

Significance
Nanobodies are a promising class of biologics that can be used to prevent or treat viral infections. Here, we describe the production and validation of a discovery library that produces single-domain nanobodies using an engineered human antibody variable gene segment. As a test case, anti-SARS-CoV-2 nanobodies were isolated from this library and pairs of complementary nanobodies were incorporated into an antibody-like molecule that targets the receptor-binding domain using a biparatopic mode of engagement. This modular bispecific format enabled the rapid testing of nanobody pairs, and we show that incorporating pairs of nanobodies with different specificities can have synergistic effects on neutralization breadth and potency.
Keywords: synthetic library, bispecific antibody, nanobody, SARS-CoV-2 neutralization
Abstract
Nanobodies bind a target antigen with a kinetic profile similar to a conventional antibody, but exist as a single heavy chain domain that can be readily multimerized to engage antigen via multiple interactions. Presently, most nanobodies are produced by immunizing camelids; however, platforms for animal-free production are growing in popularity. Here, we describe the development of a fully synthetic nanobody library based on an engineered human VH3-23 variable gene and a multispecific antibody-like format designed for biparatopic target engagement. To validate our library, we selected nanobodies against the SARS-CoV-2 receptor–binding domain and employed an on-yeast epitope binning strategy to rapidly map the specificities of the selected nanobodies. We then generated antibody-like molecules by replacing the VH and VL domains of a conventional antibody with two different nanobodies, designed as a molecular clamp to engage the receptor-binding domain biparatopically. The resulting bispecific tetra-nanobody immunoglobulins neutralized diverse SARS-CoV-2 variants with potencies similar to antibodies isolated from convalescent donors. Subsequent biochemical analyses confirmed the accuracy of the on-yeast epitope binning and structures of both individual nanobodies, and a tetra-nanobody immunoglobulin revealed that the intended mode of interaction had been achieved. This overall workflow is applicable to nearly any protein target and provides a blueprint for a modular workflow for the development of multispecific molecules.
The use of biologics for the prevention or treatment of viral disease is increasing. Palivizumab was the first monoclonal antibody approved for the prevention of respiratory syncytial virus (RSV) disease in infants, but Ebola virus outbreaks and the COVID-19 pandemic have led to the development of several other monoclonal antibodies. Antibodies have the potential to be transformative within this space due to their high specificity, long half-life, and ability to coordinate the response from the innate immune system via Fc-mediated interactions. One of the main challenges in using antibodies for antiviral indications is the ever-present risk of resistant viral variants. This was exemplified by the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOCs) during the COVID-19 pandemic that were partially or fully resistant to the therapeutic antibody countermeasures (1). Strategies to counter this viral escape generally focus on targeting functionally conserved epitopes and/or the use of a cocktail of multiple antibodies that recognize nonoverlapping epitopes, thus requiring multiple mutations for the virus to effectively evade all antibodies in the cocktail (2). Single-domain “nanobodies” are promising as antiviral biologics because they recognize their targets using a single-variable heavy (VHH) domain. One advantage of this smaller size is that nanobodies can target epitopes that are sterically inaccessible to bulkier antibodies that require both variable heavy (VH) and light (VL) domains to form the antibody paratope (3). The lack of light chain pairing greatly simplifies the expression of multiple nanobodies targeting different specificities as a single molecule. The most common method to obtain nanobodies requires immunizing camelids (llamas or alpacas) that produce VHHs as part of their Ig repertoire, but phage- and yeast-based synthetic platforms can also be used for de novo discovery. To date, neutralizing nanobodies have been developed for numerous viruses including HIV (4, 5), influenza (6), RSV (7, 8), SARS-CoV-2 (9, 10), hepatitis C (11), and rabies virus (12). In most instances, monomeric VHH exhibits moderate neutralizing potency, but when homo- or hetero-nanobodies are placed in tandem, avid interactions greatly increase the overall neutralization potency (9, 13, 14). While the value of combining multiple nanobodies into a single molecule has been clearly demonstrated (6, 9, 10, 13, 15), selecting the candidate nanobodies to include generally requires either a priori structural information, or the screening of many constructs to find synergistic nanobody combinations.
Because nanobodies from other species are often immunogenic in humans, we created a human-based nanobody library and validated this library using SARS-CoV-2 as a test antigen. We developed an on-yeast competitive selection strategy paired with deep sequencing to rapidly determine the specificity of the selected nanobodies. The epitope binning information was then used to select candidate nanobodies targeting nonoverlapping epitopes that can be incorporated into a chimeric nanobody/antibody hybrid architecture by replacing the conventional antibody VH and VL domains with different nanobodies. This modular format facilitates bispecific nanobody–based creation through the native Ig CH1/CL domain pairing and allows for the rapid production of different nanobody combinations by mixing different plasmids during the transfection. The resulting bispecific tetra-nanobody immunoglobulin (bsNb4-Ig) can achieve multivalent interactions through the two nanobodies on a single fragment antigen-binding region (Fab) and/or through the contribution of nanobodies on adjacent Fab arms of the molecule. Furthermore, nanobodies that recognize nonoverlapping epitopes may facilitate biparatopic engagement of a single target molecule, provided the linkers that connect the nanobodies to the constant domain are sufficiently long. The presence of the Fc domain also enables the use of conventional purification strategies during production and provides the benefit of Fc interactions in vivo, including long serum half-life and antibody effector functions. Lastly, we structurally characterized the selected nanobodies to confirm the mode of interaction compared to conventional antibodies.
Results
hVHH323 Construction and Validation.
The human-based nanobody library was designed to have a conserved variable domain (including CDRH1 and CDRH2) with all the diversity localized in the CDRH3 loop, mimicking the diversification of a naïve immune repertoire. Based on the high sequence identity between camelids VHHs and human VH3s (16) (Fig. 1A), a synthetic VHH scaffold was developed by incorporating common VHH mutations into the human VH3-23 heavy chain (HC) gene (Fig. 1B). Our rationale for starting with the human VH gene and adding “camelizing” mutations was that it would be more human than a camelid VHH, thus contains fewer nonself epitopes, potentially reducing the likelihood of antidrug antibody development. We looked at predicted class II binding during the design of the hVHH323 base, but rational deimmunization is poorly understood for humans and we decided to instead maximize the similarity of our starting scaffold with human VH genes. T cells should be tolerized against those peptides, thus minimizing potential epitopes. The starting scaffold (hVH323) was modified by including previously reported “camelizing” mutations that remove the need for light chain (LC) pairing (17–20), and mutations from consensus VHH sequences that removed surface-exposed hydrophobic residues (SI Appendix, Fig. S1). To further stabilize the VHH scaffold, an additional internal disulfide bond was introduced by mutating Ser49 and Ile69 to Cys (21).
Fig. 1.

Properties of the synthetic human-based hVHH323 library. (A) Overlay of the VH domain from a conventional human IgG (PDB 5I19, blue) with a representative camel VHH (PDB 5U65, orange). Structures were aligned on the VHH, and the rmsd of the alpha carbons is indicated. The VL domain is shown as surface. (B) VH domain from a human antibody that utilizes VH3-23 (PDB 5I19) with the positions of the mutations that were introduced to produce the hVHH323 scaffold highlighted as spheres. Orange spheres are “camelizing” mutations to remove the need for LC pairing. Yellow spheres indicate the additional disulfide bond that was introduced to increase stability. The variable CDRH3 is colored in red. (C) CDRH3 loop length distribution from the transformed hVHH323 library obtained by deep sequencing analysis compared to human (conventional antibodies) (22) and camelids (HC-only antibodies) (23) CDRH3 repertoires. (D) Schematic yeast display of the hVHH323 nanobodies with a V5 epitope tag. (E) Deep sequencing analysis of AA frequencies of diversified positions in CDRH3s, showing both the intended frequencies and those observed in the transformed library.
The CDRH3 loops all begin and end with the CAR and FDYW motifs, respectively. Between these conserved amino acids (AA), 5 to 15 randomized AAs were inserted, resulting in nanobodies with CDRH3 loop lengths of 10 to 20 AAs. The AA frequencies in the CDRH3 loops were designed based on the nontemplated AA frequencies found in naturally occurring antibodies and nanobodies, excluding Met and Cys to avoid producing nanobodies with unwanted chemical liabilities (SI Appendix, Text). CDRH3 lengths were distributed following a sigmoidal distribution rather than mimicking the naturally occurring normal distribution (Fig. 1C). The right-shifted distribution was selected because the theoretical diversity of the shorter CDRH3 loops is relatively small and would be oversampled if a normal distribution were used (SI Appendix, Fig. S2). We also hypothesized that longer CDRH3 loops could help better access recessed epitopes. DNA encoding the human-based nanobody library was transformed into a yeast surface display vector via homologous recombination with an estimated diversity of 3 × 109, based on a colony formation assay performed after the transformation (SI Appendix, Text). The nanobodies displayed well on the yeast surface, with ~67% of the library showing surface display and a median 180-fold increase in signal between the displaying and nondisplaying cell populations (Fig. 1D). Deep sequence analysis of nanobody-encoding DNA recovered from the transformed cells showed a good agreement with the target library specifications for both CDRH3 length distributions and AA frequencies (Fig. 1 C and E).
Discovery of Binders Using SARS-CoV-2 Receptor–Binding Domain (RBD).
Upon completion of the library, we next set out to validate it using SARS-CoV-2 RBD as a test antigen. The overall library screening strategy was performed using a combination of magnetic-activated cell sorting (MACS) followed by fluorescence-activated cell sorting (FACS) (Fig. 2A). After each round of selection, the collected cell population was expanded and reinduced prior to the next round. The naïve library was too large to be screened using conventional FACS, so three rounds of MACS were used to bulk enrich the small number of SARS-CoV-2-reactive clones. RBD-reactive cells were enriched in the first round of MACS by labeling 2 × 1011 cells from the induced library with biotinylated SARS-CoV-2 RBD. Excess antigen was removed, and the cells were then incubated with streptavidin-conjugated magnetic microbeads. Cells bound to the magnetic microbeads were magnetically enriched, expanded, and reinduced for additional rounds of selection. In this first positive MACS selection, the collected cells were enriched for SARS-CoV-2 RBD–reactive clones, but also were enriched for streptavidin- and microbead-reactive clones. All selected cells were expanded and reinduced for additional rounds of selections. To deplete the latter two populations, the second round of MACS was a counterselection, where induced cells were incubated with the streptavidin-conjugated magnetic microbeads only and the nonreactive population was collected. Immediately following the counterselection and without expanding the collected cells, a third MACS selection was used to further enrich the RBD-reactive clones.
Fig. 2.
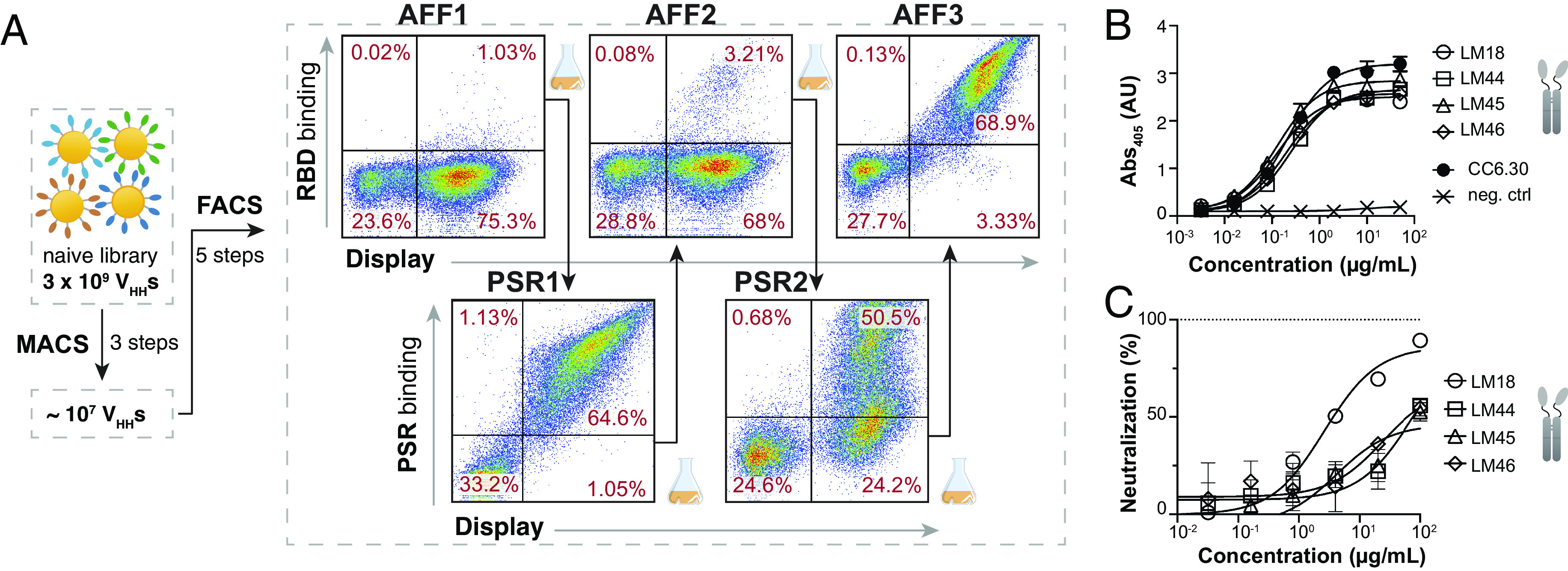
Selection of SARS-CoV-2 RBD–specific nanobodies. (A) FACS workflow to enrich populations in antigen-specific clones. Cells in AFF1, AFF2, and AFF3 are labeled with 100 nM biotinylated RBD to depict the enrichment overtime. Cells in PSR1 and PSR2 are labeled with biotinylated PSR preparation. The RBD and PSR concentrations used for sorting are indicated in SI Appendix, Text. (B) ELISA plot showing Fc-fused LM18, LM44, LM45, and LM46 binding to SARS-CoV-2 RBD. CC6.30 was used as a positive control and a nanobody-Fc from our library not selected for RBD binding was used as a negative control. The assay was run in duplicate. (C) Neutralization assay of Wuhan-1 SARS-CoV-2 PSV for the four nanobodies tested showing neutralization by LM18. Assay was run in triplicate. Error bars indicate the SD of the mean.
After MACS enrichments, five additional rounds of multicolor FACS were used to isolate clones with high binding affinity and specificity for RBD. In FACS rounds 1, 3, and 5 (AFF1, 2, and 3), cells were labeled with subsaturating concentrations of biotinylated RBD, followed by fluorescently conjugated streptavidin and an anti-V5 antibody to quantify the amount of RBD and the amount of surface-displayed nanobody, respectively. Normalizing for the level of nanobody displayed on the surface of the cell, the high RBD-binding clones were enriched. To help ensure the selected nanobodies were specific for RBD, negative selections were interspersed at FACS round 2 and 4 (PSR1 and 2) to remove polyreactive “sticky” clones (24). In this counterselection, cells were labeled with a complex preparation of biotinylated and detergent-solubilized HEK cell membrane proteins (polyspecificity reagent or PSR) and the PSR-low fraction was collected, again normalizing for the level of nanobody display on the cell surface (Fig. 2A).
As this was the first antigen screened against this library, we sought to quickly test the specificity and biochemical behavior of the selected nanobodies before conducting a more in-depth analysis of the enriched population. Sanger sequencing of 96 AFF3 clones found the library to be highly enriched, recovering a total of four unique clones. These four nanobodies were expressed in Expi293 cells as both the nanobody alone (His-tagged) and linked to a human Fc to create a homodimeric nanobody fusion protein. Nanobodies expressed well in both formats, producing a normalized expression titer of 50 to 200 mg/L and 220 to 450 mg/L for the His-tagged nanobodies and Fc fusions, respectively. Both formats were monodispersed by analytical size-exclusion chromatography (SEC). Enzyme-linked immunosorbent assay (ELISA) confirmed RBD binding of the four variants (Fig. 2B), and no polyspecific binding was observed in our standard assays (SI Appendix, Fig. S3). Finally, we tested the four nanobody–Fc fusions in a SARS-CoV-2 pseudovirus (PSV) neutralization assay and found that one of the nanobodies, LM18, neutralized with an IC50 of 2.65 µg/mL (66 nM) (Fig. 2C and SI Appendix, Table S1). Interestingly, LM18 also neutralized SARS-CoV-1 PSV with an IC50 of 0.5 µg/mL (or 12 nM) (SI Appendix, Fig. S4) as it serendipitously targeted a conserved epitope between the two variants.
FACS-Based Epitope Binning and Deep Sequencing to Determine Library Specificities.
After confirming that the base hVHH323 could produce biochemically well-behaved nanobodies that bound a target antigen with high specificity, we next set out to determine how many nanobodies were enriched during the selections and what epitopes on RBD were targeted by these nanobodies. Epitope specificity was determined using an on-yeast competition assay with three structurally characterized SARS-CoV-2 antibodies: CC12.1 (class 1), CC6.30 (class 2), and CR3022 (class 4) (Fig. 3 A–C) (25–27). Cells from the outgrowth following the FACS 4-negative selection (PSR2) were analyzed, as a large fraction of clones in this population were RBD reactive and had undergone both polyreactive negative selections, and thus should have relatively few sticky clones. The induced library was labeled with SARS-CoV-2 RBD, and then incubated with one of the three anti-SARS-CoV-2 antibodies. Fluorescent secondaries were used to measure the amount of nanobody on the surface of the cell, the amount of RBD bound, and the amount of the SARS-CoV-2 antibody bound. In this format, nanobodies that recognized overlapping epitopes with the human IgGs would block the antibody from binding. For analysis, both the competitive (C) population (nanobody competes with the human antibody for binding) and the noncompetitive (NC) population (both the nanobody and antibody engage RBD simultaneously) were sorted. The starting population and competition sorts for CC12.1, CC6.30, and CR3022 were deep sequenced to analyze the nanobodies in each population. After filtering to remove likely sequencing artifacts, 123 unique nanobodies were observed in the datasets (SI Appendix, Table S2). Epitope bins were assigned by defining an overlapping epitope with a tested SARS-CoV-2 antibody as having a C/NC ratio >10, and C/NC ratio <10 for a nonoverlapping epitope (SI Appendix, Text). From the 123 unique nanobodies identified, 100 nanobodies have a clear competitive binding profile with at least one of the three tested antibodies (Fig. 3C), whereas 13 nanobodies were identified as NC with all the three antibodies, indicating they likely bind elsewhere on the RBD. Four nanobodies seemed to compete with all the three antibodies, and six others seemed to compete with both CR3022 and CC6.30. Structurally, the CR3022 and CC6.30 epitopes are situated so that one nanobody should not be able to simultaneously overlap with both (Fig. 3C), but it is possible that nanobody binding induces a conformational change in RBD that prevents the antibodies from simultaneous binding. Ultimately, the epitopes targeted by these 10 nanobodies remain ambiguous.
Fig. 3.

Design of bispecific antibodies and neutralization of Wuhan-1 SARS-CoV-2 PSV. (A) FACS-based epitope binning on yeast cell surface. RBD was first added to the induced yeast cells and binds to the nanobodies displayed on the surface. Then, the competitive antibody (CR3022, CC12.1, or CC6.30) was incubated and binds RBD only if the nanobody does not bind a similar epitope, or approaches RBD at an angle that is not blocked by the nanobody. (B) FACS plot showing the selection of competitive (C) and noncompetitive (NC) RBD-specific clones with CC12.1. (C) Nanobody binning based on C/NC profile and NGS analysis. Graphical representation of the three antibodies bound to RBD to show the location of their respective epitopes and Venn diagram with counts of competing nanobodies. (D) Confirmation of nanobody epitope by BLI-based assay. One representative for each of the class 2 (Nb-C2-136) and class 4 (Nb-C4-240) nanobodies is displayed. RBD was first added to the BLI sensor. Then, the competitive antibody (CR3022, CC12.1, or CC6.30) was then incubated (*) until signal saturation, followed by the tested nanobodies from our library (**). (E) Design of the bsNb4-Igs with one nanobody linked to CH1 and another nanobody linked to CL (kappa chain CK). (F) IC50 values (µg/mL) of bsNb4-Igs (Nb-C2 HC/LM18 LC, Top and Nb-C4 HC/ LM18 LC, Bottom) tested for neutralization of Wuhan-1 SARS-CoV-2 PSV. The IC50 value of LM18-Fc (2.65 µg/mL) is indicated by the purple dotted line.
A total of 45 nanobodies were selected for expression and validation to further examine the biochemical behavior of nanobodies from this library, as well as to confirm the accuracy of the on-yeast epitope binning. Twenty-one nanobodies binned to the Class 2, CC6.30-compete epitope (referred to as Nb-C2) and 24 nanobodies to the Class 4, CR3022-compete epitope (referred to as Nb-C4). Nanobodies were expressed as Fc fusion constructs, their size distribution was checked by SEC (SI Appendix, Fig. S5), and specific binding to SARS-CoV-2 RBD was confirmed by ELISA (SI Appendix, Figs. S6 and S7). To evaluate the accuracy of our FACS-based epitope binning, we performed biolayer interferometry (BLI)–based competition assays with the three antibodies used for the selection (CR3022, CC12.1, or CC6.30). The results are consistent with the on-yeast binning and validate the approach used to select our nanobodies (Fig. 3D and SI Appendix, Fig. S8).
Using Epitope Information to Develop Bispecific Constructs.
To improve the functional properties of the selected nanobodies, we sought strategies to format several nanobodies into a single molecule, aided by the epitope binning data. It has been shown that nanobodies can be readily assembled into multimers using peptide linkers, which can result in improved binding, and consequently, neutralization (6, 9, 10, 13, 15). Multimerizing a single nanobody can improve neutralization by facilitating interspike and/or intraspike cross-linking (9), and incorporating nanobodies targeting nonoverlapping epitopes has the potential added benefits of facilitating biparatopic interactions with a single subunit and consequently greater resistance to neutralization escape (6, 28). Our approach to combine multiple nanobodies was to construct a bispecific tetra-nanobody-based Ig (bsNb4-Ig) by replacing the antibody VH and VL domains with two nanobodies that target distinct RBD epitopes (Fig. 3E) (29–33). Nanobodies were linked to the CH1 and CL (kappa chain CK) domains by a flexible Gly/Ser linker, ensuring a 2:2 ratio in the bsNb4-Ig that could be easily expressed and purified using standard antibody production protocols. This linkage potentially facilitated simultaneous nanobody engagement on a single RBD, on multiple RBDs with SARS-CoV-2 spike trimer, or potentially through interspike cross-linking.
Based on our preliminary analysis of LM18 and its interesting breadth of neutralization for both SARS-CoV-1 and SARS-CoV-2 PSVs, we choose to construct bispecific designs around LM18. Deep sequencing analysis found that LM18 competed with CR3022 and CC12.1 but not CC6.30, suggesting that it may target an epitope similar to ADG20 (34). This specificity indicated that class 2 nanobodies (compete with CC6.30 only) should be capable of biparatopic RBD engagement when paired with LM18, forming a “clamp” that engages the two opposite faces of RBD. We also hypothesized that some class 4 nanobodies (compete with CR3022 only) may also be able to simultaneously engage with LM18, as LM18 binned to the compete bin for both CR3022 and CC12.1 (Fig. 3D), thus likely targets the edge of the CR3022 epitope. All nanobodies showing both specific RBD binding and monodispersed size-exclusion profiles by SEC were reformatted into the bsNb4-Ig format with a class 2 or 4 nanobody as the HC, and LM18 as the LC. SARS-CoV-2 RBD binding was first confirmed by ELISA (SI Appendix, Figs. S6 and S7) and then tested in a SARS-CoV-2 PSV neutralization assay. Sixteen out of the 20 Nb-C2/LM18 bsNb4Igs and 13 out of the 23 Nb-C4/LM18 bsNb4-Igs showed at least a 10-fold (1 µg/mL, 7 nM) and up to a 470-fold (14 ng/mL, 0.14 nM) improvement in neutralization IC50 compared to LM18-Fc, demonstrating that the additional interactions can produce large gains in neutralization potency (Fig. 3F and SI Appendix, Table S3).
A selection of the 10 most potently neutralizing bsNb4-Igs (Fig. 3F) was then assessed on a panel of SARS-CoV-2 PSV variants (SI Appendix, Table S4). Initially, all 10 bsNb4-Igs were screened against PSV variants containing single L452R (present in kappa and delta variants) and E484Q (present in kappa variant) point mutations. Two out of the five Nb-C2/LM18 bsNb4-Igs and all five Nb-C4/LM18 bsNb4-Igs neutralized these variants. This neutralization profile was consistent with the epitope binning data, as the tested mutations disrupt class 2 antibody (e.g., CC6.30) binding (1), but also demonstrated that the nanobodies within an epitope have subtly different specificities that can enhance or reduce their susceptibility to antigenic variability. The seven bsNb4-Igs that neutralized the L452R and E484Q mutants were then tested against SARS-CoV-2 VOC PSVs: beta (B.1.351), gamma (P.1), kappa (B.1.617.1), and delta (B.1.617.2) variants (Fig. 4A). Although the neutralization potency varied across the bsNb4Igs in our panel, 6/7 in our panel neutralized all 5 PSVs with an IC50 < 1 µg/mL. Several of the bsNb4-Ig where LM18 was paired with a class 4 nanobody (LM18 as LC and Nb-C4-198, Nb-C4-225, Nb-C4-237, or Nb-C4-255 as HC) neutralized all the PSVs with an average IC50 of 30 ng/mL (0.2 nM).
Fig. 4.

Neutralization of PSV variants by bsNb4-Igs. (A) Neutralization of SARS-CoV-2 PSVs by LM18-based bsNb4-Igs, LM18-Nb4-Ig, and Nb-C2/Nb-C4 bsNb4-Igs. IC50 values of clinical antibody candidates (LY-CoV16, REGN10933, and REGN-10987), CC12.1, and CC6.30 from our previous study (35) were added for reference. (B) Neutralization of omicron PSV by LM18- or Nb-C2-136-based bsNb4Igs. Omicron escaped from the clinical antibody candidates LY-CoV555, REGN-10933, and REGN-10987 and the monoclonal antibodies CC12.1 and CC6.30 (36). (C) Comparison between bsNb4-Ig and the corresponding Fabs, with IC50 as molar concentration. The black and purple dotted lines indicate the absence of neutralization (≥100 µg/mL) and the IC50 value of LM18-Fc, respectively. (D) Neutralization of Wuhan-1 SARS-CoV-2 PSV by LM18-based and affinity-matured LM18-based bsNb4-Igs.
We next constructed seven bsNb4-Igs from class 2 and 4 building blocks, predicting that these should also be capable of the biparatopic clamping interaction made by the Nb-C2/LM18 bsNb4-Igs. Nb-C2-121 or Nb-C2-136 was combined with nanobodies Nb-C4-198, Nb-C4-225, Nb-C4-237, Nb-C4-240, and Nb-C4-255. All the five of Nb-C2-136-based bsNb4-Igs could neutralize the five viral variants, albeit less potently than the clinical antibodies and the LM18-based bsNb4-Igs (Fig. 4A and SI Appendix, Table S5). When the omicron variant (B.1.1.529) was reported, all bsNb4-Igs mentioned here were tested and we found that bsNb4-Igs with Nb-C2-136 on one arm and an Nb-C4 on the other could neutralize this new variant. Omicron was resistant to LM18 and all of the bispecifics that included LM18 (Fig. 4B and SI Appendix, Fig. S9 and Table S6).
Neutralization Potency Gains from Tetravalent-Ig Format.
Several of the bsNb4-Igs constructs were able to achieve neutralization breadth and potency comparable to best-in-class antibodies recovered from SARS-CoV-2 convalescent donors (37, 38), and we wanted to better understand what enabled this activity. First, we tested all eight of the nanobody-building blocks (LM18, Nb-C2-121, Nb-C2-136, Nb-C4-198, Nb-C4-225, Nb-C4-237, Nb-C4-240, and Nb-C4-255) from our most potent bsNb4-Igs expressed as homomeric Fc fusions for the ability to neutralize Wuhan-1 SARS-CoV-2 PSV. Fc-fused LM18 and Nb-C2-136 neutralized, while the other six nanobodies-Fc (Nb-C2-121, Nb-C4-198, Nb-C4-225, Nb-C4-237, Nb-C4-240, Nb-C4-255) did not at a maximum concentration of 100 µg/mL (2.5 µM), ruling out the possibility that our tetravalent bsNb4-Ig potency was primarily due to one of the nanobodies with potent neutralizing ability (SI Appendix, Fig. S10). Next, cocktails with equimolar equivalents of LM18-Fc and either Nbs-C2-Fc or Nbs-C4-Fc were tested (SI Appendix, Fig. S10). The neutralization potency of these cocktails was roughly equivalent to that of the LM18-Fc fusion alone, indicating that the increased potency was not due to synergy between the two nanobody specificities in monospecific format but required them to be on a single molecule in the bsNb4-Igs.
To test the contributions of the two nanobody specificities formatted into a single molecule, molecular “Fabs” were produced that consisted of a single arm from the bsNb4-Igs. Bispecific nanobody-based Fabs with LM18 fused to the CL1 domain and one of the class 2 nanobodies (Nb-C2-121 or Nb-C2-136) fused to the CH1 domain showed modest increase in neutralization potency compared to the homomeric nanobody-Fc format, (Fig. 4C). In contrast, four of the five bispecific Fabs with an Nb-C4 (Nb-C4-198, Nb-C4-225, Nb-C4-237, Nb-C4-240, or Nb-C4-255) fused to the CH1 domain failed to neutralize at concentrations of 100 µg/mL (1.9 µM), and the sole variant that showed any neutralization (Fab Nb-C4-237/LM18) had reduced potency compared to the LM18 homomeric Fc fusion. These findings were unexpected, as the Nb-C4/LM18 bsNb4-Igs were more potent than the Nb-C2/LM18 bsNb4-Igs (Fig. 3A) despite the fact that class 4 nanobodies and LM18 cannot engage the same RBD simultaneously (SI Appendix, Fig. S8).
Lastly, we expressed the tetravalent Igs with four copies of the same nanobody (Nb4-Igs). In this format (Fig. 4A), LM18-Nb4-Ig showed a 340-fold increase in potency compared to the LM18 bivalent homomeric Fc fusion against Wuhan-1 SARS-CoV-2 PSV (IC50 of 0.029 µg/mL or 0.2 nM). Nb-C2-136-Nb4-Ig also showed a 20-fold increase in neutralization potency over the corresponding bivalent homomeric Fc fusion with an IC50 of 0.36 µg/mL (2.4 nM) for SARS-CoV-2 PSV. Nb-C2-121-Nb4-Ig, Nb-C4-225-Nb4-Ig, Nb-C4-240-Nb4-Ig, and Nb-246-C4-Nb4-Ig did not neutralize at concentrations of 100 µg/mL (SI Appendix, Table S7). These results highlight the LM18 specific epitope and neutralization properties, but also the advantages of bispecific constructs rather than monospecific tetravalent for neutralization. Several of the Nb-C4/LM18 bsNb4-Ig neutralized more potently than LM18-Nb4-Igs across the panel of SARS-CoV-2 PSVs, despite the inability of the Nbs-C4 to neutralize independently.
To evaluate the correlation between binding affinity and neutralization potency, we determined the binding affinities of the nanobody building blocks LM18, Nb-C2-136, and Nb-C4-225 as well as the corresponding bsNb4-Igs for SARS-CoV-2 RBD by surface plasmon resonance (SPR). All three of the individual nanobodies had relatively weak binding for SARS-CoV-2 RBD, with dissociation constants (KDs) between 143 nM and 391 nM and exhibiting a rapid dissociation rate, commonly observed with synthetic nanobodies from discovery libraries (SI Appendix, Fig. S11 and Tables S8 and S9). In the tetra-Ig format, LM18-Nb4-Ig and the Nb-C4-225/LM18 bsNb4-Ig had kinetics that were similar to the individual nanobodies (SI Appendix, Fig. S11). The Nb-C2-136/LM18 bsNb4-Ig showed a 630-fold improvement in KD to 500 pM due to a nearly nonexistent off-rate, consistent with the epitope binning that showed LM18 and Nb-C2-136 bind to nonoverlapping epitopes and could be capable of biparatopic engagement (Fig. 3D). Finally, we tested the prefusion-state-stabilized SARS-CoV-2 S trimer (38) against all the three formats, all of which bound with high affinity and had a very slow dissociation rate (SI Appendix, Fig. S11). Of note, the Nb-C2-136/LM18 bsNb4-Ig had a notably slower dissociation rate compared to the other two variants, yet this construct was the least potent neutralizer of the group. The mode of binding to the S trimer for the bsNb4-Ig is unclear and potentially heterogeneous; however, it is noted that all tetra-Igs are able to achieve intraspike avidity and substantially reduce the apparent off-rate. There was nothing in the binding data to definitively suggest why Nb-C4/LM18-based constructs would neutralize as potently as they do, but this may be partially attributed to the RBD subunits being preferentially in the “down” conformation on the stabilized S trimers (39).
Affinity Maturation of Nanobody Building Blocks.
All of the above work explored how the nanobody potency can be improved through modular multivalent formats. However, it is well established that increased antibody- or nanobody-binding affinity for a target antigen can also enhance neutralization potency. We hypothesized that affinity maturation of the base nanobodies would translate to an increase in potency for the resulting bsNb4-Igs. To test this notion, an affinity maturation library of LM18 was created using our SAMPLER maturation strategy (40), where one of all single-mutation variants of each CDR loop is generated and combined into a combinatorial library (SI Appendix, Text). This library was displayed on the surface of yeast and subjected to four rounds of FACS selections–two rounds with RBD to enrich higher affinity clones, one PSR round to deplete sticky clones, and a final round with RBD to further enrich higher affinity clones (SI Appendix, Fig. S12). At the conclusion of the selections, 96 colonies were submitted for Sanger sequencing. No clear consensus sequence was present; therefore, we selected 10 constructs for recombinant production as Fc fusions. The IC50 value of the best variant, LM18.1.17-Fc, improved neutralization potency by a factor of 6 (0.41 µg/mL or 10 nM) and 13 (0.037 µg/mL or 0.9 nM) for SARS-CoV-2 and SARS-CoV, respectively (SI Appendix, Figs. S4 and S13). SPR analysis of LM18.1.17 binding to RBD also identified a 6-fold improvement in binding affinity compared to the parental LM18 (SI Appendix, Table S8). We then incorporated LM18.1.17 instead of LM18 into the bsNb4-Igs format and measured the neutralization potency against SARS-CoV-2 PSV. In all cases, LM18.1.17-based bsNb4-Igs showed more potent neutralization compared to LM18-based bsNb4-Igs (Fig. 4D and SI Appendix, Fig. S14 and Table S10). As expected, affinity maturation of the base nanobody components translated to potency gains of the bsNb4-Igs, demonstrating that the overall function can be further improved.
Structural Validation of Engineered Nanobody Design.
To understand the structural integrity and epitope propensity of our engineered nanobodies, we determined atomic structures of a selection of our lead nanobodies (LM18, Nb-C2-136, Nb-C4-225, Nb-C4-240, and Nb-C4-255) in complex with SARS-CoV-2 RBD (X-ray crystallography, SI Appendix, Table S11) and SARS-CoV-2 S protein (cryo-electron microscopy, SI Appendix, Table S12). Our structures reveal that these nanobodies adopt a VHH fold as expected (Fig. 5A). The average rmsd of protein backbone structures between our five engineered nanobodies and a representative SARS-CoV-2 camelid nanobody (PDB ID: 7KN6) is 0.63 (0.42 to 0.93) Å when the conformationally variable CDRH3 is excluded, confirming that our synthetic VHH scaffold mimic naturally occurring nanobodies in terms of overall topology. The engineered Cys49 and Cys69 disulfide designed to maintain the structural stability in these nanobodies is evident in the X-ray electron density maps (SI Appendix, Fig. S15A).
Fig. 5.

Structural validation of engineered nanobody design. (A) Our engineered nanobodies adopt the same backbone structure as representative llama nanobodies. Structures of engineered nanobodies (i.e., LM18, class 2 Nb-C2-136, class 4 Nb-C4-225, 240, and 255) were overlapped with VHH V, a SARS-CoV-2 nanobody (PDB ID: 7KN6) isolated from an immunized llama (13). The CDRH3 of these nanobodies was not considered for rmsd calculations due to its high sequence diversity. The backbone structures of these engineered nanobodies are virtually the same as the representative llama nanobodies, VHH V (rmsd<1 Å). (B) Cryo-EM reconstruction of LM18/Nb-C2-136 bsNb4-Ig in complex with SARS-CoV-2 6Pmut7 S protein colored by subunit/domain. (C) Cryo-EM focused classification and local refinement of a single RBD bound to LM18 and Nb-C2-136. The EM map is shown in transparent gray, with fitted atomic models in backbone tube representation. (D) Modeling of RBD-LM18/Nb-C2-136 Fab complex into cryo-EM reconstruction density of LM18/Nb-C2-136 bsNb4-Ig in complex with SARS-CoV-2 6Pmut7 S protein. The density allows for approximate (not atomic resolution) placement of CH1/CL. LM18 is in blue; Nb-C2-136 in green; CH1/CL in wheat color. (E) The crystal structures of class 4 Nb-C4-225, Nb-C4-240, and Nb-C4-255 share the same binding mode to the SARS-CoV-2 RBD (Top). These three nanobodies bind the conserved epitope site previously described for CR3022 and COVA1-16 (41, 42). Coincidently, these nanobodies bind SARS-CoV-2 RBD in a highly similar way as YYDRxG antibodies, e.g., COVA1-16 (Bottom) and ADI-62113 (43). (F) Nb-C4-225, Nb-C4-240, and Nb-C4-255 share similar CDRH3 motifs as COVA1-16. CDRH3 motifs are shown in color and stick representation and SARS-CoV-2 RBD in gray and ribbon representation. Dashed lines indicate internal polar interactions within the CDRH3 motif.
The cryo-EM structure of the LM18/Nb-C2-136/ bsNb4-Ig in complex with trimeric S protein helped elucidate the mode of binding that facilitated the high binding affinity and potent neutralization. The structure shows LM18 and Nb-C2-136 binding to the same RBD in the up conformation with two of the three RBDs bound on the S protein (Fig. 5 B and C). Consistent with the binding data and our molecular clamp hypothesis, it appears that the LM18 and Nb-C2-136 nanobodies are linked to the same CH1/CL domain. A small amount of density is observed in the EM map (SI Appendix, Fig. S15C), and modeling suggests that the CH1/CL domain can adopt an ensemble of conformations with the 12 AA linkers that connect the nanobodies to the CH1/CL domain (Fig. 5D and SI Appendix, Fig. S16A). No density is visible for the Fc domain; however, the orientation of the CH1/CL domains is compatible with a single bsNb4-Ig using both arms to engage the two up RBDs on S protein (SI Appendix, Fig. S16B).
Three Different SARS-CoV-2 RBD Epitopes Targeted by Engineered Nanobodies.
The X-ray structures of the three class 4 nanobodies, Nb-C4-225, Nb-C4-240, and Nb-C4-255, in complex with SARS-CoV-2 RBD, revealed that these nanobodies bind to the CR3022 epitope site in an approach angle similar to YYDRxG antibodies (e.g., COVA1-16 and ADI-62113) that we reported recently (42, 43)(Fig. 5E). The CDRH3s of these nanobodies contribute to >90% of the buried surface area on the RBD, and structural analysis showed that the CDRH3 of Nb-C4-225, Nb-C4-240, and Nb-C4-255 forms remarkably similar β-bulge structures to those in the YYDRxG antibodies (Fig. 5F). These structures use the same tyrosines (as YY in the YYDRxG motif) to form hydrophobic and π–π interaction with highly conserved residues in the SARS-CoV-2 RBD. Glycine at 100c (corresponding to G100d in COVA1-16 CDRH3) also shifts the following residue from a down to an up register as also reported in the ADI-62113 crystal structure (43). Meanwhile, similar hydrogen bonds and salt bridges are used to maintain these β-bulge structures. Structural superimposition of these nanobodies with COVA1-16 showed that these residues are positioned in the same location in binding to the highly conserved neutralizing epitope site of SARS-CoV-2 RBD (SI Appendix, Fig. S15B). Although those three nanobodies did not compete with ACE2 binding due to their smaller size compared to COVA1-16 and other YYDRxG antibodies (SI Appendix, Fig. S8), combination of these nanobodies with LM18 or Nb-C2-136 in the bsNb4-Ig format demonstrated broad neutralization against SARS-CoV-2 variants, including omicron (Fig. 4 A and B).
Structural Validation of On-Yeast Epitope-Binning Strategy.
An atomic model of LM18 and Nb-C2-136 was generated using focused classification and local refinement methods on a single RBD in the cryo-EM data (Fig. 5 B–D and SI Appendix, Fig. S15C). LM18 binds an epitope between the class 1 (CC12.1) and class 4 (CR3022) binding sites in a manner that is predicted to sterically clash with and compete with these two antibodies (Fig. 6 A, B, and E and SI Appendix, Fig. S15). Similar to the class 4 nanobodies, approximately 88% of the buried surface area is contributed by CDRH3. Nb-C2-136 binds on the opposite face of the RBD in a region that would sterically clash with class 2 antibodies (e.g., CC6.30) (Fig. 6 A, C, and E). Unlike the other nanobodies, two CDRs are involved with binding to RBD, with CDRH2 and CDRH3 contributing approximately 30% and 50%, respectively, to the buried surface area. The epitopes of all five nanobodies, as determined by high-resolution structural analysis, validate the FACS-based epitope-binning strategy used for selection and engineering (Fig. 6 B–E).
Fig. 6.

Structural validation of on-yeast epitope binning. (A) Binding of LM18 and Nb-C2-136 with respect to antibodies representing the three epitopes used for FACS-based binding. Nanobodies were displayed as ribbons and binning epitope antibodies as transparent surfaces. Comparison of (B) LM18-, (C) Nb-C2-136-, and (D) Nb-C4-255-binding sites on RBD with representative neutralizing antibodies (space-filled structures). Nanobody atoms predicted to clash with representative antibodies are colored in yellow. (E) Three different classes of nanobodies (LM18, Nb-C2-136, and Nb-C4-255) in complex with RBD. (F) NGS analysis and CDRH3 sequences from competitive sorts with CC12.1, CR3022, and CC6.30. Counts for C and NC population with the three antibodies are indicated.
Discussion
The use of recombinantly produced monoclonal antibodies for the prophylaxis and/or therapy of RSV, Ebola virus, and SARS-CoV-2 infections has provided the foundation for a novel class of antivirals. Single-domain nanobodies offer many of the same beneficial properties of monoclonal antibodies: high binding affinity, target specificity, and, when expressed as Fc fusions, long in vivo half-life via FcRn recycling and Fc effector functions. Additionally, nanobodies offer some unique opportunities owing to their small size and the ease with which they can be multimerized. Single-domain nanobodies can access cryptic epitopes on immunization-resistant viruses, such as HIV, that are sterically less accessible to two domain antibodies (4). It also has been well established that oligomerization of nanobodies to create multimers that can simultaneously engage multiple epitopes can produce substantial gains in binding affinity and neutralization potency (6, 9, 10, 13, 15). These two distinct properties of nanobodies allow for the rational design of antivirals that can overcome some of the mechanisms that viruses use to evade antibody-mediated neutralization.
Our bsNb4-Ig platform was designed to leverage the power of nanobodies—specifically the creation of heterodimers for biparatopic target engagement—in an antibody-like molecule. The modular nature of the chain pairing enabled the rapid production and functional screening of nanobody combinations to find productive pairs. In this work, all bsNb4-Igs were expressed as human IgG1; however, the VH/VL heterodimerization strategy should be compatible with any IgG isotype, so the Fc domain can be selected for the intended in vivo application and can include mutations to extend half-life and/or modulate effector functions. Our human-based nanobody library provides a source of biochemically well-behaved nanobodies with minimal modifications from a starting human antibody VH gene, potentially reducing immunogenicity compared to camelid-derived nanobodies. The resulting tetra-specific nanobody-based constructs achieved a neutralization potency on par with SARS-CoV-2 antibodies isolated from convalescent donors, some in the ng/ml range (37, 38); meanwhile, constructs with more modest IC50 values showed breadth of neutralization across all tested variants (Fig. 4 A and B). The bsNb4-Ig format also contributed significantly to overall efficacy. As an Fc fused dimer, the nanobody LM18 neutralized SARS-CoV-2 but had a 340-fold potency increase when expressed as an Nb4-Ig homotetramer and a 175- and 470-fold increase when expressed as a bsNb4-Ig heterotetramer with the best class 2 and 4 nanobodies, respectively. We had hypothesized that the best combinations would either be Nb-C2/LM18 or Nb-C2/Nb-C4, where both nanobodies could simultaneously engage RBD. Unexpectedly, Nb-C4-237/LM18 was our most potently neutralizing variant despite the two nanobodies being sterically unable to simultaneously engage the same RBD (SI Appendix, Fig. S8).
Another unexpected finding was that Nb-C4-237 does not neutralize as an Nb4-Ig homotetramer, but the combination of this nonneutralizing nanobody with the neutralizing LM18 was more effective than the LM18 Nb4-Ig homotetramer (with an average of 10-fold more potent across the panel of VOCs). It is not clear why this combination was so effective in the bsNb4-Ig format, but these results highlight the need to have a platform that allows rapid experimental validation of different combinations as they cannot necessarily be predicted. Another unexpected finding with the class 4 nanobodies 225, 240, and 255 was their convergent mode of binding (Fig. 5 and SI Appendix, Fig. S15B). The YYDRxG-like motif ([YY(D/T/S)xxG]) in the CDRH3 found in these three nanobodies was also identified in several antibodies isolated from different convalescent COVID donors (43). While the concept of a shared antibody response is not new, previous examples of structurally convergent antibody/antigen solutions could often be attributed to biases in naïve antibody repertoire due to the limited number of immune gene segments that rearrange during B cell development. Structurally conserved responses against the influenza HA stem (44), HIV CD4–binding site (45), malaria major CSP repeat region (46), and SARS-CoV-2 RBD have been reported (47), where antibodies isolated from different donors achieve a nearly identical mode of recognition through contacts encoded largely in the starting VH gene. Examples of CDRH3 convergence have also been reported, such as for HIV apex-targeting antibodies (48); for influenza hemagglutinin receptor–binding site binders (49), and more recently the SARS-CoV-2 RBD YYDRxG–targeting antibodies (43) where a shared D gene segment encodes many of the critical contacts. Neither of these biases can explain the convergence observed with the class 4 nanobodies, as the constant VHH region does not make significant contacts with RBD and the CDRH3 diversity was randomly generated using mixed trimer phosphoramidites during oligonucleotide synthesis. Here, a synthetic antibody/nanobody has converged to the same structural motif as a B cell–derived antibody/nanobody. We also note that there are additional nanobody sequences that bin with the CR3022 site that contain a YYDRxG-like motif (SI Appendix, Fig. S17). These data would suggest that the convergent binding solutions are more attributed to the geometric compatibility of this epitope, i.e., the formation of the backbone hydrogen–bonding network and aromatic packing, than to the presence of starting antibody gene segments. It also raises the possibility that epitope immunodominance may be largely dictated by the “ease of targetability,” where certain protein surfaces are just more geometrically compatible to form extended interaction networks with the spacing of AAs coming off from a normal beta-hairpin CDRH3 loop.
In summary, our platform enables the incorporation of individual nanobody binders into an IgG-like scaffold, and the resulting biparatopic constructs present an equivalent neutralization potential to canonical antibodies (37, 38). Additionally, this approach offers the possibility to further affinity mature each building block separately, and therefore increase potency. We rapidly identified nanobodies targeting specific epitopes from our naïve library using on-yeast surface epitope binning. This workflow and the resulting bsNb4-Ig provide a unique biologic that can target viruses in ways not possible with conventional antibodies.
Materials and Methods
Discovery of Specific SARS-CoV-2 RBD Binders.
We used a combination of three rounds of MACS to deplete the library of nonbinding clones, with five rounds of FACS to enrich the population in nanobodies that target SARS-CoV-2 RBD. Specific details can be found in SI Appendix, Materials and Methods.
hVHH323 Sequencing and Analysis.
Libraries were deep sequenced to determine the CDRH3 at each round of selection on an Illumina MiSeq (Illumina Inc., San Diego, CA, USA) with the paired-end MiSeq v2 500 bp kit. Paired-end fastqs were analyzed for sequence quality and forward and reverse reads were merged, then clustered in groups with fully identical sequences. Specific details can be found in SI Appendix, Materials and Methods.
Protein Expression and Purification.
All recombinant soluble proteins from SARS-CoV, SARS-CoV-2, and their truncated protein versions (RBD) were expressed and purified as previously described (38). All antibodies, nanobodies, and nanobody-based constructs were expressed and purified from Expi293F (Thermo Fisher Scientific), and methods for purification are described in SI Appendix, Materials and Methods.
PSV Assay.
PSV assays were performed according to the protocol described by Roger et al. (38).
Crystallization and X-Ray Structure Determination.
The RBD-Nb-C4-CC12.1 complexes were crystallized by vapor diffusion using the hanging drop method. All X-ray datasets were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) and the Advanced Photon Source (APS) at Argonne National Laboratory. Information regarding X-ray data indexing, processing, and model building is described in SI Appendix, Materials and Methods.
Cryo-EM Sample Preparation and Data Acquisition.
Trimeric SARS-CoV-2 6P-Mut7 S protein was incubated with a threefold molar excess of LM18/Nb-C2-136 bsNb4-Ig at room temperature for 100 min at a concentration of 0.85 mg/mL as determined by A280. n-dodecyl-β-D-maltopyranoside was added to a final concentration of 0.06 mM and the sample deposited on plasma-cleaned Quantifoil 1.2/1.3 300 mesh grids. A Thermo Fisher Vitrobot Mark IV set to 4 °C, 100% humidity, 3 s wait time, and a 3 s blot time was used to vitrify samples in liquid ethane. Data were collected on a Thermo Fisher Titan Krios operating at 300 keV and equipped with a Gatan K2 Summit direct electron detector. Information regarding processing and model building is described in SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Henry Tien for technical support with the crystallization robot and Wenli Yu for insect cell culture. We are grateful to the staff of the SSRL beamline 12-1 and 12-2 and the beamline 23-ID-B of the APS at Argonne National Laboratory for assistance. We thank Dr. Meng Yuan for assistance with protein crystallization and Dr. James Voss and Dr. Raiees Andrabi for insightful comments on the manuscript. This work was supported by the Wellcome Trust 221705/Z/20/Z (J.G.J.) and the Bill and Melinda Gates Foundation INV-004923 (I.A.W., D.R.B., A.B.W.). This research used resources of the SSRL, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393). This research also used resources of the APS, a U.S. DOE Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Extraordinary facility operations were supported in part by the DOE Office of Science through the National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on the response to COVID-19, with funding provided by the Coronavirus CARES Act.
Author contributions
L.M.M. and J.G.J. designed research; L.M.M., H.L., G.O., Q.T., J.W., S.B., F.Z., C.K., M.V., J.L., R.L.S., X.Z., H.L.T., and P.-S.H. performed research; L.M.M., H.L., G.O., I.K., J.M.S., O.L., and M.V. contributed new reagents/analytic tools; L.M.M., H.L., G.O., J.W., J.M.S., I.A.W., and J.G.J. analyzed data; and L.M.M., H.L., G.O., I.K., D.S., P.-S.H., D.R.B., A.B.W., I.A.W., and J.G.J. wrote the paper.
Competing interests
J.G.J. and L.M.M. are listed as inventors on pending patent applications describing the hVHH323 nanobody library and anti-SARS-CoV-2 nanobodies.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
X-ray coordinates and structure factors were deposited in the RCSB Protein Data Bank (PDB) under accession code 8ELO for Nb-C4-225 in complex with SARS-CoV-2 RBD and CC12.1 Fab (50), 8ELP for Nb-C4-240 in complex with SARS-CoV-2 RBD and CC12.1 Fab (51), and 8ELQ for Nb-C4-255 in complex with SARS-CoV-2 RBD and CC12.1 Fab (52). Cryo-EM maps were deposited to the Electron Microscopy Data Bank (EMDB) under accession codes EMD-27692 (focused refinement) (53) and EMD-27693 (global refinement) (54). Atomic coordinates for the focused refinement model were deposited to the PDB under accession code 8DT8 (55). Some study data are available: the plasmids described in the manuscript will be available by MTA.
Supporting Information
References
- 1.Harvey W. T., et al. , SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baum A., et al. , Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014–1018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Genst E., et al. , Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss R. A., Verrips C. T., Nanobodies that neutralize HIV. Vaccines (Basel) 7, 77 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobkov V., et al. , Nanobody-Fc constructs targeting chemokine receptor CXCR4 potently inhibit signaling and CXCR4-mediated HIV-entry and induce antibody effector functions. Biochem. Pharmacol. 158, 413–424 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Laursen N. S., et al. , Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 362, 598–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hultberg A., et al. , Llama-derived single domain antibodies to build multivalent, superpotent and broadened neutralizing anti-viral molecules. PLoS One 6, e17665 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Detalle L., et al. , Generation and characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob. Agents Chemother. 60, 6–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoof M., et al. , An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive spike. Science 370, 1473–1479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wrapp D., et al. , Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 181, 1004–1015.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarr A. W., et al. , An alpaca nanobody inhibits hepatitis C virus entry and cell-to-cell transmission. Hepatology 58, 932–939 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Terryn S., et al. , Protective effect of different anti-rabies virus VHH constructs against rabies disease in mice. PLoS One 9, e109367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koenig P.-A., et al. , Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 371, eabe6230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J., et al. , Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature 595, 278–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salvador J.-P., Vilaplana L., Marco M.-P., Nanobody: Outstanding features for diagnostic and therapeutic applications. Anal. Bioanal. Chem. 411, 1703–1713 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Nguyen V. K., Hamers R., Wyns L., Muyldermans S., Camel heavy-chain antibodies: Diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 19, 921–930 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muyldermans S., Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Vincke C., et al. , General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 284, 3273–3284 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Riechmann L., Muyldermans S., Single domain antibodies: Comparison of camel VH and camelised human VH domains. J. Immunol. Methods 231, 25–38 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann I., et al. , Synthetic single domain antibodies for the conformational trapping of membrane proteins. Elife 7, e34317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagihara Y., Mine S., Uegaki K., Stabilization of an immunoglobulin fold domain by an engineered disulfide bond at the buried hydrophobic region. J. Biol. Chem. 282, 36489–36495 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Briney B., Inderbitzin A., Joyce C., Burton D. R., Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu Z., et al. , Landscape of variable domain of heavy-chain-only antibody repertoire from alpaca. Immunology 161, 53–65 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y., et al. , Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 26, 663–670 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Chan B. M., Badh A., Berry K. A., Grauer S. A., King C. T., Flow cytometry-based epitope binning using competitive binding profiles for the characterization of monoclonal antibodies against cellular and soluble protein targets. SLAS Discov. 23, 613–623 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Hudson E. P., Uhlen M., Rockberg J., Multiplex epitope mapping using bacterial surface display reveals both linear and conformational epitopes. Sci. Rep. 2, 706 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes C. O., et al. , SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 588, 682–687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., et al. , A human Bi-specific antibody against zika virus with high therapeutic potential. Cell 171, 229–241.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi S. Y., et al. , A biparatopic agonistic antibody that mimics fibroblast growth factor 21 ligand activity. J. Biol. Chem. 293, 5909–5919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ljungars A., et al. , A bispecific IgG format containing four independent antigen binding sites. Sci. Rep. 10, 1546 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller K. M., Arndt K. M., Strittmatter W., Plückthun A., The first constant domain (CH1 and CL) of an antibody used as heterodimerization domain for bispecific miniantibodies. FEBS Lett. 422, 259–264 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Pekar L., et al. , Biophysical and biochemical characterization of a VHH-based IgG-like bi- and trispecific antibody platform. MAbs 12, 1812210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brinkmann U., Kontermann R. E., The making of bispecific antibodies. MAbs 9, 182–212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan M., et al. , A broad and potent neutralization epitope in SARS-related coronaviruses. Proc. Natl. Acad. Sci. U.S.A. 119, e2205784119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao F., et al. , Engineering SARS-CoV-2 neutralizing antibodies for increased potency and reduced viral escape pathways. iScience 25, 104914 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yunlong C., et al. , Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wec A. Z., et al. , Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 369, 731–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers T. F., et al. , Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan R., et al. , Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 31, 717–719 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao F., et al. , Broadening a SARS-CoV-1 neutralizing antibody for potent SARS-CoV-2 neutralization through directed evolution. bioRxiv [Preprint] (2021). 10.1101/2021.05.29.443900. (Accessed 9 February 2022). [DOI] [PMC free article] [PubMed]
- 41.Yuan M., et al. , A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H., et al. , Cross-neutralization of a SARS-CoV-2 antibody to a functionally conserved site is mediated by avidity. Immunity 53, 1272–1280.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu H., et al. , Human antibodies to SARS-CoV-2 with a recurring YYDRxG motif retain binding and neutralization to variants of concern including omicron. Commun. Biol. 5, 766 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen F., Tzarum N., Wilson I. A., Law M., VH1-69 antiviral broadly neutralizing antibodies: Genetics, structures, and relevance to rational vaccine design. Curr. Opin. Virol. 34, 149–159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West A. P. Jr., Diskin R., Nussenzweig M. C., Bjorkman P. J., Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc. Natl. Acad. Sci. U.S.A. 109, E2083–E2090 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pholcharee T., et al. , Diverse antibody responses to conserved structural motifs in Plasmodium falciparum circumsporozoite protein. J. Mol. Biol. 432, 1048–1063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu N. C., et al. , An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 33, 108274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrabi R., et al. , Identification of common features in prototype broadly neutralizing antibodies to HIV envelope V2 apex to facilitate vaccine design. Immunity 43, 959–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt A. G., et al. , Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H., Wilson I. A., Crystal structure of SARS-CoV-2 spike protein receptor-binding domain in complex with antibody CC12.1 Fab and nanobody Nb-C4-225. Protein Data Bank. https://www.rcsb.org/structure/8ELO. Deposited 26 September 2022. [Google Scholar]
- 51.Liu H., Wilson I. A., Crystal structure of SARS-CoV-2 spike protein receptor-binding domain in complex with antibody CC12.1 Fab and nanobody Nb-C4-240. Protein Data Bank. https://www.rcsb.org/structure/8ELP. Deposited 1 November 2022. [Google Scholar]
- 52.Liu H., Wilson I. A., Crystal structure of SARS-CoV-2 spike protein receptor-binding domain in complex with antibody CC12.1 Fab and nanobody Nb-C4-255. Protein Data bank. https://www.rcsb.org/structure/8ELQ. Deposited 27 September 2022. [Google Scholar]
- 53.Ozorowski G., Turner H. L., Ward A. B., LM18/Nb136 bispecific tetra-nanobody immunoglobulin in complex with SARS-CoV-2-6P-Mut7 S protein (focused refinement). Electron Microscopy Data Bank. https://www.ebi.ac.uk/emdb/EMD-27692. Deposited 25 July 2022. [Google Scholar]
- 54.Ozorowski G., Turner H. L., Ward A. B., LM18/Nb136 bispecific tetra-nanobody immunoglobulin in complex with SARS-CoV-2-6P-Mut7 S protein (global refinement). Electron Microscopy Data Bank. https://www.ebi.ac.uk/emdb/EMD-27693. Deposited 25 July 2022. [Google Scholar]
- 55.Ozorowski G., Turner H. L., Ward A. B., LM18/Nb136 bispecific tetra-nanobody immunoglobulin in complex with SARS-CoV-2-6P-Mut7 S protein (focused refinement). Protein Data Bank. https://www.rcsb.org/structure/unreleased/8DT8. Deposited 25 July 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
X-ray coordinates and structure factors were deposited in the RCSB Protein Data Bank (PDB) under accession code 8ELO for Nb-C4-225 in complex with SARS-CoV-2 RBD and CC12.1 Fab (50), 8ELP for Nb-C4-240 in complex with SARS-CoV-2 RBD and CC12.1 Fab (51), and 8ELQ for Nb-C4-255 in complex with SARS-CoV-2 RBD and CC12.1 Fab (52). Cryo-EM maps were deposited to the Electron Microscopy Data Bank (EMDB) under accession codes EMD-27692 (focused refinement) (53) and EMD-27693 (global refinement) (54). Atomic coordinates for the focused refinement model were deposited to the PDB under accession code 8DT8 (55). Some study data are available: the plasmids described in the manuscript will be available by MTA.