

Abstract
The detection of site-specific phosphorylation in the microtubule-associated protein tau is emerging as a means to diagnose and monitor the progression of Alzheimer’s Disease and other neurodegenerative diseases. However, there is a lack of phospho-specific monoclonal antibodies and limited validation of their binding specificity. Here, we report a novel approach using yeast biopanning against synthetic peptides containing site-specific phosphorylations. Using yeast cells displaying a previously validated phospho-tau (p-tau) single-chain variable region fragment (scFv), we show selective yeast cell binding based on single amino acid phosphorylation on the antigen. We identify conditions that allow phospho-specific biopanning using scFvs with a wide range of affinities (KD = 0.2 to 60 nM). Finally, we demonstrate the capability of screening large libraries by performing biopanning in 6-well plates. These results show that biopanning can effectively select yeast cells based on phospho-site specific antibody binding, opening doors for the facile identification of high-quality monoclonal antibodies.
Keywords: antibody binding, biopanning, phospho-tau, post-translational modification, tau
Introduction
Reversible protein phosphorylation plays a central role in how cells adapt to the environment, differentiate and coordinate. Protein phosphorylation enables regulations at timescales much faster than gene expression, by inducing conformational changes that alter enzymatic activity, allosteric regulation or protein–protein interactions (Graves and Krebs, 1999; Johnson and Lewis, 2001; Pawson, 2004). Abnormal phosphorylation is associated with many human diseases and is a source of toxicity in pathogens (Cohen, 2001). Phosphorylation patterns of the microtubule-associated protein tau are a prominent example for which association with neurodegenerative diseases has been extensively documented. In tau, increased phosphorylation at specific sites shows a strong correlation with disease stage and progression rate (Dujardin et al., 2020; Wesseling et al., 2020). Moreover, elevation in plasma concentration of tau phosphorylated at threonine 181 or 231 (T181 or T231) is a strong biomarker of Alzheimer’s disease (AD) (Arbaciauskaite et al., 2021; Ashton et al., 2021; Janelidze et al., 2020; Thijssen et al., 2020). Therefore, there is a great deal of interest in the accurate detection of site-specific protein phosphorylation.
Antibodies are widely used for detecting protein phosphorylation, but reproducible detection of protein phosphorylation at a specific site remains a challenge. We and others have shown that many existing phospho-site antibodies suffer from poor specificity, often showing cross-binding to non-phosphorylated target sites or other phosphorylation sites (Bordeaux et al., 2010; Ercan et al., 2017; Li and Cho, 2020). In addition, the irreproducibility of detection is exacerbated by the fact that most available phospho-specific antibody preparations are rabbit polyclonal (Mandell, 2003). This is primarily because rabbits are highly immunogenic to small molecules and haptens, unlike rodents (Li et al., 2000; Liu et al., 2016; Weber et al., 2017). According to a curated antibody database (Labome), 93% (15 234 out of 16 371) of existing phospho-site antibodies are rabbit immunoglobulins, and 89% are polyclonal. Immunization with synthetic peptides that contain site-specific phosphorylations leads to reagents with broad applicability, but the lack of monoclonal antibodies critically limits validation and reproducible detection of protein phosphorylation. This also limits the development of phospho-specific antibody drugs, which started to enter clinical trials (Xia et al., 2021).
In vitro library screening approaches are well-suited to address these needs, as cross-reactive clones can be eliminated through negative selections. However, there have been few reports using existing in vitro screening methods to generate phospho-specific antibodies (Feldhaus et al., 2003; Velappan et al., 2019). These studies commonly reported that the identified antibodies lacked specificity. To overcome this problem, we previously developed a yeast surface display-based screening approach that leverages multi-color flow cytometry to quantify cross-reactivity during library screening (Li et al., 2018). Although this approach is effective, fluorescence-activated cell sorting (FACS) is not well-suited for screening large antibody libraries. Therefore, other methods capable of screening large libraries such as magnetic-activated cell sorting or biopanning approaches are needed during the initial stages.
To meet this need, we sought to develop a biopanning method for stringent screening of antibody clones based on specific binding to phosphorylated target protein sites. Using peptides as antigens enables screening antibodies against defined post-translational modification sites, particularly for targeting intrinsically disordered proteins such as the human tau protein. Considering the challenge of finding high-specificity clones, we aimed to achieve quantitative discrimination based on binding specificity. Here we report the implementation of yeast display biopanning with whole-well cell counting to measure phospho-site-specific binding to the clinically relevant tau phosphorylation site T231. We show that synthetic phospho-peptides can be immobilized on a layer of human embryonic kidney (HEK) 293 cells and allow yeast biopanning based on specific antigen-single-chain variable region fragment (scFv) interaction. A bi-directional expression plasmid was introduced to enable scFv surface display and intracellular expression of fluorescent proteins in yeast. This allowed rapid cell counting using automated microscopy and the addition of in-well control yeast for detecting non-specific binding. We report improved peptide immobilization and biopanning conditions that allow phospho-specific capture of yeast displaying scFvs with moderate affinity (KD of 60 nM), well within the affinity level of antibodies identified from naïve libraries through yeast biopanning (Wang et al., 2007). The results clearly show that yeast biopanning can identify phospho-site specific antibodies and demonstrate the potential for clonal selection of phospho-site binders.
Materials and Methods
Bi-directional expression for yeast surface display of antibody fragments and intracellular production of fluorescent proteins
For bi-directional expression in yeast, plasmid vector pBEVY-GT (Addgene, RRID:Addgene_51231) containing the GAL1-10 promoter with two distinct terminators was used. Yeast-enhanced green fluorescent protein (hereafter referred to as GFP) (Huang and Shusta, 2005) was cloned into the vector pBEVY-GT between restriction sites BamHI and PstI. pT231 tau scFvs variants (pT231 scFv, pT231 scFv mutants 3.24 or Y31A) (Li et al., 2018) were cloned into the vector pBEVY-GT between restriction sites XmaI and EcoRI, resulting in pBEVY-pT231 scFv (or other mutants)-GFP). To generate yeast expressing a control scFv, the anti-fluorescein scFv 4420 (a gift from Dr Eric Shusta) (Boder and Wittrup, 1997) was inserted into pBEVY-GT using the restriction sites XmaI and NheI. The scFv constructs contain a FLAG tag (DYKDDDDK) at their N-terminus (FLAG tag-scFv-Aga2p) to detect full-length scFv expression. To distinguish the control yeast, Golden Gate assembly was used to insert a yeast codon mCherry (version 4) (Qian et al., 2012) into the vector pBEVY-GT, resulting in pBEVY-4420-mCherry.
Plasmid pAP208 was cloned for surface display of scFvs fused to GFP (FLAG tag-scFv-Aga2p-GFP). The Aga2p and secretion signal sequence were digested with restriction enzymes NheI and BsaI, and the GFP sequence was digested with restriction enzymes BsaI and XhoI. The sequences were then cloned into the pCT-4RE backbone between restriction sites NheI and XhoI. All restriction endonucleases were purchased from New England BioLabs.
Resulting plasmid constructs were transformed into Saccharomyces cerevisiae strain EBY100 (ATCC MYA-4941) (Boder and Wittrup, 1997) using frozen-EZ yeast transformation II kit (Zymo Research, Cat. No. T2001) and grown on SD-CAA agar (0.1 M sodium phosphate, pH 6.0, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 20 g/L glucose, 15 g/L agar) plates for 3–4 days. From the plates, single colonies were picked and grown in 3 ml of SD-CAA medium at 30°C with shaking at 250 rpm overnight. The cell concentration was then determined by measuring the optical density at 600 nm (OD600), and 107 yeast cells were resuspended in 3 ml of SG-CAA medium (0.1 M sodium phosphate, pH 6.0, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 20 g/L galactose) at 30°C with shaking at 250 rpm for at least 20 h to induce expression of proteins on the surface.
HEK293FT cell culture and seeding
HEK293FT cells (Invitrogen Cat. No. R70007, RRID:CVCL_6911) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Cat. No. 12320-032) supplemented with 10% fetal bovine serum (Cytiva, heat-inactivated). The cells were used for <15 passages in continuous culture from cells that were previously frozen at early passage. To coat the plate surface with Matrigel, Matrigel (Corning Cat. No. 354234) was diluted 40-fold in ice-cold DMEM, according to the thin-coat protocol recommended by the manufacturer. Wells in a 96-well plate (Thermo Scientific Cat. No. 165305) were coated with 50 μl of the diluted Matrigel, and the plate was rocked to ensure even coating. The plate was then incubated at 37°C for at least 1 h, and the Matrigel was aspirated to leave a thin layer of the coat before cells were added. About 10,000 HEK293FT cells were seeded per well in the 96-well plate the day before experiments to reach 100% confluency. The next day, the cells were used in the yeast biopanning protocol as described below.
Yeast biopanning against tau peptide ligands
To carry out yeast cell biopanning against peptide ligands, HEK293FT cells are first grown to 100% confluency in wells within a 96-well plate as described above. These wells were then washed twice with 100 μl of ice-cold PBSCMA (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2 and 1 g/L BSA, pH 7.4). Cells were then biotinylated using NHS-PEG4-Biotin (125 μM unless otherwise specified, Thermo Fisher Cat. No. A39259) diluted in PBSCMA to achieve a total volume of 50 μl per well for 30 min at room temperature. After this, wells were washed again twice with 100 μl of ice-cold PBSCMA. To quench the biotinylation reaction, wells were incubated with 50 μl of PBSCM with 0.1 M Glycine for 10 min at room temperature. Wells were washed once with 100 μl ice-cold PBSCMA after this.
Streptavidin was then added to the cells at 50 μl total volume per well diluted in PBSCMA for 30 min at room temperature. For initial experiments aiming to visualize the degree of HEK293FT cell biotinylation, streptavidin conjugated with Alexa Fluor 647 (1200, 155 nM, Invitrogen Cat. No. S32357) was used as the streptavidin reagent. For all other experiments, unconjugated streptavidin (1 mg/ml diluted to 155 nM unless otherwise stated, Sigma Cat. No. 85878) was used. After incubation, wells were washed twice with 100 μl of ice-cold PBSCMA.
The wells were then incubated with biotinylated peptide antigens (0.1 μM unless otherwise stated) diluted in PBSCMA to achieve 50 μl total volume per well for 30 min at room temperature. The peptides used were previously described biotinylated peptides containing a sequence found in the tau protein with and without the phosphorylated threonine site pT231 (Li et al., 2018). The biotinylated phospho-peptide (KKVAVVR(pT)PPK(pS)PSSAK-biotin) and the non-phospho-peptide (KKVAVVRTPPKSPSSAK-biotin) were synthesized by Peptide 2.0. The phospho-peptide contains two phosphorylation sites, but the pT231 scFv interacts only with the pT231 site (Shih et al., 2012). To test a range of peptide concentrations, the peptide was prepared at the highest concentration necessary for that day’s experiments and then continuously diluted 2-fold to cover the range of the titration curve. Wells were then washed twice with 100 μl of ice-cold PBSCMA.
For experiments using only a single yeast transformant (e.g. yeast transformed with pBEVY-pT231 scFv 3.24-GFP), the cells were separated out at a concentration of ~106 yeast cells per well (as determined from OD600). For experiments using two yeast transformants, namely those expressing pBEVY-4420-mCherry in addition to yeast cells expressing pBEVY-pT231 scFv 3.24-GFP, cells were mixed in equal amounts to the desired final concentration. These cells were washed three times with 500 μl of ice-cold PBSCMA and resuspended in PBSCMA at a volume of 50 μl per 106 yeast cells. Yeast cells were then incubated in the wells for 30 min at room temperature. After incubation, wells were washed once with 100 μl of ice-cold PBSCMA.
Wells were then washed by dispensing 100 μl of ice-cold PBSCMA to one side of a wall (for example, the east wall side) and aspirating the liquid from the opposite side (the west side). The buffer was then dispensed to a different wall side (for example, the north wall side) and aspirated from the opposite side (the south side). This was repeated for a third time with yet again a different combination of wall sides (for example, northeast to southeast well walls). This whole process was repeated twice again using different combinations of wall sides, making sure to dispense liquid to walls that were aspirated from previously. In other words, if the first round of washing included pipetting from the north to the south side, pipetting from the south to the north side needed to be accounted for. Additionally, we made sure to include wall sides that were not included initially such as the northwest and southwest sides of the well walls. At this point, each well should have been washed once plainly and three times with three different combinations of well wall sides. If this protocol is carried out, each well should have been washed (dispensing media and aspirating it) a total of 10 times, thus maximizing the washing efficiency. After washing, 100 μl of ice-cold PBSCMA was added to the wells and the plate was used for imaging and analysis.
Six-well plate yeast biopanning enrichment of yeast displaying pT231 scFv
The peptide immobilization in 6-well plates was conducted in the same way as in the 96-well experiments, except a larger volume of reagents was used and 300 000 HEK293FT cells were seeded per well. For incubation steps (diluted Matrigel coating, biotinylation, quenching, streptavidin incubation, peptide incubation), 1 ml of reagents were used in each well. All wash steps were conducted with 2 ml of ice-cold PBSCMA.
Yeast cells displaying the pT231 scFv Y31A and expressing GFP (using plasmid pBEVY-pT231 scFv Y31A-GFP) were mixed with yeast cells displaying a control scFv and expressing mCherry (using plasmid pBEVY-4420-mCherry) at ratios of 1:1000 or 1:100 000 (Y31A:4420). For both biopanning conditions, a total of 3 × 108 cells were screened. The cells were resuspended in PBSCMA at a density of 3 × 108 cells per ml for all rounds of screening for the 1:1000 ratio screen. For the 1:100 000 ratio screen, cells were resuspended at a density of 3 × 108 cells per ml in the first round of screening and at a density of 5 × 107 cells per ml in subsequent rounds. About 1 ml of cells was added to each well and incubated for 30 min at room temperature. To wash unbound yeast, cells were removed by aspiration, followed by dropwise addition of 1 ml of ice-cold PBSCMA in each well. The plate was rocked 25 times and rotated 5 times, then the buffer was aspirated. The washing steps were repeated two more times. After washing, 1 ml of PBSCMA was added to each well and the cells were scraped, followed by centrifugation at 4000 rpm (358 g) for 10 min using a swinging bucket rotor. After the first round of the 1:100 000 ratio screen, we grew a fraction of collected cells on SD-CAA agar plates to estimate the number of cells recovered. The rest of the cells were resuspended in 10 ml SD-CAA supplemented with tetracycline (10 μg/ml), carbenicillin (100 μg/ml) and streptomycin (500 μg/ml). The recovered cells were grown and the biopanning procedure was repeated for multiple rounds. The enrichment was determined by counting the number of GFP and mCherry-positive cells using the BD Biosciences LSR Fortessa X-20 flow cytometer (UConn Center for Open Research Resources and Equipment).
96-well image acquisition
The automated microscope Keyence BZ-X810 was used for imaging the wells. For each well, a minimum of four set points were made and focused within the 96-well plate setting in the BZ-X800 Viewer software. Once the set points were made, the microscope automatically took scanning fluorescence images of the wells. After the images were acquired, they were analyzed by the BZ-X800 Analyzer software. An appropriate threshold was determined manually using an image at the edge of a well and this threshold was applied to all of the images taken during the same day of experiments. All of the images from an individual well were then loaded into the software along with the threshold image, image stitching was turned on and the software counted the individual cells present. The number reported by the software was used as the ‘cell count’ number in the analysis.
Antibody Labeling of yeast displayed scFv
To quantify scFv expression, 2 × 106 yeast cells displaying scFvs were washed twice with PBSA (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 and 1 g/L BSA, pH 7.4) and resuspended in 100 μl PBSA with a chicken anti-FLAG tag antibody (Abnova PAB29056, 1:500 dilution). Cells were incubated with the primary antibody for 30 min on ice and washed once with 500 μl PBSA. Cells were then stained with either goat anti-chicken IgY Alexa Fluor 647 (Thermo Fisher, Cat. No. A21449, 1:200 dilution) or goat anti-chicken IgY Alexa Fluor 488 (Thermo Fisher, Cat. No. A11039, 1:200 dilution) in 100 μl PBSA for 30 min on ice. Cells were then washed once with 500 μl PBSA and resuspended in 500 μl PBSA before detecting fluorescence.
To evaluate scFv binding to target peptide ligands, 2 × 106 yeast cells were washed twice with PBSA and incubated with the biotinylated phospho-peptide in PBSA at room temperature for 1 h. The volume of PBSA used for incubation varied depending on the concentration of phospho-peptide that was tested to ensure the peptide to scFv ratio remained constant. After labeling with the peptide, the cells were then stained as described above with streptavidin R-phycoerythrin (Thermo Fisher, Cat. No. S866, 1:100 dilution) added along with a secondary antibody. Cells were washed and resuspended before detecting fluorescence.
Yeast cell fluorescence was detected using the BD Biosciences LSR Fortessa X-20 flow cytometer. ‘scFv expression’ and ‘GFP expression’ indicate geometric mean of background subtracted fluorescence values recorded from 10 000 cells for the FLAG tag staining and GFP fluorescence, respectively. ‘binding/expression’ refers to the background-subtracted fluorescence values from the biotinylated peptide binding (fluorescence from streptavidin R-phycoerythrin) divided by the scFv expression. Background fluorescence was determined from untransformed cells. Fluorescence bleed-through between channels was not detected since each fluorescence was recorded using separate lasers and therefore compensation of fluorescence values was not necessary.
Statistical analysis
Prism 8 (GraphPad) was used for statistical analysis. Statistical tests used for each dataset are described in the figure legends.
Results
Development of yeast biopanning for a peptide antigen
To assess the binding of yeast cells displaying antibody fragments to a peptide antigen target, we initially attempted yeast biopanning on 96-well plates coated with neutravidin (see Supporting Information). Although the T231 phosphorylated tau (p-tau) peptide was immobilized on the plate surface and accessible for antibody binding (Fig. S1a and b), it did not allow significant binding of yeast cells displaying the high affinity and specificity pT231 scFv mutant (pT231 scFv 3.24) (Li et al., 2018; Li and Cho, 2020) in biopanning experiments (Fig. S1c and d). Based on the previous finding that yeast cells displaying an anti-fluorescein scFv adhere to a monolayer of cultured mammalian cells chemically modified with fluorescein, we predicted that yeast cells displaying phospho-specific scFvs can be biopanned on a layer of mammalian cells (Wang and Shusta, 2005). Therefore, we developed a biopanning approach (Fig. 1) outlined as follows (Fig. 1a). First, wells within a 96-well plate were coated with 50-fold diluted Matrigel to allow for the attachment of HEK293FT cells, which were grown to 100% confluency. These cells were biotinylated, and then streptavidin was added to the biotinylated HEK293FT cells. Since four individual biotin molecules can bind to one streptavidin molecule, we anticipated there would be unoccupied streptavidin binding sites to capture biotinylated peptides. After biotinylated peptide was added, yeast cells expressing scFv binders were added.
Fig. 1.
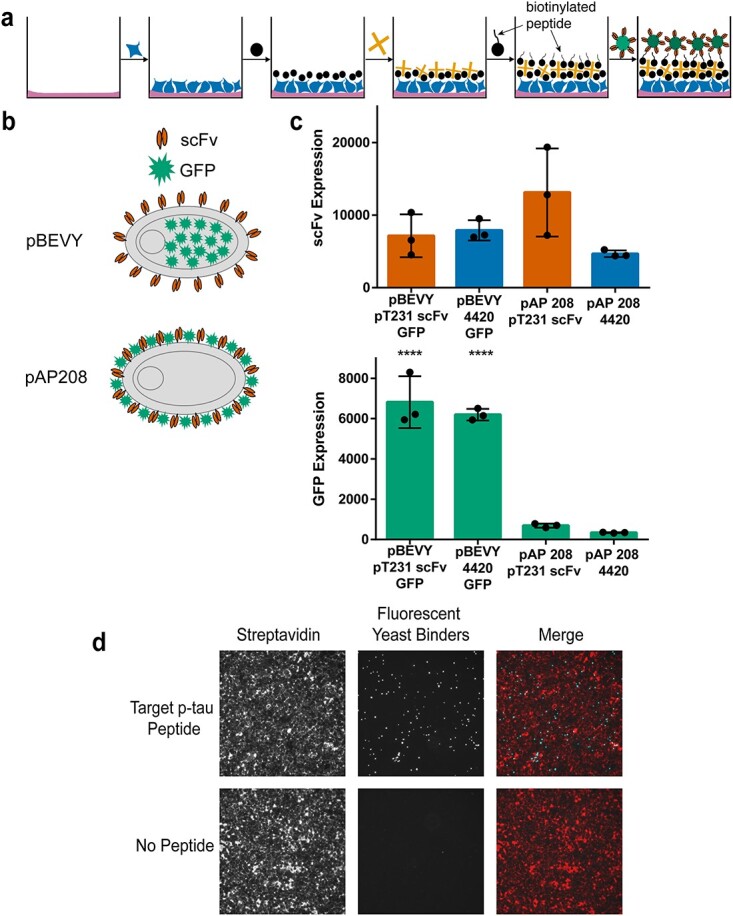
Overview of biopanning method and yeast reagent design. (a) Schematic representation of biopanning method. Wells are first coated with Matrigel, and HEK293FT cells are seeded. The cell surface is then biotinylated, and streptavidin is added. Biotinylated peptides are then added to the wells to be used as target antigens. Yeast cells displaying antibody binders and intracellularly expressing fluorescent protein are added to the wells and used as reporters. (b) Schematic representation of different reporter designs. The pBEVY backbone expresses antibody fragments (scFv) on the yeast cell surface and fluorescent protein (GFP) intracellularly. The pAP208 backbone expresses both scFv and GFP on the yeast cell surface. (c) Comparing expression levels for scFv and GFP using flow cytometry. scFv expression was measured using antibody staining of the FLAG epitope tag. Each datapoint indicates geometric mean of fluorescence from three separate experiments. Error bars indicate standard deviation of the datapoints. **** P ≤ 0.0001 using Tukey’s multiple comparisons test. Otherwise, P > 0.05. (d) Representative images indicating presence of streptavidin (labeled with Alexa Fluor 647) on HEK293FT cell surface (red) and yeast cells expressing GFP (cyan) binding to target peptide.
To facilitate the detection of yeast cells, we designed and tested two different plasmid constructs that allow surface display of scFvs and expression of fluorescent proteins (Fig. 1b). Both designs use the Aga2p system for yeast surface display of scFvs (Boder and Wittrup, 1997). The first design uses bi-directional expression based on the pBEVY backbone for intracellular expression of fluorescent proteins (e.g. GFP or mCherry) and surface display of scFvs (Fig. 1b) (Miller et al., 1998). On the other hand, in the second design (pAP208), GFP was fused to the C-terminus of Aga2p (scFv-Aga2p-GFP), enabling its expression on the yeast cell surface (Fig. 1b). To these constructs, we cloned the wild-type pT231 tau scFv, which binds a phosphorylated peptide derived from the human microtubule-associated tau protein or an anti-fluorescein scFv 4420 as a control (Boder and Wittrup, 1997; Li et al., 2018). We then compared the expression levels of both the scFv and GFP using flow cytometry. We found that there were no significant differences in scFv expression when comparing the two different backbone constructs (Fig. 1c). However, when comparing GFP expression levels, there were clear significant differences between the two backbones (Fig. 1c). The pBEVY backbone expressed GFP at about 6–7-fold higher levels than the pAP208 backbone (Fig. 1c) and was therefore used for the subsequent biopanning experiments.
To quantitatively assess binding, we performed biopanning using yeast cells displaying the high affinity pT231 scFv 3.24 in 96-well plates as described in Fig. 1a, but with or without the biotinylated p-tau peptide. After the addition of these yeast binders, the wells were thoroughly washed, following the previously described yeast biopanning protocol with some modifications (see Methods) (Wang et al., 2007). Successful biotinylation of HEK293FT cells is shown using streptavidin conjugated with Alexa 647 (Fig. 1d). We found clear binding of the yeast cells in wells containing the p-tau peptide, but nearly no binding in wells without the peptide (Fig. 1d).
To verify that the yeast cells were selectively binding to the p-tau peptide, we repeated the biopanning experiment with the phospho peptide, the phospho peptide with scrambled sequence or a non-phosphorylated peptide (with the same amino acid sequence) in addition to the no peptide control (Fig. 2a, see Table S1 for cell counts). To comprehensively assess the binding, we used an automated microscope (Keyence BZ-X810) that takes fluorescence images of the entire well and quantified the amount of yeast cells present in each well after washing. These experiments were repeated across 2 or 3 different days with triplicates each day. When analyzing the number of yeast cells in each well, it is clear that they specifically bind to the intended p-tau peptide (Fig. 2b). We attribute the relatively small number of cells present in wells with no peptide or non-phosphorylated peptide to the inability to wash out non-binding cells completely.
Fig. 2.
Testing selective biopanning of the scFv to peptide of interest. (a) Schematic representation showing different control conditions tested. Wells with target peptide (topmost image) are expected to contain more yeast cell reporters than wells with non-target peptide (middle image) and wells without peptide (bottom image). (b) Quantification of yeast cells expressing pT231 scFv 3.24 and GFP present in wells after washing. Each datapoint indicates average of values measured from three biological replicates in two to three separate experiments. Error bars indicate standard deviation of the datapoints. *P ≤ 0.05 using Tukey’s multiple comparisons test.
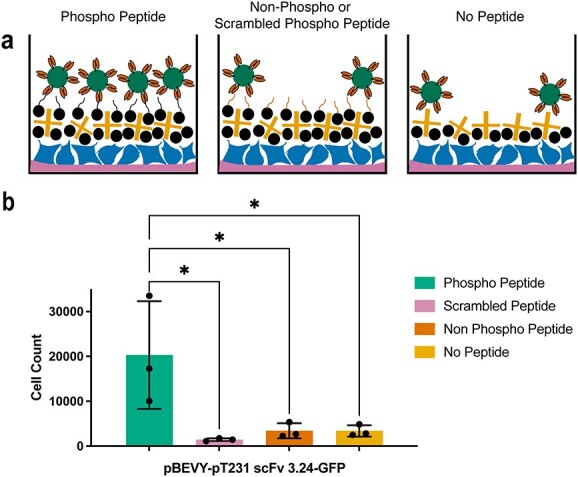
Biopanning using an internal control yeast strain
After observing non-specific signal present in wells with non-phosphorylated peptide and no peptide, we wanted to introduce a more reliable method of capturing non-specific signal. To do this, we designed a second strain of yeast cells that could be used as an internal control for the biopanning experiments (Fig. 3a). This control yeast strain expresses mCherry instead of GFP and the anti-fluorescein scFv using the pBEVY plasmid backbone. Using yeast cells expressing this plasmid (pBEVY-4420-mCherry) in conjunction with yeast cells expressing the plasmid described above (pBEVY-pT231 scFv 3.24-GFP), enabled us with the ability to quantify the non-specific signal within each well. The experimental set-up again included wells with p-tau peptide and controls (Fig. 3b). Yeast cells expressing pBEVY-4420-mCherry and yeast cells expressing pBEVY-pT231 scFv 3.24-GFP were mixed in equal numbers and added to the wells (Fig. 3b). After washing, the wells were imaged and the number of yeast cells in each well was quantified.
Fig. 3.
Design and application of internal control yeast cell reporters in biopanning. (a) Schematic representation (of control yeast cell design. Yeast cells display a control scFv on their surface and intracellularly express a fluorescent protein (mCherry). (b) Schematic representation of biopanning method with control yeast cells included. Yeast cells expressing pBEVY-4420-mCherry and pBEVY-pT231 scFv 3.24-GFP are mixed in equal ratio and added to wells containing phospho-peptide (left), non-phospho-peptide (middle) and no peptide (right). (c) Quantification of the ratio of yeast cells expressing pBEVY-pT231 scFv 3.24-GFP to yeast cells expressing pBEVY-4420-mCherry present in wells after washing. Each datapoint indicates a ratio measured from a biological replicate. Each condition was repeated in two separate experiments with three biological replicates (6 datapoints per condition). Error bars indicate standard deviation of the datapoints. *P ≤ 0.05, ****P ≤ 0.0001 using Tukey’s multiple comparisons test. Otherwise, P > 0.05.
To evaluate if the total number of yeast cells in each well affects the degree of non-specific binding, we tested different cell densities in the biopanning experiment. For each cell density condition tested, the yeast cells expressing the two different plasmid constructs were mixed in equal proportion. After washing, full-well imaging and cell counting, the ratio of yeast cells expressing pBEVY-pT231 scFv 3.24-GFP to yeast cells expressing pBEVY-4420-mCherry was determined (Fig. 3c, see Table S1 and Fig. S2 for cell counts). These experiments showed that this ratio remained relatively consistent for the control wells, indicating that non-specific binding can be accurately captured by using the internal control yeast strain. Additionally, we observed that adding an initial total number of 0.5 × 106 yeast cells yields the best ratio of specific binders to non-specific binders (Fig. 3c). Taken together, these results show that, although cell density does not affect the degree of non-specific binding, it is important to consider when looking to optimize the best specific-to non-specific binder ratio.
Improved biopanning parameters
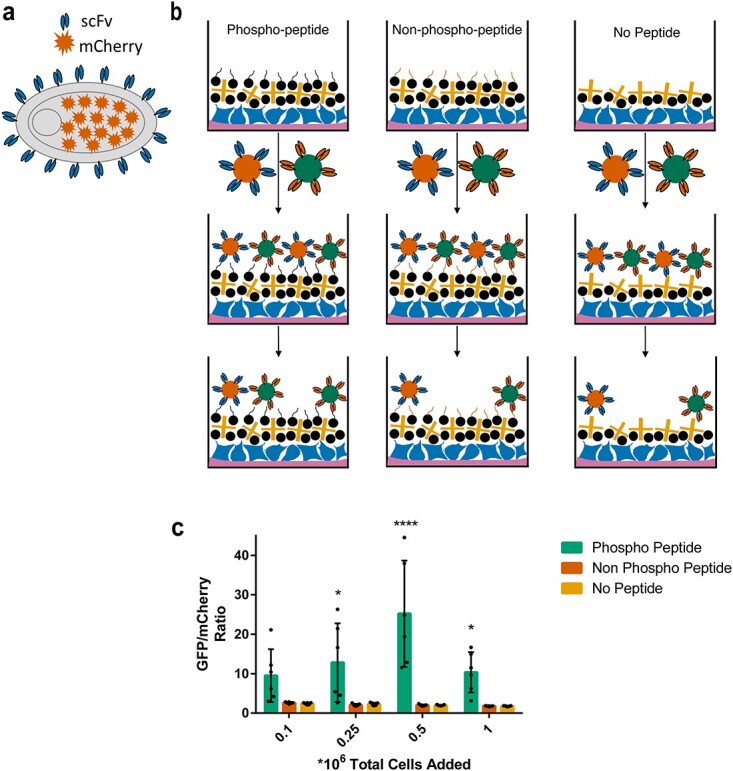
To further understand the mechanisms of this approach and to achieve the highest possible number of binding yeast cells, we aimed to test and optimize three parameters: the concentration of the biotinylation reagent (NHS-PEG4-Biotin) applied to HEK293FT cells, the concentration of the peptide antigen and the concentration of the streptavidin reagent that captures biotinylated peptide (Fig. 4).
Fig. 4.
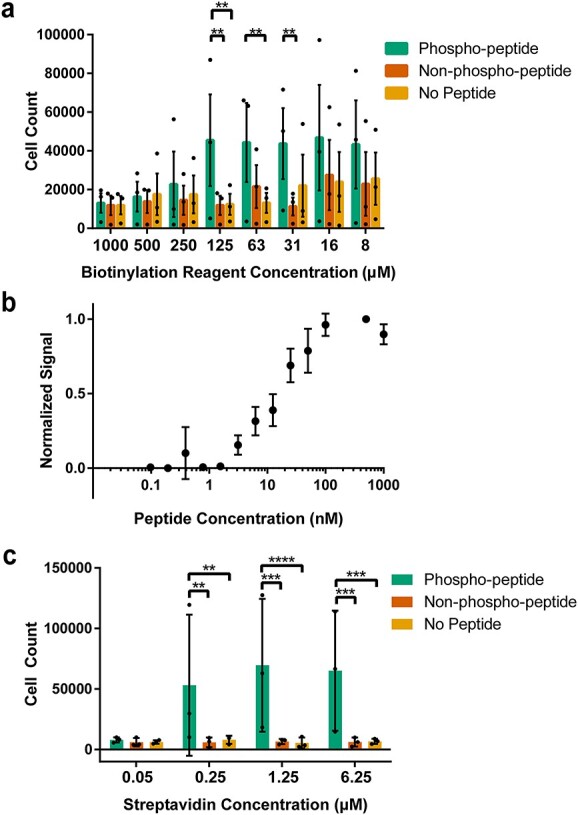
Improving biopanning parameters. (a) Quantification of number of yeast cells expressing pBEVY-pT231 scFv 3.24-GFP present in wells when tested across different biotin concentrations. Each datapoint indicates a value measured from a biological replicate in an experiment (3 separate experiments). Error bars indicate standard error of means of the datapoints. (b) Normalized biopanning ‘signal’ [(# yeast cells, phospho-peptide wells)-(# yeast cells, no peptide wells)]/(# yeast cells, highest concentration of peptide) for a range of peptide concentrations. Each datapoint indicates average of values measured from three separate experiments with three biological replicates in each experiment (3 separate experiments). Error bars indicate standard deviation. (c) Number of yeast cells present in wells when tested across different streptavidin concentrations. Each datapoint indicates average of values measured from two biological replicates in an experiment (3 separate experiments). Error bars indicate standard deviation of the datapoints. For panels (a) and (c), **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 using Tukey’s multiple comparisons test. Otherwise, P > 0.05.
To analyze the effect of the biotinylation reagent, we tested eight different concentrations ranging from 7.81 μM to 1 mM (Fig. 4a, see Table S1 for cell counts). For each of these concentrations, cells expressing pBEVY-pT231 scFv 3.24-GFP were added to wells containing p-tau peptide or controls. After washing and well imaging, the number of cells in each well was determined. Each biotinylation reagent concentration parameter was tested on 3 separate days with one sample quantified each day. Statistical testing for the differences between target peptide and control peptides for each concentration showed that only three biotin concentrations resulted in significantly higher binding compared with the controls (Fig. 4a). At a biotinylation reagent concentration of 31.25 μM, there was a significant difference in the cell count of the phosphorylated target peptide when compared with the non-phosphorylated peptide, but not when compared with the cell count of wells without peptide (Fig. 4a). At 62.5 μM, there was a significant difference in the cell counts between phosphorylated target peptide and wells containing no peptide, but not when compared with wells containing non-phosphorylated peptide (Fig. 4a). A biotinylation reagent concentration of 125 μM proved to be the most promising one, showing a significant difference in the cell count of phosphorylated target peptide when compared with both non-phosphorylated peptide and wells without peptide (Fig. 4a). For this reason, this reagent concentration was chosen as the standard in subsequent biopanning experiments. Notably, at higher biotinylation reagent concentrations ranging from 250 to 1000 μM, there was no significant difference between wells containing phospho-peptide and others (Fig. 4a). This may be due to increased density of biotin on the mammalian cell surface, leading to saturation of biotin binding sites in streptavidin and making it less available for binding to biotinylated peptides.
To assess the effect of peptide target antigen concentration on yeast biopanning, we tested a range of p-tau peptide concentrations between 0.098 nM and 1 μM (Fig. 4b, see Table S1 for cell counts). Each peptide concentration was tested on at least 3 separate days, with triplicates each day. For analysis of this data, the ‘signal’ (number of yeast cells expressing pBEVY-pT231 scFv 3.24-GFP still present after washing) was normalized. Normalizing the data involved subtracting the yeast cell count present in wells without peptide (taken as background) from the yeast cell count present in wells with target peptide. This number was then normalized to the highest signal from that day of experiments, which was always at least 0.1 μM. From these experiments, we saw no detectable signal from peptide concentrations ranging from 0.098 to 1.56 nM (Fig. 4b). However, we observed significant binding starting at a peptide concentration of 3.13 nM and apparent signal saturation beginning at a concentration of 0.1 μM (Fig. 4b). From these results, a peptide concentration of 0.1 μM was chosen for the purpose of obtaining the strongest signal possible in subsequent experiments.
To further optimize the interactions within this biopanning platform, we tested a range of streptavidin concentrations between 50 nM and 6.25 μM (Fig. 4c, see Table S1 for cell counts). For each concentration, cells expressing pBEVY-pT231 scFv 3.24-GFP were added to wells containing p-tau peptide and controls. After washing and full-well imaging, the number of cells in each well was determined, and each parameter was tested on 3 separate days with duplicates each day. Statistical analysis showed that a streptavidin concentration of 50 nM resulted in no significant difference between cells binding to target peptide and controls. However, each of the three higher streptavidin concentrations (250 nM, 1.25 μM, 6.25 μM) showed a significant difference when comparing cell count in wells with target peptide versus controls (Fig. 4c). Furthermore, when comparing the cell count of the p-tau peptide across these three streptavidin concentrations, statistical testing showed no significant differences. For these reasons, a streptavidin concentration of 1.25 μM was chosen to ensure there is enough streptavidin present for the capture of biotinylated peptides.
Biopanning with a lower affinity variant
Once the most important conditions were optimized, we were interested in testing the performance of biopanning to detect the binding of lower affinity scFvs (Fig. 5). Working toward the goal of using this platform to screen pools of antibodies, it is highly important to capture cells displaying antibody fragments with sub-optimal affinities.
Fig. 5.
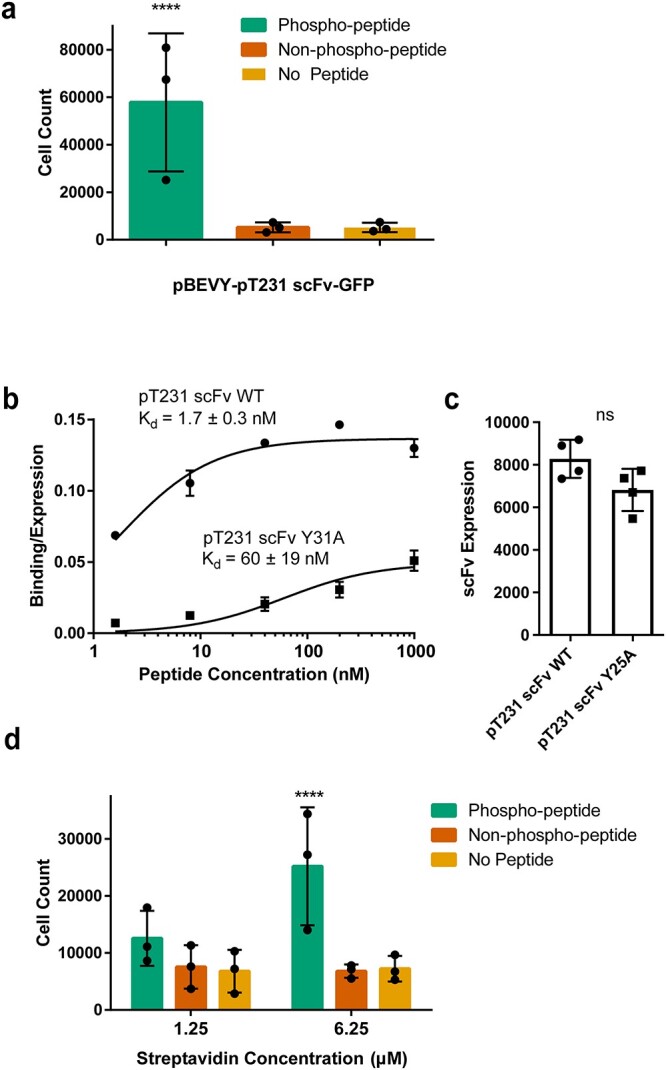
Testing selective binding for lower affinity scFvs to peptide of interest after optimizing parameters. (a) Quantification of number of yeast cells expressing pBEVY-pT231 scFv-GFP present in wells after biopanning with a biotin concentration of 125 μM, a peptide concentration of 0.1 μM and a streptavidin concentration of 1.25 μM. Each datapoint indicates the value measured from three biological replicates in an experiment (3 separate experiments). Error bars indicate standard deviation of the datapoints. (b) Binding/expression curve for wild-type scFv (pCT-4RE-pT231 scFv) and lower affinity mutant (pCT-4RE-pT231 scFv Y31A) measured using flow cytometry. Each datapoint indicates the average of values from two to four biological replicates. Error bars indicate standard deviation. (c) scFv expression of wild-type scFv and lower affinity mutant Y31A measured using flow cytometry. Each datapoint indicates the average of geometric means from two to four biological replicates. Error bars indicate standard deviation of the datapoints. (d) Quantification of yeast cells expressing pBEVY-pT231 scFv Y31A-GFP present in wells after biopanning. Each datapoint indicates the value measured from two biological replicates in an experiment (three separate experiments). Error bars indicate standard deviation of the datapoints. For panels (a) and (d), ****P ≤ 0.0001 using Tukey’s multiple comparisons test. Otherwise, P > 0.05. For panel (c), ns P > 0.05 using two-tailed Student’s t-test.
Because the pT231 scFv 3.24 used (KD = 200 pM) (Li et al., 2018) is a high affinity and specificity mutant of a previously described antibody (pT231 scFv WT, KD = 2.2 nM) (Shih et al., 2012), we first tested this wild-type scFv (Fig. 5a, see Table S1 for cell counts). The optimized conditions of biotin (125 μM), peptide (0.1 μM) and streptavidin (1.25 μM) were tested with the wild-type scFv (pBEVY-pT231 scFv-GFP) across 3 different days with triplicates each day. After the washing steps, the wells were imaged and the number of cells in each well was counted and analyzed. These experiments showed a significant difference in the number of cells in p-tau peptide wells when compared with negative controls (Fig. 5a).
To test even lower affinity antibodies, we turned our attention to a lower affinity mutant (pT231 scFv containing an alanine point mutation Y31A in the complementarity-determining region (CDR) we reported previously) (Li et al., 2018) of the original wild type. To compare the affinity of this mutant to the wild type, we performed titrations for yeast cells expressing the wild-type pT231 scFv or the mutant (Y31A) (expressed using the pCT-4RE plasmids cloned previously) (Li et al., 2018) with the p-tau peptide at concentrations ranging from 1.6 nM to 1 μM (Fig. 5b). These experiments showed that the affinity of pT231 scFv Y31A is ~35-fold less than that of pT231 scFv WT, with KD = 60 ± 19 nM (Fig. 5b). Although the binding/expression (see Materials and Methods for definition) of pT231 scFv Y31A was clearly smaller than that of pT231 scFv WT (Fig. 5b), the expression levels of the two scFvs were not significantly different (Fig. 5c), indicating that a higher fraction of displayed pT231 scFv Y31A is not functional.
The lower affinity variant was cloned into the pBEVY backbone co-expressing GFP (pBEVY-pT231 scFv Y31A-GFP) and tested in the biopanning platform (Fig. 5d, see Table S1 for cell counts). These experiments were repeated across 3 different days, with duplicates each day. These experiments showed that a streptavidin concentration of 1.25 μM, which previously appeared to be the concentration at which cell counts leveled off (Fig. 4c), no longer resulted in a significant difference between target wells and control wells. However, when we further increased the streptavidin concentration to 6.25 μM, we observed significant capture of yeast cells based on phospho-specific antibody binding (Fig. 5d). These results indicate that a high streptavidin concentration is necessary when biopanning for low to moderate affinity antibodies, which are often seen in antibody library screening.
Mock library biopanning in 6-well plates
To enable screening large libraries against phosphorylated protein sites using biopanning, we adapted the 96-well biopanning method to 6-well plates. We performed two biopanning experiments to test the ability to enrich yeast cells displaying the pT231 scFv Y31A from a background of yeast cells displaying a control scFv (4420). The ratio of yeast cells was 1:1000 and 1:100 000 (Y31A:4420), with a total of 3 × 108 yeast cells subjected to biopanning in the first round and 5 × 107 cells in subsequent rounds in both experiments. For the 1:100 000 experiment, we recovered ~2 × 106 cells after the first round, as estimated by the number of colonies obtained by growing several diluted fractions of collected cells on agar plates. We determined the ratio of cells displaying pT231 scFv Y31A versus 4420 using flow cytometry, leveraging the intracellular fluorescent proteins (Y31A cells expressing GFP and 4420 cells expressing mCherry). We observed a robust enrichment after each round of biopanning in both experiments, achieving ~600-fold enrichment in 4 rounds for the 1:1000 (Fig. 6a) and 1000-fold enrichment in 5 rounds for the 1:100 000 (Fig. 6b) experiment. The number of cells obtained in these biopanning experiments can be readily further enriched using FACS. These results demonstrate the feasibility of large library screening for p-tau binding scFvs using yeast biopanning.
Fig. 6.
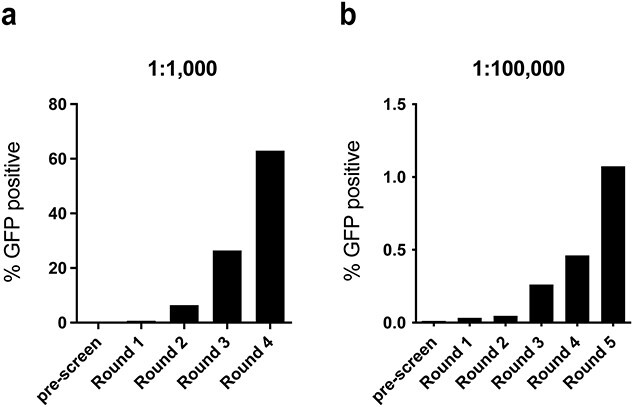
Biopanning yeast cells against p-tau in 6-well plates. Ratios of the number of yeast cells expressing pBEVY-pT231 scFv Y31A-GFP to yeast cells expressing pBEVY-4420-mCherry were measured using flow cytometry. Results from an initial ratio of Y31A:4420 = 1:1000 (a) and 1:100 000 (b). % GFP positive indicates the percentage of yeast cells expressing GFP among all yeast cells recovered after each round.
Discussion
We developed a novel method of yeast biopanning against site-specific protein phosphorylations and demonstrated its use against the human tau protein. The approach allows discriminating yeast cells displaying scFvs based on binding to peptides containing a single site-specific phosphorylation. The primary focus of the biopanning approach was on selective yeast capture based on phospho-site specific antibody interaction. We have previously demonstrated the capability to enhance the affinity of phospho-site specific antibodies without sacrificing specificity (Li et al., 2018), but clonal selection strategies based on phospho-specificity are still critically lacking. Here we demonstrated that the biopanning is highly selective to yeast cells expressing phospho-specific scFvs, and only to peptides that contain the phosphorylated residue. Using this approach, we show that yeast cells displaying scFvs with moderate affinities (KD = 60 nM), well within the range of antibodies identified from scFv libraries using yeast display biopanning(Wang et al., 2007; Zorniak et al., 2017), can be enriched from a pool of yeast cells consisting mostly of those displaying a non-binding scFv.
Although we consistently observed low cell counts from non-specific binding, we did observe large variations in the number of phospho-specific cell binding. This may originate from variations in the scFv display level, mammalian cell density and biotinylation (due to varying cell surface protein levels). In an antibody screening campaign, such variations may delay the enrichment of positive binders, but the high selectivity will ensure the specificity of clones identified. The bi-directional expression plasmids to display scFvs and intracellularly express fluorescent protein reporters allowed efficient counting of interacting yeast cells, and in-well monitoring of non-specific binding. This allowed us to improve biopanning conditions, such as streptavidin and biotinylation reagent concentrations. The expression scheme decouples fluorescent reporter expression from scFv expression, which would be particularly advantageous for validating scFvs that are challenging to produce. However, in library screening, the use of ribosomal skipping sequences such as T2A (Hinz et al., 2020; Jia et al., 2019) would allow monitoring scFv display levels during the enrichment process, since the fluorescence is correlated with the scFv production. This feature could be harnessed by using methods to screen yeast–mammalian cell interactions using FACS (Bogen et al., 2021; Yang et al., 2019).
The yield of cells expressing binders is critical for enrichment. For the 96-well experiments, the main purpose was to validate the specific capture of yeast cells based on phospho-specific scFv-antigen interaction. To this end, we used a stringent washing condition, which relies on pipetting the cells against the plate walls multiple times. Figure 2b shows that we recovered about 2% (2 × 104 out of 106 cells added) of cells expressing the high affinity pT231 scFv 3.24 after biopanning in 96 wells. Under the improved conditions described in Fig. 5a, we recovered ~5.8% (an average of 57 828 cells from ~106 cells added) albeit using the WT scFv. Using the lower affinity Y25A scFv, the recovery was about 2.5% (Fig. 5d, an average of 25 209 cells from 106 cells added). Therefore, for a successful enrichment, we would need to screen at least 40-fold excess of the library size to reliably retain a binder. Considering the large number of wells to be screened, the 96-well biopanning approach will be more useful for verifying individual clones or screening small libraries. In the 6-well experiments, we used a different washing approach that relies on rocking or tilting the plates. In the 6-well enrichment experiment that started with a 1:100 000 ratio of Y31A:4420 cells (Fig. 6b), we recovered ~2 × 106 cells after the first round. Based on the percentage of GFP-positive cells (0.0011%, Fig. 6b) in the recovered cells determined using flow cytometry, we estimate that about 225 cells were GFP-positive. Since the input was about 3000 GFP-positive cells (1 in 105 out of 3 × 108 total cells), we recovered about 7.5% (225/3000) of GFP-positive cells in the first round of biopanning. This shows that the 6-well approach allows higher recovery of p-tau expressing cells compared with the 96-well experiment.
The recovery percentage is an important guide for designing biopanning enrichment. In antibody screening, it is customary to screen 10 times the library size (or the diversity of unique clones in the pool), which implies about a 10% chance of recovering binders. Our results show that for our biopanning scheme, it is necessary to screen ~13.3 times the library size (based on the 7.5% recovery) to ensure the recovery of a binder cell. This indicates that to screen a library of 108 unique binders, at least 1.33 × 109 cells need to be screened, to recover a modest affinity binder present in the library. Based on the number of cells we used per 6-well plate (3 × 108 cells), we need to use four to five wells to comprehensively screen the library. Assuming the presence of a binder with a similar affinity and expression, this result indicates that screening large libraries containing >109 binders will require at least 40–50 wells or 7–8 6-well plates. Overall, these results show that the success of finding a binder using the biopanning will be dictated by the affinity and frequency of binders in the pool screened, as well as the number of cells screened and the washing condition.
In our experiments, biopanning using peptides immobilized directly on a neutravidin-coated surface of plates was unsuccessful (see Supporting Information), and the monolayer of HEK293FT cells on which the biotinylated peptides are immobilized seems to be required. Previous studies have demonstrated that yeast cells can be captured on flat surfaces, such as those made of single-crystal cadmium sulfide and synthetic sapphire (α-Al2O3) through peptide–solid surface interactions (Krauland et al., 2007; Peelle et al., 2005). Other studies have reported a yeast rolling assay, in which yeast cells displaying a subunit of integrin showed varying rolling velocity under controlled shear flow on a slide coated with the intercellular adhesion molecule-1 (Pepper et al., 2006; Pepper et al., 2013). The fact that yeast cell binding to surface-immobilized ligand is affected by shear flow indicates that the washing conditions used in our yeast biopanning approach may not allow yeast binding to flat surfaces based on peptide-scFv interaction. Since discriminating antibody clones based on specificity is critical, such stringent washing conditions are desirable in this application. The mammalian cell surface provides a matrix in which proteins are embedded and can be biotinylated. This complex structure may provide higher avidity interactions that allow yeast cell binding. For library screening, a negative selection step will be necessary to eliminate antibodies that bind to non-targets, including the mammalian cell surface antigens, biotin and streptavidin. Negative selections have been already demonstrated using yeast biopanning to identify cell-type selective binders (Zorniak et al., 2017). In addition, to prevent selecting antibodies that bind mammalian cell surface antigens, switching the type of mammalian cells used between rounds of biopanning would be necessary. For this purpose, mouse endothelial cells that have been used for yeast biopanning (Wang and Shusta, 2005) would be an ideal choice since it allows yeast biopanning against small antigens and reduces the chance of selecting binders against commonly expressed human cell surface proteins.
Notably, the streptavidin concentration needed to be increased by 5-fold to capture the yeast cells displaying the lower affinity scFv mutant (pT231 scFv Y31A) (Fig. 5c). In addition to having lower affinity, the Y31A mutant also showed lower binding signal normalized to expression level (Fig. 5b), indicating that a greater fraction of displayed scFv may not be functional compared with the wild-type scFv. This will reduce the surface density of available scFvs that interact with the target, which may explain why a higher streptavidin concentration was required. Such sub-optimal scFv folding is a likely scenario in heterologous antibody fragment libraries, and therefore further demonstrates the robustness of the yeast biopanning method for detecting phospho-site specific interaction. At higher streptavidin concentrations, the mass transfer rate of streptavidin to the surface may be higher, leading to an increased number of streptavidin immobilized, rather than increasing the number of biotins occupying each streptavidin molecule. This may enhance the density of phospho-peptide immobilized on the surface, leading to increased avidity and thus the capture of yeast cells displaying the modest-affinity clone.
Although we have demonstrated this capability using a single phosphorylation site in tau, we anticipate this method will be widely applicable to phospho-specific antibody characterization and screening. Due to the fact that protein phosphorylation sites tend to be located in disordered regions (Iakoucheva et al., 2004; Nicolaou et al., 2021), synthetic peptides containing phosphorylated residues have been extremely effective antigens for generating phospho-site specific antibodies suitable for a wide range of applications, including various cell and tissue labeling, immunoblotting and immunoassays. This new capability to biopan against phospho-peptides will allow rapid identification of monoclonal antibodies against protein phospho-sites after immunization. Moreover, the biopanning approach followed by FACS for screening high specificity clones is a promising strategy for identifying phospho-specific binder clones from large naïve libraries (Feldhaus et al., 2003; Li et al., 2018), camelid nanobodies(McMahon et al., 2018) and other binders (Hackel et al., 2008; Kruziki et al., 2015). This is a unique advantage of our yeast biopanning approach compared with other phospho-specific antibody screening methods, as it allows screening large libraries while quantifying the binding specificity. Using yeast biopanning, we anticipate identifying antibodies against tau phosphorylation sites that currently lack specific antibodies (Arbaciauskaite et al., 2021).
Author Contributions
Monika Arbaciauskaite (Conceptualization [lead], Investigation [lead], Methodology [lead], Resources [lead], Validation [equal], Visualization [lead], Writing—original draft [equal], Writing—review & editing [equal]), Azady Pirhanov (Data curation [supporting], Investigation [supporting], Methodology [supporting], Resources [supporting], Writing—review & editing [supporting]), Erik Ammermann (Investigation [supporting], Resources [supporting], Writing—review & editing [supporting]), Yu Lei (Funding acquisition [equal], Project administration [equal], Supervision [equal], Writing—review & editing [equal]), Yong Ku Cho (Conceptualization [equal], Funding acquisition [equal], Project administration [equal], Writing—review & editing [equal]).
Conflict of Interest
The authors declare no commercial or financial conflict of interest.
Funding
This work was funded by grants NSF 1706743, NIH 1R21NS111358-01A1 and Alzheimer’s Association 2019-AARG-NFT-640971. M.A. was also supported by the GE innovation fellowship.
Data Availability
Biopanning cell count data are included as a supplementary data file. All other data will be available upon reasonable request.
Supplementary Material
Contributor Information
Monika Arbaciauskaite, Department of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA.
Azady Pirhanov, Department of Biomedical Engineering, University of Connecticut, Storrs, CT 06269, USA.
Erik Ammermann, Department of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA.
Yu Lei, Department of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA; Department of Biomedical Engineering, University of Connecticut, Storrs, CT 06269, USA.
Yong Ku Cho, Department of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA; Department of Biomedical Engineering, University of Connecticut, Storrs, CT 06269, USA; Institute for Systems Genomics, University of Connecticut, Storrs, CT 06269, USA.
References
- Arbaciauskaite, M., Lei, Y. and Cho, Y.K. (2021) Antib. Ther., 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton, N.J., Pascoal, T.A., Karikari, T.K. et al. (2021) Acta Neuropathol., 141, 709–724. 10.1007/s00401-021-02275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boder, E.T. and Wittrup, K.D. (1997) Nat. Biotechnol., 15, 553–557. 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Bogen, J.P., Storka, J., Yanakieva, D. et al. (2021) Biotechnol. J., 16, 2000240. 10.1002/biot.202000240. [DOI] [PubMed] [Google Scholar]
- Bordeaux, J., Welsh, A.W., Agarwal, S., Killiam, E. et al. (2010) Biotechniques, 48, 197–209. 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, P. (2001) Eur. J. Biochem., 268, 5001–5010. 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Dujardin, S., Commins, C., Lathuiliere, A. et al. (2020) Nat. Med., 26, 1256–1263. 10.1038/s41591-020-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan, E., Eid, S., Weber, C. et al. (2017) Mol. Neurodegener., 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhaus, M.J., Siegel, R.W., Opresko, L.K. et al. (2003) Nat. Biotechnol., 21, 163–170. 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- Graves, J.D. and Krebs, E.G. (1999) Pharmacol. Ther., 82, 111–121. 10.1016/S0163-7258(98)00056-4. [DOI] [PubMed] [Google Scholar]
- Hackel, B.J., Kapila, A. and Dane Wittrup, K. (2008) J. Mol. Biol., 381, 1238–1252. 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, S.C., Elter, A., Grzeschik, J. et al. (2020) Methods Mol. Biol., 2070, 211. 10.1007/978-1-4939-9853-1_12. [DOI] [PubMed] [Google Scholar]
- Huang, D. and Shusta, E.V. (2005) Biotechnol. Prog., 21, 349–357. [DOI] [PubMed] [Google Scholar]
- Iakoucheva, L.M., Radivojac, P., Brown, C.J. et al. (2004) Nucleic Acids Res., 32, 1037–1049. 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze, S., Mattsson, N., Palmqvist, S. et al. (2020) Nat. Med., 26, 379–386. 10.1038/s41591-020-0755-1. [DOI] [PubMed] [Google Scholar]
- Jia, Y., Ren, P., Duan, S. et al. (2019) Biotechnol. Lett., 41, 1067–1076. 10.1007/s10529-019-02710-5. [DOI] [PubMed] [Google Scholar]
- Johnson, L.N. and Lewis, R.J. (2001) Chem. Rev., 101, 2209–2242. 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Krauland, E.M., Peelle, B.R., Wittrup, K.D. and Belcher, A.M. (2007) Biotechnol. Bioeng., 97, 1009–1020. 10.1002/bit.21341. [DOI] [PubMed] [Google Scholar]
- Kruziki, M.A., Bhatnagar, S., Woldring, D.R. et al. (2015) Chem. Biol., 22, 946–956. 10.1016/j.chembiol.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. and Cho, Y.K. (2020) J. Neurochem., 152, 122–135. 10.1111/jnc.14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Cockburn, W., Kilpatrick, J.B. and Whitelam, G.C. (2000) Biochem. Biophys. Res. Commun., 268, 398–404. 10.1006/bbrc.2000.2129. [DOI] [PubMed] [Google Scholar]
- Li, D., Wang, L., Maziuk, B.F. et al. (2018) J. Biol. Chem., 293, 12081–12094. 10.1074/jbc.RA118.003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N., Zhao, Z., Tan, Y. et al. (2016) Anal. Chem., 88, 1246–1252. 10.1021/acs.analchem.5b03637. [DOI] [PubMed] [Google Scholar]
- Mandell, J.W. (2003) Am. J. Pathol., 163, 1687–1698. 10.1016/S0002-9440(10)63525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon, C., Baier, A.S., Pascolutti, R. et al. (2018) Nat. Struct. Mol. Biol., 25, 289–296. 10.1038/s41594-018-0028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C.A., Martinat, M.A. and Hyman, L.E. (1998) Nucleic Acids Res., 26, 3577–3583. 10.1093/nar/26.15.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou, S.T., Hebditch, M., Jonathan, O.J. et al. (2021) Sci. Rep., 11, 9930. 10.1038/s41598-021-88992-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson, T. (2004) Cell, 116, 191–203. 10.1016/S0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- Peelle, B.R., Krauland, E.M., Wittrup, K.D. and Belcher, A.M. (2005) Acta Biomater., 1, 145–154. 10.1016/j.actbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Pepper, L.R., Hammer, D.A. and Boder, E.T. (2006) J. Mol. Biol., 360, 37–44. 10.1016/j.jmb.2006.04.049. [DOI] [PubMed] [Google Scholar]
- Pepper, L.R., Parthasarathy, R., Robbins, G.P. et al. (2013) Protein Eng. Des. Sel., 26, 515–521. 10.1093/protein/gzt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, W., Yang, J.-R., Pearson, N.M. et al. (2012) PLoS Genet., 8, e1002603. 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, H.H., Tu, C., Cao, W. et al. (2012) J. Biol. Chem., 287, 44425–44434. 10.1074/jbc.M112.415935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijssen, E.H., La Joie, R., Wolf, A. et al. (2020) Nat. Med., 26, 387–397. 10.1038/s41591-020-0762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velappan, N., Mahajan, A., Naranjo, L. et al. (2019) MAbs, 11, 1206–1218. 10.1080/19420862.2019.1632113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X.X. and Shusta, E.V. (2005) J. Immunol. Methods, 304, 30–42. 10.1016/j.jim.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Wang, X.X., Cho, Y.K. and Shusta, E.V. (2007) Nat. Methods, 4, 143–145. 10.1038/nmeth993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, J., Peng, H. and Rader, C. (2017) Exp. Mol. Med., 49, e305. 10.1038/emm.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling, H., Mair, W., Kumar, M. et al. (2020) Cell, 1. 10.1016/j.cell.2020.10.029. [DOI] [Google Scholar]
- Xia, Y., Prokop, S. and Giasson, B.I. (2021) Mol. Neurodegener., 16, 37. 10.1186/s13024-021-00460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., Wan, Y., Tao, P. et al. (2019) Proc. Natl. Acad. Sci. U S A, 116, 14971–14978. 10.1073/pnas.1908571116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorniak, M., Clark, P.A., Umlauf, B.J. et al. (2017) Sci. Rep., 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Biopanning cell count data are included as a supplementary data file. All other data will be available upon reasonable request.