

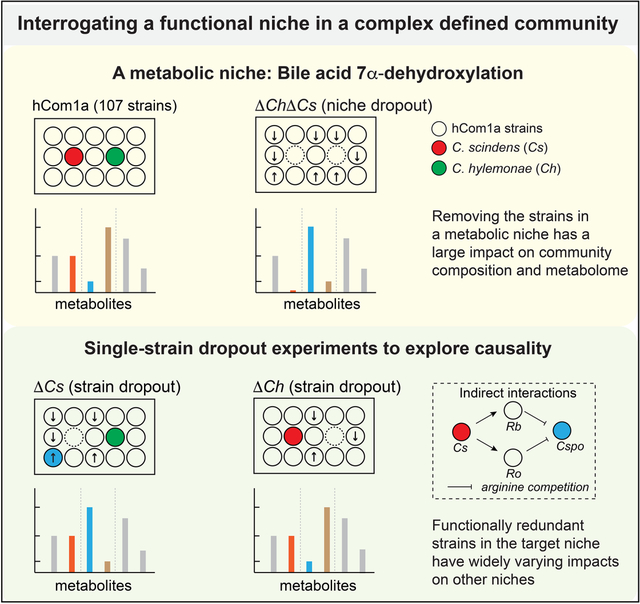
Abstract
The gut microbiome is complex, raising questions about the role of individual strains in the community. Here, we address this question by constructing variants of a complex defined community in which we eliminate strains that occupy the bile acid 7α-dehydroxylation niche. Omitting Clostridium scindens (Cs) and Clostridium hylemonae (Ch) eliminates secondary bile acid production and reshapes the community in a highly specific manner: eight strains change in relative abundance by >100-fold. In single-strain dropout communities, Cs and Ch reach the same relative abundance and dehydroxylate bile acids to a similar extent. However, Clostridium sporogenes increases >1000-fold in the ΔCs but not ΔCh dropout, reshaping the pool of microbiome-derived phenylalanine metabolites. Thus, strains that are functionally redundant within a niche can have widely varying impacts outside the niche, and a strain swap can ripple through the community in an unpredictable manner, resulting in a large impact on an unrelated community-level phenotype.
Graphical Abstract
IN BRIEF
Construction of variants of a complex defined microbial community reveals that strains that are functionally redundant within a niche can have widely varying impacts outside the niche.
INTRODUCTION
A typical gut microbiome consists of several hundred bacterial strains that span at least six orders of magnitude in relative abundance. Determining the contribution of individual strains to community ecology and host physiology is a daunting challenge. A variety of studies have characterized the functional properties of bacterial strains from the gut microbiome1–10. However, in vivo demonstrations of function typically involve mice colonized by one species or a small community; it remains difficult to study the functional contribution of a strain in the context of a native-scale community.
In thinking about where to start, two considerations led to the same idea. First, we were concerned about functional redundancy11,12. If we drop out a single strain, will we fail to see a phenotype because a strain with a similar function is present in the community? Second, the intestinal ecosystem is organized into physical and metabolic niches, which are thought to serve as functional units within the community11,12. A long-standing set of questions concerns how strains function within a niche. What is the mapping of strains to niches? Can changes in one niche propagate to others? And how do these events govern emergent behaviors such as community composition and metabolic output?
Taking these considerations into account, we decided to interrogate a niche rather than an individual strain. Given our interest in the chemistry of the microbiome, we focused on a well-studied microbial pathway—bile acid 7α-dehydroxylation—reasoning that it fits the definition of a metabolic niche since it offers a fitness advantage to a limited set of species that are specialized for bile acid utilization13,14. The products of this niche are a highly concentrated pool of metabolites with important biological activities13–15, but the interactions among its constituent species are poorly understood.
We took advantage of a recently developed model system for the gut microbiome that is composed of >100 of the most common gut bacterial species16 (Table S1). We find that the niche consists of two species, Clostridium scindens (Cs) and Clostridium hylemonae (Ch); when we drop them out of the community together (ΔCsΔCh), eight strains go up or down sharply in relative abundance. Single-strain dropout communities (ΔCs and ΔCh) reveal that either strain alone can metabolize bile acids exhaustively, and compensation within the niche keeps the relative abundance of its members within a narrow range. They also enable us to test causality in each strain-strain interaction, establishing whether it is specific to Cs or Ch or a function of both strains. ΔCs- and ΔCh-colonized mice have similar bile acid profiles but a large and unexpected difference in phenylalanine metabolism owing to a Cs-specific interaction with Clostridium sporogenes, showing that a strain swap within a niche can have a cascading effect that impacts unrelated niches. These data show that a highly controlled experiment can be performed on a complex community to elucidate strain-level causality and mechanism.
Reproducible colonization of a complex gut bacterial community in germ-free mice
The model microbiome we used in this work is based on a recently developed 104-member defined community, hCom116. We included 3 additional strains—Turicibacter sanguinis DSM 14220, Lactobacillus plantarum WCFS1, and Clostridium sp. D5—to improve the community’s metabolic potential, resulting a 107-member community (hereafter, hCom1a) (Figure S1).
To test the technical and biological reproducibility of hCom1a colonization in C57BL/6N mice, we used three groups of mice colonized by replicates of hCom1a constructed independently on different days. We gavaged germ-free C57BL/6N mice with thawed aliquots of the frozen community (Figure 1A). After four weeks of colonization, mice were sacrificed and cecal and colonic contents were analyzed by high-resolution metagenomic sequencing (Table S2). Strain abundances in all technical replicates were highly similar in the cecum (pairwise Pearson’s R=0.94±0.04). The cecal communities in three groups of biological replicates were very similar in relative abundance profiles (R >0.87 between all pairs, Figure 1B). This high degree of reproducibility supports the use of hCom1a as an experimental model in C57BL/6N mice.
Figure 1: Colonizing germ-free C57BL/6N mice with a complex gut bacterial community (hCom1a).
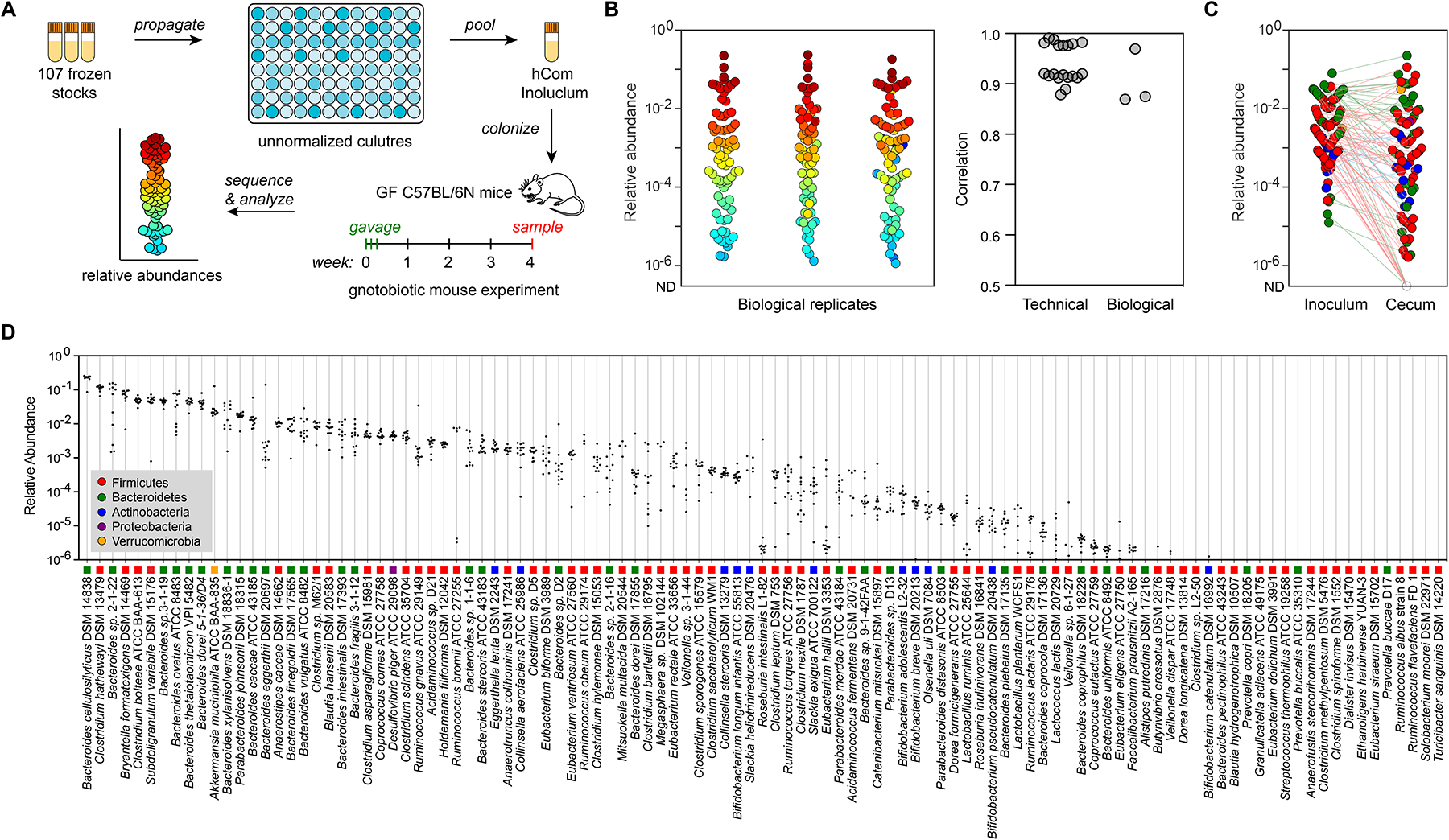
(A) Schematic of the experiment. Frozen stocks of the 107 strains were used to inoculate cultures that were sub-cultured every 24 h and then pooled after three days. The mixed culture was used to colonize germ-free C57BL/6N mice (n=3–5 mice per replicate) by oral gavage. After four weeks of colonization, mice fed ad libitum on chow diet were sacrificed and intestinal contents were collected, subjected to metagenomic sequencing, and analyzed by NinjaMap to measure the composition of the community. (B) The architecture of hCom1a in the cecum is highly reproducible. Left: community composition is highly similar across three biological replicates. Each dot is an individual strain; the collection of dots in a column represents the community at 4 weeks averaged over multiple mice receiving the same inoculum. Strains are colored according to their average rank-order relative abundance across all samples. Right: Pearson’s pairwise correlation coefficients for technical and biological replicates. The log10(relative abundance) values of all strains were used to calculate the Pearson correlation coefficient (R). For strains not detected, the relative abundance was set as 1e-8. (C) Averaged relative abundances of the inoculum versus the communities at week 4. Strains in the community span >6 orders of magnitude of relative abundance when colonizing the mouse gut. Dots are colored by phylum according to the legend in panel D. (D) Relative abundances for most strains are tightly distributed. Each column depicts the relative abundance of an individual strain across all samples at week 4. See also Figure S1 and S7, and Table S1 and S2.
Our analysis of the community composition in all C57BL/6N mice as well as the inoculum yielded four conclusions: 1) We confirmed the presence of almost all strains in the inoculum. Of these, 101 strains were detected with a mean relative abundance >1e-6. Two strains showed a low relative abundance (Anaerofustis stercorihominis DSM 17244, Blautia hydrogenotrophica DSM 10507) and four others were not detected in all three replicates (Clostridium methylpentosum DSM 5476, Dialister invisus DSM 15470, Ethanoligenens harbinense YUAN-3, Eubacterium dolichum DSM 3991) (Figure 1C). 2) Most strains in the inoculum colonized the mouse gut. 96 strains were detected in the mice cecum at least once; of these, 83 strains were detected with a mean relative abundance >1e-6 (Figure 1D). 3) Strain relative abundances were tightly distributed in the inoculum but spanned >6 orders of magnitude in the cecum with a coefficient of variation (CV, standard deviation/mean) <0.4 for nearly all strains (Figure 1C–D). The four strains with variable relative abundances are Bacteroides sp. 2-1-22, Ruminococcus bromii ATCC 27255, Slackia heliotrinireducens DSM 20476, and Mitsuokella multacida DSM 20544. 4) All 5 bacterial phyla were observed in the cecum. Bacteroidetes dominated, accounting for 58.2% of total reads, followed by Firmicutes (34.7%), Verrucomicrobia (3.1%), Proteobacteria (0.5%) and Actinobacteria (0.5%) (Figure 1C–D). Taken together, these data show that hCom1a can colonize GF C57BL/6N mice in a highly reproducible manner, facilitating the strain dropout experiments described in this manuscript.
A computational search to identify strains that carry out bile acid 7α-dehydroxylation
Of the strains in hCom1a, only Clostridium scindens ATCC 35704 (Cs) and Clostridium hylemonae DSM 15053 (Ch) were known to produce secondary bile acids14. Next, we determined whether any additional strains in hCom1a are part of the 7α-dehydroxylation niche. To address this question, we performed a multigeneblast search against a database of genome sequences from hCom1a for the eight-gene bai operon (Figure 2A), which encodes a metabolic pathway for the 7α-dehydroxylation of cholic acid (CA) and chenodeoxycholic acid (CDCA) to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively13 (Figure 2B). Of the 107 strains, only Cs and Ch harbor the bai operon.
Figure 2: The 7α-dehydroxylation niche in hCom1a is composed of Clostridium scindens (Cs) and Clostridium hylemonae (Ch).
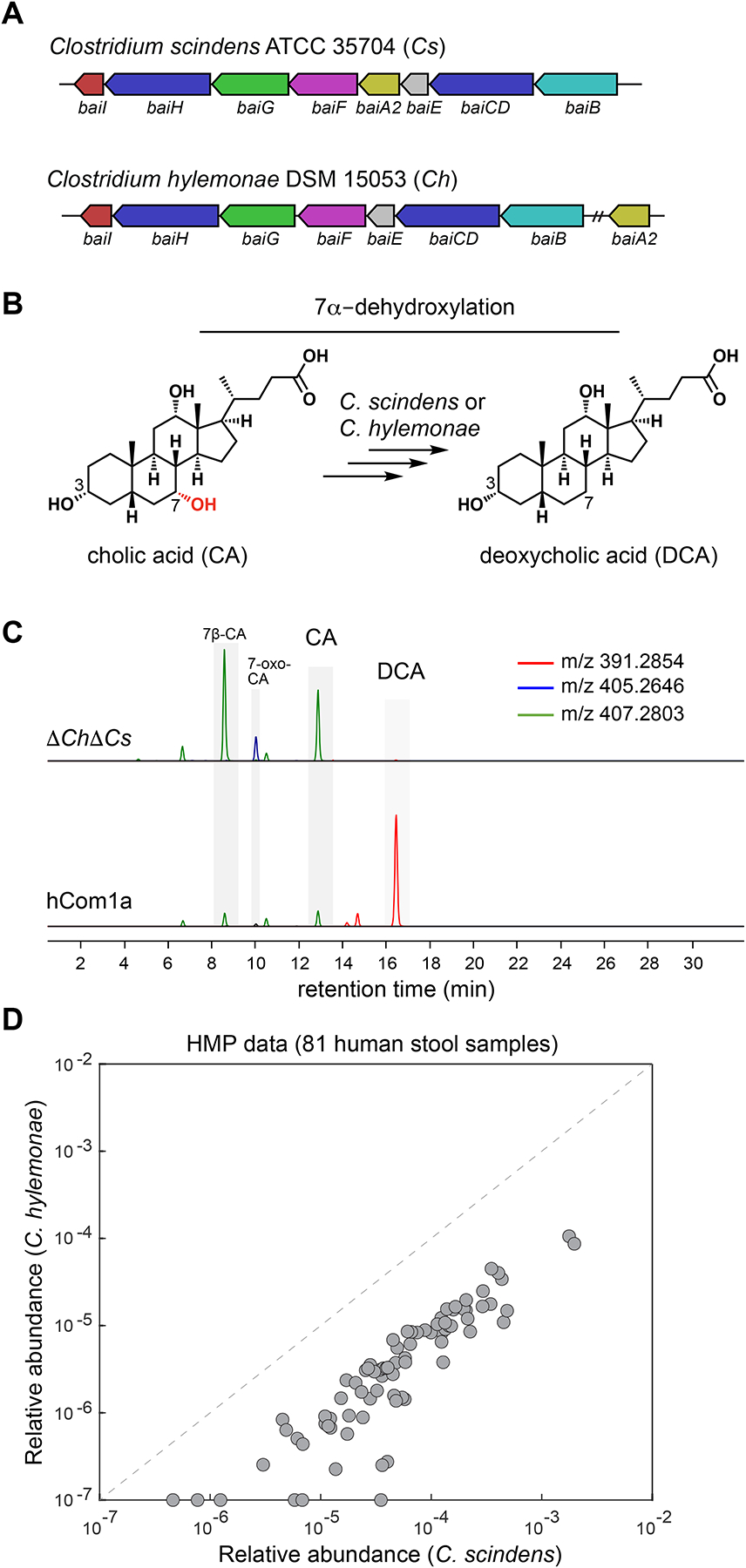
(A) A multigeneblast search of the 107 genomes in hCom1a shows that only Clostridium scindens and Clostridium hylemonae harbor the bai operon, which encodes the bile acid 7α-dehydroxylation pathway. (B) A simplified schematic showing the dehydroxylation of cholic acid (CA) to deoxycholic acid (DCA). (C) Combined extracted ion chromatogram showing that hCom1a converts CA to DCA in vitro, whereas the two-strain dropout community ΔChΔCs does not. Constituent strains were cultured separately, pooled, and subcultured (1:100) in Mega medium containing 100 μM cholic acid for 72 h. Culture supernatants were collected and analyzed by LC-MS. (D) Clostridium scindens and Clostridium hylemonae typically co-colonize the human gut. Each dot indicates one of the 81 gut metagenomic samples from the NIH HMP, and the relative abundance of Cs and Ch are indicated in the x axis and y-axis, respectively. See also Table S1.
We then sought to test whether any additional strains in the community harbor an alternative (unknown) pathway. We constructed hCom1a and a ΔChΔCs dropout community by mixing individually cultured strains, and then we grew these communities in the presence of CA and profiled their culture supernatants by LC-MS. We find that hCom1a converts CA to DCA whereas ΔChΔCs does not (Figure 2C). These data suggest that Cs and Ch are the only strains in the 7α-dehydroxylation niche, but they do not exclude the possibility that another strain in hCom1a is capable of carrying out this reaction in vivo. By analyzing publicly available human metagenomic data17, we observe that Cs and Ch often co-exist in the gut community, indicating that they are not competitively exclusive in the human gut (Figure 2D, Table S1).
Determining the occupancy of the 7α-dehydroxylation niche in vivo
To test whether Cs and Ch are the only occupants of the 7α-dehydroxylation niche, we colonized germ-free C57BL/6 mice with hCom1a or the two-strain dropout community (ΔChΔCs). After three weeks, we sacrificed the mice, harvested intestinal contents, and subjected the samples to targeted metabolomic and high-resolution metagenomic analysis16 (Figure 3A). By analyzing metagenomic data, we verified the absence of Cs and Ch in cecal samples from the ΔChΔCs-colonized mice (Figure 3B and S2A, Table S3).
Figure 3: Metabolic and ecological impacts of removing strains in the 7α-dehydroxylation niche.
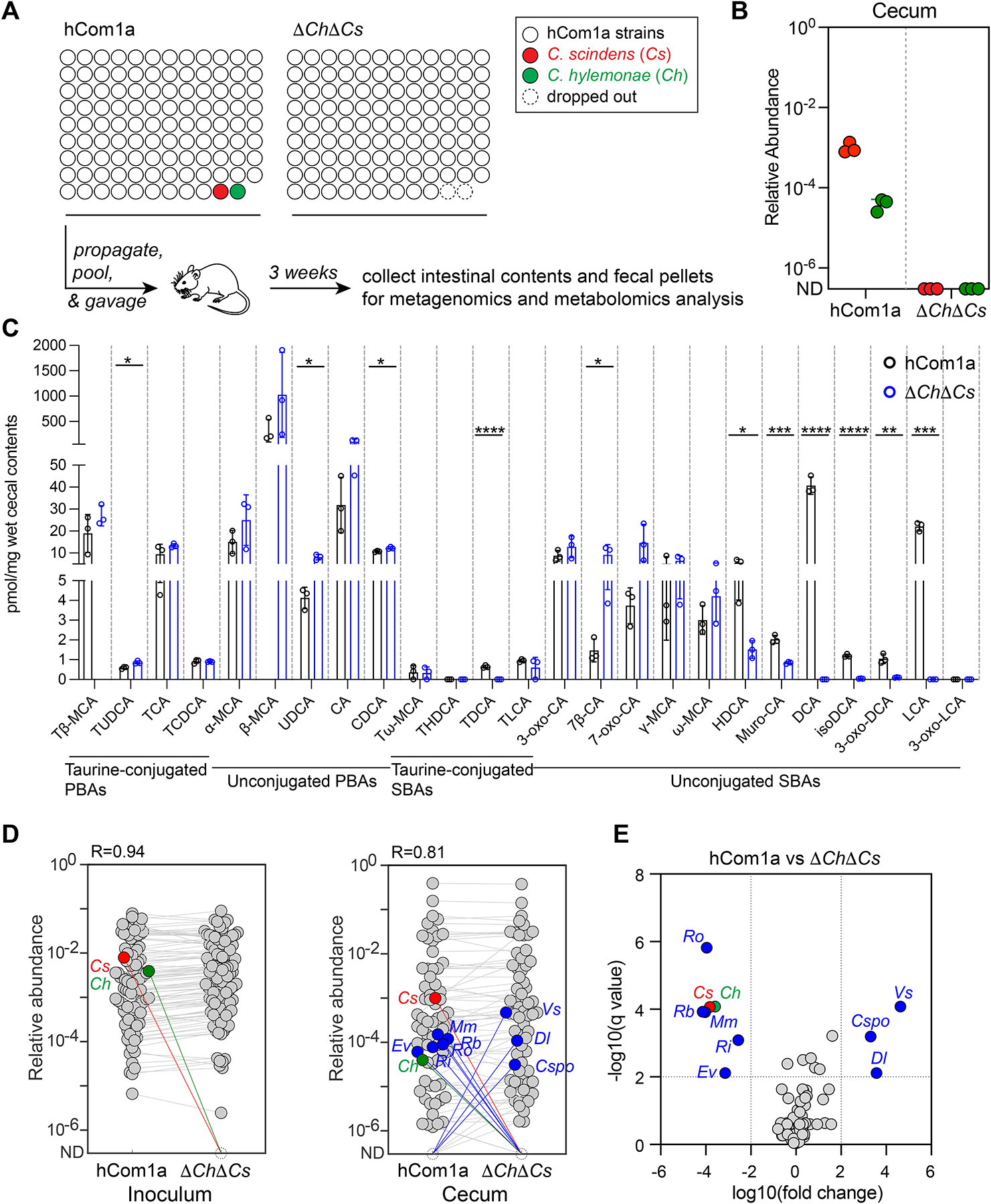
(A) Schematic of the experiment. Germ-free C57BL/6 mice (n=3 per group) were colonized with hCom1a or ΔChΔCs and housed for 3 weeks before sacrifice. Fecal pellets and intestinal contents were subjected to metagenomic analysis or targeted metabolite profiling. (B) Cs and Ch are undetectable in ΔChΔCs-colonized mice. Relative abundances were calculated through a high-resolution metagenomic analysis of the inoculum and cecal communities. (C) Secondary bile acids are eliminated in ΔChΔCs-colonized mice. Bile acids were quantified in cecal contents by targeted LC-MS-based profiling. Statistical significance was assessed using a Student’s two-tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001). PBAs: primary bile acids; SBAs: secondary bile acids. (D) Average relative abundances of the inoculum (left) versus the cecal communities at week 3 (right). Each dot is an individual strain; the collection of dots in a column represents the community averaged over 3 mice co-housed in one cage. Cs and Ch are highlighted in red and green, respectively. Strains highlighted in blue went up or down in relative abundance between hCom1a-colonized and ΔChΔCs-colonized mice (FDR < 0.01, fold change > 100). (E) Volcano plot of differential strain relative abundance. The log10(fold change) values of each strain are shown; relative abundances were set at 10−8 for strains not detected. Relative abundances were analyzed using a multiple unpaired t test, corrected with FDR (Q<1%). Strains discovered with significantly different relative abundance (FDR<0.01, fold change >100) are colored blue; the full names of the blue strains can be found in Figures 5D. See also Figure S2, and Table S3 and S4.
The removal of Ch and Cs has a large impact on the bile acid pool (Figure 3C, Table S4). The products of 7α-dehydroxylation—the secondary bile acids DCA and LCA—are absent in fecal pellets from the ΔChΔCs mice, as are prominent derivatives including isoDCA and 3-oxo-DCA. In contrast, ΔChΔCs mice have a higher level of the pathway intermediates 7β-CA and 7-oxo-CA and the CDCA epimer ursodeoxycholic acid (UDCA) (Figure 3C). Together, these data suggest Cs and Ch are the only strains in the 7α-dehydroxylation niche and that dropping them out results in the complete elimination of secondary bile acids. Previous attempts to knock out secondary bile acid production were conducted in the context of simple defined communities5,6,8,10 or by antibiotic knockdown of resident colonists followed by colonization with an antibiotic-resistant mutant, which depletes a variety of other strains in the community18. In contrast, our system is a defined knockout of 7α-dehydroxylation in the setting of a complex community.
Eliminating Cs and Ch has a large impact on a subset of strains
Next, we sought to assess the ecological impact of Cs/Ch removal on the rest of the community. Removing a species from a natural ecosystem can have consequences that are difficult to predict19,20. In mice, species have been removed from simple defined communities5,6,10,21 or from undefined communities using antibiotic pretreatment and phage22, but it remains unclear what effect species removal will have on a complex, unperturbed community in its native setting.
hCom1a and ΔChΔCs assume a broadly similar composition in mice (R=0.84) (Figure 3D, Table S3). While 97/107 of the community members have a similar relative abundance, the removal of Cs and Ch has a striking effect on a subset of eight strains, using a strict threshold of q<0.01 and fold change >100 (Figure 3D–E). Veillonella sp. 3-1-44 (Vs), Clostridium sporogenes (Cspo), and Dorea longicatena (Dl) went up sharply in relative abundance, while Ruminococcus obeum (Ro), Ruminococcus bromii (Rb), Mitsuokella multacida (Mm), Roseburia intestinalis (Ri), and Eubacterium ventriosum (Ev) decreased (Figures 3D and S2). Thus, at least in this case, the removal of two strains from a complex gut community has a large effect on a confined subset of strains that lie outside the 7α-dehydroxylation niche.
Compensation and functional redundancy within the 7α-dehydroxylation niche
To gain more resolution into the interactions within the 7α-dehydroxylation niche, we constructed two new communities—ΔCh and ΔCs—in which the 7α-dehydroxylation niche is occupied by Cs or Ch, but not both. We colonized germ-free C57BL/6 mice with each of these communities or the parental community (hCom1a) and sampled fecal pellets weekly for three weeks. We then sacrificed the mice, harvested intestinal contents, and subjected all the samples to bile acid profiling and metagenomic sequencing with high-resolution read mapping (Figure 4A).
Figure 4: Compensation and functional redundancy within the 7α-dehydroxylation niche.
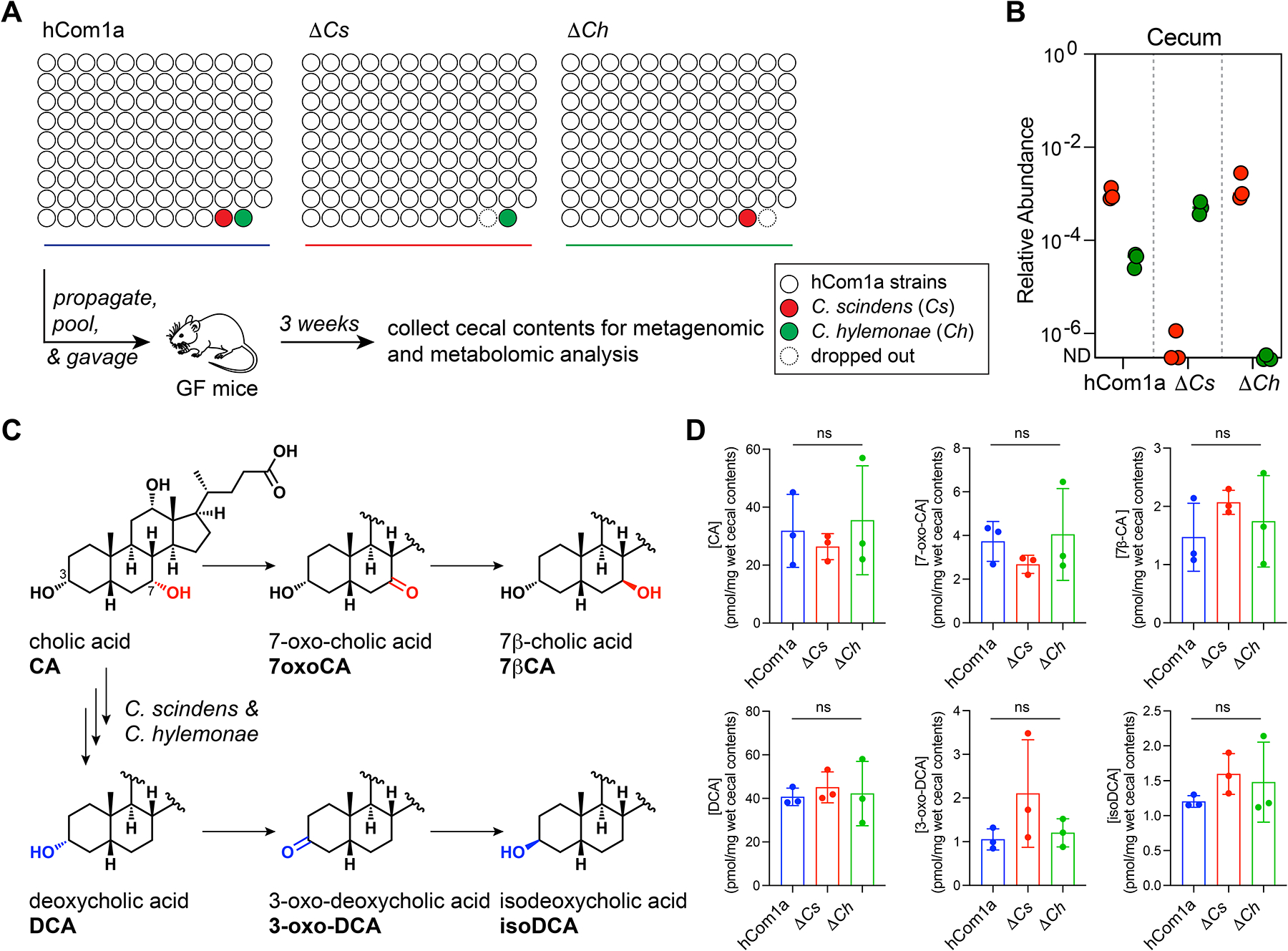
(A) Schematic of the experiment. Germ-free C57BL/6 mice (n=3 per group) were colonized with hCom1a, ΔCs, or ΔCh and housed for 3 weeks before sacrifice. Cecal contents were subjected to metagenomic and targeted metabolomic profiling. (B) Compensation within the 7α-dehydroxylation niche keeps the total relative abundance of its residents similar. Relative abundances of Cs and Ch in cecal contents from hCom1a-, ΔCh-, and ΔCs-colonized mice are shown. (C) Metabolic pathways for bile acid transformation by the gut microbiota. (D) The bile acid pools of mice colonized by hCom1a, ΔCh, and ΔCs are comparable. Thus, in the context of a complete community, Ch and Cs can carry out the core function of the niche—the conversion of primary to secondary bile acids—on its own. See also Figure S3 and S5, and Table S3 and S4.
First, we validated that Ch and Cs were absent from their respective dropout communities in the cecal samples from week 3 (Figure 4B and S2, Table S3). Next, we analyzed the interactions within the niche by metagenomic sequence analysis. In hCom1a, the relative abundances of Cs and Ch are ~10−3 and ~10−4, respectively. Consistent with the human data (Figure 2D), Cs is present at a higher relative abundance than Ch when they co-occupy the niche. In the absence of Cs, Ch goes up 12-fold in relative abundance; likewise, but to a lesser extent, Cs is more abundant in the ΔCh community (Figure 4B). Thus, Cs and Ch can co-exist, but compensation within the niche keeps the total relative abundance of its residents at a similar level (1.0 × 10−3 in hCom1a, 5.1 × 10−4 in ΔCs, and 1.5 × 10−3 in ΔCh) (Figure S2).
Finally, we quantified the cecal bile acid pool by LC-MS (Figures 4C and S3A, Table S4). The bile acid profiles of ΔCh, ΔCs, and the parental (hCom1a) community are remarkably similar: CA, DCA, and their derivatives (7-oxoCA, 7β-CA, 3-oxoDCA, and isoDCA) are all produced at comparable levels (Figure 4D and S3A). Thus, in the context of a complete community, either strain can carry out the core function of the niche—the conversion of primary to secondary bile acids—on its own.
Single-strain dropouts reveal complex interactions among Cs, Ch, and interacting strains
Next, we turned to effects outside the 7α-dehydroxylation niche. To test whether the strain-level interactions observed in the ΔChΔCs community are caused by Cs or Ch, we compared metagenomic data from hCom1a-colonized mice with that of ΔCs- and ΔCh-colonized mice to assess the relative abundances of the rest of the strains in the community (Figure 5A, Table S3). The compositions of ΔCs and ΔCh are very similar to that of hCom1a (R=0.86 and 0.88, respectively) (Figure 5B–C). Interestingly, of the eight strains whose relative abundances increased or decreased by >100-fold in the ΔChΔCs community, five respond to the absence of Cs (Vs and Cspo ↑; Ro, Rb, and Mm ↓) and three are impacted by the absence of Ch (Vs ↑; Ri and Mm ↓) (Figure 5B–C and S2). These sets overlap partially, suggesting that certain interactions share a common mechanism while others are Cs- or Ch-specific.
Figure 5: Single-strain dropouts reveal complex interactions among Cs, Ch, and interacting strains.
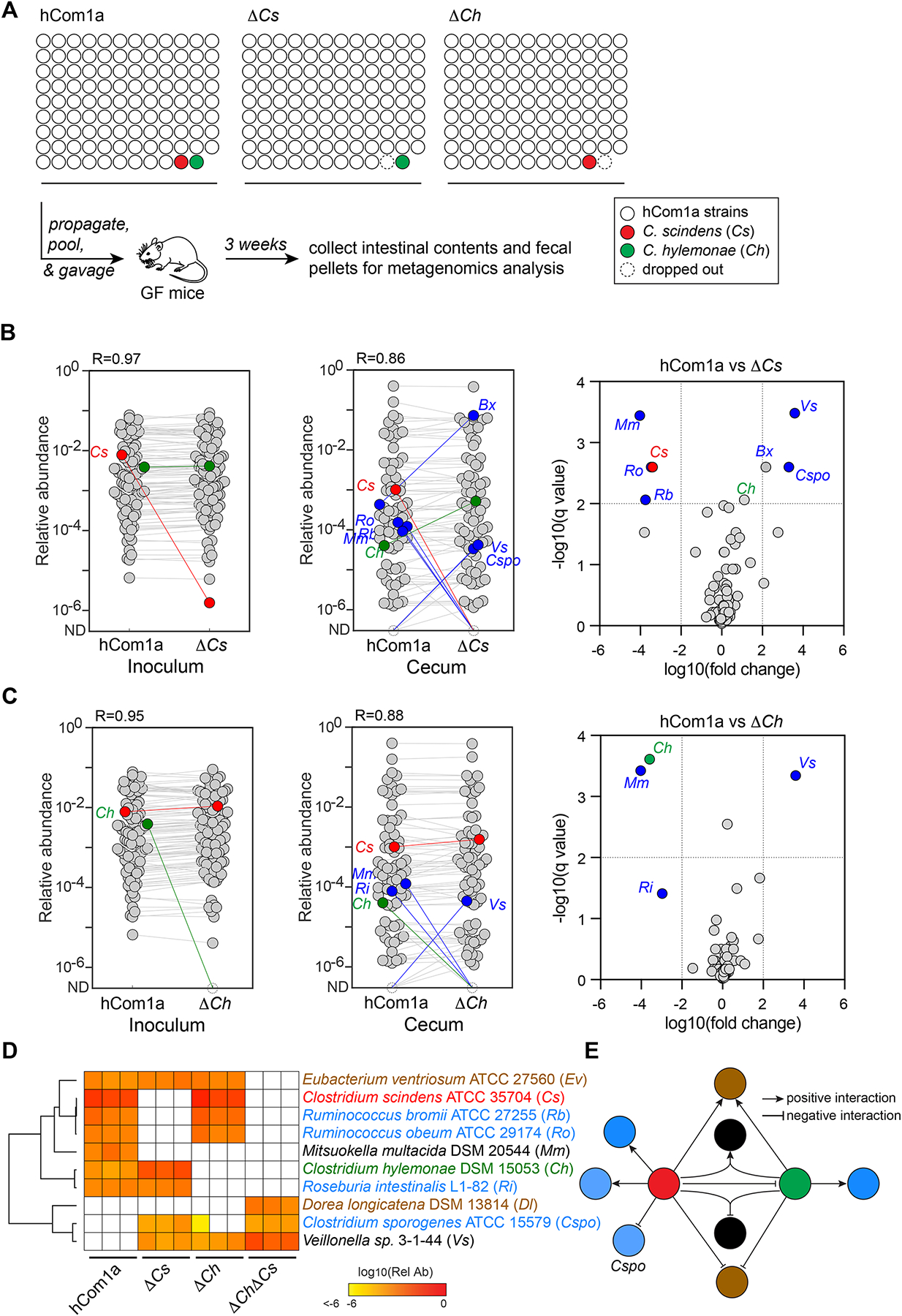
(A) Schematic of the experiment. Germ-free C57BL/6 mice (n=3 per group) were colonized with hCom1a, ΔCs, or ΔCh and housed for 3 weeks before sacrifice. Fecal pellets and intestinal contents were subjected to metagenomic analysis. (B-C) While most strains remain unchanged when dropping out Cs or Ch, a small number of strains change in relative abundance >100-fold. Left: Metagenomic analysis showing that Cs and Ch are absent in the ΔCs and ΔCh community inocula, respectively. Middle: Dropping out Cs or Ch impacts the relative abundance of five or three strains in the ΔCs and ΔCh communities, respectively. Each dot is an individual strain; the collection of dots in a column represents the community averaged over three mice co-housed in one cage. Cs and Ch are highlighted in red and green. Strains colored blue went up or down in relative abundance between hCom1a-colonized and ΔCs or ΔCh-colonized mice (FDR<0.01, fold change >100). Right: Volcano plot showing the log10(fold change) values for each strain; for strains that were not detected, relative abundances were set at 10−8. Strains with significantly different relative abundance (FDR <0.01, fold change >102) are colored blue; the full names of these strains are shown next to the heatmap in (D). The change in the relative abundance of Bacteroides xylanisolvens DSM 18836 (Bx) was close to the cutoff in ΔCs but not the other communities; this strain is not further discussed. (D) Heatmap representing the 8-strain interaction network around Cs and Ch. The relative abundance of each strain is shown in three mice colonized by hCom1a, ΔCs, ΔCh, or ΔChΔCs. Strains with relative abundance <10−6 are colored white. (E) Schematic of the interaction network. Strains are linked to the 7α-dehydroxylation niche in one of three ways: 1) Some strains are specific to Cs (Rb, Ro, and Cspo) or Ch (Ri); these interactions are presumably strain-specific and unrelated to bile acids. 2) Ev and Dl only respond to the double-strain dropout, indicating a mechanism related to secondary bile acid production. 3) The remaining strains, Mm and Vs, respond when either or both strains are missing, suggesting a requirement for the simultaneous presence of Cs and Ch. See also Figure S2 and S5, and Table S3.
To explore the interaction network around the 7α-dehydroxylation niche in more detail, we combined and clustered the metagenomics data from Cs, Ch, and each of the 8 affected strains (Figure 5D). The data are consistent with a model in which strains are linked to the niche in one of three ways: 1) Some strains are specific to Cs (Rb, Ro, and Cspo) or Ch (Ri); these interactions are presumably strain-specific and unrelated to bile acids. 2) Ev and Dl only respond to the double-strain dropout, indicating a mechanism shared by Cs and Ch, possibly related to secondary bile acid production. 3) The remaining strains, Mm and Vs, respond when either or both strains are missing, suggesting a requirement for the simultaneous presence of Cs and Ch (Figure 5E). We note that we cannot distinguish between direct interactions with Cs and/or Ch and indirect interactions that involve a third strain. Nevertheless, this analysis demonstrates the power of single-strain dropouts in discovering strain-strain interactions in a complex community.
Dl growth is inhibited by a product of 7α-dehydroxylation
Next, we sought to gain insight into the mechanisms that govern the negative (inhibitory) interactions revealed by this analysis. We focused on Dorea longicatena (Dl) and Clostridium sporogenes (Cspo) since Veillonella sp. 3-1-44 (Vs) did not grow robustly in vitro. Dl is only detectable in vivo when Cs and Ch are both absent, so we reasoned that either Cs or Ch should be capable of inhibiting its growth. We hypothesized that this growth inhibition is mediated by secondary bile acids, which are produced by both strains. To test this hypothesis, we cultured Dl (and, as a control, Cspo) in growth medium supplemented with CA and DCA at concentrations ranging from 39 μM-1.25 mM; CDCA and LCA were not considered due to poor solubility at high concentrations. The growth of Dl is completely inhibited by DCA at 625 μM, a physiological concentration, while Cspo was only partially affected (Figure S4A). In contrast, neither strain was affected by CA at concentrations as high as 1.25 mM (Figure S4B). These results suggest that DCA, which is produced by Cs and Ch, inhibits colonization by Dl.
Investigating the mechanism of the interaction between Cs and Cspo
We started by testing the hypothesis that a diffusible molecule produced by Cs inhibits the growth of C. sporogenes. We ruled out the possibility that this molecule is DCA in two ways: we observed that the interaction is specific to Cs but not Ch (Figure 5), even though both strains produce DCA; and we showed directly that DCA does not inhibit the growth of C. sporogenes (Figure S4A). Next, we considered the possibility that the antibacterial metabolite is 1-acetyl-β-carboline (AbC). C. scindens has been reported to produce AbC, which inhibits the growth of C. difficile and other gut bacterial species in vitro23. While DCA has a mild inhibitory effect at high concentrations, AbC has no effect on the growth of C. sporogenes, even at concentrations as high as 100–500 μM (higher than reported in the literature) (Figure S4C). Follow-up experiments in which C. sporogenes was grown in the presence of spent Cs culture medium (Figure 6A) suggest that—at least under the conditions tested here—Cs does not produce a diffusible molecule that inhibits the growth of C. sporogenes.
Figure 6: Investigating the mechanism of the interaction between Cs and Cspo.
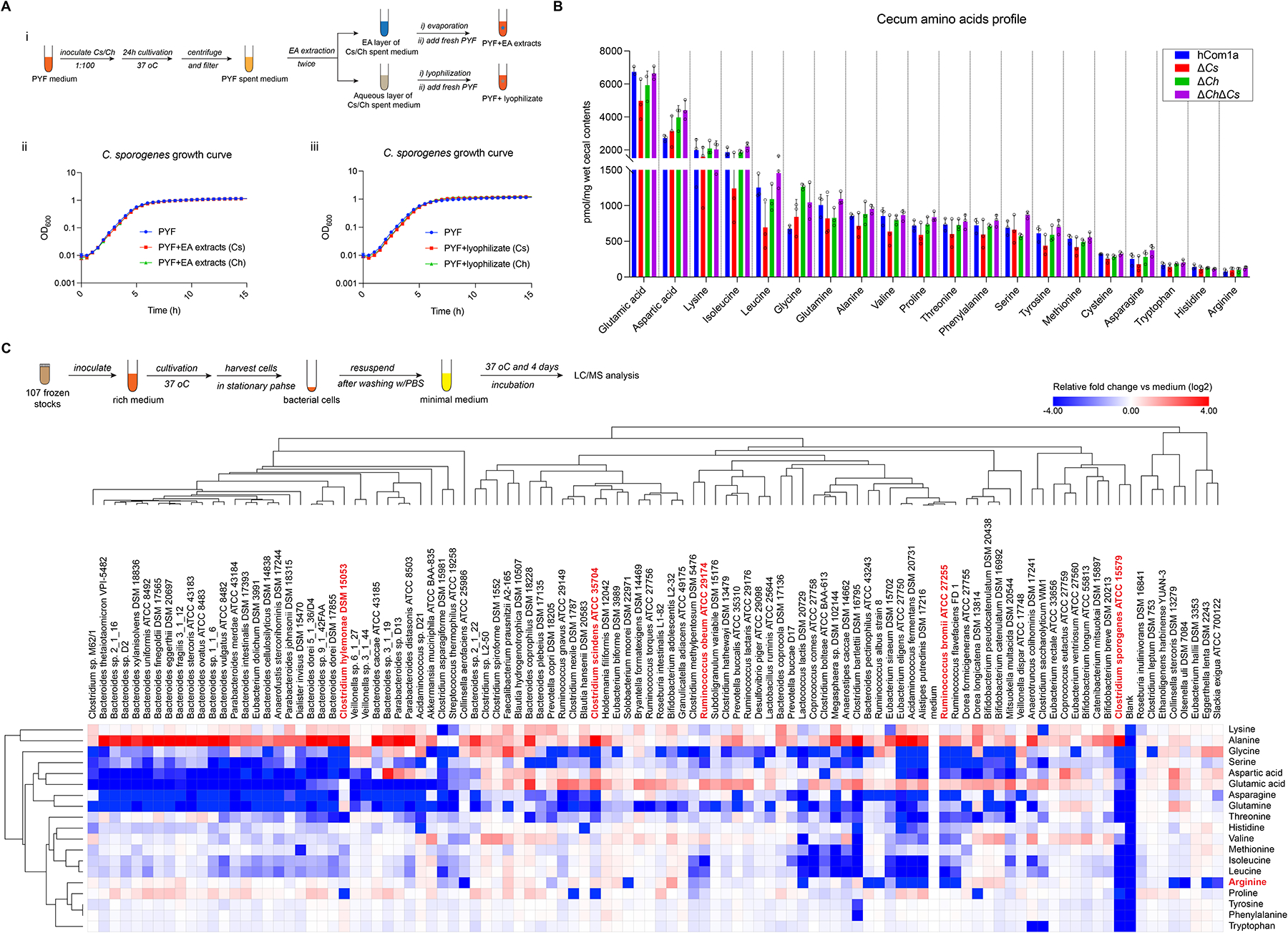
(A) No diffusible antimicrobial metabolites are found in the culture fluid of C. scindens (Cs) or C. hylemonae (Ch). (i) Schematic of preparing EA extracts and lyophilizate from the spent medium of Cs and Ch cultures. (ii) Growth curves of Cspo in fresh PYF medium without or with the EA extracts or lyophilizate from the spent medium of Cs or Ch. (B) Targeted profiling of amino acids in the cecum of hCom1a-, ΔCs-, ΔCh- and ΔChΔCs- colonized mice by LC-MS. (C) Targeted profiling of amino acid depletion by hCom1a strains in vitro. (Top) schematic of the experiment. (Bottom) heatmap of amino acid depletion by hCom1a strains, clustered by one minus pearson correlation. The abundance of each amino acid was normalized to the bacteria-free control (medium); in the log2 transformed data, red indicates an increased concentration while blue means the concentration has decrease. The data represent one of two independent experiments. See also Figure S6.
We next considered the possibility that the interaction between C. scindens and C. sporogenes results from competition for a nutrient. C. sporogenes specializes in metabolizing amino acids24,25, so we focused our efforts on amino acid utilization. We started by profiling amino acid levels in the cecum of mice colonized by hCom1a, ΔCs, ΔCh, and ΔChΔCs. Though the levels of all amino acids are largely similar among all groups, arginine, histidine, and tryptophan are at very low levels in the cecum, suggesting their limiting availabilities (Figure 6B).
Since arginine is known to be a particularly important amino acid for C. sporogenes16,26, we considered the possibility that competition for arginine underlies the interaction between C. scindens and C. sporogenes. To test this hypothesis in an unbiased way, we systematically screened amino acid consumption by every strain in the community (Figure 6C). C. scindens did not consume arginine at all, so we considered a second possibility: that the interaction between C. scindens and C. sporogenes is centered around arginine but is indirect (i.e., involves an intermediary organism).
Among the arginine consumers in our community, Ruminococcus caught our attention since all 7 Ruminococcus species depleted arginine to some extent (Figure 6C). Notably, Ruminococcus bromii (Rb) and Ruminococcus obeum (Ro) respond strongly to Cs dropout but in the opposite direction to that of C. sporogenes; i.e., like Cs, the relative abundance of Rb and Ro is strongly negatively correlated with that of C. sporogenes (Figure S2). While Ro depletes about half of the arginine in the medium, Rb completely consumes all of the arginine (Figure 6C). These data are consistent with the possibility that Rb and Ro might compete with C. sporogenes for the consumption of arginine in vivo.
An unexpected impact of Cs on aromatic amino acid metabolism
The data from ΔCs- and ΔCh-colonized mice suggest that although Ch and Cs appear functionally redundant within their niche—the two strains are interchangeable in terms of the resulting bile acid profile—they have distinct effects on the rest of the community. However, there are only six strains whose relative abundances are meaningfully different between the ΔCh and ΔCs communities (Bx, Ri, Rb, Ro, Mm, and Cspo); apart from these, the relative abundances of the rest of the community are nearly superimposable (Figure S5A, Table S3). To determine whether the ΔCh and ΔCs communities are truly indistinguishable, we compared microbiome-derived metabolites in the cecal contents and urine of the ΔCh- and ΔCs-colonized mice, expecting to find concordant profiles in light of the compositional similarities (Figure 7A).
Figure 7: An unexpected impact of Cs on aromatic amino acid metabolism.
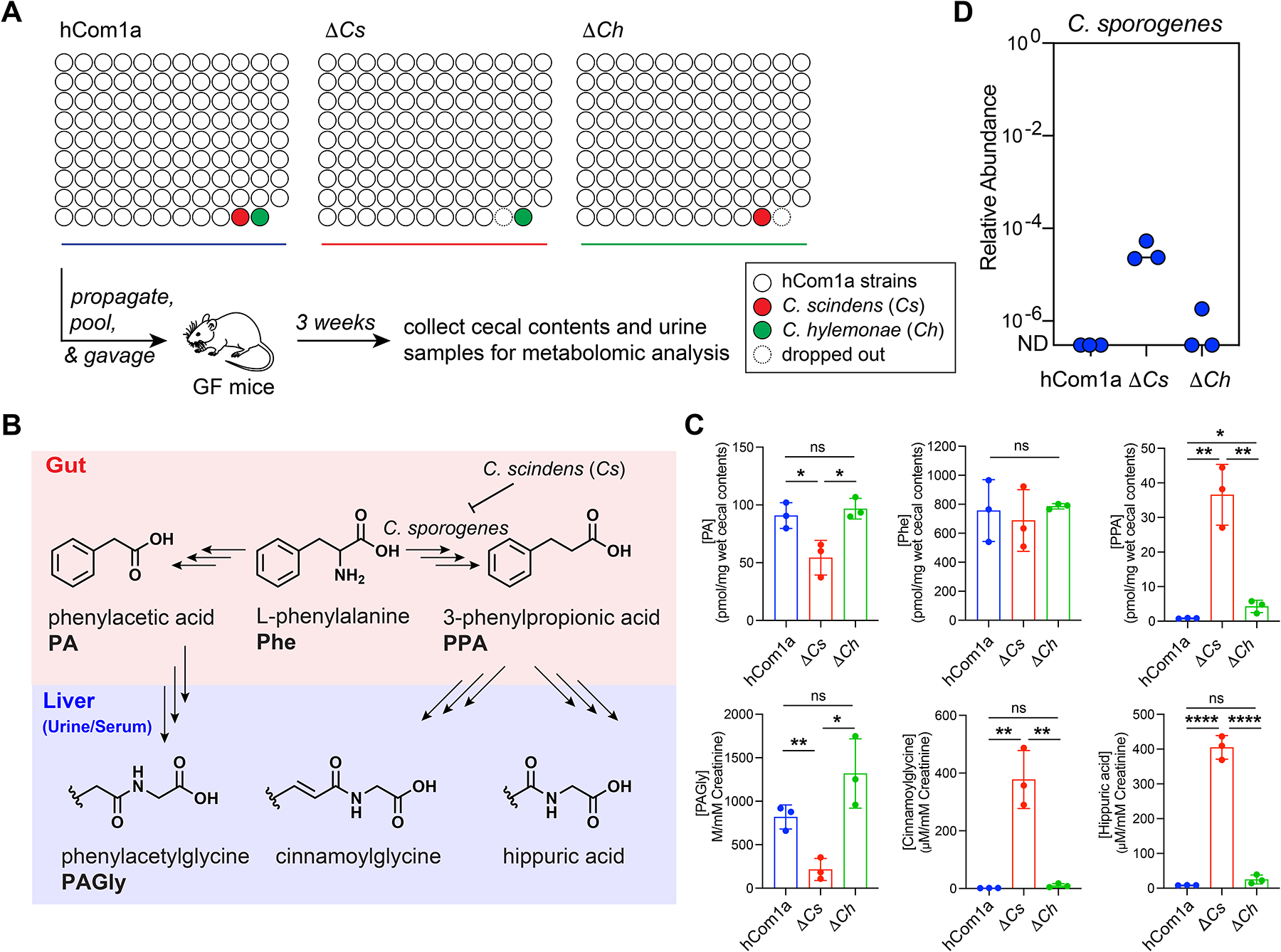
(A) Schematic of the experiment. Germ-free C57BL/6 mice (n=3 per group) were colonized with hCom1a or its single-strain dropout variant (ΔCs and ΔCh) and housed for 3 weeks before sacrifice. Fecal pellets, cecal contents, and urine samples were subjected to targeted metabolite profiling. (B) Certain gut bacteria can reduce phenylalanine (Phe) to phenylpropionic acid (PPA), which is converted to hippuric acid and cinnamoylglycine by the host. Other gut bacteria oxidize Phe to phenylacetic acid (PA), which is metabolized in the liver to phenylacetylglycine (PAGly). (C) ΔCs differs markedly from hCom1a and ΔCh in terms of AAA metabolite output. hCom1a and ΔCh convert phenylalanine almost exclusively to PA (from which the host generates PAGly); hippurate is nearly undetectable in the urine and serum (Figure S3). In contrast, ΔCs converts Phe predominantly to phenylpropionic acid (which the host metabolizes to hippurate); PAGly levels are very low in the urine and serum (Figure S3). (D) C. sporogenes, which is undetectable in hCom1a- and ΔCh-colonized mice, rises to a relative abundance of 10−5-10−4 in ΔCs-colonized mice. The relative abundance of Cspo in cecal contents from hCom1a-, ΔCs-, and ΔCh-colonized mice is shown. Statistical significance was assessed using a Student’s two tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001, n.s: no significance). See also Figure S2, S3, and S5, and Table S4.
To our surprise, there were striking differences among the profiles (Figure 7B, Table S4). First, three reductive phenylalanine metabolites—phenylpropionic acid, hippuric acid, and cinnamoylglycine—were produced abundantly in ΔCs-colonized mice, but at very low levels in ΔCh- and hCom1a-colonized mice (Figure 7C and S3B–D). Three observations suggest a causal role for Cspo: it is the only strain that is essentially undetectable in ΔCh- and hCom1a-colonized mice but up >1000-fold in ΔCs-colonized mice (Figure 5D and 7D), it is the only strain in hCom1a known to produce reductive phenylalanine metabolites in vitro and in vivo24,27, and its dropout from ΔCs-colonized mice results in a decrease in reductive phenylalanine metabolites (Figure S6).
Second, two oxidative metabolites of phenylalanine—phenylacetic acid and phenylacetylglycine—show the opposite pattern (Figure 7C and S3B–D). Given that reductive and oxidative pathways are competing since they share phenylalanine as a substrate24, the decrease in oxidative metabolites likely results from the increase in C. sporogenes-mediated reductive metabolism.
These data are consistent with a model in which a strain swap (ΔCh vs ΔCs) initiated a multi-step process that cascaded through the community: Cs was replaced with Ch, which increased the relative abundance of Cspo from nearly undetectable to ~10−4, which increased the level of hippurate (~16 fold) and cinnamoylglycine (~37 fold), which decreased the production of phenylacetate (~2 fold) and phenylacetylglycine (~6 fold) (Figure S5B).
This is especially notable in light of the biological activities of the metabolites involved. Phenylacetylglycine, which plays a causative role in cardiovascular disease28, is down substantially in ΔCs-colonized mice. It is replaced by hippuric acid and cinnamoylglycine, which are notable for their lack of toxicity29,30. Replacing Cs with Ch results in a 133-fold difference in the ratio of favorable (hippurate and cinnamoylglycine) to unfavorable (phenylacetylglycine) Phe metabolites. Thus, a strain swap in the 7α-dehydroxylation niche is ‘silent’ in terms of bile acids but yields a large, desirable effect in an unrelated compartment of the community.
DISCUSSION
A long-standing challenge in microbiome research has been to assess the impact of individual strains on community ecology and host physiology. Pioneering efforts have focused on interactions in binary culture31,32 or in simple communities33,34, where the rules and selective conditions are likely distinct from those in a complex community35. A complex defined community offers a setting in which a ‘clean’ dropout can be constructed in a physiologically relevant background. This approach, in combination with recent advances in genetic systems for the microbiome18,25,36, will greatly improve our understanding of strain-strain and strain-host interactions.
Our results emphasize the importance of focusing on a functional unit within the community—here, a metabolic niche—rather than on individual strains. Initially, our main focus was to characterize the effects of secondary bile acids on the host, and our interest in the niche was driven mainly by the concern that the phenotype induced by dropping out a single bile-acid-producing colonist would be masked by another functionally redundant strain. But as we began analyzing metagenomic and metabolomic data from the strain dropout experiments, we came to think that the main story was the connection between ecological interactions and community metabolism.
Using this model, we show that there is functional redundancy within the 7α-dehydroxylation niche. An unknown mechanism—we speculate that it could be the availability of the primary bile acids CA and CDCA—keeps the total relative abundance of strains in the niche within a 3-fold range, regardless of whether the niche is mono- or bi-colonized. Cs and Ch can co-occupy the niche rather than excluding each other. However, when the niche is mono-colonized, Cs and Ch are functionally interchangeable; either strain alone is capable of dehydroxylating the entire pool of primary bile acids, and the resulting bile acid profiles are indistinguishable.
However, changes in the occupancy of the niche lead to unexpected effects elsewhere in the community. Communities in which the niche is mono-colonized with Cs (ΔCh) vs. Ch (ΔCs) are extremely similar in terms of composition—only six strains differ significantly in relative abundance. But in ΔCs, the unexpected proliferation of Clostridium sporogenes—which is below the limit of detection in hCom1a and ΔCh—led to a large increase in reductive phenylalanine metabolites, reshaping the chemical output of a different niche in the community. This finding has two important implications. First, it highlights the functional impact of species- and strain-level variation in the microbiome. Different species that fill the same niche—even those that appear functionally redundant—may yield a large difference outside the niche.
Second, this has important implications for understanding the principles of rational community design. An emerging goal in microbiome research is to design microbial communities that are endowed with an immunologic or metabolic phenotype of interest37. If the goal is to make a community that generates hippurate and not phenylacetylglycine, the obvious strategy would be to include strains that produce the former and exclude those that generate the latter. But by dropping out a strain in an unrelated niche, we observed a comparable difference to what we would have expected through rational engineering—and through a simpler maneuver that does not reduce diversity or compromise any other niche. Thus, to alter a phenotype of interest, one needs to consider not just the strains that carry it out but their interaction partners in the community.
LIMITATIONS OF THE STUDY
As a model microbiome, hCom1a has two main limitations in the context of this study. First, it produces a smaller quantity of secondary bile acids than a human fecal community (Figure S7), indicating the possibility of missing strain(s) that could improve bile acid metabolism. hCom2, a derivative of hCom1, is more comparable to a human fecal community16 and could be used for subsequent strain dropout studies. Second, hCom1a is only one community; there is substantial strain- and species-level variation among human microbiomes, so the interactions discovered here may not exist in other communities.
Our experimental findings have two additional limitations. Although we explored the interaction between Cs and C. sporogenes in detail and present evidence consistent with an indirect competition for arginine (Figures 6 and S4), our data are not sufficient to establish the mechanism; further studies in which Ro and/or Rb are dropped out, alone and in combination with C. sporogenes, would be required, as would experiments in which arginine levels are modulated.
Our experimental setup and data interpretation may imply an overly simplistic view in which bacterial species belong to an individual niche. In reality, strains occupy multiple niches simultaneously. We hope that an approach similar to the one employed here can be used to explore the way in which strains map to niches, and strain-strain interactions yield effects across multiple niches.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Fischbach (fischbach@fischbachgroup.org).
Materials availability
The bacterial strains used in this study are available from the sources listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Bacterial and Virus Strains | |||
| Alistipes putredinis DSM 17216 | DSMZ | Chopped Meat Medium | |
| Anaerotruncus colihominis DSM 17241 | DSMZ | Mega Medium | |
| Bacteroides caccae ATCC 43185 | ATCC | Mega Medium | |
| Bacteroides coprophilus DSM 18228 | DSMZ | Mega Medium | |
| Bacteroides dorei 5_1_36/D4 | BEI | Mega Medium | |
| Bacteroides eggerthii DSM 20697 | DSMZ | Mega Medium | |
| Bacteroides finegoldii DSM 17565 | DSMZ | Mega Medium | |
| Bacteroides fragilis 3_1_12 | BEI | Mega Medium | |
| Bacteroides intestinalis DSM 17393 | DSMZ | Mega Medium | |
| Bacteroides sp. 1_1_6 | BEI | Mega Medium | |
| Bacteroides sp. 2_1_22 | BEI | Mega Medium | |
| Bacteroides sp. 3_1_19 | BEI | Mega Medium | |
| Bacteroides sp. 9_1_42FAA | BEI | Mega Medium | |
| Bacteroides sp. 2_1_16 | BEI | Mega Medium | |
| Bacteroides sp. D2 | BEI | Mega Medium | |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | Mega Medium | |
| Bacteroides xylanisolvens DSMZ 18836 | DSMZ | Mega Medium | |
| Bacteroides uniformis ATCC 8492 | ATCC | Mega Medium | |
| Bacteroides pectinophilus ATCC 43243 | ATCC | Chopped Meat Medium | |
| Bacteroides plebeius DSM 17135 | DSMZ | Chopped Meat Medium | |
| Bacteroides coprocola DSM 17136 | DSMZ | Chopped Meat Medium | |
| Bacteroides stercoris ATCC 43183 | DSMZ | Mega Medium | |
| Coprococcus eutactus ATCC 27759 | ATCC | Chopped Meat Medium | |
| Eubacterium dolichum DSM 3991 | DSMZ | Mega Medium | |
| Ruminococcus gnavus ATCC 29149 | BEI | Mega Medium | |
| Eubacterium rectale ATCC 33656 | ATCC | Mega Medium | |
| Clostridium methylpentosum DSM 5476 | DSMZ | Mega Medium | |
| Clostridium nexile DSM 1787 | DSMZ | Mega Medium | |
| Clostridium scindens ATCC 35704 | ATCC | Mega Medium | |
| Clostridium sp. L2–50 | BEI | Chopped Meat Medium | |
| Clostridium sp. M62/1 | BEI | Chopped Meat Medium | |
| Clostridium asparagiforme DSM 15981 | DSMZ | Mega Medium | |
| Clostridium bolteae ATCC BAA-613 | ATCC | Mega Medium | |
| Clostridium hathewayi DSM 13479 | DSMZ | Mega Medium | |
| Clostridium leptum DSM 753 | DSMZ | Chopped Meat Medium | |
| Dorea formicigenerans ATCC 27755 | DSMZ | Mega Medium | |
| Dorea longicatena DSM 13814 | DSMZ | Mega Medium | |
| Coprococcus comes ATCC 27758 | ATCC | Mega Medium | |
| Blautia hansenii DSM 20583 | DSMZ | Mega Medium | |
| Bryantella formatexigens DSM 14469 | DSMZ | Mega Medium | |
| Butyrivibrio crossotus DSM 2876 | DSMZ | Chopped Meat Medium | |
| Ruminococcus torques ATCC 27756 | ATCC | Mega Medium | |
| Parabacteroides merdae ATCC 43184 | DSMZ | Mega Medium | |
| Subdoligranulum variabile DSM 15176 | DSMZ | Mega Medium | |
| Parabacteroides johnsonii DSM 18315 | DSMZ | Chopped Meat Medium | |
| Roseburia intestinalis L1–82 | ATCC | Mega Medium | |
| Ruminococcus obeum ATCC 29174 | DSMZ | Mega Medium | |
| Eubacterium ventriosum ATCC 27560 | DSMZ | Mega Medium | |
| Faecalibacterium prausnitzii A2–165 | DSMZ | Chopped Meat Medium | |
| Parabacteroides sp. D13 | BEI | Mega Medium | |
| Eubacterium hallii DSM 3353 | DSMZ | Chopped Meat Medium | |
| Roseburia inulinivorans DSM 16841 | DSMZ | Chopped Meat Medium | |
| Prevotella buccalis ATCC 35310 | DSMZ | Chopped Meat Medium | |
| Ruminococcus lactaris ATCC 29176 | ATCC | Chopped Meat Medium | |
| Eubacterium eligens ATCC 27750 | DSMZ | Mega Medium | |
| Holdemania filiformis DSM 12042 | DSMZ | Mega Medium | |
| Bacteroides ovatus ATCC 8483 | ATCC | Mega Medium | |
| Bacteroides vulgatus ATCC 8482 | ATCC | Mega Medium | |
| Clostridium spiroforme DSM 1552 | DSMZ | Chopped Meat Medium | |
| Eubacterium biforme DSM 3989 | DSMZ | Mega Medium | |
| Blautia hydrogenotrophica DSM 10507 | DSMZ | Chopped Meat Medium | |
| Clostridium saccharolyticum WM1 | DSMZ | Mega Medium | |
| Parabacteroides distasonis ATCC 8503 | ATCC | Mega Medium | |
| Eubacterium siraeum DSM 15702 | DSMZ | Chopped Meat Medium | |
| Eggerthella lenta DSM 2243 | DSMZ | Chopped Meat Medium | |
| Anaerostipes caccae DSM 14662 | DSMZ | Mega Medium | |
| Bacteroides cellulosilyticus DSM 14838 | DSMZ | Mega Medium | |
| Clostridium hylemonae DSM 15053 | DSMZ | Mega Medium | |
| Acidaminococcus sp. D21 | BEI | Mega Medium | |
| Catenibacterium mitsuokai DSM 15897 | DSMZ | Mega Medium | |
| Collinsella aerofaciens ATCC 25986 | ATCC | Mega Medium | |
| Acidaminococcus fermentans DSM 20731 | DSMZ | Mega Medium | |
| Clostridium bartlettii DSM 16795 | DSMZ | Mega Medium | |
| Ethanoligenens harbinense YUAN-3 | DSMZ | Chopped Meat Medium | |
| Veillonella dispar ATCC 17748 | DSMZ | Chopped Meat Medium | |
| Collinsella stercoris DSM 13279 | DSMZ | Chopped Meat Medium | |
| Prevotella buccae D17 | BEI | Chopped Meat Medium | |
| Mitsuokella multacida DSM 20544 | DSMZ | Mega Medium | |
| Olsenella uli DSM 7084 | DSMZ | Chopped Meat Medium | |
| Slackia heliotrinireducens DSM 20476 | DSMZ | Chopped Meat Medium | |
| Bifidobacterium longum infantis ATCC 55813 | BEI | Mega Medium | |
| Dialister invisus DSM 15470 | DSMZ | Mega Medium | |
| Prevotella copri DSM 18205 | DSMZ | Chopped Meat Medium | |
| Veillonella sp. 6_1_27 | BEI | Chopped Meat Medium | |
| Slackia exigua ATCC 700122 | DSMZ | Chopped Meat Medium | |
| Streptococcus thermophilus LMD-9 | ATCC | Chopped Meat Medium | |
| Desulfovibrio piger ATCC 29098 | DSMZ | Chopped Meat Medium | |
| Lactobacillus ruminis ATCC 25644 | ATCC | Mega Medium | |
| Akkermansia muciniphila ATCC BAA-835 | DSMZ | Mega Medium | |
| Bifidobacterium adolescentis L2–32 | BEI | Mega Medium | |
| Bifidobacterium pseudocatenulatum DSM 20438 | DSMZ | Mega Medium | |
| Solobacterium moorei DSM 22971 | DSMZ | Chopped Meat Medium | |
| Anaerofustis stercorihominis DSM 17244 | DSMZ | Mega Medium | |
| Lactococcus lactis DSMZ 20729 | DSMZ | Mega Medium | |
| Granulicatella adiacens ATCC 49175 | DSMZ | Mega Medium | |
| Clostridium sporogenes ATCC 15579 | ATCC | Mega Medium | |
| Bacteroides dorei DSM 17855 | DSMZ | Mega Medium | |
| Bifidobacterium catenulatum DSM 16992 | DSMZ | Mega Medium | |
| Ruminococcus albus strain 8 | Laboratory of Robert Mackie | Chopped Meat Medium | |
| Ruminococcus flavefaciens FD 1 | Laboratory of Robert Mackie | Chopped Meat Medium | |
| Ruminococcus bromii ATCC (L2–63) | ATCC | Chopped Meat Medium | |
| Veillonella sp. 3_1_44 | BEI | Chopped Meat Medium | |
| Bifidobacterium breve DSM 20213 | DSMZ | Mega Medium | |
| Megasphaera sp. DSMZ 102144 | DSMZ | Mega Medium | |
| Clostridium sp. D5 | BEI | Mega Medium | |
| Lactobacillus plantarum WCFS1 | ATCC | Mega Medium | |
| Turicibacter sanguinis DSM 14220 | DSMZ | Chopped Meat Medium | |
| Paenibacillus barengoltzii CC33–002B | BEI | Mega Medium | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| PBS | Gibco | 10010023 | |
| Tryptone peptone | Difco | 211921 | |
| Bacto yeast extract | Difco | 212750 | |
| Magnesium sulfate heptahydrate | Sigma | M2773 | |
| Sodium bicarbonate | Sigma | S5761 | |
| Calcium chloride | Sigma | C7902 | |
| Resazurin | Sigma | R7017 | |
| Agar | Difco | DF0140-01-0 | |
| Sodium acetate | Sigma | S2889 | |
| Meat extract | Sigma | 70164 | |
| D-glucose | Sigma | 47829 | |
| L-cystine HCl | Sigma | C7477 | |
| Potassium phosphate monobasic | Sigma | P5655 | |
| Potassium phosphate dibasic | Sigma | P3786 | |
| Vitamin K3 | Sigma | M5625 | |
| Hematin | Sigma | H3281 | |
| Tween 80 | Sigma | P4780 | |
| Vitamin mix | ATCC | MD-VS | |
| Trace mineral supplement | ATCC | MD-TMS | |
| D-(+)-cellobiose | Sigma | C7252 | |
| D-(+)-maltose monohydrate | Sigma | M5885 | |
| D-(−)-fructose | Sigma | F0127 | |
| Acetic acid, glacial | Sigma | A6283 | |
| Propionic acid | Sigma | P5561 | |
| Butyric acid | Sigma | B103500 | |
| Isovaleric acid | Sigma | 129542 | |
| Sterilized rumen fluid | Bar Diamond Ranch | #SRF | |
| Chopped meat media | Hardy Diagnostics | K219 | |
| Vitamin K2 | Sigma | V9378 | |
| Ammonium sulfate | Sigma | A4418 | |
| Nitrilotriacetic acid | Sigma | N9877 | |
| Manganese(II) chloride tetrahydrate | Sigma | M5005 | |
| Cobalt (II) hexahydrate | Sigma | C8661 | |
| Calcium chloride dihydrate | Sigma | 223506 | |
| Zinc chloride | Sigma | Z0152 | |
| Copper chloride | Sigma | 451665 | |
| Sodium molybdate dihydrate | Sigma | M1651 | |
| Boric acid | Sigma | B6768 | |
| Sodium selenite | Sigma | 214485 | |
| Nickel chloride hexahydrate | Sigma | N6136 | |
| Sodium tungstate dihydrate | Sigma | 72069 | |
| L-alanine | Sigma | A7469 | |
| L-arginine | Sigma | A5006 | |
| L-asparagine | Sigma | A4159 | |
| L-aspartic Acid | Sigma | A8949 | |
| L-glutamic Acid | Sigma | 49449 | |
| L-glutamine | Sigma | 49419 | |
| L-glycine | Sigma | G7126 | |
| L-histidine | Fisher | BP382 | |
| L-isoleucine | TCI | I0181 | |
| L-leucine | TCI | L0029 | |
| L-lysine | Sigma | L5751 | |
| L-methionine | Sigma | 64319 | |
| L-phenylalanine | Sigma | P5482 | |
| L-proline | Sigma | 81709 | |
| L-serine | Sigma | S4500 | |
| L-threonine | Sigma | 89179 | |
| L-tryptophan | Sigma | T0254 | |
| L-tyrosine | Sigma | 93829 | |
| L-valine | Sigma | 94619 | |
| Columbia agar with 5% sheep blood | BD | 221165 | |
| Brain Heart Infusion broth | Fisher | CM1136B | |
| Horse blood, defibrinated | Fisher | 50863761 | |
| Peptone Yeast Extract Broth with Fructose | Anaerobe Systems | AS-840 | |
| Glycerol | Fisher | PRH5433 | |
| Potassium chloride | Sigma | P9541 | |
| Magnesium chloride | Sigma | M1028 | |
| Sodium phosphate dibasic | Sigma | S3264 | |
| Sodium chloride | Sigma | S3014 | |
| Methanol | Fisher | A456 | |
| Formic acid | Sigma | 426229 | |
| Ammonium bicarbonate | Sigma | 9830 | |
| Ammonium formate | Sigma | 70221 | |
| Acetonitrile | Fisher | A955 | |
| 4-chloro-L-phenylalanine | Carbosynth | FC13398 | |
| Taurobetamuricholic acid | Steraloids | C1899–000 | |
| Tauroursodeoxycholic acid | Sigma | 580549 | |
| Taurocholic acid | Sigma | 86339 | |
| Taurochenodeoxycholate | Sigma | T6260 | |
| Alphamuricholic acid | Steraloids | C1890–000 | |
| Betamuricholic acid | Steraloids | C1895–000 | |
| Ursodeoxycholic acid | Sigma | U5127 | |
| Cholic acid | Sigma | C1129 | |
| Chenodeoxycholic acid | Sigma | c9377 | |
| Tauroomegamuricholic acid | Steraloids | C1889–000 | |
| Taurohyodeoxycholic acid | Steraloids | C0890–000 | |
| Taurodeoxycholic acid | Sigma | T0557 | |
| Taurolithocholic acid | Sigma | T7515 | |
| 3-oxocholic acid | Steraloids | C1272–000 | |
| 7-betacholic acid | TRC | U849900 | |
| 7-oxocholic acid | Sigma | SMB00806 | |
| Gammamuricholic acid | Steraloids | C1850–000 | |
| Omegamuricholic acid | Steraloids | C1888–000 | |
| Hyodeoxycholic acid | Sigma | H3878 | |
| Murocholic acid | Steraloids | C0910–000 | |
| Deoxycholic acid | Sigma | D2510 | |
| Isodeoxycholic acid | Steraloids | C1165–000 | |
| 3-oxodeoxycholic Acid | TRC | O856870 | |
| Lithocholic acid | Sigma | L6250 | |
| 3-oxolithocholic acid | TRC | O848490 | |
| Cholic acid-2,2,4,4-d4 | Sigma | 614149 | |
| 1-acetyl-β-carboline | Aobious | CFN92122 | |
| Critical Commercial Assays | |||
| DNeasy Power Soil Kit | Qiagen | 12955–4 | |
| Illumina NextSeq Kit | Illumina | NextSeq 500/550 v2.5 | |
| Illumina NovaSeq kit | Illumina | NovaSeq 6000 S4 Reagent Kit v1.5 | |
| Pico488 dsDNA quantification reagent | Lumiprobe | 92010 | |
| Creatinine Assay Kit | Abcam | ab204537 | |
| Deposited Data | |||
| Raw metabolomics data | This study | MassIVE (ftp://massive.ucsd.edu/MSV000091763/) |
|
| Experimental Models: Organisms/Strains | |||
| Mouse: C57BL/6 GF | Taconic Biosciences | N/A | |
| Mouse: Swiss-Webster GF | Taconic Biosciences | N/A | |
| Software and Algorithms | |||
| NinjaMap | Cheng et al.16 | https://github.com/FischbachLab/ninjaMap/releases/tag/cheng_et_al | |
| Bowtie2 v. 2.3.5.1 | Langmead et al.39 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| Matlab R2022a | MathWorks | https://www.mathworks.com/products/matlab.html | |
| Prism v. 9.3.1 | GraphPad software | https://www.graphpad.com/features | |
| Morpheus | N/A | https://software.broadinstitute.org/morpheus | |
| MassHunter Qualitative Analysis Software v. 7.0 | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/qualitative-analysis | |
| MassHunter Quantitative Analysis Software v. B.09.00 | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/quantitative-analysis | |
| Other | |||
| Precellys® CK28 Hard Tissue Homogenizing Kit, Beads | VWR | 10144–494 | |
| Durapore PVDF 0.22-μm membrane | Millipore | UFC30GV00 | |
| MultiScreen Solvinert 96 Well Filter Plate | Millipore | MSRLN0410 | |
| 1.2 ml V-bottom 96-well plates | Thomas Scientific | OX1263-S | |
| 2.2 ml V-bottom 96-well deep-well plates | Thomas Scientific | OX1265-S | |
| Silicone fitted plate mat | Thomas Scientific | EK-2066 | |
| Corning 96 Well Clear Flat Bottom, Polystyrene, sterile | Corning | 3370 | |
| Vinyl Tape | Coy | 1600330w | |
| Microplate spectrophotometer | BioTeK | Epoch2 | |
| Tissue Lyser II | Qiagen | N/A | |
| ACQUITY UPLC BEH C18 Column, 130Å, 1.7 μm, 2.1 mm×100 mm | Waters | 186002352 | |
| ACQUITY UPLC BEH C18 VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 | Waters | 186003975 | |
| ACQUITY UPLC BEH Amide VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 | Waters | 186004799 | |
| Waters ACQUITY UPLC BEH Amide Column, 130Å, 1.7 μm, 2.1 mm×150 mm | Waters | 186004802 | |
| Kinetex C18 column (1.7 μm, 2.1×100 mm) | Phenomenex | N/A | |
| Agilent 1290 Infinity II UPLC | Agilent | N/A | |
| Agilent 6530 QTOF MS | Agilent | N/A | |
Data and code availability
Metagenomic sequencing datasets generated for this study have been deposited at the NCBI Sequence Read Archive and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. Metabolomics data have been deposited on the ProteomeXchange Consortium via the MassIVE database and are publicly available as of the date of publication. Accession numbers are also listed in the Key Resources Table. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code. All computational approaches and software used are described in the STAR Methods and listed Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions
All strains used in the synthetic community were obtained from American Type Culture Collection (ATCC), Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), BEI resources (BEI), and other sources as indicated in Table S1. All strains were cultured in one of two growth media: mega medium (MM) and chopped meat medium w/ rumen fluid and carbohydrates (CMM) (Table S1). Cultures were incubated at 37 °C in an anaerobic chamber (Coy Laboratories) in an atmosphere of 5% hydrogen, 10% CO2 and 85% N2. Cultures were stored in anaerobically prepared 25% glycerol/water (v/v) in 12.7 × 49 mm cryogenic vials with closures (Corning 430659) or 1.2 ml V-bottom 96-well plates (Thomas Scientific, OX1263-S) capped with a plate seal (Thomas Scientific, EK-2066) and sealed with oxygen-impervious yellow vinyl tape (Coy Laboratories, 1600330w) to ensure anoxic conditions during long-term storage. All medium and reagents used in the anaerobic chamber were pre-reduced for at least 48 h.
Synthetic community construction
Frozen stocks in a 96-well plate were thawed, and 100 μl of each thawed culture was used to inoculate 1 ml of growth medium (Table S1) in a sterile 2.2 ml 96-well plate (Thomas Scientific, OX1265-S). Strains were sub-cultured by 1:10 dilution into fresh medium daily for 2–3 days; growth was monitored by optical density at 600 nm (OD600) using a microplate spectrophotometer (BioTeK, Epoch2). For each batch of inoculum, a few strains (typically <10) had a low OD (<0.1). For these strains, additional cells were harvested and added from a 10 ml liquid culture prepared from a glycerol stock, or from single colonies scraped from solid growth medium. Finally, non-normalized cultures of all strains were pooled into a mixture. A 1 ml aliquot of the resulting mixed culture was stored at −80 °C for metagenomic sequencing. The remainder of the mixed culture was subjected to centrifugation (5000 × g, 15 min); the cell pellet was washed with an equal volume of pre-reduced sterile phosphate-buffered saline (PBS), and then resuspended in 1/10 of the initial volume of a 25% glycerol/water (v/v) solution. 1.2 ml aliquots of the resulting synthetic community were stored in 2 ml cryovials (Corning, 430659) at −80 °C until use.
Gnotobiotic mouse experiments
Germ free Swiss-Webster or C57BL/6N mice (male, 6–8 weeks of age) were originally obtained from Taconic Biosciences (Hudson, NY) and colonies were maintained in gnotobiotic isolators and fed ad libitum. The Institutional Animal Care and Use Committee (IACUC) at Stanford University approved all procedures involving animals.
Glycerol stocks of synthetic communities (~1.2 ml) were thawed and shaken well at room temperature, and mice were orally gavaged with ~200 μl of the mixed culture. To ensure efficient colonization by all strains in the community, mice were gavaged using the same procedure on three successive days for all experiments. Human stool homogenates were prepared in PBS and administered by gavage to germ-free mice using the same protocol as the synthetic community.
For experiments in which Swiss-Webster or C57BL/6N mice were fed standard chow (LabDiet 5k67), fresh fecal pellets were collected weekly at the same time of day and stored at −80 °C prior to analysis. The mice were maintained on a standard diet (LabDiet 5k67; 0.2% Trp) for 4 weeks (unless otherwise stated) before sacrifice (fed ad libitum). Mice were euthanized humanely by CO2 asphyxiation and the luminal contents of the small intestine, cecum, and colon were collected at the same time of day and stored at −80 °C until use.
METHOD DETAILS
Metagenomic sequencing
The same experimental pipeline was used for sequencing bacterial isolates and microbial communities. Bacterial cells were pelleted by centrifugation in an anaerobic environment. To estimate the absolute abundance of all strains in the communities, 100 μl of bacterial suspension containing 105 cells of Paenibacillus barengoltzii CC33–002B was added to each sample prior DNA extraction. Genomic DNA was extracted using the DNeasy PowerSoil HTP kit (Qiagen) and the quantity of extracted genomic DNA was measured in a 384-well format using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher). Sequencing libraries were generated in a 384-well format using a custom, low-volume protocol based on the Nextera XT process (Illumina). Briefly, the DNA concentration from each sample was normalized to 0.18 ng/μl using a Mantis liquid handler (Formulatrix). In cases where the concentration was below 0.18 ng/μl, the sample was not diluted further. Tagmentation, neutralization, and PCR steps of the Nextera XT process were performed on the Mosquito HTS liquid handler (TTP Labtech), creating a final volume of 4 μl per library. During the PCR amplification step, custom 12-bp dual unique indices were introduced to eliminate barcode switching, a phenomenon that occurs on Illumina sequencing platforms with patterned flow cells38. Libraries were pooled at the desired relative molar ratios and cleaned up using Ampure XP beads (Beckman) to effect buffer removal and library size selection. The cleanup process was used to remove fragments shorter than 300 bp and longer than 1.5 kb. Final library pools were quality checked for size distribution and concentration using the Fragment Analyzer (Agilent) and qPCR (BioRad). Sequencing reads were generated using the NovaSeq S4 flow cell or the NextSeq High Output kit, both in 2×150 bp configuration. 5–10 million paired-end reads were targeted for bacterial isolates and 20–30 million paired end reads for bacterial communities.
Metagenomic read mapping
Paired-end reads from each sample were aligned to the hCom1a database using Bowtie239 with maximum insert length (-maxins) set to 3000, maximum alignments (-k) set to 300, suppressed unpaired alignments (--no-mixed), suppressed discordant alignments (--no-discordant), suppressed output for unaligned reads (--no-unal), required global alignment (--end-to-end), and using the “--very-sensitive” alignment preset (command: --very-sensitive -maxinsX 3000 -k 300 --no-mixed --no-discordant --end-to-end --no-unal). The output was piped into Samtools v. 1.940, which was used to convert the alignment output from SAM output stream to BAM format and then sort and index the BAM file by coordinates. Alignments were filtered to only keep those with >99% identity for the entire length of the read.
The median percentage of unaligned reads was 4.95% (range 4.10% – 8.35%). To assess the origin of these reads, we performed a BLAST v2.11.0+ search through the ncbi/blast:latest docker image with parameters “-outfmt ‘6 std qlen slen qcovs sscinames staxids’ -dbsize 1000000, -num_alignments 100” from a representative sample against the ‘NCBI - nt’ database as on 2021-02-16. We then filtered the BLAST results to obtain the top hits for a given query. Briefly, the script defined top hits as ones that had an e-value <= 1e-30, percent identity >= 99% and were within 10 percent of the best bit score for that query. To visualize and summarize the output, we used the ktImportTaxonomy script from the Krona package with default parameters. Reads were aggregated by NCBI taxon id and separately by genus. We found that most of the hits are from taxa that are closely related to the organisms in our community, while others are from the mouse genome. We conclude that our experiments did not suffer from any appreciable level of contamination.
Sample preparation for LC/MS
For mouse fecal samples (~40 mg) or cecal contents and liver (~80 mg), wet tissues were pre-weighed into a 2 ml screw top tube containing six 6 mm ceramic beads (Precellys® CK28 Lysing Kit). 600 μL (for fecal samples) or 1 ml (for cecal contents and liver) of a mixture of ice-cold acetonitrile, methanol, and water (4/4/2, v/v/v) was then added to each tube and samples were homogenized by vigorous shaking using a QIAGEN Tissue Lyser II at 25/s for 10 min. The resulting homogenates were subjected to centrifugation for 15 min at 4 °C at 18,000 × g. 100 μl of the supernatant was then combined with 100 μl of aqueous solution of internal standards (2 μM d4-cholic acid and 20 μM 4-chloro-L-phenylalanine). The resulting mixtures were then filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), or MultiScreen Solvinert 96 Well Filter Plate (Millipore, MSRLN0410), and 5 μl was injected into the LC/MS.
For mouse urine samples, 5 μl of a urine sample was diluted 10-fold with ddH2O and then mixed with 50 μl of an aqueous internal standard solution (20 μM 4-chloro-L-phenylalanine). After centrifugation for 15 min at 4 °C at 18,000 × g, 50 μl of the resulting mixture was used for quantification of creatinine using a Creatinine Assay Kit (ab204537) as described in the manufacturer’s protocol. The remaining 50 μl of each sample was filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), and 5 μl was injected into the LC/MS.
For mouse serum or plasma samples, protein was precipitated by mixing blood samples (50 μl) with an equal volume of 6% aqueous sulfosalicylic acid, followed by incubation at room temperature for 5 min and clarification by centrifugation (18,000 × g, 10 min, room temperature). Subsequently, 50 μl of the supernatant was combined with 50 μl of aqueous solution of internal standards (2 μM d4-cholic acid and 20 μM 4-chloro-L-phenylalanine). The resulting mixtures were then filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), and 5 μl was injected into the LC/MS.
For bacterial (community) cultures, 200 μl of each sample was transferred into individual 1.5 ml microtubes or 1.2 ml 96 well plates and then centrifuged for 15 min at 4 °C at 18,000 × g or 3,700 × g. Subsequently, 100 μl of the supernatant was combined with 100 μL of an aqueous solution of the internal standard (20 μM 4-chloro-L-phenylalanine). The resulting mixtures were filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), or a MultiScreen Solvinert 96 Well Filter Plate (Millipore, MSRLN0410), and 5 μl was injected into the LC/MS.
Liquid chromatography/mass spectrometry (LC/MS)
Bile acids: compounds were separated using an Agilent 1290 Infinity II UPLC equipped with a Kinetex C18 column (1.7 μm, 2.1 × 100 mm, Phenomenex) and detected using an Agilent 6530 Q-TOF equipped with a dual Agilent jet stream electrospray ionization (AJS-ESI) source operating under extended dynamic range (EDR 1700 m/z) in negative ionization mode. The parameters of the AJS-ESI source were as follows: gas temp: 300 °C; drying gas: 7.0 l/min; nebulizer: 40 psig; sheath gas temp: 350 °C; sheath gas flow: 10.0 l/min; VCap: 3500 V; nozzle voltage: 1400V; and fragmenter: 200 V. Mobile phase A was 0.05% formic acid in H2O, and mobile phase B was 0.05% formic acid in acetone. 5 μl of each sample was injected via autosampler into mobile phase and chromatographic separation was carried out at a flow rate of 0.35 ml/min with a 32-min gradient condition (t = 0 min, 25% B; t = 1 min, 25% B; t = 25 min, 75% B, t = 26 min, 100% B, t = 30 min, 100% B, t = 32 min, 25% B).
(Aromatic) amino acid metabolites: compounds were separated using an Agilent 1290 Infinity II UPLC equipped with an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 mm × 150 mm, Waters) and detected using an Agilent 6530 Q-TOF equipped with a standard atmospheric-pressure chemical ionization (APCI) source or dual Agilent jet stream electrospray ionization (AJS-ESI) source operating under extended dynamic range (EDR 1700 m/z) in negative ionization mode. For the APCI source the parameters were as follows: gas temp: 350 °C; vaporizer: 350 °C; drying gas: 6.0 l/min; nebulizer: 60 psig; VCap: 3500 V; corona: 20 μA; and fragmenter: 135 V. For the AJS-ESI source the parameters were as follows: gas temp: 350 °C; drying gas: 10.0 l/min; nebulizer: 40 psig; sheath gas temp: 300 °C; sheath gas flow: 11.0 l/min; VCap: 3500 V; nozzle voltage: 1400V; and fragmenter: 130 V. Mobile phase A was 6.5 mM ammonium bicarbonate in H2O, and B was 6.5 mM ammonium bicarbonate in 95 % MeOH/H2O. 5 μl of each sample was injected via autosampler into mobile phase and chromatographic separation was carried out at a flow rate of 0.35 mL/min with a 10-min gradient condition (t = 0 min, 0.5% B; t = 4 min, 70% B; t = 4.5 min, 98% B; t = 5.4 min, 98% B; t = 5.6 min, 0.5% B).
Online mass calibration was performed using a second ionization source and a constant flow (5 μl/min) of reference solution (119.0363 and 966.0007 m/z). The MassHunter Quantitative Analysis Software (Agilent, version B.09.00) was used for peak integration based on retention time (tolerance of 0.2 min) and accurate m/z (tolerance of 30 ppm) of chemical standards. Quantification was based on a 2-fold dilution series of chemical standards spanning 0.098 to 200 μM (for AAA metabolites) or 0.001 to 200 μM (for bile acids) and measured amounts were normalized by weights of extracted tissue samples (pmol/mg wet tissue) or creatinine level in the urine sample (μM/mM creatinine). The linear quantification range and lower limit of detection for all metabolites are listed in Table S1. The MassHunter Qualitative Analysis Software (Agilent, version 7.0) was used for targeted feature extraction, allowing mass tolerances of 30 ppm.
Bile acids inhibition assay
D. longicatena and C. sporogenes were streaked from a glycerol stock onto Columbia Agar with 5 % Sheep Blood (BD 221165) and incubated for ~ 24 h at 37 °C. Individual colonies were picked and used to inoculate 3 ml of Mega medium and cultured for 24 h at 37 °C. Cells were diluted 100-fold into fresh Mega medium supplemented with various concentration of CA or DCA (final concentration raging from 1.25 mM, 625 μM, 312 μM, 156 μM, 78 μM, to 39 μM) or DMSO blank control. The starting OD600 for each culture was 0.1. 200 μl aliquots of each culture were taken and monitored for growth using an Epoch 2 microplate reader (Biotek) in an anaerobic chamber every 15 min until stationary phase was reached. Bacterial growth curves were performed in triplicate with each biological replicate derived from a single isolated colony. Growth curves were plotted using GraphPad.
1-Acetyl-β-carboline inhibition assay
C. sporogenes was streaked from a glycerol stock onto Columbia Agar with 5% Sheep Blood (BD 221165) and incubated for ~24 h at 37 °C. Individual colonies were picked and used to inoculate 3 ml of Mega medium and cultured for 24 h at 37 °C. Cells were diluted 100-fold into fresh Mega medium supplemented with various concentration of AbC or DCA (final concentration raging from 500 μM, 100 μM, 50 μM, 25 μM, to 5 μM) or DMSO blank control. The starting OD600 for each culture was ~0.01 after background subtraction. 200 μl aliquots of each culture were sampled to monitor growth using an Epoch 2 microplate reader (Biotek) in an anaerobic chamber every 30 min until stationary phase was reached. Bacterial growth curves were performed in one biological replicate. Growth curves were plotted using GraphPad.
C. scindens and C. hylemonae spent medium assay
Glycerol stocks of C. scindens and C. hylemonae were used to inoculate 3 ml of Mega medium and cultured for ~24 h at 37 °C. Cells were diluted 100-fold into 7 ml of Peptone Yeast Extract Broth with Fructose (PYF medium, Anaerobe system) and incubated for 24 h at 37 °C. After centrifugation (5000 rpm, 10 min), bacterial supernatants were filtered through a 0.22 μm nylon membrane to remove cell material. The resulting spent medium was extracted 2x with ethyl acetate (EA); the ethyl acetate layer was evaporated to yield the extract, while the aqueous layer was lyophilized to generate the lyophilizate. Finally, we inoculated an overnight culture of C. sporogenes into fresh PYF medium or fresh PYF medium supplemented with either the extract or lyophilizate of C. scindens or C. hylemonae. The growth of C. sporogenes was monitored using an Epoch 2 microplate reader (Biotek) in an anaerobic chamber every 30 min until stationary phase was reached. Bacterial growth curves were performed in three biological replicates. Growth curves were plotted using GraphPad.
In vitro profiling of amino acids depletion by hCom1a strains
Glycerol stocks of each strain in hCom1a were used to inoculate 3 ml of rich medium (Mega medium or modified Chopped meat medium); these cultures were incubated 1–4 days at 37 °C. After reaching stationary phase, bacterial cells were harvested by centrifugation (5,000 × g, 10 min), washed twice with an equal volume of pre-reduced sterile phosphate-buffered saline (PBS), and resuspended in 0.75 ml minimal medium with 20 amino acids (modified SAAC medium, see Table S1). The resulting cell suspensions were incubated at 37 °C for another 4 d. All bacterial cultures were stored at −80 °C prior to LC-MS analysis. These samples were used to profile amino acids, 3-phenylpropionic acid, 3-(4-hydrophenyl) propionic acid, and 3-indolepropionic acid.
QUANTIFICATION AND STATISTICAL ANALYSIS
Relative abundances were calculated from the raw output of NinjaMap-processed metagenomic data without rarefying the total number of reads across samples. After setting undetected bins to a minimum value of 10−8, all relative abundances were further transformed by log10. To evaluate the reproducibility of in vivo colonization, Pearson’s correlation coefficients (R) were calculated for the community structure between each two experiments after averaging the relative abundance of each strain across the 3–5 mice that were co-housed in the same cage/experiment. To find the potential strains significantly impacted by strain(s) dropout, multiple unpaired t test with FDR correction was performed for each strain in all mice between two groups. The statistical parameters were set as default with individual variance computed for each comparison followed by multiple comparison of false discovery rate (FDR) with two-stage step-up (Benjamini, K Krieger, and Yekutieli) and Q=0.1%. Dot plots showing relative abundance data were plotted with a conservative lower threshold of 1 × 10−6. Further details of statistical analyses can be found in the corresponding figure legends. All statistical analysis and plotting were performed in MATLAB or Prism.
For metabolomic data, metabolite concentration differences between experimental groups or conditions were evaluated using unpaired two-tailed Students’ t test for pairwise comparison, one-way ANOVA for multiple comparisons. All statistical analysis and plotting were performed in Prism.
Supplementary Material
Figure S1: Phylogenetic tree of hCom1a, the 107-member gut bacterial community used in this study, related to Figure 1. The phylogenetic tree was constructed based on a multiple sequence alignment of conserved single-copy genes.
Figure S2: Relative and absolute abundances of select strains in cecal contents from hCom1a-, ΔCs-, ΔCh-, and ΔChΔCs-colonized mice, related to Figures 3 and 5. (A) (i) Relative abundances of C. scindens and C. hylemonae. (ii) Total relative abundance of C. scindens and C. hylemonae. (iii) Relative abundances of the eight interacting strains discovered in the analysis shown in Figure 3. (B) (i) Absolute abundances of C. scindens and C. hylemonae. (ii) Total absolute abundance of C. scindens and C. hylemonae. (iii) Absolute abundances of the eight interacting strains discovered in the analysis shown in Figure 3. (iv) Cell density in cecal contents of all four groups, estimated by metagenomics with a spike-in control strain. n=3 mice per group; ND: not detected (or less than 1e-6 for A or 100 cells/mg wet cecal contents for B).
Figure S3: Targeted profiling of bile acids (cecum) and aromatic amino acid metabolites (cecum and urine) by LC-MS, related to Figures 4 and 7. (A) Germ-free mice colonized by hCom1a or the single-strain dropout communities (ΔCs and ΔCh) have similar cecal bile acid pools. (B) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their cecal contents, while ΔCs-colonized mice are skewed toward reduced aromatic amino acid metabolites (PPA and 4-HO-PPA ↑, PAA and p-cresol ↓). (C) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their urine, while ΔCs-colonized mice displayed an altered profile that is skewed toward products of amino acid reduction (hippuric acid and cinnamoylglycine ↑, phenylacetylglycine ↓). (D) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their serum, while ΔCs-colonized mice displayed an altered profile that is skewed toward products of amino acid reduction (hippuric acid and cinnamoylglycine ↑, phenylacetylglycine ↓). n=3 mice per group. Statistical significance was assessed using a Student’s two tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001; n.s.: no significance).
Figure S4: Growth curves of Dorea longicatena and Clostridium sporogenes in the presence of various concentrations of cholic acid (CA), deoxycholic acid (DCA), or 1-acetyl-β-carboline (AbC), related to Figure 6. (A) The growth of Dorea longicatena is inhibited by DCA at the physiological concentration of 625 μM, while Clostridium sporogenes is only partially inhibited. (B) Neither Dorea longicatena nor Clostridium sporogenes is affected by various concentrations of CA. (C) The growth of Clostridium sporogenes (Cspo) is not inhibited by various concentrations of DCA and 1-acetyl-β-carboline (AbC).
Figure S5: A strain swap in the 7α-dehydroxylation niche (Cs vs Ch) has a large impact on community structure and metabolic output, related to Figures 4, 5 and 7. (A) While most strains remain unchanged between the ΔCs and ΔCh dropouts, a small number of strains change in relative abundance >100-fold. Left: Metagenomic analysis showing that Cs and Ch are swapped in the ΔCh and ΔCs community inocula, respectively. Middle: The swap between Cs and Ch impacts the relative abundance of four strains in the ΔCs and ΔCh communities. Each dot is an individual strain; the collection of dots in a column represents the community averaged over 3 mice co-housed in one cage. Cs and Ch are highlighted in red and green. Strains colored blue went up or down in relative abundance between ΔCh and ΔCs-colonized mice (FDR<0.01, fold change >100). Right: Volcano plot showing the log10(relative abundance) values for each strain; for strains that were not detected, relative abundances were set at 10−8. Strains with significantly different relative abundance (FDR<0.01, fold change >100) are colored blue; the full names of these strains are shown in Figure 5D and S3. (B) Schematic showing the cascading effects of a single-strain swap within the 7α-dehydroxylation niche: the relative abundance of Cspo increased from undetectable to ~10−4, which increased the abundance of hippurate (~16 fold) and cinnamoylglycine (~37 fold), which decreased the abundance of phenylacetate (~2 fold) and phenylacetylglycine (~6 fold).
Figure S6: C. sporogenes is responsible for altering the phenylalanine metabolite profile, related to Figures 6 and 7. (A) Aromatic amino acid metabolite production by each of the 107 strains in hCom1a in vitro. C. sporogenes (Cspo) is the only strain in hCom1a that can produce 3-phenylpropionic acid, 3-(4-hydrophenyl)propionic acid, and 3-indolepropionic acid, the reductive metabolites of phenylalanine, tyrosine, and tryptophan, respectively. The in vitro screen was performed using the same assay described in Figure 6C. (B) Schematic of the in vivo dropout experiment. Germ-free C57BL/6 mice (n=3 each group) were colonized with hCom1a, ΔCs or ΔCsΔCspo and housed for 10 days before sacrifice. Fecal pellets and intestinal contents were subjected to metagenomic analysis or targeted metabolite profiling. (C-D) Cs and Cspo are undetectable in ΔCs- and ΔCsΔCspo-colonized mice, respectively. Relative abundances were calculated through a high-resolution metagenomic analysis of the inoculum and fecal communities. (E) Removal of Cspo from ΔCs (i.e., ΔCsΔCspo) restores a metabolite profile comparable to that of hCom1a-colonized mice. In the new study, the change in phenylalanine metabolites in ΔCs mice is weaker than that in Figure 7, which is consistent with the smaller impact on arginine consumers (Ro alone decreases significantly but Rb is largely unchanged in ΔCs mice, as shown in panel D). Statistical significance was assessed using a Student’s two-tailed t-test (*: p<0.05).
Figure S7: Profiling bile acids in hCom1a and human stool-colonized C57BL/6N mice, related to Figure 1. (A) Schematic of the experiment. Three biological replicates of an hCom1a inoculum and two human stool samples were used to colonize germ-free C57BL/6N mice by gavage (n=3–5 mice per group). Conventional mice of a similar age were used as controls (n=5 mice). After 4 weeks of colonization, mice were sacrificed and fecal pellets and cecal contents were subjected to LC/MS analysis. (B) hCom1a-colonized mice (hCom1a_1, hCom1a_2, and hCom1a_3) and humanized mice (Hum1 and Hum2) have broadly similar fecal bile acid pools. (C) hCom1a-colonized mice (hCom1a_1, hCom1a_2, and hCom1a_3) and humanized mice (Hum1 and Hum2) have broadly similar cecal bile acid pools. (D) hCom1a-colonized mice (n=13 total) produce a smaller quantity of the secondary bile acids DCA and LCA than humanized mice (n=10 total) and conventional mice (n=5) in both cecum and feces. Statistical significance was assessed using a Student’s two-tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
Table S1: Strains, media, and metabolites used in this study, related to STAR Methods
Table S2: Metagenomic and metabolomic data of reproducibility study for hCom1a-colonized mice, related to Figures 1 and S14
Table S3: Metagenomic data of strain dropout study on the Chow diet, related to Figures 3, 4, 5 and 7
Table S4: Metabolomic data of strain dropout study on the Chow diet, related to Figures 3, 4 and 7
HIGHLIGHTS.
Construction of variants of a complex community missing two strains in a metabolic niche In the ΔCsΔCh community, eight strains change in relative abundance by >100-fold Clostridium sporogenes increases >1000-fold in the ΔCs but not ΔCh dropout A strain swap can ripple through the community in an unpredictable manner
ACKNOWLEDGMENTS
We are deeply indebted to members of the Fischbach, Brown, and Hazen labs for helpful discussions. We are grateful to Mohamed Donia (Princeton) for helpful discussions regarding data analysis, and to Brian Yu (Chan Zuckerberg Biohub) for help with figure preparation. This work was supported by the Human Frontier Science Program LT000493/2018-L (K.N.), a Fellowship from the Astellas Foundation for Research on Metabolic Disorders (K.N.), a research grant from Kanae Fundation for the Promotion of Medical Science (K.N.), the Stanford Microbiome Therapies Initiative (M.A.F.), NIH grants DP1 DK113598 (to M.A.F.), P01 HL147823 (to M.A.F. and J.M.B.), R01 DK101674 (to M.A.F.), P50 AA024333 (J.M.B.), R01 DK130227 (J.M.B.), NSF grant EF-2125383 (M.A.F.) the Bill and Melinda Gates Foundation (to M.A.F.), an HHMI-Simons Faculty Scholars Award (M.A.F.), the Leducq Foundation (M.A.F.), the Leona M. and Harry B. Helmsley Charitable Trust (M.A.F.); and MAC3 Impact Philanthropies (M.A.F.). M.A.F. is a Chan Zuckerberg Biohub Investigator.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Stanford University and the Chan Zuckerberg Biohub have patents pending for microbiome technologies on which the authors are co-inventors. M.A.F. is a co-founder and director of Federation Bio and Kelonia, a co-founder of Revolution Medicines, an Innovation Partner at the Column Group, and a member of the scientific advisory board of NGM Bio. All of the other authors have no competing interests.
REFERENCES
- 1.Samuel BS, and Gordon JI (2006). A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc. Natl. Acad. Sci 103, 10011–10016. 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci 106, 5859–5864. 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, and Gordon JI (2010). Dissecting the in Vivo Metabolic Potential of Two Human Gut Acetogens. J. Biol. Chem 285, 22082–22090. 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2014). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schürch CM, McCoy KD, Kuehne SA, Minton NP, et al. (2016). Functional Intestinal Bile Acid 7α-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front. Cell. Infect. Microbiol 6, 1–15. 10.3389/fcimb.2016.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marion S, Studer N, Desharnais L, Menin L, Escrig S, Meibom A, Hapfelmeier S, and Bernier-Latmani R (2019). In vitro and in vivo characterization of Clostridium scindens bile acid transformations. Gut Microbes 10, 481–503. 10.1080/19490976.2018.1549420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patnode ML, Beller ZW, Han ND, Cheng J, Peters SL, Terrapon N, Henrissat B, Le Gall S, Saulnier L, Hayashi DK, et al. (2019). Interspecies Competition Impacts Targeted Manipulation of Human Gut Bacteria by Fiber-Derived Glycans. Cell 179, 59–73.e13. 10.1016/j.cell.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marion S, Desharnais L, Studer N, Dong Y, Notter MD, Poudel S, Menin L, Janowczyk A, Hettich RL, Hapfelmeier S, et al. (2020). Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res 61, 1450–1463. 10.1194/jlr.RA120001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridlon JM, Devendran S, Alves JM, Doden H, Wolf PG, Pereira GV, Ly L, Volland A, Takei H, Nittono H, et al. (2020). The ‘in vivo lifestyle’ of bile acid 7α-dehydroxylating bacteria: comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes 11, 381–404. 10.1080/19490976.2019.1618173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Streidl T, Karkossa I, Segura Muñoz RR, Eberl C, Zaufel A, Plagge J, Schmaltz R, Schubert K, Basic M, Schneider KM, et al. (2021). The gut bacterium Extibacter muris produces secondary bile acids and influences liver physiology in gnotobiotic mice. Gut Microbes 13, 1–21. 10.1080/19490976.2020.1854008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louca S, Polz MF, Mazel F, Albright MBN, Huber JA, O’Connor MI, Ackermann M, Hahn AS, Srivastava DS, Crowe SA, et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol 2, 936–943. 10.1038/s41559-018-0519-1. [DOI] [PubMed] [Google Scholar]
- 12.Tian L, Wang X-W, Wu A-K, Fan Y, Friedman J, Dahlin A, Waldor MK, Weinstock GM, Weiss ST, and Liu Y-Y (2020). Deciphering functional redundancy in the human microbiome. Nat. Commun 11, 6217. 10.1038/s41467-020-19940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, Guo C, Higginbottom S, Almo SC, and Fischbach MA (2020). A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570. 10.1038/s41586-020-2396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ridlon JM, Harris SC, Bhowmik S, Kang D-J, and Hylemon PB (2016). Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7, 22–39. 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridlon JM, Kang D-J, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259. 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Cheng AG, Ho P, Aranda-Díaz A, Jain S, Yu FB, Meng X, Wang M, Iakiviak M, Nagashima K, Zhao A, et al. (2022). Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 185, 3617–3636.e19. 10.1016/j.cell.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kraal L, Abubucker S, Kota K, Fischbach MA, and Mitreva M (2014). The Prevalence of Species and Strains in the Human Microbiome: A Resource for Experimental Efforts. PLoS One 9, e97279. 10.1371/journal.pone.0097279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin W-B, Li T-T, Huo D, Qu S, Li XV, Arifuzzaman M, Lima SF, Shi H-Q, Wang A, Putzel GG, et al. (2022). Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell 185, 547–562.e22. 10.1016/j.cell.2021.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wootton JT (2010). Experimental species removal alters ecological dynamics in a natural ecosystem. Ecology 91, 42–48. 10.1890/08-1868.1. [DOI] [PubMed] [Google Scholar]
- 20.Díaz S, Symstad AJ, Stuart Chapin F, Wardle DA, and Huenneke LF (2003). Functional diversity revealed by removal experiments. Trends Ecol. Evol 18, 140–146. 10.1016/S0169-5347(03)00007-7. [DOI] [Google Scholar]
- 21.Marion S, Desharnais L, Studer N, Dong Y, Notter MD, Poudel S, Menin L, Janowczyk A, Hettich RL, Hapfelmeier S, et al. (2020). Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res 61, 1450–1463. 10.1194/jlr.RA120001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam KN, Spanogiannopoulos P, Soto-Perez P, Alexander M, Nalley MJ, Bisanz JE, Nayak RR, Weakley AM, Yu FB, and Turnbaugh PJ (2021). Phage-delivered CRISPR-Cas9 for strain-specific depletion and genomic deletions in the gut microbiome. Cell Rep. 37, 109930. 10.1016/j.celrep.2021.109930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang JD, Myers CJ, Harris SC, Kakiyama G, Lee I-K, Yun B-S, Matsuzaki K, Furukawa M, Min H-K, Bajaj JS, et al. (2019). Bile Acid 7α-Dehydroxylating Gut Bacteria Secrete Antibiotics that Inhibit Clostridium difficile: Role of Secondary Bile Acids. Cell Chem. Biol 26, 27–34.e4. 10.1016/j.chembiol.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. 10.1038/nature24661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo C, Allen BM, Hiam KJ, Dodd D, Van Treuren W, Higginbottom S, Nagashima K, Fischer CR, Sonnenburg JL, Spitzer MH, et al. (2019). Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science. 366, eaav1282. 10.1126/science.aav1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Chen H, Van Treuren W, Hou B, Higginbottom SK, and Dodd D (2022). Clostridium sporogenes uses reductive Stickland metabolism in the gut to generate ATP and produce circulating metabolites. Nat. Microbiol 7, 695–706. 10.1038/s41564-022-01109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han S, Van Treuren W, Fischer CR, Merrill BD, DeFelice BC, Sanchez JM, Higginbottom SK, Guthrie L, Fall LA, Dodd D, et al. (2021). A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 595, 415–420. 10.1038/s41586-021-03707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, Cajka T, Mohan ML, Li L, Wu Y, et al. (2020). A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 180, 862–877.e22. 10.1016/j.cell.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoskins JA, Holliday SB, and Greenway AM (1984). The metabolism of cinnamic acid by healthy and phenylketonuric adults: A kinetic study. Biol. Mass Spectrom 11, 296–300. 10.1002/bms.1200110609. [DOI] [PubMed] [Google Scholar]
- 30.Lees HJ, Swann JR, Wilson ID, Nicholson JK, and Holmes E (2013). Hippurate: The natural history of a mammalian-microbial cometabolite. J. Proteome Res 12, 1527–1546. 10.1021/pr300900b. [DOI] [PubMed] [Google Scholar]
- 31.Medlock GL, Carey MA, McDuffie DG, Mundy MB, Giallourou N, Swann JR, Kolling GL, and Papin JA (2018). Inferring Metabolic Mechanisms of Interaction within a Defined Gut Microbiota. Cell Syst. 7, 245–257.e7. 10.1016/j.cels.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venturelli OS, Carr AV, Fisher G, Hsu RH, Lau R, Bowen BP, Hromada S, Northen T, and Arkin AP (2018). Deciphering microbial interactions in synthetic human gut microbiome communities. Mol. Syst. Biol 14, 1–19. 10.15252/msb.20178157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Gorostiaga A, Bajić D, Osborne ML, Poyatos JF, and Sanchez A (2019). High-order interactions distort the functional landscape of microbial consortia. PLOS Biol. 17, e3000550. 10.1371/journal.pbio.3000550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutiérrez N, and Garrido D (2019). Species Deletions from Microbiome Consortia Reveal Key Metabolic Interactions between Gut Microbes. mSystems 4, 1–16. 10.1128/mSystems.00185-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bairey E, Kelsic ED, and Kishony R (2016). High-order species interactions shape ecosystem diversity. Nat. Commun 7, 12285. 10.1038/ncomms12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.García-Bayona L, and Comstock LE (2019). Streamlined Genetic Manipulation of Diverse Bacteroides and Parabacteroides Isolates from the Human Gut Microbiota. MBio 10. 10.1128/mBio.01762-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischbach MA (2018). Microbiome: Focus on Causation and Mechanism. Cell 174, 785–790. 10.1016/j.cell.2018.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, Wei E, Chan CKF, Nabhan AN, Su T, Morganti RM, et al. (2017). Index Switching Causes “Spreading-Of-Signal” Among Multiplexed Samples In Illumina HiSeq 4000 DNA Sequencing. bioRxiv, 125724. 10.1101/125724. [DOI] [Google Scholar]
- 39.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup, 1000 Genome Project Data Processing (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Phylogenetic tree of hCom1a, the 107-member gut bacterial community used in this study, related to Figure 1. The phylogenetic tree was constructed based on a multiple sequence alignment of conserved single-copy genes.
Figure S2: Relative and absolute abundances of select strains in cecal contents from hCom1a-, ΔCs-, ΔCh-, and ΔChΔCs-colonized mice, related to Figures 3 and 5. (A) (i) Relative abundances of C. scindens and C. hylemonae. (ii) Total relative abundance of C. scindens and C. hylemonae. (iii) Relative abundances of the eight interacting strains discovered in the analysis shown in Figure 3. (B) (i) Absolute abundances of C. scindens and C. hylemonae. (ii) Total absolute abundance of C. scindens and C. hylemonae. (iii) Absolute abundances of the eight interacting strains discovered in the analysis shown in Figure 3. (iv) Cell density in cecal contents of all four groups, estimated by metagenomics with a spike-in control strain. n=3 mice per group; ND: not detected (or less than 1e-6 for A or 100 cells/mg wet cecal contents for B).
Figure S3: Targeted profiling of bile acids (cecum) and aromatic amino acid metabolites (cecum and urine) by LC-MS, related to Figures 4 and 7. (A) Germ-free mice colonized by hCom1a or the single-strain dropout communities (ΔCs and ΔCh) have similar cecal bile acid pools. (B) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their cecal contents, while ΔCs-colonized mice are skewed toward reduced aromatic amino acid metabolites (PPA and 4-HO-PPA ↑, PAA and p-cresol ↓). (C) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their urine, while ΔCs-colonized mice displayed an altered profile that is skewed toward products of amino acid reduction (hippuric acid and cinnamoylglycine ↑, phenylacetylglycine ↓). (D) hCom1a- and ΔCh-colonized mice have similar aromatic amino acid metabolites in their serum, while ΔCs-colonized mice displayed an altered profile that is skewed toward products of amino acid reduction (hippuric acid and cinnamoylglycine ↑, phenylacetylglycine ↓). n=3 mice per group. Statistical significance was assessed using a Student’s two tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001; n.s.: no significance).
Figure S4: Growth curves of Dorea longicatena and Clostridium sporogenes in the presence of various concentrations of cholic acid (CA), deoxycholic acid (DCA), or 1-acetyl-β-carboline (AbC), related to Figure 6. (A) The growth of Dorea longicatena is inhibited by DCA at the physiological concentration of 625 μM, while Clostridium sporogenes is only partially inhibited. (B) Neither Dorea longicatena nor Clostridium sporogenes is affected by various concentrations of CA. (C) The growth of Clostridium sporogenes (Cspo) is not inhibited by various concentrations of DCA and 1-acetyl-β-carboline (AbC).
Figure S5: A strain swap in the 7α-dehydroxylation niche (Cs vs Ch) has a large impact on community structure and metabolic output, related to Figures 4, 5 and 7. (A) While most strains remain unchanged between the ΔCs and ΔCh dropouts, a small number of strains change in relative abundance >100-fold. Left: Metagenomic analysis showing that Cs and Ch are swapped in the ΔCh and ΔCs community inocula, respectively. Middle: The swap between Cs and Ch impacts the relative abundance of four strains in the ΔCs and ΔCh communities. Each dot is an individual strain; the collection of dots in a column represents the community averaged over 3 mice co-housed in one cage. Cs and Ch are highlighted in red and green. Strains colored blue went up or down in relative abundance between ΔCh and ΔCs-colonized mice (FDR<0.01, fold change >100). Right: Volcano plot showing the log10(relative abundance) values for each strain; for strains that were not detected, relative abundances were set at 10−8. Strains with significantly different relative abundance (FDR<0.01, fold change >100) are colored blue; the full names of these strains are shown in Figure 5D and S3. (B) Schematic showing the cascading effects of a single-strain swap within the 7α-dehydroxylation niche: the relative abundance of Cspo increased from undetectable to ~10−4, which increased the abundance of hippurate (~16 fold) and cinnamoylglycine (~37 fold), which decreased the abundance of phenylacetate (~2 fold) and phenylacetylglycine (~6 fold).
Figure S6: C. sporogenes is responsible for altering the phenylalanine metabolite profile, related to Figures 6 and 7. (A) Aromatic amino acid metabolite production by each of the 107 strains in hCom1a in vitro. C. sporogenes (Cspo) is the only strain in hCom1a that can produce 3-phenylpropionic acid, 3-(4-hydrophenyl)propionic acid, and 3-indolepropionic acid, the reductive metabolites of phenylalanine, tyrosine, and tryptophan, respectively. The in vitro screen was performed using the same assay described in Figure 6C. (B) Schematic of the in vivo dropout experiment. Germ-free C57BL/6 mice (n=3 each group) were colonized with hCom1a, ΔCs or ΔCsΔCspo and housed for 10 days before sacrifice. Fecal pellets and intestinal contents were subjected to metagenomic analysis or targeted metabolite profiling. (C-D) Cs and Cspo are undetectable in ΔCs- and ΔCsΔCspo-colonized mice, respectively. Relative abundances were calculated through a high-resolution metagenomic analysis of the inoculum and fecal communities. (E) Removal of Cspo from ΔCs (i.e., ΔCsΔCspo) restores a metabolite profile comparable to that of hCom1a-colonized mice. In the new study, the change in phenylalanine metabolites in ΔCs mice is weaker than that in Figure 7, which is consistent with the smaller impact on arginine consumers (Ro alone decreases significantly but Rb is largely unchanged in ΔCs mice, as shown in panel D). Statistical significance was assessed using a Student’s two-tailed t-test (*: p<0.05).
Figure S7: Profiling bile acids in hCom1a and human stool-colonized C57BL/6N mice, related to Figure 1. (A) Schematic of the experiment. Three biological replicates of an hCom1a inoculum and two human stool samples were used to colonize germ-free C57BL/6N mice by gavage (n=3–5 mice per group). Conventional mice of a similar age were used as controls (n=5 mice). After 4 weeks of colonization, mice were sacrificed and fecal pellets and cecal contents were subjected to LC/MS analysis. (B) hCom1a-colonized mice (hCom1a_1, hCom1a_2, and hCom1a_3) and humanized mice (Hum1 and Hum2) have broadly similar fecal bile acid pools. (C) hCom1a-colonized mice (hCom1a_1, hCom1a_2, and hCom1a_3) and humanized mice (Hum1 and Hum2) have broadly similar cecal bile acid pools. (D) hCom1a-colonized mice (n=13 total) produce a smaller quantity of the secondary bile acids DCA and LCA than humanized mice (n=10 total) and conventional mice (n=5) in both cecum and feces. Statistical significance was assessed using a Student’s two-tailed t-test (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
Table S1: Strains, media, and metabolites used in this study, related to STAR Methods
Table S2: Metagenomic and metabolomic data of reproducibility study for hCom1a-colonized mice, related to Figures 1 and S14
Table S3: Metagenomic data of strain dropout study on the Chow diet, related to Figures 3, 4, 5 and 7
Table S4: Metabolomic data of strain dropout study on the Chow diet, related to Figures 3, 4 and 7
Data Availability Statement
Metagenomic sequencing datasets generated for this study have been deposited at the NCBI Sequence Read Archive and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. Metabolomics data have been deposited on the ProteomeXchange Consortium via the MassIVE database and are publicly available as of the date of publication. Accession numbers are also listed in the Key Resources Table. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code. All computational approaches and software used are described in the STAR Methods and listed Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.