

Abstract
More than 50 million adults in America suffer from chronic pain. Opioids are commonly prescribed for their effectiveness in relieving many types of pain. However, excessive prescribing of opioids can lead to abuse, addiction, and death. Non-steroidal anti-inflammatory drugs (NSAIDs), another major class of analgesic, also have many problematic side effects including headache, dizziness, vomiting, diarrhea, nausea, constipation, reduced appetite, and drowsiness. There is an urgent need for the understanding of molecular mechanisms that underlie drug abuse and addiction to aid in the design of new preventive or therapeutic agents for pain management. To facilitate pain related small-molecule signaling pathway studies and the prediction of potential therapeutic target(s) for the treatment of pain, we have constructed a comprehensive platform of a pain domain-specific chemogenomics knowledgebase (Pain-CKB) with integrated data mining computing tools. Our new computing platform describes the chemical molecules, genes, proteins, and signaling pathways involved in pain regulation. Pain-CKB is implemented with a friendly user interface for the prediction of the relevant protein targets and analysis and visualization of the outputs, including HTDocking, TargetHunter, BBB predictor, and Spider Plot. Combining these with other novel tools, we performed three case studies to systematically demonstrate how further studies can be conducted based on the data generated from Pain-CKB and its algorithms and tools. First, systems pharmacology target mapping was carried out for four FDA approved analgesics in order to identify the known target and predict off-target interactions. Subsequently, the target mapping outcomes were applied to build physiologically based pharmacokinetic (PBPK) models for acetaminophen and fentanyl to explore the drug–drug interaction (DDI) between this pair of drugs. Finally, pharmaco-analytics was conducted to explore the detailed interaction pattern of acetaminophen reactive metabolite and its hepatotoxicity target, thioredoxin reductase.
Keywords: Pain, Knowledgebase, Opioids, NSAIDs, Computational Systems Pharmacology-Target Mapping, PBPK
Graphical Abstract
INTRODUCTION
Pain is one of the most common reasons for people to seek medical help1 and is often associated with restricted daily activities and lowered mood, which will ultimately lead to a decrease in quality of life. According to a previous National Health Interview Survey data-based estimation, around 50 million U.S. adults (20.4%) suffer from chronic pain.2 With such high prevalence, pain-related health care costs are estimated to be $280 billion each year.3 Other economic losses could result due to the loss of productivity caused by pain.4 Still, approximately 79% of patients are not satisfied with their medication management5,6 due to limited effectiveness7 and severe side effects caused by currently available analgesics.8 Thus, research into the pain process and treatment is of great urgency.
Pain is defined by the International Association for the Study of Pain (IASP) as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.”9 The detailed mechanism behind pain sensation is complex.10 Nociception starts with the activation of nociceptors caused by a thermal, mechanical, or chemical stimulus that goes beyond the noxious range.11 Once those nociceptors are evoked, electrical signals are generated and transduced from the periphery to an area called the dorsal horn located in the spinal cord, where various sensory and nociceptive signals are converged and modulated12 before being passed by neurotransmitters along the nerve fiber to the brain.
Currently, there are many categories of analgesics available on the market. Non-steroidal anti-inflammation drugs (NSAIDs), such as ibuprofen, are some of the most commonly used classes of analgesics. These drugs are known to block the synthesis of prostanoids from arachidonic acids by inhibiting the prostaglandin G/H synthases (COX enzymes).13 NSAIDs are usually used to treat mild to moderate pain,14 and their side effects include ulceration and bleeding in the GI tract, stomach pain, and renal failure15,16 depending on the dose and the inhibition ratio between COX1 and COX2.17 When it comes to the relief of moderate to severe pain, the use of opioid analgesics like morphine, fentanyl, and codeine may be more dominant.18 As agonists of opioid receptors, opioid analgesics can hyperpolarize the neuronal cells19 to inhibit the neural excitability and decrease the release of neurotransmitters to derive analgesia.20 Opioids may have better therapeutic effects; however, they often have more severe side effects such as respiratory depression, dependence, and drug addiction.8,21
Though various analgesics are available, there are critical problems associated with the use of analgesics. The most important aspect is the safety issue related to analgesics, especially opioids. Opioid poisoning is reported to be involved in nearly 40% of all drug poisoning deaths22 due to the synergistic effects caused by the combination use of opioids23 (https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates).24 Even under the recommended dosage, chronic use of opioids will still cause tolerance, addiction, and severe withdrawal syndrome.25 Moreover, the lack of progress achieved by the pharmaceutical industry during the past few years is not reflective of the urgent need for new analgesics with better efficacy and side effect profile.26
Herein, we have constructed the pain domain-specific chemogenomics knowledgebase (Pain-CKB)27 based on our previously established molecular information databases28–32 in order to accelerate the research in pain-related areas. The knowledgebase assembles a large amount of analgesic information and pain-related protein targets extracted from the literature. Integrated with the algorithms previously developed by our lab, such as TargetHunter,33 HTDocking, and Spider Plot, Pain-CKB enables pain-related target identification, drug repurposing analysis, small-molecule screening, and drug–drug interaction (DDI) predictions from the view of systems pharmacology. Therefore, Pain-CKB is a valuable platform for information sharing and investigation in the pain domain in the hope of aiding research on pain and analgesics. Based on the newly established Pain-CKB, first, a computational systems pharmacology target mapping was constructed to identify the known targets and potential off-target interactions based on the output of HTDocking and TargetHunter for four common analgesics. Second, physiologically based pharmacokinetic (PBPK) models were applied to quantify the DDI between acetaminophen and fentanyl in the pharmacokinetics (PK) field. Finally, we conducted docking studies to reveal the detailed interaction pattern between acetaminophen reactive metabolite and its reported liver toxicity target, thioredoxin reductase 1. We provide these case studies as an example of the usage of our database and how our knowledge can serve as a starting point for other follow-up studies combined with novel computational methods.
RESULTS AND DISCUSSION
Development of Pain-CKB.
To facilitate pain research, we have constructed the pain domain-specific chemogenomics knowledgebase (Pain-CKB).27 Pain-CKB is also equipped with various chemoinformatics tools like HTDocking,28 Target-Hunter,33 Spider Plot,31 and BBB predictor28 to provide public users with cloud computing services for target identification and systems pharmacology research. Basically, HTDocking is an online high-throughput molecular docking algorithm powdered by idock,34 which can identify possible targets or off-target interactions for a query compound. Target-Hunter is a molecular fingerprints-based tool for calculating the similarity between the query compound and the active compounds of each target collected in our database, predicting the potential targets for user-submitted ligand(s). Spider Plot is a tool for the online visualization of the molecule–protein interaction network based on the output from both HTDocking and TargetHunter. In addition, BBB predictor is an independent tool to predict whether a compound can pass the BBB.
Workflow of Pain-CKB.
As shown in Figure 1, our platform intrinsically works on a set of conformations of pain-related crystal or cryo-EM structures and their relevant compounds. Users can submit up to five different compounds to our platform for calculations. The platform first queues a task for each query compound and afterward converts its input format such as SMILES or SDF to PDB and PDBQT format with the help of Open Babel.35 Subsequently, the BBB predictor is applied to predict the BBB penetration for each query compound based on its structure. Simultaneously, the revised idock automatically docks each query compound into the previously defined binding pockets in different conformations of a target protein and generates docking scores, while the TargetHunter calculates the similarity score based on the molecular fingerprints using the Tanimoto coefficients (from 0.0 to 1.0, totally different to 100% similar) against its known active compounds data set. Finally, a target classification is performed by docking scores and similarity score, and the classification results are then passed to Spider Plot for visualization of the drug–targets interaction network. All the output data from Pain-CKB can serve as the starting point for other follow-up studies.
Figure 1.
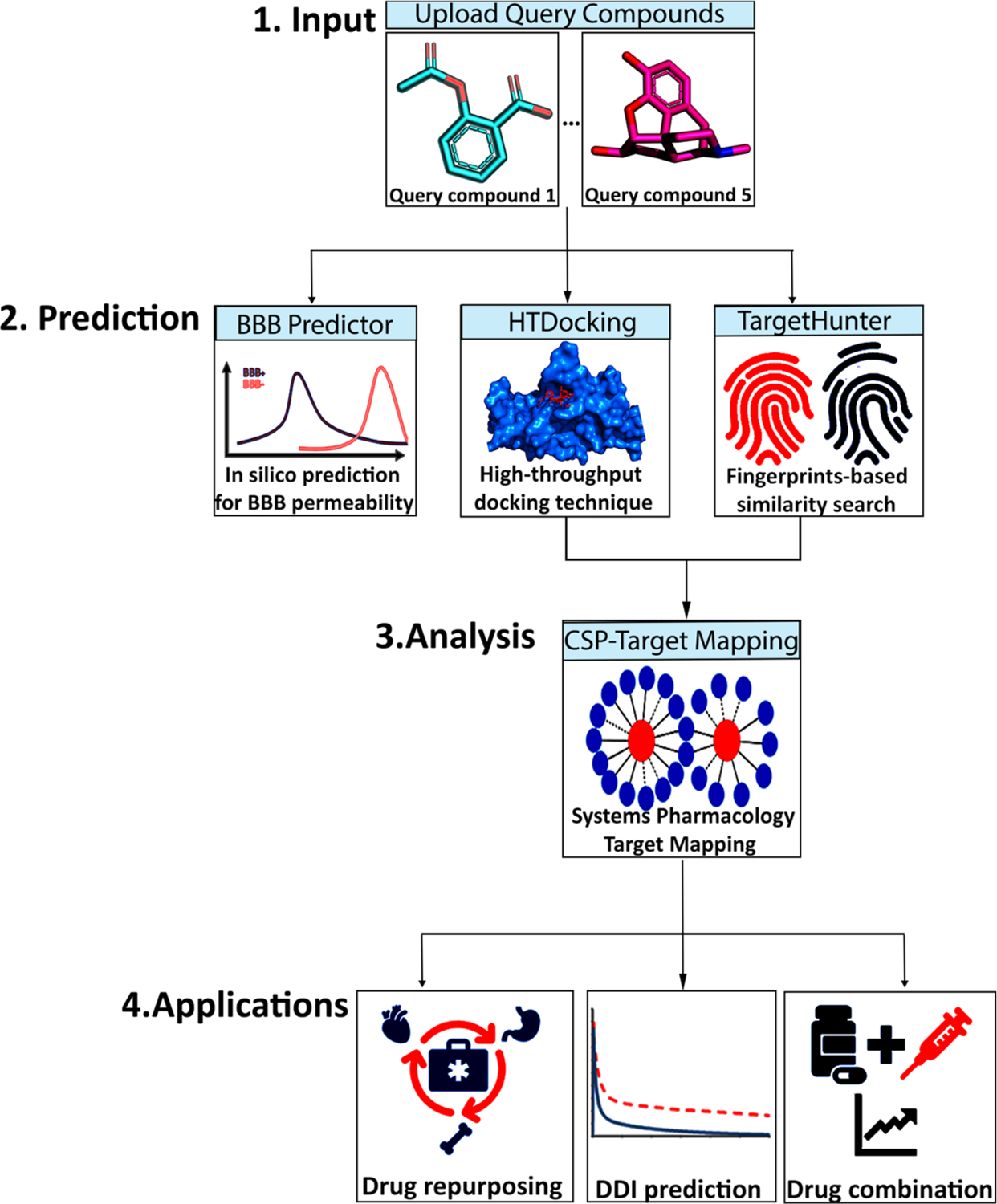
Workflow of the Pain-CKB server that is divided into three major steps: (i) input of chemical agent; (ii) in silico BBB prediction, high-throughput docking (HTDocking) with pain-related targets, and fingerprints-based similarity search (TargetHunter) by our established algorithms implemented in Pain-CKB; (iii) systems pharmacology target mapping for potential drug repurposing, DDI prediction, and drug combination.
Overview of Pain-CKB.
Our database is available at https://www.cbligand.org/g/pain-ckb. To date, Pain-CKB has archived 272 analgesics and 84 pain-related targets. As shown in Figure 2, the collected targets include (1) 28 G-protein coupled receptors (GPCR), such as mu, kappa, and delta opioid receptors (OPRM, OPRK, OPRM) and cannabinoid receptors (CNR1, CNR2); (2) 32 enzymes, such as prostaglandin G/H synthases (PGH1, PGH2) and angiotensin-converting enzyme (ACE); (3) 14 ion channels, such as transient receptor potential cation channels (TRPV1, TRPV2, TRPV3, TRPM4, and TRPM8); and (4) 2 nuclear receptors, progesterone receptor (PRGR) and estrogen receptor 1 (ESR1). The Pain-CKB will be continuously updated to maintain the validity of the information collected.
Figure 2.
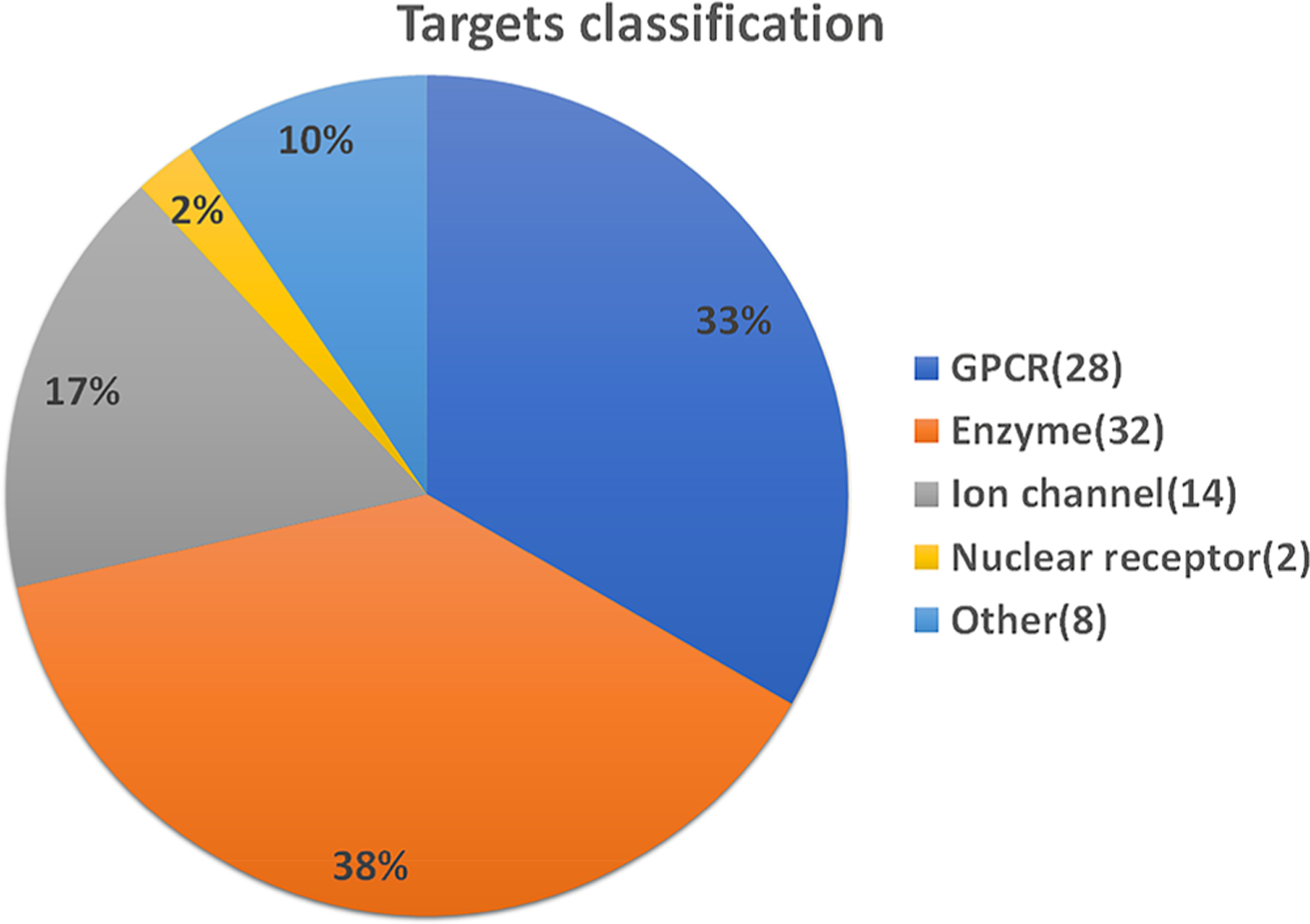
Summary of pain-related targets. A total of 84 pain-related targets are collected in the Pain-CKB: (1) 28 G-protein coupled receptors, (2) 32 enzymes, (3) 14 ion channels, (4) 2 nuclear receptors, and (5) 8 targets categorized as “other”.
Validation of Pain-CKB with Codeine (Known Drug).
As shown in Figure 3, we submitted codeine as the signal query compound to our platform. We observed that codeine connects to two green target nodes with green solid lines, including MDR1 (ATP binding cassette subfamily B member 1) and OPRM mouse (mouse Mu-type opioid receptor). These results are consistent with the fact that codeine binds to these two receptors with high affinities, which further validated that our algorithms are reliable. Interestingly, our results also showed that codeine may bind to other pain-related targets with potential synergy.
Figure 3.
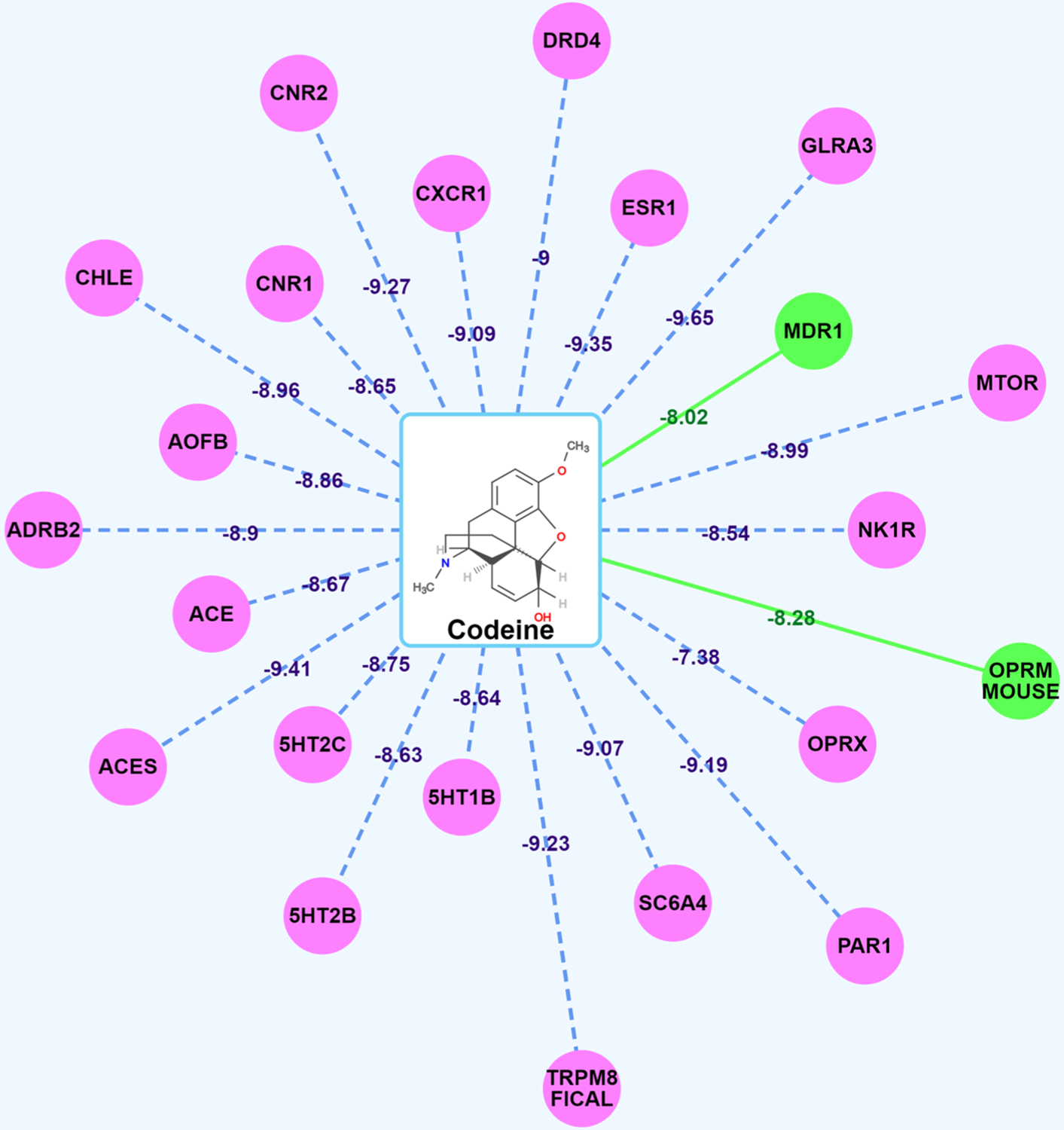
Spider Plot for codeine visualization and analysis. The average docking scores are displayed as connection labels, and the protein targets on which the query compound is active are displayed as circular discs.
Validation of Pain-CKB with Spiro[4.5]decan-8-yl Analogue 1436 (New Compound).
We submitted spiro[4.5]decan-8-yl analogue 14, which is a novel mentholbased transient receptor potential cation channel subfamily M member 8 channel (TRPM8) antagonist reported in our recent publication, into our platform for further validation.36 As shown in Figure 4, we observed that there are no green target nodes connected to spiro[4.5]decan-8-yl analogue 14. This is consistent with the fact that this compound is a new TRPM8 antagonist and the similarity with the reported compounds targeting TRPM8 calculated by TargetHunter is not higher than 0.85. However, we found that spiro[4.5]decan-8-yl analogue 14 connects to TRPM8 with pink lines (Figure 4) with a high docking score of 10.55 kcal/mol computed by HTDocking. These results further indicated that our Pain-CKB and the tools are powerful to predict the targets for new or unknown compounds.
Figure 4.
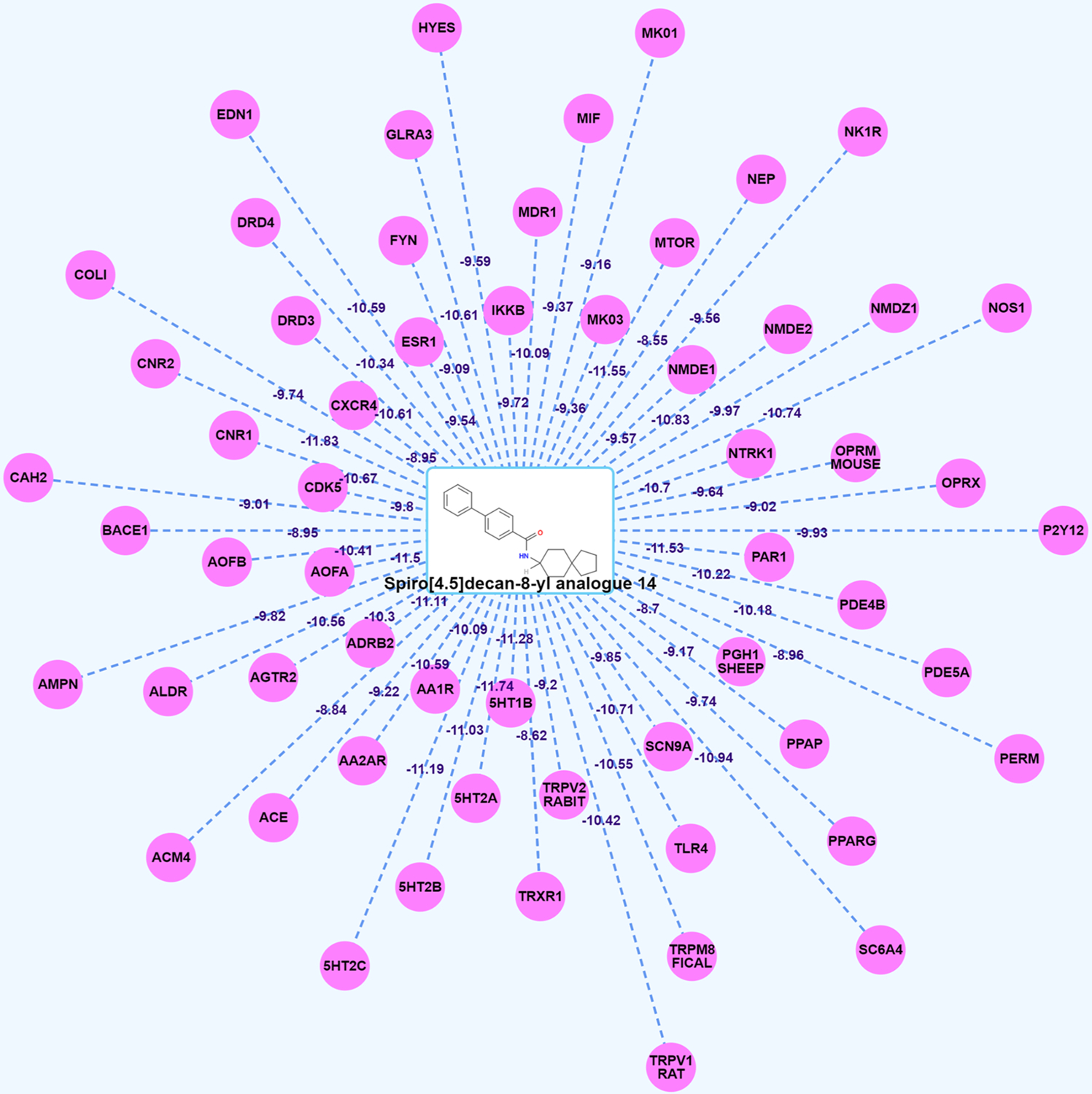
Spider Plot for spiro[4.5]decan-8-yl analogue 14 (new compound) visualization and analysis. The average docking scores are displayed as connection labels, and the protein targets on which the query compound is active are displayed as circular discs.
In the following section, we conducted three case studies to demonstrate the function of Pain-CKB and how computation algorithms can aid pain research.
Case Study 1: Computational Systems Pharmacology-Target Mapping for 4 FDA-Approved Analgesics.
The prediction of polypharmacology of known drugs is a potential method for drug repurposing and DDI prediction.37,38 Here, we conducted a computational systems pharmacology-based study using the results from both HTDocking and TargetHunter, and Spider Plot is used to generate a target mapping for four analgesics approved by the FDA (acetaminophen, diclofenac, morphine, and fentanyl). Among them, though acetaminophen works by interacting with COX enzymes,39 it is categorized with aniline analgesics because of the limited inhibition effect on COX enzymes,40 while diclofenac is a typical NSAID.41 On the other hand, morphine and fentanyl are members of the opioid analgesics family, which exhibit their pain-relieving effect by binding to opioid receptors to mimic the endogenous opioid peptides.42 As shown in Figure 5, after integrating the information included in the Pain-CKB and the results of TargetHunter and HTDocking algorithm, we were able to generate a target mapping for these four drugs by Spider Plot. The green circles and solid lines indicate known protein targets and interactions between the corresponding drug and protein targets. The purple circles and dashed lines represent the predicted protein target with interaction.
Figure 5.
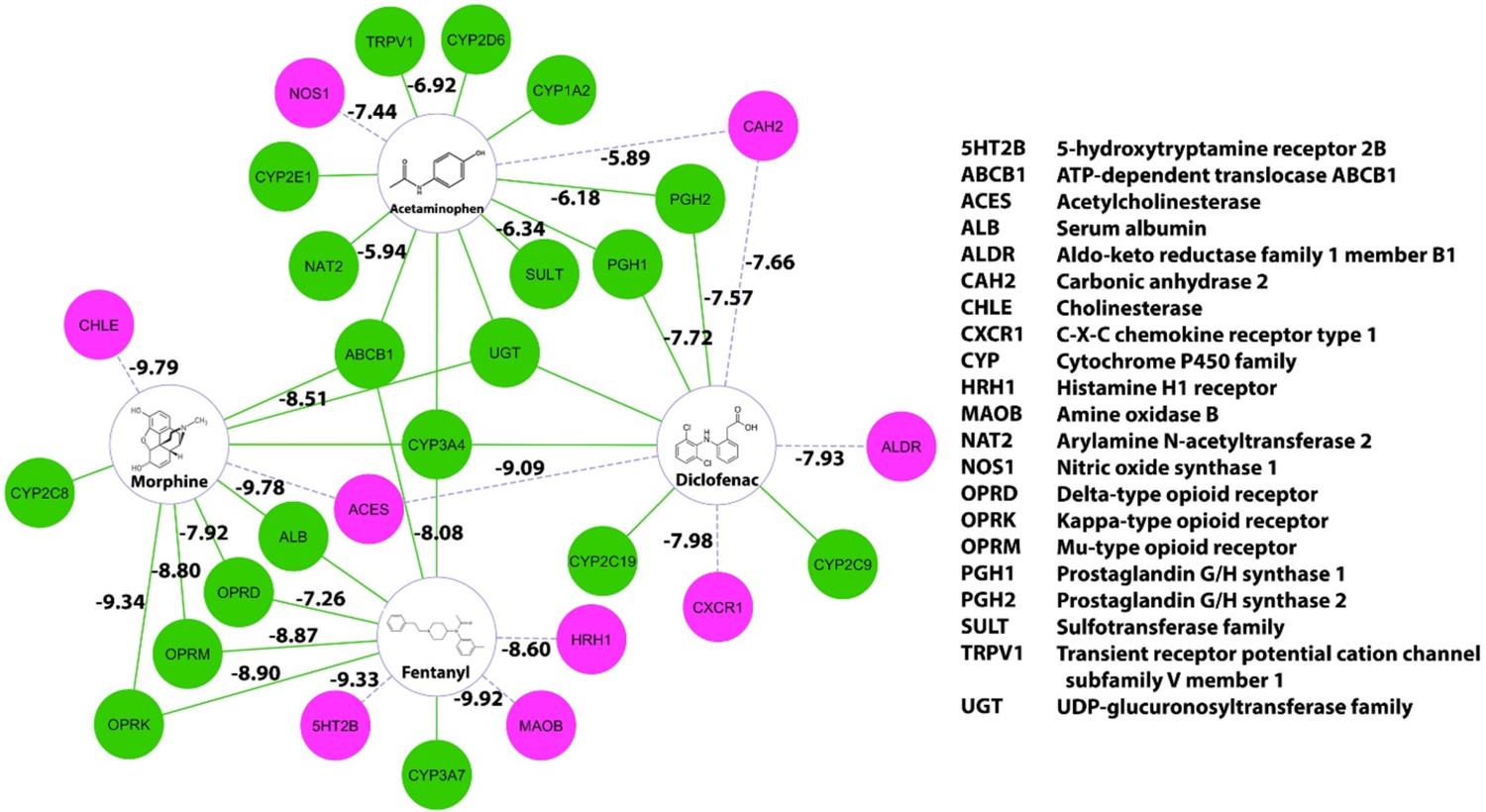
Computational systems pharmacology-target mapping (CSP-Target Mapping) for target proteins, enzymes, transporters, and potential targets for acetaminophen, diclofenac, morphine, and fentanyl. The green dots and solid lines represent the known targets and interactions. The purple dots and dashed lines represent the predicted targets and interactions.
As expected, our results showed clearly that the COX enzymes were targeted by both acetaminophen and diclofenac.43 For acetaminophen, transient receptor potential cation channel subfamily V member 1 (TRPV1), which is another known target for acetaminophen in the brain to produce antinociception,44 is also listed in our result. Moreover, Pain-CKB also predicted several acetaminophen off-target interactions such as nitric oxide synthase 1 (NOS1) and carbonic anhydrase 2 (CAH2). As to diclofenac, apart from the COX enzymes, predicted targets like acetylcholinesterase (ACES) and C-X-C chemokine receptor type 1 (CXCR1), have been reported to interact with diclofenac analogs.45,46
For morphine and fentanyl, known targets mu, kappa, and delta opioid receptors47 are all identified by Pain-CKB. Our algorithm also predicted cholinesterase (CHLE) and ACES as potential targets for morphine. Both CHLE and ACES were reported to be inhibited by morphine48 and its analogs49 in the literature. Finally, MAOB and HRH1 are predicted as potential targets for fentanyl, which is consistent with the results from ChEMBL database. This target map serves as a compelling example showing the reliability of our TargetHunter and HTDocking program. Thus, other predicted targets in this target map have the potential to become new targets for the corresponding drug, and we highly encourage other peers to experimentally validate our predictions.
Case Study 2: Virtual DDI Study between Acetaminophen and Fentanyl.
From our target mapping generated by Spider Plot, we found that two common targets including cytochrome P450 3A4 (CYP3A4) and ATP-dependent translocase (ABCB1) are shared by acetaminophen and fentanyl, suggesting there may be a DDI between these two drugs. CYP3A4 is a well-known enzyme responsible for the phase I metabolism of many drugs and compounds such as steroids, fatty acids, and xenobiotics.50 ABCB1 is a transmembrane active efflux pump for a wide range of drugs.51 We then conducted literature research and found articles reporting that acetaminophen can inhibit the metabolism of fentanyl by CYP3A4 inhibition in vitro52 and exhibit significant fentanyl-sparing effect in vivo.53 However, there is no direct evidence indicating that the DDI is related to ABCB1.
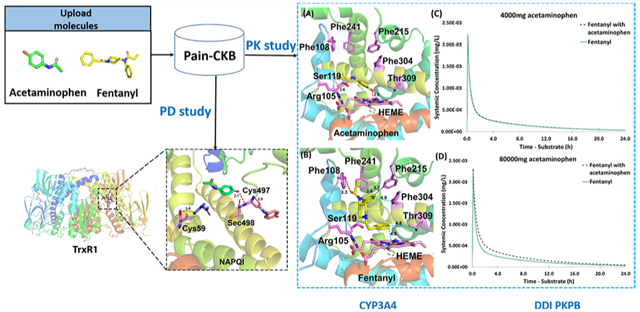
To obtain insight into the detailed interaction at the molecular level, we first conducted a docking study between CYP3A4 and these two drugs, in which the docking results are generated by revised idock algorithm implemented in Pain-CKB. As shown in Figure 6, the key residues in CYP3A4 ligand-binding pocket include Thr309, Phe304, Phe215, Phe241, Phe108, Ser119, Arg105, and HEME. As acetamino phen is a relatively small molecule, it is not likely to interact with most of the key residues. The binding pose between acetaminophen and CYP3A4 allows the formation of two strong hydrogen bonds: one is observed between the hydroxyl group of acetaminophen and the HEME structure of CYP3A4 (2.8 Å) and another is found between the acetamide group of acetaminophen and Arg105 residue (3.6 Å). The binding conformation of fentanyl presents the benzene ring structure facing the HEME structure of the enzyme, allowing the formation of hydrophobic interaction between these two structures (4.5 Å). Another hydrogen bond with a binding distance of 3.3 Å was formed between the oxygen on fentanyl and Phe108 residue of CYP3A4. Hydrophobic interactions were observed between the drug and Thr309 (4.8 Å), Phe304 (4.8 Å), Phe215 (3.7 Å), and Phe241 (3.6 Å).
Figure 6.
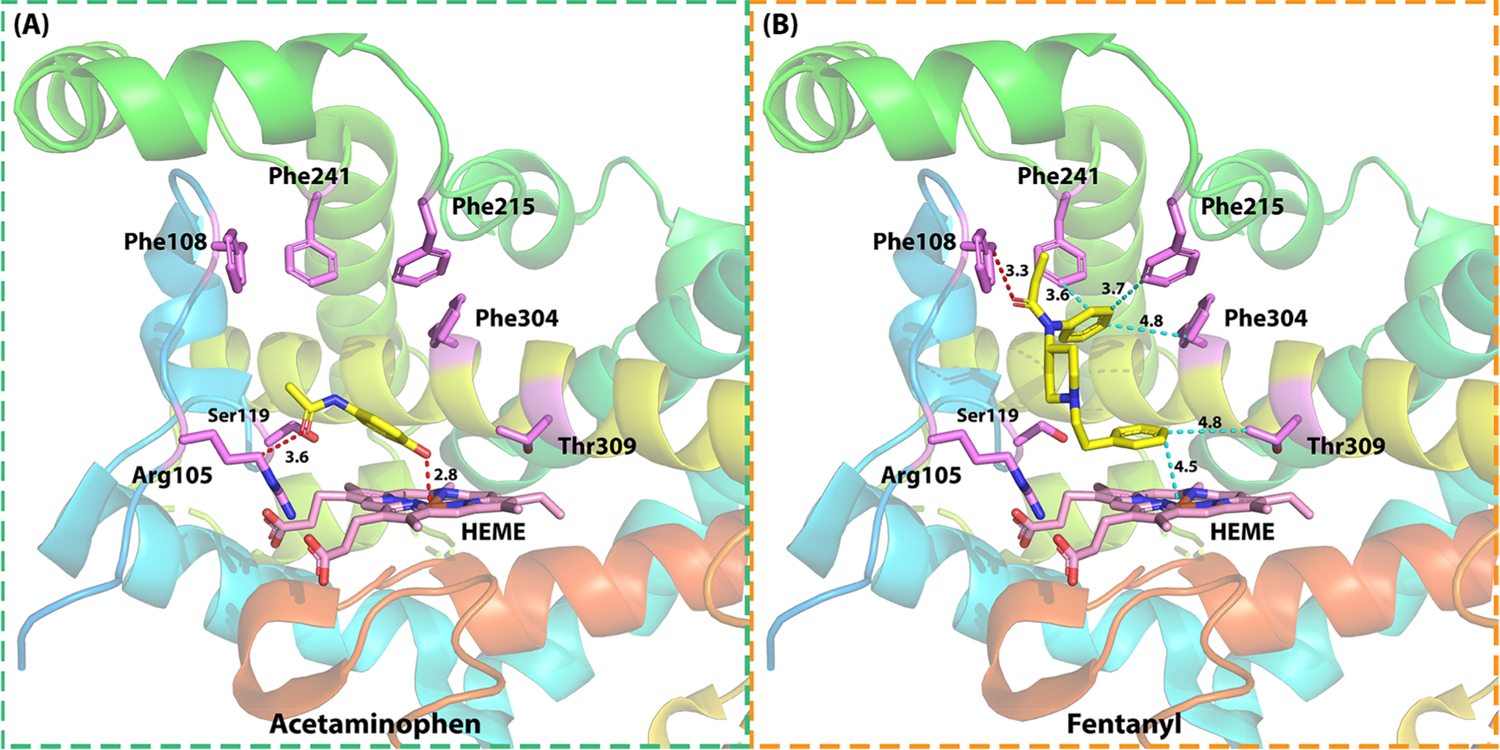
Detailed interactions of (a) acetaminophen and (b) fentanyl in CYP3A4 for their potential DDI. Two hydrogen bonds were observed between acetaminophen and Arg105 (3.6 Å) and HEME (2.8 Å). One hydrogen bond was observed between fentanyl and Phe108 (3.3 Å); five hydrophobic interactions were observed between fentanyl and Phe215 (3.7 Å), Phe241 (3.6 Å), Phe304 (4.8 Å), Thr309 (4.8 Å), and HEME (4.5 Å). PDB ID 4K9T.
Another overlapped target between acetaminophen and fentanyl is ABCB1. The docking results by HTDocking algorithm (idock) on this target are shown in Figure 7. Key residues in the ABCB1 binding pocket include Phe335, Tyr309, Tyr306, Phe727, Gln724, Phe977, and Tyr949. Acetaminophen formed two hydrogen bonds between the hydroxyl group and Tyr306 (2.7 Å) and Tyr 309 (2.3 Å). Three hydrophobic interactions were also observed at Phe335 (4.2 Å), Phe727 (3.9 Å), and Phe977 (3.8 Å). For fentanyl, during its binding with ABCB1, a hydrogen bond was formed with oxygen on the amide group of Gln632 (3.5 Å), and another hydrogen bond was observed between the nitrogen on the propenamide group and the phenyl ring of Tyr306 (4.0 Å). Five hydrophobic interactions were observed between fentanyl and Phe335 (4.2 Å), Tyr309 (3.6 Å), Phe727 (4.0 Å), Phe977 (3.1 Å), and Tyr949 (3.3 Å) as well.
Figure 7.
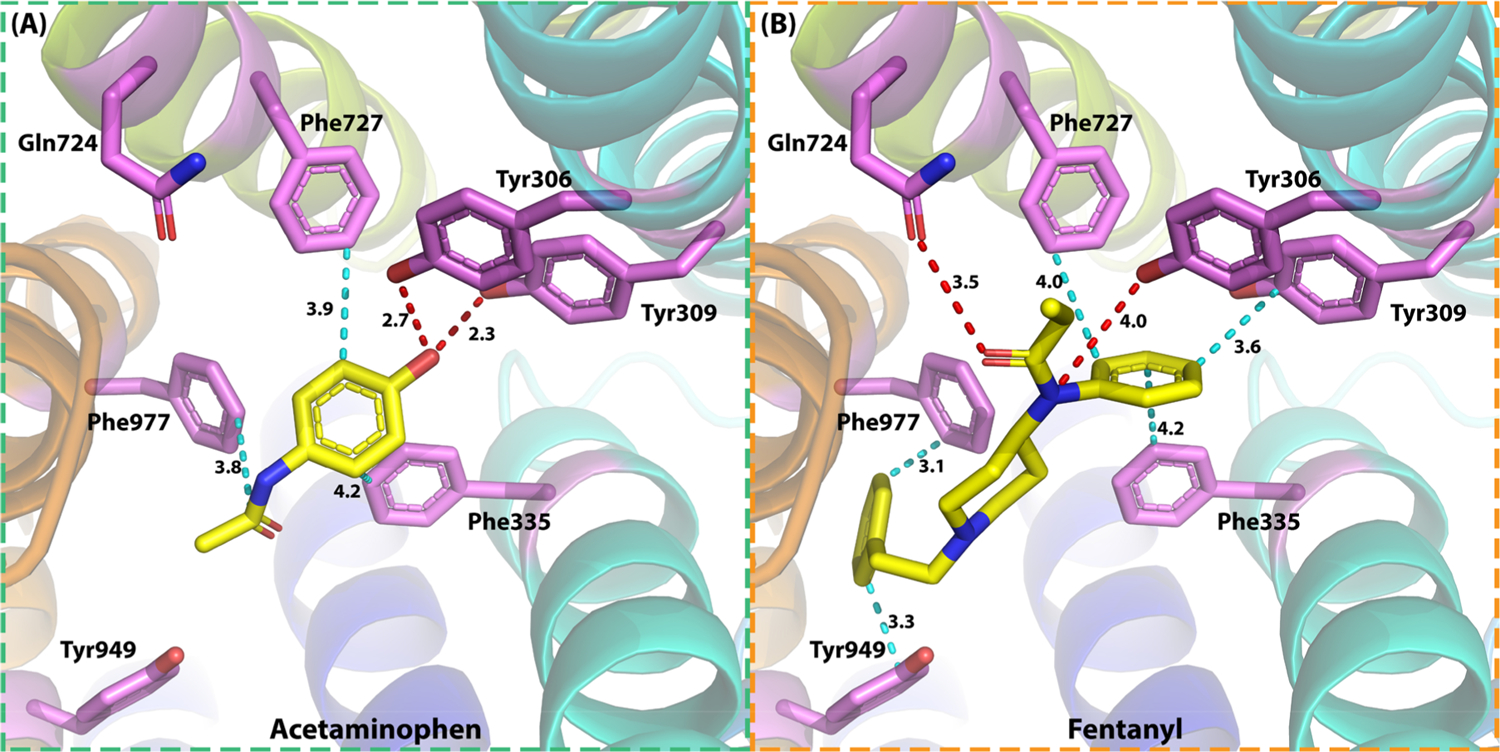
Detailed interactions of (a) acetaminophen and (b) fentanyl in P-glycoprotein (ABCB1) for their potential DDI. Two hydrogen bonds were observed between acetaminophen and Tyr306 (2.7 Å) and Tyr309 (2.3 Å); three hydrophobic interactions were observed between acetaminophen and Phe335 (4.2 Å), Phe727 (3.9 Å), and Phe977 (3.8 Å). Two hydrogen bonds were observed between fentanyl and Tyr306 (4.0 Å) and Gln724 (3.5 Å); five hydrophobic interactions were observed between fentanyl and Tyr306 (4.0 Å), Tyr309 (3.6 Å), Phe335 (4.2 Å), Phe727 (4.0 Å), and Tyr949 (3.3 Å). PDB ID 6FN1.
Based on the predicted results from Pain-CKB, we then built physiologically based pharmacokinetic (PBPK) models to quantitatively explore the metabolism changes of these two drugs, which is caused by the drug interaction on CYP3A4 when administrated simultaneously, from a pharmacometrics point of view. The parameters used to build the PBPK models for acetaminophen and fentanyl are listed in Table 1 and Table 2. All the parameters required to build the PBPK models are collected from the reported clinical studies. Using the optimized acetaminophen PBPK model, we simulated the drug plasma concentration profile after a single 1000 mg iv bolus dose in healthy volunteers. As shown in Figure 8a, our simulation is highly consistent with the observed data reported by previous papers.54 The PBPK model for fentanyl was built based on the model published by our lab before.55 The simulated drug plasma concentration profile after a single 7.0 mg iv bolus in healthy volunteers overlaps with the observed data well56 (Figure 8b). Moreover, four more validation studies for the acetaminophen and fentanyl models are presented in Figure S1–S4. We can see from the results that most of the clinical data falls within the range of arithmetic mean values and 95th/5th percentile values of simulated results. In conclusion, our PBPK model is highly consistent with the clinical data and can be extrapolated to different dosages.
Table 1.
Key Model Parameters for Acetaminophen Simulation
| parametera | value | source |
|---|---|---|
| PhysChem and Blood Binding | ||
| MW (g/mol) | 151.16 | b |
| log P | 0.46 | b |
| pKa | 9.46 | ref 87c |
| B/P | 0.98 | ref 88c |
| f u | 0.82 | ref 89c |
| Distribution | ||
| Vss (L/kg) | 1 | ref 90c |
| Elimination | ||
| ClAPAP (L/h) | 19.7 | ref 91c |
| Interaction | ||
| Ki (μM) | 2800 | ref 52c |
Abbreviations: MW, molecule weight; log P, log of the octanol–water partition coefficient for the neutral compound; pKa, dissociation constant; B/P, blood/plasma concentration ratio; fu, fraction of drug unbound in plasma; Vss, steady-state volume of distribution; ClAPAP, in vivo clearance of acetaminophen; Ki, inhibition constant of acetaminophen on CYP3A4.
From PubChem (PubChem CID 1983) (https://pubchem.ncbi.nlm.nih.gov).
Derived from published data and then optimized based on observed data.
Table 2.
Key Model Parameters for Fentanyl Simulation
| parametera | value | source |
|---|---|---|
| PhysChem and Blood Binding | ||
| MW (g/mol) | 336.47 | b |
| log P | 2.8 | b |
| pKa | 8.06 | b |
| B/P | 0.963 | ref 92c |
| f u | 0.297 | d |
| Distribution | ||
| Vss (L/kg) | 4.089 | d |
| Elimination | ||
| Clint 3A4 (μL·min–1·pmol–1) | 0.496 | ref 93c |
| C1R (L/h) | 4.6 | ref 94c |
| Interaction | ||
| Ki (μM) | 24.2 | ref 95c |
Abbreviations: MW, molecule weight; log P, log of the octanol–water partition coefficient for the neutral compound; pKa, dissociation constant; B/P, blood/plasma concentration ratio; fu, fraction of drug unbound in plasma; Vss, steady-state volume of distribution; Clint 3A4, in vivo clearance of acetaminophen; ClR, renal clearance; Ki, inhibition constant of fentanyl on CYP3A4.
From PubChem (PubChem CID 3345) (https://pubchem.ncbi.nlm.nih.gov).
Derived from published data and then optimized based on observed data.
Predicted by Simcyp.
Figure 8.
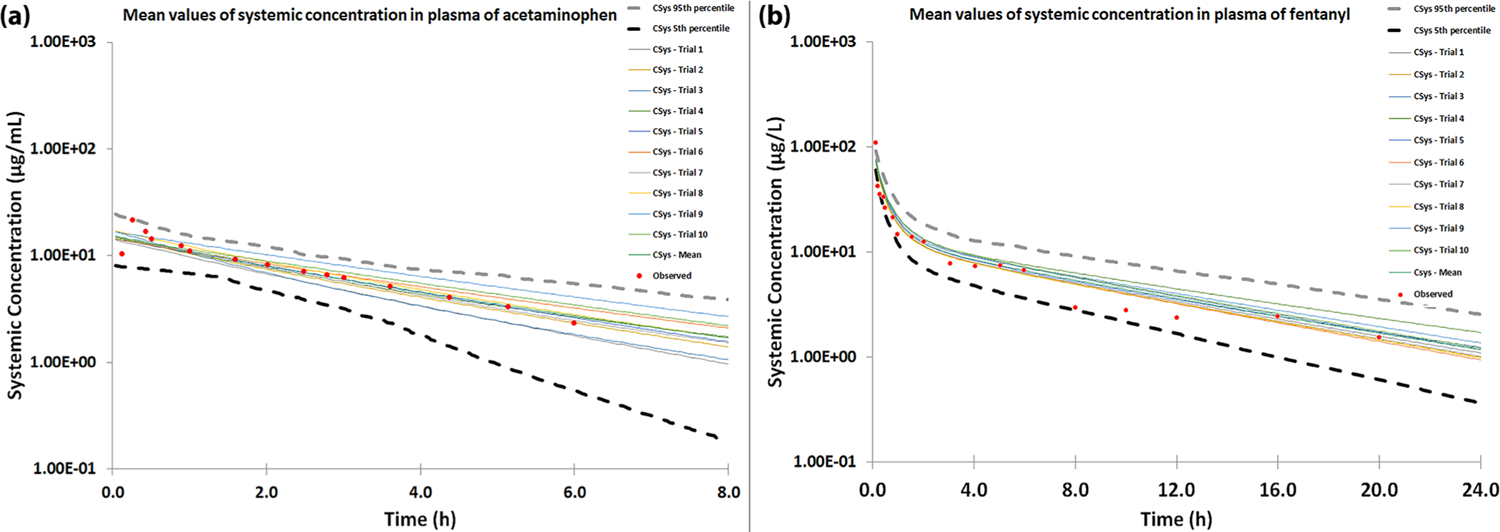
Observed and simulated concentration–time profiles of (a) acetaminophen 1000 mg iv bolus and (b) fentanyl 7 mg iv bolus. The simulated results were generated using 10 trials of 10 virtual healthy volunteers. Observed data are highlighted in red dots, while the simulated results, mean value, and 95th/5th percentile of the simulation are shown by corresponding lines.
Next, a virtual study of the DDI between acetaminophen and fentanyl was carried out based on the established PBPK models. Acetaminophen was chosen as the inhibitor substrate of fentanyl metabolism as reported by the literature.52 We first used therapeutic doses of acetaminophen (4000 mg)57 and fentanyl (0.003 mg/kg)58 to see if metabolism inhibition may occur under this dosage. As shown in Figure 9a, the systemic concentration of fentanyl with or without acetaminophen is almost identical. The AUC ratio (Figure 9b) is around 1.02, also indicating there are no significant changes in body exposure to fentanyl due to the enzyme inhibition effect of acetaminophen.
Figure 9.
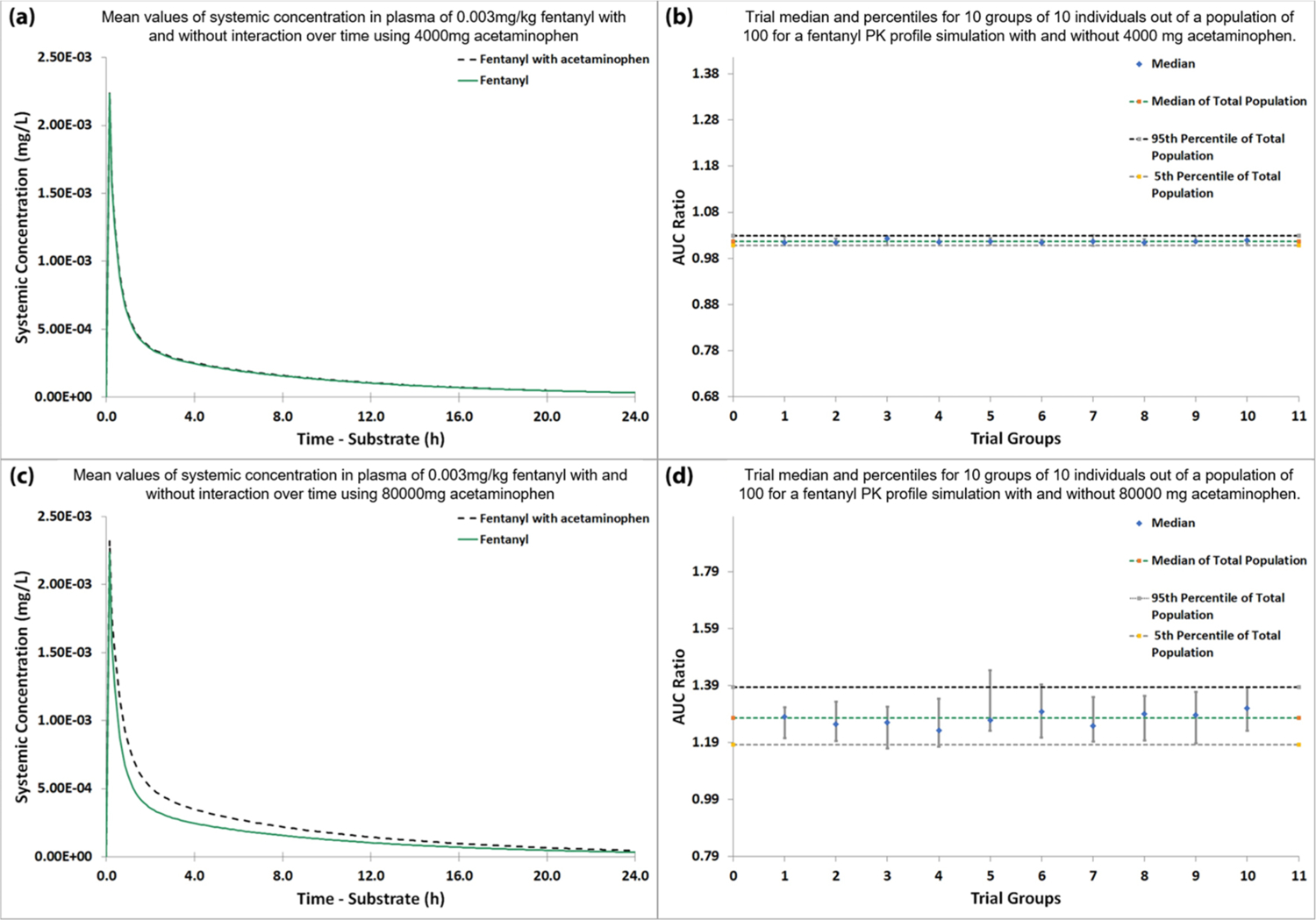
Virtual DDI studies between acetaminophen and fentanyl. (a) Systemic plasma concentration of fentanyl over time with and without 4000 mg of acetaminophen. (b) Trial arithmetic mean and standard deviation for 10 groups of 10 individuals out of a population of 100 for a fentanyl PK profile simulation with and without 4000 mg of acetaminophen. (c) Systemic plasma concentration of fentanyl over time with and without 80000 mg of acetaminophen. (d) Trial arithmetic mean and standard deviation for 10 groups of 10 individuals out of a population of 100 for a fentanyl PK profile simulation with and without 80000 mg of acetaminophen.
We then used acetaminophen concentration around 1 order of magnitude greater than the therapeutic concentration to run the simulation, where potential fentanyl–paracetamol drug interactions have been reported to occur.52 When the dosage of acetaminophen reaches 80 000 mg, which is 20-fold the therapeutic dose, we observed a significant elevation (the mean AUC ratio between administering fentanyl with acetaminophen and fentanyl alone is greater than 1.2) in the systemic concentration of fentanyl (Figure 9c,d). The simulated results indicate that when used in combination with acetaminophen at this concentration, fentanyl metabolism by CYP3A4 is likely to be affected. Considering that the lethal dose of fentanyl can be as low as 2 mg according to the Drug Enforcement Administration (DEA), more attention should be paid when administrating it with very high doses of acetaminophen. In addition, we also conducted a sensitivity analysis for the parameter reported in the range (values for Ki in the acetaminophen model were reported between 2800 and 3200 μM). The result is presented in Figure S5. Within the reported range, we can see that AUC ratio, which is a key parameter to judge the existence of DDI, is always greater than 1.2-fold, indicating any value in that range will not cause substantial difference to our results.
In this case study, we only considered the influence of enzyme inhibition on CYP3A4, and there is no literature reporting the interaction of acetaminophen and fentanyl on another overlapped target, ABCB1, so far. ABCB1 is a transmembrane active efflux pump for a wide range of drugs. If inhibition also exists, we believe the DDI will get more severe than shown here. Our results not only provide a possible explanation of the real-world observed data but also indicate that Pain-CKB is a powerful tool for studies of DDI.
Case Study 3: Molecular Docking to Study Acetaminophen Metabolite Hepatotoxicity.
Acetaminophen is one of the most common causes of acute liver failure (ALF) in the United States.59 At its therapeutic dose, around 90% of acetaminophen is metabolized in the human body into inactive glucuronide and sulfate conjugates, while the rest of the unmetabolized drug is converted to a highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI),60 primarily by CYP2E1 and CYP3A4.61 However, a supratherapeutic dose of acetaminophen will cause the conjugation pathways to become saturated and increase the amount of NAPQI in the body, which can cause hepatotoxicity.62,63 NAPQI has been reported to target thioredoxin reductase (TrxR) to induce oxidative stress and liver toxicity. In detail, LC-MS/MS analysis confirmed that NAPQI can modify the Cys59, Cys497, and Sec498 in the redox centers of TrxR to cause enzyme inhibition.64 TrxR has been collected as a potential pain target65 in our Pain-CKB. Interestingly, Pain-CKB predicts that acetaminophen is very likely to interact with TrxR. Considering the structural similarity between acetaminophen and NAPQI, it is highly possible that our algorithms would successfully predict the toxicity target for NAPQI. Indeed, Figure S6 shows that NAPQI was predicted to bind to TrxR with a moderate binding energy of −5.88 kcal/mol, indicating that our algorithms are powerful to predict the toxicity target for NAPQI.
To study the interaction pattern between NAPQI and TrxR1, the HTDocking algorithm (revised idock) implemented in Pain-CKB is applied to conduct a docking study. In perfect agreement with the observed results,64 NAPQI is likely to interact with TrxR1 in three different ways as shown in Figure 10. First, the carbonyl group of NAPQI can form a strong hydrogen bond with the Cys497 residue with a distance of 2.9 Å. Another possible hydrogen bond interaction is observed between the N-acyl amides functional group and Sec498 residue with a distance of 3.1 Å. Finally, NAPQI can also form a strong steric interaction with Cys59 (3.4 Å) because of the special spatial structure of Cys59. These possible binding poses provide a suitable spatial condition that can then facilitate the alkylation reaction on these residues and cause enzyme inhibition and further hepatotoxicity.
Figure 10.
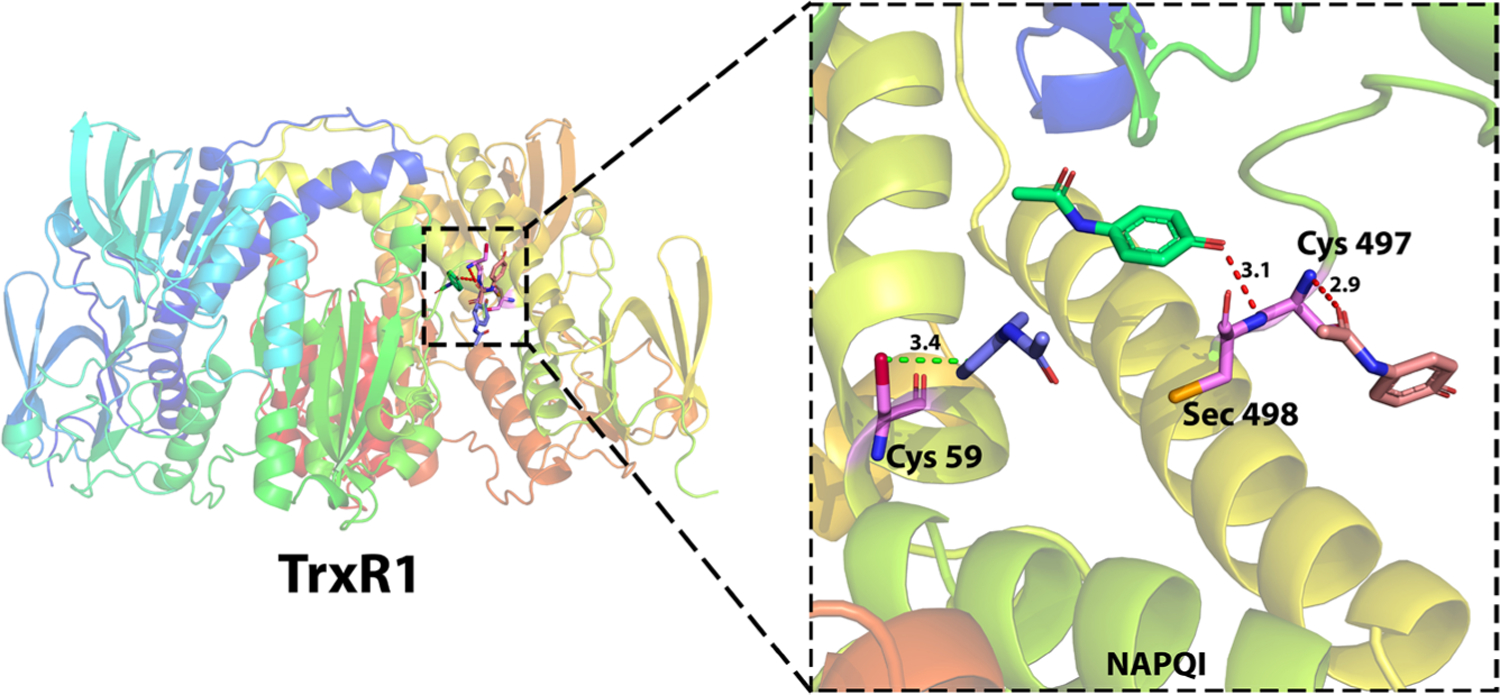
Detailed interaction of NAPQI in TrxR1 for hepatotoxicity. NAPQI has three possible binding poses with TrxR1 because of the (1) steric interaction with Cys59 (4.3 Å), (2) hydrogen bonding with Cys497 (2.9 Å), and (3) hydrogen bonding with Cys498 (3.1 Å). PDB ID 2ZZ0.
Pain-CKB and 3 Case Studies for Pain Research.
Pain is a highly complex disease66 that can cause severe challenges to patients’ daily lives.67 Efforts have been devoted to improving pain management, yet limited improvements have been achieved.68 Several databases related to pain such as PainNetworks69 have been built to study the pain-related genes and their network associations. Several other specific disease-related databases have already been developed and implemented as novel ways to explore the molecular mechanisms and pathways of the disease to facilitate studies in related areas.70,71 Here, we provided a pain domain-specific chemogenomics knowledgebase (Pain-CKB) for pain research, with a user-friendly web interface and powerful algorithms implemented, including the target structure-based HTDocking, ligand -based TargetHunter, and more.
The presented case studies serve as examples of the compound inquiry results and how follow-up studies can be performed based on the output of Pain-CKB. The first case study validated the ability of Pain-CKB to map the network between different drugs and their targets based on the results of docking score (HTDocking) and similarity score (Target-Hunter). The target map precisely identified known targets for the input analgesics. It can also make predictions based on the known target structure as well as new small-molecule structures that have been confirmed to bind to the target. The ability for Pain-CKB to identify drug targets allows users to conduct drug repurposing studies by looking for potential off-target interactions for a known compound or scanning new compounds as a starting point for new analgesics development. In addition, as shown in the second case study, based on the common targets mapped between certain drug pairs, studies can be carried out by using the docking algorithms and pharmacometrics models to explore the potential DDIs and synergistic drug pairs. In the third case study, we looked into the target mapping results and explored the relationship between acetaminophen active metabolite NAPQI and its reported hepatotoxicity target. Three case studies demonstrated the functionality and utility of our newly built database.
CONCLUSION
It is well-known that DDIs related to analgesics can cause severe adverse effects.72 This problem will become much more severe when opioids analgesics are involved. Enzyme inhibition or competition effects, if the exist, may greatly increase the blood concentration of opioids and cause unexpected opioid side effects like drug addiction, breath inhibition, and even death, On the other side, synergistic drug pairs with greater therapeutic effects and fewer side effects are of great research interest in pain-related areas.73
In the present work, we provided a Pain-CKB that can be extremely beneficial for future research. Pain-CKB is an integrative pain-domain specific knowledgebase with full access for public use. It includes pain-related target data and tools for target identification and systems pharmacology research. This knowledgebase will benefit pain research by bridging the knowledge barrier between computational, chemical, and biological areas to facilitate the identification of new DDIs and to accelerate the development of new analgesics.
However, our Pain-CKB has its limitations. First, although we conducted thorough literature research to implement as many pain-related targets as we could, a few potential pain related targets may not be included in Pain-CKB. To minimize this limitation, we will continuously update our Pain-CKB in the future. Second, the performance of the HTDocking algorithm relies largely on the quality and availability of the 3D structure of the target protein. Homology models with high accuracy will be implemented into Pain-CKB. Finally, Pain-CKB is not the first or only tool that uses integrated similarity to predict DDI interaction networks, though it is specific for pain regulating pathways.
MATERIALS AND METHODS
Genes and Proteins.
Genes and proteins related to pain were collected from public databases such as Ensembl,74 UniProt,75 KEGG,76 GPCRDB,77 and NCBI Protein Database.78 Available crystal structures or cryo-EM structures of pain-related targets were retrieved from the Protein Data Bank (PDB) (https://www.rcsb.org/). To date, we have archived 84 pain-related targets with 207 reported 3D structures in our platform.
Drugs and Chemicals.
ChEMBL database (version 23)79 and DrugBank database (version 5.1.5) were utilized in our work for searching and collecting the drugs and chemicals. The experimental data for each small molecule against its respective target proteins were collected using text mining techniques and cleaned up by manual inspection. Bioactivity data from different resources were normalized using the same standard. To date, we have collected 272 FDA-approved antipain drugs and 234 662 related chemicals, all of which have been integrated into Pain-CKB.
Database Infrastructure.
Users can submit up to five different query compounds in one task using JSME Molecular Editor v2017-03-01.80 Pain-CKB was implemented based on our established molecular database prototype DAKB-GPCRs (https://www.cbligand.org/dakb-gpcrs/) using SQLite database management system (https://sqlite.org/) and Kestrel HTTP server (https://github.com/aspnet/KestrelHttpServer) with Apache HTTP server (https://httpd.apache.org/) as its reverse proxy server. The overview of our design for Pain-CKB is depicted in Figure 1.
HTDocking.
Pain-CKB adopts our online high-throughput molecular docking technique, HTDocking,28–32 for identifying possible interactions between protein targets and small molecules. A maximum of three different reported 3D structures for each target protein can be found in our knowledgebase, depending on the availability of PDB files; if the available PDB files are less than three, then this number will change accordingly. For each query compound, HTDocking powdered by idock34 will automatically dock it into these different structures or conformations and generate docking scores. A higher docking score indicates that the protein is more likely to be the candidate target of the queried small molecule. The idock34 program can provide up to 10 predicted binding affinity values (ΔG values) from different docking poses for each compound in a binding pocket of a protein. In our HTDocking program, we only consider the best binding affinity value, which can be further transformed as a docking score. For important targets, we will collect the homology models with high accuracy from GPCRDB81 into Pain-CKB for calculations in the future.
TargetHunter.
Pain-CKB integrates our online target-identification service, TargetHunter,33 for predicting the potential off-target interactions for submitted compounds. TargetHunter exploits an important principle of medicinal chemistry: compounds with structural similarities often have similar physicochemical properties and biological profiles. For each query compound, TargetHunter calculates the similarity based on the molecular fingerprints using Tanimoto coefficients (TC) with its known active compound data set that was collected from Drugs and Chemicals (see above), in which almost all of these known ligands are orthosteric binders. As a result, TargetHunter is useful even when there are no available 3D structures of a given protein target. However, the active compound data set of this target is required.
Indeed, our program supports up to ten different kinds of molecular fingerprints, including ECFP0, ECFP10, ECFP2, ECFP4, ECFP6, ECFP8, FP2, FP3, FP4, and MACCS. Based on the feedback and statistical data from our users, FP2 is the most popular and more accurate one. So, TargetHunter uses FP2 as the default fingerprint.
Spider Plot.
Based on the target classification, our online tool Spider Plot visualizes the molecule–protein interaction network based on the output of HTDocking and TargetHunter. The average docking scores are displayed as connection labels and the protein targets on which the query compound is active are displayed as circular nodes. By default, a green node denotes a target with a high similarity score (>0.95) comparing the best matched known compound and the query compound, while a pink one denotes otherwise. With Spider Plot, the colors, font sizes, node sizes, border widths, and node shapes as well as the layout can be completely customized, and the entire network graph can be exported as an image file on particular browsers.
Blood–Brain Barrier (BBB) Predictor.
The blood–brain barrier (BBB) predictor28,31 was integrated into Pain-CKB. It predicts whether a query compound can move across the BBB to the central nervous system (CNS).82 The BBB predictor is also available for access from https://www.cbligand.org/BBB/.
Software Requirements.
The Pain-CKB Web site is compatible with modern web browsers (such as Chrome, Firefox, Microsoft Edge, and Safari) provided JavaScript and cookies are enabled. We recommend the latest release version of these web browsers for better rendering.
Simcyp Simulations.
Simcyp Population-based ADME Simulator (version 17.0, Simcyp Limited, https://www.certara.com/software/simcyp-pbpk/)83 was used to build the PBPK models for acetaminophen and fentanyl. In Simcyp, the PBPK model is separated into systems data and compound or drug data. In this study, we used healthy volunteers as our simulation population. The parameters in systems data are kept default. Parameters required from the compound models were obtained from PubChem84 (https://pubchem.ncbi.nlm.nih.gov) or from previously published in vitro and in vivo data and kept unmodified. However, the parameter Ki in the acetaminophen compound model comes in a range. For this parameter, we selected a value in that range that can reflect the influence of drug–drug interaction between acetaminophen and fentanyl to the greatest extent. We also conducted a sensitivity analysis for this parameter to show that choosing any value in the recorded value range will not cause any substantial changes to our conclusion. The acetaminophen model was built on the minimal PBPK model with four basic compartments (central, liver, gut, and single adjusting compartment (SAC)) using Simcyp because acetaminophen is mainly metabolized in the liver, which is covered by the minimal PBPK model, and here we are mainly interested in blood or plasma time–concentrations curves. The SAC is a virtual organ compartment with physiological parameters that can be adjusted arbitrarily to account for the influence caused by all the other organs on the drug PK profile.85 The total systemic clearance was used to describe the metabolism of acetaminophen, and the percentage of drug metabolized by CYP3A4 is around 5% to 10%.86
The fentanyl model was built based on the full PBPK model of Simcyp. The full PBPK distribution model simulates the concentrations in various organ compartments utilizing the time-based differential equations. Organ compartments simulated include the blood (plasma), bone, adipose, brain, heart, kidney, liver, gut, lung, muscle, skin, pancreas, and spleen. Interindividual variability is introduced through tissue volume prediction considering factors such as weight, height, sex, and age. Based on the literature, the elimination of fentanyl consisted of metabolism by CYP3A4 in a predominant way (about 90%), the renal clearance in a minor way (less than 10%), and additional clearance is negligible so that was not considered in this model.
The experimentally observed plasma concentration–time data of acetaminophen and fentanyl were extracted from a previously published paper as a validation of the reliability of PBPK models. The simulated plasma concentration profiles were generated upon 10 trials of 10 virtual healthy volunteers. The arithmetic mean values and 95th/5th percentile values of simulated results were compared with the observed data. The DDI simulation was also generated using 10 trials of 10 virtual healthy volunteers. The absorption parameters were ignored as all the drug administration routes used in this study are iv bolus in order to maintain consistency with previously published literature.
Supplementary Material
Funding
The authors acknowledge the funding support to the Xie laboratory from the National Institutes of Health, National Institute on Drug Abuse (P30 DA035778A1).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00372.
Observed and simulated concentration–time profiles of acetaminophen 650 mg iv infusion over 5 min, acetaminophen 1000 mg iv infusion over 15 min, fentanyl 100 μg/kg iv bolus, and fentanyl 5 μg/kg iv bolus, AUC ratio of fentanyl with acetaminophen when Ki value is in the range 2800–3200 μM, and computational systems pharmacology-target mapping (CSP-Target Mapping) for NAPQI (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00372
The authors declare no competing financial interest.
The Pain-CKB server is accessible at https://www.cbligand.org/g/pain-ckb.
Contributor Information
Mingzhe Shen, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Maozi Chen, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Tianjian Liang, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Siyi Wang, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Ying Xue, Department of Pharmacy and Therapeutics, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Richard Bertz, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Xiang-Qun Xie, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, and Departments of Computational Biology and Structural Biology, School of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Zhiwei Feng, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
REFERENCES
- (1).Schappert SM, and Burt CW (2006) Ambulatory care visits to physician offices, hospital outpatient departments, and emergency departments: United States, 2001–02. Vital and Health Statistics. Series 13, Data from the National Health Survey, 1–66. [PubMed] [Google Scholar]
- (2).Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L, and Helmick C (2018) Prevalence of chronic pain and high-impact chronic pain among adults—United States, 2016. Morb. Mortal. Wkly. Rep 67, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gaskin DJ, and Richard P (2012) The Economic Costs of Pain in the United States. J. Pain 13, 715–724. [DOI] [PubMed] [Google Scholar]
- (4).Leeuwen MT, Blyth FM, March LM, Nicholas MK, and Cousins MJ (2006) Chronic pain and reduced work effectiveness: The hidden cost to Australian employers. Eur. J. Pain 10, 161–161. [DOI] [PubMed] [Google Scholar]
- (5).Melnikova I (2010) Pain market. Nat. Rev. Drug Discovery 9, 589–590. [DOI] [PubMed] [Google Scholar]
- (6).Geurts JW, Willems PC, Lockwood C, van Kleef M, Kleijnen J, and Dirksen C (2017) Patient expectations for management of chronic non-cancer pain: A systematic review. Health Expect. 20, 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Innis J, Bikaunieks N, Petryshen P, Zellermeyer V, and Ciccarelli L (2004) Patient satisfaction and pain management: an educational approach. J. Nurs. Care Qual 19, 322–327. [DOI] [PubMed] [Google Scholar]
- (8).Ricardo Buenaventura M, Rajive Adlaka M, and Nalini Sehgal M (2008) Opioid complications and side effects. Pain Physician 11, S105–S120. [PubMed] [Google Scholar]
- (9).Bonica JJ (1979) The need of a taxonomy. Pain 6, 247–252. [DOI] [PubMed] [Google Scholar]
- (10).Besson JM (1997) [The complexity of physiopharmacologic aspects of pain]. Drugs 53 (Suppl 2), 1–9. [DOI] [PubMed] [Google Scholar]
- (11).Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).D’Mello R, and Dickenson AH (2008) Spinal cord mechanisms of pain. Br. J. Anaesth 101, 8–16. [DOI] [PubMed] [Google Scholar]
- (13).Cashman JN (1996) The Mechanisms of Action of NSAIDs in Analgesia. Drugs 52, 13–23. [DOI] [PubMed] [Google Scholar]
- (14).Wiebalck CA, and Van Aken H (1995) 4 Paracetamol and propacetamol for post-operative pain: contrasts to traditional NSAIDs. Bailliere’s Clin. Anaesthesiol 9, 469–482. [Google Scholar]
- (15).Henry DA (1988) Side-effects of non-steroidal anti-inflammatory drugs. Baillierè’s Clin. Rheumatol 2, 425–454. [DOI] [PubMed] [Google Scholar]
- (16).Bjarnason I, Hayllar J, Macpherson A. N. d. J., and Russell A. N. t. S. (1993) Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 104, 1832–1847. [DOI] [PubMed] [Google Scholar]
- (17).Rainsford KD (1999) Profile and mechanisms of gastrointestinal and other side effects of nonsteroidal anti-inflammatory drugs (NSAIDs). Am. J. Med 107, 27–35. [DOI] [PubMed] [Google Scholar]
- (18).Carr DB, and Goudas LC (1999) Acute pain. Lancet 353, 2051–2058. [DOI] [PubMed] [Google Scholar]
- (19).Al-Hasani R, and Bruchas MR (2011) Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 13631381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chahl LA (1996) Opioids–mechanism of action. Aust. Prescr 19, 63–65. [Google Scholar]
- (21).Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguѐre PM, Löber S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, and Shoichet BK (2016) Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Warner M, Chen LH, and Makuc DM (2009) Increase in fatal poisonings involving opioid analgesics in the United States, 1999–2006. NCHS Data Brief, 22, 1–8, https://pubmed.ncbi.nlm.nih.gov/19796521/ [PubMed] [Google Scholar]
- (23).Shah NG, Lathrop SL, Reichard RR, and Landen MG (2008) Unintentional drug overdose death trends in New Mexico, USA, 1990–2005: combinations of heroin, cocaine, prescription opioids and alcohol. Addiction 103, 126–136. [DOI] [PubMed] [Google Scholar]
- (24).Seth P, Scholl L, Rudd RA, and Bacon S (2018) Overdose deaths involving opioids, cocaine, and psychostimulants—United States, 2015–2016. Morb. Mortal. Wkly. Rep 67, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Christie MJ (2008) Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol 154, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Woolf CJ (2010) Overcoming obstacles to developing new analgesics. Nat. Med 16, 1241–1247. [DOI] [PubMed] [Google Scholar]
- (27).Feng Z, Chen M, Shen M, Liang T, Chen H, and Xie XQ (2020) Pain-CKB, A Pain Domain-specific Chemogenomics Knowledgebase for Target Identification and Systems Pharmacology Research. J. Chem. Inf. Model, DOI: 10.1021/acs.jcim.0c00633. [DOI] [PubMed] [Google Scholar]
- (28).Liu H, Wang L, Lv M, Pei R, Li P, Pei Z, Wang Y, Su W, and Xie X-Q (2014) AlzPlatform: an Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model 54, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang Y, Wang L, Feng Z, Cheng H, McGuire TF, Ding Y, Cheng T, Gao Y, and Xie X-Q (2016) StemCellCKB: an integrated stem cell-specific chemogenomics knowledgebase for target identification and systems-pharmacology research. J. Chem. Inf. Model 56, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang H, Ma S, Feng Z, Wang D, Li C, Cao Y, Chen X, Liu A, Zhu Z, Zhang J, et al. (2016) Cardiovascular disease chemogenomics knowledgebase-guided target identification and drug synergy mechanism study of an herbal formula. Sci. Rep 6, 33963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen M, Jing Y, Wang L, Feng Z, and Xie X-Q (2019) DAKB-GPCRs: An Integrated Computational Platform for Drug Abuse Related GPCRs. J. Chem. Inf. Model 59, 1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xu X, Ma S, Feng Z, Hu G, Wang L, and Xie X-Q (2016) Chemogenomics knowledgebase and systems pharmacology for hallucinogen target identification—Salvinorin A as a case study. J. Mol. Graphics Modell 70, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang L, Ma C, Wipf P, Liu H, Su W, and Xie X-Q (2013) TargetHunter: An In Silico Target Identification Tool for Predicting Therapeutic Potential of Small Organic Molecules Based on Chemogenomic Database. AAPS J. 15, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li H, Leung K-S, and Wong M-H (2012) idock: A multithreaded virtual screening tool for flexible ligand docking, 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), pp 77–84, IEEE. [Google Scholar]
- (35).O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011) Open Babel: An open chemical toolbox. J. Cheminf 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Journigan VB, Feng Z, Rahman S, Wang Y, Amin A, Heffner CE, Bachtel N, Wang S, Gonzalez-Rodriguez S, Fernández-Carvajal A, Fernández-Ballester G, Hilton JK, Van Horn WD, Ferrer-Montiel A, Xie XQ, and Rahman T (2020) Structure-Based Design of Novel Biphenyl Amide Antagonists of Human Transient Receptor Potential Cation Channel Subfamily M Member 8 Channels with Potential Implications in the Treatment of Sensory Neuropathies. ACS Chem. Neurosci 11, 268–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Anighoro A, Bajorath J, and Rastelli G (2014) Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- (38).Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KLH, Edwards DD, Shoichet BK, and Roth BL (2009) Predicting new molecular targets for known drugs. Nature 462, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Graham GG, and Scott KF (2005) Mechanism of Action of Paracetamol. Am. J. Ther 12, 46–55. [DOI] [PubMed] [Google Scholar]
- (40).Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, and Leone S (2006) Paracetamol: New Vistas of an Old Drug. CNS Drug Rev. 12, 250–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Todd PA, and Sorkin EM (1988) Diclofenac Sodium. Drugs 35, 244–285. [DOI] [PubMed] [Google Scholar]
- (42).Trescot AM, Datta S, Lee M, and Hansen H (2008) Opioid pharmacology. Pain Physician 11, S133–153. [PubMed] [Google Scholar]
- (43).Anderson BJ (2008) Paracetamol (Acetaminophen): mechanisms of action. Paediatr Anaesth 18, 915–921. [DOI] [PubMed] [Google Scholar]
- (44).Mallet C, Barrière DA, Ermund A, Jönsson BA, Eschalier A, Zygmunt PM, and Högestätt ED (2010) TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS One 5, e12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Young S, Fabio K, Guillon C, Mohanta P, Halton TA, Heck DE, Flowers RA, Laskin JD, and Heindel ND (2010) Peripheral site acetylcholinesterase inhibitors targeting both inflammation and cholinergic dysfunction. Bioorg. Med. Chem. Lett 20, 2987–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sablone MR, Cesta MC, Moriconi A, Aramini A, Bizzarri C, Giacinto CD, Bitondo RD, Gloaguen I, Aschi M, Crucianelli M, Bertini R, and Allegretti M (2009) Structure–Activity Relationship of novel phenylacetic CXCR1 inhibitors. Bioorg. Med. Chem. Lett 19, 4026–4030. [DOI] [PubMed] [Google Scholar]
- (47).Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, and Reisine T (1994) Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol 45, 330. [PubMed] [Google Scholar]
- (48).Wright CI, and Sabine JC (1943) The inactivation of cholinesterase by morphine, dilaudid, codeine and desomorphine. J. Pharmacol. Exp. Ther 78, 375. [Google Scholar]
- (49).Galli A, Renzi G, Grazzini E, Bartolini R, Aiello-Malmberg P, and Bartolini A (1982) Reversible inhibition of acetylcholinesterase by eseroline, an opioid agonist structurally related to physostigmine (eserine) and morphine. Biochem. Pharmacol 31, 1233–1238. [DOI] [PubMed] [Google Scholar]
- (50).Keshava C, McCanlies EC, and Weston A (2004) CYP3A4 Polymorphisms—Potential Risk Factors for Breast and Prostate Cancer: A HuGE Review. Am. J. Epidemiol 160, 825–841. [DOI] [PubMed] [Google Scholar]
- (51).Leschziner GD, Andrew T, Pirmohamed M, and Johnson MR (2007) ABCB1 genotype and PGP expression, function and therapeutic drug response: a critical review and recommendations for future research. Pharmacogenomics J. 7, 154–179. [DOI] [PubMed] [Google Scholar]
- (52).Feierman DE (2000) The effect of paracetamol (acetaminophen) on fentanyl metabolism in vitro. Acta Anaesthesiol. Scand 44, 560–563. [DOI] [PubMed] [Google Scholar]
- (53).Hong J-Y, Kim WO, Koo BN, Cho JS, Suk EH, and Kil HK (2010) Fentanyl-sparing Effect of Acetaminophen as a Mixture of Fentanyl in Intravenous Parent-/Nurse-controlled Analgesia after Pediatric Ureteroneocystostomy. Anesthesiology 113, 672–677. [DOI] [PubMed] [Google Scholar]
- (54).Singla NK, Parulan C, Samson R, Hutchinson J, Bushnell R, Beja EG, Ang R, and Royal MA (2012) Plasma and Cerebrospinal Fluid Pharmacokinetic Parameters After Single-Dose Administration of Intravenous, Oral, or Rectal Acetaminophen. Pain Pract. 12, 523–532. [DOI] [PubMed] [Google Scholar]
- (55).Cheng J, Wang S, Lin W, Wu N, Wang Y, Chen M, Xie X-Q, and Feng Z (2019) Computational Systems Pharmacology-Target Mapping for Fentanyl-Laced Cocaine Overdose. ACS Chem. Neurosci 10, 3486–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Björkman S (2003) Reduction and lumping of physiologically based pharmacokinetic models: prediction of the disposition of fentanyl and pethidine in humans by successively simplified models. J. Pharmacokinet. Pharmacodyn 30, 285–307. [DOI] [PubMed] [Google Scholar]
- (57).Dart RC, and Bailey E (2007) Does Therapeutic Use of Acetaminophen Cause Acute Liver Failure? Pharmacotherapy 27, 1219–1230. [DOI] [PubMed] [Google Scholar]
- (58).Peng PWH, and Sandler AN (1999) A Review of the Use of Fentanyl Analgesia in the Management of Acute Pain in Adults. Anesthesiology 90, 576–599. [DOI] [PubMed] [Google Scholar]
- (59).Chun LJ, Tong MJ, Busuttil RW, and Hiatt JR (2009) Acetaminophen Hepatotoxicity and Acute Liver Failure. J. Clin. Gastroenterol 43, 342–349. [DOI] [PubMed] [Google Scholar]
- (60).Mazaleuskaya LL, Sangkuhl K, Thorn CF, FitzGerald GA, Altman RB, and Klein TE (2015) PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genomics 25, 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Manyike PT, Kharasch ED, Kalhorn TF, and Slattery JT (2000) Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin. Pharmacol. Ther 67, 275–282. [DOI] [PubMed] [Google Scholar]
- (62).Streeter A, Dahlin D, Nelson S, and Baillie T (1984) The covalent binding of acetaminophen to protein. Evidence for cysteine residues as major sites of arylation in vitro. Chem.-Biol. Interact 48, 349–366. [DOI] [PubMed] [Google Scholar]
- (63).Hart SGE, Cartun RW, Wyand DS, Khairallah EA, and Cohen SD (1995) Immunohistochemical Localization of Acetaminophen in Target Tissues of the CD-1 Mouse: Correspondence of Covalent Binding with Toxicity. Fundam. Appl. Toxicol 24, 260–274. [DOI] [PubMed] [Google Scholar]
- (64).Jan Y-H, Heck DE, Dragomir A-C, Gardner CR, Laskin DL, and Laskin JD (2014) Acetaminophen reactive intermediates target hepatic thioredoxin reductase. Chem. Res. Toxicol 27, 882–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Xiao WH, and Bennett GJ (2012) Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. Pain 153, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Stephenson R (1999) The complexity of pain: part 1. No pain without gain: the augmentation of nociception in the CNS. Phys. Ther. Rev 4, 105–116. [Google Scholar]
- (67).Eccleston C, Morley SJ, and Williams A. C. d. C. (2013) Psychological approaches to chronic pain management: evidence and challenges. Br. J. Anaesth 111, 59–63. [DOI] [PubMed] [Google Scholar]
- (68).Gordon DB, and Dahl JL (2004) Quality improvement challenges in pain management. Pain 107, 1–4. [DOI] [PubMed] [Google Scholar]
- (69).Perkins JR, Lees J, Antunes-Martins A, Diboun I, McMahon SB, Bennett DLH, and Orengo C (2013) PainNetworks: A web-based resource for the visualisation of pain-related genes in the context of their network associations. Pain 154, 2586e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wang L, Xiong Y, Sun Y, Fang Z, Li L, Ji H, and Shi T (2010) HLungDB: an integrated database of human lung cancer research. Nucleic Acids Res. 38, D665–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gu J, Gui Y, Chen L, Yuan G, and Xu X (2013) CVDHD: a cardiovascular disease herbal database for drug discovery and network pharmacology. J. Cheminf 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hersh EV, Pinto A, and Moore PA (2007) Adverse drug interactions involving common prescription and over-the-counter analgesic agents. Clin. Ther 29, 2477–2497. [DOI] [PubMed] [Google Scholar]
- (73).Raffa RB (2001) Pharmacology of oral combination analgesics: rational therapy for pain. J. Clin. Pharm. Ther 26, 257–264. [DOI] [PubMed] [Google Scholar]
- (74).Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018) Ensembl 2018. Nucleic Acids Res. 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).The UniProt Consortium (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Kanehisa M, and Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Isberg V, Mordalski S, Munk C, Rataj K, Harpsøe K, Hauser AS, Vroling B, Bojarski AJ, Vriend G, and Gloriam DE (2017) GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 45, 2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).NCBI Resource Coordinators (2017) Database resources of the national center for biotechnology information. Nucleic Acids Res. 45, D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, and Overington JP (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ertl P (2010) Molecular structure input on the web. J. Cheminf 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsøe K, Hauser AS, Bojarski AJ, and Gloriam DE (2018) GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–d446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Kousik SM, Napier TC, and Carvey PM (2012) The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front. Pharmacol 3, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, and Rostami-Hodjegan A (2009) The Simcyp® population-based ADME simulator. Expert Opin. Drug Metab. Toxicol 5, 211–223. [DOI] [PubMed] [Google Scholar]
- (84).Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, et al. (2016) PubChem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ando H, Izawa S, Hori W, and Nakagawa I (2008) Utility of a single adjusting compartment: a novel methodology for whole body physiologically-based pharmacokinetic modelling. Theor. Biol. Med. Modell 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Mazaleuskaya LL, Sangkuhl K, Thorn CF, FitzGerald GA, Altman RB, and Klein TE (2015) PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genomics 25, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Lorphensri O, Intravijit J, Sabatini DA, Kibbey TCG, Osathaphan K, and Saiwan C (2006) Sorption of acetaminophen, 17α-ethynyl estradiol, nalidixic acid, and norfloxacin to silica, alumina, and a hydrophobic medium. Water Res. 40, 1481–1491. [DOI] [PubMed] [Google Scholar]
- (88).Taylor RR, Hoffman KL, Schniedewind B, Clavijo C, Galinkin JL, and Christians U (2013) Comparison of the quantification of acetaminophen in plasma, cerebrospinal fluid and dried blood spots using high-performance liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal 83, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Mutlib AE, Goosen TC, Bauman JN, Williams JA, Kulkarni S, and Kostrubsky S (2006) Kinetics of Acetaminophen Glucuronidation by UDP-Glucuronosyltransferases 1A1, 1A6, 1A9 and 2B15. Potential Implications in Acetaminophen–Induced Hepatotoxicity. Chem. Res. Toxicol 19, 701–709. [DOI] [PubMed] [Google Scholar]
- (90).Peterson RG, and Rumack BH (1978) Pharmacokinetics of acetaminophen in children. Pediatrics 62, 877–879. [PubMed] [Google Scholar]
- (91).Jiang X-L, Zhao P, Barrett J, Lesko L, and Schmidt S (2013) Application of Physiologically Based Pharmacokinetic Modeling to Predict Acetaminophen Metabolism and Pharmacokinetics in Children. CPT: Pharmacometrics Syst. Pharmacol 2, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Mather L (1983) Clinical pharmacokinetics of fentanyl and its newer derivatives. Clin. Pharmacokinet 8, 422–446. [DOI] [PubMed] [Google Scholar]
- (93).Feierman DE, and Lasker JM (1996) Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab. Dispos 24, 932–939. [PubMed] [Google Scholar]
- (94).Ariano RE, Duke PC, and Sitar DS (2001) Population pharmacokinetics of fentanyl in healthy volunteers. J. Clin. Pharmacol 41, 757–763. [DOI] [PubMed] [Google Scholar]
- (95).Oda Y, Mizutani K, Hase I, Nakamoto T, Hamaoka N, and Asada A (1999) Fentanyl inhibits metabolism of midazolam: competitive inhibition of CYP3A4 in vitro. Br. J. Anaesth 82, 900–903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.