

Abstract
Oncogene amplification on extrachromosomal DNA (ecDNA) is prevalent in human cancer and associated with poor patient outcomes. Clonal, megabase-sized circular ecDNAs in cancer are distinct from nonclonal, small sub-kilobase sized DNAs that may arise during normal tissue homeostasis. ecDNAs enable profound changes in gene regulation beyond copy number gains. An emerging principle of ecDNA regulation is the formation of ecDNA hubs, micron-sized nuclear structures of numerous copies of ecDNAs tethered by proteins in spatial proximity. ecDNA hubs enable cooperative and intermolecular sharing of DNA regulatory elements for potent and combinatorial gene activation. The 3D context of ecDNA shapes its gene expression potential, selection for clonal heterogeneity among ecDNAs, distribution through cell division, and reintegration into chromosomes. Technologies for studying gene regulation and structure of ecDNA are starting to answer long-held questions on the distinct rules governing cancer genes beyond chromsomes.
Introduction
The cancer genome undergoes extensive genetic alterations, often leading to dysregulation of gene expression. One important mechanism is amplification of oncogenes and drug resistance genes; this process increases the copies of genes that provide a selective advantage to cancer cells. While it has long been known that gene amplification can occur within or outside chromosomes, it is increasingly appreciated that the context and spatial architecture of gene amplification has an enormous impact on changes in gene expression which cannot solely be explained by gene copy number.
Major recent advances have been made regarding extrachromosomal DNA (ecDNA), a common mode of oncogene amplification. ecDNA was first observed in 1962 and described in detail by Cox et al. in 19651,2. Since then, ecDNA has been detected in nearly half of human cancer types, carrying oncogenes such as MYC, MYCN, EGFR, HER2 in tumor cell lines, patient-derived cell cultures and clinical tumor samples3–13. ecDNA can also integrate into chromosomes and therefore may act as an intermediate step toward stable chromosomal gene amplification in a subset of cases9,14–16. ecDNA is associated with poor patient outcomes even when compared to other forms of gene amplification5. These observations suggest that gene amplification in the context of ecDNA may have unique impacts on cellular programs that profoundly contribute to tumor pathogenesis and progression.
ecDNA refers to circular DNA molecules which are self-replicating, clonally selected and amplified, and range from 100 kilobases (kb) to several megabases (Mb) in size. They are selected in cancer cell populations as they provide a fitness advantage typically by carrying oncogenes and drug resistance genes. ecDNA was classically termed “double minutes”, which referred to paired, extrachromosomal chromatin bodies that can be microscopically observed on metaphase spreads. However, some extrachromosomal DNA molecules are submicroscopic and the majority of ecDNAs (~70–80%) appear as singletons on metaphase chromosome spreads rather than paired bodies3,4,16–18. Therefore, the classic “double minute” structure describes only a fraction of extrachromosomal DNA, leading to a shift towards the more inclusive term “ecDNA”. These large clonal ecDNAs found in cancer cells should not be confused with another class of smaller, nonclonal, extrachromosomal elements termed eccDNAs, or extrachromosomal circular DNA elements. While eccDNA is sometimes used as a broad umbrella term for circular DNA, it typically refers to DNA circles which are found in normal tissues or as byproducts of programmed cell death19,20, are not amplified or selected, are only up to ~10 kb in size, usually do not carry genic or regulatory sequences, and can span any part of the genome20–22. Given the similarity in nomenclature despite stark differences in sequence, function, behavior, and cell types in which the DNA elements are observed, we have constructed a summary table here to clarify the main distinctions (Table 1). This Review focuses on large clonal ecDNAs observed across many cancer types.
Table 1.
Differences between ecDNA and eccDNA.
| ecDNA | eccDNA |
|---|---|
| 100 kb to several megabases | Several hundred to thousand bases19,22 |
| Circular | Circular |
| Self-replicating | Not known to replicate |
| Clonally selected | Typically not amplified or selected |
| Contains oncogenes, drug resistance genes, or other genes that provide a selective advantage3–13,28,101 | Sequences cover the entire genome with some hotspots; typically does not contain genic sequences19 |
| Contains regulatory sequences such as oncogene enhancers24,46,48,53 | Usually does not contain regulatory sequences |
| Found in cancer cells | Found in many cell lines and tissues, including healthy samples20,22,102–104 |
| Can contain heavily recombined genomic sequences and specific mutations49,87 | Not known to carry mutations |
Genes encoded by ecDNA are expressed at much higher levels compared to the native chromosomal locus and linear gene amplicons, even when normalized for copy number4,5,23. In addition to copy number amplification, ecDNA shows unique structural, genetic and epigenetic features that are conducive to gene activation, suggesting that there are fundamental differences in how amplified genes are regulated on ecDNA. Given the observation that ecDNA is prevalent in cancer, is a key form of oncogene amplification, and is linked to poor clinical outcomes, there is a need for a better understanding of how genes are regulated on ecDNA. In this Review, we highlight aspects of regulation of gene expression on ecDNA which differ from chromosomal DNA. These unique ecDNA features are linked to alterations in structure, sequence, chromatin composition, and contacts with DNA regulatory elements.
ecDNA enables high levels of oncogene transcription
Copy number amplification.
ecDNA is associated with increased oncogene expression compared to linear amplifications as well as the native chromosomal locus4,5,23,24. This is partly driven by gene copy number amplification4,5. As ecDNAs lack centromeres, they are distributed randomly among daughter nuclei during cell division25–27 (Figure 1a). This random segregation results in copy number heterogeneity and selection of cells carrying ecDNAs that provide a fitness advantage (Figure 1b). This characteristic of extrachromosomal oncogene amplification can lead to up to several hundreds of ecDNAs in a single cell4,15,24 and has been linked to rapid adaptation to selective pressures and development of therapeutic resistance4,27–29. However, copy number alone does not fully explain the high level of oncogene expression observed in ecDNA+ cancer cells; patient tumors containing circular amplicons express amplified genes more highly than those containing linear or other types of rearranged amplicons even with copy number normalization5. This observation suggests that there are additional mechanisms that overexpress oncogenes on ecDNA in ways distinct from chromosomal regulation of gene expression.
Figure 1. Unique characteristics of ecDNA.
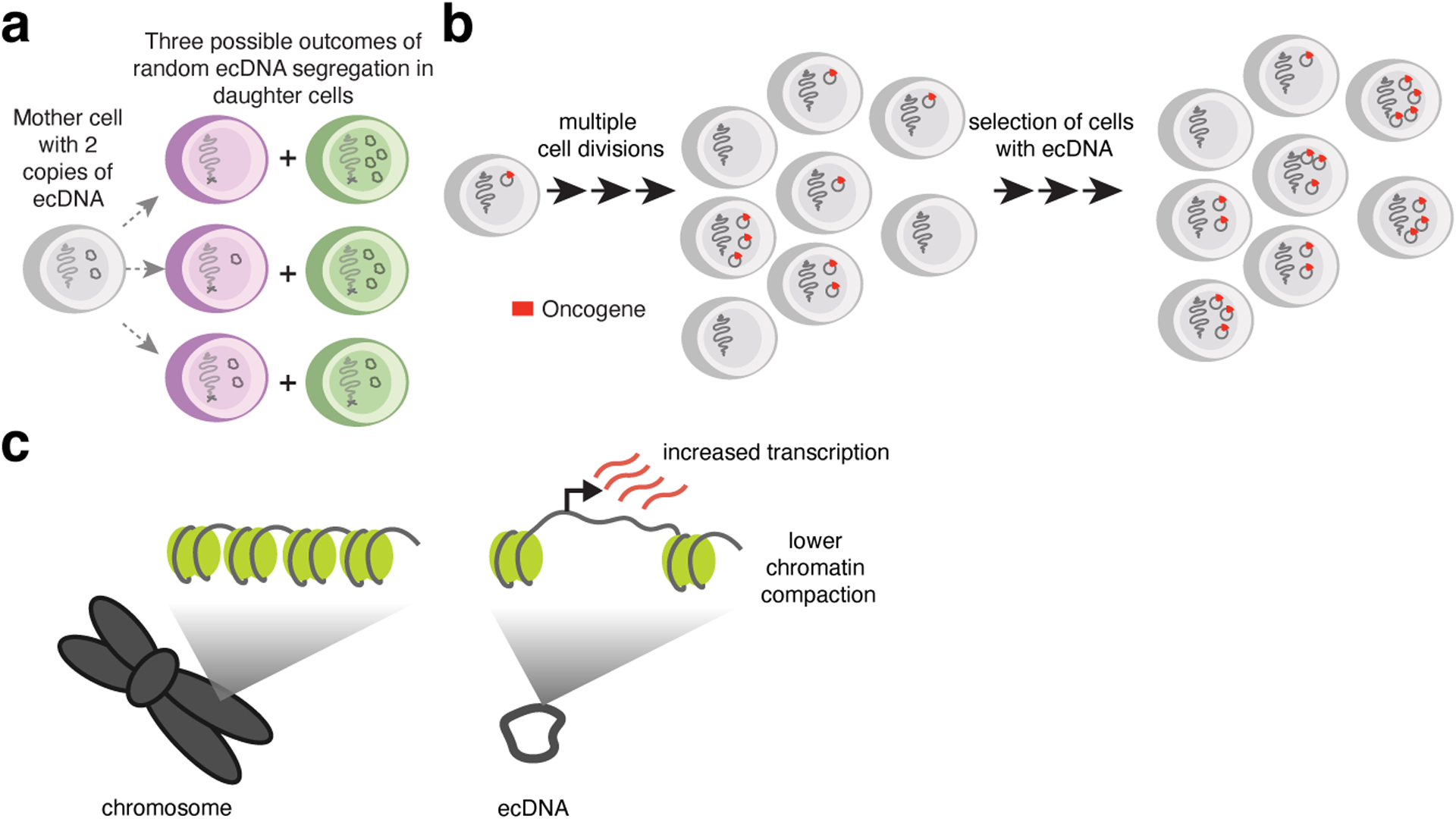
(a) Random segregation of ecDNA molecules during cell division allows multiple possible outcomes in the number of gene copies inherited. (b) Cancer cells containing ecDNAs, after undergoing multiple cell divisions with random ecDNA segregation, can have a wide range of copy numbers. ecDNAs that provide a fitness advantage to cancer cells can be selected. (c) ecDNAs have lower chromatin compaction and increased transcription.
Oncogene overexpression.
In addition to increased copy numbers, ecDNAs are associated with increased transcriptional activity and highly accessible chromatin5,23,24 (Figure 1c). ecDNAs lack higher-order compaction via depletion of large nucleosome arrays, which may allow binding by transcription factors and access to gene loci by the transcriptional machinery. There appear to be multiple mechanisms causing this oncogene overexpression. First, ecDNA molecules physically cluster with one another in the nucleus and engage in intermolecular, combinatorial enhancer-promoter interactions. Second, the circular structure of ecDNA is a stable, covalently closed structure allowing increased chromatin cis-interactions compared to chromosomes. Third, extensive genome sequence rearrangements alter the regulatory context of gene loci. This is an area of active research and there are likely additional mechanisms driving oncogene overexpression. It also remains an open question whether the ecDNA characteristics which enable oncogene overexpression are consequences of selection for transcriptionally active molecules or inherent differences between circular extrachromosomal chromatin and native chromosomes. These mechanisms driving oncogene overexpression are described in detail in the following sections of this Review.
ecDNA hubs: a new nuclear structure for intermolecular gene activation
The three-dimensional structure of mammalian chromosomes is organized at various length scales: chromosome territories, compartments A and B, topologically associating domains (TADs), and long-range enhancer-promoter interactions spanning tens to hundreds of kilobases30,31. On the finer scale, chromatin interactions, such as those occurring between enhancers and target genes, are most often found within TADs on the same DNA molecule (cis-interactions). While interchromosomal interactions have been documented, they represent exceptions rather than the norm32,33 (reviewed by Maass et al.34). In contrast, a cancer cell can have up to hundreds of ecDNA copies in the nucleus, raising the possibility that multiple ecDNA copies can interact with one another fostering new cooperative interactions.
Rather than being randomly scattered around, ecDNA molecules cluster with one another to form micron-sized hubs in the interphase nucleus24,35. ecDNA hubs, or “extrasomes”, represent a counterpart to chromosomes as units of genetic information and organization (Table 2). Chromosomes have linear arrays of genes and permit gene activation by DNA regulatory elements on the same DNA molecule. In contrast, ecDNA hubs permit intermolecular gene activation of combinatorial enhancers and promoter elements in spatial proximity24. Chromosomes are dispersed during interphase of the cell cycle and condense ~10,000-fold during mitosis. ecDNA hubs coalesce during interphase but disperse during mitosis24. These fundamental differences distinguish DNA in the form of chromosomes versus ecDNA hubs in the same nucleus.
Table 2.
Organization of ecDNA hubs and chromosomes.
| ecDNA hubs (“Extrasomes”) | Chromosomes |
|---|---|
| 10–100s of ecDNAs in proximity and interact with one another; tethered by proteins | Endogenous gene loci covalently linked; one long piece of DNA per chromosome |
| Regulatory elements such as enhancers interact with target genes in cis and in trans | Regulatory elements primarily interact with genes in cis within the same TAD |
| ecDNA hubs coalesce during interphase but can dynamically break into smaller clusters of ecDNAs during mitosis | Chromosomes are dispersed during interphase and condense during mitosis |
Previous studies also reported preferential localization of these ecDNA clusters at the nuclear periphery during G1 phase and M phase, although the significance and mechanism of this localization pattern is not well understood36. As the nuclear periphery is a transcriptionally repressive environment while ecDNAs are highly transcriptionally active37, their peripheral localization is counterintuitive and warrants further investigation into its generalizability and potential functional significance. ecDNA hubs are observed during mitosis with dynamic changes in size, suggesting that these clusters are not stable during DNA partitioning24,25,38. Finally, double-strand DNA breaks in ecDNA have also been associated with aggregation of ecDNA molecules and formation of chromosomal tandem amplifications termed homogeneously staining regions (HSRs)39, suggesting that ecDNA clustering may explain the formation of some chromosomal amplifications as well.
ecDNA hubs drive intermolecular oncogene activation.
Formation of nuclear ecDNA hubs is linked proportionally to the rate of oncogene transcription from each ecDNA molecule24,35 (Figure 2a). ecDNA hubs bring 10–100 ecDNAs into proximity and enable intermolecular enhancer-promoter interactions, increasing the level of combinatorial enhancer input to oncogenes24 (Figure 2b). Whereas genes on chromosomes are activated by enhancer and regulatory DNA elements on the same chromosome, ecDNA molecules can promiscuously engage enhancers on other ecDNAs within the spatial proximity of an ecDNA hub. Even two distinct ecDNAs derived orginally from two different chromosomes can cross activate each other via enhancer-promoter contacts24. The ability to enact intermolecular gene activation appears to be a bright dividing line between normal cellular physiology and cancer cells harboring ecDNA.
Figure 2. ecDNA hubs drive oncogene expression and may shape cancer cell evolution.

(a) ecDNA clustering in hubs is associated with oncogene transcription. (b) Enhancer-promoter contacts in trans within ecDNA hubs enable combinatorial interactions. (c) Enhancers and oncogenes may be co-selected on two levels. First, enhancer-oncogene pairs on the same molecule which drive oncogene expression can be selected together. Second, distinct ecDNA molecules containing enhancers and oncogenes that interact intermolecularly may be co-selected. (d) Dynamic ecDNA hub transcriptional activity is linked to heterogeneous oncogene expression levels in a cell population. (e) ecDNA hubs may allow correlated integration of multiple amplicon copies into chromosomes to form HSRs. (f) ecDNA-chromosome contacts allow enhancers on ecDNAs to interact with chromosomal genes and activate transcription.
Potential mechanisms of ecDNA hub formation.
Small transient transcriptional hubs are necessary for gene transcription40, and we speculate that ecDNA hubs are a kind of transcriptional hub mediated by protein-protein interactions which may be oncogene specific. This is supported by dispersal of MYC ecDNA hubs via inhibition of the bromodomain and extraterminal (BET) proteins in a colorectal cancer cell line24. MYC ecDNA hubs are not disrupted by transcriptional inhibition with alpha-amanitin nor by 1,6-hexanediol24, suggesting that ecDNA hubs do not depend on RNA polymerase II or specific interactions between intrinsically disordered regions (IDRs) which are sensitive to hexanediol such as Mediator 141. As BET proteins can normally concentrate accessible DNA, exclude heterochromatin, and mediate long-range enhancer-promoter communication42,43, it is possible that ecDNA hubs may coopt endogenous mechanisms of long-range gene looping within chromosomes to promote intermolecular chromatin interactions in ecDNA ensembles. Other than MYC which is regulated by BET proteins44,45, this model predicts that other oncogenes amplified on ecDNA may also exploit their endogenous enhancer mechanisms to operate in ecDNA hubs. As functional enhancers are co-selected with EGFR on ecDNAs in glioblastoma46, we speculate that proteins that mediate endogenous enhancer-EGFR interactions could be involved in ecDNA hub maintenance as well.
Dispersal of ecDNA hubs was associated with reduced oncogene expression in ecDNA-containing cells, suggesting that ecDNA hubs may be a vulnerability of these oncogene-addicted cells. This observation also implies that ecDNAs may depend on unique transcriptional regulators, warranting further investigation of distinct or common regulators of ecDNA hub formation and transcription across cancer cells with various ecDNA amplicons. As long noncoding RNAs (lncRNAs) are involved in the formation of interchromosomal interactions34, additional studies may address whether lncRNAs play a role in ecDNA hub formation. Finally, it is still an open question whether ecDNA hubs inhabit specific territories in the nucleus in relation to other chromosomes. Chromosome territories are non-randomly distributed in the nucleus and can even be conserved across different species, suggesting functional importance of the radial organization of chromosomes in the nucleus31,47. The radial, sub-nuclear arrangement of ecDNA hubs in relative to chromosome territories may provide novel insights into ecDNA functions.
Implications of ecDNA hubs for evolution of oncogene diversification, cooperation, and ecDNA reintegration
Given that ecDNA has been separated from the 3D genomic context of its chromosomal origin, it has been proposed that the co-selection of oncogenes and enhancers shapes ecDNA amplicon structures46,48. With the observation of intermolecular interactions among ecDNAs carrying distinct enhancer elements, we propose a two-level model for oncogene-enhancer co-selection (Figure 2c). The first level of co-selection occurs to individual ecDNAs; molecules that possess functional enhancers can promote oncogene expression and provide better fitness to cancer cells compared to ones that do not. The second level of oncogene-enhancer co-selection occurs to the repertoire of ecDNAs in hubs. We predict that each ecDNA molecule does not need to contain the full set of enhancer elements for oncogene activation; rather, they exist as part of an ecDNA hub that facilitates chromatin interactions among a diverse repertoire of regulatory elements and promotes interactions between the target oncogene and functional enhancers, which may be located on distinct molecules. This model raises the intriguing concept that winning the clonal competition among cancer cells occurs through clonal cooperation among ecDNA molecules. Furthermore, the presence of functional regulatory elements in a cooperative ecDNA hub may increase tolerance of mutations on individual molecules. Others have previously reported ecDNA mutational diversity and rapid response to environmental changes49, though further investigation is needed to measure mutational diversity in functional enhancers on ecDNAs.
ecDNA hubs are also associated with variable enhancer usage and cancer cell heterogeneity in oncogene activity24 (Figure 2d). This may be attributed to differential enhancer-promoter interactions which occur in the context of ecDNA hubs. As dozens of ecDNA molecules can cluster together in many possible spatial configurations, this may provide an opportunity for ectopic enhancer-promoter interactions which do not normally occur on linear chromosomes. We speculate that these differential interactions contribute to the highly variable enhancer activities and enhancer rewiring on ecDNAs. Furthermore, we speculate that ecDNAs markedly extend the concept of cancer genetic heterogeneity, as tumor cells can contain ecDNAs with diverse sequences and different oncogenes and regulatory sequences, which can interact with each other and even potentially combine into larger, single circular elements. The potential for driving diversity and accelerated evolution is remarkable.
ecDNA-chromosome interactions.
The formation of ecDNA hubs may provide a palatable explanation for the well-known tendency of a subset of ecDNA+ cancer cells to develop homogeneously staining regions (HSRs, a type of tandem amplifications on chromosomes). Double-strand breaks in ecDNAs can trigger aggregation, micronucleus formation, and reintegration into chromosomal HSRs39. ecDNAs that are spatially proximal in hubs could enable correlated DNA breaks50 and concentrated DNA cargo, creating a potential set up for HSR formation (Figure 2e). Hub formation may also impact ecDNA segregation. Previous work has demonstrated that ecDNAs are transmitted into daughter cells in clusters during mitosis38. Future studies may address whether an ecDNA hub serves as a unit of inheritance or merely as a transient congregation. Other cellular processes, such as DNA repair and replication, are also regulated by genome organization51,52. For example, replication units, or replicons, form clusters in which replication origins fire synchronously51. Furthermore, genomic loci located in the nuclear interior contain early-replicating domains, while the nuclear periphery is associated with late-replicating domains51. Therefore, the observation of ecDNA hubs warrants further investigation into whether this unusual 3D organization of DNA molecules impacts these cellular processes.
In addition to regulatory interactions among ecDNAs, ecDNAs also interact with specific sites within chromosomes53 (Figure 2f). These sites of ecDNA-chromosome interactions are associated with increased transcriptional activity and active histone H3K27 acetylation (H3K27ac) marks, suggesting that these may be functional regulatory interactions involving ecDNA enhancers transcriptionally active regions in chromosomes53. Therefore, in addition to amplifying oncogenes, ecDNAs may also act as “mobile enhancer carriers” that modulate chromosomal gene expression. Further investigation may address how these ecDNA-chromosome interactions affect cancer cell fitness.
Genetic basis of regulatory rewiring on ecDNA
Genetic alterations and rearrangements on ecDNAs are frequently observed5,24,48,54–58. As enhancer-promoter interactions are sensitive to physical distance between regulatory elements, sequence rearrangements can alter this physical distance and subsequently influence enhancer-promoter circuitry, leading to dysregulation of gene expression59–62. As a platform for genomic rearrangements, ecDNAs frequently contains sequences originating from the same locus as the amplified-oncogene, as well as sequences originating from distal chromosomal regions or other chromosomes of origin24,48,54–58. These rearrangements enable co-amplification of cognate as well as ectopic enhancers with oncogenes46,48. Sequence rearrangements can also result in fusion gene transcripts, leading to dysregulation of gene expression via promoter hijacking24,63. As ecDNA is associated with accelerated adaptation and selection, molecules with increased transcriptional activity due to regulatory rewiring can be selected by providing a fitness advantage to cancer cells harboring these molecules.
Enhancer hijacking.
ecDNAs, while containing sequences that originated from chromosomes, are uncoupled from their chromosomal loci of origin and the regulatory context thereof. Importantly, regulation of gene expression is tightly connected to non-coding regulatory elements, such as enhancers, which physically interact with target genes. Consistent with this idea, ecDNAs containing oncogenes typically co-amplify enhancer elements which upregulate oncogene expression46,48 (Figure 2c). Unlike the native chromosomal locus, ecDNAs containing sequence rearrangements can create new regulatory circuitry between ectopic enhancers and oncogene promoters46,48 (Figure 3a). These novel regulatory interactions include enhancers which are normally insulated from the target oncogene on the linear chromosome, or those which are normally located in distal chromosomal regions46,48. This adoption of novel enhancers is termed enhancer hijacking48,64–66. As ecDNA amplicon structures can vary greatly among tumor samples, including different amplified sequences and different structural rearrangements, there is likely a high level of diversity in enhancer-promoter circuitry among different tumors with ecDNA amplifications of a given oncogene.
Figure 3. Genetic and structural basis of the regulatory circuitry on ecDNA.
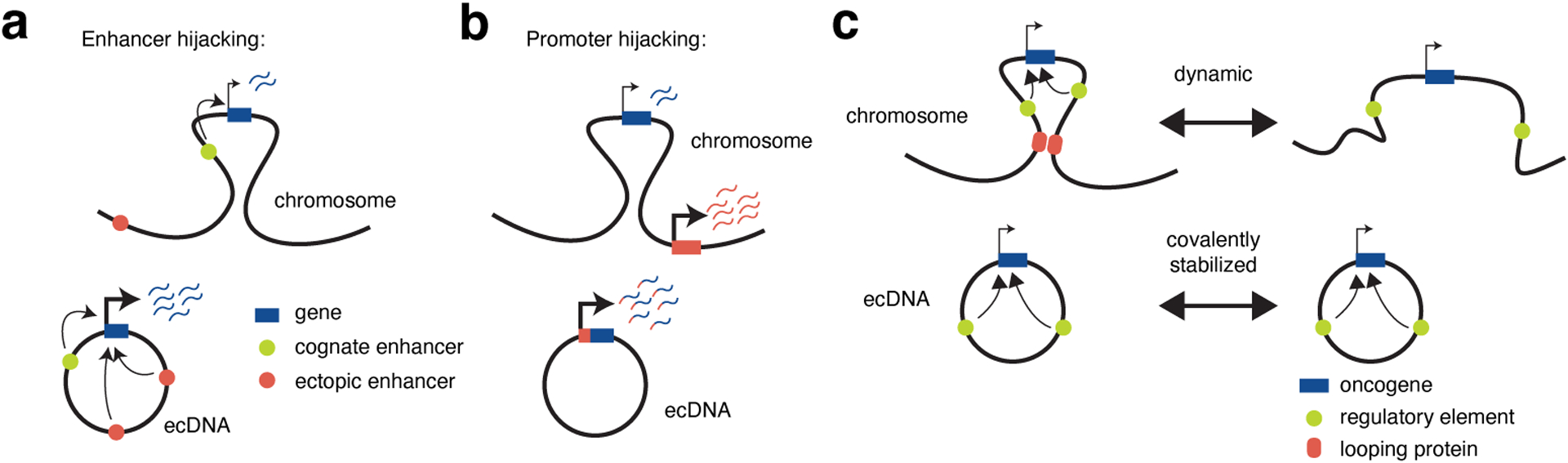
(a) Enhancer hijacking on ecDNA leads to ectopic enhancer-gene interactions by bringing distal enhancers into proximity via sequence rearrangement. (b) Promoter hijacking drives gene expression by structural rearrangement on ecDNA. (c) The circular structure of ecDNA brings regulatory elements into spatial proximity and increases their interactions, as compared to the more variable and dynamic loop structure of a chromosomal TAD.
Promoter hijacking.
Sequence rearrangements on ecDNA can also lead to gene fusions, resulting in upregulation of oncogenes via hijacking of highly active promoters (Figure 3b). An example of promoter hijacking was observed for the MYC oncogene on ecDNAs24,57,67. MYC is located ~50 kb upstream of the long non-coding RNA (lncRNA) gene PVT1, which negatively regulates MYC expression via enhancer competition with the MYC promoter normally68. Fusion of the PVT1 promoter with the coding sequence of MYC is proposed to overcome this negative regulatory feedback loop by direct linkage of the PVT1 promoter activation with MYC transcription69. PVT1-MYC fusion has been reported in multiple cancer types including breast, colon, ovary, esophagus cancers and medulloblastomas57,69–72. This rearrangement can be observed on ecDNAs24,67 and is generally associated with focal copy number amplifications57,69–72. PVT1-MYC fusion was observed in 60% of select MYC-amplified cases of Group 3 medulloblastomas69, a cancer subtype that is 18% MYC ecDNA+73. Therefore, PVT1-MYC fusion potentially accounts for a significant portion of medulloblastoma cases with ecDNA amplifications, although further systematic analyses are needed to provide more insight into how frequently promoter hijacking events are observed in the context of ecDNAs in various tumor types. A recent study showed viral-human hybrid ecDNAs containing sequences of human and human papillomaviral (HPV) origins in ~20% (6 out of 28) of HPV oropharyngeal cancer samples, including amplicons that enable high levels of viral-human fusion gene transcription driven by HPV promoters63. These observations hint at promoter hijacking as a powerful mechanism for upregulating gene expression on ecDNAs.
Ectopic self-associating chromatin domain.
Most of the genome is organized into TADs, which are self-associating chromatin domains on the scale of 100 kilobases to over 1 Mb74–78. DNA elements within a chromosomal TAD are in contact at much higher frequencies, enabling cis-interactions between regulatory elements including enhancer elements and gene promoters. These enhancer-promoter cis-interactions activate gene transcription and control cellular programs. Typically several hundred kilobases to several megabases, ecDNAs are on a similar length scale as chromosomal TADs. Nevertheless, ecDNAs differ from chromosomal TADs in two major ways. First, ecDNAs are circular as demonstrated by electron microscopy and pulsed field gel electrophoresis23,79–82. Therefore, In contrast to chromosomal TADs, ecDNAs are covalently closed rather than stabilized by looping proteins such as cohesin and CTCF. Thus, while TADs are variable from cell to cell83–85, the covalent circular structure of ecDNA is stable (Figure 3c). Consistent with this idea, gene loci and regulatory elements on a circular ecDNA interact with each other with much higher frequencies than corresponding loci on chromosomes23,53. Second, while chromosomal TADs are relatively conserved across cell types74,86, ecDNA amplicons can have variable boundaries and sequence arrangements in individual tumors, allowing non-interacting loci in the native chromosomal locus to interact with each other ectopically on the circle23,46,48 (Figure 3a). Therefore, by bringing regulatory elements into proximity on the circular structure, ecDNAs effectively serve as a type of covalently closed, ectopic “TAD”. This circular structure also serves as the basis for enhancer hijacking via incorporation of distal enhancer elements into the same DNA circle as the target gene as described above.
Methods for studying gene regulation on ecDNA
ecDNAs containing oncogene amplifications arise from chromosomes originally and, therefore, comprise sequences that heavily overlap with chromosomal DNA. As such, bulk sequencing data typically do not provide specificity to ecDNAs over the native chromosomal loci containing the corresponding genes and must therefore be carefully interpreted. Imaging-based methods can provide good separation of chromosomal and ecDNA signals depending on the approach, but suffer from limited throughput and DNA sequence resolution. We have recently demonstrated a strategy for isolating ecDNAs and separating amplicons by size, providing empirical evidence for ecDNA structures and enabling targeted profiling of their genetic sequences as well as epigenomic landscapes87.
Imaging-based methods.
ecDNAs were originally discovered by imaging of metaphase chromosome spreads from cancer cells1. Cells arrested in metaphase are fixed and condensed mitotic chromosomes are physically spread out on a microscope slide, providing excellent separation of chromosomes and ecDNAs. Metaphase spreading followed by DNA fluorescence in-situ hybridization (FISH) further allows hybridization of a sequence-specific probe and detection of oncogenes on ecDNA (Figure 4a). This method typically requires cell culturing followed by metaphase arrest. DNA FISH can also be performed on cells in interphase without arrest (Figure 4a), allowing detection of oncogene amplifications in clinical tumor sections. Interphase DNA FISH provides information about the spatial distribution of ecDNAs in intact cancer cell nuclei and led to the discovery of ecDNA hubs24. Interphase DNA FISH combined with nascent RNA FISH (using probes that target intronic sequences on pre-messenger RNA) also allows single-molecule assessment of transcriptional activity on ecDNAs24. However, interphase FISH alone does not identify ecDNA presence definitively and, therefore, should be used in combination with other methods such as metaphase FISH. Finally, live cell imaging has been used to visualize ecDNA dynamics in live cancer cells during interphase and mitosis24,27,35 (Figure 4a).
Figure 4. Technologies used to reveal ecDNA gene regulation and structure.
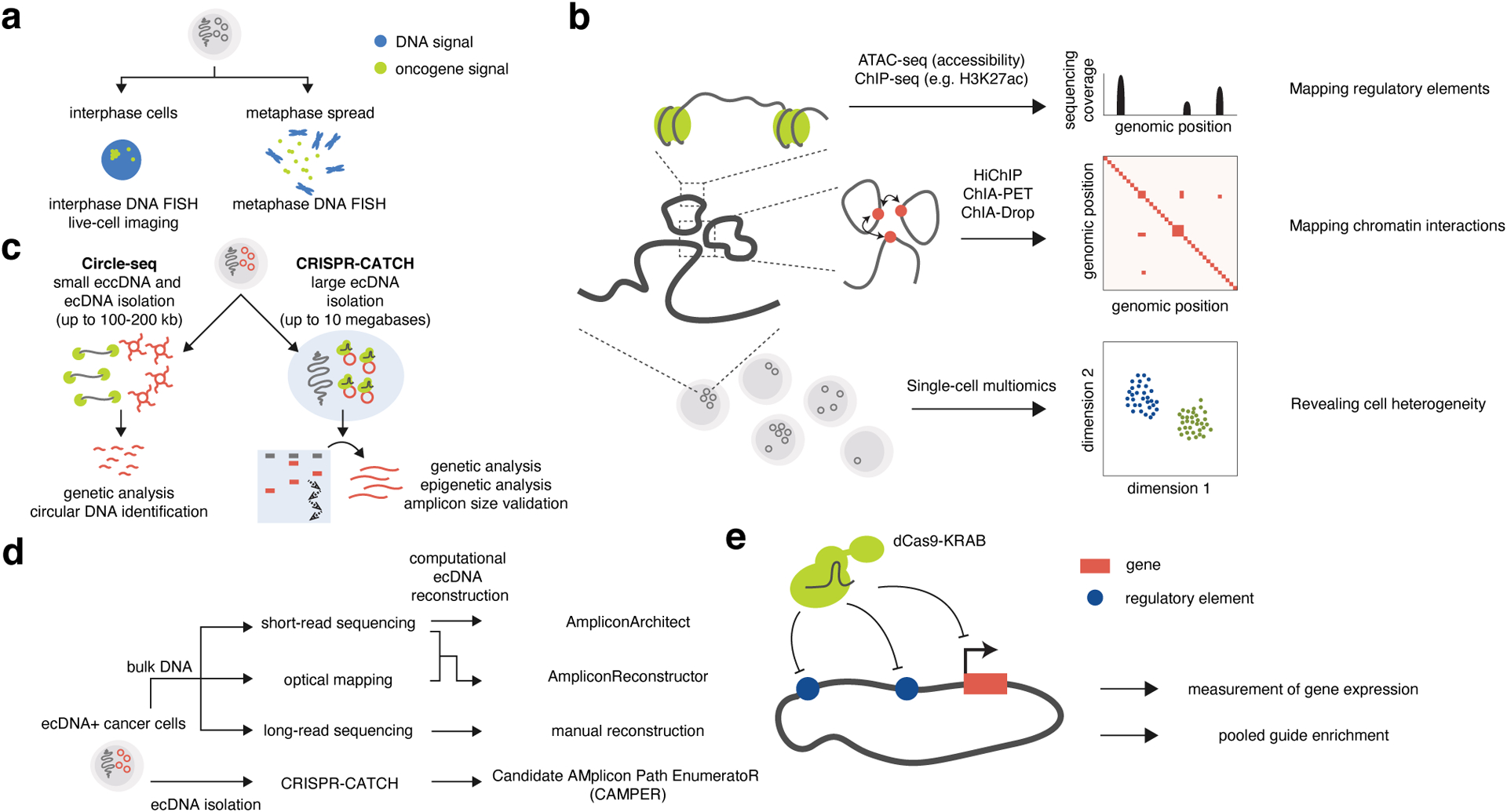
(a) Imaging of ecDNA in interphase cells or metaphase spreads. (b) Bulk and single-cell sequencing approaches to identify regulatory elements, map chromatin interactions and reveal cell heterogeneity in the context of ecDNA. (c) Isolation of ecDNA and targeted analyses. (d) Computational methods and tools for reconstruction of ecDNA sequence. (e) CRISPR interference of gene promoters and regulatory elements on ecDNA.
Bulk sequencing-based methods.
High-throughput sequencing has enabled detailed characterization of the cancer genome and epigenome and have provided novel insights into the spatial and structural bases of regulation of gene expression on ecDNA. Some examples are (1) the mapping of DNA regulatory elements using assay for transposase accessible chromatin with sequencing (ATAC-seq)88 and chromatin immunoprecipitation sequencing (ChIP-seq) targeting markers of regulatory elements such as H3K27ac89; (2) the identification of chromatin interactions associated with spatial organization involving regulatory elements using HiChIP90,91, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET)92, or chromatin interaction analysis with droplet sequencing (ChIA-Drop)93; (3) the fine-scale assessment of variable regulatory element activities at the single-cell level using single-cell multiomics, allowing simultaneous measurements of RNA expression and chromatin accessibility94–96 (Figure 4b). These various methods have been applied to studying gene regulation on ecDNAs23,24,46,48,53. Sequencing signals represent all DNA material in the samples, including both chromosomal DNA and ecDNA. In cancer cells containing high copy numbers of ecDNA, the majority of sequencing signals is assumed to originate from ecDNA molecules. However, ecDNA-focused interpretations of bulk sequencing data are more challenging when ecDNA molecules are present at lower copy numbers or in a small subset of cells in a heterogeneous cell population. Furthermore, ecDNAs typically contain extensive structural rearrangements and can be a mixture of heterogeneous amplicons containing various sequence elements within a cancer cell population5,24,48,54–58,87, thus altering the 2D distances between loci as compared to the chromosomal reference sequence. Therefore, interpretations of chromatin regulatory interactions in relation to gene loci can benefit from construction of custom rearranged ecDNA sequence maps in addition to alignment to the reference genome.
Isolation of ecDNA.
To better profile the genetic and epigenetic landscapes of ecDNAs, there is a need for new molecular methods to isolate ecDNAs from cancer cells for targeted profiling and comparisons between ecDNAs and chromosomes. A technique for unbiased isolation of DNA circles, termed Circle-seq, was previously developed for small eccDNA molecules19–21 and recently applied to oncogenic ecDNAs97 (Figure 4c). Circle-seq involves magnetic bead-based genomic DNA isolation, exonuclease digestion of linear DNA fragments followed by multiple displacement amplification (MDA) of remaining DNA. Application of this method on neuroblastoma samples enabled analysis of structural rearrangements on ecDNAs97. However, as megabase-sized ecDNAs are extremely fragile in solution and prone to breakage, Circle-seq favors small ecDNA and eccDNA species. In-solution DNA isolation is not recommended for DNA molecules that are above 100 kb in size.
To isolate ecDNAs which are megabases in size as are commonly observed in cancer cells, a method termed CRISPR-CATCH (CRISPR-Cas9-Assisted Targeting of CHromosome segments) can be used87,98 (Figure 4c). In this method, ultra high molecular weight genomic DNA is embedded in agarose to maintain DNA integrity87,99. Following agarose entrapment of genomic DNA, CRISPR-Cas9 ribonucleoprotein is used to cleave ecDNA circles in vitro and the resulting DNA is separated in pulsed field gel electrophoresis (PFGE). As linearized ecDNAs represent specific amplicon sizes, they are separated by size in PFGE and extracted from the gel. CRISPR-CATCH allows targeted profiling of the ecDNA genetic sequence and epigenomic landscape. This method serves as an empirical approach for heterogeneous amplicon separation and sequence reconstruction. It also enables physical separation of chromosomal DNA and ecDNA from the same cell sample, allowing direct comparisons. However, this targeted approach can only enrich for ecDNAs containing the CRISPR-Cas9 target sequence (e.g. a known oncogene) and therefore cannot be used as a method for unbiased ecDNA detection.
Computational inference of ecDNA structure.
In parallel to molecular techniques for ecDNA isolation, computational tools can infer ecDNA amplicon structures from bulk sequencing data (Figure 4d). Whole-genome short-read sequencing data can be analyzed by AmpliconArchitect, which constructs breakpoint graphs based on structural rearrangements detected and infer amplicon structures54. Sequence information from long DNA molecules can be used to provide more accurately phased structural arrangements. Such information can be collected using long-read sequencing or optical mapping (OM)100, both of which have been applied to ecDNA amplicons to resolve complex structures23,24,48. AmpliconReconstructor integrates short-read sequencing data and OM data for accurate reconstruction of ecDNA amplicons55. Finally, ecDNA sequence maps can be orthogonally constructed using sequence data from CRISPR-CATCH87 (Figure 4d). Analysis of ecDNA amplicon structures in the context of regulatory elements may provide a better picture of how regulatory circuitry can be rewired by structural rearrangements on ecDNA, including enhancer and promoter hijacking events described earlier.
Functional interrogation via CRISPR interference.
To interrogate the regulation of gene expression by perturbation, CRISPR interference (CRISPRi) has been used to both directly target gene promoters on ecDNA as well as target non-coding regulatory DNA elements like enhancers23,24,46 (Figure 4e). Due to elevated ecDNA copy numbers, Cas9-mediated DNA cleavage can lead to many double-strand breaks and is likely to come with unintended effects caused by DNA damage response as well as chromosomal integration of ecDNAs58. On the other hand, CRISPRi can effectively silence ecDNA promoters despite high copy numbers, though effects of enhancer targeting appear diminished potentially due to combinatorial enhancer-gene interactions and compensation by other enhancers within ecDNA hubs24. Nonetheless, enhancer effects on cell survival and oncogene expression can be detected in more sensitive, pooled assays24,46. Pooled perturbation of enhancers by CRISPRi has identified cognate as well as ectopic oncogene enhancers that upregulate oncogene expression on ecDNAs and improve ecDNA+ cancer cell survival. These studies demonstrated an enhancer hijacking mechanism as well as intermolecular cooperativity in ecDNA hubs, two driving forces of ecDNA evolution and oncogene upregulation24,46.
Conclusions
While ecDNAs have been long known to be an important mechanism of oncogene amplification in cancer, their impacts on cancer development, progression, evolution, and drug resistance are only beginning to be appreciated. With recent advances in sequencing technologies, we are now able to obtain detailed information about alterations in the cancer genome including oncogene amplifications, as well as consequences in gene expression programs. While mapping sequencing data of cancer cells to the reference sequence shows amplified oncogene sequences and structural rearrangements, it obscures differences in the spatial distribution of these oncogene amplifications. With the renewed interest in extrachromosomal oncogene amplification, we are now equipped to study a wide range of cancer biology questions in the context of ecDNA, including how gene expression is regulated.
As collective attention shifts from a sequence-oriented view to interrogating molecular topology, architecture, and structural organization in cancer, ecDNA has emerged as a challenge at the interface of cancer genetics and epigenetics. ecDNA also poses exciting new challenges for our understanding of tumor evolution. As described, the concept of ecDNA hubs and intermolecular combinatorial interactions opens a new paradigm in understanding how physical architecture regulates gene regulation, as the fundamental unit of transcription shifts from the gene to the hub.
Future development.
The discoveries of ecDNA hubs and regulatory rewiring on ecDNAs have provided a strong basis for the identification of unique aspects of transcriptional regulation of oncogenes not previously appreciated. These unique regulatory mechanisms on ecDNAs may point to unique oncogene transcriptional dependencies of ecDNA+ cancer cells. Future studies dissecting these regulatory mechanisms may provide novel insights into potential vulnerabilities of ecDNA-driven and oncogene-addicted cancers. Genome-wide genetic screens as well as small-molecule screens with a focus on ecDNA+ cancer models may identify these potential vulnerabilities. The observation that ecDNA hubs drive extrachromosomal and chromosomal gene expression warrants further investigation of the their significance in in vivo models as well as models from various cancer types. These future investigations will address whether ecDNA hubs are universal, cancer type- or oncogene-specific. In addition, Large-scale studies tracking extrachromosomal oncogene contents within primary tumors as well as metastases in cancer patients during chemotherapy and immunotherapy may also address the long-standing question of whether ecDNAs provide an additional advantage to cancer cells undergoing selective pressure.
Ongoing efforts in method development allow ecDNA characterization with ever increasing resolution. As molecular and computational tools now enable isolation of ecDNAs as well as prediction and reconstruction from bulk cancer samples, these techniques may provide important insights into the prevalence and evolution of ecDNAs in clinical tumor samples. Finally, future development of empirical ecDNA detection methods in clinical samples is urgently needed. While oncogene copy number amplifications can be detected by interphase DNA FISH and whole-genome sequencing, it is still challenging to empirically and definitively attribute these amplifications to ecDNAs in clinical tumor samples. Development of such techniques will enable systematic examination of clinical outcomes associated with ecDNAs as well as sequence and structural features of ecDNAs in patient tumors. Together, these upcoming advances will more precisely pinpoint the role of ecDNA-based oncogene amplifications in cancer and identify new ways to target oncogene-addicted tumors therapeutically.
Acknowledgement
H.Y.C is supported by R35-CA209919 and is an Investigator of the Howard Hughes Medical Institute. P.S.M. is supported in part by grants U24CA264379 and RO1 CA238249 from the NIH.
Footnotes
Disclosure
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, and advisor of 10x Genomics, Arsenal Biosciences, and Spring Discovery. P.S.M. is a co-founder of Boundless Bio, Inc. He has equity in the company and chairs the scientific advisory board, for which he is compensated.
References
- 1.Cox D, Yuncken C & Spriggs, ArthurI. MINUTE CHROMATIN BODIES IN MALIGNANT TUMOURS OF CHILDHOOD. The Lancet 286, 55–58 (1965). [DOI] [PubMed] [Google Scholar]; original description of ecDNA in tumor cells manifesting as small chromatin bodies on chromosome spreads.
- 2.Spriggs AI, Boddington MM & Clarke CM Chromosomes of Human Cancer Cells. Br Med J 2, 1431–1435 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoff DDV, Needham-VanDevanter DR, Yucel J, Windle BE & Wahl GM Amplified human MYC oncogenes localized to replicating submicroscopic circular DNA molecules. PNAS 85, 4804–4808 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner KM et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Systematic analysis of human cancer models using sequencing and cytogenetics identified ecDNAs in nearly half of human cancers and not in normal cells.
- 5.Kim H et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics 52, 891–897 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive analysis of primary patient tumors found increased oncogene transcription and worsened patient outcomes linked to ecDNA.
- 6.Kohl NE et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35, 359–367 (1983). [DOI] [PubMed] [Google Scholar]
- 7.Benner S, Wahl G & Hoff DV Double minute chromosomes and homogeneously staining regions in tumors taken directly from patients versus in human tumor cell lines. Anti-cancer Drugs 2, 11–26 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Bigner SH, Mark J & Bigner DD Cytogenetics of human brain tumors. Cancer Genetics and Cytogenetics 47, 141–154 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Storlazzi CT et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res. 20, 1198–1206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshimoto M et al. MYCN Gene Amplification: Identification of Cell Populations Containing Double Minutes and Homogeneously Staining Regions in Neuroblastoma Tumors. The American Journal of Pathology 155, 1439–1443 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vicario R et al. Patterns of HER2 Gene Amplification and Response to Anti-HER2 Therapies. PLOS ONE 10, e0129876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGill JR et al. Double minutes are frequently found in ovarian carcinomas. Cancer Genetics and Cytogenetics 71, 125–131 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Lin CC et al. Evolution of karyotypic abnormalities and C-MYC oncogene amplification in human colonic carcinoma cell lines. Chromosoma 92, 11–15 (1985). [DOI] [PubMed] [Google Scholar]
- 14.Wahl GM The Importance of Circular DNA in Mammalian Gene Amplification. Cancer Res 49, 1333–1340 (1989). [PubMed] [Google Scholar]
- 15.Quinn LA, Moore GE, Morgan RT & Woods LK Cell Lines from Human Colon Carcinoma with Unusual Cell Products, Double Minutes, and Homogeneously Staining Regions. Cancer Research 39, 4914–4924 (1979). [PubMed] [Google Scholar]
- 16.Carroll SM et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Molecular and Cellular Biology 8, 1525–1533 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurer BJ, Lai E, Hamkalo BA, Hood L & Attardi G Novel submicroscopic extrachromosomal elements containing amplified genes in human cells. Nature 327, 434–437 (1987). [DOI] [PubMed] [Google Scholar]
- 18.Pauletti G, Lai E & Attardi G Early appearance and long-term persistence of the submicroscopic extrachromosomal elements (amplisomes) containing the amplified DHFR genes in human cell lines. PNAS 87, 2955–2959 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y et al. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature 1–7 (2021) doi: 10.1038/s41586-021-04009-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Møller HD et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nature Communications 9, 1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Møller HD, Parsons L, Jørgensen TS, Botstein D & Regenberg B Extrachromosomal circular DNA is common in yeast. PNAS 112, E3114–E3122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulsen T, Kumar P, Koseoglu MM & Dutta A Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends in Genetics 34, 270–278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu S et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 1–5 (2019) doi: 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung KL et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 1–6 (2021) doi: 10.1038/s41586-021-04116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovery of ecDNA hubs that enable intermolecular activation of oncogene expression via enhancer-promoter interactions.
- 25.Levan A & Levan G Have double minutes functioning centromeres? Hereditas 88, 81–92 (1978). [DOI] [PubMed] [Google Scholar]; Conclusive evidence that ecDNAs lack centromeres, explaining their distinct mode of random segregation that results in copy number heterogeneity.
- 26.Lundberg G et al. Binomial Mitotic Segregation of MYCN-Carrying Double Minutes in Neuroblastoma Illustrates the Role of Randomness in Oncogene Amplification. PLOS ONE 3, e3099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange JT et al. Principles of ecDNA random inheritance drive rapid genome change and therapy resistance in human cancers. http://biorxiv.org/lookup/doi/10.1101/2021.06.11.447968 (2021) doi: 10.1101/2021.06.11.447968. [DOI] [Google Scholar]
- 28.Ståhl F, Wettergren Y & Levan G Amplicon structure in multidrug-resistant murine cells: a nonrearranged region of genomic DNA corresponding to large circular DNA. Molecular and Cellular Biology 12, 1179–1187 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nathanson DA et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 343, 72–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu M & Ren B The Three-Dimensional Organization of Mammalian Genomes. 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cremer T & Cremer M Chromosome Territories. Cold Spring Harb Perspect Biol 2, a003889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spilianakis CG, Lalioti MD, Town T, Lee GR & Flavell RA Interchromosomal associations between alternatively expressed loci. Nature 435, 637–645 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Apostolou E & Thanos D Virus Infection Induces NF-κB-Dependent Interchromosomal Associations Mediating Monoallelic IFN-β Gene Expression. Cell 134, 85–96 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Maass PG, Barutcu AR & Rinn JL Interchromosomal interactions: A genomic love story of kissing chromosomes. Journal of Cell Biology 218, 27–38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi E et al. Live-cell imaging shows uneven segregation of extrachromosomal DNA elements and transcriptionally active extrachromosomal DNA hubs in cancer. Cancer Discov (2021) doi: 10.1158/2159-8290.CD-21-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Itoh N & Shimizu N DNA replication-dependent intranuclear relocation of double minute chromatin. Journal of Cell Science 111, 3275–3285 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Misteli T Beyond the Sequence: Cellular Organization of Genome Function. Cell 128, 787–800 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Kanda T, Sullivan KF & Wahl GM Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Current Biology 8, 377–385 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Oobatake Y & Shimizu N Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes, Chromosomes and Cancer 59, 133–143 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Chong S et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson BA et al. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin Y et al. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 175, 1481–1491.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lovén J et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henssen A et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin Cancer Res 22, 2470–2481 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Morton AR et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 179, 1330–1341.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Discription of oncogene-enhancer co-amplification on ecDNAs, showing hijacking of both cognate and ectopic enhancers to drive oncogene expression.
- 47.Tanabe H et al. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. PNAS 99, 4424–4429 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helmsauer K et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nature Communications 11, 5823 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikolaev S et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nature Communications 5, 5690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Risca VI, Denny SK, Straight AF & Greenleaf WJ Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature 541, 237–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fragkos M, Ganier O, Coulombe P & Méchali M DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16, 360–374 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Carré-Simon À & Fabre E 3D Genome Organization: Causes and Consequences for DNA Damage and Repair. Genes 13, 7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 39, 694–707.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deshpande V et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nature Communications 10, 392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luebeck J et al. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nature Communications 11, 4374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu K et al. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol 137, 123–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.L’Abbate A et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42, 9131–9145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shoshani O et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lupiáñez DG et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uslu VV et al. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat Genet 46, 753–758 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Franke M et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Schoenfelder S & Fraser P Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Pang J et al. Extrachromosomal DNA in HPV mediated oropharyngeal cancer drives diverse oncogene transcription. Clin Cancer Res (2021) doi: 10.1158/1078-0432.CCR-21-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spielmann M, Lupiáñez DG & Mundlos S Structural variation in the 3D genome. Nat Rev Genet 19, 453–467 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Weischenfeldt J et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 49, 65–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Northcott PA et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwab M, Klempnauer KH, Alitalo K, Varmus H & Bishop M Rearrangement at the 5’ end of amplified c-myc in human COLO 320 cells is associated with abnormal transcription. Molecular and Cellular Biology 6, 2752–2755 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Northcott PA et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tolomeo D, Agostini A, Visci G, Traversa D & Storlazzi CT PVT1: A long non-coding RNA recurrently involved in neoplasia-associated fusion transcripts. Gene 779, 145497 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Kalyana-Sundaram S et al. Gene Fusions Associated with Recurrent Amplicons Represent a Class of Passenger Aberrations in Breast Cancer. Neoplasia 14, 702–IN13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson DR et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chapman OS et al. The landscape of extrachromosomal circular DNA in medulloblastoma. http://biorxiv.org/lookup/doi/10.1101/2021.10.18.464907 (2021) doi: 10.1101/2021.10.18.464907 [DOI] [Google Scholar]
- 74.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanyal A, Lajoie BR, Jain G & Dekker J The long-range interaction landscape of gene promoters. Nature 489, 109–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ganji M et al. Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terakawa T et al. The condensin complex is a mechanochemical motor that translocates along DNA. Science 358, 672–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hamkalo BA, Farnham PJ, Johnston R & Schimke RT Ultrastructural features of minute chromosomes in a methotrexate-resistant mouse 3T3 cell line. Proceedings of the National Academy of Sciences 82, 1126–1130 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Bliek AM, Lincke CR & Borst P Circular DNA of 3T6R50 double minute chromosomes. Nucleic Acids Research 16, 4841–4851 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rattner JB & Lin CC Ultrastructural organization of double minute chromosomes and HSR regions in human colon carcinoma cells. Cytogenetic and Genome Research 38, 176–181 (1984). [DOI] [PubMed] [Google Scholar]
- 82.VanDevanter DR, Piaskowski VD, Casper JT, Douglass EC & Von Hoff DD Ability of Circular Extrachromosomal DNA Molecules to Carry Amplified MYCN Protooncogenes in Human Neuroblastomas In Vivo. J Natl Cancer Inst 82, 1815–1821 (1990). [DOI] [PubMed] [Google Scholar]
- 83.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nagano T et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hung KL et al. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. 2021.11.28.470285 https://www.biorxiv.org/content/10.1101/2021.11.28.470285v1 (2021) doi: 10.1101/2021.11.28.470285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current Protocols in Molecular Biology 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mumbach MR et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature Genetics 49, 1602–1612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang Z et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng M et al. Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen S, Lake BB & Zhang K High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol 37, 1452–1457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma S et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koche RP et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nature Genetics 1–6 (2019) doi: 10.1038/s41588-019-0547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang W et al. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nature Communications 6, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Overhauser J Encapsulation of Cells in Agarose Beads. in Pulsed-Field Gel Electrophoresis: Protocols, Methods, and Theories (eds. Burmeister M & Ulanovsky L) 129–134 (Humana Press, 1992). doi: 10.1385/0-89603-229-9:129. [DOI] [PubMed] [Google Scholar]
- 100.Cao H et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. GigaScience 3, 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baskin F, Rosenberg RN & Dev V Correlation of double-minute chromosomes with unstable multidrug cross-resistance in uptake mutants of neuroblastoma cells. PNAS 78, 3654–3658 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Møller HD, Parsons L, Jørgensen TS, Botstein D & Regenberg B Extrachromosomal circular DNA is common in yeast. PNAS 112, E3114–E3122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shoura MJ et al. Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3: Genes, Genomes, Genetics 7, 3295–3303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Smith CA & Vinograd J Small polydisperse circular DNA of HeLa cells. Journal of Molecular Biology 69, 163–178 (1972). [DOI] [PubMed] [Google Scholar]