

Summary
In-frame deletion mutations can result in disease. The impact of these mutations on protein structure and subsequent functional changes remain understudied, partially due to the lack of comprehensive datasets including a structural read-out. Additionally, the recent breakthrough in structure prediction through deep learning demands an update of computational deletion mutation prediction. In this study, we deleted individually every residue of a small α-helical sterile alpha motif (SAM) domain and investigated the structural and thermodynamic changes using 2D NMR spectroscopy and differential scanning fluorimetry. Then, we tested computational protocols to model and classify observed deletion mutants. We show a method using AlphaFold2 followed by RosettaRelax performs the best overall. Additionally, a metric containing pLDDT values and Rosetta ΔΔG is most reliable in classifying tolerated deletion mutations. We further test this method on other datasets and show they hold for proteins known to harbor disease-causing deletion mutations.
Keywords: Deletion, mutation, AlphaFold, Modeling, SAM domain, indel, Rosetta, ΔΔG
Graphical Abstract:
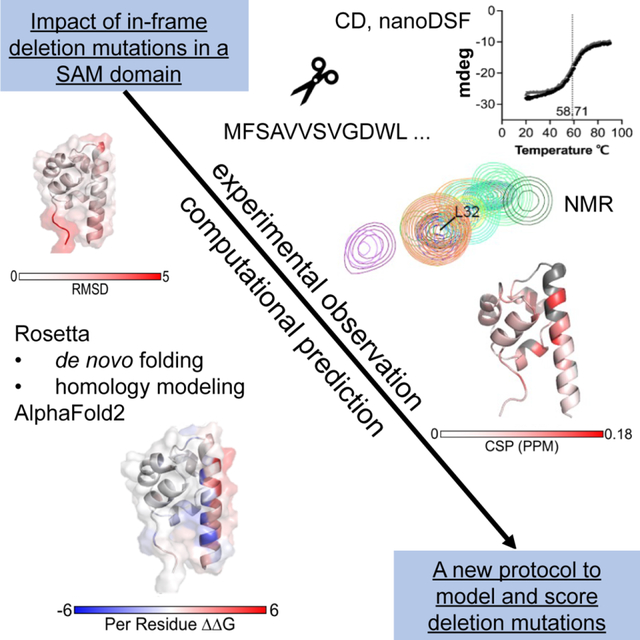
We lack datasets to develop computational tools that can help predict the effect of in-frame deletion mutations on protein structure and function. Woods et al. presents a comprehensive structural and biophysical analysis of a series of deletion mutations. The experimental results are evaluated against computational predictions using AlphaFold2 and Rosetta.
Introduction
According to the Humane Gene Mutation Database, 22% of disease-associated mutations are insertions or deletions (indels).1 The 1000 Genomes Project reports each individual carries around 200 small in-frame indels.2 In-frame deletion mutations cause genetic disease, for example, the deletion of F417 in the ferrochelatase enzyme results the enzyme being inactive causing erythropoietic protoporphyria.3 Deletion mutations also occur in the rapid viral escape, recently observed in SARS-CoV2 alpha-variant containing a del69-70 deletion.4,5 Deletions as escape mutations have also been observed in cancer. For example, the interaction of tyrosine kinase inhibitors such as erlotinib with their target epidermal growth factor receptor (EGFR).6,7
Accurate computation of the mutational impact on protein structure and function is key to predicting biological readouts of pathogenicity. However, most structural methods and protocols focus on the prediction of missense mutations8–10 and model their impact on protein folding, function, or drug interaction. Modeling how indels effect protein structure-energy relationships will progress our biophysical understanding of how deletion mutations alter structure-function mechanisms to drive disease states.
One resource that predicts the impact of indels is PROVEAN, however, it is purely sequence-based and takes no structural information into account.11,12 Structure-based methods that predict the functional and structural impact of deletions have not been updated since the recent paradigm shift in computational protein prediction provided by deep learning-based algorithms. A major limitation for modeling deletion mutations accurately is the lack of exhaustive datasets. While data for somatic mutations have become available through the increasing application of deep mutational scanning13–15, the amount of structural and functional data that can be used to test methods for deletion mutations are limited. Structural data are especially rare as deletion mutations often destabilize the protein and make it unavailable to classic structure determination methods such as X-ray crystallography or cryo-electron microscopy (cryo-EM). One of the few systematic studies on the impact of deletion mutants on the structure and function of a protein has been conducted by Arpino et al.16 who deleted individually every residue from green fluorescent protein (GFP) and monitored its functional fitness. In their study, it became apparent that GFP tolerated deletions in the loop regions, but not in the core residues that form β-strands of the β-barrel. This study falls short in that GFP is limited in secondary structural elements since it does not contain many α-helical regions and the study did not collect any direct structural data. Using this GFP dataset, Jackson et al.17 tested a computational protocol to model deletion mutants that consisted of a combined RosettaRemodel18 and Modeller19,20 approach and found weighted contact number (WCN) to be a predictive measure.
Another published computational protocol uses cyclic coordinate descent to close the gap in the protein structure and a Monte Carlo search with gradient based minimization in Rosetta.21 It was tested on a benchmark set consisting of five proteins which had both mutant and wildtype crystal structures reported in the Protein Data Bank (PDB)22 and a ricin dataset that contained data on enzyme activity.23 While this protocol was tested on deletion mutant-determined structures, it focused on predicting enzyme activity of Ricin not stability effects of mutations.
Attempts have been made to model disease causing deletion mutation such as delF508 using the respective wildtypes cryo-EM map.24 These attempts have been of a case study nature and assume that the deletion mutation undergoes only local and not global structural changes.
All these methods were created and tested before paradigm-shifting advances in the field of computational structure prediction. New deep learning structure prediction methods such as AlphaFold225,26 and RoseTTAFold27,28 dramatically outperform traditional modeling techniques such as homology modeling. However, these methods rely on their training data, which consist of the structurally resolved proteins in the PDB and make poor predictions for structures for which they were not trained29. Additionally, preliminary reports indicate that AlphaFold2 falls short in predicting the structural effects of missense mutations given an input sequence of a protein variant30. This is most likely due to the fact that AlphaFold2 training datasets are limited to determined structures and therefore, predicted models are biased towards wild-type or homologous sequences. As deletion mutant-determined structural models are rare and only cover tolerated mutations that result in similar structures, these methods may fall short in predicting structural models for deletion mutations at high accuracy.
Overall, datasets to benchmark computational methods for modeling deletion mutations are sparse and limited in structural and stability effects. Structural read-outs for multiple deletion mutants are necessary for the systematic evaluation of structural changes. In order to compare computational predictors for protein stability with a test dataset, thermostability of respective deletion mutants must be evaluated. The performance of recently reported deep learning methods, therefore, must be compared with traditional structure prediction methods.
In this study, we use a small α-helical protein and biomolecular nuclear magnetic resonance (NMR) spectroscopy for generating a deletion mutant dataset that covers the entire amino acid sequence. With this, we aim to cover the effect of mutations on α-helical proteins. We measure thermostability using 2D NMR and nanoDSF, then match our observations with four computational protocols in order to identify metrics to differentiate deleterious from tolerated deletion mutations. Using AlphaFold2 in combination with RosettaRelax31,32, we benchmark the performance of new deep-learning methods for modeling deletion mutations. In total, we were able to obtain data for seventeen deletion mutants and observed that N- and C-terminus and a loop region tolerated in-frame deletion mutations whether all other attempts resulted in insoluble protein. When applying structural modeling protocols, we observed that a combination of AlphaFold2 in combination with RosettaRelax performed best over our test case and two other reported deletion mutant studies. With this we are proposing a comprehensive method to model and score the impact of deletion mutations on protein structures of various structural compositions.
Results
The modeling of deletion mutations in an α-helical protein was studied using multiple computational protocols, reflecting protocols available for sampling of missense mutations but also emerging modeling methods using deep learning. The lack of a test dataset was overcome through the investigation of structural and biophysical changes in deletion mutants in an α-helical model protein. Solution NMR experiments were chosen to probe structural changes in the protein backbone, as it generates a structural footprint for every soluble deletion mutant. Thermal unfolding using nanoscale differential scanning fluorimetry (nanoDSF) was chosen to probe the structure-energy relationship between wild-type and deletion mutants. We compared these data to our modeling results and identified metrics capable of differentiating structurally disturbing deletion mutations.
Structural and biophysical effects of deletion mutations on a small α-helical domain
The effect of deletion mutations on the structure and biophysical properties were investigated using a small α-helical sterile-alpha-motif (SAM) domain (Figure 1A). The backbone assignment for this protein was recently published (BMRB: 51377)33,34 and allowed for a structural evaluation of every soluble deletion mutant in 1H-15N-HSQC experiments. Biomolecular NMR studies were accompanied by circular dichroism (CD) measurements35 to verify α-helical structure and melting temperature determination using both CD and nanoDSF.
Figure 1: Deletion mutants of a SAM domain.
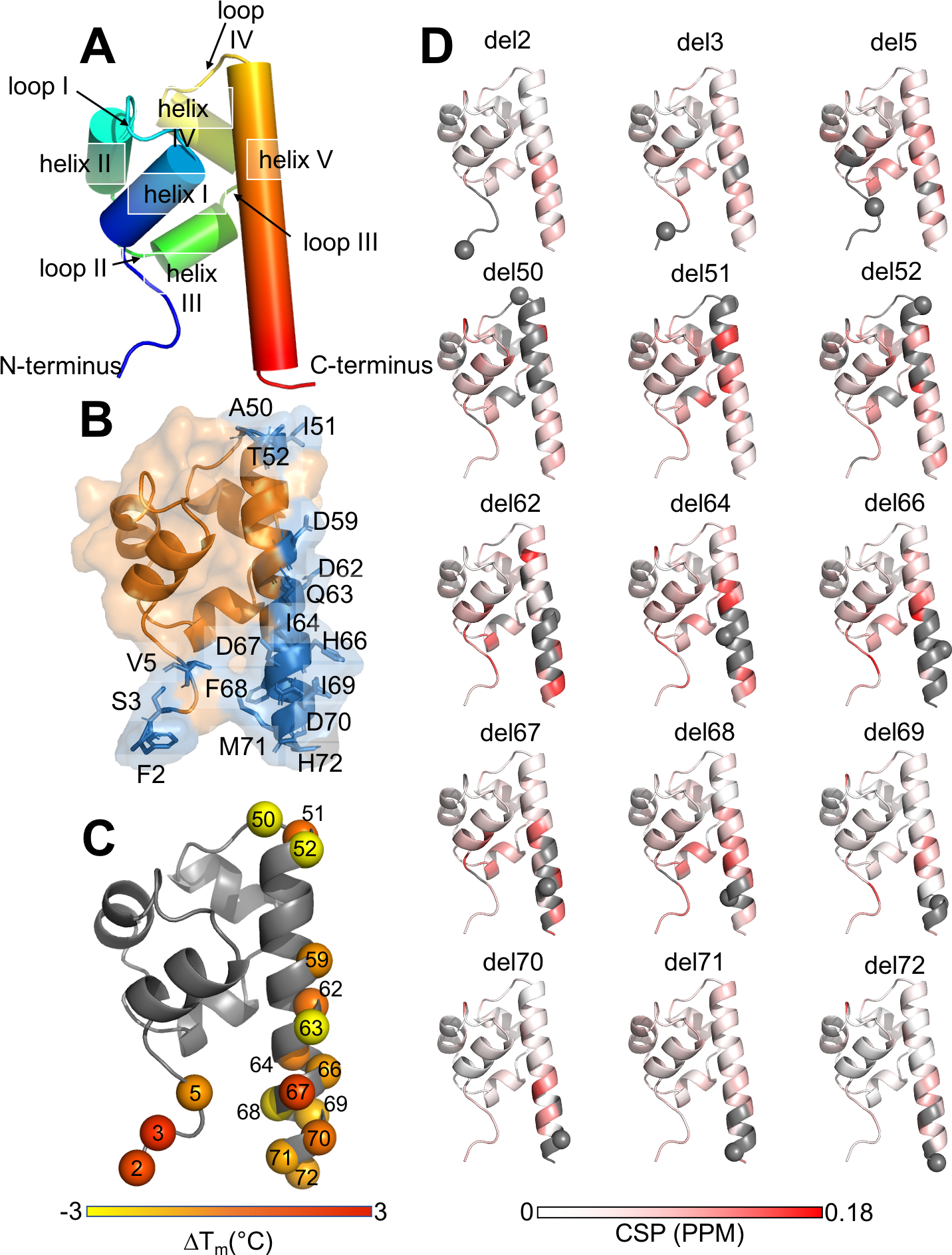
A. Structural composition of the SAM domain. B. Soluble deletion mapped onto the structure of the SAM domain labeled, shown in blue and insoluble shown in orange. C. Melting temperature difference ΔTM for deletion mutants mapped onto the SAM domain structure (from nanoDSF measurements). D. NMR data (chemical shift perturbation plots); grey indicating regions where no CSP value could be determined because the corresponding peak could not be reliably identified in the spectrum. Gray spheres indicate the deleted residue.
The SAM domain consisted of seventy-two amino acids with an N-terminal his-tag. Amino acid duplications were located at five positions in the amino acid sequence, meaning that upon amino acid removal they resulted in the same amino acid sequence (doublets: V 5-V6, A26-A27, T30-T31, D41-D42, and I64-I65). In this study, every unique amino acid was removed using site-directed mutagenesis. Excluding doublets, and the N- and C-terminal residues, sixty-five deletion constructs were tested.
All sixty-five possible deletion mutants were tested for expression in the soluble fraction of E. coli culture lysates. Seventeen of the sixty-five possible deletions were purifiable and processable for further analysis in CD and nanoDSF, all other mutants were either insoluble or adequate amounts for CD and nanoDSF analysis could not be purified. We classified these seventeen deletion mutations into four groups on the protein sequence: the N-terminal residues 2, 3 and 5, the loop IV residues 50–52 and the helix V residues 59,62–64 and the C-terminal residues 66–72 (Figure 1A). These residues were surface exposed or in flexible regions of the protein (Figure 1B). In contrast, deletion of core residues likely resulted in insoluble protein products or lacked expression levels. The seventeen soluble deletion mutants were subsequently investigated in CD spectroscopy and nanoDSF for their secondary structure composition and stability (Table 1). Overall, all deletion mutants showed an α-helical composition in CD wavelength scans (SI, Figure S1A–D), indicating that the overall composition of secondary structure elements was maintained for all soluble deletion mutants.
Table 1:
Deletion mutants of a SAM domain.
| # | deletion | Average TM (CD) [°C] |
Average TM (nanoDSF) [°C] |
|---|---|---|---|
| WT | WT | 58.9 +/− 0.2 | 60.2 +/− 0.4 |
| 1 | delF2 | 59.2 +/− 0.5 | 61.3 +/− 0.1 |
| 2 | delS3 | 58.5 +/− 0.2 | 61.9 +/− 0.7 |
| 3 | delV5 | 58.5 +/− 0.1 | 59.6 +/− 0.0 |
| 4 | delA50 | 55.5 +/− 1.1 | 56.6 +/− 0.3 |
| 5 | delI51 | 58.1 +/− 2.5 | 60.6 +/− 0.2 |
| 6 | delT52 | 62.2 +/− 0.1 | 56.0 +/− 2.6 |
| 7 | DelD59 | 59.2 +/− 0.2 | 59.5 +/− 0.0 |
| 8 | delD62 | 58.7 +/− 1.1 | 60.5 +/− 2.6 |
| 9 | delQ63 | 56.1 +/− 0.3 | 54.2 +/− 0.3 |
| 10 | delI64 | 58.8 +/− 0.63 | 60.4 +/− 0.2 |
| 11 | delH66 | 58.7 +/− 0.52 | 59.3 +/− 0.2 |
| 12 | delD67 | 61.4 +/− 1.1 | 61.4 +/− 0.1 |
| 13 | delF68 | 57.2 +/− 1.1 | 57.8 +/− 0.2 |
| 14 | delI69 | 57.8 +/− 1.3 | 58.8 +/− 1.5 |
| 15 | delD70 | 58.7 +/− 0.5 | 60.4 +/− 0.0 |
| 16 | delM71 | 57.7 +/− 0.8 | 59.2 +/− 0.0 |
| 17 | delH72 | 58.7 +/− 0.3 | 59.3 +/− 0.1 |
The deletion mutants were purified and evaluated for their stability and biophysical properties (n = 2–3). The standard deviation of the difference from the mean in circular dichroism (CD) measurements for each variant is 0.74 and for nano differential scanning fluorimetry (nanoDSF) measurements is 0.69. (Full melting curves for CD and nanoDSF in Supplementary Data, Figure S2, S3 respectively)
The protein stability change was estimated through the determination of melting temperatures from CD and nanoDSF measurements (SI Figures S2, S3). In general, the measurements agreed fairly well. The original SAM protein (called “wildtype”) displayed a TM of 58.92 °C in CD melting experiments and of 60.15 °C in nanoDSF. Most TM’s of the respective deletion mutations varied around the wildtype temperature, with exception of delA50 and delQ63, which were significantly lower for both CD and nanoDSF (Table 1). When mapping the temperature differences to the wildtype structure from nanoDSF experiments onto the structure (Figure 1C), some of the most drastic changes occurred at more occluded sites; such as delA50 that belongs to the connecting loop between α-helix IV and the C-terminal α-helix V. For delT52, which mirrors delA50 position and a similar behavior was expected, the TM measurements were not conclusive; while the nanoDSF suggests a temperature decrease, the TM from CD melting curves was elevated.
All soluble deletion mutants were labeled with 15N and assessed in 1H-15N-HSQC experiments, except for delD59, which could not be produced in sufficient yields. The resulting spectra were compared to the wildtype protein to investigate structural changes on a residue level (SI, Figures S1E, S4). The N-terminal deletions delF2, delS3 and delV5 resulted in spectra with mostly perfect overlaying peaks indicating only small and local structural changes as compared to the wildtype (Figure 1D and 2A). The deletion mutations of the loop IV between α-helix IV and α-helix V, delA50, delI51 and delT52, resulted in distinct spectra with more drastic changes as compared to N- and C-terminal deletion mutants (Figure 1D and 2B). The overall differences in the spectra become more pronounced for α-helix V deletions in closer proximity to the core of the SAM domain, e.g. delD62-64 (Figure 2C). The spectrum of delQ63 (2C and SI Figure S1E) was of poor quality and lacks a number of signals, which makes it insufficient for further analysis. However, this result is in accordance with the low TM observed in CD and nanoDSF (Table 1). We observed a similar pattern for the C-terminal deletions 66–72 as for the N-terminal deletions: the core of the protein stays largely unchanged with perfectly overlapping peaks except for the locally neighboring residues of the deletion mutations (Figure 1D and 2D). As N- and C-terminus are in proximal space some of the N-terminal residues show smaller shifts in peaks (e.g. residue A4). Overall, for deletion 66–72, the protein core is maintained and smaller structural changes occur in the N- and C-terminal regions. This result is reflected in the chemical shift perturbation (CSP) plots that were calculated for all deletion mutants except delQ63 (Figure 1D).
Figure 2: 1H-15N-HSQC spectra of the wildtype protein and respective deletion mutants, grouped by observed structural clusters.
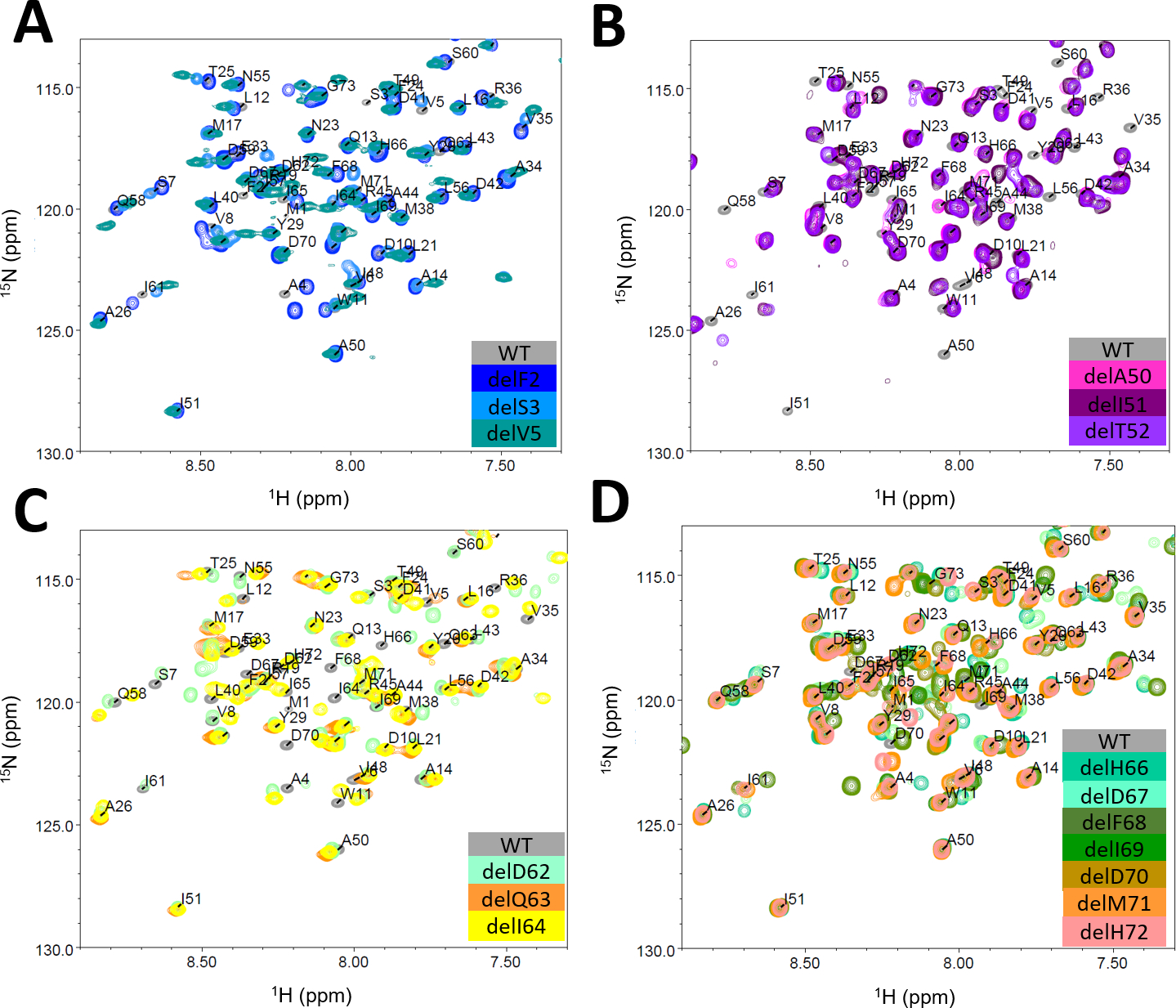
A. N-terminal deletion mutants del2, 3, 5, B. Loop IV deletion mutants del50-52, C. Structurally diverse helix V deletion mutants del62-64 and D. C-terminal deletion mutants del66-72.
In summary, seventeen deletion mutants out of a total of sixty-five were soluble and subsequently investigated for their structural and biophysical properties. This experimental dataset will serve as a test case for computational methods to model deletion mutations.
Computational methods to predict structure and stability of deletion mutations in a SAM domain
Four computational methods commonly used in structural modeling were chosen for modeling deletion mutations in an α-helical protein: 1) De novo folding using Rosetta36–39; 2) RosettaCM40-based segment hybridize as templated protocol; 3) RosettaRelax31,32,41,42 and 4) AlphaFold225,26 combined with RosettaRelax (SI Figure S7). All methods were used to generate models for all possible deletion mutants of the SAM domain and subsequently scored using the Rosetta scoring function.43,44 We applied RosettaRelax before scoring AlphaFold2 output models with the Rosetta scoring function to optimize the structure based on the Rosetta score function and to relieve any clashes that may artificially inflate the Rosetta score. For example, the distance between two atoms may appear reasonable to AlphaFold2, but be slightly too close for Rosetta causing the energy term representing repulsion forces between atoms to be high. This type of clash is easily adjusted during RosettaRelax. Detailed protocols can be found in the SI.
Firstly, we investigated whether any of the methods were able to distinguish between soluble and insoluble deletion mutants based on the ΔΔG. Out of all methods, de novo folding and AlphaFold2 predicted deletion tolerance based on score the best (Figure 3A, SI Figure S5C). Relax, and Segment Hybridize protocols all produced score distributions for soluble and insoluble mutants that overlap substantially (Figure 3A).
Figure 3: Computational protocols for the prediction of deletion mutants of the investigated SAM domain.
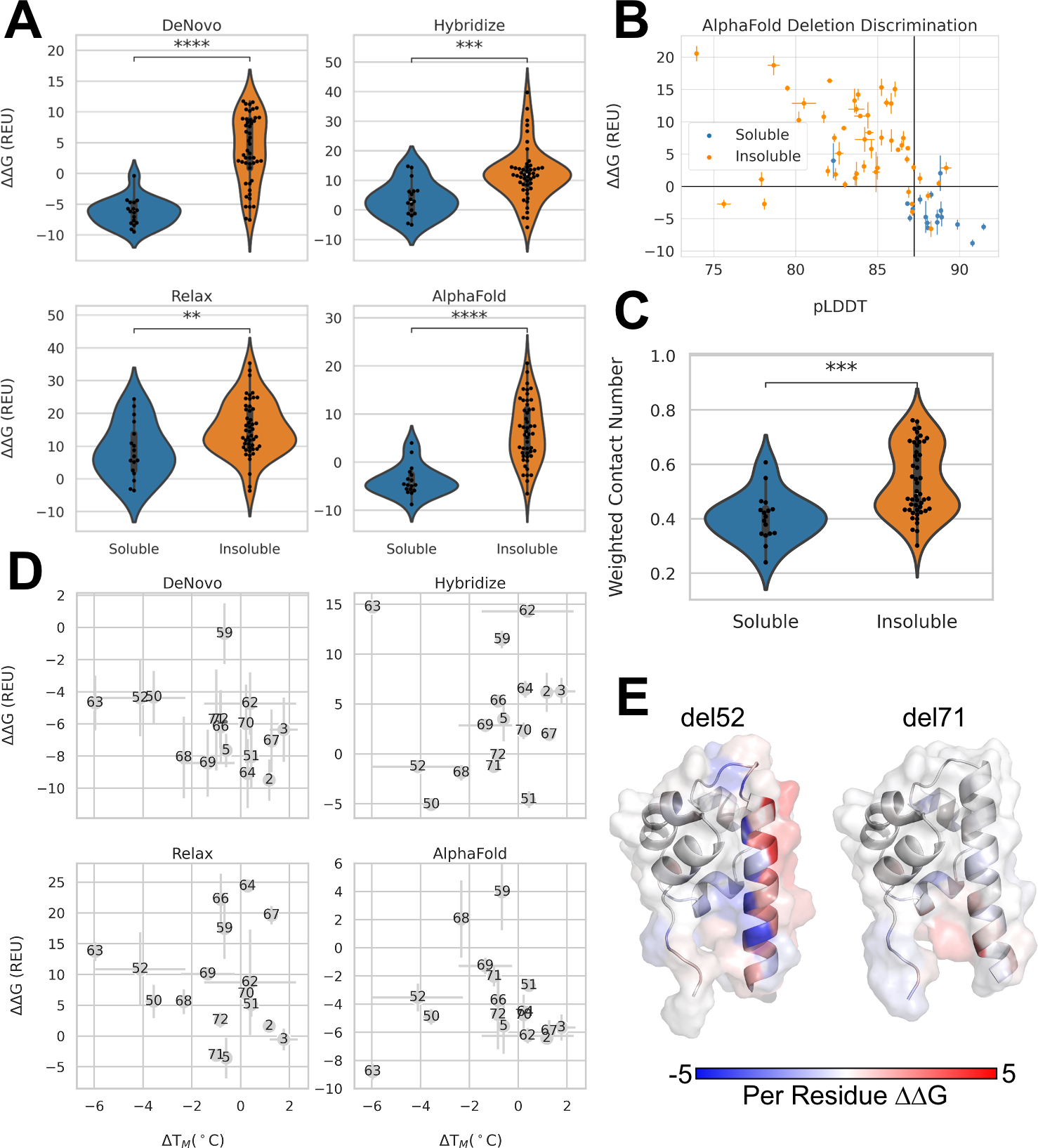
A. Distribution of ΔΔG calculated from four different computational protocols of deletions mutants that were soluble verses insoluble. Blue indicates soluble and orange indicates insoluble mutants. Stars indicate p-values calculated from Mann-Whitney test. DeNovo p-value: 4.91×10−8, Hybridize p-value: 3.28×10−4, Relax p-value: 6.12×10−3, AlphaFold p-value: 2.27×10−7. B. Average pLDDT values from AlphaFold2 verses average ΔΔG of AlphaFold2+Rosetta Relax three lowest scoring models for soluble (blue) and insoluble (orange) deletions. Vertical black line indicates wildtype average pLDDT. Horizontal black line drawn at 0 REU. Error bars depict standard error. C. Distribution of weighted contact number for soluble vs insoluble deletion mutants. P-value: 1.22×10−4. D. Melting temperatures measured with nanoDSF versus ΔΔG calculated from tested computational protocols. Higher melting temperatures indicate higher thermostability; therefore a negative correlation is expected between ΔΔG and ΔTm E. PerResidue ΔΔG scores AlphaFold2+RosettaRelax for deletion 52 and deletion 71 mapped onto the lowest scoring structures from the AlphaFold2 protocol.
Instead of using Rosetta ΔΔG scores alone, the additional information from predicted Local Distance Difference Test (pLDDT) scores from Alphafold2 were investigated for the AlphaFold2 protocol, which represent confidence of prediction per residue. When pLDDT scores averaged across all residues are plotted with ΔΔG measures from Rosetta, they show a clear separation between soluble and insoluble variants, when using 0 REU as a cut-off for ΔΔG measures and the wildtype average pLDDT score as reference (Figure 3B). Interestingly, there are few deletion mutations that pLDDT and ΔΔG measures disagree, having either high average pLDDT and high ΔΔG measures or low average pLDDT and low ΔΔG measures.
In a study by Jackson et al.17 on the tolerance of deletion mutations in GFP, side-chain weighted contact number (WCN) was found to be predictive for discriminating between tolerated and non-tolerated deletion mutations. We computed WCN as described by Jackson et al.17 for deletion mutations in our α-helical protein and observed that while there is a significant difference in the distribution of WCN, there is substantial overlap in the distributions (Figure 3C). Interestingly, the WCN metric for insoluble proteins, which also include non-expressing proteins, fall into two populations, resulting in a bimodal distribution (Figure 3C). Based on WCN alone it was not possible to group deletion mutants in the SAM domain in tolerated and non-tolerated.
Next, we compared soluble deletion mutants’ ΔTms to scores obtained from all four tested computational models (Figure 3D). Rosetta ΔΔG calculations are assumed to capture the enthalpic difference between mutants43, therefore, ΔΔG was compared against measured ΔTms from nanoDSF (Figure 3D). Overall, no protocol reliably predicted changes in melting temperature based on score alone. The segment hybridize and AlphaFold2-RosettaRelax protocol both capture the relationship between ΔTM and score insufficiently (Figure 3D). De novo folding and RosettaRelax both show a clearer correlation between ΔTM and Rosetta score, with de novo folding outperforming the other method. Interestingly, de novo is able to map deletion delA50 and delQ63 in a higher scoring range as compared to the other deletion mutants, which correctly described the measured ΔTM values (Figure 3D).
In NMR studies, some of the deletion mutants resulted in 1H-15N-HSQCs with substantially different chemical shift patterns (Figure 1D and Figure 2A–D). In order to investigate whether one of the protocols was able to match the experimentally determined chemical shift differences between deletion mutants and wildtype protein, the scores and Cα root mean squared deviation (RMSD) were broken down for every residue (Figure 3E, 4A–B). RMSD is able to partially capture some of the patterns observed in the CSP plots with lower RMSD values throughout the structure for deletions at the N and C-termini (Figure 4A). Surprisingly, we see higher ΔΔG per residue scores in the N-terminal deletions (del3-5) compared to other deletions (Figure 4B). Similar to RMSD, we see lower changes in score for C-terminal deletions (Figure 3E, 4B).
Figure 4: RMSD and Per Residue Scores on SAM Domain Deletion Structures.
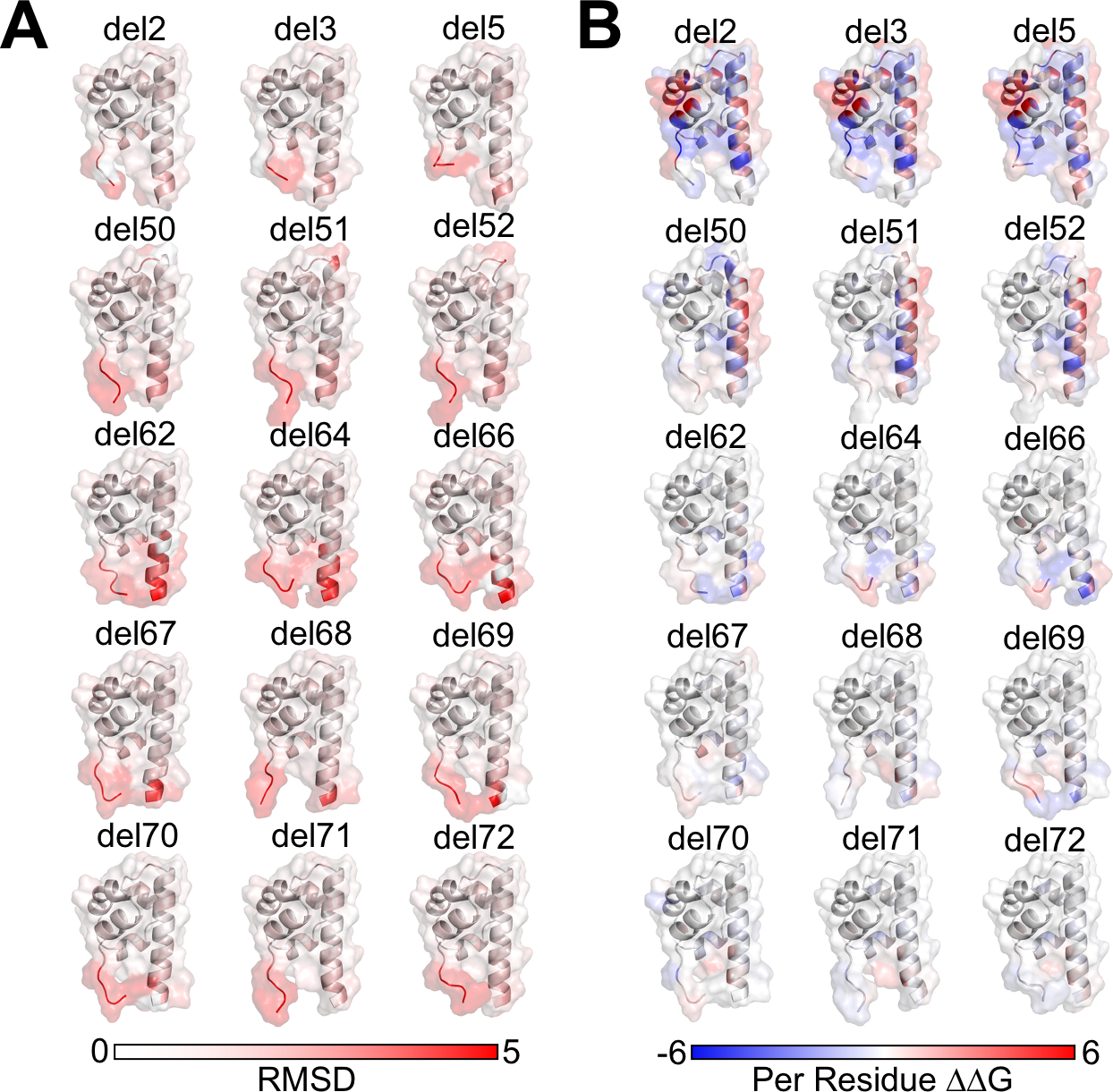
A. Per-Residue RMSD from starting structure of SAM deletion mutants mapped onto structure. B. Per-Residue Rosetta ΔΔG calculated from difference of lowest scoring mutant and lowest scoring wildtype models from AlphaFold2+RosettaRelax protocols. Negative (blue) values indicate mutant has a lower, more stable, score; positive (red) values indicate mutant has a higher, less stable, score.
In summary, the AlphaFold2-RosettaRelax protocol matches the experimental data for a small α-helical protein, both on predicting the tolerance of the deletion mutation and the structural changes in the protein. De novo folding was similar powerful in predicting deletion tolerance, but not as good in matching with experimental melting temperatures. We found using both Rosetta ΔΔG and average pLDDT together was the best way to recover the experimentally observed tolerance of deletions.
Modeling of deletion mutations in proteins with different structural composition - GFP and Ricin
In order to further test our two best methods Alphafold2 and de novo modeling, we used two reported datasets to monitor the methods’ performance: GFP and Ricin (Figure 5A–B). The dataset from Arpino et al.16 contained tolerated deletion mutants of GFP using a functional GFP fluorescence read-out comparable our the SAM domain expression, but without a method to probe structural changes.17 De novo folding failed to differentiate between tolerated and non-tolerated deletion mutants in GFP (SI, Figure S5D,E), which is expected as de novo folding is limited to small proteins with total amino acid counts of 120–150.37 The AlphaFold2 plus RosettaRelax protocol showed a significant difference for the GFP dataset, although not as pronounced as for the α-helical SAM domain (Figure 5C, SI Figure S5E). The same is true when modeling Ricin (Figure 5D, SI Figure S5F), which was probed for its enzymatic activity after expressing deletion mutants23. Additionally, WCN was calculated for GFP and Ricin deletion mutants, confirming the observation made by Jackson et al.17 that WCN is a reliable predictor of deletion mutation tolerance in GFP (Figure 5E, SI Figure S5E). However, WCN was not able to separate active and non-active deletion mutants in Ricin (Figure 5F, SI Figure S5F). Although we confirmed the WCN results from Jackson et al. for GFP, our data suggest WCN is not a reliable predictor for tolerated deletion mutations overall as both Ricin and the SAM domain results cannot be matched with WCN. The other problem with WCN is that values are not comparable between different proteins. Using both Rosetta ΔΔG and average pLDDT scores from AlphaFold2, tolerated deletion mutations could be enriched for GFP (Figure 5G). However, the usage of a reference for pLDDT values was impeded because GFP wildtype outperformed all but 10 deletion mutations with a score of 97.2. Interestingly, Rosetta ΔΔG has strong predictive power: whenever the ΔΔG is negative the deletion is tolerated, with a single misclassified deletion in the GFP dataset. Rosetta ΔΔG predicts a number of tolerated deletions as not-tolerated, however together with average pLDDT most of these deletions are stable, low ΔΔG, and confidently predicted, high average pLDDT - lower right corner of the plot (Figure 5G). For Ricin, similar behavior can be observed: the wildtype protein already contains an average pLDDT value higher than most active deletion mutants (Figure 5H). For Ricin most of the discriminative power comes from Rosetta ΔΔG. Also, average pLDDT values for different proteins are not comparable and cannot be used as a general cut-off or measure of deletion mutation tolerance.
Figure 5: Performance of computational protocols on GFP and Ricin dataset.
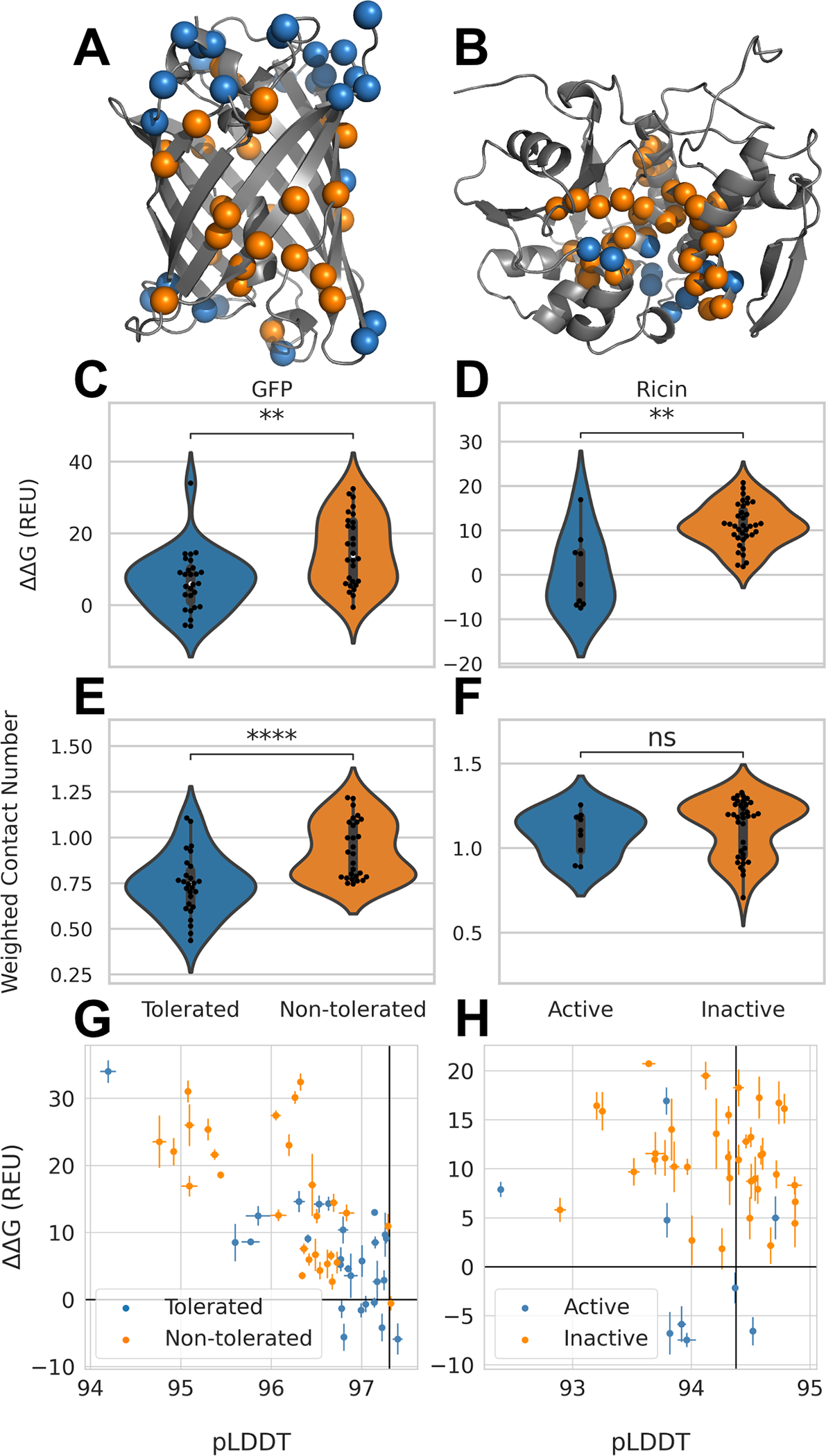
A. Tolerated deletion mutations in blue and non-tolerated in orange mapped on GFP structure17; B. Deletion mutants with remaining Ricin activity in orange and deletion mutations without activity in blue21,23; C. Distribution of ΔΔG values from AlphaFold2-RosettaRelax protocol from tolerated and non-tolerated deletion mutants in GFP; stars indicate p-values calculated from Mann-Whitney test with a p-value of 2.12×103 and D. for Ricin with a p-value of 1.8×103. E. Distribution of WCN for tolerated and non-tolerated deletion mutations in GFP with a p-value of 6.84×105 and F. in Ricin with a p-value of 0.26. G. ΔΔG values plotted against average pLDDT values from AlphaFold2 for GFP and H. for Ricin. Black lines represent values obtained for GFP and Ricin wildtype respectively. Error bars depict standard error.
In summary, the AlphaFold2 – RosettaRelax protocol outperformed de novo folding on the structurally distinct GFP, WCN has no predictive power for Ricin, and a metric using Rosetta ΔΔG versus average pLDDT reliably at differentiates tolerated deletion mutants from non-tolerated also for protein topologies distinct from the SAM domain.
Predicting pathogenic deletion mutations
We applied our computation to disease causing deletion mutations in proteins with benign and deleterious outcomes. We used the AlphaFold2 plus Rosetta Relax protocol on 22 proteins with 34 unique deletion mutations of known pathogenicity, for which the wildtype protein was available in the PDB (SI Table1). These proteins were extracted from the PROVEAN dataset11,12 and cross-referenced with the determined protein structures from the PDB22 with a resolution below 2 angstroms where the deleted residue is covered by the structure. We used ΔΔG versus average pLDDT as a metric, specifically with the ΔpLDDT from deletion mutant minus wildtype. Most reported deleterious deletion mutants, 25/34, have high ΔΔG and low ΔpLDDT and are distributed in the upper left corner, indicating they are scored both by Rosetta and AlphaFold2 as unpreferable (Figure 6). Five mutants had ΔpLDDT values greater than their respective wildtype. However, two of these are coming from the same protein structure (1EP945) and one was scored with positive ΔΔG values by Rosetta. This result indicates our protocol performs robustly on disease causing deletion cases. WCNs were also calculated for all cases and ranged between 0.4 to 1.8 (SI Figure S6). For the α-helical SAM domain, a clear differentiation could not be observed, but the soluble deletion mutants scored between 0.2 and 0.6 while non-tolerated deletions were ranged in 0.3 to 0.8. For GFP, the WCN for non-tolerated deletion mutants started at 0.6. Again, WCN had no clear separation power, as observed for Ricin and the SAM domain.
Figure 6: Modeling known pathogenic deletion mutants.
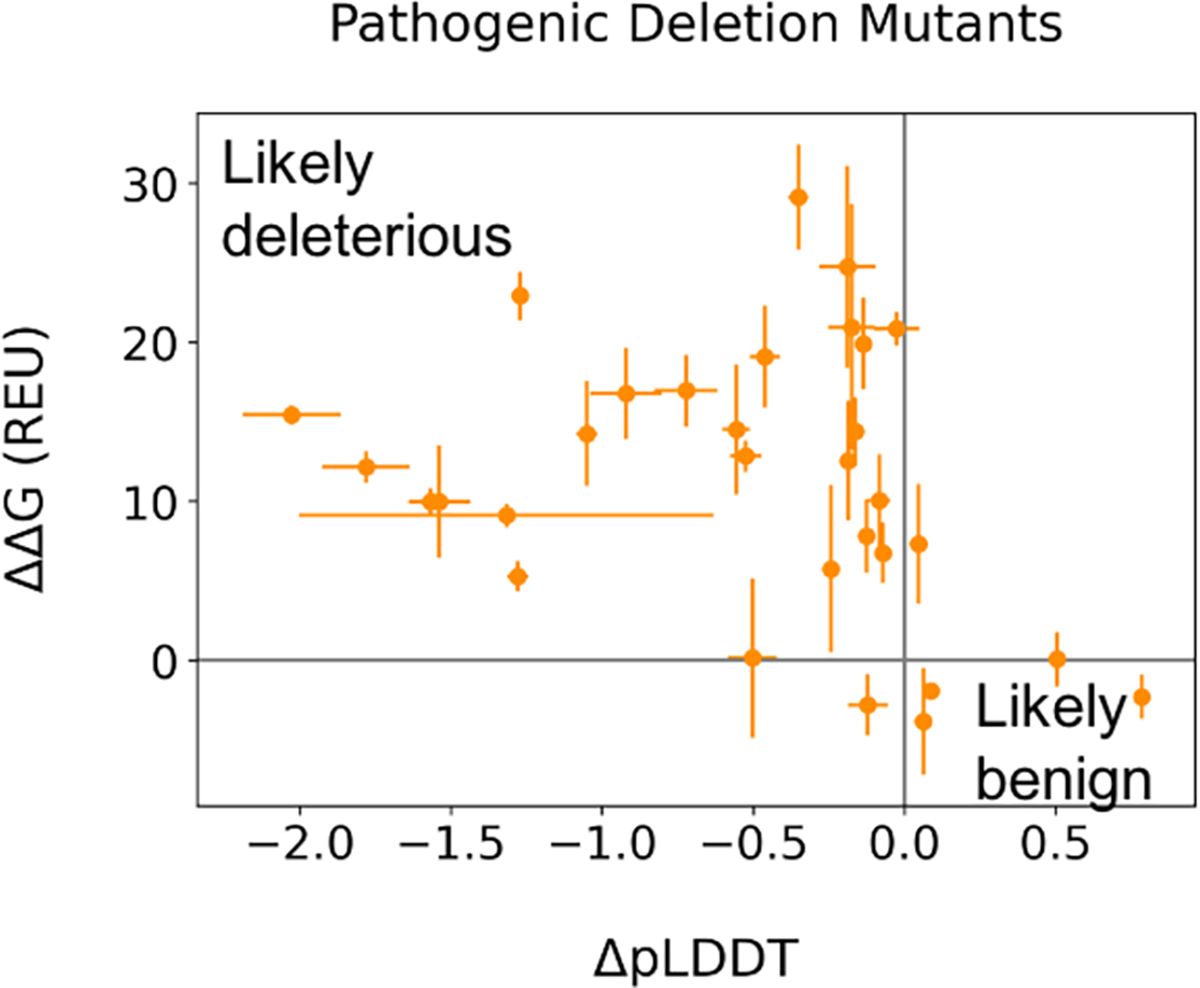
Average ΔpLDDT values from AlphaFold2 versus ΔΔG from AlphaFold+RosettaRelax for pathogenic deletion mutations listed in Table S4. Gray lines drawn at 0 to indicate how far from the wildtype value each mutant is. Error bars depict standard error.
Discussion
Understanding the structural consequences of deletion mutations is important due to the frequency of indel mutations in human disease. The small α-helical SAM domain allowed the deletion of every single amino acid and the subsequent structural investigation using 1H-15N-HSQCs for all soluble deletion mutants. The N- and C- terminal regions tolerate deletions more than the folded head of the SAM domain. The only three deletion mutants that are not in the N- or C-terminus, del50-del52, form loop IV, which may support enough structural flexibility that the SAM domain can be expressed in the soluble fraction of E. coli lysates.
Chemical shifts between wildtype and deletion mutants in 1H-15N-HSQC spectra measured structural changes for all tolerated deletion mutants. However, this method may only be reasonable on smaller proteins since it is not high-throughput; therefore, the choice of example protein is crucial to reflect as much structural space as possible. Overall, the observed melting temperatures match the structural heterogeneity of the deletion mutants well, with some exceptions (delT52).
The SAM domain is not necessarily a perfect surrogate to capture the whole structural diversity of all α-helical proteins. It contains a structured core region and more flexible N- and C termini, which reflects some structural diversity. However, the results might not be fully predictive of α-helical proteins that span the plasma membrane. Further systematic structural investigations on more diverse proteins are necessary to expand upon the dataset reported here.
Overall, four computational methods were tested in modeling deletion mutants and testing different metrics to differentiate between structurally and functionally tolerated deletion mutations. Of all tested computational methods de novo folding and AlphaFold2-RosettaRelax performed best for the small α-helical SAM domain. The de novo folding protocol has been used in the past extensively to probe energetic landscapes46–48 and characterize folding funnels but is computationally more expensive. De novo folding has been traditionally strong in predicting α-helical proteins in contrast to β-strand-rich proteins. The reason for this behavior lies in the complex long-range hydrogen bond connections that are formed between β-strands49,50. Therefore, it is not surprising that the de novo folding protocol fails to maintain the same performance when modeling GFP deletion mutants, as compared to the SAM domain. However, in rare cases de novo folding might be a good choice to model small domains.
One major challenge in computational prediction performance turned out to be the choice of metric. We showed a combination of average pLDDT scores from AlphaFold2 and Rosetta ΔΔGs was most successful in differentiating tolerated versus non-tolerated deletions (Figures 3B and 5G–H, SI Figures S5C–F). While Rosetta ΔΔGs easily assessed stabilizing or non-stabilizing effects on the protein by using the exact values, ΔpLDDT reference values remain hard to define, which substantially lowered the predictive power of the metric, e.g. in the GFP and Ricin dataset. Often, while average pLDDT scores may be similar between single deletion mutants and wildtype, one might be able to gain more insight in looking at per-residue pLDDT scores in the core of the protein. Differences at the termini may decrease average pLDDT but not have as much of an impact on the general structure. delT52 and delM71, for example, have similar average pLDDT, 87.0 and 87.6 respectively (SI, Figure S5A,B). If we break that down to the per-residue pLDDT, delT52 has a decrease in the core of the protein while delM71 has a decrease in the C-terminus, which reflects the observed changes in the chemical shift data (Figure 1D). In all datasets, it can further be found that average pLDDT are less able to differentiate tolerated versus non-tolerated mutations than Rosetta’s ΔΔGs. Overall, this might not be unsurprising, as Rosetta’s energy function is biophysically driven and has been shown to correlate with estimations of folding energy and protein stability.43 Interestingly, it was the de novo folding protocol that correlated best with measured melting temperatures from the SAM domain. Melting temperatures as measured in this study for the SAM domain, although describing the protein stability, are only a surrogate for the free energy of folding. However, the determination of the free energy of folding requires set-ups such as differential scanning calorimetry which are time and protein intensive, which might not be realizable for all deletion mutants.
The WCN metric reported by Jackson et al.17 and predictive when analyzing GFP deletion mutants was less reliable when using it on the α-helical SAM domain. Again, this might be due to the fact that protein formed by β-strand rely on long range hydrogen bond networks, while α-helical proteins contain more local hydrogen bonds to maintain the α-helix formation. Overall, we conclude that WCN is not a good metric to predict deletion mutation tolerance in structurally diverse proteins.
When evaluating our protocol in the blind modeling of deletion mutants using a set of known deleterious deletion mutations, we observed our protocol predicted higher scores for most deletions than the respective wildtype protein, suggesting mutations were unstable. The recovery in unfavorable areas of our metric indicates that our protocol and workflow is suitable to identify deletion mutations that impair the protein structure and may lead to misfolding, aggregation, instability or non-expression. Also reassuring is the fact that Rosetta ΔΔG classifies non-tolerated deletions almost always correct. It misclassifies some tolerated deletion mutations as not-tolerated in all three test cases, however, this behavior is more favorable for real-life cases. Computational tools such as this, are typically used to narrow down a large list of variants of unknown significance to a smaller list of potential pathogenic variants to investigate further with other tools. In that case it would be preferred to predict a deletion to be not-tolerated that is not impairing the structure, but not the other way around. However, if this was to be used closer to the end point in clinical genetic setting, the user needs to be aware of the limitations in classifying tolerated deletion mutations as not-tolerated.
In summary, the described work represents a state-of-the-art modeling protocol for deletion mutations that captures α-helical and structurally diverse protein geometries. With a combination of average pLDDT scores from AlphaFold2 and Rosetta ΔΔG calculations, we identified a suitable metric for the evaluation of deletion mutations also for such that have not been reported yet. It showed to hold some predictive power for all three test cases in this study, which was not true for the recently reported metric WCN17. Predictive power may be improved by further optimizing the protocol presented here by either combining metrics through machine learning methods such as linear regression or adapting new structure prediction tools as they become available.
Effects of deletion mutations on protein structure have not been well characterized in the past. There are only a handful of determined structures, naturally only on tolerated deletions. In addition, previous systematic deletion analyses on single proteins have only tested functional impact. With our dataset of deletion mutants of a small α-helical protein, we add a structurally investigated deletion mutant dataset for further studies.
With the best performing AlphaFold2-RosettaRelax protocol, we provide a method that can be easily applied when trying to predict the impact of a deletion mutation on a protein that has not been structurally resolved. In this study, all test cases were structurally resolved. The results might be different for proteins with structurally flexible regions. This study was focused on modeling single deletion mutations, it will have to be investigated whether it will also hold for deletions of multiple residues or insertions. Thus, we present a study using structural and thermodynamic data compared to computational modeling to understand deletion mutations.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact Clara T. Schoeder (clara.schoeder@medizin.uni-leipzig.de).
Materials Availability
Primers used to generate SAM domain deletion mutants can be found in Table S2.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
The Rosetta software is free for academic use (www.rosettacommons.org). All original code used in this work can be found in the Supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
BL21(DE3) cells were used for protein expression. After plasmid transformation, overnight cultures were produced in lysogeny broth (LB) media (10 mL per 1L of total target culture). The overnight cultures were transferred to 1L LB media and grown at 37 °C, 230 rpm to an OD600 of 0.6–0.8 before inducing protein expression with 1M Isopropyl-β-D-thiogalactopyranosid (IPTG). The cultures were incubated for 3–5 hours at 37 °C and 230 rpm.
METHOD DETAILS
Site-directed mutagenesis
Site-directed mutagenesis was performed using the Agilent QuickChange Lightning Site directed Mutagenesis kit according to the instruction of the supplier using the gene of the SAM domain on an expression vector (pET11a). Sequences were confirmed using Sanger Sequencing.
Expression and purification
Once sequencing was confirmed the plasmids were transformed into BL21(DE3) cells for protein expression. Conditions for cell growth and protein expression are described above. Cells were harvested by centrifugation at 6500 rpm for 20 minutes at 4 °C in an Avanti JXN26 (Beckmann, Brea, CA, USA). Subsequently, the supernatant was discarded and cell pellets stored at −80 °C. Cells were thawed, lysed in 25 mM Tris buffer, 240 mM NaCl, 20 mM imidazole, and 1 mM DTT, pH 8.0 using a homogenizer and sonication (10 min, amplitude 60%). The lysate was centrifuged at 20,000 rpm for 20 min at 4 °C and supernatant added over a column containing Ni-NTA resin (Qiagen, Venlo, Netherlands). Immobilized protein was washed with 10 column volumes and eluted with buffer containing 25 mM Tris buffer, 240 mM NaCl, 250 mM imidazole, pH 8,0. The protein was concentrated to 1 mL for further purification using Amicon filter tubes (Merck KGaA, Darmstadt, Germany) and processed using size exclusion chromatography (SEC) with a Superdex 75 column in 25 mM, 150 mM NaCl, and 1 mM DTT, pH 8.0. Fractions containing a single band of the protein of interest were pooled and concentrated.
Circular Dichroism (CD) spectroscopy
CD wavelength and temperature scans were performed using a Jasco J-810 instrument. The wavelength range covered 190–280 nm. Each deletion mutant was buffer exchanged into 10 mM potassium phosphate, pH 7.4 at a concentration of 0.18–0.21 mg/mL prior to recording CD spectra. Three scans were performed per deletion followed by three melting experiments through increasing the temperature in steps of 1°C per minute from 20 °C to 90 °C.
Nanoscale Differential Scanning Fluorimetry (nanoDSF)
NanoDSF runs were completed using the Nanotemper Prometheus Panta instrument. Similar to CD spectroscopy measurements, the protein solution contained 10 mM potassium phosphate buffer at a pH of 7.4 and a concentration of 1.0 mg/mL. Two replicates of temperature scans were completed using a temperature increase of 0.1 °C per minute covering a range of 20–90 °C and subsequently from 90–20 °C to probe protein refolding. Melting temperatures were calculated using the MoltenProt online tool, fitting with a two-state equilibrium model.52,54
Expression of 15N-labeled deletion mutants
Deletion mutants were expressed and purified as described above with the following modifications for the preparation of NMR samples. Transformed BL21(DE3) cells were grown overnight in 50 mL of LB-medium at 37°C, centrifuged at 4,000 rpm for 10 min and the pellet subsequently transferred to 1L of M9 minimal medium containing 1 g of 15N-labelled ammonium sulfate (15NH4)2SO4 (Cambridge Isotope Laboratory, Tewksbury, MA, USA), 4 g of glucose, 1 ml of a 1:1000 sodium ampicillin solution, 10 ml of MEM-vitamins, and a final concentration of 0.1 mM CaCl2, 0.1 mM ZnCl2, 1 mM MgSO4, 42 mM Na2HPO4, 22 mM KH2PO4 and 8.5 mM NaCl. After culturing in M9 medium until cell density reached an OD600 of > 0.8, protein expression was induced by adding IPTG to a final concentration of 50 μM. Cells were harvested and processed as described above. The final sample was prepared in NMR buffer containing 50 mM imidazole, 50 mM NaCl, 0.2 mM EDTA, and 7% D2O (pH 6.5).
Nuclear Magnetic Resonance (NMR)
NMR spectra were collected on uniformly labeled 15N-wildtype and 15N-deletion mutants. All samples were spiked with 5% D2O to lock the signal. All 2D-HSQC NMR experiments were performed on a 600 MHz Bruker AV-III spectrometer at 25°C equipped with an inverse broad-band probe (5 mm BBI 1H/D-BB Z-GRD) and a sample jet. All spectra were processed using the software TopSpin 3.6.2 (Bruker, Billerica, MA, USA) and analyzed using the program NMRViewJ55,56. Spectra were aligned with the assigned spectrum of 15N-wildtype using the D22 signal. Signals of deletion mutants were manually inspected and recorded. CSP values were calculated using the following equation with a scaling factor of 0.106:
Modeling deletion mutants
Four different modeling approaches were tested for modeling SAM domain deletions. Protocols Relax and Hybridize are based in Rosetta Scripts.57 De novo protocol is a command line application from Rosetta.36–39 AlphaFold2+RosettaRelax combines AlphaFold2 modeling and FastRelax, a command line application from Rosetta.41,51,58 All Rosetta protocols used the ref2015 score function.43 A description of each protocol is included below with full protocol captures in the supplement (Methods S1).
For both RosettaScript protocols, Relax, and Hybridize, the preparation included downloading the PDB file and cleaning the PDB to remove any HETATM records. Clashes are removed by running Rosetta Fast Relax on the structure, producing 100 output structures. The lowest scoring structure was then used as the starting structures for Minimize, Relax, and Segment Hybridize protocols.
The Relax protocol starts by deleting all atoms of the deleted residue followed by a gradient based minimization to close the gap left by the deleted residue. After the minimization, a dual space relax move is performed. The Hybridize protocol uses fragment-based sampling on a section 10 residues before and after the deletion to close the gap left by the deleted residue, followed by a dual space relax move. For each of these protocols, the same sampling is done with the starting structure, without deleting a residue to calculate the score for the wild-type structure.
Rosetta de novo protocol starts from a fasta file containing the sequence. Three and nine residue long fragments are picked based on secondary structure predictions from PSI-BLAST and JUFO9D. 1000 output structures are generated for both the deletion and starting sequences. The lowest three scoring models are averaged to calculate the score for both deletion and wild-type.
AlphaFold2 was also tested for its ability to model deletion mutations. AlphaFold2 was ran with both the wild-type sequence and the deletion mutant sequence. Each AlphaFold2 ran output five models. Each model was used as the input for a Rosetta relax, that output ten models, for a total of 50 models for each. The lowest three scoring models from the relax were averaged to calculate the score. The average pLDDT was calculated from all five AlphaFold2 output models.
Calculating ΔΔG values
The magnitude of Rosetta scores is dependent on the number of residues present. Therefore, the reduction in residue number for deletion mutants was taken into account for calculating the ΔΔG value. ΔΔG values were calculated with the following equation:
where is the number of residues in the wildtype protein, is the Rosetta score for the deletion and is the Rosetta score for the wildtype protein.
QUANTIFICATION AND STATISTICAL ANALYSIS
Three independent experiments were performed for CD and nanoDSF analysis and reported as means and respective standard deviation using GraphPad Prism 9.0. Statistical analysis on computational modeling is reported in the respective method sections and protocols.
Supplementary Material
Table S2: Primers used to generate SAM domain deletion mutants, related to STAR Methods. Primers were designed according to the manual of the Agilent QuickChange Lightning Site Directed Mutagenesis Kit.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| BL21(DE3) | Agilent Technologies (formerly Stratagene) | product number: 200131 |
| Chemicals, peptides, and recombinant proteins | ||
| 15N-labelled ammonium sulfate (15NH4)2SO4 | Cambridge Isotope Laboratory | NLM-713-1 |
| D2O | Cambridge Isotope Laboratory | DLM-4-99-1000 |
| SAM domain deletion mutant protein | This paper | N/A |
| 15N-labeled SAM domain deletion mutant protein | This paper | N/A |
| Critical commercial assays | ||
| QuickChangeLightning Site directed Mutagenesis kit | Agilent | #210519 |
| Oligonucleotides | ||
| Primers to generate deletion mutants, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pET11a_1b0x_BDBV | Schoeder et al.34 | N/A |
| Plasmid DNA of deletion mutants | This paper | N/A |
| Software and algorithms | ||
| Rosetta software suite 3.13 version r280 8ee4f02ac5768a8a339ffada74cb0ff5f778b3e6 | RosettaCommons | www.rosettacommons.org |
| AlphaFold2 | Jumper et al.51 | https://github.com/deepmind/alphafold |
| TopSpin 3.6.2 | Bruker | https://www.bruker.com/de/products-and-solutions/mr/nmr-software/topspin.html |
| NMRViewJ | SBGrid | https://sbgrid.org/ |
| PyMOL | SBgrid | https://sbgrid.org/ |
| GraphPad Prism 9.1 | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| MoltenProt | Kotov et al.52 | https://spc.emblhamburg.de/app/moltenprot |
| PSI-BLAST | National Center for Biotechnology Information (NCBI) | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Python 3 | Anaconda | https://www.anaconda.com/ |
| Jupyter notebook | Anaconda | https://www.anaconda.com/ |
| JUFO | Koehler-Leman et al.53 | www.meilerlab.org |
| Computational Protocol Captures | This paper | N/A |
| Other | ||
| Prometheus Standard Capillaries | Nanotemper | PR-C002 |
Highlights.
Experimental investigation of in-frame deletion mutations in an α-helical protein
Biomolecular NMR and stability tests to characterize tolerated deletion mutations
Comparison of computational protocols to model deletion mutations
A combination of Rosetta ΔΔG’s and AlphaFold2 pLDDT predicts solubility
Acknowledgements
The authors thank Alex Fisch for excellent technical assistance in supporting wetlab experiments and Nina Bozhanova for guidance on experiments and helpful discussions. We thank Chuck Sanders for use of the Nanotemper Prometheus Panta instrument. This work was supported by National Institutes of Health grant R01GM080403 (to J.M.), National Institutes of Health grant R01GM129261 (to J.M.) and a Humboldt Professorship of the Alexander von Humboldt Foundation (to J.M.). Supported in part by grants for NMR instrumentation from the National Science Foundation (NSF 0922862), National Institutes of Health (NIH S10 RR025677) and Vanderbilt University matching funds. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. E.F.M. was supported by NRSA #1F31HL162483-01A1 from the NHLBI.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Footnotes
Declaration of Interest
C.T.S. has received an unrelated research fund from Navigo Proteins GmbH, Halle (Saale), Germany. J.I.A is currently affiliated with Molecular Pharmacology and Therapeutics Graduate Program, University of Minnesota, Minneapolis, MN 55455, United States. All authors declare no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NST, and Cooper DN (2009). The Human Gene Mutation Database: 2008 update. Genome Medicine 1, 13. 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, and McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rufenacht UB, Gouya L, Schneider-Yin X, Puy H, Schafer BW, Aquaron R, Nordmann Y, Minder EI, and Deybach JC (1998). Systematic analysis of molecular defects in the ferrochelatase gene from patients with erythropoietic protoporphyria. Am J Hum Genet 62, 1341–1352. 10.1086/301870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng Q, Zhou R, Liu N, Wang H, Xu H, Zhao M, Yang D, Au KK, Huang H, Liu L, and Chen Z (2022). Naturally occurring spike mutations influence the infectivity and immunogenicity of SARS-CoV-2. Cell Mol Immunol, 1–9. 10.1038/s41423-022-00924-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scovino AM, Dahab EC, Vieira GF, Freire-de-Lima L, Freire-de-Lima CG, and Morrot A (2022). SARS-CoV-2’s Variants of Concern: A Brief Characterization. Front Immunol 13, 834098. 10.3389/fimmu.2022.834098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrews Wright NM, and Goss GD (2019). Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Transl Lung Cancer Res 8, S247–s264. 10.21037/tlcr.2019.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown BP, Zhang YK, Kim S, Finneran P, Yan Y, Du Z, Kim J, Hartzler AL, LeNoue-Newton ML, Smith AW, et al. (2022). Allele-specific activation, enzyme kinetics, and inhibitor sensitivities of EGFR exon 19 deletion mutations in lung cancer. Proc Natl Acad Sci U S A 119, e2206588119. 10.1073/pnas.2206588119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barlow KA, S ÓC, Thompson S, Suresh P., Lucas JE., Heinonen M, and Kortemme. (2018). Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein-Protein Binding Affinity upon Mutation. J Phys Chem B 122, 5389–5399. 10.1021/acs.jpcb.7b11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alford RF, and Gray JJ (2021). Membrane Protein Engineering with Rosetta. Methods Mol Biol 2315, 43–57. 10.1007/978-1-0716-1468-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strokach A, Corbi-Verge C, and Kim PM (2019). Predicting changes in protein stability caused by mutation using sequence-and structure-based methods in a CAGI5 blind challenge. Hum Mutat 40, 1414–1423. 10.1002/humu.23852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi Y, and Chan AP (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi Y, Sims GE, Murphy S, Miller JR, and Chan AP (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688. 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matreyek KA, Stephany JJ, and Fowler DM (2017). A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res 45, e102. 10.1093/nar/gkx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee AG, Kuntz CP, Ortega JT, Woods H, Most V, Roushar FJ, Meiler J, Jastrzebska B, and Schlebach JP (2021). Systematic profiling of temperature- and retinal-sensitive rhodopsin variants by deep mutational scanning. J Biol Chem 297, 101359. 10.1016/j.jbc.2021.101359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penn WD, McKee AG, Kuntz CP, Woods H, Nash V, Gruenhagen TC, Roushar FJ, Chandak M, Hemmerich C, Rusch DB, et al. (2020). Probing biophysical sequence constraints within the transmembrane domains of rhodopsin by deep mutational scanning. Sci Adv 6, eaay7505. 10.1126/sciadv.aay7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arpino JA, Reddington SC, Halliwell LM, Rizkallah PJ, and Jones DD (2014). Random single amino acid deletion sampling unveils structural tolerance and the benefits of helical registry shift on GFP folding and structure. Structure 22, 889–898. 10.1016/j.str.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson EL, Spielman SJ, and Wilke CO (2017). Computational prediction of the tolerance to amino-acid deletion in green-fluorescent protein. Plos One 12, e0164905. 10.1371/journal.pone.0164905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang PS, Ban YE, Richter F, Andre I, Vernon R, Schief WR, and Baker D (2011). RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One 6, e24109. 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, and Sali A (2003). Tools for comparative protein structure modeling and analysis. Nucleic Acids Res 31, 3375–3380. 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiser A, and Sali A (2003). Modeller: generation and refinement of homology-based protein structure models. Methods in enzymology 374, 461–491. 10.1016/s0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 21.Berrondo M, and Gray JJ (2011). Computed structures of point deletion mutants and their enzymatic activities. Proteins 79, 2844–2860. 10.1002/prot.23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, Christie CH, Dalenberg K, Di Costanzo L, Duarte JM, et al. (2021). RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 49, D437–d451. 10.1093/nar/gkaa1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munishkin A, and Wool IG (1995). Systematic deletion analysis of ricin A-chain function. Single amino acid deletions. J Biol Chem 270, 30581–30587. 10.1074/jbc.270.51.30581. [DOI] [PubMed] [Google Scholar]
- 24.McDonald EF, Woods H, Smith ST, Kim M, Schoeder CT, Plate L, and Meiler J (2022). Structural Comparative Modeling of Multi-Domain F508del CFTR. Biomolecules 12. 10.3390/biom12030471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Applying and improving AlphaFold at CASP14. Proteins 89, 1711–1721. 10.1002/prot.26257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baek M, and Baker D (2022). Deep learning and protein structure modeling. Nat Methods 19, 13–14. 10.1038/s41592-021-01360-8. [DOI] [PubMed] [Google Scholar]
- 28.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald EF, Jones T, Plate L, Meiler J, and Gulsevin A (2023). Benchmarking AlphaFold2 on peptide structure prediction. Structure 31, 111–119 e112. 10.1016/j.str.2022.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pak MA, Markhieva KA, Novikova MS, Petrov DS, Vorobyev IS, Maksimova ES, Kondrashov FA, and Ivankov DN (2021). Using AlphaFold to predict the impact of single mutations on protein stability and function. bioRxiv, 2021.2009.2019.460937. 10.1101/2021.09.19.460937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conway P, Tyka MD, DiMaio F, Konerding DE, and Baker D (2014). Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci 23, 47–55. 10.1002/pro.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nivon LG, Moretti R, and Baker D (2013). A Pareto-optimal refinement method for protein design scaffolds. PLoS One 8, e59004. 10.1371/journal.pone.0059004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ledwitch KSC,; Voehler M.; Meiler J. (2022). Backbone and side-chain chemical shift assignments for a rosetta-designed BDBV-MPER immunogen doi: 10.13018/BMR51377. [DOI] [Google Scholar]
- 34.Schoeder CT, Gilchuk P, Sangha AK, Ledwitch KV, Malherbe DC, Zhang X, Binshtein E, Williamson LE, Martina CE, Dong J, et al. (2022). Epitope-focused immunogen design based on the ebolavirus glycoprotein HR2-MPER region. PLoS Pathog 18, e1010518. 10.1371/journal.ppat.1010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenfield NJ (2006). Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1, 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonneau R, Strauss CE, Rohl CA, Chivian D, Bradley P, Malmstrom L, Robertson T, and Baker D (2002). De novo prediction of three-dimensional structures for major protein families. J Mol Biol 322, 65–78. 10.1016/s0022-2836(02)00698-8. [DOI] [PubMed] [Google Scholar]
- 37.Bender BJ, Cisneros A 3rd, Duran AM, Finn JA, Fu D, Lokits AD, Mueller BK, Sangha AK, Sauer MF, Sevy AM, et al. (2016). Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry 55, 4748–4763. 10.1021/acs.biochem.6b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonneau R, Tsai J, Ruczinski I, Chivian D, Rohl C, Strauss CE, and Baker D (2001). Rosetta in CASP4: progress in ab initio protein structure prediction. Proteins Suppl 5, 119–126. 10.1002/prot.1170. [DOI] [PubMed] [Google Scholar]
- 39.Bradley P, Misura KM, and Baker D (2005). Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871. 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 40.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, and Baker D (2013). High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742. 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popovic Z, Baker D, and Players F (2011). Algorithm discovery by protein folding game players. Proc Natl Acad Sci U S A 108, 18949–18953. 10.1073/pnas.1115898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyka MD, Keedy DA, André I, Dimaio F, Song Y, Richardson DC, Richardson JS, and Baker D (2011). Alternate states of proteins revealed by detailed energy landscape mapping. J Mol Biol 405, 607–618. 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, et al. (2017). The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 13, 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Meara MJ, Leaver-Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, Bradley P, Kortemme T, Baker D, Snoeyink J, and Kuhlman B (2015). Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J Chem Theory Comput 11, 609–622. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi D, Morizono H, Yu X, Tong L, Allewell NM, and Tuchman M (2001). Human ornithine transcarbamylase: crystallographic insights into substrate recognition and conformational changes. Biochem J 354, 501–509. 10.1042/0264-6021:3540501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norn C, Wicky BIM, Juergens D, Liu S, Kim D, Tischer D, Koepnick B, Anishchenko I, Baker D, and Ovchinnikov S (2021). Protein sequence design by conformational landscape optimization. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2017228118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam KH, Yao G, et al. (2017). Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. 10.1038/nature23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A, et al. (2017). Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357, 168–175. 10.1126/science.aan0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcos E, Basanta B, Chidyausiku TM, Tang Y, Oberdorfer G, Liu G, Swapna GV, Guan R, Silva DA, Dou J, et al. (2017). Principles for designing proteins with cavities formed by curved β sheets. Science 355, 201–206. 10.1126/science.aah7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, and Baker D (2012). Principles for designing ideal protein structures. Nature 491, 222–227. 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kotov V, Mlynek G, Vesper O, Pletzer M, Wald J, Teixeira-Duarte CM, Celia H, Garcia-Alai M, Nussberger S, Buchanan SK, et al. (2021). In-depth interrogation of protein thermal unfolding data with MoltenProt. Protein Sci 30, 201–217. 10.1002/pro.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leman JK, Mueller R, Karakas M, Woetzel N, and Meiler J (2013). Simultaneous prediction of protein secondary structure and transmembrane spans. Proteins 81, 1127–1140. 10.1002/prot.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burastero O, Niebling S, Defelipe LA, Gunther C, Struve A, and Garcia Alai MM (2021). eSPC: an online data-analysis platform for molecular biophysics. Acta Crystallogr D Struct Biol 77, 1241–1250. 10.1107/S2059798321008998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson BA (2018). From Raw Data to Protein Backbone Chemical Shifts Using NMRFx Processing and NMRViewJ Analysis. Methods Mol Biol 1688, 257–310. 10.1007/978-1-4939-7386-6_13. [DOI] [PubMed] [Google Scholar]
- 56.Norris M, Fetler B, Marchant J, and Johnson BA (2016). NMRFx Processor: a cross-platform NMR data processing program. J Biomol NMR 65, 205–216. 10.1007/s10858-016-0049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, et al. (2011). RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. Plos One 6, e20161. 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maguire JB, Haddox HK, Strickland D, Halabiya SF, Coventry B, Griffin JR, Pulavarti S, Cummins M, Thieker DF, Klavins E, et al. (2021). Perturbing the energy landscape for improved packing during computational protein design. Proteins 89, 436–449. 10.1002/prot.26030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Primers used to generate SAM domain deletion mutants, related to STAR Methods. Primers were designed according to the manual of the Agilent QuickChange Lightning Site Directed Mutagenesis Kit.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
The Rosetta software is free for academic use (www.rosettacommons.org). All original code used in this work can be found in the Supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.