

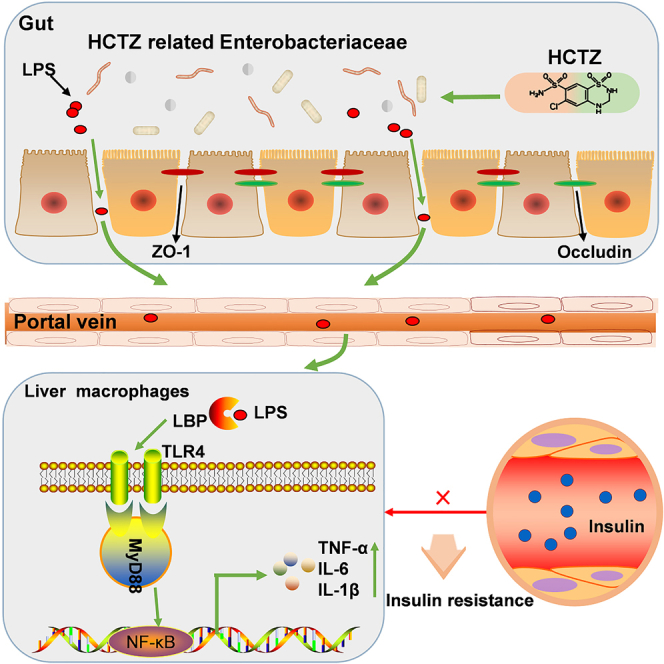
Summary
Hydrochlorothiazide (HCTZ) is reported to impair glucose tolerance and may induce new onset of diabetes, but the pharmacomicrobiomics of the adverse effect for HCTZ remains unknown. Mice-fed HCTZ exhibited insulin resistance and impaired glucose tolerance. By using FMT and antibiotic cocktail models, we found that HCTZ-induced metabolic disorder was mediated by commensal microbiota. HCTZ consumption disturbed the structure of the intestinal microbiota, causing abnormal elevation of Gram-negative Enterobacteriaceae and lipopolysaccharide (LPS) then leading to intestinal barrier dysfunction. Additionally, HCTZ activated TLR4 signaling and induced macrophage polarization and inflammation in the liver. Furthermore, HCTZ-induced macrophage polarization and metabolic disorder were abrogated by blocking TLR4 signaling. HCTZ consumption caused a significant increase in Gram-negative Enterobacteriaceae, which elevated the levels of LPS, thereby activating LPS/TLR4 pathway, promoting inflammation and macrophage polarization, and resulting in metabolic disorders. These findings revealed that the gut microbiome is the key medium underlying HCTZ-induced metabolic disorder.
Subject areas: Biological sciences, Molecular physiology, Immunology, Cell biology
Graphical abstract
Highlights
-
•
HCTZ consumption caused abnormal elevation of Enterobacteriaceae and LPS
-
•
LPS activated TLR4/MyD88/NF-κB pathways and macrophage polarization
-
•
Macrophage polarization led to metabolism disorder in TLR4-dependent manner
Biological sciences; Molecular physiology; Immunology; Cell biology
Introduction
Hydrochlorothiazide (HCTZ), a thiazide-like diuretic widely used for the long-term management of hypertension, has been proven to reduce the risk of stroke, myocardial infarction, and heart failure.1 Nevertheless, compelling evidence from numerous trials and meta-analyses indicate that thiazide diuretics potentially increase the risk of metabolic abnormalities, including hyperlipidemia, insulin resistance, and new-onset diabetes, especially in persons with abdominal obesity accompanied by metabolic abnormalities.2,3 Furthermore, the data from the European working party on hypertension in the elderly demonstrated that hyperglycemia induced by HCTZ was not an adverse effect experienced by a few patients but an impairment in glucose tolerance overall.4 This risk prevents the recommendation of HCTZ in guidelines and limits its clinical application.
Although metabolic abnormalities have long been linked to HCTZ therapy, the precise mechanisms are not well understood. Every 0.5-mEq/L decrease in serum potassium predicts a 45% increase in diabetes risk.5 One prevailing theory is that this risk attributed to thiazide-induced hypokalemia and might be compensatorily prevented by potassium-sparing diuretics.6 However, several other studies claim that there is no correlation between serum potassium and insulin sensitivity or blood glucose levels during thiazide-like diuretic treatment.7,8 Moreover, even though pharmacogenomics has advanced our knowledge about the role of genes in the HCTZ response, the mechanism underlying HCTZ-induced metabolic abnormalities is still ambiguous.9,10
In recent years, growing evidence has emphasized the critical impact of the gut microbiota on the pathologies of common chronic metabolic disorders, including obesity, insulin resistance, and type 2 diabetes mellitus.11 The probiotics Lactobacillus and Bifidobacterium contribute to glucostasis by competing with pathogens and re-establishing intestinal balance.12 Another potentially insulin-sensitizing bacterial species is Akkermansia muciniphila, which might be administered safely to improve metabolic abnormalities in individuals with obesity.13 Gram-negative Enterobacteriaceae, representing a taxonomic signature of gut microbiota dysbiosis, is enriched in individuals with diabetes.14 Microbiota-derived components and microbial metabolites, including lipopolysaccharide (LPS), bile acids (BA), trimethylamine N-oxide (TMAO), and short-chain fatty acids (SCFAs), are correlated with host metabolism.15 LPS penetrates the intestinal barrier and triggers metabolic endotoxemia, resulting in low-grade inflammation and metabolic diseases.16,17
HCTZ is able to bind the NADPH binding region of bacterial dihydrofolate reductase, which has the potential to modulate the gut microbiota.18 However, the role of HCTZ in modulating the gut microbiota has not been reported. Gut microbiota-derived metabolites or components involved in HCTZ-induced metabolic disorders are also unknown.
In this study, we aimed to explore the causal relationship between the HCTZ-driven gut microbiota and metabolic disorder in mice. Our findings showed that HCTZ-impaired glucose tolerance was mediated by the gut microbiota which activated hepatic macrophage polarization and inflammation through the LPS-TLR4 pathway. We demonstrated the effects of the gut microbiota on HCTZ-induced metabolic disorder.
Results
HCTZ-induced glucose intolerance mediated by the gut microbiota
Results from pilot study indicated that HCTZ-fed mice showed a slow and steady increase in fasting blood glucose (FBG) levels. Compared with the control group, 10 mg/kg HCTZ significantly increased FBG at week 4, whereas 5 mg/kg HCTZ significantly increased FBG at week 8 (Figures S1A–S1C). To obtain a better understanding of HCTZ-induced metabolic disorder, 10 mg/kg·d HCTZ was used for further research.
Considering persons with abdominal obesity accompanying metabolic abnormalities are more likely to develop new onset of diabetes,8 we added high-fat diet (HFD) group. Mice were exposed to HCTZ or vehicle control and were fed a normal chow diet or a high-fat diet (Figure 1A). As expected, the high-fat diet aggravated HCTZ-induced metabolic disorder. A faster body weight gain trend was observed in the HFD-HCTZ group than in the HFD group (Figure 1B). HCTZ exposure resulted in a significant increase in FBG, FINS, and HOMA-IR and an impairment in glucose tolerance compared with controls in both the NCD- and HFD-fed groups (Figures 1C–1G). Islet morphology exhibited an increased islet size induced by HCTZ on two diets (Figure 1H).
Figure 1.

HCTZ consumption led to metabolic disorder
(A) Schematic of the HCTZ treatment of mice in both the NCD- and HFD-fed groups.
(B–G) Body weight (B), FBG (C), FINS (D), HOMA-IR (E), OGTT (F), and OGTT-AUC (G) of mice treated with HCTZ or vehicle control (n = 5–8 per group). ∗p < 0.05, ∗∗p < 0.01. Data are represented as mean ± SEM.
(H) Representative immunofluorescence images of insulin in islets. Scale bars = 500 μm.
Antibiotic cocktails (Abx) were used for gut sterilization (Figure S2A), which had no impact on body weight (Figure S2B). Notably, after antibiotic treatment, the differences in metabolic abnormalities between HCTZ-treated mice and control were abolished in both the NCD- and HFD-fed groups (Figures S2C–S2G). These results suggested that HCTZ-induced glucose metabolism disturbance was mediated by the gut microbiota.
FMT experiments were carried out to directly address the effect of the HCTZ-driven gut microbiota on glucose metabolism (Figure S2H). No significant difference in weight or FBG was observed between these groups (Figures S2I and S2J). However, recipients of fecal microbiota from HCTZ-treated mice exhibited a significant increase in FINS and HOMA-IR levels and glucose intolerance compared with those of controls (Figures S2K–S2N), as did recipients of FMT of HFD-HCTZ mice.
HCTZ remodulated the composition of gut microbiota and plasma metabolome
A tendency toward decreased alpha diversity (Chao index, Shannon index, Ace index, and Simpson index) was observed after HCTZ treatment (Figure 2A). The PCoA of UniFrac distances revealed a remarkable separation of bacterial composition, suggesting that a shift in the gut microbiota was induced by HCTZ treatment in both the NCD- and HFD-fed groups (Figure 2B). Using linear discriminant analysis effect size (LEfSe), we observed a notable clustering effect in the gut microbiota induced by HCTZ (Figures S3A and S3B). HCTZ changed the family level proportional abundance in the feces of mice and caused an enrichment of Enterobacteriaceae (Figure 2C). Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis revealed that the term “lipopolysaccharide biosynthesis” was significantly enriched in HCTZ-treated mice in the HFD-fed group (Figure 2D).
Figure 2.

HCTZ consumption altered the gut microbiota profiles
(A) Alpha diversity (Chao index, Shannon index, Ace index, and Simpson index) of fecal samples (n = 5–7 per group).
(B) Principal coordinates analysis of fecal samples (n = 5–7 per group).
(C) Abundance of Enterobacteriaceae in the feces of mice treated with HCTZ or vehicle control under normal chow diet (left) and high-fat diet (right) (n = 5–7 per group). ∗∗p < 0.01. Data are represented as mean ± SEM.
(D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the most significantly changed pathways according to 16S rRNA sequencing of fecal samples.
Untargeted metabolite profiling characterized the plasma metabolome. The orthogonal projections to latent structures (OPLS) model exhibited a significant differentiation of clusters with HCTZ treatment on two diets (Figure S3C). Differential metabolites were annotated with pathway enrichment analysis using KEGG. The metabolomics map revealed that significant changes were enriched in the mTOR-signaling pathway and amino acid-related metabolic pathways such as valine, leucine, and isoleucine biosynthesis pathways; arginine and proline metabolism pathways; and D-arginine and D-ornithine metabolism pathways (Figure S3D).
HCTZ impaired the gastrointestinal barrier
We observed a significant increase in FITC-labeled dextran in serum, suggesting impaired intestinal permeability after HCTZ treatment in both the NCD- and HFD-fed groups (Figure 3A). The mRNA expression levels of tight junction proteins (occludin and ZO-1) were significantly decreased in the terminal ileum in HCTZ-treated mice (Figures 3B and 3C), while no significant difference was observed in the colon (Figures S4A and S4B). In line with the mRNA expression results, the immunofluorescence results also showed the effect of HCTZ on occludin and ZO-1 protein levels (Figure 3D). The intestinal morphology of the ileum indicated that HCTZ decreased the muscularis thickness (red arrows), the quantities of goblet cells (yellow arrows), and the villus integrity, especially in the HFD-fed group (Figure 3E). These results indicated that HCTZ treatment led to impairment in gut barrier function in the ileum.
Figure 3.

HCTZ impaired the gastrointestinal barrier
(A) HCTZ increased the appearance of FITC-dextran in serum in both the NCD- and HFD-fed groups (n = 5–8 per group). ∗∗p < 0.01.
(B and C) HCTZ decreased the expression of the tight junction proteins occludin (B) and ZO-1 (C) in the ileum (n = 5–8 per group). ∗p < 0.05, ∗∗p < 0.01. Data are represented as mean ± SEM.
(D) Representative immunofluorescence images for occludin (green) and ZO-1 (red) in the ileum are shown. Scale bar = 50 μm.
(E) Representative images of H&E staining. Red arrows indicate muscularis thickness. Yellow arrows indicate goblet cells. Scale bar = 50 μm.
LPS contributed to HCTZ-induced inflammation and insulin resistance
LPS concentrations were increased in HCTZ-treated mice both in the cecal contents (Figure 4A) and serum (Figure 4B), especially in the HFD-fed group. More importantly, a positive correlation between serum LPS and HOMA-IR in the HFD-fed group was observed (Figure 4C). The inflammatory cytokines TNF-α, IL-1β, and IL-6 were significantly elevated in serum samples from mice treated with HCTZ (Figures 4D–4F). In FMT experiment, recipients of fecal microbiota from HCTZ-treated mice exhibited an obvious increase in LPS concentrations in the cecal contents, as well as inflammatory cytokines TNF-α, IL-1β, and IL-6 in serum samples (Figures 4G–4J). These data suggested that the HCTZ-induced LPS and inflammatory cytokines were produced by the gut microbiota. To directly address the specific role of Gram-negative bacteria-derived LPS on HCTZ-induced metabolic disorder, neomycin was used to selectively target Gram-negative bacteria (Figure S5A). As expected, neomycin treatment prevented metabolic derangements induced by HCTZ in the high-fat diet setting (Figures S5B–S5F). These data demonstrated that metabolic imbalance in HCTZ treatment was mediated by intestinal Gram-negative bacteria.
Figure 4.

LPS from the microbiota contributes to HCTZ-induced inflammation and insulin resistance
(A and B) Cecal content (A) and serum (B) LPS levels in mice treated with HCTZ or vehicle control (n = 6–8 per group). ∗p < 0.05. Data are represented as mean ± SEM.
(C) Correlation between serum LPS and HOMA-IR in the HFD-fed group (n = 5–8 per group).
(D–F) Serum TNF-α (D), IL-1β (E), and IL-6 (F) in mice treated with HCTZ or vehicle control (n = 5–8 per group). ∗p < 0.05. Data are represented as mean ± SEM.
(G) Serum LPS levels in recipient mice in the FMT experiment (n = 6–8 per group). ∗p < 0.05. Data are represented as mean ± SEM.
(H–J) Serum TNF-α (H), IL-1β (I), and IL-6 (J) in recipient mice in the FMT experiment (n = 6–8 per group). ∗p < 0.05. Data are represented as mean ± SEM.
HCTZ caused hepatic macrophage polarization and inflammation through a TLR4-dependent mechanism
To explore the detailed mechanism by which HCTZ induced metabolic disorders, we investigated the induction of hepatic macrophage polarization and inflammation by HCTZ. The expression of downstream genes of the LPS/TLR4 pathway, including MyD88, NF-κB, TNF-α, IL-1β, and IL-6, were significantly increased after HCTZ treatment in both NCD- and HFD-fed groups (Figures 5A–5E). The elevated protein levels of MyD88, NF-κB, TNF-α, IL-1β, and IL-6 were further confirmed by Western blotting (Figures 5F–5K). Histological analysis revealed that HCTZ treatment facilitated hepatic pathological changes, including inflammation and necrosis, compared with controls on two diets (Figure 5L), while livers from HCTZ-treated mice had more lipid vacuoles in the HFD-fed group (Figure 5L). Consistently, the expression levels of MyD88, NF-κB, TNF-α, IL-1β, and IL-6 in the liver were higher after FMT with stool samples derived from mice treated with HCTZ than with the control in both the NCD- and HFD-fed groups (Figures S6A–S6K), suggesting that HCTZ activated the LPS/TLR4 signaling pathway in a gut microbiota-dependent manner. We further quantified the immune cell populations of M1/M2 macrophages in the liver in response to HCTZ treatment by flow cytometry. We screened F4/80+ macrophages in the liver (Figure S7). Interestingly, under two diets, the M1-specific polarization marker CD80 was induced by HCTZ in F4/80+ macrophages (Figures 5M and 5N), as well as the prominent M2 marker CD206 (Figures 5O and 5P).
Figure 5.

HCTZ caused hepatic macrophage polarization and inflammation through a TLR4-dependent mechanism
(A–E) Relative MyD88 (A), NF-κB (B), TNF-α (C), IL-1β (D), and IL-6 (E) expression in the livers of mice treated with HCTZ or vehicle control (n = 5–8 per group). ∗p < 0.05, ∗∗p < 0.01. Data are represented as mean ± SEM.
(F–K) Representative western blots (F) and quantification of MyD88 (G), NF-κB (H), TNF-α (I), IL-1β (J), and IL-6 (K) levels in the livers of mice treated with HCTZ or vehicle control (n = 3–4 per group). ∗p < 0.05. Data are represented as mean ± SEM.
(L) Representative H&E staining in the livers of mice treated with HCTZ or vehicle control.
(M and N) (M)Representative CD80 staining of hepatic F4/80+ macrophages in mice treated with HCTZ or vehicle control.
(N) Flow cytometry analysis of CD80+-F4/80+ macrophages (n = 3–4 per group). ∗p < 0.05. Data are represented as mean ± SEM.
(O and P) (O)Representative CD206 staining of hepatic F4/80+ macrophages in mice treated with HCTZ or vehicle control.
(P) Flow cytometry analysis of CD206+-F4/80+ macrophages (n = 3–4 per group). ∗p < 0.05. Data are represented as mean ± SEM.
The macrophage polarization and inflammatory effects of HCTZ were dependent on Toll-like receptor 4 (TLR4) signaling
Since macrophage polarization and inflammation could decrease hepatogenous insulin sensitivity,19 we tested the impacts of HCTZ on insulin sensitivity. Mice that were intraperitoneally treated with the pharmacological TLR4 inhibitor TAK-242 displayed lower weight gain than that of control mice (Figures S8A and S8B), consistent with the observations in HFD-fed mice. HCTZ treatment successfully increased FBG, FINS, and HOMA-IR and impaired glucose tolerance and insulin sensitivity, while TAK-242 treatment counteracted the HCTZ-induced glucose metabolism disorder (Figures 6A–6G). Accordingly, pharmacological TLR4 inhibition intercepted HCTZ-induced hepatic activation of the TLR4/MyD88/NF-κB pathway, macrophage polarization, and inflammatory cytokine expression, including TNF-α, IL-1β, and IL-6 (Figures S8C–S8H).
Figure 6.

The macrophage polarization and inflammatory effects of HCTZ were dependent on TLR4 signaling
(A–N) FBG (A), FINS (B), HOMA-IR (C), OGTT (D), OGTT-AUC (E), ITT (F), and ITT-AUC (G) in mice treated with the TLR4 inhibitor TAK-242 (n = 6–8 per group); FBG (H), FINS (I), HOMA-IR (J), OGTT (K), OGTT-AUC (L), ITT (M), and ITT-AUC (N) in WT or Tlr4−/− mice treated with HCTZ or vehicle control (n = 4–5 per group). ∗p < 0.05, ∗∗p < 0.01. Data are represented as mean ± SEM.
(O and P) (O) Representative CD80 staining of hepatic F4/80+ macrophages of WT or Tlr4−/− mice treated with HCTZ or vehicle control.
(P) Flow cytometry analysis of CD80+-F4/80+ macrophages (n = 4–5 per group). ∗∗p < 0.01. Data are represented as mean ± SEM.
(Q and R) (Q) Representative CD206 staining of hepatic F4/80+ macrophages of WT or Tlr4−/− mice treated with HCTZ or vehicle control.
(R) Flow cytometry analysis of CD206+-F4/80+ macrophages (n = 4–5 per group). ∗∗p < 0.01. Data are represented as mean ± SEM.
TLR4 deficiency increased weight gain compared with that of the wild-type (WT) controls (Figures S8I and S8J). Consistent with pharmacological inhibition, WT mice treated with HCTZ exhibited increased FBG, FINS, and HOMA-IR and impaired glucose tolerance and insulin sensitivity, while TLR4 deficiency abrogated HCTZ-induced glucose metabolism disorder (Figures 6H–6N). Similarly, HCTZ-related hepatic activation of TLR4 downstream signaling, macrophage polarization and inflammatory cytokine expression were abolished in Tlr4−/− mice (Figures 6O–6R, S8K and S8L). Together, these data suggested that the absence of TLR4 signaling was sufficient to prevent HCTZ-induced glucose metabolism disorder by effectively disturbing macrophage polarization and inflammation in the liver.
Discussion
Our data revealed that the potential mechanisms underlying HCTZ-induced metabolic disorder were as follows: (1) HCTZ consumption led to microbiota alteration, causing abnormal elevation of Gram-negative Enterobacteriaceae and its metabolite LPS; (2) the HCTZ-induced microbial dysbiosis disrupted the intestinal barrier and increased intestinal permeability, which allowed intestinal LPS to translocate into the circulation; and (3) LPS activated TLR4/MyD88/NF-κB pathways and induced macrophage polarization and inflammation in the liver, ultimately leading to metabolic disorder.
HCTZ remodulated the gut microbial composition of mice. The proportion at the family level was significantly changed by HCTZ exposure, and the main change was the enrichment of Enterobacteriaceae. Enterobacteriaceae, the main Gram-negative bacteria, is associated with gut microbiota dysbiosis and systemic inflammation.20 Previous studies have reported an enhanced ratio of Enterobacteriaceae in individuals with diabetes in both the plasma and mesenteric adipose tissue.14 In this study, neomycin, which is usually used to deplete Gram-negative bacteria in the gut,21 prevented HCTZ-induced metabolic derangements. A diet containing whole grains, prebiotics and traditional Chinese medicinal foods reduced Enterobacteriaceae levels and ameliorated inflammation and metabolic phenotypes in overweight people,22 suggesting that HCTZ-induced metabolic disorders might be controlled via dietary intervention.
Microbiota-derived components and microbial metabolites are considered pivotal mediators of host-microbiota interactions. LPS is a cell wall structural component of most Gram-negative bacteria and is released during bacterial lysis. Evidence has demonstrated that LPS contributes to low-grade inflammation, which is the hallmark of metabolic disorders.23,24 In mouse models, HFD increased serum LPS and gut microbiota that produce LPS, leading to insulin resistance, which is consistent with the changes caused by HCTZ.25 In our FMT studies, recipients of fecal microbiota from HCTZ-treated mice exhibited an obvious increase in LPS concentrations in the cecal contents at the endpoint. It proved that HCTZ-induced LPS elevation can be transferred to recipient mice through FMT. However, the deficiency lies in our negligence to detect the level of LPS-producing bacteria in recipient mice. Furthermore, the metabolomics map described the effect of HCTZ on the mTOR signaling pathway and amino acid-related metabolic pathways, which are involved in the LPS-mediated physiological changes, including macrophage polarization, immune activation, and metabolic changes.26,27
Long-term microbial dysbiosis causes a disrupted intestinal barrier, which, in turn, allows the transfer of the intestinal microbiota and its metabolites to various metabolic organs and results in metabolic inflammation.28 Ileum contains abundant anaerobic microbiota. Ileal dysbiosis was dominated by the expansion of Enterobacteriaceae which accounted for over 80% of the 16S sequences.29 Compared with the colon, the ileum, with a thinner nonstratified mucus layer, is more readily penetrable by bacteria-sized particles.30 Accordingly, HCTZ exposure increased the intestinal permeability and downregulated the tight junction proteins occludin and ZO-1 in the ileum rather than in the colon.
Intestinal LPS translocates to the systemic circulation and is then quickly recognized by Toll-like receptor 4 (TLR4).31 TLR4 recruits the intracellular adapter molecule MyD88 and then activates downstream signaling cascades, resulting in the induction and translocation of NF-κB and subsequent inflammation.32 Immune cells, such as macrophages, have long been investigated at the molecular level for the role they play in regulating metabolic homeostasis. TLR4 signaling-mediated macrophage polarization and inflammation promote insulin resistance in hepatocytes.33 This evidence prompted us to explore in more detail whether hepatic macrophage polarization plays a part in HCTZ-induced metabolic derangements.
Macrophages are often classified into two phenotypes: classically activated (M1) macrophages and alternatively activated (M2) macrophages.34 The polarization of macrophages activated by LPS is a multistep process beginning with the recognition of TLR4 and the subsequent downstream signaling cascades.35 LPS-TLR4 signaling promotes M1 macrophage polarization at the early stage of inflammation and mediates the normal defense response to infection, while M2 macrophages exert anti-inflammatory effects at the end of inflammation.34,36 Abnormal macrophage polarization, with an increase in proinflammatory M1 macrophages, enhances hepatic inflammation and liver injury in obesity induced by an HFD.35 Here, we observed simultaneous activation of M1-and M2-polarized macrophages in HCTZ-treated mouse livers, which could be explained by the distinct role of LPS as an upstream regulator of macrophage polarization. Furthermore, in in vivo and ex vivo studies, deletion of TLR4 not only reduced the secretion of proinflammatory mediators in macrophages but also counteracted obesity-associated insulin resistance.37,38 Correspondingly, by blocking TLR4 signaling pharmacologically and genetically, the HCTZ-induced TLR4 signaling activation, macrophage polarization, and increases in inflammatory cytokines in the liver, as well as the metabolic disorder, were abrogated.
In conclusion, the current study proposed an important step toward understanding how HCTZ regulates the gut microbiota and induces metabolic abnormalities. Although these data are based on murine models and still need to be verified in human trials, they provide new pathophysiological insights into the causal relationship between the gut microbiota and HCTZ and open a new avenue for the prevention and treatment of HCTZ-induced metabolic disorders.
Limitations of the study
Our study revealed that LPS derived from gut microbiota is the key medium underlying HCTZ-induced metabolic disorder. Butyrate is important for metabolic and immune health. In gut microbiome studies, depletion of butyrate and butyrate-producing taxa was recurrently documented in type 2 diabetes.39 However, the connection between butyrate-producing taxa and HCTZ-induced metabolic disorder has not been considered yet. Another potential limitation to the findings presented is that we do not offer proposes as gut microbial-based strategies to mitigate HCTZ-induced metabolic disorder.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-ZO-1 | Affinity | Cat#AF5145; RRID: AB_2837631 |
| Rabbit monoclonal anti-Occludin | Affinity | Cat# DF7504; RRID: AB_2837631 |
| Rabbit monoclonal anti- MyD88 | Abcam | Cat#ab219413; |
| Mouse monoclonal anti- NF-κB | Abcam | Cat#ab288751; |
| Rabbit polyclonal anti- TNF-α | Abcam | Cat#ab183896; |
| Rabbit monoclonal anti- IL-1β | Abcam | Cat#ab234437; |
| Rabbit monoclonal anti- IL-6 | Abcam | Cat#ab259341; |
| Rat monoclonal anti-F4/80-APC | eBioscience | Cat# 17-4801-82 RRID:AB_2784648 |
| Mouse monoclonal anti- CD80-FITC | eBioscience | Cat# 11-0801-86 RRID: AB_465135 |
| Rat monoclonal anti- CD206-PE | eBioscience | Cat# 12-2061-82 RRID: AB_2637422 |
| Chemicals, peptides, and recombinant proteins | ||
| TAK-242, Resatorvid, TLR4 inhibitor | MCE | HY-11109; CAS: 243984-11-4 |
| Hydrochlorothiazide | MCE | HY-B0252;CAS: 58-93-5 |
| Dimethyl sulfoxide | Sigma-Aldrich | D8418;CAS: 67-68-5 |
| FD4 | Sigma-Aldrich | MFCD00131092; CAS: 60842-46-8 |
| Neomycin trisulfate salt hydrate | Sangon Biotech | A610366;CAS: 1405-10-3 |
| Ampicillin sodium | Sangon Biotech | A610028;CAS: 69-52-3 |
| Metronidazole | Sangon Biotech | A600633;CAS: 443-48-1 |
| Vancomycin hydrochloride | Sangon Biotech | A600983;CAS: 1404-93-9 |
| Critical commercial assays | ||
| Mouse Insulin ELISA Kit | Mericodia | 10-1247-01 |
| Instant ELISA Kit for Lipopolysaccharide | Cloud-Clone Crop | IEB526Ge |
| QlAamp DNA Stool Mini Kit | Qiagen | ID: 51604 |
| Mouse TNF-α ELISA Kit | Elabscience | E-EL-M0044c |
| IL-1 beta Mouse ELISA Kit | Invitrogen | BMS6002TEN |
| IL-6 Mouse ELISA Kit | Invitrogen | KMC0061 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6N-Tlr4em1C/Cya | Cyagen Biosciences | S-KO-05468 |
| Oligonucleotides | ||
| Primer: ZO-1 Forward: GCCGCTAAGAGCACAGCAA Reverse: GCCCTCCTTTTAACACATCAGA | This paper | N/A |
| Primer: Occludin Forward: TGAAAGTCCACCTCCTTACAGA Reverse: CCGGATAAAAAGAGTACGCTGG | This paper | N/A |
| Primer: MyD88 Forward: TGCCGTCCTGTCTACATCTTTG Reverse: GTTGCTCAGGCCAGTCATCA | This paper | N/A |
| Primer: NF-κB Forward:GCATTCTGACCTTGCCTATCT Reverse: CTCCAGTCTCCGAGTGAAGC | This paper | N/A |
| Primer: IL-1β Forward: TCGCTCAGGGTCACAAGAAA Reverse: CATCAGAGGCAAGGAGGAAAAC | This paper | N/A |
| Primer: IL-6 Forward: TCGTGGAAATGAGAAAAGAGTTG Reverse: AGTGCATCATCGTTGTTCATACA | This paper | N/A |
| Primer: TNF-α Forward: TCGCTCAGGGTCACAAGAAA Reverse: TTCGGAAAGCCCATTTGAGT | This paper | N/A |
| Other | ||
| Rodent Diet with 60 kcal% Fat | Researchdiets | D12492 |
| RNAiso plus | Takara | D9108 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wei Zhang (csuzhangwei@csu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Mice
C57BL/6J male mice (6-7 weeks old) were provided by Hunan Slyk Jingda Experimental Animal Co., Ltd. and were raised at the Experimental Animal Center of Central South University. Tlr4−/− mice on C57BL/6N background and C57BL/6N control mice (male, 6-7 weeks old) were purchased from Cyagen Biosciences. All studies with mice were approved by the Animal Ethics Committee of Central South University and complied with the National Institutes of Health regulations for the care and use of animals in research. All mice were housed under specific pathogen-free (SPF) conditions and controlled light-dark (12 h-12 h) cycles, with free access to food and water. After a 1-week run-in period, mice were divided into different groups with similar body weights before treatment. The animals were fed a normal chow diet (NCD, MD17121, China) or a high-fat diet (HFD D12492, Research Diets, USA). For HFD studies, mice were subjected to a 60% HFD. Feces were collected for gut microbiota analysis before sacrifice.
Method details
HCTZ administration
HCTZ was supplied in sterile drinking water. HCTZ was dissolved in DMSO and then added to drinking water for mice. Add equal volume of DMSO to the control group. The drinking water was changed and measured every 3-4 days. Because there are variable doses of HCTZ reported,40,41 a pilot study was performed to determine the adequate dosage of HCTZ for this study. Animals were offered pure water or HCTZ at a low dose (L, 5 mg/kg) or high dose (H, 10 mg/kg) for 12 weeks.
Antibiotic cocktail (Abx) administration
In order to avoid interfering HCTZ intake, antibiotic cocktail (ampicillin, neomycin, metronidazole, and vancomycin; 50 mg/kg, each) were administrated by oral gavage to ablate gut microbiota. The mice were administrated with antibiotics starting 7 days before the 8-week diet and HCTZ supplementation regimens and throughout the experiment. Water intake and food intake were observed during the antibiotics treatment.
Neomycin administration
Neomycin is usually used to deplete Gram-negative bacteria in the gut.21 Neomycin (0.5 g/L) was given in the drinking water after day 7 before HCTZ administration before the 8-week diet and HCTZ supplementation regimens and throughout the experiment. Considering that mice fed a HFD are more susceptible to HCTZ-induced metabolic abnormalities, neomycin treatment was provided to mice fed a HFD only.
Fecal microbiota transplantation (FMT)
Fresh feces from donor mice were collected at the end of the experiment, then suspended in maltodextrin-trehalose solution at a 1:20 ratio .42 The suspension was passed through a 70 μm nylon filter to remove food residue. Mice were treated with the antibiotic cocktail for 7 days to deplete the gut microbiota. After a one-day washout period, 200 μL of suspended bacteria in maltodextrin-trehalose solution was transferred into gut-sterilized recipient mice intragastrically once a day for two weeks.
TAK-242 administration and Toll-like receptor 4 (Tlr4) knockout
Mice were intraperitoneally treated with 1 mg/kg/day TAK-242 (TLR4 inhibitor) once daily. Tlr4 knockout mice were only exposed to HFD due to the susceptibility to HCTZ-induced metabolic abnormalities.
Fasting blood glucose (FBG) and fasting serum insulin (FINS)
The mice were fasted for 6 h before the tests. Blood from the tail vein was collected and measured immediately by a blood glucose meter (Accu-Chek Active blood glucose meter, Roche, Switzerland). The concentration of insulin in serum was measured using a commercial insulin ELISA kit (Mercodia, Sweden) according to the manufacturer's protocols.
Oral glucose tolerance test (OGTT)
After fasting for 6 h, blood glucose was measured at 0 min. Subsequently, the animals were treated with D-glucose (2 g/kg body weight) by oral gavage, and blood glucose was measured at 15, 30, 60, 90, and 120 min.
Insulin tolerance test (ITT)
After fasting for 4 h, insulin (0.75 U/kg, Sigma Aldrich, USA) was i.p. injected, and blood glucose was measured before (0 min) and after the injection (15, 30, 60, and 120 min).
Analyses of serum LPS, TNF-α, IL-1β, and IL-6
Serum LPS (Cloud-Clone Crop, IEB526Ge), TNF-α (Elabscience, E-MSEL-M0002), IL-1β (Invitrogen, BMS6002TEN), and IL-6 (Invitrogen, KMC0061) were detected using commercial ELISA kits according to the manufacturer’s protocols.
Analyses of cecal LPS
Cecal contents were suspended in PBS and centrifuged at 1000 × g for 20 min at 4 °C. The supernatant was collected for standard LPS detection.
Homeostatic model assessment of insulin resistance (HOMA-IR)
The index of homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as fasting serum insulin (FINS, μU/mL) × fasting blood glucose (FBG, mmol/L)/22.5.
16S rRNA microbiome gene sequencing
Fecal samples were collected from each group. Total genomic DNA was extracted using the CTAB/SDS method. DNA quality and concentration was checked by 1% agarose gel electrophoresis. Then, DNA was diluted to 1ng/μL. Bacterial taxa were analyzed by amplifying the hypervariable V3–V4 region of 16S rRNA (Phusion® High-Fidelity PCR Master Mix, New England Biolabs). The main 400–450-bp amplicon was sequenced and analyzed with a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA). Quality filtering of 16S rRNA sequences was conducted on the Qubit® 2.0 Fluorometer (Thermo Scientific, USA) and Agilent Bioanalyzer 2100 system. The resulting sequences were sequenced on an Illumina NovaSeq 6000 platform, and 250-bp paired-end reads were generated.
Assessment of intestinal permeability
Gut permeability was measured by 4-kDa FITC–dextran (FD4, Sigma, USA) in PBS at a concentration of 50 mg/mL. Mice were fasted for 6 h and gavaged with FD4 (0.01 mL/g body weight). After 1 h, whole blood was collected from the canthus vein. Serum was collected and diluted in an equal volume of PBS. The fluorescence value was quantified at excitation wavelengths of 485 nm and emission wavelengths of 535 nm.
Histological evaluation and immunofluorescence staining
Segments of tissues (pancreas, liver, and ileum) were fixed in 4% paraformaldehyde and embedded in paraffin. For morphologic analysis, paraffin-embedded tissues were cut into 5-μm sections and subsequently stained with hematoxylin and eosin. For immunofluorescence, after deparaffinization and rehydration, tissues were blocked with 5% bovine serum albumin (BSA) for 30 min at room temperature, incubated with anti-Flag primary antibodies at 4°C overnight, and then were incubated with the corresponding secondary Alexa Fluor antibody for 50 min at room temperature. The nuclei were counterstained with DAPI. The tissues were imaged with Imaging Systems for Fluorescence (NIKON, Japan).
qRT‒PCR
Total RNA of tissues was extracted using TRIzol reagent (Takara, Japan) according to the manufacturer’s instructions. cDNA was synthesized from 500 ng of RNA using the PrimeScript RT Master Kit (Takara, Japan). Quantitative PCR (qPCR) was performed on a LightCycler 480 Real-Time PCR detection system (Roche, Switzerland) with a SYBR Real-Time PCR Kit (Takara, Japan). Gene expression was measured and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the 2-ΔΔCt method. The sequences of primers used in this study are listed in Table S1.
Western blots
Tissues were homogenized and lysed (30 min on ice) in RIPA buffer supplemented with protease inhibitor cocktail (Sigma, USA) to harvest total protein. The homogenate was centrifuged at 14,000 × g for 15 min. Equal amounts of protein (20 μg) were loaded on 4%–20% Ready Gels (Bio-Rad Laboratories) and then transferred to a polyvinylidene difluoride membrane. After blocking with QuickBlock Western blocking buffer (Beyotime Biotechnology), the membrane was incubated with primary antibodies against MyD88 (Abcam, ab219413), NF-κB (Abcam, ab288751), TNF-α (Abcam, ab183896), IL-1β (Abcam, ab234437), and IL-6 (Abcam, ab259341) at 4°C overnight and probed with secondary antibodies conjugated with horseradish peroxidase for 1-2 h at room temperature. The bands were visualized by enhanced ECL chemiluminescence reagent (Invitrogen, USA). Signals were analyzed with Image Lab and normalized to controls.
Flow cytometry
The liver homogenate was filtered through a 70-μm cell strainer in PBS and centrifuged at 400 × g for 5 min at 4°C. Red blood cells were removed using red blood cell lysis buffer. For extracellular staining, cells were stained with anti-F4/80, anti-CD80, and viability dye (Live/Dead) for 30 min in the dark, followed by washing with PBS. For intracellular staining of cytoplasmic proteins, after viability and surface staining, a fixation/permeabilization buffer set (BD Bioscience) solution was used according to the manufacturer’s protocol, followed by washing with Perm/Wash buffer (BD Bioscience). Then, the cells were incubated with antibodies against CD206 for 50 min in the dark. The antibodies used in this flow cytometry analysis were as follows: anti-F4/80-APC (17-4801-82, eBioscience), anti-CD80-FITC (11-0801-86, eBioscience), anti-CD206-PE (12-2061-82, eBioscience), and Live/Dead (423104, BioLegend). Data analysis was performed using FlowJo VX Software (FlowJo, Ashland, OR).
Quantification and statistical analysis
Data analysis was performed in GraphPad Prism 8 (GraphPad Software). Other than gut microbiome, other data were performed in SPSS 22.0 (SPSS Inc., USA) and shown as the means ± standard errors of the means (SEMs). The number of animals was exhibited as “n” which was indicated in figure legends. Significant differences between mean values were determined by Student’s t test and one-way ANOVA. The data for antibiotics treatment experiments were determined with two-way ANOVA. P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from National Key Research and Development Program (2021YFA1301200), National Natural Science Foundation of China (Nos. 82074000, 81903784, 82073945, 81874329 and 81703623), Project Program of National Clinical Research Center for Geriatric Disorders (Xiangya Hospital, Grant No. 2020LNJJ06), Science and technology Innovation Project of Hunan Province (2018SK2129), and Natural Science Foundation of Hunan (2023JJ30772).
Author contributions
J.L., H.R., and W.Z. conceived of the presented idea. M.C., Q.Z., N.Y., Q.L., and Y.G. helped with sample collection. J.L., H.R., H.Z., and W.H. were major contributors in writing the manuscript with input from all authors. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 15, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107130.
Contributor Information
Wei-Hua Huang, Email: endeavor34852@csu.edu.cn.
Wei Zhang, Email: csuzhangwei@csu.edu.cn.
Supplemental information
Data and code availability
Data: All data reported in this paper will be shared by the lead contact upon request.
Code: This paper does not report original code.
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Psaty B.M., Lumley T., Furberg C.D., Schellenbaum G., Pahor M., Alderman M.H., Weiss N.S. Health outcomes associated with various antihypertensive therapies used as first-line agents: a network meta-analysis. JAMA. 2003;289:2534–2544. doi: 10.1001/jama.289.19.2534. [DOI] [PubMed] [Google Scholar]
- 2.Bangalore S., Parkar S., Grossman E., Messerli F.H. A meta-analysis of 94,492 patients with hypertension treated with beta blockers to determine the risk of new-onset diabetes mellitus. Am. J. Cardiol. 2007;100:1254–1262. doi: 10.1016/j.amjcard.2007.05.057. [DOI] [PubMed] [Google Scholar]
- 3.Verdecchia P., Reboldi G., Angeli F., Borgioni C., Gattobigio R., Filippucci L., Norgiolini S., Bracco C., Porcellati C. Adverse prognostic significance of new diabetes in treated hypertensive subjects. Hypertension. 2004;43:963–969. doi: 10.1161/01.HYP.0000125726.92964.ab. [DOI] [PubMed] [Google Scholar]
- 4.Amery A., Berthaux P., Bulpitt C., Deruyttere M., de Schaepdryver A., Dollery C., Fagard R., Forette F., Hellemans J., Lund-Johansen P., et al. Glucose intolerance during diuretic therapy. Results of trial by the European Working Party on Hypertension in the Elderly. Lancet. 1978;1:681–683. doi: 10.1016/s0140-6736(78)90797-3. [DOI] [PubMed] [Google Scholar]
- 5.Shafi T., Appel L.J., Miller E.R., 3rd, Klag M.J., Parekh R.S. Changes in serum potassium mediate thiazide-induced diabetes. Hypertension. 2008;52:1022–1029. doi: 10.1161/HYPERTENSIONAHA.108.119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown M.J., Williams B., Morant S.V., Webb D.J., Caulfield M.J., Cruickshank J.K., Ford I., McInnes G., Sever P., Salsbury J., et al. Effect of amiloride, or amiloride plus hydrochlorothiazide, versus hydrochlorothiazide on glucose tolerance and blood pressure (PATHWAY-3): a parallel-group, double-blind randomised phase 4 trial. Lancet Diabetes Endocrinol. 2016;4:136–147. doi: 10.1016/S2213-8587(15)00377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriksson J.W., Jansson P.A., Carlberg B., Hägg A., Kurland L., Svensson M.K., Ahlström H., Ström C., Lönn L., Ojbrandt K., et al. Hydrochlorothiazide, but not Candesartan, aggravates insulin resistance and causes visceral and hepatic fat accumulation: the mechanisms for the diabetes preventing effect of Candesartan (MEDICA) Study. Hypertension. 2008;52:1030–1037. doi: 10.1161/HYPERTENSIONAHA.108.119404. [DOI] [PubMed] [Google Scholar]
- 8.Cooper-DeHoff R.M., Wen S., Beitelshees A.L., Zineh I., Gums J.G., Turner S.T., Gong Y., Hall K., Parekh V., Chapman A.B., et al. Impact of abdominal obesity on incidence of adverse metabolic effects associated with antihypertensive medications. Hypertension. 2010;55:61–68. doi: 10.1161/HYPERTENSIONAHA.109.139592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shahin M.H., Sá A.C., Webb A., Gong Y., Langaee T., McDonough C.W., Riva A., Beitleshees A.L., Chapman A.B., Gums J.G., et al. Genome-Wide Prioritization and Transcriptomics Reveal Novel Signatures Associated With Thiazide Diuretics Blood Pressure Response. Circ. Cardiovasc. Genet. 2017;10:e001404. doi: 10.1161/CIRCGENETICS.116.001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh S., McDonough C.W., Gong Y., Bailey K.R., Boerwinkle E., Chapman A.B., Gums J.G., Turner S.T., Cooper-DeHoff R.M., Johnson J.A. Genome wide analysis approach suggests chromosome 2 locus to be associated with thiazide and thiazide like-diuretics blood pressure response. Sci. Rep. 2019;9:17323. doi: 10.1038/s41598-019-53345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Y., Pedersen O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021;19:55–71. doi: 10.1038/s41579-020-0433-9. [DOI] [PubMed] [Google Scholar]
- 12.Proceedings of the 3rd IPLeiria's International Health Congress : Leiria, Portugal. 6-7 May 2016. Vol. 16. BMC Health Serv Res; 2016. p. 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan S., Luck H., Winer S., Winer D.A. Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat. Commun. 2021;12:2598. doi: 10.1038/s41467-021-22727-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anhê F.F., Jensen B.A.H., Varin T.V., Servant F., Van Blerk S., Richard D., Marceau S., Surette M., Biertho L., Lelouvier B., et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020;2:233–242. doi: 10.1038/s42255-020-0178-9. [DOI] [PubMed] [Google Scholar]
- 15.Meijnikman A.S., Gerdes V.E., Nieuwdorp M., Herrema H. Evaluating causality of gut microbiota in obesity and diabetes in humans. Endocr. Rev. 2018;39:133–153. doi: 10.1210/er.2017-00192. [DOI] [PubMed] [Google Scholar]
- 16.Cani P.D., Bibiloni R., Knauf C., Waget A., Neyrinck A.M., Delzenne N.M., Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 17.Okin D., Medzhitov R. The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell. 2016;165:343–356. doi: 10.1016/j.cell.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur S., Bhattacharyya R., Banerjee D. Hydrochlorothiazide and Indapamide bind the NADPH binding site of bacterial Dihydrofolate Reductase: results of an in-silico study and their implications. In Silico Pharmacol. 2020;8:5. doi: 10.1007/s40203-020-00056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen M.C., Shulman G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018;98:2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergalli J., Bodrenko I.V., Masi M., Moynié L., Acosta-Gutiérrez S., Naismith J.H., Davin-Regli A., Ceccarelli M., van den Berg B., Winterhalter M., et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 2020;18:164–176. doi: 10.1038/s41579-019-0294-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q., Ma C., Duan Y., Heinrich B., Rosato U., Diggs L.P., Ma L., Roy S., Fu Q., Brown Z.J., et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov. 2021;11:1248–1267. doi: 10.1158/2159-8290.CD-20-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao S., Fei N., Pang X., Shen J., Wang L., Zhang B., Zhang M., Zhang X., Zhang C., Li M., et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol. Ecol. 2014;87:357–367. doi: 10.1111/1574-6941.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou S.Y., Gillilland M., 3rd, Wu X., Leelasinjaroen P., Zhang G., Zhou H., Ye B., Lu Y., Owyang C. FODMAP diet modulates visceral nociception by lipopolysaccharide-mediated intestinal inflammation and barrier dysfunction. J. Clin. Invest. 2018;128:267–280. doi: 10.1172/JCI92390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilg H., Zmora N., Adolph T.E., Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020;20:40–54. doi: 10.1038/s41577-019-0198-4. [DOI] [PubMed] [Google Scholar]
- 25.Cani P.D., Amar J., Iglesias M.A., Poggi M., Knauf C., Bastelica D., Neyrinck A.M., Fava F., Tuohy K.M., Chabo C., et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 26.Lieboldt M.A., Frahm J., Halle I., Görs S., Schrader L., Weigend S., Preisinger R., Metges C.C., Breves G., Dänicke S. Metabolic and clinical response to Escherichia coli lipopolysaccharide in layer pullets of different genetic backgrounds supplied with graded dietary L-arginine. Poult. Sci. 2016;95:595–611. doi: 10.3382/ps/pev359. [DOI] [PubMed] [Google Scholar]
- 27.Turnquist H.R., Cardinal J., Macedo C., Rosborough B.R., Sumpter T.L., Geller D.A., Metes D., Thomson A.W. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood. 2010;115:4758–4769. doi: 10.1182/blood-2009-10-251488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allaire J.M., Crowley S.M., Law H.T., Chang S.Y., Ko H.J., Vallance B.A. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. 2018;39:677–696. doi: 10.1016/j.it.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Shaler C.R., Parco A.A., Elhenawy W., Dourka J., Jury J., Verdu E.F., Coombes B.K. Psychological stress impairs IL22-driven protective gut mucosal immunity against colonising pathobionts. Nat. Commun. 2021;12:6664. doi: 10.1038/s41467-021-26992-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grasberger H., Magis A.T., Sheng E., Conomos M.P., Zhang M., Garzotto L.S., Hou G., Bishu S., Nagao-Kitamoto H., El-Zaatari M., et al. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J. Clin. Invest. 2021;131:e141676. doi: 10.1172/JCI141676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoshino K., Takeuchi O., Kawai T., Sanjo H., Ogawa T., Takeda Y., Takeda K., Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 32.Liu K., Zhao E., Ilyas G., Lalazar G., Lin Y., Haseeb M., Tanaka K.E., Czaja M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olefsky J.M., Glass C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 34.Murray P.J., Wynn T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain N., Vogel V. Spatial confinement downsizes the inflammatory response of macrophages. Nat. Mater. 2018;17:1134–1144. doi: 10.1038/s41563-018-0190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapouri-Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.A., Mardani F., Seifi B., Mohammadi A., Afshari J.T., Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018;233:6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen M.T.A., Favelyukis S., Nguyen A.K., Reichart D., Scott P.A., Jenn A., Liu-Bryan R., Glass C.K., Neels J.G., Olefsky J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 38.Jia L., Vianna C.R., Fukuda M., Berglund E.D., Liu C., Tao C., Sun K., Liu T., Harper M.J., Lee C.E., et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat. Commun. 2014;5:3878. doi: 10.1038/ncomms4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui J., Ramesh G., Wu M., Jensen E.T., Crago O., Bertoni A.G., Gao C., Hoffman K.L., Sheridan P.A., Wong K.E., et al. Butyrate-producing bacteria and insulin homeostasis: the microbiome and insulin longitudinal evaluation study (MILES) Diabetes. 2022;71:2438–2446. doi: 10.2337/db22-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J., Thabet S.R., Kirabo A., Trott D.W., Saleh M.A., Xiao L., Madhur M.S., Chen W., Harrison D.G. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ. Res. 2014;114:616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolda Mariano L.N., Boeing T., da Silva R.d., Cechinel-Filho V., Niero R., Mota da Silva L., de Souza P., Faloni de Andrade S. 1,3,5,6-Tetrahydroxyxanthone, a natural xanthone, induces diuresis and saluresis in normotensive and hypertensive rats. Chem. Biol. Interact. 2019;311:108778. doi: 10.1016/j.cbi.2019.108778. [DOI] [PubMed] [Google Scholar]
- 42.Burz S.D., Abraham A.L., Fonseca F., David O., Chapron A., Béguet-Crespel F., Cénard S., Le Roux K., Patrascu O., Levenez F., et al. A guide for ex vivo handling and storage of stool samples intended for fecal microbiota transplantation. Sci. Rep. 2019;9:8897. doi: 10.1038/s41598-019-45173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: All data reported in this paper will be shared by the lead contact upon request.
Code: This paper does not report original code.
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.