


Abstract
Most studies on the structure of DNA in telomeres have been dedicated to the double-stranded region or the guanosine-rich strand and consequently little is known about the factors that may bind to the telomere cytosine-rich (C-rich) strand. This led us to investigate whether proteins exist that can recognise C-rich sequences. We have isolated several nuclear factors from human cell extracts that specifically bind the C-rich strand of vertebrate telomeres [namely a d(CCCTAA)n repeat] with high affinity and bind double-stranded telomeric DNA with a 100× reduced affinity. A biochemical assay allowed us to characterise four proteins of apparent molecular weights 66–64, 45 and 35 kDa, respectively. To identify these polypeptides we screened a λgt11-based cDNA expression library, obtained from human HeLa cells using a radiolabelled telomeric oligonucleotide as a probe. Two clones were purified and sequenced: the first corresponded to the hnRNP K protein and the second to the ASF/SF2 splicing factor. Confirmation of the screening results was obtained with recombinant proteins, both of which bind to the human telomeric C-rich strand in vitro.
INTRODUCTION
Telomeres, the extremities of eukaryotic chromosomes, have recently become the focus of intense and highly competitive biological research (1–9). Telomeres are essential for the maintenance of genome integrity (10,11). Telomeres also participate in various aspects of the functional organisation of the nucleus (12). They play a role in homologous pairing (13), timing of DNA replication (14) and recombination (15–17) and they exert positional effects on transcription (18). Yeast telomeres are also considered to be heterochromatin-like regions that serve as molecular sinks for factors involved in chromatin-mediated repression of gene expression (19).
In vertebrates and in many other organisms, the ‘natural’ ends of chromosomes are composed of tandemly repeated DNA sequences with a sequence disequilibrium leading to a guanine-rich (G-rich) strand (%G = 50% for vertebrates) oriented 5′→3′ towards the end of the chromosome. This G-rich strand is longer than the complementary cytosine-rich (C-rich) strand and a 3′-overhang of the G-rich strand is observed (20–22). At least two distinct structural domains have been described within telomeric chromatin. One reflects the binding of specific proteins to the single-stranded 3′-overhang. This domain constitutes the very end of chromosomes and is essential for chromosome capping and telomerase regulation (23,24). The second corresponds to the double-stranded telomeric repeats, which appear to be organised, at least in part, in a non-nucleosomal manner. This domain contains specific telomeric factors that play a critical role in telomere maintenance (10,25–30). The structural and functional relationships between these two domains remain largely unknown.
Most structural and protein-binding studies on telomeres have been dedicated to the duplex region or the single-stranded G-rich strand of telomeres (23,31–47). Less work has been performed on proteins that may recognise the C-rich strand (48,49). It has been shown that a fragment of four repeats of cytosines corresponding to the sequence of the C-rich strand of vertebrate telomeres may form a pH-dependent intramolecular folded structure called ‘i-DNA’ (50–54). i-DNA has been observed for a large number of C-rich DNA sequences, but not for RNA (55,56). This prompted us to determine whether nuclear factors could bind to DNA sequences mimicking the C-rich strand of telomeres. Furthermore, results obtained on Saccharomyces cerevisiae (57) and human cells (22) suggest that a shortening of the C-rich strand might be mediated by recognition of the C-rich strand. Other biologically relevant sequences may also form this motif (58,59) and thus C-rich sequence-binding proteins might not be limited to telomeres but might be shared between telomeres and other chromosomal locations. In particular, proteins that bind single-stranded C-rich sequences have been described for the c-myc promoter (60) and for the Drosophila centromeric dodeca-satellite (61,62).
MATERIALS AND METHODS
Oligonucleotides, polynucleotides and chemicals
Oligodeoxyribonucleotide and oligoribonucleotide probes were synthesised by Eurogentec (Belgium) on the 0.2 µmol scale and treated as previously described (55). All oligonucleotide concentrations were expressed in strand molarity, using calculated absorption coefficients (63) for the unfolded species. dT26 and ds26 were used as non-specific competitors (their respective sequences are reported in Table 1). The sequences of all other oligonucleotides and polynucleotides are given in Table 1. tRNA from Escherichia coli MRE600 and calf thymus DNA were obtained from Boehringer Mannheim, poly(dC) and poly(rC) from Pharmacia Biotech, molecular weight markers from Novex, New England Biolabs and Amersham and all other chemicals from Sigma. For equivalence purpose, 0.5 µg/µl of oligonucleotide or polynucleotide represents ~1.5 mM nucleotides or 60 µM 26mer.
Table 1. Sequence and competition efficacy of the different competitors.

aStructure of the oligo/polynucleotides is indicated. i-DNA/ss means that the oligonucleotide may fold into an i-motif at slightly acidic pH but remains single-stranded at basic pH. For poly(rC) and poly(dC) i-DNA structure is suspected but has not been demonstrated. The most stable i-motif resulted from folding of oligonucleotides 29h and 29i. 27h, 21i and 21h give i-DNA of intermediate stability. 17h gave a very unstable intramolecular i-DNA structure at neutral pH. ss, single stranded; ds, double-stranded.
bCompetition efficacy was characterised by the first concentration sufficient to totally compete with the probe (conditions identical to Fig. 3). For the first half of the table (27h to 27dx) competitors ranked ++++ are able to compete at a stoichiometric ratio (10 nM), competitors ranked +++ compete at 100 nM, ++ at 1 µM, + at 10 µM, +/– partially compete at 10 µM and – show no competition at 10 µM. For the second half (ds26 to tRNA) only a 0.5 µg/µl concentration of competitor was used: ++ means that at this concentration competition was complete, + competition was partial, +/– competition was weak and – no competition was observed.
cCompetition was also evaluated at high protein/probe concentrations (0.9 µg/µl and 1 µM, respectively) and the competitors were used at 20 µM (27h to R27) or 0.5 µg/µl (ds26 to tRNA). This concentration corresponds to a nucleotide concentration of 1.5 mM. The nomenclature is the same as for the second part of footnote b.
d21x3 is not able to form an intramolecular i-motif, therefore its Tm is concentration dependent and below 15°C at 1 µM strand concentration and pH 6.0.
eThe letters used designate the bands that were most efficiently competed, according to the nomenclature used in Figure 1.
f9h is not able to form an intramolecular i-motif and the Tm is lower than 10°C at pH 6 and 6 µM.
gEscherichia coli single-stranded DNA was obtained by fast cooling of boiled E.coli DNA, unable to anneal properly with this protocol.
n.d., not determined.
Nuclear extracts
HeLa nuclear extracts, transcription grade (8.5–9 mg/ml), were purchased from Promega. Primary human fibroblasts were obtained from breast biopsies (mean donor age 45 years) and cultivated in MEM medium supplemented with 10% FCS for 4–15 passages. Human fibroblast extracts were prepared according to a published protocol (64) with little modification (65). Nuclear extracts from ‘young’ primary fibroblasts (four independent preparations of cells at the fourth passage) and senescent primary fibroblasts (two independent preparations of cells at the fifteen passage) were prepared.
Antibodies
12g4, a mouse monoclonal antibody directed against the hnRNP K protein (66), a kind gift of Prof. G. Dreyfuss, was used at 1/1000 dilution. mAb 104, a mouse monoclonal antibody against the RS domain of SF2 (67), was used at 1/50 dilution.
Electrophoretic mobility shift assay (EMSA)
The C-rich strand was 32P-end-labelled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP according to the manufacturer’s protocol. The binding reaction was performed for 15 min at room temperature or 4°C with 0.017–18 µg of nuclear extract in 10 µl of 50 mM HEPES pH 7.2, 100 mM KCl, 1 mM MgCl2, 10% sucrose and 5 µg of a non-specific competitor (dT26). If required, nuclear extracts were diluted in a 50 mM Tris (pH 7.5) buffer containing 0.1 µg/µl BSA (New England Biolabs). The solution contained 0.1 pmol (10 nM) of the end-labelled probe (20 000–40 000 c.p.m.). Where indicated, the incubation mixture also contained a large excess of a double-stranded or single-stranded unlabelled oligonucleotide competitor. The binding mixtures were electrophoresed at room temperature or 4°C for 90 min (10 V/cm) on a 8% non-denaturing polyacrylamide gel (acrylamide:bisacrylamide 29:1) in 22 mM Tris, 22 mM borate, 0.1 mM EDTA, pH 8.3 (0.25× TBE) dried and analysed.
Crosslinking of radiolabelled oligonucleotides with HeLa cell extracts
A binding reaction, similar to that presented in the EMSA experiments was incubated on ice for 15 min, then crosslinked to the radiolabelled 27h probe with a germicidal lamp (6 W at 254 nm) for 1 h on ice, loaded on a denaturing 8–16% SDS–polyacrylamide gel (Novex), run at 15 V/cm for 90 min, fixed with acetic acid (10%)/ethanol (10%), dried and analysed.
Southwestern analysis
Aliquots of 1–5 µg of HeLa nuclear extracts or bacterial extracts or 50–250 ng of recombinant proteins were loaded on a denaturing 8–16% SDS–polyacrylamide gel (Novex). After a 90 min migration at 15 V/cm the gel was electrotransferred to a polyvinylidene difluoride (PVDF) or nitrocellulose membrane (Amersham). This membrane was soaked in a 6 M guanidine hydrochloride (Gn-HCl) buffer and renatured at 4°C in buffer containing decreasing concentrations of Gn-HCl (6, 4, 3, 2, 1, 0.5, 0.25 and 0 M) over a 3 h period (68). The membrane was then blocked for 1 h in 5% non-fat dried milk at 4°C and hybridised with the telomeric probe (2–20 pmol) overnight at 4°C in 10 ml of 50 mM HEPES, pH 7.2, 0.1 M KCl, 1 mM MgCl2, 1 mM DTT, 1 mM EDTA and 10% glycerol with 10 µg/ml of dT26 and ds26 as non-specific competitors. After three washes in a similar buffer, the membranes were analysed with an SP PhosphorImager (Molecular Dynamics).
Characterisation of the DNA-binding proteins
An EMSA assay was performed on a 2% agarose (1× TBE, 1.5 mm thick) vertical gel, using a fluorescein-labelled telomeric oligonucleotide (F26 5′-fluo-TTTAACCCTAACCCTAACCCT-AACCC-3′; 69) at 2 µM strand concentration and 45 µg of HNE in 50 mM MES, pH 6.5, 0.1 M NaCl, 10 mM MgCl2, 5% sucrose, 0.6 µg/µl dT26. The retarded band was cut from the gel, melted at 95°C in the presence of an equivalent amount of 2× Laemmli loading buffer and directly loaded on an 8–16% polyacrylamide denaturing gel (Novex). After a 90 min migration at 15 V/cm, the gels were either silver stained (using the Novex kit) or electrotransferred onto a nitrocellulose membrane (Amersham). This membrane was then renatured and hybridised as for a southwestern analysis.
λgt11 expression library
A total of 300 000 independent clones (30 000 per 140 mm Petri dish) of a λgt11-based HeLa library (Clontech) were tested in each experiment using either the 27h or 29i radiolabelled probe. Screening was performed as previously described (70–72) but with all binding and washing reactions performed at 4°C in 50 mM MES buffer, pH 6, containing 50 mM NaCl, 10 mM MgCl2, 1 mM DTT. The binding buffer contained 10–50 µg/ml of non-specific single-stranded DNA (dT26). After three rounds of selection, one clone was positive in each experiment. Phage DNA was sequenced directly by Eurogentec (Seraing, Belgium) and Blast searches (http://www.ncbi.nlm.nih.gov ) were performed.
Recombinant proteins
ASF/SF2 was expressed in bacterial strain TG1 transfected with a plasmid containing ASF c-DNA. ASF/SF2 deletion mutants (197C, 215C, Δ197, Δ207, 197N and 210N) were generated by PCR amplification (73). GST–hnRNP K fusion constructs (74) were overexpressed in E.coli XL1 blue carrying an IPTG-inducible plasmid and crude extracts were obtained using the following protocol. Pellets of bacteria from induced or non-induced culture were resuspended in cold phosphate-buffered saline (PBS) containing 0.1% Triton X100 in the presence of lysozyme (0.5 mg/ml) and kept on ice for 30 min. The samples were then sonicated, centrifuged and the surpernatant supplemented with glycerol (10% final concentration). Protein concentration was estimated using the Bio-Rad Protein Assay kit and the protein extracts were stored at –80°C.
Western blot analysis
Western blots on membranes following SDS gel electrophoresis were performed as follows. The membrane was incubated in PBS, 0.05% Tween 20, 5% non-fat dried milk for 6 h at 4°C, washed in PBS, 0.05% Tween 20 (PBST) and incubated overnight at 4°C with the primary antibody in PBST, 5% non-fat dried milk. After three washes in PBST, the membrane was incubated for 2–3 h at room temperature with a 1/1000 dilution of the horseradish peroxidase-conjugated secondary antibody [anti-Ig (Amersham) for 12g4 and anti-IgM (Sigma) for mAb 104] and then washed with PBST. After a final wash in PBS detection was with the ECL-plus kit (Amersham).
We also performed western blot analysis of non-denaturing gels (EMSA). Briefly, gel shift assays were performed as usual (using 10 pmol of the specific probe and 9 µg of HNE). The gel was then transferred overnight by capillarity to a nitrocellulose membrane (Amersham) in 1× TBE buffer containing 0.5 mM DTT. The membrane was then blocked and revealed as a classic western blot.
RESULTS
Gel retardation assay
A 27mer oligonucleotide (27h) containing four repeats of the telomeric sequence (CCCTAA) was used as a probe for protein binding. In the absence of competitors, HeLa nuclear extract contained several factors that bound to the telomeric (CCCTAA)n probe (27h) at 4°C (Fig. 1, lane 2; see Table 1 for sequence). Depending on the quality of the gel, the protein:probe ratio and the absolute amount of probe used, up to five shifted bands were observed and labelled A, B, C, K and D, with A referring to the material remaining in the wells (see Fig. 1). Adding a single-stranded competitor (dT26) abolished some, but not all, of these binding activities; two major shifted bands (C and K) were still observed (Fig. 1, lane 3). A double-stranded competitor (ds26) did not compete. When both competitors were added together the profile was identical to the lane with the single-stranded competitor alone (Fig. 1, lanes 4 and 5). The binding was salt-resistant (up to 1 M KCl) and magnesium-independent (up to 0.1 M MgCl2), temperature-sensitive (incubation >55°C abolished the retarded complexes), RNase-A-insensitive and proteinase K-sensitive (data not shown). These results are in good agreement with recognition by proteins which do not require RNA cofactors. When incubation and electrophoresis were performed at room temperature, the profiles remained qualitatively the same, but the binding activity was generally reduced (data not shown). The Tm of the i-motif of the 27h probe is ~20°C at pH 7.2. Therefore, the oligonucleotide is expected to be folded when incubation is performed at 4°C. However, we cannot exclude that proteins might thus be able to unfold the i-structure and bind the single strand.
Figure 1.
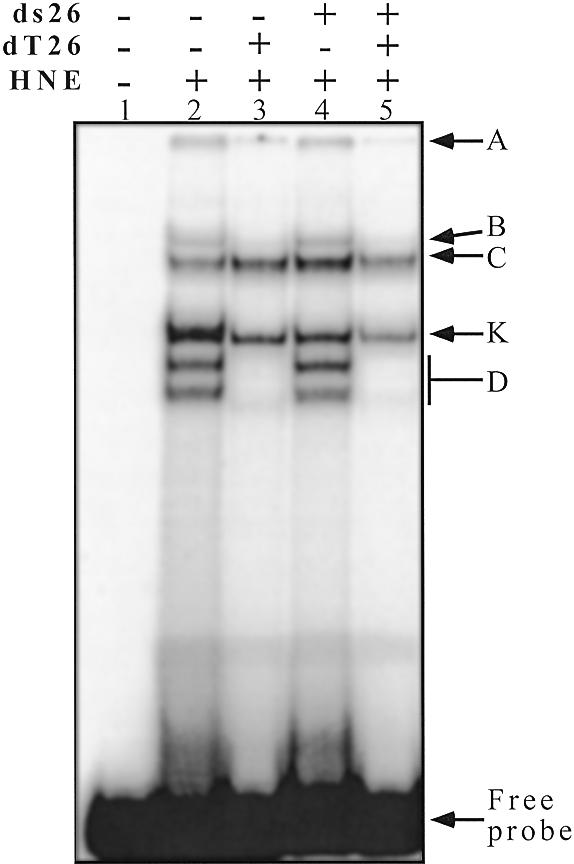
EMSA with a radiolabelled telomeric C-rich strand (27h) and HeLa nuclear extracts. The probe (1 µM 27h) was incubated in 50 mM HEPES buffer pH 7.2, containing 0.1 M KCl, 1 mM MgCl2 and 5% sucrose (15 min on ice) and run at 4°C on an 8% polyacrylamide gel (see Materials and Methods). Lane 1, 27h probe alone; lanes 2–5, HNE 0.9 µg/µl. The competitors dT26 (lanes 3 and 5) and ds26 (lanes 4 and 5) were added at 0.5 µg/µl each. A, B, C, K and D refer to the names of the bands used in the text.
Several EMSA were carried out to evaluate the affinity of the proteins in the different retarded complexes. We first tested the effect of protein concentration on the EMSA profile, using a small amount (0.1 pmol) of the radiolabelled 27h probe with a large excess (6000×) of single-stranded competitor (Fig. 2). Small amounts of crude HeLa nuclear extracts were sufficient to observe the retarded complexes (8.5 ng/µl in Fig. 2, lane 4). The K band was the first to appear. The C band showed the same variation (but the signal was less intense in Fig. 2 compared to Fig. 1). Only 0.85 µg/µl of HNE were necessary to produce complete disappearance of the free probe, showing that telomeric C-rich strand-binding proteins are relatively abundant in these nuclear extracts. At high protein:probe ratios, both the K and C bands disappeared and the D band predominated (Fig. 2, lanes 8–10). These experiments led us to conclude that the best EMSA signal with our HeLa nuclear extract was obtained with a protein:probe ratio of 4 µg/pmol. The proteins in the D band were bound to the probe when nuclear extract was in large excess (>2 µg/pmol), whereas the K band encompassed proteins that bound to the probe for protein:probe ratios between 1 and 10 µg/pmol.
Figure 2.
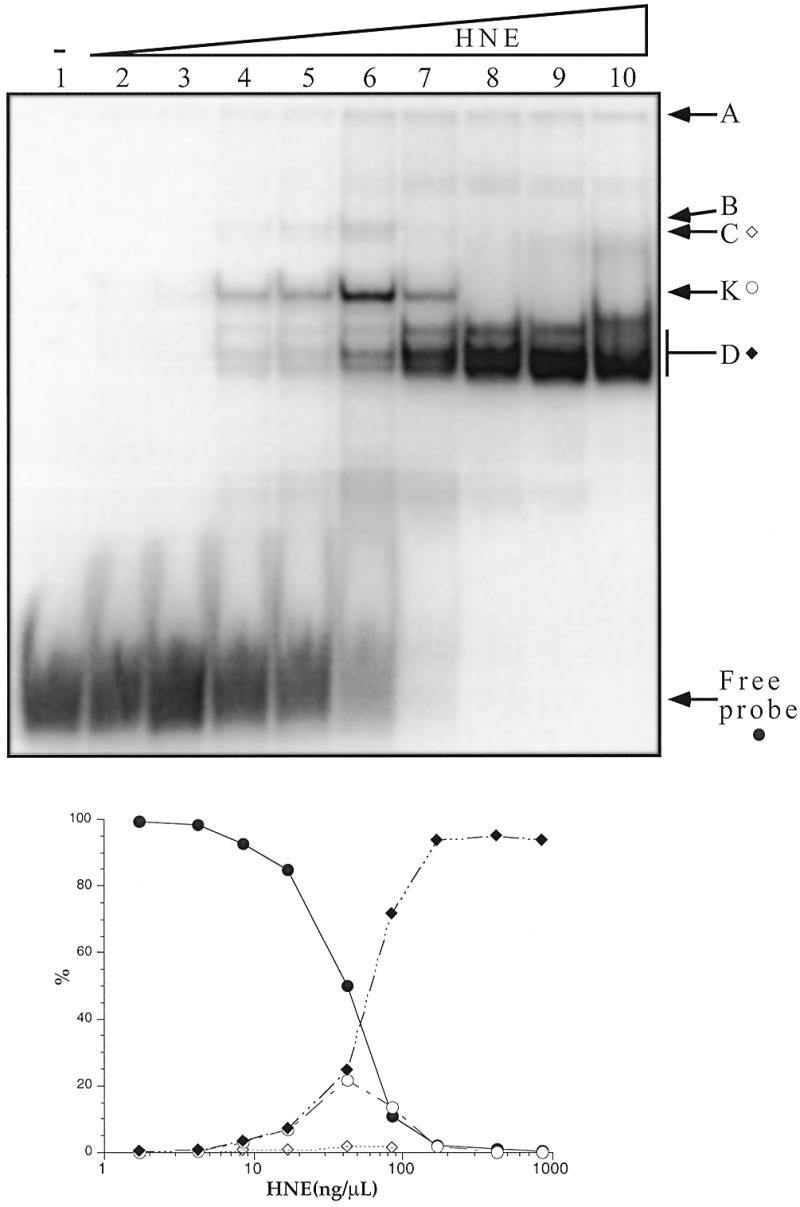
(Upper) EMSA with the 27h probe and increasing concentrations of HeLa nuclear extracts. The radiolabelled probe (10 nM) was incubated in 50 mM HEPES buffer pH 7.2, containing 0.1 M KCl, 1 mM MgCl2 and 5% sucrose, in the presence of ds26 and dT26 (0.5 µg/µl each) and various concentrations of HNE for 15 min on ice and run at 4°C on an 8% polyacrylamide gel (see Materials and Methods). Lane 1, no protein; lanes 2–10, increasing concentrations of HNE (1.7, 4.25, 8.5, 17, 42.5, 85, 170, 425 and 850 ng/µl). (Lower) Quantification of the radioactivity in the different bands divided by the total radioactivity in the lane. Closed circle, free probe; closed diamond, band D; open circle, band K; open diamond, band C.
Competitive binding experiments were performed to assess the sequence specificity of these binding activities. A list and the sequences of the oligonucleotides used is reported in Table 1. This analysis was carried out at low protein/probe concentration to allow weak competitors to be revealed. As expected we observed specific competition upon increasing the concentration of 27h (from 10 nM to 10 µM). A 10-fold excess (100 nM) was sufficient to displace most of the radiolabelled probe (Fig. 3). We classified the competitors according to the minimal oligonucleotide concentration sufficient to compete with the 27h probe. Competitors ranked ++++ were able to compete at a stoichiometric level (10 nM), competitors ranked +++ competed at 100 nM, ++ at 1 µM, + at 10 µM, +/– partially competed at 10 µM and – show no competition at 10 µM. The results of the classification are reported in Table 1. The best competitors, 29h and 29i, both possessed four repeats of five cytosines. Oligonucleotides with four repeats of three cytosines were next in competition efficacy. Weak or no competition was observed with non-C-rich sequences (27sc, 27sg and 21ct), RNA oligoribonucleotides (R27 and 21Rh) or DNA sequences that did not contain four blocks of cytosines (21x3, 21mix, 9h and 17h). An intramolecular telomeric duplex (27dx) competed weakly, but led to the appearance of a new band of lower mobility suggesting that 27dx interacts directly with the 27h probe. Competition experiments performed at high protein/probe concentrations (0.9 µg/µl and 1 µM, respectively) confirmed the results obtained at low concentrations (Table 1). The conclusion from these competitions is that DNA-binding activities detected in this study have a preference for C-rich sequences that contain several repeats of at least three cytosines.
Figure 3.

Competition assays with different oligonucleotides. The 27h probe (10 nM) was incubated with 42.5 ng/µl of HNE in 50 mM HEPES buffer pH 7.2, containing 0.1 M KCl, 1 mM MgCl2, 5% sucrose, 0.5 µg/µl dT26 and 0.5 µg/µl ds26 for 15 min on ice and run at 4°C on an 8% polyacrylamide gel (see Materials and Methods). For each competitor, concentrations of 10 nM, 100 nM, 1 µM and 10 µM (from left to right) were used. The sequences of the competitors are reported in Table 1. It should be noted that oligonucleotides that compete poorly, such as 17h and 21ct, are not recognised by other nuclear HeLa proteins (not shown).
Competition experiments were also performed with higher molecular weight nucleic acids in the presence of dT26 (Table 1, lower part). Poly(dC) and dC26 were very efficient competitors. This confirmed the specificity of these nucleic acid-binding activities for C-rich sequences [dC26, poly(dC) and poly(rC)].
Determination of the protein molecular weights
Different approaches were tested to obtain information on C-rich DNA-binding proteins.
UV crosslinking approach. An estimate of the protein molecular weight was obtained using UV crosslinking experiments (48,49). At low probe concentration (10 nM), a major covalent complex was observed with a migration distance corresponding to a molecular weight of 50 kDa. Without UV irradiation, no retarded complex was observed, showing that the interactions were effectively disrupted in denaturing SDS gels. At a higher probe concentration, the major crosslinked complex migrated around 70–75 kDa. The free oligonucleotide itself migrated as an ~10 kDa peptide under these conditions and the profile was not altered by UV irradiation (data not shown). Adding a large excess of the non-radiolabelled probe or a specific competitor (10 µM) abolished the crosslinked bands, whereas non-specific competitors had no effect, which confirmed the specificity of binding (data not shown). As the molecular weight given by this method corresponds to the migration of a polypeptide–DNA covalent complex, it is somewhat difficult to deduce the precise molecular weight of the polypeptide alone.
Southwestern approach. To circumvent this problem, a southwestern approach was used. Briefly, a crude nuclear extract was loaded on a denaturing SDS gel, transferred to a membrane, renatured and hybridised with radiolabelled 27h in the presence of a large excess of single- and double-stranded competitors. Four major radioactive bands (Fig. 4) were revealed with this method (molecular weights 66, 64, 45 and 32 kDa). These bands were not observed when unrelated radiolabelled DNA oligonucleotides were tested (dT26, 27sc and 27sg). In contrast, other C-rich probes (29i, 29h, 21h and 45h) revealed the same pattern of hybridisation. In this experiment the position of the radioactive band is in direct relation to the molecular weight of the protein itself. On the other hand, this method does not provide an exhaustive list of 27h-binding proteins as some proteins may not be renaturated properly. Furthermore, in the case of multiprotein complexes gel electrophoresis followed by transfer to the membrane should prevent any reconstitution of the complex, even if the proteins are renatured properly. Putative 27h-binding multiprotein complexes are therefore lost in this experiment. To assess if the proteins revealed by the 27h* probe bound selectively to the telomeric C-rich strand, parallel southwestern experiments were performed (Fig. 4) with labelled telomeric C-rich strand (21h*), its complementary G-rich strand [21g*, (GGGTTA)3GGG] or a duplex formed by these two sequences (21h/21g*). For comparison purposes the intensities were adjusted: the signal was 10× weaker with 21g* and 100× in the case of 21h/21g*. The southwestern pattern obtained with the G-rich strand (Fig. 4, 21g*) was different from that obtained with the C-rich probes (Fig. 4, 27h* and 21h*), showing that G- and C-rich strand-binding activities were distinct. Surprisingly, the pattern obtained with the duplex (Fig. 4, 21h/21g*) shared similarities with that obtained with the C-rich strand, but with a 100× weaker affinity, suggesting that some of the C-rich strand-binding proteins may also recognise a telomeric duplex sequence, but with a strongly reduced affinity (at least 100×).
Figure 4.
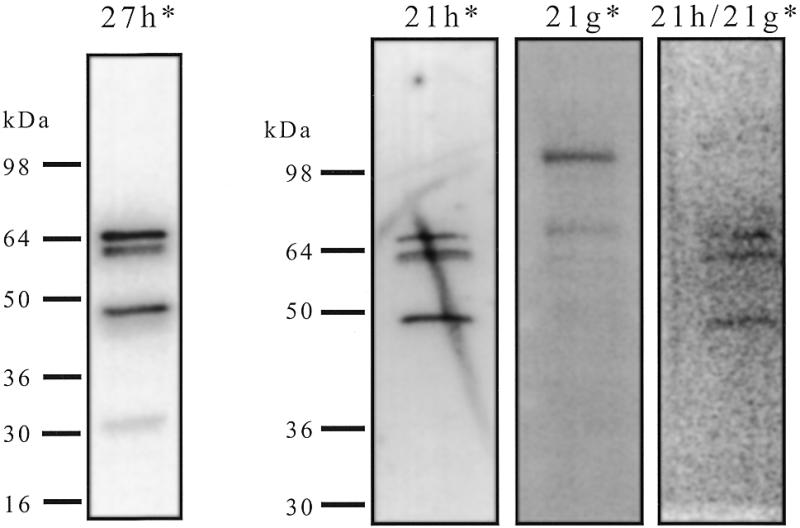
Southwestern analysis of HNE with different probes. Aliquots of 4.5 µg of HNE were run on an 8–16% SDS–PAGE gel and transferred onto a PVDF membrane. After blocking and renaturation each strip of the membrane was hybridised with a different radiolabelled probe (0.4 nM) (27h*, 21h*, 21g* or a preformed duplex 21h/21g*) in HEPES buffer pH 7.2, in the presence of single- and double-stranded competitors, washed three times and exposed on a phosphorimager screen. *, the radiolabelled strand. The position of molecular weight markers was obtained using coloured markers transferred to the membrane.
Gel slice approach. Affinity purification was attempted to confirm the molecular weight of the proteins. A fluorescent oligonucleotide (F26; 69) was incubated in the presence of 45 µg of nuclear extract and loaded on a 2% agarose gel (Fig. 5A). Only one retarded band was observed as a result of the poor resolution of this agarose gel. A slice corresponding to the fluorescent retarded band was cut, melted at 95°C, mixed with Laemmli buffer and run on a 8–16% SDS–polyacrylamide gel. Silver staining revealed several proteins. Among them, at least two (66 and 45 kDa) were absent in the control lane (Fig. 5B, lane 3) and a third band (35 kDa) was more intense in the specific lane (Fig. 5B, lane 2). To confirm that these proteins have C-rich strand-binding activity a duplicate of this gel was transferred to a nitrocellulose membrane, renatured and tested in a southwestern assay. As shown in Figure 5C, these three polypeptides bound 27h after renaturation (Fig. 5C, lane 2) and correspond to the bands found in the nuclear extract (Fig. 5C, lane 1) whereas no binding activity was revealed in the control lane (Fig. 5C, lane 3).
Figure 5.

Partial characterisation of the C-rich strand-binding proteins. (A) EMSA with a fluorescein labelled C-rich strand telomeric oligonucleotide (F26) in a 2% vertical agarose gel. Incubation in MES buffer and migration were performed at 4°C (see Materials and Methods). Fluorescence was revealed with a UV table (312 nm). Lane 1, F26 alone; lane 2, F26 + HNE; lane 3, HNE. (B) Agarose slices cut from the previous EMSA corresponding to the retarded band and to the control lane were loaded on an 8–16% SDS–PAGE gel. The gel was then silver stained. Lane 1, molecular weight marker; lane 2, F26 + HNE from the EMSA; lane 3, HNE from the EMSA. (C) Southwestern analysis on a duplicate of the gel presented in (B). Hybridisation overnight with labelled 27h in 50 mM MES pH 6.0, containing dT26 (10 µg/ml) at 4°C. Lane 1, crude HNE; lane 2, F26 + HNE from the EMSA; lane 3, HNE from the EMSA.
Fraction purification. Fraction Purification by gel filtration on Superdex 75 was also attempted but the binding activity (tested by EMSA) of all positive fractions was extremely labile and lost upon concentration or precipitation (data not shown). Nevertheless, this approach revealed three different retarded bands corresponding to different fractions: a weak band corresponding to a native apparent molecular weight of 36 ± 4 kDa, another band corresponding to 45 ± 5 kDa and the strongest band split between several fractions (molecular weight 65 ± 7 kDa).
Taken together, these results show that several distinct proteins are able to bind to the telomeric C-rich strand probe. We could identify at least three different proteins that recognise the 27h oligonucleotide. Their respective molecular weights are 35, 45 and 65 kDa. The similarities between the native weights (as inferred from the Superdex gel filtration data) and the weights of the individual polypeptides (deduced from southwestern analysis) suggest that all three proteins exist in solution as monomers. This does not exclude that they bind to DNA as dimers or multimers. Size fractionation and affinity-based separation did not give sufficiently pure material for protein sequencing. Therefore we decided to use a different approach to identify these proteins.
Screening of a λgt11 expression library
Based on the fact that several individual renatured proteins were able to bind specifically to the 27h probe we decided to screen an expression library with cloned cDNAs from HeLa cells. Two clones were positive and sequencing of the cDNAs revealed that both clones contained a single open reading frame in-phase with the N-terminus of the β-galactosidase protein. Searches for homologies revealed a cDNA expressing the full-length hnRNP K protein and a cDNA for the ASF/SF2 splicing factor. Four different approaches were attempted to test: (i) if these proteins could bind specifically to the telomeric probe in vitro; (ii) if one of the retarded complexes shown in Figures 1 and 2 actually involves these proteins.
DNA-binding properties of hnRNP K
EMSA with the recombinant protein. EMSA was performed with a radiolabelled 27h probe and bacterial protein extracts overexpressing hnRNP K deleted for the first 36 amino acids (74) but fused with GST (Fig. 6A). Uninduced bacterial protein extracts did not show any C-rich strand-binding activity (Fig. 6A, lane 6). Induced extracts possessed a single binding activity (Fig. 6A, lane 7) that could be competed by 27h and 29h but not by 17h (Fig. 6A, lanes 8–10), as observed with HNE (Fig. 6A, lanes 3–5).
Figure 6.
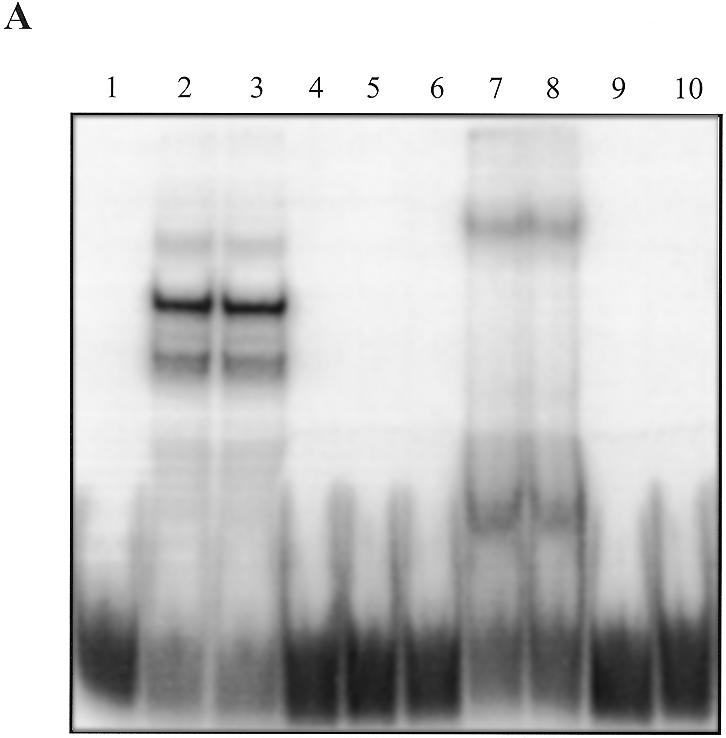
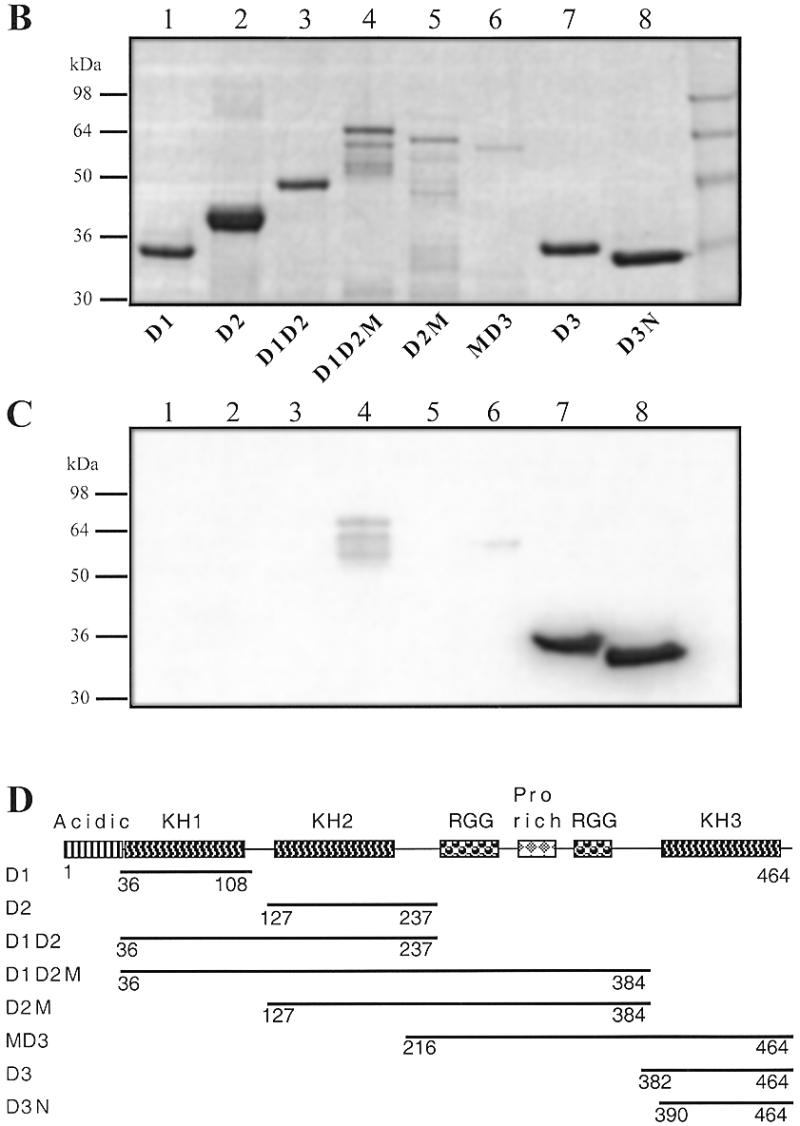
(A) Binding of GST–hnRNP K to the 27h probe. Classical EMSA was performed as reported in Figure 3. Lane 1, no protein; lanes 2–5, 42.5 ng/µl HNE; lane 6, protein extract (50 ng/µl) from non-induced bacteria with an inducible GST–hnRNP K (construct B; 74) plasmid (GST–hnRNP K –); lanes 7–10, protein extract (50 ng/µl) from induced bacteria (GST–hnRNP K +). Lanes 3 and 8, 10 µM 17h; lanes 4 and 9, 10 µM 27h; lanes 5 and 10, 10 µM 29h. (B) SDS–PAGE of the bulk purified GST–hnRNP K construct. Aliquots of 3–6 µl of the GST fusion constructs attached to Sepharose beads were loaded in a 14% SDS gel and Coomassie stained. Lane 1, D1 (amino acids 36–108); lane 2, D2 (amino acids 127–237); lane 3, D1D2 (amino acids 36–237); lane 4, D1D2M (amino acids 36–384); lane 5, D2M (amino acids 127–384); lane 6, MD3 (amino acids 216–464); lane 7, D3 (amino acids 384–464); lane 8, construct D3N (amino acids 390–464). (C) Southwestern analysis of a duplicate of the preceding gel transferred to PVDF and hybridised with 2 pmol of 27h in the presence of dT26 (50 µg/ml) and ds26 (10 µg/ml) in HEPES buffer pH 7.2. (D) Scheme of the different hnRNP K constructs (74).
Southwestern analysis with the recombinant protein. Constructs containing different domains of hnRNP K (74) fused with GST were overexpressed in bacteria (see Fig. 6D for the nomenclature of the different polypeptides). GST fusion proteins from bacterial extracts were bound to glutathione–Sepharose. Aliquots from these beads were loaded onto two denaturing SDS gels. One of the gels was Coomassie stained to assess the purity of the proteins (Fig. 6B). The other was transferred to a PVDF membrane, renaturated and hybridised with radiolabelled probe 27h (Fig. 6C). D3N and D3 bound to 27h with high affinity (Fig. 6C, lanes 7 and 8); D1D2M and MD3 bound to 27h with intermediate/low affinity (Fig. 6C, lanes 4 and 6); D1D2 bound with a very low affinity (visible only after a long exposure). Three of these five positive constructs (D3, D3N and MD3) contain the third K-homology RNA-binding (KH) motif, which has already been shown to be sufficient for poly(rC) binding (74). The two other weakly positive constructs include the first two KH domains. For the KH3 domain-containing constructs (D3N, D3 and MD3) and D1D2, the observed bands corresponded to the expected molecular weight of the GST fusion construct and to the main band observed by Coomassie staining (Fig. 6B and C, lanes 4 and 6–8). For D1D2M several bands were observed (Fig. 6B and C, lane 4) as a result of partial degradation.
Western analysis of the EMSA with crude nuclear extracts. Making use of an available monoclonal antibody specific for the hnRNP K protein (12g4), we tested whether this protein takes part in one or several of the retarded bands. This antibody gave no supershift when added to the binding reaction, but addition of massive amounts of 12g4 led to disappearance of the K band (not shown). To circumvent this problem, a classical binding reaction was prepared, using 10 pmol of radiolabelled 27h and 9 µg of HNE. After migration on a non-denaturing gel, a short exposure allowed accurate determination of the position of the retarded bands (Fig. 7A). The gel was transferred by capillarity to a nitrocellulose membrane and subjected to a classical western analysis. No band was detected with the 12g4 antibody in the absence of the DNA probe, whereas in the presence of 27h a major band corresponding to K was readily visible (Fig. 7B, lanes 2–4). As expected, this band was not competed out by dT26 or ds26. The additional slow migrating band (Fig. 7B, lanes 3 and 4) was also detected when these competitors were used in the absence of the specific probe. This experiment showed that hnRNP K was indeed present in the K band and its migration in the gel was dependent on the presence of the specific oligonucleotide.
Figure 7.

(A) EMSA with 27h and HNE. Binding and migration were performed as described for previous EMSA, with the 27h probe (20 nM radiolabelled and 1 µM cold). HNE, 0.9 µg/µl; dT26, 0.5 µg/µl; ds26, 0.5 µg/µl. The wet gel was briefly exposed to a PhosphorImager screen at 4°C. Lane 1, 27h alone; lane 2, 27h + HNE; lane 3, 27h + HNE + dT26; lane 4, 27h + HNE + dT26 + ds26. (B) Western blot with an anti-hnRNP K antibody on the same gel transferred to a nitrocellulose membrane. Lanes 1–4, as in (A). (C) Southwestern blot of HNE (4.5 µg) hybridised overnight at 4°C with 4 pmol of radiolabelled 27h in the presence of dT26 (10 µg/ml) and ds26 (10 µg/ml) in HEPES buffer pH 7.2. (D) Western blot on the previous membrane with an anti-hnRNP K antibody (12g4).
Western analysis of the southwestern assay with crude nuclear extracts. The 12g4 antibody stained a single protein band of HeLa nuclear extracts that had an apparent molecular weight, on SDS–PAGE, of 66 kDa. This protein has the same electrophoretic mobility as the band revealed by southwestern analysis with the 27h probe (Fig. 7C), suggesting that this protein is hnRNP K. Thus, we performed a western analysis on the membrane used for the southwestern analysis, and the exact same band was revealed (Fig. 7D).
DNA binding properties of ASF/SF2
EMSA with the recombinant proteins. Recombinant ASF/SF2 proteins with different deletions (73) were tested for their abilities to bind to the telomeric probe by EMSA. Unfortunately, none of the DNA–protein complexes entered the gel. However, these retarded complexes were observed with the 27h probe but not with a dT26 or 27sg probe. Among the deleted ASF/SF2 constructs, 197N and 210N did not bind to the probe and 197C bound weakly. As a control, EMSA was also performed on the R27 probe, which is an aptamer sequence selected to bind to ASF/SF2 (E.Labourier, unpublished results); the same results as with the 27h probe but with a weaker retarded signal were observed.
Southwestern blot analysis with the recombinant proteins. Recombinant ASF/SF2 proteins were loaded on denaturing SDS gels and Coomassie stained (Fig. 8A). A duplicate of this gel was transferred to a nitrocellulose membrane, renaturated and hybridised with a radiolabelled probe. Strong signals were observed with the 27h probe corresponding to the position of the ASF/SF2 bands on the gel (Fig. 8B). Full-length ASF/SF2 and the four truncated proteins containing the RNA recognition motif (RRM) domains (Fig. 8B, lanes 1–5), but not 197N and 210N (Fig. 8B, lanes 6 and 7), bound to the telomeric probe. Binding to 197C was weak, as already observed in the EMSA. The binding activity of the different truncated proteins was qualitatively the same with the R27 probe, but the extent of binding was weaker compared with that obtained with the telomeric probe. For the other control radiolabelled (dT26, 27sg, 27sc and R27) strands no signal was observed with dT26 and 27sg and a very weak signal was obtained with 27sc (data not shown).
Figure 8.
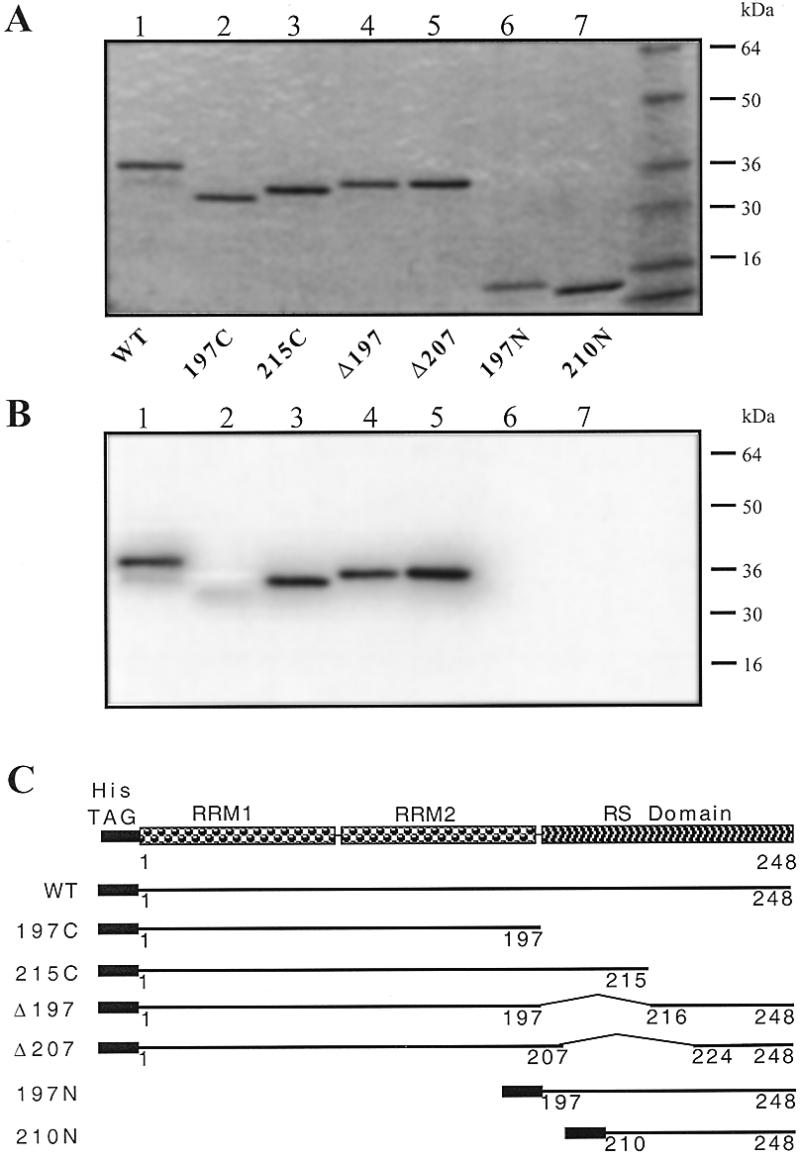
(A) SDS–PAGE of the recombinant ASF/SF2 proteins. Aliquots of 250 ng of each deleted form of ASF/SF2 were loaded on an 8–16% SDS gel and Coomassie stained. Lane 1, wild-type ASF/SF2; lane 2, 197C; lane 3, 215C; lane 4, Δ197; lane 5, Δ207; lane 6, 197N; lane 7, 210N. (B) Southwestern analysis of a duplicate of the gel transferred onto nitrocellulose and hybridised with 10 pmol of 27h in the presence of dT26 (10 µg/ml) and ds26 (5 µg/ml) in HEPES buffer pH 7.2. (C) Scheme of the different ASF/SF2 constructs (73).
Western blot analysis of the EMSA with crude nuclear extracts. We used an antibody directed against the RS domain of ASF/SF2 (mAb 104). This antibody is not specific for ASF/SF2 only, but also reveals other RS domain-containing proteins. This antibody gave no supershift when added to the binding reaction. Thus, we used the same approach as with hnRNP K. In the absence of 27h no signal was observed in western analysis, showing that ASF/SF2 did not enter the gel. In contrast, in the presence of 27h the western blot revealed ASF/SF2 in the well (data not shown). This confirms that ASF/SF2 binds effectively to the telomeric probe and that the DNA–protein complex remains in the well under our EMSA conditions.
DISCUSSION
Identification of proteins that bind to the C-rich strand of telomeric DNA
In this work we have identified two proteins that bind specifically to a C-rich oligonucleotide that mimics the C-rich strand of vertebrate telomeres. A few groups have reported the existence of C-rich-specific factors in Trypanosoma brucei (75), in HeLa and various mammalian cells (48,49) and in Drosophila (61,62). Unfortunately, the identity of these proteins was not determined for the two former groups. Marsich et al. have reported that HeLa cells contain a single nuclear factor that specifically recognises the C-rich telomeric repeat (48). Their estimate of the protein–DNA complex molecular weight was 50 kDa but the identity of the protein remained elusive. However, our data suggest the existence of several (three or more) different complexes. Their EMSA was performed at low probe concentration and a high protein:probe ratio (the equivalent of 40 µg/pmol in our definition), which gave a single retarded band, our D band. Our UV crosslinking experiment with a high protein:probe ratio showed a major crosslinked product with an apparent molecular weight of 50 kDa. This is in good agreement with the molecular mass reported in a later paper (49) with an equivalent protein:probe ratio and probe concentrations. Our results are therefore in accordance with those of Marsich et al., but they also reveal the existence of other C-rich strand-binding proteins in HeLa nuclear extracts.
Several biochemical methods were used to obtain information on the properties of the DNA-binding proteins detected by EMSA. An estimate of the molecular weight of the proteins was given by several independent experiments (64–66, 45 and 30–35 kDa; the apparent molecular weight of the smallest protein could not be measured precisely). Unfortunately, we could not deduce the identity of these molecules from these experiments. An affinity-based method using magnetic beads was tried without success (data not shown) and a traditional biochemical purification failed.
Therefore, we decided to screen a cDNA expression library with a C-rich probe. Two clones were selected: one coded for full-length hnRNP K and the other for the splicing factor ASF/SF2 (>98% identity). Multiple experiments later confirmed that both polypeptides bind to the 27h oligonucleotide in vitro. Our screening cannot be considered as exhaustive: the gel shift assays as well as the southwestern approach suggested that at least three or four specific complexes are present in HeLa cells. According to the EMSA and the above discussion, neither hnRNP K nor ASF/SF2 correspond to the single band observed by Marsich et al.
It is rather striking that both proteins are known as RNA-binding proteins and play a role in the splicing and transport of mRNA. It should also be noted that the in vitro affinities for these DNA sequences are higher than those for RNA single strands. As shown in Table 1, the two RNA oligomers (21Rh and R27) are weak competitors, even though the R27 sequence is the result of a SELEX experiment using ASF/SF2 as the target (E.Labourier et al., unpublished results). tRNA does not compete at all (Table 1). Overall these results show that hnRNP K and ASF/ASF2 have a preference for DNA over RNA. Tomonaga and Levens have obtained the same DNA versus RNA preference for hnRNP K (76).
hnRNP K and ASF/ASF2 do not share a common RNA-binding domain: the hnRNP K protein contains three KH domains, whereas ASF/SF2 binds to RNA via its RRM. There is no homology between the two proteins and the two binding motifs.
ASF/SF2 is a member of a conserved family of splicing factors known as SR proteins (77,78). These proteins, which are necessary for splicing in vitro, contain one or two N-terminal RRM(s) and an extensively phosphorylated C-terminal region enriched in repeating Arg-Ser dipeptides (RS domain). ASF/SF2 is required for cell viability (79) and may be phosphorylated by topoisomerase I (80). Phosphorylation may modulate the interaction of this protein with nucleic acids (81). The molecular weight of the dephosphorylated protein is 30 kDa. Our results suggest that the RRM domains are required but not sufficient for the C-rich sequence-binding activity. It could also be expected that the RS domain plays a role in regulation of this binding activity.
hnRNP K was originally identified as a component of hnRNP particles (82). hnRNP K is in fact a family composed of four major proteins (hnRNPs K A, B, C and D) and their modified forms (74). This abundant protein binds tenaciously to poly(rC) (66) and binding depends on the phosphorylation level of the protein (74). Nevertheless, belying its name, this protein binds more strongly to DNA than to RNA (76) and may activate transcription (83,84). Recently hnRNP K has been also identified as a (TC)n-binding protein (85). Deletion analysis showed that hnRNP K possesses several non-overlapping DNA-binding domains, composed of at least one KH motif (76). Our results suggest that the KH3 domain may be sufficient for a strong C-rich sequence-binding activity and that the combination of the first two domains, KH1 and KH2, has weak binding activity. Nevertheless, the remaining portions of the protein (the RGG domains in particular) are not able to bind to 27h, but may have a role in the sequence specificity of binding.
A parallel observation was made with the opposite strand: several proteins that bind to the telomeric DNA G-rich strand d(GGGTTA) were identified first as RNA-binding proteins and specifically as components of the hnRNP complex (35). hnRNP A1 as well as hnRNP A2/B1 and hnRNP D have been shown to bind to GGGTTA repeats in vitro (31,35,86) and hnRNP A1 modulates telomerase activity in vivo (87). Therefore, hnRNP A1 was established as the first single-strand DNA-binding protein involved in mammalian telomere biogenesis. It is also interesting to note that hnRNP A1 (the G-rich strand binder) and ASF/SF2 (the C-rich strand binder) regulate alternative splice site selection in an antagonistic manner (88). Interestingly, a recent model for regulation of the P1 promoter of c-myc proposed binding of factors on both single strands: hnRNP K on the C-rich strand and hnRNP A1 on the G-rich strand (89). Furthermore, the telomeric repeat (CCCTAA)n fits the consensus for the splice site YNCURAY (Y, pyrimidine base; R, purine base; N, any base).
Structure of the telomeric oligonucleotides
The i-motif structure is an attractive model for specific structural recognition of DNA by proteins, because of its geometry and charge distribution (90–92). However, the simple fact that a protein can bind to an oligonucleotide having the capacity to form the i-motif is not proof that the oligonucleotide, once bound to the protein, is still in the i-motif configuration, even if binding is performed under physicochemical conditions strongly favouring the folded form. In the range pH 6–9.2 binding to 27h was not pH sensitive. Therefore, either the protein binds to the folded i-motif structure and is able to stabilise it even under basic conditions or the protein binds to the unfolded sequence and is able to open the i-motif at acidic pH. The latter hypothesis would explain the observed salt-insensitive binding. Further structural studies of the complexes are required to solve this question.
Putative telomeric role of these proteins in vivo
Currently we do not have hard evidence that these proteins play a functional role in telomere architecture and function. Immunostaining with antibodies against hnRNP K showed a dotted nucleoplasmic staining that excluded the nucleolus (74). The ASF/SF2 factor is concentrated in 20–40 distinct nuclear domains called speckles, but is also present in a diffuse nucleoplasmic pool (93). This nuclear localisation does not exclude or favour a role for these proteins in the telomeres.
A possible role for these factors would be in modulating telomerase activity. One could imagine that C-rich strand-binding proteins may help to provide an accessible free 3′-end for the G-rich strand, by trapping its complementary strand in a specific DNA–protein complex. Another possible role can be proposed based on results obtained for S.cerevisiae (57). These experiments suggest a new step in telomere maintenance, i.e. cell cycle-regulated degradation of the C-rich strand which can generate a potential substrate for telomerase. In fact, the ends of mammalian chromosomes consist of an overhang on the G-rich strand and these overhangs may be considerably larger than previously anticipated (21,22). Nevertheless, we did not observe any C-rich strand-specific nuclease activity in HeLa cell nuclear extracts.
C-rich strand-binding activities are also present in non-transformed human fibroblasts and keratinocytes (data not shown) and have already been reported in rat and pig liver (49). We performed EMSA with nuclear extracts from primary fibroblasts after a varying number of cell divisions in vitro. Preliminary results indicate that C-rich strand-binding activities are dependent on the age of the fibroblasts and differ from established cell lines: C-rich strand-binding activities are especially abundant in young primary cells.
It is also interesting to note that hnRNP K might have a telomeric role in vivo even if it does not bind directly to telomeric DNA. hnRNP K may act as a transcription factor for the myc gene (60,83,84,94,95) and, in turn, the Myc protein has been shown to play a role in telomerase activity (96,97). Our findings support the possibility that some RNA-binding proteins might bind to the C-rich strand of telomeres in addition to having a role in pre-mRNA metabolism (35). Much work remains to be done to assess the telomeric function of these genes in vivo. An investigation of cell lines expressing various levels of ASF/SF2 (98) or hnRNP K proteins might provide some answers to these questions. We are also currently exploring the binding ability of individual DNA-binding domains with the aim of obtaining unambiguous NMR data on the solution structure of the DNA in the protein–DNA complex.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. T. Garestier, P.B. Arimondo, F. Guillonneau, M. Mills, K. Dejgaard, J.-C. François, A.-L. Guieyesse-Peugeot, J.-L. Leroy and M. Lum for helpful discussions and R. Ternac for technical support. The gel filtration experiments were performed with the help of J.-F. Cornuel at the Groupe de Biophysique of the Ecole Polytechnique (Palaiseau, France).
REFERENCES
- 1.Blackburn E.H. (1994) Cell, 77, 621–623. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V.A. (1995) Science, 270, 1601–1607. [DOI] [PubMed] [Google Scholar]
- 3.Greider C.W. (1996) Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 4.Holt S.E., Shay,J.W. and Wright,W.E. (1996) Nature Biotechnol., 14, 836–839. [DOI] [PubMed] [Google Scholar]
- 5.Olovnikov A.M. (1996) Exp. Gerontol., 31, 443–448. [DOI] [PubMed] [Google Scholar]
- 6.Greider C.W. (1998) Curr. Biol., 8, R178–R181. [Google Scholar]
- 7.Shore D. (1998) Curr. Biol., 8, R192–R195. [Google Scholar]
- 8.Gottschling D.E. and Stoddard,B. (1999) Curr. Biol., 9, R164–R167. [DOI] [PubMed] [Google Scholar]
- 9.O’Reilly M., Teichmann,S.A. and Rhodes,D. (1999) Curr. Opin. Struct. Biol., 9, 56–65. [DOI] [PubMed] [Google Scholar]
- 10.van Steensel B., Smogorzewska,A. and de Lange,T. (1998) Cell, 92, 401–413. [DOI] [PubMed] [Google Scholar]
- 11.Li L., Lejnine,S., Makarov,V. and Langmore,J.P. (1998) Nucleic Acids Res., 26, 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palladino F., Laroche,T., Gilson,E., Axelrod,A., Pillus,L. and Gasser,S.M. (1993) Cell, 75, 543–555. [DOI] [PubMed] [Google Scholar]
- 13.Scherthan H., Weich,S., Schwegler,H., Heyting,C., Harle,M. and Cremer,T. (1996) J. Cell Biol., 134, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevenson J.B. and Gottschling,D.E. (1999) Genes Dev., 13, 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper J.P., Watanabe,Y. and Nurse,P. (1998) Nature, 392, 828–831. [DOI] [PubMed] [Google Scholar]
- 16.Nimmo E.R., Pidoux,A.L., Perry,P.E. and Allshire,R.C. (1998) Nature, 392, 825–828. [DOI] [PubMed] [Google Scholar]
- 17.Stavenhagen J.B. and Zakian,V.A. (1998) Genes Dev., 12, 3044–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyrion G., Liu,K., Liu,C. and Lustig,A.J. (1993) Genes Dev., 7, 1146–1159. [DOI] [PubMed] [Google Scholar]
- 19.Marcand S., Buck,S.W., Moretti,P., Gilson,E. and Shore,D. (1996) Genes Dev., 10, 1297–1309. [DOI] [PubMed] [Google Scholar]
- 20.Wellinger R.J., Wolf,A.J. and Zakian,V.A. (1993) Cell, 72, 51–60. [DOI] [PubMed] [Google Scholar]
- 21.Wright W.E., Tesmer,V.M., Huffman,K.E., Levene,S.D. and Shay,J.W. (1997) Genes Dev., 11, 2801–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarov V.L., Hirose,Y. and Langmore,J.P. (1997) Cell, 88, 657–666. [DOI] [PubMed] [Google Scholar]
- 23.Nugent C.I., Hughes,T.R., Lue,N.F. and Lundblad,V. (1996) Science, 274, 249–252. [DOI] [PubMed] [Google Scholar]
- 24.Grandin N., Reed,S.I. and Charbonneau,M. (1997) Genes Dev., 11, 512–527. [DOI] [PubMed] [Google Scholar]
- 25.Conrad M.N., Wright,J.H., Wolf,A.J. and Zakian,V.A. (1990) Cell, 63, 739–750. [DOI] [PubMed] [Google Scholar]
- 26.Boulton S.J. and Jackson,S.P. (1996) Nucleic Acids Res., 24, 4639–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter S.E., Greenwell,P.W., Ritchie,K.B. and Petes,T.D. (1996) Nucleic Acids Res., 24, 582–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broccoli D., Smogorzewska,A., Chong,L. and de Lange,T. (1997) Nature Genet., 17, 231–235. [DOI] [PubMed] [Google Scholar]
- 29.Bilaud T., Brun,C., Ancelin,K., Koering,C.E., Laroche,T. and Gilson,E. (1997) Nature Genet., 17, 236–239. [DOI] [PubMed] [Google Scholar]
- 30.van Steensel B. and de Lange,T. (1997) Nature, 385, 740–743. [DOI] [PubMed] [Google Scholar]
- 31.McKay S.J. and Cooke,H. (1992) Nucleic Acids Res., 20, 6461–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gualberto A., Patrick,R.M. and Walsh,K. (1992) Genes Dev., 6, 815–824. [DOI] [PubMed] [Google Scholar]
- 33.Brigati C., Kurtz,S., Balderes,D., Vidali,G. and Shore,D. (1993) Mol. Cell. Biol., 13, 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang G.W. and Cech,T.R. (1993) Biochemistry, 32, 11646–11657. [DOI] [PubMed] [Google Scholar]
- 35.Ishikawa F., Matunis,M.J., Dreyfuss,G. and Cech,T.R. (1993) Mol. Cell. Biol., 13, 4301–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang G.W. and Cech,T.R. (1993) Cell, 74, 875–885. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z.P., Frantz,J.D., Gilbert,W. and Tye,B.K. (1993) Proc. Natl Acad. Sci. USA, 90, 3157–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giraldo R., Suzuki,M., Chapman,L. and Rhodes,D. (1994) Proc. Natl Acad. Sci. USA, 91, 7658–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin J.J. and Zakian,V.A. (1994) Nucleic Acids Res., 22, 4906–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schierer T. and Henderson,E. (1994) Biochemistry, 33, 2240–2246. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z.P. and Gilbert,W. (1994) Cell, 77, 1083–1092. [DOI] [PubMed] [Google Scholar]
- 42.Giraldo R. and Rhodes,D. (1994) EMBO J., 13, 2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frantz J.D. and Gilbert,W. (1995) J. Biol. Chem., 270, 20692–20697. [DOI] [PubMed] [Google Scholar]
- 44.Lin J.J. and Zakian,V.A. (1996) Proc. Natl Acad. Sci. USA, 93, 13760–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eid J.E. and Sollner-Webb,B. (1997) J. Biol. Chem., 272, 14927–14936. [DOI] [PubMed] [Google Scholar]
- 46.Harrington C., Lan,Y. and Akman,S.A. (1997) J. Biol. Chem., 272, 24631–24636. [DOI] [PubMed] [Google Scholar]
- 47.Santori F. and Donini,P. (1994) Res. Microbiol., 145, 519–530. [DOI] [PubMed] [Google Scholar]
- 48.Marsich E., Piccini,A., Xodo,L.E. and Manzini,G. (1996) Nucleic Acids Res., 24, 4029–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marsich E., Xodo,L.E. and Manzini,G. (1998) Eur. J. Biochem., 258, 93–99. [DOI] [PubMed] [Google Scholar]
- 50.Gehring K., Leroy,J.L. and Guéron,M. (1993) Nature, 363, 561–565. [DOI] [PubMed] [Google Scholar]
- 51.Leroy J.L., Gehring,K., Kettani,A. and Guéron,M. (1993) Biochemistry, 32, 6019–6031. [DOI] [PubMed] [Google Scholar]
- 52.Ahmed S., Kintanar,A. and Henderson,E. (1994) Nature Struct. Biol., 1, 83–88. [DOI] [PubMed] [Google Scholar]
- 53.Manzini G., Yathindra,N. and Xodo,L.E. (1994) Nucleic Acids Res., 22, 4634–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leroy J.L., Guéron,M., Mergny,J.L. and Hélène,C. (1994) Nucleic Acids Res., 22, 1600–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mergny J.L., Lacroix,L., Han,X., Leroy,J.L. and Hélène,C. (1995) J. Am. Chem. Soc., 117, 8887–8898. [Google Scholar]
- 56.Lacroix L., Mergny,J.L., Leroy,J.L. and Hélène,C. (1996) Biochemistry, 35, 8715–8722. [DOI] [PubMed] [Google Scholar]
- 57.Wellinger R.J., Ethier,K., Labrecque,P. and Zakian,V.A. (1996) Cell, 85, 423–433. [DOI] [PubMed] [Google Scholar]
- 58.Gallego J., Chou,S.H. and Reid,B.R. (1997) J. Mol. Biol., 273, 840–856. [DOI] [PubMed] [Google Scholar]
- 59.Catasti P., Chen,X., Deaven,L.L., Moyzis,R.K., Bradbury,E.M. and Gupta,G. (1997) J. Mol. Biol., 272, 369–382. [DOI] [PubMed] [Google Scholar]
- 60.Michelotti E.F., Tomonaga,T., Krutzsch,H. and Levens,D. (1995) J. Biol. Chem., 270, 9494–9499. [DOI] [PubMed] [Google Scholar]
- 61.Ferrer N., Azorin,F., Villasante,A., Gutierrez,C. and Abad,J.P. (1995) J. Mol. Biol., 245, 8–21. [DOI] [PubMed] [Google Scholar]
- 62.Cortes A., Huertas,D., Fanti,L., Pimpinelli,S., Marsellach,F.X., Pina,B. and Azorin,F. (1999) EMBO J., 18, 3820–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cantor C.R., Warshaw,M.M. and Shapiro,H. (1970) Biopolymers, 9, 1059–1077. [DOI] [PubMed] [Google Scholar]
- 64.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Djavaheri-Mergny M., Gras,M.P., Mergny,J.L. and Dubertret,L. (1999) Biochem. J., 338, 607–613. [PMC free article] [PubMed] [Google Scholar]
- 66.Matunis M.J., Michael,W.M. and Dreyfuss,G. (1992) Mol. Cell. Biol., 12, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roth M.B., Murphy,C. and Gall,J.G. (1990) J. Cell Biol., 111, 2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kohtz J.D., Jamison,S.F., Will,C.L., Zuo,P., Luhrmann,R., Garcia-Blanco,M.A. and Manley,J.L. (1994) Nature, 368, 119–124. [DOI] [PubMed] [Google Scholar]
- 69.Mergny J.L. (1999) Biochemistry, 38, 1573–1581. [DOI] [PubMed] [Google Scholar]
- 70.Singh H., LeBowitz,J.H., Baldwin,A.S.,Jr and Sharp,P.A. (1988) Cell, 52, 415–423. [DOI] [PubMed] [Google Scholar]
- 71.Lacoste J., Codani-Simonart,S., Best-Belpomme,M. and Peronnet,F. (1995) Nucleic Acids Res., 24, 5073–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cowell I.G. (1997) In Cowell,I.G. and Austin,C.A. (eds), cDNA Library Protocols. Humana Press, Totowa, NJ, Vol. 69, pp. 161–170.
- 73.Labourier E., Rossi,F., Gallouzi,I., Allemand,E., Divita,G. and Tazi,J. (1998) Nucleic Acids Res., 26, 2955–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dejgaard K. and Leffers,H. (1996) Eur. J. Biochem., 241, 425–431. [DOI] [PubMed] [Google Scholar]
- 75.Eid J.E. and Sollner-Webb,B. (1995) Mol. Cell. Biol., 15, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomonaga T. and Levens,D. (1995) J. Biol. Chem., 270, 4875–4881. [DOI] [PubMed] [Google Scholar]
- 77.Krainer A.R., Conway,G.C. and Kozak,D. (1990) Genes Dev., 4, 1158–1171. [DOI] [PubMed] [Google Scholar]
- 78.Ge H., Zuo,P. and Manley,J.L. (1991) Cell, 66, 373–382. [DOI] [PubMed] [Google Scholar]
- 79.Wang J., Takagaki,Y. and Manley,J.L. (1996) Genes Dev., 10, 2588–2599. [DOI] [PubMed] [Google Scholar]
- 80.Rossi F., Labourier,E., Forne,T., Divita,G., Derancourt,J., Riou,J.F., Antoine,E., Cathala,G., Brunel,C. and Tazi,J. (1996) Nature, 381, 80–82. [DOI] [PubMed] [Google Scholar]
- 81.Xiao S.H. and Manley,J.L. (1997) Genes Dev., 11, 334–344. [DOI] [PubMed] [Google Scholar]
- 82.Swanson M.S. and Dreyfuss,G. (1988) Mol. Cell. Biol., 8, 2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tomonaga T. and Levens,D. (1996) Proc. Natl Acad. Sci. USA, 93, 5830–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee M.H., Mori,S. and Raychaudhuri,P. (1996) J. Biol. Chem., 271, 3420–3427. [DOI] [PubMed] [Google Scholar]
- 85.Garcia-Bassets I., Ortiz-Lombardia,M., Pagans,S., Romero,A., Canals,F., Avils,F.X. and Azorin,F. (1999) Nucleic Acids Res., 27, 3267–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dempsey L.A., Sun,H., Hanakahi,L.A. and Maizels,N. (1999) J. Biol. Chem., 274, 1066–1071. [DOI] [PubMed] [Google Scholar]
- 87.La Branche H., Dupuis,S., Ben-David,Y., Bani,M.R., Wellinger,R.J. and Chabot,B. (1998) Nature Genet., 19, 199–202. [DOI] [PubMed] [Google Scholar]
- 88.Mayeda A. and Krainer,A.R. (1992) Cell, 68, 365–375. [DOI] [PubMed] [Google Scholar]
- 89.Simonsson T., Pecinka,P. and Kubista,M. (1998) Nucleic Acids Res., 26, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sen D. and Gilbert,W. (1988) Nature, 334, 364–366. [DOI] [PubMed] [Google Scholar]
- 91.Williamson J.R. (1994) Annu. Rev. Biophys. Biomol. Struct., 23, 703–730. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y. and Patel,D.J. (1993) Structure, 1, 263–282. [DOI] [PubMed] [Google Scholar]
- 93.Misteli T., Caceres,J.F. and Spector,D.L. (1997) Nature, 387, 523–527. [DOI] [PubMed] [Google Scholar]
- 94.Michelotti G.A., Michelotti,E.F., Pullner,A., Duncan,R.C., Eick,D. and Levens,D. (1996) Mol. Cell. Biol., 16, 2656–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Michelotti E.F., Michelotti,G.A., Aronsohn,A.I. and Levens,D. (1996) Mol. Cell. Biol., 16, 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J., Xie,L.Y., Allan,S., Beach,D. and Hannon,G.J. (1998) Genes Dev., 12, 1769–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Greenberg R.A., O’Hagan,R.C., Deng,H., Xiao,Q., Hann,S.R., Adams,R.R., Lichtsteiner,S., Chin,L., Morin,G.B. and DePinho,R.A. (1999) Oncogene, 18, 1219–1226. [DOI] [PubMed] [Google Scholar]
- 98.Shih S.R. and Krug,R.M. (1996) EMBO J., 15, 5415–5427. [PMC free article] [PubMed] [Google Scholar]