

Abstract
Multiple anticancer drugs have been proposed to cause cell death, in part, by increasing the steady-state levels of cellular reactive oxygen species (ROS). However, for most of these drugs exactly how the resultant ROS function and are sensed is poorly understood. It remains unclear which proteins the ROS modify and their roles in drug sensitivity/resistance. To answer these questions, we examined 11 anticancer drugs with an integrated proteogenomic approach identifying many unique targets but also shared ones–including ribosomal components, suggesting common mechanisms by which drugs regulate translation. We focus on CHK1 which we find is a nuclear H2O2 sensor that launches a cellular program to dampen ROS. CHK1 phosphorylates the mitochondrial-DNA binding protein SSBP1 to prevent its mitochondrial localization, which in turn decreases nuclear H2O2. Our results reveal a druggable nucleus-to-mitochondria ROS sensing pathway–required to resolve nuclear H2O2 accumulation and mediate resistance to platinum-based agents in ovarian cancers.
Keywords: Nuclear ROS, CHK1, mitochondrial translation, chemical proteomics, chemoresistance, nuclear-to-mitochondria signaling
Graphical Abstract
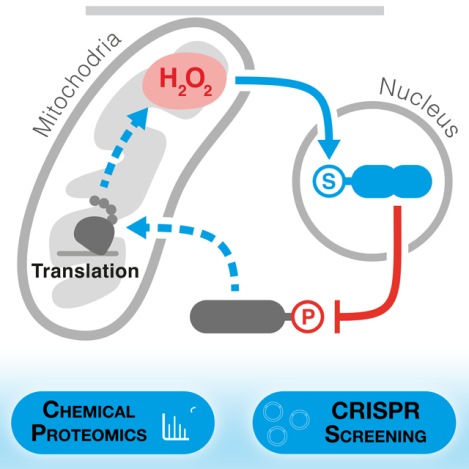
Anticancer drug mechanism studies using an integrated proteogenomic framework reveal a nucleus-to-mitochondria ROS sensing pathway that couples DNA damage response to control of mitochondrial translation and may serve as a mechanism of resistance to platinum-based agents.
Introduction
ROS represent a distinct family of reactive molecules that arise during normal cellular metabolism and are further generated in the context of disease states or toxin exposure11. The reactive nature of these molecules allows them to exert substantial control over multiple cellular pathways through the direct modification of proteins, nucleic acids, or lipids1,2. Mitochondria are best appreciated for their functions as intracellular ROS generators and sinks3,4 and cells have evolved numerous pathways to neutralize these reactive metabolites1. ROS levels are thought to dictate the scope of their cellular targets. At low levels, ROS are required to maintain normal cellular homeostasis, functioning in numerous signaling capacities through the modification of phosphatases and metabolic enzymes. At high levels, ROS damage nucleic acids, inactivate proteins and induce lipid peroxidation, leading to ferroptotic cell death5. In this regard, anticancer drugs that increase the steady-state levels of ROS are currently used in the treatment of multiple cancers, including leukemia (arsenic trioxide), ovarian (cisplatin), bladder (doxorubicin) and pancreatic cancers (5-fluorouracil)1,6,7. However, the mechanisms underlying ROS increase following anticancer drug treatment, their sensing and targets are poorly described8. As a result of this knowledge gap, there is limited understanding of chemoresistance, one of the greatest clinical challenges in modern cancer treatment and the promise of harnessing ROS as a therapeutic modality remains to be fully realized.
Whereas the non-specific nature of heightened ROS following anticancer drug treatment and its destruction of DNA has been proposed to underlie much of the activity of these agents8, there is a growing appreciation that ROS modification of specific proteins involved in key cellular pathways may also contribute to anticancer drug cytotoxicity9. Elucidating the mechanism of action of these ROS and in particular their molecular targets, has historically been problematic given the transient nature of ROS, the high concentrations required to observe phenotypic changes and the use of non-specific readouts, which have greatly limited functional insights. The global dissection of ROS-target proteins has been advanced using chemical proteomic technologies to profile changes in the reactivity of the amino acid, cysteine10. The unique chemical property of cysteine makes this residue a primary ROS target with central roles in regulating protein function. Using electrophilic cysteine-reactive probes, recent studies have categorized the direct cysteine targets of H2O2 and electrophilic lipids in vitro, revealing that ROS-regulated cysteines exist in kinases and metabolic enzymes11. Moreover, these platforms have been used to define the mechanisms by which cells reprogram their redox environment to protect essential pathways12–14.
While these chemical proteomic studies provide a list of ROS targets, a key challenge is to functionally understand how these protein targets contribute to the phenotypic consequences of ROS following anticancer drug treatment. To address these challenges, we describe an integrated approach comprised of cysteine-based chemical proteomics and functional genomic CRISPR screening. Using this framework, we provide a comprehensive portrait of functional protein targets of 11 different anticancer agents that have been previously found to regulate the steady-state levels of ROS in cells. We identified distinct proteins targeted by each cytotoxic agent, but also common targets including ribosomal proteins, which we connect as regulatory sites for ROS controlled translation. By leveraging our integrated approach, we uncover a nucleus-based ROS sensor that controls compartmentalized H2O2 levels through regulation of mitochondrial translation. We demonstrate that nuclear H2O2 modifies a functional and conserved cysteine within the CHK1 kinase. This modification leads to CHK1 activation through a conformational change in its autoinhibitory domain, resulting in the activation of the kinase which launches a cellular program required to decrease nuclear H2O2 levels. Functional studies using a clinical CHK1 inhibitor (MK-8776, an agent in clinical trials as a mono/combination therapy15–17), delineated a nuclear-to-mitochondria ROS sensing pathway that couples DNA damage response to control of mitochondrial translation, through the regulation of the mitochondrial DNA (mtDNA) binding protein SSBP1. Loss of SSBP1 decreases nuclear H2O2 levels and provides resistance to platinum-based agents in ovarian cancer models. Our findings underscore the value of integrating distinct read outs of ROS activity to systematically characterize the cellular response to broad acting anticancer agents and precision-oncology medicines.
Results
Many anticancer drugs regulate cysteine reactivity
Our studies focused on multiple anticancer drugs at different stages of clinical evaluation that have been shown to increase steady-state ROS levels (Table S1). These include arsenic trioxide18,19 (ATO), used for treating acute promyelocytic leukemia that functions in part through the degradation of the aberrant PML-retinoic acid receptor alpha fusion protein; β-lapachone20,21 (LAP), which has pleiotropic effects and functions by increasing steady-state ROS through NAD(P)H: quinone oxidoreductase-1; doxorubicin22 (DOXO), a DNA topoisomerase II inhibitor; elesclomol23–25 (ELC), a copper chelator which raises steady-state ROS and blocks mitochondria metabolism; cisplatin26,27 (DDP), a DNA crosslinker; bleomycin28,29 (BLE), which oxidatively cleaves DNA; 5-fluorouracil30 (5FU), an antimetabolite drug, exerting its anticancer effects through inhibition of thymidylate synthase and incorporation of its metabolites into RNA and DNA; sulfasalazine31–33 (SUL), an anti-inflammatory agent that functions as an xCT1 inhibitor resulting in an antioxidant imbalance; 2-methoxyestradiol34,35 (2ME), a microtubule stabilizer with pleotropic effects; Auranofin36 (AUR), which inhibits TXNRD1/2 and antioxidant imbalance; and NOV-00237,38 (NOV), a glutathione disulfide mimetic, which disrupts cellular redox balance. These drugs potently blocked the growth of the K562 cell line at 4 days (Figure S1A, Table S1). Testing these agents at ~5X their IC50 concentrations revealed minimal loss of cell viability at the 24 hrs time period, providing a context for exploring ROS signaling independent of secondary effects resulting from proliferation arrest (Figure S1B). We characterized changes in steady-state ROS levels of K562 cells following treatment with the above mentioned agents by measuring the intensity of 2’,7’-dicholorodihydrofluourscien diacetate (DCF) and the levels of metabolites and pathways required for ROS detoxification including: NAD+/NADH39 NADP+/NADPH40 and NRF2 signaling (Figures 1A, S1C). This analysis revealed both time and compound specific differences in ROS response pathways following drug treatment.
Figure 1: Defining cysteine targets of anticancer drugs with chemical proteomics.
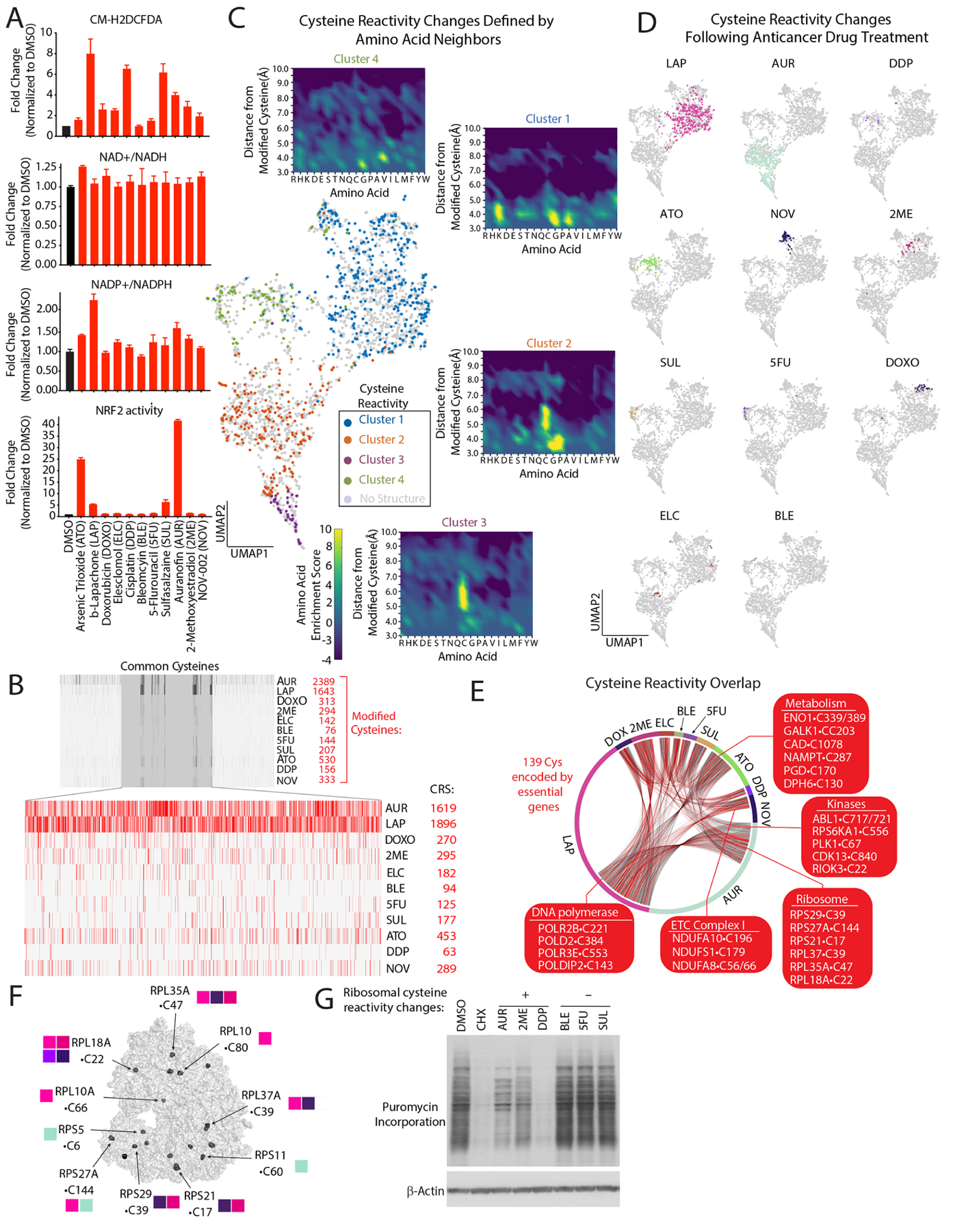
(A) Anticancer drugs regulate steady-state levels of ROS and antioxidant response pathways (see also Figure S1B). (B) Barcode plot of modified cysteines following treatment with the indicated agents and corresponding cysteine reactivity score (CRS) (see also methods, Table S2A). (C) UMAP representation of commonly detected cysteines regulated by anticancer drugs reveals they are localized to four distinct clusters which are color-coded based on available structures. Insets, enrichments plots of residues within a 10Å radial-sphere of reactive cysteines (see methods, Tables S2C). (D) UMAP of cysteine reactivity changes following treatment with indicated agents. (E) Connectivity diagram for shared cysteine targets of anticancer agents. (F) Ribosomal cysteines regulated by anticancer drugs. Adapted from PDB ID: 5LKS97. (G) Anticancer treatments blocks protein synthesis. Immunoblot analysis of puromycin incorporation into nascent proteins following treatment with the indicated compounds. Data are represented as mean ± SEM.
We next sought to discover proteins targeted by anticancer agents by monitoring changes in cysteine reactivity using the iso-TMT platform13,41,42. We rationalized that using this unbiased approach would allow us to capture the largest number of cysteine modifications, including oxidation, direct modification by a compound and adduction by ROS-related metabolites (e.g., lipid peroxides). These modifications are collectively read out as changes in cysteine reactivity that reflect primary and secondary ROS activities. We analyzed cells treated at early time points (2–3 hrs) to minimize changes in cysteine reactivities brought about by expression changes. We define cysteine reactivity changes regulated by these drugs as those showing an iso-TMT Ratio (R) with ≥1.5-fold change in reactivity compared to vehicle control. Out of 35656 cysteines and 8297 proteins identified, we found that 4980 cysteines within 2910 proteins had changes in their reactivity (Table S2A). K-means clustering of a subset (2498) of reactive cysteines detected in all proteomic experiments resulted in four distinct clusters of reactive cysteines (Figure 1C–D). To characterize the structural features underlying this clustering, we analyzed protein structures containing 4000+ commonly detected reactive and non-reactive cysteines (Table S2A). By identifying the nearest amino acid neighbors within a 10Å sphere centered on each cysteine of interest, we found specific amino acids enriched in each cluster (Figures 1C, S2A–B). For example, proximal cysteines are strongly selected for in clusters 2 and 3, suggesting the presence of disulfide bonds upon oxidation of the corresponding reactive cysteine. Interestingly, we identified a proximal lysine in cluster 1, suggesting the presence of the recently described lysine–cysteine redox switch43 (Figures 1C, S2B). We found that amino acids identified by our structural analysis were not encapsulated in the primary sequence surrounding a cysteine of interest, implying that the chemical properties of cysteine reactivity changes may be missed by analyzing primary sequences alone (Figure S2C). Using commonly detected cysteines, we developed a ‘cysteine reactivity score’, finding that AUR- and LAP had the greatest changes in reactivity among all treatments (Figures 1B). By concentrating our analysis on cysteines identified in all treatments, we found 500+ common cysteine targets regulated by two or more agents (Figure 1E) in addition to cysteines that were distinctly targeted by each drug (Figure 1C). The cysteine reactivity score correlated with DCF staining, with the notable exception of SUL (Figure S2D). This suggests that the cysteine reactivity score may provide a faithful representation of the cellular ROS status.
We found that modified cysteines mapped to multiple pathways, including protein synthesis, glycolysis, DNA replication, and mTORC1 nutrient sensing (Figure S2E, Table S3A). Multiple ribosomal proteins contained cysteines regulated by anticancer agents (Figures 1F, S2E), suggesting a mechanistic link between changes in cysteine reactivity and translational control. Treatment of cells with compounds that regulated ribosomal cysteine reactivity revealed that AUR, DDP, and 2ME to a lesser extent, decreased protein synthesis, whereas other agents that do not modify ribosomal cysteines reactivity (e.g., BLE, 5FU and SUL) did not have a measurable impact (Figure 1G). Among the regulated ribosomal cysteines, we found that C22 in RPL18A and C39 in RPLA37A were altered following treatment with multiple agents, implicating these proteins as potential ribosomal sensors of ROS imbalance (Figure S2F). We determined that 82% of cysteine reactivity changes reflected bona-fide changes in reactivity, while we attribute the remaining changes to alterations in protein expression (Table S2A). For AUR treatment, we monitored reactivity changes at 2 and 6 hrs to test if any early changes in cysteine reactivity would translate to changes in protein abundance. To this end, we overlaid modified cysteines with known ubiquitination sites on proteins and discovered that 41% of proteins whose expression was reduced contain a ubiquitination site within ≤20 residues of a modified cysteine (Figure S2G). For example, in AKT2, a critical regulator of cell growth and metabolism, C297 is the only cysteine modified at early time points. However, at later time points multiple cysteines on AKT2 are modified, decreasing expression by 1.5-fold, an observation that we confirmed by immunoblot (Figure S2H)–suggesting the existence of degrons specific to ROS-based cysteine modification. Collectively, these findings provide a comprehensive portrait of anticancer drug regulated cysteines and the immediate cellular pathways that are impacted by these agents.
Defining mechanisms of sensitivity and resistance to ROS regulated by an anticancer agent
Because cysteine reactivity changes can encompass many different modifications, we sought to prioritize agents that operate via an explicit ROS-based mechanism. For each compound, we compared its cysteine reactivity changes to those mediated by H2O2 (Table S2B) to the change in cytotoxicity following treatment with n-acetyl cysteine (NAC), a molecule with ROS scavenging abilities44. This comparison highlighted AUR as having the greatest overlap with H2O2 targets and rescue by NAC (Figure 2A), suggesting that it could be relevant to cellular pathways that sense and respond to an increase in H2O2. AUR is an FDA-approved drug for rheumatoid arthritis and is currently under experimental investigation for the treatment of chronic lymphocytic leukemia45 and ovarian cancer in combination with Sirolimus46. Although the established targets of this gold thiolate are TXNRD1/247, enzymes which are critical in the cellular antioxidant response, we suspected that given the diversity of its cysteines targets additional pathways may contribute to the cellular response to AUR. Importantly, we also suspected that AUR can be used as a tool to help understand the ROS-based pathways which are targeted by other anticancer drugs.
Figure 2: Functional genomic characterization of AUR-sensitizing and resistance pathways.
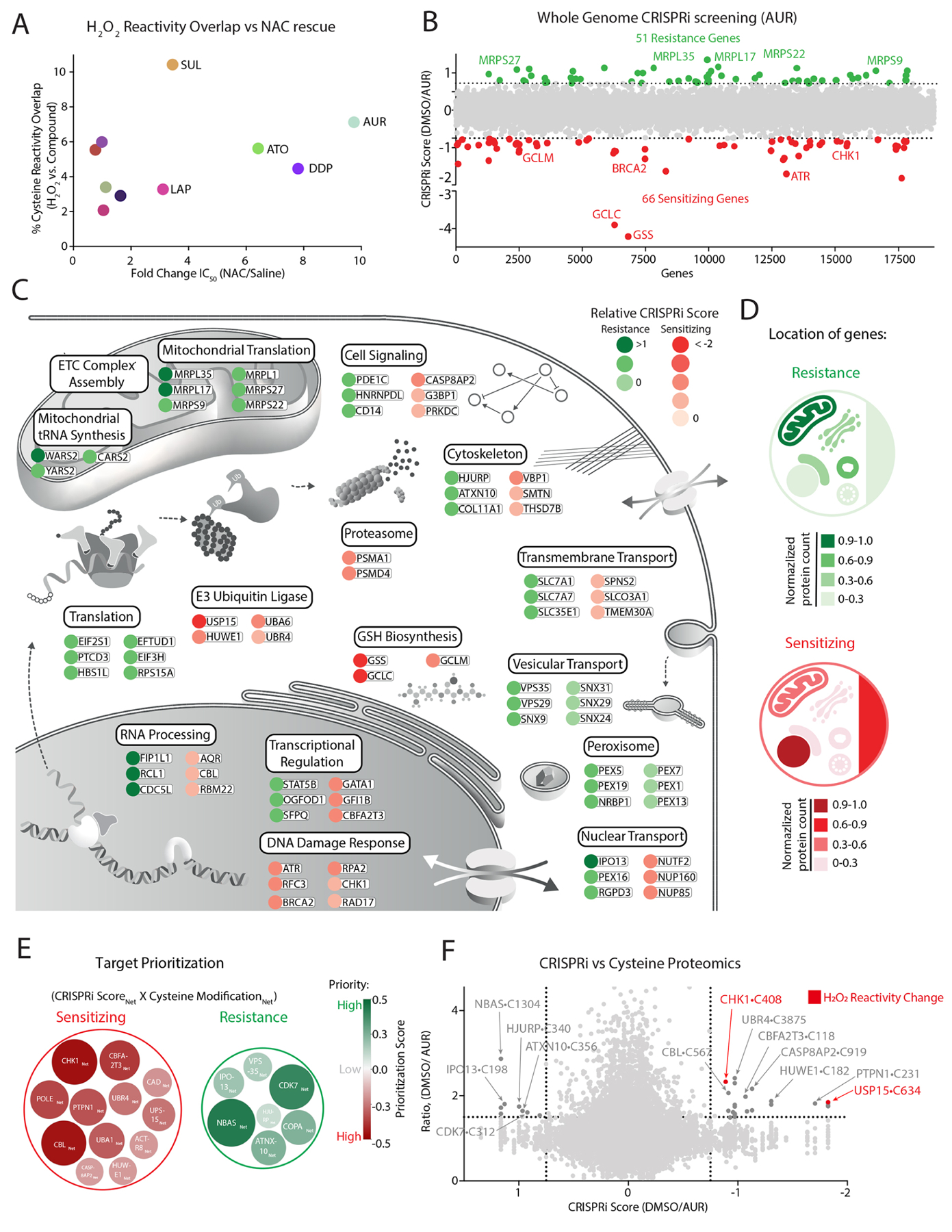
(A) Comparison of each agent’s overlap with H2O2 cysteine reactivity and fold-change in IC50 following NAC treatment (see also Figure S1C, Table S2A–B). (B) Genome-wide CRISPRi screen in K562 cells identifies genes that mediate sensitivity and resistance to AUR (see also Table S4). (C-D) Summary of top-scoring genes and corresponding pathways (C) and their cellular location (D) that promote resistance or sensitivity to AUR treatment. (E-F) Prioritization scheme for selecting targets (E) based on comparing their CRISPRi score and cysteine reactivity following AUR and H2O2 treatments (F) (see also methods, Tables S2–5).
To this end, we performed a genome-wide CRISPRi screen to identify mechanisms of sensitivity and resistance to AUR treatment. K562 cells expressing dCas9-KRAB48 were infected with a genome-wide sgRNA library and grown for 11 population doublings in the presence of vehicle or 1 μM of AUR (Figure 1B, Table S4). For each gene, we calculated a CRISPRi score by comparing the relative fold change between corresponding sgRNAs enriched in AUR vs. vehicle. This analysis identified 51 genes mediating resistance (e.g., whose corresponding sgRNAs were enriched in the treated cells) and 66 genes mediating sensitivity (e.g., sgRNAs depleted in the treated cells). The subcellular localization of hits from this screen highlighted the mitochondria and the nucleus as hubs of resistance and sensitivity, respectively (Figure 2D). We found multiple cellular pathways correlating with resistance, including those required for mitochondrial translation and ETC complex assembly. In contrast, genetic disruption of the DNA damage response (DDR) or the glutathione (GSH) biosynthetic pathway with buthionine sulfoximine (BSO)49, strongly sensitized cells to AUR treatment (Figures 2C, S3A–B) with the identification of GSH biosynthesis confirming the robustness of this study to identify key regulatory pathways.
To prioritize AUR targets for mechanistic characterization, we calculated a ‘prioritization score’ by integrating the CRISPRi score and cysteine reactivity change for a given target (Figure 2E, Table S5). We reasoned that relevant targets would belong to cellular networks whose members are likely to mediate AUR cytotoxicity or become modified following treatment. Thus, we incorporated the 10 closest interactors as defined by STRING50 into our prioritization score. This approach revealed multiple target proteins and corresponding networks, with an enrichment for high priority AUR targets/networks belonging to stress response or E3-ubiqutin ligase pathways (Figures 2E, S3F). Many of these pathways have been previously connected to ROS control51–55 and localize to the nucleus (Tables S3B,5). To further focus our analysis on response to ROS, we overlaid H2O2-mediated changes in cysteine reactivity within the AUR response networks, revealing that the DNA damage kinase CHK156 is both required for mediating sensitivity to AUR and is further modified by H2O2 (Figures 2F, S3C–E). This finding suggests that CHK1 may participate in nuclear ROS sensing and response.
Identification of CHK1•C408 as a nuclear ROS sensor
CHK1 is modified at C408, a highly conserved cysteine that lies within the C terminal KA1 domain (Figure 3A). KA1 functions in an autoinhibitory capacity by binding and blocking the activity of the N terminal CHK1 kinase domain57. Short-term treatment with AUR (3 hrs) led to CHK1 activation as measured by CHK1 autophosphorylation at S296, prior to a change in the phosphorylation of H2AX•S139 (Figure 3B), an established marker of DNA damage. Importantly, AUR activation of CHK1 was blocked by treatment with the antioxidant NAC (Figure 3B). To test the hypothesis that nuclear H2O2 may be involved in CHK1 activation, we first confirmed that AUR treatment increased nuclear H2O2 levels using two different nuclear localized H2O2 reporters, HyPer7 and roGFP2-Orp158,59 (Figure S4A). Both reporters were oxidized following treatment with AUR, and this oxidation was completely rescued following treatment with NAC (Figures 3C, S4B–C). Treatment of cells with H2O2 led to a decrease in IA-DTB labeling of CHK1•C408 (Figures 3D, S4D) and a concomitant increase in binding of the sulfinic acid specific probe, nitroso-desthiobiotin (NO-DTB)60–62, to CHK1•C408 (Figure 3E) suggesting that H2O2 directly oxidizes CHK1. To determine if CHK1•C408 oxidation by H2O2 leads to kinase activation, we targeted D-amino acid oxidase (DAAO) to the nucleus (Figure S4E). DAAO is an enzyme which oxidizes D-amino acids to their corresponding α-keto acids producing H2O263 and we found that short-term treatment with D-Ala but not L-Ala leads to an increase in CHK1 activity in a NAC-dependent manner (Figure 3F). At early time points, activation of nuclear DAAO did not increase H2AX•S139 phosphorylation (Figure 3F), suggesting that H2O2 activation of CHK1 precedes activation of canonical DNA damage markers. To demonstrate that H2O2 in cells is sufficient to activate CHK1, we fused DAAO to the N-terminus of CHK1 and localized the chimeric protein to the mitochondrial outer membrane, finding H2O2 activates CHK1 in this context (Figures 3G–H). Finally, in vitro treatment of CHK1 with H2O2 led to a dose-dependent increase in CHK1 activity (Figures 3I, S4F). Using a CHK1 in vitro binding assay, we found that addition of H2O2 strongly diminished the interaction between the KA1 and CHK1 kinase domain (Figure 3J), providing a mechanism by which CHK1•C408 oxidation activates CHK1.
Figure 3: CHK1 functions as a sensor of nuclear H2O2 levels.
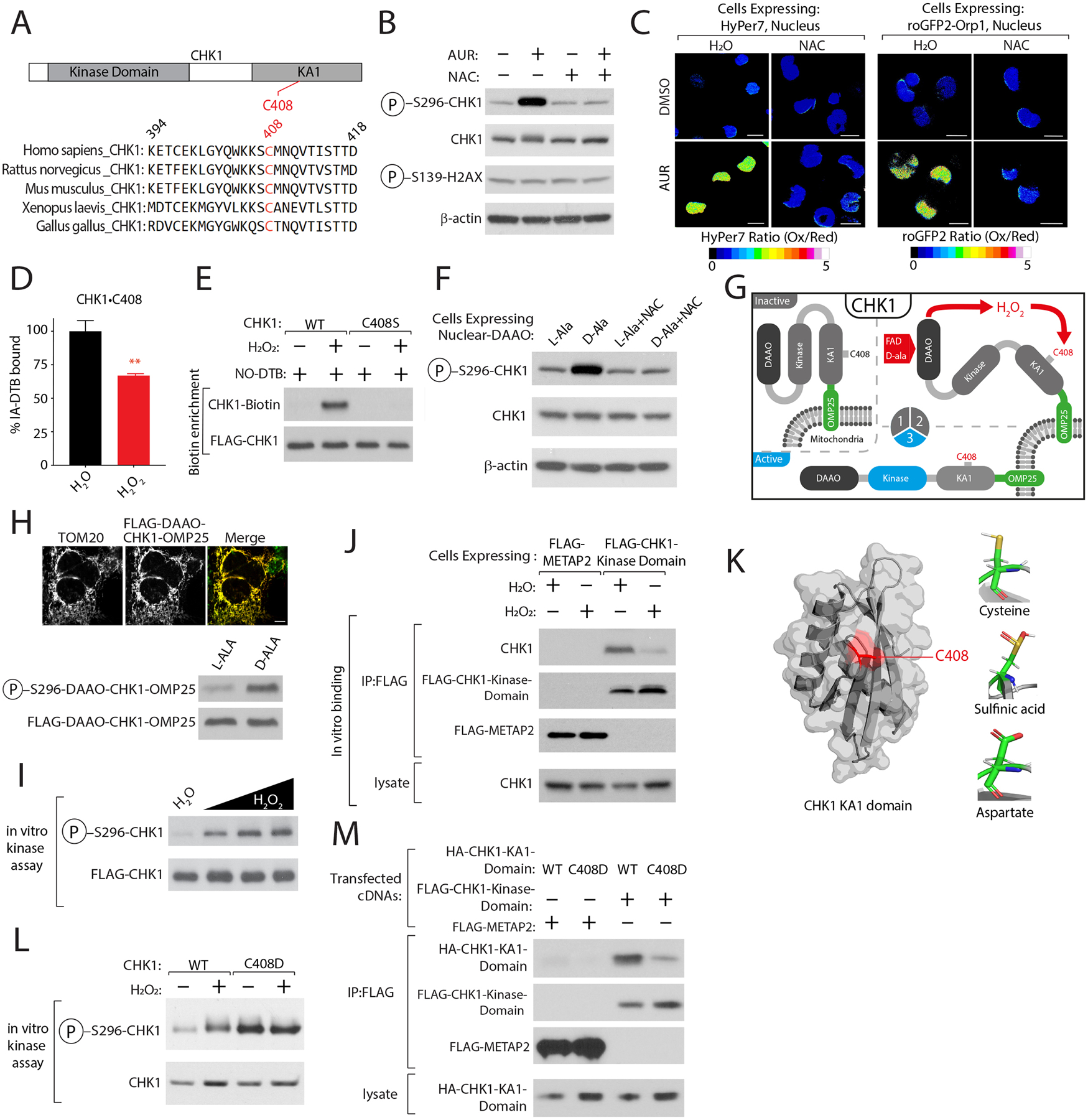
(A) C408 is highly conserved. (B) AUR activates CHK1 in a NAC-dependent manner. K562 cells were treated with AUR in the presence of NAC or vehicle control CHK1 activity was determined by immunoblotting for the indicated proteins. (C) AUR increases the steady-state levels of nuclear H2O2. Ratiometric images of HyPer7 or roGFP2-ORP1 H2O2 reporters localized to the nucleus following indicated treatments (see also Figures S5A–C). (D) H2O2 treatment regulates CHK1•C408 oxidation (see also Table S2B). (E) Oxidation of CHK1•C408 results in sulfinic acid formation (see methods). (F) Nuclear H2O2 activates CHK1. K562 cells stably expressing D-amino acid oxidase (DAAO) localized to the nucleus were treated with either L-Ala or D-Ala or NAC and CHK1 kinase activity was determined as described in (B). (G) Schematic depicting the localization of DAAO-CHK1 to the mitochondrial outer membrane. (H) H2O2 is sufficient to activate CHK1. Top, Immunofluorescence analysis of mitochondrial localization of DAAO-CHK1-OMP25. Bottom, DAAO-CHK1-OMP25 kinase activity was as described in (F). (I) H2O2 directly activates CHK1. CHK1 in vitro kinase assay following treatment with increasing amounts of H2O2. (J) H2O2 reduces the interaction between endogenous CHK1 and the CHK1 kinase domain. (K) Right, Crystal structure of C-terminal Kinase Associate 1 (KA1) domain of CHK1 highlighting the location of C408 in red, adapted from PDB ID: 5WI298. Left, Modeling of CHK1•C408 interactions as a sulfinic acid or when mutated to Asp. (L) CHK1•C408D has elevated kinase activity. (M) CHK1•C408D-mutation in KA1 domain blocks interaction with CHK1 kinase domain. Scale Bar=10 μm. Data are represented as mean ± SEM. **p < 0.001, ***p< 0.0001. Student’s t-test (two-tailed, unpaired) were used to determine statistical significance.
Although no amino acid mutation can mimic C408 oxidation, we mutated this residue to Asp to model an oxidized form of cysteine, sulfinic acid64 (Figure 3K). In vitro, the CHK1•C408D mutant and H2O2 -treated CHK1 demonstrated similar activation. However, the mutant could not be further activated, indicating that C408 is the target of H2O2 in the kinase (Figure 3L). Using a modified binding assay that monitors KA1 and kinase domain interaction, we confirmed that the KA1 domain harboring the C408D mutant poorly interacted with the N-terminal kinase domain of CHK1 (Figure 3M). Following AUR treatment, we found that K562 cells stably expressing FLAG-CHK1•C408D proliferate to a greater degree than cells expressing a control protein (METAP2) or WT CHK1, with a concomitant reduction in H2AX•S139 phosphorylation (Figures S4G–H). These data strongly suggest that C408 in CHK1 is a nuclear H2O2 sensor, which functions by disrupting the association of KA1 domain with the kinase domain leading to CHK1 activation.
CHK1 inhibition increases steady-state levels of nuclear H2O2
Given our identification of CHK1 as a nuclear H2O2 sensor, we wondered whether CHK1 might have a broader role in controlling ROS levels within this compartment. Treatment of K562 cells with a CHK1-inhibitor (MK-8776 herein referred to as CHK1i)65 or depletion of CHK1 resulted in a substantial increase in nuclear H2O2 levels as determined by both the HyPer7 and roGFP2-Orp1 reporters (Figures 4A, D-E, S4I–L). Accordingly, reintroduction of CHK1 into CHK1 depleted cells decreased nuclear H2O2 levels (Figure 4B–C). Treatment with antioxidants including NAC, GSH ethyl ester (GSHee), and L-ergothionine rescued CHK1i mediated nuclear H2O2 (Figure 4D). Interestingly, NAC treatment also partially rescued the proliferation defects and H2AX•S139 phosphorylation incurred by this inhibitor (Figure 4F–G), further supporting the role of CHK1 in nuclear ROS response. Consistent with previous reports that demonstrate that high levels of nuclear H2O2 can inhibit cell growth66, we found that targeted generation of nuclear H2O2 with nuclear DAAO enhanced CHK1i-mediated cytotoxicity (Figure 4H). These results suggest that CHK1 is not simply an oxidized bystander, but rather is part of a dedicated sensing pathway that integrates nuclear H2O2 levels to combat a lethal rise in peroxide levels in the nucleus.
Figure 4: CHK1 regulates nuclear H2O2 levels.
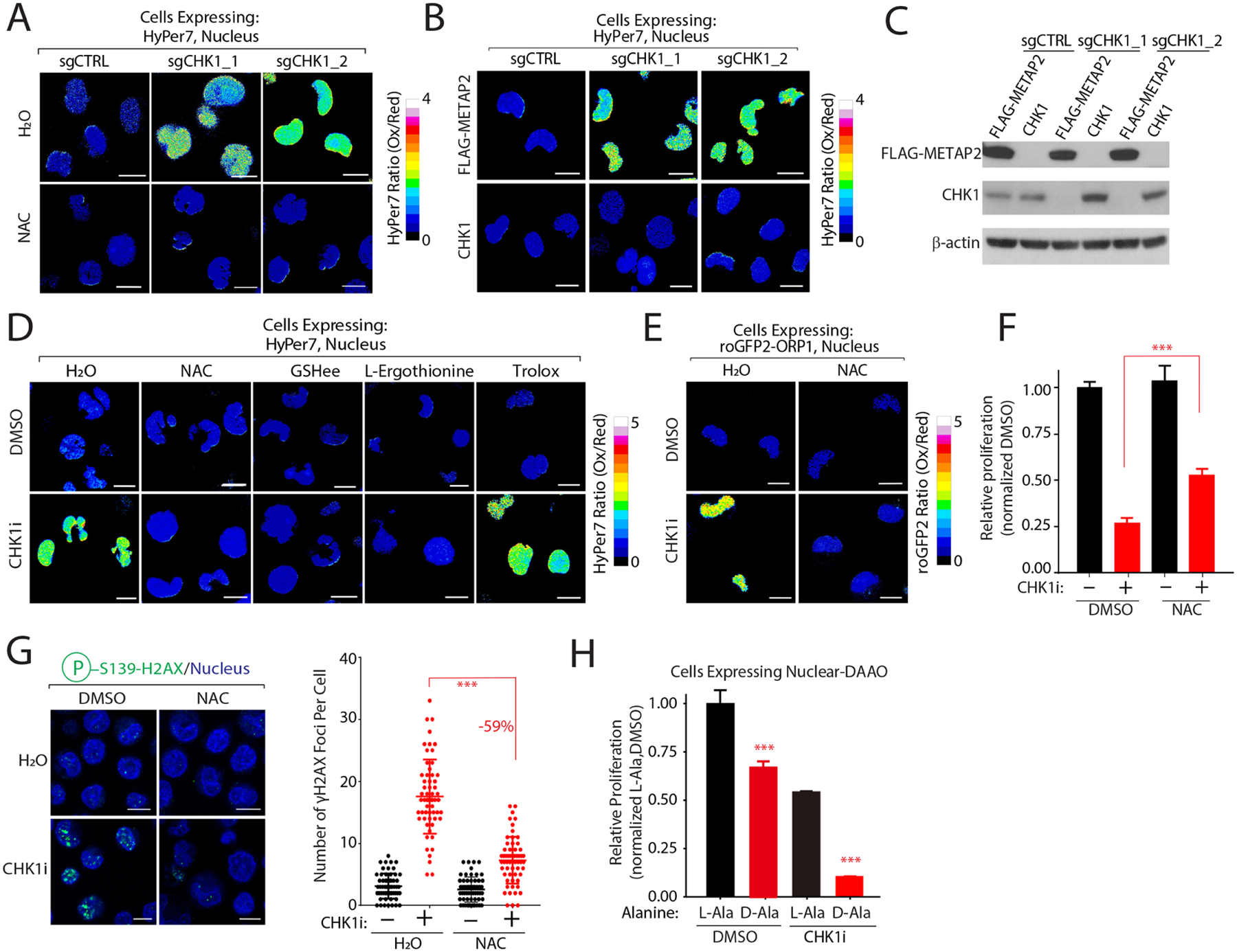
(A) CHK1 depletion increases steady-state nuclear H2O2 levels in a NAC-dependent manner. Nuclear H2O2 levels were measured in K562 co-expressing the indicated sgRNAs and nuclear HyPer7. (B-C) Re-introduction of CHK1 restores nuclear H2O2 levels. H2O2 levels were determined by nuclear HyPer7 (B) and levels of the indicated proteins by immunoblot (C) following reintroduction of CHK1 into K562 cells depleted of CHK1. (D-E) CHK1 inhibition increases nuclear H2O2 in an antioxidant-dependent manner as measured by HyPer7 (D) and roGFP2-ORP1 (E). (F) NAC treatment partially rescues CHK1i cytotoxicity. Relative proliferation was determined after 96 hrs by measuring cellular ATP concentrations. (G) NAC protects cells from CHK1i mediated DNA damage. Immunofluorescence analysis of H2AX•S139 staining (left) and quantification (right). (H) Nuclear H2O2 increases CHK1i cytotoxicity to block cell proliferation. K562 cells stably expressing nuclear localized DAAO were pretreated with L-Ala or D-Ala prior to treatment with CHK1i. Relative proliferation was determined as described in (F). Scale bar=10 μm. Data are represented as mean ± SEM. ***P < 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
CHK1 phosphorylation of SSBP1 restricts its mitochondrial localization and decreases nuclear H2O2 level
Because CHK1 inhibition raises nuclear H2O2 levels, we hypothesized that a CHK1 substrate might be directly involved in ROS regulation. To identify the substrate, we overlaid CHK1 phosphoproteomics data67 with our CRISPRi screen to find substrates whose depletion may revert or exacerbate AUR cytotoxicity (Figure 5A). Using this approach, we found that SSBP1, a mtDNA binding protein, met the criteria of a potential candidate. SSBP1 is active in the mitochondria, where it facilitates the interaction between mtDNA polymerase pol γ and the mtDNA helicase at the mtDNA replication fork68. We verified that a reduction in SSBP1 levels led to a pronounced rescue of AUR cytotoxicity (Figure S5A). To mechanistically dissect SSBP1 regulation by CHK1, we first established that CHK1 phosphorylates SSBP1 in vitro at S67 (Figures 5B, S5F). We found an H2O2-dependent increase in SSBP1 phosphorylation but did not detect any phosphorylation in a SSBP1•S67A mutant (Figure 5B). We also observed the CHK1•C408D mutant had heightened phosphorylation of SSBP1 (Figure 5C), consistent with the hyperactive state of this mutant. CHK1 and SSBP1 exist in an epistatic relationship, because nuclear H2O2 levels were significantly lowered following CHK1i treatment in cells depleted of SSBP1 in addition to a partial rescue of the cytotoxicity following CHK1 inhibition (Figures 5D, S5B–D).
Figure 5: CHK1 phosphorylates SSBP1 blocking its mitochondrial localization to decrease nuclear H2O2 levels.
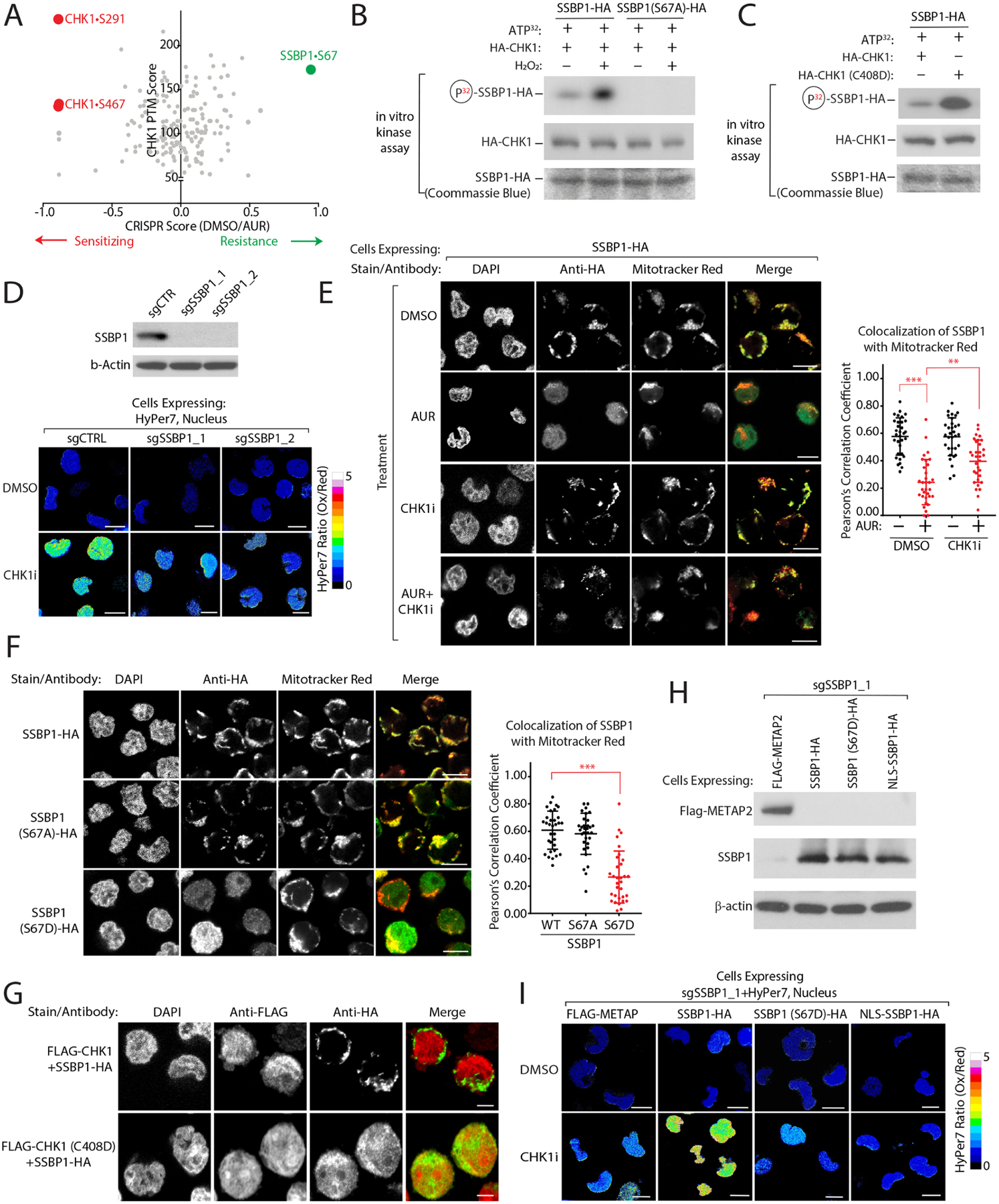
(A) Comparison of CRISPRi scores following AUR treatment with CHK1 phosphorylation sites characterized in Blasius et al.67 identifies SSBP1•S67 as a potential CHK1 target mediating resistance to AUR. (B) SSBP1 is a direct target of CHK1 that is phosphorylated in a H2O2-dependent manner in vitro (see methods). (C) CHK1•C408D has heightened levels of activity towards SSBP1. (D) SSBP1 regulates nuclear H2O2 levels downstream of CHK1. Left, immunoblot of SSBP1 levels in K562-dCas9-KRAB cells expressing the indicated sgRNAs. Right, Measurement of nuclear H2O2 with HyPer7 in K562 cells depleted of the indicated genes. (E) SSBP1 phosphorylation by CHK1 blocks its mitochondrial localization. Left, the localization of SSBP1-HA was determined by immunofluorescence analysis of K562 cells following the indicated treatments. Right, quantification of mitochondrial colocalization of HA-SSBP1. (F) SSBP1•S67D phosphomimetic mutant does not localize to the mitochondria. (G) CHK1•C408D is sufficient to drive SSBP1 re-localization. (H-I) SSBP1•S67D phosphomimetic mutant decreases nuclear H2O2 following CHK1 inhibition. Immunoblot analysis of the indicated proteins reintroduced into SSBP1-depleted cells (H) and corresponding levels of H2O2 levels measured with nuclear HyPer7 (I). Scale Bar=10 μm. Data are represented as mean ± SEM. **p < 0.001, ***p< 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
The mitochondrial localization of SSBP1 is required for its cellular activity69. Strikingly, we found that CHK1 activation, following AUR treatment, results in the redistribution of SSBP1 away from the mitochondria which could be rescued following treatment with CHK1i (Figure 5E, S5H). A SSBP1•S67D phosphomimetic mutant mirrored the redistribution of WT SSBP1 following AUR treatment (Figure 5F, S5I). The SSBP1•S67A phospho-deficient mutant constitutively localized to the mitochondria even following AUR treatment (Figure S5J), suggesting that CHK1 directly regulates SSBP1 localization through CHK1•S67 phosphorylation. Importantly, expression of constitutively active CHK1•C408D resulted in the cytosolic localization of SSBP1, in comparison to cells expressing wildtype CHK1 (Figure 5G), indicating that CHK1 activity is sufficient to direct the localization of SSBP1. To evaluate the impact of SSBP1 localization on nuclear H2O2 levels, we depleted endogenous SSBP1 and added back WT SSBP1, SSBP1•S67D or a nuclear localized SSBP1 (SSBP1-NLS), finding that in comparison to WT SSBP1, expression of SSBP1•S67D or SSBP1-NLS significantly reduced nuclear H2O2 levels following CHK1i or AUR treatment (Figures 5H–I, S5E). Cells expressing SSBP1•S67D were additionally protected from CHK1i and AUR cytotoxicity relative to cells expressing WT SSBP1 (Figures S5K–L), demonstrating that CHK1 regulates nuclear H2O2 levels through the phosphorylation and subsequent cytosolic retention of SSBP1.
CHK1-SSBP1 modulates mitochondrial translation to control nuclear H2O2 levels
Given its role in mitochondrial function, we suspected that SSBP1 may impact mitochondrial ROS, one of the major sites of cellular H2O2 production4. We found that depletion of SSBP1 led to a significant decrease in mitochondrial matrix H2O2 and superoxide following treatment with CHK1i (Figures 6A, S6A–B), suggesting that CHK1 regulates nuclear H2O2 levels through mitochondrial ROS generation. These results pointed to a nucleus-to-mitochondria signaling pathway, and we found that treatment of cells with Mito-TEMPO, a mitochondrially localized redox modulator70, lowered nuclear H2O2 levels following CHK1i treatment (Figure 6B). Targeted generation of H2O2 within the mitochondrial matrix was only cytotoxic in the presence of CHK1i (Figure 6C, S6C), suggesting that cytotoxicity imparted by CHK1 inhibition is partially dependent on mitochondrial H2O2. To determine which ETC complex might contribute to CHK1-mediated mitochondrial ROS, we co-treated cells with CHK1i and S1QEL (a complex I superoxide suppressor71,72) and S3QEL (a complex III superoxide suppressor73), finding a decrease in nuclear H2O2 only after complex I superoxide suppression (Figure 6D). These epistasis experiments indicate that SSBP1 functions downstream of CHK1 to regulate mitochondrial H2O2 levels which in turn control nuclear H2O2 levels.
Figure 6: CHK1-SSBP1 regulates mitochondrial translation to control mitochondrial/nuclear H2O2 levels.
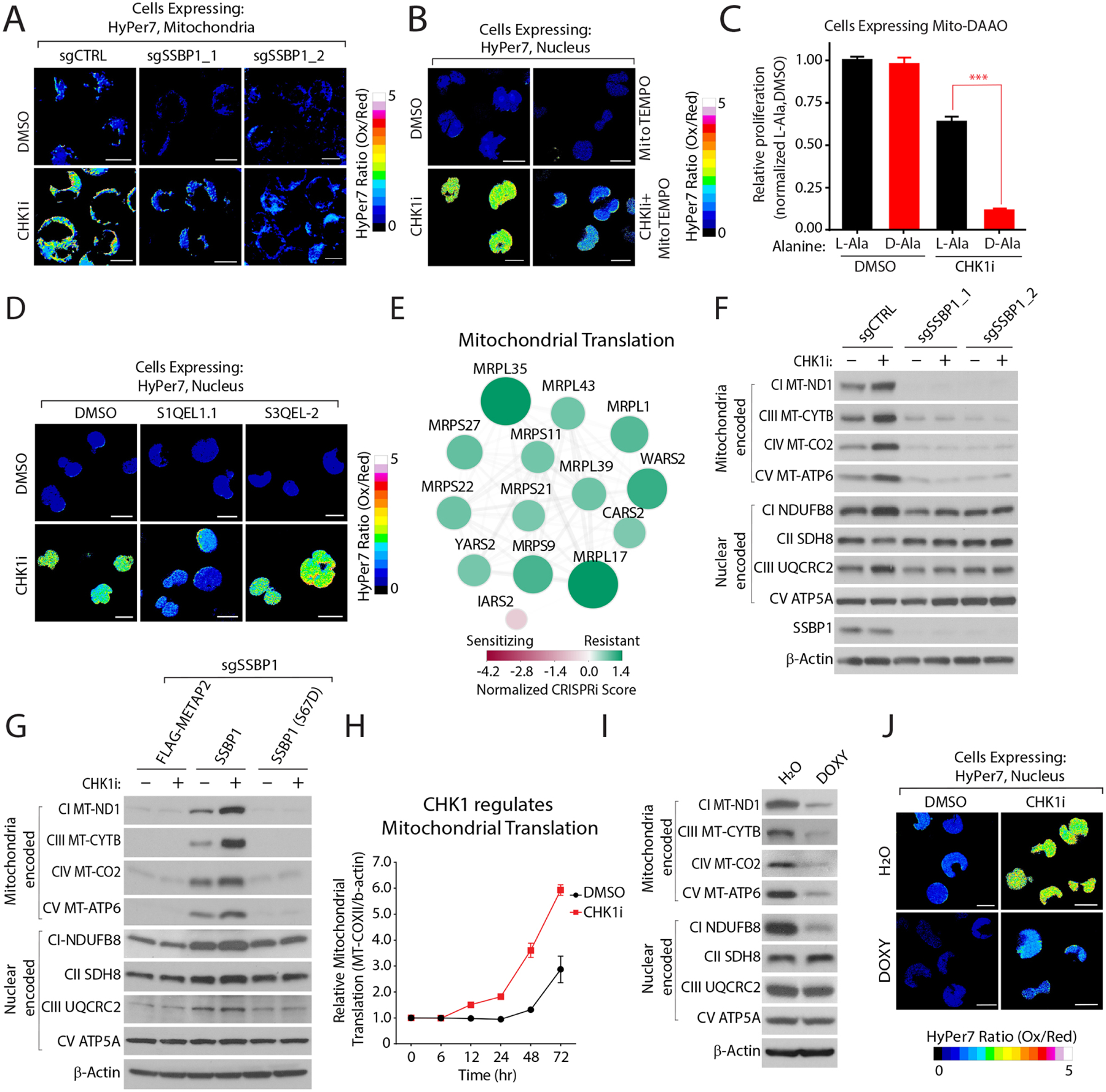
(A) CHK1 regulates mitochondrial H2O2 levels in a SSBP1-dependent manner. H2O2 levels were determined with HyPer7 localized to the mitochondrial membrane in K562 cells depleted of the indicated genes. (B) Mitochondrial H2O2 levels regulate nuclear H2O2 levels. Nuclear H2O2 was measured with HyPer7 in K562 cells treated with mitoTEMPO and CHK1i. (C) Mitochondrial H2O2 increases CHK1i cytotoxicity. Proliferation was determined by measuring relative ATP levels. (D) Suppression of complex I (S1QEL1.1) but not complex III (S3QEL-2) superoxide partially reverts CHK1i-mediated nuclear H2O2. (E) CRISPRi scores of the indicated genes involved in mitochondrial translation. (F) CHK1 regulates mitochondrial translated proteins in a SSBP1-dependent manner. Immunoblot analysis of the indicated proteins in cells depleted of SSBP1 and treated with CHK1i. (G) CHK1-regulation of mitochondrial translation depends on SSPB1 localization. Immunoblot analysis of the indicated mitochondrial proteins as described in (F) in cells expressing SSBP1 phosphorylation mutants in K562 cells depleted of SSBP1. (H) Inhibition of CHK1 increases mitochondrial translation. Expression of MT-CO2 was determined by immunoblot and normalized to β-actin following CHK1i treatment (see also methods, Figure S6H). (I) Doxycycline (DOXY) decreases mitochondrially translated proteins. (J) DOXY treatment reduces CHK1i-mediated nuclear H2O2 levels. Scale Bar=10 μm. Data are represented as mean ± SEM. ***p< 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Recalling that top-scoring resistance genes to AUR toxicity per our CRISPRi screen were involved in mitochondrial translation (Figure 6E), we wondered whether the regulation of nuclear H2O2 by SSBP1 occurs at the level of mitochondrial translation. Consistent with its regulation of mtDNA74, depletion of SSBP1 reduced mtDNA (Figures S6D–E) and led to a strong downregulation of ETC proteins encoded by mtDNA, including MT-ND1, MT-CO2, MT-CYTB, and MT-ATP6 (Figure 6F, S6F). Other ETC components encoded by genomic DNA did not show a similar decrease (Figure 6F, S6F). Interestingly, inhibition of CHK1 resulted in an increase in the expression of mitochondrially encoded proteins and further increased mitochondrial translation rates in comparison to vehicle control (Figures 6F–H, S6G–H). The regulation of mitochondrial translation by CHK1 was directly dependent on SSBP1, as SSBP1 depletion or expression of the SSBP1•S76D cytosolic mutant prevented a corresponding increase in mitochondrially-encoded protein expression following CHK1i treatment (Figure 6G). To directly demonstrate that mitochondrial translation is necessary to regulate nuclear H2O2 levels downstream of CHK1, we treated cells with doxycycline (DOXY), an inhibitor of mitochondrial translation75, finding a significant rescue of nuclear H2O2 levels following CHK1 inhibition (Figures 6I–J, S6I). Importantly, DOXY treatment reduced total mitochondrial H2O2 levels following CHK1i treatment (Figure S6J). Finally, we observed a significant decrease in CHK1i-mediated H2AX•S139 phosphorylation following DOXY treatment or SSBP1 depletion (Figures S6K–N), supporting our finding that DNA damage following CHK1 inhibition is, in part, due to dysregulation of mitochondrial translation.
We next asked whether SSBP1 could alter nuclear H2O2 levels following treatment with 9 additional anticancer agents, finding that depletion of this protein substantially decreased nuclear H2O2 levels following treatment with ATO, LAP and DDP (Figure 7A). Interestingly, cells depleted of SSBP1 were partially protected from the cytotoxicity of agents that raised nuclear H2O2 levels (Figures 7B, S6P). Because inhibition of GSH biosynthesis increased sensitivity to AUR treatment (Figures S3A–B), we wondered whether this GSH-dependency would also extend to other agents that increase nuclear H2O2 levels. Indeed, we find that depletion of glutathione increased the cytotoxicity of AUR, LAP and DDP (Figure S7A), and further complements previous findings demonstrating the requirement of GSH to neutralize DDP preventing its adduction to DNA76,77. In contrast, GSH depletion did not affect the cytotoxicity of drugs such as 5FU or 2ME that do not alter nuclear H2O2 (Figures 7B, S6P, S7A). Mechanistically, we find that treatment with agents that result in higher levels of nuclear H2O2 also have an increased GSSG:GSH ratio, whereas compounds such as 5FU or 2ME did not alter this ratio (Figure S7B). Because H2O2 generated within the mitochondria must traverse the cytosol to reach the nucleus overcoming high concentrations of GSH78,79, we elected to measure cytosolic GSH/GSSG following treatment with these agents using a cytosolic GSH/GSSG reporter (GRX1-roGFP2)80. Anticancer agents which raise nuclear H2O2 (e.g. AUR, LAP and DDP) decreased the cytosolic GSH/GSSG ratio (Figure S7C–D). These results suggest the presence of an ‘AND gate’ required to regulate nuclear ROS by anticancer drugs: CHK1/SSBP1 control mitochondrial H2O2 and anticancer agents contribute to the regulation of GSH/GSSG ratio. Together, they work in concert to permit mitochondrial H2O2 to travel to the nucleus, raising peroxide levels in this compartment.
Figure 7: SSBP1 regulates nuclear H2O2 levels and mediates cisplatin resistance in ovarian cancer cells.
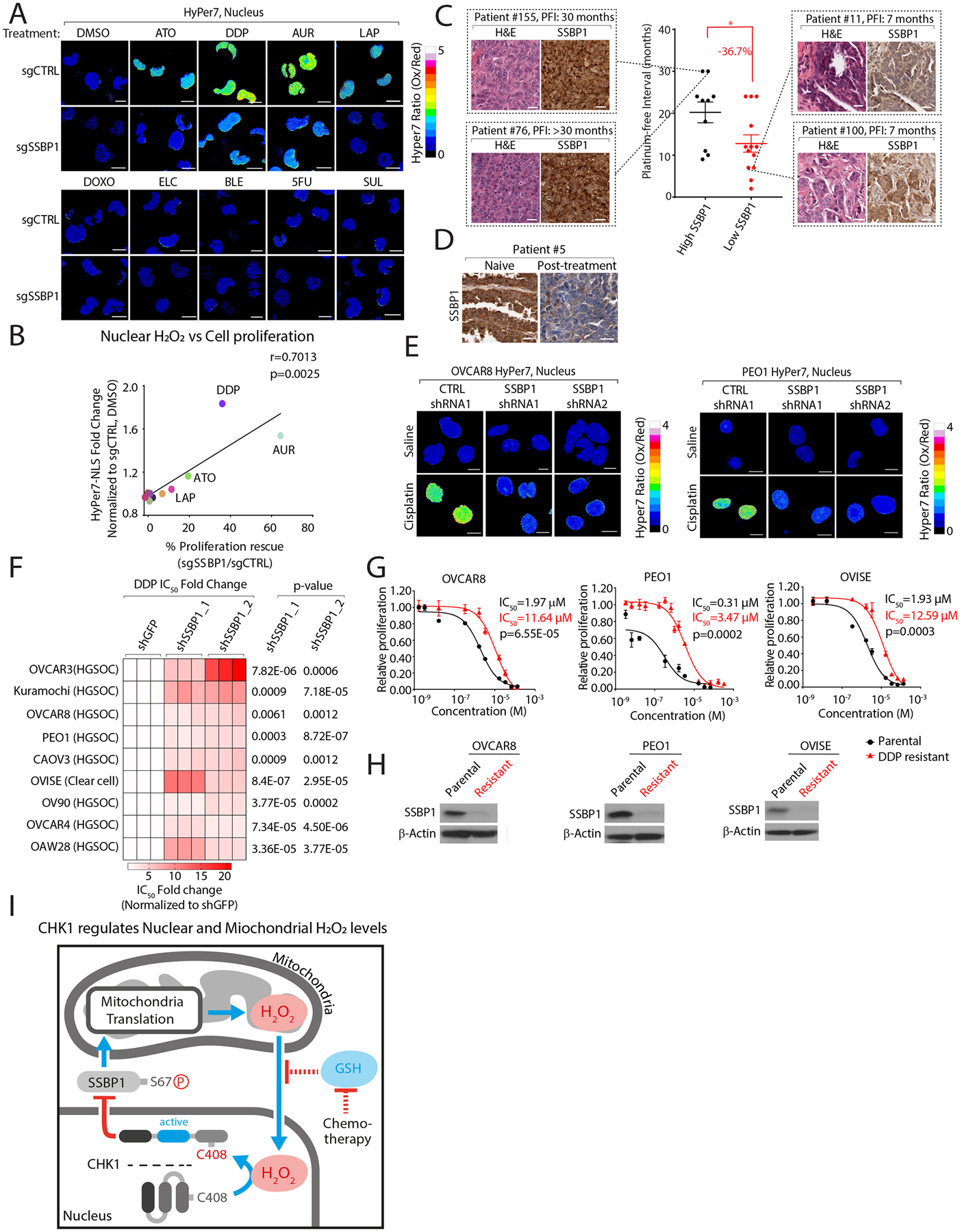
(A) SSBP1 depletion decreases nuclear H2O2 levels following treatment with anticancer agents in K562 cells. (B) Comparison of fold-change in nuclear H2O2 levels with proliferation rescue in K562 cells depleted of SSBP1 following treatment with the indicated compounds. (C) Lower SSBP1 levels correlate with shorter platinum free intervals (PFI) in high-grade serous ovarian cancer (HGSOC) tumors. (D) SSPB1 levels are decreased in platinum-refractory HGSOC tumors. (E) Knockdown of SSBP1 decreases cisplatin regulated nuclear H2O2 levels in ovarian cancer cell lines. (F) Heatmap depicting fold change in DDP IC50 values in ovarian cancer cell lines expressing the indicated shRNAs targeting SSBP1. (G-H) DDP-resistant ovarian cancers have decreased SSBP1 expression. (G) DDP IC50 values were measured in parental or DDP-resistant ovarian cancer cell lines. (H) Immunoblot analysis of SSBP1 in the indicated cell lines. (I) Model. Nuclear H2O2 activates CHK1 leading to the phosphorylation and cytosolic retention of SSBP1. Cytosolic SSBP1 cannot promote mitochondrial translation which generates H2O2. Mitochondrial H2O2 is transmitted to the nucleus following a decrease in GSH:GSSH ratio by certain anticancer drugs. Data are represented as mean ± SEM. * p < 0.05, ***p< 0.0001. Statistical significance was determined by one-way ANOVA with Sidak’s post-hoc correction.
Depletion of SSBP1 mediates resistance to cisplatin cytotoxicity
Platinum-based chemotherapies are common adjuvant treatments for women with high grade serous ovarian cancers (HGSOCs). When we stratified patients based on SSBP1 mRNA levels, we found that patients with lower levels of SSBP1 transcripts had a shorter duration to tumor recurrence following platinum-based chemotherapy than patients with higher levels of SSBP1 (Figure S7E). In a cohort of HGSOC patients treated at the Massachusetts General Hospital Cancer Center (n=23) with differing platinum free intervals (PFIs) we queried SSBP1 protein expression in corresponding tumors, finding lower levels of SSBP1 correlated with a shorter PFI (Figure 7C). For a subset of patients (n=3), tumor tissue was available at the time of diagnosis and from biopsies of recurrent disease in the platinum resistant setting. In one patient of this subset, we also observed a decrease in SSBP1 staining in a recurrent post-treatment tumor compared treatment naive tumor (Figure 7D, Table S6). To probe the role of ROS in cisplatin cytotoxicity, we treated nine ovarian cancer models of different histological subtypes (HGSOC and clear cell carcinoma) with NAC, finding a decrease in nuclear H2O2 and a corresponding increase in the IC50 of this drug (Figure S7F–G). As we observed in K562 cells, loss of SSBP1 decreased nuclear H2O2 levels in ovarian cancer models, which could be reverted by reintroduction of the protein in SSBP1-deficient cells (Figure 7E, S7H, J–K). Consistent with previous reports demonstrating a decrease in ROS production in ETC depleted cells4,81, we found decreased nuclear H2O2 levels in cells depleted of SSBP1 at baseline (Figure S7I). Importantly, SSBP1 depleted cells demonstrated a reduced sensitivity to cisplatin cytotoxicity across the various ovarian cancer models we examined (Figures 7F, S7L). Finally, we generated three cisplatin resistant ovarian cancer cell lines, finding SSBP1 protein decreased in resistant cells compared to their non-resistant counterparts (Figures 7G–H). Collectively, these finding suggest that loss of SSBP1 may be selected for during the acquisition of platinum resistance.
Here, we define a nuclear-to-mitochondria ROS sensing circuit that illustrates how nuclear H2O2 sensing controls mitochondrial translation which in turn regulates the steady-state levels of nuclear ROS, revealing an unexpected connection between DNA damage sensing compartmentalized ROS regulation and platinum resistance in ovarian cancers (Figure 7I).
Discussion
The majority of cancer patients succumb to disease following the onset of chemoresistance, which we now appreciate arises through both genetic and non-genetic mechanisms82–86. While proteins have long been appreciated as targets of ROS, the identity and functional significance of these targets following treatment with anticancer drugs is not established. This not only limits our understanding of resistance and normal tissue cytotoxicity, but hampers expanded use of these drugs in the clinic. Herein, using cysteine-focused chemical proteomics and functional genomics, we generated a global portrait of targets for 11 anticancer agents–information that is necessary to understand the cellular response to these drugs. Using this approach, we uncovered a nucleus-to-mitochondria ROS sensing pathway that may play a role in resistance to platinum-based therapies.
Our results suggest that mitochondrial translation is a major determinant of nuclear H2O2 levels and demonstrate how some anticancer drugs collaborate with mitochondrial H2O2 to increase nuclear H2O2 and DNA damage. Given that high ROS levels damage nucleic acids87,88 it is perhaps not surprising that the nucleus has evolved its own pathway to dynamically respond to this stressor. Crosstalk between these two organelles has been extensively studied in the context of anterograde signaling and corresponding transcriptional regulation89,90. However, our findings provide a direct posttranslational mechanism by which the nucleus leverages the DDR pathway to respond to high levels of nuclear H2O2 through the concomitant downregulation of mitochondrial translation. The canonical framework for ROS-based activation of DDR relies on direct DNA damage56. Our multiomic analysis of ROS-regulated targets following anticancer drug treatment focused our attention on CHK1•C408, suggesting a parallel mechanism, by which the cell can respond to changes in altered nuclear H2O2 levels. While CHK1•C408 was a moderate-to-strong hit in both our chemical proteomic and functional genomic analysis, combining these orthogonal approaches elevated CHK1•C408 above other targets, prompting us to explore its role in nuclear ROS response. Our cellular and in vitro characterization of nuclear H2O2 indicate that this ROS is both necessary and sufficient to oxidize C408. Whether other species of ROS or electrophilic compounds can modify C408 remains an open question, however, the conservation of C408 in CHK1 suggests that this residue is likely to be important in multiple kingdoms of life and may function as a general nuclear ROS sensor.
Given the role of ROS in DNA damage, the downregulation of mitochondrial translation and resultant decrease in nuclear H2O2 provides a safety check mechanism to preserve genomic integrity. These findings suggest that reducing mitochondrial translation by regulating SSBP1 levels, may be a general mechanism of resistance to anticancer agents that increase steady-state levels of nuclear H2O2 levels. Indeed, we find that HGSOC patients with lower levels of tumoral SSBP1 have shorter platinum-free interval and directly demonstrate that lowering SSBP1 in multiple models of ovarian cancer confers resistance to cisplatin. Thus, our study suggests that co-treatment of tumors with platinum-based chemotherapies and CHK1 inhibitors, which restore SSBP1 localization to the mitochondria and increase ROS, may be an approach to overcome platinum resistance. Moreover, they suggest a general mechanism of resistance for therapeutic agents whose cytotoxicity is mediated in part by increasing the steady-state levels of nuclear H2O2 and is in accordance with the growing body of evidence that mitochondrial function is a critical determinant of therapeutic response91–94.
By combining functional genomics with chemical proteomics, we have defined the mechanisms of sensitivity and resistance to AUR, a drug that is being explored for its anticancer properties and has been at the forefront of efforts to interrogate and interpret ROS biology. Our findings that AUR is sufficient to regulate nuclear H2O2 levels underscores the importance of its canonical targets TXNRD1/2 in the antioxidant response. Given the high levels of nuclear H2O2 following AUR treatment, it is perhaps surprising that depletion of one protein, SSBP1, can dramatically decrease H2O2 at this organelle and corresponding cytotoxicity. Thus, it illustrates the central role of mitochondrial translation in controlling ROS levels at other organelles and its centrality in the response to anticancer agents.
Our previous studies have indicated a strong enrichment for ROS-sensitive cysteines as targets of covalent inhibitors13. Thus, ROS targets defined as essential by functional genomic studies may offer an attractive starting point for the future development of powerful therapeutics that will be more specific than broadly cytotoxic drugs studied herein.
Limitations of Study
In this study we identified cysteines whose reactivity changes following treatment with anticancer drugs. Hierarchal clustering for cysteine reactivity changes identified four distinct clusters and using structural modeling we found distinct amino acids patterns were enriched near reactive cysteines in each cluster, that may be important in controlling cysteine reactivity. However, our study does not address the root feature(s) that drives this clustering. These additional drivers could be: 1) the cellular location of ROS following anticancer drug treatment, 2) detoxification mechanisms unique to each agent; 3) the generation of additional cysteine reactive molecules (e.g. lipid peroxides); and 4) direct adduction of cysteines by these agents as has been previously reported for arsenic trioxide and auranofin95,96. We still know very little about what governs cysteine reactivity changes at both the structural and cellular levels. Thus, more studies are needed to understand the mechanisms by which anticancer drugs increase ROS levels and the particular species which alter cysteines.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to the Lead Contact, Liron Bar-Peled (LBAR-PELED@mgh.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Proteomics data have been deposited in PRIDE: PXD041138 and is publicly available.
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper is available from the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cells were maintained at 37°C with 5% CO2. HEK293T were grown in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS, Corning), Penicillin-Streptomycin (100 mg/ml, Millipore) and L-glutamine (2 mM, Corning). K562, K562-dCAS9-KRAB, OVCAR8, Kuramochi, OVISE, OVCAR4, PEO1 were grown in RPMI-1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Corning), Penicillin-Streptomycin (100 mg/ml, Millipore) and 1% GlutaMax (Millipore). OVCAR3 were grown in RPMI-1640 (Invitrogen) supplemented with 20% fetal bovine serum (FBS, Corning), Penicillin-Streptomycin (100 mg/ml, Millipore) and 1% GlutaMax (Millipore). OV90, CAOV3 and OAW28 were grown in DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Corning), Penicillin-Streptomycin (100 mg/ml, Millipore) and 1% GlutaMax (Millipore). All cell lines were routinely tested for Mycoplasma and if not noted elsewhere were obtained from American Tissue Type Collection (ATCC). Whenever thawed, cells were passaged at least three times before being used in experiments.
METHOD DETAILS
Compound treatment
Anticancer drugs were formatted in the following manner: ATO and DDP were dissolved in saline solution, LAP, DOXO, ELC, BLE, 5FU, SUL, AUR, 2ME and NOV were dissolved in DMSO. Cells were treated as described below. Figure 1A: Changes in DCF intensity, NAD+/NADH, NADP+/NADPH, and NRF2 activity in K562 cells following treatment with 5.1 μM ATO, 1.5 μM LAP, 0.1 μM DOXO, 30 nM ELC, 8.3 μM DDP, 7.1 μM BLE, 6.0 μM 5FU, 0.5 mM SUL, 2.5 μM AUR, 1 μM 2ME or 0.5 mM NOV for 24 hrs; Figure 1G: K562 cells were treated with 2.5 μM AUR, 1 μM 2ME, 8.3 μM DDP, 7.1 μM BLE, 6.0 μM 5FU, 0.5 mM SUL or cycloheximide (CHX, 1hr) for 12 hrs; Figure 3B: K562 cells were treated with 1.5 μM AUR in the presence of 5mM NAC or vehicle control for 3 hrs; Figure 3C: K562 cells expressing the indicated reporters were treated with DMSO or 1.5 μM AUR in the presence or absence of 5mM NAC; Figure 3D: K562 cells were treated with 100 μM H2O2 for 1 hr; Figure 3E: HEK-293T cells expressing FLAG-CHK1 or CHK1•S408S and treated with 100 μM H2O2 for 1 hr; Figure 3F: K562 cells stably expressing D-amino acid oxidase (DAAO) localized to the nucleus were treated for 3 hrs. with either 10 mM L-Ala or D-Ala or 5 mM NAC; Figure 3H: HEK293T cells expressing the indicated proteins were treated with 10 mM L-Ala or D-Ala as described in 3F; Figure 3I: Purified CHK1 was treated with 0–1 mM H2O2; Figure 3J: K562 cell lysates were treated H2O or 100 μM H2O2; Figure 3L: Purified CHK1 was treated with 1000 μM H2O2; Figure 4A: K562-dCas9-KRAB cells expressing sgCTRL or sgCHK1 were treated with saline buffer or 5 mM NAC for 6hrs; Figure 4D: K562 cells expressing HyPer7-NLS were treated with 2 μM CHK1 inhibitor MK-8776 (CHK1i) and the following antioxidants: NAC (5 mM), GSH ethyl ester (GSHee, 1 mM), Ergothioneine (1 mM) or Trolox (50 uM) for 48 hrs; Figure 4E: K562 cells expressing nuclear roGFP2-ORP1 were treated with CHK1i (2 μM) and NAC (5 mM) for 48 hrs; Figure 4F: K562 cells were co-treated with 1 μM CHK1i and 5 mM NAC for 96 hrs; Figure 4G: CHK1i (2 μM) and NAC (5 mM) treatment in K562 for 24 hrs; Figure 4H: K562 cells stably expressing nuclear localized DAAO were pretreated for 72 hrs. with 10mM L-Ala or D-Ala prior to treatment with vehicle or 0.5 μM CHK1i; Figure 5B: Purified CHK1 was treated with 1000 μM H2O2; Figure 5D: K562-dCas9-KRAB cells expressing the indicated sgRNAs targeting SSBP1 and treated with DMSO or 2 μM CHK1i for 48hrs; Figure 5E: K562-dCas9-KRAB cells treated with vehicle, 1.5μM AUR, 2μM CHK1i or AUR/CHK1i for 6 hrs; Figure 5I: Reintroduction of SSBP1-HA, SSBP1•S67D-HA SSBP1-NLS-HA into K562 depleted of SSBP1 were treated with DMSO or 2 μM CHK1i for 48hrs; Figure 6A: K562-dCas9-KRAB cells expressing the indicated sgRNAs and treated with DMSO or 2 μM CHK1i for 48hrs; Figure 6B: K562 cells expressing HyPer7-NLS were treated with 10μM mitoTEMPO and 2 μM CHK1i for 48hrs; Figure 6C: K562 cells expressing mitochondrial matrix DAAO were treated with 0.5 μM CHK1 or 5 mM L-Ala or D-Ala for 96hrs; Figure 6D: K562 cells expressing HyPer7-NLS were co-treated with 5 μM S1QEL1.1 or 50 μM S3QEL-2 for 24 hrs followed by 2 μM CHK1i for 48 hrs; Figure 6F: K562-dCas9-KRAB cells stably expressing the indicated sgRNAs and treated with 2 μM CHK1i for 72 hrs; Figure 6G: K562 cells expressing the indicated sgRNA targeting SSBP1 and SSBP1, SSBP1•S67D or METAP2 (control) were treated as described in 6F; Figure 6H: K562 cells were pre-treated for 72 hrs. with 2.3 μM Doxycycline (DOXY) at which time the drug was removed and cells were treated with 2 μM CHK1i or vehicle control; Figure 6I: K562 cells treated with 2.3 μM doxycycline (DOXY) for 72 hrs; Figure 6J: K562 cells expressing HyPer7 localized to nucleus were pre-treated for 72 hrs., with 2.3 μM DOXY followed by 2 μM CHK1i treatment for 48 hrs; Figure 7A: K562 cells expressing HyPer7 localized to nucleus were treated with the indicated agents as described in Figure S1A; Figure 7G: Cell lines expressing nuclear HyPer7 were co-treated with 5 mM NAC or vehicle control and 8.3 μM cisplatin for 24 hrs.
In experiments where multiple anticancer drugs were compared, we added DMSO to the ATO or DDP treated samples and saline to the rest including vehicle control. The final concentration of DMSO in cell culture medium was maintained at 0.1% during small molecule treatment unless otherwise noted.
cDNA cloning and mutagenesis
cDNAs were amplified using Q5 High-Fidelity 2X master mix (NEB) and subcloned into the pRK5 (Addgene), pLJM1 (Addgene) or pLenti CMV (Addgene) by T4 ligation or Gibson cloning. Site directed mutants were generated using QuikChange XLII site-directed mutagenesis (Agilent), using primers containing the desired mutations. All constructs were verified by DNA sequencing. Primer sequences can be found in Table S7.
H2DCFDA and MitoSox measurements
For H2DCFDA (ThermoFisher Scientific, Wlatham, MA) staining in K562 cells, cells were treated as indicated in the text and were washed with prewarmed PBS and harvested by centrifugation at 1200 g at room temperature for 2 mins. The cell pellet was resuspended in PBS with 1 μM of CM-H2DCFDA and incubated for 45 min in a 37°C incubator with controlled CO2 levels (5%). Cells were subsequently washed with PBS. Changes in CM-H2DCFDA fluorescence were determined via flow cytometry using Aurora (Cytek) or CytoFLEX (Beckman Coulter). Data was analyzed using Flowjo v10.6 for FITC intensity. For MitoSox™ Red (ThermoFisher Scientific) staining in K562 cells, 0.5 03BCzM MitoSox™ Red was added directly to the culture medium and incubated for 20 min in a 37°C incubator with controlled CO2 levels (5%). Changes in MitoSox™ Red fluorescence were determined by flow cytometry using Aurora or CytoFLEX. PE intensity was analyzed using Flowjo v10.6.
GSSG/GSH, NADH/NAD+ and NADPH/NADP+ measurement
K562 cells were treated with different anticancer drugs in 6-well plates per the timepoints indicated in the text. The ratio of GSSG/GSH, NADH/NAD+ and NADPH/NADP+ was determined using the GSH/GSSG-Glo™ Assay Kit (Promega, Madison, WI), NAD/NADH-Glo™ Assay Kit (Promega) and NADP/NADPH-Glo™ Assay Kit (Promega), respectively, following the manufacturer’s protocol. Absorbance was measured using a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA).
Confocal imaging of cell lines expressing HyPer7/roGFP2-Orp1 reporters
K562 or K562-dCas9-KRAB cells expressing the indicated HyPer7/ roGFP2-Orp1 reporters with a concentration of approximately 5×105 cells/ml were seeded on poly-lysine coated glass bottom dish (ThermoFisher Scientific) and treated with compounds as indicated in the methods sections. Dishes were firmly mounted on the stage adaptor of the Zeiss 710 Laser Scanning Confocal microscope (Carl Zeiss Inc.). Constant temperature (37 °C), humidity, and 5% CO2 atmosphere were maintained throughout the duration of cell imaging. Images were acquired using a 63X oil objective. The oxidized form of the HyPer7 reporter was detected by exciting HyPer7 expressing cells with a 488-nm laser and measuring emission in the 500–520 nm range. Reduced HyPer7 was detected by exciting HyPer7 expressing cells with a 405-nm laser and measuring emission in the 500–545 nm range. The oxidized form of the roGFP2-Orp1 reporter was measured by exciting roGFP2-Orp1 expressing cells with a 405-nm laser and measuring emission in the 500–520 nm range. The reduced form was measured by exciting roGFP2-Orp1 expressing cells with a 488-nm laser and measuring emission in the 500–545 nm range. Acquisition parameters were identical between samples. Images were processed using the ZEN 2.6 Image software (Carl Zeiss Inc.). Ratiometric images of HyPer7 were processed using ImageJ (NIH). Threshold images after subtraction of background were split into two different channels, divided with Image Calculator. 32-bit ratiometric images were generated and presented in the 16 color mode using Lookup Tables.
Flow cytometry analysis of cell lines expressing HyPer7/roGFP2-Orp1 reporters
Sixteen thousand K562 or K562-dCas9-KRAB cells expressing the indicated HyPer7/ roGFP2-Orp1 reporters were seeded in a 96-well plate for 24 hrs and treated as indicated in the methods section. HyPer7/ roGFP2-Orp1 oxidation and reduction was determined by flow cytometry using an Aurora (Cytek) or CytoFLEX (Beckman Coulter) measuring emission at 530 nm following excitation at 405 nm or 488 nm. The ratio of λex = 488 nm/ λem = 530 nm to λex = 405 nm/ λem = 530 nm signal for HyPer7 and ratio of λex = 405 nm/ λem = 530 nm to λex = 488 nm/ λem = 530 nm signal for roGFP2-Orp1 was determined using Flowjo v10.6.
Immunofluorescence
One million K562 or K562-dCas9-KRAB cells expressing the indicated HyPer7/ roGFP2-Orp1 reporters or SSBP1-HA/ SSBP1-FLAG were fixed with 4% PFA (EMS) for 15 min and resuspend with PBS to a concentration of approximately 5×105 cells/ml. Cells spun onto coverslips using a ThermoFisher Scientific cytospin cytocentrifuge. The slides were then rinsed with PBS and cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min. The slides were rinsed with PBS and incubated with primary antibodies in 4% BSA overnight at 4°C. Following three PBS washes, the slides were incubated with secondary antibodies conjugated to the Alexa Fluor® 488 and 594 fluorophores (Invitrogen) for 2 hrs at room temperature. The slides were rinsed and mounted on glass slides using ProLong™ gold antifade mount without or with DAPI (ThermoFisher Scientific). For HEK293T stably expressing FLAG-DAAO (nuclear or mitochondria matrix localized), cells were plated on poly-lysine coated glass coverslips in 12-well tissue culture plates. 48 hrs later, the culture media was removed, and cells were fixed with 4% paraformaldehyde (Electron microscopy services) for 15min. Staining was performed as described above. Cells were imaged on Zeiss LSM 710 laser scanning confocal microscope or Olympus under 63x oil objective. Images were processed using ZEN 2.6 Image software (Carl Zeiss Inc.) and ImageJ (NIH). For high resolution imaging, cells were imaged on Nikon CSU-W1 SoRa Spinning Disk Microscope (Nikon Instruments Inc.) under 63x oil objective, images were deconvoluted and processed using Nikon elements software.
Cell lysis and FLAG-Immunoprecipitations
K562 or HEK-293T cells expressing the indicated proteins were washed once with ice-cold PBS and lysed using a chilled bath sonicator (Q700, QSonica) in Triton IP buffer (1% Triton X-100 (sigma), 5 mM MgCl2, 40 mM HEPES pH 7.4, 10 mM KCl) supplemented with protease inhibitors (Roche), phosphatase inhibitors (Roche, Boston, MA) and Benzonase (Santa Cruz Biotechnology, Santa Cruz, CA). Lysates were clarified by centrifugation at 13000rpm for 10 min. Samples were normalized to 1 mg/ml and boiled following the addition of sample buffer. For FLAG immunoprecipitations, anti-FLAG M2 resin (Sigma-Aldrich, St Louis, MO) was added to the pre-cleared lysates and incubated 3 hrs at 4°C. Following immunoprecipitation, beads were washed once with Triton IP buffer followed by 3 times with Triton IP buffer supplemented with 500 mM NaCl. In the in vitro binding assay of CHK1 kinase domain with endogenous CHK1, K562 cells expressing FLAG-CHK1 kinase domain was lysed using IP buffer supplemented with 0.3% CHAPS (Sigma-Aldrich) instead of 1 % Triton. Clarified lysate was pretreated with DMSO or 100 μM H2O2 for 2 hrs and then incubated with anti-FLAG M2 resin for immunoprecipitation. Following bead washes, loading buffer was added to the immunoprecipitated proteins which were subsequently denatured by boiling for 5 min. Proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
IA-DTB labeling of CHK1
K562 cells stably expressing FLAG-CHK1 or FLAG-C408S•CHK1 were treated with H2O or 100 μM H2O2 for 1 hr and then incubated with 1 mM IA-DTB for 1 hr. Cells were washed with ice-cold PBS and lysed using a chilled bath sonicator (Q700, QSonica) in PBS buffer supplemented with Benzonase (Santa Cruz Biotechnology). Samples were adjusted to 2 mg/mL. IA-DTB modified proteins were enriched by the addition of streptavidin beads (ThermoFisher Scientific) and following 2 hrs incubation, beads were washed twice with 0.1% IGEPAL (Sigma-Aldrich), PBS and Triton IP buffer supplemented with 500 mM NaCl. Proteins were then resolved by SDS-PAGE and analyzed by immunoblotting of FLAG-CHK1 or FLAG-C408S•CHK1.
Synthesis of NO-DTB probe
Synthesis of phenyl 4-((2-(6-((4S,5R)-5-methyl-2-oxoimidazolidin-4-yl)hexanamido)ethyl)carbamoyl)-2-nitrosobenzoate (NO-DTB)42,99–101 is summarized below and reaction schematic can be found in Method S1A. To a solution of 2-aminoterephthalic acid (1.0 g, 5.53 mmol) in 1,4-Dioxane (80 mL) was added triphosgene (1.64 g, 5.53 mmol) at room temperature. The resulting reaction mixture was stirred for 6 hr. at room temperature. The reaction mixture was poured in H2O (200 mL) and extracted with ethyl acetate (3 × 75mL). The organic layers were combined, washed with brine and concentrated under reduced pressure to afford 4-carboxylic isatoic anhydride as an off white solid (97%).
Phenol (681 mg, 7.25 mmol) (anhydrous; prior to addition: solubilized in EtOAc, dried with MgSO4, concentrated, then placed under vacuum to dry) and triethylamine (1.34 mL, 9.66 mmol) were added to a solution of 4-carboxylic isatoic anhydride (1000 mg, 4.83 mmol) in anhydrous 1,4-dioxane (40 mL). The mixture was stirred under N2 and refluxed for 6 h. The crude mixture was concentrated, then diluted with water (15 mL), and the pH was adjusted to 3 with conc. HCl. The solution was extracted with EtOAc (3 × 20 mL) to yield the 3-amino-4-(phenoxycarbonyl)benzoic acid as a bright yellow solid (98%).
To a flame dried 3-necked flask, 3-amino-4-(phenoxycarbonyl)benzoic acid (166 mg, 0.578 mmol), COMU (292 mg, 0.682 mmol), DIPEA (220 mg, 1.70 mmol), and DMF (5 mL) were added. The mixture was stirred under N2 for 15 minutes, then N-(2-aminoethyl)-6-((4R,5S)-5-methyl-2-oxoimidazolidin-4-yl) hexanamide (166 mg, 0.625 mmol) was added. The mixture was stirred for 12 hr. at RT, then it was concentrated and purified via column chromatography with DCM/MeOH to afford phenyl 2-amino-4-((2-(6-((4S,5R)-5-methyl-2-oxoimidazolidin-4-yl)hexanamido)ethyl)carbamoyl)benzoate (35%) (See Method MS1B).
A solution of oxone (46.6 mg, 0.303 mmol) in water (3.0 mL) was added to a solution of 1-phenyl2-aminoterephthalate (50.0 mg, 0.101 mmol) in CHCl3 (1.0 mL). The reaction was vigorously stirred for 12 hrs. The solution was concentrated and purified via column chromatography with DCM/MeOH to afford phenyl 4-((2-(6-((4S,5R)-5-methyl-2-oxoimidazolidin-4-yl)hexanamido)ethyl)carbamoyl)-2-nitrosobenzoate (nitroso-dtb) (21%). 1H NMR (MeOH-d4, 400 MHz, mixture of monomer and dimer 1:1): δ 8.34 (d, J = 8.33 Hz, 0.5H), 8.17–8.04 (m, 2H), 7.63 (s, 1H), 7.49–7.16 (m, 6H), 3.77–3.42 (m, 6H), 2.18 (t, J = 2.18 Hz, 2H), 1.60–1.19 (m, 10H), 1.03 (d, J = 1.03 Hz, 3H). 13C NMR (MeOH-d4, 100 MHz, mixture of monomer and dimer): δ 175.5, 150.9, 133.4, 130.0, 129.4, 129.2, 126.2, 121.8, 121.3, 113.77, 113.11, 56.0, 51.3, 38.45, 35.7, 29.3, 28.9, 25.8, 25.5, 14.27. ESI-LCMS calcd. for C26H31N5O6 (M-H) 510.2, found 510.2 (see Method MS1C).
Detection of CHK1 Sulfinylation
Detection of CHK1 Sulfinylation by NO-DTB labeling was performed as previously reported61,62. In brief, K562 cells stably expressing FLAG-CHK1 or FLAG-C408S•CHK1 were treated with H2O or 100 μM H2O2 for 1hr. Cells were washed with ice-cold PBS and lysed using a chilled bath sonicator (Q700, QSonica) in Triton IP buffer (1% Triton X-100 (Sigma-Aldrich), 5 mM MgCl2, 40 mM HEPES pH 7.4, 10 mM KCl) supplemented with protease inhibitors (Roche), phosphatase inhibitors (Roche), Benzonase (Santa Cruz Biotechnology) and 5 mM DTT. Lysates were clarified by centrifugation at 13000 rpm for 10 min. Free thiols were trapped by incubation with 2 mM of 4,4’-dithiodipyridine (4-DPS) at room temperature for 1 hr and subsequently the buffer was exchanged using one Micro Bio-Spin column pre-equilibrated with 100 mM HEPES, pH 8.5, 100 mM NaCl. DPS-free lysates were then reacted with 500 μM NO-DTB in the dark at room temperature with rotation for 1 hr. Total protein was purified by the addition of a chloroform-methanol solution (4:4:1, methanol, water, chloroform) to each sample and precipitated following centrifugation at 4200 RPM for 10 min. The protein disc was isolated, washed once in methanol, resuspended in Buffer X1 (9 M Urea, 10 mM DTT, 50 mM tetramethylammonium bicarbonate (TEAB) and incubated for 20 min at 65°C. NO-DTB modified proteins were then enriched by the addition of streptavidin beads (ThermoFisher Scientific) and following 2 hrs incubation, beads were washed twice with 0.1% IGEPAL (Sigma-Aldrich), PBS and Triton IP buffer supplemented with 500mM NaCl. Proteins were then resolved by SDS-PAGE and analyzed by immunoblotting of FLAG-CHK1 or FLAG-C408S•CHK1.
In vitro CHK1 kinase assay
K562 cells stably expressing FLAG-CHK1 or FLAG-C408D•CHK1 were lysed by sonication in Triton IP buffer and immunoprecipitated using anti-FLAG M2 beads as described above. Immobilized FLAG-CHK1 was dephosphorylated by treating the protein with calf alkaline phosphatase (NEB) for 1 hr at 37°C. Immobilized FLAG-CHK1 was subsequently washed 3 times in CHK1 kinase buffer (10 mM HEPES, pH 7.5, 10 mM MgCl2, 10 mM MnCl2) and the kinase assay was initiated by adding 1 mM ATP (Sigma-Aldrich) and the indicated compounds and incubating at 37°C for 45 min. The reaction was stopped by washing the samples once with ice-cold CHK1 kinase buffer and adding loading buffer. Proteins were resolved by SDS-PAGE and analyzed by immunoblotting. For in vitro phosphorylation of SSBP1 by CHK1, HEK293T cells transiently expressed SSBP1-HA or SSBP1 (S67A)-HA were lysed by sonication in Triton IP buffer and immunoprecipitated using anti-HA magnetic beads. Immobilized SSBP1-HA was dephosphorylated by treating the protein with calf alkaline phosphatase (NEB) for 1 hr. as described above. Immobilized SSBP1-HA or SSBP1 (S67A)-HA was subsequently incubated with HA-CHK1 or HA-METAP2 and washed 3 times in CHK1 kinase buffer (10 mM HEPES, pH 7.5, 10 mM MgCl2, 10 mM MnCl2). The kinase assay was initiated by adding 100 μM ATP (Sigma-Aldrich), 2 μCi [γ−32P] ATP (PerkinElmer, Hopkinton, MA), incubated at 30°C for 30 min, and then boiled at 95°C for 5 min in 5X sample buffer. Phosphorylation was assessed by 16% SDS-PAGE by autoradiography.
Mitochondria translation measurement
K562 cells were treated with 2.3 μM Doxycycline to inhibit mitochondrial translation. After 72 hrs treatment, the cells were refreshed with RPMI medium with DMSO or 2 μM CHK1i. Aa final concentration of 0.1% DMSO was used. Cell were harvested at different timepoints as indicated in the text and lysed as described above. Proteins were resolved by SDS-PAGE and analyzed by immunoblotting as described in the text.
isoTOP-TMT sample preparation
isoTMT samples were prepared as described in13, with the modifications noted below. Briefly, K562-dCas9-KRAB cells were treated at 37°C with the indicated compounds for the noted time in the text. Cells were harvested by centrifugation at 1200 g for 2 min and then washed once with ice-cold PBS and lysed in PBS with Benzonase (Santa Cruz Biotechnology) by using a chilled bath sonicator. Samples were clarified by centrifugation for 3 min at 300 g. Samples were adjusted to 2 mg/mL and incubated with 100 μM of iodoacetamide-desthiobiotin (IA-DTB, Santa Cruz Biotechnology) for 1 hr at room temperature. Alkylation was terminated by the addition of a chloroform-methanol solution (4:4:1, methanol, water, chloroform) to each sample and proteins were precipitated following centrifugation at 4200 RPM for 10 min. The protein disc was isolated, washed once in methanol, resuspended in Buffer X1 (9 M Urea, 10 mM DTT, 50 mM tetramethylammonium bicarbonate (TEAB) and incubated for 20 min at 65°C. Samples were subsequently alkylated with 500 mM Iodoacetamide for 30 min at 37°C and digested for 3 hrs with Trypsin (Promega). IA-DTB modified peptides were enriched by the addition of streptavidin beads (ThermoFisher Scientific) and following 2 hrs incubation, beads were washed twice with 0.1% IGEPAL (Sigma-Aldrich), PBS and H2O. Peptides were eluted with a mixture of 50:50:0.1 (Acetonitrile: H2O: Formic Acid) and subsequently dried.
TMT-Labeling
Samples were prepared as previously described102. Briefly, 20μg of peptides from each sample were labeled with isobaric tandem-mass-tag (TMT) reagents (Thermo Fisher Scientific, San Jose, CA) in 20 mM pH 8.5 HEPES with 30% acetonitrile (v/v) with 50 ug of TMT reagent. The reaction was quenched for 15 min by adding hydroxylamine to a final concentration of 0.3% (v/v). Samples were combined, dried, purified over SepPak C18 columns, and dried again. Samples were then resuspended in 40 μL of basic reverse phase (bRP) buffer A (10 mM NH4HCO2, pH10, 5% ACN) and separated on a Zorbax Extended C18 column (2.1 × 150 mm, 3.5 μm, no. 763750–902, Agilent) using a gradient of 10–40% bRP buffer B (10 mM NH4HCO2, pH 10, 90% ACN). 96 fractions were collected before concatenation to 12 or 24 fractions. Each fraction was dried and desalted over a C18 STAGE-Tip prior to analysis by mass spectrometry.
LC–MS Analysis of Total Protein Fractions
Samples were analyzed on an Orbitrap Fusion Lumos or Orbitrap Eclipse mass spectrometer (ThermoFisher Scientific) coupled with a Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (ThermoFisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~40 cm of Accucore150 resin (2.6 μm, 150 Å, Thermo Fisher Scientific, San Jose, CA). For each analysis, we loaded approximately 1 μg onto the column. Peptides were separated using a 2.5 hr. gradient of 6–30% acetonitrile in 0.125% formic acid with a flow rate of 550 nL/min. Each analysis used an SPS-MS3-based TMT method103–105, which has been shown to reduce ion interference compared to MS2 quantification. The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000; 350–1400 m/z, automatic gain control (AGC) target 4.0 × 105, maximum injection time 50 ms). Precursors for MS2/MS3 analysis were selected using a Top10 method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap; AGC 2.0 × 104; normalized collision energy (NCE) 35; maximum injection time 120 ms). Following acquisition of each MS2 spectrum, we collected an MS3 spectrum a method in which multiple MS2 fragment ions are captured in the MS3 precursor population using isolation waveforms with multiple frequency notches. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (NCE 65, AGC 3.5 × 105, maximum injection time 150 ms, isolation window 1.2 Th, resolution was 50,000 at 200 Th).
MS Data Processing and Analysis
MS spectra were evaluated using Comet and the GFY-Core platform (Harvard University)106–109. Searches were performed against the most recent update of the Uniprot Homo sapiens database with a mass accuracy of +/−50ppm for precursor ions and 0.02 Da for product ions. Static modification of lysine and N-termini with TMT (229.1629 Da) and carbamidomethylation (57.0215 Da) of cysteine were allowed, along with oxidation (15.9949 Da) of methionine residues and modification (398.2529 Da) of cysteine residues as variable modifications. Results were filtered to a 1% peptide-level FDR with mass accuracy +/−5ppm on precursor ions and presence of a modified cysteine residue for Cys-Mod samples. Results were further filtered to a 1% protein level false discovery rate. TMT quantitative results were generated in GFY-Core. For TMT-based reporter ion quantitation, we extracted the summed signal-to-noise (S/N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. MS3 spectra with TMT reporter ion summed signal-to-noise ratios less than 100 were excluded from quantitation.
Ratio and Median Calculation
Abundances for each peptide corresponding to a site on a canonical protein were totaled and normalized to the median for each agent and control. The ratio (R) between the abundances for the control and agent was calculated. For each protein and agent, the median was calculated among the ratios for each site on the protein if there were 3 or more sites identified.
Circos plot
The R package “circlize” was used to create a chord diagram where each point around the circle represented a site with R > 1.5 in a particular treatment and edges were drawn between any sites shared between two treatments (https://cran.r-project.org/web/packages/circlize/citation.html).
Clustering Analysis
The UMAP embedding was calculated with the umap. UMAP function of the Python umap.umap_package (n_neighbors = 15 and random_state = 42). Reactive cysteines were clustered using K-means clustering.
Structural Analysis
PDB files mapping to 4199/15165 commonly detected cysteines were downloaded (Table S2C)110. Centroids for each amino acid within a structure were computed, and all residues with Euclidean distance ≤ 10Å of a cysteine of interest were included for further analysis. The distances of these neighbors were then rank ordered in ascending fashion, and these sorted lists were used to generate pLogo motifs111. The neighbors of cysteines with 0.7 ≤ max(R) ≤ 1.3 were used to calculate background probabilities in the pLogo algorithm. To obtain spatial enrichment of residues near cysteines of interest we binned the 10Å radius around cysteines of interest into 0.5Å intervals111. In order to ascertain the concordance between neighbors in the primary sequence (derived from UP000005640_9606.fasta) and 3-dimensional space, we tabulated the frequency of agreement between the nth-nearest neighbor in 3D space and the ±nth-nearest neighbor in linear sequence on all reactive cysteines. All scenes were generated in PyMOL (PyMOL Molecular Graphics System, version 2.5.2, Schrödinger).
Reactive Cysteine Signature Score
The score was calculated by multiplying the number of cysteines with a ratio above 1.5 for all agents by the median of those ratios above 1.5, only including cysteines which had observations for all agents.
Protein Localization Analysis
The most common annotation among the five cell lines (A431, H322, HCC827, MCF7, and U251) from SubCellBarCode (SCBC) was calculated for each protein. Annotated localizations from UniProt and Protein Atlas were compared to extract matching terms and prefixes. Terms, prefixes, SCBC neighborhoods were mapped to a discrete list of subcellular localizations, and the consensus localization was calculated from any localization found in two of the three datasets or SCBC if there were no matching localizations between UniProt and Protein Atlas. Additionally, UniProt annotations tagged with “ECO:0000269” (indicating manually curated annotations derived from published experimental evidence) were extracted and mapped to the same list of subcellular localizations112.
Gene Ontology Analysis
The R package “topGO” was used to perform a gene ontology analysis on the set of proteins containing any cysteine with R > 1.5 for a single anticancer drug. Molecular function, cellular compartment, or biological process terms were derived from Bioconductor’s org.Hs.eg.db database, and enrichments were computed using the classic method and Fisher’s exact test113.
Annotations
Ubiquitination sites from PhosphoSitePlus114 were matched to nearby sites identified by mass spectrometry. Essentiality scores were derived from DepMap115,116. Ribosomal proteins were annotated from the Ribosomal Protein Gene Database. Domain annotations are from UniProt. Functional annotations were taken from the Gene Ontology Annotation database117. BiomaRt118 was used to map proteins between UniProt, ensembl, entrez, and PDB IDs and gene names.
Genome-wide CRISPRi screen
The CRISPRi screen in K562 cells was conducted as previously described48. Briefly, K562 cells stably expressing dCas9-KRAB were infected with a genome-wide CRISPRi library cloned into the pU6-BFP vector, ensuring a multiplicity of infection ~0.3 following 3 days puromycin selection. Cells were allowed to recover for 1 day and an initial input was taken with the number of infected cells corresponding to 1000X the size of the library (~200×106 cells). The screen was initiated by treating 240×106 with DMSO or 1 μM Auranofin, maintaining this cell number and compound for 10 population doublings. At the end of the screen, cells were harvested, and genomic DNA was extracted using Macherey Nagel Blood XL kit (Macherey-Nagel). Libraries were generated from each sample by PCR based amplification of the sgRNA amplicon from 200 μg of genomic DNA using custom PCR primers harboring an index primer and illumina 5’ and 3’ adaptors. Libraries were pooled and analyzed on a NextSeq500 (Illumina) use single end 75bp reads. sgRNAs were mapped and quantified using the Screen Processing pipeline48. The enrichment for each sgRNA was calculated by taking the log2 ratio of (sgRNA counts, treatment/sgRNA counts input). The CRISPR score for each gene was calculated by subtracting the enrichment score for ARU treated samples from the enrichment score for DMSO treated samples.
Cluster analysis
To identify functionally related genetic clusters from CRISPR-Cas9 based essentiality screens, we utilized the DEPMAP resource (https://depmap.org/portal/download/; access: 03/23/2020). The Achilles_gene_effect.csv (Version: DepMap Public 20Q1) matrix summarizes essentiality scores obtained from genome-wide loss-of-function screens from a set of 18,333 genes among 741 different cell lines. Gene-gene correlations across cell lines were computed in R using the cor() function (parameters: method “pearson”; use = “complete.obs”) generating a matrix with 18,333 × 18,333 entries representing correlations of each gene pair. We extracted correlations with values > 0.25 and listed them together with its corresponding gene pair. These list entries were considered as edges of an undirected graph and were imported as network in Cytoscape (Version 3.6.0). The gene network was further dissected using the random walk Markov Clustering algorithm within the Cytoscape plugin clusterMaker (parameters: “granularity” = 1.8; “number of iterations” = 16; “input” = edgelist with gene-gene-correlation as weight). This operation created gene groups with 1 gene up to 695 genes with each gene only occurring in 1 group (= cluster_ID). We proceeded with clusters that contained 3 or more genes resulting in 1028 distinct groups containing a total of 10474 genes. Genes were assigned to list entries in R by their cluster_ID.
Competition scores for cysteines of individual peptides from chemical proteomics experiments were transformed in a binary matrix (1 for competition values ≥ 1.5 = “hit” and 0 for < 1.5 = “no hit”) in R. All peptide hits for each condition were collapsed per protein/gene. Hits were summed up for all genes of a cluster (cluster score) and normalized by a cluster size factor ((size cluster of interest/size largest cluster)^2.03). Hits on the cluster level were summarized by summing up hits for each gene within the cluster for each condition.
Clusters were plotted as circles in Cytoscape (= nodes with no edges) and scaled by cluster scores. Proteomics hits of genes in the cluster were depicted as ratios in pie chart superimposed on the cluster node.
Target Prioritization analysis
For identifying top scoring gene/protein candidates from chemical proteomics and CRISPRi screens, we combined both data sets. All data processing steps were performed in R. The chemical proteomics data matrix was imported and NA containing rows were removed. We collapsed chemical proteomics competition values per protein/gene name by only keeping the top scoring peptide and ignoring others.
CRISPR scores from loss-of-function screens were imported in R and NA containing cells were removed. The chemical proteomics and CRISPR matrices were merged using merge() in R by the gene/protein name as identifier. We calculated priority scores by multiplying CRISPR scores with proteomics competition values for each gene/protein.
To identify functionally related protein groups, we utilized the STRING resource (https://string-db.org/; access 01/08/2022). We considered genes/proteins as hits if they scored with a CRISPR value of < − 0.75 or > + 0.75 and with a proteomics competition value of > 1.5. For these genes/proteins we extracted the 10 top scoring interactors from the STRING network and considered these 11 genes/proteins as a functional group. For each group we averaged priority scores and corrected them with a penalty score of 1% (multiplication with 0.99^(number of missing genes in the group)) per missing gene in the priority score matrix. These optimized priority scores were plotted as nodes in Cytoscape (= nodes with no edges) and scaled by priority scores (color, node size, label size).
CRISPRi-mediated knockdown in K562 cells
sgRNAs targeting the promoters of CHK1 and SSBP1 were cloned into pU6-BFP (Addgene: #60955). sgRNA-encoding plasmids were co-transfected with pspAX2 envelope and CMV VSV-G packaging plasmids into 1.8×106 HEK293T cells using the Xtremegene 9 transfection reagent (Sigma-Aldrich). Virus-containing supernatants were collected 48 hrs after transfection and used to infect K562- dCas9-KRAB cells in the presence of 10 mg/ml polybrene (Santa Cruz). Twenty-four hours post-infection, fresh media was added to the infected cells which were allowed to recover for an additional 24 hrs. Puromycin was then added to cells, which were analyzed after 3 days after selection was added.
Cell Proliferation assays
2.5 × 103 K562 cells were seeded in 96-well plates per well in 100 μl medium. K562 cells were subsequently treated with the indicated compounds in 100 μl medium, whereas adherent cells were treated 24 hrs after seeding. Ninety-six hrs after compound treatment, 50 μl Cell Titer Glo™ (Promega) was added to each well sample and the luminescence was monitored on a SpectraMax M5 plate reader (Molecular Devices). For cell counting using trypan blue, 0.5 ml of a cell suspension (dilute cells in complete medium without serum to an approximate concentration of 1 × 105 to 2 × 105 cells per ml) was mixed with 0.1 ml of 0.4% trypan blue staining and counted using a hemocytometer. To calculate half maximal inhibitory concentrations (IC50) cells were cultured at 2.5 × 103 cells per well in 100 μL RPMI media and compounds were added the following day. Cell viability was assessed on day four of treatment by measuring relative ATP concentrations. IC50 values were calculated using log(inhibitor) vs % normalized response formula in Prism v7.0 (GraphPad).
Generation of Cisplatin resistance cell lines
The IC50 and IC10 values for cisplatin in OVCAR8, PEO1 and OVISE were determined as above following four days treatment. Cell lines were treated with cisplatin at a starting concentration of IC10. Once confluent, the cell lines were maintained for at least 4 generations in drug or vehicle control. After that, the concentration of cisplatin was gradually increased to IC50 for each cell line. Each cell line was maintained at the IC50 concentration for at least 4 passages. Resistant cell lines were maintained in cisplatin containing media for a totality of 3 months.
Immunohistochemical (IHC) staining
Immunohistochemistry for SSBP1 was conducted using a two-step protocol (GTVisionTMIII). Briefly, tumor microarray (TMA) sections were washed with phosphate-buffered saline (PBS) after rehydration and then the antigens were retrieved by boiling the TMA slides in citrate buffer (pH 6.2) at 100°C for 10 min. The TMAs were permeabilized with 0.1% Triton-X-100 for 10 min. and then treated with 3% hydrogen peroxide for 10 min. to block endogenous peroxidase activity. The TMAs were blocked with 6% BSA for 1 hr at room temperature (RT) and incubated in a humid chamber with SSBP1 primary antibody (1:50) for 1 hr. Following PBS washes, all the TMAs were incubated with secondary antibody (HRP-labeled anti-rabbit antibody 1:50) at RT for 1hr. The sections were counterstained with hematoxylin and mounted after clearing with xylene. For hematoxylin and eosin staining, TMA sections were washed with phosphate-buffered saline after rehydration and stained for 1min in hematoxylin solution. The slides were then washed in running tap water. After washing, the slides were dipped in Eosin solution for 1 min and rinsed with absolute alcohol. The TMA slides were dipped two times in Xylene solution for 1 min. each and mounted with the DPX mount (Sigma). SSBP1 immunohistochemistry were semi-quantitatively scored via light microscopy by a board-certified pathologist (Z.O.) using a 3-point index scoring system for relative expression intensity. When a specimen had heterogenous scoring the higher score was reported.
TCGA analysis
RNA-seq count data was downloaded from TCGA and parsed for patients that were treated with platinum-based chemotherapies (cisplatin or carboplatin). Patients were censored if clinical notes indicated confounding treatment or inconclusive progression. For each patient normalized SSBP1 RNA-counts were extracted, and patients were separated into an SSBP1-high and SSBP1-low expression cohort. A Kaplan-Meyer curve was generated in PRISM (version 7.0) comparing time to tumor recurrence as determined for each patient cohort. In the first line therapy, tumor recurrence following platinum chemotherapy is calculated as the difference between the platinum chemotherapy start date and date of relapse; time to progression, recurrence, or death; or time to last follow-up. The difference between the start date of a numbered chemotherapy regimen and the start date of next chemotherapy regimen is defined as the tumor recurrence following platinum chemotherapy for second line or third line therapy135–136.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with Excel (Microsoft) and Prism (GraphPad). Error bars represent mean ± SEM. Statistical comparisons were analyzed using unpaired two-tailed Student’s t-test or one-way ANOVA with Sidak’s post-hoc correction with p values indicated in figure legends and source data. Data were considered statistically different at P < 0.05. P < 0.05 is indicated with single asterisks, P < 0.001 with double asterisks, and P < 0.0001 with triple asterisks.
Supplementary Material
Figure S1: Anticancer drug treatment regulates cellular antioxidant levels, related to Figure 1. (A) Half-maximal inhibitory effect (IC50) of anticancer agents used in this study. Proliferation of K562 was determined after 96 hrs. by measuring cellular ATP concentrations. (B) Short-term treatment with anticancer agents does not alter K562 cell proliferation. Heatmaps depicting changes in K562 proliferation following treatment of the indicated compounds at 5-fold their IC50 concentrations as determined in (A). Concentrations of the compounds are: 5.1 μM ATO, 1.5 μM LAP, 0.1 μM DOXO, 30 nM ELC, 8.3 μM DDP, 7.1 μM BLE, 6.0 μM 5FU, 0.5 mM SUL, 2.5 μM AUR, 1 μM 2ME or 0.5 mM NOV for 0.5, 3, 6 and 24 hrs. Proliferation was determined by measuring cellular ATP concentrations (left) or trypan blue staining (right). (C) Characterization of changes in DCF fluorescence and antioxidant response following anticancer drug treatments. K562 cells treated with the indicated compounds at the noted time points and heatmaps illustrating changes were generated for DCF intensity, NAD+/NADH ratio, NADP+/NADPH ratio, and NRF2 activation. (D) Supplementation with NAC increases the IC50 for some anticancer agents. K562 cells were co-treated with the indicated concentrations of each agent and either 5 mM NAC or vehicle control. Relative proliferation was determined as described in (A). Data are represented as mean ± SEM.
Figure S2: Anticancer drug treatment regulates cysteine reactivity, related to Figure 1. (A) pLOGO plots of residues found within 10Å of cysteines from different clusters. (B) Examples of cysteines (red) targeted by anticancer drugs and their nearest residue neighbors. HSPA8: 1FE0137; TK1:1W4R138; ATOX1: 4KBQ138; SPR:1Z6Z. Enriched amino acids were colored based on corresponding cluster (see also Figure 1B). (C) Bar chart showing disagreement between primary sequence neighbors and 3D neighbors of reactive cysteines. (D) Correlation analysis between DCF staining and cysteine reactivity score. (E) Analysis of cysteine reactivity changes following anticancer drug treatment localized to corresponding cellular pathways. Pathways were determined by clustering genes based on co-essentiality. (F) C22 in RPL18A and C39 in RPL37A are modified following treatment with multiple anticancer drugs. (G) AUR reduces protein expression following cysteine modification. Left, plot of cysteine-reactivity changes in K562 following AUR treatment. Center, expression of AKT2 is decreased following AUR treatment. Right, crystal structure of AKT2 (PDB ID: 3D0E) highlighting the location of C297 and K298. K298 has been previously annotated as ubiquitinated. (H) Immunoblot analysis of AKT2 levels following AUR treatment. Data are represented as mean ± SEM. *P < 0.05, ***P < 0.0001, Student’s t-test (two-tailed, unpaired) were used to determine statistical significance.
Figure S3: Proteogenomic prioritization of AUR targets, related to Figure 2. (A-B) Inhibition of GSH biosynthesis sensitizes cells to AUR. (A) K562-dCas9-KRAB cells expressing the indicated sgRNAs were treated with 0.5 μM AUR or vehicle control. (B) K562 cells were co-treated with 0.5 μM AUR and 50 μM BSO. Relative proliferation was determined after 96 hrs. by measuring cellular ATP concentrations. (C) Immunoblot analysis of CHK1 levels in K562-dCas9-KRAB cells expressing the indicated sgRNAs. (D) Depletion of CHK1 potentiates AUR-mediated proliferation. Relative proliferation of K562-dCas9-KRAB cells stably expressing the indicated sgRNAs following treatment with 0.5 μM AUR was determined by measuring cellular ATP concentrations following 96 hrs. post treatment. (E) CHK1 inhibition potentiates AUR cytotoxicity. Proliferation of K562 cells treated with 0.5 μM CHK1i or 1 μM AUR was measured after 96 hrs. as described in (A). (F) Network analysis of functional targets for AUR and corresponding pathways and cellular locations. Data are represented as mean ± SEM. ***p< 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S4: Cysteine 408 in CHK1 functions as a nuclear H2O2 sensor, Related to Figure 3. (A) Left, Immunofluorescence analysis of K562 cells stably expressing the HyPer7 or roGFP2-ORP1 reporters in the nucleus, co-stained with Lamin A/C. Right, Immunofluorescence analysis of K562 cells stably expressing the HyPer7 or roGFP2-ORP1 reporters in the nucleus following H2O or 100 μM H2O2 treatment for 5 min. The ratiometric image was constructed by taking the ratio of the emission intensity of Oxidized (Ox) vs Reduced (Red) HyPer7 or roGFP2-ORP1. (B-C) Addition of NAC decreases nuclear H2O2 levels following AUR treatment. K562 cells expressing nuclear HyPer7 (B) or roGFP2-ORP1 (C) were treated with the indicated compounds for 6 hrs (5 mM NAC, 1.5 μM AUR) and the Ox/Red ratio of HyPer7/roGFP2-Orp1 was determined by flow cytometry (see also Figure 3C). (D) H2O2 treatment blocks alkylation of CHK1•C408. HEK-293T cells stably expressing CHK1 or CHK1•C408S were treated with 100 μM H2O2. IA-DTB modified CHK1 was determined by immunoblot following enrichment from cell lysates treated with a cysteine-specific IA-DTB. (E) Immunofluorescence analysis of FLAG-DAAO localized to nucleus in HEK-293T cells. (F) Characterization of CHK1 in vitro kinase assay. Immunoblot analysis of CHK1 in-vitro kinase assay following the addition of 5 μM CHK1 kinase inhibitor (CHK1i). (G) CHK1-C408D protects cells from AUR cytotoxicity. Relative proliferation of K562 cells stably expressing the indicated proteins was determined by measuring the changes in cellular ATP concentrations following treatment with 0.5 μM AUR for 96 hrs. (H) Left, immunofluorescence analysis of K562 cells stained with H2AX•S139 and DAPI. K562 cells stably expressing the indicated proteins were treated with 0.5 μM AUR for 24 hrs. Right, quantification of H2AX•S139 in the indicated samples. (I) CHK1 inhibition increases nuclear H2O2. K562 cells expressing nuclear HyPer7 (left) or roGFP2-ORP1 (right) were treated for 48 hrs. with 2 μM CHK1i. The Ox/Red ratio for each reporter was determined by flow cytometry. (J) Inactivation of CHK1 increases DCF fluorescence. Flow cytometry was used to monitor changes in DCF fluorescence in K562-dCas9-KRAB cells treated with CHK1i (2 μM) or expressing the indicated sgRNAs. (K) NAC treatment reverts CHK1-mediated nuclear H2O2 levels. K562 cells expressing nuclear localized HyPer7 were treated with 2 μM CHK1i and 5 mM NAC. Nuclear H2O2 levels were determined following 48 hrs. of treatment as described in (A). (L) Flow cytometry analysis of nuclear localized roGFP2-ORP1 (Ox/Red) ratio following inhibition of CHK1. K562 cells expressing nuclear roGFP2-ORP1 were treated as in (K) and analyzed as in (I). Scale bar=10 μm. Data are represented as mean ± SEM. *P < 0.05, **P < 0.001, ***P < 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S5: CHK1 regulates nuclear H2O2 levels through SSBP1 phosphorylation, related to Figure 5. (A-B) SSBP1 depletion rescues AUR and CHK1i cytotoxicity. K562 cells expressing the indicated sgRNAs were treated with 1.5 μM AUR (A), 1 μM CHK1i (B) or vehicle and proliferation was determined by measuring cellular ATP concentrations following 96 hrs. of treatment. (C-D) SSBP1 functions downstream of CHK1 to regulate DCF fluorescence. K562-dCas9-KRAB cells expressing the indicated sgRNAs were treated with CHK1i or vehicle control and analyzed as described above. (E) SSBP1•S67D phosphomimetic mutant decreases nuclear H2O2. Reintroduction of SSBP1-HA or SSBP1•S67D-HA into K562 cells depleted of SSBP1. Cells were treated with 1.5 μM AUR or vehicle and nuclear H2O2 levels were measured with nuclear localized HyPer7 (see also Figure 5I). (F) CHK1i blocks the phosphorylation of SSBP1. CHK1 in vitro kinase assay was conducted with SSBP1-HA and purified HA-CHK1 or HA-METAP2 (control protein). CHK1 was treated with 5 μM CHK1i and its phosphorylation of SSBP1 was monitored autoradiogram analysis. (G) Forced localization of SSBP1 to the nucleus. Immunofluorescence analysis of NLS-SSBP1 in K562 cells co-stained with Lamin. (H) SSBP1 localization is dependent on CHK1 activity. High-resolution spinning disk confocal microscopy immunofluorescence analysis of K562 cells expressing SSBP1-HA and co-stained with MitoTracker™ Red following the indicated treatments (see also Figures 5H–I). (I) The SSBP1 phospho-mimetic mutant is not localized to the mitochondria. High-resolution spinning disk confocal microscopy immunofluorescence analysis of K562 cells expressing the indicated SSBP1 WT or mutant proteins and co-stained with MitoTracker™ Red (see also Figure 5F). (J) The localization of SSBP1•S67A phosphodeficient mutant is not regulated by CHK1. Immunofluorescence analysis of K562 cells expressing SSBP1•S67A-HA following treatment with 1.5 μM AUR for 6 hrs. (K-L) The SSBP1•S67D mutant partial rescues AUR and CHK1i cytotoxicity. Cell lines described in (E) were treated with 1.5 μM AUR (K) or 1 μM CHK1i (L) and proliferation was determined as described in (A). Scale bar=10 μm. Data are represented as mean ± SEM.***P < 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S6: Mitochondrial translation regulates nuclear H2O2 levels, related to Figure 6. (A) Left, immunofluorescence analysis of K562 cells stably expressing the HyPer7 reporters in the mitochondrial matrix, co-stained with MitoTracker™ Red. Right, immunofluorescence analysis of K562 cells stably expressing the HyPer7 in the mitochondrial matrix following H2O or 100 μM H2O2 treatment for 5 min. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized HyPer7 over reduced HyPer7. (B) CHK1 regulates mitochondrial superoxide in a SSBP1-dependent manner. Mitochondrial superoxide was determined by monitoring changes in MitoSOX™ fluorescence with flow cytometry in K562-dCas9-KRAB cells expressing the indicated sgRNAs and treated with 2 μM of CHK1i for 48 hrs. (C). Immunofluorescence analysis of FLAG-DAAO in K562 cells. (D-E) SSBP1 depletion decreases mtDNA content. (D) Immunofluorescence analysis of mtDNA content in K562-dCas9-KRAB cells expressing the indicated sgRNAs. (E) Quantification of mtDNA content following depletion of SSBP1. (F) SSBP1 depletion decreases expression of mitochondrially encoded genes. Immunoblot analysis of the indicated genes in K562-dCas9-KRAB genes expressing the indicated sgRNAs. (G) CHK1 inhibition increases the expression of mitochondrially translated proteins. Immunoblot analysis of the indicated proteins in K562 cells treated with 2 μM CHK1i or vehicle control for 72 hrs. (H) CHK1 regulates mitochondrially encoded proteins in a time-dependent manner. K562 cells were treated with 2.3 μM doxycycline (DOXY) for 72 hrs., at which point the media was exchanged and replaced with 2 μM CHK1i or vehicle control. MT-CO2 protein levels were determined by immunoblot (see also Figure 6H). (I) CHK1 functions upstream of mitochondrial translation to regulate nuclear H2O2 levels. K562 cells expressing nucleus localized HyPer7 were treated with 2.3 μM DOXY for 24 hrs. followed by 2 μM CHK1i for 48 hrs. The HyPer7 Ox/Red ratio was determined by flow cytometry. (J) CHK1 functions upstream of mitochondrial translation to regulate mitochondrial H2O2 levels. K562 cells expressing HyPer7 localized to the mitochondrial matrix were treated with 2.3 μM DOXY for 24 hrs. followed by 2 μM CHK1i for 48 hrs. Mitochondria H2O2 levels were determined as in (L). (K-N) Genetic or pharmacological inhibition of mitochondrial translation blunts CHK1i mediated DNA-damage. Immunofluorescence analysis of H2AX•S139 staining in K562-dCas9-KRAB cells expressing the indicated sgRNAs targeting SSBP1 (K, L) or treated with 2.3 μM doxycycline (DOXY) (M, N) were treated with 2 μM CHK1i for 48 hrs. (n= 60 cells counted per condition). (O) Doxycycline (DOXY) partially rescues AUR growth inhibition. K562 cells were co-treated with 2.3 μM DOXY and 1.5 μM AUR. Proliferation was determined by measuring relative ATP concentrations following 96 hrs. of treatment.. (P) SSBP1 depletion partially rescues the proliferation of select anticancer agents which increase nuclear H2O2 levels as determined in (O). Data are represented as mean ± SEM. ***P < 0.0001. Scale bar=10 μm. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S7: SSBP1 levels regulate platinum-based nuclear H2O2 and resistance in ovarian cancers, related to Figure 7. (A) GSH depletion sensitizes cells to anticancer drug treatments that increases nuclear H2O2. K562 cells were co-treated with 50 μM BSO and the indicated compounds (1 μM LAP, 2.5 μM DDP, 0.25 μM AUR, 0.5 μM 2ME, 1 μM 5FU). Proliferation was determined by measuring relative ATP concentrations following 96 hrs. of treatment. (B) The GSSG:GSH ratio increases following treatment with anticancer drugs that increase nuclear H2O2. K562 cells were treated with the indicated compounds (1.5 μM LAP, 8.3 μM DDP, 2.5 μM AUR, 1 μM 2ME or 6.0 μM 5FU) for 24 hrs., and the GSSG:GSH ratio was determined. (C) Left, immunofluorescence analysis of K562 cells stably expressing the GRX1-roGFP2-NES GSH:GSSG reporter localized to the cytosol, co-stained with MEK1/2. Right, immunofluorescence analysis of K562 cells stably expressing GRX1-roGFP2 in the cytosol matrix following H2O2 or 1 mM BSO treatment for 72 hrs. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized roGFP2 over reduced roGFP2. (D) Regulation of cytosolic GSH:GSSG by anticancer drugs. K562 cells stably expressing GRX1-roGFP2-NES were treated with the 1.5 μM LAP, 8.3 μM DDP, 2.5 μM AUR, 1 μM 2ME or 6.0 μM 5FU for 24 hrs. and cytosolic GSH:GSSG ratio was determined as described in (C). (E) Patients with lower SSBP1 expression have shorter duration to tumor recurrence following platinum-based chemotherapy. Left, high-grade serous ovarian patients were stratified based on SSBP1 mRNA expression. Clinical data were obtained from135,136. (F) NAC treatment decreases cisplatin cytotoxicity in ovarian cancer cell lines. Cell lines were co-treated with 5 mM NAC or vehicle control and the indicated concentrations of cisplatin. Relative proliferation was determined as described in (A). Heatmap depicts fold change in IC50 between treatments, normalized to saline. (G) Cisplatin increases steady-state levels of nuclear H2O2 in ovarian cancer cells, which can be rescued by NAC treatment. Cell lines expressing nuclear HyPer7 were co-treated with 5 mM NAC or vehicle control and 8.3 μM cisplatin for 24 hrs. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized HyPer7 over reduced HyPer7. (H) Depletion of SSBP1 in ovarian cancer cell lines decreases cisplatin regulated nuclear H2O2 levels. (I) Steady-state levels of nuclear H2O2 decrease following depletion of SSBP1. Relative nuclear H2O2 levels were measured in cell lines co-expressing nuclear HyPer7 and the indicated shRNAs by determining the Ox/Red HyPer7 ratio by flow cytometry. (J-K) Re-introduction of SSBP1 increases nuclear H2O2 levels following DPP treatment. Immunofluorescence analysis of OVCAR3 cells expressing nuclear HyPer7, the indicated shRNAs and shRNA-resistant SSBP1 or METAP2 (control)] (J). The ratiometric image was constructed as described in (C). Immunoblot analysis (K) of the indicated proteins in OVCAR3 co-expressing the indicated shRNAs and shRNA resistant SSBP1 or METAP2. (L) Immunoblot analysis of SSBP1 in ovarian cancer cell lines expressing the indicated shRNAs. Scale bar=10 μm. Data are represented as mean ± SEM. *P < 0.05, **P < 0.001, ***P < 0.0001. Statistical significance was determined by one-way ANOVA with Sidak’s post-hoc correction (E) and Student’s t-test (two-tailed, unpaired) (A, F, I).
Table S1. Description of anticancer drugs analyzed in this study, related to Figure 1.
Table S2. Iso-TMT analysis of cysteine reactivity changes following treatment with anti-cancer drugs and structural files used in this study, related to Figure 1.
Table S3. Co-essentiality clustering and annotated protein localizations, related to Figure 1.
Table S4. Genome-wide CRISPRi screen identifies pathways that mediate resistance and sensitivity to Auranofin, related to Figure 2.
Table S5. Target prioritization based on chemical proteomics and functional genomic analysis, related to Figure 2.
Table S6. Clinical synopsis of patients, related to Figure 7.
Table S7. Primers used in this study, related to STAR Methods.18–20,22,28,30,32,37,119–125,126–134
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||||
|---|---|---|---|---|---|---|---|
| Antibodies | |||||||
| Total OXPHOS | Abcam | Cat#: ab110411; RRID:AB 2756818 | |||||
| CHK1 | Abcam | Cat#: ab40866; RRID:AB 726820 | |||||
| MT-ND1 | Elabscience | Cat#: E-AB-32173; RRID:AB 2881266 | |||||
| MT-CYTB | ProteinTech | Cat#: 55090–1-AP | |||||
| FLAG | Sigma | Cat#: F7425; RRID:AB_439687 | |||||
| Anti-Puromycin | Sigma | Cat#: MABE343; RRID:AB 2566826 | |||||
| AKT2 | Cell Signaling Tech | Cat#: 3063; RRID:AB 2225186 | |||||
| CHK1-S296 | Cell Signaling Tech | Cat#: 2349; RRID:AB 2080323 | |||||
| Anti-biotin, HRP-linked | Cell Signaling Tech | Cat#: 7075; RRID:AB 10696897 | |||||
| YH2AX | Cell Signaling Tech | Cat#: 2577; RRID:AB 2118010 | |||||
| FLAG | Cell Signaling Tech | Cat#: 14793; RRID:AB 2572291 | |||||
| LAMIN A/C | Cell Signaling Tech | Cat#: 4777; RRID:AB 10545756 | |||||
| HA | Cell Signaling Tech | Cat#: 3724; RRID:AB 1549585 | |||||
| TOM20 | Cell Signaling Tech | Cat#: 13929; RRID:AB 2631994 | |||||
| p-ACTIN | Cell Signaling Tech | Cat#: 4970; RRID:AB 2223172 | |||||
| MT-ATP6 | ProteinTech | Cat#: 55313–1-AP; RRID:AB 2881305 | |||||
| SSBP1 | ProteinTech | Cat#:12212–1-AP; RRID:AB 2195320 | |||||
| DNA | Progen | Cat#: 61014; RRID:AB 2750935 | |||||
| CHK1 | Santa Cruz | Cat#: SC-8408; RRID:AB 627257 | |||||
| HRP-labeled anti-mouse | Santa Cruz | Cat#:SC-2005; RRID:AB 631736 | |||||
| HRP-labeled anti-rabbit | Santa Cruz | Cat#: SC-2030; RRID:AB 631747 | |||||
| Anti-mouse IgG (H+L), F(ab’)2 Fragment (Alexa Fluor® 594 Conjugate) | Cell Signaling Tech | Cat#: 8890; RRID:AB 2714182 | |||||
| Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor(R) 647 Conjugate) | Cell Signaling Tech | Cat#: 4414; RRID:AB 10693544 | |||||
| Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor(R) 488 Conjugate) | Cell Signaling Tech | Cat#: 4412; RRID:AB 1904025 | |||||
| Bacterial and virus strains | |||||||
| DH5a | Fisher Scientific | Cat#:18265017 | |||||
| Chemicals, peptides, and recombinant proteins | |||||||
| RPMI-160 | Corning | Cat#: ICN1646454 | |||||
| FBS | Corning | Cat#: 35–010-CV | |||||
| DMEM | Corning | Cat#: 10–013-CV | |||||
| L-Glutamine | Corning | Cat#: 25–005-CI | |||||
| Glutamax I | Invitrogen | Cat#: 35050061 | |||||
| Penicillin-Streptomycin | Millipore | Cat#: P0781 | |||||
| Blasticidin | Fisher Scientific | Cat#: 50712728 | |||||
| Puromycin | Sigma-Aldrich | Cat#: P8833 | |||||
| Streptavidin agarose resin | Fisher Scientific | Cat#: 20347 | |||||
| Sequencing grade modified trypsin | Promega | Cat#: V5111 | |||||
| 2-Iodoacetamide | Fisher Scientific g | Cat#: ICN10035125 | |||||
| Iodoacetamide-desthiobiotin | Santa Cruz | Cat#: sc-300424 | |||||
| Nitroso-DTB | This Study | N/A | |||||
| DMSO | Fisher Scientific | Cat#: MT- 25950CQC | |||||
| Urea | VWR Intl | Cat#: 97063–798 | |||||
| CHAPS hydrate | Sigma-Aldrich | Cat#: C3023 | |||||
| DTT | Fisher Scientific | Cat#: BP1725 | |||||
| Micro bio-spin column | Bio-rad | Cat#: 7326204 | |||||
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#: 5892791001 | |||||
| PhosSTOP™ | Sigma-Aldrich | Cat#: 4906845001 | |||||
| Polybrene | Santa Cruz | Cat#: 134220 | |||||
| X-tremegene 9 transfection reagent | Sigma-Aldrich | Cat#: 6365787001 | |||||
| X-tremegene HP transfection reagent | Sigma-Aldrich | Cat#: 6366236001 | |||||
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat#: A2220 | |||||
| Anti-HA Magnetic Beads | Fisher Scientific | Cat#: 88836 | |||||
| Cell Titer Glo™ reagent | Promega | Cat#: G7570 | |||||
| Prolong Gold Antifade Mount | Life Technologies | Cat#: P36930 | |||||
| Formaldehyde | Sigma | Cat#: UN3334 | |||||
| Arsenic Trioxide | Fisher Scientific | Cat#: R0817000500 | |||||
| β-Lapachone | Cayman Chemical | Cat#: 15021 | |||||
| Doxorubicin | Cayman Chemical | Cat#: 15007 | |||||
| Elesclomol | Selleckchem | Cat#: S1052 | |||||
| Cisplatin | Cayman Chemical | Cat#: 13119 | |||||
| Bleomycin | Cayman Chemical | Cat#: 13877 | |||||
| 5-Fluorouracil | Cayman Chemical | Cat#: 14416 | |||||
| Sulfasalazine | Cayman Chemical | Cat#: 15025 | |||||
| Auranofin | Cayman Chemical | Cat#: 15316 | |||||
| 2-Methoxyestradiol | Cayman Chemical | Cat#: 13021 | |||||
| NOV-002 | MedKoo | Cat#: 202030 | |||||
| Hydrogen peroxide solution | Sigma | Cat#: 216763 | |||||
| Doxycycline | Sigma | Cat#: D3447 | |||||
| MitoTEMPO | Sigma | Cat#: SML0737 | |||||
| N-acetyl-L-Cysteine | Sigma | Cat#: 20261 | |||||
| Carboxy-DCFDA (5-(and-6)-Carboxy-2’,7’- Dichlorofluorescein Diacetate) | Fisher Scientific | Cat#: C369 | |||||
| MitoSOX™ Red | Fisher Scientific | Cat#: M36008 | |||||
| L-ALA | TCI America | Cat#: A0179 | |||||
| D-ALA | TCI America | Cat#: A0177 | |||||
| L-Buthionine-sulfoximine | Sigma | Cat#: B2515 | |||||
| Trolox | Cayman | Cat#: 10011659 | |||||
| Glutathione reduced ethyl ester | Sigma | Cat#: G1404 | |||||
| L -(+)-Ergothioneine | Sigma | Cat#: 497-30-3 | |||||
| S1QEL1.1 | Sigma | Cat#: SML1948 | |||||
| S3QEL-2 | Sigma | Cat#: SML1554 | |||||
| ATP, [Y-32P] | Perkin Elmer | Cat#: BLU002Z250UC | |||||
| Critical commercial assays | |||||||
| NADP/NADPH-Glo™ Assay kit | Promega | Cat#: G9081 | |||||
| NAD/NADH-Glo™ Assay kit | Promega | Cat#: G9071 | |||||
| GSH/GSSG-Glo™ Assay | Promega | Cat#: V6611 | |||||
| Deposited data | |||||||
| Proteomics data | This study | PRIDE: PXD041138 | |||||
| Experimental models: Cell lines | |||||||
| K562 | ATCC | CCL-243 | |||||
| HEK293T | ATCC | CRL-11268 | |||||
| K562-dCas9-KRAB | Gilbert et al., 2014 | N/A | |||||
| OVCAR3 | ATCC | HTB-161 | |||||
| Kuramochi | JCRB | JCRB0098 | |||||
| OVCAR8 | ATCC | CMT-929 | |||||
| PEO1 | ECACC | 10032308 | |||||
| CAOV3 | ATCC | HTB-75 | |||||
| OVISE | JCRB | JCRB1043 | |||||
| OV90 | ATCC | CRL-11732 | |||||
| OVCAR4 | NCI/DTP | N/A | |||||
| OAW28 | ECACC | 85101601 | |||||
| Recombinant DNA | |||||||
| Plenti: Hyper7-NLS | This study | N/A | |||||
| Plenti: Mito-Hyper7 | This study | N/A | |||||
| Plenti: roGFP2-Orp1-NLS | This study | N/A | |||||
| Plenti: Mito-roGFP2-Orp1 | This study | N/A | |||||
| Plenti: FLAG-DAAO-NLS | This study | N/A | |||||
| Plenti: FLAG-MITO-DAAO | This study | N/A | |||||
| Plenti: FLAG-Mito-DAAO-CHK1 | This study | N/A | |||||
| Plenti: CHK1 | This study | N/A | |||||
| pLJM1: FLAG-METAP2 | This study | N/A | |||||
| pLJM1: FLAG-CHK1 | This study | N/A | |||||
| pLJM1: FLAG-CHK1 (C408D) | This study | N/A | |||||
| pLJM1: FLAG-CHK1 (C408S) | This study | N/A | |||||
| pRK5: FLAG-METAP2 | This study | N/A | |||||
| pRK5: FLAG-CHK1 | This study | N/A | |||||
| pRK5: FLAG-CHK1(C408D) | This study | N/A | |||||
| pRK5: FLAG-CHK1-Kinase domain | This study | N/A | |||||
| pRK5: HA-CHK1-KA1 | This study | N/A | |||||
| pRK5: HA-CHK1-KA1 (C408D) | This study | N/A | |||||
| Lenti-CRISPRv2: sgSSBP1–1 | This study | N/A | |||||
| Lenti-CRISPRv2: sgSSBP1–2 | This study | N/A | |||||
| Lenti-CRISPRv2: sgCTRL | This study | N/A | |||||
| Pu6-BFP: sgCONTROL | This study | N/A | |||||
| pU6-BFP: sgSSBP1–1 | This study | N/A | |||||
| pU6-BFP: sgSSBP1–2 | This study | N/A | |||||
| pU6-BFP: sgCHK1–1 | This study | N/A | |||||
| pU6-BFP: sgCHK1–2 | This study | N/A | |||||
| pLKO: shCTRL | This study | N/A | |||||
| pLKO: shSSBP1_1 | This study | N/A | |||||
| pLKO: shSSBP1_2 | This study | N/A | |||||
| Plenti: SSBP1-HA | This study | N/A | |||||
| Plenti: NLS-SSBP1-HA | This study f ^ | N/A | |||||
| Plenti: SSBP1-FLAG | This study | N/A | |||||
| Plenti: SSBP1-shRNA mutant | This study | N/A | |||||
| Plenti: SSBP1 (S67A)-HA | This study | N/A | |||||
| Plenti: SSBP1 (S67D)-HA | This study | N/A | |||||
| Plenti: SSBP1-gRNA mutant | This study | N/A | |||||
| Plenti: SSBP1 (S67A)-gRNA mutant | This study | N/A | |||||
| Plenti: SSBP1 (S67D)-gRNA mutant | This study | N/A | |||||
| Software and algorithms | |||||||
| Prism (v7.0) | GraphPad | https://www.graphpad.com/scientific-software/prism/ | |||||
| FlowJo (v10.0.7) | Treestar Inc. | https://www.flowjo.com/ | |||||
| ZEN 2.6 | Zeiss | https://www.zeiss.com | |||||
| Floview 3.1 | Olympus | https://www.olympus-lifescience.com/ | |||||
| ImageJ | NIH | https://imagej.net/software/fiji/ | |||||
Highlights.
Integrated chemical proteomic and CRISPRi screens identify ROS-target proteins
Nuclear H2O2 oxidizes C408 in CHK1’s autoinhibitory domain leading to its activation
CHK1 regulates mitochondrial translation by inhibiting mtDNA binding protein SSBP1
SSBP1 promotes resistance to platinum-based agents and nuclear H2O2 in ovarian cancers
Acknowledgements
We thank Thomas Michel for providing DAAO-HyPer7 plasmid. We thank all members of the Bar-Peled Lab, David Sabatini and Lee Zou for helpful suggestions. This work was supported by the Damon Runyon Cancer Research Foundation (62-20), the American Association for Cancer Research (19-20-45-BARP), the American Cancer Society, the Melanoma Research Alliance, the Ludwig Cancer Center of Harvard Medical School, Lungevity, ALK-Positive, V-Foundation, Mary Kay Foundation, Paula and Rodger Riney Foundation, Nile Albright Research Foundation (BRR), Vincent Memorial Hospital Foundation (BRR) and the NIH/NCI (1R21CA226082-01, R37CA260062 to L.B-P).
INCLUSION AND DIVERSITY
We support inclusive, diverse and equitable conduct in research.
Footnotes
DECLARATION OF INTERESTS
A.P.P., C.M.S. M.P.S., and S.A.B. are employees of Cell Signaling Technology, INC. L.B-P. is a founder, consultant and holds privately held equity in Scorpion Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chio IIC, and Tuveson DA (2017). ROS in Cancer: The Burning Question. Trends in molecular medicine 23, 411–429. 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reczek CR, and Chandel NS (2015). ROS-dependent signal transduction. Current opinion in cell biology 33, 8–13. 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chouchani ET, Kazak L, and Spiegelman BM (2017). Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. The Journal of biological chemistry 292, 16810–16816. 10.1074/jbc.R117.789628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins Y, Chouchani ET, James AM, Menger KE, Cocheme HM, and Murphy MP (2012). Mitochondrial redox signalling at a glance. Journal of cell science 125, 801–806. 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 5.Dixon SJ, and Stockwell BR (2019). The Hallmarks of Ferroptosis. Annual Review of Cancer Biology 3, 35–54. 10.1146/annurev-cancerbio-030518-055844. [DOI] [Google Scholar]
- 6.Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, and Liang X (2018). The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res 37, 266. 10.1186/s13046-018-0909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G, and Migliaccio A (2020). ROS in cancer therapy: the bright side of the moon. Exp Mol Med 52, 192–203. 10.1038/s12276-020-0384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chabner B, and Longo DL (2011). Cancer chemotherapy and biotherapy : principles and practice (Wolters Kluwer Health/Lippincott Williams & Wilkins). [Google Scholar]
- 9.Liou GY, and Storz P (2010). Reactive oxygen species in cancer. Free Radic Res 44, 479–496. 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Backus KM (2019). Applications of Reactive Cysteine Profiling. Curr Top Microbiol Immunol 420, 375–417. 10.1007/82_2018_120. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014). A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nature methods 11, 79–85. 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, et al. (2016). NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 166, 963–976. 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu TA, Svensson RU, Olucha J, et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 e623. 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, Heppner DE, Li J, Long J, Mills EL, Szpyt J, et al. (2020). A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 180, 968–983 e924. 10.1016/j.cell.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, Smith BD, Flatten KS, Peterson K, Schneider P, et al. (2012). Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 6723–6731. 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webster JA, Tibes R, Morris L, Blackford AL, Litzow M, Patnaik M, Rosner GL, Gojo I, Kinders R, Wang L, et al. (2017). Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk Res 61, 108–116. 10.1016/j.leukres.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, Venook AP, Loechner S, Rosen LS, Shanahan F, et al. (2015). Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33, 1060–1066. 10.1200/JCO.2014.57.5027. [DOI] [PubMed] [Google Scholar]
- 18.Xie SL, Yang MH, Chen K, Huang H, Zhao XW, Zang YS, and Li B (2015). Efficacy of Arsenic Trioxide in the Treatment of Malignant Pleural Effusion Caused by Pleural Metastasis of Lung Cancer. Cell Biochem Biophys 71, 1325–1333. 10.1007/s12013-014-0352-3. [DOI] [PubMed] [Google Scholar]
- 19.Miller WH Jr., Schipper HM, Lee JS, Singer J, and Waxman S (2002). Mechanisms of action of arsenic trioxide. Cancer research 62, 3893–3903. [PubMed] [Google Scholar]
- 20.Gerber DE, Beg MS, Fattah F, Frankel AE, Fatunde O, Arriaga Y, Dowell JE, Bisen A, Leff RD, Meek CC, et al. (2018). Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. British journal of cancer 119, 928–936. 10.1038/s41416-018-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong Q, Hu J, Wang P, Li X, and Zhang X (2021). A comprehensive review on beta-lapachone: Mechanisms, structural modifications, and therapeutic potentials. Eur J Med Chem 210, 112962. 10.1016/j.ejmech.2020.112962. [DOI] [PubMed] [Google Scholar]
- 22.Rivankar S (2014). An overview of doxorubicin formulations in cancer therapy. J Cancer Res Ther 10, 853–858. 10.4103/0973-1482.139267. [DOI] [PubMed] [Google Scholar]
- 23.Nagai M, Vo NH, Shin Ogawa L, Chimmanamada D, Inoue T, Chu J, Beaudette-Zlatanova BC, Lu R, Blackman RK, Barsoum J, et al. (2012). The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med 52, 2142–2150. 10.1016/j.freeradbiomed.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 24.Gohil VM (2021). Repurposing elesclomol, an investigational drug for the treatment of copper metabolism disorders. Expert Opin Investig Drugs 30, 1–4. 10.1080/13543784.2021.1840550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng P, Zhou C, Lu L, Liu B, and Ding Y (2022). Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res 41, 271. 10.1186/s13046-022-02485-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lugones Y, Loren P, and Salazar LA (2022). Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules 12. 10.3390/biom12101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranasinghe R, Mathai ML, and Zulli A (2022). Cisplatin for cancer therapy and overcoming chemoresistance. Heliyon 8, e10608. 10.1016/j.heliyon.2022.e10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecht SM (2000). Bleomycin: new perspectives on the mechanism of action. J Nat Prod 63, 158–168. 10.1021/np990549f. [DOI] [PubMed] [Google Scholar]
- 29.Dorr RT (1992). Bleomycin pharmacology: mechanism of action and resistance, and clinical pharmacokinetics. Seminars in oncology 19, 3–8. [PubMed] [Google Scholar]
- 30.Longley DB, Harkin DP, and Johnston PG (2003). 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews. Cancer 3, 330–338. 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang J, Liu X, Yang Y, Zhang Y, and Guan G (2021). Sulfasalazine, a potent suppressor of gastric cancer proliferation and metastasis by inhibition of xCT: Conventional drug in new use. J Cell Mol Med 25, 5372–5380. 10.1111/jcmm.16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shitara K, Doi T, Nagano O, Fukutani M, Hasegawa H, Nomura S, Sato A, Kuwata T, Asai K, Einaga Y, et al. (2017). Phase 1 study of sulfasalazine and cisplatin for patients with CD44v-positive gastric cancer refractory to cisplatin (EPOC1407). Gastric Cancer 20, 1004–1009. 10.1007/s10120-017-0720-y. [DOI] [PubMed] [Google Scholar]
- 33.Mushtaq S, and Sarkar R (2020). Sulfasalazine in dermatology: A lesser explored drug with broad therapeutic potential. Int J Womens Dermatol 6, 191–198. 10.1016/j.ijwd.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakhani NJ, Sarkar MA, Venitz J, and Figg WD (2003). 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy 23, 165–172. 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- 35.Mueck AO, and Seeger H (2010). 2-Methoxyestradiol--biology and mechanism of action. Steroids 75, 625–631. 10.1016/j.steroids.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Abdalbari FH, and Telleria CM (2021). The gold complex auranofin: new perspectives for cancer therapy. Discov Oncol 12, 42. 10.1007/s12672-021-00439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montero AJ, Diaz-Montero CM, Deutsch YE, Hurley J, Koniaris LG, Rumboldt T, Yasir S, Jorda M, Garret-Mayer E, Avisar E, et al. (2012). Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res Treat 132, 215–223. 10.1007/s10549-011-1889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsend DM, He L, Hutchens S, Garrett TE, Pazoles CJ, and Tew KD (2008). NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer research 68, 2870–2877. 10.1158/0008-5472.CAN-07-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu J, Jin Z, Zheng H, and Yan LJ (2016). Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes 9, 145–153. 10.2147/DMSO.S106087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, Suski JM, et al. (2017). The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. 10.1038/nature22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, et al. (2020). An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 182, 1009–1026 e1029. 10.1016/j.cell.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuljanin M, Mitchell DC, Schweppe DK, Gikandi AS, Nusinow DP, Bulloch NJ, Vinogradova EV, Wilson DL, Kool ET, Mancias JD, et al. (2021). Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nature biotechnology 39, 630–641. 10.1038/s41587-020-00778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wensien M, von Pappenheim FR, Funk LM, Kloskowski P, Curth U, Diederichsen U, Uranga J, Ye J, Fang P, Pan KT, et al. (2021). A lysine-cysteine redox switch with an NOS bridge regulates enzyme function. Nature 593, 460–464. 10.1038/s41586-021-03513-3. [DOI] [PubMed] [Google Scholar]
- 44.Zhitkovich A (2019). N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem Res Toxicol 32, 1318–1319. 10.1021/acs.chemrestox.9b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.NCT01419691 (2011). Phase I and II Study of Auranofin in Chronic Lymphocytic Leukemia (CLL).
- 46.NCT03456700 (2018). Auranofin and Sirolimus in Treating Participants With Ovarian Cancer.
- 47.Stafford WC, Peng X, Olofsson MH, Zhang X, Luci DK, Lu L, Cheng Q, Tresaugues L, Dexheimer TS, Coussens NP, et al. (2018). Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Science translational medicine 10. 10.1126/scitranslmed.aaf7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris IS, Endress JE, Coloff JL, Selfors LM, McBrayer SK, Rosenbluth JM, Takahashi N, Dhakal S, Koduri V, Oser MG, et al. (2019). Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell metabolism 29, 1166–1181 e1166. 10.1016/j.cmet.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. (2009). STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic acids research 37, D412–416. 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zarei M, Du H, Nassar AH, Yan RE, Giannikou K, Johnson SH, Lam HC, Henske EP, Wang Y, Zhang T, et al. (2019). Tumors with TSC mutations are sensitive to CDK7 inhibition through NRF2 and glutathione depletion. The Journal of experimental medicine 216, 2635–2652. 10.1084/jem.20190251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, and Barford D (2003). Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773. 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 53.Niederkorn M, Ishikawa C, K MH, Bartram J, Stepanchick E, J RB, A EC-C, Bolanos LC, Uible E, Choi K, et al. (2022). The deubiquitinase USP15 modulates cellular redox and is a therapeutic target in acute myeloid leukemia. Leukemia 36, 438451. 10.1038/s41375-021-01394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gajewska KA, Lescesen H, Ramialison M, Wagstaff KM, and Jans DA (2021). Nuclear transporter Importin-13 plays a key role in the oxidative stress transcriptional response. Nature communications 12, 5904. 10.1038/s41467-021-26125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnandis T, Monteiro P, Adams SD, Bridgeman VL, Rajeeve V, Gadaleta E, Marzec J, Chelala C, Malanchi I, Cutillas PR, and Godinho SA (2018). Oxidative Stress in Cells with Extra Centrosomes Drives Non-Cell-Autonomous Invasion. Developmental cell 47, 409–424 e409. 10.1016/j.devcel.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Molecular cell 40, 179–204. 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katsuragi Y, and Sagata N (2004). Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Molecular biology of the cell 15, 1680–1689. 10.1091/mbc.e03-12-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, and Dick TP (2009). Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. The Journal of biological chemistry 284, 31532–31540. 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pak VV, Ezerina D, Lyublinskaya OG, Pedre B, Tyurin-Kuzmin PA, Mishina NM, Thauvin M, Young D, Wahni K, Martinez Gache SA, et al. (2020). Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell metabolism 31, 642–653 e646. 10.1016/j.cmet.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reddie KG, Seo YH, Muse Iii WB, Leonard SE, and Carroll KS (2008). A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst 4, 521–531. 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang J, Carroll KS, and Liebler DC (2016). The Expanding Landscape of the Thiol Redox Proteome. Molecular & cellular proteomics : MCP 15, 1–11. 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo Conte M, Lin J, Wilson MA, and Carroll KS (2015). A Chemical Approach for the Detection of Protein Sulfinylation. ACS chemical biology 10, 1825–1830. 10.1021/acschembio.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinhorn B, Sorrentino A, Badole S, Bogdanova Y, Belousov V, and Michel T (2018). Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nature communications 9, 4044. 10.1038/s41467-018-06533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Permyakov SE, Zernii EY, Knyazeva EL, Denesyuk AI, Nazipova AA, Kolpakova TV, Zinchenko DV, Philippov PP, Permyakov EA, and Senin II (2012). Oxidation mimicking substitution of conservative cysteine in recoverin suppresses its membrane association. Amino acids 42, 1435–1442. 10.1007/s00726-011-0843-0. [DOI] [PubMed] [Google Scholar]
- 65.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, Taricani L, Wiswell D, Seghezzi W, Penaflor E, et al. (2011). Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Molecular cancer therapeutics 10, 591–602. 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- 66.Tan R, Nakajima S, Wang Q, Sun H, Xue J, Wu J, Hellwig S, Zeng X, Yates NA, Smithgall TE, et al. (2017). Nek7 Protects Telomeres from Oxidative DNA Damage by Phosphorylation and Stabilization of TRF1. Molecular cell 65, 818–831 e815. 10.1016/j.molcel.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, and Jackson SP (2011). A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol 12, R78. 10.1186/gb-2011-12-8-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Copeland WC, and Longley MJ (2003). DNA polymerase gamma in mitochondrial DNA replication and repair. ScientificWorldJournal 3, 34–44. 10.1100/tsw.2003.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rajala N, Gerhold JM, Martinsson P, Klymov A, and Spelbrink JN (2014). Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic acids research 42, 952–967. 10.1093/nar/gkt988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, and Dikalov SI (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107, 106–116. 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brand MD (2020). Riding the tiger - physiological and pathological effects of superoxide and hydrogen peroxide generated in the mitochondrial matrix. Critical reviews in biochemistry and molecular biology 55, 592–661. 10.1080/10409238.2020.1828258. [DOI] [PubMed] [Google Scholar]
- 72.Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT, et al. (2016). Suppressors of Superoxide-H(2)O(2) Production at Site I(Q) of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell metabolism 24, 582–592. 10.1016/j.cmet.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, et al. (2015). Suppressors of superoxide production from mitochondrial complex III. Nat Chem Biol 11, 834–836. 10.1038/nchembio.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang M, Xie X, Zhu X, Jiang S, Milenkovic D, Misic J, Shi Y, Tandukar N, Li X, Atanassov I, et al. (2021). The mitochondrial single-stranded DNA binding protein is essential for initiation of mtDNA replication. Sci Adv 7. 10.1126/sciadv.abf8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chatzispyrou IA, Held NM, Mouchiroud L, Auwerx J, and Houtkooper RH (2015). Tetracycline antibiotics impair mitochondrial function and its experimental use confounds research. Cancer research 75, 4446–4449. 10.1158/0008-5472.CAN-15-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, and Menck CFM (2018). DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 73, e478s. 10.6061/clinics/2018/e478s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rocha CR, Garcia CC, Vieira DB, Quinet A, de Andrade-Lima LC, Munford V, Belizario JE, and Menck CF (2014). Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis 5, e1505. 10.1038/cddis.2014.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forman HJ, Zhang H, and Rinna A (2009). Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 30, 1–12. 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Go YM, and Jones DP (2008). Redox compartmentalization in eukaryotic cells. Biochimica et biophysica acta 1780, 1273–1290. 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP (2008). Real-time imaging of the intracellular glutathione redox potential. Nature methods 5, 553–559. 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, Wang J, Liu Z, Yang H, Zhu Y, Zhao M, Liu Y, and Yan M (2016). Mitochondrial DNA depletion causes decreased ROS production and resistance to apoptosis. Int J Mol Med 38, 1039–1046. 10.3892/ijmm.2016.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vasan N, Baselga J, and Hyman DM (2019). A view on drug resistance in cancer. Nature 575, 299–309. 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 84.Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, Almazyadi HAM, Kallmeyer K, Dandara C, Pepper MS, et al. (2017). The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int J Mol Sci 18. 10.3390/ijms18071586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng HC (2017). The molecular mechanisms of chemoresistance in cancers. Oncotarget 8, 59950–59964. 10.18632/oncotarget.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yeldag G, Rice A, and Del Rio Hernandez A (2018). Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers (Basel) 10. 10.3390/cancers10120471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Juan CA, Perez de la Lastra JM, Plou FJ, and Perez-Lebena E (2021). The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int J Mol Sci 22. 10.3390/ijms22094642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chao MR, Rossner P Jr., Haghdoost S, Jeng HA, and Hu CW (2013). Nucleic acid oxidation in human health and disease. Oxid Med Cell Longev 2013, 368651. 10.1155/2013/368651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lechuga-Vieco AV, Justo-Mendez R, and Enriquez JA (2021). Not all mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life 73, 511–529. 10.1002/iub.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guaragnella N, Coyne LP, Chen XJ, and Giannattasio S (2018). Mitochondria-cytosol-nucleus crosstalk: learning from Saccharomyces cerevisiae. FEMS Yeast Res 18. 10.1093/femsyr/foy088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, Hurbain I, Raposo G, Popova T, Stern MH, et al. (2019). PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell metabolism 29, 156–173 e110. 10.1016/j.cmet.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu Y, Ricciardiello F, Yang G, Qiu J, Huang H, Xiao J, Cao Z, Zhao F, Liu Y, Luo W, et al. (2021). The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells 10. 10.3390/cells10030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen L, Xia M, Zhang Y, Luo H, Dong D, and Sun L (2021). Mitochondrial integration and ovarian cancer chemotherapy resistance. Experimental cell research 401, 112549. 10.1016/j.yexcr.2021.112549. [DOI] [PubMed] [Google Scholar]
- 94.Xie L, Zhou T, Xie Y, Bode AM, and Cao Y (2021). Mitochondria-Shaping Proteins and Chemotherapy. Front Oncol 11, 769036. 10.3389/fonc.2021.769036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang HN, Yang L, Ling JY, Czajkowsky DM, Wang JF, Zhang XW, Zhou YM, Ge F, Yang MK, Xiong Q, et al. (2015). Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proceedings of the National Academy of Sciences of the United States of America 112, 15084–15089. 10.1073/pnas.1521316112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abhishek S, Sivadas S, Satish M, Deeksha W, and Rajakumara E (2019). Dynamic Basis for Auranofin Drug Recognition by Thiol-Reductases of Human Pathogens and Intermediate Coordinated Adduct Formation with Catalytic Cysteine Residues. ACS Omega 4, 9593–9602. 10.1021/acsomega.9b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Myasnikov AG, Kundhavai Natchiar S, Nebout M, Hazemann I, Imbert V, Khatter H, Peyron JF, and Klaholz BP (2016). Structure-function insights reveal the human ribosome as a cancer target for antibiotics. Nature communications 7, 12856. 10.1038/ncomms12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen P, Luo C, Deng Y, Ryan K, Register J, Margosiak S, Tempczyk-Russell A, Nguyen B, Myers P, Lundgren K, et al. (2000). The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 100, 681–692. 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 99.El-Faham A, and Albericio F (2010). COMU: a third generation of uronium-type coupling reagents. J Pept Sci 16, 6–9. 10.1002/psc.1204. [DOI] [PubMed] [Google Scholar]
- 100.Shi Y, and Carroll KS (2020). Activity-Based Sensing for Site-Specific Proteomic Analysis of Cysteine Oxidation. Acc Chem Res 53, 20–31. 10.1021/acs.accounts.9b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lo Conte M, and Carroll KS (2012). Chemoselective ligation of sulfinic acids with aryl-nitroso compounds. Angewandte Chemie 51, 6502–6505. 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Possemato AP, Paulo JA, Mulhern D, Guo A, Gygi SP, and Beausoleil SA (2017). Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events. J Proteome Res 16, 1506–1514. 10.1021/acs.jproteome.6b00905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8, 937–940. 10.1038/nmeth.1714 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paulo JA, O’Connell JD, and Gygi SP (2016). A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom 27, 1620–1625. 10.1007/s13361-016-1434-910.1007/s13361-016-1434-9 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. 10.1016/j.cell.2010.12.001S0092-8674(10)01379-6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 108.Eng JK, Jahan TA, and Hoopmann MR (2013). Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 109.Villen J, Beausoleil SA, Gerber SA, and Gygi SP (2007). Large-scale phosphorylation analysis of mouse liver. Proceedings of the National Academy of Sciences of the United States of America 104, 1488–1493. 0609836104 [pii] 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martin AC (2005). Mapping PDB chains to UniProtKB entries. Bioinformatics 21, 4297–4301. 10.1093/bioinformatics/bti694. [DOI] [PubMed] [Google Scholar]
- 111.O’Shea JP, Chou MF, Quader SA, Ryan JK, Church GM, and Schwartz D (2013). pLogo: a probabilistic approach to visualizing sequence motifs. Nature methods 10, 1211–1212. 10.1038/nmeth.2646. [DOI] [PubMed] [Google Scholar]
- 112.Orre LM, Vesterlund M, Pan Y, Arslan T, Zhu Y, Fernandez Woodbridge A, Frings O, Fredlund E, and Lehtio J (2019). SubCellBarCode: Proteome-wide Mapping of Protein Localization and Relocalization. Molecular cell 73, 166–182 e167. S1097–2765(18)31005–0 [pii] 10.1016/j.molcel.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 113.Alexa A, Rahnenfuhrer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. btl140 [pii] 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 114.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research 43, D512–520. 10.1093/nar/gku1267 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nature genetics 49, 1779–1784. 10.1038/ng.3984 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, Martin MJ, and O’Donovan C (2015). The GOA database: gene Ontology annotation updates for 2015. Nucleic acids research 43, D1057–1063. 10.1093/nar/gku1113 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature protocols 4, 1184–1191. 10.1038/nprot.2009.97 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tevaarwerk AJ, Holen KD, Alberti DB, Sidor C, Arnott J, Quon C, Wilding G, and Liu G (2009). Phase I trial of 2-methoxyestradiol NanoCrystal dispersion in advanced solid malignancies. Clin Cancer Res 15, 1460–1465. 10.1158/1078-0432.CCR-08-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, Fife K, Armstrong V, Treston A, Sidor C, and Wilding G (2005). A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res 11, 6625–6633. 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- 121.Rosenberg B, VanCamp L, Trosko JE, and Mansour VH (1969). Platinum compounds: a new class of potent antitumour agents. Nature 222, 385–386. 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 122.O’Day SJ, Eggermont AM, Chiarion-Sileni V, Kefford R, Grob JJ, Mortier L, Robert C, Schachter J, Testori A, Mackiewicz J, et al. (2013). Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol 31, 1211–1218. 10.1200/JCO.2012.44.5585. [DOI] [PubMed] [Google Scholar]
- 123.Qu Y, Wang J, Sim MS, Liu B, Giuliano A, Barsoum J, and Cui X (2010). Elesclomol, counteracted by Akt survival signaling, enhances the apoptotic effect of chemotherapy drugs in breast cancer cells. Breast Cancer Res Treat 121, 311–321. 10.1007/s10549-009-0470-6. [DOI] [PubMed] [Google Scholar]
- 124.Pardee AB, Li YZ, and Li CJ (2002). Cancer therapy with beta-lapachone. Curr Cancer Drug Targets 2, 227–242. 10.2174/1568009023333854. [DOI] [PubMed] [Google Scholar]
- 125.Fang Y, and Zhang Z (2020). Arsenic trioxide as a novel anti-glioma drug: a review. Cell Mol Biol Lett 25, 44. 10.1186/s11658-020-00236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, and De Camilli P (2012). Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci U S A 109, E2316–2323. 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sun Q, Westphal W, Wong KN, Tan I, and Zhong Q (2010). Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 107, 19338–19343. 10.1073/pnas.1010554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ray GJ, Boydston EA, Shortt E, Wyant GA, Lourido S, Chen WW, and Sabatini DM (2020). A PEROXO-Tag Enables Rapid Isolation of Peroxisomes from Human Cells. iScience 23, 101109. 10.1016/j.isci.2020.101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pan D, Klare K, Petrovic A, Take A, Walstein K, Singh P, Rondelet A, Bird AW, and Musacchio A (2017). CDK-regulated dimerization of M18BP1 on a Mis18 hexamer is necessary for CENP-A loading. Elife 6. 10.7554/eLife.23352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, and Gadella TW Jr. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14, 53–56. 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- 131.Malinouski M, Zhou Y, Belousov VV, Hatfield DL, and Gladyshev VN (2011). Hydrogen peroxide probes directed to different cellular compartments. PLoS One 6, e14564. 10.1371/journal.pone.0014564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee JE, Cathey PI, Wu H, Parker R, and Voeltz GK (2020). Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science 367. 10.1126/science.aay7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, and Sabatini DM (2015). Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343. 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, and Lukyanov KA (2006). A genetically encoded photosensitizer. Nat Biotechnol 24, 95–99. 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- 135.Villalobos VM, Wang YC, and Sikic BI (2018). Reannotation and Analysis of Clinical and Chemotherapy Outcomes in the Ovarian Data Set From The Cancer Genome Atlas. JCO Clin Cancer Inform 2, 1–16. [DOI] [PubMed] [Google Scholar]
- 136.Cancer Genome Atlas Research, N. (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, and Rosenzweig AC (2000). Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat Struct Biol 7, 766–771. 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 138.Birringer MS, Claus MT, Folkers G, Kloer DP, Schulz GE, and Scapozza L (2005). Structure of a type II thymidine kinase with bound dTTP. FEBS Lett 579, 1376–1382. 10.1016/j.febslet.2005.01.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Anticancer drug treatment regulates cellular antioxidant levels, related to Figure 1. (A) Half-maximal inhibitory effect (IC50) of anticancer agents used in this study. Proliferation of K562 was determined after 96 hrs. by measuring cellular ATP concentrations. (B) Short-term treatment with anticancer agents does not alter K562 cell proliferation. Heatmaps depicting changes in K562 proliferation following treatment of the indicated compounds at 5-fold their IC50 concentrations as determined in (A). Concentrations of the compounds are: 5.1 μM ATO, 1.5 μM LAP, 0.1 μM DOXO, 30 nM ELC, 8.3 μM DDP, 7.1 μM BLE, 6.0 μM 5FU, 0.5 mM SUL, 2.5 μM AUR, 1 μM 2ME or 0.5 mM NOV for 0.5, 3, 6 and 24 hrs. Proliferation was determined by measuring cellular ATP concentrations (left) or trypan blue staining (right). (C) Characterization of changes in DCF fluorescence and antioxidant response following anticancer drug treatments. K562 cells treated with the indicated compounds at the noted time points and heatmaps illustrating changes were generated for DCF intensity, NAD+/NADH ratio, NADP+/NADPH ratio, and NRF2 activation. (D) Supplementation with NAC increases the IC50 for some anticancer agents. K562 cells were co-treated with the indicated concentrations of each agent and either 5 mM NAC or vehicle control. Relative proliferation was determined as described in (A). Data are represented as mean ± SEM.
Figure S2: Anticancer drug treatment regulates cysteine reactivity, related to Figure 1. (A) pLOGO plots of residues found within 10Å of cysteines from different clusters. (B) Examples of cysteines (red) targeted by anticancer drugs and their nearest residue neighbors. HSPA8: 1FE0137; TK1:1W4R138; ATOX1: 4KBQ138; SPR:1Z6Z. Enriched amino acids were colored based on corresponding cluster (see also Figure 1B). (C) Bar chart showing disagreement between primary sequence neighbors and 3D neighbors of reactive cysteines. (D) Correlation analysis between DCF staining and cysteine reactivity score. (E) Analysis of cysteine reactivity changes following anticancer drug treatment localized to corresponding cellular pathways. Pathways were determined by clustering genes based on co-essentiality. (F) C22 in RPL18A and C39 in RPL37A are modified following treatment with multiple anticancer drugs. (G) AUR reduces protein expression following cysteine modification. Left, plot of cysteine-reactivity changes in K562 following AUR treatment. Center, expression of AKT2 is decreased following AUR treatment. Right, crystal structure of AKT2 (PDB ID: 3D0E) highlighting the location of C297 and K298. K298 has been previously annotated as ubiquitinated. (H) Immunoblot analysis of AKT2 levels following AUR treatment. Data are represented as mean ± SEM. *P < 0.05, ***P < 0.0001, Student’s t-test (two-tailed, unpaired) were used to determine statistical significance.
Figure S3: Proteogenomic prioritization of AUR targets, related to Figure 2. (A-B) Inhibition of GSH biosynthesis sensitizes cells to AUR. (A) K562-dCas9-KRAB cells expressing the indicated sgRNAs were treated with 0.5 μM AUR or vehicle control. (B) K562 cells were co-treated with 0.5 μM AUR and 50 μM BSO. Relative proliferation was determined after 96 hrs. by measuring cellular ATP concentrations. (C) Immunoblot analysis of CHK1 levels in K562-dCas9-KRAB cells expressing the indicated sgRNAs. (D) Depletion of CHK1 potentiates AUR-mediated proliferation. Relative proliferation of K562-dCas9-KRAB cells stably expressing the indicated sgRNAs following treatment with 0.5 μM AUR was determined by measuring cellular ATP concentrations following 96 hrs. post treatment. (E) CHK1 inhibition potentiates AUR cytotoxicity. Proliferation of K562 cells treated with 0.5 μM CHK1i or 1 μM AUR was measured after 96 hrs. as described in (A). (F) Network analysis of functional targets for AUR and corresponding pathways and cellular locations. Data are represented as mean ± SEM. ***p< 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S4: Cysteine 408 in CHK1 functions as a nuclear H2O2 sensor, Related to Figure 3. (A) Left, Immunofluorescence analysis of K562 cells stably expressing the HyPer7 or roGFP2-ORP1 reporters in the nucleus, co-stained with Lamin A/C. Right, Immunofluorescence analysis of K562 cells stably expressing the HyPer7 or roGFP2-ORP1 reporters in the nucleus following H2O or 100 μM H2O2 treatment for 5 min. The ratiometric image was constructed by taking the ratio of the emission intensity of Oxidized (Ox) vs Reduced (Red) HyPer7 or roGFP2-ORP1. (B-C) Addition of NAC decreases nuclear H2O2 levels following AUR treatment. K562 cells expressing nuclear HyPer7 (B) or roGFP2-ORP1 (C) were treated with the indicated compounds for 6 hrs (5 mM NAC, 1.5 μM AUR) and the Ox/Red ratio of HyPer7/roGFP2-Orp1 was determined by flow cytometry (see also Figure 3C). (D) H2O2 treatment blocks alkylation of CHK1•C408. HEK-293T cells stably expressing CHK1 or CHK1•C408S were treated with 100 μM H2O2. IA-DTB modified CHK1 was determined by immunoblot following enrichment from cell lysates treated with a cysteine-specific IA-DTB. (E) Immunofluorescence analysis of FLAG-DAAO localized to nucleus in HEK-293T cells. (F) Characterization of CHK1 in vitro kinase assay. Immunoblot analysis of CHK1 in-vitro kinase assay following the addition of 5 μM CHK1 kinase inhibitor (CHK1i). (G) CHK1-C408D protects cells from AUR cytotoxicity. Relative proliferation of K562 cells stably expressing the indicated proteins was determined by measuring the changes in cellular ATP concentrations following treatment with 0.5 μM AUR for 96 hrs. (H) Left, immunofluorescence analysis of K562 cells stained with H2AX•S139 and DAPI. K562 cells stably expressing the indicated proteins were treated with 0.5 μM AUR for 24 hrs. Right, quantification of H2AX•S139 in the indicated samples. (I) CHK1 inhibition increases nuclear H2O2. K562 cells expressing nuclear HyPer7 (left) or roGFP2-ORP1 (right) were treated for 48 hrs. with 2 μM CHK1i. The Ox/Red ratio for each reporter was determined by flow cytometry. (J) Inactivation of CHK1 increases DCF fluorescence. Flow cytometry was used to monitor changes in DCF fluorescence in K562-dCas9-KRAB cells treated with CHK1i (2 μM) or expressing the indicated sgRNAs. (K) NAC treatment reverts CHK1-mediated nuclear H2O2 levels. K562 cells expressing nuclear localized HyPer7 were treated with 2 μM CHK1i and 5 mM NAC. Nuclear H2O2 levels were determined following 48 hrs. of treatment as described in (A). (L) Flow cytometry analysis of nuclear localized roGFP2-ORP1 (Ox/Red) ratio following inhibition of CHK1. K562 cells expressing nuclear roGFP2-ORP1 were treated as in (K) and analyzed as in (I). Scale bar=10 μm. Data are represented as mean ± SEM. *P < 0.05, **P < 0.001, ***P < 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S5: CHK1 regulates nuclear H2O2 levels through SSBP1 phosphorylation, related to Figure 5. (A-B) SSBP1 depletion rescues AUR and CHK1i cytotoxicity. K562 cells expressing the indicated sgRNAs were treated with 1.5 μM AUR (A), 1 μM CHK1i (B) or vehicle and proliferation was determined by measuring cellular ATP concentrations following 96 hrs. of treatment. (C-D) SSBP1 functions downstream of CHK1 to regulate DCF fluorescence. K562-dCas9-KRAB cells expressing the indicated sgRNAs were treated with CHK1i or vehicle control and analyzed as described above. (E) SSBP1•S67D phosphomimetic mutant decreases nuclear H2O2. Reintroduction of SSBP1-HA or SSBP1•S67D-HA into K562 cells depleted of SSBP1. Cells were treated with 1.5 μM AUR or vehicle and nuclear H2O2 levels were measured with nuclear localized HyPer7 (see also Figure 5I). (F) CHK1i blocks the phosphorylation of SSBP1. CHK1 in vitro kinase assay was conducted with SSBP1-HA and purified HA-CHK1 or HA-METAP2 (control protein). CHK1 was treated with 5 μM CHK1i and its phosphorylation of SSBP1 was monitored autoradiogram analysis. (G) Forced localization of SSBP1 to the nucleus. Immunofluorescence analysis of NLS-SSBP1 in K562 cells co-stained with Lamin. (H) SSBP1 localization is dependent on CHK1 activity. High-resolution spinning disk confocal microscopy immunofluorescence analysis of K562 cells expressing SSBP1-HA and co-stained with MitoTracker™ Red following the indicated treatments (see also Figures 5H–I). (I) The SSBP1 phospho-mimetic mutant is not localized to the mitochondria. High-resolution spinning disk confocal microscopy immunofluorescence analysis of K562 cells expressing the indicated SSBP1 WT or mutant proteins and co-stained with MitoTracker™ Red (see also Figure 5F). (J) The localization of SSBP1•S67A phosphodeficient mutant is not regulated by CHK1. Immunofluorescence analysis of K562 cells expressing SSBP1•S67A-HA following treatment with 1.5 μM AUR for 6 hrs. (K-L) The SSBP1•S67D mutant partial rescues AUR and CHK1i cytotoxicity. Cell lines described in (E) were treated with 1.5 μM AUR (K) or 1 μM CHK1i (L) and proliferation was determined as described in (A). Scale bar=10 μm. Data are represented as mean ± SEM.***P < 0.0001. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S6: Mitochondrial translation regulates nuclear H2O2 levels, related to Figure 6. (A) Left, immunofluorescence analysis of K562 cells stably expressing the HyPer7 reporters in the mitochondrial matrix, co-stained with MitoTracker™ Red. Right, immunofluorescence analysis of K562 cells stably expressing the HyPer7 in the mitochondrial matrix following H2O or 100 μM H2O2 treatment for 5 min. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized HyPer7 over reduced HyPer7. (B) CHK1 regulates mitochondrial superoxide in a SSBP1-dependent manner. Mitochondrial superoxide was determined by monitoring changes in MitoSOX™ fluorescence with flow cytometry in K562-dCas9-KRAB cells expressing the indicated sgRNAs and treated with 2 μM of CHK1i for 48 hrs. (C). Immunofluorescence analysis of FLAG-DAAO in K562 cells. (D-E) SSBP1 depletion decreases mtDNA content. (D) Immunofluorescence analysis of mtDNA content in K562-dCas9-KRAB cells expressing the indicated sgRNAs. (E) Quantification of mtDNA content following depletion of SSBP1. (F) SSBP1 depletion decreases expression of mitochondrially encoded genes. Immunoblot analysis of the indicated genes in K562-dCas9-KRAB genes expressing the indicated sgRNAs. (G) CHK1 inhibition increases the expression of mitochondrially translated proteins. Immunoblot analysis of the indicated proteins in K562 cells treated with 2 μM CHK1i or vehicle control for 72 hrs. (H) CHK1 regulates mitochondrially encoded proteins in a time-dependent manner. K562 cells were treated with 2.3 μM doxycycline (DOXY) for 72 hrs., at which point the media was exchanged and replaced with 2 μM CHK1i or vehicle control. MT-CO2 protein levels were determined by immunoblot (see also Figure 6H). (I) CHK1 functions upstream of mitochondrial translation to regulate nuclear H2O2 levels. K562 cells expressing nucleus localized HyPer7 were treated with 2.3 μM DOXY for 24 hrs. followed by 2 μM CHK1i for 48 hrs. The HyPer7 Ox/Red ratio was determined by flow cytometry. (J) CHK1 functions upstream of mitochondrial translation to regulate mitochondrial H2O2 levels. K562 cells expressing HyPer7 localized to the mitochondrial matrix were treated with 2.3 μM DOXY for 24 hrs. followed by 2 μM CHK1i for 48 hrs. Mitochondria H2O2 levels were determined as in (L). (K-N) Genetic or pharmacological inhibition of mitochondrial translation blunts CHK1i mediated DNA-damage. Immunofluorescence analysis of H2AX•S139 staining in K562-dCas9-KRAB cells expressing the indicated sgRNAs targeting SSBP1 (K, L) or treated with 2.3 μM doxycycline (DOXY) (M, N) were treated with 2 μM CHK1i for 48 hrs. (n= 60 cells counted per condition). (O) Doxycycline (DOXY) partially rescues AUR growth inhibition. K562 cells were co-treated with 2.3 μM DOXY and 1.5 μM AUR. Proliferation was determined by measuring relative ATP concentrations following 96 hrs. of treatment.. (P) SSBP1 depletion partially rescues the proliferation of select anticancer agents which increase nuclear H2O2 levels as determined in (O). Data are represented as mean ± SEM. ***P < 0.0001. Scale bar=10 μm. Statistical significance was determined by Student’s t-test (two-tailed, unpaired).
Figure S7: SSBP1 levels regulate platinum-based nuclear H2O2 and resistance in ovarian cancers, related to Figure 7. (A) GSH depletion sensitizes cells to anticancer drug treatments that increases nuclear H2O2. K562 cells were co-treated with 50 μM BSO and the indicated compounds (1 μM LAP, 2.5 μM DDP, 0.25 μM AUR, 0.5 μM 2ME, 1 μM 5FU). Proliferation was determined by measuring relative ATP concentrations following 96 hrs. of treatment. (B) The GSSG:GSH ratio increases following treatment with anticancer drugs that increase nuclear H2O2. K562 cells were treated with the indicated compounds (1.5 μM LAP, 8.3 μM DDP, 2.5 μM AUR, 1 μM 2ME or 6.0 μM 5FU) for 24 hrs., and the GSSG:GSH ratio was determined. (C) Left, immunofluorescence analysis of K562 cells stably expressing the GRX1-roGFP2-NES GSH:GSSG reporter localized to the cytosol, co-stained with MEK1/2. Right, immunofluorescence analysis of K562 cells stably expressing GRX1-roGFP2 in the cytosol matrix following H2O2 or 1 mM BSO treatment for 72 hrs. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized roGFP2 over reduced roGFP2. (D) Regulation of cytosolic GSH:GSSG by anticancer drugs. K562 cells stably expressing GRX1-roGFP2-NES were treated with the 1.5 μM LAP, 8.3 μM DDP, 2.5 μM AUR, 1 μM 2ME or 6.0 μM 5FU for 24 hrs. and cytosolic GSH:GSSG ratio was determined as described in (C). (E) Patients with lower SSBP1 expression have shorter duration to tumor recurrence following platinum-based chemotherapy. Left, high-grade serous ovarian patients were stratified based on SSBP1 mRNA expression. Clinical data were obtained from135,136. (F) NAC treatment decreases cisplatin cytotoxicity in ovarian cancer cell lines. Cell lines were co-treated with 5 mM NAC or vehicle control and the indicated concentrations of cisplatin. Relative proliferation was determined as described in (A). Heatmap depicts fold change in IC50 between treatments, normalized to saline. (G) Cisplatin increases steady-state levels of nuclear H2O2 in ovarian cancer cells, which can be rescued by NAC treatment. Cell lines expressing nuclear HyPer7 were co-treated with 5 mM NAC or vehicle control and 8.3 μM cisplatin for 24 hrs. The ratiometric image was constructed by taking the ratio of the emission intensity of oxidized HyPer7 over reduced HyPer7. (H) Depletion of SSBP1 in ovarian cancer cell lines decreases cisplatin regulated nuclear H2O2 levels. (I) Steady-state levels of nuclear H2O2 decrease following depletion of SSBP1. Relative nuclear H2O2 levels were measured in cell lines co-expressing nuclear HyPer7 and the indicated shRNAs by determining the Ox/Red HyPer7 ratio by flow cytometry. (J-K) Re-introduction of SSBP1 increases nuclear H2O2 levels following DPP treatment. Immunofluorescence analysis of OVCAR3 cells expressing nuclear HyPer7, the indicated shRNAs and shRNA-resistant SSBP1 or METAP2 (control)] (J). The ratiometric image was constructed as described in (C). Immunoblot analysis (K) of the indicated proteins in OVCAR3 co-expressing the indicated shRNAs and shRNA resistant SSBP1 or METAP2. (L) Immunoblot analysis of SSBP1 in ovarian cancer cell lines expressing the indicated shRNAs. Scale bar=10 μm. Data are represented as mean ± SEM. *P < 0.05, **P < 0.001, ***P < 0.0001. Statistical significance was determined by one-way ANOVA with Sidak’s post-hoc correction (E) and Student’s t-test (two-tailed, unpaired) (A, F, I).
Table S1. Description of anticancer drugs analyzed in this study, related to Figure 1.
Table S2. Iso-TMT analysis of cysteine reactivity changes following treatment with anti-cancer drugs and structural files used in this study, related to Figure 1.
Table S3. Co-essentiality clustering and annotated protein localizations, related to Figure 1.
Table S4. Genome-wide CRISPRi screen identifies pathways that mediate resistance and sensitivity to Auranofin, related to Figure 2.
Table S5. Target prioritization based on chemical proteomics and functional genomic analysis, related to Figure 2.
Table S6. Clinical synopsis of patients, related to Figure 7.
Table S7. Primers used in this study, related to STAR Methods.18–20,22,28,30,32,37,119–125,126–134
Data Availability Statement
Proteomics data have been deposited in PRIDE: PXD041138 and is publicly available.
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper is available from the lead contact.