

Abstract
This study aimed to design anticancer theobromine derivatives inhibiting VEGFR-2. The new compounds were tested in vitro to evaluate their effectiveness against MCF-7 and HepG2 cancer cell lines. Among these compounds, 15a showed the highest cytotoxicity against HepG2, with an IC50 value of 0.76 μM, and significant anti-proliferative effects on MCF-7, with an IC50 value of 1.08 μM. Notably, the selectivity index of 15a against the two cancer cells was 98.97 and 69.64, respectively. Moreover, 15a demonstrated potent VEGFR-2 inhibitory activity (IC50 = 0.239 μM). Further investigations revealed that 15a induced apoptosis in HepG2 cells, significantly increasing early-stage and late-stage apoptosis percentages from 3.06% and 0.71% to 29.49% and 9.63%, respectively. It also upregulated caspase-3 and caspase-9 levels by 3.45-fold and 2.37-fold, respectively compared to control HepG2 cells. Additionally, 15a inhibited the migration and wound healing ability of HepG2 cells. Molecular docking confirmed the binding affinities of the semi-synthesized compounds to VEGFR-2, consistent with in vitro results. Several computational analyses (DFT, MD simulations, MM-GBSA, PLIP, and essential dynamics) supported the stability of the 15a-VEGFR-2 complex. Overall, the biological and computational findings suggest that compound 15a could be a promising lead compound for the development of a novel apoptotic anticancer agent.
This study aimed to design anticancer theobromine derivatives inhibiting VEGFR-2.
1. Introduction
Despite the discovery of a vast array of anticancer drugs, cancer incidence, and prevalence continue to rise significantly.1 Developing targeted chemotherapeutic agents that can interact effectively with the molecular targets of cancer cells to inhibit their growth remains a major challenge for cancer researchers.2 This task gives hope to developing precise lead compounds that can selectively target cancer cells while minimizing harm to healthy tissues.3–5
The vascular endothelial growth factor receptor-2 (VEGFR-2) is one of the most promising targets for cancer therapy. As a type of tyrosine kinase receptor, VEGFR-2 plays a crucial role in various cellular processes such as cell proliferation, adhesion, division, and angiogenesis.6 VEGFR-2 is usually expressed much more in cancer cells than in normal cells, providing an opportunity to develop drugs that selectively target angiogenesis in tumor cells while sparing normal cells.7
Throughout human history, nature has been a vital source of essential resources, including medicine, food, and cosmetics.8,9 Between the years 1981 and 2014, nearly one-third of all drugs approved by the FDA were either based on or derived from natural sources.10 The utilization of semi-synthesis techniques on natural products enables the production of diverse analogs, leading to the discovery of more potent drugs and the repurposing of existing ones.11 This approach also facilitates the investigation of the structure–activity relationship, which is often easier and more accessible in natural product-based drug discovery, providing a great opportunity for obtaining novel bioactive compounds, improving drug-likeness, and enhancing pharmacokinetic and pharmacodynamic properties.12
Theobromine, a naturally occurring alkaloid, has emerged as a promising candidate for the treatment of cancer, with evidence from both in vitro and in vivo studies suggesting its potential therapeutic value. Specifically, theobromine has been found to induce apoptosis in colorectal cancer cells,13 and to inhibit DNA synthesis and hinder the growth of glioblastoma multiform cells.14 Furthermore, theobromine has demonstrated anti-angiogenic properties by suppressing vascular endothelial growth factor (VEGF) in ovarian15 and lung16 cancers, indicating its potential to serve as a potent therapeutic agent against several malignancies.
2. Rationale
VEGFR-2 inhibitors have four pharmacophoric features that are crucial for the correct binding in the active site of VEGFR-2 (Fig. 1). Hetero aromatic system, linker group, pharmacophore moiety, and hydrophobic tail are the main required features. The hetero-aromatic system exerts high fitting in the hinge region of the active site. The linker group can occupy the gatekeeper region to enable the pharmacophore moiety to be oriented into the DFG motif region. The DFG motif region has two crucial amino acids (Asp1044 and Glu833) that should be activated via hydrogen bonding to give maximal fitting. The pharmacophore moiety should comprise at least one hydrogen bond donor and one hydrogen bond acceptor. The hydrophobic tail has a crucial role to block the allosteric pocket of the binding site.17–19
Fig. 1. (A) Pharmacophoric requirements for VEGFR-2 inhibitors. (B) Reported VEGFR-2 inhibitors.

Sorafenib I,20 sunitinib II,21 regorafenib III, and altiratinib IV are reported VEGFR-2 inhibitors possessing the main pharmacophoric features. Sorafenib I, regorafenib III, and altiratinib IV have a pyridine moiety as a hetero-aromatic system. On the other hand, sunitinib II has isatin moiety as a hetero-aromatic system (Fig. 1).
Our research group has discovered compounds V,22VI,22VII,23 and VIII24 as potential VEGFR-2 inhibitors having promising anti-proliferative activities (Fig. 1). Such compounds possess a pyridine system to occupy the hinge region VEGFR-2. Compounds V, VI, and VII have a para-di-substituted phenyl ring as a linker group while compound VIII has a meta-di-substituted ring. Compounds V, VI, and VIII have a formyl hydrazone moiety as a pharmacophore system while compound VII possesses a hydrazone moiety. Furthermore, all the discovered compounds have substituted phenyl rings as a hydrophobic tail.
In this work, the previously synthesized compounds (V, VI, VII, and VIII) were used as lead compounds for the design and synthesis of new VEGFR-2 inhibitors. As shown in Fig. 2, the newly designed compounds have a xanthine moiety as a bioisostere for the pyridine moiety of the lead compounds. The xanthine moiety was selected for some consideration. (i) It has a large size, which is convenient for the hinge region of the VEGFR-2.25 (ii) It has many hydrogen bond acceptor atoms, which may facilitate the hydrogen bonding interaction with the biological target. (iii) Its naturally derived nucleus increases its safety margin. For the linker group, some compounds were designed to have a di-para-substituted phenyl ring (compounds 15a, 15b, and 17) and the other compound 16 has a meta di-substituted phenyl ring. For the pharmacophore moiety, all the designed compounds have a formyl hydrazone moiety as a pharmacophore system. Finally, different substituted phenyl rings were used as a hydrophobic tail to occupy the allosteric pocket.
Fig. 2. Rationale design of the work.

3. Results
3.1. Chemistry
The synthetic pathways for the final target compounds 15a,b, 16, and 17 are summarized in Schemes 1, 2, and 3. Theobromine, 1, was first converted to its potassium salt 2 by refluxing it with alcoholic KOH. N-(3-Acetylphenyl)-2-chloroacetamide 4 and N-(4-acetylphenyl)-2-chloroacetamide 6 were produced by the reaction of p-aminoacetophenone 3 and m-aminoacetophenone 5, respectively with chloroacetylchloride in DMF using NaHCO3 as a base. Equimolar amounts of theobromine potassium salt 2 reacted with 4 and 6 in DMF using potassium iodide as a catalyst to afford the key intermediates compound 7 and 8, respectively (Scheme 1).
Scheme 1. Synthesis of the key intermediates 7 and 8.

Scheme 2. Synthesis of the key intermediates 11a–d and 14.

Scheme 3. Synthesis of the target compounds 15a,b, 16, and 17.

Moreover, benzohydrazide derivatives 11a–d and 14 were readily prepared by refluxing the appropriate benzoate derivatives 10a–c and 13, respectively with hydrazine hydrate in absolute ethanol (Scheme 2).
Condensation of compound 7 with benzohydrazide derivatives 11a,b, afforded the final target compounds 15a,b. On the other hand, condensation of compound 8 with 11c afforded the final target derivative 16. Additionally, the condensation of compound 7 with 14 produced the final target compound 17 (Scheme 3).
The IR spectra of compounds 15a,b, 16, and 17 exhibited the appearance of absorption bands at the ranges of 3196–3265 cm−1 and 1667–1704 cm−1 attributable to the NH and C O groups, respectively. 1H NMR spectra of compounds 15a,b, 16, and 17 showed the appearance of the two amidic protons at δ ranges of 10.89–10.93 ppm and 10.42–10.45 ppm. Moreover, their 1H NMR spectra revealed the presence of shielded singlet signals for the methyl protons at a δ range of 2.29–2.36 ppm, and singlet signals around δ 4.70 ppm for the methylene protons. In addition, 13C NMR spectra showed the presence of four shielded signals at around 43.94, 33.68, 29.93, and 14.85 ppm attributed to the methylene carbon and the three-methyl carbons, respectively. Moreover, the carbonyl carbons were displayed at their expected regions.
3.2. Biology
3.2.1. In vitro cytotoxicity and safety
The current study used the MTT assay utilizing sorafenib as a positive control to assess the anti-proliferative effects of the synthesized derivatives 15a,b, 16, and 17 against HepG2 and MCF-7 cell lines. Interestingly, as summarized in Table 1, all compounds exhibited stronger IC50 values than those of sorafenib (2.24 ± 0.06, 3.17 ± 0.01, against HepG2 and MCF-7 cell lines, respectively). Compound 15a was the strongest cytotoxic member against HepG2 with an IC50 value of 0.76 ± 0.01 μM. Also, it demonstrated notable cytotoxicity against MCF-7 cells with an IC50 value of 1.08 ± 0.02 μM. Additionally, compounds 15b, 16, and 17 demonstrated considerable cytotoxic action against the same cancer cell line with IC50 values of 0.78 ± 0.02, 1.23 ± 0.03 μM, and 1.05 ± 0.01 μM, respectively. Also, compounds 15b and 17 displayed strong cytotoxic action against MCF-7 cells with an IC50 value of 0.65 ± 0.01 μM and 0.8 ± 0.01 μM, respectively. Additionally, compound 16 demonstrated a notable cytotoxicity against the MCF-7 cells with an IC50 value of 2.47 ± 0.04 μM.
In vitro anti-proliferative assessment of the target compounds 15a,b, 16, and 17.
| Comp. | Structure | In vitro cytotoxicity IC50 (μM)a | |
|---|---|---|---|
| HepG2 | MCF-7 | ||
| 15a |
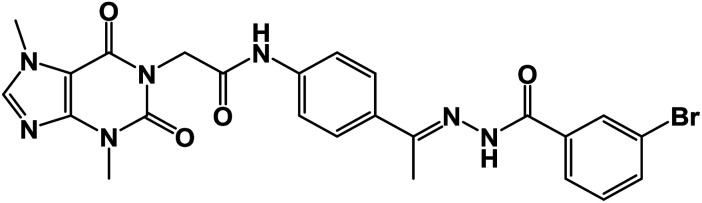
|
0.76 ± 0.01 | 1.08 ± 0.02 |
| 15b |
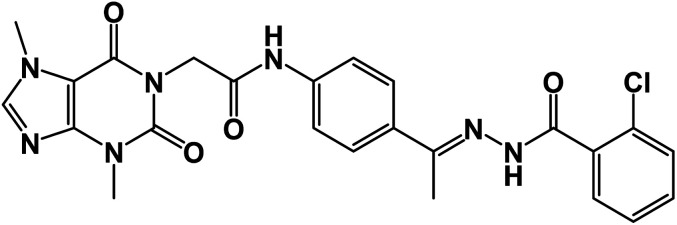
|
0.78 ± 0.02 | 0.65 ± 0.01 |
| 16 |
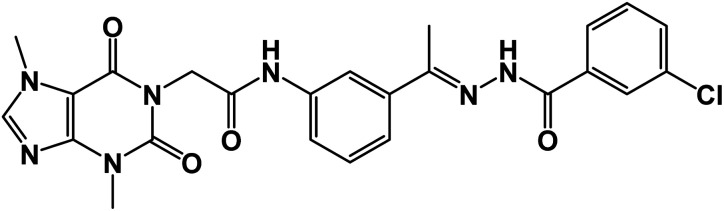
|
1.23 ± 0.03 | 2.47 ± 0.04 |
| 17 |
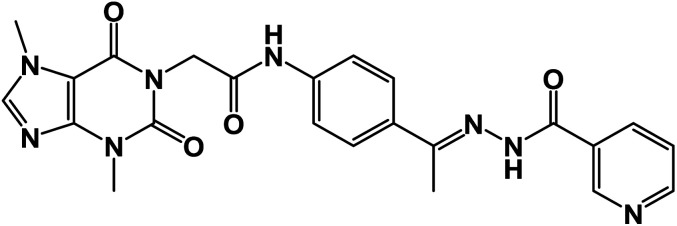
|
1.05 ± 0.01 | 0.8 ± 0.01 |
| Sorafenib |
|
2.24 ± 0.06 | 3.17 ± 0.01 |
The data were presented as the mean of three experiments.
Next, the safety of compound 15a against Vero cells was examined to determine its selectivity against cancer cells. Based on the IC50 value of 75.22 μM, the selectivity index values of compound 15a against HepG2 and MCF7 cell lines were calculated to be 98.97 and 69.64, respectively. Overall, our results imply that compound 15a has a promising future as a secure anticancer drug and call for additional research.
3.2.2. In vitro VEGFR-2 enzyme assay inhibition
From the cytotoxicity results, it was concluded that compound 15a was the strongest cytotoxic member against HepG2 (the most sensitive cancer cell line). Therefore, compound 15a was assessed for its in vitro VEGFR-2 inhibitory activity using sorafenib as a reference. The results showed that compound 15a has potent VEGFR-2 inhibitory activity with an IC50 value of 0.239 μM against 0.056 μM for sorafenib. These intriguing findings gave us further motivation to investigate compound 15a 's cellular mechanistic studies.
3.2.3. Apoptosis assay
To examine the apoptotic effects of compound 15a on HepG2 cells, a flow cytometry technique was applied. The test utilizes the Annexin V and PI double staining. According to Table 2 and Fig. 3, compound 15a significantly increased the proportion of HepG2 cells from 3.06% to 29.49% in the early-stage apoptosis and from 0.71% to 9.63% in the late-stage apoptosis. These results imply that compound 15a has the potential to be cytotoxic and may be linked to programmed cell death in the HepG2 cells.
Effect of compound 15a on stages of the cell death process in HepG2 cells.
| Comp. | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| Compound 15a/HepG2 | 29.49 | 9.63 | 12.17 | 7.69 |
| Cont. HepG2 | 3.06 | 0.71 | 0.13 | 2.22 |
Fig. 3. Flow cytometry chart of apoptosis in HepG2 cells exposed to compound 15a.

3.2.4. Effects on caspases
This study examined the effects of compound 15a at a concentration of 0.76 μM (IC50 value) on the expression levels of caspase-3 and caspase-9 in HepG2 cells. In comparison to the control, the results showed that compound 15a increased the level of caspase-3 by 3.45-fold, compared to the control. Furthermore, compound 15a exhibited a significant increase in the level of caspase-9 by 2.37-fold, compared to the control (Table 3).
Effect of compound 15a on the levels of active caspases-3, and active caspases-9.
| Sample | Protein expression | |
|---|---|---|
| Caspase-3 | Caspase-9 | |
| Cont. HepG2 | 1 | 1 |
| Compound 15a/HepG2 | 3.451 | 2.378 |
3.2.5. The effect of compound 15a on HepG2's migration and healing
To assess the impact of compound 15a on the migration and healing of HepG2 cancer cells, a wound healing assay was applied. This technique involves making a scratch on a cancer cell monolayer, measuring the initial diameter, and monitoring the closure of the scratch at specific time intervals for both treated and untreated cells. The images of the scratch areas of treated and untreated cell lines were compared after 0 and 48 hours. The results (shown in Table 4 and Fig. 4) indicated that the scratch of the untreated HepG2 cells closed significantly within 48 hours, reducing the width by 65.9%. On the other hand, the scratch width of HepG2 cells treated with compound 15a decreased by only 34.17%, indicating that the treatment significantly inhibited the closure of the scratch.
The effect of compound 15a on HepG2's migration and healing after 48 hours.
| At 0 h | At 48 h | RM (μm) | Wound closure (% μm2) | Area difference (%) | |||
|---|---|---|---|---|---|---|---|
| Area | Width | Area | Width | ||||
| Cont. HepG2 | 1000.333 | 999.3853 | 340.5 | 339.5385 | 13.74681 | 65.96135 | 659.8333 |
| Compound 15a/HepG2 | 1000.667 | 999.7462 | 658.6667 | 657.7792 | 7.124313 | 34.17722 | 342 |
Fig. 4. The effect of compound 15a on HepG2's migration and healing after 48 hours.

3.3. Computational studies
3.3.1. Molecular docking
The crystal structure of VEGFR-2 (PDB ID: 2OH4) was downloaded from the protein data bank and prepared for docking using MOE2019 software and sorafenib as a reference molecule. The binding pattern of the synthesized compounds as well as sorafenib were clarified in the following figures. The binding energies (ΔG) of the tested compounds were summarized in Table 5.
The binding energies (ΔG) of the theobromine derivatives and sorafenib against VEGFR-2.
| Comp. | 15a | 15b | 16 | 17 | Sorafenib |
|---|---|---|---|---|---|
| ΔG [kcal mol−1] | −23.29 | −24.74 | −23.09 | −23.11 | −21.20 |
At first, the validation process was carried out by docking the co-crystallized ligand in the active pocket of VEGFR-2. The co-crystallized ligand has a general name of benzimidazole-urea inhibitor. In addition it has an IUPAC name of methyl N-[5-[4-[[2-fluoro-5-(trifluoromethyl) phenyl]carbamoylamino]phenoxy]-1-methylbenzimidazol-2-yl]carbamate. The results indicated the high validity of the docking process since the RMSD value between the docked and original ligand was (1.19 Å) less than 2.00 Å (Fig. 5).
Fig. 5. Validation of the docking process. The RMSD value between the original ligand (pink) and the docked one (orange) is 1.19 Å.

The reference molecule, sorafenib, revealed a binding mode similar to the reported results.22–24 It had an energy score of −21.20 kcal mol−1. It formed one hydrogen bond with Cys917 and three hydrophobic interactions with Leu838, Ala864, and Leu1033 at the hinge region. In addition, its linker moiety formed six hydrophobic interactions with Val914, Val846, Cys1043, and Phe1045. Furthermore, the pharmacophore moiety was engaged in three hydrogen bonds with Glu883 and Asp1044. The terminal hydrophobic tail formed five hydrophobic interactions with Leu887, Cys1043, His1024, and Leu1017. Also, it formed an electrostatic interaction with Asp104 (Fig. 6).
Fig. 6. 2D interaction of sorafenib with the active site of VEGFR-2.

Compound 15a has a binding energy of −23.29 kcal mol−1. The different features occupied the different sub-pockets of the active site. The heterocyclic system (xanthine moiety) was oriented into the hinge region forming one hydrogen bond and two hydrophobic interactions with Cys917, Leu838, and Phe916. The linker (phenyl) moiety formed one hydrophobic interaction with Val914 and one electrostatic attraction with Cys1043. The N-ethylideneformohydrazide moiety occupied the DFG motif region and formed two hydrogen bonds and three hydrophobic interactions with Glu883, Asp1044, Lys866, Val914, and Leu887. The terminal hydrophobic tail (3-bromophenyl moiety) occupied the allosteric pocket forming two hydrophobic interactions with Ile886 and Ile890. In addition, it formed one electrostatic interaction with Asp1044 (Fig. 7A).
Fig. 7. 3D interactions of the synthesized compounds in the VEGFR-2 binding site. (A) Compound 15a, (B) compound 15b, (C) compound 16, (D) compound 17.

Compound 15b showed a binding score of −24.74 kcal mol−1. The xanthine moiety occupied the hinge region of VEGFR-2 forming one hydrogen bond and two hydrophobic interactions with Cys917, Leu838, and Phe916. The phenyl moiety formed three hydrophobic interactions with Val914, Ala864, and Val846. In addition, it formed one electrostatic attraction with Cys1043. The N-ethylideneformohydrazide moiety occupied the DFG motif region forming two hydrogen bonds and three hydrophobic interactions with Glu883, Asp1044, Lys866, Val914, and Leu887. The 2-chlorophenyl moiety occupied the allosteric pocket forming five hydrophobic interactions with Ile886, Val879, Leu887, and Ile890. In addition, it formed one electrostatic interaction with Asp1044 (Fig. 7B).
Compound 16 showed a binding score of −23.09 kcal mol−1. The xanthine moiety formed one hydrogen bond with Cys917 and five hydrophobic interactions with Leu838, Leu1033, and Phe1045. The central phenyl moiety formed four hydrophobic interactions with Val914, Ala864, Lys866, and Val846. The N-ethylideneformohydrazide moiety occupied the DFG motif region forming two hydrogen bonds and one hydrophobic interaction with Glu883, Asp1044, and Lys866. The 3-chlorophenyl moiety occupied the allosteric pocket forming two hydrophobic interactions with Ile886 and Ile890. In addition, it formed one electrostatic interaction with Asp1044 (Fig. 7C).
Compound 17 showed a binding score of −23.11 kcal mol−1. The xanthine moiety was engaged in one hydrogen bond and three hydrophobic interactions with Cys917, Leu838 and Phe916. The central phenyl ring occupied the central region forming three hydrophobic interactions with Val914, Ala864, and Val846. The N-ethylideneformohydrazide moiety occupied the DFG motif region forming two hydrogen bonds and three hydrophobic interactions with Glu883, Asp1044, Lys866, Val914, and Leu887. The terminal pyridine moiety occupied the allosteric pocket forming two hydrophobic interactions with Ile886 and Leu887. In addition, it formed one electrostatic interaction with Asp1044 (Fig. 7D).
3.3.2. Correlation of docking studies and biological results
Sorafenib has ideal binding mode against the active site of VEGFR-2. It formed two hydrogen bonds at the hinge region with Cys917. In addition, it formed three hydrogen bonds at the DFG motif region with Asp1044 and Glu883. These hydrogen bonds give a high stability at the active site for long time. This indicates the high affinity of sorafenib with VEGFR-2 and reflects the high inhibitory activity against VEGFR-2. The tested compounds showed binding energies higher than that of sorafenib ranging from-23.09 to −24.74 kcal mol−1. The higher scores of the synthesized compounds may be related to the hydrophobic interactions and van der Waals forces.
On the other hand, although compound 15a has good binding mode against VEGFR-2, it showed number of hydrogen bond interactions less than that sorafenib. It formed one hydrogen bond ay the hinge region and two hydrogen bonds at the DFG motif region. Such results reflect the higher activity of sorafenib as VEGFR-2 (IC50 = 0.056 μM) than compound 15a (IC50 = 0.239 μM).
Investigating the binding modes of the four synthesized compounds indicated that all of them have the same number of hydrogen bonds at the hinge region (one bond) and the DFG motif region (two bonds).
Observing the cytotoxicity of compounds 15a and 15b against the most sensitive cells (HepG2), these two compounds have non-significant differences with IC50 values of 0.76 and 0.78 μM, respectively. Such biological results match with the docking results, as these two compounds have the same number of hydrogen bonds with non-significant difference in the binding energy (ΔG = −23.29 and −24.74 kcal mol−1, respectively).
Regarding compounds 16 and 17, although these compounds have the same binding mode as compounds 15a and 15b, both of them have less cytotoxic activity against HepG2. The reduced activity of compound 16 may be due to the change in the configuration of the linker moiety, as this compound has a meta-disubstituted phenyl linker while compounds 15a, 15b, and sorafenib have a para-disubstituted phenyl linker. Moreover, the reduced activity of compound 17 may be due to the absence of halogen atoms at the hydrophobic tail in contrast to that of compounds 15a, 15b, and sorafenib. It is well-known that halogen atoms have a high participation in hydrophobic interaction in the active site.
3.3.3. In silico ADME analysis
In order to detect the ADMET parameters of the synthesized compounds, Discovery Studio 4.0 software was used. Sorafenib was used as a reference molecule. As shown in Table 6, the synthesized molecules have very low BBB penetration levels expecting the absence of CNS toxicity. For the predicted aqueous solubility, compounds 15b and 17 showed good levels while compounds 15a and 16 showed low levels. Regarding the expected intestinal absorption of the tested molecules, all of them showed moderate levels. The tested molecules were expected to be devoid of cytochrome P450 (CYP-2D6) inhibitory activity indicating the non-hepatotoxicity except for compound 17. Finally, all the examined molecules were expected to bind plasma protein less than 90% (Fig. 8).
ADMET screening of the synthesized compounds.
| Comp. | BBB level | Solubility level | Absorption level | Hepatotoxic prediction | CYP2D6 prediction | PPB prediction |
|---|---|---|---|---|---|---|
| 15a | Very low | Low | Moderate | Non-toxic | Non-inhibitor | <90% |
| 15b | Good | |||||
| 16 | Low | |||||
| 17 | Good | Toxic | ||||
| Sorafenib | Very low | Good | >90% |
Fig. 8. The executed ADMET study.

3.3.4. Toxicity studies
Additional in silico study was carried out for the synthesized molecules to predict their toxicity potential against different validated models in Discovery studio software. Sorafenib was used as a reference molecule. The expected toxicity results were summarized in Table 7.
Toxicity study of the synthesized compounds.
| Comp. | Ames prediction | Rat-male-FDA | Carcinogenic potency TD50 (mouse)a | Rat oral LD50b | Rat chronic LOAELb | Skin irritancy | Ocular irritancy |
|---|---|---|---|---|---|---|---|
| 15a | Non-mutagen | Non-carcinogen | 23.973 | 1.785 | 0.046 | Non-irritant | Mild |
| 15b | 18.268 | 1.580 | 0.108 | ||||
| 16 | 24.482 | 1.305 | 0.046 | ||||
| 17 | 27.036 | 0.768 | 0.062 | ||||
| Sorafenib | 19.236 | 0.823 | 0.005 |
Unit: mg kg−1 body weight per day.
Unit: g kg−1 body weight.
At first, the mutagenic potential of a particular molecule can be assessed using the Ames prediction model. Depending on this model, all the synthesized molecules were predicted to be non-mutagenic. Furthermore, the FDA rodent carcinogenicity model can predict the carcinogenicity of a particular compound. This model indicated that the synthesized compounds are non-carcinogens. In addition, the carcinogenic potency TD50 values of the synthesized compounds ranged from 23.973 to 27.036 g kg−1. This range of carcinogenic potency TD50 values was higher than that of sorafenib (19.236 g kg−1) indicating the safety margin of the synthesized compounds. An additional sigh for the safety of the tested compounds was reached by the rat oral LD50 model. This model predicted that compounds 15a, 15b, and 16 have rat oral LD50 values ranging from 0.768 to 1.785 g kg−1, which were higher than that of sorafenib (0.823 g kg−1). For the rat chronic LOAEL model which predicts the rat chronic lowest observed adverse effect level value of a chemical, all compounds (0.046–0.108 g kg−1) have higher values than sorafenib (0.005 g kg−1). All compounds were predicted to be non-irritant with mild irritancy against the skin and the eyes, respectively (Table 7).
3.3.5. Molecular dynamic (MD) simulation
The conformation of compound 15a and the reference compound, sorafenib, and their distance from the protein's center of mass were both stable throughout the production run. After around 20 ns, the RMSD values for apo (blue line) and VEGFR-2 & 15a complex (red line) had equilibrated at about 2.7 Å and 4 Å, respectively. In addition, the reference compound (VEGFR-2 & sorafenib) shows more stable values around 2 Å throughout the simulation as shown by the green line (Fig. 9A). In Fig. 9B, we can see that the RMSD of 15a (blue line) has stabilized around 4 Å while the sorafenib compound (red line) shows a more stable fluctuation with an average of around 2 Å. As can be seen in Fig. 9C, the average apo (blue line), VEGFR-2 & 15a complex (red line), and VEGFR-2 & sorafenib complex (green line) systems show a radius of gyration with values near each other with the VEGFR-2 & sorafenib as the most stable one followed by the apo protein then the VEGFR-2 & 15a complex. As shown in Fig. 9D, apo protein, VEGFR-2 & 15a complex, and VEGFR-2 & sorafenib complex had average SASA values of 17 656 Å2, 18 076 Å2, and 17 180 Å2, respectively. Fig. 9E showed that the average number of H-bonds formed between each compound and the VEGFR-2 protein differs across the two systems. The 15a compound (blue line) forms 1 H-bond during a low number of frames with zero H-bond as a common number. On the other hand, sorafenib compound (pale red line) shows that at least there is a constant 1 H-bond with 2 H-bonds as less common. Overall, this demonstrates that the protein structure is rather stable. A comparison between the three systems RMSF shows that no significant fluctuation is happening (Fig. 9F). The separation between the 15a and the protein centers of mass is stable with an average distance of 7 Å which is slightly smaller than the distance of sorafenib 7.8 Å, indicating a stable interaction (Fig. 9G).
Fig. 9. (A) RMSD values from the trajectory for the VEGFR-2 protein in apo form (blue line) and holo forms (red line for 15a system and green for sorafenib system). (B) Ligand RMSD values (15a: blue line, sorafenib: green line). (C) Radius of gyration for the VEGFR-2 protein in apo form (blue line) and holo forms (red line). (D) SASA for the VEGFR-2 protein in apo form (blue line) and holo forms (red line for 15a system and green for sorafenib system). (E) Change in the number of hydrogen bonds between 15a and VEGFR-2 protein (blue line) and sorafenib and VEGFR-2 protein (red line). (F) RMSF for the VEGFR-2 protein in apo form (blue line) and holo forms (red line for 15a system and green for sorafenib system). (G) Distance from the center of mass of compound 15a or sorafenib and VEGFR-2 protein.

3.3.6. MM-GBSA studies
The MM-GBSA method's breakdown of the binding free energy into its component was shown in Fig. 10. The binding energy of compound 15a is −36.9 kcal mol−1 which is slightly smaller than the reference compound by 8.5 kcal mol−1, indicating a slightly weaker interaction compared to the reference. The electrostatic interactions (−3.41 kcal mol−1) are less significant than the van der Waals interactions (−56.79 kcal mol−1) in determining binding stability for 15a. On the other hand, the reference compound shows a larger average electrostatic contribution (−20 kcal mol−1) and slightly smaller van der Waals interactions (−52.5 kcal mol−1). Using decomposition analysis, the contributions of amino acids within 1 nm of compound 15a or sorafenib were calculated and compared (Fig. 11). Leu838 (−3.05 kcal mol−1 for 15avs. 1.25 kcal mol−1 for sorafenib), Val846 (−1.33 kcal mol−1 for 15avs. −1.22 kcal mol−1 for sorafenib), Leu887 (−0.7 kcal mol−1 for 15avs. −1.51 kcal mol−1 for sorafenib), Val897 (−1.74 kcal mol−1 for 15avs. −0.99 kcal mol−1 for sorafenib), Val914 (−0.82 kcal mol−1 for 15avs. −1.15 kcal mol−1 for sorafenib), Phe916 (−0.22 kcal mol−1 for 15avs. −1.53 kcal mol−1 for sorafenib), Cys917 (−0.17 kcal mol−1 for 15avs. 1.11 kcal mol−1 for sorafenib), Leu1033 (−1.24 kcal mol−1 for 15avs. −1.16 kcal mol−1 for sorafenib), Cys1043 (−3.17 kcal mol−1 for 15avs. −2.71 kcal mol−1 for sorafenib), Asp1044 (−0.98 kcal mol−1 for 15avs. −1.21 kcal mol−1 for sorafenib), and Phe1045 (−4.71 kcal mol−1 for 15avs. −1.43 kcal mol−1 for sorafenib) are the amino acids that have a contribution less than −1 kcal mol−1.
Fig. 10. Different energetic components of MM-GBSA. Bars represent the standard deviations.

Fig. 11. Comparison between the binding free energy decomposition of the VEGFR-2_15a complex and VEGFR-2_sorafenib complex.

Extremely long-lasting hydrophobic interactions (90.2% occurrence or greater) are present between compound 15a and Leu838, Val846, Leu887, Val897, Leu1017, Cys1022, His1024, Leu1033, Cys1043, Asp1044, and Phe1045 amino acids (Fig. 12A–C). Phe1045 also often formed Pi-stacking, with a rate of 90.6%. Out of these amino acids, Leu838, Val846, Val897, Leu1033, Cys1043, and Phe1045 show a good contribution to the binding energy and high incidence. Comparing these amino acids with the interacting amino acids from docking, we can see that Leu838, Val846, and Cys1043 remained in contact with the 15a compound for more than 90% of the time and showed a good contribution of the binding. After clustering, PLIP was used to extract 3D binding interactions as .pse files using representative frames (Fig. 13). Similarly, the reference compound shows that there are 15 amino acids (Leu838, Val846, Ala864, Lys866, Ile886, Leu887, Ile890, Val897, Val914, Leu1017, His1024, Leu1033, Cys1043, Asp1044, and Phe1045) that interact with sorafenib compound using hydrophobic interaction with a proportion of at least 90%. In addition, Asp1044 was found to form a H-bond with a proportion of 81.3% and Phe1045 forms a Pi-stacking of 83% (Fig. 14A–C). Leu838, Val846, Leu887, Val914, Leu1033, Cys1043, Asp1044, and Phe1045 are the amino acids that show high persistence and high contribution to the binding energy. Out of the most common amino acids, Val846, Ala864, Leu887, Val914, Leu1017, Leu1033, Cys1043, and Asp1044 remained in contact with the sorafenib from docking. The representative frames obtained from clustering each trajectory were used with PLIP to identify the interactions and get the 3D binding conformation as .pse files (Fig. 15).
Fig. 12. The amino acids, the types of interactions with 15a, and their occurrence during the whole simulation time using the ProLIF python library. (A) Amino acids panel from HSD814 to VAL897, (B) amino acids panel from VAL912 to ILE1023 and (C) amino acids panel from HES1024 to TYR1052.

Fig. 13. The three clusters representative obtained from TTClust and their 3D interactions with 15a. Grey dashed lines: hydrophobic interactions, blue solid lines: H-bonds, green dashed line: Pi-stacking interaction, orange sticks: 15a, blue sticks: amino acids of VEGFR-2 protein.

Fig. 14. The amino acids, the types of interactions with compound sorafenib, and their occurrence during the whole simulation time using the ProLIF python library. (A) Amino acids panel from HSD814 to LEU887, (B) amino acids panel from ILE890 to CYS1022 and (C) amino acids panel from ILE1023 to LEU1047.

Fig. 15. The five clusters representative obtained from TTClust and their 3D interactions with sorafenib. Grey dashed lines: hydrophobic interactions, blue solid lines: H-bonds, green dashed line: Pi-stacking interaction, cyan solid line: halogen bond, orange sticks: sorafenib, blue sticks: amino acids of VEGFR-2 protein.

3.3.7. Principle component analysis (PCA)
With the use of PCA, the trajectory's high-amplitude, and coordinated motion was identified. As mentioned in the methods section, we used the scree plot to determine the number of PCs that contains large variance. We can see that the slope at the 2nd PC showed a large decrease. Moreover, 81.8% of the entire variance was accounted for by the first eigenvector alone, and almost 89.5% of the total variance was accounted for by the first three eigenvectors (Fig. 16).
Fig. 16. The change in the eigenvalues with increasing the eigenvectors (blue line) and the cumulative variance retained in the eigenvectors is shown (red line).

3.3.8. Bi-dimensional projection calculations
In Fig. 17, 18, 19, we see the results of projecting each trajectory onto the first three eigenvectors of the combined C matrix. The bigger marker in these figures represents the average structure of the trajectories. Fig. 17 (a projection on the first two eigenvectors) demonstrated how the two trajectories differ in their average structure and showed sampling that rarely coincides. Fig. 18 showed that the two average structures projected onto the first and third eigenvectors still seem distinct, but the overlap between the two trajectories was much larger. Finally, projection on the second and third eigenvectors (Fig. 19) revealed that the two trajectories are quite dissimilar and have minimal overlap. The motion associated with the first three eigenvectors was shown using porcupine diagrams (Fig. 20). The Gly1046:Leu1065 loop was the biggest motion represented by these three eigenvectors. The first eigenvector of both trajectories describes the same motion (loop opening). The second eigenvector indicated that the loop opens in the green apo structure with a loop rotation while it closes in the red holo structure. The movement captured for the apo protein in the third eigenvector showed the loop opening as the protein becomes more compact, whereas the motion captured for the holo protein showed just the loop closing.
Fig. 17. The projection of each trajectory on the first two eigenvectors.

Fig. 18. The projection of each trajectory on the first and third eigenvectors.

Fig. 19. The projection of each trajectory on the second and third eigenvectors.

Fig. 20. The porcupine figures of each of the first three eigenvectors for both the apo and VEGFR-2_15a systems. Green cartoon: apo protein trajectory, red cartoon: VEGFR-2_15a trajectory.

3.3.9. DFT calculations
After optimizing the neutral singlet molecular structure under DFT/B3LYP/6-311++G(d,p) theory level, the charge distribution function was analyzed using Mulliken analysis. The labeled chemical system of 15a includes 282 electrons and 58 atoms as shown in the labeled molecular scheme, Fig. 21a. The outcomes of the Mulliken population study of compound 15a at DFT-B3LYP/6-311G++(d,p) level, which was used to determine the atomic charge values, are displayed in Fig. 21b. All anticipated values show that the molecule under investigation has significant charge delocalization. Over the hydrogen atoms, the positive charges are concentrated and the most positively charged carbon atoms in the suggested molecule are C21 and C29, which are ready to be attacked by nucleophilic targets. The most negative carbons are C12 and C26 as demonstrated in Fig. 21b. The estimated dipole moment (Dm, 11.59Debye) in Table 8 suggested that the designed structure is reactive with high internal interaction.26
Fig. 21. The optimized geometry (a), the Mullikan atomic charge distribution (b), the frontier molecular orbitals (c), the electrostatic potential (d), the total density of states (e), and the QTAIM maps (f and g) at B3LYB/6-311++G(d,p) for 15a.

The DFT calculated global reactivity parameters for 15a.
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | D m (Debye) | TE (eV) | ΔNmax | ΔE (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 5.953 | −1.783 | −2.085 | 2.085 | 3.868 | 0.259 | 8.406 | 11.596 | −113923.2 | 0.539 | −8.406 |
To shed more light on the reactivity of 15a, frontier molecular orbital analysis has been explored. As demonstrated in Fig. 21c, HOMO is localized all over most parts of the molecule while LUMO is localized over the middle part of the structure. The energy gap value, Egap, between HOMO and LUMO is 7.73 eV predicting a small energy is needed for electronic transition within the molecule. Such findings confirm the potential biological reactivity of 15a.
The computational indices that depend on HOMO and LUMO energies such as global softness (σ), global hardness (η), chemical potential (μ), electron affinity (EA), and electrophilicity index (ω) are calculated for further shedding light on the chemical reactivity of the designed molecule. The results in Table 8 revealed that the molecule is soft and potentially active.27
The molecular electrostatic potential estimations have been carried out to understand the electronic charge is distributed within compound 15a and hence the behavior of binding with VEGFER-2 can be predicted. The 3D potential surface map is presented in Fig. 21d. It is noticed that the negative potential areas (red) are localized over oxygen atoms and are ready for electrophilic attacks. Nucleophilic attacks will occur over a positive potential surface that is localized over hydrogen atoms (blue areas). The green zones over the resonance system are ready for hydrophobic interactions.
The total density of states (TDOS) has been carried out and the spectrum is depicted in Fig. 21e. As seen in the TDOS spectrum, the highest density is noticed for vacant orbitals over LUMO.
Topological analysis has been performed using the Multiwfn program to study the quantum theory of atoms in molecules (QTAIM) based on bond bathes, bond critical points (BCP), and QTAIM parameters. AIMALL software was also utilized to calculate the QTAIM parameters such as electron density (ρ), Laplacian (∇2ρ), and energy density H(r) as the estimated values are represented in ESI.† while the molecular graph and main generated QTAIM bonding bath with relative critical points are shown in Fig. 21f and g, while the detailed bond critical points (BCP) are resented in ESI.† The calculated (ρ), (∇2ρ), and H(r) revealed that the designed drug is stable with internal noncovalent bonding which grants the molecule its stability.
4. Conclusion
In conclusion, this study focused on the design of theobromine derivatives as potential anticancer and VEGFR-2 inhibitors. The investigation involved in vitro assessments of the new compounds' effectiveness against MCF-7 and HepG2 cancer cell lines. Compound 15a emerged as the most cytotoxic member, exhibiting strong inhibitory effects on both HepG2 and MCF-7 cancer cells, with notable selectivity indices. Moreover, compound 15a demonstrated potent VEGFR-2 inhibitory activity and induced apoptosis in HepG2 cells, as evidenced by increased caspase levels and enhanced apoptotic cell percentages. Furthermore, it significantly hindered the migration and healing abilities of HepG2 cancer cells. Molecular docking MD simulations, PLIP, ED, and DFT studies confirmed the binding affinity of the synthesized compounds to VEGFR-2. Finally, computational ADMET and toxicity assessments supported the potential for drug development of the synthesized compounds. Overall, the combination of biological findings and the CADD methodologies positions compound 15a as a promising candidate for the development of a novel anti-apoptotic lead anticancer medication.
5. Experimental
5.1. Chemistry
All solvents, reagents, and devices were illustrated in ESI.†
5.1.1. General procedure for the semi-synthesis of compounds 15a,b, 16, and 17
N-(4-Acetylphenyl)-2-(3,7-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl) acetamide 7 (0.001 mol, 0.355 g) and/or N-(3-acetylphenyl)-2-(3,7-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl)acetamide 8 (0.001 mol, 0.355 g) were mixed and refluxed with the appropriate benzohydrazide derivatives 11a–c and/or 14 (0.001 mol) in absolute ethanol (25 mL) in the presence of catalytic amount of glacial acetic acid (3 drops). The mixtures were then concentrated and cooled. The obtained yields were filtered and purified via crystallization from methanol to produce the final compounds 15a,b, 16, and 17.
5.1.1.1. N-(4-(1-(2-(3-Bromobenzoyl)hydrazono)ethyl)phenyl)-2-(3,7-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl)acetamide 15a
White powder (yield = 0.38 g, 68%); mp = 210–212 °C; IR (KBr) ν cm−1: 3196 (NH), 1704, 1667, 1649 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H, NH), 10.45 (s, 1H, NH), 8.08 (d, J = 4.6 Hz, 1H, Ar-H), 7.83 (d, J = 8.5 Hz, 1H, Ar-H), 7.72–7.62 (m, 2H, Ar-H), 7.56–7.43 (m, 3H, Ar-H), 7.39 (m, 2H, Ar-H), 4.71 (s, 2H, CH2), 3.91 (s, 3H, CH3), 3.46 (s, 3H, CH3), 2.29 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 170.64, 166.41, 164.22, 154.66, 151.37, 148.99, 143.73, 140.45, 139.94, 133.11, 132.35, 130.74, 129.99, 129.05, 128.03, 127.64, 126.93, 120.06, 119.04, 107.07, 43.94, 33.68, 29.93, 14.85; mass (m/z): 554 (M++2, 8.32%), 552 (M+, 22.99%), 411 (100%, base peak); Anal. calcd for C24H22BrN7O4 (552.39): C, 52.18; H, 4.01; N, 17.75; found: C, 52.42; H, 4.23; N, 17.98%.
5.1.1.2. N-(4-(1-(2-(2-Chlorobenzoyl)hydrazono)ethyl)phenyl)-2-(3,7-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl)acetamide 15b
White powder (yield = 0.38 g, 75%); mp = 207–209 °C; IR (KBr) ν cm−1: 3265 (NH), 1745,1696, 1667 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H, NH), 10.43 (s, 1H, NH), 8.03 (d, 1H, Ar-H), 7.84 (d, J = 8.1 Hz, 1H, Ar-H), 7.65 (d, J = 8.1 Hz, 1H, Ar-H), 7.60–7.35 (m, 6H, Ar-H), 4.72 (s, 2H, CH2), 3.91 (s, 3H, CH3), 3.47 (s, 3H, CH3), 2.30 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 169.98, 166.42, 163.37, 154.67, 151.49, 151.39, 149.01, 143.74, 143.27, 130.96, 130.20, 130.04, 129.28, 129.04, 127.66, 126.93, 119.07, 107.09, 43.95, 33.68, 29.93, 14.85; Anal. calcd for C24H22ClN7O4 (507.94): C, 56.75; H, 4.37; N, 19.30; found: C, 56.31; H, 4.10; N, 19.69%.
5.1.1.3. N-(3-(1-(2-(3-Chlorobenzoyl)hydrazono)ethyl)phenyl)-2-(3,7-dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl)acetamide 16
White powder (yield = 0.39 g, 77%); mp = 200–202 °C; IR (KBr) ν cm−1: 3258 (NH), 1707, 1666 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H, NH), 10.44 (s, 1H, NH), 8.32–8.01 (m, 2H, Ar-H), 7.89 (d, 2H, Ar-H), 7.57 (m, 5H, Ar-H), 4.70 (s, 2H, CH2), 3.89 (s, 3H, CH3), 3.46 (s, 3H, CH3), 2.36 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 166.41, 154.65, 151.38, 148.96, 143.69, 139.34, 139.03, 136.51, 133.57, 131.78, 130.77, 129.33, 128.11, 127.15, 122.19, 120.71, 117.48, 107.06, 43.89, 33.67, 29.92, 15.22; Anal. calcd for C24H22ClN7O4 (507.94): C, 56.75; H, 4.37; N, 19.30; Found: C, 56.91; H, 4.52; N, 19.57%.
5.1.1.4. 2-(3,7-Dimethyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-1-yl)-N-(4-(1-(2-nicotinoylhydrazono)ethyl)phenyl)acetamide 17
White crystal (yield = 0.31 g, 65%); mp = 230–232 °C; IR (KBr) ν cm−1: 3265 (NH), 1745,1695, 1666 (C
O); 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H, NH), 10.42 (s, 1H, NH), 9.04 (s, 1H, Ar-H), 8.75 (s, 1H, Ar-H), 8.15 (d, 2H, Ar-H), 7.91 (d, 2H, Ar-H), 7.60 (d, 3H, Ar-H), 4.71 (s, 2H, CH2), 3.90 (s, 3H, CH3), 3.34 (s, 3H, CH3), 2.36 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 166.42, 154.64, 151.36, 148.98, 143.74, 143.26, 133.21, 127.71, 123.92, 119.07, 107.06, 43.94, 33.69, 29.93, 14.95; Anal. calcd. for C23H22N8O4 (474.48): C, 58.22; H, 4.67; N, 23.62; Found: C, 58.59; H, 4.42; N, 23.91%.
5.2. Biological examinations
5.2.1. In vitro anti-proliferative activity
To assess the inhibitory potential of the synthesized theobromine derivatives 15a, 15b, 16 and 17 against the proliferation of MCF-7 and HepG2 cancer cells an in vitro MTT assay was applied.28 The research's ESI† file goes into more insight into this experiment.
5.2.2. In vitro VEGFR-2 inhibition
The synthesized theobromine derivatives 15a, 15b, 16 and 17 were subjected to an in vitro evaluation against VEGFR-2 via the implementation of a VEGFR-2 ELISA kit.29 The research's ESI† file goes into more insight into this experiment.
5.2.3. Flow cytometry assay
The potential inhibition of compound 15a on HepG2 apoptosis was evaluated using flow cytometry.30 The research's ESI† file goes into more insight into this experiment.
5.2.4. Caspase 3 and 9 determinations
The potential inhibition of compound 15a on caspases level in HepG2 cells was evaluated using RT PCR analysis. The research's ESI† file goes into more insight into this experiment.
5.3. In silico studies
5.3.1. Docking studies
Utilizing docking investigations with MOE2019 software, the potential inhibitory effects of the synthesized theobromine derivatives 15a, 15b, 16 and 17 against VEGFR-2 [PDB ID: 2OH4, resolution: 2.05] were assessed. The research's ESI† file goes into more insight into this experiment.31–33
5.3.2. ADMET studies
ADMET profiles of the synthesized theobromine derivatives 15a, 15b, 16 and 17 were evaluated computationally by Discovery Studio 4.0. The research's ESI† file goes into more insight into this experiment.34,35
5.3.3. Toxicity studies
The toxicity profiles of the synthesized theobromine derivatives 15a, 15b, 16 and 17 were evaluated computationally by Discovery Studio 4.0. The research's ESI† file goes into more insight into this experiment.36–38
5.3.4. MD simulations
Compound 15a's capacity to inhibit the VEGFR-2 was examined computationally through MD simulations that were run by the CHARMM-GUI web server39 and the MD engine GROMACS 2021.40,41 The research's ESI† file goes into more insight into this experiment.
5.3.5. MM-GBSA analysis
MM-GBSA analysis of the VEGFR-2-compound 15a complex was evaluated by the Gmx_MMPBSA package.42,43 The research's ESI† file goes into more insight into this experiment.
5.3.6. ProLIF analysis
ProLIF analysis of VEGFR-2-15a complex was evaluated by the GROMACS package.44–46 The research's ESI† file goes into more insight into this experiment.
5.3.7. Essential dynamics (ED) analysis
Essential Dynamics (ED) analysis of VEGFR-2-15a complex was evaluated by GROMACS by utilizing the gmx covar command.47 The research's ESI† file goes into more insight into this experiment.
5.3.8. Cosine Content analysis
Cosine Content analysis of VEGFR-2-15a complex was evaluated by GROMACS.48 The research's ESI† file goes into more insight into this experiment.
5.3.9. Bidimensional Projections analysis
Bidimensional Projections analysis of the VEGFR-2-15a complex was evaluated by GROMACS49,50 The research's ESI† file goes into more insight into this experiment.
5.3.10. Density function theory (DFT) calculations
DFT analyses were evaluated for 15a by Gaussian software. The research's ESI† file goes into more insight into this experiment.51,52
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Research Center at AlMaarefa University for funding this work.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04007k
References
- Bray F. Ferlay J. Soerjomataram I. Siegel R. L. Torre L. A. Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Belal A. Abdel Gawad N. M. Mehany A. B. Abourehab M. A. Elkady H. Al-Karmalawy A. A. Ismael A. S. Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3,2-d] pyrimidines and pyrrolo [3,2-e][1,4] diazepine derivatives as potent EGFR/CDK2 inhibitors. J. Enzyme Inhib. Med. Chem. 2022;37(1):1884–1902. doi: 10.1080/14756366.2022.2096019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifa M. M. Al-Karmalawy A. A. Elkaeed E. B. Nafie M. S. Tantawy M. A. Eissa I. H. Mahdy H. A. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies. J. Enzyme Inhib. Med. Chem. 2022;37(1):299–314. doi: 10.1080/14756366.2021.2007905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita Jr V. T. Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- Fidler I. J. Ellis L. M. Chemotherapeutic drugs—more really is not better. Nat. Med. 2000;6(5):500–502. doi: 10.1038/74969. [DOI] [PubMed] [Google Scholar]
- Shah A. A. Kamal M. A. Akhtar S. Tumor angiogenesis and VEGFR-2: mechanism, pathways and current biological therapeutic interventions. Curr. Drug Metab. 2021;22(1):50–59. doi: 10.2174/1389200221666201019143252. [DOI] [PubMed] [Google Scholar]
- Elrazaz E. Z. Serya R. A. Ismail N. S. Albohy A. Abou El Ella D. A. Abouzid K. A. Discovery of potent thieno [2,3-d] pyrimidine VEGFR-2 inhibitors: Design, synthesis and enzyme inhibitory evaluation supported by molecular dynamics simulations. Bioorg. Chem. 2021;113:105019. doi: 10.1016/j.bioorg.2021.105019. [DOI] [PubMed] [Google Scholar]
- Metwaly A. M. Lianlian Z. Luqi H. Deqiang D. Black ginseng and its saponins: Preparation, phytochemistry and pharmacological effects. Molecules. 2019;24(10):1856. doi: 10.3390/molecules24101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metwaly A. M. Ghoneim M. M. Eissa I. H. Elsehemy I. A. Mostafa A. E. Hegazy M. M. Afifi W. M. Dou D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021;28(10):5823–5832. doi: 10.1016/j.sjbs.2021.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D. J. Cragg G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016;79(3):629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- da Rosa R. Schenkel E. P. Bernardes L. S. C. Semisynthetic and newly designed derivatives based on natural chemical scaffolds: moving beyond natural products to fight Trypanosoma cruzi. Phytochem. Rev. 2020;19(1):105–122. [Google Scholar]
- Sánchez-Recillas A. Navarrete-Vázquez G. Hidalgo-Figueroa S. Rios M. Y. Ibarra-Barajas M. Estrada-Soto S. Semisynthesis, ex vivo evaluation, and SAR studies of coumarin derivatives as potential antiasthmatic drugs. Eur. J. Med. Chem. 2014;77:400–408. doi: 10.1016/j.ejmech.2014.03.029. [DOI] [PubMed] [Google Scholar]
- Carla Cadoná F. Kolinski Machado A. Farina Azzolin V. Barbisan F. Bortoluzzi Dornelles E. Glanzner W. Bayard Gonçalves P. D. Elias Assmann C. Esteves Ribeiro E. Beatrice Mânica da Cruz I. Guaraná a caffeine-rich food increases oxaliplatin sensitivity of colorectal HT-29 cells by apoptosis pathway modulation. Anti-Cancer Agents Med. Chem. 2016;16(8):1055–1065. doi: 10.2174/1871520616666151217121138. [DOI] [PubMed] [Google Scholar]
- Sugimoto N. Miwa S. Katakura M. Matsuzaki K. Shido O. Tsuchiya H. Yachie A. Theobromine, the primary methylxanthine found in Theobroma cacao, inhibits malignant glioblastoma cell growth by negatively regulating Akt/mammalian target of rapamycin kinase (LB836) FASEB J. 2014;28:LB836. doi: 10.1080/01635581.2013.877497. [DOI] [PubMed] [Google Scholar]
- Barcz E. Sommer E. Sokolnicka I. Gawrychowski K. Roszkowska-Purska K. Janik P. Skopinska-Rózewska E. The influence of theobromine on angiogenic activity and proangiogenic cytokines production of human ovarian cancer cells. Oncol. Rep. 1998;5(2):517–537. doi: 10.3892/or.5.2.517. [DOI] [PubMed] [Google Scholar]
- Gil M. Skopińska-Rózewska E. Radomska D. Demkow U. Skurzak H. Rochowska M. Beuth J. Roszkowski K. Effect of purinergic receptor antagonists suramin and theobromine on tumor-induced angiogenesis in BALB/c mice. Folia Biol. 1993;39(2):63–68. [PubMed] [Google Scholar]
- Alanazi M. M. Mahdy H. A. Alsaif N. A. Obaidullah A. J. Alkahtani H. M. Al-Mehizia A. A. Alsubaie S. M. Dahab M. A. Eissa I. H. New bis ([1,2,4]triazolo)[4,3-a: 3′,4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021;112:104949. doi: 10.1016/j.bioorg.2021.104949. [DOI] [PubMed] [Google Scholar]
- Alanazi M. M. Elkady H. Alsaif N. A. Obaidullah A. J. Alkahtani H. M. Alanazi M. M. Alharbi M. A. Eissa I. H. Dahab M. A. New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies. RSC Adv. 2021;11(48):30315–30328. doi: 10.1039/d1ra05925d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef R. G. Sakr H. M. Eissa I. H. Mehany A. B. Metwaly A. M. Elhendawy M. A. Radwan M. M. ElSohly M. A. Abulkhair H. S. El-Adl K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021;45(36):16949–16964. [Google Scholar]
- Adnane L. Trail P. A. Taylor I. Wilhelm S. M. Sorafenib (BAY 43-9006, Nexavar®), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. doi: 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- Peng F.-W. Liu D.-K. Zhang Q.-W. Xu Y.-G. Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012-2016) Expert Opin. Ther. Pat. 2017;27(9):987–1004. doi: 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- Yousef R. G. Elwan A. Gobaara I. M. Mehany A. B. Eldehna W. M. El-Metwally S. A. Alsfouk B. A. Elkaeed E. B. Metwaly A. M. Eissa I. H. Anti-cancer and immunomodulatory evaluation of new nicotinamide derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: in vitro and in silico studies. J. Enzyme Inhib. Med. Chem. 2022;37(1):2206–2222. doi: 10.1080/14756366.2022.2110868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef R. G. Ibrahim A. Khalifa M. M. Eldehna W. M. Gobaara I. M. Mehany A. B. Elkaeed E. B. Alsfouk A. A. Metwaly A. M. Eissa I. H. Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: Virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies. J. Enzyme Inhib. Med. Chem. 2022;37(1):1389–1403. doi: 10.1080/14756366.2022.2070744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef R. G. Elkady H. Elkaeed E. B. Gobaara I. M. Al-Ghulikah H. A. Husein D. Z. Ibrahim I. M. Metwaly A. M. Eissa I. H. (E)-N-(3-(1-(2-(4-(2,2,2-Trifluoroacetamido) benzoyl) hydrazono) ethyl) phenyl) nicotinamide: A Novel Pyridine Derivative for Inhibiting Vascular Endothelial Growth Factor Receptor-2: Synthesis, Computational, and Anticancer Studies. Molecules. 2022;27(22):7719. doi: 10.3390/molecules27227719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T. Zhu L. Chen L. Xu X. Mapping the binding site of a large set of quinazoline type EGF-R inhibitors using molecular field analyses and molecular docking studies. J. Chem. Inf. Comput. Sci. 2003;43(1):273–287. doi: 10.1021/ci025552a. [DOI] [PubMed] [Google Scholar]
- Husein D. Z. Hassanien R. Khamis M. Cadmium oxide nanoparticles/graphene composite: Synthesis, theoretical insights into reactivity and adsorption study. RSC Adv. 2021;11(43):27027–27041. doi: 10.1039/d1ra04754j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. Husein D. Z. Novel synthesis of multicomponent porous nano-hybrid composite, theoretical investigation using DFT and dye adsorption applications: Disposing of waste with waste. Environ. Sci. Pollut. Res. 2023;30(4):8928–8955. doi: 10.1007/s11356-022-20050-2. [DOI] [PubMed] [Google Scholar]
- Van de Loosdrecht A. Beelen R. Ossenkoppele G. Broekhoven M. Langenhuijsen M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods. 1994;174(1–2):311–320. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Elkaeed E. B. Yousef R. G. Elkady H. Mehany A. B. Alsfouk B. A. Husein D. Z. Ibrahim I. M. Metwaly A. M. Eissa I. H. In silico, in vitro VEGFR-2 inhibition, and anticancer activity of a 3-(hydrazonomethyl) naphthalene-2-ol derivative. J. Biomol. Struct. Dyn. 2022:1–16. doi: 10.1080/07391102.2022.2127907. [DOI] [PubMed] [Google Scholar]
- Eldehna W. M. El Hassab M. A. Elsayed Z. M. Al-Warhi T. Elkady H. Abo-Ashour M. F. Abourehab M. A. Eissa I. H. Abdel-Aziz H. A. Design, synthesis, in vitro biological assessment and molecular modeling insights for novel 3-(naphthalen-1-yl)-4,5-dihydropyrazoles as anticancer agents with potential EGFR inhibitory activity. Sci. Rep. 2022;12(1):12821. doi: 10.1038/s41598-022-15050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghour M. S. Elkady H. Eldehna W. M. El-Deeb N. M. Kenawy A. M. Elkaeed E. B. Alsfouk A. A. Alesawy M. S. Metwaly A. M. Eissa I. H. Design and synthesis of thiazolidine-2,4-diones hybrids with 1,2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: In-vitro anticancer evaluation and in-silico studies. J. Enzyme Inhib. Med. Chem. 2022;37(1):1903–1917. doi: 10.1080/14756366.2022.2085693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkaeed E. B. Yousef R. G. Elkady H. Gobaara I. M. Alsfouk A. A. Husein D. Z. Ibrahim I. M. Metwaly A. M. Eissa I. H. The assessment of anticancer and VEGFR-2 inhibitory activities of a new 1 H-indole derivative: In silico and in vitro approaches. Processes. 2022;10(7):1391. [Google Scholar]
- Ma C. Taghour M. S. Belal A. Mehany A. B. Mostafa N. Nabeeh A. Eissa I. H. Al-Karmalawy A. A. Design and synthesis of new quinoxaline derivatives as potential histone deacetylase inhibitors targeting hepatocellular carcinoma: in silico, in vitro, and SAR studies. Front. Chem. 2021;9:725135. doi: 10.3389/fchem.2021.725135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkaeed E. B. Yousef R. G. Khalifa M. M. Ibrahim A. Mehany A. B. Gobaara I. M. Alsfouk B. A. Eldehna W. M. Metwaly A. M. Eissa I. H. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules. 2022;27(19):6203. doi: 10.3390/molecules27196203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alesawy M. S. Abdallah A. E. Taghour M. S. Elkaeed E. B. Eissa I. H. Metwaly A. M. In silico studies of some isoflavonoids as potential candidates against COVID-19 targeting human ACE2 (hACE2) and viral main protease (Mpro) Molecules. 2021;26(9):2806. doi: 10.3390/molecules26092806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkady H. Elwan A. El-Mahdy H. A. Doghish A. S. Ismail A. Taghour M. S. Elkaeed E. B. Eissa I. H. Dahab M. A. Mahdy H. A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzyme Inhib. Med. Chem. 2022;37(1):403–416. doi: 10.1080/14756366.2021.2015343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa I. H. Khalifa M. M. Elkaeed E. B. Hafez E. E. Alsfouk A. A. Metwaly A. M. In silico exploration of potential natural inhibitors against SARS-CoV-2 nsp10. Molecules. 2021;26(20):6151. doi: 10.3390/molecules26206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissa I. H. Dahab M. A. Ibrahim M. K. Alsaif N. A. Alanazi A. Eissa S. I. Mehany A. B. Beauchemin A. M. Design and discovery of new antiproliferative 1,2,4-triazin-3 (2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site. Bioorg. Chem. 2021;112:104965. doi: 10.1016/j.bioorg.2021.104965. [DOI] [PubMed] [Google Scholar]
- Abraham M. J. Murtola T. Schulz R. Páll S. Smith J. C. Hess B. Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- Jo S. Cheng X. Islam S. M. Huang L. Rui H. Zhu A. Lee H. S. Qi Y. Han W. Vanommeslaeghe K. CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv. Protein Chem. Struct. Biol. 2014;96:235–265. doi: 10.1016/bs.apcsb.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Cheng X. Jo S. MacKerell A. D. Klauda J. B. Im W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 2016;110(3):641a. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuccinardi T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discovery. 2021;16(11):1233–1237. doi: 10.1080/17460441.2021.1942836. [DOI] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S. Valdés-Tresanco M. E. Valiente P. A. Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021;17(10):6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Bouysset C. Fiorucci S. ProLIF: a library to encode molecular interactions as fingerprints. J. Cheminf. 2021;13:1–9. doi: 10.1186/s13321-021-00548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S. Schreiber S. Haupt V. J. Adasme M. F. Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubiana T. Carvaillo J.-C. Boulard Y. Bressanelli S. TTClust: a versatile molecular simulation trajectory clustering program with graphical summaries. J. Chem. Inf. Model. 2018;58(11):2178–2182. doi: 10.1021/acs.jcim.8b00512. [DOI] [PubMed] [Google Scholar]
- Amadei A. Linssen A. B. Berendsen H. J. Essential dynamics of proteins. Proteins: Struct., Funct., Bioinf. 1993;17(4):412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- Papaleo E. Mereghetti P. Fantucci P. Grandori R. De Gioia L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: the myoglobin case. J. Mol. Graphics Modell. 2009;27(8):889–899. doi: 10.1016/j.jmgm.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Maisuradze G. G. Leitner D. M. Free energy landscape of a biomolecule in dihedral principal component space: Sampling convergence and correspondence between structures and minima. Proteins: Struct., Funct., Bioinf. 2007;67(3):569–578. doi: 10.1002/prot.21344. [DOI] [PubMed] [Google Scholar]
- Hess B. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2000;62(6):8438. doi: 10.1103/physreve.62.8438. [DOI] [PubMed] [Google Scholar]
- Elkaeed E. B. Yousef R. G. Elkady H. Gobaara I. M. Alsfouk B. A. Husein D. Z. Ibrahim I. M. Metwaly A. M. Eissa I. H. Design, synthesis, docking, DFT, MD simulation studies of a new nicotinamide-based derivative: In vitro anticancer and VEGFR-2 inhibitory effects. Molecules. 2022;27(14):4606. doi: 10.3390/molecules27144606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkaeed E. B. Eissa I. H. Elkady H. Abdelalim A. Alqaisi A. M. Alsfouk A. A. Elwan A. Metwaly A. M. A multistage in silico study of natural potential inhibitors targeting SARS-CoV-2 main protease. Int. J. Mol. Sci. 2022;23(15):8407. doi: 10.3390/ijms23158407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.