

Abstract
The misfolding and aggregation of α-synuclein is the general hallmark of a group of devastating neurodegenerative pathologies referred to as synucleinopathies, such as Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. In such conditions, a range of different misfolded aggregates, including oligomers, protofibrils, and fibrils, are present both in neurons and glial cells. Growing experimental evidence supports the proposition that soluble oligomeric assemblies, formed during the early phases of the aggregation process, are the major culprits of neuronal toxicity; at the same time, fibrillar conformers appear to be the most efficient at propagating among interconnected neurons, thus contributing to the spreading of α-synuclein pathology. Moreover, α-synuclein fibrils have been recently reported to release soluble and highly toxic oligomeric species, responsible for an immediate dysfunction in the recipient neurons. In this review, we discuss the current knowledge about the plethora of mechanisms of cellular dysfunction caused by α-synuclein oligomers and fibrils, both contributing to neurodegeneration in synucleinopathies.
Key Words: amyloid aggregation, neurodegeneration, Parkinson’s disease, protein aggregation, protein misfolding
Introduction
α-Synucleinopathies are a group of devastating neurodegenerative disorders characterized by the abnormal deposition of insoluble aggregates, both in neurons and glial cells, whose major filamentous component is the α-synuclein (αS) protein. This group includes Parkinson’s disease (PD), dementia with Lewy bodies (DLB), multiple system atrophy (MSA), and other rare neuroaxonal dystrophies (McCann et al., 2014). Among these conditions, PD is the most widespread, affecting 1–2% of the global population aged 65 years, and more than 4% of the population aged 85 years or older; thus, age can be considered the most relevant risk factor for PD (GBD 2016, 2018). This pathology is characterized by the death of dopaminergic neurons in the substantia nigra pars compacta; their loss results in a severe depletion of the neurotransmitter dopamine, which is in turn responsible for the cardinal motor dysfunctions of the disease, represented by bradykinesia, muscular rigidity, resting tremor, postural and gait impairment (Kalia et al., 2015). DLB is the second most common neurodegenerative dementia, characterized by progressive memory loss, visual hallucinations, parkinsonism, and cognitive decline, which have been linked to the presence of insoluble αS assemblies in the cortex (McKeith et al., 2017). Finally, MSA is a rare and fatal neurodegenerative condition defined by the presence of filamentous αS inclusions in oligodendrocytes, referred to as Papp-Lantos bodies (Roncevic et al., 2014). It is characterized by motor disturbances such as akinesia, rigidity, and postural tremor, but also by autonomic failure ad cerebellar ataxia (Wenning et al., 1994). Importantly, aggregated forms of αS were also found to be present in a high percentage (more than 50%) of Alzheimer’s disease (AD) patients (Lippa et al., 1998; Koehler et al., 2013; Mattson et al., 2013; Robinson et al., 2018; Twohig et al., 2019), with pure AD representing a minority of cases (Robinson et al., 2018). Lewy body (LB) pathology in the brain of AD patients does not show the same pattern of accumulation observed in PD (De Ture et al., 2019), occurring in the olfactory bulb, amygdala, and other limbic structures (Serrano-Pozo et al., 2011). Its presence has been associated with faster cognitive decline, but also with visual hallucinations and extrapyramidal signs (Twohig et al., 2019). Similarly, αS accumulation and toxicity have also been linked to lysosomal storage disorders such as Gaucher Disease (GD), caused by biallelic mutations in the GBA1 gene, encoding the lysosomal acid β-glucocerebrosidase (GCase), an enzyme deputed to the degradation of lysosomal glucosylceramide. GD is clinically characterized by hepatosplenomegaly, anemia, and bone crises (Daykin et al., 2021). In the last two decades, accumulated evidence indicated that people carrying GBA1 mutations possess a high probability to develop PD (Horowitz et al., 2022), with some GD patients showing forms of parkinsonism concomitantly with the occurrence of LB pathology (Blanz et al., 2016), which is also present in other lysosomal storage disorders such as GM1/GM2 gangliosidosis, Sanfilippo syndrome, Niemann-Pick type C and α- and β-Mannosidosis (Shachar et al., 2011). αS is a 14-kDa intrinsically disordered protein first discovered in the electric organ of the cartilaginous fish Torpedo californica (Maroteaux et al., 1988), that became the object of intense investigation upon its association with PD. The primary structure of αS is reported in Figure 1, and consists of three main portions: the positively charged and amphipathic N-terminal region (residues 1–60), that mediates the association with phospholipid membranes and, after binding, forms α-helical structures (Chandra et al., 2003; Ferreon et al., 2009); the hydrophobic non-Aβ-amyloid component (NAC) region (residues 61–95), responsible for αS aggregation (Giasson et al., 2001), and the negatively charged highly acidic C-terminal region (residues 96–140), that remains largely unstructured and is involved in many different interactions, such as those with ions, polycations, and polyamines (Brown, 2007; Lautenschläger et al., 2018). Interestingly, the NAC region was observed in senile plaques isolated from a single AD patient (Ueda et al., 1993), and its presence was lately confirmed in a larger number of AD-affected subjects (Masliah et al., 1996). αS populates a dynamic equilibrium between the soluble, monomeric, and unfolded state acquired in the cytosol of neuronal cells, and a membrane-bound one, in which it assumes a partial α-helical secondary structure (Chandra et al., 2003; Bussell et al., 2005; Burré et al., 2013, 2018). In contrast to the physiological conformations, a persistent β-sheet amyloid secondary structure is acquired in pathological conditions and is associated with the formation of oligomers and fibrils, ultimately deposited into Lewy bodies (Conway et al., 1998; Lashuel et al., 2002; Uversky, 2007), as represented in Figure 2. Such conformations, having different structures, sizes and morphologies, co-exist and convert each other depending on environmental conditions (Uversky, 2007). A growing body of evidence suggests that protein aggregation, including that of αS, is responsible for a toxic-gain-of-function mediated by misfolded aggregates, and plays a cardinal role in the pathogenesis of the associated diseases (Chiti et al., 2017; Tofaris, 2022). Over the last decades, the relationship between structure and toxicity of the different αS aggregates and the specific contribution of these conformers to neurodegeneration have been a matter of intense research. Mounting evidence suggests that prefibrillar oligomeric species, formed during the very early phases of the aggregation process, rather than mature fibrils or deposits, are by far the most neurotoxic (Bengoa-Vergniory et al., 2017; Bigi et al., 2021; Cascella et al., 2021, 2022). In particular, such assemblies were reported to have a central pathogenic role in the onset and progression of synucleinopathies, impairing the functionality of neuronal cells and ultimately leading to their death. Simultaneously, other researchers proposed that fibrillar αS species could also have an important role in neurodegeneration, by seeding the self-assembly of soluble endogenous αS into aggregated forms (Luk et al., 2009; Volpicelli-Daley et al., 2011; Cascella et al., 2022), and because of their ability to interact aberrantly with the plasma membrane, thus inducing its permeabilization, with subsequent ionic alterations and impairment of cytosolic organelles (Pemberton et al., 2011; Pieri et al., 2012). Here, we describe the main structural features of oligomeric and fibrillar αS conformers and their detrimental effects on neuronal cells.
Figure 1.

Schematic representation of the primary structure of wild-type full-length monomeric αS.
NAC: Non-Aβ-amyloid component. Created with BioRender.com.
Figure 2.
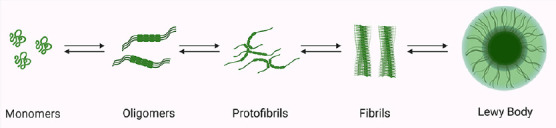
Under specific conditions, the natively unstructured monomer can convert into oligomers, which can further aggregate into protofibrils and finally in highly ordered cross-β-sheet fibrils.
Created with BioRender.com.
Alpha-Synuclein Oligomers
An oligomer is a form of soluble aggregated αS that has not still raised the fibrillar conformation, and the term refers to a range of considerably different species varying in size, structural and biophysical properties, such as β-sheet content, and solvent-exposed hydrophobicity (Lashuel et al., 2002; Bengoa-Vergniory et al., 2017; Cremades et al., 2017). αS oligomers are usually described as on-pathway when they further aggregate forming mature fibrils, or off-pathway, if they are kinetically stable and remain in the oligomeric state without converting into fibrillar assemblies (Gosh et al., 2015). Small oligomeric conformers, intermediates within the aggregation pathway, are actually considered to be the most neurotoxic species in synucleinopathies (Bengoa-Vergniory et al., 2017), and, as a consequence, promising biomarkers for the early diagnosis and relevant putative disease-modifying therapeutic targets for such diseases (Majbour et al., 2016; Kulenkampff et al., 2021). Despite considerable effort, methods for the in vivo identification and isolation of αS oligomers are still lacking, given their metastable nature and very low concentration in human samples, such as the cerebrospinal fluid and plasma (Blömeke et al., 2022). For these reasons, studies aimed at identifying their specific role in neurodegeneration actually take advantage of oligomeric conformers synthesized in vitro from the monomeric protein. A large variety of preparation methods is described in the literature, giving rise to a plethora of species with extremely high levels of heterogeneity in terms of size, secondary structure, and solvent-exposed hydrophobic regions (Cremades et al., 2017); the formation of αS oligomers in solution can be promoted by incubating the monomeric protein with a range of different molecules, such as lipids, metal ions, or other small molecules (Cremades et al., 2017; Alam et al., 2019). Given the metastable and transient nature of these species, numerous approaches have been used to stabilize them and to generate “kinetically-trapped” conformers remaining stable for time intervals that allow their structural, biophysical, and biological characterization (Cremades et al., 2017; Alam et al., 2019). Lyophilization is one of the most commonly used methods to stabilize oligomers, promoting at the same time the generation of conformers that are recognized by the oligomer-specific A11 antibody (Kayed et al., 2003); these species were reported not to further aggregate forming fibrils (Celej et al., 2012; Chen et al., 2015). Moreover, a range of natural compounds with the ability to bind specific oligomeric conformations was reported to block their elongation; among them, there are polyphenols such as curcumin, rosmarinic acid, baicalein and epigallocatechin gallate (Cremades et al., 2017). Oligomers have been shown to form upon αS interaction with brain-derived membranes (Ding et al., 2002) or other phospholipid bilayers and vesicles resembling biological membranes; their interactions give rise to conformational modifications that lead to oligomerization (Eliezer et al., 2001; Volles et al., 2001). Among the plethora of oligomeric conformers that could assemble during αS aggregation, single molecule techniques have suggested that the neurotoxic capability is associated with proteinase K resistance, which is, in turn, correlated with a β-sheet-rich structure (Cremades et al., 2012; Cascella et al., 2022). Consistently, two distinct oligomeric populations, referred to as Type A* and Type B* oligomers (OA* and OB*, respectively), with significantly different toxic properties, have been identified (Cremades et al., 2012); such species, presenting similar dimensions and molecular configuration, exhibit a considerable different secondary structure: while OA* are largely disordered, negative to the conformation-sensitive A11 antibody and non-toxic to neuronal cells, OB* possess a relevant β-sheet secondary structure, are positive to the A11 antibody and highly toxic to neuronal cells (Chen et al., 2015; Fusco et al., 2017; Cascella et al., 2021). These species closely match those of the previously identified unstable forms of nontoxic (type A) and toxic (type B) αS oligomers (Cremades et al., 2012). The structural determinants of their differential neurotoxicity have been identified by using the solution and solid-state nuclear magnetic resonance (ssNMR). While OA* remain largely unstructured and only binds to the membrane surface, OB* expose the highly lipophilic N-terminal region, responsible for the interaction with the membrane surface, and a structured and rigid core rich in β-sheet able to insert into the lipid bilayer, thus disrupting the neuronal membrane integrity (Fusco et al., 2017). Accordingly, the use of a specific antibody that targets the N-terminus of αS significantly reduced OB* toxicity when they were incubated with neuronal cells (Cascella et al., 2019).
Alpha-Synuclein Fibrils
Highly stable and ordered fibrils are the end product of αS aggregation, and represent the major filamentous components of the intracellular inclusions found in the brain of people affected by synucleinopathies (Arima et al., 1998; Spillantini et al., 1998; Goedert et al., 2017). However, whether the formation of such inclusions is cause or consequence of neurodegeneration is still a matter of intense debate. αS fibrils formed in vitro possess considerably different morphological and biological properties, depending on the various aggregation conditions (Kim et al., 2016; De Giorgi et al., 2020; Shrivastava et al., 2020). Despite such heterogeneities, these mature fibrillar conformers share cardinal amyloid properties, including an elongated and filamentous morphology, the capability to bind Thioflavin T, and a persistent β-sheet structure (Serpell et al., 2000; Sulatskaya et al., 2018; Gracia et al., 2020). The filament core approximately includes amino acidic residues 30–100 of αS (Miake et al., 2002); a range of different techniques has been employed for the structural characterization of αS fibrils, including ssNMR, micro-electron diffraction, electron paramagnetic resonance, circular dichroism, hydrogen/deuterium exchange NMR, and cryo-electron microscopy (cryo-EM) (Vilar et al., 2008; Comellas et al., 2011; Gath et al., 2012; Tuttle et al., 2016; Cremades et al., 2017). In this way, the existence of different fibril polymorphs at the molecular level has been elucidated. Their heterogeneity is essentially associated with differences in the number of protofilaments, in their organization, as well as in the strength of the amino acid interactions determining the β-sandwich fold (Vilar et al., 2008; Comellas et al., 2011; Tuttle et al., 2016; Cremades et al., 2017; Li et al., 2018; Guerrero-Ferreira et al., 2019; Gracia et al., 2020), and directly related to the differential toxicity they evoke when applied to neuronal cells (Peelaerts et al., 2015). The heterogeneity of αS strains needs, however, to be validated by isolating ex vivo fibrils from patients’ specimens. In this context, Strohäker and coworkers recently investigated the structural characteristics of αS fibrils amplified from brain extracts of both PD and MSA patients. Such filaments were reported not to possess markedly distinct structural properties, but to be substantially different from an array of fibrillar polymorphs synthesized in vitro, thus suggesting the existence of potentially considerable dissimilarities between in vitro and in vivo fibrils, and highlighting the urgency to use brain-derived conformers in future studies (Strohäker et al., 2019). In a subsequent study, Schweighauser and collaborators used cryo-EM to explore αS filaments, revealing that fibrils from the brain of MSA patients are structurally different from those present in individuals with DLB; moreover, they revealed that DLB-derived fibrils do not appear to twist, and are thinner than those present in the brain of MSA-affected subjects (Schweighauser et al., 2020). Again, despite some topological similarities, the structure of fibrils arising from MSA patients resulted to be different from in vitro assembled conformers (Schweighauser et al., 2020). In a very recent and elegant work, Yang et al. (2022) reported the cryo-EM microscopy structures of αS fibers extracted from the brain of PD, PDD, and DLB patients. Such conformers were composed of a single protofilament (referred to as Lewy fold) remarkably different from the protofilaments observed in MSA subjects, thus probing the existence of different αS conformations in equally different neurodegenerative conditions. Even though the increasing relevance given to αS oligomers as the major culprits of neurodegeneration in synucleinopathies, important contributions of fibrillar species have also been reported. Such conformers are actually considered to be prion-like seeds able to transfer from neuron-to-neuron and propagate between interconnected vulnerable brain areas, thus driving the pathogenesis of α-synucleinopathies (Kordower et al., 2008; Li et al., 2008). Specifically, the addition of αS preformed fibrils (PFFs) to the extracellular medium of cultured neuronal cells was reported to corrupt the endogenous monomeric protein, that aggregates acquiring a pathological misfolded conformation finally culminating in the formation of inclusions morphologically and biochemically comparable to LBs (Volpicelli-Daley et al., 2011). The presence of these LB-like structures has been reported to evoke the loss of dopaminergic neurons in the substantia nigra (SN), as well the development of PD-like motor phenotypes in relevant animal models (Luk et al., 2012; Masuda-Suzukake et al., 2014; Osterberg et al., 2015). In a recent study, Bassil et al. (2020) revealed that the injection of αS PFFs in AD 5xFAD mice induced a significant increase in Aβ plaque burden by growing β-amyloid production from the amyloid precursor protein, in good agreement with previously reported evidence (Roberts et al., 2017).
Alpha-Synuclein Oligomers and Fibrils: a Complex Interplay
Since both αS oligomers and fibrils have been described as neurotoxic agents in synucleinopathies, the identification of the main culprit starting neurodegeneration is still a matter of debate. Although the presence of large intracellular αS inclusions in the brain of affected people is a cardinal hallmark of synucleinopathies, their abundance does not directly correlate with the disease phenotype observed in patients (Colosimo et al., 2003). We have recently demonstrated that fibrils can release highly toxic prefibrillar oligomeric species that promptly cause a massive dysfunction in neighboring neurons (Bigi et al., 2021, 2022; Cascella et al., 2021). Taking advantage of specific conformation-sensitive antibodies, we revealed that αS fibrils interacting with neuronal membranes progressively release A11-reactive oligomers resembling on-pathway oligomers formed during αS aggregation (Bigi et al., 2021; Cascella et al., 2021). Such conformers were internalized by neurons, where they induced ionic dyshomeostasis and oxidative stress, finally culminating in cell death (Cascella et al., 2021). Thus, we propose that both oligomeric and fibrillar aggregates of αS play a crucial role in neurodegeneration: fibrillar conformers act as a reservoir of soluble oligomeric species, which are the toxic counterpart promptly triggering neuronal dysfunction. In order to probe the nature of the aggregated species that penetrated into the cytosolic compartment, we performed immunofluorescence experiments using the A11 antibody in human induced pluripotent stem cells (iPSC)-derived dopaminergic neurons exposed to in vitro-synthetized fibrils (Figure 3; Bigi et al., 2021; Cascella et al., 2021). Confocal images revealed a remarkable A11-positive signal in cells treated for 24 hours with the previously described OB* and fibrils as compared to untreated ones, thus pointing out the presence of αS oligomeric species similar to OB* inside dopaminergic neurons upon treatment with extracellularly-added fibrillar assemblies (Figure 3).
Figure 3.
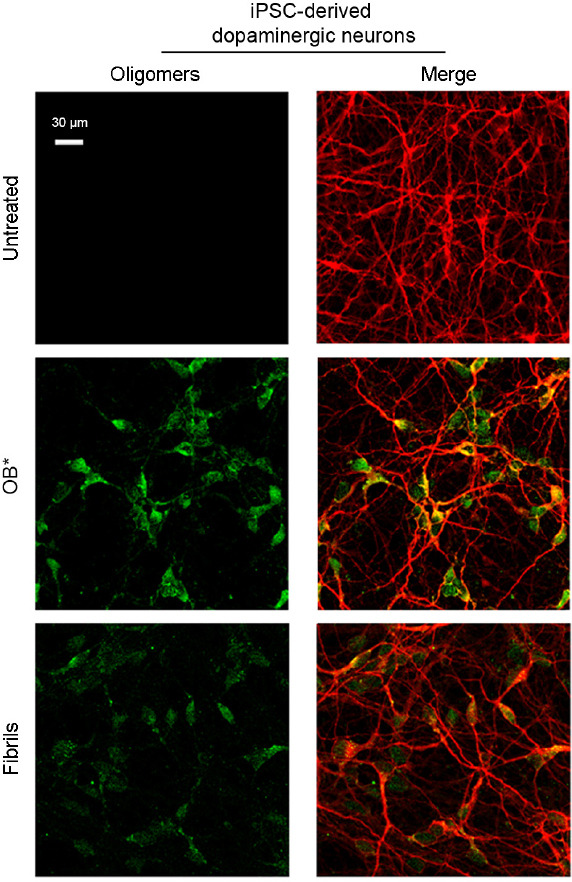
Representative confocal scanning microscope images showing human induced pluripotent stem cell (iPSC)-derived dopaminergic neurons treated for 24 hours with type B* oligomers (OB*) and fibrils of α-synuclein at 0.3 μM.
Red and green fluorescence indicates mouse anti-MAP-2 antibodies and the A11-positive prefibrillar oligomers, respectively. Images adapted and reprinted from Cascella et al. (2021), licensed under Creative Commons Attribution 4.0 International Public License (CC BY 4.0, https://creativecommons.org/licenses/ by/4.0/).
The Pathological Spreading of Alpha-Synuclein
Our evidence clearly suggests the existence of a complex interplay between the different aggregated forms of αS, which can continuously convert each other under specific environmental conditions, as extensively reviewed in Bigi et al. (2022). The release of toxic oligomers from mature fibrils is one of the possible mechanisms explaining the fiber neurotoxicity, together with their widely explored capability to diffuse among neurons, thus contributing to LB pathology spreading between synaptically-connected brain areas. Importantly, αS fibrils are also uptaken by microglial cells, that preferentially phagocytose such conformers with respect to monomeric and oligomeric αS (Hoffmann et al., 2016). Fibers promptly activate microglia, inducing the production of pro-inflammatory cytokines such as tumor necrosis factor-alpha and interleukin-1 beta (TNF-α and IL-1β, respectively; Hoffmann et al., 2016). A detailed description of all the mechanisms by which neuron-to-neuron and neuron-to-glia spreading occurs was recently provided by Cascella et al. (2022) and Bigi et al. (2022) with their collaborators in two distinct reviews. The hypothesis of a prion-like spreading of αS strains through neuroanatomically connected areas in PD patients first emerged from the Braak hypothesis (Braak et al., 2002, 2003a). This theory postulates that Lewy pathology is induced by an unknown pathogen or toxin penetrated in the nasal cavity that subsequently reaches the gut and then spreads into the brain via the vagus nerve, finally joining the substantia nigra pars compacta (Braak et al., 2003a, b). This hypothesis was lately corroborated by Holmqvist et al. (2014) through the intestinal injection of lysates of human PD brains containing monomeric, oligomeric, and fibrillar αS in animal models, demonstrating the existence of a retrograde transport of such conformers through the vagal nerve, finally reaching the brainstem in a time-dependent manner. Consistently, further experimental evidence supported the notion that αS aggregates are present in the enteric system before they occur in the brain (Chandra et al., 2017). An alternative scenario, characterized by the bidirectional spreading of misfolded αS (from the brain to the gut and vice versa) has been recently speculated by Arotcarena et al. (2020) upon striatal or enteric injection of PD-brain extracts in non-human primates. Even if the Braak’s theory does not fit with all PD cases, this hypothesis is supported by several clinical observations, such as the presence of hyposmia (Doty, 2012), as well as a range of gastrointestinal problems (Del Tredici et al., 2016) in PD patients carrying LB pathology both in olfactory neurons and in the enteric system (Lebouvier er al., 2010). Thus, it is of remarkable importance to shed light on the gut involvement in PD, with particular attention to the crosstalk with the brain, commonly referred to as the “gut-brain axis” (Carabotti et al., 2015). Indeed, fecal samples from PD patients revealed relevant alterations of gut microbiota, that were found to be associated with specific motor phenotypes (Scheperjans et al., 2015), raising the hypothesis that microbial alterations could have a role in PD pathogenesis. More recently, abnormal concentrations of stool immune factors were revealed in the feces of early PD patients, confirming the existence of a massive inflammatory intestinal process (Houser et al., 2018; Schwiertz et al., 2018). Many in vivo studies on animal models of PD have shown that chronic intestinal inflammation hastens and increases neuroinflammation and dopaminergic neuropathology (Garrido-Gil et al., 2018; Kishimoto et al., 2019).
Alpha-Synuclein Pathogenic Pathways
The neurotoxicity of αS misfolded aggregates involves a range of distinct cellular pathways and organelles, such as the plasma membrane, the endoplasmic reticulum (ER) and Golgi, mitochondria, nucleus, and synaptic vesicles.
Perturbation of the Plasma Membrane Integrity
The maintenance of the plasma membrane integrity is of fundamental importance for every cell type, for normal functionality and survival. Its loss has been widely described as a cardinal pathogenic mechanism of PD and proteinopathies in general (Cenini et al., 2010; Evangelisti et al., 2016). αS aggregates, and in particular oligomers, were reported to exert their cytotoxic effects upon an abnormal interaction with lipid membranes (Iyer et al., 2019). Specifically, they can induce changes in the permeability and fluidity of the lipid bilayer, and influence the normal distribution of exposed proteins (Shrivastava et al., 2017). Their binding is generally mediated by their positively charged core, which establishes electrostatic interactions with negatively charged lipids present in the plasma membrane (Stöckl et al., 2012). This step is then followed by the permeabilization of the lipid bilayer, occurring through different mechanisms; the first one is the pore formation, or “poration” (Zakharov et al., 2007; Tosatto et al., 2012). This model assumes that annular αS oligomers with a central pore embed into lipid bilayers forming transbilayer proteins that allow the passage of small molecules and ions (Stöckl et al., 2013). Specifically, such pores were reported to possess an ion channel-like activity, resulting in an abnormal calcium influx (Figure 4; Danzer et al., 2007; Kim et al., 2009). Another possible mechanism is that in which αS oligomers cause the thinning of the lipid bilayer upon their binding, thus inducing an increase of membrane permeability and causing its leakage. According to this model, αS oligomers establish electrostatic interactions between their positively charged core and the negatively charged lipid headgroups; then, they are incorporated between the tightly packed lipids, thus evoking the thinning of the hydrophobic core of the bilayer and increasing its permeability (Stöckl et al., 2013). Finally, αS oligomers were also reported to bind to existing packing defects present in the bilayer, and extract phospholipids (Chaudhary et al., 2016); specifically, the increased exposure of lipid acyl chains at the edges of defects was reported to facilitate the interaction between oligomers and membranes, resulting in the growth of fractal domains in which there are no lipids (Chaudhary et al., 2016). Consistently, Reynolds and coworkers reported that the addition of monomeric wild-type and mutated A53T and E57K αS on the surface of supported lipid bilayers induced the protein adsorption to the membrane, whose subsequent aggregation caused both membrane thinning and lipid extraction in the near proximity of the growing aggregates (Reynolds et al., 2011). The previously described OB* and, to a minor extent, short fibrils (SF), were recently reported to alter the permeability of the membrane of synthetic synaptic-like small unilamellar vesicles and cultured neuronal cells, by inducing the release of the calcein-AM fluorescent probe (Cascella et al., 2021). A significant leakage of intracellular calcein, corresponding to the low green fluorescent signal in Figure 5A, was shown in rat primary cortical neurons following the addition to the culture medium of OB* and SF (Cascella et al., 2021), suggesting a permanent dysfunction of neuronal bilayers following the aggregates interaction with the lipid bilayer. Specifically, the propensity of OB* to permeabilize plasma membranes has been associated with their ability to anchor to the lipids through the N-terminus α-helical structure and to later insert their rigid β-sheet structure into the hydrophobic core of the membrane (Fusco et al., 2017). By contrast, all other αS species, such as αS monomer, OA* and long fibrils (LF), caused negligible calcein release (Figure 5A), indicating a low propensity to permeabilize neurons (Cascella et al., 2021).
Figure 4.

An overview of the main neurotoxic cascades evoked by αS oligomers and PFFs.
BAX: Bcl-2-associated X protein; Bcl2: B-cell lymphoma 2; Ca2+: calcium; ΔΨm: mitochondrial membrane potential; ER: endoplasmic reticulum; HMGB1: high mobility group box 1; mGluR5: metabotropic glutamate receptor 5; mtDNA: mitochondrial DNA; NMDA: N-methyl-D-aspartate receptors; PFFs: pre formed fibrils; PrPC: cellular prion protein; PTP: permeability transition pore; ROS: reactive oxygen species; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; TADA2a: transcriptional adapter 2-alpha. Created with BioRender.com.
Figure 5.

Disruption of the plasma membrane integrity and Ca2+ dyshomeostasis evoked by αS oligomers and fibrils.
(A) Representative confocal microscope images showing primary rat cortical neurons loaded with the calcein-AM probe for 10 minutes and then treated for 1 hour with αS monomer (M), type A* oligomers (OA*), type B* oligomers (OB*), short fibrils (SF), and long fibrils (LF) at 0.3 μM. Semi-quantitative analyses of the calcein-derived fluorescence signal in primary rat cortical neurons and SH-SY5Y cells. (B) Representative confocal microscope images showing the Ca2+-derived fluorescence in primary rat cortical neurons treated for 15 minutes with the indicated 0.3 μM αS species and then loaded with the Fluo-4 AM probe. Semi-quantitative analysis of the intracellular Ca2+-derived fluorescence in primary rat cortical neurons and SH-SY5Y cells. Images reprinted from Cascella et al. (2021), licensed under Creative Commons Attribution 4.0 International Public License (CC BY 4.0, https://creativecommons.org/licenses/ by/4.0/).
Calcium Dyshomeostasis
Calcium (Ca2+) is an essential metal ion and the most ubiquitous intracellular second messenger, involved in the modulation of almost all neuronal processes, such as gene expression, synaptogenesis, synaptic transmission, energy production, membrane excitability, neuronal plasticity, learning and memory (Berridge, 1998; Kawamoto et al., 2012). Neuronal Ca2+ signaling depends both on highly transient and reversible elevation of cytosolic concentrations via an influx from the extracellular space through specific plasma membrane channels, as well as from its release from intracellular stores (Brini et al., 2014). αS aggregates, and particularly oligomers, were reported to enhance the plasma membrane permeability to Ca2+ ions through the perforation of the lipid bilayer by forming pore-like complexes (Danzer et al., 2007). Several observations have been performed after the addition of different αS species to the external medium of cultured cells. Angelova and coworkers revealed that both monomeric and oligomeric αS can induce a massive influx of Ca2+ ions in neurons and astrocytes, but only oligomers evoke neuronal cell death (Angelova et al., 2016). Consistently, we recently observed that a 15 min treatment with OB* and SF and, to a lesser extent, LF, evoked an extensive influx of Ca2+ both in cortical neurons and SH-SY5Y cells, whereas OA* and monomer caused a negligible elevation of intracellular calcium (Figure 5B; Cascella et al., 2021). Simultaneously, the interaction with a range of membrane channels and receptors, with the consequent modulation of their activity, has also been revealed. A prolonged treatment of rat hippocampal slices with αS oligomers, but not with the monomeric protein or fibrillar conformers, was demonstrated to cause a massive activation of N-methyl-D-aspartate receptors (NMDARs), which triggered in turn an increased activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), so promoting the insertion into the postsynaptic membrane of higher conductance Ca2+-permeable versus GluR2-containing AMPARs, involved in basal synaptic transmission, ultimately leading to impaired long-term potentiation (Diógenes et al., 2012). More recently, Ferreira and coworkers revealed that the αS oligomers-induced impairment of long-term potentiation was due to the interaction with the cellular prion protein (PrPC), with the consequent phosphorylation of the Fyn kinase via metabotropic glutamate receptors 5, finally culminating in the phosphorylation and activation of NMDARs, through which a massive increase of intracellular Ca2+ levels occurs (Figure 4), ultimately triggering synaptic dysfunction in hippocampal neurons and synaptic and cognitive deficits in mice (Ferreira et al., 2017). More recently, Trudler et al. (2021) demonstrated that both oligomeric and fibrillar αS species evoke a rise in astrocytic intracellular Ca2+, which is responsible for a massive release of glutamate, that activates aberrantly extra-synaptic NMDARs on neighboring neurons, directly contributing to synapse loss. In the same work, αS oligomers were also reported to directly activate extra-synaptic NMDARs on neurons, thus exacerbating their damage (Trudler et al., 2021). Aggregated - but not monomeric - αS has also been shown to lead to Ca2+ dyshomeostasis upon intracellular accumulation; once in the cytoplasm, αS aggregates bind to the sarco/endoplasmic reticulum Ca2+-ATPase, thus stimulating its activity and triggering an initial reduction of cytosolic Ca2+ concentration and an overload in the ER (Figure 4). This phase is thought to be characterized by a significant imbalance of Ca2+-dependent processes, that is responsible for cellular dysfunction, and it is followed by a later increase in cytosolic Ca2+ levels, that precedes cell death (Betzer et al., 2018).
Lysosomal and Autophagy Impairment
Cell-to-cell propagation of neurotoxic and pathogenic αS conformers has been widely described as a cardinal mechanism in the progression of synucleinopathies. This necessarily implies that such species are released from donor cells and then uptaken by neighboring neurons, where they induce the aggregation of endogenous αS (Luk et al., 2012; Volpicelli-Daley et al., 2014). Thus, it is likely that internalized aggregates circumvent, or either impair, cellular degradation pathways, most importantly the autophagy-lysosome one (Bae et al., 2015; Sacino et al., 2017), which is actually considered to be the elective mechanism for the degradation of αS assemblies by neuronal cells (Cuervo et al., 2004; Mak et al., 2010). This process encompasses a range of distinct pathways, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), to name the most relevant. In particular, aggregated forms of αS were reported to colocalize with both early and late endo/lysosomal markers (Lee et al., 2008; Brahic et al., 2016; Karpowicz et al., 2017; Bayati et al., 2022). Human αS overexpression was observed to impair macroautophagy in mammalian cells and in transgenic mice, where it affects Rab1a-specific secretory pathways, hampering omegasome (autophagosome precursor) formation (Winslow et al., 2010), as well as to damage the autophagic flux in aging adult neurons through the inhibition of mitophagosome maturation and autophagosome-mediated αS clearance (Figure 4; Sarkar et al., 2021). CMA was also reported to be affected by aggregated αS. This specific type of autophagy implies protein recognition by the cytosolic chaperone heat shock cognate protein 70 (HSC70), with the subsequent delivery to the lysosomal-associated membrane protein 2A (LAMP2A), and the final translocation into lysosomes (Cuervo et al., 2004). While wild-type αS is efficiently degraded by CMA, the pathogenic mutants A53T and A30P are poorly degraded, despite their higher binding ability to LAMP2A with respect to the wild-type protein. In this way, such mutants also block the lysosomal uptake and degradation of other target substrates (Cuervo et al., 2004). Accordingly, reductions in LAMP2 and LAMP2A have been observed in the brain tissue of PD patients (Alvarez et al., 2010; Murphy et al., 2014). CMA impairment was also revealed in the presence of dopamine-modified αS, which also blocked the degradation of other substrates by the same pathway (Martinez-Vicente et al., 2008). As previously reported, a range of mutations of the GBA1 gene are important genetic risk factors for PD onset (Horowitz et al., 2022). Among these mutations, those responsible for the GCase mislocalization on the lysosomal surface, instead of in the lumen, have been associated with a significant inhibition of CMA, with subsequent intracellular accumulation of CMA protein targets, including αS (Kuo et al., 2022). This evidence is substantially in line with previous reports revealing that the reduction of the GCase physiological activity can be responsible for the accumulation of high molecular weight αS aggregates (Kim et al., 2018; Zunke et al., 2018). Recently, the inhibition of GCase activity has been reported to be evoked by extracellularly added PFFs, but not by the monomeric protein (Gegg et al., 2020; Henderson et al., 2020a).
Endoplasmic Reticulum and Golgi Functionality
ER plays an essential role in protein synthesis, folding and secretion, as well as in lipid synthesis and in the regulation of intracellular Ca2+ homeostasis (Baumann et al., 2001). The misfolding and accumulation of disease-related proteins, including αS, is responsible for the impairment of ER functionality, and evoke a spectrum of detrimental effects, finally culminating in cell death. In such conditions, the ER initiates a multi-signaling pathway called unfolded protein response (UPR), aiming at restoring its functionality (Hetz et al., 2011). The UPR is orchestrated by three major ER stress sensors, including the activating transcription factor 6, inositol-requiring kinase 1α, and protein kinase RNA-activated-like ER kinase (PERK) (Manié et al., 2014).
During UPR, the cell attenuates protein synthesis, increases that of chaperones to facilitate the proper protein folding, and enhances the translation of proteins implicated in the ER-associated protein degradation, in order to increase the elimination of misfolded proteins (Shröder et al., 2005). An overactivation of the UPR response has been observed in the SN of PD patients as compared to control cases (Hoozemans et al., 2007), as well as in postmortem brain tissue of people affected by DLB and PDD (Baek et al., 2016), strongly suggesting that the activation of this process should be evoked by αS accumulation (Figure 4). Consistently, Bellucci and coworkers (2011) showed that the overexpression of human full-length and truncated αS both in cultured cells and in a transgenic mouse model of PD resulted in the accumulation of the protein within the ER and in the activation of the PERK-dependent pathway of the UPR. Accordingly, the induction of ER-stress and UPR has been demonstrated in a synucleinopathy-affected transgenic mouse model expressing a range of human αS variants (Colla et al., 2012), with αS oligomers accumulating in the ER lumen (Colla et al., 2012; Kalia et al., 2013). Moreover, the subsequent inhibition of the vesicular trafficking between the ER and the Golgi apparatus has been demonstrated in vitro (Cooper et al., 2006). More recently, Liu et al. (2018) showed that the overexpression of both wild-type and mutant αS (A30P and A53T) was able to induce the dysfunction of the ER-Golgi compartment in astrocytes, thus leading to a massive reduction of the glial cell line-derived neurotrophic factor levels, which is, in turn, responsible for the suppression of neurite outgrowth. The addition of oligomeric, but not monomeric and fibrillar forms of αS, to the extracellular medium of cultured human neuroblastoma cells was reported to evoke the activation of the ER stress response factor XBP1 (X-box binding protein 1), which exerts a protective effect: this evidence clearly indicates the prominent capacity of oligomeric αS to compromise the cellular homeostasis, including that of ER (Castillo-Carranza et al., 2012).
Mitochondrial Dysfunction
Mitochondrial dysfunction is actually considered to be a prominent toxic event in PD and synucleinopathies in general. Mitochondria are abundant organelles where adenosine 5′-triphosphate (ATP) synthesis takes place; they are also involved in the regulation of Ca2+ storage, lipid metabolism, and neuronal survival (Nunnari et al., 2012). Neuronal cells, and in particular dopaminergic neurons, due to their high synaptic connectivity, need high amounts of ATP and, as consequence, elevated mitochondrial activity. In physiological conditions, ATP synthesis occurs through an electron transport chain composed of four different complexes (I–IV), responsible for the formation of a protonic gradient over the mitochondrial inner membrane, required for ATP production. αS is a predominantly cytosolic protein but, in specific conditions, can interact and localize in mitochondria-associated ER membranes (Guardia-Laguarta et al., 2014); the occurrence of pathogenic point mutations in αS resulted in a reduced association with mitochondria-associated ER membranes, causing, in turn, a significant decrease of mitochondria-associated ER membranes functionality, as well as in an increase in mitochondrial fragmentation, thus pointing out a prominent role of αS in the control and modulation of mitochondrial morphology (Guardia-Laguarta et al., 2014). Concomitantly, an α-helical-mediated binding of monomeric αS with the inner mitochondrial membranes has been described by Robotta and collaborators (2014). Not surprisingly, the existence of a mitochondrial targeting sequence, comprising the first N-terminal 32 amino acids has been suggested (Devi et al., 2008). Specifically, such sequence is recognized by the translocase of the outer membrane (TOM) receptors. Upon recognition, target proteins are translocated through the TOM complex to the inner membrane and finally into the matrix (Devi et al., 2008). When applied exogenously, monomeric αS was reported to localize to mitochondria, to interact with ATP synthase subunit α, and increase its activity, thus playing a role in the physiological regulation of mitochondrial bioenergetics (Figure 4; Ludtmann et al., 2016). In this regard, Di Maio et al. (2016) revealed that oligomeric, dopamine-modified, and S129 phosphorylated (but not monomeric and nitrated) αS was able to bind with high affinity the TOM20 receptor of the mitochondrial protein import machinery. Such interaction inhibited the binding of TOM20 with its co-receptor, TOM22, and impaired mitochondrial protein import. Consequently, deficient mitochondrial respiration, enhanced production of reactive oxygen species (ROS), and loss of mitochondrial membrane potential were observed (Di Maio et al., 2016). Consistently, the concomitant overexpression of human αS and TOM20 in the SN of Lewis rats was reported to prevent the mitochondrial damage evoked by αS (De Miranda et al., 2020).
Importantly, defects in mitochondrial protein import determine the dysfunction of complex I, which is a cardinal pathological hallmark of PD (Franco-Iborra et al., 2018); consistently, a reduction in complex I activity is responsible for ROS formation, and has been observed in the SN of patients with sporadic PD (Hattingen et al., 2009), as well as in the skeletal muscle (Bindoff et al., 1991) and platelets of PD patients (Haas et al., 1995), supporting the notion that systemic mitochondrial alterations are implicated in PD. The αS oligomers-linked complex I inhibition also leads to slow depolarisation of the mitochondrial membrane potential (Ludtmann et al., 2018), to a rapid accumulation of mitochondrial DNA (mtDNA) mutations (Helley et al., 2017), as well as to the selective oxidation of the beta subunit of ATP synthase, and to mitochondrial lipid peroxidation. Such oxidation events dramatically increase the probability of permeability transition pore opening (Figure 4), triggering mitochondrial swelling, and ultimately leading to cell death (Ludtmann et al., 2018). Oligomers formed upon αS overexpression were reported to reduce axonal mitochondrial transport in human neurons derived from induced pluripotent stem cells of a PD patient, in turn responsible for reduced axonal density and severe synaptic degeneration (Prots et al., 2018). Also, extracellularly-added oligomers were described for their ability to impair mitochondrial biosynthesis due to the parkin-dependent reduction of the protein levels of the peroxisome proliferator-activated receptor-gamma coactivator (Wilkaniec et al., 2021). Other studies investigated the effect of αS PFFs on mitochondrial functionality; to this aim, Tapias et al. (2017) added PFFs to the culture medium of primary ventral midbrain neurons, revealing a significant increase of mitochondrial ROS, together with a massive impairment of mitochondrial respiration. Concomitantly, a significant depletion of optic atrophy-1 expression was observed, with consequent perturbation of the mitochondrial fission-fusion cycle (Tapias et al., 2017). The mitochondrial accumulation of phosphorylated αS in a similar neuronal model was confirmed by Wang and coworkers, that reported that PFFs possess a high binding capability to purified mitochondria with respect to the monomeric protein, revealing a preferential mitochondrial binding by aggregated αS (Wang et al., 2019).
Nuclear Function
The nuclear localization of αS has been posited since its discovery (Maroteaux et al., 1988), but its occurrence here, as well as the effect of its nuclear localization, are still a matter of investigation (Nishie et al., 2004; Huang et al., 2011; Davidi et al., 2020). Importantly, the presence of nuclear αS inclusions has been observed in MSA patients (Nishie et al., 2004). Extracellularly added αS was reported to evoke ROS production, to decrease mitochondrial membrane potential and to activate programmed cell death through the induction of the expression of the pro-apoptotic protein Bax and the reduction of the activity of the anti-apoptotic protein Bcl2 (Figure 4; Motyl et al., 2018). The largest majority of studies on the effects of αS in the nucleus has been performed by injecting PFFs in the mouse brain, or adding such conformers to the extracellular medium of cultured cells; their administration to various cell types such as dopaminergic N27 cells, primary cortical neurons, microglia, and astrocytes, as well as their injection in mouse brain tissues, resulted in a substantial reduction in the levels of Lamin B1 and high mobility group box 1, both established markers of cellular senescence, together with an increase in the levels of p21, a cell cycle-arrester and senescence marker, in both reactive astrocytes and microglia of mouse brains (Figure 4; Verma et al., 2021). In a recent study, Lee et al. (2021) revealed that the injection of PFFs in mice also resulted in a significant decrease in transcriptional adapter 2-alpha in the striatum and SN; the block of the activity of this component of the p300/CBP-associated factor led to a reduced histone H3 acetylation. Therefore, histone modification through αS-transcriptional adapter 2-alpha interaction may be associated with αS neurotoxicity in PD pathology.
Synaptic-Vesicle Trafficking and Neurotransmitter Release
Since its discovery, αS has been reported to be mainly localized at presynaptic terminals (Maroteaux et al., 1988); this observation, together with its reported capability to bind synaptic vesicles (Man et al., 2021), strongly suggests a regulatory function in synaptic activity and plasticity, neurotransmitter release, and in the management of the synaptic vesicle pool and trafficking, as reviewed in Burré et al. (2015). Neurotransmitter release is a complex mechanism firstly requiring the mobilization and clustering of the reserve pool of synaptic vesicles near synapses, and then the docking at active zones of such vesicles, that become the readily releasable pool. This step is followed by the formation of the synaptic soluble NSF attachment protein receptor (SNARE) complex, constituted by the proteins syntaxin-1, SNAP-25, and VAMP2/synaptobrevin-2 (Söllner et al., 1993), then by the fusion of synaptic vesicles with the presynaptic plasma membrane upon Ca2+ signaling, and finally by the disassembly of the SNARE complex and endosomal recycling of synaptic vesicles (Sudhof, 2004). αS has been reported to chaperone the SNARE complex assembly through its binding to the VAMP2/synaptobrevin-2 protein, thus promoting synaptic vesicle fusion at the presynaptic terminal (Burré et al., 2010). Choi et al. (2013) reported that large αS oligomers bind the N-terminal domain of synaptobrevin-2, thus blocking SNARE-mediated vesicle docking (Figure 4). More recently, the same group pointed out a cooperative pathological effect in an in vitro liposome fusion setting, occurring when αS monomers at submicromolar concentrations were co-incubated with oligomers. In such condition, oligomeric αS exclusively interacted with synaptobrevin-2, without binding vesicle lipids. The addition of small peptide fragments from the C-terminal domain of αS blocked αS oligomers binding to synaptobrevin-2, thus restoring the formation of the SNARE complex and vesicle fusion (Yoo et al., 2021). Consistently, Volpicelli-Daley and coworkers previously revealed a significant reduction in the level of many synaptic proteins, such as the complex SNARE proteins SNAP25 and VAMP2, as well as other proteins involved in SNARE complex assembly or in the exoendocytic synaptic vesicle cycle, such as synapsin II (which regulates the motility of synaptic vesicle pools) and cysteine string protein α (that chaperones the SNARE complex assembly very similarly to αS), 2 weeks after the addition of αS PFFs to primary neuronal cultures (Volpicelli-Daley et al., 2011).
Aging and Its Impact on Neurodegeneration
It is well-known that motor performance declines with age (Seidler et al., 2010; Hoogendam et al., 2014). Several age-dependent changes, including alterations in dopamine metabolism, uptake and synthesis, receptor sensitivity, mitochondrial function, calcium homeostasis, and proteostasis make dopaminergic neurons more susceptible to dysfunction and senescence with respect to other neuronal cells (Reeve et al., 2014; Zucca et al., 2017; Surmeier, 2018). The manifestation of PD is complex and heterogeneous, however, several features that differently contribute to brain aging might help the differential diagnosis with other synucleinopathies (Chen et al., 2022). Although it is well-known that age is the highest risk factor in the development of neurodegenerative diseases, including PD, the mechanisms and resulting effects have only been explored to a limited extent. In the last few years, experimental studies on animal models generated conflicting results, generally due to an increased vulnerability of aged animals (Klæstrup et al., 2022). The first animal models have been focused mainly on reflecting the disease in the CNS with different motor phenotypes. However, most studies failed to induce pathology in the CNS and/or neurodegeneration and a motor phenotype. In addition, different effects on treatment efficacy have been reported between young and old animals, indicating that the models actually used in preclinical research may be suboptimal and that the translational outcomes are limited. As reported above, it has been postulated that PD may start in the gut through a pre-motor phase (Reichmann, 2017). Indeed, gut dysbiosis has been shown in PD patients (Romano et al., 2021; Klann et al., 2022) and potential alterations in our microbiome during aging might also contribute to PD risk. Recent studies on gut-seeding properties demonstrated that old age is a crucial factor to obtain effective gut-to-brain propagation of αS pathology (Challis et al., 2020; Van Den Berge et al., 2021). Importantly, gut microbiota also influences the immune environment of the brain, contributing to the inflammation both in the brain and in the peripheral body and to immunosenescence, with functional consequences both in neurons and microglia (Ogrodnik et al., 2021). Overall, identifying age-associated features in disease presentation and progression could be useful both on the individual level, impacting positively the patient management, and for clinical research developing new therapeutic strategies.
Alpha-Synuclein-Targeting Therapeutic Approaches
Disease-modifying therapies aimed at targeting αS pathology actually include various approaches to prevent and mitigate its neurotoxicity; such strategies involve: (I) the reduction of αS expression; (II) the inhibition of its misfolding and aggregation; (III) the enhancement of cellular degradation mechanisms; (IV) the clearance of αS pathogenic aggregates from the brain; (V) the prevention of cell-to-cell transmission of αS aggregates. A potential strategy to diminish the level of αS aggregates implies the reduction of SNCA expression; several experimental evidences obtained in cultured cells, mouse models and human primates revealed that the use of small interfering RNA, as well as short hairpin RNA, significantly decreased SNCA expression and accumulation of misfolded αS (Lewis et al., 2008; Khodr et al., 2014; Zharikov et al., 2019). However, questionable results emerged from in vivo evidence, suggesting potential detrimental effects on dopaminergic neurons upon total abolition of SNCA expression (Menon et al., 2022). Thus, partial silencing is actually suggested, considering that moderate αS reduction is enough to prevent misfolding and pathology spreading (Menon et al., 2022). The reduction of αS mRNA transcription with beta-2 adrenoreceptor antagonists such as metaproterenol, clenbuterol, and salbutamol, has been proposed as a disease-modifying strategy (Mittal et al., 2017); more recently, small molecules able to bind the iron-responsive element of SNCA mRNA, a region involved in translation regulation, were designed to reduce its translation (Zhang et al., 2020). αS aggregation can be inhibited taking advantage of small molecules as epigallocatechin gallate, previously reported to reduce the formation of oligomeric αS and their associated cytotoxicity (Kurnik et al., 2018). Consistently, epigallocatechin gallate has been recently tested in a Phase III trial in MSA patients, revealing no clinical efficacy, and adverse effects in some patients (Levin et al., 2019). Other compounds, referred to as naturally occurring aminosterols, such as squalamine (ENT-01) and trodusquemine were recently reported to modulate αS aggregation and to inhibit the binding of toxic oligomers to the plasma membrane of neuronal cells (Perni et al., 2017, 2018; Limbocker et al., 2021). Thanks to promising results obtained in animal models, ENT-01 has been tested in the phase IIa RASMET study, which was conducted on 50 PD patients with constipation. Its administration was safe, and bowel function resulted to be rapidly improved, as well as neuropsychiatric symptomatology, confirming that targeting enteric αS might be a beneficial approach in PD therapy (Hauser et al., 2019). The IIb study KARMET, a randomized and placebo-controlled trial involving another 150 PD patients, was conducted to determine the safety and efficacy of up to 25 days of ENT-01 treatment, confirming its ability to improve both bowel motility and gut-brain axis (Camilleri et al., 2022). Another promising therapeutic approach consists of the enhancement of the degradation of αS misfolded aggregates. To this aim, rapamycin was proposed as potential disease-modifying therapy given its ability to enhance macroautophagy through the inhibition of the mechanistic target of rapamycin. Despite promising results obtained both in cellular and animal models (Moors et al., 2017), the low specificity of this drug, together with the severe adverse effects it evoked, limited its potential therapeutic use (Brundin et al., 2017). A valid alternative to enhance the autophagic process through the downregulation of the mechanistic target of rapamycin pathway is represented by tyrosine kinase inhibitors. Specifically, the tyrosine kinase c-Abl has been identified as a potent inducer of αS aggregation with subsequent neuronal cell death (Brahmachari et al., 2016); consistently, its inhibition has been reported to be neuroprotective in PD animal models (Brahmachari et al., 2017; Lee et al., 2018). Despite some promising results observed in nonblinded safety trials of the c-Abl inhibitor nilotinib in DLB and PD patients (Pagan et al., 2016, 2019), a subsequent efficacy trial conducted in PD subjects did not reveal any significant improvement with respect to the placebo group (Simuni et al., 2021). Immunoregulatory approaches are based on the ability of specific antibodies to bind and clear both intracellular and extracellular αS aggregates (Bhatt et al., 2013; Spencer et al., 2017; Messer et al., 2020), and many clinical trials employing both passive and active immunization are being developed and tested (Valera et al., 2016; Wang et al., 2019; Kwon et al., 2020). In particular, active immunization consists of vaccination through the administration of a specific antigen; this treatment requires rare and low-cost administrations, at the same time the immune response elicited does not follow the same pattern in different individuals, as the immune response pattern cannot be predicted (Schneeberger et al., 2016). Passive immunization is based on the administration of antibodies by specialized personnel; at the same time, this procedure guarantees the possibility to control systemic antibody concentrations and to interrupt treatment when side effects occur (Bergström et al., 2016). The largest majority of such antibodies are raised against small epitopes predominantly localized at the C-terminal domain of αS (Knecht et al., 2022). These approaches, targeting various αS epitopes, have shown neuroprotection in transgenic mouse models of PD, DLB, and MSA (Masliah et al., 2011; Mandler et al., 2015; Tran et al., 2014; Spencer et al., 2017; Henderson et al., 2020b; Lemos et al., 2020), thus leading to reduced accumulation of toxic oligomers and limited neurodegeneration. Nevertheless, the existence of potential detrimental effects against physiological αS, as well as the logistical challenges related to the crossing of the blood-brain barrier must be considered (Knecht et al., 2022). Very recently, the first two phase II trials of therapies aimed at targeting αS in early PD did not pursue the primary endpoints. Specifically, the humanized monoclonal immunoglobulin G1 antibody Prasimezumam, targeting an epitope in the C-terminal domain of αS and previously reported to easily penetrate the brain and to evoke the reduction of serum αS levels (Schenk et al., 2017; Jankovc et al., 2018) did not produce any significant positive effect on global or imaging measures of PD progression as compared with placebo (Pagano et al., 2022). Similarly, the human-derived monoclonal antibody Cinpanemab, previously reported to target aggregated αS (Brys et al., 2019) and to reduce αS spreading and motor dysfunction in PD mice (Weihofen et al., 2019), failed in modifying disease progression in early PD subjects as compared to the placebo group (Lang et al., 2022). Finally, given the prominent role of receptor-mediated endocytosis in the uptake of αS misfolded aggregates (Mao et al., 2016; Thom et al., 2022) the inhibition of their membrane receptors could be a valuable strategy to prevent their cell-to-cell transmission, as extensively reviewed by Henderson et al. (Henderson et al., 2019).
Concluding Remarks
αS misfolding and aggregation in specific cellular subpopulations is the cardinal hallmark of a range of devastating neurodegenerative conditions, collectively referred to as synucleinopathies. During this process, a vast range of distinct aggregates is formed, including oligomers, protofibrils, and fibrils. Such species differentially exert detrimental effects on neuronal cells: while oligomeric aggregates seem to be directly implicated in the induction of neurotoxicity, given their ability to directly affect multiple cellular pathways, fibrillar assemblies not only play a crucial role in disease spreading and progression, but they also release harmful oligomeric conformers, promptly evoking cellular dysfunction in the recipient neurons. Actual research attempts to identify possible disease-modifying therapeutic strategies aiming at reducing the neurotoxicity of αS misfolded aggregates, by inhibiting their formation from the monomeric protein, by reducing their accumulation, or by enhancing their degradation. To this aim, a deeper understanding of the triggers of αS misfolding and aggregation, as well as of the molecular mechanisms of their spreading, and of the plethora of neurotoxic pathways they evoke will be of fundamental importance.
Additional file: Open peer review reports 1 (89.2KB, pdf) and 2 (89.1KB, pdf) .
Footnotes
Funding: The work was supported by the University of Florence (Fondi Ateneo to RC and CC), and the Ministry of Education, Universities and Research of Italy (Progetto Dipartimento di Eccellenza to CC).
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement: Not applicable.
Open peer reviewers: Shady Rahayel, Montreal Neurological Institute-Hospital Department of Neurology and Neurosurgery, Canada; Nathalie Van Den Berge, Aarhus University: Aarhus Universitet Clinical and Nuclear Medicine, Denmark.
P-Reviewers: Rahayel S, Van Den Berge N; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Alam P, Bousset L, Melki R, Otzen DE. α-synuclein oligomers and fibrils:a spectrum of species, a spectrum of toxicities. J Neurochem. 2019;150:522–534. doi: 10.1111/jnc.14808. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AHV. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 3.Angelova PR, Ludtmann MH, Horrocks MH, Negoda A, Cremades N, Klenerman D, Dobson CM, Wood NW, Pavlov EV, Gandhi S, Abramov AY. Ca2+is a key factor in α-synuclein-induced neurotoxicity. J Cell Sci. 2016;129:1792–1801. doi: 10.1242/jcs.180737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arima K, Ueda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka-Uehara H, Kawai M. Immunoelectron-microscopic demonstration of NACP/α-synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson's disease and in dementia with Lewy bodies. Brain Res. 1998;808:93–100. doi: 10.1016/s0006-8993(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 5.Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, Thiolat ML, Tasselli M, Aubert P, Kruse N, Mollenhauer B, Trigo Damas I, Estrada C, Garcia-Carrillo N, Vaikath NN, El-Agnaf OMA, Herrero MT, Vila M, Obeso JA, Derkinderen P, Dehay B, Bezard E. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. 2020;143:1462–1475. doi: 10.1093/brain/awaa096. [DOI] [PubMed] [Google Scholar]
- 6.Bae EJ, Yang NY, Lee C, Kim S, Lee HJ, Lee SJ. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis. 2015;6:e1901. doi: 10.1038/cddis.2015.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baek JH, Whitfield D, Howlett D, Francis P, Bereczki E, Ballard C, Hortobagyi T, Attems J, Aarsland D. Unfolded protein response is activated in Lewy body dementias. Neuropathol Appl Neurobiol. 2016;42:352–365. doi: 10.1111/nan.12260. [DOI] [PubMed] [Google Scholar]
- 8.Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ, Lee VM. Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβpathology. Neuron. 2020;105:260–275. doi: 10.1016/j.neuron.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol. 2001;205:149–214. doi: 10.1016/s0074-7696(01)05004-5. [DOI] [PubMed] [Google Scholar]
- 10.Bayati A, Banks E, Han C, Luo W, Reintsch WE, Zorca CE, Shlaifer I, Del Cid Pellitero E, Vanderperre B, McBride HM, Fon EA, Durcan TM, McPherson PS. Rapid macropinocytic transfer of α-synuclein to lysosomes. Cell Rep. 2022;40:111102. doi: 10.1016/j.celrep.2022.111102. [DOI] [PubMed] [Google Scholar]
- 11.Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, Battistin L, Spillantini M, Missale C, Spano P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson's disease. J Neurochem. 2011;116:588–605. doi: 10.1111/j.1471-4159.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- 12.Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J. Alpha-synuclein oligomers:a new hope. Acta Neuropathol. 2017;134:819–838. doi: 10.1007/s00401-017-1755-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 14.Betzer C, Lassen LB, Olsen A, Kofoed RH, Reimer L, Gregersen E, Zheng J, Calì T, Gai WP, Chen T, Moeller A, Brini M, Fu Y, Halliday G, Brudek T, Aznar S, Pakkenberg B, Andersen JP, Jensen PH. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018;19:e44617. doi: 10.15252/embr.201744617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatt MA, Messer A, Kordower JH. Can intrabodies serve as neuroprotective therapies for Parkinson's disease?Beginning thoughts. J Parkinsons Dis. 2013;3:581–591. doi: 10.3233/JPD-130252. [DOI] [PubMed] [Google Scholar]
- 16.Bigi A, Ermini E, Chen SW, Cascella R, Cecchi C. Exploring the release of toxic oligomers from α-synuclein fibrils with antibodies and STED microscopy. Life (Basel) 2021;11:431. doi: 10.3390/life11050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bigi A, Cascella R, Chiti F, Cecchi C. Amyloid fibrils act as a reservoir of soluble oligomers, the main culprits in protein deposition diseases. Bioessays. 2022;44:e2200086. doi: 10.1002/bies.202200086. [DOI] [PubMed] [Google Scholar]
- 18.Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD, Jr, Turnbull DM. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson's disease. J Neurol Sci. 1991;104:203–208. doi: 10.1016/0022-510x(91)90311-t. [DOI] [PubMed] [Google Scholar]
- 19.Blömeke L, Pils M, Kraemer-Schulien V, Dybala A, Schaffrath A, Kulawik A, Rehn F, Cousin A, Nischwitz V, Willbold J, Zack R, Tropea TF, Bujnicki T, Tamgüney G, Weintraub D, Irwin D, Grossman M, Wolk DA, Trojanowski JQ, Bannach O, Chen-Plotkin A, Willbold D. Quantitative detection of α-synuclein and Tau oligomers and other aggregates by digital single particle counting. NPJ Parkinsons Dis. 2022;8:68. doi: 10.1038/s41531-022-00330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages) J Neurol. 2002;249(Suppl 3):III/1–5. doi: 10.1007/s00415-002-1301-4. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003a;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease:possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003b;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 23.Brahic M, Bousset L, Bieri G, Melki R, Gitler AD. Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol. 2016;131:539–548. doi: 10.1007/s00401-016-1538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brahmachari S, Ge P, Lee SH, Kim D, Karuppagounder SS, Kumar M, Mao X, Shin JH, Lee Y, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Ko HS. Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J Clin Invest. 2016;126:2970–2988. doi: 10.1172/JCI85456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brahmachari S, Karuppagounder SS, Ge P, Lee S, Dawson VL, Dawson TM, Ko HS. c-Abl and Parkinson's disease:mechanisms and therapeutic potential. J Parkinsons Dis. 2017;7:589–601. doi: 10.3233/JPD-171191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling:function and dysfunction. Cell Mol Life Sci. 2014;71:2787–2814. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown DR. Interactions between metals and a-synuclein—Function or artefact? FEBS J. 2007;274:3766–3774. doi: 10.1111/j.1742-4658.2007.05917.x. [DOI] [PubMed] [Google Scholar]
- 28.Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. 2017;298:225–235. doi: 10.1016/j.expneurol.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, Welch M, Koenig E, David E, Fox T, Makh S, Aldred J, Goodman I, Pepinsky B, Liu Y, Graham D, Weihofen A, Cedarbaum JM. Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Mov Disord. 2019;34:1154–1163. doi: 10.1002/mds.27738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native brain α-synuclein. Nature. 2013;498:E4–7. doi: 10.1038/nature12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burré J. The synaptic function of α-synuclein. J Parkinsons Dis. 2015;5:699–713. doi: 10.3233/JPD-150642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burré J, Sharma M, Südhof TC. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med. 2018;8:a024091. doi: 10.1101/cshperspect.a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bussell R, Jr, Ramlall TF, Eliezer D. Helix periodicity, topology, and dynamics of membrane-associated α-synuclein. Protein Sci. 2005;14:862–872. doi: 10.1110/ps.041255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Camilleri M, Subramanian T, Pagan F, Isaacson S, Gil R, Hauser RA, Feldman M, Goldstein M, Kumar R, Truong D, Chhabria N, Walter BL, Eskenazi J, Riesenberg R, Burdick D, Tse W, Molho E, Robottom B, Bhatia P, Kadimi S, et al. Oral ENT-01 targets enteric neurons to treat constipation in Parkinson disease:a randomized controlled trial. Ann Intern Med. 2022;175:1666–1674. doi: 10.7326/M22-1438. [DOI] [PubMed] [Google Scholar]
- 36.Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis:interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–209. [PMC free article] [PubMed] [Google Scholar]
- 37.Cascella R, Perni M, Chen SW, Fusco G, Cecchi C, Vendruscolo M, Chiti F, Dobson CM, De Simone A. Probing the origin of the toxicity of oligomeric aggregates of α-synuclein with antibodies. ACS Chem Biol. 2019;14:1352–1362. doi: 10.1021/acschembio.9b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cascella R, Chen SW, Bigi A, Camino JD, Xu CK, Dobson CM, Chiti F, Cremades N, Cecchi C. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat Commun. 2021;12:1814. doi: 10.1038/s41467-021-21937-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cascella R, Bigi A, Cremades N, Cecchi C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell Mol Life Sci. 2022;79:174. doi: 10.1007/s00018-022-04166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castillo-Carranza DL, Zhang Y, Guerrero-Muñoz MJ, Kayed R, Rincon-Limas DE, Fernandez-Funez P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem Res. 2012;37:1707–1717. doi: 10.1007/s11064-012-0780-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Celej MS, Sarroukh R, Goormaghtigh E, Fidelio GD, Ruysschaert JM, Raussens V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem J. 2012;443:719–726. doi: 10.1042/BJ20111924. [DOI] [PubMed] [Google Scholar]
- 42.Cenini G, Cecchi C, Pensalfini A, Bonini SA, Ferrari-Toninelli G, Liguri G, Memo M, Uberti D. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids. 2010;38:1101–1106. doi: 10.1007/s00726-009-0319-7. [DOI] [PubMed] [Google Scholar]
- 43.Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA, Gradinaru V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. 2020;23:327–336. doi: 10.1038/s41593-020-0589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken α-helix in folded α- Synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 45.Chandra R, Hiniker A, Kuo YM, Nussbaum RL, Liddle RA. α-Synuclein in gut endocrine cells and its implications for Parkinson's disease. JCI Insight. 2017;2:e92295. doi: 10.1172/jci.insight.92295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaudhary H, Iyer A, Subramaniam V, Claessens MM. α-Synuclein oligomers stabilize pre-existing defects in supported bilayers and propagate membrane damage in a fractal-like pattern. Langmuir. 2016;32:11827–11836. doi: 10.1021/acs.langmuir.6b02572. [DOI] [PubMed] [Google Scholar]
- 47.Chen CL, Kuo MC, Wu WC, Hsu YC, Wu RM, Tseng WI. Advanced brain aging in multiple system atrophy compared to Parkinson's disease. Neuroimage Clin. 2022;34:102997. doi: 10.1016/j.nicl.2022.102997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TP, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM, Cremades N. Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc Natl Acad Sci U S A. 2015;112:E1994–E2003. doi: 10.1073/pnas.1421204112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiti F, Dobson CM. Protein misfolding, amyloid, formation and human disease:a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. doi: 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- 50.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A. 2013;110:4087–4092. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colosimo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2003;74:852–856. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. Structured regions of alpha-synuclein fibrils include the early-onset Parkinson's disease mutation sites. J Mol Biol. 2011;411:881–895. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 55.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cremades N, Chen SW, Dobson CM. Structural characteristics of α-synuclein oligomers. Int Rev Cell Mol Biol. 2017;329:79–143. doi: 10.1016/bs.ircmb.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Crowther RA, Daniel SE, Goedert M. Characterisation of isolated alphasynuclein filaments from substantia nigra of Parkinson's disease brain. Neurosci Lett. 2000;292:128–130. doi: 10.1016/s0304-3940(00)01440-3. [DOI] [PubMed] [Google Scholar]
- 59.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 60.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davidi D, Schechter M, Elhadi SA, Matatov A, Nathanson L, Sharon R. α-Synuclein translocates to the nucleus to activate retinoic-acid-dependent gene transcription. iScience. 2020;23:100910. doi: 10.1016/j.isci.2020.100910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daykin EC, Ryan E, Sidransky E. Diagnosing neuronopathic Gaucher disease:new considerations and challenges in assigning Gaucher phenotypes. Mol Genet Metab. 2021;132:49–58. doi: 10.1016/j.ymgme.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Giorgi F, Laferrière F, Zinghirino F, Faggiani E, Lends A, Bertoni M, Yu X, Grélard A, Morvan E, Habenstein B, Dutheil N, Doudnikoff E, Daniel J, Claverol S, Qin C, Loquet A, Bezard E, Ichas F. Novel self-replicating α-synuclein polymorphs that escape ThT monitoring can spontaneously emerge and acutely spread in neurons. Sci Adv. 2020;6:eabc4364. doi: 10.1126/sciadv.abc4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Miranda BR, Rocha EM, Castro SL, Greenamyre JT. Protection from α-synuclein induced dopaminergic neurodegeneration by overexpression of the mitochondrial import receptor TOM20. NPJ Parkinsons Dis. 2020;6:38. doi: 10.1038/s41531-020-00139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Del Tredici K, Braak H. Review:sporadic Parkinson's disease:development and distribution of α-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42:33–50. doi: 10.1111/nan.12298. [DOI] [PubMed] [Google Scholar]
- 66.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. 2016;8:342ra78. doi: 10.1126/scitranslmed.aaf3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ding TT, Lee SJ, Rochet JC, Lansbury PT Jr. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 70.Diógenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Näsström T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergström J, Ingelsson M, Quintas A, Sebastião AM, Lopes LV, Outeiro TF. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci. 2012;32:11750–11762. doi: 10.1523/JNEUROSCI.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doty RL. Olfaction in Parkinson's disease and related disorders. Neurobiol Dis. 2012;46:527–552. doi: 10.1016/j.nbd.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eliezer D, Kutluay E, Bussell R, Browne G. Conformational properties of α- synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 73.Evangelisti E, Cascella R, Becatti M, Marrazza G, Dobson CM, Chiti F, Stefani M, Cecchi C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci Rep. 2016;6:32721. doi: 10.1038/srep32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferreira DG, Temido-Ferreira M, Vicente Miranda H, Batalha VL, Coelho JE, SzegöÉ M, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes LV, Outeiro TF. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017;20:1569–1579. doi: 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- 75.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci U S A. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franco-Iborra S, Cuadros T, Parent A, Romero-Gimenez J, Vila M, Perier C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson's disease. Cell Death Dis. 2018;9:1122. doi: 10.1038/s41419-018-1154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM, De Simone A. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science. 2017;358:1440–1443. doi: 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- 78.Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson's disease models. Mol Neurobiol. 2018;55:7297–7316. doi: 10.1007/s12035-018-0937-8. [DOI] [PubMed] [Google Scholar]
- 79.Gath J, Habenstein B, Bousset L, Melki R, Meier BH, Böckmann A. Solid-state NMR sequential assignments of α-synuclein. Biomol NMR Assign. 2012;6:51–55. doi: 10.1007/s12104-011-9324-3. [DOI] [PubMed] [Google Scholar]
- 80.GBD 2016; Parkinson's Disease Collaborators. Global, regional, and national burden of Parkinson's disease, 1990-2016:a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:939–953. doi: 10.1016/S1474-4422(18)30295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gegg ME, Verona G, Schapira AHV. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum Mol Genet. 2020;29:1716–1728. doi: 10.1093/hmg/ddaa085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, Kumar A, Riek R, Maji SK. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci Rep. 2015;5:9228. doi: 10.1038/srep09228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 84.Goedert M, Jakes R, Spillantini MG. The synucleinopathies:twenty years on. J Parkinsons Dis. 2017;7:S51–69. doi: 10.3233/JPD-179005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon EA, Przedborski S. α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34:249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gracia P, Camino JD, Volpicelli-Daley L, Cremades N. Multiplicity of α-synuclein aggregated species and their possible roles in disease. Int J Mol Sci. 2020;8043;21 doi: 10.3390/ijms21218043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guerrero-Ferreira R, Taylor NM, Arteni AA, Kumari P, Mona D, Ringler P, Britschgi M, Lauer ME, Makky A, Verasdonck J, Riek R, Melki R, Meier BH, Böckmann A, Bousset L, Stahlberg H. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife. 2019;8:e48907. doi: 10.7554/eLife.48907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol. 1995;37:714–22. doi: 10.1002/ana.410370604. [DOI] [PubMed] [Google Scholar]
- 89.Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C, Steinmetz H, Zanella F, Hilker R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson's disease. Brain. 2009;132((Pt 12)):3285–3297. doi: 10.1093/brain/awp293. [DOI] [PubMed] [Google Scholar]
- 90.Hauser RA, Sutherland D, Madrid JA, Rol MA, Frucht S, Isaacson S, Pagan F, Maddux BN, Li G, Tse W, Walter BL, Kumar R, Kremens D, Lew MF, Ellenbogen A, Oguh O, Vasquez A, Kinney W, Lowery M, Resnick M, et al. Targeting neurons in the gastrointestinal tract to treat Parkinson's disease. Clin Park Relat Disord. 2019;1:2–7. doi: 10.1016/j.prdoa.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Helley MP, Pinnell J, Sportelli C, Tieu K. Mitochondria:a common target for genetic mutations and environmental toxicants in Parkinson's disease. Front Genet. 2017;8:177. doi: 10.3389/fgene.2017.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response:integrating stress signals through the stress sensor IRE1α. Physiol Rev. 2011;91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- 93.Henderson MX, Trojanowski JQ, Lee VM. α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies. Neurosci Lett. 2019;709:134316. doi: 10.1016/j.neulet.2019.134316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, Li HL, Zhang B, Brown HJ, Olufemi MF, Bassett DS, Trojanowski JQ, Lee VMY. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein Insult. Neuron. 2020a;105:822–836. doi: 10.1016/j.neuron.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henderson MX, Covell DJ, Chung CH, Pitkin RM, Sandler RM, Decker SC, Riddle DM, Zhang B, Gathagan RJ, James MJ, Trojanowski JQ, Brunden KR, Lee VMY, Luk KC. Characterization of novel conformation-selective α-synuclein antibodies as potential immunotherapeutic agents for Parkinson's disease. Neurobiol Dis. 2020b;136:104712. doi: 10.1016/j.nbd.2019.104712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J, Winkler J, Xiang W, Schlachetzki JCM. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun. 2016;479:881–886. doi: 10.1016/j.bbrc.2016.09.109. [DOI] [PubMed] [Google Scholar]
- 97.Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;28:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- 98.Hoogendam YY, Hofman A, van der Geest JN, van der Lugt A, Ikram MA. Patterns of cognitive function in aging:the Rotterdam Study. Eur J Epidemiol. 2014;29:133–140. doi: 10.1007/s10654-014-9885-4. [DOI] [PubMed] [Google Scholar]
- 99.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 100.Horowitz M, Braunstein H, Zimran A, Revel-Vilk S, Goker-Alpan O. Lysosomal functions and dysfunctions:Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv Drug Deliv Rev. 2022;187:114402. doi: 10.1016/j.addr.2022.114402. [DOI] [PubMed] [Google Scholar]
- 101.Houser MC, Chang J, Factor SA, Molho ES, Zabetian CP, Hill-Burns EM, Payami H, Hertzberg VS, Tansey MG. Stool immune profiles evince gastrointestinal inflammation in Parkinson's disease. Mov Disord. 2018;33:793–804. doi: 10.1002/mds.27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iyer A, Claessens MMAE. Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta Proteins Proteom. 2019;1867:468–482. doi: 10.1016/j.bbapap.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 103.Huang Z, Xu Z, Wu Y, Zhou Y. Determining nuclear localization of alpha-synuclein in mouse brains. Neuroscience. 2011;199:318–332. doi: 10.1016/j.neuroscience.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE. α-Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol. 2013;73:155–169. doi: 10.1002/ana.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 106.Karpowicz RJ, Jr, Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM. Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem. 2017;292:13482–13497. doi: 10.1074/jbc.M117.780296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 108.Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012;3:61. doi: 10.3389/fphar.2012.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim C, Lv G, Lee JS, Jung BC, Masuda-Suzukake M, Hong CS, Valera E, Lee HJ, Paik SR, Hasegawa M, Masliah E, Eliezer D, Lee SJ. Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci Rep. 2016;6:30891. doi: 10.1038/srep30891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, Rhee P, Karuppagounder SS, Kwon SH, Lee H, Mao X, Kim D, Pandey A, Lee G, Dawson VL, Dawson TM, Ko HS. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A. 2018;115:798–803. doi: 10.1073/pnas.1700465115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kishimoto Y, Zhu W, Hosoda W, Sen JM, Mattson MP. Chronic mild gut inflammation accelerates brain neuropathology and motor dysfunction in α-synuclein mutant mice. Neuromol Med. 2019;21:239–249. doi: 10.1007/s12017-019-08539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Klann EM, Dissanayake U, Gurrala A, Farrer M, Shukla AW, Ramirez-Zamora A, Mai V, Vedam-Mai V. The gut-brain axis and its relation to Parkinson's disease:a review. Front Aging Neurosci. 2022;13:782082. doi: 10.3389/fnagi.2021.782082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khodr CE, Becerra A, Han Y, Bohn MC. Targeting alpha synuclein with a microRNA-embedded silencing vector in the rat substantia nigra:positive and negative effects. Brain Res. 2014;1550:47–60. doi: 10.1016/j.brainres.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klæstrup IH, Just MK, Holm KL, Alstrup AKO, Romero-Ramos M, Borghammer P, Van Den Berge N. Impact of aging on animal models of Parkinson's disease. Front Aging Neurosci. 2022;14:909273. doi: 10.3389/fnagi.2022.909273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Koehler NK, Stransky E, Shing M, Gaertner S, Meyer M, Schreitmüller B, Leyhe T, Laske C, Maetzler W, Kahle P, Celej MS, Jovin TM, Fallgatter AJ, Batra A, Buchkremer G, Schott K, Richartz-Salzburger E. Altered serum IgG levels to α-synuclein in dementia with Lewy bodies and Alzheimer's disease. PLoS One. 2013;8:e64649. doi: 10.1371/journal.pone.0064649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 117.Knecht L, Folke J, Dodel R, Ross JA, Albus A. Alpha-synuclein immunization strategies for synucleinopathies in clinical studies:a biological perspective. Neurotherapeutics. 2022;19:1489–1502. doi: 10.1007/s13311-022-01288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kulenkampff K, Wolf Perez AM, Sormanni P, Habchi J, Vendruscolo M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Na Rev Chem. 2021;5:277–294. doi: 10.1038/s41570-021-00254-9. [DOI] [PubMed] [Google Scholar]
- 119.Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv. 2022;8:eabm6393. doi: 10.1126/sciadv.abm6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kurnik M, Sahin C, Andersen CB, Lorenzen N, Giehm L, Mohammad-Beigi H, Jessen CM, Pedersen JS, Christiansen G, Petersen SV, Staal R, Krishnamurthy G, Pitts K, Reinhart PH, Mulder FAA, Mente S, Hirst WD, Otzen DE. Potent α-synuclein aggregation inhibitors, identified by high-throughput screening, mainly target the monomeric state. Cell Chem Biol. 2018;25:1389–1402. doi: 10.1016/j.chembiol.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 121.Kwon S, Iba M, Kim C, Masliah E. Immunotherapies for aging-related neurodegenerative diseases-emerging perspectives and new targets. Neurotherapeutics. 2020;17:935–954. doi: 10.1007/s13311-020-00853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lang AE, Siderowf AD, Macklin EA, Poewe W, Brooks DJ, Fernandez HH, Rascol O, Giladi N, Stocchi F, Tanner CM, Postuma RB, Simon DK, Tolosa E, Mollenhauer B, Cedarbaum JM, Fraser K, Xiao J, Evans KC, Graham DL, Sapir I, et al. Trial of cinpanemab in early Parkinson's disease. N Engl J Med. 2022;387:408–420. doi: 10.1056/NEJMoa2203395. [DOI] [PubMed] [Google Scholar]
- 123.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson's disease-associated mutants, form pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 124.Lautenschläger J, Stephens AD, Fusco G, Ströhl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, Pinotsi D, Zago W, Fraser P, Tandon A, St George-Hyslop P, Rees E, Phillips JJ, De Simone A, Kaminski CF, Schierle GSK. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat Commun. 2018;9:712. doi: 10.1038/s41467-018-03111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N'Guyen JM, Chaumette T, Tasselli M, Paillusson S, Flamand M, Galmiche JP, Damier P, Derkinderen P. Colonic biopsies to assess the neuropathology of Parkinson's disease and its relationship with symptoms. PLoS One. 2010;5:e12728. doi: 10.1371/journal.pone.0012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 127.Lee JY, Kim H, Jo A, Khang R, Park CH, Park SJ, Kwag E, Shin JH. α-Synuclein A53T binds to transcriptional adapter 2-alpha and blocks histone H3 acetylation. Int J Mol Sci. 2021;22:5392. doi: 10.3390/ijms22105392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee S, Kim S, Park YJ, Yun SP, Kwon SH, Kim D, Kim DY, Shin JS, Cho DJ, Lee GY, Ju HS, Yun HJ, Park JH, Kim WR, Jung EA, Lee S, Ko HS. The c-Abl inhibitor, Radotinib HCl, is neuroprotective in a preclinical Parkinson's disease mouse model. Hum Mol Genet. 2018;27:2344–2356. doi: 10.1093/hmg/ddy143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lemos M, Venezia S, Refolo V, Heras-Garvin A, Schmidhuber S, Giese A, Leonov A, Ryazanov S, Griesinger C, Galabova G, Staffler G, Wenning GK, Stefanova N. Targeting α-synuclein by PD03 AFFITOPE®and Anle138b rescues neurodegenerative pathology in a model of multiple system atrophy:clinical relevance. Transl Neurodegener. 2020;9:38. doi: 10.1186/s40035-020-00217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Levin J, Maaß S, Schuberth M, Giese A, Oertel WH, Poewe W, Trenkwalder C, Wenning GK, Mansmann U, Südmeyer M, Eggert K, Mollenhauer B, Lipp A, Löhle M, Classen J, Münchau A, Kassubek J, Gandor F, Berg D, Egert-Schwender S, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA):a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2019;18:724–735. doi: 10.1016/S1474-4422(19)30141-3. [DOI] [PubMed] [Google Scholar]
- 131.Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, Braithwaite A, He Z, Ogholikhan S, Hinkle K, Kent C, Toudjarska I, Charisse K, Braich R, Pandey RK, Heckman M, Maraganore DM, Crook J, Farrer MJ. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. 2008;3:19. doi: 10.1186/1750-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, Eisenberg DS, Zhou ZH, Jiang L. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9:3609. doi: 10.1038/s41467-018-05971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 134.Limbocker R, Staats R, Chia S, Ruggeri FS, Mannini B, Xu CK, Perni M, Cascella R, Bigi A, Sasser LR, Block NR, Wright AK, Kreiser RP, Custy ET, Meisl G, Errico S, Habchi J, Flagmeier P, Kartanas T, Hollows JE, et al. Squalamine and its derivatives modulate the aggregation of amyloid-βand α-synuclein and suppress the toxicity of their oligomers. Front Neurosci. 2021;15:680026. doi: 10.3389/fnins.2021.680026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O'Connell B, Pollen DA, St George-Hyslop P, Ghetti B, Nochlin D, Bird TD, Cairns NJ, Lee VM, Iwatsubo T, Trojanowski JQ. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu M, Qin L, Wang L, Tan J, Zhang H, Tang J, Shen X, Tan L, Wang C. α-synuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulum-Golgi compartment. Mol Med Rep. 2018;18:322–332. doi: 10.3892/mmr.2018.9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J Neurosci. 2016;36:10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, et al. 2018 α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nat Commun. 9:2293. doi: 10.1038/s41467-018-04422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Majbour NK, Vaikath NN, van Dijk KD, Ardah MT, Varghese S, Vesterager LB, Montezinho LP, Poole S, Safieh-Garabedian B, Tokuda T, Teunissen CE, Berendse HW, van de Berg WD, El-Agnaf OM. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson's disease. Mol Neurodegener. 2016;11:7. doi: 10.1186/s13024-016-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of α-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Man WK, Tahirbegi B, Vrettas MD, Preet S, Ying L, Vendruscolo M, De Simone A, Fusco G. The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition. Nat Commun. 2021;12:927. doi: 10.1038/s41467-021-21027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, Adame A, Schmidhuber S, Santic R, Schneeberger A, Schmidt W, Mattner F, Masliah E. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10. doi: 10.1186/s13024-015-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Manié SN, Lebeau J, Chevet E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis:an update. Am J Physiol Cell Physiol. 2014;307:C901–907. doi: 10.1152/ajpcell.00292.2014. [DOI] [PubMed] [Google Scholar]
- 146.Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353:aah3374. doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maroteaux L, Campanelli JT, Scheller RH. Synuclein:a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neurodegeneration in Alzheimer's disease. Am J Pathol. 1996;148:201–210. [PMC free article] [PubMed] [Google Scholar]
- 149.Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 150.Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun. 2014;2:88. doi: 10.1186/s40478-014-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mattsson N, Insel P, Tosun D, Zhang J, Jack CR, Jr, Galasko D, Weiner M. Alzheimer's Disease Neuroimaging Initiative. Effects of baseline CSF α-synuclein on regional brain atrophy rates in healthy elders, mild cognitive impairment and Alzheimer's disease. PLoS One. 2013;8:e85443. doi: 10.1371/journal.pone.0085443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McCann H, Stevens CH, Cartwright H, Halliday GM. α-Synucleinopathy phenotypes. Parkinsonism Relat Disord. 2014;20:S62–S67. doi: 10.1016/S1353-8020(13)70017-8. [DOI] [PubMed] [Google Scholar]
- 153.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, et al. Diagnosis and management of dementia with Lewy bodies:Fourth consensus report of the DLB Consortium. Neurology. 2017;89:88–100. doi: 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mehra S, Gadhe L, Bera R, Sawner AS, Maji SK. Structural and functional insights into α-synuclein fibril polymorphism. Biomolecules. 2021;11:1419. doi: 10.3390/biom11101419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Menon S, Armstrong S, Hamzeh A, Visanji NP, Sardi SP, Tandon A. Alpha-synuclein targeting therapeutics for Parkinson's disease and related synucleinopathies. Front Neurol. 2022;13:852003. doi: 10.3389/fneur.2022.852003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Messer A, Butler DC. Optimizing intracellular antibodies (intrabodies/nanobodies) to treat neurodegenerative disorders. Neurobiol Dis. 2020;134:104619. doi: 10.1016/j.nbd.2019.104619. [DOI] [PubMed] [Google Scholar]
- 157.Miake H, Mizusawa H, Iwatsubo T, Hasegawa M. Biochemical characterization of the core structure of alpha-synuclein filaments. J Biol Chem. 2002;277:19213–19219. doi: 10.1074/jbc.M110551200. [DOI] [PubMed] [Google Scholar]
- 158.Mittal S, Bjørnevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, Xiang YK, Rochet JC, Engeland A, Rizzu P, Heutink P, Bartels T, Selkoe DJ, Caldarone BJ, Glicksman MA, Khurana V, et al. Beta2- Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson's disease. Science. 2017;357:891–898. doi: 10.1126/science.aaf3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moors TE, Hoozemans JJM, Ingrassia A, Beccari T, Parnetti L, Chartier-Harlin M-C, van de Berg WDJ. Therapeutic potential of autophagy-enhancing agents in Parkinson's disease. Mol Neurodegener. 2017;12:11. doi: 10.1186/s13024-017-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Motyl J, Wencel PL, Cieślik M, Strosznajder RP, Strosznajder JB. Alpha-synuclein alters differently gene expression of Sirts, PARPs and other stress response proteins:implications for neurodegenerative disorders. Mol Neurobiol. 2018;55:727–740. doi: 10.1007/s12035-016-0317-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson's disease. Brain. 2014;137:834–848. doi: 10.1093/brain/awt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nishie M, Mori F, Fujiwara H, Hasegawa M, Yoshimoto M, Iwatsubo T, Takahashi H, Wakabayashi K. Accumulation of phosphorylated alpha-synuclein in the brain and peripheral ganglia of patients with multiple system atrophy. Acta Neuropathol. 2004;107:292–298. doi: 10.1007/s00401-003-0811-1. [DOI] [PubMed] [Google Scholar]
- 163.Nunnari J, Suomalainen A. Mitochondria:in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, Weigand BM, Patel AD, Pirtskhalava T, Inman CL, Johnson KO, Dickinson SL, Rocha A, Schafer MJ, Zhu Y, Allison DB, von Zglinicki T, LeBrasseur NK, Tchkonia T, Neretti N, et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell. 2021;20:e13296. doi: 10.1111/acel.13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–1260. doi: 10.1016/j.celrep.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, Wilmarth BM, Howard H, Dunn C, Carlson A, Lawler A, Rogers SL, Falconer RA, Ahn J, Li Z, Moussa C. Nilotinib Effects in Parkinson's disease and Dementia with Lewy bodies. J Parkinsons Dis. 2016;6:503–517. doi: 10.3233/JPD-160867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, Yusuf N, Starr NJ, Arellano J, Howard HH, Peyton M, Matar S, Liu X, Fowler AJ, Schwartz SL, Ahn J, Moussa C. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson's disease. Pharmacol Res Perspect. 2019;7:e00470. doi: 10.1002/prp2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, Simuni T, Marek K, Postuma RB, Pavese N, Stocchi F, Azulay JP, Mollenhauer B, López-Manzanares L, Russell DS, Boyd JT, Nicholas AP, Luquin MR, Hauser RA, Gasser T, Poewe W, Ricci B, et al. Trial of prasinezumab in early-stage Parkinson's disease. N Engl J Med. 2022;387:421–432. doi: 10.1056/NEJMoa2202867. [DOI] [PubMed] [Google Scholar]
- 169.Pemberton S, Madiona K, Pieri L, Kabani M, Bousset L, Melki R. Hsc70 protein interaction with soluble and fibrillar alpha-synuclein. J Biol Chem. 2011;286:34690–34699. doi: 10.1074/jbc.M111.261321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MB, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SI, Cascella R, Chen SW, Limbocker R, Sormanni P, Heller GT, Aprile FA, Cremades N, Cecchi C, Chiti F, Nollen EA, Knowles TP, et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci U S A. 2017;114:E1009–E1017. doi: 10.1073/pnas.1610586114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, Heller GT, Meisl G, Chen SW, Kumita JR, Challa PK, Kirkegaard JB, Cohen SIA, Mannini B, Barbut D, Nollen EAA, Cecchi C, Cremades N, Knowles TPJ, Chiti F, et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem Biol. 2018;13:2308–2319. doi: 10.1021/acschembio.8b00466. [DOI] [PubMed] [Google Scholar]
- 172.Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, Hannappel C, Krach F, Krumbiegel M, Schütz O, Reis A, Wrasidlo W, Galasko DR, Groemer TW, Masliah E, Schlötzer-Schrehardt U, Xiang W, Winkler J, Winner B. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci U S A. 2018;115:7813–7818. doi: 10.1073/pnas.1713129115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL. Cerebellar and parkinsonian phenotypes in multiple system atrophy:similarities, differences and survival. J Neural Transm. 2014;121:507–512. doi: 10.1007/s00702-013-1133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Reeve A, Simcox E, Turnbull D. Ageing and Parkinson's disease:why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14:19–30. doi: 10.1016/j.arr.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reichmann H. Premotor diagnosis of Parkinson's disease. Neurosci Bull. 2017;33:526–534. doi: 10.1007/s12264-017-0159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Reynolds NP, Soragni A, Rabe M, Verdes D, Liverani E, Handschin S, Riek R, Seeger S. Mechanism of membrane interaction and disruption by α-synuclein. J Am Chem Soc. 2011;133:19366–19375. doi: 10.1021/ja2029848. [DOI] [PubMed] [Google Scholar]
- 178.Roberts HL, Schneider BL, Brown DR. α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PLoS One. 2017;12:e0171925. doi: 10.1371/journal.pone.0171925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141:2181–2193. doi: 10.1093/brain/awy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, Leist M, Subramaniam V, Drescher M. Alpha-synuclein binds to the inner membrane of mitochondria in an α-helical conformation. Chembiochem. 2014;15:2499–2502. doi: 10.1002/cbic.201402281. [DOI] [PubMed] [Google Scholar]
- 181.Romano S, Savva GM, Bedarf JR, Charles IG, Hildebrand F, Narbad A. Meta-analysis of the Parkinson's disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 2021;7:27. doi: 10.1038/s41531-021-00156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe-Kaufmann L, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy:similarities, differences and survival. J Neural Transm (Vienna) 2014;121:507–512. doi: 10.1007/s00702-013-1133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, Giasson BI. Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem. 2017;140:662–678. doi: 10.1111/jnc.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sarkar S, Olsen AL, Sygnecka K, Lohr KM, Feany MB. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Gen. 2021;17:e1009359. doi: 10.1371/journal.pgen.1009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, Soto J, Atiee G, Ostrowitzki S, Kinney GG. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32:211–218. doi: 10.1002/mds.26878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, Kinnunen E, Murros K, Auvinen P. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord. 2015;30:350–358. doi: 10.1002/mds.26069. [DOI] [PubMed] [Google Scholar]
- 187.Schneeberger A, Tierney L, Mandler M. Active immunization therapies for Parkinson's disease and multiple system atrophy. Mov Disord. 2016;31:214–224. doi: 10.1002/mds.26377. [DOI] [PubMed] [Google Scholar]
- 188.Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 189.Schröder M. The unfolded protein response. Mol Biotechnol. 2006;34:279–290. doi: 10.1385/MB:34:2:279. [DOI] [PubMed] [Google Scholar]
- 190.Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Murayama S, Yoshida M, Hasegawa M, Scheres SHW, Goedert M. Structures of α-synuclein filaments from multiple system atrophy. Nature. 2020;585:464–469. doi: 10.1038/s41586-020-2317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schwiertz A, Spiegel J, Dillmann U, Grundmann D, Bürmann J, Faßbender K, Schäfer KH, Unger MM. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson's disease. Parkinsonism Relat Disord. 2018;50:104–107. doi: 10.1016/j.parkreldis.2018.02.022. [DOI] [PubMed] [Google Scholar]
- 192.Seidler RD, Bernard JA, Burutolu TB, Fling BW, Gordon MT, Gwin JT, Kwak Y, Lipps DB. Motor control and aging:links to age-related brain structural, functional, and biochemical effects. Neurosci Biobehav Rev. 2010;34:721–733. doi: 10.1016/j.neubiorev.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson's disease:Gaucher disease and beyond. Mov Disord. 2011;26:1593–604. doi: 10.1002/mds.23774. [DOI] [PubMed] [Google Scholar]
- 196.Shrivastava AN, Aperia A, Melki R, Triller A. Physico-pathologic mechanisms involved in neurodegeneration:misfolded protein-plasma membrane interactions. Neuron. 2017;95:33–50. doi: 10.1016/j.neuron.2017.05.026. [DOI] [PubMed] [Google Scholar]
- 197.Shrivastava AN, Bousset L, Renner M, Redeker V, Savistchenko J, Triller A, Melki R. Differential membrane binding and seeding of distinct α-synuclein fibrillar polymorphs. Biophys J. 2020;118:1301–1320. doi: 10.1016/j.bpj.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, Lafontant DE, Matthews H, Wyse RK, Brundin P, Simon DK, Schwarzschild M, Weiner D, Adams J, Venuto C, Dawson TM, Baker L, Kostrzebski M, Ward T, Rafaloff G, et al. Efficacy of nilotinib in patients with moderately advanced Parkinson disease:a randomized clinical trial. JAMA Neurol. 2021;78:312–320. doi: 10.1001/jamaneurol.2020.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 200.Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, Zago W, Seubert P, Barbour R, Schenk D, Games D, Rissman RA, Masliah E. Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun. 2017;5:7. doi: 10.1186/s40478-016-0410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Stöckl MT, Claessens MM, Subramaniam V. Kinetic measurements give new insights into lipid membrane permeabilization by α-synuclein oligomers. Mol Biosyst. 2012;8:338–345. doi: 10.1039/c1mb05293d. [DOI] [PubMed] [Google Scholar]
- 203.Stöckl MT, Zijlstra N, Subramaniam V. α-Synuclein oligomers:an amyloid pore?Insights into mechanisms of α-synuclein oligomer-lipid interactions. Mol Neurobiol. 2013;47:613–621. doi: 10.1007/s12035-012-8331-4. [DOI] [PubMed] [Google Scholar]
- 204.Strohäker T, Jung BC, Liou SH, Fernandez CO, Riedel D, Becker S, Halliday GM, Bennati M, Kim WS, Lee SJ, Zweckstetter M. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat Commun. 2019;10:5535. doi: 10.1038/s41467-019-13564-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 206.Sulatskaya AI, Rodina NP, Sulatsky MI, Povarova OI, Antifeeva IA, Kuznetsova IM, Turoverov KK. Investigation of α-synuclein amyloid fibrils using the fluorescent probe thioflavin T. Int J Mol Sci. 2018;19:2486. doi: 10.3390/ijms19092486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Surmeier DJ. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 2018;285:3657–3668. doi: 10.1111/febs.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tapias V, Hu X, Luk KC, Sanders LH, Lee VM, Greenamyre JT. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol Life Sci. 2017;74:2851–2874. doi: 10.1007/s00018-017-2541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Thom T, Schmitz M, Fischer AL, Correia A, Correia S, Llorens F, Pique AV, Möbius W, Domingues R, Zafar S, Stoops E, Silva CJ, Fischer A, Outeiro TF, Zerr I. Cellular prion protein mediates α-synuclein uptake, localization, and toxicity in vitro and in vivo. Mov Disord. 2022;37:39–51. doi: 10.1002/mds.28774. [DOI] [PubMed] [Google Scholar]
- 210.Tofaris GK. Initiation and progression of α-synuclein pathology in Parkinson's disease. Cell Mol Life Sci. 2022;79:210. doi: 10.1007/s00018-022-04240-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tosatto L, Andrighetti AO, Plotegher N, Antonini V, Tessari I, Ricci L, Bubacco L, Dalla Serra M. Alpha-synuclein pore forming activity upon membrane association. Biochim Biophys Acta. 2012;1818:2876–2883. doi: 10.1016/j.bbamem.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 212.Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014;7:2054–2065. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Trudler D, Sanz-Blasco S, Eisele YS, Ghatak S, Bodhinathan K, Akhtar MW, Lynch WP, Piña-Crespo JC, Talantova M, Kelly JW, Lipton SA. α-Synuclein oligomers induce glutamate release from astrocytes and excessive extrasynaptic NMDAR activity in neurons, thus contributing to synapse loss. J Neurosci. 2021;41:2264–2273. doi: 10.1523/JNEUROSCI.1871-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Twohig D, Nielsen HM. Alpha-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. 2019;14:23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 218.Valera E, Masliah E. Therapeutic approaches in Parkinson's disease and related disorders. J Neurochem. 2016;139(Suppl 1(Suppl 1)):346–352. doi: 10.1111/jnc.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, Tsung-Pin P, Riess O, Nyengaard JR, Tamgüney G, Jensen PH, Borghammer P. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138:535–550. doi: 10.1007/s00401-019-02040-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Van Den Berge N, Ferreira N, Mikkelsen TW, Alstrup AKO, Tamgüney G, Karlsson P, Terkelsen AJ, Nyengaard JR, Jensen PH, Borghammer P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain. 2021;144:1853–1868. doi: 10.1093/brain/awab061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Verma DK, Seo BA, Ghosh A, Ma SX, Hernandez-Quijada K, Andersen JK, Ko HS, Kim YH. Alpha-synuclein preformed fibrils induce cellular senescence in Parkinson's disease models. Cells. 2021;10:1694. doi: 10.3390/cells10071694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vilar M, Chou HT, Lührs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek The fold of α-synuclein fibrils. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein:implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- 224.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wang X, Becker K, Levine N, Zhang M, Lieberman AP, Moore DJ, Ma J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol Commun. 2019;7:41. doi: 10.1186/s40478-019-0696-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang Z, Gao G, Duan C, Yang H. Progress of immunotherapy of anti-α-synuclein in Parkinson's disease. Biomed Pharmacother. 2019;115:108843. doi: 10.1016/j.biopha.2019.108843. [DOI] [PubMed] [Google Scholar]
- 228.Weihofen A, Liu Y, Arndt JW, Huy C, Quan C, Smith BA, Baeriswyl JL, Cavegn N, Senn L, Su L, Marsh G, Auluck PK, Montrasio F, Nitsch RM, Hirst WD, Cedarbaum JM, Pepinsky RB, Grimm J, Weinreb PH. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiol Dis. 2019;124:276–288. doi: 10.1016/j.nbd.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 229.Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835–845. doi: 10.1093/brain/117.4.835. [DOI] [PubMed] [Google Scholar]
- 230.Wilkaniec A, Lenkiewicz AM, Babiec L, Murawska E, Jęśko HM, Cieślik M, Culmsee C, Adamczyk A. Exogenous alpha-synuclein evoked parkin downregulation promotes mitochondrial dysfunction in neuronal cells. Implications for Parkinson's disease pathology. Front Aging Neurosci. 2021;13:591475. doi: 10.3389/fnagi.2021.591475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. α-Synuclein impairs macroautophagy:implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yang Y, Shi Y, Schweighauser M, Zhang X, Kotecha A, Murzin AG, Garringer HJ, Cullinane PW, Saito Y, Foroud T, Warner TT, Hasegawa K, Vidal R, Murayama S, Revesz T, Ghetti B, Hasegawa M, Lashley T, Scheres SHW, Goedert M. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature. 2022;610:791–795. doi: 10.1038/s41586-022-05319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yoo G, Yeou S, Son JB, Shin YK, Lee NK. Cooperative inhibition of SNARE-mediated vesicle fusion by α-synuclein monomers and oligomers. Sci Rep. 2021;11:10955. doi: 10.1038/s41598-021-90503-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zakharov SD, Hulleman JD, Dutseva EA, Antonenko YN, Rochet JC, Cramer WA. Helical α-synuclein forms highly conductive ion channels. Biochemistry. 2007;46:14369–14379. doi: 10.1021/bi701275p. [DOI] [PubMed] [Google Scholar]
- 235.Zhang P, Park HJ, Zhang J, Junn E, Andrews RJ, Velagapudi SP, Abegg D, Vishnu K, Costales MG, Childs-Disney JL, Adibekian A, Moss WN, Mouradian MM, Disney MD. Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc Natl Acad Sci U S A. 2020;117:1457–1467. doi: 10.1073/pnas.1905057117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zharikov A, Bai Q, De Miranda BR, Van Laar A, Greenamyre JT, Burton EA. Long-term RNAi knockdown of alpha-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol Dis. 2019;125:146–153. doi: 10.1016/j.nbd.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol. 2017;155:96–119. doi: 10.1016/j.pneurobio.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97:92–107. doi: 10.1016/j.neuron.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.