

Abstract
This white paper is the outcome of the seventh UC Davis Cardiovascular Research Symposium on Systems Approach to Understanding Cardiovascular Disease and Arrhythmia. This biannual meeting aims to bring together leading experts in subfields of cardiovascular biomedicine to focus on topics of importance to the field. The theme of the 2022 symposium was “Cell Diversity in the Cardiovascular System, cell-autonomous and cell-cell signaling”. Experts in the field contributed their experimental and mathematical modelling perspectives and discussed emerging questions, controversies, and challenges in examining cell and signal diversity, coordination, and interrelationships involved in cardiovascular function. This paper originates from the topics of formal presentations and informal discussions from the symposium, which aimed to develop a holistic view of how the multiple cell types in the cardiovascular system integrate to influence cardiovascular function, disease progression, and therapeutic strategies. The first section describes the major cell types (e.g., cardiomyocytes, vascular smooth muscle and endothelial cells, fibroblasts, neurons, immune cells, etc.) and the signals involved in cardiovascular function. The second section emphasizes the complexity at the subcellular, cellular, and system levels in the context of cardiovascular development, aging, and disease. Finally, the third section surveys the technological innovation that allows interrogating this diversity and advancing our understanding of the integrated cardiovascular function and dysfunction.
Keywords: Omics, ion channels, ion transporters, animal models, signal transduction, modelling
Graphical Abstract
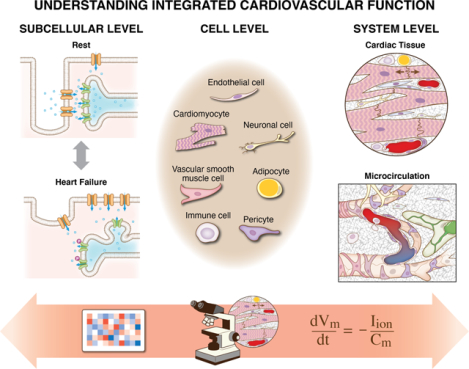
The white paper discusses the cell diversity, coordination, and interaction patterns that are critical for robust cardiovascular function. We identify the major cell types and signals involved in cardiovascular function and emphasize the complexity at the subcellular, cellular, and system levels that motivate both challenges and opportunities for researchers. A survey of recent advances enabled by innovative experimental and computational modeling approaches serve to guide researchers moving forward.
Motivation for Understanding Cell Diversity and Dynamics Involved in Cardiovascular Function
The cardiovascular system is a highly organized, closed, and efficient circuit consisting of a pump (i.e., the heart) and pipes (i.e., the blood vessels). The heart pumps oxygen-rich blood and nutrients to the rest of the body via blood vessels, which also deliver deoxygenated blood to the lungs. This relatively straightforward process of pumping, delivering, and return of blood throughout the body belies the hidden complexity of an incredibly diverse array of molecular-cellular elements of the heart and the vasculature (Figure 1). These components coordinate via a complex network of signals to sustain roughly 1 billion heartbeats over a lifetime (Levine, 1997).
Figure 1:
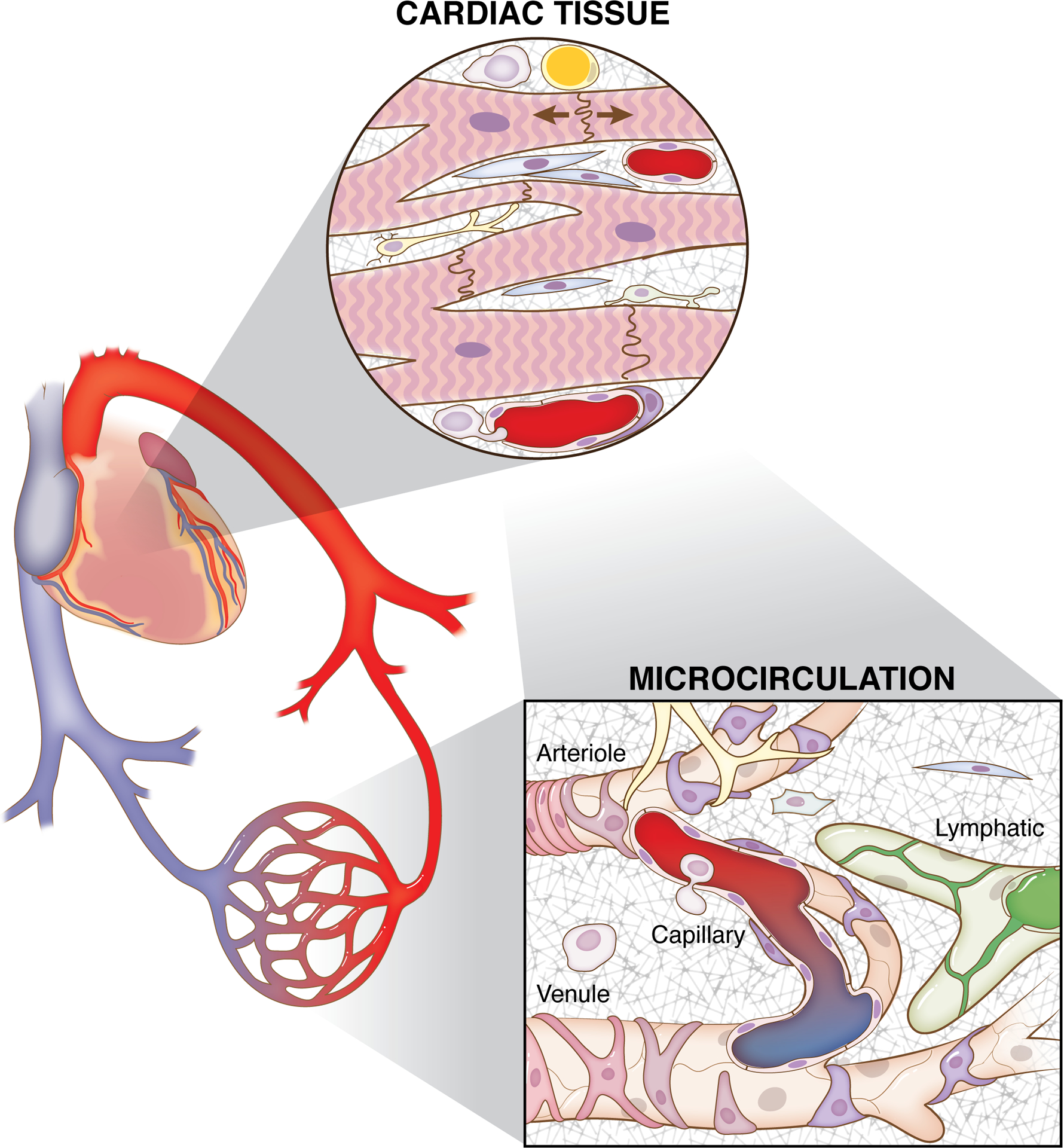
Cardiovascular function depends on integrated cell dynamics across scales. The purpose of this review is to emphasize the involved cellular diversity, coordination, and interrelationships. The concert of interactions is appreciated through consideration of the specific cell types in cardiac tissues and the embedded microcirculation.
Given the interdependence of heart muscle and vascular function, it is important to appreciate the common and integrated cell dynamics between cardiac tissue and microcirculation (Figure 1). Often, cell dynamics are considered in isolation. While reductionistic, mechanistic studies have provided a foundation for our physiological understanding, emerging evidence of overlapping cellular phenotypes, common cell function mechanisms, structural interactions, and paracrine interactions motivate the conceptual and experimental integration of the diverse cell dynamics. What are the cell types involved in cardiovascular function in health and disease? Do the diverse cell populations highlight common cell lineages? Do cell contributions vary temporally? How do we dissect and interpret specific cell functions in a diverse tissue scenario? Based on the recent UC Davis Cardiovascular Research Symposium on Systems Approach to Understanding Cardiovascular Disease and Arrhythmia, the purpose of this white paper is to emphasize the cell diversity, coordination, and interaction patterns critical for robust cardiovascular function. By identifying the major cell types and signals, the article will highlight the complexity across cell and system levels that motivate both challenges and opportunities for researchers. A foundation for future research and related knowledge gaps will be framed in the context of alterations during development, aging, and pathological scenarios. The complexity will be further highlighted by considering how specific cell functions are influenced by energetic costs, cell topology, and sub-cellular mechanisms. Finally, examples of recent advances made possible by innovative experimental and computational modeling approaches will serve to guide researchers moving forward. Rather than providing a comprehensive compilation of specific cell functions, this article integrates multiple research areas spanning cardiovascular physiology, immunology, stem cell biology, microvascular physiology, imaging, and computational modeling to generate new important yet underrecognized questions.
1. Cell Types and Signals in Cardiovascular Function
Appreciation of the multiple cell types across the cardiac and vascular systems is realized by considering system-level dynamics in cardiac tissue and the embedded microcirculation (Figure 2). The major cell types in the heart include cardiomyocytes (CMs), endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and fibroblasts (FBs). Neuronal and immune cells, adipocytes, and pericytes are also found in the heart (Doll et al., 2017; Litvinukova et al., 2020).
Figure 2:
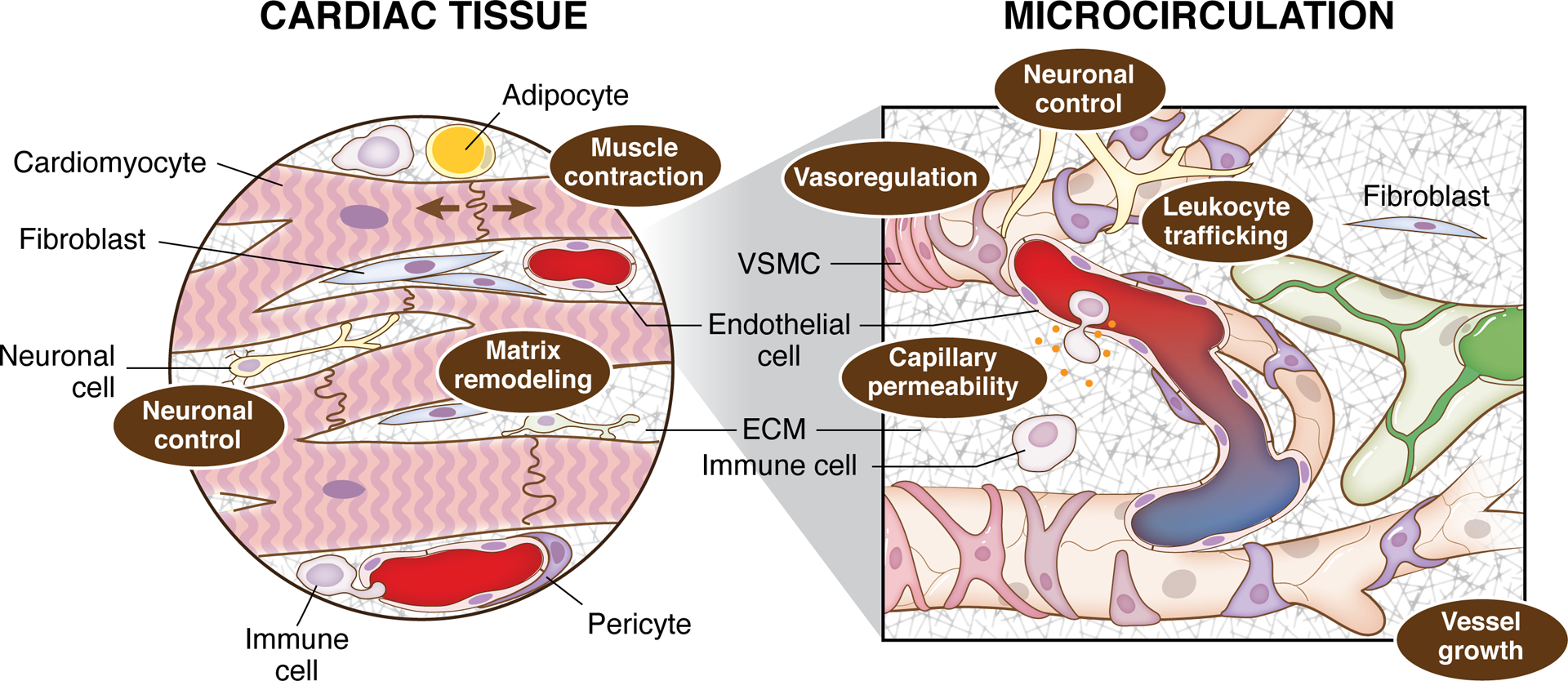
Cardiac tissue function depends on multiple cell types across the cardiac and microvascular systems. The major cell types include cardiomyocytes, endothelial cells, vascular smooth muscle cells, and fibroblasts, neuronal, immune cells, adipocytes, and vascular pericytes. Interactions between the cell types influence multiple dynamics: muscle contraction, extracellular matrix (ECM) remodeling and neuronal control (cardiac tissue level) and neuronal control, vasoregulation, leukocyte trafficking and permeability dynamics (microcirculation level). The dynamics across the system level scales influence each other and contribute to the local microenvironment.
The relative abundance of these various cell types is controversial and may depend on technical differences among studies (based on transcriptomic approaches and spectroscopy-based proteomics) (Doll et al., 2017; Litvinukova et al., 2020). Species differences are also important for the relative prevalence of the various cell types (e.g., rodents have no substantial adipose tissue, and FBs are less abundant in mouse vs. human).
Cardiac tissue level dynamics resulting from integrated cell interactions include muscle contraction, matrix remodeling, and neuronal control. Contributing to the complexity, cardiac tissue function is influenced by microvascular level dynamics such as additional neuronal control, vasoregulation, leukocyte trafficking, and permeability dynamics. Given their large size, CMs occupy most of the heart volume. Importantly, transcriptional differences between atrial and ventricular CM populations indicate the potential for different developmental origins, distinctive hemodynamic forces, and specialized functions in cardiac chambers. Also, the cellular composition may differ locally in different heart muscle regions (i.e., specific chambers) and between male and female hearts (Litvinukova et al., 2020).
The major classes of cells along the microvasculature include ECs and mural cells (i.e., VSMCs and pericytes). The ECs face the lumen of the vessel, and mural cells cover the vascular endothelium to different degrees depending on the vessel and localization along the arterial tree. Accordingly, vascular smooth muscle wraps around the artery and arteriole endothelium, whereas pericytes have distinct levels of coverage in small arterioles and the capillary endothelial network. Importantly, vascular smooth muscle contains the contractile machinery to control arterial/arteriole diameter. Emerging evidence also suggests that pericytes may be contractile cells that help modulate capillary diameter (Gonzales et al., 2020). Extracellular matrix (ECM), perivascular macrophages, and perivascular FB-like cells are also present and/or reside along the vascular wall. These cells help to facilitate vascular function by providing stability to the blood vessels, and contribute to (inter)cellular communication that is essential for the modulation of vessel diameter and, therefore, control of arterial tone, peripheral resistance, and tissue blood flow. Intriguingly, all major classes of vascular cells show remarkable heterogeneity in terms of gene expression, morphology, and even function, among other properties. Like CM heterogeneity, vascular cell heterogeneity may result from developmental origins, structural requirements, shear stress, microenvironmental factors, and/or adaptation to specific functions or pathological conditions, such as hypertension, diabetes, and aneurism, within the vascular network.
CMs are the basic cellular units of cardiac excitability and contractility. Their structure/function characteristics are reviewed first and compared with that of VSMC. CMs are electrically and mechanically coupled with each other and communicate with other cell types via both direct physical interaction and paracrine and autocrine signaling. Other cell types are required to provide structural support and blood supply for efficient contraction and long-term survival and are described in subsequent paragraphs.
To appreciate the diversity and, at the same time, the perplexing interrelationships between the major cardiac tissue cell types and microvascular cell types, this section will focus on key known cell functions that substantiate their contributions. Importantly, excitation-contraction coupling (EC-coupling) in CMs and VSMCs is detailed to showcase both the divergent and overlapping mechanisms. The key roles of the other cell types are highlighted to introduce their underappreciated importance.
1.1. Excitation-Contraction Coupling in Cardiomyocytes vs. Vascular Smooth Muscle Cells
Nobel laureate Otto Loewi made the infamous statement: “Ja Kalzium, das ist alles” - “Yes calcium, that is all”. The universal nature of Ca2+ signaling is well established, including in CMs and VSMCs (Amberg & Navedo, 2013; Parekh, 2016). One key biological function that is tightly regulated by Ca2+ is muscle contraction. Ringer showed that heart contraction depends on Ca2+ (Ringer, 1883), and these foundational findings brought Ca2+ signaling research to the forefront for many years to follow. It is evident that all muscle types (smooth, cardiac, skeletal) contract in a Ca2+-dependent fashion through a process that is widely referred to as EC-coupling. Understanding cardiac tissue function requires consideration of both the common and distinct Ca2+ signaling processes observed in CM and smooth muscle cells.
Cardiomyocytes (CMs)
Membrane depolarization during an action potential (AP) activates voltage-dependent L-type Ca2+ channels (LTCCs) in the sarcolemma, allowing for Ca2+ entry into the cell. This Ca2+ influx activates Ca2+ release from ryanodine receptor type-2 channels (RyR2s) in the membrane of the sarcoplasmic reticulum (SR) in a process termed Ca2+-induced Ca2+ release (CICR). RyR2 activation and release across each CM provide enough cytosolic Ca2+ to trigger contraction upon binding to troponin C (Bers, 2002). The location of these Ca2+ channels (LTCCs and RyR2s) is critical for CM function, as the Ca2+ entering the cell through LTCCs rapidly diffuses in the cytoplasm, and thus [Ca2+]i rapidly decreases with distance from the entry point. These intracellular Ca2+ gradients ensure that RyR2s are closed during diastolic periods, and RyR2s located in the vicinity of the LTCCs are activated during cardiac EC-coupling. The RyR2s are clustered in Ca2+-release units, i.e., domains including the LTCC and the SR membrane with RyR2s which openings can be visualized with fluorescent dyes and confocal microscopy as localized, rapid, and brief elevations in [Ca2+]i termed Ca2+ sparks (Cheng & Lederer, 2008). The [Ca2+]i transient produced by the CICR results from the spatial and temporal summation of Ca2+ sparks. In ventricular CMs, the sarcolemma periodically invaginates at intervals roughly coincident with the sarcomeric Z-lines, forming a network of transverse t-tubules that plunge into the depth of these wide (~100–150 μm), thick (~5–15 μm) cells, serving to juxtapose the LTCC-containing sarcolemma within 12–15 nm of the junctional SR at specialized membrane contact sites called dyads. Ca2+-release units are maintained and supported by junctional SR-anchored junctophilin-2, which tethers t-tubules to the junctional SR, traversing the dyadic cleft and interacting with sarcolemmal phospholipids at the t-tubule side. Junctophilin-2 binds to both LTCCs and RyR2 Ca2+ channels (Lehnart & Wehrens, 2022), and its alteration has been implicated in cardiac pathology (Guo et al., 2018; Yin et al., 2021). Because of this architecture, ventricular CMs respond to the AP with a fast and homogenous [Ca2+]i transient. Atrial CMs possess few or no t-tubules, depending on the species. In those atrial CMs without t-tubules, Ca2+ release originates on the surface, where RyR2s are close to LTCCs, and then propagates to the central RyR2s from the periphery (Blatter et al., 2003). However, recent work has revealed that atrial CMs have axial tubules of sarcolemma in the center of the cell, with a hypersensitive RyR2 phosphorylation state, which counteracts the scarcity of t-tubules (Brandenburg et al., 2022; Zhang et al., 2022a).
Mathematical models of CM electrophysiology, consisting of coupled, nonlinear differential equations, have been developed with increasing biophysical detail and complexity for more than 50 years (Fink et al., 2011) and have inarguably increased our understanding of cellular electrophysiology and Ca2+ signaling in health and disease, including arrhythmia syndromes (Rudy & Silva, 2006; Roberts et al., 2012; Winslow et al., 2016). Computational and theoretical approaches have been central to establishing what we know of the fundamental structure-function relationships in EC-coupling. Mathematical models have attempted to reconcile measurable single protein function and membrane structure with observable changes in cytosolic and SR [Ca2+] that occur in larger surrounding volumes. These efforts generated a large family of models that include an increasingly sophisticated representation of the gradients in intracellular Ca2+ concentration occurring during a normal cardiac cycle (Greenstein & Winslow, 2002; Shannon et al., 2004; Restrepo et al., 2008; Winslow et al., 2016). Models in which Ca2+ dynamics are governed by the underlying cell ultrastructure (Nivala et al., 2015; Colman et al., 2017; Shiferaw et al., 2020; Zhang et al., 2022a; Zhang et al., 2022b) further inform how the fundamental EC-coupling relationships are altered in atrial vs. ventricular CMs or in CMs with varying t-tubule density and organization. These models have developed interactively with experimental techniques to establish the current state of knowledge describing how nanoscale Ca2+ signaling determines macroscopic (cell-scale) EC-coupling in the heart.
Atrial and ventricular CMs are excitable cells that respond with an AP to an electrical impulse that propagates via gap junctions and initiates heart contraction (Bers, 2002). The primary pacemaker cells located in the sinoatrial node (SAN) exhibit automaticity and originate this electrical impulse, which is then transmitted through the atria and into the atrioventricular node (AVN), with AVN myocytes also being automatic but firing APs at a lower intrinsic rate than SAN myocytes. The AVN can induce ventricular contraction when the SAN fails or slow ventricular rate with fibrillating atria. From the AVN, the electrical impulse then propagates to the ventricles via the conducting myocytes of the His and Purkinje fibers. As discussed at the symposium, nodal cell automaticity is governed by a complex coupled system of cellular “clocks” that integrates ion channels and transporters on the cell membrane surface (“membrane clock”) with subcellular Ca2+-handling machinery (referred to as an intracellular “Ca2+ clock”) (Rubenstein & Lipsius, 1989; Lakatta & DiFrancesco, 2009). While lacking t-tubules, caveolae may provide these cells with structural support for automaticity. Sophisticated nodal cell models have contributed to our understanding of how the different types of ion channels and ion channel isoforms act together with subcellular and global Ca2+-handling mechanisms to produce stable pacemaker activity (Kharche et al., 2011; Maltsev et al., 2011; Yaniv et al., 2013). Nodal cell firing is driven by gradual changes in the membrane potential (Vm) (namely, diastolic depolarization) that are mediated by the concomitant action of these clocks: nodal-specific hyperpolarization-activated current (If) and a low voltage-activated T-type Ca2+ current (ICa,T) contribute to early diastolic depolarization, while SR Ca2+ release via RyR2s occurs late during the pacemaker potential (Rubenstein & Lipsius, 1989; Lakatta & DiFrancesco, 2009). This Ca2+ release is spontaneous and/or synchronized by the L-type CaV1.3 channels that have an activation range more negative than the predominant CaV1.2 channels (Zhang et al., 2002; Torrente et al., 2016). These local Ca2+ releases activate an inward Na+/Ca2+ exchange (NCX) carried current (INCX), contributing to the late phase of diastolic depolarization and subsequent activation of L-type Ca2+ current (ICa,L; CaV1.2 as in ventricular CMs, and CaV1.3), which initiates the rapid AP upstroke and global CICR, with [Ca2+]i transients originating from the periphery and propagating towards the center as in atrial CMs (Bers, 2002).
Cardiac EC-coupling is physiologically regulated by the autonomic nervous system. Under stress conditions, catecholamines are secreted from sympathetic neurons to cause β-adrenoceptor (β-AR) stimulation that activates adenylyl cyclase, producing cAMP, and initiates a signaling cascade allowing the heart to increase its output by increasing the beating rate (positive chronotropy), accelerating conduction (positive dromotropy), increasing strength of contraction (positive inotropy), and ensuring appropriate filling of the chambers by accelerating relaxation (positive lusitropy). The parasympathetic nerves secrete acetylcholine to cause opposite effects, by activation of the muscarinic receptors that suppress adenylyl cyclase activity, thus limiting cAMP levels. Modulation of cAMP levels has several downstream signaling components and functional effects depending on the cell type. In CMs, the main cAMP effect is through activation of the protein kinase A (PKA) (Bers et al., 2019), which phosphorylates several proteins that are key to EC-coupling. The LTCC regulation by β-ARs is mediated by the small GTP-binding protein Rad (Ahern et al., 2019; Liu et al., 2020) and results in a gain of ICa,L function and subsequent increase in Ca2+ content (Liu et al., 2020), along with phosphorylation of phospholamban, which hastens Ca2+ reuptake in the SR, and accelerates relaxation (positive lusitropic effect) (Bers, 2002). These effects are also achieved by phosphorylation of the contractile myofibrils by altering cross-bridge cycling and modulating Ca2+ affinity. RyR2 is also phosphorylated by PKA, which increases channel open probability. Various sites of RyR2 phosphorylation by PKA have been identified (e.g. S2808 and S2030), though the functional roles of this modulation are debated, especially regarding their importance concerning CaMKII phosphorylation (Dobrev & Wehrens, 2014). Indeed, the increased [Ca2+]i facilitated by PKA activation downstream of β-AR stimulation binds to calmodulin (CaM) and activates CaMKII, which phosphorylates several of the same targets as PKA (at different sites), and is itself further activated by Epac - another cAMP effector. Nodal cell AP firing rate acceleration occurs via a direct effect of cAMP on the HCN channels (i.e. via a shift of the If activation curve to more negative potentials (Wainger et al., 2001)), PKA phosphorylation of If-carrying hyperpolarization-activated cyclic nucleotide-gated channels (Liao et al., 2010), and effects on several Ca2+ handling proteins that increase the frequency of diastolic SR Ca2+ releases (Vinogradova & Lakatta, 2009).
To probe the functional interactive consequences of protein phosphorylation in an integrative cellular environment, many computational modeling studies incorporate steady-state effects of β-AR activation on the various ion channels and Ca2+-handling proteins (Winslow et al., 2016). Saucerman et al. were the first to develop and validate a functionally integrated system coupling a biochemically detailed model of β-AR signaling with models of Ca2+ handling and electrophysiology (Saucerman et al., 2003). This framework was later expanded to study the synergy between PKA and CaMKII signaling and adopted in several contemporary descriptions of EC-coupling in various species (Soltis & Saucerman, 2010; Morotti et al., 2021) and cardiac regions, providing quantitative insights into the individual and combined roles of these kinases in regulating EC-coupling (Winslow et al., 2016).
Vascular Smooth Muscle Cells (VSMCs)
Spindle-shaped VSMCs play an important role in determining vascular diameter and, therefore, control of blood flow and blood pressure. Arteries and arterioles are surrounded by multiple layers or a single layer of SMCs, respectively. In distinction from CMs, each VSMC has only one central nucleus. Importantly, VSMCs allow blood vessels to constrict and dilate and therefore play crucial roles in controlling blood flow, peripheral resistance, and blood pressure.
The mechanisms underlying VSMC contraction are uniquely distinct from those in other muscle types. Canonically, SMC contraction is triggered by a physiological stimulus that increases intracellular Ca2+ concentration ([Ca2+]i). Different stimuli achieve this outcome through various mechanisms. For instance, an increase in intravascular pressure elevates the vascular wall stress, depolarizing the smooth muscle plasma membrane. VSMC membrane potential (VM) depolarization is mediated by transient receptor potential (TRP) channels and subsequently leads to the graded opening of voltage-gated Ca2+ channels at the VSMC plasma membrane (Knot & Nelson, 1998; Welsh et al., 2002; Inoue et al., 2009). A steady-state change in VM and Ca2+ influx from the extracellular space through voltage-gated Ca2+ channels establish the vascular tone. The LTCC is the primary voltage-activated Ca2+ influx pathway in VSMCs (Knot & Nelson, 1998). Pharmacological inhibition of LTCCs lowers peripheral resistance in the vasculature and therefore is widely used therapeutically to treat hypertension.
Like CMs, the LTCCs in VSMCs are the target of post-translational modifications that alter their function (Keef et al., 2001). Intriguingly, CaV1.2 activity can be modified by β-AR/PKA signaling, but the functional implications are unclear. More recently, it was found that elevations in extracellular glucose (e.g. hyperglycemia) potentiate LTCC current density by direct PKA-mediated phosphorylation of the CaV1.2 pore-forming subunit α1C at serine 1928, which led to increased vasoconstriction (Nystoriak et al., 2017; Prada et al., 2019; Nieves-Cintron et al., 2021).
Recent data highlight a clear difference in how PKA regulates CaV1.2 channels in the heart versus the vasculature. Accordingly, PKA-dependent regulation of CaV1.2 channels in the heart proceeds via Rad proteins, while in the vasculature, β-AR regulation of LTCC occurs via direct phosphorylation of the channel (Liu et al., 2020; Nieves-Cintron et al., 2021). Intriguingly, the cardiac CaV1.2 pore-forming subunit can still be directly phosphorylated by PKA, although the precise functional implications of this pos-translational modification are still unclear (De Jongh et al., 1996; Hulme et al., 2006; Lemke et al., 2008; Katchman et al., 2017). These observations highlight clear tissue-specific differences in CaV1.2 regulation. Yet, the detailed mechanisms are unclear, thus representing an area for further exploration.
Other physiological stimuli that evoke VSMC contraction and vasoconstriction include hormone or neurotransmitter release. Several vasoactive substances (e.g., vasoconstrictors such as norepinephrine) are released from sympathetic nerves near the vascular wall and act through G-protein coupled receptors in VSMCs. Receptor agonists that transduce through Gq-coupled receptors ultimately elevate cytosolic [Ca2+]i, due to the generation of inositol 1,4,5-trisphosphate (IP3) and the downstream activation of IP3-receptors which facilitate Ca2+ release from the SR. Alternatively, the formation of second messengers such as diacylglycerol upon vasoactive receptor activation can stimulate TRP channels and increase [Ca2+]i due to direct Ca2+ influx or due to vascular SMC depolarization and subsequent Ca2+ influx (Earley & Brayden, 2015).
When a stimulus elevates cytosolic [Ca2+]i in VSMCs, the “contraction” component of EC-coupling ensues. Unlike CMs, where Ca2+ binds to troponin C, Ca2+ binds to CaM in VSMCs, activating light chain kinase and phosphoryl transfer to myosin (Eisner et al., 2017). Phosphorylation of myosin triggers myosin/actin binding and cross-bridge cycling and ultimately leads to SMC contraction. Notably, the actin-to-myosin ratio is higher in smooth muscle averaging 15:1, compared to 6:1 in CMs (Murakami & Uchida, 1985). Another distinction is that VSMCs have no intercalated disks or z-disks like those found in CMs. Rather, SMC dense bodies are considered to be analogous to z-disks (Cole & Welsh, 2011; Eisner et al., 2017). Activation of myosin light chain phosphatase (e.g., via β2-AR stimulation) directly dephosphorylates myosin regulatory light chain and drives the reaction away from cross-bridge cycling, leading to smooth muscle relaxation (Cole & Welsh, 2011; Eisner et al., 2017). An additional key difference between VSMCs and CMs is how RyRs are activated, the role of CaV1.2 channels in controlling the RyR-mediated Ca2+ sparks, and their functional role in controlling EC-coupling (discussed below).
As in the cardiac field, detailed mathematical models have been developed that integrate plasma membrane electrophysiology and Ca2+ dynamics in VSMCs to investigate EC-coupling responses to various stimuli (Kapela et al., 2008; Karlin, 2015). These models, validated against broad experimental data sets, have provided mechanistic and quantitative insights about how different ion channels and distinct signaling pathways interact to alter VSMC electrophysiology and global [Ca2+]i, in health, in response to stress and in disease (Morotti et al., 2017; Syed et al., 2019).
The further refinement of VSMC computational models to include, for example, detailed signaling and regulatory processes, as well as their use to discover new mechanisms that may lay the foundation for the formulation of new hypotheses, represent areas of opportunity.
1.2. Other Cell Types in the Cardiovascular System
The cardiovascular system is highly dependent on intercellular communication for cardiac and vascular development, normal cardiovascular function, as well as in the evolution of disease-causing cardiovascular remodeling. Both myocytes and non-myocytes in the heart and vasculature respond to physiological and pathological stress, and maladaptive changes in non-myocytes, such as cardiac fibrosis or reduced capillary density, are key elements in the pathogenesis of cardiovascular disease. The role of myocytes in normal and pathological heart and vessel function is well documented. Despite the importance of non-myocytes in cardiovascular health and disease, we are only just starting to dissect the interaction patterns between these various cell types and to understand their physiological and pathophysiological significance. In this section, we review the main non-myocyte cell types in the heart and the vasculature (Figure 2) and discuss how the non-myocytes could influence cardiovascular function in health and disease (Figure 3).
Figure 3:
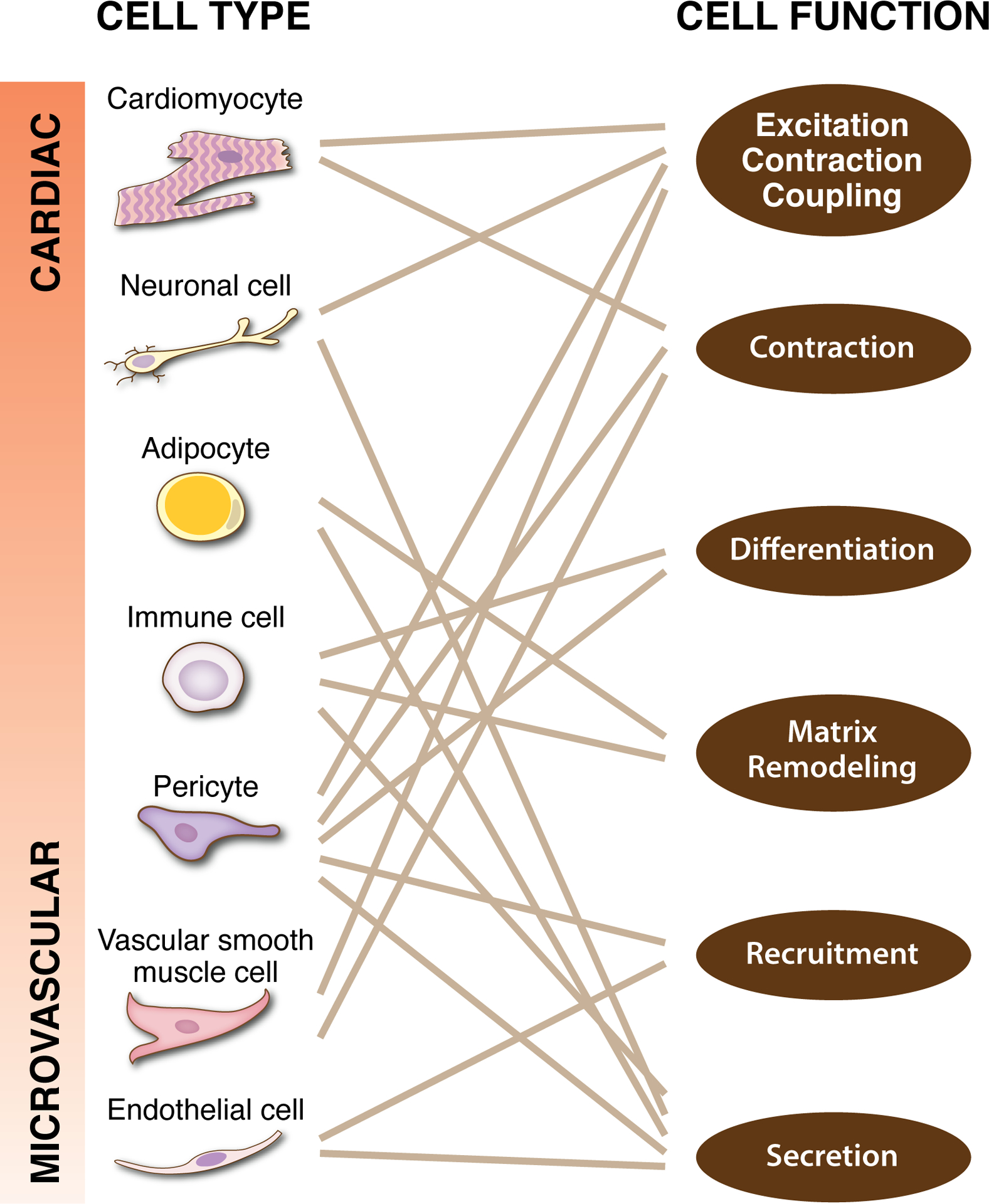
Understanding cardiac tissue function is complicated by the common functions across the diverse cell types. Cell types associated with the cardiac and microvascular systems are often considered to be distinct, yet common cell functions highlight their potential overlaps along a cellular continuum. Each specific cell function can be attributed to or influenced by multiple cell types. The solid lines represent functional links and are not inclusive of all overlaps. The potential for shared mechanisms across cell types and cell-cell interactions motivates future research focused on novel models and approaches that enable multi-scale integration.
Fibroblasts (FBs)
FBs are non-excitable cells that may originate from primary mesenchymal cells, local epithelial-mesenchymal transition, or bone marrow-derived precursors (Stenmark et al., 2013; Saljic et al., 2022). FBs are genetically, morphologically, and functionally distinct from smooth muscle, myo-FBs, and pericytes (Di Carlo & Peduto, 2018; Muhl et al., 2020; Rajan et al., 2020; Lendahl et al., 2022). The key function of FBs is to produce constituents of the extracellular matrix (ECM). Intriguingly, FBs show both common and distinctive signature markers in different organs and pathological conditions (Muhl et al., 2020), thus highlighting their heterogeneity and ability to produce diverse ECM in various tissues. In the heart, FBs are interspersed between CMs throughout the myocardium and interconnected by connexin junctions to form a sheet-like cellular layer that supports cardiac structures (reviewed in (Ongstad & Kohl, 2016)). In addition, FBs reside proximal to blood vessels, where they contribute to vessel structure, formation, and stabilization (Newman et al., 2011; Rajan et al., 2020). FBs possess a large surface area with sheet-like extensions, elongated cytoplasmic processes, and irregular folds that allow for long-distance signaling.
In the heart, most cardiac FBs originate from the epithelial cells of the proepicardium through epithelial-mesenchymal transition. Cardiac FBs may also originate from the epicardium before migrating into atria and ventricles and differentiating into cardiac FBs. In the vasculature, single-cell transcriptomic analysis and lineage studies suggest that FBs most likely arise from cells of mesenchymal origin in different animal models (Muhl et al., 2020; Rajan et al., 2020). Endothelial-mesenchymal transformation has also been described, along with other less frequent origins, such as bone marrow-derived cells, hematopoietic cells, and mesangioblasts.
FBs in cardiac and vascular tissue respond to a wide range of (patho)physiological stimuli. Various interactions between CMs, vascular cells, and FBs have been described in the heart, blood vessels, and lung vasculature and include biochemical, electrical, and/or biomechanical signals (Stenmark et al., 2013; Pellman et al., 2016). Paracrine mediators allow for indirect communication as they are secreted from one cell type and diffuse onto the other cell types. Activation of paracrine TGF-β1 and angiotensin II signaling have been widely studied and shown to affect CM-FB communication in the context of cardiac fibrosis, hypertrophy, arrhythmias, hypertension, atherosclerosis, and vascular injury (Stenmark et al., 2013). Recent data suggest that FB Nox2 signaling plays a role in the development of vascular remodeling and hypertension, and this may be driven by paracrine signaling between FBs and VSMCs (Harrison et al., 2021). The impact of these signals on fibrotic and arrhythmogenic pathways might differ in atria, and this may involve both effects on CM-FB paracrine interaction and direct effects on CMs. Likewise, given the role of FBs in different vascular beds, regulatory signals, such as TGF-β1 and angiotensin II, may have a distinct impact on vascular SMC excitability, migration, proliferation, and growth (Stenmark et al., 2013). Additionally, interleukins have more recently been involved in CM-FB communication in the context of cardiac hypertrophy, and Wnt signaling has been implicated in fibrosis and cardiac repair. Interleukins also influence FB-VSMC communication to promote vascular smooth muscle and ECM remodeling via non-coding RNA modifications, thus contributing to hypertension (Melton & Qiu, 2021). CMs and FBs can also be directly coupled via gap junctions (De Maziere et al., 1992) and membrane nanotubes (He et al., 2011), which provide support for electrical communication. While FBs are unable to generate APs, they can alter myocardial conduction via an electrotonic load through effectively increasing the cardiac cell capacitance and altering the resting membrane potential.
Computational models linking CMs and FBs, initially represented as electrically passive and eventually incorporating active experimentally-determined ion currents (MacCannell et al., 2007), have been useful for examining the role of cardiac FBs in electrical conduction and tissue remodeling in the heart (reviewed in (Zeigler et al., 2016)). FBs may alter vascular tone or vascular reactivity by modulating the contractility of VSMCs or remodeling the structure of the vessel wall (Melton & Qiu, 2021). Moreover, FB activation in response to pathological stress may lead to excess production of ECM components (e.g. collagen and fibronectin) and secretion of enzymes that degrade ECM components (e.g. matrix metalloproteinases, MMPs, and tissue inhibitors of MMPs), which can be sensed by CMs and VSMCs, thus altering their electrophysiology, organization and contractile state (Melton & Qiu, 2021). Several multi-scale computational models that incorporate FB-vascular cell interactions have been developed and have been used to acquire insight into mechanisms underlying atherosclerosis, restenosis, and vascular remodeling (Corti et al., 2021).
The exact molecular pathways controlling FB activation, proliferation/differentiation, and those regulating transcription of ECM proteins remain largely unknown. To address these unresolved questions, large-scale computational models of cardiac FB signaling have been developed, accounting for several context-specific regulators of fibrosis (Zeigler et al., 2016) and virtual drug screening (Zeigler et al., 2021). New modeling approaches are required to deepen our understanding of how FB-VSMC interaction patterns alter vascular reactivity in health and disease. Moreover, a key question that remains to be addressed is the extent to which FB-myocyte electrical coupling alters the properties of native cardiac and vascular tissue (Ongstad & Kohl, 2016).
Endothelial Cells (ECs)
ECs originate from the mesoderm, and their renewal and replacement are mediated by resident cells in the local environment (Aquino et al., 2021). ECs are critical regulators of the cardiovascular system. In the vasculature, the influence of ECs in the control of vascular function is well documented (King et al., 2022). These cells line the lumen of arteries and arterioles and are the principal cell type forming capillaries. EC expression profile may be distinct depending on the vascular bed, which may lead to diverse functions. For example, ECs in the central nervous system have low expression of leukocyte adhesion molecules that may limit the movement of immune cells into the brain (Rossler et al., 1992). Another example is the unique ability of capillary ECs, compared with arterial/arteriolar ECs, to initiate retrograde hyperpolarization leading to vasodilation and increased cerebral blood flow, with arteries and arterioles presumably exhibiting faster signal attenuation (Longden et al., 2017). ECs communicate with underlying VSMC via diffusible molecules (e.g. nitric oxide, ATP, K+) and/or electrical signals via gap junctions in myoendothelial projections. These endothelial-derived signals act on VSMCs to alter their contractile state, thus influencing vascular reactivity, tissue perfusion, and blood pressure in health and disease. Capillaries are critical for gas and nutrient exchange and, more recently, have been proposed as a sensory web for the regulation of neurovascular coupling and cerebral blood flow (Longden et al., 2017; Alvarado et al., 2021). The communication between ECs and VSMCs was examined in further detail in a recent comprehensive review (King et al., 2022). Biophysically detailed models of EC plasma membrane electrophysiology allowed analyzing the interplay between Vm and intracellular Ca2+ dynamics (Silva et al., 2007). This integrative model substantially improved our understanding of the vascular EC-VSMC interaction and how this communication affects vasoreactivity (Kapela et al., 2010).
ECs have been described as the most abundant non-myocyte population in the heart (Pinto et al., 2016). Most ECs in the heart are in the microcirculation, where they form the capillaries and serve a structural role as a barrier between blood and the myocardial tissue. ECs also communicate with adjacent CMs by producing paracrine factors that play essential roles in cardiovascular homeostasis, including modulation of CM contractility, growth, and survival (Lim et al., 2015). In the heart, nitric oxide, endothelin-1, neuregulin, and apelin produced by ECs have been shown to regulate the functions of neighboring CMs, and disturbances in these pathways have been shown to contribute to the development of heart failure (Lim et al., 2015). VEGFR2-ErbB signaling has recently emerged as a key pathway for the regulation of cardiac growth and homeostasis, whereby activation of VEGFR2 signaling in cardiac ECs induces angiogenesis and release of ErbB receptor ligands, which in turn activate growth signaling in CMs (Kivela et al., 2019).
Neuronal cells
Neuronal control of the cardiovascular system is well appreciated. Both heart and vascular tissue are surrounded by neurons to control their function. Neuronal activity may also influence the structural organization of the tissue, including vascular networks (Lacoste et al., 2014). The intrinsic cardiac nervous system includes all neurons located in the heart within the pericardium and those in the ganglionated plexi primarily located on the posterior atrial wall and fat pads. The intrinsic cardiac nervous system contains complex networks of neurons and interconnecting nerves that include afferent neurons (i.e., sensory), efferent neurons (i.e., parasympathetic and sympathetic), and interconnecting local circuit neurons (Hanna et al., 2017). Likewise, blood vessels are innervated by afferent (e.g., sensory fibers) and efferent (e.g., autonomic sympathetic fibers) fibers to control vascular function (Westcott & Segal, 2013; Shoemaker et al., 2015). Intriguingly, autonomic innervation increases with a decrease in the size of blood vessels, with arterioles being highly innervated by these fibers. Afferent neurons are mechano- and/or chemoreceptors that sense cardiovascular function on a beat-to-beat basis (Hadaya & Ardell, 2020). These signals are transmitted locally as well as to higher centers, which in turn modulates efferent outflow. Even minor alterations in R-R interval and/or activation sequence (as occurs during premature ventricular contractions) increase afferent activity and efferent outflow, leading to electrical instability on subsequent beats (Hamon et al., 2017). During ischemia, the release of metabolites, including bradykinin, prostaglandins, and protons, stimulates afferent neurons and leads to reflex increases in sympathetic outflow (Longhurst et al., 2001). Afferent signaling also plays a role in chronic disease remodeling, as ablation of sensory neurons improves cardiac function after myocardial infarction (MI) and slows the progression of heart failure (Wang et al., 2014), as well as leads to a reduction in blood pressure in spontaneously hypertensive rats (Li et al., 2022).
Efferent fibers communicate with CMs and blood vessels via neurotransmitters (primarily norepinephrine for sympathetic and acetylcholine for parasympathetic neurons). Norepinephrine produces prototypical cardiac fight-or-flight responses (e.g., CM and VSMC contractility leading to increased inotropy and vasoconstriction), and acetylcholine has the opposite effects (e.g., VSMC relaxation leading to vasodilation). Sympathetic neuron-tissue signaling is likely to occur in a quasi-synaptic fashion, with relatively limited diffusion of norepinephrine outside the junctional cleft, thus facilitating the stimulation of only those β-ARs on cells that are in close contact with the neuron (Prando et al., 2018). Efferent sympathetic neurons can modulate CM size via regulation of cellular proteolytic machinery, with increased neuron density correlating with increased CM size (Pianca et al., 2019). Moreover, sympathetic neuronal activity distinctively influences different vascular beds, with effects in smaller arteries leading to increased vascular resistance and alterations in blood flow, and in larger arteries resulting in alterations in arterial stiffness (Bruno et al., 2012). Sex differences may also play a key role in how sympathetic neuronal activity modulate cardiovascular function (Hissen & Taylor, 2020). In addition to the primary neurotransmitters, efferent neurons also release abundant co-transmitters, including neuropeptide Y, galanin, and ATP for sympathetic neurons, and vasoactive intestinal peptide, nitric oxide, and ATP for parasympathetic neurons (Burnstock, 2009). Co-transmitter releases affect many cell types, including CMs, VSMCs, ECs, and also feedback to the neurons themselves to impact subsequent neurotransmitter release (Tan et al., 2018).
Immune cells
Macrophages are essential components of the immune system and play a role in the normal functioning of the cardiovascular system (Moskalik et al., 2022). Macrophages may derive from embryonic precursors, blood monocytes, or hemogenic endocardium and increase in number by in situ proliferation (Moskalik et al., 2022). Macrophages are in close association with the vasculature, where they can serve many roles as part of the immune response to injury or pathological state, including in vascular repair and regeneration and modulation of vascular reactivity leading to changes in blood flow and tissue perfusion (Corliss et al., 2016; Hong & Tian, 2020; Barkaway et al., 2022). In the adult heart, resident cardiac macrophages represent ~6–8% of the non-myocyte population (Litvinukova et al., 2020), with their proportion changing with aging, cardiovascular disease state and progression, and depending on sex. The various functions of macrophage populations in the cardiovascular tissue depend on their origin and phenotype.
Macrophages in the cardiovascular system maintain tissue homeostasis by defending the body from pathogens and eliminating apoptotic cells. Macrophages also help to regulate extracellular matrix turnover, remove debris shed by local cells, and adapt to altered and increased tissue stress and strain. It has been suggested (but not yet tested) that macrophages may therefore play an important role in physiological hypertrophy that occurs with exercise or pregnancy (Swirski & Nahrendorf, 2018). According to a recent report, resident macrophages located in the atrioventricular node directly modulate electrical properties of CMs owing to macrophage coupling with CMs via Cx43-containing gap junctions (Hulsmans et al., 2017). Specifically, macrophages depolarize the resting membrane potential of CMs and, according to computational modeling, accelerate their repolarization, thereby potentially leading to arrhythmias (Hulsmans et al., 2017). Likewise, macrophages can modulate vascular function, blood flow, and tissue perfusion by altering the contractile state of VSMCs and pericytes via the release of vasoactive molecules (Barkaway et al., 2022). Mechanisms by which macrophages could modulate the contractility of mural cells include changes in intracellular Ca2+, ROS signaling, reduced nitric oxide bioavailability, and inhibition or hyperactivation of ion channels.
Direct electrical coupling between VSMCs and macrophages, and the in vivo significance of electrical coupling of CMs and macrophages remain to be determined and thus represent areas for promising research.
Within minutes of ischemia, the leukocyte population of the cardiac and vascular network changes dramatically, with neutrophils infiltrating first and peaking within ~24 hours, followed by the influx of monocyte-derived macrophages (Frodermann & Nahrendorf, 2018; Hong & Tian, 2020). This process is critical for proper wound healing, vascular repair, and angiogenesis (Shirai et al., 2015). However, dysregulated cardiac and vascular inflammation may play a role in arrhythmias (e.g., atrial fibrillation (Dobrev et al., 2022)) and heart failure progression (Dick & Epelman, 2016), as well as atherosclerosis and vasculitis (Shirai et al., 2015). Indeed, recent studies suggest that infiltrating neutrophils following MI increase arrhythmia burden via ROS production, whereas infiltrating macrophages reduce arrhythmia incidence by removing dead or damaged cells and by supporting CM mitochondrial function (Grune et al., 2022). Infiltrating macrophages post-MI, after inflammatory condition or pathology may also secrete a variety of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, which may have deleterious effects on CM and mural cell function (Francis Stuart et al., 2016; Heijman et al., 2020; Barkaway et al., 2022; Dobrev et al., 2022). In this issue of the Journal of Physiology, Chowkwale et al. developed a computational model of diverse immune cell types (neutrophils, monocytes, macrophages, FBs) and cytokine dynamics post-MI. The model predicted that neutrophil-IL-1β positive feedback recruited/activated monocytes/macrophages, which amplified TGFβ-mediated fibrosis (Chowkwale et al., 2022). The authors termed this cascade inflammation-fibrosis coupling, drawing parallels with the regulatory structure of excitation-contraction coupling. Further, the model predicted that the ultrasensitive amplification of fibrosis by inflammation was driven primarily by FB proliferation rather than collagen expression per cell (Chowkwale et al., 2022).
Adipocytes
Adipose tissue regulates cardiovascular health by producing and releasing several active molecules that may impact cardiovascular function. Adipose tissue that directly surrounds the heart is known as epicardial adipose tissue. Adipocytes originate from adult cardiac progenitor cells in the epicardium that can undergo epithelial-to-mesenchymal transition. Epicardial adipose tissue is present in both healthy and diseased individuals, and its volume increases during the first 40 years of life, as well as in the presence of inflammation or cardiomyopathy. Its main function is to protect the heart from mechanical deformation and provide storage for free fatty acids (Ernault et al., 2021). Likewise, most blood vessels are surrounded by adipose tissue, known as perivascular adipose tissue (Chang et al., 2020). The developmental origins of adipocytes of the perivascular adipose tissue are unclear but seem to vary depending on the location of the blood vessel and could even share similar precursor cells as those leading to vascular smooth muscle in the particular vascular bed.
Adipocytes secrete numerous adipokines, which modulate (myo)FB and myocyte physiology (Chang et al., 2020; Ernault et al., 2021). These adipokines may be protective in healthy hearts, preventing inflammation and fibrosis. However, adipokines secreted from adipocytes may switch to pro-inflammatory and pro-fibrotic, leading to reactive oxygen species generation. Pro-fibrotic adipokines stimulate myoFB differentiation, causing pronounced fibrosis in the epicardial adipose tissue and the myocardium and promoting arrhythmogenic cardiac remodeling (Gawalko et al., 2022). Inflamed perivascular adipose tissue in response to pathological conditions may also influence neointima formation by triggering phenotypic switching of VSMCs from contractile to synthetic (Chang et al., 2020).
Adipose tissue may also influence cardiac and vascular electrophysiology via adipokines and/or through electrotonic interactions, which could have an impact on cardiac function and vascular tone. However, whether adipocytes in can electrically couple to CMs or VSMCs remains unknown, and thus an area for further exploration.
Pericytes
Pericytes are emerging as significant regulators of the cardiovascular system. They may originate from mesenchymal and/or neural crest cells, depending on their location in the body, but may also be derived from bone marrow cells during tumor-induced angiogenesis (Kelly-Goss et al., 2014; Stapor et al., 2014; Trost et al., 2016). The genetic profile of pericytes seems to be heterogenous between different tissues but less variable within the same tissue (Trost et al., 2016). In the vasculature, pericytes wrap around ECs. Pericyte morphology is diverse, with recent examples showing their characteristic “bump on a log” shape with differences in extension and projection as the vessel transition from the post-arteriole to the higher-order capillary regions (Gonzales et al., 2020; Hariharan et al., 2020). Pericytes communicate with ECs via gap junction and more recently was shown that they could even communicate among themselves via interpericyte tunneling nanotubes (Alarcon-Martinez et al., 2020). Although the functional role of pericytes is still under intense investigation, they have been clearly shown to contribute to the stabilization of blood vessels, angiogenesis, regulation of EC function, and maintenance of the blood-brain barrier (Stapor et al., 2014; Trost et al., 2016; Hartmann et al., 2022). Recent data also suggest that pericytes contribute to the regulation of blood flow by contracting in capillary junctions to direct flow (Gonzales et al., 2020).
In addition to their well-established association with ECs, pericytes play an essential role in cardiac physiology and pathology by interacting with other adjacent cell types, such as CMs, FBs, and adipocytes (Su et al., 2021). Several lines of evidence suggest that cardiac pericytes are critical to sustaining cardiac function during myocardial injury, as reviewed in (Yin et al., 2021). Specifically, VEGF and miR-132 from pericytes were found to induce survival responses in CMs, and through secreting stromal cell-derived factor-1, pericytes were shown to activate the mobilization of stem cells, thus benefiting cardiomyogenesis. Furthermore, some pericytes sharing the same origin of epicardium with CMs exhibit myogenic capacity and were found to function as (and replace) CMs. In addition, some pericytes can differentiate into immature CM-like cells.
In many ways, cell diversity in cardiac tissue is confounded by overlapping and interconnected cell behaviors (Figure 3). The complexity and emerging knowledge gaps are highlighted by the overlapping cell phenotypes and the potential for direct cell coupling. CMs, FBs, pericytes, macrophages, ECs, and neuronal cells share phenotypical and potential lineage characteristics. Future studies are needed to elucidate the implications of common cell origins and how the differentiated cell populations interact. Throughout this section, we have highlighted several unresolved questions, which focus on the local cardiac tissue microenvironment, investigation of multi-cell interactions, and understanding how these interactions are temporally and spatially regulated.
2. Contribution of Cell Diversity to Cardiovascular Remodeling Associated with Development, Disease, and Aging
The diverse cell types and signals described in the previous sections distinctly contribute to the development of disease-promoting cardiovascular remodeling with molecular, cellular, and interstitial changes manifesting clinically as alterations in the structure and function of the heart and vessels after injury. The remodeling process involves myocyte growth and death, fibrosis, inflammation, and electrophysiological alterations, which are the result of subcell-, cell- and tissue-level responses to pathophysiological stimuli. In the following paragraphs, we describe the multi-level aspects of cardiovascular remodeling during development, aging, and diseased states, as well as the contribution of different cell populations and signals.
2.1. Cellular Energetic Costs of Cardiac Function
Due to the immense energetic requirements of mechanical pump function, the heart is a major contributor to whole body oxygen consumption (Rolfe & Brown, 1997; Knaapen et al., 2007). At an organ level, cardiac consumption rates for oxygen and catabolic substrates are dynamic and determined largely by the systolic parameters of contractile force generation, wall stress, and heart rate. With each contractile cycle, CMs expend ATP in biochemical reactions that maintain transmembrane ionic gradients and drive actin-myosin cross-bridge cycling, thereby orchestrating AP generation and repolarization, CICR, contraction, and relaxation. Recent work using single-cell RNA sequencing (scRNA-seq) has indicated subpopulations of CMs in adult human myocardium that are enriched in nuclear-encoded mitochondrial genes (NDUFB11, NDUFA4, COX7C, and COX5B), suggesting regional heterogeneities in cellular workload capacities (Litvinukova et al., 2020).
Whether specialized subgroups of CMs represent stable cell populations or are modified in number or spatial distribution throughout development or in response to environmental or pathological stimuli is still unclear. If so, modulation of regional CM heterogeneities may drive large-scale changes in cardiac metabolic requirements during development and in homeostasis and disease progression.
Cell types in the heart other than CMs contribute directly and indirectly to balancing oxygen and nutrient delivery, ATP generation, and CM contractility. While the oxygen and substrate input needed to support cellular functions in VSMCs and ECs, FBs, and immune cells are low relative to that in CMs (Wagner et al., 2011), these cell populations influence CM workloads and, thus, energetic demand. For instance, cardiac FBs proliferate and activate to a contractile myoFB state in response to mechanical tension or injury-associated stimuli (cytokines and chemokines) (Frangogiannis, 2019). Once activated, cardiac FBs remodel the structure and composition of the ECM. This ECM remodeling can occur within days of ischemic injury to replace dead or dying CMs to prevent wall rupture or can develop more slowly in the interstitium and perivascular spaces to provide structural support as an adaptation to hemodynamic stress. Regardless of the stimulus, ECM replacement, collagen deposition, and structural modifications (i.e., crosslinking) can alter myocardial wall compliance and influence the relationship between energy expenditure and cardiac output (Lopaschuk et al., 2021). Thus, ECM-dependent changes in wall stress contribute to CM remodeling that ultimately impacts metabolic pathway flux and may contribute to energetic deficits in the failing heart.
The immune system also exerts profound effects on myocardial function and metabolism. Macrophage populations include those of embryonic origin (tissue resident) and monocyte-derived, which differ with respect to inflammatory patterns and metabolic profiles (Patel et al., 2018; Dick et al., 2019). Differential consumption of nutrient metabolites by distinct populations of macrophages may regulate the local availability of substrates for other cell types (Kedia-Mehta & Finlay, 2019). Tissue-resident macrophages have also been shown to eliminate dysfunctional mitochondria that are expelled by otherwise healthy proximal CMs (Nicolas-Avila et al., 2020). Conversely, proinflammatory macrophages may play a key role in altered redox conditions that are linked to age-associated disease states. In advanced age, accumulated senescent cells, via secretion of cytokines, can promote the expansion of CD38+ M1-like macrophages and reductions in tissue NAD levels (Covarrubias et al., 2020), as well as CM activation leading to the production of IL-1β and consequent arrhythmia (Heijman et al., 2020). Activation of inflammatory pathways and secreted inflammatory cytokines have also been shown to impact repolarizing currents and cause Ca2+-mediated voltage instabilities, such as delayed afterdepolarizations, in CMs (Heijman et al., 2020), which may produce a substrate for arrhythmia and remodeling of the conduction system (Lazzerini et al., 2022).
Much attention has focused on how cardiac metabolism adapts under physiologic and pathologic states and how altered metabolic states influence overall function. The heart is thought of as a metabolic omnivore in that it is capable of processing multiple substrates to produce ATP. While the healthy heart relies on β-oxidation of fatty acids for the majority of ATP generated, it can also derive energy from other sources, including glucose, pyruvate, lactate, branched-chain amino acids, and ketone bodies (Lopaschuk & Ussher, 2016). The balance of substrate utilization becomes altered toward greater reliance on glucose in the setting of heart failure and is associated with decreased mechanical efficiency and adverse outcomes (Lopaschuk et al., 2021). Moreover, advanced age is associated with a reduction in the mitochondrial machinery required for fatty acid oxidation and an increase in glycolytic enzymes and intermediates in CMs (Lesnefsky et al., 2016).
The upstream mechanisms that drive metabolic remodeling are poorly understood. Whether these changes occur because of direct signaling at the level of CMs or from external factors such as vascular dysfunction and insufficient oxygen delivery relative to demand remains to be clarified. Nonetheless, changes in vascular structure or function may be a key contributor (Anversa et al., 1994). Oxygen delivery via the blood supply is inherently linked to the metabolic demand of the myocardium (Feigl, 1983). Changes in vascular structure and function with age may suppress perfusion with the consequence of ischemia-related remodeling and contractile dysfunction (Heusch, 2022). Further work to determine the underlying causes of disease-related metabolic and functional remodeling is warranted.
Metabolic adaptations are also important for postnatal heart development. Fetal and neonatal cardiac physiology is fundamentally different from that of an adult, and a number of modifications are required to transition from the intra- to extrauterine environment (Hew & Keller, 2003; Morton & Brodsky, 2016; Prada et al., 2020). This dynamic process is most evident at the cellular level, as CMs undergo a series of morphological and metabolic changes during their transformation into mature CMs (Chang, 1988; Kannan & Kwon, 2020; Karbassi et al., 2020). To adapt to increased workload and energy demand after birth, immature CMs undergo physiological hypertrophy, which is accompanied by a withdrawal from the cell cycle and loss of proliferative capacity (Li et al., 1996; Soonpaa & Field, 1998). Hypertrophic growth transforms these small, circular cells into large rod-shaped CMs with a massive 10-fold increase in cell volume (Mollova et al., 2013; Pohjoismaki et al., 2013; Velayutham et al., 2020). Developing CMs also undergo a metabolic switch, wherein cells become less dependent on glucose and more dependent upon fatty acid oxidation for efficient energy production (Lopaschuk & Jaswal, 2010). To accommodate this switch, mitochondria increase in number and size - occupying nearly 20–40% of the total cell volume in adult CMs (Barth et al., 1992). As mitochondria mature, densely organized cristae are also formed and the mitochondrial network aligns with myofibrils to efficiently supply ATP for contractile function and ion pump activity (Kaasik et al., 2001; Saks et al., 2001; Andrienko et al., 2003; Bleck et al., 2018). The overall increase in cell size is accompanied by a large-scale expansion and alignment of myofibrils (Bishop et al., 1990; Lundy et al., 2013), and maturation of sarcomeric proteins from fetal to adult isoforms (e.g., troponin, titin, myosin heavy chain, myosin light chain; (Opitz et al., 2004; Talman et al., 2018)).
2.2. Cellular Topologies and Impact on Cardiovascular Function in Health and Disease
Maintenance of optimal cardiac function depends on complex heterocellular signaling paradigms that serve to maintain homeostasis, as discussed above. Likewise, aberrant communication among CMs and interstitial cells is a central player in the development and progression of declining cardiac function in disease. A primary example is the regulation of cardiac FB activation by CMs and macrophages that contributes to ECM remodeling. In the setting of acute myocardial injury due to ischemia, death of CMs triggers leukocyte infiltration and an acute inflammatory response (Prabhu & Frangogiannis, 2016). Resultant elevation of macrophage-derived inflammatory cytokines (e.g., TGF-β, IL-6, IL-4, and IL-1) promotes the proliferation and activation of cardiac FBs, transforming them into a profibrotic phenotype (Guo et al., 2012; Kanellakis et al., 2012; Saxena et al., 2013; Khalil et al., 2017). Activated FBs degrade existing ECM and deposit altered matrix proteins that ultimately result in scar formation in the infarct region that prevents wall rupture but also increases wall stiffness and reduces compliance (Nielsen et al., 2019). Changes in wall tension may also alter the availability of latent ECM-localized growth factors and cytokines to CMs (Bowers et al., 2022), thus promoting hypertrophic growth and, in turn, altering wall mechanics and CM-derived proteins to the ECM that could influence immune cell and FB activation states. Even in the absence of ischemia, prolonged activation of several molecular processes and increased FB activation contributes to age-related collagen accumulation, interstitial and perivascular fibrotic remodeling, and functional decline (Biernacka & Frangogiannis, 2011). Further research that elucidates molecular interactions between cells of the heart and the ECM is needed to advance the development of more effective antifibrotic therapies.
Recent work has also advanced our knowledge of the local intercellular signaling that underlies local control of oxygen delivery to the myocardium. Zhao et al. investigated the molecular processes that mediate coupling between CM metabolic demand and local and regional blood flow, which have remained elusive for decades. The authors provided evidence that a population of capillary ECs is electrically coupled to CMs, such that hyperpolarizing signals may be transmitted from the myocardium to the vasculature (Zhao et al., 2020). In the case of declining ATP in CMs, activation in ATP-sensitive K+ channels could thereby be transmitted to the local capillary network to induce upstream arteriolar vasodilation and increase oxygen delivery to the affected CMs. At the level of coronary arteries and arterioles, other work has shown that acute increases in CM workload and oxygen demand are transmitted by an unknown mechanism to proximal VSMCs, culminating in altered vascular redox pairs (Dwenger et al., 2022). As discussed at the symposium, acute elevation of cytosolic NADH levels in VSMCs as a result of enhanced myocardial oxygen demand is, in turn, sensed by smooth muscle Kv1 channels, which are differentially regulated by cellular redox via associating heteromeric β subunit assemblies (Dwenger et al., 2018; Ohanyan et al., 2021).
To date, the mechanisms of local blood flow regulation have been identified/investigated under homeostatic conditions. Further work is necessary to determine how altered myocardial metabolism in aging and specific cardiovascular disease states may disrupt the balance of oxygen supply-demand balance via newly identified heterocellular signaling axes.
In a similar manner, biochemical cues including oxygen levels, glucose/fatty acid availability, glucocorticoid or thyroid hormone signaling (Burridge et al., 2014; Neary et al., 2014; Yang et al., 2014b; Rog-Zielinska et al., 2015; Parikh et al., 2017; Yang et al., 2019) and biophysical cues including electromechanical conditioning, micropatterning, or substrate stiffness also help to guide CM development (Bakunts et al., 2008; Heidi Au et al., 2009; Bhana et al., 2010; Kim et al., 2010; Feaster et al., 2015; Ronaldson-Bouchard et al., 2018; Herron et al., 2016). As an example, hypoxia signaling has been identified as a key regulator of CM maturation. Fetal CMs are well adapted to the intrauterine hypoxic environment, but with birth, the change in ambient oxygen levels initiates a reduction in hypoxia signaling (e.g., oxygen-sensitive transcription factors HIF1α and HAND1), which is accompanied by subsequent adaptations in cardiac bioenergetics and mitochondrial remodeling (Breckenridge et al., 2013; Neary et al., 2014). Of interest, chronic hypoxia can extend the window of CM immaturity, as evident by smaller CM size, continued CM proliferation, increased HIF1α signaling, reduced mitochondrial cristae density, and metabolism – while hyperoxemia promotes oxygen-dependent mitochondrial metabolism and cell cycle arrest (Puente et al., 2014). Hypoxic signaling is being explored as a potential therapeutic target, which could allow CMs to reenter the cell cycle, promote cardiac regeneration, and improve myocardial recovery following injury (e.g., myocardial infarction (Jopling et al., 2012; Kimura et al., 2016; Kimura et al., 2017; Nakada et al., 2017; Savla et al., 2018).
CM maturation is also guided through interactions with non-CM cells and the tissue microenvironment. When co-cultured with adult FBs (but not neonatal FBs), neonatal CMs display multiple markers of maturation, including withdrawal from the cell cycle, aligned myofibrils, formation of t-tubules, and increased expression of the gap junctional protein connexin-43 (Wang et al., 2020). Similarly, human stem cell derived-CMs (hiPSC-CM) hypertrophy and develop well-organized myofibrils when engrafted into an adult rat heart, while the maturation rate is significantly slowed when engrafted into younger neonatal hearts (Kadota et al., 2017). Co-culturing hiPSC-CMs with epicardial cells (precursor to cardiac FBs and ECs) or ECs alone also produces a more mature electrophysiological phenotype (Dunn et al., 2019), improved Ca2+ handling and contractility, and promotes vascularization when implanted (Bargehr et al., 2019). Similarly, when 3D multicell-type microtissues are constructed, intercellular interactions with FBs and ECs promote the maturation of CMs – with the development of sarcomeric structures, enhanced contractility, increased oxidative metabolism, and a more mature electrophysiological phenotype (Giacomelli et al., 2020). Taken together, these findings indicate that CMs are highly responsive to cell-cell interactions with non-myocytes and that both FBs and ECs play a supportive role in guiding the CM maturation process.
2.3. Subcellular Structural and Functional Heterogeneity in Cardiovascular in Health and Disease
While we have so far focused on the diversity of cell types that govern cardiovascular function, it is important to note that heterogeneity also exists within any given cell type, including molecular structural and functional diversity that play important roles in normal function and can importantly contribute to disease induction and progression. Ion channel remodeling is a hallmark of cardiovascular development, disease, and aging. The number and functional availability of ion channels at the sarcolemma limit the magnitude ion fluxes across it. In the heart and vasculature, these fluxes impact contraction and relaxation of the muscles. The abundance of ion channels at the sarcolemma is dictated by the lifetime of the channels and the balance between channel insertion, removal, and degradation. A further regulatory influence over ion flux across the plasma membrane is the clustering state of the channels. The idea that the nanoscale arrangement of ion channels in the plasma membrane can impact their function is an emerging concept in several fields, including cardiac and vascular smooth muscle physiology (Del Villar et al., 2021; Dixon et al., 2022). At the symposium, it was discussed that this concept that the clustering state of ion channels affects their activity also extends to SR-localized RyR2, where heterogeneity of cluster sizes has been found to support arrhythmogenic Ca2+-wave propagation (Galice et al., 2018; Xie et al., 2019). Control of transmembrane ion fluxes is also determined by the expression of distinct pore-forming and accessory subunit isoform heterogeneity, which may evolve during maturation to achieve optimal fine-tuning of cardiovascular signaling and function and can be dysregulated in disease. We discuss the state of knowledge for each of these channel types below, considering how maturation, aging, and disease processes, including heart failure and hypertension, affect channel arrangements with functional implications and highlighting open questions, controversies, and challenges in the field.
Trafficking, recycling, and nanostructural rearrangement of L-Type Ca2+ Channels (LTCCs)
The first important question to answer is how can clustering of LTCCs affect their activity. Work from multiple labs has revealed that physical proximity between adjacent CaV channels in clusters facilitates their dynamic, reciprocal, and allosteric interactions (recently reviewed in (Dixon et al., 2022)). Groups of channels within clusters communicate with each other to coordinate their gating behavior so that the opening of one channel can drive the opening of other adjoined channels. The interactions are initiated by the binding of incoming Ca2+ to CaM and proceed through Ca2+/CaM binding to the CaV1.2 pre-IQ domain (Dixon et al., 2015; Moreno et al., 2016). This ‘functional coupling’ or ‘cooperative gating’ facilitates and amplifies Ca2+ influx because physically interacting channels within clusters adopt the open probability of the most active channel, as demonstrated in optogenetic experiments in which stimulated interactions between CaV1.2 channel C-termini resulted in enhanced whole-cell ICa,L and EC-coupling in ventricular CMs (Dixon et al., 2012). Experimental (Dixon et al., 2012) and computational approaches (Sato et al., 2018) have further demonstrated that excessive CaV1.2 channel cooperativity can become pathogenic and promote cardiac arrhythmias, including alternans.
In the heart, at least two physiological signaling cascades have been demonstrated to trigger nanoscale rearrangements of CaV1.2 channels, affecting their clustering and activity. Acute (2 – 8 min long) activation of β-ARs with isoprenaline (ISO) leads to CaV1.2 channel cluster enlargement in mouse ventricular CMs (Ito et al., 2019; Del Villar et al., 2021) while more prolonged (30 min) activation of type 1 angiotensin receptors (AT1R) with angiotensin II favors channel endocytosis (Hermosilla et al., 2017), thereby reducing whole-cell ICa,L. In the case of the β-AR stimulated cluster enlargement, the mechanism involves mobilization of an endosomal reservoir of CaV1.2 channels to the t-tubule sarcolemma via Rab4-dependent fast and Rab11-dependent slow recycling pathways (Del Villar et al., 2021; Westhoff & Dixon, 2021). The enhanced channel recycling and insertion reinforces the CaV1.2 population at the sarcolemma, favoring enhanced EC-coupling and the positive inotropic response associated with fight-or-flight (Figure 4).
Figure 4: Nanoscale arrangement and rearrangement of dyadic calcium channels.
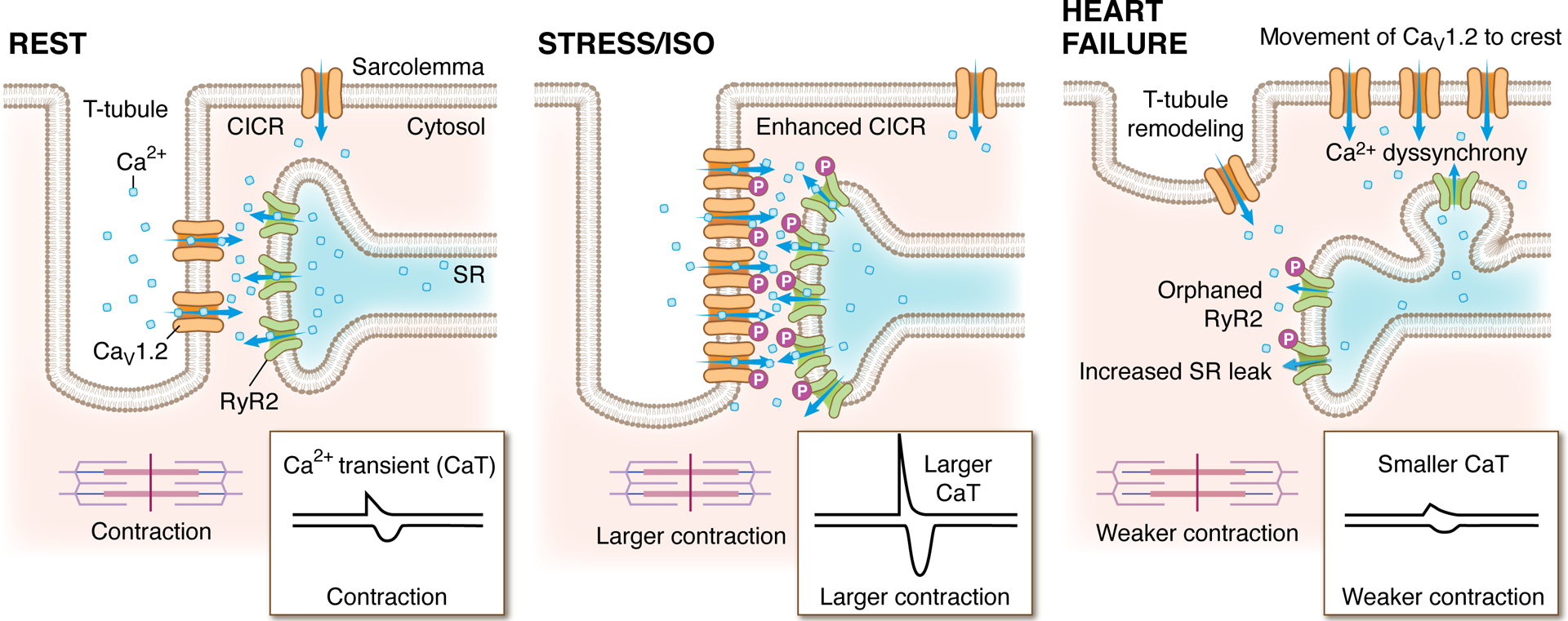
The localization and degree of clustering of cardiac CaV1.2 and RyR2 channels influences functional output. These cartoon depictions of the cardiac dyad, illustrate the clustering state and localization of CaV1.2 channels and juxtaposed RyR2 in resting (left), acutely stressed (middle), and heart failure conditions (right). At rest, clusters of CaV1.2 channels are located on the t-tubules opposite clusters of RyR2 on the jSR. During exercise, acute stress, or isoprenaline (ISO) application, the ensuing β-AR/Gs/adenylyl cyclase/cAMP/PKA signaling cascade results in phosphorylation of both CaV1.2 channels and RyR2, increasing their activity (Po). In parallel, PKA stimulates enhanced recycling of CaV1.2 from an endosomal reservoir to augment CaV1.2 number and cluster size at the t-tubules. On the jSR, RyR2 clusters also undergo remodeling, becoming enlarged during stress. Altogether, the combination of enhanced channel Po, increased levels of cooperative gating, and larger numbers of CaV1.2 channels within enlarged clusters leads tο greater Ca2+ entry which stimulates more CICR. Larger RyR2 clusters also generate larger, more frequent sparks which summate to form larger Ca2+ transients and stronger contractions, creating the characteristic positive inotropic response during acute stress. Right: In heart failure, cardiomyocytes undergo architectural remodeling and ion channel cluster rearrangements. RyR2 channel cluster fragmentation/dispersal and orphaning occur as jSR elements are left behind while t-tubules recede. CaV1.2 channels migrate to the crest but the reduced proximity to RyR2 leads to an inefficient CICR that results in Ca2+ dyssynchrony. Hyperphosphorylation of RyR2 increases their activity leading to SR leak which elevates diastolic Ca levels in the cytosol and reduces the releasable pool in the SR that can be accessed during CICR. The result is smaller Ca2+ transients and weaker contractions.
Whether CaV1.2 clustering is altered in heart failure and/or in aging remains to be fully elucidated. In failing hearts, nanostructural rearrangements of CaV1.2 have been described with channel localization shifting away from the t-tubules toward the sarcolemmal crest (Bryant et al., 2015; Sanchez-Alonso et al., 2016; Sanchez-Alonso et al., 2020) (Figure 4). As yet, there have been no reports of how these rearrangements impact channel clustering, although constitutive phosphorylation of CaV1.2 by PKA reportedly enhances cooperative gating behavior of t-tubules localized channels in failing right ventricular CMs in an ischemic cardiomyopathy model inviting speculation that CaV1.2 channel cluster size may be augmented in those cells (Medvedev et al., 2021).
Hypertension (Navedo et al., 2010a), diabetes, and hyperglycemia (Navedo et al., 2010b; Nystoriak et al., 2017; Prada et al., 2019; Syed et al., 2019; Prada et al., 2020) have been linked with alterations in CaV1.2 channel function that favor enhanced cooperative gating behavior in VSMCs. Given that enhanced proximity between CaV1.2 channels facilitates cooperative gating, these studies hint that these pathophysiological stimuli may also be driven by CaV1.2 channel cluster area augmentation, but this hypothesis remains to be experimentally explored.
Trafficking and recycling of CaV1.2 channels in VSMCs is a largely understudied area, although it is known that Rab25-dependent recycling is important for CaV1.2 expression at the sarcolemma of VSMCs (Bannister et al., 2016). Future work should explore whether VSMCs have an endosomal reservoir of pre-formed CaV1.2 channels analogous to that seen in CMs, and if so, whether CaV1.2 cargo can be dispatched to the sarcolemma to reinforce LTCC clusters or sequestered to endosomes for storage during various signaling pathways.
Ryanodine Receptor Type-2 (RyR2) clustering
As discussed at the symposium, there is an accumulating body of experimental and in silico evidence suggesting that RyR2 cluster size can impact the magnitude of SR Ca2+ release and muscle contraction in CMs. The larger clusters of RyR2 in CMs increase frequency and size of Ca2+ sparks, and cause greater Ca2+ transients and stronger contractions, while smaller RyR2 clusters have the opposite effect (Sobie et al., 2002; Galice et al., 2018). Like CaV1.2 channels, RyR2 clustering plasticity has been reported in response to activation of physiological signaling pathways with their acute phosphorylation downstream of β-AR stimulation or treatment with a phosphorylation-promoting cocktail linked to cluster enlargement (Fu et al., 2016; Asghari et al., 2020). Given the association between RyR2 cluster size and functional output, these cluster enlargements would be predicted to favor enhanced contractility, supporting the positive inotropic response observed during fight-or-flight. Recent work has revealed that more prolonged β-AR stimulation favors RyR2 cluster fragmentation after ~60 mins of stimulation with ISO (Shen et al., 2022) (Figure 4). This fragmentation was accompanied by decreased Ca2+-transient amplitude and was dependent on CaMKII and PKA-mediated RyR2 phosphorylation. Prolonged β-AR stimulation is a feature of heart failure and indeed, several investigators have reported similar RyR2 cluster or Ca2+ release unit (a functionally grouped collection of RyR2 clusters) fragmentation/dispersal in models of heart failure (Kolstad et al., 2018; Sheard et al., 2019), and in AF (MacQuaide et al., 2015) (Figure 4). Notably, similar RyR2 cluster fragmentation and smaller Ca2+ transients were observed in CMs isolated from patient samples with idiopathic dilated cardiomyopathy compared to healthy controls (Hou et al., 2021). However, the phenomenon was not observed in atrial CMs from AF patients suggesting that dispersion of RyR2 is not an essential feature of that pathology (Munro et al., 2021). Careful analysis of RyR2 cluster size may reveal new insight moving forward since the report that a heterogeneous mixture of large and small RyR2 clusters can facilitate Ca2+ wave propagation and associated arrhythmias (Xie et al., 2019).
RyR2 are also present in VSMCs where Ca2+ sparks are not coupled to a contractile process, but instead promote hyperpolarization and vasodilation (relaxation) due to their proximity and functional coupling to large conductance Ca2+-activated BK channels. There is evidence however, that RyR2 clustering plasticity also affects this physiological process with RyR2 cluster enlargement favoring enhanced BK channel activity and reduced cerebral artery tone and vascular dysfunction in Duchenne muscular dystrophy mdx mice compared to WT controls (Pritchard et al., 2018). Thus, it appears nanostructural RyR2 arrangement also impacts smooth muscle function.
While RyR2 cluster enlargement and dispersal have been observed in various cell types, the mechanisms of trafficking and recycling of RyR2 that contribute the to their dynamic clustering responses are poorly understood.
Heterogeneity and Overlap of K+ Channel Activity in Cardiovascular Health and Disease
K+ channel activity universally mediates repolarization in cardiovascular tissues. Accordingly, K+ channel distribution and expression are tightly controlled to coordinate chamber and tissue-specific electrical activity. Several mechanisms have been identified that help to ensure the appropriate balance of repolarizing (K+) and depolarizing (Na+ and Ca2+) channels at the surface membrane. For example, the inward rectifier Kir2.1 expression is reciprocal to NaV1.5 (Milstein et al., 2012). The Kir2.1/NaV1.5 interaction ensures appropriate conduction velocity when Kir2.1 is upregulated (Varghese, 2016), as while IK1 upregulation is expected to lower excitability and conduction velocity, reciprocal modulation leads to INa enhancement and consequent increase in conduction velocity. Similarly, KV11.1 subunit transcripts are co-translated at the ribosome with NaV1.5-encoding transcripts (Eichel et al., 2019). How either of these mechanisms is affected in cases of cardiac disease has yet to be studied directly.
AF is associated with complex remodeling in several K+ currents (Dobrev & Ravens, 2003; Gaborit et al., 2005; Heijman et al., 2014). Chronic AF in human tissue down was shown to downregulate Ito, upregulate IK1, IKACh, IKs, and IK2P, and has mixed or no effects on IKr and IKur (Dobrev et al., 2001; Workman et al., 2001; Dobrev et al., 2005; Caballero et al., 2010; Schmidt et al., 2015). However, works in animal models have shown that the effects on K+ currents in the context of AF are dependent on the duration of AF and the underlying cardiac disease (Li et al., 2000; Gaborit et al., 2005; Sridhar et al., 2009). The identification of the specific triggers of K+-channel dysregulation in AF is still onging. Most recently Aromolaran et al. demonstrated using a rat lipotoxicity model of AF that mTOR signaling increases IKr magnitude by selectively promoting surface expression of the KV11.1 channel subunit, KV11.1b (Martinez-Mateu et al., 2019; Aromolaran et al., 2022).
In the failing ventricle, the major K+ currents Ito, IKr, and IKs, and IK1 are downregulated and cause homeostatic responses that lead to arrhythmogenesis (Michael et al., 2009). The specific molecular mechanisms pertaining to each current continue to be dissected. In healthy canine heart, the IKr conducting KV11.1 channel is distributed within the lateral membrane along the Z-lines, with particular enrichment within the t-tubules (Jones et al., 2004). As t-tubule organization is altered in heart failure (Lipsett et al., 2019), KV11.1 organization could be similarly disrupted. Indeed, KV11.1 organization is highly sporadic in stem cell-derived CMs, which lack any discernable t-tubule structure (Jones et al., 2014). The E3 ubiquitin ligase Nedd4–2 was recently shown to be upregulated in the failing heart (Wang et al., 2021). Nedd4–2 mediates KV11.1 degradation and could contribute to reduced KV11.1 in the failing heart (Guo et al., 2012). Holzem et al. used human tissue to demonstrate that while IKr is downregulated in the failing human heart, KV11.1 subunit composition is also shifted towards a more fetal-like composition (Crotti et al., 2013; Holzem et al., 2015). The mechanism by which this shift occurred remains unclear. A cis-regulatory element was recently identified as a subunit-specific regulator in mouse (van den Boogaard et al., 2019), but its role in the failing heart has not been explored.
Reduced Ito contributes to the diminished contractility of the failing heart, where the reduced phase 1 repolarization leads to diminished ICa,L and reduced Ca2+ transient. It was shown in failing human CMs that the fast component of Ito, ItoF, is selectively downregulated, whereas the slow component (ItoS) is unaffected (Johnson et al., 2018b). Nassal et al. demonstrated that KChIP2 function is disrupted in the failing heart, triggering increased miR-34 activity that then disrupts the production of the ItoF conducting channel, Kv4.3 (Nassal et al., 2017). KV4.3 was also shown to promote KV11.1 expression in HEK293 cells by upregulating hsp70 (Zhao et al., 2017). This finding highlights the complex crosstalk between distinct K+ channel subtypes in cardiac tissue.
Accessory proteins (i.e., minK, miRP1–4) were originally thought to be relatively exclusive to a single family of pore-forming K+ channel subunits. The recently established knowledge that accessory subunits interact with multiple ion channel groups with distinct effects provides the potential for targeted therapies that could benefit multiple K+ currents. The accessory K+-channel protein minK is upregulated in heart failure. Xu et al showed that phosphorylation at S102 drives KV7.1/minK channel internalization during sustained PKC activity (Xu Parks et al., 2020). Because minK interacts with KV11.1, minK could represent a singular target to restore IKs and IKr activity in the failing heart. Phosphatidylinositol 4,5 bisphosphate (PIP2) represents an additional target for improving K+-channel function in failing CMs. PIP2 is decreased in spontaneously hypertensive rats (Zhou et al., 2021). PIP2 depletion has been known for years to diminish IKs, IKr, and IK1. A recent study in stem cell-derived CMs demonstrated that PIP2 depletion using HIV-Tat reduced IKr and IKs in tandem (Es-Salah-Lamoureux et al., 2016). Identifying the networks of interactions between ion channel proteins across distinct cardiovascular tissues will help to identify future targets for broad therapeutic effects.
Ion channel remodeling also occurs throughout development, which helps to produce a more mature electrophysiological and contractile phenotype. Adult ventricular CMs are quiescent, while immature CMs display pronounced automaticity (Sun et al., 2017) that can be attributed to a less negative resting membrane potential with reduced expression of the inwardly rectifying K+ channel (Wahler, 1992; Masuda & Sperelakis, 1993; Vaidyanathan et al., 2016; Goversen et al., 2018), increased pacemaker current (Cerbai et al., 1999; Giannetti et al., 2021), and/or spontaneous SR Ca2+ leak from the sarcoplasmic reticulum that can trigger depolarizations via NCX activation (Kim et al., 2015; Carmeliet, 2019). The shape of the cardiac AP also differs between immature and adult CMs, albeit the details of these differences are species dependent (Wang et al., 1996; Mummery et al., 2003; Hamaguchi et al., 2013; Swift et al., 2020; Cooper et al., 2021). Likewise, EC-coupling becomes more efficient in mature CMs (Escobar et al., 2004; Lundy et al., 2013; Louch et al., 2015), as these large rod-shaped cells contain t-tubules or sarcolemmal invaginations that allow an AP to quickly reach the interior of the myocyte. This structural adaptation improves CICR in adult CMs (Haddock et al., 1999; Hamaguchi et al., 2013) by juxtaposing LTCCs in the plasma membrane with RyR2s in the SR (Seki et al., 2003; Ziman et al., 2010). In comparison, neonatal CMs are less dependent on LTCC-mediated CICR, and instead rely more heavily on reverse-mode NCX for Ca2+ influx and EC-coupling (Wetzel et al., 1995; Huang et al., 2005). Developmental maturation also includes the formation of specialized intercellular junctions called intercalated discs (Peters et al., 1994; Angst et al., 1997), which facilitate electrical and mechanical communication between neighboring CMs (Angst et al., 1997; Vreeker et al., 2014). Structurally, the intercalated discs connect the actin cytoskeleton and intermediate filament network between CMs and also establish intercellular gap junctions, which permit the cell-cell transmission of ions and metabolites. Several ion channels have also been found to reside within the intercalated disc (e.g., KV, Kir, NaV; (Vermij et al., 2017)). Collectively, these structures allow an AP to quickly propagate through the myocardium syncytium to coordinate contraction.
K+ channels are also critical in VSMC regulation. Accordingly, K+ channels, including KV, KATP, BK and Kir, control VSMC membrane potential and, therefore, LTCC activity, [Ca2+]i, myocyte contractility, myogenic tone, and vascular reactivity (Tykocki et al., 2017). The expression of VSMC K+ channel subunits is heterogeneous and depends on the vessel location (Tykocki et al., 2017). K+ channel subunits have a tremendous influence on channel activity and cellular function (Tykocki et al., 2017). For example, KVβ subunits can interact with the KV1 channels to link excitability and metabolic state to channel activity and VSMC contractile state (Ohanyan et al., 2021). This mechanism can help modulate myocardial blood flow. Another well-known example is BK β1, which contributes to BK channel voltage and Ca2+ sensitivity and modulates vascular tone (Tykocki et al., 2017). The expression of VSMC K+ channel subunits can also be modified during pathological conditions, such as diabetes and hypertension (Eichhorn & Dobrev, 2007; Tykocki et al., 2017; Nieves-Cintron et al., 2018; Nieves-Cintron et al., 2021). For example, KV2.1 and BK β1 subunits are downregulated in diabetic and hypertensive VSMC (Nieves-Cintron et al., 2018; Nieves-Cintron et al., 2021). This KV2.1 and BK β1 downregulation is mediated by activation of the transcription factor NFATc3 (Nystoriak et al., 2014; Nieves-Cintron et al., 2015). Downregulation of these subunits was correlated with an increase in VSMC contractility, myogenic tone, and mean arterial pressure. VSMC K+ channel functional expression is also modulated by the dynamic trafficking of different K+ channel subunits (Leo & Jaggar, 2016). This dynamic trafficking of VSMC K+ channel subunits such as KV1.5 and BK β1 has been shown to be influenced by membrane potential, stimulation of angiotensin II signaling, and pathological conditions such as hypertension to control VSMC contractility and vascular reactivity (Leo & Jaggar, 2016). Because of the diversity of K+ channel subunits, the impact of each of the subunits is unclear and thus represents an area for future research.
Action potential and ionic current variability
The concerted action of several ionic channels and transporters shapes the cardiac AP (see section 1). There are large species differences in ion channel expression patterns, especially considering K+ channels that govern the repolarization dynamics (Grandi et al., 2017). Small animal models of cardiovascular diseases can inform about changes in ventricular Ito, IK1 and INaL, and the signaling mechanisms involved. However, they provide little or no information about IKr and IKs (the physiological role of IKr and IKs is unclear in small animals) and phase 3 repolarization dynamics (small animals have different AP morphology). Therefore, large animal models with ion channel expression patterns and AP morphology resembling the human heart have a pivotal role in understanding exact arrhythmia mechanisms relevant to human. Guinea pig has been a highly useful small animal model in which cardiac repolarization mechanisms are more alike those in larger mammals. However, the use of guinea pig has decreased due to the rise of much more sophisticated tools for genetic manipulation of mice.
A potential alternative for interspecies translation of electrophysiology and Ca2+ handling from mouse to human is the use of a powerful computational framework as recently established (Morotti et al., 2021), and discussed below in section 3.3. It is also still unknown whether a central genetic or epigenetic mechanism(s) responsible for pathogenic ionic remodeling exists and whether this central mechanism is cell-specific, all shared among different cell lines (CM-FB-EC), and emerging areas for future research.
In traditional voltage-clamp experiments, only one ionic current can be reliably measured at a time. Using novel approaches such as sequential dissection of ionic currents under AP-clamp using selective ion channel inhibitors (Hegyi et al., 2018) or complex voltage protocols optimized by a genetic algorithm (Clark et al., 2022) may inform about multiple ionic current magnitudes in CMs yielding rich data from a limited number of diseased cells/animals/patients. These same approaches could be extended to examine ionic conductances in VSMCs. Moreover, these methods may help to better understand the co-expression and co-regulation of multiple ion channels within the same cell and their variability among cells, including VSMCs. The presence of significant cell-cell variability in ionic current densities, even in the same cardiac region in the very same heart, has been long recognized. Moreover, similar AP duration can be achieved by various combinations of ionic current densities (see “good enough solutions” (Weiss et al., 2012)). Increasing data show that the large variability in each ionic current density/expression is not random but that there is a strong correlation between distinct ion channels in their expression, trafficking, membrane targeting, functional coherent translation, and regulation. Such coordinated interactions were demonstrated between IK1 and INa (Milstein et al., 2012; Ponce-Balbuena et al., 2018), IKr and ICaL (Ballouz et al., 2021; Horvath et al., 2021), and INa and IKr (Eichel et al., 2019; Hegyi et al., 2020). Interestingly, this mutual regulation between ionic currents may also be altered in disease (Hegyi et al., 2020), which may represent a new avenue of understanding disease mechanisms and discovering novel therapeutic targets.
There exist many sources of cell-cell variability in cardiovascular physiology, which encompass multiple spatial and temporal scales, and may play important roles in the development and progression of cardiovascular diseases and could impact the outcome of therapeutic interventions. We have highlighted here some critical sources of natural physiological diversity, and in the next section, we will overview novel technologies that allow for interrogating this diversity.
3. Technical innovations to assess biological diversity
There is a long history of new technologies leading to breakthroughs in scientific discovery. Here, we review selected technologies that are providing new insights into biological diversity. We highlight new technical developments in animal models, omics, mathematical modeling, and subcellular imaging. All four of these areas have enabled substantial discoveries in biological discovery yet realizing their full benefit towards biological diversity will require a closer interaction between biologists, electrophysiologists, and engineers. Technical innovations work together in an iterative cycle to bring greater and deeper insight into cardiovascular diversity (Figure 5).
Figure 5:
Iterative cycling between technical innovations to measure and model cardiovascular function. High throughput characterization of genome, proteome, transcriptome, etc. (“omics”) and microscopy allow scientists to capture rich tapestries of cardiovascular variation in space, time, and molecule. Both animal model systems and mathematical models provide abstractions from reality that enable new insights. Used together, these and other technologies are accelerating the discovery of novel biological mechanisms and therapeutics.Vm: transmembrane voltage; Iion: sum of the ionic currents; Cm: membrane capacitance.
3.1. Experimental models of cardiovascular disease
Duality in animal models of cardiovascular diseases: Disease as outcome vs. etiology
Although cardiovascular diseases are frequently named under an umbrella term, e.g., hypertension and heart failure, which reflect critical functional changes in the cardiovascular system, these diseases have many different etiologies. In turn, current clinical guidelines recommend therapies for a disease and not the pathophysiology (e.g., treatment recommendations for heart failure do not differentiate between ischemic and non-ischemic origin, sex, or genetic background). With the emergence of personalized medicine and patient-specific therapies, there is hope that patient-specific pathophysiology and genetics can be targeted in the future. At the same time, basic cardiovascular research that uses animal models for cardiovascular diseases is inherently pathophysiology-based. When creating an animal model of a disease, researchers usually start with a healthy animal and make an intervention (e.g., surgery, treatment, or genetic manipulation) that is associated with only one cardiovascular risk factor among the many that contribute to the clinical evolution of disease in human patients.
Complementary use of small and large animals in cardiovascular research provides important insights about impaired signaling mechanisms in disease and may identify and immediately test novel therapeutic targets. The use of small animals (mouse, rat) has important advantages, including easy genetic manipulations, faster disease progression and reproductive times, and lower costs, but their hemodynamics, as well as protein expression and metabolic profiles, may markedly differ from the human (Dobrev & Wehrens, 2018; Riehle & Bauersachs, 2019). Larger animals provide more closely translatable results, but the exponentially higher cost, long disease progression times, and difficult genetic manipulations limit their use. See recent reviews comparing small vs. large animal models of hypertension (Lerman et al., 2019) and heart failure with reduced ejection fraction (Pilz et al., 2022). Moreover, naturally occurring diseases in animals and data from veterinary practice can also provide additional insights, e.g., due to frequent hypertrophic cardiomyopathy occurrence in cats (Luis Fuentes et al., 2020), and degenerative mitral valve disease and dilated cardiomyopathy in dogs (Keene et al., 2019).
In the past decade, the exponential increase in the use of hiPSC-CMs has revolutionized cardiovascular research. The clear advantages of hiPSC-CMs are that these cells carry the patients-specific genetics and have a virtually unlimited source that is more easily applicable in high-throughput screenings for drugs. While hiPSC-CMs are still limited in their usually immature cellular phenotype, as noted in section 2.2 above, notable progress towards a more adult-like phenotype has been achieved by maturation approaches, including hormonal stimulation (Parikh et al., 2017), providing oxidative metabolic substrates (Feyen et al., 2020), and co-culturing with ECs and FBs in 3-D microtissues with biophysical stimulation (Giacomelli et al., 2020). Engineered heart tissues (Zimmermann et al., 2002) and cardiac organoids (Kupfer et al., 2020) can further improve cell maturation and physiological cell function. Conditional immortalization of human atrial CMs constitutes a novel methodology to generate functional cell lines for the development of in vitro models of human disease (Harlaar et al., 2022).
Heart failure with preserved ejection fraction: a truly heterogeneous disease
Heart failure with preserved ejection fraction (HFpEF) is set to become the leading form of heart failure due to the rapid populational growth of disease-associated conditions (e.g., aging) and co-morbidities, including obesity, diabetes, hypertension, kidney disease, depression, etc. Patients with HFpEF represent a highly heterogeneous group with variable presence and severity of different co-morbidities. Animal models of HFpEF were initially focused only on one major aspect of the disease but fell short of recapitulating the clinical characteristics and complex phenotype of HFpEF patients. Recently, two- or multi-hit models that better recapitulate human HFpEF have emerged (Withaar et al., 2021). These small and large animal models of HFpEF are reviewed and compared by Roh et al. (Roh et al., 2022). However, none of the more translational animal models is expected to cover the whole spectrum of HFpEF characteristics. Thus, creating and investigating multiple different disease models that recapitulate specific sub-phenogroups of HFpEF patients, with distinct phenotypical characteristics and underlying pathological mechanisms, may deepen our understanding of HFpEF biology and improve therapeutic testing. Assessing extracardiac functional impairments (lungs, kidney, skeletal muscle, etc.) and their contributions to HFpEF are key elements in developing animal models for HFpEF. HFpEF is considered a cardio-immuno-metabolic disease (Schiattarella et al., 2022) in which the function of different cell types in the heart must be carefully evaluated. There is an expectation that a better understanding of the various cell-to-cell and subcellular signaling networks by omics approaches may identify specific patient subgroups and may uncover novel therapeutic targets. A detailed cardiac transcriptomic analysis (Hahn et al., 2021) matched with clinical characteristics in human HFpEF identified three patient subgroups using a non-negative matrix factorization (NMF) (Gaujoux & Seoighe, 2010) and weighted gene co-expression network analysis (WGCNA) (Langfelder & Horvath, 2008). Such approaches can be of value when comparing different HFpEF animal models and potentially translating the findings toward human HFpEF subgroups. The disease pathophysiology is likely similarly diverse and includes oxidative and nitrosative stresses, inflammation, volume or high salt-sensitive hypertension, obesity, diabetes, and other metabolic derangements, etc. (Mishra & Kass, 2021).
3.2. Omics technologies to assess molecular and cellular diversity
Genomics
Some of the most transformative biotechnologies are those for comprehensive molecular profiling, collectively known as omics. The development of high-throughput DNA sequencers enabled in 2001 the sequencing of the human genome (Venter et al., 2001), which turned a $3.8 billion investment by the U.S. government into a genome-enabled industry with $796 billion economic impact by 2011 (Tripp & Grueber, 2011). However, this initial genome sequence contained only euchromatin, with the remaining heterochromatin regions sequenced only recently (Nurk et al., 2022). Further technological advances enabled the decreasing cost of sequencing at a rate even exceeding Moore’s law. For example, the UK Biobank has developed a database containing genomes and deep phenotyping from 500,000 individuals from the United Kingdom, 22,000 of which reported ethnic backgrounds originating outside of Europe (Bycroft et al., 2018). Such resources and studies enable the quantification of ancestry diversity at greater scale (Prive, 2022).
Measures of genomic diversity are now being combined with phenotypic data to provide important insights into the genetic basis of cardiac disease. Accordingly, a genome-wide association study (GWAS) for features derived from cardiac magnetic resonance imaging in 36K UK Biobank participants for genetic variants found a number of loci near genes well-established for Mendelian cardiomyopathies, as well as 45 new loci that enabled a polygenic risk score for dilated cardiomyopathy (Pirruccello et al., 2020). In a follow-up study, deep learning approaches were implemented using the UK Biobank participants with magnetic resonance imaging to characterize features of the right heart (Pirruccello et al., 2022). Results revealed key genes known in congenital heart disease (e.g. NKX2–5, TBX5) and also a powerful new polygenic predictor of right heart function.
Transcriptomics
Comprehensive measurements of the transcriptome have also had a major impact on our ability to assess biomolecular diversity. One of the most influential methods is RNA sequencing (RNA-seq) (Wang et al., 2009). RNA-seq has had an important impact on many cardiovascular studies, such as profiling the transcriptome in response to Gq-overexpression (Matkovich et al., 2010) or discovering noncoding RNAs in failing human hearts. Transcriptome measurements capture a much broader, more diverse range of transcripts than would be possible with RT-qPCR (Yang et al., 2014a). However, a limitation of RNA-seq in bulk samples, such as a tissue, is that it does not capture the cellular diversity of gene expression within a sample.
Because the heart and vasculature contain many cell types and states, profiling bulk tissue may not adequately capture a complex disease. To study congenital heart disease in human hearts at the single-cell level, Reichart et al. performed single-nucleus RNA-sequencing of patients with cardiomyopathies, with or without known genetic causes (Reichart et al., 2022). They found important differences in intercellular signaling between genotypes, such as endothelin signaling in Lamin A/C gene (LMNA)-related dilated cardiomyopathy and TNFα in plakophilin-2 gene (PKP2)-related cardiomyopathy. Network models demonstrated genotype-specific heart failure pathways. Such insights are leading toward precision medicine approaches.
Single-cell RNA-seq is also having an impact on fundamental cardiac biology. The process of cardiac differentiation from a stem cell involves transition through multiple states, and the mechanisms for robust differentiation are unclear. As presented at the UC Davis Cardiovascular Research Symposium, the gene Brm was critical for cardiac differentiation (Hota et al., 2022). Single-cell RNA-seq revealed that Brm-deleted cells instead became neural precursors, which were rescued with BMP4. Mathematical modeling of the single-cell RNA-seq data showed how Brm deletion disrupted hysteresis, a pair of saddle-node bifurcations needed for a robust cardiac differentiation (Hota et al., 2022). Audience members drew parallels to bifurcations in other cardiovascular processes, such as arrhythmia.
A variety of new methods are being developed to not only provide single-cell transcriptomes but also provide spatial resolution to contextualize cell niches or tissues (Moffitt et al., 2022). These spatial omics methods may be particularly valuable for investigating structural remodeling in development or disease. An impressive example was recently reported of spatial omics of the human heart after myocardial infarction (Kuppe et al., 2022). Accordingly, a profile of the post-MI heart with single-cell transcriptomes and chromatin accessibility with a resolution of ~100 μm revealed the spatial relationships of disease-specific cell states, including a continuum of states of CMs and myeloid cells. Such spatially detailed datasets will be invaluable for later multi-scale computational models that combine molecular and cellular heterogeneity. In addition, the roles of non-coding RNA including microRNAs and long non-coding RNAs in the regulation of cardiac excitability are well recognized (Johnson et al., 2018a; Yang et al., 2022).
Proteomics
Global measures of protein abundance and modifications (proteomics) provide a complementary view to that of transcriptomics, with advantages including proximity to cellular function and the utility of proteins as clinical biomarkers. A variety of proteomics technologies have been applied for heart failure proteomics, including mass spectrometry, protein microarray, aptamers, and proximity extension arrays, each of which has its advantages (Michelhaugh & Januzzi, 2020). In a wide survey of the field, an American Heart Association consensus statement noted the broad potential of proteomics to establish cause-and-effect mechanisms and signaling networks of cardiovascular disease (Lindsey et al., 2015).
Proteomics methods have been effectively used to identify the diverse protein responses to distinct stimuli. With their previous studies implicating MMP-12 in caspase-3 activity and cardiac wound healing, Chalise et al. asked what proteomic responses may mediate this effect (Chalise et al., 2022). They subjected neutrophils to MMP-12, IL-1β, or IL-4 and measured the intracellular signaling state via a 304-plex antibody array and 28 secreted cytokines. They found that these stimuli induced diverse proteomic responses, with a specific signature of MMP-12 signaling including increased FOXO1, apoptotic proteins, and decreased Wnt signaling with MMP-12. This provides a mechanistic basis for how MMP-12 modulates neutrophil polarization and apoptosis.
Clinical proteomics has demonstrated a substantial ability to identify new biomarkers and, when paired with other approaches, mechanisms of disease. As part of the TOPMed Consortium, Katz et al. used aptamer methods to profile 1301 plasma proteins in 1852 adults of African ancestry (Katz et al., 2022), who have considerable genetic diversity (Genomes Project et al., 2015). Paired proteomic and genomic profiling revealed 114 novel locus-protein relationships, including a locus at the HPX gene, associations between APOE, ZAP70 and MMP-3, and association between ATTR amyloidosis and RBP4 expression. Importantly, these results were validated with complementary proteomics methods and replicated in other studies such as the Multi-Ethnic Study of Atherosclerosis (Katz et al., 2022).
Multi-omics and other omics
While we have focused on genomics, transcriptomics, and proteomics, there are a number of additional profiling technologies that assess other molecular states. Some notable examples include metabolomics (Li et al., 2011; McGarrah et al., 2018), lipidomics (Tabassum & Ripatti, 2021), and epigenomics (Rosa-Garrido et al., 2018). All of these methods have made substantial contributions to understanding cardiovascular disease. One of the major frontiers is integrating multiple omics technologies (Joshi et al., 2021). As one recent example, a single-nucleus RNA sequencing from human hearts with multiple congenital heart diseases found diverse, disease-specific CM and FB states (Hill et al., 2022). Applying imaging mass cytometry, a spatial proteomics approach, Hill et al. identified a distinct immunosuppressed perivascular microenvironment, which was further characterized by immune cell profiling. mRNA expression, ligand-receptor analysis, and plasma protein profiling all reinforced a role for THBS1 in CM pathology of congenital heart disease (Hill et al., 2022). The battery of different approaches in this study reinforced and validated their findings. With the ability to perform such multi-modal experiments, a rising challenge is to develop computational methods to integrate these datasets rigorously (Leon-Mimila et al., 2019; Joshi et al., 2021).
Omics technologies have provided nearly comprehensive profiling of molecular states. However, it is also important to recognize these methods’ limitations. In particular, molecular profiles may correlate with developmental or disease states, but are not sufficient to characterize underlying mechanisms or predict the effect of new perturbations. Omics also do not elucidate the causal relationships between molecular components, cellular states, and biological function. The high throughput omics technologies are outstanding at characterizing molecular diversity, but complementary techniques are needed to understand how molecular/cellular diversity arises and how it can be harnessed for therapeutic purposes. As described in the next section, mathematical modeling has significant potential for complementing omics technologies toward these goals.
Current methods for omic data analyses focus on statistical methods to identify patterns. New computational modeling frameworks are needed to integrate and distill mechanistic insight from omic data and integrate it with prior knowledge.
3.3. Mathematical modeling of population heterogeneity
In recent years, increased focus on the significance of cellular heterogeneity and diversity, especially among CMs, has prompted an evolution of in silico modeling from single cells to heterogeneous populations. One approach to such modeling is to generate a large set of models by randomly sampling values of select parameters and subsequently discard models that generate non-physiological output. For example, in pioneering this approach for CM electrophysiology modeling, Britton et al. constructed a population of 10,000 candidate models through Latin hypercube sampling of values for 11 different ionic current conductances and kinetics parameters (Britton et al., 2013). Candidate models whose simulated behavior was not consistent with experimental AP recordings were eliminated, reducing the population down to 213 models. When simulating IKr block in this calibrated population, the range of variability in AP prolongation seen in the models agreed with that measured experimentally with the IKr blocker dofetilide.
In general, this approach can be helpful in determining why some cells/organs/individuals are more prone to pathological outcomes than others and what quantitative feature(s) of a subpopulation is associated with specific disease patterns (Sarkar & Sobie, 2011; Passini et al., 2017; Ni et al., 2018). In a recent study, a model was constructed with four subpopulations of ventricular CMs based on sex and disease state (male/female, healthy/heart failure) and found that the female heart failure group was the most susceptible to proarrhythmic events when simulating effects of drugs proposed for COVID-19 treatment (Varshneya et al., 2021). These results suggested that women with heart disease are particularly vulnerable to cardiac arrhythmias if treated with the drugs examined in the study. In addition to this subgroup-specific prediction, logistic regression was used to identify three specific ionic currents (IKr, ICaL, INCX) as most important for arrhythmogenesis, thus helping determine the relative roles of individual parameters in identifying ionic mechanisms underlying arrhythmogenesis. The population-of-models approach has also been used in making quantitative predictions on the role of combined inhibition of the atrial-dominant currents IK2P, IKCa, and IKur in different chronic AF subpopulations, demonstrating the advantage of computer power in assessing a number of drug combinations across multiple subpopulations (Ni et al., 2020).
Outside of the heart, heterogeneity in the density of inward rectifier K+ channels among capillary ECs has been included to simulate electrical responses in microvascular networks, specifically to predict the amount of hyperpolarization in response to local K+ stimulation as part of the neurovascular coupling (Moshkforoush et al., 2020). Population-based methods have significant potential for biochemical networks and mechanical modeling as well.
Cell-type translation
Simulated data from a population of models can form the basis for statistical or machine learning-based analyses allowing translation of the behavior in one cell type into quantitative predictions of outcomes in a different cell type. This approach has been applied to derive predictions of drug effects on electrophysiological metrics in adult human CMs based on measurements or simulations in hiPSC-CMs or animal CMs (Gong & Sobie, 2018; Aghasafari et al., 2021; Morotti et al., 2021). When subsequently testing predictions of human responses to different ion-channel blockers based on animal experiments, translators were found to closely reproduce drug-induced changes seen in human data (Morotti et al., 2021). While these approaches have focused mainly on translating across cell differences that limit the usefulness of experimental cells as models of adult human physiology, this methodology is directly applicable to investigate differences along disease axes (e.g. from healthy state to heart failure (Gong & Sobie, 2018; Morotti et al., 2021)), or between different non-CM cell types.
Cell- and patient-specific modeling
The populations of models derived by randomizing parameter values should be understood to be hypothetical populations, as they do not build on specific information regarding parameter values in individual cells. An alternative approach to modeling a heterogeneous population is to generate models that are calibrated to data from individual cells/organs/subjects, thereby creating cell-, organ, or subject-specific models. For CM electrophysiology models, such an approach has been demonstrated for determining ionic current conductances in guinea pig ventricular CMs (Groenendaal et al., 2015) and tailored models have been developed for hiPSC-CMs (Lei et al., 2017). In both cases, the reparameterized models improved predictive power over the non-tailored baseline models.
If accurately calibrated, a cell-specific model population could help identify population heterogeneity both across healthy tissue (e.g., left vs. right ventricle, atrial vs. ventricular cells) and heterogeneity resulting from pathological ionic remodeling. A cell-specific model population should also capture correlations among ionic currents observed experimentally, in particular positive correlations between inward and outward currents arising due to co-expression of ion channels or co-regulation at the translational level (Rees et al., 2018; Eichel et al., 2019; Hegyi et al., 2020), which we have discussed in section 2.3 above. Such co-variations could also be built into the population-of-models approach as a next step for development (Ballouz et al., 2021) and applied across different disease and cell-types, to potentially help to address whether there are central genetic or epigenetic mechanisms underlying ionic remodeling in CM, ECs, and VSMCs.
At the organ level, patient-specific cardiac models may be constructed based on personalized geometries, fiber orientation, infarct scars, fibrosis, and/or electrophysiology (Niederer et al., 2019; Corral-Acero et al., 2020). Patient-specific models are expected to contribute increasingly to clinical decision-making and have been utilized in predicting arrhythmia risk in patients with hypertrophic cardiomyopathy (O’Hara et al., 2022) or myocardial infarction (Arevalo et al., 2016). Still, challenges remain in developing methodologies and pipelines for personalized model creation at scale, including building frameworks for high-quality clinical data acquisition, automating image segmentation, and overcoming computational costs of model development and simulation of predictions (Costa et al., 2020).
Beyond conductances
The electrophysiology modeling approaches discussed above have prioritized varying conductance parameters, which have traditionally been based on data from voltage clamp experiments. More recently, expression data have been utilized to inform conductance levels between subpopulations (female vs. male, (Yang et al., 2017)) or individual patients (Smirnov et al., 2020)). An open question remains as to how to go beyond conductance parameters and consider variations in kinetics, channel subunit distribution, and signaling processes and pathways. One issue is obtaining measurements that can inform the estimation of the many parameters involved in, e.g., kinetics for multiple ion channels. Using phenomenological models with relatively few parameters rather than biophysically detailed models may therefore provide a helpful starting point for cell-specific modeling of kinetics (Cairns et al., 2017).
Validation
Although the need for model validation has been highlighted in recent years and generally called for by experimentalists at the meeting, there is no existing framework for validation. The process of validation involves comparing model predictions to experimental or clinical data not used in building the model. As an example, a recent optimization of the much-used O’Hara-Rudy human ventricular CM model carefully separated validation from calibration data in a multi-objective optimization using complex data sets, including AP and ionic current recordings, Ca2+ transients, drug block data, rate dependencies, and conduction velocity (Tomek et al., 2019).
However, a model can only be validated for a certain context of used – a general sticker of “validated model” cannot be meaningfully applied (Pathmanathan & Gray, 2018). If the intended use of a model is to gain insights into the biophysical underpinnings of an observed phenomenon, larger differences between model output and validation data would be acceptable than if the model is intended for quantitatively accurate predictions. Therefore, the selection of validation data should also be done within the context of use, with the process of validation benefitting from synergistic collaborations between experimentalists and modelers in protocol design to generate information-rich data sets for calibration and validation (Carusi et al., 2012; Pathmanathan & Gray, 2018; Whittaker et al., 2020).
3.4. What tools are needed for us to understand subcellular cardiovascular function in greater detail?
Over the last few decades, sub-cellular heterogeneities from nano through micro scales have emerged as key determinants of cellular and tissue scale physiology. Perhaps the most illustrative example is that of EC-coupling, where heterogeneous organization of t-tubules, SR structures, and dyadic nanodomains enables local control and governs cellular-scale behavior in ways that cannot be predicted without accounting for subcellular heterogeneities (Qu et al., 2013; Winslow et al., 2016; Radwanski et al., 2018). Indeed, our understanding of other areas of cardiovascular physiology and biology are undergoing similar paradigm shifts, underscoring the need for experimental and computational tools to investigate subcellular heterogeneity. Here, we highlight some recently developed and emerging approaches that meet this need.
Experimental Imaging Approaches
The ability to assess subcellular structural and functional heterogeneities has been greatly enhanced by the advent of novel sample labeling and interrogation strategies. Super-resolution imaging techniques such as single molecule localization microscopy (stochastic optical reconstruction microscopy [STORM], photoactivation localization microscopy [PALM] etc) and stimulated emission depletion (STED) have pushed light microscopy to molecular-scale resolution. Although these techniques have been applied to live cells, their live-cell friendliness is limited by the massive photon fluxes involved. Methods like structured illumination microscopy (SIM) and fluctuation-based super-resolution methods (super-resolution optical fluctuation imaging [SOFI], super-resolution radial fluctuations [SRRF], etc) enable sub-diffraction imaging in live cells with minimal phototoxicity. Also, of note here are expansion microscopy methods (Wassie et al., 2019; Gallagher & Zhao, 2021), which enable sub-diffraction resolutions to be attained using simple hardware and emerging techniques like minFlux (Gwosch et al., 2020) and minSTED (Weber et al., 2021), which are achieving near-atomic scale resolutions comparable to electron microscopy. The impressive power of these light microscopy techniques to reveal the molecular workings of cells is further enhanced by the addition of ultrastructural context through correlative light and electron microscopy (CLEM) techniques (Dillard et al., 2018; Mezache et al., 2019; Heiligenstein & Lucas, 2022; Jeong & Kim, 2022; van den Dries et al., 2022). For a more detailed review of current super-resolution methods, readers are referred to the excellent recent review by Jacquemet et al (Jacquemet et al., 2020). As discussed at the symposium, some of these methods have been applied to cardiac cells and tissues (Agullo-Pascual et al., 2013; Veeraraghavan et al., 2015; Leo-Macias et al., 2016; Veeraraghavan & Gourdie, 2016; Dixon et al., 2017; Ghosh et al., 2018; Veeraraghavan et al., 2018; Mezache et al., 2019), but we are still in the early days of leveraging them for cardiovascular research. It should also be noted here that the advent of scanning ion conductance microscopy (SICM) –guided “smart” patch clamp paralleled the aforementioned developments in microscopy and enabled direct interrogation of electrophysiology within sub-cellular niches (Gorelik et al., 2002; Bhargava et al., 2013; Schobesberger et al., 2017).
Of equal importance to imaging techniques discussed above have been advances in the ability to label specific types of molecules and structures within cells. In addition to the development of nanobodies (de Beer & Giepmans, 2020; Erreni et al., 2020), which lower linkage error in protein labeling, new methods have been developed to visualize other types of biomolecules and sub-cellular structures, such as single molecule fluorescence in situ hybridization (smFISH) to visualize mRNA (Savulescu et al., 2021; Piskadlo et al., 2022), OligoSTORM/OligoDNA-PAINT to image genomic DNA (Beliveau et al., 2017; Nir et al., 2018; Nguyen et al., 2020), and single molecule Förster resonance energy transfer (smFRET) to study protein-protein interactions / protein conformational dynamics and subcellular domain function (Lerner et al., 2021; Ha et al., 2022). Likewise, the expanding toolbox for label-free imaging is enabling unprecedented interrogation of live cell dynamics with minimal perturbation (Ojaghi et al., 2020; Borile et al., 2021; Morgan et al., 2021).
The investigative power of sample labeling and imaging techniques is ultimately contingent upon the ability to robustly quantify structural and/or functional properties from the resulting data, particularly when it comes to capturing heterogeneity. Thus, recent advances in artificial intelligence / machine learning –powered image segmentation (Vicar et al., 2019; Durkee et al., 2021; Liu et al., 2021), cluster analysis for single molecule localization data (Levet et al., 2015; Chamma et al., 2016; Veeraraghavan & Gourdie, 2016), and spatial analysis of image data (Bogdanov et al., 2021; Bao et al., 2022) merit attention here.
Multiscale Modeling Approaches
Directly and indirectly based on innovations in subcellular imaging, the integration of subcellular structural detail within and between CMs has facilitated simulations of multiscale models, enabling predictions of the role of subcellular structure on cellular- to tissue-scale function. Early multiscale modeling studies investigated the role of subcellular structure in EC-coupling and Ca2+ handling, representing ~10,000s distinct, coupled Ca2+ release units, each with stochastic gating of LTCCs and RyR2s and distinct subcellular Ca2+ compartments (Gaur & Rudy, 2011; Nivala & Qu, 2012). These early generation subcellular models often assumed homogeneous subcellular structure. Yet simulations predicted that even in the absence of spatial heterogeneity, stochasticity and channel refractoriness can drive spatial heterogeneity of subcellular Ca2+ signaling and importantly, can link local Ca2+-spark properties with pro-arrhythmic whole-cell alternans behavior (Nivala & Qu, 2012). To further connect subcellular structure with disease, disease-related structural disruption required model representation of subcellular spatial heterogeneity. Multiscale simulations investigating heart failure-induced arrhythmia showed that both RyR2 redistribution and alteration of the t-tubule membrane structure could alter Ca2+-release properties, which in turn can drive Ca2+ alternans and, in some conditions, triggered activity (Nivala et al., 2015; Shiferaw et al., 2018; Song et al., 2018; Shiferaw et al., 2020). High-resolution structural electron micrograph imaging has further been integrated into modeling frameworks to directly address the impact of membrane geometry relative to channel distribution (Colman et al., 2017).
The inclusion of highly detailed 3-dimensional subcellular geometry comes at a cost, specifically a large computational cost, such that simulations extending the multiscale model to include hundreds to thousands of cells, each with ~10,000s stochastic Ca2+-release units, are generally computational prohibitive. Thus, the question arises of how to directly investigate the role of subcellular structure and heterogeneity at the tissue-level? As presented at UC Davis Cardiovascular Research Symposium, one innovative solution applied to this question has been the use of phenomenological relationships. Specifically, key statistical properties of the subcellular dynamics (e.g. Ca2+-spark kinetics) define a phenomenological relationship that can be simulated at the whole-cell level, which in turn is integrated into a multicellular tissue framework (Colman, 2019; Greene et al., 2022). Thus, the key properties of the subcellular structure are captured and represented at the tissue level. Two excellent recent reviews have highlighted advances in subcellular Ca2+ modeling and the integration of image-based subcellular structure as described in section 2.3 (Colman et al., 2022; Louch et al., 2022).
In addition to Ca2+ signaling, recent imaging-driven simulation studies have suggested that subcellular structure can directly impact electrical conduction via interactions at the cell-cell junctions, specifically at the intercalated disk. Two critical structural properties governing these interactions are the localization of high-density clusters of voltage-gated Na+ channels at the intercalated disk, in close proximity to electrical and mechanical junctional proteins, and the narrow cleft spacing between adjacent cells, both identified with electron and super-resolution imaging (Veeraraghavan et al., 2014a, b; Veeraraghavan et al., 2015). Model predictions incorporating these key structural features predict additional cell-cell coupling mechanisms beyond direct electrical coupling via gap junctions, which have been termed ‘ephaptic coupling’ (i.e. interactions occurring in the narrow extracellular space between cells) (Lin & Keener, 2010; Weinberg, 2017; Nowak et al., 2021b). Recent modeling studies have predicted that subcellular structural properties, including the localization and density of Na+ channels within the intercalated disk, can have significant effects on conduction (Kucera et al., 2002; Hichri et al., 2018; Ivanovic & Kucera, 2021). Further, multiscale models integrating intercalated disk structure, based on electron microscopy measurements, predict that heterogeneity within the intercalated disk can also impact conduction (Moise et al., 2021). In addition to conduction, simulations have predicted that the intercalated disk structure can impact repolarization in arrhythmias such as long QT syndrome (Greer-Short et al., 2017; Nowak et al., 2021a), spontaneous ectopy (Poelzing et al., 2021; Ly & Weinberg, 2022; Yu et al., 2022), and reentry formation (Tsumoto et al., 2020). Thus, the combination of imaging-based details of subcellular structure and multiscale modeling provides a powerful framework for investigating functional relationships. While most such studies, to our knowledge, have focused on CM subcellular or CM-CM interactions, generalization to subcellular and cell-cell interactions with other diverse cell types, including FBs, vascular cells, and immune cells, is an area for much fruitful future investigation.
4. Summary and Opportunities for Future Research
It is becoming increasingly evident that a complex interplay of signaling events among various cell types plays key roles in cardiovascular function and that abnormal cell-autonomous and cell-cell signaling is involved in the development and progression of cardiovascular disease. In this white paper, stemming from the seventh UC Davis Cardiovascular Research Symposium, we illustrated how biological diversity in structure-function (electrical, Ca2+, and regulatory signaling) within myocytes and non-myocytes (e.g., FBs, ECs, neuronal and immune cells, etc.) integrate to influence normal cardiovascular function, and disease development and progression. Moreover, we highlight the unmet need to quantitatively characterize and model additional cell types (e.g., stem cells, neutrophils, T cells) and systems (e.g. lymphatics) that have had lesser attention to date. Technical innovations in animal models, omics, mathematical modeling, and subcellular imaging are discussed as tools to illuminate and provide insight into the diversity of subcellular, cellular, and tissue-level processes. Integration of these diverse technologies and data types is challenging but offers unique opportunities to interpret the rich datasets and translate findings to human health and disease, with the ultimate goal to better our mechanistic understanding and gain a holistic view of disease progression and therapy.
Supplementary Material
Funding:
National Institutes of Health:
R13HL162430 (EG, DMB, MFN)
R01HL131517, R01HL141214, 1OT2OD026580-01 (EG)
P01HL141084 (DMB, EG, CMR)
R01HL161872, R01HL121059, R01HL149127, R01HL162825 (MFN)
R01HL162925 and R01HL160665 (JJS)
R01HL085844, R01HL085727, R01HL137228 (NC)
R01AG063796 and R01HL159304 (RED)
R01HL136389, R01HL163277, R01HL089598 (DD)
P20GM135007 (OFH)
U01HL136297 (TKM)
R21HL159501 (WLM)
R01 HL142710 (MAN)
R01HD108839 (NGP)
R01HL111600 (CMR)
R01HL138003 (SHW)
European Union Horizon 2020
large-scale integrative project MAESTRIA, No. 965286 (DD)
Agence National de la Recherche
ANR-19-CE14-0031-01 (AMG)
AHA
20CDA35310097 (OFH)
Biographies
Ele Grandi, PhD, FHRS, is a Professor in the Department of Pharmacology, the Chair of the Biophysics Graduate Group, and a Chancellor’s Fellow at the University of California Davis. Her research utilizes mathematical modeling and simulation as an interpretative and predictive science to investigate the mechanisms of cardiac arrhythmias.

Manuel F. Navedo, PhD, FAHA is a Professor in the Department of Pharmacology and the Chair of the Molecular, Cellular, and Integrative Physiology Graduate Group at UC Davis. He is an established physiologist and vascular biologist examining mechanisms regulating vascular smooth muscle function in health and disease.

Jeffrey J. Saucerman, PhD, FAHA, FAIMBE, is a Professor of Biomedical Engineering and Cardiovascular Medicine at the University of Virginia. His research develops experimental and computational methods to discover molecular networks that regulate cardiac remodeling.

Footnotes
Competing Interests:
The authors have no competing interests to disclose.
References
- Aghasafari P, Yang PC, Kernik DC, Sakamoto K, Kanda Y, Kurokawa J, Vorobyov I & Clancy CE. (2021). A deep learning algorithm to translate and classify cardiac electrophysiology. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyo D, Rothenberg E & Delmar M. (2013). Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res 100, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern BM, Levitan BM, Veeranki S, Shah M, Ali N, Sebastian A, Su W, Gong MC, Li J, Stelzer JE, Andres DA & Satin J. (2019). Myocardial-restricted ablation of the GTPase RAD results in a pro-adaptive heart response in mice. J Biol Chem 294, 10913–10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon-Martinez L, Villafranca-Baughman D, Quintero H, Kacerovsky JB, Dotigny F, Murai KK, Prat A, Drapeau P & Di Polo A. (2020). Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 585, 91–95. [DOI] [PubMed] [Google Scholar]
- Alvarado MG, Thakore P & Earley S. (2021). Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC & Navedo MF. (2013). Calcium dynamics in vascular smooth muscle. Microcirculation 20, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrienko T, Kuznetsov AV, Kaambre T, Usson Y, Orosco A, Appaix F, Tiivel T, Sikk P, Vendelin M, Margreiter R & Saks VA. (2003). Metabolic consequences of functional complexes of mitochondria, myofibrils and sarcoplasmic reticulum in muscle cells. J Exp Biol 206, 2059–2072. [DOI] [PubMed] [Google Scholar]
- Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI & Gourdie RG. (1997). Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res 80, 88–94. [DOI] [PubMed] [Google Scholar]
- Anversa P, Li P, Sonnenblick EH & Olivetti G. (1994). Effects of aging on quantitative structural properties of coronary vasculature and microvasculature in rats. Am J Physiol 267, H1062–1073. [DOI] [PubMed] [Google Scholar]
- Aquino JB, Sierra R & Montaldo LA. (2021). Diverse cellular origins of adult blood vascular endothelial cells. Dev Biol 477, 117–132. [DOI] [PubMed] [Google Scholar]
- Arevalo HJ, Vadakkumpadan F, Guallar E, Jebb A, Malamas P, Wu KC & Trayanova NA. (2016). Arrhythmia risk stratification of patients after myocardial infarction using personalized heart models. Nat Commun 7, 11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromolaran KA, Do J, Bernardi J & Aromolaran AS. (2022). mTOR Modulation of IKr through hERG1b-Dependent Mechanisms in Lipotoxic Heart. Int J Mol Sci 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari P, Scriven DR, Ng M, Panwar P, Chou KC, van Petegem F & Moore ED. (2020). Cardiac ryanodine receptor distribution is dynamic and changed by auxiliary proteins and post-translational modification. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakunts K, Gillum N, Karabekian Z & Sarvazyan N. (2008). Formation of cardiac fibers in Matrigel matrix. Biotechniques 44, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballouz S, Mangala MM, Perry MD, Heitmann S, Gillis JA, Hill AP & Vandenberg JI. (2021). Co-expression of calcium and hERG potassium channels reduces the incidence of proarrhythmic events. Cardiovasc Res 117, 2216–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister JP, Bulley S, Leo MD, Kidd MW & Jaggar JH. (2016). Rab25 influences functional Cav1.2 channel surface expression in arterial smooth muscle cells. American journal of physiology Cell physiology 310, C885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Deng Y, Wan S, Shen SQ, Wang B, Dai Q, Altschuler SJ & Wu LF. (2022). Integrative spatial analysis of cell morphologies and transcriptional states with MUSE. Nat Biotechnol 40, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Bargehr J, Ong LP, Colzani M, Davaapil H, Hofsteen P, Bhandari S, Gambardella L, Le Novere N, Iyer D, Sampaziotis F, Weinberger F, Bertero A, Leonard A, Bernard WG, Martinson A, Figg N, Regnier M, Bennett MR, Murry CE & Sinha S. (2019). Epicardial cells derived from human embryonic stem cells augment cardiomyocyte-driven heart regeneration. Nat Biotechnol 37, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkaway A, Attwell D & Korte N. (2022). Immune-vascular mural cell interactions: consequences for immune cell trafficking, cerebral blood flow, and the blood-brain barrier. Neurophotonics 9, 031914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth E, Stammler G, Speiser B & Schaper J. (1992). Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24, 669–681. [DOI] [PubMed] [Google Scholar]
- Beliveau BJ, Boettiger AN, Nir G, Bintu B, Yin P, Zhuang X & Wu CT. (2017). In Situ Super-Resolution Imaging of Genomic DNA with OligoSTORM and OligoDNA-PAINT. Methods Mol Biol 1663, 231–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM, Xiang YK & Zaccolo M. (2019). Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology (Bethesda) 34, 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhana B, Iyer RK, Chen WL, Zhao R, Sider KL, Likhitpanichkul M, Simmons CA & Radisic M. (2010). Influence of substrate stiffness on the phenotype of heart cells. Biotechnol Bioeng 105, 1148–1160. [DOI] [PubMed] [Google Scholar]
- Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M & Gorelik J. (2013). Super-resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res 112, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A & Frangogiannis NG. (2011). Aging and Cardiac Fibrosis. Aging Dis 2, 158–173. [PMC free article] [PubMed] [Google Scholar]
- Bishop SP, Anderson PG & Tucker DC. (1990). Morphological development of the rat heart growing in oculo in the absence of hemodynamic work load. Circ Res 66, 84–102. [DOI] [PubMed] [Google Scholar]
- Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J & Lipsius SL. (2003). Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol 546, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleck CKE, Kim Y, Willingham TB & Glancy B. (2018). Subcellular connectomic analyses of energy networks in striated muscle. Nat Commun 9, 5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov V, Soltisz AM, Moise N, Sakuta G, Orengo BH, Janssen PML, Weinberg SH, Davis JP, Veeraraghavan R & Gyorke S. (2021). Distributed synthesis of sarcolemmal and sarcoplasmic reticulum membrane proteins in cardiac myocytes. Basic Res Cardiol 116, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borile G, Sandrin D, Filippi A, Anderson KI & Romanato F. (2021). Label-Free Multiphoton Microscopy: Much More Than Fancy Images. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers S, Meng Q & Molkentin J. (2022). Fibroblasts orchestrate cellular crosstalk in the heart through the ECM. Nature Cardiovascular Research 1, 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg S, Pawlowitz J, Steckmeister V, Subramanian H, Uhlenkamp D, Scardigli M, Mushtaq M, Amlaz SI, Kohl T, Wegener JW, Arvanitis DA, Sanoudou D, Sacconi L, Hasenfuss G, Voigt N, Nikolaev VO & Lehnart SE. (2022). A junctional cAMP compartment regulates rapid Ca(2+) signaling in atrial myocytes. J Mol Cell Cardiol 165, 141–157. [DOI] [PubMed] [Google Scholar]
- Breckenridge RA, Piotrowska I, Ng KE, Ragan TJ, West JA, Kotecha S, Towers N, Bennett M, Kienesberger PC, Smolenski RT, Siddall HK, Offer JL, Mocanu MM, Yelon DM, Dyck JR, Griffin JL, Abramov AY, Gould AP & Mohun TJ. (2013). Hypoxic regulation of hand1 controls the fetal-neonatal switch in cardiac metabolism. PLoS Biol 11, e1001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ & Rodriguez B. (2013). Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci U S A 110, E2098–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Ghiadoni L, Seravalle G, Dell’oro R, Taddei S & Grassi G. (2012). Sympathetic regulation of vascular function in health and disease. Front Physiol 3, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SM, Kong CH, Watson J, Cannell MB, James AF & Orchard CH. (2015). Altered distribution of ICa impairs Ca release at the t-tubules of ventricular myocytes from failing hearts. J Mol Cell Cardiol 86, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G (2009). Autonomic neurotransmission: 60 years since sir Henry Dale. Annu Rev Pharmacol Toxicol 49, 1–30. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD & Wu JC. (2014). Chemically defined generation of human cardiomyocytes. Nat Methods 11, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P & Marchini J. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero R, de la Fuente MG, Gomez R, Barana A, Amoros I, Dolz-Gaiton P, Osuna L, Almendral J, Atienza F, Fernandez-Aviles F, Pita A, Rodriguez-Roda J, Pinto A, Tamargo J & Delpon E. (2010). In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J Am Coll Cardiol 55, 2346–2354. [DOI] [PubMed] [Google Scholar]
- Cairns DI, Fenton FH & Cherry EM. (2017). Efficient parameterization of cardiac action potential models using a genetic algorithm. Chaos 27, 093922. [DOI] [PubMed] [Google Scholar]
- Carmeliet E (2019). Pacemaking in cardiac tissue. From IK2 to a coupled-clock system. Physiol Rep 7, e13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carusi A, Burrage K & Rodriguez B. (2012). Bridging experiments, models and simulations: an integrative approach to validation in computational cardiac electrophysiology. Am J Physiol Heart Circ Physiol 303, H144–155. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Pino R, Sartiani L & Mugelli A. (1999). Influence of postnatal-development on I(f) occurrence and properties in neonatal rat ventricular myocytes. Cardiovasc Res 42, 416–423. [DOI] [PubMed] [Google Scholar]
- Chalise U, Becirovic-Agic M, Konfrst SR, Rodriguez-Paar JR, Cook LM & Lindsey ML. (2022). MMP-12 polarizes neutrophil signalome towards an apoptotic signature. J Proteomics 264, 104636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamma I, Levet F, Sibarita JB, Sainlos M & Thoumine O. (2016). Nanoscale organization of synaptic adhesion proteins revealed by single-molecule localization microscopy. Neurophotonics 3, 041810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Garcia-Barrio MT & Chen YE. (2020). Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 40, 1094–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang ZH. (1988). [Latent WPW syndrome diagnosed in Valsalva’s method]. Kokyu To Junkan 36, 873–874. [PubMed] [Google Scholar]
- Cheng H & Lederer WJ. (2008). Calcium sparks. Physiol Rev 88, 1491–1545. [DOI] [PubMed] [Google Scholar]
- Chowkwale M, Lindsey ML & Saucerman JJ. (2022). Intercellular model predicts mechanisms of inflammation-fibrosis coupling after myocardial infarction. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AP, Wei S, Kalola D, Krogh-Madsen T & Christini DJ. (2022). An in silico-in vitro pipeline for drug cardiotoxicity screening identifies ionic pro-arrhythmia mechanisms. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole WC & Welsh DG. (2011). Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys 510, 160–173. [DOI] [PubMed] [Google Scholar]
- Colman MA. (2019). Arrhythmia mechanisms and spontaneous calcium release: Bi-directional coupling between re-entrant and focal excitation. PLoS Comput Biol 15, e1007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman MA, Alvarez-Lacalle E, Echebarria B, Sato D, Sutanto H & Heijman J. (2022). Multi-Scale Computational Modeling of Spatial Calcium Handling From Nanodomain to Whole-Heart: Overview and Perspectives. Front Physiol 13, 836622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman MA, Pinali C, Trafford AW, Zhang H & Kitmitto A. (2017). A computational model of spatio-temporal cardiac intracellular calcium handling with realistic structure and spatial flux distribution from sarcoplasmic reticulum and t-tubule reconstructions. PLoS Comput Biol 13, e1005714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BL, Gloschat C, Swift LM, Prudencio T, McCullough D, Jaimes R & Posnack NG. (2021). KairoSight: Open-Source Software for the Analysis of Cardiac Optical Data Collected From Multiple Species. Front Physiol 12, 752940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corliss BA, Azimi MS, Munson JM, Peirce SM & Murfee WL. (2016). Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation 23, 95–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral-Acero J, Margara F, Marciniak M, Rodero C, Loncaric F, Feng Y, Gilbert A, Fernandes JF, Bukhari HA, Wajdan A, Martinez MV, Santos MS, Shamohammdi M, Luo H, Westphal P, Leeson P, DiAchille P, Gurev V, Mayr M, Geris L, Pathmanathan P, Morrison T, Cornelussen R, Prinzen F, Delhaas T, Doltra A, Sitges M, Vigmond EJ, Zacur E, Grau V, Rodriguez B, Remme EW, Niederer S, Mortier P, McLeod K, Potse M, Pueyo E, Bueno-Orovio A & Lamata P. (2020). The ‘Digital Twin’ to enable the vision of precision cardiology. Eur Heart J 41, 4556–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti A, Colombo M, Migliavacca F, Rodriguez Matas JF, Casarin S & Chiastra C. (2021). Multiscale Computational Modeling of Vascular Adaptation: A Systems Biology Approach Using Agent-Based Models. Front Bioeng Biotechnol 9, 744560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MC, Roney C, Strocchi M, Razeghi O & Niederer S. (2020). Clinical translation of patient-specific organ level cardiac models. In Modeling and Simulating Cardiac Electrical Activity, ed. Krogh-Madsen T & Christini DJ. IOP Publishing Ltd. [Google Scholar]
- Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG, Schmidt MS, Heckenbach I, Kwok R, Wiley CD, Wong HS, Gibbs E, Iyer SS, Basisty N, Wu Q, Kim IJ, Silva E, Vitangcol K, Shin KO, Lee YM, Riley R, Ben-Sahra I, Ott M, Schilling B, Scheibye-Knudsen M, Ishihara K, Quake SR, Newman J, Brenner C, Campisi J & Verdin E. (2020). Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab 2, 1265–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti L, Tester DJ, White WM, Bartos DC, Insolia R, Besana A, Kunic JD, Will ML, Velasco EJ, Bair JJ, Ghidoni A, Cetin I, Van Dyke DL, Wick MJ, Brost B, Delisle BP, Facchinetti F, George AL, Schwartz PJ & Ackerman MJ. (2013). Long QT syndrome-associated mutations in intrauterine fetal death. JAMA : the journal of the American Medical Association 309, 1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer MA & Giepmans BNG. (2020). Nanobody-Based Probes for Subcellular Protein Identification and Visualization. Front Cell Neurosci 14, 573278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M & Catterall WA. (1996). Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3’,5’-cyclic monophosphate-dependent protein kinase. Biochemistry 35, 10392–10402. [DOI] [PubMed] [Google Scholar]
- De Maziere AM, van Ginneken AC, Wilders R, Jongsma HJ & Bouman LN. (1992). Spatial and functional relationship between myocytes and fibroblasts in the rabbit sinoatrial node. J Mol Cell Cardiol 24, 567–578. [DOI] [PubMed] [Google Scholar]
- Del Villar SG, Voelker TL, Westhoff M, Reddy GR, Spooner HC, Navedo MF, Dickson EJ & Dixon RE. (2021). beta-Adrenergic control of sarcolemmal CaV1.2 abundance by small GTPase Rab proteins. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Carlo SE & Peduto L. (2018). The perivascular origin of pathological fibroblasts. J Clin Invest 128, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick SA & Epelman S. (2016). Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res 119, 159–176. [DOI] [PubMed] [Google Scholar]
- Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS & Epelman S. (2019). Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard RS, Hampton CM, Strauss JD, Ke Z, Altomara D, Guerrero-Ferreira RC, Kiss G & Wright ER. (2018). Biological Applications at the Cutting Edge of Cryo-Electron Microscopy. Microsc Microanal 24, 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF & Santana LF. (2015). Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Navedo MF, Binder MD & Santana LF. (2022). Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol Rev 102, 1159–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Vivas O, Hannigan KI & Dickson EJ. (2017). Ground State Depletion Super-resolution Imaging in Mammalian Cells. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RE, Yuan C, Cheng EP, Navedo MF & Santana LF. (2012). Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci U S A 109, 1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M & Ravens U. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Graf E, Wettwer E, Himmel HM, Hala O, Doerfel C, Christ T, Schuler S & Ravens U. (2001). Molecular basis of downregulation of G-protein-coupled inward rectifying K(+) current (I(K,ACh) in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced I(K,ACh) and muscarinic receptor-mediated shortening of action potentials. Circulation 104, 2551–2557. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Heijman J, Hiram R, Li N & Nattel S. (2022). Inflammatory signalling in atrial cardiomyocytes: a novel unifying principle in atrial fibrillation pathophysiology. Nat Rev Cardiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D & Ravens U. (2003). Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol 98, 137–148. [DOI] [PubMed] [Google Scholar]
- Dobrev D & Wehrens XH. (2014). Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res 114, 1311–1319; discussion 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D & Wehrens XHT. (2018). Mouse Models of Cardiac Arrhythmias. Circ Res 123, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Dressen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, Meier F, Deutsch MA, Lahm H, Lange R, Krane M & Mann M. (2017). Region and cell-type resolved quantitative proteomic map of the human heart. Nat Commun 8, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KK, Reichardt IM, Simmons AD, Jin G, Floy ME, Hoon KM & Palecek SP. (2019). Coculture of Endothelial Cells with Human Pluripotent Stem Cell-Derived Cardiac Progenitors Reveals a Differentiation Stage-Specific Enhancement of Cardiomyocyte Maturation. Biotechnol J 14, e1800725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkee MS, Abraham R, Clark MR & Giger ML. (2021). Artificial Intelligence and Cellular Segmentation in Tissue Microscopy Images. Am J Pathol 191, 1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwenger MM, Ohanyan V, Navedo MF & Nystoriak MA. (2018). Coronary microvascular Kv1 channels as regulatory sensors of intracellular pyridine nucleotide redox potential. Microcirculation 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwenger MM, Raph SM, Reyzer ML, Lisa Manier M, Riggs DW, Wohl ZB, Ohanyan V, Mack G, Pucci T, Moore JBt, Hill BG, Chilian WM, Caprioli RM, Bhatnagar A & Nystoriak MA. (2022). Pyridine nucleotide redox potential in coronary smooth muscle couples myocardial blood flow to cardiac metabolism. Nat Commun 13, 2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S & Brayden JE. (2015). Transient receptor potential channels in the vasculature. Physiol Rev 95, 645–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel CA, Rios-Perez EB, Liu F, Jameson MB, Jones DK, Knickelbine JJ & Robertson GA. (2019). A microtranslatome coordinately regulates sodium and potassium currents in the human heart. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn B & Dobrev D. (2007). Vascular large conductance calcium-activated potassium channels: functional role and therapeutic potential. Naunyn Schmiedebergs Arch Pharmacol 376, 145–155. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Caldwell JL, Kistamas K & Trafford AW. (2017). Calcium and Excitation-Contraction Coupling in the Heart. Circ Res 121, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernault AC, Meijborg VMF & Coronel R. (2021). Modulation of Cardiac Arrhythmogenesis by Epicardial Adipose Tissue: JACC State-of-the-Art Review. J Am Coll Cardiol 78, 1730–1745. [DOI] [PubMed] [Google Scholar]
- Erreni M, Schorn T, D’Autilia F & Doni A. (2020). Nanobodies as Versatile Tool for Multiscale Imaging Modalities. Biomolecules 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Es-Salah-Lamoureux Z, Jouni M, Malak OA, Belbachir N, Al Sayed ZR, Gandon-Renard M, Lamirault G, Gauthier C, Baró I, Charpentier F, Zibara K, Lemarchand P, Beaumelle B, Gaborit N & Loussouarn G. (2016). HIV-Tat induces a decrease in I(Kr) and I(Ks)via reduction in phosphatidylinositol-(4,5)-bisphosphate availability. J Mol Cell Cardiol 99, 1–13. [DOI] [PubMed] [Google Scholar]
- Escobar AL, Ribeiro-Costa R, Villalba-Galea C, Zoghbi ME, Perez CG & Mejia-Alvarez R. (2004). Developmental changes of intracellular Ca2+ transients in beating rat hearts. Am J Physiol Heart Circ Physiol 286, H971–978. [DOI] [PubMed] [Google Scholar]
- Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, Bloodworth N, Merryman WD, Lim CC, Wu JC, Knollmann BC & Hong CC. (2015). Matrigel Mattress: A Method for the Generation of Single Contracting Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res 117, 995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigl EO. (1983). Coronary physiology. Physiol Rev 63, 1–205. [DOI] [PubMed] [Google Scholar]
- Feyen DAM, McKeithan WL, Bruyneel AAN, Spiering S, Hormann L, Ulmer B, Zhang H, Briganti F, Schweizer M, Hegyi B, Liao Z, Polonen RP, Ginsburg KS, Lam CK, Serrano R, Wahlquist C, Kreymerman A, Vu M, Amatya PL, Behrens CS, Ranjbarvaziri S, Maas RGC, Greenhaw M, Bernstein D, Wu JC, Bers DM, Eschenhagen T, Metallo CM & Mercola M. (2020). Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep 32, 107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink M, Niederer SA, Cherry EM, Fenton FH, Koivumaki JT, Seemann G, Thul R, Zhang H, Sachse FB, Beard D, Crampin EJ & Smith NP. (2011). Cardiac cell modelling: observations from the heart of the cardiac physiome project. Prog Biophys Mol Biol 104, 2–21. [DOI] [PubMed] [Google Scholar]
- Francis Stuart SD, De Jesus NM, Lindsey ML & Ripplinger CM. (2016). The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol 91, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. (2019). Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 65, 70–99. [DOI] [PubMed] [Google Scholar]
- Frodermann V & Nahrendorf M. (2018). Macrophages and Cardiovascular Health. Physiol Rev 98, 2523–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Shaw SA, Naami R, Vuong CL, Basheer WA, Guo X & Hong T. (2016). Isoproterenol Promotes Rapid Ryanodine Receptor Movement to Bridging Integrator 1 (BIN1)-Organized Dyads. Circulation 133, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Leger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S & Demolombe S. (2005). Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 112, 471–481. [DOI] [PubMed] [Google Scholar]
- Galice S, Xie Y, Yang Y, Sato D & Bers DM. (2018). Size Matters: Ryanodine Receptor Cluster Size Affects Arrhythmogenic Sarcoplasmic Reticulum Calcium Release. J Am Heart Assoc 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher BR & Zhao Y. (2021). Expansion microscopy: A powerful nanoscale imaging tool for neuroscientists. Neurobiol Dis 154, 105362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R & Seoighe C. (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur N & Rudy Y. (2011). Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J 100, 2904–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawalko M, Saljic A, Li N, Abu-Taha I, Jespersen T, Linz D, Nattel S, Heijman J, Fender A & Dobrev D. (2022). Adiposity-associated atrial fibrillation: molecular determinants, mechanisms and clinical significance. Cardiovasc Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA & Abecasis GR. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Nieves-Cintron M, Tajada S, Brust-Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF & Navedo MF. (2018). Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomelli E, Meraviglia V, Campostrini G, Cochrane A, Cao X, van Helden RWJ, Krotenberg Garcia A, Mircea M, Kostidis S, Davis RP, van Meer BJ, Jost CR, Koster AJ, Mei H, Miguez DG, Mulder AA, Ledesma-Terron M, Pompilio G, Sala L, Salvatori DCF, Slieker RC, Sommariva E, de Vries AAF, Giera M, Semrau S, Tertoolen LGJ, Orlova VV, Bellin M & Mummery CL. (2020). Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 26, 862–879 e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannetti F, Benzoni P, Campostrini G, Milanesi R, Bucchi A, Baruscotti M, Dell’Era P, Rossini A & Barbuti A. (2021). A detailed characterization of the hyperpolarization-activated “funny” current (If) in human-induced pluripotent stem cell (iPSC)-derived cardiomyocytes with pacemaker activity. Pflugers Arch 473, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JQX & Sobie EA. (2018). Population-based mechanistic modeling allows for quantitative predictions of drug responses across cell types. NPJ Syst Biol Appl 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales AL, Klug NR, Moshkforoush A, Lee JC, Lee FK, Shui B, Tsoukias NM, Kotlikoff MI, Hill-Eubanks D & Nelson MT. (2020). Contractile pericytes determine the direction of blood flow at capillary junctions. Proc Natl Acad Sci U S A 117, 27022–27033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik J, Shevchuk A, Ramalho M, Elliott M, Lei C, Higgins CF, Lab MJ, Klenerman D, Krauzewicz N & Korchev Y. (2002). Scanning surface confocal microscopy for simultaneous topographical and fluorescence imaging: application to single virus-like particle entry into a cell. Proc Natl Acad Sci U S A 99, 16018–16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goversen B, van der Heyden MAG, van Veen TAB & de Boer TP. (2018). The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on IK1. Pharmacol Ther 183, 127–136. [DOI] [PubMed] [Google Scholar]
- Grandi E, Sanguinetti MC, Bartos DC, Bers DM, Chen-Izu Y, Chiamvimonvat N, Colecraft HM, Delisle BP, Heijman J, Navedo MF, Noskov S, Proenza C, Vandenberg JI & Yarov-Yarovoy V. (2017). Potassium channels in the heart: structure, function and regulation. J Physiol 595, 2209–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene D, Kaboudian A, Wasserstrom JA, Fenton FH & Shiferaw Y. (2022). Voltage-mediated mechanism for calcium wave synchronization and arrhythmogenesis in atrial tissue. Biophys J 121, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein JL & Winslow RL. (2002). An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J 83, 2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer-Short A, George SA, Poelzing S & Weinberg SH. (2017). Revealing the Concealed Nature of Long-QT Type 3 Syndrome. Circ Arrhythm Electrophysiol 10, e004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendaal W, Ortega FA, Kherlopian AR, Zygmunt AC, Krogh-Madsen T & Christini DJ. (2015). Cell-specific cardiac electrophysiology models. PLoS Comput Biol 11, e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune J, Lewis AJM, Yamazoe M, Hulsmans M, Rohde D, Xiao L, Zhang S, Ott C, Calcagno DM, Zhou Y, Timm K, Shanmuganathan M, Pulous FE, Schloss MJ, Foy BH, Capen D, Vinegoni C, Wojtkiewicz GR, Iwamoto Y, Grune T, Brown D, Higgins J, Ferreira VM, Herring N, Channon KM, Neubauer S, Oxford Acute Myocardial Infarction S, Sosnovik DE, Milan DJ, Swirski FK, King KR, Aguirre AD, Ellinor PT & Nahrendorf. (2022). Neutrophils incite and macrophages avert electrical storm after myocardial infarction. Nat Cardiovasc Res 1, 649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Wang Y, Chen B, Wang Y, Yuan J, Zhang L, Hall D, Wu J, Shi Y, Zhu Q, Chen C, Thiel WH, Zhan X, Weiss RM, Zhan F, Musselman CA, Pufall M, Zhu W, Au KF, Hong J, Anderson ME, Grueter CE & Song LS. (2018). E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wang T, Li X, Shallow H, Yang T, Li W, Xu J, Fridman MD, Yang X & Zhang S. (2012). Cell surface expression of human ether-a-go-go-related gene (hERG) channels is regulated by caveolin-3 protein via the ubiquitin ligase Nedd4–2. J Biol Chem 287, 33132–33141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwosch KC, Pape JK, Balzarotti F, Hoess P, Ellenberg J, Ries J & Hell SW. (2020). MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat Methods 17, 217–224. [DOI] [PubMed] [Google Scholar]
- Ha T, Kaiser C, Myong S, Wu B & Xiao J. (2022). Next generation single-molecule techniques: Imaging, labeling, and manipulation in vitro and in cellulo. Mol Cell 82, 304–314. [DOI] [PubMed] [Google Scholar]
- Hadaya J & Ardell JL. (2020). Autonomic Modulation for Cardiovascular Disease. Front Physiol 11, 617459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddock PS, Coetzee WA, Cho E, Porter L, Katoh H, Bers DM, Jafri MS & Artman M. (1999). Subcellular [Ca2+]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ Res 85, 415–427. [DOI] [PubMed] [Google Scholar]
- Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, Bader JS, Kass DA & Sharma K. (2021). Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation 143, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi S, Kawakami Y, Honda Y, Nemoto K, Sano A, Namekata I & Tanaka H. (2013). Developmental changes in excitation-contraction mechanisms of the mouse ventricular myocardium as revealed by functional and confocal imaging analyses. J Pharmacol Sci 123, 167–175. [DOI] [PubMed] [Google Scholar]
- Hamon D, Rajendran PS, Chui RW, Ajijola OA, Irie T, Talebi R, Salavatian S, Vaseghi M, Bradfield JS, Armour JA, Ardell JL & Shivkumar K. (2017). Premature Ventricular Contraction Coupling Interval Variability Destabilizes Cardiac Neuronal and Electrophysiological Control: Insights From Simultaneous Cardioneural Mapping. Circ Arrhythm Electrophysiol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna P, Rajendran PS, Ajijola OA, Vaseghi M, Andrew Armour J, Ardell JL & Shivkumar K. (2017). Cardiac neuroanatomy - Imaging nerves to define functional control. Auton Neurosci 207, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan A, Weir N, Robertson C, He L, Betsholtz C & Longden TA. (2020). The Ion Channel and GPCR Toolkit of Brain Capillary Pericytes. Front Cell Neurosci 14, 601324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlaar N, Dekker SO, Zhang J, Snabel RR, Veldkamp MW, Verkerk AO, Fabres CC, Schwach V, Lerink LJS, Rivaud MR, Mulder AA, Corver WE, Goumans M, Dobrev D, Klautz RJM, Schalij MJ, Veenstra GJC, Passier R, van Brakel TJ, Pijnappels DA & de Vries AAF. (2022). Conditional immortalization of human atrial myocytes for the generation of in vitro models of atrial fibrillation. Nat Biomed Eng 6, 389–402. [DOI] [PubMed] [Google Scholar]
- Harrison CB, Trevelin SC, Richards DA, Santos CXC, Sawyer G, Markovinovic A, Zhang X, Zhang M, Brewer AC, Yin X, Mayr M & Shah AM. (2021). Fibroblast Nox2 (NADPH Oxidase-2) Regulates ANG II (Angiotensin II)-Induced Vascular Remodeling and Hypertension via Paracrine Signaling to Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 41, 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann DA, Coelho-Santos V & Shih AY. (2022). Pericyte Control of Blood Flow Across Microvascular Zones in the Central Nervous System. Annu Rev Physiol 84, 331–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Shi X, Zhang X, Dang S, Ma X, Liu F, Xu M, Lv Z, Han D, Fang X & Zhang Y. (2011). Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res 92, 39–47. [DOI] [PubMed] [Google Scholar]
- Hegyi B, Bossuyt J, Griffiths LG, Shimkunas R, Coulibaly Z, Jian Z, Grimsrud KN, Sondergaard CS, Ginsburg KS, Chiamvimonvat N, Belardinelli L, Varro A, Papp JG, Pollesello P, Levijoki J, Izu LT, Boyd WD, Banyasz T, Bers DM & Chen-Izu Y. (2018). Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc Natl Acad Sci U S A 115, E3036–E3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi B, Chen-Izu Y, Izu LT, Rajamani S, Belardinelli L, Bers DM & Banyasz T. (2020). Balance Between Rapid Delayed Rectifier K(+) Current and Late Na(+) Current on Ventricular Repolarization: An Effective Antiarrhythmic Target? Circ Arrhythm Electrophysiol 13, e008130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidi Au HT, Cui B, Chu ZE, Veres T & Radisic M. (2009). Cell culture chips for simultaneous application of topographical and electrical cues enhance phenotype of cardiomyocytes. Lab Chip 9, 564–575. [DOI] [PubMed] [Google Scholar]
- Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Kunzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens XHT & Dobrev D. (2020). Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ Res 127, 1036–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Nattel S & Dobrev D. (2014). Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 114, 1483–1499. [DOI] [PubMed] [Google Scholar]
- Heiligenstein X & Lucas MS. (2022). One for All, All for One: A Close Look at In-Resin Fluorescence Protocols for CLEM. Front Cell Dev Biol 10, 866472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosilla T, Encina M, Morales D, Moreno C, Conejeros C, Alfaro-Valdes HM, Lagos-Meza F, Simon F, Altier C & Varela D. (2017). Prolonged AT1R activation induces CaV1.2 channel internalization in rat cardiomyocytes. Sci Rep 7, 10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron TJ, Rocha AM, Campbell KF, Ponce-Balbuena D, Willis BC, Guerrero-Serna G, Liu Q, Klos M, Musa H, Zarzoso M, Bizy A, Furness J, Anumonwo J, Mironov S & Jalife J. (2016). Extracellular Matrix-Mediated Maturation of Human Pluripotent Stem Cell-Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ Arrhythm Electrophysiol 9, e003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G (2022). Coronary blood flow in heart failure: cause, consequence and bystander. Basic Res Cardiol 117, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hew KW & Keller KA. (2003). Postnatal anatomical and functional development of the heart: a species comparison. Birth Defects Res B Dev Reprod Toxicol 68, 309–320. [DOI] [PubMed] [Google Scholar]
- Hichri E, Abriel H & Kucera JP. (2018). Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J Physiol 596, 563–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MC, Kadow ZA, Long H, Morikawa Y, Martin TJ, Birks EJ, Campbell KS, Nerbonne J, Lavine K, Wadhwa L, Wang J, Turaga D, Adachi I & Martin JF. (2022). Integrated multi-omic characterization of congenital heart disease. Nature 608, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hissen SL & Taylor CE. (2020). Sex differences in vascular transduction of sympathetic nerve activity. Clin Auton Res 30, 381–392. [DOI] [PubMed] [Google Scholar]
- Holzem KM, Gomez JF, Glukhov AV, Madden EJ, Koppel AC, Ewald GA, Trenor B & Efimov IR. (2015). Reduced response to I blockade and altered hERG1a/1b stoichiometry in human heart failure. J Mol Cell Cardiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H & Tian XY. (2020). The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath B, Kiss D, Dienes C, Hezso T, Kovacs Z, Szentandrassy N, Almassy J, Magyar J, Banyasz T & Nanasi PP. (2021). Ion current profiles in canine ventricular myocytes obtained by the “onion peeling” technique. J Mol Cell Cardiol 158, 153–162. [DOI] [PubMed] [Google Scholar]
- Hota SK, Rao KS, Blair AP, Khalilimeybodi A, Hu KM, Thomas R, So K, Kameswaran V, Xu J, Polacco BJ, Desai RV, Chatterjee N, Hsu A, Muncie JM, Blotnick AM, Winchester SAB, Weinberger LS, Huttenhain R, Kathiriya IS, Krogan NJ, Saucerman JJ & Bruneau BG. (2022). Brahma safeguards canalization of cardiac mesoderm differentiation. Nature 602, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Bai J, Shen X, de Langen O, Li A, Lal S, Dos Remedios CG, Baddeley D, Ruygrok PN, Soeller C & Crossman DJ. (2021). Nanoscale Organisation of Ryanodine Receptors and Junctophilin-2 in the Failing Human Heart. Front Physiol 12, 724372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Hove-Madsen L & Tibbits GF. (2005). Na+/Ca2+ exchange activity in neonatal rabbit ventricular myocytes. American journal of physiology Cell physiology 288, C195–203. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Westenbroek RE, Scheuer T & Catterall WA. (2006). Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci U S A 103, 16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT & Nahrendorf M. (2017). Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y & Ito Y. (2009). Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res 104, 1399–1409. [DOI] [PubMed] [Google Scholar]
- Ito DW, Hannigan KI, Ghosh D, Xu B, Del Villar SG, Xiang YK, Dickson EJ, Navedo MF & Dixon RE. (2019). beta-adrenergic-mediated dynamic augmentation of sarcolemmal CaV 1.2 clustering and co-operativity in ventricular myocytes. J Physiol 597, 2139–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic E & Kucera JP. (2021). Localization of Na(+) channel clusters in narrowed perinexi of gap junctions enhances cardiac impulse transmission via ephaptic coupling: a model study. J Physiol 599, 4779–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemet G, Carisey AF, Hamidi H, Henriques R & Leterrier C. (2020). The cell biologist’s guide to super-resolution microscopy. J Cell Sci 133. [DOI] [PubMed] [Google Scholar]
- Jeong D & Kim D. (2022). Recent Developments in Correlative Super-Resolution Fluorescence Microscopy and Electron Microscopy. Mol Cells 45, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EK, Matkovich SJ & Nerbonne JM. (2018a). Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci Rep 8, 13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EK, Springer SJ, Wang W, Dranoff EJ, Zhang Y, Kanter EM, Yamada KA & Nerbonne JM. (2018b). Differential Expression and Remodeling of Transient Outward Potassium Currents in Human Left Ventricles. Circ Arrhythm Electrophysiol 11, e005914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC & Robertson GA. (2014). hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci U S A 111, 18073–18077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EM, Roti Roti EC, Wang J, Delfosse SA & Robertson GA. (2004). Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem 279, 44690–44694. [DOI] [PubMed] [Google Scholar]
- Jopling C, Sune G, Faucherre A, Fabregat C & Izpisua Belmonte JC. (2012). Hypoxia induces myocardial regeneration in zebrafish. Circulation 126, 3017–3027. [DOI] [PubMed] [Google Scholar]
- Joshi A, Rienks M, Theofilatos K & Mayr M. (2021). Systems biology in cardiovascular disease: a multiomics approach. Nat Rev Cardiol 18, 313–330. [DOI] [PubMed] [Google Scholar]
- Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A & Ventura-Clapier R. (2001). Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res 89, 153–159. [DOI] [PubMed] [Google Scholar]
- Kadota S, Pabon L, Reinecke H & Murry CE. (2017). In Vivo Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Neonatal and Adult Rat Hearts. Stem Cell Reports 8, 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellakis P, Ditiatkovski M, Kostolias G & Bobik A. (2012). A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc Res 95, 77–85. [DOI] [PubMed] [Google Scholar]
- Kannan S & Kwon C. (2020). Regulation of cardiomyocyte maturation during critical perinatal window. J Physiol 598, 2941–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapela A, Bezerianos A & Tsoukias NM. (2008). A mathematical model of Ca2+ dynamics in rat mesenteric smooth muscle cell: agonist and NO stimulation. Journal of theoretical biology 253, 238–260. [DOI] [PubMed] [Google Scholar]
- Kapela A, Nagaraja S & Tsoukias NM. (2010). A mathematical model of vasoreactivity in rat mesenteric arterioles. II. Conducted vasoreactivity. Am J Physiol Heart Circ Physiol 298, H52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X & Murry CE. (2020). Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol 17, 341–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A (2015). Membrane potential and Ca2+ concentration dependence on pressure and vasoactive agents in arterial smooth muscle: A model. J Gen Physiol 146, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchman A, Yang L, Zakharov SI, Kushner J, Abrams J, Chen BX, Liu G, Pitt GS, Colecraft HM & Marx SO. (2017). Proteolytic cleavage and PKA phosphorylation of alpha1C subunit are not required for adrenergic regulation of CaV1.2 in the heart. Proc Natl Acad Sci U S A 114, 9194–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DH, Tahir UA, Bick AG, Pampana A, Ngo D, Benson MD, Yu Z, Robbins JM, Chen ZZ, Cruz DE, Deng S, Farrell L, Sinha S, Schmaier AA, Shen D, Gao Y, Hall ME, Correa A, Tracy RP, Durda P, Taylor KD, Liu Y, Johnson WC, Guo X, Yao J, Ida Chen YD, Manichaikul AW, Jain D, Bouchard C, Sarzynski MA, Rich SS, Rotter JI, Wang TJ, Wilson JG, Natarajan P, Gerszten RE, National Heart L & Blood Institute TC. (2022). Whole Genome Sequence Analysis of the Plasma Proteome in Black Adults Provides Novel Insights Into Cardiovascular Disease. Circulation 145, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedia-Mehta N & Finlay DK. (2019). Competition for nutrients and its role in controlling immune responses. Nat Commun 10, 2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keef KD, Hume JR & Zhong J. (2001). Regulation of cardiac and smooth muscle Ca(2+) channels (Ca(V)1.2a,b) by protein kinases. American journal of physiology Cell physiology 281, C1743–1756. [DOI] [PubMed] [Google Scholar]
- Keene BW, Atkins CE, Bonagura JD, Fox PR, Haggstrom J, Fuentes VL, Oyama MA, Rush JE, Stepien R & Uechi M. (2019). ACVIM consensus guidelines for the diagnosis and treatment of myxomatous mitral valve disease in dogs. J Vet Intern Med 33, 1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Goss MR, Sweat RS, Stapor PC, Peirce SM & Murfee WL. (2014). Targeting pericytes for angiogenic therapies. Microcirculation 21, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J & Molkentin JD. (2017). Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127, 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharche S, Yu J, Lei M & Zhang H. (2011). A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am J Physiol Heart Circ Physiol 301, H945–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Lipke EA, Kim P, Cheong R, Thompson S, Delannoy M, Suh KY, Tung L & Levchenko A. (2010). Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc Natl Acad Sci U S A 107, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Yang L, Lin B, Zhu X, Sun B, Kaplan AD, Bett GC, Rasmusson RL, London B & Salama G. (2015). Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J Mol Cell Cardiol 81, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura W, Nakada Y & Sadek HA. (2017). Hypoxia-induced myocardial regeneration. J Appl Physiol (1985) 123, 1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, Abderrahman Y, Chen R, Garcia JA, Shelton JM, Richardson JA, Ashour AM, Asaithamby A, Liang H, Xing C, Lu Z, Cheng Zhang C & Sadek HA. (2016). Corrigendum: Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 532, 268. [DOI] [PubMed] [Google Scholar]
- King DR, Sedovy MW, Eaton X, Dunaway LS, Good ME, Isakson BE & Johnstone SR. (2022). Cell-To-Cell Communication in the Resistance Vasculature. Compr Physiol 12, 1–35. [DOI] [PubMed] [Google Scholar]
- Kivela R, Hemanthakumar KA, Vaparanta K, Robciuc M, Izumiya Y, Kidoya H, Takakura N, Peng X, Sawyer DB, Elenius K, Walsh K & Alitalo K. (2019). Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation 139, 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaapen P, Germans T, Knuuti J, Paulus WJ, Dijkmans PA, Allaart CP, Lammertsma AA & Visser FC. (2007). Myocardial energetics and efficiency: current status of the noninvasive approach. Circulation 115, 918–927. [DOI] [PubMed] [Google Scholar]
- Knot HJ & Nelson MT. (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508 (Pt 1), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, Norden ES, Cataliotti A, Sjaastad I, Sejersted OM, Edwards AG, Lines GT & Louch WE. (2018). Ryanodine receptor dispersion disrupts Ca(2+) release in failing cardiac myocytes. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucera JP, Rohr S & Rudy Y. (2002). Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res 91, 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer ME, Lin WH, Ravikumar V, Qiu K, Wang L, Gao L, Bhuiyan DB, Lenz M, Ai J, Mahutga RR, Townsend D, Zhang J, McAlpine MC, Tolkacheva EG & Ogle BM. (2020). In Situ Expansion, Differentiation, and Electromechanical Coupling of Human Cardiac Muscle in a 3D Bioprinted, Chambered Organoid. Circ Res 127, 207–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppe C, Ramirez Flores RO, Li Z, Hayat S, Levinson RT, Liao X, Hannani MT, Tanevski J, Wunnemann F, Nagai JS, Halder M, Schumacher D, Menzel S, Schafer G, Hoeft K, Cheng M, Ziegler S, Zhang X, Peisker F, Kaesler N, Saritas T, Xu Y, Kassner A, Gummert J, Morshuis M, Amrute J, Veltrop RJA, Boor P, Klingel K, Van Laake LW, Vink A, Hoogenboezem RM, Bindels EMJ, Schurgers L, Sattler S, Schapiro D, Schneider RK, Lavine K, Milting H, Costa IG, Saez-Rodriguez J & Kramann R. (2022). Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste B, Comin CH, Ben-Zvi A, Kaeser PS, Xu X, Costa Lda F & Gu C. (2014). Sensory-related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 83, 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG & DiFrancesco D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol 47, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P & Horvath S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzerini PE, Laghi-Pasini F, Boutjdir M & Capecchi PL. (2022). Inflammatory cytokines and cardiac arrhythmias: the lesson from COVID-19. Nat Rev Immunol 22, 270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE & Wehrens XHT. (2022). The role of junctophilin proteins in cellular function. Physiol Rev 102, 1211–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei CL, Wang K, Clerx M, Johnstone RH, Hortigon-Vinagre MP, Zamora V, Allan A, Smith GL, Gavaghan DJ, Mirams GR & Polonchuk L. (2017). Tailoring Mathematical Models to Stem-Cell Derived Cardiomyocyte Lines Can Improve Predictions of Drug-Induced Changes to Their Electrophysiology. Front Physiol 8, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F & Moosmang S. (2008). Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J Biol Chem 283, 34738–34744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Muhl L & Betsholtz C. (2022). Identification, discrimination and heterogeneity of fibroblasts. Nat Commun 13, 3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo MD & Jaggar JH. (2016). Ion Channel Trafficking and Control of Arterial Contractility. In Vascular Ion Channels in Physiology and Disease, ed. Levitan PI & Dopico MDPAM, pp. 153–168. Springer International Publishing, Cham. [Google Scholar]
- Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, Keegan S, Lin X, Arcos T, Feng Xia L, Korchev YE, Gorelik J, Fenyo D, Rothenberg E, Rothenberg E & Delmar M. (2016). Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun 7, 10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Mimila P, Wang J & Huertas-Vazquez A. (2019). Relevance of Multi-Omics Studies in Cardiovascular Diseases. Front Cardiovasc Med 6, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C & Coffman TM. (2019). Animal Models of Hypertension: A Scientific Statement From the American Heart Association. Hypertension 73, e87–e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner E, Barth A, Hendrix J, Ambrose B, Birkedal V, Blanchard SC, Borner R, Sung Chung H, Cordes T, Craggs TD, Deniz AA, Diao J, Fei J, Gonzalez RL, Gopich IV, Ha T, Hanke CA, Haran G, Hatzakis NS, Hohng S, Hong SC, Hugel T, Ingargiola A, Joo C, Kapanidis AN, Kim HD, Laurence T, Lee NK, Lee TH, Lemke EA, Margeat E, Michaelis J, Michalet X, Myong S, Nettels D, Peulen TO, Ploetz E, Razvag Y, Robb NC, Schuler B, Soleimaninejad H, Tang C, Vafabakhsh R, Lamb DC, Seidel CA & Weiss S. (2021). FRET-based dynamic structural biology: Challenges, perspectives and an appeal for open-science practices. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Chen Q & Hoppel CL. (2016). Mitochondrial Metabolism in Aging Heart. Circ Res 118, 1593–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levet F, Hosy E, Kechkar A, Butler C, Beghin A, Choquet D & Sibarita JB. (2015). SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods 12, 1065–1071. [DOI] [PubMed] [Google Scholar]
- Levine HJ. (1997). Rest heart rate and life expectancy. J Am Coll Cardiol 30, 1104–1106. [DOI] [PubMed] [Google Scholar]
- Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A & Nattel S. (2000). Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation 101, 2631–2638. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM & Gerdes AM. (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28, 1737–1746. [DOI] [PubMed] [Google Scholar]
- Li N, Liu JY, Qiu H, Harris TR, Sirish P, Hammock BD & Chiamvimonvat N. (2011). Use of metabolomic profiling in the study of arachidonic acid metabolism in cardiovascular disease. Congest Heart Fail 17, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu B, Wu X, Lu Y, Qiu M, Shen Y, Tian Y, Liu C, Chen X, Yang C, Deng M, Wang Y, Gu J, Su Z, Chen X, Zhao K, Sheng Y, Zhang S, Sun W & Kong X. (2022). Perirenal adipose afferent nerves sustain pathological high blood pressure in rats. Nat Commun 13, 3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Lockhead D, Larson ED & Proenza C. (2010). Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J Gen Physiol 136, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SL, Lam CS, Segers VF, Brutsaert DL & De Keulenaer GW. (2015). Cardiac endothelium-myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur Heart J 36, 2050–2060. [DOI] [PubMed] [Google Scholar]
- Lin J & Keener JP. (2010). Modeling electrical activity of myocardial cells incorporating the effects of ephaptic coupling. Proc Natl Acad Sci U S A 107, 20935–20940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Mayr M, Gomes AV, Delles C, Arrell DK, Murphy AM, Lange RA, Costello CE, Jin YF, Laskowitz DT, Sam F, Terzic A, Van Eyk J, Srinivas PR, American Heart Association Council on Functional G, Translational Biology CoCDitYCoCCCoC, Stroke Nursing CoH & Stroke C. (2015). Transformative Impact of Proteomics on Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association. Circulation 132, 852–872. [DOI] [PubMed] [Google Scholar]
- Lipsett DB, Frisk M, Aronsen JM, Nordén ES, Buonarati OR, Cataliotti A, Hell JW, Sjaastad I, Christensen G & Louch WE. (2019). Cardiomyocyte substructure reverts to an immature phenotype during heart failure. J Physiol 597, 1833–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N & Teichmann SA. (2020). Cells of the adult human heart. Nature 588, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben-Johny M, Kalocsay M & Marx SO. (2020). Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Jin L, Chen J, Fang Q, Ablameyko S, Yin Z & Xu Y. (2021). A survey on applications of deep learning in microscopy image analysis. Comput Biol Med 134, 104523. [DOI] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D & Nelson MT. (2017). Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhurst JC, Tjen ALSC & Fu LW. (2001). Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann N Y Acad Sci 940, 74–95. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD & Jaswal JS. (2010). Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 56, 130–140. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Karwi QG, Tian R, Wende AR & Abel ED. (2021). Cardiac Energy Metabolism in Heart Failure. Circ Res 128, 1487–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk GD & Ussher JR. (2016). Evolving Concepts of Myocardial Energy Metabolism: More Than Just Fats and Carbohydrates. Circ Res 119, 1173–1176. [DOI] [PubMed] [Google Scholar]
- Louch WE, Koivumaki JT & Tavi P. (2015). Calcium signalling in developing cardiomyocytes: implications for model systems and disease. J Physiol 593, 1047–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Perdreau-Dahl H & Edwards AG. (2022). Image-Driven Modeling of Nanoscopic Cardiac Function: Where Have We Come From, and Where Are We Going? Front Physiol 13, 834211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis Fuentes V, Abbott J, Chetboul V, Cote E, Fox PR, Haggstrom J, Kittleson MD, Schober K & Stern JA. (2020). ACVIM consensus statement guidelines for the classification, diagnosis, and management of cardiomyopathies in cats. J Vet Intern Med 34, 1062–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundy SD, Zhu WZ, Regnier M & Laflamme MA. (2013). Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 22, 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly C & Weinberg SH. (2022). Automaticity in ventricular myocyte cell pairs with ephaptic and gap junction coupling. Chaos 32, 033123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCannell KA, Bazzazi H, Chilton L, Shibukawa Y, Clark RB & Giles WR. (2007). A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J 92, 4121–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQuaide N, Tuan HT, Hotta J, Sempels W, Lenaerts I, Holemans P, Hofkens J, Jafri MS, Willems R & Sipido KR. (2015). Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res 108, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev AV, Maltsev VA, Mikheev M, Maltseva LA, Sirenko SG, Lakatta EG & Stern MD. (2011). Synchronization of stochastic Ca(2)(+) release units creates a rhythmic Ca(2)(+) clock in cardiac pacemaker cells. Biophys J 100, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Mateu L, Saiz J & Aromolaran AS. (2019). Differential Modulation of IK and ICa,L Channels in High-Fat Diet-Induced Obese Guinea Pig Atria. Front Physiol 10, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H & Sperelakis N. (1993). Inwardly rectifying potassium current in rat fetal and neonatal ventricular cardiomyocytes. Am J Physiol 265, H1107–1111. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Zhang Y, Van Booven DJ & Dorn GW 2nd. (2010). Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Galphaq. Circ Res 106, 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarrah RW, Crown SB, Zhang GF, Shah SH & Newgard CB. (2018). Cardiovascular Metabolomics. Circ Res 122, 1238–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev RY, Sanchez-Alonso JL, Mansfield CA, Judina A, Francis AJ, Pagiatakis C, Trayanova N, Glukhov AV, Miragoli M, Faggian G & Gorelik J. (2021). Local hyperactivation of L-type Ca(2+) channels increases spontaneous Ca(2+) release activity and cellular hypertrophy in right ventricular myocytes from heart failure rats. Sci Rep 11, 4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton E & Qiu H. (2021). Interleukin-1beta in Multifactorial Hypertension: Inflammation, Vascular Smooth Muscle Cell and Extracellular Matrix Remodeling, and Non-Coding RNA Regulation. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezache L, Struckman HL, Greer-Short A, Hund TJ & Veeraraghavan R. (2019). Indirect CLEM Identifies Proarrhythmic Remodeling of Intercalated Disk Nanodomains in Murine Atria Following Acute VEGF Treatment. Microscopy and Microanalysis 25, 1136–1137. [Google Scholar]
- Michael G, Xiao L, Qi XY, Dobrev D & Nattel S. (2009). Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res 81, 491–499. [DOI] [PubMed] [Google Scholar]
- Michelhaugh SA & Januzzi JL Jr. (2020). Finding a Needle in a Haystack: Proteomics in Heart Failure. JACC Basic Transl Sci 5, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR & Jalife J. (2012). Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109, E2134–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S & Kass DA. (2021). Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 18, 400–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Lundberg E & Heyn H. (2022). The emerging landscape of spatial profiling technologies. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- Moise N, Struckman HL, Dagher C, Veeraraghavan R & Weinberg SH. (2021). Intercalated disk nanoscale structure regulates cardiac conduction. J Gen Physiol 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S & Kuhn B. (2013). Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A 110, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno CM, Dixon RE, Tajada S, Yuan C, Opitz-Araya X, Binder MD & Santana LF. (2016). Ca(2+) entry into neurons is facilitated by cooperative gating of clustered CaV1.3 channels. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan ML, Brideau C, Teo W, Caprariello AV & Stys PK. (2021). Label-free assessment of myelin status using birefringence microscopy. J Neurosci Methods 360, 109226. [DOI] [PubMed] [Google Scholar]
- Morotti S, Liu C, Hegyi B, Ni H, Fogli Iseppe A, Wang L, Pritoni M, Ripplinger CM, Bers DM, Edwards AG & Grandi E. (2021). Quantitative cross-species translators of cardiac myocyte electrophysiology: Model training, experimental validation, and applications. Sci Adv 7, eabg0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotti S, Nieves-Cintron M, Nystoriak MA, Navedo MF & Grandi E. (2017). Predominant contribution of L-type Cav1.2 channel stimulation to impaired intracellular calcium and cerebral artery vasoconstriction in diabetic hyperglycemia. Channels (Austin) 11, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton SU & Brodsky D. (2016). Fetal Physiology and the Transition to Extrauterine Life. Clin Perinatol 43, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshkforoush A, Ashenagar B, Harraz OF, Dabertrand F, Longden TA, Nelson MT & Tsoukias NM. (2020). The capillary Kir channel as sensor and amplifier of neuronal signals: Modeling insights on K(+)-mediated neurovascular communication. Proc Natl Acad Sci U S A 117, 16626–16637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalik A, Niderla-Bielinska J & Ratajska A. (2022). Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail Rev 27, 1413–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl L, Genove G, Leptidis S, Liu J, He L, Mocci G, Sun Y, Gustafsson S, Buyandelger B, Chivukula IV, Segerstolpe A, Raschperger E, Hansson EM, Bjorkegren JLM, Peng XR, Vanlandewijck M, Lendahl U & Betsholtz C. (2020). Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun 11, 3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R & Tertoolen L. (2003). Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation 107, 2733–2740. [DOI] [PubMed] [Google Scholar]
- Munro ML, van Hout I, Aitken-Buck HM, Sugunesegran R, Bhagwat K, Davis PJ, Lamberts RR, Coffey S, Soeller C & Jones PP. (2021). Human Atrial Fibrillation Is Not Associated With Remodeling of Ryanodine Receptor Clusters. Front Cell Dev Biol 9, 633704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami U & Uchida K. (1985). Contents of myofibrillar proteins in cardiac, skeletal, and smooth muscles. J Biochem 98, 187–197. [DOI] [PubMed] [Google Scholar]
- Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W & Sadek HA. (2017). Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227. [DOI] [PubMed] [Google Scholar]
- Nassal DM, Wan X, Liu H, Maleski D, Ramirez-Navarro A, Moravec CS, Ficker E, Laurita KR & Deschênes I. (2017). KChIP2 is a core transcriptional regulator of cardiac excitability. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD & Santana LF. (2010a). Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Takeda Y, Nieves-Cintron M, Molkentin JD & Santana LF. (2010b). Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. American journal of physiology Cell physiology 298, C211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary MT, Ng KE, Ludtmann MH, Hall AR, Piotrowska I, Ong SB, Hausenloy DJ, Mohun TJ, Abramov AY & Breckenridge RA. (2014). Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol 74, 340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC, Nakatsu MN, Chou W, Gershon PD & Hughes CC. (2011). The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell 22, 3791–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HQ, Chattoraj S, Castillo D, Nguyen SC, Nir G, Lioutas A, Hershberg EA, Martins NMC, Reginato PL, Hannan M, Beliveau BJ, Church GM, Daugharthy ER, Marti-Renom MA & Wu CT. (2020). 3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing. Nat Methods 17, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Fogli Iseppe A, Giles WR, Narayan SM, Zhang H, Edwards AG, Morotti S & Grandi E. (2020). Populations of in silico myocytes and tissues reveal synergy of multiatrial-predominant K(+) -current block in atrial fibrillation. Br J Pharmacol 177, 4497–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Morotti S & Grandi E. (2018). A Heart for Diversity: Simulating Variability in Cardiac Arrhythmia Research. Front Physiol 9, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, Martinez-de-Mena R, Castejon-Vega B, Pun-Garcia A, Traves PG, Bonzon-Kulichenko E, Garcia-Marques F, Cusso L, N AG, Gonzalez-Guerra A, Roche-Molina M, Martin-Salamanca S, Crainiciuc G, Guzman G, Larrazabal J, Herrero-Galan E, Alegre-Cebollada J, Lemke G, Rothlin CV, Jimenez-Borreguero LJ, Reyes G, Castrillo A, Desco M, Munoz-Canoves P, Ibanez B, Torres M, Ng LG, Priori SG, Bueno H, Vazquez J, Cordero MD, Bernal JA, Enriquez JA & Hidalgo A. (2020). A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94–109 e123. [DOI] [PubMed] [Google Scholar]
- Niederer SA, Lumens J & Trayanova NA. (2019). Computational models in cardiology. Nat Rev Cardiol 16, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SH, Mouton AJ, DeLeon-Pennell KY, Genovese F, Karsdal M & Lindsey ML. (2019). Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol 75–76, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves-Cintron M, Flores-Tamez VA, Le T, Baudel MM & Navedo MF. (2021). Cellular and molecular effects of hyperglycemia on ion channels in vascular smooth muscle. Cell Mol Life Sci 78, 31–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves-Cintron M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell’Acqua ML, Scott JD & Navedo MF. (2015). Selective downregulation of Kv2.1 function contributes to enhanced arterial tone during diabetes. Journal of Biological Chemistry 290, 7918–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieves-Cintron M, Syed AU, Nystoriak MA & Navedo MF. (2018). Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir G, Farabella I, Perez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj S, Erceg J, AlHaj Abed J, Martins NMC, Nguyen HQ, Hannan MA, Russell S, Durand NC, Rao SSP, Kishi JY, Soler-Vila P, Di Pierro M, Onuchic JN, Callahan SP, Schreiner JM, Stuckey JA, Yin P, Aiden EL, Marti-Renom MA & Wu CT. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala M & Qu Z. (2012). Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol 303, H341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala M, Song Z, Weiss JN & Qu Z. (2015). T-tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol 79, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MB, Poelzing S & Weinberg SH. (2021a). Mechanisms underlying age-associated manifestation of cardiac sodium channel gain-of-function. J Mol Cell Cardiol 153, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MB, Veeraraghavan R, Poelzing S & Weinberg SH. (2021b). Cellular Size, Gap Junctions, and Sodium Channel Properties Govern Developmental Changes in Cardiac Conduction. Front Physiol 12, 731025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, Aganezov S, Hoyt SJ, Diekhans M, Logsdon GA, Alonge M, Antonarakis SE, Borchers M, Bouffard GG, Brooks SY, Caldas GV, Chen NC, Cheng H, Chin CS, Chow W, de Lima LG, Dishuck PC, Durbin R, Dvorkina T, Fiddes IT, Formenti G, Fulton RS, Fungtammasan A, Garrison E, Grady PGS, Graves-Lindsay TA, Hall IM, Hansen NF, Hartley GA, Haukness M, Howe K, Hunkapiller MW, Jain C, Jain M, Jarvis ED, Kerpedjiev P, Kirsche M, Kolmogorov M, Korlach J, Kremitzki M, Li H, Maduro VV, Marschall T, McCartney AM, McDaniel J, Miller DE, Mullikin JC, Myers EW, Olson ND, Paten B, Peluso P, Pevzner PA, Porubsky D, Potapova T, Rogaev EI, Rosenfeld JA, Salzberg SL, Schneider VA, Sedlazeck FJ, Shafin K, Shew CJ, Shumate A, Sims Y, Smit AFA, Soto DC, Sovic I, Storer JM, Streets A, Sullivan BA, Thibaud-Nissen F, Torrance J, Wagner J, Walenz BP, Wenger A, Wood JMD, Xiao C, Yan SM, Young AC, Zarate S, Surti U, McCoy RC, Dennis MY, Alexandrov IA, Gerton JL, O’Neill RJ, Timp W, Zook JM, Schatz MC, Eichler EE, Miga KH & Phillippy AM. (2022). The complete sequence of a human genome. Science 376, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell’acqua ML, Scott JD, Santana LF & Navedo MF. (2014). AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res 114, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, Sasse KC, Scott JD, Ward SM, Hell JW & Navedo MF. (2017). Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara RP, Binka E, Prakosa A, Zimmerman SL, Cartoski MJ, Abraham MR, Lu DY, Boyle PM & Trayanova NA. (2022). Personalized computational heart models with T1-mapped fibrotic remodeling predict sudden death risk in patients with hypertrophic cardiomyopathy. Elife 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanyan V, Raph SM, Dwenger MM, Hu X, Pucci T, Mack G, Moore JBt, Chilian WM, Bhatnagar A & Nystoriak MA. (2021). Myocardial Blood Flow Control by Oxygen Sensing Vascular Kvbeta Proteins. Circ Res 128, 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaghi A, Carrazana G, Caruso C, Abbas A, Myers DR, Lam WA & Robles FE. (2020). Label-free hematology analysis using deep-ultraviolet microscopy. Proc Natl Acad Sci U S A 117, 14779–14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongstad E & Kohl P. (2016). Fibroblast-myocyte coupling in the heart: Potential relevance for therapeutic interventions. J Mol Cell Cardiol 91, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz CA, Leake MC, Makarenko I, Benes V & Linke WA. (2004). Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res 94, 967–975. [DOI] [PubMed] [Google Scholar]
- Parekh AB. (2016). Advances in intracellular Ca(2+) signalling. J Physiol 594, 2811–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE & Knollmann BC. (2017). Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res 121, 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini E, Britton OJ, Lu HR, Rohrbacher J, Hermans AN, Gallacher DJ, Greig RJH, Bueno-Orovio A & Rodriguez B. (2017). Human In Silico Drug Trials Demonstrate Higher Accuracy than Animal Models in Predicting Clinical Pro-Arrhythmic Cardiotoxicity. Front Physiol 8, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M & Prabhu SD. (2018). CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci 3, 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathmanathan P & Gray RA. (2018). Validation and Trustworthiness of Multiscale Models of Cardiac Electrophysiology. Front Physiol 9, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellman J, Zhang J & Sheikh F. (2016). Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J Mol Cell Cardiol 94, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH & Green CR. (1994). Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation 90, 713–725. [DOI] [PubMed] [Google Scholar]
- Pianca N, Di Bona A, Lazzeri E, Costantini I, Franzoso M, Prando V, Armani A, Rizzo S, Fedrigo M, Angelini A, Basso C, Pavone FS, Rubart M, Sacconi L, Zaglia T & Mongillo M. (2019). Cardiac sympathetic innervation network shapes the myocardium by locally controlling cardiomyocyte size through the cellular proteolytic machinery. J Physiol 597, 3639–3656. [DOI] [PubMed] [Google Scholar]
- Pilz PM, Ward JE, Chang WT, Kiss A, Bateh E, Jha A, Fisch S, Podesser BK & Liao R. (2022). Large and Small Animal Models of Heart Failure With Reduced Ejection Fraction. Circ Res 130, 1888–1905. [DOI] [PubMed] [Google Scholar]
- Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA & Tallquist MD. (2016). Revisiting Cardiac Cellular Composition. Circ Res 118, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello JP, Bick A, Wang M, Chaffin M, Friedman S, Yao J, Guo X, Venkatesh BA, Taylor KD, Post WS, Rich S, Lima JAC, Rotter JI, Philippakis A, Lubitz SA, Ellinor PT, Khera AV, Kathiresan S & Aragam KG. (2020). Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat Commun 11, 2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello JP, Di Achille P, Nauffal V, Nekoui M, Friedman SF, Klarqvist MDR, Chaffin MD, Weng LC, Cunningham JW, Khurshid S, Roselli C, Lin H, Koyama S, Ito K, Kamatani Y, Komuro I, BioBank Japan P, Jurgens SJ, Benjamin EJ, Batra P, Natarajan P, Ng K, Hoffmann U, Lubitz SA, Ho JE, Lindsay ME, Philippakis AA & Ellinor PT. (2022). Genetic analysis of right heart structure and function in 40,000 people. Nat Genet 54, 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskadlo E, Eichenberger BT, Giorgetti L & Chao JA. (2022). Design, Labeling, and Application of Probes for RNA smFISH. Methods Mol Biol 2537, 173–183. [DOI] [PubMed] [Google Scholar]
- Poelzing S, Weinberg SH & Keener JP. (2021). Initiation and entrainment of multicellular automaticity via diffusion limited extracellular domains. Biophys J 120, 5279–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjoismaki JL, Kruger M, Al-Furoukh N, Lagerstedt A, Karhunen PJ & Braun T. (2013). Postnatal cardiomyocyte growth and mitochondrial reorganization cause multiple changes in the proteome of human cardiomyocytes. Mol Biosyst 9, 1210–1219. [DOI] [PubMed] [Google Scholar]
- Ponce-Balbuena D, Guerrero-Serna G, Valdivia CR, Caballero R, Diez-Guerra FJ, Jimenez-Vazquez EN, Ramirez RJ, Monteiro da Rocha A, Herron TJ, Campbell KF, Willis BC, Alvarado FJ, Zarzoso M, Kaur K, Perez-Hernandez M, Matamoros M, Valdivia HH, Delpon E & Jalife J. (2018). Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ Res 122, 1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu SD & Frangogiannis NG. (2016). The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119, 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, Simo S, Sato D, Sasse KC, Ward SM, Santana LF, Xiang YK, Hell JW, Nieves-Cintron M & Navedo MF. (2019). A Gs-coupled purinergic receptor boosts Ca(2+) influx and vascular contractility during diabetic hyperglycemia. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada MP, Syed AU, Reddy GR, Martin-Aragon Baudel M, Flores-Tamez VA, Sasse KC, Ward SM, Sirish P, Chiamvimonvat N, Bartels P, Dickson EJ, Hell JW, Scott JD, Santana LF, Xiang YK, Navedo MF & Nieves-Cintron M. (2020). AKAP5 complex facilitates purinergic modulation of vascular L-type Ca(2+) channel CaV1.2. Nat Commun 11, 5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prando V, Da Broi F, Franzoso M, Plazzo AP, Pianca N, Francolini M, Basso C, Kay MW, Zaglia T & Mongillo M. (2018). Dynamics of neuroeffector coupling at cardiac sympathetic synapses. J Physiol 596, 2055–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard HAT, Pires PW, Yamasaki E, Thakore P & Earley S. (2018). Nanoscale remodeling of ryanodine receptor cluster size underlies cerebral microvascular dysfunction in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 115, E9745–E9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prive F (2022). Using the UK Biobank as a global reference of worldwide populations: application to measuring ancestry diversity from GWAS summary statistics. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI & Sadek HA. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Nivala M & Weiss JN. (2013). Calcium alternans in cardiac myocytes: order from disorder. J Mol Cell Cardiol 58, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwanski PB, Johnson CN, Gyorke S & Veeraraghavan R. (2018). Cardiac Arrhythmias as Manifestations of Nanopathies: An Emerging View. Front Physiol 9, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan AM, Ma RC, Kocha KM, Zhang DJ & Huang P. (2020). Dual function of perivascular fibroblasts in vascular stabilization in zebrafish. PLoS Genet 16, e1008800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees CM, Yang JH, Santolini M, Lusis AJ, Weiss JN & Karma A. (2018). The Ca(2+) transient as a feedback sensor controlling cardiomyocyte ionic conductances in mouse populations. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichart D, Lindberg EL, Maatz H, Miranda AMA, Viveiros A, Shvetsov N, Gartner A, Nadelmann ER, Lee M, Kanemaru K, Ruiz-Orera J, Strohmenger V, DeLaughter DM, Patone G, Zhang H, Woehler A, Lippert C, Kim Y, Adami E, Gorham JM, Barnett SN, Brown K, Buchan RJ, Chowdhury RA, Constantinou C, Cranley J, Felkin LE, Fox H, Ghauri A, Gummert J, Kanda M, Li R, Mach L, McDonough B, Samari S, Shahriaran F, Yapp C, Stanasiuk C, Theotokis PI, Theis FJ, van den Bogaerdt A, Wakimoto H, Ware JS, Worth CL, Barton PJR, Lee YA, Teichmann SA, Milting H, Noseda M, Oudit GY, Heinig M, Seidman JG, Hubner N & Seidman CE. (2022). Pathogenic variants damage cell composition and single cell transcription in cardiomyopathies. Science 377, eabo1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo JG, Weiss JN & Karma A. (2008). Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J 95, 3767–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle C & Bauersachs J. (2019). Small animal models of heart failure. Cardiovasc Res 115, 1838–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer S (1883). A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J Physiol 4, 29–42 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BN, Yang PC, Behrens SB, Moreno JD & Clancy CE. (2012). Computational approaches to understand cardiac electrophysiology and arrhythmias. Am J Physiol Heart Circ Physiol 303, H766–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog-Zielinska EA, Craig MA, Manning JR, Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes MC, Hardie DG, Smith GL & Chapman KE. (2015). Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC-1alpha. Cell Death Differ 22, 1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J, Hill JA, Singh A, Valero-Munoz M & Sam F. (2022). Heart Failure With Preserved Ejection Fraction: Heterogeneous Syndrome, Diverse Preclinical Models. Circ Res 130, 1906–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DF & Brown GC. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77, 731–758. [DOI] [PubMed] [Google Scholar]
- Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M & Vunjak-Novakovic G. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa-Garrido M, Chapski DJ & Vondriska TM. (2018). Epigenomes in Cardiovascular Disease. Circ Res 122, 1586–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossler K, Neuchrist C, Kitz K, Scheiner O, Kraft D & Lassmann H. (1992). Expression of leucocyte adhesion molecules at the human blood-brain barrier (BBB). J Neurosci Res 31, 365–374. [DOI] [PubMed] [Google Scholar]
- Rubenstein DS & Lipsius SL. (1989). Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res 64, 648–657. [DOI] [PubMed] [Google Scholar]
- Rudy Y & Silva JR. (2006). Computational biology in the study of cardiac ion channels and cell electrophysiology. Q Rev Biophys 39, 57–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks VA, Kaambre T, Sikk P, Eimre M, Orlova E, Paju K, Piirsoo A, Appaix F, Kay L, Regitz-Zagrosek V, Fleck E & Seppet E. (2001). Intracellular energetic units in red muscle cells. Biochem J 356, 643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saljic A, Grandi E & Dobrev D. (2022). TGF-beta1-induced endothelial-mesenchymal transition: a potential contributor to fibrotic remodeling in atrial fibrillation? J Clin Invest 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alonso JL, Bhargava A, O’Hara T, Glukhov AV, Schobesberger S, Bhogal N, Sikkel MB, Mansfield C, Korchev YE, Lyon AR, Punjabi PP, Nikolaev VO, Trayanova NA & Gorelik J. (2016). Microdomain-Specific Modulation of L-Type Calcium Channels Leads to Triggered Ventricular Arrhythmia in Heart Failure. Circ Res 119, 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alonso JL, Loucks A, Schobesberger S, van Cromvoirt AM, Poulet C, Chowdhury RA, Trayanova N & Gorelik J. (2020). Nanoscale regulation of L-type calcium channels differentiates between ischemic and dilated cardiomyopathies. EBioMedicine 57, 102845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA. (2011). Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: a computational analysis. Heart Rhythm 8, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Dixon RE, Santana LF & Navedo MF. (2018). A model for cooperative gating of L-type Ca2+ channels and its effects on cardiac alternans dynamics. PLoS Comput Biol 14, e1005906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Brunton LL, Michailova AP & McCulloch AD. (2003). Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem 278, 47997–48003. [DOI] [PubMed] [Google Scholar]
- Savla JJ, Levine BD & Sadek HA. (2018). The Effect of Hypoxia on Cardiovascular Disease: Friend or Foe? High Alt Med Biol 19, 124–130. [DOI] [PubMed] [Google Scholar]
- Savulescu AF, Brackin R, Bouilhol E, Dartigues B, Warrell JH, Pimentel MR, Beaume N, Fortunato IC, Dallongeville S, Boulle M, Soueidan H, Agou F, Schmoranzer J, Olivo-Marin JC, Franco CA, Gomes ER, Nikolski M & Mhlanga MM. (2021). Interrogating RNA and protein spatial subcellular distribution in smFISH data with DypFISH. Cell Rep Methods 1, 100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N & Frangogiannis NG. (2013). IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol 191, 4838–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiattarella GG, Alcaide P, Condorelli G, Gillette TG, Heymans S, Jones EAV, Kallikourdis M, Lichtman A, Marelli-Berg F, Shah S, Thorp EB & Hill JA. (2022). Immunometabolic Mechanisms of Heart Failure with Preserved Ejection Fraction. Nat Cardiovasc Res 1, 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Wiedmann F, Voigt N, Zhou XB, Heijman J, Lang S, Albert V, Kallenberger S, Ruhparwar A, Szabo G, Kallenbach K, Karck M, Borggrefe M, Biliczki P, Ehrlich JR, Baczko I, Lugenbiel P, Schweizer PA, Donner BC, Katus HA, Dobrev D & Thomas D. (2015). Upregulation of K(2P)3.1 K+ Current Causes Action Potential Shortening in Patients With Chronic Atrial Fibrillation. Circulation 132, 82–92. [DOI] [PubMed] [Google Scholar]
- Schobesberger S, Wright P, Tokar S, Bhargava A, Mansfield C, Glukhov AV, Poulet C, Buzuk A, Monszpart A, Sikkel M, Harding SE, Nikolaev VO, Lyon AR & Gorelik J. (2017). T-tubule remodelling disturbs localized beta2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc Res 113, 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki S, Nagashima M, Yamada Y, Tsutsuura M, Kobayashi T, Namiki A & Tohse N. (2003). Fetal and postnatal development of Ca2+ transients and Ca2+ sparks in rat cardiomyocytes. Cardiovasc Res 58, 535–548. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Wang F, Puglisi J, Weber C & Bers DM. (2004). A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J 87, 3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheard TMD, Hurley ME, Colyer J, White E, Norman R, Pervolaraki E, Narayanasamy KK, Hou Y, Kirton HM, Yang Z, Hunter L, Shim JU, Clowsley AH, Smith AJ, Baddeley D, Soeller C, Colman MA & Jayasinghe I. (2019). Three-Dimensional and Chemical Mapping of Intracellular Signaling Nanodomains in Health and Disease with Enhanced Expansion Microscopy. ACS Nano 13, 2143–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, van den Brink J, Bergan-Dahl A, Kolstad TR, Norden ES, Hou Y, Laasmaa M, Aguilar-Sanchez Y, Quick AP, Espe EKS, Sjaastad I, Wehrens XHT, Edwards AG, Soeller C & Louch WE. (2022). Prolonged beta-adrenergic stimulation disperses ryanodine receptor clusters in cardiomyocytes and has implications for heart failure. Elife 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiferaw Y, Aistrup GL, Louch WE & Wasserstrom JA. (2020). Remodeling Promotes Proarrhythmic Disruption of Calcium Homeostasis in Failing Atrial Myocytes. Biophys J 118, 476–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiferaw Y, Aistrup GL & Wasserstrom JA. (2018). Synchronization of Triggered Waves in Atrial Tissue. Biophys J 115, 1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T, Hilhorst M, Harrison DG, Goronzy JJ & Weyand CM. (2015). Macrophages in vascular inflammation--From atherosclerosis to vasculitis. Autoimmunity 48, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JK, Badrov MB, Al-Khazraji BK & Jackson DN. (2015). Neural Control of Vascular Function in Skeletal Muscle. Compr Physiol 6, 303–329. [DOI] [PubMed] [Google Scholar]
- Silva HS, Kapela A & Tsoukias NM. (2007). A mathematical model of plasma membrane electrophysiology and calcium dynamics in vascular endothelial cells. American journal of physiology Cell physiology 293, C277–293. [DOI] [PubMed] [Google Scholar]
- Smirnov D, Pikunov A, Syunyaev R, Deviatiiarov R, Gusev O, Aras K, Gams A, Koppel A & Efimov IR. (2020). Genetic algorithm-based personalized models of human cardiac action potential. PLoS One 15, e0231695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ & Jafri MS. (2002). Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J 83, 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis AR & Saucerman JJ. (2010). Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J 99, 2038–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Liu MB & Qu Z. (2018). Transverse tubular network structures in the genesis of intracellular calcium alternans and triggered activity in cardiac cells. J Mol Cell Cardiol 114, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa MH & Field LJ. (1998). Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res 83, 15–26. [DOI] [PubMed] [Google Scholar]
- Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, Nakayama T, Gyorke S, Cardounel AJ & Carnes CA. (2009). Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res 84, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapor PC, Sweat RS, Dashti DC, Betancourt AM & Murfee WL. (2014). Pericyte dynamics during angiogenesis: new insights from new identities. J Vasc Res 51, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR & Frid MG. (2013). The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 75, 23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Cantrell AC, Zeng H, Zhu SH & Chen JX. (2021). Emerging Role of Pericytes and Their Secretome in the Heart. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Timofeyev V, Dennis A, Bektik E, Wan X, Laurita KR, Deschenes I, Li RA & Fu JD. (2017). A Singular Role of IK1 Promoting the Development of Cardiac Automaticity during Cardiomyocyte Differentiation by IK1 -Induced Activation of Pacemaker Current. Stem Cell Rev Rep 13, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift LM, Burke M, Guerrelli D, Reilly M, Ramadan M, McCullough D, Prudencio T, Mulvany C, Chaluvadi A, Jaimes R & Posnack NG. (2020). Age-dependent changes in electrophysiology and calcium handling: implications for pediatric cardiac research. Am J Physiol Heart Circ Physiol 318, H354–H365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK & Nahrendorf M. (2018). Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol 18, 733–744. [DOI] [PubMed] [Google Scholar]
- Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, Grandi E, Sirish P, Chiamvimonvat N, Hell JW, Santana LF, Xiang YK, Nieves-Cintron M & Navedo MF. (2019). Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Invest 129, 3140–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum R & Ripatti S. (2021). Integrating lipidomics and genomics: emerging tools to understand cardiovascular diseases. Cell Mol Life Sci 78, 2565–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talman V, Teppo J, Poho P, Movahedi P, Vaikkinen A, Karhu ST, Trost K, Suvitaival T, Heikkonen J, Pahikkala T, Kotiaho T, Kostiainen R, Varjosalo M & Ruskoaho H. (2018). Molecular Atlas of Postnatal Mouse Heart Development. J Am Heart Assoc 7, e010378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CMJ, Green P, Tapoulal N, Lewandowski AJ, Leeson P & Herring N. (2018). The Role of Neuropeptide Y in Cardiovascular Health and Disease. Front Physiol 9, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomek J, Bueno-Orovio A, Passini E, Zhou X, Minchole A, Britton O, Bartolucci C, Severi S, Shrier A, Virag L, Varro A & Rodriguez B. (2019). Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente AG, Mesirca P, Neco P, Rizzetto R, Dubel S, Barrere C, Sinegger-Brauns M, Striessnig J, Richard S, Nargeot J, Gomez AM & Mangoni ME. (2016). L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc Res 109, 451–461. [DOI] [PubMed] [Google Scholar]
- Tripp S & Grueber M. (2011). Economic Impact of the Human Genome Project.
- Trost A, Lange S, Schroedl F, Bruckner D, Motloch KA, Bogner B, Kaser-Eichberger A, Strohmaier C, Runge C, Aigner L, Rivera FJ & Reitsamer HA. (2016). Brain and Retinal Pericytes: Origin, Function and Role. Front Cell Neurosci 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumoto K, Ashihara T, Naito N, Shimamoto T, Amano A, Kurata Y & Kurachi Y. (2020). Specific decreasing of Na(+) channel expression on the lateral membrane of cardiomyocytes causes fatal arrhythmias in Brugada syndrome. Sci Rep 10, 19964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykocki NR, Boerman EM & Jackson WF. (2017). Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr Physiol 7, 485–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, January CT & Eckhardt LL. (2016). IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: an improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol 310, H1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard M, van Weerd JH, Bawazeer AC, Hooijkaas IB, van de Werken HJG, Tessadori F, de Laat W, Barnett P, Bakkers J & Christoffels VM. (2019). Identification and Characterization of a Transcribed Distal Enhancer Involved in Cardiac Kcnh2 Regulation. Cell Rep 28, 2704–2714 e2705. [DOI] [PubMed] [Google Scholar]
- van den Dries K, Fransen J & Cambi A. (2022). Fluorescence CLEM in biology: historic developments and current super-resolution applications. FEBS Lett. [DOI] [PubMed] [Google Scholar]
- Varghese A (2016). Reciprocal Modulation of I(K1)-I(Na) Extends Excitability in Cardiac Ventricular Cells. Front Physiol 7, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshneya M, Irurzun-Arana I, Campana C, Dariolli R, Gutierrez A, Pullinger TK & Sobie EA. (2021). Investigational Treatments for COVID-19 may Increase Ventricular Arrhythmia Risk Through Drug Interactions. CPT Pharmacometrics Syst Pharmacol 10, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R & Gourdie RG. (2016). Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol Biol Cell 27, 3583–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Hoeker GS, Alvarez-Laviada A, Hoagland D, Wan X, King DR, Sanchez-Alonso J, Chen C, Jourdan J, Isom LL, Deschenes I, Smyth JW, Gorelik J, Poelzing S & Gourdie RG. (2018). The adhesion function of the sodium channel beta subunit (beta1) contributes to cardiac action potential propagation. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG & Poelzing S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch 467, 2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Poelzing S & Gourdie RG. (2014a). Intercellular electrical communication in the heart: a new, active role for the intercalated disk. Cell Commun Adhes 21, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Poelzing S & Gourdie RG. (2014b). Old cogs, new tricks: a scaffolding role for connexin43 and a junctional role for sodium channels? FEBS Lett 588, 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayutham N, Alfieri CM, Agnew EJ, Riggs KW, Baker RS, Ponny SR, Zafar F & Yutzey KE. (2020). Cardiomyocyte cell cycling, maturation, and growth by multinucleation in postnatal swine. J Mol Cell Cardiol 146, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Deslattes Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A & Zhu X. (2001). The sequence of the human genome. Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Vermij SH, Abriel H & van Veen TA. (2017). Refining the molecular organization of the cardiac intercalated disc. Cardiovasc Res 113, 259–275. [DOI] [PubMed] [Google Scholar]
- Vicar T, Balvan J, Jaros J, Jug F, Kolar R, Masarik M & Gumulec J. (2019). Cell segmentation methods for label-free contrast microscopy: review and comprehensive comparison. BMC Bioinformatics 20, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova TM & Lakatta EG. (2009). Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol 47, 456–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreeker A, van Stuijvenberg L, Hund TJ, Mohler PJ, Nikkels PG & van Veen TA. (2014). Assembly of the cardiac intercalated disk during pre- and postnatal development of the human heart. PLoS One 9, e94722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BA, Venkataraman S & Buettner GR. (2011). The rate of oxygen utilization by cells. Free Radic Biol Med 51, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahler GM. (1992). Developmental increases in the inwardly rectifying potassium current of rat ventricular myocytes. Am J Physiol 262, C1266–1272. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA & Tibbs GR. (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810. [DOI] [PubMed] [Google Scholar]
- Wang HJ, Wang W, Cornish KG, Rozanski GJ & Zucker IH. (2014). Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 64, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng ZP, Kondo CS, Sheldon RS & Duff HJ. (1996). Developmental changes in the delayed rectifier K+ channels in mouse heart. Circ Res 79, 79–85. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pan W, Bai X, Wang X, Wang Y & Yin Y. (2021). microRNA-454-mediated NEDD4–2/TrkA/cAMP axis in heart failure: Mechanisms and cardioprotective implications. J Cell Mol Med 25, 5082–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yao F, Wang L, Li Z, Ren Z, Li D, Zhang M, Han L, Wang SQ, Zhou B & Wang L. (2020). Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat Commun 11, 2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M & Snyder M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassie AT, Zhao Y & Boyden ES. (2019). Expansion microscopy: principles and uses in biological research. Nat Methods 16, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Leutenegger M, Stoldt S, Jakobs S, Mihaila TS, Butkevich AN & Hell SW. (2021). MINSTED fluorescence localization and nanoscopy. Nat Photonics 15, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SH. (2017). Ephaptic coupling rescues conduction failure in weakly coupled cardiac tissue with voltage-gated gap junctions. Chaos 27, 093908. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y & Lusis AJ. (2012). “Good enough solutions” and the genetics of complex diseases. Circ Res 111, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DG, Morielli AD, Nelson MT & Brayden JE. (2002). Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90, 248–250. [DOI] [PubMed] [Google Scholar]
- Westcott EB & Segal SS. (2013). Perivascular innervation: a multiplicity of roles in vasomotor control and myoendothelial signaling. Microcirculation 20, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff M & Dixon RE. (2021). Mechanisms and Regulation of Cardiac CaV1.2 Trafficking. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel GT, Chen F & Klitzner TS. (1995). Na+/Ca2+ exchange and cell contraction in isolated neonatal and adult rabbit cardiac myocytes. Am J Physiol 268, H1723–1733. [DOI] [PubMed] [Google Scholar]
- Whittaker DG, Clerx M, Lei CL, Christini DJ & Mirams GR. (2020). Calibration of ionic and cellular cardiac electrophysiology models. Wiley Interdiscip Rev Syst Biol Med 12, e1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow RL, Walker MA & Greenstein JL. (2016). Modeling calcium regulation of contraction, energetics, signaling, and transcription in the cardiac myocyte. Wiley Interdiscip Rev Syst Biol Med 8, 37–67. [DOI] [PubMed] [Google Scholar]
- Withaar C, Lam CSP, Schiattarella GG, de Boer RA & Meems LMG. (2021). Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J 42, 4420–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AJ, Kane KA & Rankin AC. (2001). The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res 52, 226–235. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yang Y, Galice S, Bers DM & Sato D. (2019). Size Matters: Ryanodine Receptor Cluster Size Heterogeneity Potentiates Calcium Waves. Biophys J 116, 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Parks X, Qudsi H, Braun C & Lopes CMB. (2020). The auxiliary subunit KCNE1 regulates KCNQ1 channel response to sustained calcium-dependent PKC activation. PLoS One 15, e0237591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Deschenes I & Fu JD. (2022). Multilayer control of cardiac electrophysiology by microRNAs. J Mol Cell Cardiol 166, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL & Nerbonne JM. (2014a). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 129, 1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC, Perissinotti LL, Lopez-Redondo F, Wang Y, DeMarco KR, Jeng MT, Vorobyov I, Harvey RD, Kurokawa J, Noskov SY & Clancy CE. (2017). A multiscale computational modelling approach predicts mechanisms of female sex risk in the setting of arousal-induced arrhythmias. J Physiol 595, 4695–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, Sniadecki NJ, Ruohola-Baker H & Murry CE. (2014b). Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol 72, 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC Jr., Pabon L, Reinecke H, Sniadecki NJ, Tian R, Ruohola-Baker H, Xu H & Murry CE. (2019). Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Reports 13, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaniv Y, Stern MD, Lakatta EG & Maltsev VA. (2013). Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca(2)(+) cycling dynamics. Biophys J 105, 1551–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Zahradnikova A Jr., Rizzetto R, Boncompagni S, Rabesahala de Meritens C, Zhang Y, Joanne P, Marques-Sule E, Aguilar-Sanchez Y, Fernandez-Tenorio M, Villejoubert O, Li L, Wang YY, Mateo P, Nicolas V, Gerbaud P, Lai FA, Perrier R, Alvarez JL, Niggli E, Valdivia HH, Valdivia CR, Ramos-Franco J, Zorio E, Zissimopoulos S, Protasi F, Benitah JP & Gomez AM. (2021). Impaired Binding to Junctophilin-2 and Nanostructural Alteration in CPVT Mutation. Circ Res 129, e35–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JK, Liang JA, Weinberg SH & Trayanova NA. (2022). Computational modeling of aberrant electrical activity following remuscularization with intramyocardially injected pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 162, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler AC, Chandrabhatla AS, Christiansen SL, Nelson AR, Holmes JW & Saucerman JJ. (2021). Network model-based screen for FDA-approved drugs affecting cardiac fibrosis. CPT Pharmacometrics Syst Pharmacol 10, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler AC, Richardson WJ, Holmes JW & Saucerman JJ. (2016). Computational modeling of cardiac fibroblasts and fibrosis. J Mol Cell Cardiol 93, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ni H, Morotti S, Smith CER, Sato D, Louch WE, Edwards AG & Grandi E. (2022a). Mechanisms of spontaneous Ca(2+) release-mediated arrhythmia in a novel 3D human atrial myocyte model: I. Transverse-axial tubule variation. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Smith CER, Morotti S, Edwards AG, Sato D, Louch WE, Ni H & Grandi E. (2022b). Mechanisms of spontaneous Ca(2+) release-mediated arrhythmia in a novel 3D human atrial myocyte model: II Ca(2+) -handling protein variation. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS & Chiamvimonvat N. (2002). Functional Roles of Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res 90, 981–987. [DOI] [PubMed] [Google Scholar]
- Zhao G, Joca HC, Nelson MT & Lederer WJ. (2020). ATP- and voltage-dependent electro-metabolic signaling regulates blood flow in heart. Proc Natl Acad Sci U S A 117, 7461–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XJ, Zhu C, Tian LY, Fu YC, Zhang Y, Chen X, Huang Y & Li Y. (2017). Kv4.3 Modulates the Distribution of hERG. Sci Rep 7, 17757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Singh N, Monnier C, Marszalec W, Gao L, Jin J, Frisk M, Louch WE, Verma S, Krishnamurthy P, Nico E, Mulla M, Aistrup GL, Kishore R & Wasserstrom JA. (2021). Phosphatidylinositol-4,5-Bisphosphate Binding to Amphiphysin-II Modulates T-Tubule Remodeling: Implications for Heart Failure. Front Physiol 12, 782767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman AP, Gomez-Viquez NL, Bloch RJ & Lederer WJ. (2010). Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol 48, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, Heubach JF, Kostin S, Neuhuber WL & Eschenhagen T. (2002). Tissue engineering of a differentiated cardiac muscle construct. Circ Res 90, 223–230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.