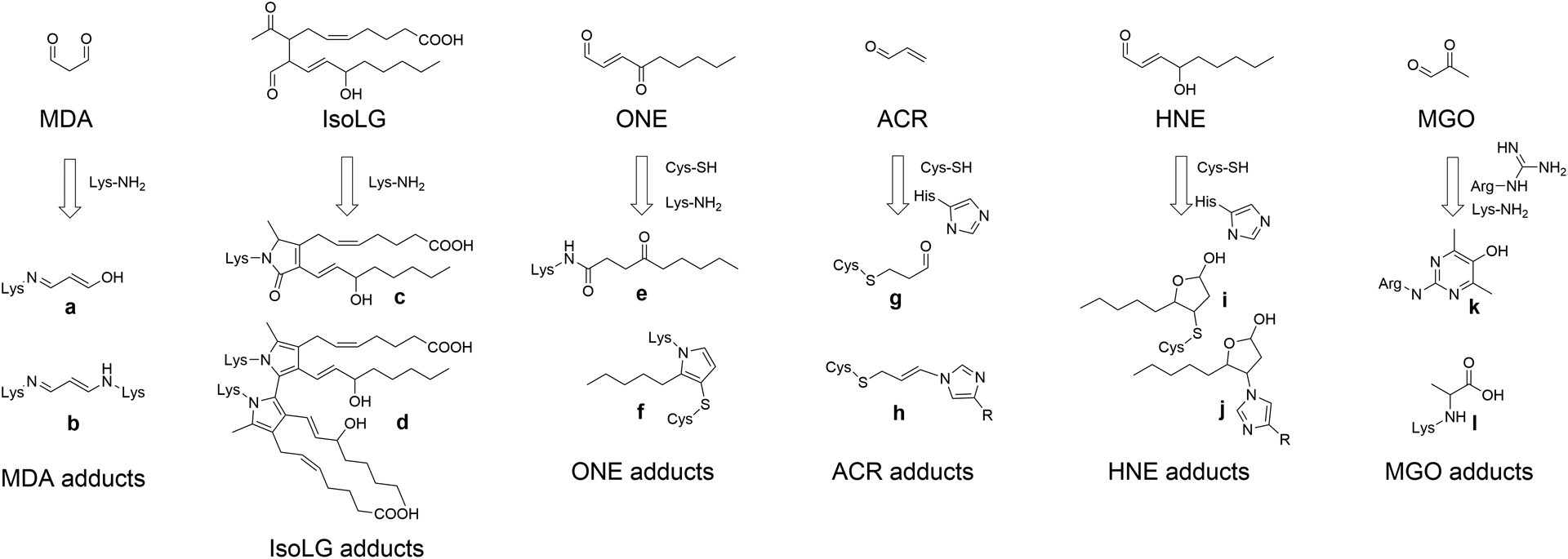
MDA (Malondialdehyde), IsoLG (isolevuglandins), ONE (4-oxo-nonenal), ACR (acrolein), HNE (4-hydroxy-nonenal), and MGO (methylglyoxal) react with their preferred amino acid targets including Lys-NH2 (lysine), Cys-SH (cysteine), His-imidazole (histidine), or Arg-guanidine (arginine) to form various adducts and crosslinks. Crosslinks form when the same lipid carbonyl reacts with two closely adjacent amino acids. In general, multiple species of adducts can form for each lipid carbonyls, two of the most important for each reactive lipid carbonyl are shown here. Adducts shown are: a. MDA N-propenal-Lys monoadduct, b. MDA Lys-1-amino-3-iminopropene-Lys crosslink, c. IsoLG-lactam-Lys monoadduct, d. IsoLG-dipyrrole-Lys crosslink, e. ONE N-4-ketoamide-Lys monoadduct, f. ONE-Lys-pyrrole-Cys crosslink, g. ACR-S-propanol-Cys monoadduct, h. ACR-His-propenamine-Cys crosslink, i. HNE-S-hemiacetal-Cys monoadduct, j. HNE-N-hemiacetal-His monoadduct, k. MGO Argpyrimidine monoadduct, and l. MGO-carboxy-ethyl-Lys.
HDL Reverse Cholesterol Transport (RCT)
First described by Glomset,16 RCT is the process by which HDL removes excess cholesterol from peripheral tissues for transport through the plasma and delivery to the liver for either direct excretion or conversion into bile acids and subsequent secretion into bile (Figure 2). Studies have established the atheroprotective effects of HDL in mediating RCT in atherosclerotic animal models.17 However, the lack of effects of HDL-C raising treatments on reducing CAD risks in humans have called into question the atheroprotective effects of HDL RCT in humans. In this regard, the heightened oxidative stress in CAD likely impairs every step of RCT by promoting oxidative modifications of HDL thereby negating any benefit of raising HDL-C. Here, we discuss the steps of HDL RCT and their relevance to atherosclerosis and the role of oxidative stress.
Figure 2. Critical steps of the HDL reverse cholesterol transport (RCT) pathway.
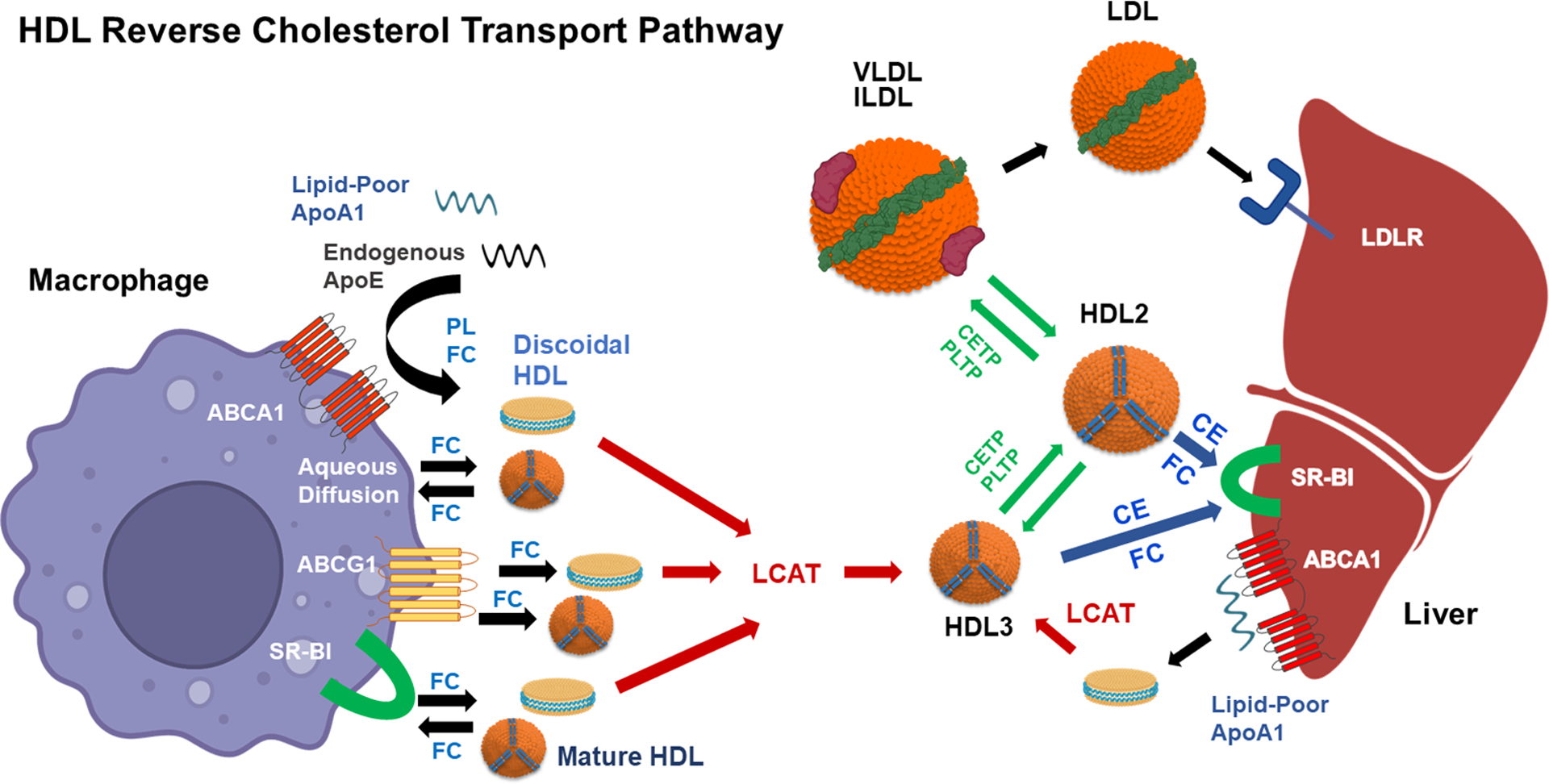
The first step, which is rate limiting, is the removal of free cholesterol (FC) from macrophage foam cells. FC is released from macrophages by four mechanisms. ABCA1 (ATP-binding cassette transporter A1) releases PL (phospholipid) and FC to lipid-poor apoAI or endogenous apoE that is secreted by macrophage foam cells. FC is released to the discoidal HDL formed by ABCA1 and to mature HDL by ABCG1, SR-BI (scavenger receptor-class BI), and aqueous diffusion. The flux of FC between cells and HDL is bidirectional. FC influx occurs by SR-BI and aqueous diffusion. The FC in discoidal HDL particles is esterified by LCAT (lecithin:cholesterol acyltransferase) to form mature HDL3. Plasma HDL is remodeled by CETP (cholesteryl ester transfer protein) and PLTP (phospholipid transfer protein). PLTP transfers PL between VLDL and HDL and among HDL particles. CETP transfers triglyceride from VLDL/IDL to HDL3 to form larger HDL2 particles. CETP also transfers HDL CE to VLDL and IDL to form LDL, which is then internalized by the hepatic LDLR (LDL receptor) for cholesterol routing into bile. An alternative pathway for hepatic delivery of cholesterol and routing to bile is the selective uptake of HDL CE and the influx of HDL FC via SR-BI. CETP also transfers oxidized lipids from LDL to HDL for delivery to the liver by SR-BI. Nascent HDL is synthesized by interaction of hepatocyte or enterocyte (not shown) derived apoAI with hepatic or intestinal ABCA1, and then nascent HDL is routed to peripheral tissue to act as a FC acceptor. Created with BioRender.com.
Cholesterol Efflux
The rate-limiting step in HDL RCT for protection against atherosclerosis is the efflux of free cholesterol (FC) to HDL from macrophages. Alleviation of the macrophage cholesterol burden is critical to the inhibition of plaque progression as excess membrane FC promotes inflammatory signaling and cell death (Discussed later). Cholesterol transport from macrophages to HDL/apoAI occurs by four mechanisms (Figure 2). ATP-binding cassette transporter A1 (ABCA1) mediates the efflux of phospholipid and FC to lipid-poor apoAI or pre-β HDL.17–19 In addition, endogenous apoE is an efficient acceptor for ABCA1-mediated cholesterol efflux, and the absence of macrophage apoE markedly accelerates atherosclerosis.19, 20 Cholesterol efflux to discoidal and mature spherical HDL occurs via ATP-binding cassette transporter G1 (ABCG1) and scavenger receptor-class BI (SR-BI).18, 21 In vitro studies have shown that, while a large part of the total cholesterol efflux from mouse macrophages to serum or the apoB-depleted serum fraction occurs via ABCA1 (35%) versus ABCG1 (21%) or SR-BI (9%), a large portion (35%) also occurs by aqueous diffusion and/or unknown mechanisms.17, 22 In contrast, cholesterol efflux from human macrophage foam cells is largely independent of ABCG1, and SR-BI plays a major role.17 The finding that apoAI in normal human arteries is mostly lipid-poor, suggests that ABCA1 mediated cholesterol efflux is a major mechanism in preventing atherosclerotic lesion formation.23 However, studies suggest that the lipid-poor apoAI has a limited capacity to sustain net cholesterol efflux via ABCA1.24 In this regard, ABCG1, SR-BI, and aqueous diffusion are likely important for driving cholesterol efflux to the discoidal particles formed via ABCA1, and all the mechanisms may act in a synergistic fashion.18, 21 In mediating cholesterol efflux, ABCA1 interacts with apoAI and recycles back and forth from the plasma membrane to endo/lysosomal compartments to facilitate release of FC derived from hydrolysis of lipid droplet CE by either neutral esterase or acid lipase in the process of autophagy.25,26 In contrast, ABCG1 does not interact with HDL, but localizes to intracellular vesicles to traffic FC from the golgi and ER to the plasma membrane for release to the HDL.27 SR-BI acts intracellularly to facilitate CE clearance by operating directly as part of the VPS34/Beclin-1 complex to stimulate autophagosome formation, suggesting that ABCA1 and SR-BI cooperate to enhance lipophagy.28 The flux of FC to HDL is bi-directional, and the net flux is determined by the direction of the FC gradient, which is influenced by the FC and phospholipid subspecies content of the HDL and plasma membrane.17, 29 In this regard, HDL with an increased FC/phospholipid ratio, such as HDL from SR-BI deficient mice, promotes the accumulation of cholesterol in tissues.30 In addition, oxidative modifications of HDL/apoAI, including MDA, carbamylation, and IsoLG, that occur in subjects with FH or CAD, not only can inhibit cholesterol efflux but enhance HDL recognition by scavenger receptors, thereby promoting cholesterol influx via endocytic mechanisms.31–38 In vivo RCT studies in C57BL/6 mice have shown that cholesterol efflux from macrophage foam cells via ABCA1 and ABCG1 is routed to the liver for excretion.39 Consistent with a role for ABCA1 in alleviating macrophage cholesterol burden in plaques, deletion of bone marrow ABCA1 accelerates atherosclerosis in mice, and humans heterozygous for loss of cholesterol efflux function in the ABCA1 gene are at increased CVD risk.18, 40 In contrast, deletion of bone marrow ABCG1 in Ldlr−/− and Apoe−/− mice decreases atherosclerosis, which may stem from compensatory upregulation of macrophage apoE and ABCA1.41, 42 Indeed, a number of variants of the ABCG1 gene in humans have been implicated in enhancing the risk of CAD.43 In contrast to ABCA1 and ABCG1, a study using an in vivo RCT assay with cholesterol-normal cells in C57BL/6 mice suggested that macrophage cholesterol released via SR-BI is not preferentially transported to the liver for processing and excretion.39 However, another study by Zanotti and colleagues showed that cholesterol released via SR-B1 is routed for excretion in mice treated with an LXR agonist.22 These conflicting results are likely due to differences in assays, as the study by Zanotti et. al. used macrophage foam cells to examine RCT, which would increase the net cholesterol efflux potential via SR-BI, and also treatment of mice with the LXR agonist increased the number of enlarged α-migrating HDL particles, which are a preferred acceptor for SR-BI.22, 29 It should also be noted that the contribution of ABCA1, ABCG1, and SR-BI mediated cholesterol efflux to RCT in vivo has never been tested in atherosclerotic mouse models (Ldlr−/−, Apoe−/−), where oxidative stress clearly plays a role. In this regard, studies have established that cholesterol efflux via both ABCA1 and SR-BI to oxidized forms of HDL can be severely impaired, but some modifications (i.e. carbamylation) impair SR-BI cholesterol efflux without impacting ABCA1 activity.32, 38 In addition, in vivo MPO-mediated oxidation impairs in vivo RCT from macrophage foam cells in wildtype C57BL/6 mice and reduces HDL CEC.44 Furthermore, the premise that cholesterol efflux via SR-BI is atheroprotective is substantiated by studies showing that deletion of macrophage SR-BI accelerates plaque development in Ldlr−/− and Apoe−/− mice, and that macrophages in Scarb1−/− plaques are severely engorged with lipid inclusions with only 3% of their cytoplasm lipid-free.45–47 In addition, SR-BI deficient macrophages exhibit lipid engorged lysosomes which are enriched in FC, which is consistent with the role of SR-BI in autophagy.28, 47 Elucidation of the role of macrophage cholesterol efflux via SR-BI in human atherosclerosis is complicated in that SR-BI is a multifunctional receptor that is expressed in multiple tissues. However, carriers of the P376L variant in SR-BI are at increased risk of CAD, whereas carriers of the P297S variant do not have enhanced CAD risk.48–50 These differences are likely due to the degree of loss of SR-BI function and HDL dysfunction.48, 50 Indeed, P376L variant carriers have increased plasma levels of oxidized HDL.49 Interestingly, macrophage SR-BI internalizes oxidized LDL as well as oxidized forms of HDL for degradation rather than mediating efflux, thereby promoting cholesterol accumulation.32, 38 At present it is unknown, whether, similar to other scavenger receptors (i.e. CD36, LOX-1, SRA1) that mediate an inflammatory response to oxidized lipoproteins, SR-BI internalization of oxidized HDL is proinflammatory.31, 51, 52 It is possible that SR-BI provides an anti-inflammatory signaling pathway for removal of toxic HDL/LDL as it does in the efferocytosis of plaque apoptotic cells.53
Lecithin:cholesterol acyltransferase (LCAT)
Once in plasma, HDL FC is esterified by LCAT (Figure 2). ApoAI activates LCAT to drive the net movement of cholesterol from the periphery to plasma. The importance of LCAT in RCT was substantiated in studies showing in vivo that there is enhanced macrophage cholesterol efflux and subsequent cholesterol delivery to the liver and biliary excretion in atherosclerotic Ldlr−/− mice and cynomolgus monkeys treated with a novel LCAT activator.54 In addition, tracer studies in humans suggested that the CE formed by LCAT is preferentially routed for excretion.55 Nonetheless, the association of LCAT activity with CAD risk in humans is controversial. While there is a strong association between HDL-C levels and LCAT variants in humans, there is no link to increased CAD risk.56 Similar results were observed in a large Mendelian randomization study.57 In addition, humans that have complete loss of LCAT activity are not at increased risk for the development of CAD, which is likely due to their extremely low levels of LDL-C.58 In this regard, subjects that are heterozygous for LCAT loss of function mutations exhibit higher LDL-C and lower HDL-C levels and have increased incidence of CAD.17 Interestingly, studies have also demonstrated that a number of atherosclerotic mouse models that have increased oxidative stress naturally, and/or from consuming a western diet, have impaired LCAT activity resulting from oxidation of apoAI or LCAT.59–61 Importantly, overexpression of LCAT or increased activation of LCAT reduces atherosclerosis in a number of mouse models including Ldlr−/−, Scarb1−/−, and Ldlr−/−;ob/ob mice.54, 61, 62 It is also worth noting that the plasma and plaques of humans with CAD contain oxidized forms of apoAI that are known to inhibit LCAT, including nitrated-apoAI, acrolein-apoAI, and carbamylated-apoAI.32, 63–66 In this regard, studies are consistent with impaired LCAT activity being critical to CAD in humans as LCAT activity is significantly decreased in humans with CAD compared to controls67. In a Phase 2a study, MEDI6012, an active recombinant human (rh)LCAT, showed safety and increased HDL-C, apoAI, non-ABCA1 mediated CEC and reduced LDL particles, suggesting enhanced RCT.68 However, in a recent Phase 2b study, treatment of patients with acute ST-segment–elevation myocardial infarction with MEDI6012 did not result in a significant reduction in infarct size or noncalcified plaque volume at 12 weeks.69 Limitations of this study included a smaller than anticipated infarct size that may have limited achievement of a significant outcome. Further studies will be required to demonstrate whether treatment with rhLCAT can promote RCT and improve ASCVD outcomes.
Plasma phospholipid transfer protein (PLTP)
PLTP transfers phospholipids among HDL subpopulations and between VLDL and HDL (Figure 2). Remodeling of HDL in interstitial fluid by PLTP results in the formation of lipid-poor apoAI or pre-β HDL to act as acceptors of cellular cholesterol.70 Despite the increased production of pre-β HDL by interstitial PLTP, increased plasma PLTP activity is associated with enhanced atherosclerosis in humans and mice, effects that are likely due to reduced production of nascent HDL by hepatic ABCA1 and increased VLDL production.70 Thus far, little is known about the effects of HDL oxidation on PLTP activity. However, in vitro studies found that HOCl− prevents PLTP mediated dissociation of pre-β HDL from HDL3, which could impact RCT.71
CE Transfer Protein (CETP)
HDL is further remodeled in humans by CE transfer protein (CETP), which is absent in mice. CETP transfers CEs from HDL to VLDL and triglycerides from VLDL to HDL resulting in the conversion of VLDL to IDL and LDL(Figure 2).9 Importantly, CETP activity improves the ability of HDL to prevent oxidation of LDL by transferring peroxidized lipids from LDL to HDL for inactivation by HDL, as well as by delivery to the liver directly or via SR-BI.72, 73 Indeed, humans with homozygous CETP deficiency have increased MDA-LDL74 and oxidized HDL enriched with modified phospholipids.75 In addition, CETP expression in mice enhances the hepatic uptake of HDL CE via SR-BI in vivo, which may be due to its transfer activity as well as by CETP activity enhancing SR-BI expression.76, 77 Furthermore, studies suggested an independent mechanism for direct delivery of CE to the liver by CETP.78 However, it is worth noting that other studies showed that transgenic expression of CETP in mice increases RCT by promoting clearance of cholesterol via the hepatic LDL receptor.79 In addition, studies have reported variable results on the effects of transgenic expression of CETP on atherosclerosis in mice, which is likely due to differences in the mouse model tested.76, 80 In this regard, CETP likely enhances atherosclerosis in mice under conditions where hepatic clearance of LDL/remnant particles is impaired (i.e. Ldlr−/− and Apoe−/−). Similarly, the impact of CETP on atherosclerosis in humans has been variable, which may depend upon the populations examined (sex, CETP variants, alcohol consumption) and/or the degree of CETP activity (for an in depth review see,81). A number of studies showed that CETP activity was inversely associated with CAD and/or that lower activity was associated with CVD events or mortality including the Framingham Heart Study, the LURIC Study, and KAROLA.81, 82 In contrast, a recent large Mendelian randomization study in humans with CETP genetic variants found that a decrease in CETP activity was associated with reduced CAD risk, but the effect was suggested to be due more to the decrease in apoB levels (−1.4 mg/dL) than the increase in HDL-C levels (+4.6 mgd/L).83 However, other studies on CETP genetic variants suggested a higher risk of CAD.81, 82, 84 In addition, humans with complete loss of function due to mutations in the CETP gene have 3 to 6-fold higher HDL-C levels compared to controls, but they have enlarged HDL that is enriched with apoCIII, apoAII, and apoE and has reduced PON1 activity and CEC.82, 85 Regardless of the controversy over the atherogenic effects of CETP, the premise that CETP inhibition would raise HDL-C, lower LDL-C, and presumably increase direct delivery of HDL-C to the liver, led to the development of CETP inhibitors such as torcetrapib, dalcetrapib, anacetrapib, and evacetrapib as potential therapeutic treatment for CAD in humans. However, these clinical trials have been largely disappointing. Administration of torcetrapib in humans with enhanced CVD risk, increased CVD events and mortality while raising HDL-C by 72%.86 Despite raising HDL-C by 31% and 130%, respectively, dalcetrapib and evacetrapib had no effect on CVD events.87, 88 Although anacetrapib reduced CVD events, the effect was attributed to lower LDL-C levels and not an increase in HDL, and the sponsor decided not to pursue approval.89 Similar to CETP deficiency, CETP inhibition in humans led to the accumulation of apoC3 and apoE enriched HDL particles, which have impaired CEC and are associated with increased CAD risk.90 Importantly, CETP inhibition in humans failed to enhance in vivo HDL RCT.91 These observations suggest that uptake via the LDL receptor is the preferred route for delivery of cholesterol to the liver in humans. However, the findings that CETP can stimulate HDL CE selective uptake via SR-BI and that loss of CETP decreases hepatic SR-BI expression raises the possibility that CETP inhibition may also have impacted cholesterol delivery via the SR-BI pathway.76, 77 Importantly, CETP inhibition in humans decreases the proportion of HDL particles containing PON1 and in vitro studies have shown that CETP inhibition markedly impairs PON1 activity.90, 92 Taken together, these studies substantiate that raising HDL-C is not beneficial without improving the number of functional HDL particles and suggest that CETP inhibition will exacerbate the effects of the heightened oxidative stress in individuals with CAD, thereby worsening HDL dysfunction in promoting RCT and preventing oxidation. Indeed, Hine et al.73 proposed that the failure of CETP inhibitors to reduce atherosclerosis maybe due to the loss of CETP activity reducing LDL oxidation. In contrast, Schmidt et al. suggested that the failures of CETP inhibitors are likely compound and due to the heterogeneity of effects of the candidate drugs tested.93 Furthermore, they performed Mendelian randomization analyses that support CETP as an effective target for CHD prevention.93 A potential limitation of CETP inhibition is that it promotes accumulation of mature HDL; whereas treatment with dicarbonyl scavengers has the potential to block the modification of HDL in vivo and to protect the function of newly formed HDL.
Delivery of HDL-C to the Liver
In humans, the cholesteryl esters transferred from HDL to LDL by CETP are internalized by the hepatic LDL receptor for processing and/or trafficking of the cholesterol into bile, comprising a major route of delivery of cholesterol from HDL to the liver in humans (Figure 2). The importance of this pathway is exemplified in humans with FH who have markedly elevated levels of LDL-C due to mutations in the Ldlr gene9. The lack of this route in FH subjects leads to oxidation of LDL and HDL resulting in HDL dysfunction due in part to highly reactive dicarbonyls, including MDA, ONE, and IsoLG.34–37 In mice, the main route of hepatic delivery is selective uptake of HDL CE and influx of HDL FC via SR-BI (Figure 2).94, 95 Remodeling of HDL by CETP and hepatic triglyceride lipase enhances HDL CE uptake via SR-BI.96 Although less substantial than the LDLR route, SR-BI also mediates direct delivery of HDL-C in humans (Figure 2) as substantiated by markedly elevated levels of HDL-C in subjects with loss of CE selective uptake function mutations in the Scarb1 gene.48–50 As shown recently, human carriers of the P376L Scarb1 variant also have elevated oxidized HDL.49 In addition, human carriers of a number of the Scarb1 gene variants have markedly elevated levels of lipoprotein (a) Lp(a), an atherogenic LDL particle that is heavily enriched in oxidized lipids, suggesting that SR-BI is the preferential route for selective uptake of Lp(a) cholesterol.97 In this regard, studies have shown that an in vivo function of SR-BI is to detoxify both HDL and Lp(a) via the selective uptake of oxidized phospholipids and CEs, thereby reducing the atherogenicity of Lp(a) and preserving HDL antioxidant status.98, 99 Regardless of the pathway, it is likely that the increased oxidative stress in humans with CAD would impair both delivery routes for hepatic processing of cholesterol to the feces as the selective uptake of HDL CE via SR-BI is severely impaired by both HDL and LDL oxidation, and oxidation of LDL/HDL promotes uptake by nonparenchymal cells and macrophages in other tissues.100, 101
Measurements of HDL RCT in Humans and Relevance to Atherosclerosis
As the most easily measured step in RCT, emphasis has been placed on measurements of HDL CEC as it relates to CAD and CVD events and mortality in humans. Most studies have used assays geared toward measurement of ABCA1-mediated HDL CEC involving upregulation of ABCA1 with cAMP in cholesterol-normal mouse macrophages. Using this system, Khera and colleagues were the first to report an inverse relationship between CAD and HDL CEC that was independent of HDL-C, suggesting that HDL function is more relevant to CAD risk.102 This premise was then bolstered by other studies demonstrating an inverse association between HDL CEC and incidents of CVD events.103, 104 However, in patients with high C reactive protein (CRP), there was no association of HDL CEC with incident CVD events at baseline, but after statin treatment and lowering of CRP, an inverse association was detected.105 Similar results were observed in a CKD population.106 These studies raise the possibility that measurements of ABCA1 CEC do not provide a reliable assessment of CVD event risk in populations with heightened inflammation. However, studies using cholesterol loaded THP-1 cells, which would test the overall ability of HDL to alleviate the macrophage cholesterol burden via multiple mechanisms and are likely more representative of human plaque cells, also found no association between HDL CEC and CAD and/or CVD events in subjects with either Type 2 Diabetes Mellitus or normal glucose metabolism.107 Similar results were observed in subjects with end stage renal disease.108 Taken together, the relationship between HDL CEC and CVD events is likely lost in populations with more vulnerable plaques with increased inflammation and oxidative stress. Indeed, other studies using the ABCA1 CEC system have shown positive associations between HDL CEC and major CVD events, including in populations with either CKD or enriched with smokers.104, 109 Similarly, earlier studies with smaller populations showed a positive association with major CVD events, which is consistent with studies showing that plasma pre-β HDL levels are positively associated with CAD and CVD events.110–112 Indeed, a number of populations with increased inflammation and oxidative stress (i.e. diabetics, hypertriglyceridemic, renal impaired, postmenopausal women with CAD) have increased pre-β HDL levels and ABCA1 CEC compared to controls.104, 113–116 Taken together, these observations highlight that in certain populations with systemic inflammation, HDL RCT is impaired in vivo either via decreased expression or function of ABCA1 and/or decreased maturation of HDL (i.e. decreased LCAT). In this regard, a number of these populations have impaired LCAT activity.67, 116–118 In addition, novel single cell analyses determined that proinflammatory macrophage populations (IL-1β+) in advanced plaques of humans express lower levels of lipid handling genes including ABCA1, ABCG1, and apoE.119 These observations also stress that measuring whole body HDL RCT may be a better predictor of CAD and CVD events in humans. The novel method recently developed by Cuchel and colleagues using [H3]-cholesterol-labeled nanoparticles to track the movement of macrophage cholesterol to feces in humans will hopefully make this examination feasible.120
HDL Anti-inflammatory Function
The CANTOS trial supports the importance of inflammation in atherosclerotic cardiovascular disease as treatment of humans with an IL-1β monoclonal antibody lowered recurrent cardiovascular events independent of cholesterol lowering.121 Abundant evidence supports that HDL/ apoAI impairs inflammatory signaling, and the CANTOS trial substantiates the need to preserve HDL anti-inflammatory signaling pathways in order to reduce the residual risk of CVD events. Increased oxidative modifications of HDL/ApoAI not only impair anti-inflammatory signaling but generate extremely proinflammatory HDL particles.31, 32, 34, 36, 51, 122, 123 HDL exerts anti-inflammatory effects on all cell types relevant to atherosclerosis, which has been detailed in recent reviews.124 Here, we discuss the effects of HDL/ApoAI on endothelial activation, monocytosis, and macrophage inflammation as it relates to atherosclerosis and oxidative stress.
Effects of HDL/ApoAI on Endothelial Cell Activation
One of the early events in atherosclerosis is endothelial cell activation and a compromised endothelial barrier.9 Triggers such as oxidized LDL and TNF-α activate nuclear factor kappa B (NF‐κB) transcriptional activation leading to increased expression of inflammatory monocyte adhesion molecules and cytokines and downregulation of endothelial nitric oxide synthase (eNOS), thereby promoting increased monocyte infiltration (Figure 3). HDL is critical to reducing the inflammatory response by impairing endothelial cell activation and maintaining endothelial barrier integrity via activation of eNOS (see detailed review,125). Interaction of endothelial cell SR-BI with HDL activates eNOS by stimulating a signaling cascade involving Src Tyrosine kinase, PI-3K, Akt kinase, and Erk1/2 MAPK and phosphorylation of eNOS.125 Cholesterol efflux via SR-BI is required for activation of eNOS.126 ABCG1 also promotes NO production by stimulating efflux of cholesterol and oxidized cholesterol and relocating eNOS away from caveolin-1.127 In addition, the HDL bioactive lipid, sphingosine-1-phosphate (S1P), which is complexed to apoM on HDL, also mediates activation of eNOS by interaction with S1P1/3 receptors to induce PI-3K, Akt signaling.125 The importance of the HDL/apoM/S1P complex in maintaining vascular integrity has been demonstrated in vivo where deletion and/or transgenic expression of apoM have opposite effects on barrier function.128, 129 It is also worth noting that both of the HDL SR-BI and S1P1/3 receptor signaling pathways stimulate endothelial cell survival and migration.125 Importantly, the HDL induced production of NO in endothelial cells reduces NF‐κB transcriptional activation, thereby decreasing expression of monocyte adhesion molecules (VCAM-1, ICAM-1, and P-selectin) (Figure 3) and proinflammatory receptors (toll-like receptor 2, TLR2) and cytokines (MCP-1 and IL-8).125 In addition, HDL/ApoAI binding to SR-BI inhibits adhesion molecule expression by activating Akt to upregulate anti-inflammatory heme oxygenase-1 expression and 3-beta-hydroxysteroid-delta reductase.125, 130 The HDL signaling pathways via S1P1/3 receptors and ABCG1 also prevent activation of endothelial cell nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 (NLRP3) inflammasome and pyroptosis.131, 132 A number of studies have suggested that HDL induced endothelial cell NO production is relevant to atherosclerosis in humans. HDL from subjects with Type 2 Diabetes have reduced ability to activate eNOS and HDL bound S1P is inversely associated with the severity of CAD.133, 134 HDL from CAD versus control subjects has reduced capacity to stimulate eNOS and prevent inflammatory/adhesion gene expression in endothelial cells.51, 135 Importantly, studies have shown that infusion of functional HDL into hypercholesterolemic or diabetic humans and subjects with low HDL due to heterozygous ABCA1 mutations stimulates eNOS activation and vasodilation.125, 136, 137 In addition, a recent study demonstrated that HDL endothelial cell anti-inflammatory capacity was inversely associated with incident CVD events in a general population, effects that were independent of HDL-C levels and HDL CEC.135 Interestingly, studies have shown that HDL from CAD subjects activates protein kinase CβII via LOX-1 leading to inhibition of eNOS activity via a mechanism involving reduced HDL PON1 activity and oxidative modification by endothelial cells.51 In addition, inhibition of PON1 in HDL from control subjects leads to MDA modification of HDL and activation of the LOX-1/PKCβII pathway, showing that oxidative modification impairs HDL endothelial cell anti-inflammatory capacity.51 Furthermore, a recent study demonstrated that the plasma levels of HDL with LOX-1 binding capability in a Japanese population were associated with coronary artery calcification independent of HDL-C and particle number.138 Taken together, these studies highlight that HDL endothelial cell anti-inflammatory capacity and/or HDL oxidative modifications may be a viable marker of incident CVD risk.
Figure 3. The anti-inflammatory functions of HDL.
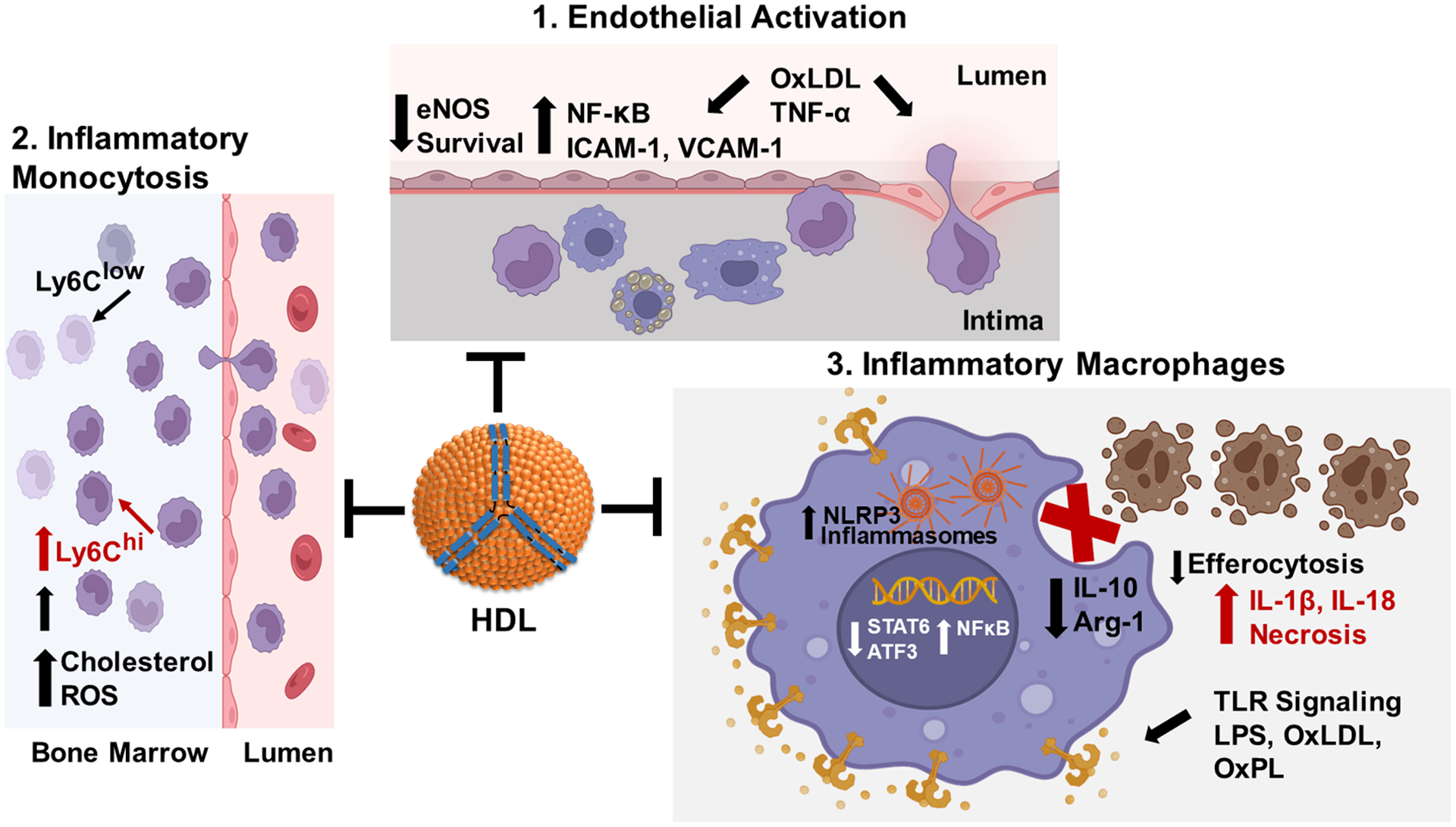
1. HDL suppresses endothelial activation in response to proinflammatory stimuli such as TNF-α and oxLDL. Proinflammatory stimuli activate NF‐κB (nuclear factor kappa B), which increases expression of monocyte adhesion proteins and chemotactic/inflammatory cytokines, thereby increasing monocyte recruitment and endothelial cell death. HDL signaling pathways stimulate nitric oxide production to decrease NF‐κB activation and maintain endothelial barrier integrity. 2. HDL prevents inflammatory monocytosis. Increased LDL and oxLDL, resulting from hypercholesterolemia, induce inflammatory signaling and ROS (reactive oxygen species) leading to expansion of bone marrow granulocyte monocyte progenitors that give rise to inflammatory Ly6Chigh monocytes. HDL signaling pathways suppress ROS production and inflammatory signaling and expansion of Ly6Chigh monocytes. 3. HDL prevents the macrophage proinflammatory phenotype. LPS, oxLDL, and other TLR (toll-like receptor) ligands activate NF‐κB leading to increased expression of inflammatory cytokines and NLRP3 (nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3) inflammasome components resulting in decreased efferocytosis of apoptotic cells and necrotic death. HDL suppresses TLR signaling and NF‐κB activation via cholesterol efflux dependent and independent pathways to promote activation of Atf3 (activating transcription factor 3) and STAT6 (signal transducer and activator of transcription 6), thereby reducing activation of inflammasomes and enhancing efferocytosis of apoptotic cells by promoting expression of Arg-1 (arginase-1) and IL-10. Created with BioRender.com.
Effects of HDL/ApoAI on Monocytosis
The number of blood monocytes are increased in humans with hypercholesterolemia and CAD (Figure 3).139 Studies in mice have shown that inflammatory Ly6Chi versus anti-inflammatory, patrolling Ly6Clow monocytes dominate hypercholesterolemia-associated monocytosis in mice and give rise to inflammatory macrophages in the atheroma (Figure 3).140 Human monocytes are heterogenous, but have been divided into three major subsets based on the differential expression of the lipopolysaccharide (LPS) receptor (CD14) and the FcγIII receptor (CD16) (for in depth reviews see139, 141). The classical CD14++CD16− and non-classical CD14+CD16++ cells subsets are the human equivalent of the mouse Ly6Chi and Ly6Clow monocytes.139 However, it was later determined that the CD16+ positive monocytes possess both anti-inflammatory and inflammatory properties, and this subset was further divided into non-classical CD14+CD16++ and intermediate CD14++CD16+ cells. CD14++CD16+ cells share some properties with both classical and non-classical monocytes but are distinguished by CCR5 (RANTES, MIP-1α, and MIP-1β receptor) expression and by the ability to produce inflammatory cytokines (IL-1β and TNF-α) and ROS.139 Indeed, studies have shown that the numbers of intermediate CD14++CD16+ monocytes are independently associated with CVD events in subjects with CKD and CAD and in a randomized population.139, 142, 143 The increased number of blood monocytes in humans and mouse models with CAD results from enhanced proliferation of bone marrow hematopoietic stem and progenitor cells (HSPCs) due to hypercholesterolemia induced lipid raft signaling and ROS production (Figure 3).141, 144 LDL and oxidized LDL induce expansion of granulocyte monocyte progenitors (GMPs), whereas functional HDL prevents the expansion via a number of signaling pathways, including ABCA1, ABCG1, and SR-BI.141, 145, 146 ABCA1 and ABCG1 expressed on HSPCs promote cholesterol efflux to lipid-poor apoAI or HDL, thereby reducing plasma membrane lipid rafts and decreasing cell surface levels of the common β subunit of the IL-3/granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor.141 In addition, HSPCs express abundant apoE, which promotes phospholipid and cholesterol efflux via ABCA1 and ABCG1 to reduce proliferative IL-3/GM-CSF receptor signaling.141 SR-BI interaction with HDL limits proliferation of HSPCs by reducing activation of Akt and p38MAPK resulting in less ROS production.146 The importance of these HDL/apoAI/apoE signaling pathways to HSPCs expansion is substantiated in studies demonstrating that mice with bone marrow deficient in ABCA1, ABCG1, and/or SR-BI have markedly enhanced inflammatory Ly6Chi monocytosis, which exacerbates lesion progression.141, 146 It is also worth noting that HDL from humans with CAD, diabetes, FH, and CKD and subjects who smoke have oxidative modifications, which are known to impair the ability of HDL to promote cholesterol efflux and prevent cellular ROS production, which likely contributes to the enhanced monocytosis and accelerated atherosclerosis in these populations.31–38, 138, 147, 148
Effects of HDL/ApoAI on Macrophages
Studies have established that macrophage inflammation is a key driver of atherosclerosis progression and that inflammatory resolution is critical to preventing vulnerable plaque formation and promoting lesion remodeling and/or regression.9 Macrophages can exhibit pro-inflammatory versus anti-inflammatory properties depending upon the environment, and HDL modulates the macrophage response to inflammatory stimuli. In the plaque, TLR ligands such as LPS and oxidized LDL and phospholipids promote inflammatory cytokine production.9 Intestinally derived HDL3 binds LPS, thereby preventing entry into the plaque.149 HDL/apoAI also directly inhibits TLR signaling by promoting cholesterol efflux via ABCA1/ABCG1 to reduce plasma membrane lipid raft content of TLRs, which results in decreased myeloid differentiation primary response 88 (MyD88) and TIR-domain–containing adapter-inducing interferon-β (TRIF) signaling, culminating in suppression of NF-κB and type I interferon (IFN) activity.150, 151 In addition, HDL reduces inflammatory signaling independently from its CEC function by relocating the TRIF-related adaptor molecule from the plasma membrane as well as by enhancing expression of activating transcription factor 3 to reduce inflammatory gene expression downstream of NF-κB (Figure 3).151, 152 Furthermore, HDL interacts with SR-BI to activate Akt and suppress p38, Jnk, and NF-κB activation and TLR expression.94, 153, 154 The suppression of NF-κB activation by HDL/ApoAI reduces expression of the NLRP3 inflammasome components, thereby reducing TLR priming of inflammasome activation and secretion of IL-1β and IL-18 in response to other signals such as excess FC (Figure 3).155, 156 One of the most important processes to reduce uncontrolled inflammation, necrotic death, and formation of the vulnerable plaque is the efficient efferocytosis of apoptotic cells,9 and evidence is accumulating that HDL facilitates macrophage efferocytosis. HDL induces an anti-inflammatory macrophage phenotype via JAK1 signaling to activate STAT6 leading to enhanced expression of arginase-1, which metabolizes apoptotic cell arginine, resulting in increased activation of Rac1 to facilitate continual rounds of efferocytosis (Figure 3).157, 158 In addition, HDL interacts with T regulatory cell-SR-BI to promote survival and IL-13 secretion, which propagates efferocytosis signaling via stimulation of macrophage IL-10 production (Figure 3).159, 160 As impaired autophagy facilitates plaque inflammasome activation and uncontrolled death, thereby impeding efferocytosis, the roles of ABCA1 and SR-BI in alleviating the efferocyte lysosomal cholesterol burden is also critical to vulnerable lesion formation (Figure 3).26, 28, 161 The importance of the HDL anti-inflammatory pathways has been demonstrated in atherosclerotic mice, where deletion of bone marrow ABCA1, ABCG1, and/or SR-BI markedly increases systemic inflammation and plaque necrosis.28, 53, 150, 156 Similar findings have been demonstrated in mice that are deficient in HDL (i.e. Apoe−/−, Ldlr−/−Apoa1−/−) or that have dysfunctional HDL (Scarb1−/−Ldlr−/−, Scarb1−/−ApoeR61h/h).162–164 Furthermore, humans that are heterozygous for loss of ABCA1 function have enhanced systemic and plaque inflammation165. It is also worth noting that HDL from humans with CAD versus controls have decreased ability to prevent macrophage inflammation in response to TLR ligands, and that oxidative modifications of the HDL heighten proinflammatory signaling via other pathways (Discussed in detail later).31, 32, 34, 36, 37
HDL antioxidant function
Similar to its role in removing cholesterol from peripheral tissues via RCT, HDL also serves to protect other lipoproteins and cells from oxidative damage by removing and inactivating lipid hydroperoxides from other lipoproteins and cells. The mechanisms of HDL’s antioxidant activity have been primarily studied using its effects on LDL oxidation as a model.166 HDL significantly inhibits the peroxidation of LDL lipids by Cu2+, while only undergoing minimal lipid peroxidation itself.167 Importantly, HDL’s antioxidant activity requires enzyme activity rather than its chelation of redox-metals.168 CETP facilitates the transfer of phospholipid and CE hydroperoxides from oxidized LDL to HDL, where these lipid hydroperoxides are reduced to lipid hydroxides, terminating peroxidation chain reactions.72, 73 PON1 appears to be the most important antioxidant enzyme associated with HDL. Other key proteins with antioxidant activity associated with HDL include apoAI, apoA2, apoA4, apoE, apoM, serum amyloid A, and potentially others.166
PON1 associates with HDL through its cooperative binding to both apoAI and adjacent phospholipids (Figure 4).169 Purified PON1 effectively inhibits Cu2+-induced peroxidation of LDL.167 PON1 catalyzes the hydrolysis of lipid peroxides170 as well as catalyzing lipolactonase, paraoxonase, and arylesterase activities, with all of these activities being strongly correlated.171 Importantly, the enzymatic activity of PON1 is markedly increased by its association with apoAI.172 For this reason, apoAI modifications that decrease the ability of PON1 to bind have the net effect of decreasing PON1 activity.34, 173 Besides its direct antioxidant enzyme activity, PON1 may also exert antioxidant effects indirectly by inhibiting MPO activity (Figure 4). Both PON1 and MPO bind to apoAI, and PON1 binding markedly inhibits MPO’s activity (and vice versa).169 Both animal and clinical studies support an important role for PON1 in protecting against atherosclerosis. Transgenic expression of human PON1 reduces atherosclerosis in Ldlr−/−; ob/ob mice174, while deleting Pon1 exacerbates atherosclerosis in Apoe−/− mice175. Two common polymorphisms, L55M and Q192R, are found in the human PON1 gene. These polymorphisms alter PON1 activity, as well as the risk for cardiovascular disease.176–179 Importantly, PON1 activity appears to be markedly reduced in patients with verified cardiovascular disease.180 One potential mechanism for reduced PON1 activity is oxidative modification of PON1 and apoAI.
Figure 4. The antioxidant functions of HDL.
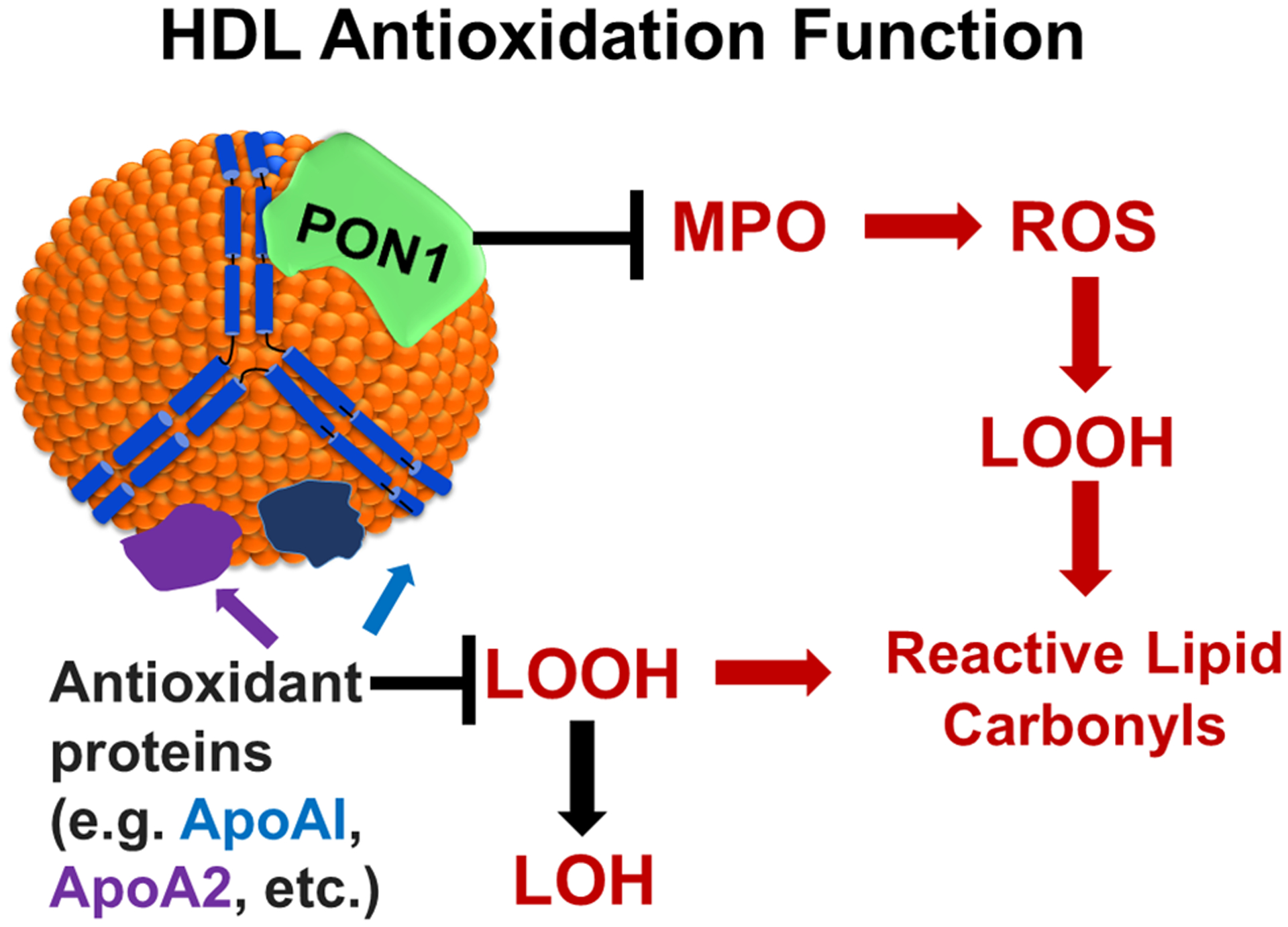
A number of proteins associated with HDL including apoAI and apoA2 prevent the formation of reactive lipid carbonyls by reducing LOOH (lipid hydroperoxides) to LOH (lipid hydroxides). MPO (myeloperoxidase) generates ROS (reactive oxygen species) leading to sequential formation of LOOH and reactive lipid carbonyls. The association of PON1 (paraoxonase 1) with apoAI both inhibits MPO activity and also directly inhibits formation of lipid hydroperoxides and reactive lipid carbonyls.
ApoA1 directly contributes its own antioxidant activity to HDL, in addition to enhancing PON1 activity (Figure 4). Mature, processed human apoAI includes three methionine (Met) residues, Met86, Met112, and Met148. Incubation of either HDL or isolated apoAI with CE hydroperoxides or phospholipid hydroperoxides reduces these lipid hydroperoxides to lipid hydroxides with concomitant oxidation of Met residues.181 In canine apoAI, valine and leucine are substituted for Met112 and Met148, respectively, and canine HDL shows far weaker antioxidant activity than human HDL.181 Interestingly, oxidation of the Met residues of apoAI enhances, rather than diminishes, its capacity to catalyze cholesterol and phospholipid efflux.182
ApoA2 can replace one or more apoAI molecules on an individual HDL lipoprotein particle, with apoA2 contributing 12% to 32% of the total apoAI/2 depending on the individual HDL subfraction examined.183 Recombinant HDL particles generated exclusively with apoA2 show greater capacity to reduce lipid hydroperoxides to hydroxides than those generated with exclusively apoAI (Figue 4).181 However, transgenic mice overexpressing apoA2 show significantly worsened atherosclerosis.184 The effect of apoA2 overexpression may be the result of apoA2 association with native HDL displacing PON1, leading to an overall net negative impact on HDL antioxidant activity.184
ApoA4 is synthesized in the intestine and initially associates with chylomicrons in lymph. In plasma, about 25% of this apoA4 transfers to HDL, while the remaining circulates in a lipoprotein-free fraction.185, 186 Purified apoA4 inhibits lipid peroxidation when Cu2+ is used to oxidize lymph and LDL.187 Overexpression of apoA4 reduces oxidative damage and atherosclerosis in vivo,188 but whether these protective effects depend on its association with HDL remains to be demonstrated.
ApoE associates with chylomicron remnants, VLDL, IDL, and HDL. ApoE has three isoforms (ε2,ε3,and ε4) with ε3 being the most common isoform in human populations. Compared to the ε3, the ε2 and ε4 isoforms increase risk for cardiovascular disease and the ε4 isoform increases risk for Alzheimer’s disease.189 In vitro, all three isoforms provide some measure of protection against against H2O2 induced cytotoxicity or Cu2+ induced LDL oxidation, with rank order of efficacy being ε2>ε3>ε4,190, 191 but when antioxidant activity is measured by protection against macrophage-induced LDL oxidation, the ε4 isoform shows greater efficacy.191 These modest and contradictory differences suggest that the antioxidant activity of apoE is not as relevant to protection against atherogenesis as its effects on lipoprotein clearance and/or the impact of macrophage endogenous apoE effects on cholesterol efflux and inflammation.9
ApoM associates with all lipoprotein particles, but is most enriched in HDL, with about 5% of HDL particles carrying apoM.192 HDL containing apoM more efficiently inhibits Cu2+-mediated LDL oxidation than HDL lacking apoM.192 One potential mechanism for this antioxidant effect is the ability of apoM to bind and sequester oxidized phospholipids193. Ldlr−/− mice with transgenic expression of apoM have significantly less atherosclerosis than non-transgenic Ldlr−/− mice;194 however, whether this anti-atherosclerosis effect is the result of HDL antioxidant activity is unclear.
In summary, the antioxidant activities of HDL derive from multiple components associated with these lipoproteins. For some of these proteins, association with HDL may not be essential to exert their protective effects, but it seems likely that HDL acts as an organizing scaffold to coordinate their activities and thereby maximize their effectiveness.
Oxidative modification of HDL proteins in vivo
HDL isolated from patients with cardiovascular disease or directly from atherosclerotic lesions shows strong evidence of enhanced oxidative modification both by amino acid oxidation and adduction of lipid carbonyls, but whether the extent of modification is sufficient to fully account for HDL dysfunction remains an open question. One challenge to capturing the full extent of HDL modification in vivo is the number of different modifications, especially in terms of those formed by reactive lipid carbonyls. For instance, when IsoLG reacts with Lys, it can form six stable monoadducts (including the IsoLG-lactam-Lys adduct shown in Fig. 1c) and multiple crosslinked Lys adduct species (including the simplest IsoLG-dipyrrole crosslinked adduct shown in Figure 1d). Furthermore, IsoLGs initially form esterified to phospholipids and then undergo hydrolysis by an as-yet-uncharacterized phospholipase, but esterified adducts are difficult to quantitatively convert to their unesterified forms due to their lability to even mild saponification conditions.195 While MDA primarily forms N-propenal-Lys monoadducts (Fig.1a) and Lys-1-amino-3-iminopropene-Lys crosslinks (Figure 1b), it can also form N-dihydropyridine-Lys monoadducts, and small amounts of N-propenal-His monoadducts and N-propenal-Arg monoadducts. ACR can form S-propanal-Cys monoadducts (Figure 1g) and His-propenamine-Cys crosslinks (Fig. 1h), but also N-3-formyl-3,4-dehydropiperidino-Lys monoadducts, N,N-Propano-Arg monoadducts, N-propanal-His monoadducts, and a number other propenamine crosslinks. ONE forms N-4-ketoamide-Lys monoadducts (Fig. 1e) and Lys-pyrrole-Cys crosslinks (Fig. 1f), but also Lys-pyrrole-His crosslinks and Lys-pyrrole-Arg crosslinks. Thus, monitoring all possible post-translational modifications of apoAI or other HDL protein is highly challenging. Given these limitations, typically only levels of the most readily measurable of the oxidative modifications are reported. While this may substantially underestimate the total extent of modification, it does allow for comparison between different patient populations or tissue sites.
Serum HDL isolated from patients with coronary artery disease showed increased levels of 3-Cl-Tyr, NO2-Tyr, and carbamyl-Lys compared to control individuals.12, 196 ApoAI isolated from patients with CAD compared to controls also showed increased 3-Cl-Tyr, NO2-Tyr, and carbamyl-Lys modification196–198. Levels of Lys modified by IsoLG, ONE, ACR, HNE, and MDA are increased in serum HDL isolated from individuals with CAD and/or FH, who are at increased risk of early onset of atherosclerotic cardiovascular disease.34–36, 199 In addition, HNE and MDA adducts as well as carbamyl-apoAI are increased in HDL from individuals with CKD, which is associated with increased risk of ASCVD32, 200. Urinary apoAI from individuals with CKD shows higher levels of IsoLG modification.201
Another challenge to understanding the actual extent of HDL modification in vivo and whether this correlates with disease progression is that modification of HDL proteins such as apoAI appears to primarily occur outside the plasma compartment. This conclusion is based on the findings that levels of apoAI modification are significantly greater for apoAI/HDL isolated from atherosclerotic lesions compared to that isolated from circulation. For example, average levels of apoAI oxTrp72 are 0.07 mmol/mol apoAI in plasma, but are 204 mmol/mol apoAI in aortic plaques.122 Average oxidized apoAI Tyr levels in apoAI isolated from plasma of CVD patients were 0.63 mmol NO2-Tyr/mol Tyr and 0.5 mmol Cl-Tyr /mol Tyr; while average levels in apoAI isolated from human aortic atherosclerotic lesions were 2.3 mmol NO2-Tyr/mol Tyr and 3.9 mmol Cl-Tyr/mol Tyr196. Average levels of carbamyl-Lys in HDL isolated from plasma were approximately 5 mmol/mol apoAI, while in HDL isolated from lesions it is 45 mmol/mol apoAI.32 Using an ELISA specific for MDA-modification, MDA epitopes were 3.6-fold higher in HDL isolated from atherosclerotic lesions than HDL isolated from plasma.33 The ONE-ketoamide-Lys and IsoLG-Lactam-Lys in HDL isolated from plasma of patients with FH are 2.0 mmol/mol apoAI and 0.6 mmol/mol apoAI, respectively, but their levels in atherosclerotic lesions have not been determined. Whether apoAI oxidatively modified in vascular beds enters lymphatics and returns to circulation or is instead trapped by macrophages and dendritic cells in these beds or draining lymph nodes is unknown, but a better understanding of the fate of oxidatively modified HDL would further our understanding of its contribution to disease.
Oxidative modification of HDL causes HDL dysfunction
Effects of HDL oxidiative modifications on RCT
The most direct evidence that oxidative modifications alter HDL function comes from studies where isolated HDL or HDL proteins are exposed to ROS-generating enzymes such as MPO, specific ROS or individual reactive lipid dicarbonyls. The capacity of HDL to facilitate ABCA1-mediated cholesterol efflux from macrophages is markedly reduced after exposure to MPO and cofactors (H2O2, Cl−, and NO2).66, 196 Exposure of HDL to MPO leads to oxidation of Met, Tyr, and Trp residues, as well as significantly increasing the levels of IsoLG-, MDA- and ACR-Lys adducts on HDL.34, 36, 202.
The precise mechanisms whereby oxidative modification of HDL inhibits cholesterol efflux are poorly understood. Although oxidative modification of specific apoAI residues have been implicated in at least some studies, other types of oxidative modification to the same residue have no effect. Specific oxidative modifications may therefore give rise to altered protein structures that in turn alter interactions with key proteins like ABCA1, SR-BI, and CD36, but future studies are needed to assess this hypothesis.. A series of in vitro studies found that mutation of the four apoAI Trp residues to phenylalanine (4WF apoAI) provided significant protection against MPO-induced inhibition of ABCA1-mediated cholesterol efflux and that mutating Trp72 alone was sufficient to provide significant protection.122, 203 These same investigators found that mutating apoAI Met residues to Val, mutating Tyr residues to phenylalanine (Phe), or chemically methylating Lys to make its less reactive with lipid carbonyls, all failed to protect apoAI from MPO-induced inhibition of cholesterol efflux via ABCA1.122, 203, 204 Injection of recombinant apoAI engineered to have oxidized Trp only at Trp72 into Apoa1−/− mice showed that this engineered apoAI exhibited reduced lipidation and HDL biogenesis.205 While these studies suggested that oxidation of apoAI Trp reduces HDL efflux capacity, subsequent in vivo studies with Ldlr−/− mice that overexpressed transgenic 4WF apoAI showed no greater lesion regression than Ldlr−/− mice overexpressing transgenic wild-type human apoAI.206 Furthermore, 4WF apoAI transgenic mice showed less reverse cholesterol transport to the plasma compartment than mice expressing transgenic WT human apoAI, although they did show similar overall reverse cholesterol transport to liver and feces.207 Other studies found that in vitro oxidation of HDL3 with either HOCl or MPO reduced HDL LCAT activity in direct correlation to the extent that apoAI Met148 and Trp72 were oxidized, but only Leu mutations of the three apoAI Met residues protected against MPO-induced LCAT inhibition, while deletion of the four apoAI Trp residues provided no protection.208 Thus, apoAI Met oxidation may play a role in the overall loss of cholesterol efflux capacity in vivo.
Modification of HDL or apoAI with IsoLG, MDA, ACR, or KODA-PC results in significantly reduced ABCA1-mediated cholesterol efflux, while modification of apoAI with other lipid carbonyls including HNE, ONE, and MGO does not inhibit ABCA1-mediated efflux.33, 36, 37, 202, 209, 210 MDA modifies a number of apoAI Lys including Lys118, Lys133, Lys 182, Lys195, Lys206, Lys226, and Lys239.33 ACR modifies Lys12, Lys23, Lys77, Lys88, Lys118, Lys140, Lys182, and Lys226, with ACR modification of Lys226 showing the greatest correlation with loss of efflux activity.202 The Lys modified by IsoLG have not yet been published. Although KODA-PC and ONE are both γ-keto-alkenals with presumably similar adduction chemistry, ONE modification of apoAI does not alter cholesterol efflux, while KODA-PC inhibits efflux37, 210 ONE modifies Lys12, Lys23, Lys96, and Lys22637, while KODA-PC was found to form Lys-pyrrole-His crosslinks between a number of Lys/His pairs (His155/Lys106, His162/Lys94, His162/Lys96, His193,Lys195,His199/Lys195, His135/Lys182, His155/Lys12, His199/Lys133, His199/Lys140, His193/Lys140, and His193/Lys208).210 Thus, while apoAI Lys modification by specific lipid carbonyls result in reduced ABCA1-medidated efflux, further studies are needed to determine the mechanisms underlying this reduction.
Like ABCA1-mediated efflux, how oxidative modification of HDL alters SR-BI-mediated efflux remains poorly understood. Carbamylation of apoAI Lys inhibits SR-BI mediated efflux and carbamylation of HDL increases its binding affinity to SR-BI, causing carbamylated-HDL to be internalized and promote foam cell formation.32 Interestingly, carbamylation of HDL does not inhibit ABCA1-mediated cholesterol efflux.32 In vitro oxidation of lipid poor apoAI using MPO produces carbamylation of Lys40, Lys45, Lys118, and Lys 195, while using CNO- also produces carbamylation on Lys23, Lys40, Lys59, Lys94, Lys96, Lys106,Lys107, Lys118, Lys195, and Lys226.198 Interestingly, HDL treatment with CNO- that produced less than one carbamyl-Lys per apoAI was still sufficient to inhibit SR-BI cholesterol efflux and CE selective uptake RCT functions, thereby converting SR-BI into a nonproductive and potentially proatherogenic receptor.32 A similar mechanism may be at play with ACR modification, as ACR modification of HDL inhibits SR-BI-mediated cholesterol efflux and macrophages become foam cells in the presence of ACR-HDL.209 Similar results have been seen for MPO-mediated modification of other plasma proteins (i.e. BSA) that bind SR-BI with higher affinity than native HDL, thereby impairing SR-BI/HDL RCT functions. In this regard, studies are needed to determine how specific modifications of apoAI residues alter SR-BI interactions.
CD36 is another scavenger receptor that may play a role in modulating cholesterol handling. HDL from humans with CAD interacts with CD36 for uptake and degradation resulting in foam cell formation and studies have shown that both HNE- and MDA-HDL interact with platelet CD36.31, 200 However, to date, no studies have determined the effects of specific modifications of HDL on interaction with macrophage CD36 or the effects such modifications have on cholesterol flux or inflammation.
In RCT, the esterification of HDL FC by LCAT is critical for maintaining the flux of cholesterol from the periphery to the liver, and humans with CAD and CKD have reduced LCAT activity, and a number of apoAI oxidative modifications impair activation of LCAT. NO2-Tyr166-apoA-I markedly decreases LCAT activity as a result of decreased LCAT/apoAI interaction.63 In addition, MGO modification of reconstituted apoAI particles results in glycation of ApoAI Lys, Trp, and Arg residues, which decreases the interaction of ApoAI with LCAT and lipid.211 Furthermore, studies have shown that ACR and HNE modifications of free Cys and His residues present in the catalytic pocket of LCAT markedly inhibit its activity. In addition, MDA, ACR, and HNE modified HDL particles are poor substrates for LCAT, which is likely due to altered interaction with apoAI.64, 212
Effects of HDL oxidative modifications on Inflammation
Oxidative modifications of HDL not only cause it to lose its ability to suppress inflammation, but can convert HDL into proinflammatory HDL. HDL is critical to reducing endothelial cell activation and maintaining endothelial barrier integrity via NO production in response to inflammatory stimuli, and specific MPO-mediated oxidative modifications of HDL/apoAI impair eNOS activation and increase inflammatory activation and death even in the absence of other stimuli (i.e. TNF-α).125 Indeed, oxTrp72- apoAI isolated from human atheroma enhances expression of proinflammatory VCAM-1 and activates NF-κB without other stimuli.122 In addition, MDA modification of apoAI-lys causes HDL to activate protein kinase CβII via interaction with LOX-1 leading to decreased activation of eNOS and increased endothelial cell activation.51 Interestingly, carbamyl-HDL inhibits SR-BI-mediated eNOS activation and promotes apoptosis, which is consistent with carbamyl-apoAI inhibiting SR-B-mediated cholesterol efflux that is required for activation of eNOS.123, 213 Unoxidized HDL suppresses VCAM-1 surface expression in endothelial cells normally induced by TNFa, but when HDL is oxidized with MPO, it enhances VCAM-1 expression.214 Similarly, unmodified HDL suppresses expression of Tnfα and Il-1β by macrophages that is induced by LPS, but HDL modified by IsoLG greatly enhances the expression of these cytokines.36 HDL modified with ONE fails to suppress LPS-induced Tnfa expression, but does not further enhance Tnfa expression, which demonstrates that ONE modification impacts anti-inflammatory function independent of ABCA1-mediated cholesterol efflux signaling pathways that impact TLR signaling.37 In this regard, ONE modification of HDL could impact its interaction with SR-BI, which also mediates inflammatory signaling in response to LPS.94, 153, 154 Interestingly, MDA-HDL enhances LPS induced expression of IL-1β and IL-6, but it is not as proinflammatory as IsoLG-HDL.34, 36 The proinflammatory nature of Iso-LG- and MDA-HDL suggests interaction with receptors that mediate inflammatory signaling pathways such as CD36 and LOX-1. Studies have shown that HDL from CAD subjects interact with both CD36 and LOX-1 to activate NF-κB and enhance proinflammatory gene expression. Studies are needed to determine the mechanisms by which MDA- and IsoLG-HDL are proinflammatory.
Effects of HDL oxidative modifications on antioxidant function.
HDL oxidized by MPO has reduced PON1 activity.169 Addition of SIN-1 (a peroxynitrite generator) to HDL also reduces PON1 activity.215 Treatment of HDL with IsoLG, MDA, or ACR reduces PON1 activity,34, 173 although the mechanisms by which each of these lipid carbonyls inhibit PON1 may differ. MPO-induced oxidation of HDL leads to IsoLG modification of PON1, and IsoLG added directly to PON1 inhibits its activity.173 MDA added to HDL modifies apoAI so that it has reduced ability to activate PON1.34 The mechanism by which ACR modification of HDL inhibits PON1 has not be elucidated. Information as to which specific residues of PON1 may be important for the loss of activity when HDL is oxidized is limited. PON1 Tyr71 is oxidized to 3-Cl-Tyr by MPO and mutation of Tyr71 to alanine, aspartic acid or lysine caused a loss of PON1 activity.169 The PON1 Lys modified by IsoLG have not been determined.
More definitive evidence that oxidative modifications of HDL proteins such as apoAI and PON1 are a root cause of HDL dysfunction seen in cardiovascular disease is still needed. Studies examining correlation between specific amino acid modifications of HDL proteins and HDL function are limited. One study in individuals with coronary artery disease found no correlation between HDL oxidized Tyr residues and cholesterol efflux.216 In contrast, a second study reported an inverse correlation between levels of oxidized apoAI Tyr residues and cholesterol efflux.196 More intriguingly, these investigators immunopurified apoAI from six patients using an antibody that recognized apoAI modified by Trp72 oxidation. This immunopurified apoAI showed poor CEC and when apoAI oxTrp72 levels were examined in 627 individuals, there was a strong correlation between their oxTrp72 levels and coronary artery disease.122 However, these studies did not attempt to measure individual cholesterol efflux capacities in these samples, so whether there were robust inverse correlations between oxTrp72 levels and efflux capacity is unknown. To the best of our knowledge, there have been no studies that have examined whether there are correlations between specific reactive lipid dicarbonyl modifications and HDL functionality. Clearly, more studies examining the relationship between the extent of oxidative modification and HDL function are needed. If oxidative modification of HDL proteins is an important driver of HDL dysfunction, then blocking or reversing oxidative modification of HDL proteins must be shown to restore HDL function and reduce cardiovascular disease in vivo. A potential approach to test this will be addressed in the next section.
Small molecule scavengers of lipid dicarbonyls as inhibitors of oxidative modification.
A number of small molecules have been identified that are excellent scavengers of reactive lipid monocarbonyls and dicarbonyls and therefore have potential for use to protect lipoproteins from oxidative modification. These include thiol-based scavengers such as 2-mercaptoethanesulfonate (MESNA) and amifostine, imidazole-based scavengers like carnosine and its derivatives, and 2-aminomethylphenol-based scavengers of reactive dicarbonyls (Figure 5).
Figure 5. Current classes of small molecule scavengers available to target reactive lipid carbonyls.
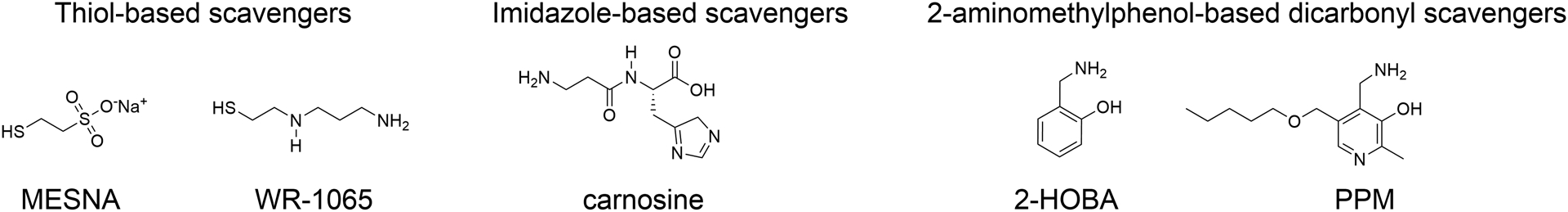
Thiol-based scavengers capture ACR (acrolein), ONE (4-oxo-nonenal), and HNE (4-hydroxy-nonenal). Imidazole-based scavengers capture ACR, ONE, HNE, and MGO (methylglyoxal). 2-aminomethylphenol-based dicarbonyl scavengers capture IsoLG (isolevuglandins), ONE, MDA (malondialdehyde), and MGO.
ACR is efficiently captured and inactivated by thiol-based scavengers including MESNA and the active form (WR-1065) of the prodrug amifostine.217 Formation of ACR is a byproduct of caner therapies such as cyclophosphamide treatments or radiation therapy and both MESNA and amifostine are widely used as adjuvants during cancer treatment to limit off-target toxicities. In addition to their use in cancer treatments, MESNA and amifostine have shown efficacy in animal models of other disease conditions with increased oxidative damage including ulcerative colitis, liver injury, Parkinson’s disease, and ischemia/reperfusion.218–222 Given their efficacy at scavenging ACR, it is surprising that to the best of our knowledge there have been no animal or clinical trials examining their effects in cardiovascular disease.
Imidazole-based scavengers effectively capture ACR and other α,β-unsaturated carbonyls like ONE and HNE. In animal models of disease, carnosine or its derivatives, have shown efficacy to protect against complications of diabetes, ischemia/reperfusion injury, and atherosclerosis.223–229 Very limited clinical studies have looked at the efficacy of carnosine or its derivatives in cardiometabolic diseases, these include a 12-week supplementation study in overweight/obese individuals that showed improved glucose tolerance,230 and a six month supplementation study that showed improved exercise tolerance in individuals with stable chronic heart failure.231
More recently, an entirely new class of scavengers directed against reactive lipid dicarbonyls have been identified and characterized, all of which contain a 2-aminomethylphenol (2-AMP) structural backbone. These novel dicarbonyl scavengers include 2-hydroxybenzylamine (2HOBA) and 5’-O-pentyl-pyridoxamine (PPM), as well as a number of other analogs.232 The compounds effectively scavenge reactive dicarbonyls including IsoLG, ONE, and MDA, and to a lesser extent some monocarbonyls like ACR. They have shown remarkable efficacy in a number of animal models of oxidative damage including Alzheimer’s Disease233, hypertension232, 234, 235, proteinuric kidney disease201 lupus236, cardiac ischemia/reperfusion injury237 and gastric cancer.238
Dicarbonyl Scavengers Reduce Atherosclerosis
FH is a common autosomal codominant disorder associated with severely elevated levels of LDL-C and increased risk of premature atherosclerotic cardiovascular disease. Pathogenic mutations in the LDLR gene are the most common cause of FH, resulting in delayed clearance of LDL by the liver. Although increased levels of LDL-C and oxidatively modified LDL have long been recognized as critical mediators of the pathogenesis of atherosclerosis in FH, there is substantial evidence that HDL metabolism and function are abnormal in FH.239, 240 HDL in subjects with FH have reduced capacity to promote RCT, impaired anti-inflammatory and antioxidant activities.239, 240 Indeed, we have found that HDL from subjects with FH shows increased modification by reactive dicarbonyl species, including MDA and IsoLG, compared to HDL from individuals with normal cholesterol levels.34–37 Furthermore, HDL from humans with severe FH showed impaired CEC and promote cholesterol loading of macrophages.35 Therefore, we examined the hypothesis that treatment of Ldlr−/− mice, a murine model of FH, with the dicarbonyl scavenger, 2-hydroxybenzylamine (2-HOBA) would reduce modification of HDL and LDL, improve HDL function, and reduce the development of atherosclerosis. Treatment of female Ldlr−/− mice fed a western-type diet for 16 weeks with 2-HOBA reduced the extent of atherosclerosis in sections of the proximal aorta by 31% and by 60% in en face analysis of the aortas compared to controls treated with water or a geometric isomer 4-HOBA that is an ineffective scavenger.35 Similar reductions in atherosclerosis were also seen in male Ldlr−/− mice treated with 2-HOBA. The reductions in the extent of atherosclerosis were especially impressive given that they occurred in the absence of changes in plasma cholesterol or triglyceride levels or lipid profiles.35 Importantly, treatment with 2-HOBA dramatically reduced the amount of MDA-protein adducts in the proximal aorta as determined by immunofluorescence and the amount of MDA- and IsoLG-lysyl adducts in the whole aorta as quantitated by LC/MS/MS, compared to controls treated with 4-HOBA. Treatment with 2-HOBA promoted features of plaque stabilization with increased collagen content and fibrous cap thickness and reduced the percentage of necrotic area by 75%. Inadequate efferocytosis of dead cells can promote inflammation and necrotic core formation. Treatment with 2-HOBA was associated with a 72% reduction in the numbers of apoptotic cells and increased efferocytosis in the atherosclerotic lesions compared to treatment with vehicle or 4-HOBA.35 Furthermore, serum levels of inflammatory cytokines, including IL-1β, IL-6, TNF-α, and serum amyloid A were reduced by 2-HOBA.35 In vitro studies showed that 2-HOBA treatment reduced the expression of inflammatory cytokines produced by macrophages exposed to OxLDL or H2O2. Taken together these results support the ability of dicarbonyl scavenging with 2-HOBA to reduce oxidative stress, inflammation, cell death, destabilization of the plaque, and the extent of atherosclerosis (Figure 6).
Figure 6. Dicarbonyl scavengers protect HDL functionality.
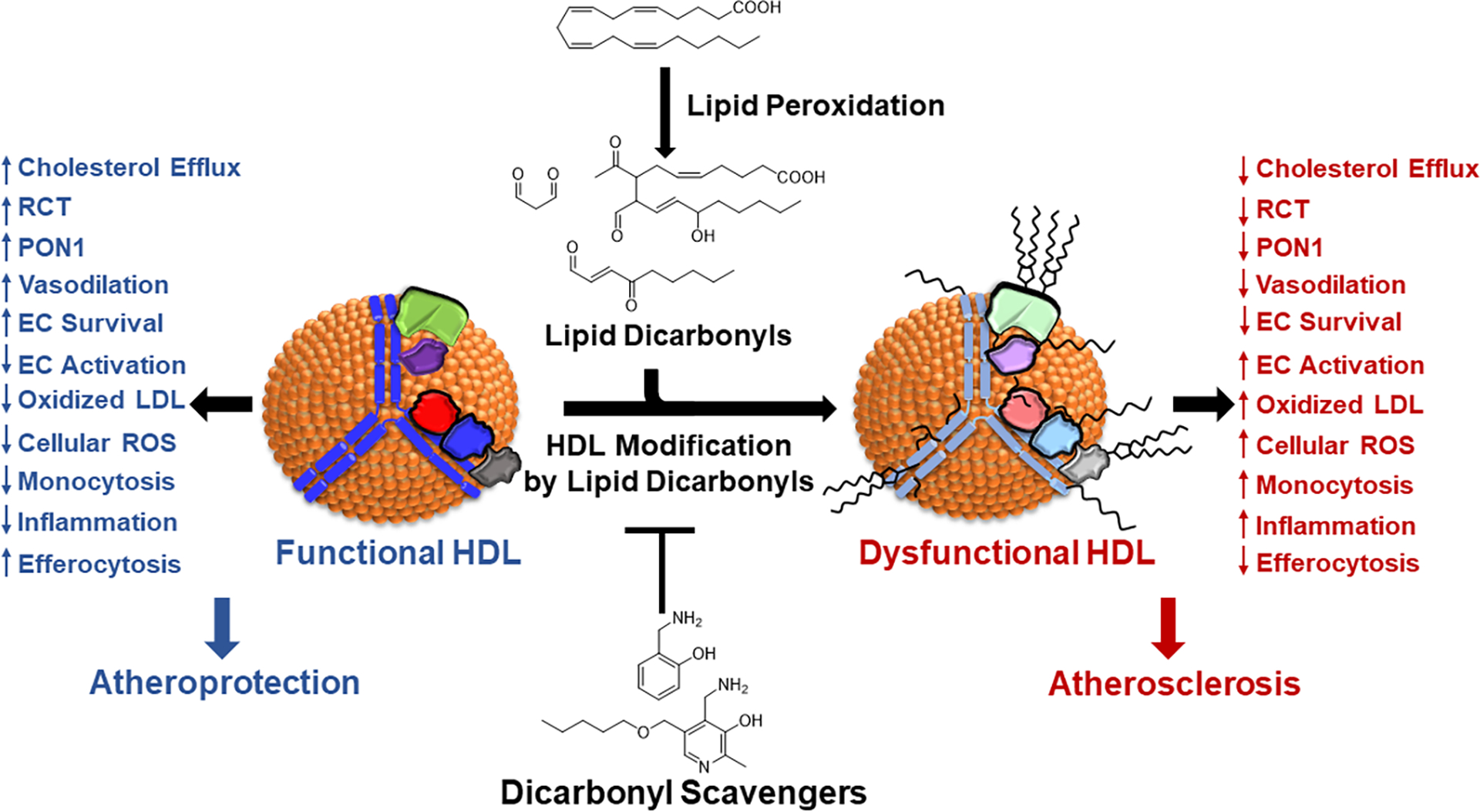
Under oxidative conditions, lipid peroxidation generates lipid dicarbonyls that react with HDL proteins to render HDL dysfunctional. Dicarbonyl scavengers can intercept these lipid dicarbonyls because the primary amines of these scavengers is even more reactive with dicarbonyls than the lysyl residues of proteins are reactive with the dicarbonyls. Thus, these scavengers protect HDL proteins from modification and thereby prevent HDL dysfunction. Shown are the atheroprotective functions of HDL that dicarbonyl scavengers can protect against dysfunction including EC (endothelial cell) activation and survival.
Consistent with evidence for increased modification of HDL by reactive dicarbonyls in humans with FH, Ldlr−/− mice fed a western diet have increased levels of MDA-apoAI adducts compared to chow fed mice.35 Most importantly, 2-HOBA treatment of Ldlr−/− mice substantially reduced MDA modification of both apoAI and HDL, and HDL isolated from the 2-HOBA treated mice had improved ability to promote macrophage cholesterol efflux compared to HDL from vehicle or 4-HOBA treated controls (Figure 6). Furthermore, 2-HOBA treated Ldlr−/− mice had reduced MDA-modification of LDL. Thus, reduced modification of both HDL and LDL by reactive dicarbonyls, including MDA and IsoLG, likely contribute to the antiatherogenic properties of 2-HOBA (Figure 6). In addition, 2-HOBA may prevent other MPO-induced oxidative modifications of HDL (i.e. apoA1 oxTrp72) as in vitro treatment with 2-HOBA negated the MPO-mediated impairment of HDL CEC.and prevented HOCl− induced crosslinked of lipid-free apoAI.34, 241 While this possibility needs to examined in vivo, it is worth noting that studies have shown that the scavenger PM prevents in vivo oxidation of Trp192 in renal collagen in a diabetic rat model.242 Therefore, dicarbonyl scavengers offer a potential new approach to reduce the residual risk of atherosclerotic cardiovascular events that remains in patients treated with statins (Figure 6).243, 244 Phase 1 studies of treating humans with 2-HOBA have demonstrated its safety,245 and we are initiating a Phase 2 trial (NCT#04941599) in humans with heterozygous FH to examine the impact of 2-HOBA on modification of HDL and HDL CEC.
Dicarbonyl Scavenger PPM Reduces Insulin Resistance and Hepatic Fat Accumulation
We have recently extended our studies of the therapeutic potential of dicarbonyl scavengers for treating atherosclerosis by examining the impact of another potent dicarbonyl scavenger, 5’-O-pentyl-pyridoxamine (PPM), on the development of atherosclerosis in Ldlr−/− mice fed a western diet for 16 weeks.241 Similar to our results with 2-HOBA, treatment of both male and female Ldlr−/− mice resulted in substantial reductions in the extent of atherosclerosis in the absence of changes in plasma cholesterol and triglyceride levels.241 In addition, PPM reduced blood Ly6Chi monocytosis, decreased lesion pro-inflammatory macrophages, enhanced efferocytosis, promoting features of plaque stabilization with dramatic reductions in plaque necrotic area (Figure 6).241 Furthermore, HDL from the PPM treated mice showed improved CEC compared to mice treated with vehicle.241
Mounting evidence supports antidiabetic functions of HDL. HDL can increase glucose uptake of by skeletal muscle and HDL can improve blood glucose levels in animal models by increasing the synthesis and secretion of insulin from pancreatic β cells.246, 247 HDL can prevent oxLDL-induced and ER stress-induced apoptosis of pancreatic β cells.248, 249 Interestingly, a metanalysis of the CETP inhibitor trials revealed a 12% reduction in the development of T2DM.250 Furthermore, infusion of reconstituted HDL was found to increase plasma insulin levels and decrease plasma glucose in patients with T2D by activating AMP-activated protein kinase.247, 251 Therefore, we examined the impact of PPM treatment on insulin sensitivity in male Ldlr−/− mice fed a western diet. Interestingly, treatment of Ldlr−/− mice with PPM promoted reductions in fasting glucose and insulin levels, reduced the HOMA-IR, and improved glucose and insulin tolerance.241 Furthermore, PPM protected male Ldlr−/− mice fed a western diet from developing hepatic steatosis.241 Thus, treatment with the dicarbonyl scavenger PPM reduces insulin resistance and hepatic fat accumulation in a model of diet-induced metabolic syndrome. Hence, reactive dicarbonyl scavengers have attractive therapeutic potential for treating multiple features of cardiometabolic disease.
Conclusions and Future Directions
Numerous in vitro and in vivo studies with atherosclerotic animal models support the role of functional HDL/apoAI in limiting atherosclerosis progression and stimulating plaque regression (Figure 6). However, unlike findings in atherosclerotic mouse models, therapeutics which either raise HDL-C or increase apoAI levels in humans have disappointingly produced no benefit to reducing atherosclerotic plaques. The heightened oxidative stress in CAD leads to extensive oxidative modifications of HDL that not only impair its atheroprotective functions, but produce proinflammatory/proatherogenic HDL particles. Indeed, treatment of mouse models with oxidized versus native HDL/apoAI promotes plaque development and inflammation.10, 11 The findings that human atherosclerotic plaques contain lipid-poor apoAI, which is mostly oxidatively modified and associated with MPO, and that human atherosclerotic lesions have markedly increased MPO levels compared to mouse lesions,12, 252 likely explains the lack of benefit of HDL/apoAI raising treatments in humans as any HDL particles entering the intima are likely oxidatively modified. However, a limitation to the studies thus far on the role of HDL oxidative modifications in atherosclerosis is that they are largely correlative. In this regard, in vivo studies are needed on the mechanisms by which specific oxidative modifications of HDL proteins and lipids impact atherosclerosis as well as the protein structural changes resulting from specific oxidation modifications that promote HDL dysfunction in atherosclerosis in order to develop more targeted therapeutic strategies. In particular, the impact of HDL modification with specific reactive carbonyls on existing atherosclerotic plaque extent and inflammatory composition needs to be examined, which could lead to the development of functional ApoAI mutants that are resistant to dicarbonyl modification. It is worth noting that despite the abundant evidence supporting the role oxidative modifications in atherosclerosis, clinical studies assessing the impact of dietary anti-oxidant in humans have shown no impact on lipoprotein modifications and atherosclerosis. However, these dietary anti-oxidants also failed to reduce LOOH levels and thus, prevent toxic reactive carbonyl modifications. The finding that neutralization of reactive lipid carbonyls with 2-AMPs effectively reduces atherosclerosis and improves HDL function in atherosclerotic mouse models offers promise of beneficial treatment of CAD in humans. The observation that 2-AMPs are atheroprotective in the setting of hypercholesterolemia suggests that dicarbonyl scavenging has the potential to alleviate the residual inflammatory risk of CAD that exists even with LDL cholesterol lowering in humans. Furthermore, the atheroprotective effects of 2-AMPs likely go beyond preserving HDL function as AMPs almost certainly prevent carbonyl modification of other plasma and cellular proteins and lipids, which could impact atherosclerosis. In this regard, 2-AMPs offer great potential for targeting CAD in humans.
Acknowledgments and Sources of Funding
This work was supported by National Institutes of Health Grants: HL116263.
List of Abbreviations
- ACR
acrolein
- ABCA1/G1
ATP-binding cassette proteins A1 and G1
- Atf3
activating transcription factor 3
- 2-AMPs
2-aminomethylphenols
- apo
Apolipoprotein
- ASCVD
atherosclerotic cardiovascular disease
- CAD
coronary artery disease
- CE
cholesteryl ester
- CEC
cholesterol efflux capacity
- CETP
cholesteryl ester transfer protein
- CVD
cardiovascular disease
- eNOS
endothelial nitric oxide synthase
- ER
endoplasmic reticulum
- FH
familial hypercholesterolemia
- FC
free cholesterol
- GM-CSF
IL-3/granulocyte-macrophage colony-stimulating factor
- HSPC
hematopoietic stem and progenitor cells
- HDL-C
high density lipoprotein cholesterol
- HNE
4-hydroxy-nonenal
- 2-HOBA
2-hydroxybenzylamine
- IsoLG
Isolevuglandins
- IL
interleukin
- LCAT
lecithin:cholesterol acyltransferase
- LPS
lipopolysaccharide
- LDLR
low density lipoprotein receptor
- MDA
malondialdehyde
- MGO
methylglyoxal
- MPO
myeloperoxidase
- NLRP3
nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3
- OxLDL
oxidized low density lipoprotein
- ONE
4-oxo-nonenal
- PPM
5’-O-pentyl-pyridoxamine
- PL
phospholipid
- PON1
paraoxonase 1
- ROS
reactive oxygen species
- RCT
reverse cholesterol transport
- S1P
sphingosine-1-phosphate
- SR-B1
scavenger receptor class B type 1
- STAT6
signal transducer and activator of transcription 6
- TLR
Toll-like receptor
Footnotes
Disclosures
Drs. Linton and Davies are inventors on a patent for the use of 2-HOBA and related dicarbonyl scavengers for the treatment of cardiovascular disease (US 11,389,414). All the other authors declare no financial conflicts of interest.
References
- 1.Gordon T, Castelli WP, Hjortland MC, Kannel WB and Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–14. [DOI] [PubMed] [Google Scholar]
- 2.Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR Jr., Bangdiwala S and Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. [DOI] [PubMed] [Google Scholar]
- 3.Rohatgi A, Westerterp M, von Eckardstein A, Remaley A and Rye KA. HDL in the 21st Century: A Multifunctional Roadmap for Future HDL Research. Circulation. 2021;143:2293–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D and Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karjalainen MK, Holmes MV, Wang Q, Anufrieva O, Kahonen M, Lehtimaki T, Havulinna AS, Kristiansson K, Salomaa V, Perola M, Viikari JS, Raitakari OT, Jarvelin MR, Ala-Korpela M and Kettunen J. Apolipoprotein A-I concentrations and risk of coronary artery disease: A Mendelian randomization study. Atherosclerosis. 2020;299:56–63. [DOI] [PubMed] [Google Scholar]
- 6.Madsen CM, Varbo A and Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J. 2017;38:2478–2486. [DOI] [PubMed] [Google Scholar]
- 7.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD and Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature cell biology. 2011;13:423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen RM, Zhao S, Ramirez Solano MA, Zhu W, Michell DL, Wang Y, Shyr Y, Sethupathy P, Linton MF, Graf GA, Sheng Q and Vickers KC. Bioinformatic analysis of endogenous and exogenous small RNAs on lipoproteins. J Extracell Vesicles. 2018;7:1506198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF, Doran AC and Vickers KC. The Role of Lipids and Lipoproteins in Atherosclerosis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F and Vinik A, eds. Endotext South Dartmouth (MA); 2019. [Google Scholar]
- 10.Ru D, Zhiqing H, Lin Z, Feng W, Feng Z, Jiayou Z, Yusheng R, Min F, Chun L and Zonggui W. Oxidized high-density lipoprotein accelerates atherosclerosis progression by inducing the imbalance between treg and teff in LDLR knockout mice. APMIS. 2015;123:410–21. [DOI] [PubMed] [Google Scholar]
- 11.Hewing B, Parathath S, Barrett T, Chung WKK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MSS, DiDonato JA, Huang Y, Buffa J, Berisha SZ, Smith JD, Hazen SL and Fisher EA. Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsche G, Stadler JT, Kargl J and Holzer M. Understanding Myeloperoxidase-Induced Damage to HDL Structure and Function in the Vessel Wall: Implications for HDL-Based Therapies. Antioxidants (Basel). 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies SS, May-Zhang LS, Boutaud O, Amarnath V, Kirabo A and Harrison DG. Isolevuglandins as mediators of disease and the development of dicarbonyl scavengers as pharmaceutical interventions. Pharmacology & Therapeutics. 2020;205:107418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brame CJ, Boutaud O, Davies SS, Yang T, Oates JA, Roden D and Roberts LJ, 2nd. Modification of proteins by isoketal-containing oxidized phospholipids. J Biol Chem. 2004;279:13447–51. [DOI] [PubMed] [Google Scholar]
- 15.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG and Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–23. [DOI] [PubMed] [Google Scholar]
- 16.Glomset JA, Janssen ET, Kennedy R and Dobbins J. Role of plasma lecithin:cholesterol acyltransferase in the metabolism of high density lipoproteins. J Lipid Res. 1966;7:638–48. [PubMed] [Google Scholar]
- 17.Rosenson RS, Brewer HB, Jr., Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR and Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oram JF and Vaughan AM. ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99:1031–43. [DOI] [PubMed] [Google Scholar]
- 19.Yancey PG, Yu H, Linton MF and Fazio S. A pathway-dependent on apoE, ApoAI, and ABCA1 determines formation of buoyant high-density lipoprotein by macrophage foam cells. Arterioscler Thromb Vasc Biol. 2007;27:1123–31. [DOI] [PubMed] [Google Scholar]
- 20.Linton MF, Atkinson JB and Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–7. [DOI] [PubMed] [Google Scholar]
- 21.Zannis VI, Chroni A and Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med (Berl). 2006;84:276–94. [DOI] [PubMed] [Google Scholar]
- 22.Zanotti I, Poti F, Pedrelli M, Favari E, Moleri E, Franceschini G, Calabresi L and Bernini F. The LXR agonist T0901317 promotes the reverse cholesterol transport from macrophages by increasing plasma efflux potential. J Lipid Res. 2008;49:954–60. [DOI] [PubMed] [Google Scholar]
- 23.DiDonato JA, Huang Y, Aulak KS, Even-Or O, Gerstenecker G, Gogonea V, Wu Y, Fox PL, Tang WH, Plow EF, Smith JD, Fisher EA and Hazen SL. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation. 2013;128:1644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weibel GL, Drazul-Schrader D, Shivers DK, Wade AN, Rothblat GH, Reilly MP and de la Llera-Moya M. Importance of evaluating cell cholesterol influx with efflux in determining the impact of human serum on cholesterol metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Wang N and Tall AR. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem. 2005;280:29277–81. [DOI] [PubMed] [Google Scholar]
- 26.Ouimet M, Franklin V, Mak E, Liao X, Tabas I and Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarling EJ and Edwards PA. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci U S A. 2011;108:19719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao H, Yancey PG, Blakemore JL, Zhang Y, Ding L, Jerome WG, Brown JD, Vickers KC and Linton MF. Macrophage SR-BI modulates autophagy via VPS34 complex and PPARalpha transcription of Tfeb in atherosclerosis. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yancey PG, de la Llera-Moya M, Swarnakar S, Monzo P, Klein SM, Connelly MA, Johnson WJ, Williams DL and Rothblat GH. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275:36596–604. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Gillard BK, Yelamanchili D, Gotto AM, Jr., Rosales C and Pownall HJ. High Free Cholesterol Bioavailability Drives the Tissue Pathologies in Scarb1(−/−) Mice. Arterioscler Thromb Vasc Biol. 2021;41:e453–e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sini S, Deepa D, Harikrishnan S and Jayakumari N. High-density lipoprotein from subjects with coronary artery disease promotes macrophage foam cell formation: role of scavenger receptor CD36 and ERK/MAPK signaling. Mol Cell Biochem. 2017;427:23–34. [DOI] [PubMed] [Google Scholar]
- 32.Holzer M, Gauster M, Pfeifer T, Wadsack C, Fauler G, Stiegler P, Koefeler H, Beubler E, Schuligoi R, Heinemann A and Marsche G. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid Redox Signal. 2011;14:2337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao B, Pennathur S, Pagani I, Oda MN, Witztum JL, Oram JF and Heinecke JW. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. 2010;285:18473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang J, Yancey PG, Tao H, Borja MS, Smith LE, Kon V, Davies SS and Linton MF. Reactive Dicarbonyl Scavenging Effectively Reduces MPO-Mediated Oxidation of HDL and Restores PON1 Activity. Nutrients. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao H, Huang J, Yancey PG, Yermalitsky V, Blakemore JL, Zhang Y, Ding L, Zagol-Ikapitte I, Ye F, Amarnath V, Boutaud O, Oates JA, Roberts LJ, 2nd, Davies SS and Linton MF. Scavenging of reactive dicarbonyls with 2-hydroxybenzylamine reduces atherosclerosis in hypercholesterolemic Ldlr(−/−) mice. Nature Communications. 2020;11:4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.May-Zhang LS, Yermalitsky V, Huang J, Pleasent T, Borja MS, Oda MN, Jerome WG, Yancey PG, Linton MF and Davies SS. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J Biol Chem. 2018;293:9176–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.May-Zhang LS, Yermalitsky V, Melchior JT, Morris J, Tallman KA, Borja MS, Pleasent T, Amarnath V, Song W, Yancey PG, Davidson WS, Linton MF and Davies SS. Modified sites and functional consequences of 4-oxo-2-nonenal adducts in HDL that are elevated in familial hypercholesterolemia. J Biol Chem. 2019;294:19022–19033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsche G, Hammer A, Oskolkova O, Kozarsky KF, Sattler W and Malle E. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J Biol Chem. 2002;277:32172–9. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR and Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Steffensen R and Tybjaerg-Hansen A. Genetic variation in ABCA1 predicts ischemic heart disease in the general population. Arterioscler Thromb Vasc Biol. 2008;28:180–6. [DOI] [PubMed] [Google Scholar]
- 41.Out R, Hoekstra M, Hildebrand RB, Kruit JK, Meurs I, Li Z, Kuipers F, Van Berkel TJ and Van Eck M. Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2295–300. [DOI] [PubMed] [Google Scholar]
- 42.Baldan A, Pei L, Lee R, Tarr P, Tangirala RK, Weinstein MM, Frank J, Li AC, Tontonoz P and Edwards PA. Impaired development of atherosclerosis in hyperlipidemic Ldlr−/− and ApoE−/− mice transplanted with Abcg1−/− bone marrow. Arterioscler Thromb Vasc Biol. 2006;26:2301–7. [DOI] [PubMed] [Google Scholar]
- 43.Rozhkova AV, Dmitrieva VG, Nosova EV, Dergunov AD, Limborska SA and Dergunova LV. Genomic Variants and Multilevel Regulation of ABCA1, ABCG1, and SCARB1 Expression in Atherogenesis. J Cardiovasc Dev Dis. 2021;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, van der Giet M and Tietge UJ. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J Lipid Res. 2010;51:743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S and Linton MF. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2003;108:2258–63. [DOI] [PubMed] [Google Scholar]
- 46.Covey SD, Krieger M, Wang W, Penman M and Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2003;23:1589–94. [DOI] [PubMed] [Google Scholar]
- 47.Yancey PG, Jerome WG, Yu H, Griffin EE, Cox BE, Babaev VR, Fazio S and Linton MF. Severely altered cholesterol homeostasis in macrophages lacking apoE and SR-BI. J Lipid Res. 2007;48:1140–9. [DOI] [PubMed] [Google Scholar]
- 48.Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, DerOhannessian S, Kontush A, Surendran P, Saleheen D, Trompet S, Jukema JW, De Craen A, Deloukas P, Sattar N, Ford I, Packard C, Majumder A, Alam DS, Di Angelantonio E, Abecasis G, Chowdhury R, Erdmann J, Nordestgaard BG, Nielsen SF, Tybjaerg-Hansen A, Schmidt RF, Kuulasmaa K, Liu DJ, Perola M, Blankenberg S, Salomaa V, Mannisto S, Amouyel P, Arveiler D, Ferrieres J, Muller-Nurasyid M, Ferrario M, Kee F, Willer CJ, Samani N, Schunkert H, Butterworth AS, Howson JM, Peloso GM, Stitziel NO, Danesh J, Kathiresan S, Rader DJ, Consortium CHDE, Consortium CAE and Global Lipids Genetics C. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351:1166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samadi S, Farjami Z, Hosseini ZS, Ferns GA, Mohammadpour AH, Tayefi M, Fal-Soleiman H, Moohebati M, Ghayour-Mobarhan M, Esmaily H and Avan A. Rare P376L variant in the SR-BI gene associates with HDL dysfunction and risk of cardiovascular disease. Clin Biochem. 2019;73:44–49. [DOI] [PubMed] [Google Scholar]
- 50.Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, Hoekstra M, Sierts JA, Dallinga-Thie GM, Motazacker MM, Holleboom AG, Van Berkel TJ, Kastelein JJ, Van Eck M and Kuivenhoven JA. Genetic variant of the scavenger receptor BI in humans. N Engl J Med. 2011;364:136–45. [DOI] [PubMed] [Google Scholar]
- 51.Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, Chroni A, Yonekawa K, Stein S, Schaefer N, Mueller M, Akhmedov A, Daniil G, Manes C, Templin C, Wyss C, Maier W, Tanner FC, Matter CM, Corti R, Furlong C, Lusis AJ, von Eckardstein A, Fogelman AM, Luscher TF and Landmesser U. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chistiakov DA, Melnichenko AA, Orekhov AN and Bobryshev YV. How do macrophages sense modified low-density lipoproteins? Int J Cardiol. 2017;230:232–240. [DOI] [PubMed] [Google Scholar]
- 53.Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, Fazio S and Linton MF. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015;56:1449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasaki M, Delawary M, Sakurai H, Kobayashi H, Nakao N, Tsuru H, Fukushima Y, Honzumi S, Moriyama S, Wada N, Kaneko T, Yamada K, Terasaka N and Kubota K. Novel LCAT (Lecithin:Cholesterol Acyltransferase) Activator DS-8190a Prevents the Progression of Plaque Accumulation in Atherosclerosis Models. Arterioscler Thromb Vasc Biol. 2021;41:360–376. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz CC, VandenBroek JM and Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–607. [DOI] [PubMed] [Google Scholar]
- 56.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkila K, Hypponen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikainen LP, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Muller-Nurasyid M, Nolte IM, O’Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Doring A, Elliott P, Epstein SE, Ingi Eyjolfsson G, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimaki T, Lin SY, Lindstrom J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Muller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancakova A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrieres J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Jarvelin MR, Jula A, Kahonen M, Kaprio J, Kesaniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, Marz W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njolstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR and Global Lipids Genetics C. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haase CL, Tybjaerg-Hansen A, Qayyum AA, Schou J, Nordestgaard BG and Frikke-Schmidt R. LCAT, HDL cholesterol and ischemic cardiovascular disease: a Mendelian randomization study of HDL cholesterol in 54,500 individuals. J Clin Endocrinol Metab. 2012;97:E248–56. [DOI] [PubMed] [Google Scholar]
- 58.Oldoni F, Baldassarre D, Castelnuovo S, Ossoli A, Amato M, van Capelleveen J, Hovingh GK, De Groot E, Bochem A, Simonelli S, Barbieri S, Veglia F, Franceschini G, Kuivenhoven JA, Holleboom AG and Calabresi L. Complete and Partial Lecithin:Cholesterol Acyltransferase Deficiency Is Differentially Associated With Atherosclerosis. Circulation. 2018;138:1000–1007. [DOI] [PubMed] [Google Scholar]
- 59.Forte TM, Subbanagounder G, Berliner JA, Blanche PJ, Clermont AO, Jia Z, Oda MN, Krauss RM and Bielicki JK. Altered activities of anti-atherogenic enzymes LCAT, paraoxonase, and platelet-activating factor acetylhydrolase in atherosclerosis-susceptible mice. J Lipid Res. 2002;43:477–85. [PubMed] [Google Scholar]
- 60.Zhao XJ, Liu LC, Guo C, Shen WW, Cao J, Du F, Wu DF and Yu H. Hepatic paraoxonase 1 ameliorates dysfunctional high-density lipoprotein and atherosclerosis in scavenger receptor class B type I deficient mice. Ann Transl Med. 2021;9:1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mertens A, Verhamme P, Bielicki JK, Phillips MC, Quarck R, Verreth W, Stengel D, Ninio E, Navab M, Mackness B, Mackness M and Holvoet P. Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation. 2003;107:1640–6. [DOI] [PubMed] [Google Scholar]
- 62.Thacker SG, Rousset X, Esmail S, Zarzour A, Jin X, Collins HL, Sampson M, Stonik J, Demosky S, Malide DA, Freeman L, Vaisman BL, Kruth HS, Adelman SJ and Remaley AT. Increased plasma cholesterol esterification by LCAT reduces diet-induced atherosclerosis in SR-BI knockout mice. J Lipid Res. 2015;56:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DiDonato JA, Aulak K, Huang Y, Wagner M, Gerstenecker G, Topbas C, Gogonea V, DiDonato AJ, Tang WHW, Mehl RA, Fox PL, Plow EF, Smith JD, Fisher EA and Hazen SL. Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J Biol Chem. 2014;289:10276–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCall MR, Tang JY, Bielicki JK and Forte TM. Inhibition of lecithin-cholesterol acyltransferase and modification of HDL apolipoproteins by aldehydes. Arterioscler Thromb Vasc Biol. 1995;15:1599–606. [DOI] [PubMed] [Google Scholar]
- 65.Holzer M, Zangger K, El-Gamal D, Binder V, Curcic S, Konya V, Schuligoi R, Heinemann A and Marsche G. Myeloperoxidase-derived chlorinating species induce protein carbamylation through decomposition of thiocyanate and urea: novel pathways generating dysfunctional high-density lipoprotein. Antioxid Redox Signal. 2012;17:1043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shao B, O’Brien KD, McDonald TO, Fu X, Oram JF, Uchida K and Heinecke JW. Acrolein modifies apolipoprotein A-I in the human artery wall. Ann N Y Acad Sci. 2005;1043:396–403. [DOI] [PubMed] [Google Scholar]
- 67.Gebhard C, Rhainds D, He G, Rodes-Cabau J, Lavi S, Spence JD, Title L, Kouz S, L’Allier PL, Gregoire J, Ibrahim R, Cossette M, Guertin MC, Beanlands R, Rheaume E and Tardif JC. Elevated level of lecithin:cholesterol acyltransferase (LCAT) is associated with reduced coronary atheroma burden. Atherosclerosis. 2018;276:131–139. [DOI] [PubMed] [Google Scholar]
- 68.George RT, Abuhatzira L, Stoughton SM, Karathanasis SK, She D, Jin C, Buss N, Bakker-Arkema R, Ongstad EL, Koren M and Hirshberg B. MEDI6012: Recombinant Human Lecithin Cholesterol Acyltransferase, High-Density Lipoprotein, and Low-Density Lipoprotein Receptor-Mediated Reverse Cholesterol Transport. J Am Heart Assoc. 2021;10:e014572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonaca MP, Morrow DA, Bergmark BA, Berg DD, Lima JAC, Hoffmann U, Kato Y, Lu MT, Kuder J, Murphy SA, Spinar J, Oude Ophuis T, Kiss RG, Lopez-Sendon J, Averkov O, Wheatcroft SB, Kubica J, Carlos Nicolau J, Furtado RHM, Abuhatzira L, Hirshberg B, Omar SA, Vavere AL, Chang YT, George RT and Sabatine MS. Randomized, Placebo-Controlled Phase 2b Study to Evaluate the Safety and Efficacy of Recombinant Human Lecithin Cholesterol Acyltransferase in Acute ST-Segment-Elevation Myocardial Infarction: Results of REAL-TIMI 63B. Circulation. 2022;146:907–916. [DOI] [PubMed] [Google Scholar]
- 70.Jiang XC. Phospholipid transfer protein: its impact on lipoprotein homeostasis and atherosclerosis. J Lipid Res. 2018;59:764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pussinen PJ, Metso J, Keva R, Hirschmugl B, Sattler W, Jauhiainen M and Malle E. Plasma phospholipid transfer protein-mediated reactions are impaired by hypochlorite-modification of high density lipoprotein. Int J Biochem Cell Biol. 2003;35:192–202. [DOI] [PubMed] [Google Scholar]
- 72.Christison J, Rye K-A and Stocker R. Exchange of oxidized cholesteryl linoleate between LDL and HDL mediated by cholesteryl ester transfer protein. J Lipid Res. 1995;36:2017–2026. [PubMed] [Google Scholar]
- 73.Hine D, Mackness B and Mackness M. Cholesteryl-ester transfer protein enhances the ability of high-density lipoprotein to inhibit low-density lipoprotein oxidation. IUBMB Life. 2011;63:772–4. [DOI] [PubMed] [Google Scholar]
- 74.Chiba H, Akita H, Kotani K, Kanno T and Manabe M. [Complete cholesteryl ester transfer protein deficiency increases oxidized-LDL in plasma]. Rinsho Byori. 1997;45:55–7. [PubMed] [Google Scholar]
- 75.Okada T, Sumida M, Ohama T, Katayama Y, Saga A, Inui H, Kanno K, Masuda D, Koseki M, Nishida M, Sakata Y and Yamashita S. Development and Clinical Application of an Enzyme-Linked Immunosorbent Assay for Oxidized High-Density Lipoprotein. J Atheroscler Thromb. 2021;28:703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Foger B, Chase M, Amar MJ, Vaisman BL, Shamburek RD, Paigen B, Fruchart-Najib J, Paiz JA, Koch CA, Hoyt RF, Brewer HB, Jr. and Santamarina-Fojo S. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J Biol Chem. 1999;274:36912–20. [DOI] [PubMed] [Google Scholar]
- 77.Huang Z, Inazu A, Kawashiri MA, Nohara A, Higashikata T and Mabuchi H. Dual effects on HDL metabolism by cholesteryl ester transfer protein inhibition in HepG2 cells. Am J Physiol Endocrinol Metab. 2003;284:E1210–9. [DOI] [PubMed] [Google Scholar]
- 78.Zhou H, Li Z, Silver DL and Jiang XC. Cholesteryl ester transfer protein (CETP) expression enhances HDL cholesteryl ester liver delivery, which is independent of scavenger receptor BI, LDL receptor related protein and possibly LDL receptor. Biochim Biophys Acta. 2006;1761:1482–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanigawa H, Billheimer JT, Tohyama J, Zhang Y, Rothblat G and Rader DJ. Expression of cholesteryl ester transfer protein in mice promotes macrophage reverse cholesterol transport. Circulation. 2007;116:1267–73. [DOI] [PubMed] [Google Scholar]
- 80.Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL and Tall AR. Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol. 1999;19:1105–10. [DOI] [PubMed] [Google Scholar]
- 81.Hovingh GK, Ray KK and Boekholdt SM. Is Cholesteryl Ester Transfer Protein Inhibition an Effective Strategy to Reduce Cardiovascular Risk? CETP as a Target to Lower CVD Risk: Suspension of Disbelief? Circulation. 2015;132:433–40. [DOI] [PubMed] [Google Scholar]
- 82.Yamashita S and Matsuzawa Y. Re-evaluation of cholesteryl ester transfer protein function in atherosclerosis based upon genetics and pharmacological manipulation. Curr Opin Lipidol. 2016;27:459–72. [DOI] [PubMed] [Google Scholar]
- 83.Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, Packard CJ, Laufs U, Brook RD, Oliver-Williams C, Butterworth AS, Danesh J, Smith GD, Catapano AL and Sabatine MS. Association of Genetic Variants Related to CETP Inhibitors and Statins With Lipoprotein Levels and Cardiovascular Risk. JAMA. 2017;318:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borggreve SE, Hillege HL, Wolffenbuttel BH, de Jong PE, Zuurman MW, van der Steege G, van Tol A, Dullaart RP and Group PS. An increased coronary risk is paradoxically associated with common cholesteryl ester transfer protein gene variations that relate to higher high-density lipoprotein cholesterol: a population-based study. J Clin Endocrinol Metab. 2006;91:3382–8. [DOI] [PubMed] [Google Scholar]
- 85.Ishigami M, Yamashita S, Sakai N, Arai T, Hirano K, Hiraoka H, Kameda-Takemura K and Matsuzawa Y. Large and cholesteryl ester-rich high-density lipoproteins in cholesteryl ester transfer protein (CETP) deficiency can not protect macrophages from cholesterol accumulation induced by acetylated low-density lipoproteins. J Biochem. 1994;116:257–62. [DOI] [PubMed] [Google Scholar]
- 86.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B and Investigators I. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22. [DOI] [PubMed] [Google Scholar]
- 87.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS and dal OI. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99. [DOI] [PubMed] [Google Scholar]
- 88.Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, Gibson CM, Granger C, Menon V, Montalescot G, Rader D, Tall AR, McErlean E, Wolski K, Ruotolo G, Vangerow B, Weerakkody G, Goodman SG, Conde D, McGuire DK, Nicolau JC, Leiva-Pons JL, Pesant Y, Li W, Kandath D, Kouz S, Tahirkheli N, Mason D, Nissen SE and Investigators A. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N Engl J Med. 2017;376:1933–1942. [DOI] [PubMed] [Google Scholar]
- 89.Group HTRC, Writing C, Sammons E, Hopewell JC, Chen F, Stevens W, Wallendszus K, Valdes-Marquez E, Dayanandan R, Knott C, Murphy K, Wincott E, Baxter A, Goodenough R, Lay M, Hill M, Macdonnell S, Fabbri G, Lucci D, Fajardo-Moser M, Brenner S, Hao D, Zhang H, Liu J, Wuhan B, Mosegaard S, Herrington W, Wanner C, Angermann C, Ertl G, Maggioni A, Barter P, Mihaylova B, Mitchel Y, Blaustein R, Goto S, Tobert J, DeLucca P, Chen Y, Chen Z, Gray A, Haynes R, Armitage J, Baigent C, Wiviott S, Cannon C, Braunwald E, Collins R, Bowman L, Landray M and Group RC. Long-term safety and efficacy of anacetrapib in patients with atherosclerotic vascular disease. Eur Heart J. 2022;43:1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Furtado JD, Ruotolo G, Nicholls SJ, Dullea R, Carvajal-Gonzalez S and Sacks FM. Pharmacological Inhibition of CETP (Cholesteryl Ester Transfer Protein) Increases HDL (High-Density Lipoprotein) That Contains ApoC3 and Other HDL Subspecies Associated With Higher Risk of Coronary Heart Disease. Arterioscler Thromb Vasc Biol. 2022;42:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brousseau ME, Diffenderfer MR, Millar JS, Nartsupha C, Asztalos BF, Welty FK, Wolfe ML, Rudling M, Bjorkhem I, Angelin B, Mancuso JP, Digenio AG, Rader DJ and Schaefer EJ. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25:1057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gugliucci A Activation of paraoxonase 1 is associated with HDL remodeling ex vivo. Clinica Chimica Acta. 2014;429:38–45. [DOI] [PubMed] [Google Scholar]
- 93.Schmidt AF, Hunt NB, Gordillo-Maranon M, Charoen P, Drenos F, Kivimaki M, Lawlor DA, Giambartolomei C, Papacosta O, Chaturvedi N, Bis JC, O’Donnell CJ, Wannamethee G, Wong A, Price JF, Hughes AD, Gaunt TR, Franceschini N, Mook-Kanamori DO, Zwierzyna M, Sofat R, Hingorani AD and Finan C. Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat Commun. 2021;12:5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Linton MF, Tao H, Linton EF and Yancey PG. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol Metab. 2017;28:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL and Tall AR. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem. 1999;274:33398–402. [DOI] [PubMed] [Google Scholar]
- 96.Collet X, Tall AR, Serajuddin H, Guendouzi K, Royer L, Oliveira H, Barbaras R, Jiang XC and Francone OL. Remodeling of HDL by CETP in vivo and by CETP and hepatic lipase in vitro results in enhanced uptake of HDL CE by cells expressing scavenger receptor B-I. J Lipid Res. 1999;40:1185–93. [PubMed] [Google Scholar]
- 97.Yang XP, Amar MJ, Vaisman B, Bocharov AV, Vishnyakova TG, Freeman LA, Kurlander RJ, Patterson AP, Becker LC and Remaley AT. Scavenger receptor-BI is a receptor for lipoprotein(a). J Lipid Res. 2013;54:2450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sharma M, Von Zychlinski-Kleffmann A, Porteous CM, Jones GT, Williams MJ and McCormick SP. Lipoprotein (a) upregulates ABCA1 in liver cells via scavenger receptor-B1 through its oxidized phospholipids. J Lipid Res. 2015;56:1318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fluiter K, Sattler W, De Beer MC, Connell PM, van der Westhuyzen DR and van Berkel TJ. Scavenger receptor BI mediates the selective uptake of oxidized cholesterol esters by rat liver. J Biol Chem. 1999;274:8893–9. [DOI] [PubMed] [Google Scholar]
- 100.Bourret G, Brodeur MR, Luangrath V, Lapointe J, Falstrault L and Brissette L. In vivo cholesteryl ester selective uptake of mildly and standardly oxidized LDL occurs by both parenchymal and nonparenchymal mouse hepatic cells but SR-BI is only responsible for standardly oxidized LDL selective uptake by nonparenchymal cells. Int J Biochem Cell Biol. 2006;38:1160–70. [DOI] [PubMed] [Google Scholar]
- 101.Fluiter K and van Berkel TJ. Scavenger receptor B1 (SR-B1) substrates inhibit the selective uptake of high-density-lipoprotein cholesteryl esters by rat parenchymal liver cells. Biochem J. 1997;326 ( Pt 2):515–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH and Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA and Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li XM, Tang WH, Mosior MK, Huang Y, Wu Y, Matter W, Gao V, Schmitt D, Didonato JA, Fisher EA, Smith JD and Hazen SL. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol. 2013;33:1696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Khera AV, Demler OV, Adelman SJ, Collins HL, Glynn RJ, Ridker PM, Rader DJ and Mora S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation. 2017;135:2494–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bauer L, Kern S, Rogacev KS, Emrich IE, Zawada A, Fliser D, Heinemann A, Heine GH and Marsche G. HDL Cholesterol Efflux Capacity and Cardiovascular Events in Patients With Chronic Kidney Disease. J Am Coll Cardiol. 2017;69:246–247. [DOI] [PubMed] [Google Scholar]
- 107.Josefs T, Wouters K, Tietge UJF, Annema W, Dullaart RPF, Vaisar T, Arts ICW, van der Kallen CJH, Stehouwer CDA, Schalkwijk CG, Goldberg IJ, Fisher EA and van Greevenbroek MMJ. High-density lipoprotein cholesterol efflux capacity is not associated with atherosclerosis and prevalence of cardiovascular outcome: The CODAM study. J Clin Lipidol. 2020;14:122–132 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kopecky C, Ebtehaj S, Genser B, Drechsler C, Krane V, Antlanger M, Kovarik JJ, Kaltenecker CC, Parvizi M, Wanner C, Weichhart T, Saemann MD and Tietge UJ. HDL Cholesterol Efflux Does Not Predict Cardiovascular Risk in Hemodialysis Patients. J Am Soc Nephrol. 2017;28:769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chindhy S, Joshi P, Khera A, Ayers CR, Hedayati SS and Rohatgi A. Impaired Renal Function on Cholesterol Efflux Capacity, HDL Particle Number, and Cardiovascular Events. J Am Coll Cardiol. 2018;72:698–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kane JP and Malloy MJ. Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol. 2012;23:367–71. [DOI] [PubMed] [Google Scholar]
- 111.Guey LT, Pullinger CR, Ishida BY, O’Connor PM, Zellner C, Francone OL, Laramie JM, Naya-Vigne JM, Siradze KA, Deedwania P, Redberg RF, Frost PH, Seymour AB, Kane JP and Malloy MJ. Relation of increased prebeta-1 high-density lipoprotein levels to risk of coronary heart disease. Am J Cardiol. 2011;108:360–6. [DOI] [PubMed] [Google Scholar]
- 112.Chirinos JA, Zambrano JP, Chakko S, Schob A, Goldberg RB, Perez G and Mendez AJ. Ability of serum to decrease cellular acylCoA:cholesterol acyl transferase activity predicts cardiovascular outcomes. Circulation. 2005;112:2446–53. [DOI] [PubMed] [Google Scholar]
- 113.de Vries R, Groen AK, Perton FG, Dallinga-Thie GM, van Wijland MJ, Dikkeschei LD, Wolffenbuttel BH, van Tol A and Dullaart RP. Increased cholesterol efflux from cultured fibroblasts to plasma from hypertriglyceridemic type 2 diabetic patients: roles of pre beta-HDL, phospholipid transfer protein and cholesterol esterification. Atherosclerosis. 2008;196:733–41. [DOI] [PubMed] [Google Scholar]
- 114.Attia N, Ramaharo A, Paul JL, Cambillau M, Beaune P, Grynberg A, Simon A and Fournier N. Enhanced removal of cholesterol from macrophage foam cells to serum from type IV hypertriglyceridemic subjects. Atherosclerosis. 2008;198:49–56. [DOI] [PubMed] [Google Scholar]
- 115.Vogt L, Laverman GD, van Tol A, Groen AK, Navis G and Dullaart RP. Cellular cholesterol efflux to plasma from proteinuric patients is elevated and remains unaffected by antiproteinuric treatment. Nephrol Dial Transplant. 2006;21:101–6. [DOI] [PubMed] [Google Scholar]
- 116.Sethi AA, Sampson M, Warnick R, Muniz N, Vaisman B, Nordestgaard BG, Tybjaerg-Hansen A and Remaley AT. High pre-beta1 HDL concentrations and low lecithin: cholesterol acyltransferase activities are strong positive risk markers for ischemic heart disease and independent of HDL-cholesterol. Clin Chem. 2010;56:1128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vaziri ND, Liang K and Parks JS. Down-regulation of hepatic lecithin:cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int. 2001;59:2192–6. [DOI] [PubMed] [Google Scholar]
- 118.Freeman DJ, Caslake MJ, Griffin BA, Hinnie J, Tan CE, Watson TD, Packard CJ and Shepherd J. The effect of smoking on post-heparin lipoprotein and hepatic lipase, cholesteryl ester transfer protein and lecithin:cholesterol acyl transferase activities in human plasma. Eur J Clin Invest. 1998;28:584–91. [DOI] [PubMed] [Google Scholar]
- 119.Vallejo J, Cochain C, Zernecke A and Ley K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-Seq. Cardiovasc Res. 2021;117:2537–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cuchel M, Raper AC, Conlon DM, Pryma DA, Freifelder RH, Poria R, Cromley D, Li X, Dunbar RL, French B, Qu L, Farver W, Su CC, Lund-Katz S, Baer A, Ruotolo G, Akerblad P, Ryan CS, Xiao L, Kirchgessner TG, Millar JS, Billheimer JT and Rader DJ. A novel approach to measuring macrophage-specific reverse cholesterol transport in vivo in humans. J Lipid Res. 2017;58:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ and Group CT. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 122.Huang Y, DiDonato JA, Levison BS, Schmitt D, Li L, Wu Y, Buffa J, Kim T, Gerstenecker GS, Gu X, Kadiyala CS, Wang Z, Culley MK, Hazen JE, Didonato AJ, Fu X, Berisha SZ, Peng D, Nguyen TT, Liang S, Chuang CC, Cho L, Plow EF, Fox PL, Gogonea V, Tang WH, Parks JS, Fisher EA, Smith JD and Hazen SL. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med. 2014;20:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA and Hazen SL. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–84. [DOI] [PubMed] [Google Scholar]
- 124.Trakaki A and Marsche G. Current Understanding of the Immunomodulatory Activities of High-Density Lipoproteins. Biomedicines. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kratzer A, Giral H and Landmesser U. High-density lipoproteins as modulators of endothelial cell functions: alterations in patients with coronary artery disease. Cardiovasc Res. 2014;103:350–61. [DOI] [PubMed] [Google Scholar]
- 126.Assanasen C, Mineo C, Seetharam D, Yuhanna IS, Marcel YL, Connelly MA, Williams DL, de la Llera-Moya M, Shaul PW and Silver DL. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Invest. 2005;115:969–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Terasaka N, Westerterp M, Koetsveld J, Fernandez-Hernando C, Yvan-Charvet L, Wang N, Sessa WC and Tall AR. ATP-binding cassette transporter G1 and high-density lipoprotein promote endothelial NO synthesis through a decrease in the interaction of caveolin-1 and endothelial NO synthase. Arterioscler Thromb Vasc Biol. 2010;30:2219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Christensen PM, Liu CH, Swendeman SL, Obinata H, Qvortrup K, Nielsen LB, Hla T, Di Lorenzo A and Christoffersen C. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J. 2016;30:2351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Del Gaudio I, Rubinelli L, Sasset L, Wadsack C, Hla T and Di Lorenzo A. Endothelial Spns2 and ApoM Regulation of Vascular Tone and Hypertension Via Sphingosine-1-Phosphate. J Am Heart Assoc. 2021;10:e021261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGrath KC, Li XH, Puranik R, Liong EC, Tan JT, Dy VM, DiBartolo BA, Barter PJ, Rye KA and Heather AK. Role of 3beta-hydroxysteroid-delta 24 reductase in mediating antiinflammatory effects of high-density lipoproteins in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:877–82. [DOI] [PubMed] [Google Scholar]
- 131.Liu Y and Tie L. Apolipoprotein M and sphingosine-1-phosphate complex alleviates TNF-alpha-induced endothelial cell injury and inflammation through PI3K/AKT signaling pathway. BMC Cardiovasc Disord. 2019;19:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xue S, Wang J, Zhang X, Shi Y, Li B, Bao Q, Pang W, Ai D, Zhu Y and He J. Endothelial ATP-binding cassette G1 in mouse endothelium protects against hemodynamic-induced atherosclerosis. Biochem Biophys Res Commun. 2016;477:247–54. [DOI] [PubMed] [Google Scholar]
- 133.Vaisar T, Couzens E, Hwang A, Russell M, Barlow CE, DeFina LF, Hoofnagle AN and Kim F. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS One. 2018;13:e0192616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sattler K, Lehmann I, Graler M, Brocker-Preuss M, Erbel R, Heusch G and Levkau B. HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell Physiol Biochem. 2014;34:172–84. [DOI] [PubMed] [Google Scholar]
- 135.Jia C, Anderson JLC, Gruppen EG, Lei Y, Bakker SJL, Dullaart RPF and Tietge UJF. High-Density Lipoprotein Anti-Inflammatory Capacity and Incident Cardiovascular Events. Circulation. 2021;143:1935–1945. [DOI] [PubMed] [Google Scholar]
- 136.Bisoendial RJ, Hovingh GK, Levels JH, Lerch PG, Andresen I, Hayden MR, Kastelein JJ and Stroes ES. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation. 2003;107:2944–8. [DOI] [PubMed] [Google Scholar]
- 137.Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS and Kingwell BA. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–71. [DOI] [PubMed] [Google Scholar]
- 138.Hirata A, Kakino A, Okamura T, Usami Y, Fujita Y, Kadota A, Fujiyoshi A, Hisamatsu T, Kondo K, Segawa H, Sawamura T, Miura K, Ueshima H and Group SR. The relationship between serum levels of LOX-1 ligand containing ApoAI as a novel marker of dysfunctional HDL and coronary artery calcification in middle-aged Japanese men. Atherosclerosis. 2020;313:20–25. [DOI] [PubMed] [Google Scholar]
- 139.Idzkowska E, Eljaszewicz A, Miklasz P, Musial WJ, Tycinska AM and Moniuszko M. The Role of Different Monocyte Subsets in the Pathogenesis of Atherosclerosis and Acute Coronary Syndromes. Scand J Immunol. 2015;82:163–73. [DOI] [PubMed] [Google Scholar]
- 140.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R and Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chistiakov DA, Grechko AV, Myasoedova VA, Melnichenko AA and Orekhov AN. The role of monocytosis and neutrophilia in atherosclerosis. J Cell Mol Med. 2018;22:1366–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Grosse-Dunker G, Heisel I, Hornof F, Jeken J, Rebling NM, Ulrich C, Scheller B, Bohm M, Fliser D and Heine GH. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. 2012;60:1512–20. [DOI] [PubMed] [Google Scholar]
- 143.Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, Nilsson J and Bjorkbacka H. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122–31. [DOI] [PubMed] [Google Scholar]
- 144.Tie G, Messina KE, Yan J, Messina JA and Messina LM. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J Am Heart Assoc. 2014;3:e000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Feng Y, Schouteden S, Geenens R, Van Duppen V, Herijgers P, Holvoet P, Van Veldhoven PP and Verfaillie CM. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS One. 2012;7:e47286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gao M, Zhao D, Schouteden S, Sorci-Thomas MG, Van Veldhoven PP, Eggermont K, Liu G, Verfaillie CM and Feng Y. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression. Arterioscler Thromb Vasc Biol. 2014;34:1900–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Park KH, Shin DG and Cho KH. Dysfunctional lipoproteins from young smokers exacerbate cellular senescence and atherogenesis with smaller particle size and severe oxidation and glycation. Toxicol Sci. 2014;140:16–25. [DOI] [PubMed] [Google Scholar]
- 148.Schill RL, Knaack DA, Powers HR, Chen Y, Yang M, Schill DJ, Silverstein RL and Sahoo D. Modification of HDL by reactive aldehydes alters select cardioprotective functions of HDL in macrophages. FEBS J. 2020;287:695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Han YH, Onufer EJ, Huang LH, Sprung RW, Davidson WS, Czepielewski RS, Wohltmann M, Sorci-Thomas MG, Warner BW and Randolph GJ. Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science. 2021;373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N and Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, de Beer FC, Miyake K, Oram JF and Heinecke JW. High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122:1919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, Vogelhuber J, Kraut M, Ulas T, Kerksiek A, Krebs W, Bode N, Grebe A, Fitzgerald ML, Hernandez NJ, Williams BR, Knolle P, Kneilling M, Rocken M, Lutjohann D, Wright SD, Schultze JL and Latz E. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 2014;15:152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dunigan-Russell K, Yaeger MJ, Hodge MX, Kilburg-Basnyat B, Reece SW, Birukova A, Guttenberg MA, Novak C, Chung S, Ehrmann BM, Wallace ED, Tokarz D, Majumder N, Xia L, Christman JW, Shannahan J, Ballinger MN, Hussain S, Shaikh SR, Tighe RM and Gowdy KM. Scavenger receptor BI attenuates oxidized phospholipid-induced pulmonary inflammation. Toxicol Appl Pharmacol. 2023;462:116381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Darabi M, Lhomme M, Dahik VD, Guillas I, Frisdal E, Tubeuf E, Poupel L, Patel M, Gautier EL, Huby T, Guerin M, Rye KA, Lesnik P, Le Goff W and Kontush A. Phosphatidylserine enhances anti-inflammatory effects of reconstituted HDL in macrophages via distinct intracellular pathways. FASEB J. 2022;36:e22274. [DOI] [PubMed] [Google Scholar]
- 155.Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, Demosky SJ Jr., and Remaley AT. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology. 2016;149:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, Welch CL, Reilly MP, Stroes ES, Moore KJ and Tall AR. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation. 2018;138:898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sanson M, Distel E and Fisher EA. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One. 2013;8:e74676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yurdagul A Jr., Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, Kolluru GK, Rymond CC, Gerlach BD, Zheng Z, Kuriakose G, Kevil CG, Koomen JM, Cleveland JL, Muoio DM and Tabas I. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020;31:518–533 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, Islam MN, Rymond CC, Du J, Hook J, Kuriakose G, Bhattacharya J and Tabas I. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity. 2018;49:666–677 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rueda CM, Rodriguez-Perea AL, Moreno-Fernandez M, Jackson CM, Melchior JT, Davidson WS and Chougnet CA. High density lipoproteins selectively promote the survival of human regulatory T cells. J Lipid Res. 2017;58:1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB and Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rivera K, Salas-Perez F, Echeverria G, Urquiaga I, Dicenta S, Perez D, de la Cerda P, Gonzalez L, Andia ME, Uribe S, Tejos C, Martinez G, Busso D, Irarrazaval P and Rigotti A. Red Wine Grape Pomace Attenuates Atherosclerosis and Myocardial Damage and Increases Survival in Association with Improved Plasma Antioxidant Activity in a Murine Model of Lethal Ischemic Heart Disease. Nutrients. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fuller M, Dadoo O, Serkis V, Abutouk D, MacDonald M, Dhingani N, Macri J, Igdoura SA and Trigatti BL. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol. 2014;34:2394–403. [DOI] [PubMed] [Google Scholar]
- 164.Zabalawi M, Bharadwaj M, Horton H, Cline M, Willingham M, Thomas MJ and Sorci-Thomas MG. Inflammation and skin cholesterol in LDLr−/−, apoA-I−/− mice: link between cholesterol homeostasis and self-tolerance? J Lipid Res. 2007;48:52–65. [DOI] [PubMed] [Google Scholar]
- 165.Bochem AE, van der Valk FM, Tolani S, Stroes ES, Westerterp M and Tall AR. Increased Systemic and Plaque Inflammation in ABCA1 Mutation Carriers With Attenuation by Statins. Arterioscler Thromb Vasc Biol. 2015;35:1663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brites F, Martin M, Guillas I and Kontush A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clinical. 2017;8:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mackness MI, Arrol S and Durrington PN. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991;286:152–4. [DOI] [PubMed] [Google Scholar]
- 168.Mackness MI, Abbott C, Arrol S and Durrington PN. The role of high-density lipoprotein and lipid-soluble antioxidant vitamins in inhibiting low-density lipoprotein oxidation. Biochemical Journal. 1993;294:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, Fu X, Wagner MA, Besler C, Gerstenecker G, Zhang R, Li XM, DiDonato AJ, Gogonea V, Tang WH, Smith JD, Plow EF, Fox PL, Shih DM, Lusis AJ, Fisher EA, DiDonato JA, Landmesser U and Hazen SL. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. The Journal of clinical investigation. 2013;123:3815–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aviram M, Hardak E, Vaya J, Mahmood S, Milo S, Hoffman A, Billicke S, Draganov D and Rosenblat M. Human Serum Paraoxonases (PON1) Q and R Selectively Decrease Lipid Peroxides in Human Coronary and Carotid Atherosclerotic Lesions. Circulation. 2000;101:2510–2517. [DOI] [PubMed] [Google Scholar]
- 171.Gaidukov L and Tawfik DS. The development of human sera tests for HDL-bound serum PON1 and its lipolactonase activity. J Lipid Res. 2007;48:1637–46. [DOI] [PubMed] [Google Scholar]
- 172.Gaidukov L and Tawfik DS. High affinity, stability, and lactonase activity of serum paraoxonase PON1 anchored on HDL with ApoA-I. Biochemistry. 2005;44:11843–54. [DOI] [PubMed] [Google Scholar]
- 173.Aggarwal G, May-Zhang LS, Yermalitsky V, Dikalov S, Voynov MA, Amarnath V, Kon V, Linton MF, Vickers KC and Davies SS. Myeloperoxidase-induced modification of HDL by isolevuglandins inhibits paraoxonase-1 activity. J Biol Chem. 2021;297:101019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mackness B, Quarck R, Verreth W, Mackness M and Holvoet P. Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:1545–50. [DOI] [PubMed] [Google Scholar]
- 175.Shih DM, Xia YR, Wang XP, Miller E, Castellani LW, Subbanagounder G, Cheroutre H, Faull KF, Berliner JA, Witztum JL and Lusis AJ. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. The Journal of biological chemistry. 2000;275:17527–35. [DOI] [PubMed] [Google Scholar]
- 176.Mackness Mackness, Durrington Arrol, Evans McMaster, Ferrières Ruidavets, Williams and Howard. Paraoxonase activity in two healthy populations with differing rates of coronary heart disease. European Journal of Clinical Investigation. 2000;30:4–10. [DOI] [PubMed] [Google Scholar]
- 177.Hernández-Díaz Y, Tovilla-Zárate CA, Juárez-Rojop IE, González-Castro TB, Rodríguez-Pérez C, López-Narváez ML, Rodríguez-Pérez JM and Cámara-Álvarez JF. Effects of paraoxonase 1 gene polymorphisms on heart diseases: Systematic review and meta-analysis of 64 case-control studies. Medicine. 2016;95:e5298–e5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu T, Zhang X, Zhang J, Liang Z, Cai W, Huang M, Yan C, Zhu Z and Han Y. Association between PON1 rs662 polymorphism and coronary artery disease. European journal of clinical nutrition. 2014;68:1029–35. [DOI] [PubMed] [Google Scholar]
- 179.Godbole C, Thaker S, Kerkar P, Nadkar M, Gogtay N and Thatte U. Association of PON1 gene polymorphisms and enzymatic activity with risk of coronary artery disease. Future Cardiology. 2021;17:119–126. [DOI] [PubMed] [Google Scholar]
- 180.Khalil A, Fulop T and Berrougui H. Role of Paraoxonase1 in the Regulation of High-Density Lipoprotein Functionality and in Cardiovascular Protection. Antioxid Redox Signal. 2021;34:191–200. [DOI] [PubMed] [Google Scholar]
- 181.Garner B, Waldeck AR, Witting PK, Rye KA and Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. The Journal of biological chemistry. 1998;273:6088–95. [DOI] [PubMed] [Google Scholar]
- 182.Panzenböck U, Kritharides L, Raftery M, Rye KA and Stocker R. Oxidation of methionine residues to methionine sulfoxides does not decrease potential antiatherogenic properties of apolipoprotein A-I. J Biol Chem. 2000;275:19536–44. [DOI] [PubMed] [Google Scholar]
- 183.Cheung MC and Albers JJ. Distribution of high density lipoprotein particles with different apoprotein composition: particles with A-I and A-II and particles with A-I but no A-II. Journal of Lipid Research. 1982;23:747–753. [PubMed] [Google Scholar]
- 184.Ribas V, Sánchez-Quesada JL, Antón R, Camacho M, Julve J, Escolà-Gil JC, Vila L, Ordóñez-Llanos J and Blanco-Vaca F. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: a new mechanism linking HDL protein composition and antiatherogenic potential. Circ Res. 2004;95:789–97. [DOI] [PubMed] [Google Scholar]
- 185.Ghiselli G, Krishnan S, Beigel Y and Gotto AM Plasma metabolism of apolipoprotein A-IV in humans. J Lipid Res. 1986;27:813–27. [PubMed] [Google Scholar]
- 186.Bisgaier CL, Sachdev OP, Megna L and Glickman RM. Distribution of apolipoprotein A-IV in human plasma. J Lipid Res. 1985;26:11–25. [PubMed] [Google Scholar]
- 187.Qin X, Swertfeger DK, Zheng S, Hui DY and Tso P. Apolipoprotein AIV: a potent endogenous inhibitor of lipid oxidation. Am J Physiol. 1998;274:H1836–40. [DOI] [PubMed] [Google Scholar]
- 188.Ostos MA, Conconi M, Vergnes L, Baroukh N, Ribalta J, Girona J, Caillaud JM, Ochoa A and Zakin MM. Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1023–8. [DOI] [PubMed] [Google Scholar]
- 189.Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl). 2016;94:739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Miyata M and Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet. 1996;14:55–61. [DOI] [PubMed] [Google Scholar]
- 191.Mabile L, Lefebvre C, Lavigne J, Boulet L, Davignon J, Lussier-Cacan S and Bernier L. Secreted apolipoprotein E reduces macrophage-mediated LDL oxidation in an isoform-dependent way. Journal of Cellular Biochemistry. 2003;90:766–776. [DOI] [PubMed] [Google Scholar]
- 192.Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH and Dahlbaäck B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. Journal of Lipid Research. 2006;47:1833–1843. [DOI] [PubMed] [Google Scholar]
- 193.Elsøe S, Ahnström J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW, Binder CJ, Björkbacka H, Dahlbäck B and Nielsen LB. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221:91–7. [DOI] [PubMed] [Google Scholar]
- 194.Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlbäck B and Nielsen LB. Effect of Apolipoprotein M on High Density Lipoprotein Metabolism and Atherosclerosis in Low Density Lipoprotein Receptor Knock-out Mice*. Journal of Biological Chemistry. 2008;283:1839–1847. [DOI] [PubMed] [Google Scholar]
- 195.Yermalitsky VN, Matafonova E, Tallman K, Li Z, Zackert W, Roberts LJ, 2nd, Amarnath V and Davies SS. Simplified LC/MS assay for the measurement of isolevuglandin protein adducts in plasma and tissue samples. Anal Biochem. 2019;566:89–101. [DOI] [PubMed] [Google Scholar]
- 196.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M and Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. The Journal of clinical investigation. 2004;114:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bergt C, Pennathur S, Fu X, Byun J, O’Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF and Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101:13032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Battle S, Gogonea V, Willard B, Wang Z, Fu X, Huang Y, Graham LM, Cameron SJ, DiDonato JA, Crabb JW and Hazen SL. The pattern of apolipoprotein A-I lysine carbamylation reflects its lipidation state and the chemical environment within human atherosclerotic aorta. J Biol Chem. 2022;298:101832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Guo L, Chen Z, Amarnath V, Yancey PG, Van Lenten BJ, Savage JR, Fazio S, Linton MF and Davies SS. Isolevuglandin-type lipid aldehydes induce the inflammatory response of macrophages by modifying phosphatidylethanolamines and activating the receptor for advanced glycation endproducts. Antioxid Redox Signal. 2015;22:1633–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Florens N, Calzada C, Lemoine S, Boulet MM, Guillot N, Barba C, Roux J, Delolme F, Page A, Poux JM, Laville M, Moulin P, Soulère L, Guebre-Egziabher F, Juillard L and Soulage CO. CKD Increases Carbonylation of HDL and Is Associated with Impaired Antiaggregant Properties. Journal of the American Society of Nephrology. 2020;31:1462–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhong J, Yang HC, Shelton EL, Matsusaka T, Clark AJ, Yermalitsky V, Mashhadi Z, May-Zhang LS, Linton MF, Fogo AB, Kirabo A, Davies SS and Kon V. Dicarbonyl-modified lipoproteins contribute to proteinuric kidney injury. JCI Insight. 2022;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF and Heinecke JW. Acrolein Impairs ATP Binding Cassette Transporter A1-dependent Cholesterol Export from Cells through Site-specific Modification of Apolipoprotein A-I. Journal of Biological Chemistry. 2005;280:36386–36396. [DOI] [PubMed] [Google Scholar]
- 203.Peng DQ, Wu Z, Brubaker G, Zheng L, Settle M, Gross E, Kinter M, Hazen SL and Smith JD. Tyrosine modification is not required for myeloperoxidase-induced loss of apolipoprotein A-I functional activities. J Biol Chem. 2005;280:33775–84. [DOI] [PubMed] [Google Scholar]
- 204.Peng DQ, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL and Smith JD. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol. 2008;28:2063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zamanian-Daryoush M, Gogonea V, DiDonato AJ, Buffa JA, Choucair I, Levison BS, Hughes RA, Ellington AD, Huang Y, Li XS, DiDonato JA and Hazen SL. Site-specific 5-hydroxytryptophan incorporation into apolipoprotein A-I impairs cholesterol efflux activity and high-density lipoprotein biogenesis. J Biol Chem. 2020;295:4836–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Opoku E, Berisha S, Brubaker G, Robinet P and Smith JD. Oxidant resistant human apolipoprotein A-I functions similarly to the unmodified human isoform in delaying atherosclerosis progression and promoting atherosclerosis regression in hyperlipidemic mice. PLoS One. 2022;17:e0259751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Berisha SZ, Brubaker G, Kasumov T, Hung KT, DiBello PM, Huang Y, Li L, Willard B, Pollard KA, Nagy LE, Hazen SL and Smith JD. HDL from apoA1 transgenic mice expressing the 4WF isoform is resistant to oxidative loss of function. J Lipid Res. 2015;56:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shao B, Cavigiolio G, Brot N, Oda MN and Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci U S A. 2008;105:12224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chadwick AC, Holme RL, Chen Y, Thomas MJ, Sorci-Thomas MG, Silverstein RL, Pritchard KA Jr. and Sahoo D Acrolein impairs the cholesterol transport functions of high density lipoproteins. PLoS One. 2015;10:e0123138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gao D, Ashraf MZ, Zhang L, Kar NS, Byzova TV and Podrez EA. Cross-linking modifications of HDL apoproteins by oxidized phospholipids: Structural characterization, in vivo detection, and functional implications. J Biol Chem. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nobecourt E, Davies MJ, Brown BE, Curtiss LK, Bonnet DJ, Charlton F, Januszewski AS, Jenkins AJ, Barter PJ and Rye KA. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia. 2007;50:643–53. [DOI] [PubMed] [Google Scholar]
- 212.Maziere JC, Myara I, Salmon S, Auclair M, Haigle J, Santus R and Maziere C. Copper- and malondialdehyde-induced modification of high density lipoprotein and parallel loss of lecithin cholesterol acyltransferase activation. Atherosclerosis. 1993;104:213–9. [DOI] [PubMed] [Google Scholar]
- 213.Sun JT, Yang K, Lu L, Zhu ZB, Zhu JZ, Ni JW, Han H, Chen N and Zhang RY. Increased carbamylation level of HDL in end-stage renal disease: carbamylated-HDL attenuated endothelial cell function. Am J Physiol Renal Physiol. 2016;310:F511–7. [DOI] [PubMed] [Google Scholar]
- 214.Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA and Hazen SL. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem. 2009;284:30825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ahmed Z, Ravandi A, Maguire GF, Emili A, Draganov D, Du BNL, Kuksis A and Connelly PW. Apolipoprotein A-I Promotes the Formation of Phosphatidylcholine Core Aldehydes That Are Hydrolyzed by Paraoxonase (PON-1) during High Density Lipoprotein Oxidation with a Peroxynitrite Donor*. Journal of Biological Chemistry. 2001;276:24473–24481. [DOI] [PubMed] [Google Scholar]
- 216.Wang G, Mathew AV, Yu H, Li L, He L, Gao W, Liu X, Guo Y, Byun J, Zhang J, Chen YE and Pennathur S. Myeloperoxidase mediated HDL oxidation and HDL proteome changes do not contribute to dysfunctional HDL in Chinese subjects with coronary artery disease. PLOS ONE. 2018;13:e0193782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tacka KA, Dabrowiak JC, Goodisman J and Souid AK. Kinetic analysis of the reactions of 4-hydroperoxycyclophosphamide and acrolein with glutathione, mesna, and WR-1065. Drug Metab Dispos. 2002;30:875–82. [DOI] [PubMed] [Google Scholar]
- 218.Amirshahrokhi K and Khalili AR. Gastroprotective effect of 2-mercaptoethane sulfonate against acute gastric mucosal damage induced by ethanol. Int Immunopharmacol. 2016;34:183–188. [DOI] [PubMed] [Google Scholar]
- 219.Triantafyllidis I, Poutahidis T, Taitzoglou I, Kesisoglou I, Lazaridis C and Botsios D. Treatment with Mesna and n-3 polyunsaturated fatty acids ameliorates experimental ulcerative colitis in rats. Int J Exp Pathol. 2015;96:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Arai T, Koyama R, Yuasa M, Kitamura D and Mizuta R. Acrolein, a highly toxic aldehyde generated under oxidative stress in vivo, aggravates the mouse liver damage after acetaminophen overdose. Biomed Res. 2014;35:389–95. [DOI] [PubMed] [Google Scholar]
- 221.Kheradmand A, Nayebi AM, Jorjani M, Khalifeh S and Haddadi R. Effects of WR1065 on 6-hydroxydopamine-induced motor imbalance: Possible involvement of oxidative stress and inflammatory cytokines. Neurosci Lett. 2016;627:7–12. [DOI] [PubMed] [Google Scholar]
- 222.Chronidou F, Apostolakis E, Papapostolou I, Grintzalis K, Georgiou CD, Koletsis EN, Karanikolas M, Papathanasopoulos P and Dougenis D. Beneficial effect of the oxygen free radical scavenger amifostine (WR-2721) on spinal cord ischemia/reperfusion injury in rabbits. Journal of cardiothoracic surgery. 2009;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Regazzoni L, de Courten B, Garzon D, Altomare A, Marinello C, Jakubova M, Vallova S, Krumpolec P, Carini M, Ukropec J, Ukropcova B and Aldini G. A carnosine intervention study in overweight human volunteers: bioavailability and reactive carbonyl species sequestering effect. Sci Rep. 2016;6:27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee YT, Hsu CC, Lin MH, Liu KS and Yin MC. Histidine and carnosine delay diabetic deterioration in mice and protect human low density lipoprotein against oxidation and glycation. Eur J Pharmacol. 2005;513:145–50. [DOI] [PubMed] [Google Scholar]
- 225.Peters V, Riedl E, Braunagel M, Höger S, Hauske S, Pfister F, Zschocke J, Lanthaler B, Benck U, Hammes HP, Krämer BK, Schmitt CP, Yard BA and Köppel H. Carnosine treatment in combination with ACE inhibition in diabetic rats. Regul Pept. 2014;194–195:36–40. [DOI] [PubMed] [Google Scholar]
- 226.Barski OA, Xie Z, Baba SP, Sithu SD, Agarwal A, Cai J, Bhatnagar A and Srivastava S. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2013;33:1162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Menini S, Iacobini C, Ricci C, Blasetti Fantauzzi C and Pugliese G. Protection from diabetes-induced atherosclerosis and renal disease by D-carnosine-octylester: effects of early vs late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia. 2015;58:845–53. [DOI] [PubMed] [Google Scholar]
- 228.Min J, Senut MC, Rajanikant K, Greenberg E, Bandagi R, Zemke D, Mousa A, Kassab M, Farooq MU, Gupta R and Majid A. Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J Neurosci Res. 2008;86:2984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhang X, Song L, Cheng X, Yang Y, Luan B, Jia L, Xu F and Zhang Z. Carnosine pretreatment protects against hypoxia-ischemia brain damage in the neonatal rat model. Eur J Pharmacol. 2011;667:202–7. [DOI] [PubMed] [Google Scholar]
- 230.de Courten B, Jakubova M, de Courten MP, Kukurova IJ, Vallova S, Krumpolec P, Valkovic L, Kurdiova T, Garzon D, Barbaresi S, Teede HJ, Derave W, Krssak M, Aldini G, Ukropec J and Ukropcova B. Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity. 2016;24:1027–34. [DOI] [PubMed] [Google Scholar]
- 231.Lombardi C, Carubelli V, Lazzarini V, Vizzardi E, Bordonali T, Ciccarese C, Castrini AI, Dei Cas A, Nodari S and Metra M. Effects of oral administration of orodispersible levo-carnosine on quality of life and exercise performance in patients with chronic heart failure. Nutrition. 2015;31:72–8. [DOI] [PubMed] [Google Scholar]
- 232.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd and Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Davies SS, Bodine C, Matafonova E, Pantazides BG, Bernoud-Hubac N, Harrison FE, Olson SJ, Montine TJ, Amarnath V and Roberts LJ 2nd. Treatment with a γ-ketoaldehyde scavenger prevents working memory deficits in hApoE4 mice. J Alzheimers Dis. 2011;27:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ, 2nd, Madhur MS and Harrison DG. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. The Journal of clinical investigation. 2016;126:50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Pitzer A, Elijovich F, Laffer CL, Ertuglu LA, Sahinoz M, Saleem M, Krishnan J, Dola T, Aden LA, Sheng Q, Raddatz MA, Wanjalla C, Pakala S, Davies SS, Patrick DM, Kon V, Ikizler TA, Kleyman T and Kirabo A. DC ENaC-Dependent Inflammasome Activation Contributes to Salt-Sensitive Hypertension. Circ Res. 2022;131:328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Patrick DM, de la Visitación N, Krishnan J, Chen W, Ormseth MJ, Stein CM, Davies SS, Amarnath V, Crofford LJ, Williams JM, Zhao S, Smart CD, Dikalov S, Dikalova A, Xiao L, Van Beusecum JP, Ao M, Fogo AB, Kirabo A and Harrison DG. Isolevuglandins disrupt PU.1-mediated C1q expression and promote autoimmunity and hypertension in systemic lupus erythematosus. JCI Insight. 2022;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Guo J, Xu F, Ji H, Jing Y, Shen L, Weng X and Hu L. Isolevuglandins Scavenger Ameliorates Myocardial Ischemic Injury by Suppressing Oxidative Stress, Apoptosis, and Inflammation. Front Cell Dev Biol. 2022;10:836035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Gobert AP, Asim M, Smith TM, Williams KJ, Barry DP, Allaman MM, McNamara KM, Hawkins CV, Delgado AG, Blanca Piazuelo M, Rathmacher JA and Wilson KT. The nutraceutical electrophile scavenger 2-hydroxybenzylamine (2-HOBA) attenuates gastric cancer development caused by Helicobacter pylori. Biomed Pharmacother. 2023;158:114092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ganjali S, Momtazi AA, Banach M, Kovanen PT, Stein EA and Sahebkar A. HDL abnormalities in familial hypercholesterolemia: Focus on biological functions. Prog Lipid Res. 2017;67:16–26. [DOI] [PubMed] [Google Scholar]
- 240.Escola-Gil JC, Rotllan N, Julve J and Blanco-Vaca F. Reverse Cholesterol Transport Dysfunction Is a Feature of Familial Hypercholesterolemia. Curr Atheroscler Rep. 2021;23:29. [DOI] [PubMed] [Google Scholar]
- 241.Huang J, Tao H, Yancey PG, Leuthner Z, May-Zhang LS, Jung JY, Zhang Y, Ding L, Amarnath V, Liu D, Collins S, Davies SS and Linton MF. Scavenging dicarbonyls with 5’-O-pentyl-pyridoxamine increases HDL net cholesterol efflux capacity and attenuates atherosclerosis and insulin resistance. Mol Metab. 2023;67:101651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Madu H, Avance J, Chetyrkin S, Darris C, Rose KL, Sanchez OA, Hudson B and Voziyan P. Pyridoxamine protects proteins from damage by hypohalous acids in vitro and in vivo. Free Radic Biol Med. 2015;89:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sampson UK, Fazio S and Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016;37:1720–2. [DOI] [PubMed] [Google Scholar]
- 245.Pitchford LM, Driver PM, Fuller JC Jr., Akers WS, Abumrad NN, Amarnath V, Milne GL, Chen SC, Ye F, Roberts LJ 2nd, Shoemaker MB, Oates JA, Rathmacher JA and Boutaud O. Safety, tolerability, and pharmacokinetics of repeated oral doses of 2-hydroxybenzylamine acetate in healthy volunteers: a double-blind, randomized, placebo-controlled clinical trial. BMC Pharmacol Toxicol. 2020;21:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Siebel AL, Heywood SE and Kingwell BA. HDL and glucose metabolism: current evidence and therapeutic potential. Front Pharmacol. 2015;6:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Dalla-Riva J, Stenkula KG, Petrlova J and Lagerstedt JO. Discoidal HDL and apoA-I-derived peptides improve glucose uptake in skeletal muscle. Journal of Lipid Research. 2013;54:1275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY, Regazzi R, Widmann C and Waeber G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia. 2007;50:1304–14. [DOI] [PubMed] [Google Scholar]
- 249.Yalcinkaya M, Kerksiek A, Gebert K, Annema W, Sibler R, Radosavljevic S, Lutjohann D, Rohrer L and von Eckardstein A. HDL inhibits endoplasmic reticulum stress-induced apoptosis of pancreatic beta-cells in vitro by activation of Smoothened. J Lipid Res. 2020;61:492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Masson W, Lobo M, Siniawski D, Huerin M, Molinero G, Valero R and Nogueira JP. Therapy with cholesteryl ester transfer protein (CETP) inhibitors and diabetes risk. Diabetes Metab. 2018;44:508–513. [DOI] [PubMed] [Google Scholar]
- 251.Drew BG, Duffy SJ, Formosa MF, Natoli AK, Henstridge DC, Penfold SA, Thomas WG, Mukhamedova N, de Courten B, Forbes JM, Yap FY, Kaye DM, van Hall G, Febbraio MA, Kemp BE, Sviridov D, Steinberg GR and Kingwell BA. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;119:2103–11. [DOI] [PubMed] [Google Scholar]
- 252.McMillen TS, Heinecke JW and LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111:2798–804. [DOI] [PubMed] [Google Scholar]