

For cells to initiate and sustain a differentiated state, it is necessary that a “memory” of this state is transmitted through mitosis to the daughter cells1–3. Mammalian SWItch/ Sucrose Non- Fermentable (SWI/SNF) complexes, also called Brg1/ Brg- associated factors (BAF), control cell identity by modulating chromatin architecture to regulate gene expression4–7, but whether they participate in cell fate memory is unclear. Here, we provide evidence that subunits of SWI/SNF act as mitotic bookmarks to safeguard cell identity during cell division. The SWI/SNF core subunits SMARCE1 and SMARCB1 are displaced from enhancers but bound on promoters during mitosis and we show that this binding is required for appropriate reactivation of bound genes after mitotic exit. Ablation of SMARCE1 during a single mitosis in mouse embryonic stem cells is sufficient to disrupt gene expression, impair the occupancy of several established bookmarks at a subset of their targets, and cause aberrant neural differentiation. Thus, SWI/SNF subunit SMARCE1 plays a mitotic bookmarking role and is essential for heritable epigenetic fidelity during transcriptional reprogramming.
Perturbation of SWI/SNF is a frequent cause of disease: ~20% of cancers contain mutation of a SWI/SNF subunit and mutation of SWI/SNF subunits has also been identified as the basis for several types of neurodevelopmental disease4. With respect to mechanistic understanding of SWI/SNF function, these complexes have been shown to serve roles in the control of lineage specification by modulating chromatin accessibility at enhancers and promoters. During mitosis, there are substantial alterations in the nuclear milieu that present a challenge for maintaining cell-type-specific transcription programs8–11. The ATPase subunits of SWI/SNF complexes have been shown to be evicted from chromatin during cell division12,13, leading to the assumption that SWI/SNF has no role in mitosis. However, this presents a conundrum as to how SWI/SNF activity results in somatically heritable phenotypes that underlie fate commitment and, when perturbed, disease.
To evaluate the binding of SWI/SNF complexes on chromatin across the cell cycle, we synchronized mouse embryonic stem cells (ES cells) at various cell cycle stages (Extended Data Fig. 1a). We then performed biochemical fractionation to analyze the chromatin-bound fraction in asynchronous, S phase, G2 phase, and mitotic cells using immunoblotting to detect SWI/SNF subunits (Extended Data Fig. 1b). Mitotic synchronization was also monitored via phosphorylation of histone H3 Serine 10 (S10P H3), a mark enriched in mitosis14 (Fig. 1a and Extended Data Fig. 1c), and analysis of mitotic DNA indices (Extended Data Fig.1d, e). SOX2, which binds mitotic chromatin15–18, was retained on mitotic chromatin (Fig. 1a), whereas RNA polymerase II (RNA POLII) and SWI/SNF ATPase subunit SMARCA4 were dissociated from mitotic chromatin, as previously reported8,12,13. While most of the cBAF, PBAF, and ncBAF SWI/SNF complex subunits were either undetectable or present at markedly reduced levels in mitotic cell chromatin, subunits SMARCE1 and SMARCB1, common to cBAF and PBAF, were readily detectable (Fig. 1a). To evaluate the subcellular localization of these factors during mitosis, we subjected cells to differential extraction (Extended Data Fig. 1f). SMARCE1 and SMARCB1 as well as EZH2, and SOX2, were retained on mitotic chromatin, whereas other SWI/SNF subunits, such as SMARCA4, were evicted from mitotic chromatin, but still resident in the cytoplasmic fraction (Fig. 1b and Extended Data Fig. 1g, h). A recent study showed that SMARCA4 is phosphorylated in mitosis12, however, we found that this was not the determinant of its lack of chromatin binding as phosphatase treatment had no effect (Extended Data Fig. 1i to k).
Fig 1. Select SWI/SNF subunits bound on mitotic chromatin.
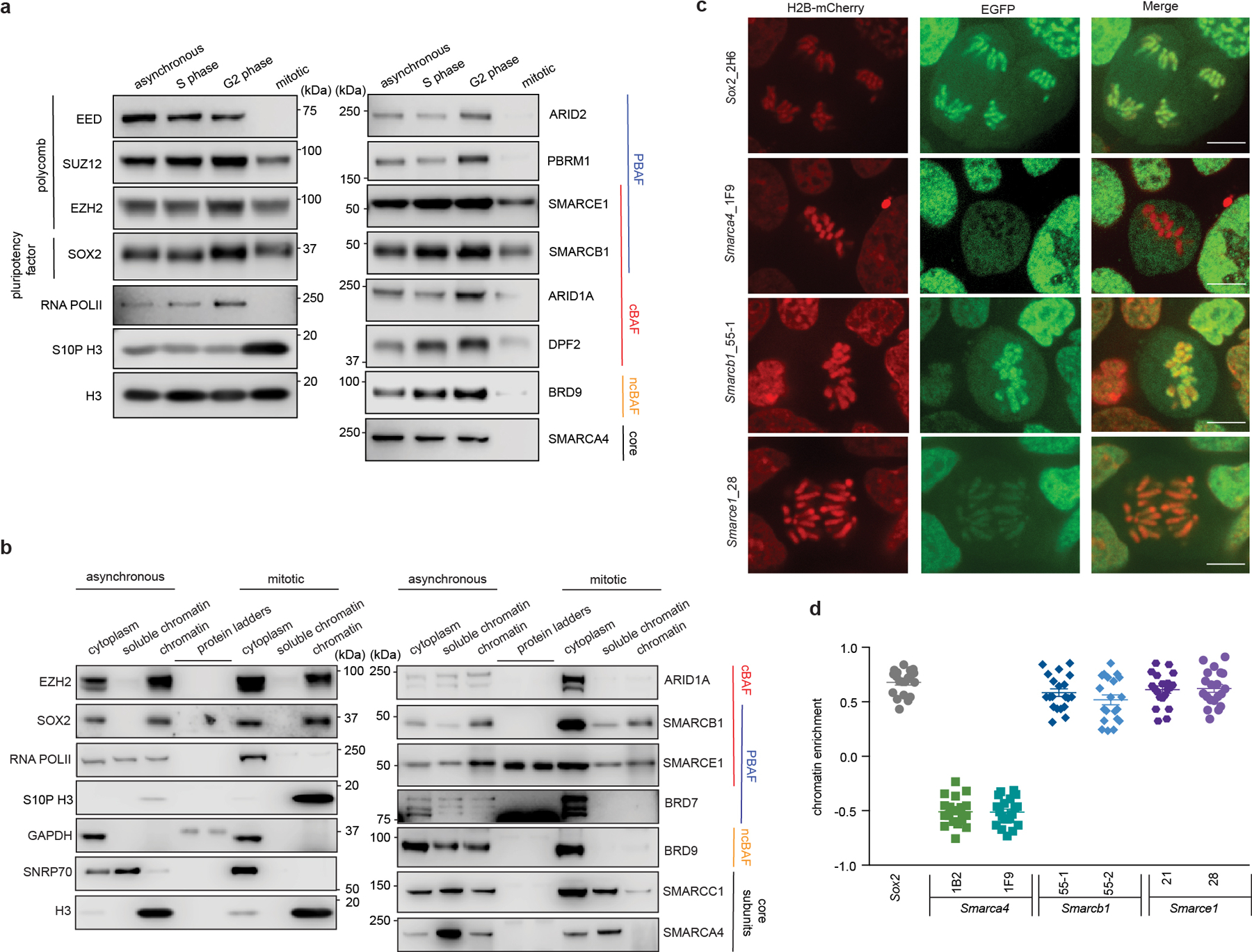
a, Extraction of chromatin fraction from asynchronous mouse ES cells and synchronized populations at S, G2, and mitosis. Histones, RNA POLII, SOX2, PRC2 components, and SWI/SNF subunits were examined by western blot analysis. b, Representative western blot analysis of histones, SNRP70, GAPDH, RNA POLII, SOX2, EZH2, and SWI/SNF subunits in cytoplasm, soluble chromatin and chromatin fractions extracted from asynchronous and mitotic mouse ES cells. c, Representative live-cell imaging of asynchronous mouse ES cells stably expressing H2B- mCherry with endogenously tagged EGFP at the C-terminal of SOX2, SMARCA4, SMARCB1, and SMARCE1, respectively. Scale bar: 10 μm. d, Quantification of SOX2, SMARCA4, SMARCB1, and SMARCE1 chromatin enrichment in mitotic cells (c and Extended Data Fig. 2k). Data are representative of two (a, b) or one (c, d) independent experiments. Data are shown as mean± s.e.m. N = 20 cells per cell line (d).
To directly visualize SWI/SNF subunit localization during mitosis, we generated mouse ES cells stably expressing H2B-mCherry to mark chromatin (Extended Data Fig. 2a) and knocked-in enhanced green fluorescent protein (EGFP) at the C-terminal ends of SMARCE1, SMARCB1, SMARCA4, and the mitotic chromatin bound protein SOX215,17,18 as a positive control (Extended Data Fig. 2b to g). The tagged subunits were efficiently incorporated into SWI/SNF complexes (Extended Data Fig. 2h to j). Live-cell imaging revealed that SOX215,17,18, SMARCB1, and SMARCE1 were enriched on chromatin in mitotic cells, while SMARCA4 was absent (Fig. 1c, 1d and Extended Data Fig. 2k). Together, these data demonstrate that SWI/SNF complex subunits SMARCE1 and SMARCB1, but not the ATPase subunit SMARCA4, are bound on mitotic chromosomes.
To analyze the genomic localization of the mitotically-retained SWI/SNF subunits, we used chromatin immunoprecipitation (ChIP) of SOX215,17,18 and ESRRB19 as controls. While ESRRB binding was readily detectable, only a small number of SOX2 peaks were detected in mitosis (Extended Data Fig. 3a, b), consistent with previous studies15. The SOX2 peaks nonetheless corresponded with high confidence to SOX2/SOX2:POU5F1 binding motifs suggesting that these are true sites (Extended Data Fig. 3c). Chemical crosslinking biases evaluation of dynamically bound factors in mitotic cells, which lack a nuclear membrane, by preferentially trapping them off chromatin16. We therefore used a formaldehyde-free method, Cut&Run20, to investigate binding. Cut&Run analysis of SOX2 revealed 11805 asynchronous (asyn)-specific peaks, 3382 peaks common to both asynchronous and mitotic cells, and 1913 mitotic (mit)-specific peaks, which account for 69%, 20%, and 11% of total peaks, respectively (Fig. 2a and Extended Data Fig. 3d, e). We found a highly significant overlap in the binding for both ESRRB and SOX2 between data obtained by ChIP-seq and data obtained by Cut&Run (Extended Data Fig. 3f), and the incremental SOX2 binding sites detected solely by Cut&Run were strongly enriched for SOX2/SOX2: POU5F1 binding motifs (Extended Data Fig. 3c, g, h) and motif score (Extended Data Fig. 3i), collectively validating the enhanced sensitivity of Cut&Run.
Fig 2. SWI/SNF subunits preferentially bind at promoters in mitosis.
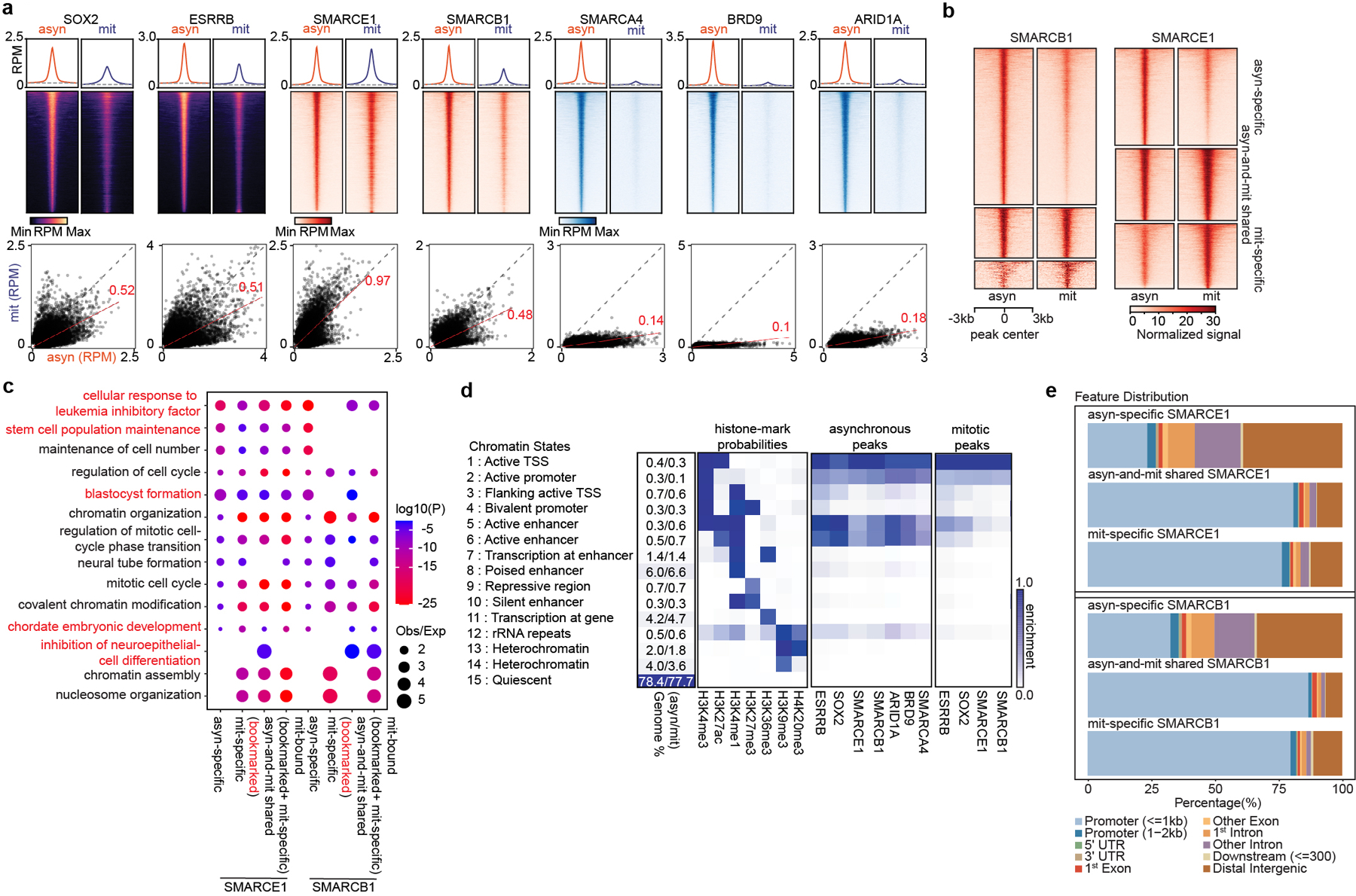
a, (top) Average binding profile of Cut&Run signal at the indicated binding regions (± 3000-bp peak summit) identified in asynchronous (asyn) and mitotic (mit) mouse ES cells. Average binding profiles represent reads per million (RPM); the y-axis is scaled by median asynchronous binding. (bottom) Scatter plots of Cut&Run signal in reads per million at the designated regions (± 250-bp peak summit) in asynchronous and mitotic mouse ES cells. Linear regression of RPM value for asynchronous cells divided by the value for mitotic cells were estimated and regression slopes are shown in red. Grey dashed lines indicate the random background. b, Heatmap of SMARCB1 and SMARCE1 peaks in asynchronous and mitotic mouse ES cells (four replicates were pooled together). c, GO analysis of genes bound by asynchronous-specific, mitosis-specific, asynchronous+ mitosis shared (bookmarked), and mitosis bound at any time (bookmarked+ mitosis-specific) SMARCE1 and SMARCB1 peaks using GREAT in asynchronous and mitotic mouse ES cells. Size of the circle represents ratio of observed (Obs) versus expected (Exp) frequency, and p-value was calculated by two- sided Fisher’s exact test. d, Fifteen chromatin states were defined by ChromHMM using seven histone marks. Genome coverage, histone-mark possibilities and the feature distributions of SWI/SNF subunits, ESRRB and SOX2 on chromatin are shown for each chromatin state in both asynchronous and mitotic mouse ES cells. e, Genomic distribution of asyn- specific, asyn- and mit- shared, and mit- specific peaks of SMARCE1 and SMARCB1. Data are compiled from four (Cut&Run for SMARCE1 and SMARCB1) and two (Cut&Run for SOX2, ESRRB, SMARCA4, BRD9, and ARID1A) replicates respectively.
We performed Cut&Run for SMARCE1, SMARCB1, SMARCA4, BRD9, and ARID1A. There were thousands of SMARCE1 and SMARCB1 binding sites in both asynchronous and mitotic cells (Fig. 2b and Extended Data Fig. 3e, j). In contrast, while there were thousands of SMARCA4, BRD9, and ARID1A peaks in asynchronous cells, there were only 18 SMARCA4, 25 BRD9 and 53 ARID1A binding sites in mitotic cells (Fig. 2a and Extended Data Fig. 3e). For SMARCB1, we identified 7635 asynchronous-specific peaks (77% of total peaks called), 1466 peaks common to both mitotic and asynchronous cells (15%) and 833 mitotic-specific peaks (8%). For SMARCE1, we identified 4864 (42%) asynchronous-specific peaks, 3501 peaks common to both mitotic and asynchronous cells (30%), and 3179 mitotic-specific peaks (27.5%) (Fig. 2b and Extended Data Fig. 3e). There was strong co-localization of mitotic SMARCB1 binding with mitotic SMARCE1 (91% binding site overlap) (Extended Data Fig. 3k). Gene ontology (GO) analysis showed that the mitotically bound peaks were significantly enriched for lineage specific gene sets (e.g., “cellular response to leukemia inhibitory factor”, “blastocyst formation”, “chordate embryonic development”) as well as gene sets associated with completion of mitotic cell division that are activated within mitosis and very early upon transition to G11 (Fig. 2c). Bookmarked peaks (those present in both asynchronous and mitosis) were enriched for “inhibition of neuroepithelial cell differentiation” (Fig. 2c). The relative binding strength of mitotic peaks and asynchronous peaks was similar for both SMARCE1 and SMARCB1 (Extended Data Fig. 3l, 3m) demonstrating that mitotic peaks represent robust binding of SMARCE1 and SMARCB1.
In unsynchronized cells, SWI/SNF complexes localize to enhancers (cBAF and ncBAF) and promoters (PBAF and ncBAF)21,22. In mitosis, higher order chromatin structures are temporarily disrupted10 raising the question of where SWI/SNF subunits bind during mitosis. We therefore analyzed the location of SMARCE1 and SMARCB1 in mitosis in comparison to histone modifications associated with enhancers and promoters. We performed ChIP-seq for H3K4me3, H3K27ac, H3K4me1, H3K27me3, H3K36me3, H3K9me3, and H4K20me3 to characterize chromatin states and executed Cut&Run-seq for SWI/SNF components ARID1A, BRD9, SMARCA4, SMARCB1, SMARCE1, as well as ESRRB and SOX2 (Fig. 2d). In asynchronous cells, we observed somewhat stronger binding of SOX215,17,18, ESRRB and ARID1A (canonical BAF/cBAF subunit) at enhancers than promoters while BRD9 (non-canonical BAF/ncBAF component) bound at both (Fig. 2d (left)), consistent with published results23–26. In mitosis, SOX215,17,18, ESRRB, and SWI/SNF components were tethered almost exclusively at proximal gene regions with little binding at enhancers (Fig. 2d (right) and Extended Data Fig. 3n), similar to the binding patterns of some other bookmarks23,27,28 (Extended Data Fig. 3o). While SMARCE1 and SMARCB1 binding at promoters accounted for 25 to 35% of the asynchronous-specific peaks, the fraction of binding at promoters was markedly higher in mitotic cells (80% of asynchronous-and-mitotic shared peaks and mitotic-specific peaks) (Fig. 2e and Extended Data Fig. 4a). The binding sites of transcription factors retained in mitosis such as ESRRB15,23 and KLF418 significantly overlapped with mitotically bound SMARCE1 and SMARCB1 (Extended Data Fig. 4b, c). In contrast, there was not significant overlap between CTCF27 and SMARCE1/B1 (Extended Data Fig. 4b). The mitotic binding of SMARCE1/B1 was associated with promoters of genes whose expression is activated earliest and most strongly following mitotic exit28 (Extended Data Fig. 4d, e). In contrast, asynchronous-specific SMARCE1/B1, ESRRB, and SOX215,17,18 most significantly correlated with the reactivation of enhancers28 (Extended Data Fig. 4d). While long-range enhancer-promoter interactions are generally disrupted during mitosis9,29, relatively open chromatin is maintained at promoters29, allowing low levels of transcription of select genes during mitosis1. Thus, the high levels of SMARCE1 and SMARCB1 present at a subset of promoters could play a role in the maintenance and inheritance of transcriptome patterns through mitosis. Of note, while genes bound by SMARCE1 specifically in mitosis (mit-specific) were activated early in G1 they retained expression later in the cell cycle suggesting SMARCE1 is not essential for sustained transcription.
To understand the role SWI/SNF subunits play during mitosis, we evaluated whether mitotic SMARCE1 binding is required for the reactivation of the bound genes following cell division. To induce SMARCE1 degradation at the metaphase-anaphase transition, we fused the mitosis-specific degradation domain (MD) of cyclin B130 to the endogenous SMARCE1 locus31. An MD carrying a R42A substitution that inactivates the destruction box constituted a control17,32 (Extended Data Fig. 5a to j). To validate degradation, we blocked cells at prometaphase with nocodazole for 6 hours followed by wash out to release the synchronized cells (Fig. 3a and Extended Data Fig. 6a). SMARCE1-MD was fully depleted by 45 min following mitotic release and wild-type levels were restored by 180 min, whereas SMARCE1-MD (R42A) levels were indistinguishable from untargeted SMARCE1 (Extended Data Fig. 6b). Despite the cyclic degradation of SMARCE1-MD in mitosis, steady-state levels of SMARCE1-MD and MD (R24A) proteins were comparable, and similar to wild-type SMARCE1 in parental cells (Extended Data Fig. 5e and Extended Data Fig. 6c).
Fig 3. SMARCE1 reactivates bookmarked genes.
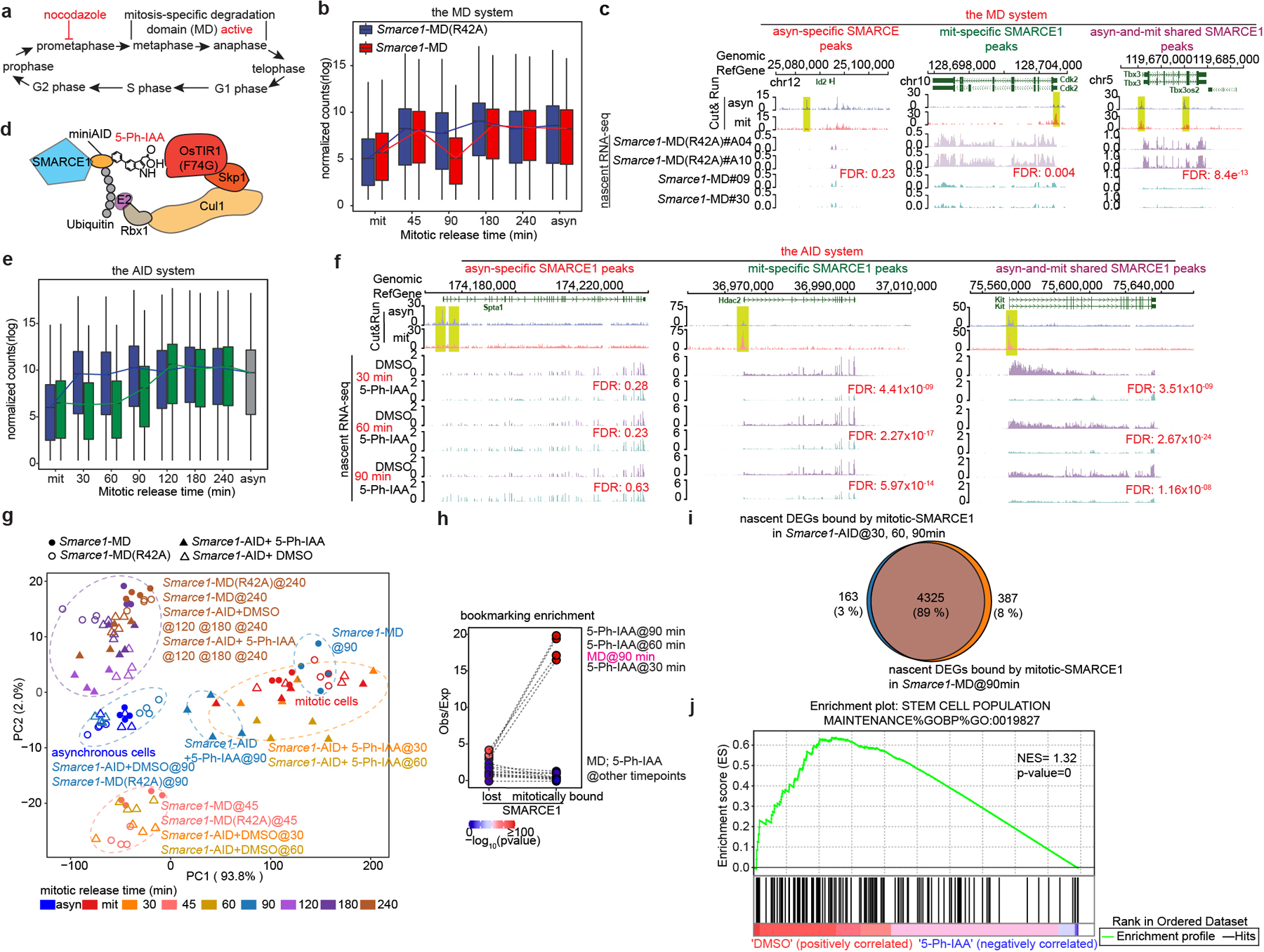
a, Schematic showing MD degron activity. b, Expression levels of genes bound by SMARCE1 in mitosis from Smarce1-MD (R42A) (clone A04 and A10) and Smarce1-MD (clone 09 and 30) in asynchronous, mitotic and released cells over time. c, Representative SMARCE1 peaks and nascent RNA profiles in Smarce1- MD (R42A) and Smarce1- MD cells 90 min after mitotic release. d, Diagram of auxin-inducible degron model. e, Expression levels of genes bound by SMARCE1 in mitosis from DMSO and 5-Ph-IAA treated Smarce1-AID (clones 6 and 23) cells in asynchronous, mitotic and mitotic-released cells. f, Representative SMARCE1 peaks and nascent RNA in DMSO and 5-Ph-IAA treated Smarce1-AID (clones 6 and 23) cells at 30 min, 60 min and 90 min after mitotic release. g, PCA of nascent transcripts from Smarce1-MD (R42A), Smarce1-MD, DMSO and 5-Ph-IAA treated Smarce1-AID cells at each time point. Colors indicate time; shapes show different cell lines and treatments. h, Enrichment of mitotically bound and mitotically lost SMARCE1 Cut&Run peaks at promoters of downregulated genes in the MD and AID systems by nascent RNA-seq. Each point represents observed/excepted frequency of DEGs to be associated with mitotically lost SMARCE1 binding (left) or with mitotically bound SMARCE1 (right). Color indicates p values. i, Overlap between genes bound by mitotic SMARCE1 and differentially expressed (DEGs) in the AID and MD systems. j, GSEA illustrating the enrichment score (ES) for the selected set (stem cell population maintenance GO: 0019827) of downregulated genes in Smarce1-AID treated with 5-Ph-IAA for 30 minutes (blue) compared to the DMSO control (red) (nominal P= 0, nonparametric permutation test). NES, normalized enrichment score. N=2 biological replicates for each clone (b, c, e, f, g, h, i, j). Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile (b, e).
The presence of homozygous Smarce1-MD and Smarce1-MD (R42A) alleles did not grossly alter the stem cell nature of mouse ES cells as there was continued high expression of pluripotency factors SOX2, NANOG, OCT4, and ESRRB, cells remained alkaline phosphatase positive, and proliferation remained unchanged (Extended Data Fig. 5f to i). We propagated the MD and MD (R42A) cell lines for over 100 generations, and both maintained typical mouse ES cell morphologies (Extended Data Fig. 5j). To evaluate the effects of mitotic degradation of SMARCE1 upon gene expression, we performed RNA-sequencing on four independent subclones each of Smarce1-MD and control Smarce1-MD (R42A) cells. Principal component analysis and a volcano plot showed significant differences in gene expression (Extended Data Fig. 6d, e). While transcription of the core pluripotency regulatory network33 was not disrupted, as indicated by western, immunofluorescence, and transcriptome analyses (Extended Data Fig. 5e, g and Extended Data Fig. 6f), GO analysis showed that neural differentiation associated terms were upregulated in Smarce1-MD cells (Extended Data Fig. 6g). Of note, one of the genes downregulated upon mitotic loss of SMARCE1 is Bmp4 (Extended Data Fig. 6h), a negative regulator of neural differentiation.
To examine the effect of mitosis-specific degradation of SMARCE1 on transcription reactivation following exit from mitosis, we used 5-ethynyluridine (EU) to pulse-label nascent transcripts1,34 in Smarce1-MD (clones #MD09, #MD30) and Smarce1-MD (R42A) (clones #A04, #A10) cells. We pulse-labeled transcripts at 0, 45, 90, 180, and 240 min after nocodazole washout, and in asynchronous cells (Extended Data Fig. 6a). We used two different biotinylated RNAs as spike-in controls (Extended Data Fig. 6i to k). Analysis of newly synthesized transcripts revealed that the lowest levels of transcripts were present in mitotically blocked cells (0 min). Following nocodazole washout, overall transcription was rapidly restored at 45 min (Fig. 3b), consistent with published results35. We associated SMARCE1 peaks with nearest genes within the same CTCF-bound domain and found that newly synthesized genes showed a significant reduction in transcript level at 90 min after mitotic release in Smarce1-MD cells (Fig. 3b, c and Extended Data Fig.6l). Overall, 45.8% of downregulated genes were bound by SMARCE1 in mitosis (Extended Data Fig. 6m). Collectively, these results demonstrate that mitotic SMARCE1 facilitates the reactivation of genes after mitotic exit.
While the MD-degron system enabled generation of cells in which SMARCE1 is depleted in mitosis, the levels of SMARCE1 remained below normal early in G1 phase raising the possibility that the transcriptional phenotypes might result from loss of function in early G1 rather than in mitosis. To address this, we next utilized the auxin-inducible degradation (AID) system 2.036 to tag endogenous SMARCE1 (Fig. 3d and Extended Data Fig. 7a, b). Treatment with 1 μM 5-Ph-IAA inducing agent led to rapid and efficient degradation of SMARCE1-AID. The large majority of SMARCE1 was degraded by 30 minutes and it became undetectable by 1 hour (Extended Data Fig. 7c, d). Importantly, the degradation was rapidly reversible as SMARCE1 levels recovered substantially by 10 minutes following 5-Ph-IAA washout (Extended Data Fig. 7e) although re-binding to chromatin might take longer. Using this system with nocodazole arrest we could confine degradation of SMARCE1 to mitosis (Extended Data Fig. 7f, g).
We performed nascent RNA-seq and again found that genes bound by SMARCE1 in mitosis displayed significantly delayed reactivation after mitotic exit (Fig. 3e, f and Extended Data Fig. 7h). PCA of the nascent transcripts revealed that Smarce1-MD cells at 90 min post mitotic release and 5-Ph-IAA treated Smarce1-AID cells at 30 min, 60 min, and 90 min post mitotic release clustered together (Fig. 3g). To evaluate whether there was a correlation between SMARCE1 mitotic binding and nascent transcription of bound genes, we compared the transcriptional effects on genes mitotically bound by SMARCE1 versus those that lose SMARCE1 during mitosis. Down-regulated differentially expressed genes (DEGs) at 30 min, 60 min, and 90 min in the AID system and down-regulated DEGs at 90 min in the MD system were highly significantly overlapping and enriched for mitotically bound SMARCE1 peaks (Fig. 3h, i). In both systems, mitotic degradation of SMARCE1 resulted in the genes that gain SMARCE1 occupancy in mitosis being downregulated in mitotic exit and early G1, with their expression increasing later in the cell cycle as SMARCE1 re-accumulated (Extended Data Fig. 7i). Depletion of SMARCE1 in mitosis significantly delayed RNA synthesis of ES cell identity genes (Fig. 3j). Collectively, results from the AID system were highly similar to those from the MD system.
We next sought to evaluate the extent to which mitotic binding of SMARCE1 contributes to overall SWI/SNF function. We utilized asynchronous Smarce1-MD cells, in which SMARCE1 is solely missing during mitosis, and compared the transcriptional consequences to cells with prolonged degradation of SMARCE1 using the AID system. We exposed asynchronous Smarce1-AID cells to the 5-Ph-IAA inducing agent for varying lengths of time: 4 hours, 8 hours, 1 day, 3 days, or 7 days (Extended Data Fig. 8a, b). The transcriptional impact of the loss of SMARCE1 progressively increased over time (Extended Data Fig. 8c, d). There was a significant correlation of genes downregulated in asynchronous Smarce1-MD cells to those downregulated in asynchronous Smarce1-AID cells treated with 5-Ph-IAA (Extended Data Fig. 8e). At 4 hours, 25% of the genes downregulated in the Smarce1-AID cells were also downregulated in Smarce1-MD cells. At 8 hours, there was 31.4% overlap, at 1 day it was 10.8%, at 3 days 16.8% and at 7 days 17.9% (p-value at 7 days < 2.2×10−16) (Extended Data Fig. 8e). Thus, absence of SMARCE1 solely in mitosis causes transcriptional effects that significantly overlap with the transcriptional consequences of complete absence of SMARCE1. We further evaluated this by comparing mitotic loss of SMARCE1 to constitutive loss of SMARCA4/BRG1. There was significant overlap between genes downregulated in asynchronous Smarce1-MD cells with genes downregulated due to SMARCA4/BRG1 knockout37 (20.8%, p-value< 2.2×10−16) (Extended Data Fig. 8f). As a control, we also compared genes downregulated by mitotic loss of SMARCE1 to those downregulated by knockdown of BRD938, a member of sole SWI/SNF family that does not contain SMARCE1 (ncBAF). The overlap was not significant (Extended Data Fig. 8g). Collectively, these results demonstrate that the bookmarking activity of SMARCE1 is a significant contributing factor to SWI/SNF complex function. Importantly, while there was a significant overlap between the genes bookmarked by SMARCE1 and the complete knockout of SMARCE1, the effects were not identical, as mitotic loss of SMARCE1 accounted for 10 to 30% of the genes downregulated by the absence of SMARCE1 throughout the cell cycle (Extended Data Fig. 8e). This is consistent with prior studies of mitotic bookmarks in which restoration of bookmark function in G1 results in a substantial degree of restoration of the transcriptome1,23,28,32,39. Collectively, while expression of SMARCE1 outside of mitosis plays important roles in facilitating transcription, it is not sufficient to rescue the impaired control of cell fate that occurs with loss of the mitotic bookmarking function of SMARCE1.
The bulk RNA-seq raised the possibility of a neural differentiation bias resulting from mitotic loss of SMARCE1. Previous studies reported that priming of neuronal pathways is present at mitotic exit with robust activation of these genes occurring in late G128,40,41. We therefore utilized nascent RNA-seq to evaluate whether there was biased lineage priming in Smarce1-MD cells. Indeed, there was enrichment for neural pathways by 240 minutes (late G1) after mitotic release (Extended Data Fig. 8h), which could also be detected, albeit more weakly, in asynchronous cells (Extended Data Fig. 8h).
To further evaluate the contributions of mitotic SMARCE21 to transcriptional activation, we analyzed nascent transcription changes. We clustered genes into two groups based upon the timing of maximal downregulation (30 minutes vs. 60 minutes) (Extended Data Fig. 8i, j, k). As a control, we also identified a group of genes (n=2401) that were constantly expressed at low levels (Extended Data Fig. 8i, j). There was an excellent correlation between the AID and MD systems (Extended Data Fig. 8i). Genes affected at 30 minutes were highly expressed and most strongly enriched for pathways associated with mitotic cell division while genes enriched at 60 minutes were more moderately expressed and most strongly enriched for cell fate (Extended Data Fig. 8l, m). Thus, at targets bound both in asynchronous cells and in mitosis, SMARCE1 bookmarks lineage-identity genes related to cell fate thus facilitating their activation shortly after exit from mitosis (Extended Data Fig. 8l). Additionally, exclusively during mitosis, SMARCE1 also binds to genes required for completion of mitotic cell division (Fig. 2c and Extended Data Fig. 8l). Both sets of genes (30 min timing> 60 min timing) were highly significantly enriched with genes previously reported to have mitotically retained ATAC signal and other retained bookmarks15,17,18 consistent with interactions between SMARCE1 and other bookmarks (Extended Data Fig. 8n). Collectively, this establishes SMARCE1 binding in mitosis as a key mechanism to both bookmark lineage identity genes to be activated upon exit from mitosis and to complete processes associated with cell division.
We investigated the mechanism by which mitotic SMARCE1 binding affects transcription. While SMARCE1 and SMARCB1 are members of SWI/SNF complexes, they lack ATPase domains and are unable to mobilize nucleosomes37,42,43. One possibility is that in early G1, SWI/SNF complexes assemble at, or are recruited to, loci bound by SMARCE1/B1 in mitosis. To test this, we performed Cut&Run for the ATPase subunit SMARCA4. Mitotic degradation of SMARCE1 markedly reduced binding of SMARCA4 in G1 (Extended Data Fig. 8o), significantly overlapping with the sites bound by SMARCE1 in mitosis (Extended Data Fig. 8p).
To test the contribution of SMARCA4 we treated Smarce1-MD and Smarce1-MD(R42A) cells with 1 μM BRM014, a SMARCA4 inhibitor44. ATAC-seq revealed a considerable reduction of accessible regions after BRM014 treatment in control Smarce1-MD(R42A) cells but more modest alterations in Smarce1- MD cells (Extended Data Fig. 9a). Nascent RNA-seq revealed that BRM014 led to a significant reduction in the expression of 537 genes in control Smarce1-MD(R42A) cells at 90 min after mitotic release but only 23 genes in Smarce1-MD cells (Extended Data Fig. 9b). We next compared the ATAC-seq changes caused by Smarce1-MD at 90 minutes to those of treatment with BRM014. There was a highly significant correlation (p-value< 1.01×10−322) (Extended Data Fig. 9c). These results are consistent with SMARCE1 binding as a bookmark during mitosis that enables subsequent assembly or recruitment of SWI/SNF complexes to activate transcription early in G1.
Given that SWI/SNF complexes can interact with other chromatin regulators and modulate the binding of transcription factors7,43,45,46, we asked whether loss of mitotic SMARCE1 impaired the binding of other bookmarker proteins during mitosis. Cut&Run analysis of SOX215,17,18, ESRRB, and EZH2 in Smarce1-MD and Smarce1-MD (R42A) cells revealed a loss of 590 SOX2 peaks and a significant reduction of an additional 602 SOX2 peaks in Smarce1-MD cells at 90 min after mitotic release, accounting for 4.9% of total SOX2 peaks (Extended Data Fig. 9d). At the same time point, 3166 ESRRB peaks were lost, and 3768 significantly decreased, accounting for 10.3% of total ESRRB peaks (Extended Data Fig. 9d). With respect to Polycomb complexes, which often antagonize SWI/SNF function, 487 EZH2 peaks showed a significant increase, and a small number revealed de novo EZH2 binding in Smarce1-MD cells, accounting for 2.05% of total EZH2 peaks (Extended Data Fig. 9d). SMARCE1 loss did not interfere with the expression of other bookmarker proteins (Extended Data Fig. 9e). Using the AID system, the effects were similar (Extended Data Fig. 9f). Comparing sites displaying reduced ESRRB/SOX215,17,18 binding to sites bound by SMARCE1 in mitosis, we found that 55% of decreased/depleted SOX2 (p=1.93×10−233) and 25.1% of reduced/depleted ESRRB peaks (p=2.28×10−124) overlapped with mitotically retained SMARCE1 (Extended Data Fig. 9g). The affected sites that do not overlap with SMARCE1 binding could represent either secondary effects or, given the fairly small number of affected sites, intrinsic variability in the binding of these factors. We next compared the sites that lost SOX2 or gained EZH2 peaks to gene expression changes and found that 22% of lost SOX2 sites corresponded to genes whose expression was downregulated in Smarce1-MD cells. The EZH2 overlap was smaller at 5.2% (Extended Data Fig. 9h). Next, we examined SOX2 binding following treatment with BRM014. ATPase inhibition also affected a relatively small percentage of SOX2 binding sites in both Smarce1-MD(R42A) (5.15%) and Smarce1-MD (4.58%) cells at 90 min after mitotic release (Extended Data Fig. 9i), reminiscent of a prior study which found a minority of SOX2 sites were affected by genetic deletion of SMARCA4/BRG137. Notably, however, when we compared the SOX2 binding sites lost upon mitotic degradation of SMARCE1 to SOX2 sites lost upon treatment with BRM014, there was a highly significant correlation (p=1×10−28.6) (Extended Data Fig. 9j). These results demonstrate that loss of mitotic SMARCE1 results in downregulation of genes, some of which are also bookmarked by SOX215,17,18 but many not. While EZH2 may contribute to downregulation of some targets, loss of SMARCE1 alone is sufficient to cause down-regulation.
As both bulk and nascent RNA-seq of Smarce1-MD mouse ES cells suggested that loss of SMARCE1 in mitosis might bias cells towards neural differentiation, we directly tested this. We induced Smarce1-MD and Smarce1-MD (R42A) mouse ES cells to undergo neural differentiation47. By culture day 6, we observed that mitotic degradation of SMARCE1 resulted in marked differences in cell morphology. Control Smarce1-MD (R42A) cells grew as aggregates and formed neural rosette-like structures, characteristic of neural progenitors. In contrast, Smarce1-MD derived cells did not aggregate and exhibited morphology suggestive of terminally differentiated glial cells and neurons with multi-directional extended dendrites or axons (Extended Data Fig. 10a). Immunohistochemistry (IHC) showed that Smarce1-MD cultures had many more gamma-aminobutyric acid (GABA) and glial fibrillary acidic protein (GFAP) positive neural cells (Fig. 4a, b and Extended Data Fig. 10b, c), and fewer SOX1 and NES positive cells (Extended Data Fig. 10b, c). To better understand the difference in neural fates, we analyzed gene expression. Mitotic degradation of SMARCE1 resulted in higher expression of GABA receptors (Gabra2 and Gabrg2) and hyper-activation of synaptic signaling on neural induction (Fig. 4c, d and Extended Data Fig. 10d), consistent with the distinct cellular morphologies. We further evaluated the effect using a spontaneous differentiation model (EB formation). Similar to results with the neural induction model, after six days following withdrawal of LIF Smarce1-MD cells, but not the control Smarce1-MD(R42A) cells, were adherent and developed branch-like structures (Extended Data Fig.10e). Gene pathways most upregulated in Smarce1-MD cultures were again neural pathway gene sets. Specifically, of the eight top pathways, five related to axon or synapse formation (Extended Data Fig. 10f). To more rigorously examine the contribution of SMARCE1 to memory, we investigated the consequences of mitotic depletion of SMARCE1 in during a single cell cycle using Smarce1-AID mouse ES cells (clones#06 and #23) followed by neural induction (Extended Data Fig. 10g). Absence of SMARCE1 from a single mitotic cell cycle was sufficient to trigger abnormal neural differentiation (Extended Data Fig. 10h, i).
Fig 4. Mitotic SMARCE1 and neural differentiation.
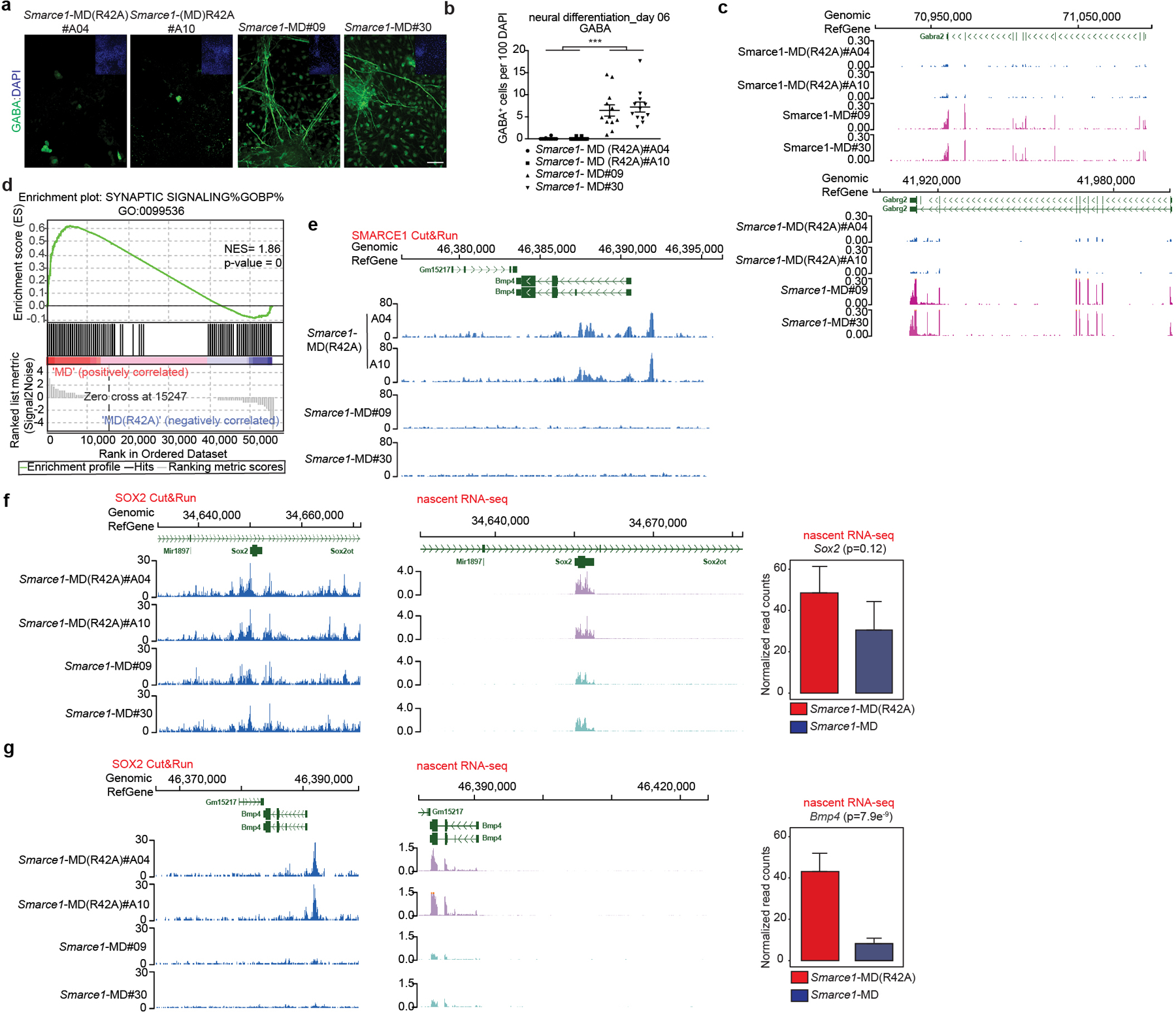
a, Representative immunohistochemical staining of GABA, with DAPI counter-stain in Smarce1-MD (R42A) and Smarce1-MD cultures at day six after neural induction. Scale bar: 50 μm. b, Statistical analysis of the data in (a). c, d, Differential gene expression profiles in Smarce1-MD (R42A) and Smarce1-MD cultures at day six after neural induction. (c) Representative browser track showing Gabra2 and Gabrg2 expression. (d) Representative example of GSEA enrichment plot of genes in synaptic signaling pathway. (nominal P= 0, nonparametric permutation test). NES, normalized enrichment score. e, Representative browser track for SMARCE1 binding at the Bmp4 locus in control Smarce1-MD (R42A) and Smarce1-MD mouse ES cells at 90 min after mitotic release. f, g, Representative browser tracks for SOX2 binding and nascent transcripts at Sox2 (f) and Bmp4 (g) loci in Smarce1-MD (R42A) and Smarce1-MD mouse ES cells at 90 min after mitotic release. Data are representative of two biological replicates for each clone (Smarce1-MD (R42A) #A04 and #A10, Smarce1-MD#09 and #30) (a, c, e, f, g); or compiled from two independent experiments each assessing two clones of each genotype (b, d) and test is performed in DESeq2 Wald test (f, g). Data are shown as mean± s.e.m., n=12 images pooled from two independent experiments / each sample; significance is calculated using two- tailed unpaired Student’s t-Test (*** p= 3.85e−8) (b).
To explore the basis for the premature neural differentiation, we compared the RNA profiles of Smarce1- MD and control cells. Expression of Bmp4, a key negative regulatory factor in neural cell fate commitment48, was significantly decreased in Smarce1-MD cells (Extended Data Fig. 6h). Further analysis showed that SMARCE1 was bound at the Bmp4 locus (Fig. 4e). Mitotic degradation of SMARCE1 had minimal effect upon SOX2 binding at the Sox2 locus itself but SOX2 binding at the Bmp4 locus was substantially reduced (Fig. 4f, g). Examining nascent transcripts at 90 min after mitosis revealed Bmp4 transcript levels were decreased in Smarce1- MD cells (Fig. 4f, g). To directly evaluate the contribution of loss of Bmp4, we supplemented Smarce1-MD cultures with BMP4. Provision of BMP4 circumvented the effects of mitotic degradation of SMARCE1, consistent with loss of BMP4 as the mechanism by which mitotic loss of SMARCE1 contributes to premature neural differentiation (Extended Data Fig. 10j, k, l).
The substantial alterations in chromosome architecture and the nuclear milieu during mitosis present a challenge for maintaining cell-type-specific transcription programs. By facilitating the binding and activity of transcription factors, SWI/SNF complexes have been shown to serve essential roles in the control of cell fate5,7,49. As the ATPase subunits of SWI/SNF were identified as evicted from chromatin in mitosis12,13, there has been little evaluation of SWI/SNF function during cell division. Here, we demonstrate that SWI/SNF complex subunits SMARCE1 and SMARCB1, but not the whole complex, bind to chromatin during mitosis. We find two groups of genes enriched for mitotic binding by SMARCE1. The first consists of genes related to chromatin assembly and are enriched for SMARCE1-binding in mitosis compared to asynchronous cells. This may reflect that genes encoding proteins required to support the newly duplicated genome, including nucleosomes, are among the earliest and most strongly activated upon emergence from mitosis1. The second group relates to cell fate specificity and are bound by SMARCE1 also in interphase cells. Binding of SMARCE1 during mitosis is required to reinitiate expression following cell division (Fig. 3) demonstrating a role for SMARCE1 in mitotic bookmarking. The mitotic-specific degradation of SMARCE1 results in loss of Bmp4 expression, a gene that suppresses neural fate commitment, and causes premature neural differentiation of ESCs upon direct neural induction (Fig. 4). We observed that SMARCE1 and SMARCB1, while bound at enhancers and promoters in interphase cells, are bound nearly exclusively at promoters in mitosis. We also found this to be the case for other mitotic bookmarks (ESRRB15,23, H3K27ac18,28). This is consistent with the observation that accessibility is generally maintained at proximal promoters during mitosis29 and supports a key role for promoter binding over enhancer binding in the function of mitotic bookmarks.
A challenge to defining the contributions of transcription factors and chromatin regulators to mitotic cell function is intrinsic to fundamental cell biology as dissolution of the nuclear membrane in mitosis can impact the results of biochemical assays. This is likely a contributing factor to discrepancies as to whether certain factors, including SOX2 and SMARCA4, are bound to chromatin12,13,15–17,50,51. It is for this reason that we utilized three independent approaches, biochemical fractionation, live-cell imaging, and Cut&Run to characterize and validate our findings. We find that Cut&Run yields increased sensitivity over ChIP-seq for the detection of mitotic binding by SOX2, supported by the incrementally detected sites correlating to SOX2 recognition motifs, and consistent with the findings for other factors52. It remains to be determined the degree to which benefits of Cut&Run for mitotic factors is generalizable.
Our work reveals that the mitotic bookmarking role of SWI/SNF is carried out by individual subunits rather than by intact mSWI/SNF complexes. SMARCE1 contains a DNA-binding HMG domain and structural studies have revealed that SMARCB1 directly tethers to nucleosomes via its C-terminal alpha-helical domain53,54. These studies also revealed SMARCE1 and SMARCB1 constitute two early assembling components of the BAF family of SWI/SNF. Our findings demonstrate that following mitotic docking of SMARCE1 and SMARCB1, the ATPase core is recruited in G1, and SWI/SNF function established.
Our results establish that SWI/SNF subunits serve key roles in mitotic cells, with mitotic bookmarking by SMARCE1 contributing to heritable memory and lineage commitment. These findings broaden our understanding of SWI/SNF complexes, which are frequently mutated in cancer and neurodevelopmental diseases, and reveal mechanisms central to chromatin-mediated control of cell fate.
MATERIALS AND METHODS
Cell Culture and Cell Line Derivation
Mouse ES cell lines AB2.2 (ATCC SCRC-1023) and E14TG2a (ATCC CRL-1821) were purchased from ATCC and tested negative for mycoplasma (Gelantis, MY01100). ES cells were cultured on 0.1% gelatin/PBS coated petri-dishes. ES cells were grown in Knock-out DMEM (Gibco, 10829018) supplemented with 15% ES-qualified FBS (Gibco, 10439024), 1% GlutaMAX (Gibco, 35050061), 1% MEM Non-Essential Amino Acids (Gibco, 11140050), 0.1 mM beta- mercaptoethanol (Gibco, 21985023), and 100 ng/ml mouse LIF (ThermoFisher, A35935 or R&D systems, 8878-LF-500/CF). ES cells were fed daily and split every two days by TrypLE (12605010, Gibco) dissociation.
5-Ph-IAA (HY-134653, MedChemExpress) was used at a final concentration of 1 μM. BRM014 (HY-119374, MedChemExpress) was used at a final concentration of 1 μM. CK2 inhibitor TBB was used at a final concentration of 10 or 50 μM as indicated. ERK1 inhibitor PD0325901 was used at a final concentration of 1 μM.
To derive H2B-mCherry stably expressed mouse ES cells: H2b was amplified by PCR from mouse cDNA (5’- ATGCCTGAGCCTGCGAAGTC-3’; 5’- CTTGGAGCTGGTGTACTTGGTGACG-3’), mCherry was amplified from dCas9-VPR_P2A_mCherry (a gift from Anna Obenauf (Addgene plasmid # 154193; http://n2t.net/addgene:154193; RRID:Addgene_154193) (5’- GATCCACCGGTCGCCACCATGGTGAGCAAGGGCGAGGAGG-3’; 5’- TTACTTGTACAGCTCGTCCATGCCGCC-3’). To assemble the H2b and mCherry fusion, overlap regions were generated by PCR from H2b (5’- ATGCCTGAGCCTGCGAAGTC-3” 5’- GACCGGTGGATCCTTGGAGCTGGTGTACTTGGTGACGGCCTTGG-3’) and mCherry (5’- CCAGCTCCAAGGATCCACCGGTCGCCACCATGGTGAGCAAGGG-3’; 5’- TTACTTGTACAGCTCGTCCATGCCGC-3’). The H2b-mCherry fusion was assembled using NEBuilder® HiFi DNA Assembly Master Mix (NEB #E2621S). The H2b-mCherry fragment was amplified with XbaI and NotI cloning sites (5’- ccccTCTAGAATGCCTGAGCCTGCGAAGTC-3’; 5’- aaaaGCGGCCGCTTACTTGTACAGCTCGTCCATGCCG-3’) and inserted into PB-EF1-MCS-IRES-Neo cDNA Cloning and Expression Vector (System Biosciences #PB533A-2). AB2.2 mESCs were transfected with PB-533A-2-H2b-mCherry and Super piggyBac Transposase expression vector (System Biosciences # PB210PA-1) using Lipofectamine 3000 (Invitrogen # L3000001) and mCherry positive cells were sorted three days after transfection.
To derive endogenous C-terminal EGFP tagged SOX2, SMARCA4, SMARCE1, and SMARCB1 in H2b-mCherry mouse ES cell lines, gRNAs (sgSox2: 5’ TGCCCCTGTCGCACATGTGA 3’; sgSmarca4: 5’ GGTCAGGACTCAGGAATGTC 3’; sgSmarce1: 5’ GTTTTAGGTCACATAAAACA 3’; sgSmarcb1: 5’ TCACCAGGCTGGGGCAGTGT 3’) were inserted in pSpCas9(BB)-2A-Puro (PX459) V2.0 (a gift from Feng Zhang (Addgene plasmid # 62988; http://n2t.net/addgene:62988 ; RRID: Addgene_62988)) to induce a double-strand break at the relevant gene. Synthetic donor constructs were generated for each gene in pUC19 with 800 base pairs of sequence from both upstream and downstream of the stop codon flanking EGFP (BioBasic. Inc). For transfection, 2.5 μg of each gRNA plasmid and donor plasmid was diluted in 250 μl Opti-MEM medium supplemented with 10 μl of P3000 reagent. This was mixed with 15 μl Lipofectamine 3000 (Invitrogen, L3000008) diluted in 250 μl Opti-MEM medium (Gibco, 31985070), and the DNA-lipid complexes were incubated at room temperature for 10 to 15 minutes. 1× 106 H2b-mCherry AB2.2 cells were then added, and the transfection suspension was seeded on the gelatin-coated 6-well plates. Twenty-four hours after transfection, cells were selected by addition of 2 μg/ml puromycin (Gibco, A1113803) for 24 hours. Single cells were then sorted into gelatin-coated 96-well plates and expanded. Homozygous clones were identified by genotyping PCR using primers (Sox2: 5’- TGATCAGCATGTACCTCCCC -3’; 5’-CCTCCAGATCTATACATGGTCCG-3’; Smarca4: 5’-ATCCCTTAAGGATCTAGGCC-3’; 5’-TGCATGTGTACAGTTCTCCAAG-3’; Smarce1: 5’-AATGGTGAAGAAGGCACGTC-3’; 5’- TTAGGCTATGATGCTGAGGG-3’; Smarcb1: 5’-CCACACCTAGCTCCTCTAGG-3’; 5’- GGACAGTGTTGGGGTTTAGC-3’).
To derive Smarce1- MD (R42A) and Smarce1- MD mouse ES cells, the DNA sequence (gcagaaaataaggccaaggtcagtatggcaggcgccaagcgtgtgcctgtgacagttactgctgcttccaagcccgggctgagaccgagaactgctcttggagacattggtaataaagtcagcgaagagctacaggcaagagtgcctctgaaaagggaagcaaaaacgctaggtactggaaaaggtactgttaaagccctaccaaaacctgtagagaaggtgcctgtgtgtgaacca) encoding the peptide corresponding to residues 13– 91 of murine cyclin B1 (mitosis-specific degradation degron, MD degron) and loxP surrounded PGK promoter driven neomycin cassette with 800 base pairs of left and right homology arms flanking the C-terminus of Smarce1 gene (Extended Data Fig. 5a). The identical donor vector was built but with the critical R42 residue (aga) mutated to alanine (gca) as the MD (R42A) control. The same guide RNA used for the establishment of Smarce1- EGFP mESCs was used to derive Smarce1- MD (R42A) and Smarce1- MD mESCs. AB2.2 mESCs were transfected with guide RNA and donor vector by Lipofectamine 3000 as previous described. Twenty-four hours after transfection, cells were selected by 2 μg/ml puromycin for 24 hours, and 2 days later, treated with 350 μg/ml neomycin (Sigma, N1142–20ML) for 2 days, then 150 μg/ml neomycin for another 5 days. Primers (5’-AATGGTGAAGAAGGCACGTC-3’; 5’-TTAGGCTATGATGCTGAGGG-3’) were used to determine homozygous clones by PCR.
To derive Smarce1-AID mouse ES cells, the miniAID sequence55 cassette containing 800 base pairs of left and right homology arms flanking the C-terminus of the Smarce1 gene was inserted into the pUC19 plasmid as the donor vector. The same guide RNA used to establish the Smarce1- EGFP cell lines was used to derive Smarce1-AID mouse ES cells. AB2.2 cells were transfected with guide RNA and donor vector by Lipofectamine 3000 as previous described. Twenty-four hours after transfection, cells were selected by 2 μg/ml puromycin for 24 hours. Then cells were seeded in singlet to 0.1% gelatin-coated 96-well plates. Primers (5’-ACTCCTGAGGACAAGGAGAG-3’; 5’-AGGTTTCAGACGTTGAGAGC-3’) were used to determine homozygous clones by PCR. The selected clones were infected with pSin-OsTIR1 (F74G)55 lentivirus particles (MOI: 10) supplemented with 6 μg/ml polybrene (Santa Cruz, 134220) for 24 hours, after which 2 μg/ml puromycin selection was used for 7 days to establish the stable cell lines.
Cell Synchronization and Cell Cycle Analysis
Mitotic ES cells were synchronized by supplementing with 100 ng/ml nocodazole (Sigma, 487929) for 6 hours followed by gentle shake-off. Any mitotic shake-off experiment with less than 95% purity of 4N cells was discarded. For S phase synchronization, mouse ES cells plated at 50%-60% confluency were incubated in 2 mM thymidine for 6 hours followed by 6 hours with fresh ES culture media. This regime was repeated twice (double thymidine block) and followed by the treatment with 1 μg/ml aphidicolin for 10 hours, and cells synchronized in S phase were collected. For G2 phase synchronization, the double thymidine block was followed by treating with 100 ng/ml nocodazole for 6 hours, and mitotic cells depleted by shake-off and by rinsing with PBS twice to remove the residual M-phase cells, then the remaining adherent cells were harvested as G2-phase cells. For cell cycle analysis, 3×106 cells/sample were collected and washed twice in cell suspension buffer (PBS+ 2% FBS) and fixed with 5 ml cold 70% ethanol for at least 1 hour at 4°C. Cells were then washed twice in cold PBS and resuspended in 1 ml of 50 μg/ml propidium iodide (PI)/PBS buffer supplemented with 0.5 μg/ml RNase A and incubated overnight at 4°C. Samples were collected and analyzed by 17-color LSR Fortessa (4 Lasers) and BD FACSDiva software in the St. Jude Flow Core. Data were analyzed by FlowJo (Version 10. 7. 1).
Phospho S10 Histone 3 Staining
1×106 cells were collected and fixed in the dark for 20 minutes at room temperature with 0.5 ml fixation buffer (BioLegend #420801). Then cells were treated with 1 ml of Intracellular Staining Perm Wash Buffer (BioLegend #421002) twice. Cells were resuspended in 100 μl of Intracellular Staining Perm Wash Buffer supplemented with 5 μl of PE anti-Histone H3 Phospho (Ser10) Antibody (BioLegend #650808) or PE Mouse IgG2b, κ Isotype Ctrl Antibody (BioLegend #400314) and incubated for 20 min in the dark at room temperature. Next, cells were washed twice with 2 ml of Intracellular Staining Perm Wash Buffer, resuspended in 0.5 ml of cell suspension buffer, and analyzed by 17-color LSR Fortessa (4 Lasers). Data were analyzed by FlowJo (Version 10. 7. 1).
Cell Proliferation
Cells were analyzed following the protocol for the BD Pharmingen BrdU flow kit (APC-BrdU) (552598). Briefly, 1×106 cells were cultured in 1 ml mESC culture medium and supplemented with 10 μl of 1 mM BrdU solution. The treated cells were incubated for 30 min at 37°C, 5% CO2 in the incubator. Cells were fixed by BD Cytofix/Cytoperm Buffer and Permeabilized by BD Cytoperm Permeabilization Buffer, and then labelled with APC-conjugated anti-BrdU antibody. After washing, cells were resuspended in 7-AAD solution and analyzed by 17-color LSR Fortessa (4 Lasers). Data were analyzed by FlowJo (Version 10. 7. 1).
Cell Fractionation
mESCs were fractionated according to a previously published procedure56. Briefly, twenty million cells were collected and washed with PBS, then centrifuged at 300 g to collect the cell pellet. 2700 μl cold hypotonic buffer (10 mM HEPES-NaOH, pH 7.9, 10 mM KCl, 0.5 mM β-mercaptoethanol, 1.5 mM MgCl2) supplemented with 1x proteinase inhibitor cocktail (PIC) (Thermo Fisher #78446) was added to resuspend and lyse the cells and incubated on ice for 20 minutes. Then 54 μl of 10% NP-40 (Thermo Scientific #28324) was added to the cell suspension, briefly vortexed, and incubated on ice for another 2 minutes and centrifuged at 4°C at 2,400 g for 15 minutes. The supernatant was collected as cytoplasmic fraction. The pellet was washed twice by cold hypotonic buffer and then resuspended in nuclei lysis buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, 2 mM MgCl2, 0.5 mM EDTA) with PIC for 1 hour at 4°C on the rotor. The lysed nuclei were pelleted at 16,000 g for 15 minutes at 4°C, and the supernatant was collected as the soluble chromatin fraction. The pellet was resuspended in 1 ml Shearing Buffer D3 (1 mM EDTA, 10 mM Tris-HCl, pH 7.6, 0.1% SDS; from Covaris, Inc.) with PIC and sonicated for 720 s in a Covaris E220. The lysate was centrifuged at 16,000 g for 15 min, and the supernatant was collected as the chromatin fraction.
Cellular Fractionation Coupled with Immunoprecipitation (IP)
Cytoplasmic and soluble fractions were isolated as previous description (Cell Fractionation). Then, the nuclear pellets were resuspended in 1 ml of high salt buffer (HSB) containing 50 mM Tris-HCl (pH 7.5), 300 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1% NP-40 supplemented with 1 mM DTT, 1 mM PMSF and 1x PIC and incubated on rotator at 4°C for 1 hour. Nuclei were pelleted by centrifugation and the supernatant was collected as the insoluble chromatin fraction. To adjust the salt concentration, 2160 μl of hypotonic buffer extraction was supplemented with 66.8 μl of 5 M NaCl and then combined with 214 μl of 125 mM buffer extraction to make the final cytoplasmic+ soluble chromatin fractions for IP; 800 μl of insoluble chromatin fraction was supplemented with 800 μl high salt dilution buffer (50 mM Tris-HCl, pH 7.6, 1 mM EDTA) for IP experiments. Cellular fractions were incubated at 4°C overnight with 3 μg of the following antibodies: anti-SMARCA4 (ab110641, Abcam), anti-SMARCB1 (A301–087A, Bethyl) and rabbit mAb IgG XP isotype control (3900, Cell Signaling Technologies). The samples were then incubated with Dynabeads Protein G (10003D, Thermo Fisher Scientific) for 1 hour at 4°C. Beads were washed three times in Co-IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, 0.02% NaN3+ 1 mM PMSF+ 1x PIC) and eluted with 1x sample buffer (NP0007, Thermo Fisher Scientific) supplemented with 10 mM DTT (20290, Thermo Fisher Scientific).
Dephosphorylation Experiment
One hundred micrograms of protein lysate was treated with indicated units of Lambda protein phosphatase (#P0753, New England Biolabs) supplemented with 1 mM MnCl2. The reaction was incubated at 30°C for 30 min. Dephosphorylated products were used for downstream applications, such as western blot and Co-IP experiments in this study.
Western Blotting
Western blot analysis was performed using standard procedures. Samples were separated using a 4%- 12% Bis-Tris PAGE gel (Thermo Fisher Scientific, NP0336BOX), and transferred onto a PVDF membrane (Millipore, SE1M003M00). Membranes were blocked with 5% milk in TBST and incubated with primary antibody (H3 (CST, 9715, 1: 10,000); S10P H3 (Millipore, 06–570, 1:1,000); RNA POLII (Bethyl, A300–653A, 1: 500); SOX2 (R&D Systems, AF2018, 1: 200); EZH2 (CST, 5246, 1: 1000); SUZ12 (CST, 3737, 1: 1000); EED (Millipore, 17–10034, 1: 1000); ARID2 (SIGMA, SAB2702340–100UL, 1: 1000); PBRM1 (Bethyl, A700–019, 1: 1000); SMARCE1 (Bethyl, A300–810A, 1: 1000); SMARCB1 (Bethyl, A301–087A, 1: 1000); ARID1A (CST, 12354, 1: 1000), DPF2 (Invitrogen, PA5–21079, 1: 1000); BRD9 (Active Motif, 61537, 1: 1000); SMARCA4 (CST, 49360, 1: 1000); SNRP70 (Abcam, ab83306, 1: 1000); BRD7 (CST, 14910, 1: 1000); SMARCC1 (CST, 11956, 1: 1000)) overnight at 4°C. Membranes were washed 3 times with TBST and then incubated with Peroxidase-conjugated AffiniPure Donkey Anti-Goat IgG (Jackson Lab, 705–035–003, 1: 10,000) or Peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson Lab, 111–035–003, 1: 10,000) or Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson Lab, 115–035–003, 1: 10,000) for 1 hour at room temperature. Following secondary, membranes were then washed 3 times with TBST, once with TBS, and then imaged using a Li-Cor Odyssey CLx imaging system (LI-COR).
Live-Cell Imaging
For live-cell imaging experiments, cells were grown on gelatin-coated μ-Slide 8 Well high (ibidi, 80806). Live-cell imaging was performed using a Zeiss Axio Observer microscope equipped with temperature and CO2 control. Quantification of chromosome enrichment was performed using Fiji. Chromatin enrichment was calculated by log2 [Chr Intensity/WC intensity]57.
Density Sedimentation Gradients
Cells were harvested (40 million), vortexed with Hypotonic Buffer (10 mM HEPES-NaOH (pH 7.9), 10 mM KCl, 0.5 mM β-mercaptoethanol, 1.5 mM MgCl2), and incubated on ice for 20 min. Cells were further lysed by addition of 10% NP-40 and incubated for 2 minutes on ice. Nuclei were pelleted by centrifugation (2400x g for 15 minutes, 4°C) and the supernatant containing cytoplasmic proteins was removed. Nuclei were washed twice with Hypotonic Buffer and subsequently incubated in 150 mM Buffer (10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.5% NP-40, 1mM DTT, 1mM PMSF, 2 mM MgCl2) for 1 hour at 4°C with rotation. The nuclei were pelleted by centrifugation (16000x g for 15 minutes, 4°C) and the supernatant containing soluble nuclear proteins was removed. Nuclear pellets were resuspended in High Salt Buffer (50 mM Tris-HCl (pH 7.5), 300 mM KCl, 1 mM MgCl2, 1 mM EDTA, 1% NP-40, 1 mM DTT, 1 mM PMSF, 1x PIC). Homogenate was incubated for 1 hour at 4°C with rotation and the supernatant was collected following centrifugation (16000x g for 15 minutes, 4°C). 1 mg of nuclear extract was layered over a 14 mL 10–30% glycerol gradient prepared in a 14x 95 mm centrifuge tube (Beckman Coulter Cat. # 344060). Gradients were placed in a SW-40 Ti 40000 RPM rotor (Beckman Coulter) and centrifuged (40000x RPM for 16 hours, 4°C). Fractions were collected, subjected to gel electrophoresis, and western blotting analysis.
Alkaline Phosphatase Staining
Alkaline phosphatase expression in cells was examined using VECTOR Red Alkaline Phosphatase (Red AP) Substrate Kit (Vector Laboratories, SK-5100) according to the manufacturer’s protocol. Briefly, 2 drops of Reagent 1, Reagent 2, and Reagent 3 were sequentially added to 5 ml of 200 mM Tris-HCl, pH 8.2– 8.5, 0.1% Tween-20, and mixed gently between additions to generate the stain. Cells were rinsed once with PBS and then incubated with the stain for 20– 30 min at 37°C in the dark. The staining solution was removed, and samples were kept in PBS prior to imaging.
Direct Neural Differentiation
For monoculture differentiation, mouse ES cells were dissociated and plated onto 0.1% gelatin-coated 6-well plates at 2×104 cells/cm2 in RHB-A medium (TAKARA, Y40001). Medium was renewed daily. Samples were recorded and collected at Day 06 after induction.
Embryoid bodies (EBs) formation
For the formation of well-defined embryoid bodies (EBs), 1.8×106 cells resuspended in 5 ml EB culture medium (Knock-out DMEM (Gibco, 10829018) supplemented with 15% ES-qualified FBS (Gibco, 10439024), 1% GlutaMAX (Gibco, 35050061), 1% MEM Non-Essential Amino Acids (Gibco, 11140050), 0.1 mM beta- mercaptoethanol (Gibco, 21985023), 10 μM all-trans retinoic acid (R2625–50MG, Sigma-Aldrich)) and were seeded into a well of AggreWell™ 800 plate (34825, STEMCELL Technologies) following pretreatment instructions. Every two days, removed and replaced with 3.75 ml of fresh EB culture medium by slowly pipetting down the wall of the well.
RNA Isolation and Preparation
RNA was collected and purified using TRIzol according to the manufacturer’s protocol. RNA-seq libraries were prepared with KAPA RNA HyperPrep Kit with RiboErase (HMR) (KK8561) using standard protocols. Libraries were analyzed for insert size distribution using the 2100 BioAnalyzer High Sensitivity kit (Agilent), 4200 TapeStation D1000 ScreenTape assay (Agilent), or 5300 Fragment Analyzer NGS fragment kit (Agilent). RNA was sequenced at the Genome Sequencing Facility at St. Jude Children’s Research Hospital on Illumina HiSeq platform in paired-end mode with 100 bp per read.
Immunocytochemistry
Cells were fixed, permeabilized, incubated, stained and imaged as described previously56. Briefly, cells were washed twice with PBS and fixed with 4% PFA/PBS for 15 min at room temperature. Samples were permeabilized with 0.2% Triton X-100/PBS for 10 min at room temperature, then blocked with 3% BSA/PBS for 30 min at room temperature. Cells were incubated with primary antibody (OCT4 (Santa Cruz, sc-5279, 1: 50); SOX2 (R&D Systems, AF2018, 1: 20); NANOG (Active Motif, 61419, 1: 200), ESRRB (R&D Systems, PP-H6705–00, 1: 100); SOX1 (R&D Systems, AF3369, 1: 20), NES (MILLIPORE, MAB353, 1: 50), GABA (Invitrogen, PA5–32241, 1: 100), GFAP (Abcam, ab7260, 1: 1000)) overnight at 4°C. Fluorescence-conjugated species-specific secondary antibodies (Invitrogen) were used for visualization.
SMARCE1 Depletion/ Recovery Using 5-Ph-IAA
Using the AID 2.0 system, cells were arrested using 100 ng/ml nocodazole for 6 hours and 1 μM 5-Ph-IAA was added for the last 30 minutes of nocodazole exposure to induce the degradation of SMARCE1. After 6 hours, nocodazole was then washed out. At that point, SMARCE1 was barely detectable. Treatment with 5-Ph-IAA was continued for an additional 30 minutes at which point SMARCE1 was no longer detectable. Flow cytometry confirmed that the cells were still in mitosis (over 90%) when 5-Ph-IAA was removed. Following the removal of 5-Ph-IAA, SMARCE1 rapidly reaccumulated such that by 10 minutes, substantial levels of SMARCE1 were present. Leveraging this new system informed us that the absence of SMARCE1 was confined to mitosis. For each time point, cells were treated as indicated or collected for downstream experiments including nascent RNA-seq, western blot, cell cycle analysis as previously described in this methods section.
Mitotic depletion of SMARCE1 during a single cell cycle
Using the AID 2.0 system, cells were arrested using 100 ng/ml nocodazole for 6 hours and 1 μM 5-Ph-IAA was added for the last 60 minutes to induce the degradation of SMARCE1. Nocodazole and 5-Ph-IAA were then removed via three washing with warm (37°C) ES culture medium. Cells were then exposed to RHB-A medium to induce differentiation. Cells were harvested in TRIzol at the indicated times (Extended Data Fig. 10g) for RNA extraction. Methods for q-PCR has been described previously56. Q-PCR data are presented as the mean of four independent experiments with error bars indicating S.E.M. qPCR primer sequences are listed in Supplementary Table 2.
ChIP-seq
We performed mSWI/SNF, SOX2, and ESRRB chromatin immunoprecipitation using a modified protocol from the truChIP® Chromatin Shearing Kit (Covaris # PN 520154). In brief, 20 million cells were washed once with 1.5 ml of cold PBS, resuspended in 1.5 ml of room temperature Fixing Buffer A and supplemented with 3 μl of 1 M DSG (dissolved in DMSO) to fix cells for 30 minutes following by adding 150 μl of freshly prepared 11.1% formaldehyde solution to a final concentration of 1% to fix cells for an additional 5 minutes at room temperature, quenched using 87 μl of Quenching Buffer E at room temperature for another 5 min. Cells were lysed in Lysis Buffer B and washed in Wash Buffer C. The prepared nuclei were resuspended in 1 ml Shearing Buffer D3 and placed in a Covaris milliTUBE 1ml AFA Fiber and sonicated for 12 min on a Covaris E220 sonicator at standard setting (140 peak incident power (PIP), 5 W, 10% duty factor). The equivalent of 5 million cells was used in each immunoprecipitation. Spike-in Drosophila chromatin (Active Motif # 53083) and spike-in antibody (Active Motif # 61751) were used following the manufacturer’s protocols. Five micrograms of antibodies to the following were used: H3K4me3 (CST, 9751S); H3K27ac (Abcam, ab4729); H3K4me1 (Abcam, ab8895); H3K27me3 (CST, 9733); H3K36me3 (CST, 4909); H3K9me3 (Active Motif, 39161); H4K20me3 (Active Motif, 39671); SOX2 (R&D systems, AF2018), CTCF (Abcam, ab70303). Immunoprecipitates were washed in 150 and 500 mM NaCl wash buffer as well as a lithium chloride wash. Protein-DNA fragments were eluted using 200 ul of elution buffer (1% SDS, 0.1 M NaHCO3 in RNase, DNase-free water) shaking at 65°C for 30 minutes. Eluted protein-DNA complexes were de-crosslinked following addition of 8 μl of 5M NaCl, and 1 μl of RNase A (10 mg/ml) at 65°C for at least 6 hours and no longer than 16 hours. Samples were protease digested following addition of 4 μl of 0.5 M EDTA, 8 μl of 1 M Tris-HCl (pH 6.5) and 1 μl of 20 mg/ml Proteinase K at 45°C for another 2 hours. ChIP DNA was purified with MinElute PCR Purification Kit (Qiagen #28006). Library preparation was performed by using KAPA Hyper Prep Kit (KK8504) according to the manufacturer’s protocols. Sequencing was performed at the Genome Sequencing Facility at St. Jude Children’s Research Hospital on the Illumina HiSeq platform in single-end mode with 50 bp per read.
Cut& Run sequencing
SOX2, ESRRB, EZH2, ARID1A, SMARCA4 (BRG1), BRD9, SMARCB1, SMARCE1 CUT&RUN was performed as previously described58, with a few modifications. Briefly, 0.5 million mESCs were attached to concanavalin A-coated magnetic meads (Bangs Laboratories inc. #BP531) for each experiment. Cells were permeabilized with 0.01% digitonin (Millipore #300410–1GM) in a Wash Buffer (20 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 0.5 mM Spermidine and PIC), and then incubated with 0.5 ug primary antibodies against SOX2 (R&D systems, AF2018), ESRRB (R&D systems, PP-H6705–00), EZH2 (CST, 5246), ARID1A (SIGMA, HPA005456–100UL), SMARCA4 (Abcam, ab110641), BRD9 (Active Motif, 61537), SMARCB1 (Bethyl, A301–087A), SMARCE1 (Bethyl, A300–810A) at 4°C overnight on a rotator. The beads-cells mixture was washed three times with Digitonin Buffer (20 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 0.01% digitonin, and PIC), and resuspended in 50 ul Digitonin Buffer supplemented with 2.5 ul EpiCypher CUTANA pAG-MNase (EpiCypher, SKU: 15–1116) at room temperature for 10 min on a rotator. After two rounds of washing, beads were resuspended in 50 μl cold Digitonin Buffer. Next, 1 μl of cold 100 mM CaCl2 was added into the suspension and incubated on nutator for 2 hours at 4°C. Then, 33 μl Stop Buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/ml RNase A, 50 μg/ml glycogen) containing 0.5 ng of E. Coli Spike-in DNA (EpiCypher, 18–1401) was added while gently vortexing. Beads were incubated at 37°C for 10 minutes to release chromatin to supernatant and degrade RNA and then placed on a magnet stand for 2 minutes. The supernatant was collected, and target DNA was purified by the NEB Monarch DNA Cleanup Kit (TL1030L) as per manufacturer’s instructions. The sequencing library was prepared by using KAPA Hyper Prep Kit (KK8504) according to the manufacturer’s protocols. Sequencing was performed at the Genome Sequencing Facility in St. Jude Children’s Research Hospital on the Illumina HiSeq platform in paired-end mode with 75 bp per read.
Cut& Run- qPCR
DNA was extracted and purified as per the previous description. Subsequently, DNA was quantified by Qubit™ dsDNA HS and BR Assay Kits (Q32851, Invitrogen). The enrichment of DNA pulled down by IgG (Rabbit (DA1E) mAb IgG XP® Isotype Control, CST) or SMARCE1 (A300–810A, Bethyl) was determined by iTaq Universal SYBR Green Supermix (1725121, Bio-Rad Laboratories). Cut& Run- qPCR primer sequences are listed in Supplementary Table 2.
Custom Spike-in Control Sequence
We designed and generated our own biotinylated spike-in controls to normalize nascent-RNA populations since commercial spike-in are not biotinylated and thus would not be pulled down in our experiment. Two endogenous loci were chosen (Chr16= mm10: chr16:10188072–10188427 and ChrX= mm10: chrX: 7506352–7506647) that we found not to be transcribed in AB2.2 mouse ES cells, and that are unique in the genome. These sequences were synthesized by BioBasic, Sanger-sequenced to confirm their sequence, cloned into a TOPO vector, and transcribed in vitro (Ambion) with biotin-UTP.
EU Pulse-Labeling of mESCs and EU-RNA-Seq Library Construction
Mouse ES cells were pulse labeled with 1 mM EU in prewarmed ES culture medium for 45 min at 37°C, 5% CO2. The media was removed, and the cells were collected with TRIzol Reagent (Invitrogen). Total RNA was isolated using the standard TRIzol RNA extraction protocol (Invitrogen). Biotin-azide was then conjugated to EU with the Click-iT Nascent RNA Capture Kit (Thermo Fisher Scientific, C10365). Both custom biotinylated, spike-in control RNAs were then added to 1.7 μg of each biotinylated sample (5e-5 μg of Chr16 and 5e-4 μg of ChrX). Biotin-EU-RNA and spike-in controls, where applicable, were pulled down from total RNA using streptavidin-coated magnetic beads (Invitrogen) and purified by TRIzol. The sequencing library was prepared by using KAPA RNA HyperPrep Kit with RiboErase (HMR) (KK8561) according to the manufacturer’s protocols. Sequencing was performed at the Genome Sequencing Facility at St. Jude Children’s Research Hospital on the Illumina HiSeq platform in single-end mode with 75 bp per read.
ATAC-seq
ATAC-seq experiments were performed following the manufacturer protocol (Active Motif, 53150). In brief, 0.1 million Smarce1-MD(R42A) or Smarce1-MD cells released from mitosis and treated with either DMSO or BRM014 for 90 minutes were lysed, washed and tagmented at 37°C for 30 minutes shaking at 800 rpm. Tagmented DNA was purified and amplified for 10 cycles to generate the library, according to manufacturer guidelines. Sequencing was performed at the Genome Sequencing Facility at St. Jude Children’s Research Hospital on the Illumina HiSeq platform in paired-end mode with 75 bp per read.
Sequencing Analysis of ChIP-seq, Cut&Run and ATAC-seq
51bp single-end reads of ChIP-seq or 51bp paired-end reads for Cut&Run or 75 bp paired-end reads for ATAC were collected and aligned to mouse genome assembly mm10 using an internal AutoMapper pipeline. Briefly, the reads were trimmed by trim_galore (v0.4.4) and then aligned by BWA (version 0.7.17, default parameters). Duplicate reads were marked by bamsormadup (biobambam2, V2.0.87). Uniquely mapped reads for ChIP-seq were obtained using samtools (V1.10), with parameter “-q 10 -F1024”. For Cut&Run and ATAC-seq reads, properly paired uniquely mapped reads were obtained using samtools (V1.10, with parameters “-q 10 -F 1804”). We only kept Cut&Run fragments with length shorter than 2000 bp and nucleosome-free fragments for ATAC-seq with length smaller than 109 bp (100+ Tn5 adjustment) for further analysis. To generate high-confidence peaks for asynchronous and mitotic mouse ESCs, stringent peaks for each replicate were called by MACS2 (V 2.1.1, with default parameters using IgG (for Cut&Run) and Input (for ChIP-seq) as control) and filtered with fold enrichment cutoff: 5 and p-value threshold: 1e-9. Peaks that overlapped within replicates were merged (bedtools, V2.17.0, overlap cutoff: 1bp) and the merged peaks for the two cell stages were kept as high-confidence peaks for further analysis. Bigwig tracks were generated by RPM normalization and then substracted to the input/IgG control using bamCoverage and bamCompare from Deeptool (V3.2.1). It was also used to generate heatmaps with average signal sampled in 50 bp windows and 3kb surrounding flanking region. Asyn-and-mit-shared peaks were defined if high-confidence peaks in asynchronized and mitotic cells had 1 bp overlap, otherwise they were called Asyn-specific or mit-specific peaks. ChIPseeker (V1.26.2) was used to analyze genomic features of SMARCE1 and SMARCB1 peaks. Homer (V4.9.1) was employed for motif analysis with default parameters. Motif score was calculated using FIMO (V4.11.2) with parameters “—thresh 1e-3 –max-stored-scores 50000000”. We Scanned ESRRB motif MA0141.3 and SOX2 motif with a combination of MA0142.1 and MA0143 and then intersected these motifs with our ESRRB and SOX2 peaks. Gene ontology was done using online tool GREAT V4.0.4 with default parameters. Enrichment analysis of previously published data in ES cells was performed with LOLA (1.20.0), with all accessible sites (aggregated from mouse ENCODE ESC DNase I hypersensitive sites and all ATAC-seq peaks produced from this study) as a custom background.
Custom enhancers were identified through the utilization of predicted H3K27ac peaks59, which were pooled from replicate samples and subsequently stitched together within a 12.5kb proximity of one another. These enhancers were not permitted to overlap with the promoter regions (+-2kb) of genes. The E1 and E2 enhancers were further characterized by their overlap with predicted H3K27me3 peaks, with E1 enhancers defined as H3K27ac+/H3K27me3- and E2 enhancers defined as H3K27ac+/H3K27me3+. Additionally, H3K27me3 and H3K4me3 peaks were used to classify the status of promoters, with bivalent promoters defined as H3K4me3+/H3K27me3+, poised promoters defined as H3K4me3-/H3K27me3+ and active promoters defined as H3K4me3+/H3K27me3-.
ChromHMM Analysis
De novo chromatin states modeling was performed using chromHMM (V1.20) with eight histone modifications (H3K4me1, H3K4me3, H3K9me3, H3K27me3, H3K27ac, H3K36me3, H3K9me3 and H4K20me3) from ChIP-seq data for asynchronous and mitotic mouse ESCs. Non-duplicated aligned reads were processed with default parameters, and the read counts were calculated in non-overlapping 200 bp bins across the genome. The 15-state model was then chosen for downstream analysis as it represented major characteristics of chromatin states while keeping minimal redundancy. The enrichment of CUT&RUN peaks within each chromatin states was computed and plotted by OverlapEnrichment with default parameters.
Sequencing Analysis of Bulk RNA-seq
Total stranded RNA sequencing data were mapped to mouse genome assembly mm10 using an internal AutoMapper pipeline. Briefly, the raw reads were trimmed with trim galore (V0.60) and mapped to mouse genome mm10 using STAR V2.7.1a. Then the read counts were computed using RSEM (V1.3.0) based on GENCODE vM23. Counts were further normalized and differentially expressed genes were calculated using DESeq2 (V1.30.1). Regularized log transformation of normalized read counts (rlog) value was calculated to represent genes’ expression. Genes with absolute fold change ≥2.0 and p-value <0.05 were considered as differentially expressed genes. Gene ontology analysis was conducted by R package ClusterProfiler (V3.18.1) and Gene set enrichment analysis (GSEA, V4.1.0) was performed on gene sets from Gene Ontology (GO:0099536 and GO:0019827) using the pre-ranked option default parameters. RPM (Reads Per Million) was applied to normalize read counts in bigwig tracks.
Sequencing Analysis of Nascent RNA-seq
Nascent RNA-seq was also mapped to mouse genome assembly mm10 as described above in the bulk RNA-seq analysis section. Spike-in reads were extracted from designed genomic regions ChrX:7505977–7506984 and Chr16:10188072–10188427. Spike-in reads were used to normalize sequencing depth for each sample to generate sample-specific scalars (Supplementary Table 1)60. Scalars were normalized to 5 if the value was even larger than 5. Read counts for each nascent RNA-seq library were then multiplied by their spike-in scalar prior to further analysis. Principal component analysis (PCA) was conducted after removing the batch effect between the MD and the AID experiments using ComBat from R package sva (V3.38.0). RPM was applied to the normalized read counts to generate bigwig tracks.
Quantification and Statistical Analysis
At least three biological replicates were used in each experiment unless otherwise stated. P values less than 0.05 were considered significant. Data are presented as mean± s.e.m. or SD. Two- tailed paired or unpaired Student’s t- Tests were applied for calculating the P value indicated in the figure legends to assess the statistical significance of differences between groups. Fisher exact tests were applied for calculating the P value to assess the relative enrichment for overlap analysis. One-way ANOVA was performed to compare the cell cycle phase percentages among cell clones in each genotype. For box plots, the center line represents the median, the box limits show the upper and lower quartiles, whiskers represent 1.5x the interquartile range. To evaluate the scatter of the data points around the fitted regression line, R2 is used in our scatter plots which indicates the percentage of the dependent variation that a linear model explains. Pearson correlation coefficients (PCC) were calculated to assess correlation.
Extended Data
Extended Data Figure 1. Cell cycle analysis and subcellular SWI/SNF interactions in asynchronous and mitotic mouse ES cells.
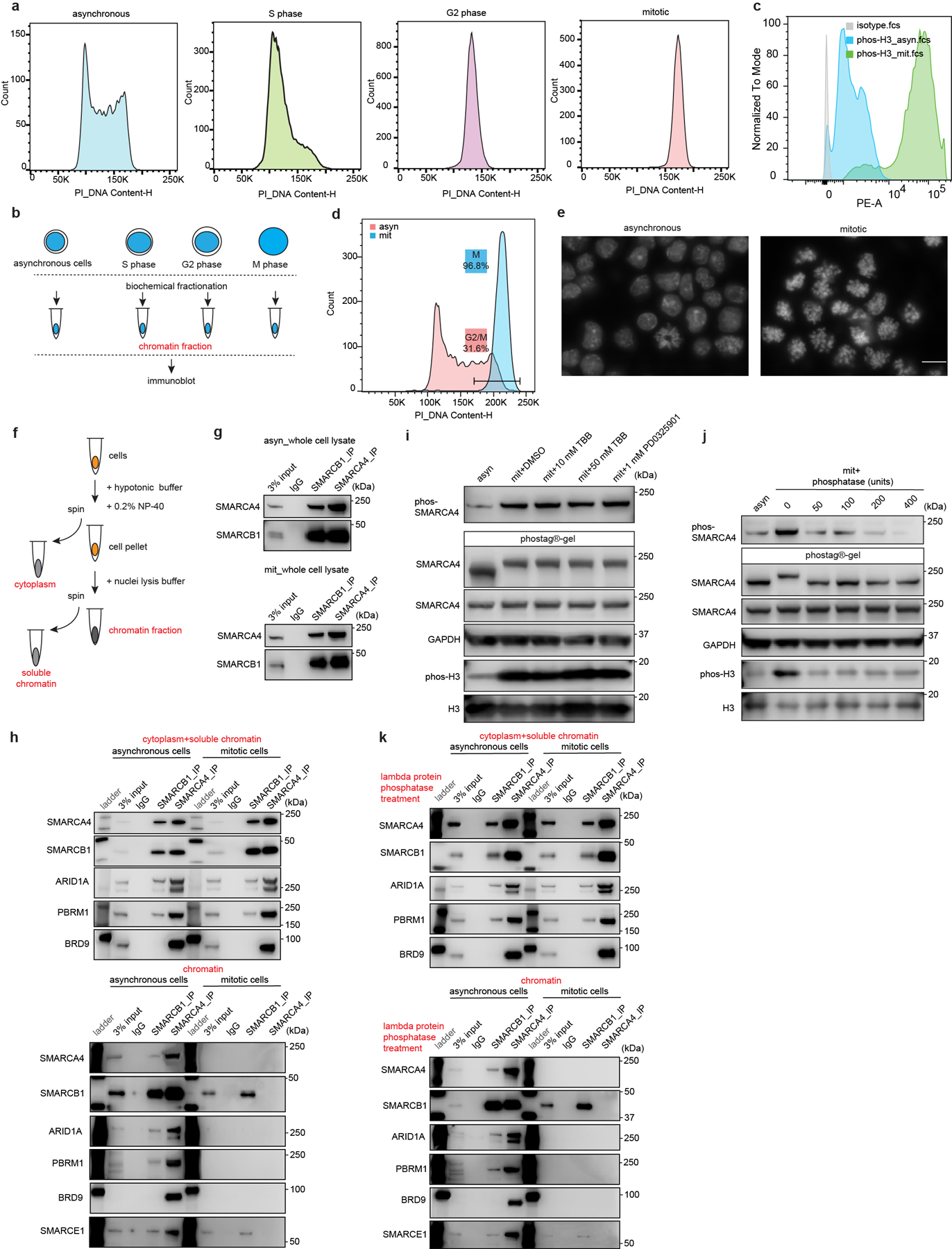
a, Representative flow cytometry analysis showing DNA contents of asynchronous mouse ES cells and cells synchronized at S, G2, and mitotic phase. b, Schematic for extraction of chromatin fraction from asynchronous mouse ES cells and populations synchronized at S, G2, and mitosis. c, Representative flow cytometry showing the distribution of phosphorylated Serine 10 of histone H3 between asynchronous and synchronized mitotic mouse ES cells. d, Representative flow cytometry analysis showing DNA contents and purity of synchronized mitotic mouse ES cells. e, Representative DAPI staining confirming the high purity of synchronized mitotic cells. Scale bar: 10 μm. f, Schematic showing the isolation of cytoplasm, soluble chromatin, and chromatin fractions from mouse ES cells. g, Immunoprecipitation (IP) of SMARCA4 and SMARCB1 in whole cell lysate from asynchronous (top) and mitotic (bottom) mouse ES cells. h, IP of SMARCB1 and SMARCA4 in the cytoplasmic+ soluble chromatin fractions (top), and chromatin fraction (bottom) isolated from asynchronous and mitotic mouse ES cells. i, Immunoblots in whole cell lysate of asynchronous (lane 1) and mitotic (other lanes) mouse ES cells treated with TBB (CK2 inhibitor) and PD0325901 (ERK1 inhibitor) for 6 hours, as indicated. Samples were separated either in NuPAGE™ 4 to 12% Bis-Tris gels or in the case of the labeled SMARCA4 sample in Phos-tag™ (50 μmol/L) precast gels. j, Immunoblot in whole cell lysate of asynchronous (lane 1) or mitotic (other lanes) mouse ES cells. Lysates were treated with increasing doses of lambda protein phosphatase as indicated. Samples were separated in NuPAGE™ 4 to 12% Bis-Tris gels or in the case of the labeled SMARCA4 sample in Phos-tag™ (50 μmol/L) precast gels. k, IP of SMARCB1 and SMARCA4 in the lambda protein phosphatase treated cytoplasmic+ soluble chromatin fractions (top), and chromatin fraction (bottom) isolated from asynchronous and mitotic mouse ES cells. Data are representative of two (a, c, d, e, h, i, j, k) and three (g) independent experiments.
Extended Data Figure 2. Generation and live-cell imaging of endogenous C-terminal EGFP-tagged SOX2, SMARCA4, SMARCB1, and SMARCE1 mouse ES cells.
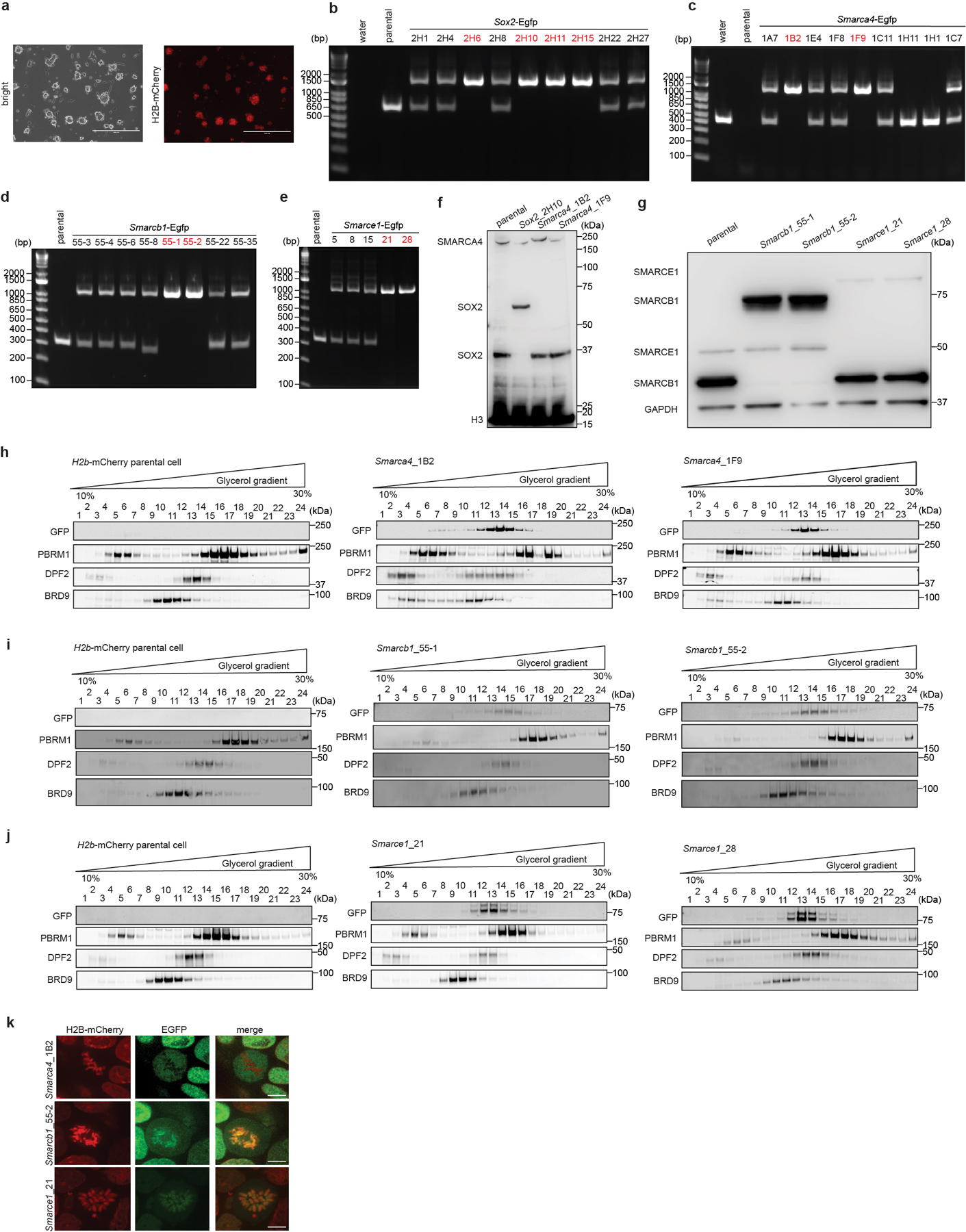
a, Images showing mouse ES cells stably expressing H2B-mCherry. b, c, d, e, PCR-based genotyping assay validating the generation of the Sox2-Egfp (b), Smarca4-Egfp (c), Smarcb1-Egfp (d), and Smarce1-Egfp (e) homozygous knock-in mouse ES cells. f, g, Western blot validating SOX2-EGFP and SMARCA4-EGFP (f), and SMARCB1-EGFP and SMARCE1-EGFP (g) protein expression, with H3 or GAPDH as loading control. h, i, j, Glycerol gradient and immunoblot performed on H2B-mCherry parental cells and Smarca4-Egfp (h), Smarcb1-Egfp (i), and Smarce1-Egfp (j) clones of mouse ES cells which were used for live-cell imaging. k, Representative live-cell imaging of asynchronous mouse ES cells stably expressing H2B- mCherry with endogenously tagged EGFP at the C-terminal of SMARCA4, SMARCB1, and SMARCE1, respectively. Scale bar: 10 μm. Data are representative of single experiments that assess several independent clones for genotype (b, c, d, e, f, g) and live-cell imaging (k). Data are representative of two independent replicates (h, i, j).
Extended Data Figure 3. Binding of SOX2, ESRRB and SWI/SNF subunits in asynchronous and mitotic cells.
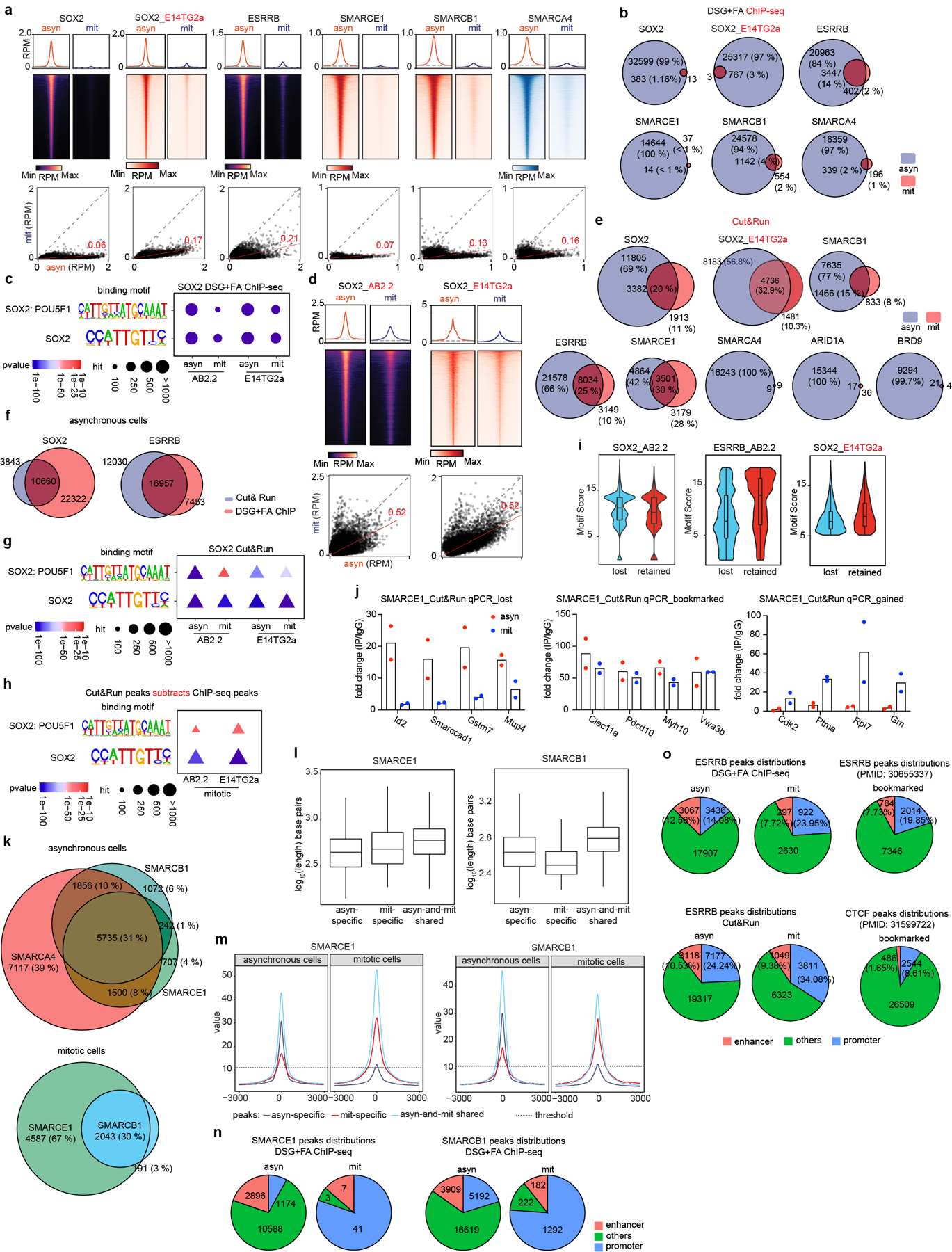
a, (top) Average binding profile of DSG+ FA cross-linked ChIP-seq signal at the indicated binding regions (± 3000-bp peak summit) identified in asynchronous (asyn) and mitotic (mit) AB2.2 and E14TG2a (as indicated) mouse ES cells. (bottom) Scatter plots of DSG+ FA ChIP-seq signal in reads per million at the designated regions (± 250-bp peak summit) for asynchronous and mitotic mouse ES cells. Linear regression of RPM value of asynchronous cells divided by the value for mitotic cells were estimated and regression slopes are shown in red. Grey dashed lines indicate the random background. b, Venn diagrams showing the overlap of DSG+ FA ChIP-seq peaks in asynchronous (blue) and mitotic (red) AB2.2 and E14TG2a mouse ES cells as indicated. c, SOX2: POU5F1 co-binding motifs and SOX2 binding motifs identified from SOX2 DSG+ FA ChIP-seq in asynchronous and mitotic AB2.2 and E14TG2a mouse ES cells as indicated. P-value was calculated by two- sided Fisher’s exact test. d, (top) Average binding profile of Cut&Run signal at SOX2 binding regions (± 3000-bp peak summit) identified in asynchronous and mitotic AB2.2 or E14TG2a cells. (bottom) Scatter plots of Cut&Run signal in reads per million at the designated regions (± 500-bp peak summit) for asynchronous and mitotic mouse ES cells. Linear regression of RPM value of asynchronous cells divided by the value for mitotic cells were estimated and regression slopes are shown in red. Grey dashed lines indicate the random background. e, Venn diagrams showing the overlap of Cut&Run peaks of designated factors in asynchronous (blue) and mitotic (red) in AB2.2 or E14TG2a mouse ES cells as indicated. f, Venn diagram showing the overlaps of SOX2 peaks (left) and ESRRB peaks (right) identified from Cut&Run (blue) and DSG+ FA ChIP-seq (red) in asynchronous mouse ES cells. g, SOX2: POU5F1 co-binding motifs and SOX2 binding motifs identified from SOX2 Cut&Run in asynchronous and mitotic AB2.2 and E14TG2a mouse ES cells as indicated. P-value was calculated by two- sided Fisher’s exact test. h, SOX2: POU5F1 co-binding motifs and SOX2 binding motifs identified from SOX2 Cut&Run peaks subtracting SOX2 DSG+ FA ChIP-seq peaks in mitotic AB2.2 and E14TG2a cells as indicated. P-value was calculated by two- sided Fisher’s exact test. i, Violin plots depicting the FIMO-called best motif score per SOX2 or ESRRB peak in sites losing binding in mitosis (lost) or retaining binding (retained), performed by Cut&Run in the indicated mouse ES cell lines. j, Cut&Run-qPCR validating the lost (asyn-specific), bookmarked (asyn- and mit- shared), and gained (mit-specific) SMARCE1 peaks in asynchronous and mitotic mouse ES cells. k, Venn diagram showing the overlap of SMARCA4, SMARCB1, and SMARCE1 Cut&Run peaks in asynchronous cells (top) and the overlap between SMARCE1 and SMARCB1 Cut&Run peaks in mitotic mouse ES cells (bottom). l, SMARCE1 (left) and SMARCB1 (right) peak length for asyn-specific, mit-specific, and asyn- and mit- shared peaks in asynchronous and mitotic mouse ES cells. m, Average SMARCE1 (left) and SMARCB1 (right) binding profiles at asyn-specific, mit-specific, and asyn- and mit- shared regions. n, To confirm localization of SMARCE1 and SMARCB1 as detected by Cut&Run, ChIP-seq was utilized. The distributions of SMARCE1 (left) and SMARCB1 (right) via ChIP-seq (using DSG+ FA fixation) at promoter and enhancer regions in asynchronous and mitotic mouse ES cells. o, The distributions of ESRRB peaks from DSG+ FA ChIP-seq and Cut&Run in asynchronous and mitotic AB2.2 mouse ES cells. Bookmarked ESRRB and CTCF peaks are as previously reported. All data are compiled from two biological replicates. Statistical analysis was performed using two- sided Fisher’s exact test (c, g, h). Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile (i, l).
Extended Data Figure 4. The comprehensive profiles of SMARCE1 and SMARCB1 bindings in asynchronous and mitotic ES cells.
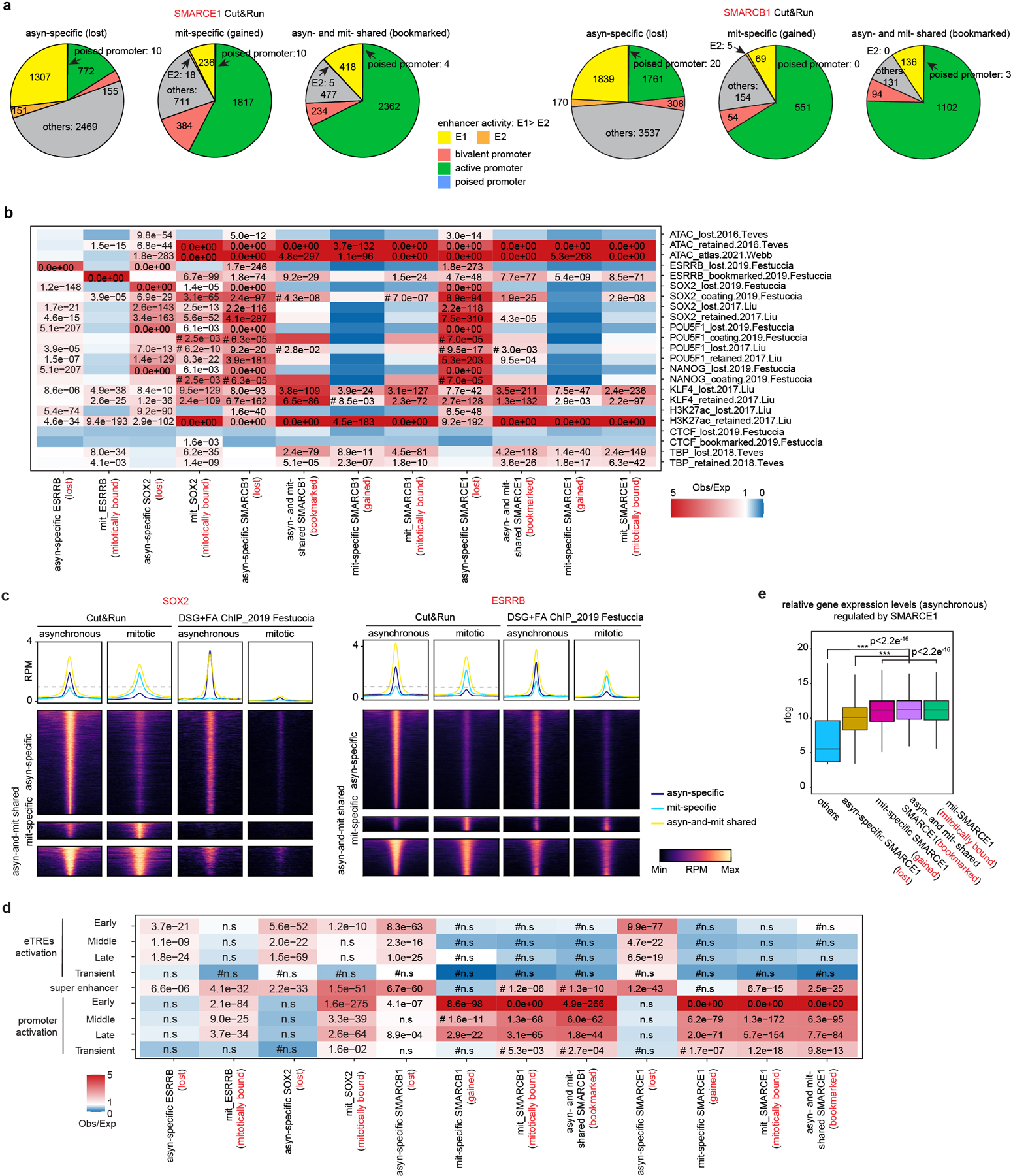
a, The distributions of asyn-specific, mit-specific, asyn- and mit-shared SMARCE1 (left) and SMARCB1 (right) peaks identified from Cut&Run at enhancer and promoter regions in asynchronous and mitotic AB2.2 cells. b, Relative enrichment or depletion of the lost (asynchronous-specific), retained (gained+ bookmarked), gained (mitosis-specific), and bookmarked (present in both asynchronous and mitosis) peaks for ESRRB, SOX2, SMARCB1 and SMARCE1 Cut&Run at the lost, retained, bookmarked, or coating regions of published ATAC signal and indicated factors. Color indicates ratio of observed (Obs) to expected (Exp) frequency, and p-value (two-sided Fisher’s exact test) is indicated if significant (p<0.01, 0.0e00 means p-value<e−350). Comparison using <100 overlapping peaks are denoted with a hash mark (#). c, Average binding profile of Cut&Run and DSG+FA ChIP-seq signal at the indicated binding regions (± 3000-bp peak summit) identified in asynchronous and mitotic mouse ES cells. Average binding profiles represent reads per million (RPM); the y-axis is scaled by median asynchronous binding. d, Relative enrichment or depletion of the lost (asynchronous-specific), retained (gained+ bookmarked), gained (mitosis-specific), and bookmarked (present in both asynchronous and mitosis) peaks for ESRRB, SOX2, SMARCB1 and SMARCE1 Cut&Run at early, middle, late, and transient reactivated enhancer transcriptional regulatory elements (eTREs), super enhancers and promoters. Color indicates ratio of observed (Obs) to expected (Exp) frequency, and p-value (two-sided Fisher’s exact test) is indicated if significant (p<0.01, 0.0e00 means p-value<e−350). Comparison using <100 overlapping peaks are denoted with a hash mark (#). e, Boxplot showing the relative expression levels in asynchronous cells of genes based upon the indicating binding status of SMARCE1. Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile. Data are compiled from two (a, b, c, d) and eight (e) replicates. Statistical analysis was performed using two-tailed unpaired Student’s t- Test, *** p-value< 0.001 (e).
Extended Data Figure 5. Generation of endogenous MD and MD (R42A) degron fused SMARCE1 mouse ES cells.
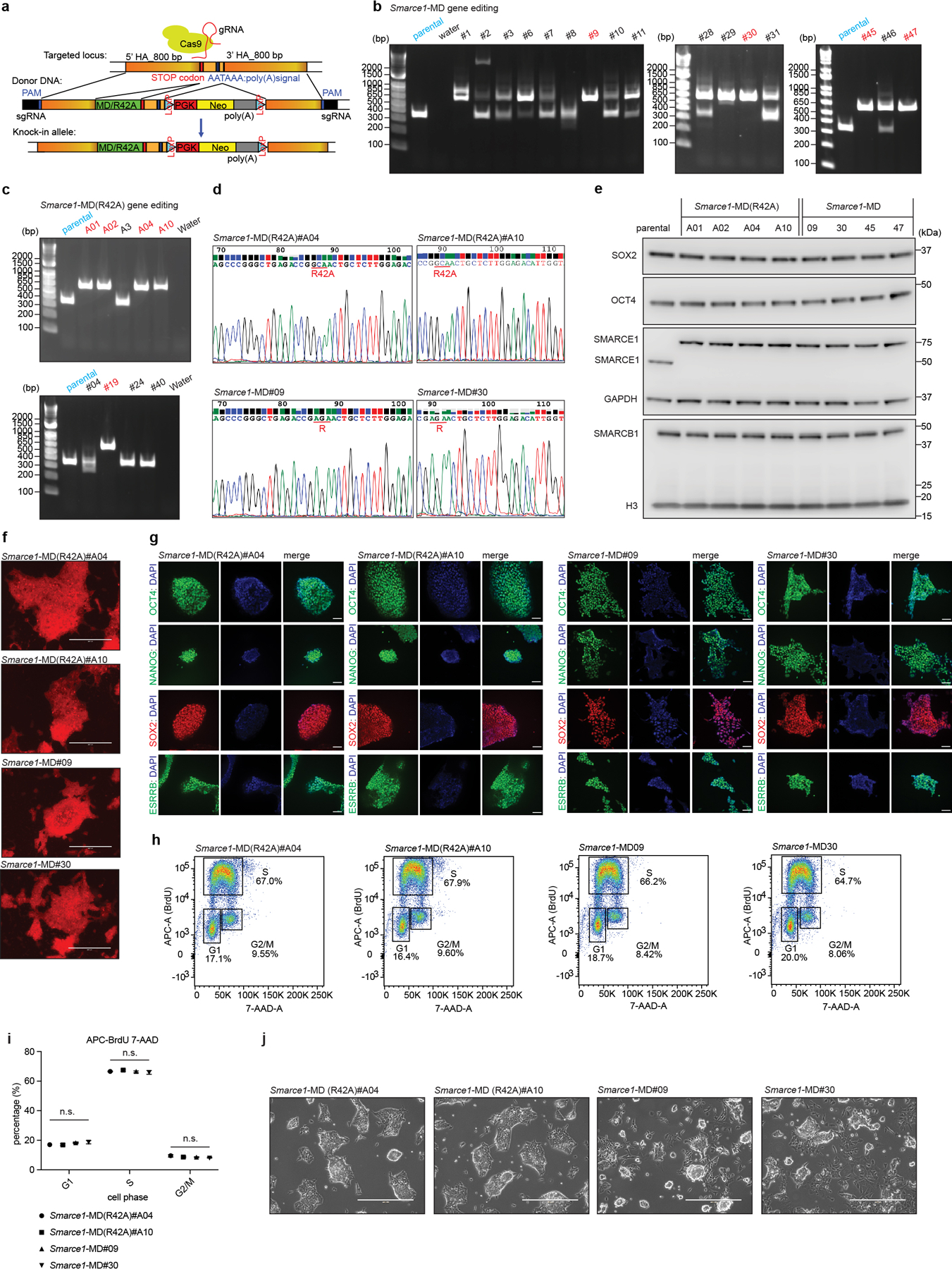
a, Schematic showing the strategy for MD and MD (R42A) knock-in at the C-terminal end of SMARCE1 in mouse ES cells by CRISPR-Cas9. b, c, PCR-based genotyping assay validating the generation of Smarce1-MD (b) and Smarce1-MD (R42A) (c) mouse ES cell lines. d, Sanger-sequencing validating the mutant MD degron (R42A: AGA to GCA) and the wild-type MD degron (R: AGA) sequences in Smarce1- MD(R42A) #A04, #A10 and Smarce1-MD#09, #30 mouse ES cells, respectively. e, Representative immunoblot showing the protein levels of SMARCE1, SMARCB1, SOX2, OCT4 in parental, Smarce1- MD(R42A) #A01, #A02, #A04, #A10, and Smarce1- MD#09, #30, #45, #47 mouse ES cells. GAPDH and H3 were used as loading controls. f, Alkaline phosphatase staining in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. g, IHC staining of core pluripotency factors (OCT4, NANOG, SOX2, ESRRB) in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. DAPI was used to indicate nuclei. h, Cell proliferation of Smarce1- MD(R42A) and Smarce1-MD mouse ES cells measuring by BrdU and 7-AAD. i, Statistics of cell proliferation from (h). j, Cell morphologies of Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. Data are representative from one (b, c, d, f, g,) or two replicates (e,); or compiled from three biological replicates (h, i). Data are shown as mean± s.e.m, n= 3 biologically independent experiments. Differences are not statistically significant (n.s.) determined by one-way ANOVA (i). Scale bars: 400 μm (f, g, j).
Extended Data Figure 6. Characterization of the MD degron cells and development of the custom biotin-RNA spike-in for nascent RNA-seq.
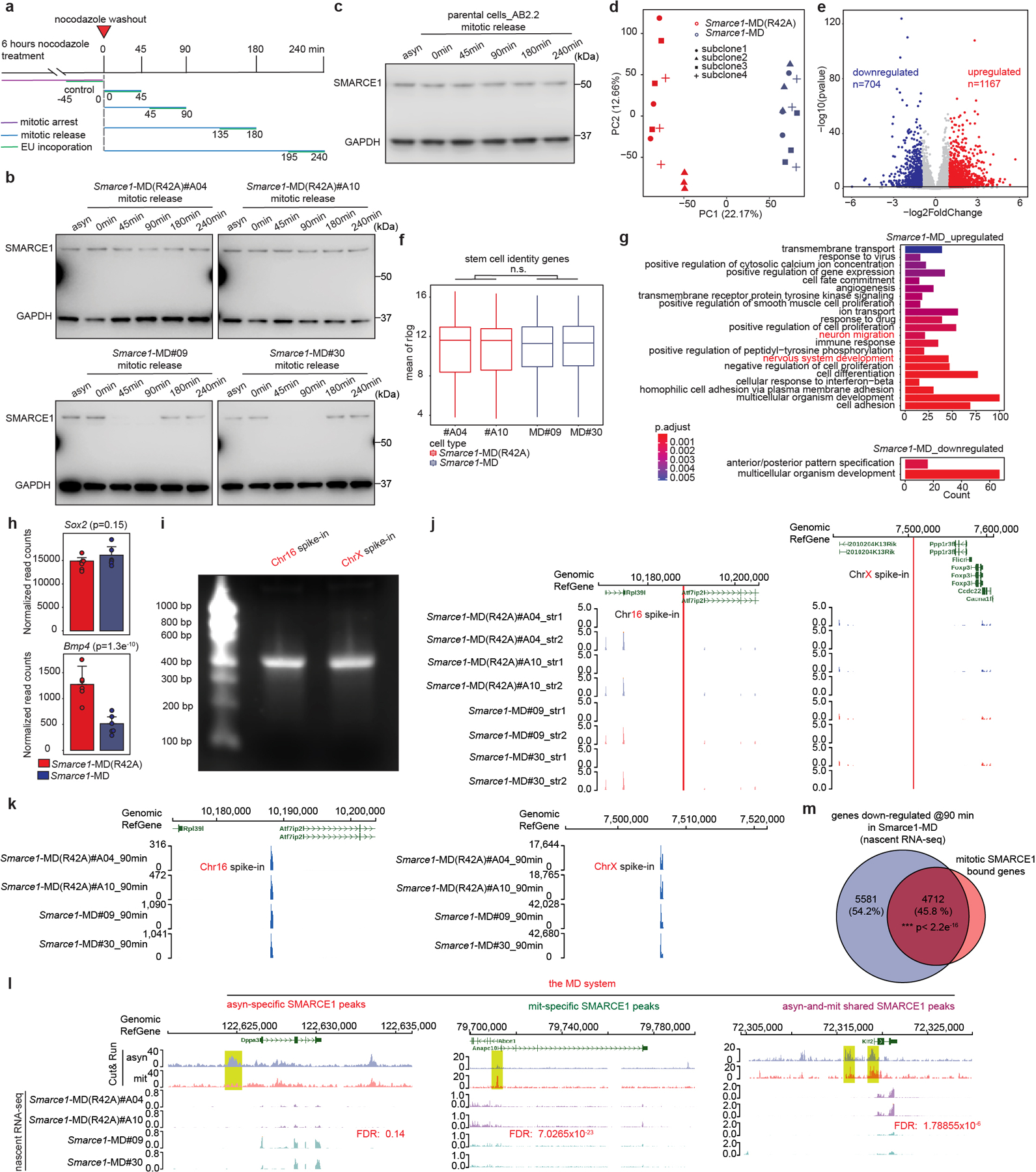
a, Schematic for EU-pulse-labeling of newly synthesized transcripts during mitosis and mitotic release in the MD degron system. b, Representative immunoblot measuring SMARCE1 levels in mouse ES cells expressing endogenous SMARCE1- MD(R42A) (left) or SMARCE1-MD (right) in asynchronous cells, mitotic cells and after release from mitotic arrest, with GAPDH as loading control. c, Representative immunoblot measuring SMARCE1 levels in parental AB2.2 mouse ES cells in asynchronous and after release from mitotic arrest, with GAPDH as loading control. d, PCA plot showing RNA-seq data from Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. e, Volcano plot showing differentially expressed genes in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. P-value in differentially expressed genes test was calculated using Wald test from DESeq2. f, Box plot showing stem cell associated (GO: 0019827) gene expression levels in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile. g, GO analysis of differentially expressed genes in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells. Over-representation test was used to calculated GO term enrichment with FDR for multiple test correction. h, Bar plot showing the expression levels of Sox2 (top) and Bmp4 (bottom) in Smarce1- MD(R42A) and Smarce1- MD mouse ES cells. i, Agarose gel showing the highly purified Chr16 spike-in and ChrX spike-in for nascent RNA-seq experiments. j, Red lines indicating the locations and reads of custom designed nascent RNA spike-ins on chromosome 16 (chr16: 10188072– 10188427) (left) and chromosome X (ChrX: 7506352– 7506647) (right) in mouse ES RNA-seq, respectively. k, Representative examples of nascent RNA-seq reads indicating high enrichment of custom designed RNA spike-in reads on chromosome 16 (left) and chromosome X (right) in the nascent RNA-seq samples at 90 min after mitotic release. l, Representative SMARCE1 peaks and associated nascent RNA profiles in Smarce1- MD (R42A) and Smarce1- MD cells at 90 min after mitotic release. m, Venn diagram showing the overlap between mitotic SMARCE1 bound genes and down-regulated genes identified at 90 min in Smarce1-MD cells by nascent RNA-seq. Data were processed with ClusterProfiler with default parameters (g). Data are representative of one (i), two (b, c, j, k, l, m) or three (d, e, f, g, h) independent replicates. Not significant: n.s., with a two-tailed unpaired Student’s t-Test (f). Data are presented as the mean expression (rlog)± SD, n= 3 biologically independent experiments; p-value was calculated using two-tailed unpaired Student’s t-Test (h). P-value determined by two-sided Fisher’s exact test is indicated (m).
Extended Data Figure 7. Generation and characterization of endogenous auxin-inducible degradation (AID) degron fused to SMARCE1 in mouse ES cells.
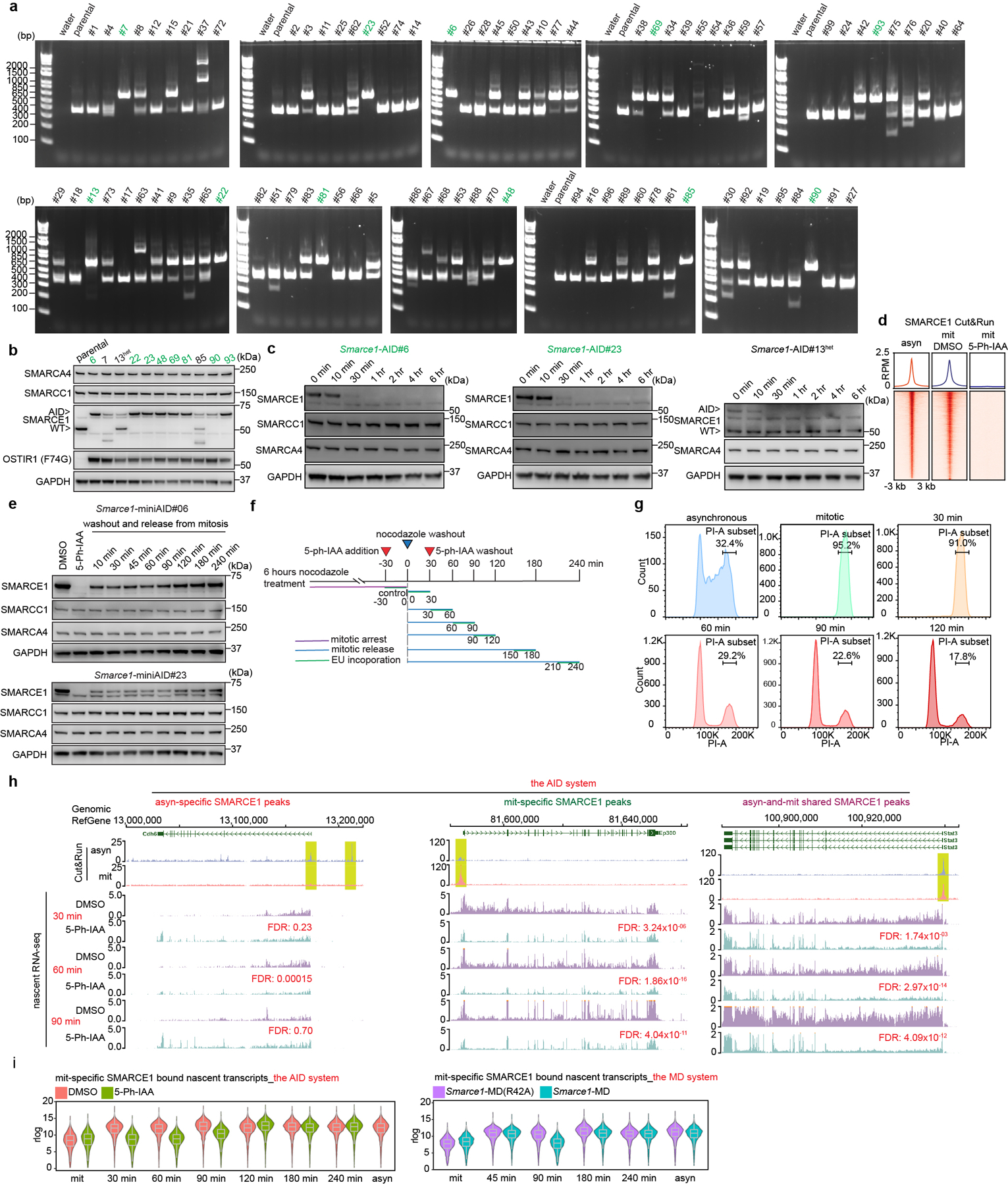
a, PCR-base genotyping assay validating the generation of Smarce1-AID homozygous knock-in mouse ES cell lines (green). b, Representative immunoblot showing the protein levels in the homozygous knock-in clones from (a); clone 13 is a heterozygous one (#13het), clone 7 and clone 85 are off-target edited. c, Representative immunoblot in Smarce1-AID clones #6, #23, and #13het treated with 1 μM 5-Ph-IAA for the indicated time following release from mitotic arrest. GAPDH was used as a loading control. d, Average binding profiles of SMARCE1 Cut&Run signal in asynchronous Smarce1-AID cells (clones #6 and #23) and mitotic cells treated with DMSO or 1 μM 5-Ph-IAA for 1 hour. e, Representative immunoblot in mitotic Smarce1-AID cells (clones #6 and #23) treated with DMSO or 1 μM 5-Ph-IAA for 1 hour (lanes 1 and 2) and in cells that underwent washout and release from mitotic arrest for the indicated time periods. f, Schematic for EU-pulse-labeling of newly synthesized transcripts and 5-Ph-IAA treatment during mitosis and mitotic release in the AID degron system. g, Representative flow cytometry plot showing DNA contents of asynchronous cells, synchronized mitotic cells, and cells released from mitotic arrest for the indicated time. h, Representative SMARCE1 peaks and associated nascent RNA profiles in DMSO and 1μM 5-Ph-IAA treated Smarce1-AID (clones 6 and 23) mouse ES cells at 30 min, 60 min and 90 min after mitotic release. i, Violin plot showing the nascent transcriptional levels of genes bound by mitosis-specific (gained) SMARCE1 in asynchronous and mitotic cells and cells released from mitotic arrest in the AID system (left) and in the MD system (right). Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile. Data are representative or compiled from one (a) or two replicates (b, c, d, e, g, h, i).
Extended Data Figure 8.
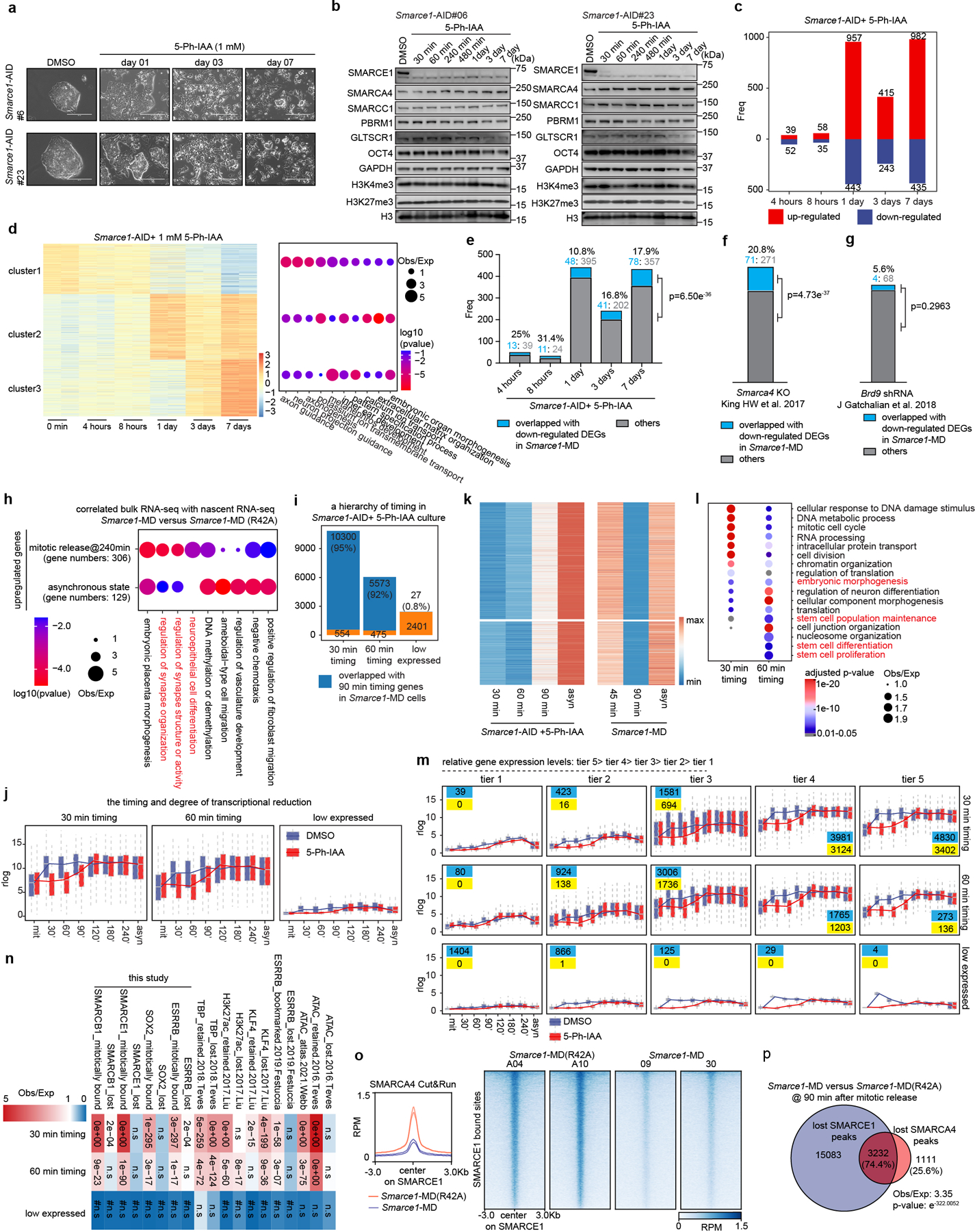
The timing and degree of nascent transcription changes in mitotic SMARCE1 depleted mouse ES cells. a, Cell morphologies of DMSO treated Smarce1-AID cells (clones #6 and #23) and the cells treated with 1 μM 5-Ph-IAA for the indicated days. b, Representative immunoblot of Smarce1-AID cells (clones #6 and #23) treated with DMSO or 1 μM 5-Ph-IAA. c, Bar plot showing the numbers of differentially expressed genes (DEGs) identified from Smarce1-AID cells (clones #6 and #23) treated with 1 μM 5-Ph-IAA for the indicated time. d, (left) Heatmap showing DEGs identified from Smarce1-AID cells (clones #6 and #23) treated with 1 μM 5-Ph-IAA for the indicated time. The DEGs were classified into three clusters by k-means clustering. Color bar indicates scaled z score of gene expression. (right) GO analysis indicating pathways enriched for the three clusters. Size of the circle represents ratio of observed (Obs) versus expected (Exp) frequency, and p-value was calculated by two- sided Fisher’s exact test. e, Bar plot showing the contributing number of genes identified from down-regulated DEGs (bulk RNA-seq) in asynchronous Smarce1-MD cells (in which SMARCE1 is solely missing in mitotic cells) to the down-regulated genes identified from Smarce1-AID cells treated with 5-Ph-IAA (in which SMARCE1 is missing throughout the cell cycle). f, Bar plot showing the contributing number of genes identified from down-regulated DEGs (bulk RNA-seq) in asynchronous Smarce1-MD cells to the down-regulated genes identified from Smarca4 knockout (KO) mouse ES cells (King HW et al., 2017). g, Bar plot showing the contributing number of genes identified from down-regulated DEGs (bulk RNA-seq) in asynchronous Smarce1-MD cells to the down-regulated genes identified from Brd9 shRNA transfected mouse ES cells (J Gatchalian et al., 2018). h, GO analysis of relative enrichment or depletion of upregulated genes identified from bulk RNA-seq in the scenario of nascent RNA-seq at 240 min after mitotic release and in asynchronous MD cells. Size of circles indicates ratio of observed (Obs) versus expected (Exp) frequency, and color presents p-value calculated by two-sided Fisher’s exact test. i, The downregulated genes that reached the lowest expression levels in 5-Ph-IAA treated Smarce1-AID mouse ES cells at the indicated time point were defined as “30 min timing” and “60 min timing” genes, respectively; “low expressed” refers to the genes that were consistently lowly expressed (rlog< 4) at all the time points. Genes within the “30 min timing” and “60 min timing” groups from the nascent RNA-seq of 5-Ph-IAA treated Smarce1-AID cells that overlapped with the genes in “90 min timing” group from the nascent transcripts of Smarce1-MD at 90 min are shown in blue. Gene numbers are shown within the bar plot. j, Box plot showing the nascent transcript levels of “30 min timing”, “60 min timing”, and “low expressed” genes at all time points examined in Smarce1-AID cells treated with DMSO or 5-Ph-IAA. k, Heatmap of the DEGs identified from nascent RNA-seq in both the AID system (left) and the MD system (right). Color bar indicates z scaled rlog value. l, GO analysis of “30 min timing” and “60 min timing” genes. Size of the circle represents ratio of observed (Obs) versus expected (Exp) frequency, and p-value was calculated by two- sided Fisher’s exact test. m, Box plot of “30 min timing”, “60 min timing”, and “low expressed” genes separated into 5 tiers based upon nascent gene expression levels. Box plots depict the median transcriptional activity across the time course; in each cluster, the total number of genes is listed in blue and the number of genes that differ significantly between 5-Ph-IAA and DMSO conditions is listed in yellow. n, Relative enrichment or depletion of the lost, retained, bookmarked, and mitotically bound peaks for chromatin accessibility and the indicated factors at “30 min timing”, “60 min timing”, and “low expressed” genes (y-axis). Color indicates ratio of observed (Obs) versus expected (Exp) frequency, and p-value (two-sided Fisher’s extract test) is indicated. Comparisons using <100 overlapping peaks are denoted with a hash mark (#). o, (left) Average binding profiles of SMARCA4 Cut&Run signal on SMARCE1 binding sites in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells at 90 min after mitotic release. (right) Heat maps of SMARCA4 Cut&Run at SMARCE1 binding sites in Smarce1- MD(R42A) and Smarce1-MD mouse ES cells at 90 min after mitotic release. p, Venn diagram showing the overlap between lost SMARCE1 peaks and lost SMARCA4 peaks identified from Smarce1-MD versus Smarce1-MD(R42A) cells at 90 min after mitotic release. Data are compiled from two biological replicates for each clone (Smarce1-AID#06, #23) (c, d, e, f, g, i, j, m). Center lines denote medians; box limits 25th- 75th percentile; whiskers 5th- 95th percentile (j, m). Statistical analysis was performed using two-sided Fisher exact test (e, f, g). The ratio of observed (Obs) versus expected (Exp) frequency is shown, and p-value determined by two-sided Fisher’s exact test is indicated (p).
Extended Data Figure 9. Subsequent Activation of Genes Bookmarked by SMARCE1 is ATPase Dependent.
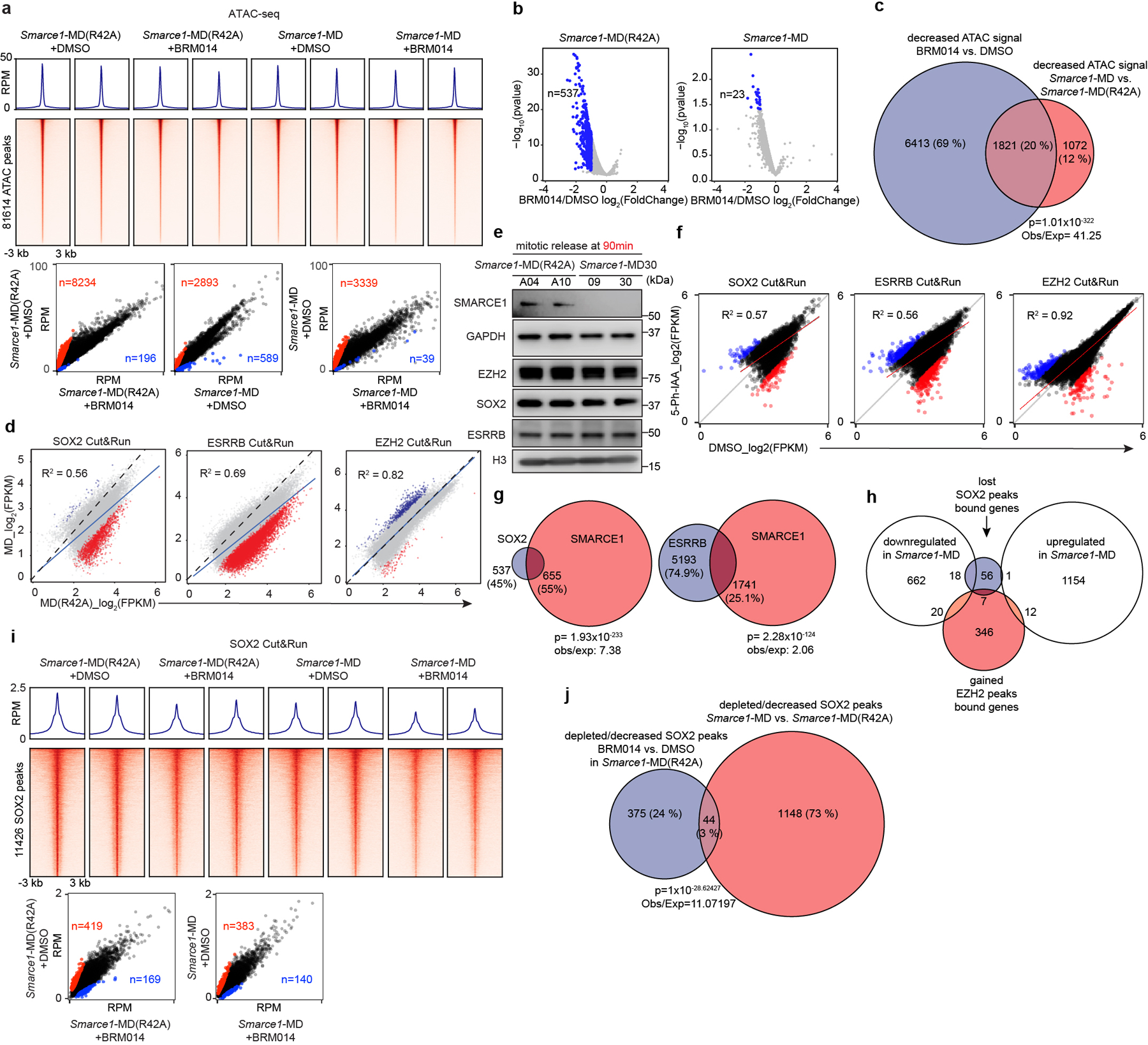
a, (top) Average binding profile of ATAC-seq signal identified in Smarce1-MD (R42A) and Smarce1-MD cells at 90 min following release from mitotic arrest. Cells were treated with either DMSO or 1 μM BRM014 (SMARCA4 ATPase inhibitor). (middle) Heat maps of ATAC-seq signal. (bottom) Scatter plots of ATAC-seq signal in reads per million in the indicated samples. Dots indicate significant loss (red) or gain (blue) of ATAC-signal. b, Volcano plots showing genes with the significantly decreased nascent RNA transcriptional levels under the treatment of BRM014 versus DMSO in Smarce1-MD(R42A) (left) and Smarce1-MD (right) cells at 90 min after releasing from mitotic arrest. P-value in differentially expressed genes test was calculated using Wald test from DESeq2. c, Venn diagram showing the overlap of decreased ATAC-seq sites affected by ATPase inhibitor BRM014 to those affected by the loss of SMARCE1 in mitosis. d, Scatter plots of SOX2 (left), ESRRB (middle), and EZH2 (right) peak changes between Smarce1-MD (R42A) and Smarce1-MD cells at 90 min after mitotic release. Dots indicate significant binding loss (red) or gain (blue) following mitotic loss of SMARCE1. e, Representative immunoblot of Smarce1- MD (R42A) clones A04 and A10 and Smarce1- MD clones 09 and 30 at 90 min after mitotic release. f, Scatter plots of SOX2 (left), ESRRB (middle), and EZH2 (right) peak changes in mitotic Smarce1-AID mouse ES cells (clones 06 and 23) treated with DMSO or 5-Ph-IAA for 1 hour. Dots indicate significant binding loss (red) or gain (blue) following mitotic loss of SMARCE1. g, Venn diagram showing the overlap between decreased/depleted SOX2 (left)/ESRRB (right) peaks and retained SMARCE1 peaks identified from Smarce1-MD versus Smarce1-MD (R42A) cells at 90 min after mitotic release. h, Overlap of genes with lost/decreased SOX2 peaks or gained EZH2 peaks and genes upregulated or downregulated in Smarce1-MD mouse ES cells. i, (top) Average binding profile of SOX2 Cut&Run signal identified in Smarce1-MD (R42A) and Smarce1- MD cells at 90 min following release from mitotic arrest. Cells were treated with either DMSO or 1 μM BRM014. (middle) Heat maps of SOX2 signal. (bottom) Scatter plots of SOX2 signal in reads per million in the indicated samples. Dots indicate significant loss (red) or gain (blue) of SOX2 signal. j, Venn diagram showing the overlap of depleted/decreased SOX2 peaks identified from Smarce1-MD (R42A) cells treated with BRM014 versus DMSO and Smarce1-MD versus Smarce1-MD (R42A) cells at 90 min after mitotic release. All data are compiled from two replicates. Correction values were obtained from Pearson’s product moment correlation (d, f). The ratio of observed (Obs) versus expected (Exp) frequency is shown, and p-value determined by two-sided Fisher’s exact test is indicated (c, g, j).
Extended Data Figure 10. Mitotic SMARCE1 is required for the appropriate neural differentiation.
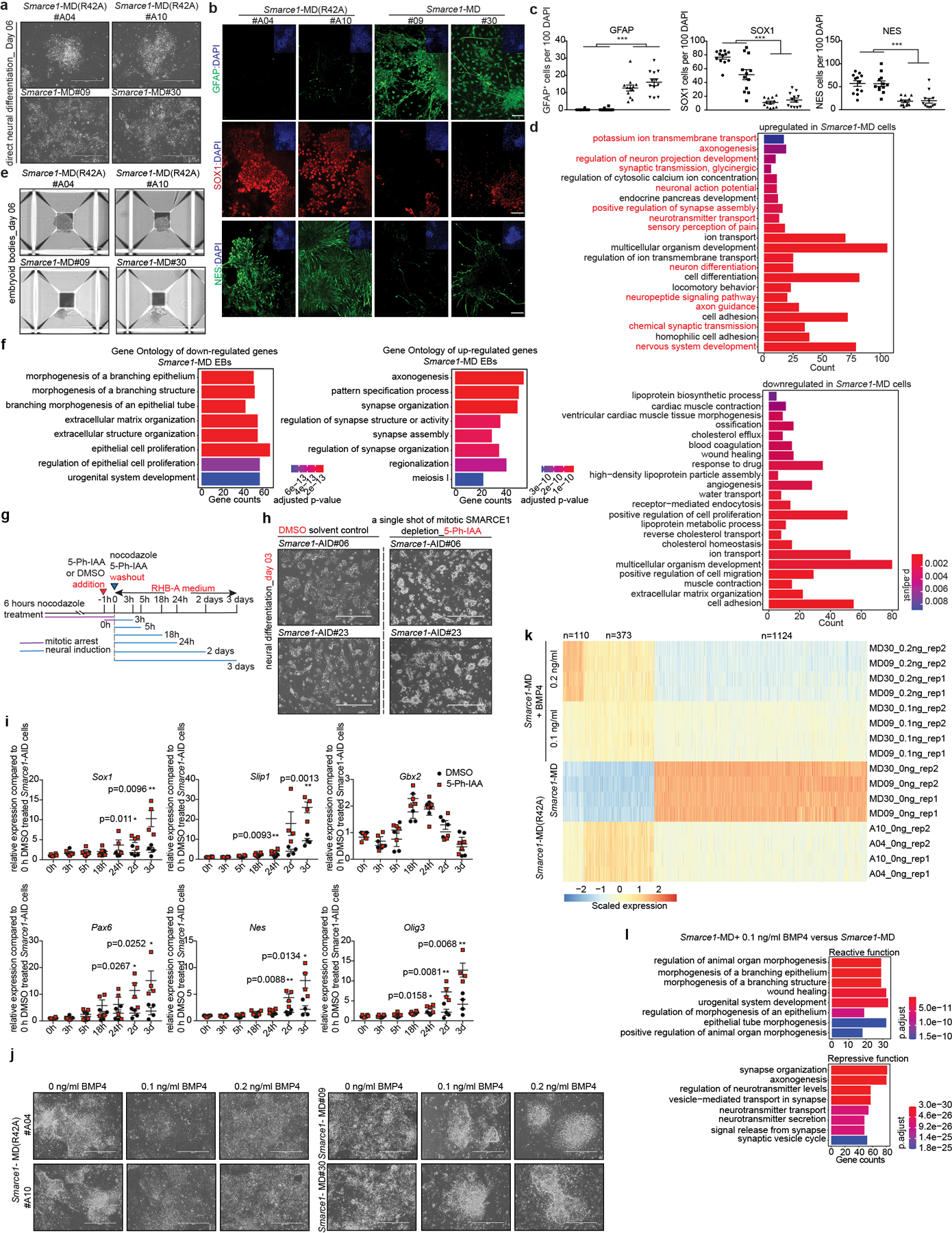
a, Bright field images presenting cell morphologies of Smarce1-MD(R42A) and Smarce1-MD derived neural cells at day six after induction. b, c, (b) IHC staining of DAPI and GFAP (top), SOX1 (middle), and NES (bottom) in Smarce1-MD (R42A) and Smarce1-MD derived neural cells at day six after induction. (c) Statistical analysis of (b) (p value for GFAP= 2.72e−10, p value for SOX1= 3.01e−11, p value for NES= 4.62e−9). d, GO analysis of differentially expressed genes between Smarce1- MD (R42A) and Smarce1- MD cultures at day six after neural induction. Over-representation test was used to calculated GO term enrichment with FDR for multiple test correction. e, Representative morphologies of embryoid bodies derived from Smarce1- MD(R42A) and Smarce1- MD mouse ES cells at day six. f, GO analysis of differentially expressed genes identified in (e). Over-representation test was used to calculated GO term enrichment with FDR for multiple test correction. g, Schematic for direct neural induction and 5-Ph-IAA treatment during mitosis and mitotic release in the AID degron system. h, Bright field images presenting cell morphologies of DMSO or 5-Ph-AA treated mitotic Smarce1-AID derived cultures at day three after induction. i, Quantitative PCR (qPCR) for neuroectoderm markers in DMSO or 5-Ph-AA treated mitotic Smarce1-AID (clones #6 and #23). j, Representative morphologies of Smarce1- MD(R42A) and Smarce1- MD cultures supplemented with indicated dose of BMP4 at day six after neural induction. k, Heat map showing the differentially expressed genes in (j). l. GO analysis of (k). Over-representation test was used to calculated GO term enrichment with FDR for multiple test correction. All data are representative and compiled from two independent experiments. N= 2 biological replicates for each clone (Smarce1-AID#06, #23) (i). Data are shown as mean± s.e.m., n= 12 images/sample collected from 2 biologically independent experiments (c). Scale bars: 400 μm (a, b, e, j), 1000 μm (h). P-value was calculated using two-tailed unpaired Student’s t- Test (* p<0.05, ** p<0.01, *** p< 0.001) (c, i).
Supplementary Material
Acknowledgments:
The authors thank all members of the Roberts lab for insightful discussions and Guy Riddihough for help with the manuscript; the Hartwell Center Core for sequencing; St. Jude Center for Applied Bioinformatics (CAB) for help with alignment of RNA-seq, ChIP-seq and CUT&RUN-seq reads; St. Jude flow cytometry and cell sorting core for cell sorting. The St. Jude core facilities are supported by National Cancer Institute Cancer Center Support Grant (NCI CCSG 2 P30 CA021765) and by American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital. The Center for Applied Bioinformatics is supported by the National Cancer Institute, Cancer Center Support Grant P30 CA21765 and ALSAC. This study was supported by the National Cancer Institute (NCI) R01 CA113794 and R01 CA172152 to C.W.M.R., National Cancer Institute Cancer Center support grant (NCI CCSG 2 P30 CA021765), ALSAC of St. Jude Children’s Research Hospital, CURE AT/RT Now to C.W.M.R., Garrett B. Smith Foundation to C.W.M.R., St. Jude Children’s Research Hospital Collaborative Research Consortium on Chromatin Regulation in Pediatric Cancer to C.W.M.R., R01 CA216391 to J. Z., National Institutes of Health (NIH) grant R01-AI123322 to D.R.G. S.R-J. is supported by the St. Jude Graduate School of Biomedical Sciences and the Ruth L. Kirschstein National Research Service Award (F31 CA261150). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing Interests: C.W.M.R. is a scientific advisory board (SAB) member of Exo Therapeutics, unrelated to this manuscript.
Code Availability
The code for analyzing the data has been deposited in GitHub (https://github.com/xiaolongchen0627/SWISNF.mitotic.bookmarking.code).
Material Availability
Plasmids and cell lines generated in this study are available upon request following completion of a material transfer agreement.
Data Availability
The ChIP-seq, Cut&Run, nascent RNA-seq, RNA-seq and ATAC-seq data that support the findings of this study have been deposited in the GEO database under the accession number GSE189563.
References
- 1.Palozola KC et al. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122, doi: 10.1126/science.aal4671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinberg D & Vales LD Chromatin domains rich in inheritance. Science 361, 33–34, doi:doi: 10.1126/science.aat7871 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Ming X et al. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res 30, 980–996, doi: 10.1038/s41422-020-0359-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson BG & Roberts CW SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11, 481–492, doi: 10.1038/nrc3068 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Lessard J et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215, doi: 10.1016/j.neuron.2007.06.019 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho L, R. J, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proceedings of the National Academy of Sciences of the United States of America 106, 5181–5186, doi: 10.1073/pnas.0812889106. (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alver BH et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nature Communications 8, doi: 10.1038/ncomms14648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.PARSONS GG & SPENCER CA Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. MOLECULAR AND CELLULAR BIOLOGY 17, 5791–5802, doi: 10.1128/MCB.17.10.5791 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naumova N et al. Organization of the Mitotic Chromosome. Science 342, 948–953, doi: 10.1126/science.1236083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H et al. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature, doi: 10.1038/s41586-019-1778-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonin W & Neumann H Chromosome condensation and decondensation during mitosis. Curr Opin Cell Biol 40, 15–22, doi: 10.1016/j.ceb.2016.01.013 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Muchardt C, Reyes JC, Bourachot B, Leguoy E & Yaniv M The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J 15, 3394–3402, doi: 10.1002/j.1460-2075.1996.tb00705.x (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sif S, Stukenberg PT, Kirschner MW & Kingston RE Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes&Development 12, 2842–2851, doi: 10.1101/gad.12.18.2842 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurley LR, Walters RA & Tobey RA Cell cycle-specific changes in histone phosphorylation associated with cell proliferation and chromosome condensation. Journal of Cell Biology 60, 356–364, doi: 10.1083/jcb.60.2.356 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Festuccia N et al. Transcription factor activity and nucleosome organization in mitosis. Genome Research 29, 250–260, doi: 10.1101/gr.243048.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teves SS et al. A dynamic mode of mitotic bookmarking by transcription factors. Elife 5, doi: 10.7554/eLife.22280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deluz C et al. A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes Dev 30, 2538–2550, doi: 10.1101/gad.289256.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y et al. Widespread Mitotic Bookmarking by Histone Marks and Transcription Factors in Pluripotent Stem Cells. Cell Rep 19, 1283–1293, doi: 10.1016/j.celrep.2017.04.067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festuccia N, Gonzalez I, Owens N & Navarro P Mitotic bookmarking in development and stem cells. Development 144, 3633–3645, doi: 10.1242/dev.146522 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protocols 13, 1006–1019, doi: 10.1038/nprot.2018.015 (2018). [DOI] [PubMed] [Google Scholar]
- 21.de Dieuleveult M et al. Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530, 113–116, doi: 10.1038/nature16505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magana-Acosta M & Valadez-Graham V Chromatin Remodelers in the 3D Nuclear Compartment. Front Genet 11, 600615, doi: 10.3389/fgene.2020.600615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Festuccia N et al. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat Cell Biol 18, 1139–1148, doi: 10.1038/ncb3418 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Lodato MA et al. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9, e1003288, doi: 10.1371/journal.pgen.1003288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun 10, 1881, doi: 10.1038/s41467-019-09891-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michel BC et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol 20, 1410–1420, doi: 10.1038/s41556-018-0221-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owens N et al. CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife 8, doi: 10.7554/eLife.47898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelham-Webb B et al. H3K27ac bookmarking promotes rapid post-mitotic activation of the pluripotent stem cell program without impacting 3D chromatin reorganization. Mol Cell 81, 1732–1748 e1738, doi: 10.1016/j.molcel.2021.02.032 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiung CC et al. Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res 25, 213–225, doi: 10.1101/gr.180646.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glotzer M, Murray AW & Kirschner MW Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138, doi: 10.1038/349132a0 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadauke S et al. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150, 725–737, doi: 10.1016/j.cell.2012.06.038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Chu J, Shen X, Wang J & Orkin SH An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132, 1049–1061, doi: 10.1016/j.cell.2008.02.039 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jao CY & Salic A Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105, 15779–15784, doi: 10.1073/pnas.0808480105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsiung CC-S et al. A hyperactive transcriptional state marks genome reactivation at the mitosis–G1 transition. Genes & Development 30, 1423–1439, doi: 10.1101/gad.280859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yesbolatova A et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat Commun 11, 5701, doi: 10.1038/s41467-020-19532-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King HW & Klose RJ The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife 6, doi: 10.7554/eLife.22631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gatchalian J et al. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat Commun 9, 5139, doi: 10.1038/s41467-018-07528-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caravaca JM et al. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev 27, 251–260, doi: 10.1101/gad.206458.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asenjo Helena G., G. A, López-Onieva Lourdes, Tejada Irene, Martorell-Marugán Jordi, Carmona-Sáez Pedro and Landeira David. Polycomb regulation is coupled to cell cycle transition in pluripotent stem cells. Science Advances 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pauklin S & Vallier L The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147, doi: 10.1016/j.cell.2013.08.031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodges HC et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol 25, 61–72, doi: 10.1038/s41594-017-0007-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanton BZ et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat Genet 49, 282–288, doi: 10.1038/ng.3735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schick S et al. Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat Genet, doi: 10.1038/s41588-021-00777-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosen OR et al. The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc Natl Acad Sci U S A 95, 4152–4157, doi: 10.1073/pnas.95.8.4152 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsiao PW, Fryer CJ, Trotter KW, Wang W & Archer TK BAF60a Mediates Critical Interactions between Nuclear Receptors and the BRG1 Chromatin-Remodeling Complex for Transactivation. Molecular and Cellular Biology 23, 6210–6220, doi: 10.1128/mcb.23.17.6210-6220.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abranches E et al. Neural differentiation of embryonic stem cells in vitro: a road map to neurogenesis in the embryo. PLoS One 4, e6286, doi: 10.1371/journal.pone.0006286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawasaki Hiroshi, M. K, Nishikawa Satomi, Kaneko Satoshi, Kuwana Yoshihisa, Nakanishi Shigetada, Nishikawa Shin-Ichi, and Sasai Yoshiki. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28, 31–40, doi: 10.1016/S0896-6273(00)00083-0 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Panamarova M et al. The BAF chromatin remodelling complex is an epigenetic regulator of lineage specification in the early mouse embryo. Development 143, 1271–1283, doi: 10.1242/dev.131961 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginno PA, Burger L, Seebacher J, Iesmantavicius V & Schubeler D Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat Commun 9, 4048, doi: 10.1038/s41467-018-06007-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Djeghloul D et al. Identifying proteins bound to native mitotic ESC chromosomes reveals chromatin repressors are important for compaction. Nat Commun 11, 4118, doi: 10.1038/s41467-020-17823-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442 e417, doi: 10.1016/j.cell.2018.03.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valencia AM et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell, doi: 10.1016/j.cell.2019.10.044 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mashtalir N et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 183, 802–817 e824, doi: 10.1016/j.cell.2020.09.051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yesbolatova A et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat Commun 11, 5701, doi: 10.1038/s41467-020-19532-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Z et al. PHB Associates with the HIRA Complex to Control an Epigenetic-Metabolic Circuit in Human ESCs. Cell Stem Cell 20, 274–289 e277, doi: 10.1016/j.stem.2016.11.002 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Teves SS et al. A dynamic mode of mitotic bookmarking by transcription factors. Elife 5, doi: 10.7554/eLife.22280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protocols 13, 1006–1019, doi: 10.1038/nprot.2018.015 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947, doi: 10.1016/j.cell.2013.09.053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palozola KC et al. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122, doi: 10.1126/science.aal4671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-seq, Cut&Run, nascent RNA-seq, RNA-seq and ATAC-seq data that support the findings of this study have been deposited in the GEO database under the accession number GSE189563.