

Abstract
Deficiency in the Conserved Oligomeric Golgi (COG) complex that orchestrates SNARE-mediated tethering/fusion of vesicles that recycle the Golgi’s glycosylation machinery results in severe glycosylation defects. Although two major Golgi v-SNAREs, GS28/GOSR1, and GS15/BET1L, are depleted in COG-deficient cells, the complete knockout of GS28 and GS15 only modestly affects Golgi glycosylation, indicating the existence of an adaptation mechanism in Golgi SNARE. Indeed, quantitative mass-spectrometry analysis of STX5-interacting proteins revealed two novel Golgi SNARE complexes - STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6. These complexes are present in wild-type cells, but their usage is significantly increased in both GS28- and COG-deficient cells. Upon GS28 deletion, SNAP29 increased its Golgi residency in a STX5-dependent manner. While STX5 depletion and Retro2-induced diversion from the Golgi severely affect protein glycosylation, GS28/SNAP29 and GS28/VTI1B double knockouts alter glycosylation similarly to GS28 KO, indicating that a single STX5-based SNARE complex is sufficient to support Golgi glycosylation. Importantly, co-depletion of three Golgi SNARE complexes in GS28/SNAP29/VTI1B TKO cells resulted in severe glycosylation defects and a reduced capacity for glycosylation enzyme retention at the Golgi. This study demonstrates the remarkable plasticity in SXT5-mediated membrane trafficking, uncovering a novel adaptive response to the failure of canonical intra-Golgi vesicle tethering/fusion machinery.
Graphical Abstract
Introduction
The Golgi is the central highly dynamic organelle in the secretory pathway that continuously receives cargo molecules (proteins and lipids) from the endoplasmic reticulum and the endosomal system. Golgi integrity and compartmental identity are essential for intracellular membrane transport. According to the cisternal maturation model for Golgi trafficking, cargo delivered to cis-Golgi, is carried forward by gradual maturation of the cis-Golgi to trans-Golgi network (TGN), without leaving the Golgi (1, 2). As the cisterna matures, its resident proteins and enzymes must recycle back by vesicular retrograde trafficking. The Golgi’s trafficking machinery includes small GTPases, coat proteins, cargo adaptors, tethers, and SNAREs. The COG (Conserved Oligomeric Golgi), the major multi-subunit tethering complex (MTC) at the Golgi. Its 8 subunits interact with members from all classes of the Golgi’s trafficking machinery and play a critical role in intra-Golgi trafficking (3-5). The COG complex and Golgi SNAREs drive tethering, docking and fusion of vesicles recycling glycosylation enzymes at the Golgi cisternae (4). Human mutations in COG subunits and Golgi t-SNARE STX5 disturb Golgi integrity and physiology resulting in severe glycosylation defects (6, 7).
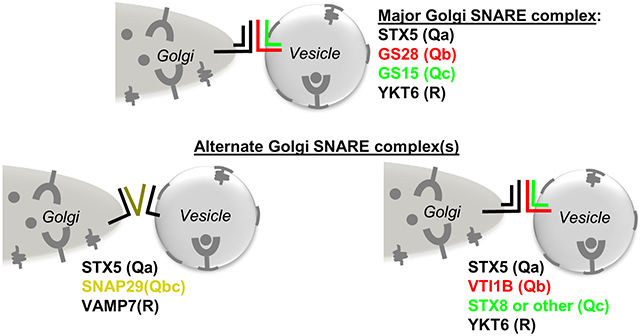
The pairing and formation of a SNARE complex is highly specific. A fusogenic SNARE complex consists of 4 SNARE domains classified as Qa, Qb, Qc, and R present on the target (t-SNARE) or vesicle (v-SNARE) membrane (for review see (8)). The major SNARE complex that controls intra-Golgi trafficking is STX5/GS28/GS15/YKT6 (Figure 1A) (9), while the STX5/GS27/BET1/SEC22B operates in ER-Golgi anterograde cargo delivery (10) and STX16/VTI1A/STX6/VAMP4 or STX16/VTI1A/STX10/VAMP3 complexes drive fusion of endosome-derived vesicles with TGN (11, 12).
Figure 1: HEK293T cells lacking SNAREs GS28 and GS15 are viable and maintain normal Golgi morphology.

(A) Cartoon depicting the role of canonical Golgi SNAREs in vesicle fusion. (B) WB analysis of GS28 KO and GS15 KO in HEK293T-Cas9 cell line. (C, D) Airyscan images of WT and GS28 KO (C) or GS15 KO (D) cells stained for GS28 and GS15, respectively, show complete loss of Golgi localized GS28 or GS15 in the KO cell lines. The Golgi is stained with Giantin (B) or GM130 (C). Scale bars are 5 μm
COG-SNARE interactions have been well established by several in vivo and in vitro studies (13-18). The COG complex is predicted to tether COPI-formed intra-Golgi carriers (4, 19). Both GS28 and GS15 are present in COPI vesicles (9, 20, 21). COG-subunit deletions/mutations affect the steady-state levels of GS28 and GS15. Furthermore, depletion of COG3 by siRNA-mediated knockdown (KD) as well as acute depletion of COG4 results in the accumulation of small trafficking intermediates termed COG-complex dependent (CCD) vesicles which contain GS28 and GS15 (14, 22), indicating that both molecules act as v-SNAREs to form a SNARE complex with STX5 and YKT6 during vesicle fusion at the Golgi. Importantly, the abundance of STX5 and YKT6 is not regulated by the COG complex (23), and these SNAREs are not accumulated on CCD vesicles (22), indicating their role as t-SNAREs in the intra-Golgi trafficking. The COG complex not only maintains the steady-state abundance and Golgi localization GS28 and GS15 but is also required for SNARE complex formation. Upon COG3 or COG7 KD in HeLa cells, the amount and/or stability of STX5 complex containing GS28 and GS15 is dramatically reduced (24). COG4 physically interacts with STX5 and STX5’s partner hSLY1/SCFD1 via its N-terminal domain (15). Over-expression of a COG4 N-terminal 153aa-long fragment as well as expression of E53/71A COG4 mutant disrupts the Golgi morphology, mislocalizes GS15, and dramatically decreases GS28/STX5 interaction (15, 25).
While the exact temporal events in COG and SNARE-driven vesicle tethering and fusion at the Golgi is yet to be fully understood, the evidence so far indicates that the COG complex orchestrates intra-Golgi trafficking by capturing vesicles and modulating/proof-reading SNARE alignment that precedes fusion (5, 22, 25). Therefore, we hypothesized that impairment in Golgi SNARE interactions due to COG malfunction is the major contributor to trafficking and glycosylation defects seen in COG mutants. Indeed, GS28 along with COG subunits are frequently identified as hits in CRISPR screens for Golgi trafficking and glycosylation players (26-30). We reasoned that knocking out either GS28 or GS15 would phenocopy COG KD/KO glycosylation defects because Golgi enzymes will be trapped in transport vesicles unable to fuse with the Golgi and will subsequently be degraded in GS28/GS15-depleted cells. In this study, we successfully knocked out GS28 and GS15 in HEK293T cells and characterized trafficking and glycosylation defects associated with the deletion of Golgi v-SNAREs. Surprisingly, we discovered that both GS28 and GS15 are mostly dispensable for Golgi glycosylation. To accommodate retrograde trafficking of Golgi enzymes, mutant cells compensate for the absence of Golgi v-SNAREs by increased utilization of auxiliary STX5-containing SNARE complexes formed by post-Golgi SNAREs SNAP29, VAMP7 and VTI1B.
Results
HEK293T cells lacking SNAREs GS28 and GS15 are viable and maintain normal Golgi morphology.
Using a CRISPR-Cas9 approach, we successfully knocked out (KO) GS28/GOSR1, the Golgi Qb SNARE and GS15/BET1L, the Qc SNARE from the STX5/GS28/GS15/YKT6 SNARE complex. Individual HEK293T clones lacking the targeted SNAREs were selected by western blotting (WB) (Figure 1B). GS28 and GS15 are Golgi localized SNARES and immunofluorescence (IF) was used for further confirmation of KO (Figure 1C, D). The resultant GS28 and GS15 KO clones were viable and did not show either altered cell morphology or growth delays (Z.D., unpublished observation). Giantin/GOLGB1 and GM130/GOLGA2 staining showed that the cis/medial-Golgi morphology was unaffected by the deletion of either GS28 or GS15. The Golgi continuity and size in the SNARE KO cells appeared like WT Golgi. Sanger sequencing of genomic DNA from GS28 KO and GS15 KO cells further validated deletion of target proteins. In GS28 KO cells a single nucleotide insertion in exon 8 altered the amino acid sequence after Gln194 leading to disruption of the SNARE domain (Figure S1A). Since antibodies to GS28 were developed against amino acids 3-108 lack of GS28 detection on WB indicates that the altered protein was degraded. GS15 KO cells are heterozygotes with a 5-nucleodie deletion in exon 3 of one allele and a single nucleotide insertion in exon 3 (Figure S1B). The deletion introduces a premature stop codon producing a 40 amino acid peptide and the single nucleotide insertion results in an altered amino acid sequence after Leu40. Since antibodies to GS15 were raised against the first 93 amino acids of the protein lack of WB signal indicates complete degradation of altered protein. We thus obtained HEK293T clones deficient for GS28 or GS15 and evaluated the effect of their deletion on Golgi physiology. STX5 is an essential protein, and STX5/Sed5p knockout is lethal for mammalian cells (31) and yeast (32). Of note, we also tried to obtain YKT6 KO cells but failed to recover any viable YKT6 KO colonies in HEK293T cells, confirming the essential role of YKT6 in multiple intracellular fusion events (33).
GS28 KO alters the stability and localization of GS15 but does not affect STX5 and YKT6.
Thereon, we measured the protein levels of GS28/GS15 SNARE partners using WB (Figure 2A, B). In GS28 and GS15 KO cells, the total protein levels of STX5 and YKT6 were unaffected. GS15 deletion did not affect the abundance of GS28. However, upon GS28 deletion, the level of GS15 was significantly decreased (Figure 2A, B). STX5 is expressed as long (STX5 L) and short (STX5 S) isoforms. STX5 S interacts with Golgi SNAREs GS28 and GS15 (34). GS15’s interaction with its SNARE partners STX5 S and YKT6 was also reduced in GS28 KO cells (Figure S1C, D). IF also showed that the signal intensity of Golgi localized GS15 was significantly reduced in GS28 KO cells, but GS15 KO did not affect the Golgi localization of GS28 (Figure 2C, D). We have also created a GS28/GS15 double KO (DKO) HEK293T cells line, and for all tested parameters, DKO behaved identically to GS28 KO (Z.D., unpublished observation). Comparing COG KO effects on the STX5 SNARE partners, GS28 KO partially phenocopied the COG KO. In other words, GS28 and GS15 were significantly depleted in COG KOs similar to the GS28 KO phenotype. We then sought to address whether deleting SNAREs in the Golgi’s core fusion machinery will phenocopy any of the effects of COG malfunction.
Figure 2: GS28 KO alters the stability and localization of GS15 but does not affect STX5 and YKT6.

(A) WB of Golgi SNAREs in WT, GS28 KO, GS15 KO and COG4 KO cells. (B) Quantification of fold changes in the total levels of tested SNAREs. GS15 is significantly depleted in GS28 KO cell lysates. In COG4 KO cell lysates, both GS28 and GS15 are significantly depleted. The amount of STX5 and YKT6 is unaffected by either SNARE or COG KO. (C) Airyscan images of STX5, GS28, GS15 and YKT6 in WT, GS28 KO and GS15 KO cell lines show all remaining STX5 partners are mostly Golgi localized in KO cells. The Golgi is stained with GM130. Scale bars are 5 μm. (D) The total intensity of Golgi localized GS15 is significantly reduced in GS28 KO cells but GS15 KO does not have any effect on GS28’s intensity at the Golgi. Quantification of GS15’s and GS28’s signal intensities in the Golgi region in GS28 KO and GS15 KO cells, respectively. n≥30, *p<0.05, **p<0.001, ***p<0.001
Knocking out GS28 impairs retrograde trafficking.
Subtilase cytotoxin (SubAB), an AB5 toxin exploits the cell’s retrograde trafficking pathway. It gets transported from the plasma membrane to the ER via the Golgi where it cleaves an ER-resident protein, GRP78/BiP. Cells with defects in the retrograde trafficking machinery are protected from BiP cleavage by SubAB (35-37). We used GRP78 cleavage by SubAB as a readout for Golgi retrograde trafficking in SNARE-depleted cells. In WT cells, GRP78 was completely cleaved 180 min after the exposure toxin exposure (Figure 3A, B). A similar dynamic of GRP78 cleavage was observed in GS15 KO cells, indicating that this SNARE is disposable for SubAB trafficking. However, in GS28 KO cells, only 50% of GRP78 was cleaved, similar to incomplete GRP78 cleavage observed in COG4 KO cells (Figure 3A, B) (35). Altered SubAB trafficking may also indicate changes in Golgi integrity. As we have shown previously (Figure 1C, D; figure 2C, D), GS28 and GS15 KO did not change the integrity of the cis-Golgi compartment (Figure 1C, D). To investigate whether there is any alteration in the overall Golgi morphology, we stained cells for cis (GM130) and trans (TGN46/TGOLN2) Golgi markers and measured their colocalization using super-resolution Airyscan microscopy. We found a significant reduction in the colocalization between GM130 and TGN46 in the GS28 KO compared to WT cells. Furthermore, there was a reduced intensity of TGN46 staining in the GS28 KO compared to WT cells. In GS15 KO cells GM130-TGN46 colocalization was not significantly reduced, while in COG4 KO cells relative cis-trans Golgi colocalization was slightly increased, probably due to a severe fragmentation and intermixing of both compartments (Figure 3C, D) (35). Since GS28 KO phenocopied the COG KO retrograde trafficking defects but had a rather intact Golgi morphology, we wondered whether Golgi glycosylation would also be affected in SNARE-deficient cells.
Figure 3: GS28 KO phenocopies COG4 KO retrograde trafficking defects. GS28 KO alters endosome-to-Golgi delivery of SubAB toxin and TGN46.

(A) WB showing GRP78 levels in lysates from WT, GS28 KO, GS15 KO and COG4 KO cells treated with SubAB toxin for indicated time periods. (B) Quantification from GRP78 levels (n=3). After 180 mins of treatment with SubAB, 100% GRP78 is cleaved in WT and GS15 KO cells. However, in this time, only 50% is cleaved in GS28 KO and COG4 KO cells. (C) Airyscan images of the cis and trans Golgi compartments GM130 and TGN46 in SNARE-depleted cells show that TGN46 staining intensity in GS28 KO cells is reduced. (D) Compared to WT, there is a significant decrease in the colocalization (Pearson’s colocalization coefficient) between GM130 and TGN46 in GS28 KO cells **p<0.005, n≥30
GS28 is partially needed for proper Golgi glycosylation.
Glycosylation is a template-independent process which is entirely dependent on the sequence of enzyme-substrate interactions (6). Anterograde cargo molecules delivered from the ER traverse the Golgi by cisternal maturation, and encounter glycosylation enzymes localized in each of the Golgi cisternae. The Golgi is tasked with transporting cargo forward and ensuring its glycosylation machinery including glycosylation enzymes are properly compartmentalized. The retention and recycling of enzymes are intimately tied to the balance in anterograde and retrograde trafficking at the Golgi. Mutations or KO of COG subunits severely impairs Golgi retrograde trafficking (6). Acute knock-down (KD) or KO of any individual COG subunit results in mislocalization and depletion of every tested Golgi (4, 14, 22, 35, 38). We used WB to measure the total cellular level of four endogenous glycosylation enzymes involved in N- (MGAT1 and B4GALT) and O- (GALNT2 and GALNT3) glycosylation. Measurement of the total levels of glycosyltransferases in WT and SNARE KO cells, showed that the abundance of the tested enzymes was similar to WT in GS15 KO cells and somewhat reduced in GS28 KO, but this reduction was not as dramatic as in COG4 KO cells (Figure 4A, B). Since expression of GS28 was found to be needed in maintaining the normal levels of B4GALT1, MGAT1 and GALNT3 we wondered whether partial depletion of these Golgi enzymes would affect Golgi glycosylation in GS28 KO cells. First, we examined changes in electrophoretic mobility of Golgi-glycosylated LAMP2, SDF4 and TMEM165 proteins (Figure 4C) (39-41). In GS28 KO, but not in GS15 KO cells, there was a subtle but highly reproducible smearing down of LAMP2 signal, likely related to incomplete protein glycosylation (Figure 4C). LAMP2 has 16 potential N-glycosylation and 10 O-glycosylation sites. Our previous studies in cells deficient for N- or/and O-glycosylation indicate that aberrant O-glycosylation affects the total abundance of LAMP2, suggesting its contribution to LAMP2’s stability (42). On the other hand, aberrant N-glycosylation does not affect the abundance of LAMP2 but results in a dramatic shift in LAMP2’s electrophoretic mobility as observed in COG deficient cells (42) (Figure 4C). TMEM165 is a putative ion channel localized in the Golgi with three predicted O-glycosylation sites. Mutations/deletions in TMEM165 result in congenital disorders of glycosylation (43, 44). The contribution of glycosylation to its abundance and function is unknown, but COG-deficient cells demonstrate altered electrophoretic mobility of TMEM165 (22). SDF4, also known as Cab45, is a soluble TGN protein that is thought to function in the sorting and retention of soluble cargo at the TGN in a Ca2+-dependent manner (45). Since it has only one putative N-glycosylation site, alterations in glycosylation may not produce a detectable shift in electrophoretic mobility, but in COG-deficient cells the cellular abundance of SDF4 is significantly reduced (22). GS28 KO did not show any effect on the abundance of SDF4 but affected the electrophoretic mobility and stability of TMEM165 (Figure 4C, D). These results indicate that GS28 is partially needed for efficient Golgi glycosylation, but its deletion does not affect glycosylation as severely as COG’s deletion.
Figure 4: The abundance of several Golgi enzymes is reduced in GS28 KO cells but to a lesser degree than in COG depleted cells. Enzyme reduction in SNARE-depleted cells did not significantly alter glycosylation of lysosomal and Golgi glycoproteins.

(A) WB analysis of Golgi glycosylation enzymes in WT, GS28 KO, GS15 KO and COG4 KO cells. While the total amount of B4GALT1, MGAT1, GALNT3 and GALNT2 is not as severely depleted in GS28 KOs compared to COG4 KOs, their levels are significantly reduced compared to the WT. (B) Quantification of fold changes in enzyme levels with respect to WT from biological triplicates. (C) WB showing electrophoretic mobility and abundance of glycoproteins; (D) Quantification of the fold change in glycoprotein levels from biological triplicates with respect to WT. (E) Lectin blot detects the total amount of intermediate O-glycan - Tn antigen labeled by HPA-647, and N-glycan – high mannose labeled by GNL-647, in cell lysates. (F) Quantification in the fold change in HPA and GNL staining intensity with respect to WT shows a significant increase in the accumulation of immature glycoproteins in GS28 KOs, but this is not as dramatic as in COG4 KOs. **p<0.001, ***p<0.0001
We further assessed glycosylation defects in SNARE KOs by utilizing fluorescently labelled lectins. GNL and HPA are lectins that recognize early N- and O- linked glycans. GNL binds high-mannose residues on N-glycans and HPA binds alpha N-acetylgalactosamine residues also known as Tn-antigen of O-glycans (46-48). Both these glycans are intermediates of the Golgi’s N- and O- glycosylation pathways. There was a slight but significant increase in GNL-647 and HPA-647 binding to the total cellular glycoproteins in the GS28 KO cell lysate (Figure 4E, F). This suggests an increase in the accumulation of immature glycans. However, compared to GS28 KO, COG KO resulted in more than a 5-fold increase of immature N- and O- linked glycans. Importantly, knock-down (KD) of other two Golgi SNARE proteins, STX5 and YKT6, was as detrimental for Golgi O-glycosylation as KD of COG3 subunit of the COG complex (Figure S2).
We were surprised that impairing the STX5 Golgi SNARE complex by deleting a key SNARE, GS28, from the Golgi fusion machinery, did not severely affect the maintenance of Golgi glycosylation machinery and Golgi glycosylation. Since in vitro binding studies have shown SNAREs could be promiscuous in their binding preferences (49, 50), we entertained the possibility that in GS28 KO cells, GS28 (and possibly GS15) is substituted by other SNAREs to accommodate sufficient vesicular recycling of Golgi resident proteins. To test this possibility, we looked for potential changes in STX5’s partners in GS28 KO cells.
GS28 KO impairs the partnering of the Golgi STX5 complex, leading to an increased usage of post Golgi SNAREs by STX5.
Immunoprecipitation (IP) with affinity-purified antibodies to STX5 followed by quantitative label-free DIA analysis was performed to determine the effect of GS28 KO on STX5’s partnering with other SNARE proteins. In this approach, cells were briefly pretreated with N-ethylmaleimide (NEM) to inhibit the activity of NSF and thereby prevent dissociation of SNARE complexes during isolation (51-53). Proteomic analysis and WB validation revealed the presence of both known and novel STX5 SNARE partners (Figure 5A-C). In addition to the expected reduction in STX5/GS15 interaction, we also observed a 2-fold reduction in STX5’s interactions with both GS27/GOSR2 and BET1 in GS28 KO cells (Figure 5A), indicating that GS28 KO not only impairs the intra-Golgi STX5/GS28/GS15/YKT6 complex but also affects the ER-Golgi STX5/GS27/BET1/SEC22B SNARE complex. On the other hand, compared to WT, GS28 KO significantly increased STX5’s partnering with SNAP29, VTI1B and VAMP7 (Figure 5A-C). The amount of the endosomal Qc SNARE STX8 was also increased. The unbiased mass spectrometry (MS) results suggest that GS28 deletion is compensated for by the increased usage of one or more “non-canonical” STX5-containing complexes, which potentially operate at the Golgi under “SNARE-stress” conditions. Likely, GS15 is also partially substituted by another Qc SNARE since its total abundance and interaction with STX5 are decreased in GS28 KO cells (Figure 2A; 5A-C; S1C, D). MS results were validated by quantitative WB, confirming a significant increase in STX5’s partnering with SNAP29 and VTI1B in GS28 KO cells (Figure 5B, C). This was also confirmed by SNAP29 IP and VTI1B IP (Figure 5D-F). Both long (STX5 L) and short (STX5S) isoforms of STX5 are present in the SNAP29 and VTI1B IP. There is a significant increase in STX5 S coIPed with SNAP29 in GS28 KO cells compared to WT (Figure 5D, E). In the VTI1B IP both isoforms of STX5 are significantly increased in GS28 KO compared to WT (Figure 5F, G).
Figure 5: Label-free mass spectrometry (DIA MS) analysis of STX5 binding proteins reveals an increase in non-canonical STX5 SNARE partners.

(A) Volcano plot showing fold changes in SNAREs coimmunoprecipitated (coIPed) with STX5 in GS28 KO vs WT based on DIA MS of four biological replicates. Magenta and green dots indicate SNARE proteins whose fold changes increased or decreased significantly in the STX5 IP in GS28 KO vs WT. The volcano plot for identified SNARE proteins was obtained from 4 biological replicates. A fold-change >2 and p<0.05 was considered statistically significant. (B) WB analysis of endogenous STX5 IP in WT and GS28 KO cells. (C) Bar graph represents fold changes of SNAREs coIPed with STX5 in GS28 KO vs WT. (D) WB analysis of endogenous SNAP29 IP in WT and GS28 KO cells. (E) Bar graph represents fold changes of coIPed SNAREs with SNAP29 in GS28 KO vs WT. (F) WB analysis of endogenous VTIB IP in WT and GS28 KO cells. Note an appreciable increase in the amount of STX5 and YKT6 coIPed with VTI1B and SNAP29 in the GS28 KO compared to WT. (G) Bar graph represents fold changes of coIPed SNAREs with SNAP29 in GS28 KO vs WT. n=3, *p<0.05, **p<0.001, ***p<0.0001
We proposed that the absence of GS28 increased the usage of two different “non-canonical” SNARE assemblies. Qb SNARE VTI1B may substitute for GS28 to form STX5/VTI1B/Qc-SNARE/YKT6 complex (Figure 5F-G). Qbc SNARE SNAP29 may substitute both GS28 and GS15 whose total abundance, Golgi localization and pairing with STX5 is reduced in GS28 KO cells (Figure 2, 5A-C; S1C, D) resulting in STX5/SNAP29/R-SNARE complex (Figure 6A). In the first scenario, canonical Golgi Qc SNARE GS15 is likely to be substituted by the endosomal Qc SNARE STX8, since the amount of STX8 was increased in STX5 IP in GS28 KO cells and STX8 is a known partner of VTI1B (54) (Figure 5A). Moreover, GS15 was not detected in VTI1B native IP (Figure 5F, G). In the latter scenario, the canonical Golgi R-SNARE YKT6 is likely to be substituted with VAMP7, since SNAP29 IP showed a preference for that R-SNARE as a partner (Figure 5D, E). WB analysis of the STX5 IP, SNAP29 IP and VTI1B IP suggests that both “non-canonical” complexes exist in cells under normal conditions and their abundance significantly increased upon GS28 deletion (Figure 5B-F).
Figure 6: Model for alternate Golgi SNARE complexes.

(A) SNAP29 and VTI1B are GS28 substitutes. SNAP29 operates in a complex with STX5 and VAMP7, while VTI1B operates in a complex with STX5, STX8 and YKT6. (B-D) In vitro interactions between GST-STX5, SNAP29-His6 and GFP-VAMP7. (B) Purified bacterially expressed GST-STX5 and SNAP29-His6 were mixed in equal amounts and the complex was isolated using purification on Talon affinity column. Individual proteins and the eluate from Talon affinity resin were loaded on 4-15% SDS gradient gel and stained with Coomassie G250. (C, D) STX5/SNAP29 Q-SNARE complex is specifically pulled-down by R SNARE GFP-VAMP7. (C) Purified GST-STX5 (lane 1) and SNAP29-His6 (lane 2) were mixed and incubated on ice for 6 h; the resulting complex was sequentially purified on Glutathione and Talon affinity columns (lanes 3, 4).The STX5/SNAP29 Q-SNARE complex was recovered with GFP-VAMP7, GFP-YKT6 or GFP immobilized on GBP (GFP binding protein)-Sepharose beads after overnight incubation at 4°C. Proteins were identified by WB and the recovery of individual SNAREs was quantified. The picture underneath lanes 5-7 depict Coomassie-stained GFP-tagged proteins. (D) Bar graphs represent relative recovery of STX5 and SNAP29 on GBP beads. Note that GFP-VAMP7 was the preferred R-SNARE partner for STX5/SNAP29 Q-SNARE complex. (E, F) STX5/VTI1B/STX8 Q-SNARE complex is specifically pulled down by GFP-YKT6 and GFP-VAMP7. (E) Purified bacterially expressed GST-STX5 (lane 1), VTI1B-His6 (lane 2) and STX8-His6 (lane 3) were mixed and incubated on ice for 6 h; the resulting complex was sequentially purified on Glutathione and Talon affinity columns (lanes 4, 5). Purified STX5/VTI1B/STX8 Q-SNARE complex was mixed with GFP-YKT6, GFP or GFP-VAMP7 (lanes 6-8) immobilized on GBP-Sepharose beads and incubated overnight at 4°C. Proteins were identified by WB with antibodies as indicated and the recovery of individual SNAREs was quantified. The picture underneath lanes 6-8 represents Coomassie-stained GFP-tagged proteins. (F) Bar graphs represent relative recovery of STX5, VTI1B and STX8 on GBP beads.
We further validated the interaction between STX5 and its non-canonical SNARE partners in an in vitro setup. First, we found that bacterially expressed purified GST-STX5 and His6-SNAP29 form 1:1 complex in vitro (Figure 6B). Subsequent incubation of purified GST-STX5/His6-SNAP29 Q-SNARE complex with R-SNAREs GFP-VAMP7, GFP-YKT6 or GFP immobilized on agarose beads revealed a preferential formation of STX5/SNAP29/VAMP7 complex (Figure 6C, D). Similarly, the STX5/VTI1B/STX8 Q-SNARE complex was assembled in vitro using purified GST-STX5, His6-VTI1B and His6-STX8. Incubation of preassembled Q-SNARE complex with GFP-YKT6, GFP-VAMP7 or immobilized on agarose beads revealed the efficient formation of STX5/VTI1B/STX8/YKT6 SNARE complex (Figure 6E, F). Interestingly, STX5/VTI1B/STX8/VAMP7 complex was also formed in this setup, indicating that at least in vitro YKT6 and VAMP7 can substitute for each other. In summary, these in vitro experiments support the existence of STX5-based SNARE complexes with VTI1B or SNAP29.
STX5 recruits SNAP29 to the Golgi.
SNAP29 has been previously implicated in several SNARE-mediated fusion events (55-58), but not in intra-Golgi trafficking. We used Airyscan microscopy to analyze the localization of SNAP29 in WT and GS28 KO cells. As expected, SNAP29 in WT cells demonstrated a wide cellular distribution with limited perinuclear/Golgi localization (Figure 7A top panel). In contrast, a significant increase in SNAP29 colocalization with STX5 and GM130 in the Golgi area was observed in GS28 KO cells (Figure 7A, C). Fluorescence Resonance Energy Transfer (FRET) between CFP-SNAP29 and YFP-STX5 co-expressed in GS28/SNAP29 DKO cells confirmed the proximity between STX5 and SNAP29 in the Golgi region (Figure S3) confirming that the increased pairing between STX5 and SNAP29 in GS28 KO cells is occurring at the Golgi.
Figure 7: SNAP29 is dramatically relocated to the Golgi upon GS28 depletion.

(A, C) Airyscan images of WT and GS28 KO cells stained for SNAP29, GM130 and STX5 show redistribution of SNAP29 to the Golgi region in GS28 KO cells. There is a significant increase in the colocalization between SNAP29 and GM130 in GS28 KO compared to WT. (B, C) Retro2-induced displacement of STX5 from the Golgi fails to recruit SNAP29 to the Golgi in GS28 KO cells. Airyscan images of WT and GS28 KO cells treated with Retro2 and stained for SNAP29, GM130 and STX5. Scale bars are 10μm, **p<0.001
What recruits SNAP29 to the Golgi? Retro-2 has been described as a compound that causes STX5 relocalization from the Golgi to ER without affecting the total cellular abundance of STX5 (59, 60). A short treatment with Retro-2 displaces STX5 from the Golgi without affecting its SNARE partners GS28 and GS15 (60). Anterograde trafficking from the Golgi to the plasma membrane, endocytosis, as well as early and late endosomal trafficking were also unaffected by Retro-2 treatment (60). Moreover, prolonged treatment with this compound did not affect cell viability or protein synthesis and the total levels of STX5 were largely unchanged (60). To test if Golgi-localized STX5 was required for SNAP29 recruitment, WT, and GS28 KO cells were treated with Retro2 and stained for SNAP29, GM130 and STX5. In Retro-2 treated WT and GS28 KO cells, STX5 was displaced from the Golgi (Figure 7B). Importantly, SNAP29 was no longer localized to the Golgi in GS28 KO cells (Figure 7B, C), indicating that STX5 is necessary for SNAP29’s recruitment to the Golgi. Furthermore, GS28 KO cells but not WT cells have a fragmented Golgi when treated with Retro-2 for 24 h (Z.D., unpublished observation). This indicates that the lack of STX5 and SNAP29 at the Golgi are detrimental to the Golgi structure in GS28 KO HEK293T cells. Overall, this suggests that GS28 removal renders the STX5/SNAP29/VAMP7 complex as a crucial factor in Golgi physiology.
Displacement of STX5 from the Golgi exacerbates glycosylation defects in GS28 KO cells.
Upon confirming that SNAP29’s Golgi localization is dependent on Golgi-located STX5, we assessed the effect of STX5’s mislocalization on Golgi glycosylation. Prolonged treatment of WT and GS28 KO cells with Retro-2 did not affect cell viability (unpublished observation) but led to significant N-glycosylation defects, as revealed by the GNL-647 lectin staining of both the plasma membrane (Figure 8A, B) and total (Figure 8E (left blot)) glycoproteins. Importantly, Retro2-induced N- and O-glycosylation defects were more severe in GS28 KO cells (Figure 8A-D), confirming that STX5-dependent recruitment of “alternative” SNARE partners is critical for the process of glycosylation in the Golgi. We also found that the levels of Golgi glycosylation enzymes were depleted in Retro-2 treated cells (Figure 8F, G).
Figure 8: Displacement of STX5 from the Golgi impairs Golgi glycosylation more significantly in GS28 KO compared to WT.

(A-D) Flow-cytometry analysis of Retro2 treated WT and GS28 KO cells surface stained with GNL-647 and HPA-488. The histograms are representative of biological triplicates and at least 30,000 cells per replicate. GNL and HPA staining intensities are higher in WT and GS28 KO cells treated with Retro2 compared to their untreated counterparts indicating N-glycosylation and O-glycosylation defects. (E) Staining the total cell lysate with fluorescently labeled GNL and HPA indicates accumulation of underpocessed N- but not O- glycans in cells treated with Retro2. (F, G) Treatment with Retro2 affects the abundance of Golgi glycosylation enzymes ***p<0.0001
Deletion of SNAP29 and VTI1B in GS28 KO cells affects Golgi glycosylation.
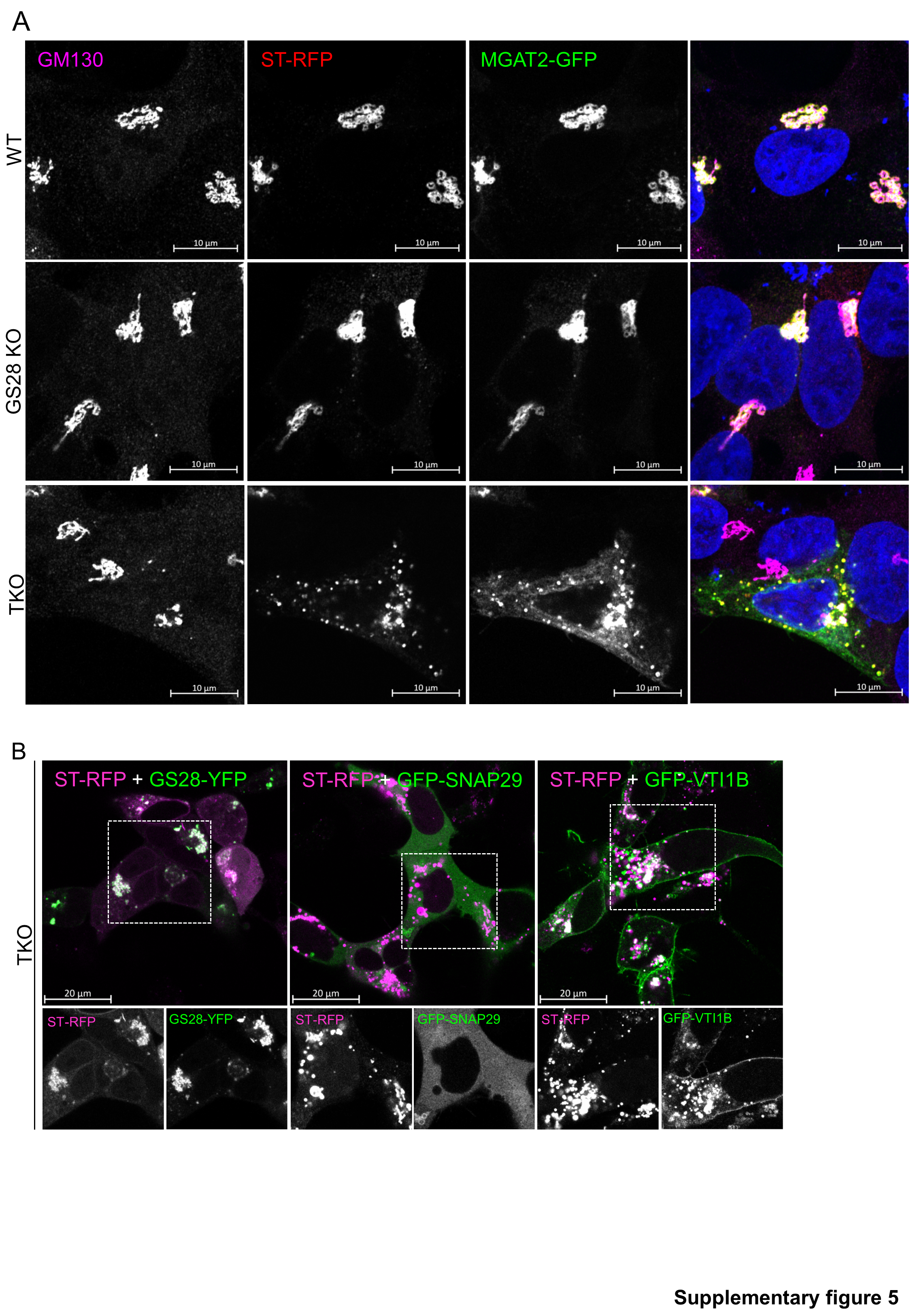
To assess the functional involvement of SNAP29 and VTI1B in recycling/retention of Golgi enzymes, we assessed the Golgi’s glycosylation capacity of cells double and triple deleted for GS28, SNAP29 and VTI1B (Figure S4A). Deleting SNAP29 or VTI1B individually in GS28 KO cells did not produce additive glycosylation defects and the cells had the glycosylation phenotype of the parent GS28 KO cell line (Z.D., unpublished observation). However, knocking out both SNAP29 and VTI1B in GS28 KO cells (GS28/SNAP29/VTI1B TKO) resulted in additive glycosylation defects (Figure 9). Flow-cytometry analysis of WT, GS28 KO and TKO cells surface-labelled with fluorescent lectins GNL, WGA and RCA-I revealed a significant increase in lectin binding to plasma membrane glycoconjugates in impermeabilized TKO cells, indicating severe glycosylation defects (Figure 9A, B). Significantly, exogenous expressions of GS28-YFP, GFP-SNAP29 or GFP-VTI1B partially rescued glycosylation abnormalities in TKO cells (Figure 9B, C). In line with these results, TKO cells were unable to retain exogenously expressed MGAT2-GFP or ST6Gal1-RFP (ST-RFP) in the Golgi region (Figure S5A) similar to the COG KO phenotype (38). MGAT2-GFP, which is normally Golgi localized in both WT and GS28 KO cells, was mislocalized in puncta throughout the cytosol in addition to its peri-nuclear density in TKO cells (Figure S5A). Similarly, ST-RFP was also present in puncta all throughout the cytosol (Figure S5A). Interestingly, only expression of GS28-YFP, but not its substitutes, GFP-SNAP29 or GFP-VTI1B, rescued the Golgi distribution of ST-RFP in the TKO cells (Figure S5B). TKO cells also show additive retrograde trafficking defects. In GS28 KO cells SubAB cleaved about 50% of GRP78 3 h after toxin treatment (Figure 3A, B), whereas in TKO cells less than 10% of GRP78 was cleaved after the same treatment (Figure 9D, E).
Figure 9: Triple knockout (TKO) of GS28, SNAP29 and VTI1B results in severe glycosylation defects.

(A) Flow cytometry analysis of WT, GS28 KO and TKO cells using fluorescent lectins. Histograms showing the fluorescence intensity of intact wild type and mutant cells labeled with the indicated lectins for 30 mins on ice (B) Histograms showing the fluorescence intensity of WT, TKO and TKO rescued cells surface labeled with the GNL for 30mins on ice. (C) Bar graphs showing the mean fluorescence intensity for GNL indicate a decrease in GNL binding in the TKO cells expressing either GS28-YFP or GFP-SNAP29 or GFP-VTI1B suggesting that each of these SNAREs can partially rescue N-glycosylation defects in the TKO cells. Monosaccharide symbols follow the Symbol Nomenclature for Glycans (SNFG). (107)
COG defects increase the usage of alternate Golgi SNARE complexes.
GS28 and GS15 are sensitive to COG subunit deletions. We reasoned that the increased usage of “alternative” SNARE complexes might be also increased in COG deficient cells. To test this possibility, the abundance of the “alternative” partners was measured in STX5 IPs from COG4 KO and COG7 KO cells. SNAP29 was significantly relocalized to the Golgi (Figure 10A, B) and its pairing with STX5 was significantly increased compared to WT cells (Figure 10C, D). There is also an appreciable increase in the levels of VTI1B coIPed with STX5 in COG4 and COG7 KO cells (Figure 10C, D). This indicates that the increased usage of SNAP29 and VTI1B as STX5 partners in Golgi trafficking could be a common feature in cells depleted for canonical STX5 partners and COG subunits.
Figure 10: Usage of SNAP29 and VTI1B is increased in COG4 KO cells.

(A) Airyscan images of WT, GS28 KO and COG4 KO cells stained for GM130, SNAP29, and STX5 show redistribution of SNAP29 to the Golgi region in GS28 KO and COG4 KO cells. (B) There is a significant increase in the colocalization between SNAP29 and STX5 in GS28 KO and COG4 KO cells compared to WT cells. (C) WB analysis of STX5 IP in WT, GS28 KO, COG4 KO and COG7 KO cells shows increased coIP of alternate partners SNPA29 and VTI1B in COG depleted cells. (D) There is more than two-fold increase of SNAP29 coIPed with STX5 in COG4 depleted cells. *p<0.05, **p<0.001, ***p<0.0001
Discussion
STX5/GS28/GS15/YKT6 is the evolutionarily conserved Golgi SNARE complex that is thought to be essential for intra-Golgi and endosome-to-Golgi vesicle trafficking steps (9, 61, 62). These SNAREs physically and functionally interact with the key Golgi MTC - the COG complex (24, 25). Consistent with the observed intimate collaboration between the COG complex and SNARE proteins in driving vesicular trafficking at the Golgi, individuals harboring mutations in COG subunits or the Golgi Qa-SNARE STX5 exhibit severe defects in protein glycosylation (6, 7). Deficiencies in the COG complex have been shown to reduce the abundance of GS28 and GS15 proteins (Figure 2A, B) (22, 24, 25, 63, 64). Specifically, the deletion of GS28 resulted in a decrease in the overall levels of GS15 (Figure 2A, B) and disrupted its interaction with STX5 (Figure 5A-C; S1C, D). Additionally, there was a notable reduction in the localization of GS15 to the Golgi. Overall, this scenario parallels the effect of COG subunit mutations, KD, or KO on the STX5 SNARE complex, where GS28 and GS15 levels are diminished. However, our findings suggest that the deletion of Golgi v-SNARE proteins does not phenocopy the severe glycosylation defects observed in COG mutants. The deletion of GS28 resulted in a delayed retrograde trafficking of SubAB toxin comparable to the effects of COG subunit deletion (Figure 3A, B), and GS28/GS15 DKO cells exhibited a phenotype similar to GS28 KO cells without any additional effects (Z.D., unpublished observation). Moreover, RPE1 cells lacking either GS28 or GS15 were viable (65).
There are several explanations for the limited impact of GS28/GS15 deletion on Golgi functions. Although Golgi glycosyltransferases are present in GS28/GS15 containing Golgi-derived COPI vesicles (22, 66-68), enzymes could be recycled back to the Golgi independently of the STX5 SNARE complex. In support of this theory, a CDG-causing mutation in STX5, that abolishes expression of the short isoform of STX5, did not result in a dramatic mislocalization of Golgi enzymes despite producing lethal glycosylation abnormalities (7). However, acute downregulation of STX5 and YKT6 has a dramatic effect on Golgi glycosylation (Figure S2) (69). Another possibility is that the COG complex regulates multiple Golgi SNARE complexes and, other Golgi-operating SNAREs could be more important than the GS28-containing complex for recycling and maintenance of Golgi glycosylation machinery. Indeed, the COG complex, in addition to the STX5 SNARE complex, is also shown to regulate the STX16/STX6/VTI1A/VAMP4 assembly (18, 25, 63), but this TGN SNARE complex has never been implicated in the recycling of Golgi enzymes, and VTI1A KO has no impact on Golgi glycosylation (Z.D., unpublished data). The third possibility is that in GS28 KO cells, retrograde trafficking of Golgi enzymes is rescued by cell adaptation via an increased utilization of an alternative non-canonical STX5-containing SNARE complex that normally plays a minor role in Golgi function. Indeed, the usage of two novel SNARE complexes, STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6, which exist in WT cells, is significantly increased in response to the deletion of canonical Golgi v-SNARE GS28. Importantly, the usage these two Golgi SNARE complexes was also increased in COG4 and COG7 deficient cells (Figure 10C, D), indicating the GS28/GS15 depletion in COG deficient cells triggers a similar SNARE compensatory response.
Since both novel SNARE complexes exist in WT cells, they might be involved in specialized Golgi-directed membrane trafficking processes. Individual deletion of VTI1B or SNAP29 did not produce glycosylation defects detectable by methods applied in this work, so additional investigations should be performed to identify cargo molecules that use STX5/VTI1B/STX8/YKT6- and STX5/SNAP29/VAMP7-dependent pathways. Interestingly, we have previously shown that in COG4 KO cells, trans-Golgi enzyme ST6GAL1 was found in VAMP7-positive enlarged endolysosomal structures (38), supporting the involvement of VAMP7 in the recycling of a subset of Golgi resident proteins. STX8 and VAMP7 have a transmembrane domain; therefore, isolation and characterizations of STX8- and VAMP7- containing membrane trafficking intermediates could help in the deciphering of novel Golgi trafficking pathways.
VTI1p was first implicated in retrograde Golgi trafficking in yeasts (70). It was found to interact with Sed5p (70, 71) and Pep12p (71). von Mollard and colleagues also found genetic interactions between VTI1 and YKT6 in yeast and proposed the existence of the Sed5p-Vt1ip-Sft1p-YKT6p SNARE complex at the Golgi (72). SNARE assemblies are usually evolutionarily conserved; therefore, the STX5/VTI1B/Qc-SNARE/YKT6 complex is highly plausible in human cells. Native VTI1B IP confirmed its interaction with STX5 and YKT6 (Figure 5F, G). However, classical in vitro studies indicated that the positioning of Sft1p (yeast GS15 homologue) on donor liposomes was not sufficient for the fusion with Sed5p-Vti1p-Ykt6p acceptor liposomes (73). Since VTI1B is transported through the Golgi on the way to endosomal compartments and is already a perinuclear localized protein with partial colocalization with STX5 in WT cells, its increased involvement in Golgi trafficking may not lead to a visible change in VTI1B intracellular distribution. Our data clearly demonstrated the increased formation of STX5/VTI1B complex in GS28 KO cells, but GS15 was not detected in this complex. Instead, we found STX8 in both VTI1B and STX5 IPs in GS28 KO cells and STX5-STX8 interaction was increased upon GS28 deletion. In vitro experiments confirmed the possibility of STX5/VTI1B/STX8/YKT6 complex formation (Figure 6E, F), indicating that increased usage of this endosomal Qb/Qc SNARE pair can accomplish intra-Golgi trafficking needs in GS28 KO cells. In vitro binding studies also supported the possibility for STX5/VTI1B/STX8/VAMP7 complex at the Golgi but the exact composition of this SNARE complex and its role in Golgi trafficking certainly requires additional investigation.
SNAP29, a Qbc SNARE has been described as a “promiscuous” SNARE, and its interaction with STX5, as well as many other SNAREs, has been previously reported (55, 58, 74-77). It was also found to have various cellular distributions including cytosolic, perinuclear, endolysosomal and plasma membrane possibly because of its engagement in multiple SNARE complexes (58, 78, 79). Intracellular SNAP29’s localization is flexible and could vary upon changes in the expression levels of its interacting partners. Overexpression of SNAP29 interactor Rab3a redistributes SNAP29 from the cytosol to the perinuclear region (80). This Qbc SNARE, would be the perfect substitute for deleted GS28 (Qb) and depleted GS15 (Qc). Structurally, it lacks a transmembrane domain, allowing immense flexibility in intracellular localization (79). Purified STX5 and SNAP29 form a 1:1 complex in vitro (Figure 6B). Although SNAP29 has been reported to form a complex with both YKT6 and VAMP7 (56, 81-85), our data indicate that in HEK293T cells, the endosomal R-SNARE VAMP7 but not YKT6 interacts with SNAP29 (Figure 5D, E; 6C, D). In agreement with our data, STX5-SNAP29 interaction was also detected in a high throughput study of endogenously tagged proteins (86). Furthermore, we show, for the first time, that STX5 is necessary to recruit SNAP29 for intra-Golgi trafficking since Retro-2-stimulated exclusion of STX5 from Golgi resulted in aggravated Golgi-glycosylation defects in GS28 KO cells (Figure 8A-E). The remarkable redistribution of SNAP29 to the Golgi bolsters our prediction that increased STX5/SNAP29/VAMP7 complex formation compensates for the loss of STX5/GS28/GS15/YKT6 fusion machinery.
If the SNARE substitution model is correct, we reasoned that knocking-out SNAP29 or VTI1B in GS28 KO cells would disrupt their respective non-canonical STX5-bearing SNARE complexes that, in turn, would further decrease intra-Golgi recycling of Golgi enzymes and glycosylation. To evaluate this possibility, GS28/VTI1B DKO, GS28/SNAP29 DKO and GS28-SNAP29-VTI1B triple knockout (TKO) cell lines were created. Both DKOs were viable, and they proliferated at WT rates (Z.D., unpublished observation). Surprisingly, the DKOs did not have additive glycosylation defects, and their glycosylation status was similar to GS28 KO (Z.D., unpublished observation). This suggests that there could be another Qb SNARE, either GS27 or VTI1A as a substitute in this scenario. Alternatively, the increased usage of any one of the two new STX5 SNARE complexes was sufficient to accommodate Golgi membrane trafficking. Indeed, we found that GS28-SNAP29-VTI1B TKO cells had additive glycosylation and retrograde trafficking defects (Figure 9). Lectins binding to immature glycans in the N-glycosylation pathway had an increased binding to TKO cells compared to WT or GS28 KO cells (Figure 9A). TKO cells were also unable to transport SubAB toxin and to retain in the Golgi exogenously expressed MGAT2-GFP and ST-RFP. Exogenous expressions of GS28-YFP, GFP-SNAP29, or GFP-VTI1B partially rescued N-glycosylation abnormalities, but interestingly, only GS28-YFP could rescue the diminished ability of the Golgi to retain overexpressed Golgi resident proteins (Figure 9B, C). This indicates that each novel Golgi SNARE complex can only partially substitute the function of canonical STX5/GS28/GS15/YKT6 complex in Golgi physiology. The transmission electron microscopy (TEM) analysis of the Golgi in Golgi TKO cells showed an accumulation of vesicles in the perinuclear region around the Golgi. (Figure S4B). These vesicles most likely accumulate because of their inability to fuse with the Golgi membrane in the absence of GS28, VTI1B and SNAP29. Presently it is unclear what regulates formation and usage of VTI1B and SNAP29 containing Golgi SNARE complexes. The COG complex is still present on the Golgi in both GS28 KO and TKO cells (Figure S6A), indicating that COG localization is independent of the expression Golgi Qb and Qc SNAREs. In making different DKO mutant combinations, we discovered that GS28/COG4 DKO is very detrimental to cell growth (Figure S6B), suggesting that the increased usage of non-canonical Golgi SNARE complexes is not sufficient to overcome COG deficiency.
Adapting the Golgi trafficking system to the loss of GS15 is another question. While GS15 yeast homolog Sft1p is an essential protein (62), KO of GS15 in human HEK293T or RPE1 cells did not produce any detectable cell growth or glycosylation phenotypes. The MS analysis of STX5-associated proteins did not reveal any significantly increased SNAREs in GS15 KO cells, but two Qc SNAREs, STX8 and STX6, were modestly (>1.3 fold) increased in STX5 IPs from GS15-deficient cells (Supplementary table 1). Perhaps the increased usage of these SNARE proteins with remaining STX5/GS28/YKT6 is sufficient to substitute canonical Qc-SNARE GS15 for Golgi trafficking needs.
The potential impact of Golgi SNARE deletion on endocytic trafficking is an important concern since our model implies that several post-Golgi SNAREs, including VTI1B, VAMP7, and STX8 are used for the intra-Golgi needs instead of the endocytic ones. Since our STX5 IP was quantitative, we estimated that ~35% of SNAP29 and less than 25 % of cellular VTI1B and VAMP7 were present in STX5 complexes in GS28 KO cells, indicating that the majority of these SNAREs were still available for endocytic needs.
In summary, our experimental data allows us to postulate that deletion of Golgi SNARE GS28 in mammalian cells abolished the formation of the canonical STX5 SNARE complex but did not significantly affect Golgi biogenesis and vesicular trafficking. We speculate that STX5 Golgi localization is critical for its Golgi function and serves as a hub to compensate for GS28 deletion by increased usage of two non-canonical SNARE complexes, STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6. We propose that increased usage of STX5-based alternative SNARE complexes is a common adaptive mechanism to the failure of canonical intra-Golgi vesicle tethering/fusion machinery.
Materials and Methods:
List of primary antibodies and dilutions
| Antibody | Source, Catalog # | Species | WB dilution | IF dilution |
|---|---|---|---|---|
| B4GALT1 | R&D Systems, AF-3609 | Goat | 1:500 | - |
| GALNT2 | R&D Systems, AF7507 | Sheep | 1:500 | - |
| GALNT3 | R&D Systems, AF7174 | Sheep | 1:500 | - |
| Giantin | Covance PRB- 114C | Rabbit | - | 1:1000 |
| GM130 | BD Biosciences, 610823 | Mouse | - | 1:500 |
| GS15 | This lab | Rabbit | 1:1000 | - |
| GS15 | BD Biosciences, 610961 | Mouse | - | 1:500 |
| GS28 | BD Biosciences, 611184 | Mouse | 1:500 | 1:500 |
| LAMP2 | DSHB, H4B4 | Mouse | 1:1000 | - |
| MGAT1 | Abcam, ab180578 | Rabbit | 1:500 | - |
| SDF4 | PtnTech, 10517-1-AP | Rabbit | 1:1000 | - |
| SNAP29 | Synaptic Systems, 111303 | Rabbit | 1:1000 | - |
| SNAP29 | R&D, AF7869 | Sheep | 1:1000 | 1:300 |
| SNAP29 | Abcam, ab181151 | Rabbit | 1:1000 | 1:500 |
| STX5 | This lab | Rabbit | 1:1000 | 1:1500 |
| STX5 | Santacruz, sc-365124 | Mouse | 1:500 | 1:100 |
| TGN46 | Bio-Rad, AHP500G | Sheep | 1:2000 | 1:500 |
| TMEM16 | Sigma, HPA038299 | Rabbit | 1:500 | - |
| VAMP7 | CST, 14811 | Rabbit | 1:1000 | - |
| VAMP7 | CST, 13876 | Rabbit | - | 1:300 |
| VT1IB | BD Biosciences, 611404 | Mouse | 1:250 | 1:300 |
| YKT6 | Santacruz, sc-365732 | Mouse | 1:300 | - |
| YKT6 | Bethyl, A305-479A | Rabbit | 1:500 | - |
| YKT6 | Bethyl A305-480A | Rabbit | - | 1:300 |
| β-actin | Sigma, A5441 | Mouse | 1:5000 | - |
Secondary antibodies used for WB or IF were as follows: fluorescent dye conjugated AffiniPure Donkey anti-mouse, anti-rabbit, or anti-sheep (IF 1:1000, Jackson Laboratories) and infrared dye IRDye 680RD or IRDye 800CW anti-mouse or anti-rabbit (WB 1:20,000, LI-COR).
List of plasmids
| Plasmid | Source | Reference |
|---|---|---|
| CFP-SNAP29 | R. Duden | (24) |
| GFP-VAMP7 | Addgene plasmid # 45922 | (87) |
| GFP-VTI1B | Addgene plasmid # 45920 | (87) |
| Golgi-CFP | Addgene plasmid # 14873 | (88) |
| GS28-YFP | R. Duden | (18) |
| hYKT6 in EGFP-C1 | This lab | This study |
| MGAT2-GFP | L. Lu | (89) |
| rSTX5 (55-333) in pGEX-KG | This lab | This study |
| SNAP29 in pET23a | This lab | This study |
| ST6GAL1-RFP | J. Rothman | (90) |
| STX8 in pET22b | F. Paumet | (91) |
| VTI1B in pET28a | F. Paumet | (91) |
| YFP-STX5 | R. Duden | (24) |
Production of affinity purified STX5 and GS15 antibodies.
Rabbits were injected with purified His6-STX5 (amino acids 55-286) or with His6-GS15-GST (amino acids 1-93). Antibodies were affinity-purified using the antigen coupled to agarose beads and antibodies were validated in KD and KO cell lines.
Cell culture
All experiments used HEK293T cells stably expressing Cas9 as described in (92). HEK293T cells were cultured in DMEM F/12 (Corning) supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat # 26140079). Cells were incubated in a 37°C incubator with 5% CO2 and 90% humidity. Cells were passaged by 3 min trypsinization (0.25% trypsin EDTA, Gibco) at 37°C and resuspended in media with 10% FBS. HEK293T COG4 KO cells were previously described in (38, 93). Where indicated, cells were treated with 25 μM Retro-2 (Sigma, Cat # SML1085). Retro-2 was reconstituted in DMSO and diluted to 25 μM in DMEM F/12 supplemented with 10% FBS. The cells were fed with fresh Retro-2 daily.
Creation of SNARE KO stable cell lines
Plasmids encoding dual gRNAs were purchased from Transomics with the following target sequences:
| GOSR1 (GS28): | 1a) AAAAGAAAATATGACTTCACAGAGAGGAAT |
| 1b) AGCGGCGGGACTCGCTCATCCTAGGGGGTG | |
| BET1L (GS15): | 1a) AACAGAGACTCCATGGTGTTGTGCTGGACA |
| 1b) AGACTATCATTCCGGACGTAGACGTGGCAC |
Plasmids encoding single gRNAs were purchased from Genecopia with the following target sequences.
SNAP29: ACAATCCGTTCGACGACGAC
VTI1B: GAAGGGGTCCTCCATGGACA
Plasmids were isolated from bacteria using the QIAprep Spin Miniprep Kits (Qiagen). HEK293T-Cas9 cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific). To create GS28 KO or GS15 KO, HEK293T-Cas9 cells were transfected with a cocktail of three plasmids containing dual gRNA targets. To create GS28/GS15 DKO, GS28 KO cells were transfected with a cocktail of three plasmids containing dual gRNA targets to GS15. GS28/SNAP29 DKOs or GS28/VTI1B DKOs, GS28 KO cells were transfected with single plasmids containing one gRNA target. After 16-18 h of transfection, untransfected cells were killed using 5 μg/ml Puromycin for 48 h. Surviving cells were then single-cell plated on 96 well plates to obtain individual colonies depleted for the target protein.
Sequencing CRISPR-Cas9 KOs
SNARE KOs were sequenced by SANGER sequencing. Primers were designed to amplify a 500 bp region containing the gRNA target region. The PCR product was purified using the QIAquick PCR purification (Qigen, Cat # 28104) and sequenced with appropriate primers. The web application “Indigo” hosted online at https://www.gear-genomics.com/ was used to deconvolute Sanger chromatograms (94).
Preparation of cell lysates and western blotting
For the preparation of cell lysates, HEK293T WT and KO cells were grown on tissue culture dishes to a confluency of 90-100%. They were washed thrice with PBS and lysed in 2% hot SDS, heated for 10 min at 70°C. The total protein concentration was measured using the BCA protein assay (Pierce). Samples were prepared so that they were at a concentration of 20-30μg and 6X Laemmli sample buffer containing 5% β-mercaptoethanol (Sigma) was added. Samples were further heated at 70°C for 10 min. Bio-Rad (4–15%) gradient gels were used for gel electrophoresis Proteins were blotted onto 0.2 μm nitrocellulose membranes (Amersham Protran, GE Healthcare) using the Thermo Scientific Pierce G2 Fast Blotter. Membranes were rinsed in PBS, blocked in EveryBolt blocking buffer (BioRAD, Cat # 12010020) for 30 min, and incubated with primary antibodies overnight at 4°C. Membranes were washed with PBS and incubated with secondary fluorescently tagged antibodies diluted in BioRAD blocking buffer for 60 min. All the primary and secondary antibodies are listed above. Blots were then washed and imaged using the Odyssey Imaging System. Images were processed using the LI-COR Image Studio software. WBs were quantified by densitometry using the LI-COR Image Studio software. For quantification proteins were normalized to β-Actin which was used as the loading control. Then, fold changes were calculated with respect to the WT.
Lectin staining
Gel electrophoresis and lectin blotting were performed as described above. After transfer, the membrane was blocked with 3% bovine serum albumin (BSA) for 30 min. The lectins HPA (Invitrogen, Cat # L32454) or GNL (92) conjugated to Alexa 647 fluorophore were diluted 1:1000 in 3% BSA from their stock concentration of 1 and 5 μg/μl, respectively. Blots were incubated with lectin solutions for 30 min and then washed in PBS four times for 4 min each and imaged using the Odyssey Imaging System.
SubAB trafficking assay
HEK WT or SNARE KO cells were treated with 5 μg/ml purified SubAB toxin for the indicated time periods. Following treatment, the cells were lysed with 2% SDS and the amount of GRP78 quantified by WB. Percent cleavage of GRP78 at each time point was calculated from the signal intensity of GRP78 band and plotted on a graph.
Flow Cytometry
Cells were grown to 80–90% confluency on a 12-well plate. On the day of the experiment, cells were detached by incubating with 10 mM EDTA in 1X PBS at 37°C. After three PBS washes, to get rid of the EDTA, cells were resuspended in ice-cold 0.1% BSA. The lectins used were GNL (92), HPA (Invitrogen, Cat # L32454), CTX (Invitrogen, Cat # C34776). They were diluted at 1:500 in ice-cold 0.1% BSA and incubated with the cells for 30 min on ice. DAPI was added to the samples just before the analysis and were run on the Attune NxT flow cytometer (Thermo Fisher Scientific). Dead cells were excluded by gating on live (DAPI) cells. Singlets were gated by side scatter height (SSC) vs SSC area density plots. Final gating was done in an SSC area vs FCS area density plot. Stop conditions were set to obtain at least 30,000 cells in the final gate. The mean or median fluorescence (of the lectin’s fluorophore) intensity for this population was obtained from a histogram plot.
Immunoprecipitation assay
WT and KO cells were plated on 15 cm dishes. Cells were grown to 100% confluency. Prior to lysis, cells were treated with 1 mM NEM for 15min at 37°C in order to inhibit NSF activity and thereby preserve SNARE complexes (12, 63, 95). Thereafter, cells were washed thrice with DPBS and lysed for 1 h on ice in 1% Triton lysis buffer made in 50 mM Tris pH 7.5, 150 mM NaCl, 0.1 mM PMSF and 1X Protease inhibitor cocktail. Lysates were centrifuged at 20,000 r.c.f for 10 min at 4°C. The supernatant was collected. A small volume of the supernatant was set aside as the “input.” For STX5 IP, STX5 (Lab) antibody was added to the remaining supernatant at a 1:100 dilution. For SNAP29 (Abcam) or VTI1B (BD Biosciences) IP, antibodies were added at a 1:100 dilution. Non-targeting IgGs (1:100) were used for all the control IPs. Lysates were incubated with the antibody at 4°C, for 16 h on a rotor. Antigen-antibodies complexes were pulled down using Protein G- Agarose (Roche, Lot #70470320). First, the beads were washed thrice in the wash buffer composed of 0.05% Triton in the Tris-NaCl buffer described above and then introduced into the lysates. The beads were incubated with the lysates on a rotor for 90 min at RT. The beads were collected by centrifugation and washed thrice in the 0.05% Triton wash buffer. Bound protein was eluted by heating the beads in 2X Laemmli sample buffer containing 10% β-mercaptoethanol at 95°C for 10 min. WB analysis for the IP samples was performed as described above. Fold changes in IP samples probed by WB were also quantified as described above. For the loading control, the target protein was used. For example, in STX5 IP, STX5 was used as the loading control.
Mass-spectrometry of STX5 IP and data analysis
The STX5 IP was performed as described above. For MS, before eluting, the Protein-G Agarose beads were washed three more times in 1X PBS to get rid of the excess detergent. Elution was performed as described above. Sample preparation for MS was described in (96). Briefly, purified proteins were reduced, alkylated, and digested using filter-aided sample preparation. Tryptic peptides were then separated by reverse-phase XSelect CSH C18 2.5 um resin (Waters) on an in-line 150 x 0.075 mm column using an UltiMate 3000 RSLCnano system (Thermo). Peptides were eluted using a 60 min gradient from 98:2 to 65:35 buffer A:B ratio. Eluted peptides were ionized by electrospray (2.2 kV) followed by mass spectrometric analysis on an Orbitrap Exploris 480 mass spectrometer (Thermo). To assemble a chromatogram library, six gas-phase fractions were acquired on the Orbitrap Exploris with 4 m/z DIA spectra (4 m/z precursor isolation windows at 30,000 resolution, normalized AGC target 100%, maximum inject time 66 ms) using a staggered window pattern from narrow mass ranges using optimized window placements. Precursor spectra were acquired after each DIA duty cycle, spanning the m/z range of the gas-phase fraction (i.e., 496-602 m/z, 60,000 resolution, normalized AGC target 100%, maximum injection time 50 ms). For wide-window acquisitions, the Orbitrap Exploris was configured to acquire a precursor scan (385-1015 m/z, 60,000 resolution, normalized AGC target 100%, maximum injection time 50 ms) followed by 50x 12 m/z DIA spectra (12 m/z precursor isolation windows at 15,000 resolution, normalized AGC target 100%, maximum injection time 33 ms) using a staggered window pattern with optimized window placements. Precursor spectra were acquired after each DIA duty cycle.
Following data acquisition, spectra were searched using an empirically corrected library and a quantitative analysis was performed to obtain a comprehensive proteomic profile. Proteins will be identified and quantified using EncyclopeDIA (97). Scaffold DIA was used for visualization. The False discovery rate (FDR) thresholds, at both the protein and peptide levels, were set at 1%. Protein exclusive intensity values were assessed for quality using ProteiNorm (98). Popular normalization methods were evaluated including log2 normalization (Log2), median normalization (Median), mean normalization (Mean), variance stabilizing normalization (VSN) (99), quantile normalization (Quantile) (100) cyclic loess normalization (Cyclic Loess) (101), global robust linear regression normalization (RLR) (102), and global intensity normalization (Global Intensity) (102). The individual performance of each method was evaluated by comparing the following metrices: total intensity, pooled intragroup Coefficient of Variation (PCV), Pooled intragroup Median Absolute Deviation (PMAD), Pooled intragroup estimate of variance (PEV), intragroup correlation, sample correlation heatmap (Pearson), and log2-ratio distributions. The VSN normalized data was used to perform statistical analysis using Linear Models for Microarray Data (limma) with empirical Bayes (eBayes) smoothing to the standard errors (101). Proteins showing a fold change ≥2 with an FDR adjusted p-value ≤0.05 were considered significant. For the MS data, n=4
Immunofluorescence (IF) microscopy
12 mm glass coverslips (#1, 0.17 mm thickness) were collagen-coated. WT and KO cells were plated to be 60 - 70% confluent at the time of processing. Cells were washed with Dulbecco’s phosphate-buffered saline (DPBS) and stained according to the protocol described previously (14). Briefly, freshly prepared 4% paraformaldehyde (PFA) (16% stock solution diluted in DPBS; Electron Microscopy Sciences) was used as a fixative. After fixation, cells were permeabilized with 0.1% Triton X-100 for 1 min followed by treatment with 50 mM ammonium chloride for 5 min to quench free aldehydes. Cells were then washed three times with DPBS and blocked twice for 10 min each with 1% BSA, and 0.1% saponin in DPBS. Antibodies were diluted in DPBS with 1% cold fish gelatin and 0.1% saponin. Cells were incubated with the primary antibody for 1 h at room temperature. Cells were washed four times with DPBS and incubated for 30 min with fluorescently tagged secondary antibodies diluted in antibody buffer. Cells were washed four times with DPBS. Hoechst diluted 1:10000 in DPBS was used to stain the nucleus. Coverslips were then washed four times with DPBS, rinsed with ddH2O, and mounted on glass microscope slides using Prolong Gold anti-fade reagent (Life Technologies).
Cells were imaged with the 63X oil 1.4 numerical aperture (NA) objective of the LSM880 using the Airyscan module. Pearson’s colocalization analysis was performed in ZenBlue version 2.6. Images were converted to TIF format and assembled using Adobe Photoshop CS6.
Förster Resonance Energy Transfer (FRET) microscopy
FRET was performed as described previously (24) with some modifications. Here, GS28/SNAP29 DKO HEK293T cells were co-transfected with plasmids encoding YFP-STX5 and CFP-SNAP29 or Golgi-CFP and plated in collagen coated glass-bottom dishes. 24 h after transfection cells (n=20) with the Golgi-localized YFP-STX5 were imaged. For the acceptor-bleached protocol, cells were excited at 2 s intervals at 458 nm for CFP detection and at 514 nm for YFP detection. After five excitation cycles, YFP was bleached at 514 nm. This procedure resulted in >90% of YFP photobleached within 10 s. The ratio of CFP fluorescence before and after photobleaching was used as a measure of the CFP-donor/YFP-acceptor FRET signal.
Transmission Electron Microscopy (TEM)
Samples were processed for TEM according to Valdivia’s lab protocol (103) with some modifications (104). Briefly, excess growth media was removed from the dish with cells and an equal volume of 1X fixative was added for 5 min at RT, effectively bringing the fixative to 0.5X concentration. 0.5X fixative was then replaced with 1X fixative for 10 min at RT. 1X fixative was composed of 4% PFA (EMS) and 1% GA (EMS) in 0.1 M Phosphate buffer, pH 7.4. Finally, cells were fixed for 20 min on ice with 2.5% GA and 0.05% malachite green (EMS) in 0.1 M sodium cacodylate buffer, pH 6.8. Cells were washed 4 x 5min with 0.1 M sodium cacodylate buffer and post-fixed for 30 min at RT with 0.5% osmium tetroxide and 0.8% potassium ferricyanide in 0.1 M sodium cacodylate buffer, washed again and incubated for 20 min on ice in 1% tannic acid. After washing in buffer and H2O samples were incubated for 1 h in 1% uranyl acetate at RT. Specimens were gradually dehydrated in a graded series of increasing ethanol concentrations, washed with Propylene Oxide (EMS), and incubated in 50% PO/resin mixture before embedding in Araldite 502/Embed 812 resins (EMS). Ultrathin sections were imaged at 80 kV on the FEI Technai G2 TF20 transmission electron microscope. Digital images were acquired with FEI Eagle 4kX USB Digital Camera.
In vitro SNARE binding
GST-STX5, His6-tagged SNAP29, VTI1B, STX8 were expressed in bacteria and purified on Glutathione Sepharose (GE Healthcare) beads and Talon Metal affinity resin (Takara) as previously described (105). In brief, His6-tagged proteins were expressed in E. coli BL21(DE3) cells. GST-STX5 was expressed in E. coli XL-10 Gold (Stratagene). All bacterial cultures were grown at 37 °C with shaking until they reached mid-log (OD600 = 0.5–0.6). Expression of proteins was induced by the addition of 0.2 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG), and then the culture was grown overnight at 22 °C. The cells were collected by centrifugation at 6000 g for 30 min at 4 °C and lysed in lysis buffer (50 mM Tris, 0.3 M NaCl, pH 8.0, 40 μg/ml lysozyme, 1 mM PMSF). His6-tagged proteins were purified by TALON metal affinity resin (Clontech) following a standard protocol. GST was purified by glutathione Sepharose 4B beads following a standard protocol. Fractions with the highest concentrations of proteins were pooled together and extensively dialyzed against the buffer (25 mM HEPES, pH 7.4, 150 mM KCl, 1 mM DTT, 0.05% Tween-20). After dialysis, proteins were frozen in liquid nitrogen and stored at −80 °C.
To purify GFP-tagged proteins, HEK293T cells from of 10 cm dishes were transfected with plasmids encoding GFP, GFP-YKT6 or GFP-VAMP7 using Lipofectamine 3000, 24 h after transfection, cells were lysed in a 1% Triton X-100 IP buffer (50mM Tris pH 7.4, 150mM NaCl) with 5 μL/mL of 100X Halt protease inhibitor cocktail for 30 min on ice. Post-nuclear supernatant was cleared and 90% was added to 100 μL of 50% beads conjugated to GFP binding protein (GBP)-beads (105, 106). After 90 min incubation, beads were washed 5 times with IP buffer with 0.1% Triton X-100, transferred to a new tube and used in binding studies. To assemble the STX5-SNAP29 complex, purified proteins were mixed in 100 μl of IP buffer at equal concentrations of 1 mg/ml and incubated on ice for 6 h. The complex was then purified sequentially on Glutathione Sepharose and Talon beads. Thereafter, the eluate from the Talon column was incubated with GFP-VAMP7, GFP-YKT6 or GFP immobilized on GBP conjugated to Sepharose beads at 4°C overnight on a rotor. Next day, the beads were washed in IP buffer containing 0.1% Triton X-100, eluted in 2X Laemmli sample buffer and analyzed by WB. Similarly, the STX5-VTI1B-STX8 complex was assembled in vitro by mixing purified proteins together in 100 μl of IP buffer at equal concentrations of 1 mg/ml for 6 h followed by sequential purification of resulted protein complexes on Glutathione Sepharose and Talon beads. The final eluate from the Talon beads was incubated overnight with GFP-VAMP7, GFP-YKT6 or GFP bound to GBP-Sepharose beads and processed the same way as in the STX5/SNAP29/GFP-SNARE complex.
Cell proliferation assay
To estimate the rate of proliferation of WT and KO cells over a 72 h period, the cells were seeded at 10% density on 24 well plates. 24 h after plating the nuclei were stained with Hoechst 33258 (Invitrogen, H1398). Cells were viewed and imaged under the Zeiss Axiovert 200M microscope driven by iVision 4.5. The 32X objective and the DAPI filter were used. Nuclei were counted using the Muti-point tool in ImageJ (NIH). The experiment was performed in triplicates with three fields imaged for each condition. To obtain the growth curve, the mean cell count vs time was plotted on a graph.
Statistical analysis
All results are representatives of at least three biological replicates. For statistical analysis, an unpaired student’s t-test was performed in Microsoft Excel 2010. All graphs were plotted in Microsoft Excel 2010. Error bars represent standard deviation.
Supplementary Material
Figure S2 (Related to figure 4): Knock-down (KD) of Golgi SNAREs STX5 or YKT6 causes glycosylation defects like downregulation of the COG complex. (A) WB analysis of protein lysates from HEK293T cells 6 days after transfection with siRNA to COG3, STX5 and YKT6. (B, C) Representative histogram and bar graph from flow-cytometry analysis of HPA-488 binding to impermeabilized WT and KD cells. An increase in HPA-488 intensity indicates the presence of under-processed O-glycans on the plasma membrane. The flow-cytometry data is a representation of biological triplicates. (D) Lectin blot detects the total amount of immature O-glycan labelled by HPA-647 in WT and KD cells. Monosaccharide symbols follow the Symbol Nomenclature for Glycans (SNFG) (107).
Figure S1 (Related to figure 1): Molecular and biochemical analysis of SNARE KO cells. (A) Sequence alignment of GS28 KO and WT chromosomal DNA. (B) Sequence alignment of GS15 KO and WT chromosomal DNA. Red boxes indicate the gRNA target sequences. (C) GS28 KO reduces GS15’s interaction with partner SNAREs. GS15-interacting SNAREs were coIPed from WT and GS28 KO cells and western blotted with anti-SNARE antibodies as indicated. (D) Graph represents fold changes in GS15 IP in GS28 KO compared to WT. **p<0.001, ***p<0.0001.
Figure S3 (Related to figure 7): Fluorescence Resonance Energy Transfer (FRET) analysis of SNAP29-STX5 interaction in vivo. (A) GS28/SNAP29 DKO HEK293T cells were co-transfected with plasmids encoding CFP-SNAP29 and YFP-STX5 (top panel) or Golgi-CFP and YFP-STX5 (bottom panel). After 24 h expression, physical interaction between YFP-STX5 and CFP-SNAP29 or Golgi-CFP was analyzed by the FRET protocol using LSM880 confocal microscope. (B) The graph represents the relative signal intensity of CFP and YFP fluorescence at indicated time points, n=20, Scale bars are 20 μm, error bars indicate SD.
Figure S4 (Related to figure 9): Simultaneous deletion of canonical and alternative STX5 partners disrupts Golgi morphology. (A) Lysates from WT, single, double, and triple SNARE KO cells were analyzed by WB with antibody indicated. (B) Transmission electron microscopy analysis of the Golgi region demonstrated unaltered Golgi morphology in GS28 KO cells and shows severe accumulation of Golgi bound vesicles in GS28/SNAP29/VTI1B TKO cells. Arrows point to vesicles. Scale bars are 500 nm
Figure S5 (Related to figure 9): TKO cells are unable to retain MGAT2-GFP or ST6GAL1-RFP (ST-RFP) in the Golgi. (A) WT, GS28 KO or TKO cells transiently co-expressing fluorescently tagged Golgi enzymes MGAT2-GFP and ST-RFP were immunostained for Golgi marker GM130. Both MGAT2 and ST6GAL1 are Golgi localized in WT and GS28 KO cells whereas TKO cells mislocalized both enzymes into multiple cytoplasmic puncta. (B) Expression of GS28-YFP rescues Golgi localization of ST-RFP in TKO cells. TKO cells were co-transfected with ST-RFP and FP-tagged SNAREs as indicated. Note that cells expressing GS28-YFP rescued Golgi localization of ST-RFP, while the expression of GFP-SNAP29 or GFP-VTI1B did not.
{kind=link}
Figure S6 (Related to figure 10): COG subunits are Golgi localized in SNARE KO cells and co-depletion of COG4 and GS28 is detrimental to cell growth. (A) IF of WT, GS28 KO, GS28/SNAP29/VTI1B TKO, COG4 KO and COG4/GS28 DKO cells stained for GM130 and COG8. (B) Growth curves of GS28 KO, COG4/GS28 DKO and GS28/SNAP29/VTI1B TKO cells over a period of 72 h.
Acknowledgments
We are thankful to all our colleagues who provided reagents. We are thankful to Wei Wang and Rose Willett for SNARE purification, Tetyana Kudlyk for technical support as well as Farhana Taher Sumya and Amrita Khakurel for critical discussion. We would also like to thank the UAMS Digital Microscopy, Proteomics and DNA sequencing cores for the use of their facilities and expertise.
Funding
This work was supported by the National Institute of Health grant R01GM083144 for Vladimir Lupashin
Footnotes
Conflict of Interest
No conflict of interests was declared.
Data availability
The mass spectrometry proteomics data for figure 5A have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD036741. The data can be accessed using the following credentials:
Username: reviewer_pxd036741@ebi.ac.uk
Password: 2kyixYK5
References:
- 1.Glick BS, Luini A. Models for Golgi traffic: a critical assessment. Cold Spring Harbor perspectives in biology 2011;3(11):a005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurokawa K, Osakada H, Kojidani T, Waga M, Suda Y, Asakawa H, Haraguchi T, Nakano A. Visualization of secretory cargo transport within the Golgi apparatus. The Journal of cell biology 2019;218(5):1602–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn JB, D'Souza Z, Lupashin VV. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS letters 2019;593(17):2466–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shestakova A, Zolov S, Lupashin V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 2006;7(2):191–204. [DOI] [PubMed] [Google Scholar]
- 5.Willett R, Ungar D, Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochemistry and cell biology 2013;140(3):271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Souza Z, Taher FS, Lupashin VV. Golgi inCOGnito: From vesicle tethering to human disease. Biochimica et biophysica acta General subjects 2020;1864(11):129694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linders PTA, Gerretsen ECF, Ashikov A, Vals MA, de Boer R, Revelo NH, Arts R, Baerenfaenger M, Zijlstra F, Huijben K, Raymond K, Muru K, Fjodorova O, Pajusalu S, Ounap K, et al. Congenital disorder of glycosylation caused by starting site-specific variant in syntaxin-5. Nature communications 2021;12(1):6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nature reviews Molecular cell biology 2006;7(9):631–643. [DOI] [PubMed] [Google Scholar]
- 9.Xu Y, Martin S, James DE, Hong W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Molecular biology of the cell 2002;13(10):3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu D, Joglekar AP, Williams AL, Hay JC. Subunit structure of a mammalian ER/Golgi SNARE complex. The Journal of biological chemistry 2000;275(50):39631–39639. [DOI] [PubMed] [Google Scholar]
- 11.Ganley IG, Espinosa E, Pfeffer SR. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. The Journal of cell biology 2008;180(1):159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mallard F, Tang BL, Galli T, Tenza D, Saint-Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. The Journal of cell biology 2002;156(4):653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suvorova ES, Duden R, Lupashin VV. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. The Journal of cell biology 2002;157(4):631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. The Journal of cell biology 2005;168(5):747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laufman O, Kedan A, Hong W, Lev S. Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. The EMBO journal 2009;28(14):2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohda M, Misumi Y, Yamamoto A, Nakamura N, Ogata S, Sakisaka S, Hirose S, Ikehara Y, Oda K. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic 2010;11(12):1552–1566. [DOI] [PubMed] [Google Scholar]
- 17.Kudlyk T, Willett R, Pokrovskaya ID, Lupashin V. COG6 interacts with a subset of the Golgi SNAREs and is important for the Golgi complex integrity. Traffic 2013;14(2):194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V. COG complexes form spatial landmarks for distinct SNARE complexes. Nature communications 2013;4:1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cottam NP, Wilson KM, Ng BG, Korner C, Freeze HH, Ungar D. Dissecting functions of the conserved oligomeric Golgi tethering complex using a cell-free assay. Traffic 2014;15(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fusella A, Micaroni M, Di Giandomenico D, Mironov AA, Beznoussenko GV. Segregation of the Qb-SNAREs GS27 and GS28 into Golgi vesicles regulates intra-Golgi transport. Traffic 2013;14(5):568–584. [DOI] [PubMed] [Google Scholar]
- 21.Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, Lesimple S, Nagaya H, Roy L, Gosline SJ, Hallett M, Paiement J, Kearney RE, Nilsson T, Bergeron JJ. Quantitative proteomics analysis of the secretory pathway. Cell 2006;127(6):1265–1281. [DOI] [PubMed] [Google Scholar]
- 22.Sumya FT, Pokrovskaya ID, D'Souza Z, Lupashin VV. Acute COG complex inactivation unveiled its immediate impact on Golgi and illuminated the nature of intra-Golgi recycling vesicles. Traffic 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Molecular biology of the cell 2004;15(5):2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shestakova A, Suvorova E, Pavliv O, Khaidakova G, Lupashin V. Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. The Journal of cell biology 2007;179(6):1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laufman O, Hong W, Lev S. The COG complex interacts with multiple Golgi SNAREs and enhances fusogenic assembly of SNARE complexes. Journal of cell science 2013;126(Pt 6):1506–1516. [DOI] [PubMed] [Google Scholar]
- 26.Diep J, Ooi YS, Wilkinson AW, Peters CE, Foy E, Johnson JR, Zengel J, Ding S, Weng KF, Laufman O, Jang G, Xu J, Young T, Verschueren E, Kobluk KJ, et al. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nature microbiology 2019;4(12):2523–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK, Haney MS, Hess GT, Han K, Jeng EE, Li A, Snyder MP, Greenleaf WJ, Kundaje A, Bassik MC. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nature communications 2017;8:15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian S, Muneeruddin K, Choi MY, Tao L, Bhuiyan RH, Ohmi Y, Furukawa K, Furukawa K, Boland S, Shaffer SA, Adam RM, Dong M. Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation. PLoS biology 2018;16(11):e2006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaji T, Hanamatsu H, Sekizuka T, Kuroda M, Iwasaki N, Ohnishi M, Furukawa JI, Yahiro K, Hanada K. A CRISPR Screen Using Subtilase Cytotoxin Identifies SLC39A9 as a Glycan-Regulating Factor. iScience 2019;15:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaji T, Sekizuka T, Tachida Y, Sakuma C, Morimoto K, Kuroda M, Hanada K. A CRISPR Screen Identifies LAPTM4A and TM9SF Proteins as Glycolipid-Regulating Factors. iScience 2019;11:409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, Marceau C, Janssen H, Carette JE, Bennett KL, Colinge J, et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015;350(6264):1092–1096. [DOI] [PubMed] [Google Scholar]
- 32.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999;285(5429):901–906. [DOI] [PubMed] [Google Scholar]
- 33.Shirakawa R, Goto-Ito S, Goto K, Wakayama S, Kubo H, Sakata N, Trinh DA, Yamagata A, Sato Y, Masumoto H, Cheng J, Fujimoto T, Fukai S, Horiuchi H. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. The EMBO journal 2020;39(8):e104120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linders PT, Horst CV, Beest MT, van den Bogaart G. Stx5-Mediated ER-Golgi Transport in Mammals and Yeast. Cells 2019;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin VV. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Frontiers in cell and developmental biology 2016;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paton AW, Paton JC. Escherichia coli Subtilase Cytotoxin. Toxins 2010;2(2):215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith RD, Willett R, Kudlyk T, Pokrovskaya I, Paton AW, Paton JC, Lupashin VV. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic 2009;10(10):1502–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D'Souza Z, Blackburn JB, Kudlyk T, Pokrovskaya ID, Lupashin VV. Defects in COG-Mediated Golgi Trafficking Alter Endo-Lysosomal System in Human Cells. Frontiers in cell and developmental biology 2019;7:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stribny J, Thines L, Deschamps A, Goffin P, Morsomme P. The human Golgi protein TMEM165 transports calcium and manganese in yeast and bacterial cells. The Journal of biological chemistry 2020;295(12):3865–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scherer PE, Lederkremer GZ, Williams S, Fogliano M, Baldini G, Lodish HF. Cab45, a novel (Ca2+)-binding protein localized to the Golgi lumen. The Journal of cell biology 1996;133(2):257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. The Journal of biological chemistry 1991;266(32):21327–21330. [PubMed] [Google Scholar]
- 42.Blackburn JB, Kudlyk T, Pokrovskaya I, Lupashin VV. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 2018;19(6):463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lebredonchel E, Houdou M, Potelle S, de Bettignies G, Schulz C, Krzewinski Recchi MA, Lupashin V, Legrand D, Klein A, Foulquier F. Dissection of TMEM165 function in Golgi glycosylation and its Mn(2+) sensitivity. Biochimie 2019;165:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morelle W, Potelle S, Witters P, Wong S, Climer L, Lupashin V, Matthijs G, Gadomski T, Jaeken J, Cassiman D, Morava E, Foulquier F. Galactose Supplementation in Patients With TMEM165-CDG Rescues the Glycosylation Defects. The Journal of clinical endocrinology and metabolism 2017;102(4):1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Blume J, Alleaume AM, Kienzle C, Carreras-Sureda A, Valverde M, Malhotra V. Cab45 is required for Ca(2+)-dependent secretory cargo sorting at the trans-Golgi network. The Journal of cell biology 2012;199(7):1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bojar D, Meche L, Meng G, Eng W, Smith DF, Cummings RD, Mahal LK. A Useful Guide to Lectin Binding: Machine-Learning Directed Annotation of 57 Unique Lectin Specificities. ACS chemical biology 2022;17(11):2993–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks SA. The involvement of Helix pomatia lectin (HPA) binding N-acetylgalactosamine glycans in cancer progression. Histology and histopathology 2000;15(1):143–158. [DOI] [PubMed] [Google Scholar]
- 48.Shibuya N, Goldstein IJ, Van Damme EJ, Peumans WJ. Binding properties of a mannose-specific lectin from the snowdrop (Galanthus nivalis) bulb. The Journal of biological chemistry 1988;263(2):728–734. [PubMed] [Google Scholar]
- 49.Bethani I, Lang T, Geumann U, Sieber JJ, Jahn R, Rizzoli SO. The specificity of SNARE pairing in biological membranes is mediated by both proof-reading and spatial segregation. The EMBO journal 2007;26(17):3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsui MM, Banfield DK. Yeast Golgi SNARE interactions are promiscuous. Journal of cell science 2000;113 (Pt 1):145–152. [DOI] [PubMed] [Google Scholar]
- 51.Rothman JE. Mechanisms of intracellular protein transport. Nature 1994;372(6501):55–63. [DOI] [PubMed] [Google Scholar]
- 52.Wilson DW, Whiteheart SW, Wiedmann M, Brunner M, Rothman JE. A multisubunit particle implicated in membrane fusion. The Journal of cell biology 1992;117(3):531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proceedings of the National Academy of Sciences of the United States of America 1988;85(21):7852–7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Antonin W, Holroyd C, Fasshauer D, Pabst S, Von Mollard GF, Jahn R. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. The EMBO journal 2000;19(23):6453–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smeele PH, Vaccari T. Snapshots from within the cell: Novel trafficking and non trafficking functions of Snap29 during tissue morphogenesis. Seminars in cell & developmental biology 2022. [DOI] [PubMed] [Google Scholar]
- 56.Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 2021;17(10):2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Q, Gao P, Arzberger T, Hollerhage M, Herms J, Hoglinger G, Koeglsperger T. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell death & disease 2021;12(10):854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morelli E, Speranza EA, Pellegrino E, Beznoussenko GV, Carminati F, Garre M, Mironov AA, Onorati M, Vaccari T. Activity of the SNARE Protein SNAP29 at the Endoplasmic Reticulum and Golgi Apparatus. Frontiers in cell and developmental biology 2021;9:637565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Forrester A, Rathjen SJ, Daniela Garcia-Castillo M, Bachert C, Couhert A, Tepshi L, Pichard S, Martinez J, Munier M, Sierocki R, Renard HF, Augusto Valades-Cruz C, Dingli F, Loew D, Lamaze C, et al. Functional dissection of the retrograde Shiga toxin trafficking inhibitor Retro-2. Nature chemical biology 2020;16(3):327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stechmann B, Bai SK, Gobbo E, Lopez R, Merer G, Pinchard S, Panigai L, Tenza D, Raposo G, Beaumelle B, Sauvaire D, Gillet D, Johannes L, Barbier J. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010;141(2):231–242. [DOI] [PubMed] [Google Scholar]
- 61.Tai G, Lu L, Wang TL, Tang BL, Goud B, Johannes L, Hong W. Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the trans-Golgi network. Molecular biology of the cell 2004;15(9):4011–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banfield DK, Lewis MJ, Pelham HR. A SNARE-like protein required for traffic through the Golgi complex. Nature 1995;375(6534):806–809. [DOI] [PubMed] [Google Scholar]
- 63.Laufman O, Freeze HH, Hong W, Lev S. Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes. Traffic 2013;14(10):1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steet R, Kornfeld S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Molecular biology of the cell 2006;17(5):2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khakurel A, Kudlyk T, Pokrovskaya I, D'Souza Z, Lupashin VV. GARP dysfunction results in COPI displacement, depletion of Golgi v-SNAREs and calcium homeostasis proteins. Frontiers in cell and developmental biology 2022;10:1066504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adolf F, Rhiel M, Hessling B, Gao Q, Hellwig A, Bethune J, Wieland FT. Proteomic Profiling of Mammalian COPII and COPI Vesicles. Cell reports 2019;26(1):250–265 e255. [DOI] [PubMed] [Google Scholar]
- 67.Lin CC, Love HD, Gushue JN, Bergeron JJ, Ostermann J. ER/Golgi intermediates acquire Golgi enzymes by brefeldin A-sensitive retrograde transport in vitro. The Journal of cell biology 1999;147(7):1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love HD, Lin CC, Short CS, Ostermann J. Isolation of functional Golgi-derived vesicles with a possible role in retrograde transport. The Journal of cell biology 1998;140(3):541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suga K, Hattori H, Saito A, Akagawa K. RNA interference-mediated silencing of the syntaxin 5 gene induces Golgi fragmentation but capable of transporting vesicles. FEBS letters 2005;579(20):4226–4234. [DOI] [PubMed] [Google Scholar]
- 70.Lupashin VV, Pokrovskaya ID, McNew JA, Waters MG. Characterization of a novel yeast SNARE protein implicated in Golgi retrograde traffic. Molecular biology of the cell 1997;8(12):2659–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.von Mollard GF, Nothwehr SF, Stevens TH. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. The Journal of cell biology 1997;137(7):1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dilcher M, Kohler B, von Mollard GF. Genetic interactions with the yeast Q-SNARE VTI1 reveal novel functions for the R-SNARE YKT6. The Journal of biological chemistry 2001;276(37):34537–34544. [DOI] [PubMed] [Google Scholar]
- 73.Parlati F, Varlamov O, Paz K, McNew JA, Hurtado D, Sollner TH, Rothman JE. Distinct SNARE complexes mediating membrane fusion in Golgi transport based on combinatorial specificity. Proceedings of the National Academy of Sciences of the United States of America 2002;99(8):5424–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smeele PH, Vaccari T. Snapshots from within the cell: Novel trafficking and non trafficking functions of Snap29 during tissue morphogenesis. Seminars in cell & developmental biology 2023;133:42–52. [DOI] [PubMed] [Google Scholar]
- 75.Mastrodonato V, Morelli E, Vaccari T. How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health. Cell stress 2018;2(4):72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morelli E, Ginefra P, Mastrodonato V, Beznoussenko GV, Rusten TE, Bilder D, Stenmark H, Mironov AA, Vaccari T. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy 2014;10(12):2251–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hohenstein AC, Roche PA. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochemical and biophysical research communications 2001;285(2):167–171. [DOI] [PubMed] [Google Scholar]
- 78.Wong SH, Xu Y, Zhang T, Griffiths G, Lowe SL, Subramaniam VN, Seow KT, Hong W. GS32, a novel Golgi SNARE of 32 kDa, interacts preferentially with syntaxin 6. Molecular biology of the cell 1999;10(1):119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH. Three novel proteins of the syntaxin/SNAP-25 family. The Journal of biological chemistry 1998;273(51):34171–34179. [DOI] [PubMed] [Google Scholar]
- 80.Schardt A, Brinkmann BG, Mitkovski M, Sereda MW, Werner HB, Nave KA. The SNARE protein SNAP-29 interacts with the GTPase Rab3A: Implications for membrane trafficking in myelinating glia. Journal of neuroscience research 2009;87(15):3465–3479. [DOI] [PubMed] [Google Scholar]
- 81.Tian X, Zheng P, Zhou C, Wang X, Ma H, Ma W, Zhou X, Teng J, Chen J. DIPK2A promotes STX17- and VAMP7-mediated autophagosome-lysosome fusion by binding to VAMP7B. Autophagy 2020;16(5):797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yong CQY, Tang BL. Another longin SNARE for autophagosome-lysosome fusion-how does Ykt6 work? Autophagy 2019;15(2):352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saleeb RS, Kavanagh DM, Dun AR, Dalgarno PA, Duncan RR. A VPS33A-binding motif on syntaxin 17 controls autophagy completion in mammalian cells. The Journal of biological chemistry 2019;294(11):4188–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takats S, Glatz G, Szenci G, Boda A, Horvath GV, Hegedus K, Kovacs AL, Juhasz G. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS genetics 2018;14(4):e1007359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. The Journal of cell biology 2018;217(8):2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cho NH, Cheveralls KC, Brunner AD, Kim K, Michaelis AC, Raghavan P, Kobayashi H, Savy L, Li JY, Canaj H, Kim JYS, Stewart EM, Gnann C, McCarthy F, Cabrera JP, et al. OpenCell: Endogenous tagging for the cartography of human cellular organization. Science 2022;375(6585):eabi6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012;151(6):1256–1269. [DOI] [PubMed] [Google Scholar]
- 88.Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. The Journal of biological chemistry 2006;281(41):30947–30956. [DOI] [PubMed] [Google Scholar]
- 89.Sun X, Mahajan D, Chen B, Song Z, Lu L. A quantitative study of the Golgi retention of glycosyltransferases. Journal of cell science 2021;134(20). [DOI] [PubMed] [Google Scholar]
- 90.Lavieu G, Zheng H, Rothman JE. Stapled Golgi cisternae remain in place as cargo passes through the stack. eLife 2013;2:e00558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman HA, Subtil A, Rothman JE. Intracellular bacteria encode inhibitory SNARE-like proteins. PloS one 2009;4(10):e7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Khakurel A, Kudlyk T, Bonifacino JS, Lupashin VV. The Golgi-associated retrograde protein (GARP) complex plays an essential role in the maintenance of the Golgi glycosylation machinery. Molecular biology of the cell 2021;32(17):1594–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blackburn JB, Lupashin VV. Creating Knockouts of Conserved Oligomeric Golgi Complex Subunits Using CRISPR-Mediated Gene Editing Paired with a Selection Strategy Based on Glycosylation Defects Associated with Impaired COG Complex Function. Methods in molecular biology 2016;1496:145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rausch T, Fritz MH, Untergasser A, Benes V. Tracy: basecalling, alignment, assembly and deconvolution of sanger chromatogram trace files. BMC genomics 2020;21(1):230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perez-Victoria FJ, Bonifacino JS. Dual roles of the mammalian GARP complex in tethering and SNARE complex assembly at the trans-golgi network. Molecular and cellular biology 2009;29(19):5251–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods 2009;6(5):359–362. [DOI] [PubMed] [Google Scholar]
- 97.Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villen J, MacCoss MJ. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nature communications 2018;9(1):5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Graw S, Tang J, Zafar MK, Byrd AK, Bolden C, Peterson EC, Byrum SD. proteiNorm - A User-Friendly Tool for Normalization and Analysis of TMT and Label-Free Protein Quantification. ACS omega 2020;5(40):25625–25633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002;18 Suppl 1:S96–104. [DOI] [PubMed] [Google Scholar]
- 100.Bolstad B. preprocessCore: A collection of pre-processing functions. 1.58.0 ed: Bioconductor; 2022. [Google Scholar]
- 101.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chawade A, Alexandersson E, Levander F. Normalyzer: a tool for rapid evaluation of normalization methods for omics data sets. Journal of proteome research 2014;13(6):3114–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cocchiaro JL, Valdivia RH. New insights into Chlamydia intracellular survival mechanisms. Cellular microbiology 2009;11(11):1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pokrovskaya ID, Szwedo JW, Goodwin A, Lupashina TV, Nagarajan UM, Lupashin VV. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cellular microbiology 2012;14(5):656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Willett R, Blackburn JB, Climer L, Pokrovskaya I, Kudlyk T, Wang W, Lupashin V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Scientific reports 2016;6:29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Willett R, Pokrovskaya I, Kudlyk T, Lupashin V. Multipronged interaction of the COG complex with intracellular membranes. Cellular logistics 2014;4(1):e27888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T, Prestegard JJ, Schnaar RL, Freeze HH, Marth JD, Bertozzi CR, et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015;25(12):1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S2 (Related to figure 4): Knock-down (KD) of Golgi SNAREs STX5 or YKT6 causes glycosylation defects like downregulation of the COG complex. (A) WB analysis of protein lysates from HEK293T cells 6 days after transfection with siRNA to COG3, STX5 and YKT6. (B, C) Representative histogram and bar graph from flow-cytometry analysis of HPA-488 binding to impermeabilized WT and KD cells. An increase in HPA-488 intensity indicates the presence of under-processed O-glycans on the plasma membrane. The flow-cytometry data is a representation of biological triplicates. (D) Lectin blot detects the total amount of immature O-glycan labelled by HPA-647 in WT and KD cells. Monosaccharide symbols follow the Symbol Nomenclature for Glycans (SNFG) (107).
Figure S1 (Related to figure 1): Molecular and biochemical analysis of SNARE KO cells. (A) Sequence alignment of GS28 KO and WT chromosomal DNA. (B) Sequence alignment of GS15 KO and WT chromosomal DNA. Red boxes indicate the gRNA target sequences. (C) GS28 KO reduces GS15’s interaction with partner SNAREs. GS15-interacting SNAREs were coIPed from WT and GS28 KO cells and western blotted with anti-SNARE antibodies as indicated. (D) Graph represents fold changes in GS15 IP in GS28 KO compared to WT. **p<0.001, ***p<0.0001.
Figure S3 (Related to figure 7): Fluorescence Resonance Energy Transfer (FRET) analysis of SNAP29-STX5 interaction in vivo. (A) GS28/SNAP29 DKO HEK293T cells were co-transfected with plasmids encoding CFP-SNAP29 and YFP-STX5 (top panel) or Golgi-CFP and YFP-STX5 (bottom panel). After 24 h expression, physical interaction between YFP-STX5 and CFP-SNAP29 or Golgi-CFP was analyzed by the FRET protocol using LSM880 confocal microscope. (B) The graph represents the relative signal intensity of CFP and YFP fluorescence at indicated time points, n=20, Scale bars are 20 μm, error bars indicate SD.
Figure S4 (Related to figure 9): Simultaneous deletion of canonical and alternative STX5 partners disrupts Golgi morphology. (A) Lysates from WT, single, double, and triple SNARE KO cells were analyzed by WB with antibody indicated. (B) Transmission electron microscopy analysis of the Golgi region demonstrated unaltered Golgi morphology in GS28 KO cells and shows severe accumulation of Golgi bound vesicles in GS28/SNAP29/VTI1B TKO cells. Arrows point to vesicles. Scale bars are 500 nm
Figure S5 (Related to figure 9): TKO cells are unable to retain MGAT2-GFP or ST6GAL1-RFP (ST-RFP) in the Golgi. (A) WT, GS28 KO or TKO cells transiently co-expressing fluorescently tagged Golgi enzymes MGAT2-GFP and ST-RFP were immunostained for Golgi marker GM130. Both MGAT2 and ST6GAL1 are Golgi localized in WT and GS28 KO cells whereas TKO cells mislocalized both enzymes into multiple cytoplasmic puncta. (B) Expression of GS28-YFP rescues Golgi localization of ST-RFP in TKO cells. TKO cells were co-transfected with ST-RFP and FP-tagged SNAREs as indicated. Note that cells expressing GS28-YFP rescued Golgi localization of ST-RFP, while the expression of GFP-SNAP29 or GFP-VTI1B did not.
Figure S6 (Related to figure 10): COG subunits are Golgi localized in SNARE KO cells and co-depletion of COG4 and GS28 is detrimental to cell growth. (A) IF of WT, GS28 KO, GS28/SNAP29/VTI1B TKO, COG4 KO and COG4/GS28 DKO cells stained for GM130 and COG8. (B) Growth curves of GS28 KO, COG4/GS28 DKO and GS28/SNAP29/VTI1B TKO cells over a period of 72 h.
Data Availability Statement
The mass spectrometry proteomics data for figure 5A have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD036741. The data can be accessed using the following credentials:
Username: reviewer_pxd036741@ebi.ac.uk
Password: 2kyixYK5