

Abstract
Geroscience posits that cardiovascular disease (CVD) and other chronic diseases result from progressive erosion of the effectiveness of homeostatic mechanisms that oppose age-related accumulation of molecular damage. This hypothetical common root to chronic diseases explains why patients with CVD are often affected by multimorbidity and frailty and why older age negatively affects CVD prognosis and treatment response. Gerotherapeutics enhance resilience mechanisms that counter age-related molecular damage to prevent chronic diseases, frailty, and disability, thereby extending healthspan. Here, we describe the main resilience mechanisms of mammalian aging, with a focus on how they can affect CVD pathophysiology. We next present novel gerotherapeutic approaches, some of which are already used in management of CVD, and explore their potential to transform care and management of CVD. The geroscience paradigm is gaining traction broadly in medical specialties, with potential to mitigate premature aging, reduce health care disparities, and improve population healthspan.
Keywords: frailty, geroscience, hallmarks, inflammation, multimorbidity
Aging and cardiovascular disease (CVD) are interconnected. As far back as the 15th century, Leonardo da Vinci (1452–1519) wrote: “vessels in the elderly restrict the transit of blood through thickening of the tunics.” Aging as a powerful risk factor for chronic disease and the population aging contribute to the expansion of CVD in the world population. There is evidence that chronic diseases result from the failure of mechanisms that oppose molecular damage with aging and that enhancing these mechanisms extends lifespan and healthspan. This research has led to the concept of geroscience,1 which posits that therapeutics that target the fundamental mechanisms of aging (gerotherapeutics) delay or avert age-related chronic diseases and increase healthspan (Table 1). As CVD is the most prevalent cause of morbidity and mortality in the elderly, the hypothesis that the biology of aging contributes to CVD as well as other chronic diseases should interest cardiovascular clinicians for 3 main reasons.
TABLE 1.
Glossary of Relevant Terms
| Geroscience | Research focus premised on the concept that the physiology of aging plays a determinant role in chronic diseases as well as geriatric syndromes. Related assumptions are that are common therapeutic approaches to prolong life and moderate chronic diseases as well as geriatric syndromes. |
| Lifespan | Duration of life for a species or subgroup within a species. |
| Healthspan | Duration of life spent in good health, free from the chronic diseases and disabilities of aging. |
| Hallmarks of aging | Cellular biologic processes that are manifest with aging and that interact in various combinations within each adult to modify lifespan and healthspan. |
| Resilience | The intrinsic capacity to tolerate stresses including molecular, cellular, and systems mechanisms. |
| Geriatric syndrome | Health impairments that do not fit into discrete disease categories. Multimorbidity and frailty are common geriatric syndromes, with high prevalence in older adults with cardiovascular disease. |
First, although CVD prevalence and mortality fell in recent decades because of aggressive control of risk factors, geroscience-based therapies that target mechanisms upstream of CVD may enhance this trend, even in individuals free of CVD risk factors.
Second, by targeting the drivers of aging, gerotherapeutics might prevent or ameliorate comorbidities and frailty often present in patients with CVD and improve prognosis and overall health (Central Illustration).
CENTRAL ILLUSTRATION.
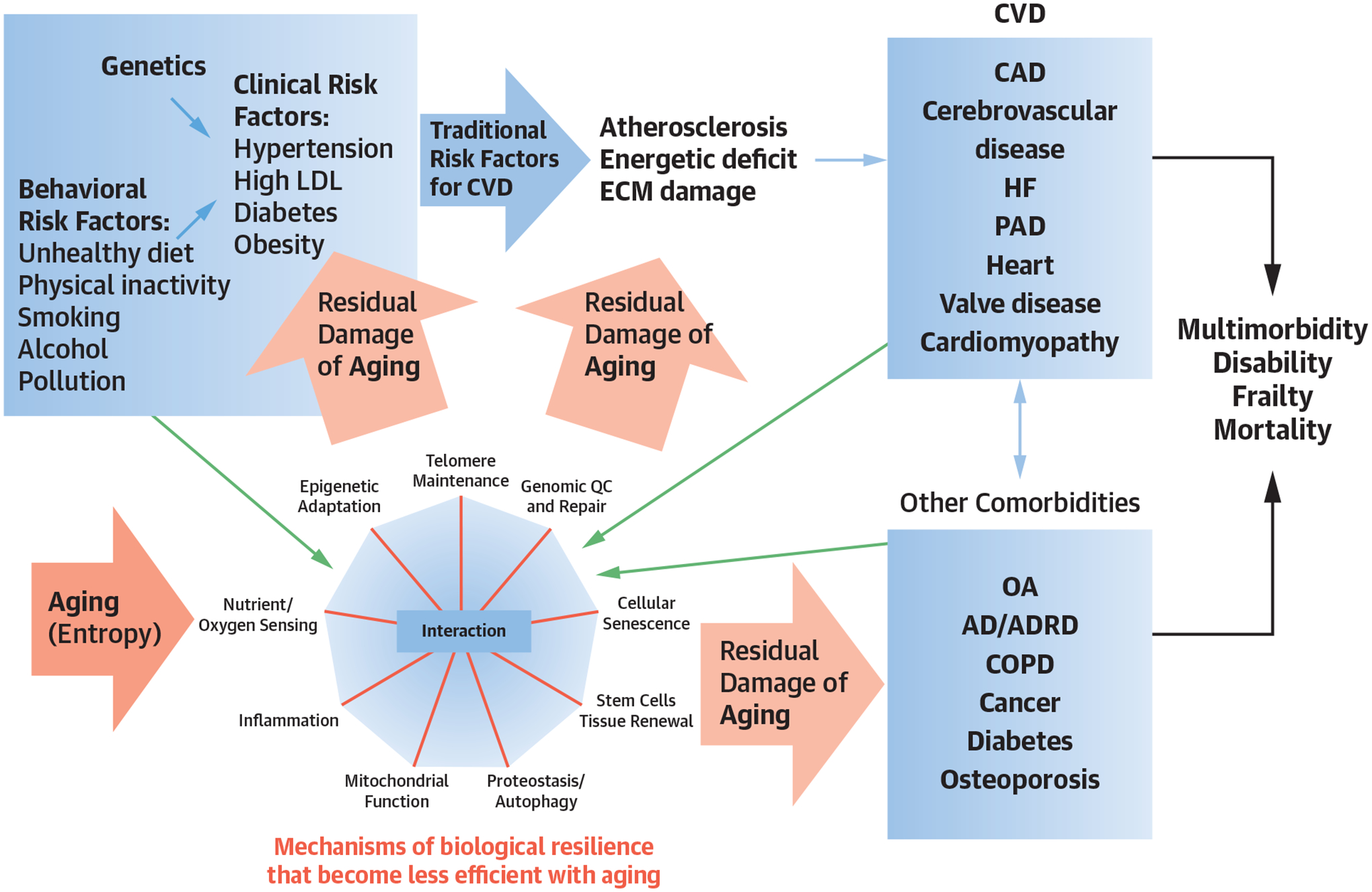
The Geroscience Hypothesis in the Context of Cardiovascular Disease
Genetic predisposition and unhealthy behaviors (blue arrows) contribute to major CVD risk factors and CVD in older persons. Aging itself is associated with stochastic damage to molecules and organelles (pink arrows) and accelerates risks of CVD, as well as multimorbidity, frailty, disability, and premature death. Whereas resilience signifies capacity to repair or replace such damaged components, resilience often diminishes over time, especially amid behavioral risk factors (eg, unhealthy diet) and chronic diseases (green arrows). The geroscience hypothesis poses that enhancing resilience reduces the burden of CVD and other chronic diseases and related susceptibility to frailty and disability. AD/ADRD = Alzheimer disease and Alzheimer disease-related dementias; CAD = coronary heart disease; COPD = chronic obstructive pulmonary disease; CVD = cardiovascular disease; ECM = extracellular matrix; HF = heart failure; LDL = low-density lipoprotein; OA = osteoarthritis; PAD = peripheral arterial disease; QC = quality control.
Third, identification of individuals with accelerated aging may allow targeting the early stages of CVD and other development of disease, when gerotherapeutics may be more effective.
CVD, FRAILTY, MULTIMORBIDITY, AND CLINICAL COMPLEXITY AMONG OLDER ADULTS
Frailty is a state of exhausted functional reserve that is thought to be caused by a failure of the resilience mechanisms of aging.2,3 Although many different definitions of frailty exist (Table 2), the syndrome of physical frailty is a distinctive high-risk clinical state characterized by 3 or more of 5 key criteria: weakness, slow walking speed, low physical activity, fatigue or exhaustion, and unintentional weight loss. Multimorbidity4 is defined as 2 or more medical diseases or conditions that persist for >1 year. The erosion of resilience mechanisms with aging prevent or repair macromolecular and cellular damage leading to frailty, multimorbidity, and other geriatric syndromes. Resilience mechanisms decline faster in some individuals, causing them to develop multimorbidity and frailty earlier in life than the average population. CVD in older patients5 typically develops in the context of frailty and multimorbidity,6 with negative implication for prognosis and management complexity. Gerotherapeutics address CVD, frailty, and multimorbidity simultaneously.
TABLE 2.
Geriatric Syndromes
| Geriatric Syndromes | Definition |
|---|---|
| Multimorbidity | ≥2 chronic medical conditions4 |
| Frailty | 2 prevailing models |
| Falls | Episodes of sudden, involuntary transfer of body to the ground and at a lower level than the previous one |
| Cognitive impairment | Mild cognitive impairment (MCI): declining and disturbance of cognition, minimal impairment of complex activities, ability to perform regular daily functions, and absence of dementia |
| Dementia: loss of cognitive functions of thinking, remembering, and reasoning to the extent that it interferes with doing everyday activities | |
| Multisensory impairment | Loss of visual acuity, hearing, olfaction, taste, and tactile sensitivity that affect everyday functioning among older adults |
| Sarcopenia | Sarcopenia is a progressive and generalized skeletal muscle disorder that is associated with an increased likelihood of adverse outcomes including falls, fractures, physical disability, and mortality |
EWGSOP definition167: 3 criteria used for sarcopenia assessment:
| |
| Criterion 1 identifies probable sarcopenia, diagnosis is confirmed by criterion 2 and if all 3 criteria are met, severe sarcopenia is diagnosed | |
| Polypharmacy | Use of ≥5 medications |
CVD AND GERIATRIC SYNDROMES SHARE AGING HALLMARKS
The impairment of mechanisms that prevent damage accumulation with aging, referred to as the “hallmarks”7 of aging, contributes to multimorbidity, frailty, and loss of physical and cognitive functions. Environmental stress and CVD risk factors,8 such as obesity and smoking, accelerate the failure of these mechanisms (Central Illustration). Here we focus on aging resilience mechanisms that targeted by gerotherapeutics may both extend healthspan and reduce CVD (Figure 1).
FIGURE 1.
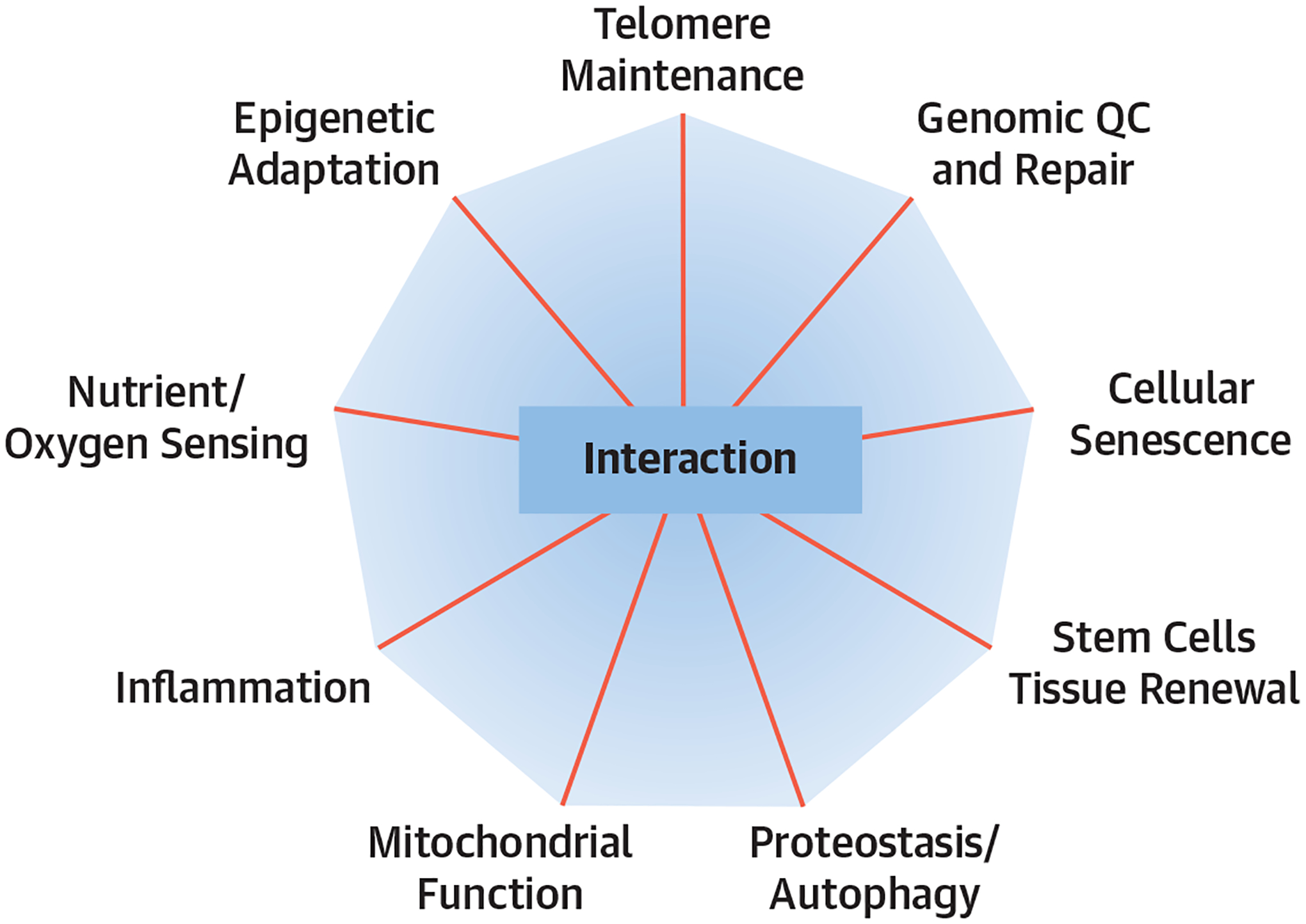
The Hallmarks of Aging
Biological processes that become impaired with aging can be potentially targeted by gerotherapeutics. The intersecting lines depict how targeting each pillar can affect the others. QC = quality control.
GENOMIC DAMAGE.
DNA, the molecule that carries all genetic information, is continuously damaged and repaired throughout life. Insufficient or inaccurate DNA repair activity leads to corruption of the genetic code, generally referred to as “genomic instability.” The accumulation of DNA mutations or chromosome aberrations drives may cause cell death or senescence: 2 drivers of diseases and aging.9 Mice with mutations that affect DNA repair proteins exhibit aging-like features including endothelial dysfunction, increased vascular stiffness, hypertension, and accumulation of senescent cells.10 In humans, mutations of mitochondria DNA are associated with atherogenesis. Studies have shown a significant association of DNA damage and genomic instability with cancer, frailty, and unsuccessful aging.11
Clonal hematopoiesis of indeterminate potential (CHIP) reflects genomic instability. With aging, hematopoietic stem cells acquire somatic mutations in genes that promote the clonal expansion of myeloid cells. Mutated leukocytes occur at a frequency of 10% to 20% in individuals by age 70, with ≥2% of cells carrying a CHIP genetic variant associated with high risk of CVD, cancer, and mortality. Certain types of CHIP-related mutations accelerate atherosclerosis and cause activation of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, a main inflammatory pathway that stimulates the production of interleukin (IL)-1β and IL-18 as well as apoptotic and pyroptotic cell death.12 CHIP, because of mutations in DNA damage response genes, promotes peripheral artery disease.13 Aging-related loss of chromosome Y in some somatic cells, but not others (mosaicism), is the most commonly acquired mutation in the male genome and is associated with cancer, Alzheimer disease, and CVD.14
EPIGENETIC ALTERATIONS.
Epigenetic mechanisms regulate gene expression through chemical modifications of chromatin, particularly histone proteins that provide structural support to chromosomes, or changes in the superstructure of DNA. Methylation of DNA—specifically, the transfer of a methyl group to a cytosine in a cytosine-guanidine (CpG) sequence—is the most studied epigenetic mechanism in aging.15
An extensive literature connects DNA methylation with CVDs and other age-related health outcomes.16 DNA from atherosclerotic plaques tends to be hypomethylated, with hypermethylation in regulatory regions of genes linked to atherosclerosis.17 DNA methylation at specific CpG islands in circulating cells is associated with increased risk of CVD.18 In the CARDIA (Coronary Artery Risk Development in Young Adults) study cohort, participants who maintained better CVD health with aging showed a slower change in GrimAge, an aggregate blood methylation-based biomarker that predicts healthspan and lifespan.19 Epigenetic indexes (clocks) built from DNA methylation are associated with CVD, multimorbidity, and frailty. Methylation levels of specific histones (ie, H3K9 and H3K27) are significantly lower in smooth muscle cells and lymphocytes of patients with carotid artery stenosis than controls.20
LOSS OF PROTEOSTASIS.
Proteins are damaged throughout cells lives, and such damage can affect their architecture, functionality, and solubility. Protein aggregation and precipitation adversely affect their function. Protein folding guides exist (chaperones) that work in combination with the ubiquitin-proteasome system (eliminates misfolded proteins) and the lysosome-autophagy system (eliminates damaged organelles and pathogens) to ensure that proteins and organelles maintain their optimal structure and function. In a process called “autophagy,” large molecular aggregates and organelles are engulfed in vacuoles that are then fused with lysosomes: that is, intracellular sachets that contain enzymes that degrade the content of autophagic vacuoles. Mitophagy, a specialized form of autophagy, recycles damaged mitochondria. Defective mitophagy causes the persistence of fragmented, dysfunctional mitochondria that trigger inflammation and deranged energy metabolism in CVDs.21
During stress conditions, protein synthesis is suppressed to shift energy to active protein folding, and genes that support survival pathways and inflammation are overexpressed. Failure to reestablish protein homeostasis causes pathology, including in CVD and sarcopenia.22,23 Accumulation of “toxic” proteins has been associated with endothelial and cardiomyocyte senescence, atrial fibrillation, cardiac hypertrophy, and cardiomyopathy.24 Abnormal transthyretin amyloid folding contributes to cardiomyopathy and preserved ejection fraction heart failure (HF).25 Impaired chaperone-mediated autophagy is also associated with the formation of atherosclerotic plaques.26
DEREGULATED NUTRIENT SENSING.
Cells sense energy availability to make decisions about growth, metabolism, and proliferation, and—in some cases—apoptosis and regulate autophagy, mitochondrial, and ribosomal biogenesis.27 The phosphatidylinositol-3-kinase (PI3K)/AKT kinase (AKT)/mammalian target of rapamycin (mTOR) pathway regulates signaling mechanisms that are central to such regulation. Altered mTOR signaling is involved in many age-associated diseases including restenosis after angioplasty, cardiac hypertrophy, HF, type 2 diabetes mellitus (T2DM) and obesity, neurodegenerative diseases, cancer, and immunosenescence. AMP-activated protein kinase (AMPK) detects a discrepancy between energy demand and availability caused by either mitochondrial dysfunction (eg, oxygen deprivation) or increased consumption (eg, during exercise). Activated AMPK increases energy production by stimulating mitochondrial biogenesis, fatty-acid oxidation, glycolysis, autophagy, angio-genesis, and nitric oxide bioavailability and by inhibiting biosynthetic processes such as gluconeo-genesis and protein synthesis. Abnormal AMPK signaling is associated with hypertension, atherosclerosis, stroke, obesity and diabetes.28 Sirtuins, a family of nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases or mono-adenosine diphosphate (ADP) ribosyltransferases, sense and activate several energy preserving pathways.29
MITOCHONDRIAL DYSFUNCTION.
Mitochondria generate most of the energy required for all biological processes including resilience mechanisms that combat aging. A decline of mitochondrial function occurs with aging and is a prominent characteristic of endothelial dysfunction, arterial stiffness, atherosclerosis, ischemia-reperfusion injury, hypertension, cardiac hypertrophy, diastolic dysfunction and HF, metabolic diseases (eg, T2DM and metabolic syndrome), and geriatric syndromes such as sarcopenia and frailty.30,31 The release of reactive oxygen species (ROS), oxidized cardiolipin and mitochondrial DNA by damaged mitochondria, drive inflammation by triggering nuclear factor kappa light chain enhancer of activated B cells (NF-κB), NLRP3 inflammasome, and the cyclic guanosine monophosphate-adenosine monophosphate synthase with the adaptor stimulator of interferon genes (cGAS-STING) pathways and stimulate expression of proinflammatory cytokines such as IL-1, IL-6, IL-18, tumor necrosis factor (TNF)α, and Type-I interferons that contribute to a state of “inflammaging.”32
CELLULAR SENESCENCE.
Cellular senescence involves cell cycle arrest triggered by developmental and stress signals and is considered a cancer-suppression mechanism. Triggers of cellular senescence include irradiation, oxidative and genotoxic agents, epigenetic changes, perturbed proteostasis, mitochondrial dysfunction, and chemotherapeutic agents. Senescent cells change morphology, often express defining biomarkers, such as p21WAF1/CIP1 and p16INK4A, exhibit signs of persistent DNA damage response and show enhanced lysosomal activity. An important feature is the secretion of bioactive molecules, known as the senescence-associated secretory phenotype (SASP), which includes senescence triggers, proinflammatory cytokines (eg, IL-6), chemokines, proapoptotic factors, growth modulators, angiogenic factors, proteases, bioactive lipids, extracellular matrix components, and metalloproteinases.33
Senescent cells accumulate with aging in multiple tissues, including the heart and vasculature,34 and many SASP factors increase in the plasma of healthy individuals with aging.35 Senescent cells contribute to pathology by obstructing tissue repair and regeneration. The SASP promotes chronic inflammation, paracrine spread of senescence, and support neoplastic transformation.36 Senescent cells accumulate in several age-related diseases, such as atherosclerosis, diabetes, lung disease, Alzheimer disease, vascular dementia, and sarcopenia.37,38 In humans, the accumulation of senescent cells in sub-cutaneous fat is associated with many age-related changes, including frailty.
The National Institutes of Health (NIH) Common Fund’s Cellular Senescence Network Program (SenNet) aims to identify and characterize senescent cells throughout the body (including the heart) during aging and chronic diseases.39
STEM-CELL EXHAUSTION.
Stem cells maintain functionality of most tissues by replacing cells lost through wear and tear or injury. With aging, the renewal, proliferation, and differentiation capacity of stem cells decline, contributing to aging-associated disorders such as CVD. Some evidence suggests that circulating stem cells can enhance the regenerative capacity of multiple tissues, including those specific to the cardiovascular system.40
DYSBIOSIS.
Dysbiosis refers to the reduction of microbial diversity of the resident microbiome. The gut is the most studied microbiome in aging, and its changes have been connected to human pathologies, including cardiometabolic diseases. In healthy adults, the gut microbiome is extremely diverse, with Bacteroidetes and Firmicutes being the most prevalent phyla. With aging, the Firmicutes to Bacteroidetes ratio and the overall diversity of the microbiome declines, possibly because of the expansion of distinct groups of bacteria. These changes are associated with declining immunocompetence and a higher risk of diabetes mellitus, atherosclerosis, neurodegenerative and liver diseases, and frailty.41,42 Dysbiosis of the gut microbiome impairs the integrity of the gut barrier, leading to leakage of bacteria and associated components into the circulation promoting chronic inflammation. Intestinal bacteria and the liver transform red meat components into trimethylamine N-oxide, whose blood level has been associated with CVD and CVD mortality.43
TELOMERE SHORTENING.
Telomeres are repetitive nucleotide sequences (TTAGGG) at the end of chromosomes that preserve chromosome integrity. In somatic cells, telomeric DNA is shortened at each cell division. The progressive shortening of telomeres in vitro causes a finite replicative lifespan, suggesting an important role in aging. Shorter telomeres in circulating leukocytes have been associated with higher risk of incident CVD such as myocardial infarction and stroke; however, other studies have failed to confirm such associations.44,45 Despite much focus on telomeres in aging, the relevance of telomere length as a marker of biological aging remains uncertain.
INFLAMMAGING
Inflammaging is the increase of proinflammatory molecules, such as IL-6 and c-reactive protein (CRP), with aging of and other in blood or tissues, paralleled by a blunted immune response to acute stimulation, such as vaccination. Consistent with the postulated interdependence between the mechanisms of aging, dysregulation of any reliance mechanisms of aging causes inflammation. For example, inflammation can be induced by dysfunction of mitochondria and defective mitophagy that leads to the release of oxidated cardiolipin and mitochondrial DNA, proteotoxicity that activates specific stress responses, or persistent DNA damage that activates responses that consume NAD+. In addition, inflammation curtails the regenerative capacity of stem cells. Thus, inflammation is a collective biomarker of the failure of biological resilience against damage accumulation with aging.46
Inflammation participates in atherosclerosis and affects multiple CVDs including myocardiopathy, late-life valvular dysfunction, and HF. Higher blood inflammatory markers predict heart attacks, cerebrovascular events, peripheral artery diseases, as well as multimorbidity and a steeper increase of multimorbidity and frailty.47 Finally, taming inflammation prevents cardiovascular events.
MEASURING BIOLOGICAL AGING
The geroscience hypothesis posits that slowing aging is maximally effective before the development of its clinical manifestations. Metrics of aging have been developed that combine various biomarkers and identify “fast aging” individuals and track the effect of interventions, such as antihypertensive medications, on the rate of aging.48 Ideal “aging clocks” correlate with chronologic age yet discriminate individuals that are aging “faster” or “slower” biologically.
The epigenetic clocks pioneered by Hannum49 and Horvath,50 as a weighted average of DNA methylation at specific CpGs, correlate with chronologic age and identify diverse adverse health outcomes, including frailty, CVD, CVD mortality, and all-cause mortality. Recent platforms, such as DNAm PhenoAge and GrimAge, were developed using health outcomes and age for reference, whereas pace of aging calculated from the epigenome (DunedinPACE) was tuned on longitudinal data.51 These DNA methylation-based tools also predict age-related outcomes, including CVD, with the most recent versions showing the highest reliability.
Aging clocks that use plasma proteomics can predict age-related outcomes with performances similar to those based on the methylome.35,52 A recently developed inflammatory clock of aging (iAge) tracked multimorbidity, immune system aging, frailty, and cardiovascular aging, with the strongest contributor being the chemokine CXCL9, which is involved in cardiac aging, adverse cardiac remodeling, and poor vascular function.53
GEROSCIENCE-GUIDED THERAPEUTIC APPROACHES TO CVD
This section considers some of the most promising nonpharmacologic and pharmacologic gerotherapeutics for the treatment of CVD, citing research in humans whenever possible. Figure 2 highlights the association of aging hallmarks to CVD. Figure 3 highlights that gerotherapeutic interventions may lead to pleotropic effects that vary from one person to another. Given the rapid growth of geroscience and the absence of U.S. Food and Drug Administration (FDA)-approved clinical therapies, we have prioritized interventions meeting 4 criteria: robust biological rationale, rooted in sound geroscience principles, relevance to CVDs, and early-stage support in human studies.
FIGURE 2.
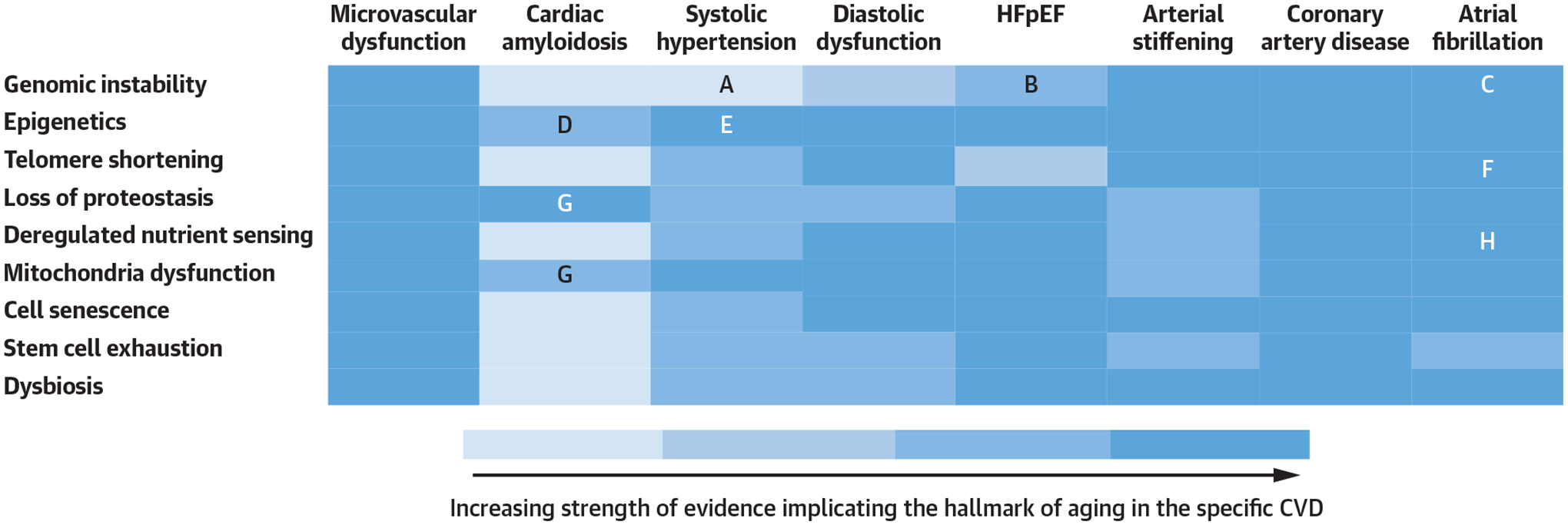
Aging Hallmarks and Cardiovascular Disease
Heat map providing a visual representation of the strength of evidence in the literature implicating hallmarks of aging in cardiovascular disease (CVD). See Supplemental Figure 1 for added detail regarding the associated references. A, Genomic stability and systolic hypertension. B, Genomic stability and heart failure with preserved ejection fraction (HFpEF). C, Genomic instability and atrial fibrillation. D, Epigenetics and amyloid. E, Epigenetics and systolic hypertension. F, Telomere modification and atrial fibrillation. G, Loss of proteostasis, mitochondrial dysfunction, and amyloid. H, Deregulated nutrient sensing and atrial fibrillation.
FIGURE 3.
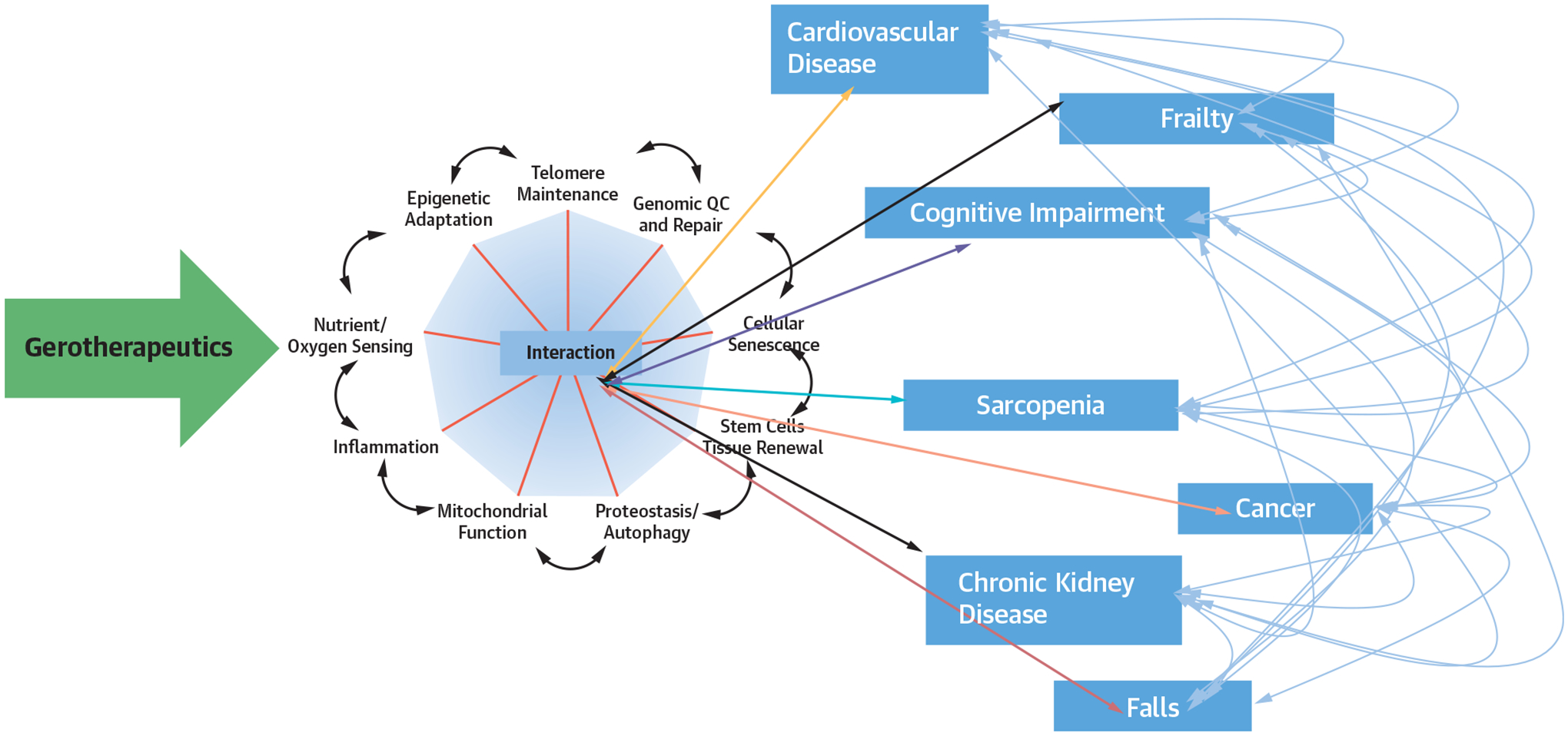
Pleotropic Effects of Gerotherapeutics
Aging mechanisms are interconnected such that an intervention targeting a single hallmark of aging or multiple hallmarks of aging also exerts downstream effects on varied other hallmarks of aging. As a result of such interactions among varied biological hallmarks of aging, an intervention targeting a single hallmark can exert variable effects on multiple chronic diseases and geriatric syndromes. An ideal gerotherapeutic intervention moderates multiple hallmarks to prevent disease and geriatric conditions. QC = quality control.
NONPHARMACOLOGIC INTERVENTIONS
EXERCISE AND PHYSICAL ACTIVITY.
Physical activity prevents or ameliorates chronic diseases, including atherosclerosis, hypertension, diabetes, CVD, and sarcopenia54,55; this is not surprising, as physical activity enhances multiple resilience mechanisms and affects core mechanisms of aging.
The molecular mechanisms by which exercise improves health are complex and are currently being determined by the NIH-funded Molecular Transducers of Physical Activity Consortium (MoTrPAC).56 In brief, acute aerobic exercise induces mild stress that increases the production ROS by mitochondria and triggers an inflammatory response, followed by a homeostatic response that enhances defense mechanisms. Aerobic exercise activates AMPK, sirtuin 1 (SIRT1), peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1-α, and Ak strain transforming (AKT) signaling in skeletal and cardiac muscle, thereby promoting mitochondrial biogenesis.57 Exercise also activates PI3-kinase, improving insulin and anabolic signaling (AKT/mTOR) in younger and older adults,58,59 and stimulating cardiac cell proliferation.57 Aerobic exercise reduces the SASP and improves production of nitric oxide and endothelial function, consequently lowering blood pressure and left ventricular load.57,60,61 Aerobic exercise may also reduce NF-κB, receptor for advanced glycation end products, TNFα, nicotinamide adenine dinucleotide phosphate (NADPH), monocyte chemo-attractant protein-1, and inducible nitric oxide synthase, whereas the effect on IL-6 is uncertain.
Acute resistance exercise activates satellite cells, mTOR signaling, and muscle protein synthesis,62 and long-term training improves hallmarks of senescence.63 Resistance training induces muscle hypertrophy by activating mTOR signaling, muscle protein anabolism, and stimulating satellite cell proliferation and activation. The positive effects of resistance exercise are reduced but not eliminated in old age.63
DIET.
A balanced diet is essential to prevent and treat CVD, obesity, diabetes, and sarcopenia in older adults. More than 41% of older Americans are obese, and approximately one-third of them have T2DM, a disease that significantly increases the risk of CVD.64,65 Diabetes and obesity accelerate aging and negatively affect most hallmarks of aging. Visceral adiposity promotes cellular senescence directly and through the production of adipokines and inflammatory mediators.66 In obese and overweight older individuals, weight loss lowers inflammatory biomarkers; improves glycemic control; and limits the risk of CVD, disability, and frailty. Adequate protein intake and resistance training should be part of any weight-loss regimen to mitigate lean mass loss and avoid disability and frailty.67
In animal models, caloric restriction (CR) in the absence of obesity is the most effective intervention at counteracting aging and promoting healthspan and increased longevity, eliciting positive effects on the core mechanisms of aging.68 Preliminary data in younger humans from the CALERIE (Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy) study suggest positive effects of CR on immune function, muscle quality, and cardiovascular risk profiles, as well as the ability to retard biological aging.69 In obese and overweight persons and in persons with or without CVD—including older individuals—a CR diet has many benefits, including increased longevity.70 In older persons with HF with preserved ejection fraction, CR improves function and quality of life.71 CR remains controversial for nonobese older adults because of insufficient evidence and malnutrition concerns.
Intermittent fasting and time-restricted eating have been proposed as alternative dietary approaches to CR to increase adherence and reduce risks of malnutrition.72 There is early evidence that intermittent fasting and time-restricted eating improve molecular markers of aging and cardiovascular risk factors73,74 such as low-density lipoprotein (LDL) cholesterol and systolic and diastolic blood pressure.75 A Mediterranean-type diet that includes a high proportion of fruit, vegetables, nuts, legumes, whole grains, fish, lean meats, moderate amounts dairy products, moderate intake of wine, and olive oil may reduce cardiovascular risk by limiting oxidative stress, inflammation, and LDL; improving insulin sensitivity and immune function; positively affecting the microbiome and genome stability76; and possibly inducing epigenetic rejuvenation.77
PHARMACOLOGIC INTERVENTIONS
The list of potential gerotherapeutic molecules grows every day, but their translation to clinical trials presents challenges that were recently addressed by the National Institute on Aging (NIA) Translational Geroscience Network (TGN).78 More than 40 clinical trials are underway of pharmacologic, lifestyle, and nutraceutical interventions that target aging hallmarks.79 Progress in the geroscience field has been hampered by the paucity of validated biomarkers that are easily measurable, reliably track biological aging,80 and dependably respond to interventions. Authenticated “gerodiagnostics” are currently being pursued, based on data from the TGN clinical trials through the Facility for Geroscience Analysis. As aging mechanisms are interconnected, an intervention targeting a single hallmark likely affects other hallmarks with mechanistic pleiotropism (Figure 3).
NICOTINAMIDE ADENINE DINUCLEOTIDE (NAD+).
NAD+ participates in DNA repair, mitochondrial biogenesis and function, and other facets of metabolism. NAD+ levels decline with aging, and such a decline contributes to several chronic conditions, including CVD. Raising NAD+ in mice improves both lifespan and healthspan and protects against vascular dysfunction and ischemic heart damage.81 Human studies testing the effectiveness of NAD precursors—such as niacinamide, nicotinamide mononucleotide, and nicotinamide riboside—in preventing or alleviating CVDs have recently begun. Early proof-of-concept clinical studies have shown good safety and tolerability of NAD precursors, with some studies showing improvement in muscle insulin sensitivity, blood pressure, and arterial compliance, as well as suppression of inflammatory activation of peripheral blood mononuclear cells from patients with HF.82 Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin.83 These promising early-stage human findings require replication in larger clinical trials, and questions remain about the optimal target population(s), type of “NAD booster,” dose regimens, length of treatment, and whether supplementation results in intracellular NAD+ repletion.
SIRTUINS AND REVERATROL.
Resveratrol, a natural phenol, and phytoalexin, which is found in the skin of grapes, blueberries, raspberries, mulberries, and peanuts, are leading modulators of sirtuin mechanisms.84 The presence of resveratrol in red wine has been proposed to explain the “French Paradox”: that is, that France has an incidence of CVDs lower than expected based on their high consumption of saturated fats. Clinical trials with either resveratrol or phytoalexin have shown little effect on CVD,85 although some benefits on blood pressure have been reported in subjects with diabetes.86
SENOLYTICS AND SENOMORPHICS.
Senolytic and senomorphic drugs may offset the deleterious effects of accumulation of senescent cells, thereby reducing CVD burden and mortality.79 Senolytics eliminate 30% to 70% of senescent cells that are proapoptotic, proinflammatory, and tissue damaging.87 Senolytics delay the onset; prevent, alleviate, or treat a range of age-associated disease features in cell culture models and animal experiments79,88; and selectively eliminate proinflammatory senescent cells in humans.89
First-generation senolytics include dasatinib, quercetin, fisetin, procyanidin C1, geldanamycin and related HSP-90 inhibitors, and navitoclax.90–93 Dasatinib, approved for adult leukemias and lymphomas, inhibits tyrosine kinases and ephrin receptors. Both mesenchymal and endothelial cell effects are pertinent to posited CVD-reducing benefits. Dasatinib is mostly active on senescent mesenchymal cells. In contrast, quercetin and fisetin are plant flavonols that mainly target senescent endothelial cells such as cultured human umbilical vein endothelial cells. Genome-wide association studies suggest that flavonoid consumption associates strongly with extreme longevity. Like the plant flavonols, navitoclax and the BCL-2 family inhibitors—A1331852 and A1155463—target mostly senescent endothelial cells.91,92 Dasatinib and quercetin used together are effective against a range of senescent mesenchymal and endothelial cells, including some senescent cell types that respond to neither alone.94
Studies have shown that senolytics enhance ejection fraction and vascular relaxation after nitroprus-side or acetylcholine administration in aged mice or ApoE−/− atherosclerotic mice fed a high fat diet95; reduce intimal calcification in high fat-fed ApoE−/− mice95; improve brain small vessel perfusion in a 23-month-old Tau+ Alzheimer mouse model96; decrease vascular senescent cells in mice with chronic kidney disease97; and improve regenerative capacity of the aged heart by ablating senescent cardiac pro-genitor cells.98 Elimination of senescent cells may slow down atherogenesis.99,100
Senomorphics inhibit release of SASP factors by senescent cells, suppressing or modulating their damaging effects.79 First-line senomorphics include rapamycin, metformin, resveratrol, and related drugs such as sirolimus and aspirin. Although they all suppress the SASP in cell lines and animal models and have health effects that—in some cases—resemble those of senolytics, the precise mechanism of action is unknown.101
METABOLIC DRUG THERAPIES.
Several of the most promising gerotherapeutics are repurposed drugs originally developed as hypoglycemic agents for T2DM.102 This is not surprising, as insulin-like growth factor (IGF) signaling is among the most deeply conserved and robustly validated mechanisms regulating aging. Insulin resistance and impaired glucose utilization are features of T2DM, among other diseases, and conditions of aging ranging from HF103 to neurodegeneration104 and frailty.3,105 Emerging evidence of benefits for CVD and chronic kidney diseases (CKD) in patients without T2DM, together with contrary data for other hypoglycemic agents, such as insulin and sulfonylureas, supports the hypothesis that hypoglycemia is not solely responsible for the beneficial effects and that other aging mechanisms beyond nutrient signaling pathways are likely more significant.106
Metformin.
Metformin is widely used to treat T2DM107 yet exerts beneficial effects on health well beyond a typical oral hypoglycemic.108 Metformin alters energy metabolism of the cell by inhibiting mitochondrial complex I and activating AMPK. Independent of its effect on AMPK, metformin affects mitochondrial function, cellular senescence, proteostasis, and autophagy via mammalian target of rapamycin complex (mTORC1) inhibition, and chronic inflammation via NFκB inhibition.108 The NIA Interventions Testing Program (ITP), which evaluates agents that extend lifespan or attenuate/delay age-related diseases by altering fundamental processes of aging, found that metformin increased mouse lifespan synergistically with rapamycin.109
In large observational studies, metformin reduced all-cause mortality of patients with diabetes compared with both nondiabetic patients and patients with diabetes not taking metformin,110 including in subgroups with CKD, HF, or chronic liver disease.111 Secondary analyses and observational studies in T2DM consistently found reduced mortality and improved cardiovascular outcomes, ranging from decreased HF hospitalizations and hypertension to reduced progression of aortic aneurysms, among patients taking metformin compared with alternatives.112 A small interventional study found that metformin diminishes left ventricular hypertrophy in patients with coronary artery disease without diabetes.113 Observational data also suggest, albeit less robustly, that metformin reduces cognitive decline114 and cancer risk,115 consistent with a broad gerotherapeutic effect.
The proposed TAME (Targeting Aging by Metformin) trial is the first randomized controlled trial (RCT) to test the geroscience hypothesis directly by examining whether a single agent can slow the incidence of age-related multimorbidity in older adults.106,116 Metformin was selected for this trial, as well as for the ongoing VA-IMPACT (Investigation of Metformin in Pre-Diabetes on Atherosclerotic Cardiovascular Outcomes Trial; NCT02915198), because of its safety profile and defined mechanism of action.116
SODIUM-GLUCOSE COTRANSPORTER 2 INHIBITORS.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors prevent hyperglycemia by blocking reuptake of glomerular filtered glucose in the renal tubule, thereby promoting glycosuria. SGLT2 inhibitors likely have pleiotropic effects on CVD that are independent of glycosuria.117 SGLT2 inhibitors received a high rank as gerotherapeutics in a systematic assessment of FDA-approved drugs based on published preclinical and clinical evidence.102 Experimentally, SGLT2 inhibitors enhance autophagy in renal glomeruli, improve mitochondrial function, activate AMPK, improve calcium homeostasis in cardiomyocytes, inhibit NLRP3 inflammatory activation, and suppress mTORC1 activity.102,118 Thus, by affecting global energy sensing, SGLT2 inhibitors promote a nutrient deprivation state that affects downstream, interconnected, aging mechanisms.117 Consistent with SGLT2 inhibitors serving as nutrient-signaling gerotherapeutics, canagliflozin was reported to extend median male mouse lifespan by 14% in the ITP.119
Clinical use of SGLT2 inhibitors has expanded rapidly from T2DM to HF and CKD. SGLT2 inhibitors reduce CKD progression in patients with120 or without121 coexisting T2DM, reduce HF hospitalizations in patients with HF but not T2DM,122 and reduce cardiovascular events in patients with T2DM.123 SGLT2 inhibitors also reduce all-cause mortality in patients with T2DM, CKD,121 or HF with reduced ejection fraction.124
Acarbose.
The α-glucosidase inhibitor acarbose impairs digestion of carbohydrates on the small intestine brush border and thereby reduces postprandial glucose absorption. It was among the first agents tested in the ITP, on the hypothesis that excessive postprandial hyperglycemia contributes to biological aging. Acarbose extends median lifespan in both male and female mice (by 22% and 5%, respectively)125 and increases lifespan synergistically with rapamycin.126 Acarbose likely functions as a nutrient-signaling regulator by modulating insulin/IGF pathways but might also interact with the proteostatic and epigenetic hallmarks of aging.102
In STOP-NIDDM (Study to Prevent Non-insulin Dependent Diabetes Mellitus), acarbose reduced the progression from impaired glucose tolerance to T2DM127 and lowered the incidence of both hypertension and major cardiovascular events. Observational studies support a reduction in cardiovascular events in patients with T2DM treated with acarbose.128 Although the more recent Acarbose Cardiovascular Evaluation trial in patients with both coronary artery disease and impaired glucose tolerance replicated the effect on prevention of diabetes,129 the study found no difference in cardiovascular events or all-cause mortality, and a recent meta-analysis of trials involving administration of acarbose were inconclusive for cardiovascular events and all-cause mortality.130 Overall, acarbose has strong preclinical data and a mechanistic basis as a gerotherapeutic but is comparatively understudied in the clinic compared with metformin and SLGT2 inhibitors.
GLP-1 agonists.
Similar to metformin and SGLT2 inhibitors, GLP-1 agonists improve insulin sensitivity and metabolic function and act as broad gerotherapeutics by interfering with the nutrient signaling mechanisms. Both preclinical and clinical evidence for GLP-1 agonists as gerotherapeutics are less well developed but are expanding rapidly.131 In animal models, GLP-1 agonists reduce oxidative stress, improve mitochondrial function, inhibit cellular senescence, and modulate mTOR activity.132
Trials of GLP-1 agonists consistently report reductions in cardiovascular events and mortality in patients with T2DM,133–135 and a few studies found improved secondary outcomes related to CKD as well.133,135 In a Cochrane meta-analysis, GLP-1 agonists, as well as SGLT2 inhibitors but not dipeptidyl peptidase (DPP)-4 inhibitors, reduced cardiovascular events, cardiovascular mortality, and all-cause mortality in patients with both T2DM and CVD.136 There are not yet similar data on GLP-1 targeting in patients without T2DM, and it is currently unknown whether these drugs positively affect other age-related outcomes.
Ketone body-related therapies.
Ketone bodies furnish energy that bypasses the insulin/glucose axis and provide a fat-derived, alternative substrate for cellular ATP generation. Thus, exogenously administered ketone bodies may help compensate for aging-or disease-related impairments in glucose utilization, as seen in neurodegenerative disease104 or HF.137 Ketone bodies affect several mechanisms of aging including reducing chronic inflammation via NLRP3 inhibition138 and microbiome modulation139; regulating epigenetic modifications via histone deacetylase inhibition; preventing endothelial senescence by stabilizing an Oct4 RNA-binding protein; and enhancing stem-cell function.140,141 Promoting endogenous ketogenesis via a carbohydrate-restricted ketogenic diet increased lifespan in 2 independent mouse studies.142 However, the ketogenic small molecule 1,3-butanediol produced indeterminate results when tested in the ITP because of exceptionally reduced longevity in the control animals.126
The recent development of ketogenic small molecules enables ketone body administration without dietary changes. For example, acute ketone body administration improved cardiac hemodynamic measures in both patients with HF143 and healthy adults.144 Long-term studies are not yet completed, and ketone body therapies remain investigational for CVD.
TOR inhibitors.
TOR inhibition has garnered the most rigorous experimental support as a gerotherapeutic intervention, showing the most robust and reproducible effect of any pharmacologic agent in extending lifespan in animals. However, clinical trial data relevant to aging applications are at an earlier stage: for example compared with metformin and SGLT2 inhibitors. TOR emerged early as a critical nutrient-sensing complex that mediates the longevity benefits of CR.145 TOR inhibition reduces protein synthesis and activates autophagy, whereas downstream effects include enhancement of many of the resilience mechanisms of aging.145,146 The TOR inhibitor rapamycin was among the first drugs found to extend healthy lifespan in the ITP, in both male and female mice.147 This lifespan extension was associated with reduced age-related changes in several tissues, including cardiac hypertrophy and other cardiovascular outcomes.146 Replicative ITP studies showed a dose-dependent median lifespan extension of up to 23% in males and 26% in females,148 with efficacy starting as late as 20 months old (equivalent to approximately the seventh decade in humans),149 and synergistic effects were observed with both metformin109 and acarbose.126 Preliminary findings of a rapamycin study show an improvement in age-related cardiac function in middle-aged companion dogs.150
TOR inhibitors such as sirolimus and everolimus are FDA approved as post-transplantation immuno-suppressants and as therapies for certain inherited disorders and cancers associated with TOR hyper-activation. However, the doses used for immuno-suppression or cancer therapy are 10-fold to 80-fold higher than those relevant to aging studies. This limitation is unlike the situation with metformin and SGLT2 inhibitors in which the doses for T2DM or CVD treatment resemble those required for geroprotection.
Anti-inflammatory therapies.
The CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) proved that inflammation participates causally in CVD pathogenesis. In CANTOS, neutralizing IL-1β with the monoclonal antibody canakinumab for over a median of 3.7 years in >10,000 patients reduced the risk of CVD events, especially in participants who showed a greater than median decline in the inflammatory markers CRP and IL-6. Canakinumab caused a small but statistically significant increase in infections,151 consistent with the role of IL-6 in immunity. Exploratory results from CANTOS also revealed reduced lung cancer mortality,152 anemia,153 gout, hospitalizations for HF,154 and replacements of the hip or knee.
The anti-inflammatory drug colchicine has shown effectiveness in prevention of secondary CVD. Among patients with recent myocardial infarctions, colchicine at a dose of 0.5 mg daily significantly lowered the risk of ischemic cardiovascular events. In a randomized trial involving patients with chronic coronary artery disease, colchicine also lowered the risk of cardiovascular events. Similar benefits of colchicine have been demonstrated for other CVDs as well.155 Whether colchicine is effective against additional age-related outcomes is unknown. Anti-inflammatory benefits have also been associated with angiotensin receptor blockers, with decreased endothelial and vascular senescence.156 Notably, other anti-inflammatory treatments were not very effective in preventing age-related outcomes. For example, low-dose weekly administration of methotrexate did not improve cardiovascular outcomes in a large trial.157 In the Aspirin in Reducing Events in the Elderly trial, the daily administration of aspirin in healthy older adults did not lower all-cause mortality and had no substantial effect on the risk of mobility loss.
NOVEL BIOMARKERS FOR GUIDING RESEARCH FOR CVD IN RELATION TO AGING
Clinical trials that target the effects of biological aging on CVD would be maximally informative if biomarkers specific for each aging hallmark could be measured. However, the development of effective biomarkers is still a work in progress, with few being validated in humans and none having entered routine clinical use. The investigators for the TAME trial80 evaluated the potential of 258 serum biomarkers using as criteria measurement reliability and feasibility; relevance to aging; ability to robustly predict all-cause mortality and clinical and functional outcomes; and responsiveness to the intervention. The final list generated included IL-6, TNFα-receptor I or II, CRP, growth differentiation factor (GDF) 15, insulin, IGF1, cystatin C, N-terminal pro–B-type natriuretic peptide (NT-proBNP), and hemoglobin A1c. These 9 biomarkers have now been incorporated into ongoing gerotherapeutic trials.
Another approach is to develop composite scores of biomarkers or “gerodiagnostics” rather than focusing on individual biomarkers. Such composite scores incorporate assays of multiple hallmarks of aging and appear to respond to interventions such as senolytics in humans.158 Ideally, biomarkers of composite gerodiagnostic scores would not just reflect “biological age” but would predict age-related health outcomes independent of chronologic age; be measurable in body fluids such as urine, saliva, or blood; be inexpensive, reliable, and reproducible; and be sensitive to the effect(s) of geroscience interventions.
Research on biomarkers for use in clinical trials that target the mechanisms of aging and CVD has benefitted from technologic advances, such as proteomic methods that measure thousands of bio-molecules in blood and tissue samples. The ability to assess stored biospecimens with increasing numbers of markers permits continued expansion and refinement of candidates that may reveal “ideal” biomarkers that report on the state or function of the aging hallmarks. The TGN strives to standardize geroscience biomarker analyses across disparate clinical trials and provides a centralized biospecimen repository.159 Similarly important, it strives to standardize clinical outcome measures that are relevant in aging such as activities of daily living and independent activities of daily living, gait speed, 6-minute walking test, short form physical battery, and frailty indices.160
THE TRANSFORMATIONAL IMPLICATIONS OF GEROSCIENCE-BASED CVD MANAGEMENT
Geroscience-based interventions are supposed to slow or delay aging-related chronic diseases (eg, CVD) and geriatric syndromes and improve general health.78,161 However, this hypothesis requires rigorous testing on a large-scale RCT basis. Geroscience interventions may substantially affect social determinants of health (SDOH),162 as income, wealth, and ZIP codes can powerfully predict longevity in the United States.163 SDOH disparities manifest as increased rates of chronic diseases, such as CVD, as well as an earlier presentation of geriatric syndromes like falls and impairment of mobility. As SDOH disparities can strongly influence biological mechanisms of aging,164 geroscience-based pharmacologic interventions that target prevention of CVD may prevent other SDOH-accelerated chronic diseases as well and with an earlier effect relative to individuals with favorable SDOH.
SUMMARY, GAPS, AND FUTURE DIRECTION
As we are faced with a growing aging population worldwide, gerotherapeutics have the ambitious goal of transforming health care. If we continue to intervene when pathology becomes clinically evident, we may only extend the period of life characterized by disease and disability but fail to extend healthspan: the period of life most enjoyable and productive. Success in promoting healthspan not only addresses the burden of aging on human life but may also come with enormous economic benefits. Compression of morbidity because of gerotherapeutics is estimated to provide a $37 trillion per year return.165 Medical practitioners focused on CVD, as well as other specialties, should strive to implement a more fundamental approach to prevent or mitigate multiple diseases, frailty, and other health impairments at all stages of life. Cardiovascular specialists have a privileged role in this process, as they have already embraced concepts of primary and secondary prevention, and cardiovascular conditions contribute enormously to the challenges of older age.
Supplementary Material
HIGHLIGHTS.
Reduced efficiency of mechanisms opposing age-related molecular damage increases susceptibility to cardiovascular disease.
Key resilience mechanisms associated with aging have been identified as potential therapeutic targets.
By targeting these mechanisms, geroscience could transform the diagnosis, prevention, treatment, and prognosis of cardiovascular disease in an aging population.
ACKNOWLEDGMENT
The authors are very grateful to David M. Wilson III for the many suggestions and meticulous editing of this manuscript.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
Dr Forman has received funding support from National Institute on Aging (NIA) 1R01 AG058883, R01 AG060499, U19AG065188, R01 AG073633, R01 AG077179, and P30AG024827; VA RR&D 1I21RX004409 and HSR&D1 I01 HX003518; and PCORI IHS-2021C3-24147. Dr Kuchel has received funding support from NIA P30 AG067988, U54 AG075941, R25AG073119, R33AG061456, R01AG058814, R01AG051647, and R01AG075271; NIAID U01 AI165452 and R01AI142086; and PCORI IHS-1502-27171. Dr Newman is cofounder and shareholder in BHB Therapeutics Ltd and Selah Therapeutics, Ltd, which develop products related to ketone bodies; and has received funding support from NIA R01 AG067333, NIA R01 AG068025, NIA R25AG073119, Longevity Impetus, Department of Defense PRMRP W81XWH2210867, and Buck institutional funds. Dr Kirkland has financial interest related to this research, including patents and pending patents covering senolytic drugs and their uses that are held by Mayo Clinic (this research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic conflict of interest policies); and has received funding support from NIH R37AG013925, R33AG061456, R01AG68048, R01AG 64165, and P01AG062413, the Connor Fund, Robert J. and Theresa W. Ryan Fund, and the Noaber Foundation. Dr Volpi has served on the Longeveron Scientific Advisory Board; has received funding from NIH P30 AG024832, R01 AG049611, R01 AG057732, and the Dairy Research Institute DMI 2859; has served as site PI for NIH R01AG078153, U19 AG062682, R01 AG0688, PCORI PCS-2017C1-6534, and Metro International Biotech, LLC; and has served as co-I for NIH UL1 TR001439, U01 AR071150, R01 AG064092. Dr Taffet has received honoraria from Boehringer Ingelheim and Novartis (Switzerland); holds intellectual property in Animatus Biosciences, and Uptodate; and has received funding support from NIA and is co-Investigator on R01AG068260, R01AG059599, and R01AG054131. Dr Barzilai has received funding from the NIA (P30AG038072) and the American Federation for Aging Research (Scientific Director). Dr Pandey has received honoraria from Applied Therapeutics, Roche, SC Pharmaceuticals, and Gilead Sciences; has served in advisory and consultant roles for Tricog Health Inc, Lilly USA, Rivus, Cytokinetics, Emmi Solutions, Axon Therapies, Sarfez Pharmaceuticals, Alleviant Medical, Palomarin Inc, Pieces Technologies, and Roche Diagnostics; has received nonfinancial support from Pfizer and Merck; and has received funding support from the NIA GEMSSTAR Grant (1R03AG067960-01), and the National Institute on Minority Health and Disparities (R01MD017529). Dr Kitzman has received honoraria as a consultant for Bayer, Merck, Corvia Medical, Boehringer Ingelheim, Ketyo, Rivus, NovoNordisk, AstraZeneca, Pfizer, and Novartis; has received grant funding from Novartis, Bayer, NovoNordisk, Rivus, Pfizer, and AstraZeneca; has stock ownership in Gilead Sciences; and has received funding support from the Kermit Glenn Phillips II Chair in Cardiovascular Medicine, and NIH grants U01AG076928, R01AG078153, R01AG045551, R01AG18915, P30AG021332, U24AG059624, and U01HL160272. Dr Libby is an unpaid consultant to or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Moderna, Novo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron; is a member of the scientific advisory board for Amgen, Caristo Diagnostics, Cartesian Therapeutics, CSL Behring, DalCor Pharmaceuticals, Eulicid Bioimaging, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, TenSixteen Bio, Soley Thereapeutics, and XBiotech, Inc; his laboratory has received research funding in the last 2 years from Novartis, Novo Nordisk and Genentech; is on the Board of Directors of XBiotech, Inc; has a financial interest in Xbiotech, a company developing therapeutic human antibodies, in TenSixteen Bio, a company targeting somatic mosaicism and clonal hematopoiesis of indeterminate potential (CHIP) to discover and develop novel therapeutics to treat age-related diseases, and in Soley Therapeutics, a biotechnology company that is combining artificial intelligence with molecular and cellular response detection for discovering and developing new drugs, currently focusing on cancer therapeutics; his interests were reviewed and are managed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their conflict-of-interest policies; and receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892 and 1R01HL163099-01), the RRM Charitable Fund, and the Simard Fund. Dr Ferrucci has received funding support through the Intramural Research Program of the National Institute on Aging, NIH.
ABBREVIATIONS AND ACRONYMS
- AMPK
AMP-activated protein kinase
- CHIP
clonal hematopoiesis of indeterminate potential
- CKD
chronic kidney disease
- CpG
cytosine-guanidine sequence
- CVD
cardiovascular disease
- FDA
U.S. Food and Drug Administration
- HF
heart failure
- ITP
Interventions Testing Program
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- NAD
nicotinamide adenine dinucleotide
- NF-κB
nuclear factor kappa light chain enhancer of activated B cells
- NLRP3
NACHT, LRR, and PYD domains-containing protein 3
- SASP
secretory phenotype
- SDOH
social determinants of health
- SGLT2
sodium-glucose co-transporter 2
- T2DM
type 2 diabetes mellitus
- TGN
Translational Geroscience Network
- TNF
tumor necrosis factor
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
APPENDIX For the supplemental figure, please see the online version of this paper.
REFERENCES
- 1.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afilalo J, Alexander KP, Mack MJ, et al. Frailty assessment in the cardiovascular care of older adults. J Am Coll Cardiol. 2014;63(8):747–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fried LP, Cohen AA, Xue QL, Walston J, Bandeen-Roche K, Varadhan R. The physical frailty syndrome as a transition from homeostatic symphony to cacophony. Nat Aging. 2021;1(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forman DE, Maurer MS, Boyd C, et al. Multimorbidity in older adults with cardiovascular disease. J Am Coll Cardiol. 2018;71(19):2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forman DE, Rich MW, Alexander KP, et al. Cardiac care for older adults: time for a new paradigm. J Am Coll Cardiol. 2011;57(18):1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Damluji AA, Chung SE, Xue QL, et al. Physical frailty phenotype and the development of geriatric syndromes in older adults with coronary heart disease. Am J Med. 2021;134(5):662–671.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–278. [DOI] [PubMed] [Google Scholar]
- 8.Oude Voshaar RC, Jeuring HW, Borges MK, et al. Course of frailty stratified by physical and mental multimorbidity patterns: a 5-year follow-up of 92,640 participants of the LifeLines cohort study. BMC Med. 2021;19(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiwari V, Wilson DM 3rd. DNA damage and associated DNA repair defects in disease and premature aging. Am J Hum Genet. 2019;105(2):237–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durik M, Kavousi M, van der Pluijm I, et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation. 2012;126(4):468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Flores M, Marcos-Perez D, Lorenzo-Lopez L, et al. Frailty syndrome and genomic instability in older adults: suitability of the cytome micronucleus assay as a diagnostic tool. J Gerontol A Biol Sci Med Sci. 2018;73(7):864–872. [DOI] [PubMed] [Google Scholar]
- 12.Natarajan P Genomic aging, clonal hematopoiesis, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2023;43(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zekavat SM, Viana-Huete V, Matesanz N, et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat Cardiovasc Res. 2023;2:144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sano S, Horitani K, Ogawa H, et al. Hematopoietic loss of Y chromosome leads to cardiac fibrosis and heart failure mortality. Science. 2022;377(6603):292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–499. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Song M, Qu J, Liu GH. Epigenetic modifications in cardiovascular aging and diseases. Circ Res. 2018;123(7):773–786. [DOI] [PubMed] [Google Scholar]
- 17.Turunen MP, Aavik E, Yla-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta. 2009;1790(9):886–891. [DOI] [PubMed] [Google Scholar]
- 18.Westerman K, Sebastiani P, Jacques P, Liu S, DeMeo D, Ordovas JM. DNA methylation modules associate with incident cardiovascular disease and cumulative risk factor exposure. Clin Epigenetics. 2019;11(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2): 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greissel A, Culmes M, Burgkart R, et al. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. 2016;25(2):79–86. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Li T, Li Z, Liu N, Yan Y, Liu B. Role of mitophagy in cardiovascular disease. Aging Dis. 2020;11(2):419–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips SM. Physiologic and molecular bases of muscle hypertrophy and atrophy: impact of resistance exercise on human skeletal muscle (protein and exercise dose effects). Appl Physiol Nutr Metab. 2009;34(3):403–410. [DOI] [PubMed] [Google Scholar]
- 23.Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368(6489):eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brundel BJ, Shiroshita-Takeshita A, Qi X, et al. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99(12):1394–1402. [DOI] [PubMed] [Google Scholar]
- 25.Rigopoulos AG, Ali M, Abate E, et al. Advances in the diagnosis and treatment of transthyretin amyloidosis with cardiac involvement. Heart Fail Rev. 2019;24(4):521–533. [DOI] [PubMed] [Google Scholar]
- 26.Madrigal-Matute J, de Bruijn J, van Kuijk K, et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc Natl Acad Sci U S A. 2022;119(14):e2121133119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson SC, Sangesland M, Kaeberlein M, Rabinovitch PS. Modulating mTOR in aging and health. Interdiscip Top Gerontol. 2015;40:107–127. [DOI] [PubMed] [Google Scholar]
- 28.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–241. [DOI] [PubMed] [Google Scholar]
- 29.Kane AE, Sinclair DA. Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ Res. 2018;123(7): 868–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zampino M, Spencer RG, Fishbein KW, Simonsick EM, Ferrucci L. Cardiovascular health and mitochondrial function: testing an association. J Gerontol A Biol Sci Med Sci. 2021;76(2):361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker KA, Basisty N, Wilson DM 3rd, Ferrucci L. Connecting aging biology and inflammation in the omics era. J Clin Invest. 2022;132(14):e158448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basisty N, Kale A, Jeon OH, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1):e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100(1):15–26. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka T, Basisty N, Fantoni G, et al. Plasma proteomic biomarker signature of age predicts health and life span. Elife. 2020;9:e61073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21(6):354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. [DOI] [PubMed] [Google Scholar]
- 38.McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217(1):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee PJ, Benz CC, Blood P, et al. NIH SenNet Consortium to map senescent cells throughout the human lifespan to understand physiological health. Nat Aging. 2022;2(12):1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fadini GP, Mehta A, Dhindsa DS, et al. Circulating stem cells and cardiovascular outcomes: from basic science to the clinic. Eur Heart J. 2020;41(44):4271–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haran JP, McCormick BA. Aging, frailty, and the microbiome-how dysbiosis influences human aging and disease. Gastroenterology. 2021;160(2): 507–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim MY, Hong S, Kim JH, Nam YD. Association between gut microbiome and frailty in the older adult population in Korea. J Gerontol A Biol Sci Med Sci. 2021;76(8):1362–1368. [DOI] [PubMed] [Google Scholar]
- 43.Senthong V, Wang Z, Li XS, et al. Intestinal microbiota-generated metabolite trimethylamine-n-oxide and 5-year mortality risk in stable coronary artery disease: the contributory role of intestinal microbiota in a COURAGE-like patient cohort. J Am Heart Assoc. 2016;5(6):e002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moslehi J, DePinho RA, Sahin E. Telomeres and mitochondria in the aging heart. Circ Res. 2012;110(9):1226–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Meyer T, Nawrot T, Bekaert S, De Buyzere ML, Rietzschel ER, Andres V. Telomere length as cardiovascular aging biomarker: JACC Review Topic of the Week. J Am Coll Cardiol. 2018;72(7):805–813. [DOI] [PubMed] [Google Scholar]
- 46.Varadhan R, Yao W, Matteini A, et al. Simple biologically informed inflammatory index of two serum cytokines predicts 10 year all-cause mortality in older adults. J Gerontol A Biol Sci Med Sci. 2014;69(2):165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrucci L, Gonzalez-Freire M, Fabbri E, et al. Measuring biological aging in humans: a quest. Aging Cell. 2020;19(2):e13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2): 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10): R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belsky DW, Caspi A, Corcoran DL, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife. 2022;11:e73420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehallier B, Gate D, Schaum N, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med. 2019;25(12):1843–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sayed N, Huang Y, Nguyen K, et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat Aging. 2021;1:598–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neufer PD, Bamman MM, Muoio DM, et al. Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. 2015;22(1):4–11. [DOI] [PubMed] [Google Scholar]
- 55.Lee DC, Artero EG, Sui X, Blair SN. Mortality trends in the general population: the importance of cardiorespiratory fitness. J Psychopharmacol. 2010;24(4 Suppl):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanford JA, Nogiec CD, Lindholm ME, et al. Molecular Transducers of Physical Activity Consortium (MoTrPAC): mapping the dynamic responses to exercise. Cell. 2020;181(7):1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bo B, Zhou Y, Zheng Q, Wang G, Zhou K, Wei J. The molecular mechanisms associated with aerobic exercise-induced cardiac regeneration. Bio-molecules. 2020;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujita S, Rasmussen BB, Cadenas JG, et al. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes. 2007;56(6):1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Timmerman KL, Dhanani S, Glynn EL, et al. A moderate acute increase in physical activity enhances nutritive flow and the muscle protein anabolic response to mixed nutrient intake in older adults. Am J Clin Nutr. 2012;95(6):1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cartee GD, Hepple RT, Bamman MM, Zierath JR. Exercise promotes healthy aging of skeletal muscle. Cell Metab. 2016;23(6):1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adams V, Reich B, Uhlemann M, Niebauer J. Molecular effects of exercise training in patients with cardiovascular disease: focus on skeletal muscle, endothelium, and myocardium. Am J Physiol Heart Circ Physiol. 2017;313(1):H72–H88. [DOI] [PubMed] [Google Scholar]
- 62.Walker DK, Fry CS, Drummond MJ, et al. PAX7+ satellite cells in young and older adults following resistance exercise. Muscle Nerve. 2012;46(1):51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lavin KM, Roberts BM, Fry CS, Moro T, Rasmussen BB, Bamman MM. The importance of resistance exercise training to combat neuromuscular aging. Physiology (Bethesda). 2019;34(2): 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prevalence of Both Diagnosed and Undiag-nosed Diabetes. Centers for Disease Control and Prevention. 2021. https://www.cdc.gov/diabetes/data/statistics-report/diagnosed-undiagnosed-diabetes.html [Google Scholar]
- 65.Nutrition, Physical Activity, and Obesity: Data, Trends and Maps. Centers for Disease Control and Prevention. 2021. https://nccd.cdc.gov/dnpao_dtm/rdPage.aspx?rdReport=DNPAO_DTM.ExploreByTopic&islClass=OWS&islTopic=&go=GO [Google Scholar]
- 66.Koster A, Stenholm S, Alley DE, et al. Body fat distribution and inflammation among obese older adults with and without metabolic syndrome. Obesity (Silver Spring). 2010;18(12):2354–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim JE, O’Connor LE, Sands LP, Slebodnik MB, Campbell WW. Effects of dietary protein intake on body composition changes after weight loss in older adults: a systematic review and meta-analysis. Nutr Rev. 2016;74(3):210–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattison JA, Colman RJ, Beasley TM, et al. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waziry R, Ryan CP, Corcoran DL, et al. Effect of long-term caloric restriction on DNA methylation measures of biological aging in healthy adults from the CALERIE trial. Nat Aging. 2023;3(3):248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kritchevsky SB, Beavers KM, Miller ME, et al. Intentional weight loss and all-cause mortality: a meta-analysis of randomized clinical trials. PLoS One. 2015;10(3):e0121993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kitzman DW, Brubaker P, Morgan T, et al. Effect of caloric restriction or aerobic exercise training on peak oxygen consumption and quality of life in obese older patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2016;315(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duregon E, Pomatto-Watson L, Bernier M, Price NL, de Cabo R. Intermittent fasting: from calories to time restriction. Geroscience. 2021;43(3):1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stekovic S, Hofer SJ, Tripolt N, et al. Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 2019;30(3):462–476.e6. [DOI] [PubMed] [Google Scholar]
- 74.Guo Y, Luo S, Ye Y, Yin S, Fan J, Xia M. Intermittent fasting improves cardiometabolic risk factors and alters gut microbiota in metabolic syndrome patients. J Clin Endocrinol Metab. 2021;106(1):64–79. [DOI] [PubMed] [Google Scholar]
- 75.Kraus WE, Bhapkar M, Huffman KM, et al. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7(9):673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tosti V, Bertozzi B, Fontana L. Health benefits of the Mediterranean diet: metabolic and molecular mechanisms. J Gerontol A Biol Sci Med Sci. 2018;73(3):318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gensous N, Garagnani P, Santoro A, et al. One-year Mediterranean diet promotes epigenetic rejuvenation with country- and sex-specific effects: a pilot study from the NU-AGE project. Geroscience. 2020;42(2):687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sierra F, Caspi A, Fortinsky RH, et al. Moving geroscience from the bench to clinical care and health policy. J Am Geriatr Soc. 2021;69(9):2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28(8):1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Justice JN, Ferrucci L, Newman AB, et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience. 2018;40(5–6):419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotin-amide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou B, Wang DD, Qiu Y, et al. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J Clin Invest. 2020;130(11): 6054–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vreones M, Mustapic M, Moaddel R, et al. Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell. 2023;22(1):e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jasinski M, Jasinska L, Ogrodowczyk M. Resveratrol in prostate diseases: a short review. Cent European J Urol. 2013;66(2):144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tome-Carneiro J, Gonzalvez M, Larrosa M, et al. Resveratrol in primary and secondary prevention of cardiovascular disease: a dietary and clinical perspective. Ann N Y Acad Sci. 2013;1290: 37–51. [DOI] [PubMed] [Google Scholar]
- 86.Fogacci F, Tocci G, Presta V, Fratter A, Borghi C, Cicero AFG. Effect of resveratrol on blood pressure: a systematic review and meta-analysis of randomized, controlled, clinical trials. Crit Rev Food Sci Nutr. 2019;59(10):1605–1618. [DOI] [PubMed] [Google Scholar]
- 87.Tripathi U, Misra A, Tchkonia T, Kirkland JL. Impact of senescent cell subtypes on tissue dysfunction and repair: importance and research questions. Mech Ageing Dev. 2021;98:111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wyles SP, Tchkonia T, Kirkland JL. Targeting cellular senescence for age-related diseases: path to clinical translation. Plast Reconstr Surg. 2022;150:20S–26S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hickson LJ, Langhi Prata LGP, Bobart SA, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Q, Fu Q, Li Z, et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat Metab. 2021;3(12):1706–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9(3):955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles heel of senescent cells: from tran-scriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020;288(5):518–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vaso-motor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6): e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nath KA, O’Brien DR, Croatt AJ, et al. The murine dialysis fistula model exhibits a senescence phenotype: pathobiological mechanisms and therapeutic potential. Am J Physiol Renal Physiol. 2018;315(5):F1493–F1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. 2019;18(3):e12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garrido AM, Kaistha A, Uryga AK, et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc Res. 2022;118(7):1713–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J. 2022;290(5):1362–1383. [DOI] [PubMed] [Google Scholar]
- 102.Kulkarni AS, Aleksic S, Berger DM, Sierra F, Kuchel GA, Barzilai N. Geroscience-guided repurposing of FDA-approved drugs to target aging: a proposed process and prioritization. Aging Cell. 2022;21(4):e13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Honka H, Solis-Herrera C, Triplitt C, Norton L, Butler J, DeFronzo RA. Therapeutic manipulation of myocardial metabolism: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;77(16):2022–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cunnane SC, Trushina E, Morland C, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19(9):609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146–M156. [DOI] [PubMed] [Google Scholar]
- 106.Justice JN, Niedernhofer L, Robbins PD, et al. Development of clinical trials to extend healthy lifespan. Cardiovasc Endocrinol Metab. 2018;7(4): 80–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Flory J, Lipska K. Metformin in 2019. JAMA. 2019;321(19):1926–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020;32(1):15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Strong R, Miller RA, Antebi A, et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell. 2016;15(5): 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. 2017;40:31–44. [DOI] [PubMed] [Google Scholar]
- 111.Crowley MJ, Diamantidis CJ, McDuffie JR, et al. Clinical outcomes of metformin use in populations with chronic kidney disease, congestive heart failure, or chronic liver disease: a systematic review. Ann Intern Med. 2017;166(3):191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang K, Yang W, Dai H, Deng Z. Cardiovascular risk following metformin treatment in patients with type 2 diabetes mellitus: results from meta-analysis. Diabetes Res Clin Pract. 2020;160: 108001. [DOI] [PubMed] [Google Scholar]
- 113.Mohan M, Al-Talabany S, McKinnie A, et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: the MET-REMODEL trial. Eur Heart J. 2019;40(41):3409–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Samaras K, Makkar S, Crawford JD, et al. Metformin use is associated with slowed cognitive decline and reduced incident dementia in older adults with type 2 diabetes: the Sydney Memory and Ageing Study. Diabetes Care. 2020;43(11): 2691–2701. [DOI] [PubMed] [Google Scholar]
- 115.Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res (Phila). 2014;7(9):867–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Newman JC, Milman S, Hashmi SK, et al. Strategies and challenges in clinical trials targeting human aging. J Gerontol A Biol Sci Med Sci. 2016;71(11):1424–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Packer M Critical reanalysis of the mechanisms underlying the cardiorenal benefits of SGLT2 inhibitors and reaffirmation of the nutrient deprivation signaling/autophagy hypothesis. Circulation. 2022;146(18):1383–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a State-of-the-Art Review. J Am Coll Cardiol Basic Trans Science. 2020;5(6):632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miller RA, Harrison DE, Allison DB, et al. Canagliflozin extends life span in genetically heterogeneous male but not female mice. JCI Insight. 2020;5(21):e140019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–2306. [DOI] [PubMed] [Google Scholar]
- 121.Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–1446. [DOI] [PubMed] [Google Scholar]
- 122.Packer M, Anker SD, Butler J, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383(15):1413–1424. [DOI] [PubMed] [Google Scholar]
- 123.Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644–657. [DOI] [PubMed] [Google Scholar]
- 124.McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381(21):1995–2008. [DOI] [PubMed] [Google Scholar]
- 125.Harrison DE, Strong R, Allison DB, et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014;13(2):273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Strong R, Miller RA, Cheng CJ, et al. Lifespan benefits for the combination of rapamycin plus acarbose and for captopril in genetically heterogeneous mice. Aging Cell. 2022;21(12):e13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet. 2002;359(9323):2072–2077. [DOI] [PubMed] [Google Scholar]
- 128.Zhang XL, Yuan SY, Wan G, et al. The effects of acarbose therapy on reductions of myocardial infarction and all-cause death in T2DM during 10-year multifactorial interventions (The Beijing Community Diabetes Study 24). Sci Rep. 2021;11(1):4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Holman RR, Coleman RL, Chan JCN, et al. Effects of acarbose on cardiovascular and diabetes outcomes in patients with coronary heart disease and impaired glucose tolerance (ACE): a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5(11):877–886. [DOI] [PubMed] [Google Scholar]
- 130.Mannucci E, Gallo M, Pintaudi B, et al. All-cause mortality and cardiovascular events in patients with type 2 diabetes treated with alpha-glucosidase inhibitors: a meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. 2022;32(2):511–514. [DOI] [PubMed] [Google Scholar]
- 131.Brown E, Heerspink HJL, Cuthbertson DJ, Wilding JPH. SGLT2 inhibitors and GLP-1 receptor agonists: established and emerging indications. Lancet. 2021;398(10296):262–276. [DOI] [PubMed] [Google Scholar]
- 132.Peng W, Zhou R, Sun ZF, Long JW, Gong YQ. Novel insights into the roles and mechanisms of GLP-1 receptor agonists against aging-related diseases. Aging Dis. 2022;13(2):468–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19): 1834–1844. [DOI] [PubMed] [Google Scholar]
- 135.Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394(10193):121–130. [DOI] [PubMed] [Google Scholar]
- 136.Kanie T, Mizuno A, Takaoka Y, et al. Dipeptidyl peptidase-4 inhibitors, glucagon-like peptide 1 receptor agonists and sodium-glucose co-transporter-2 inhibitors for people with cardiovascular disease: a network meta-analysis. Cochrane Database Syst Rev. 2021;10(10):CD013650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yurista SR, Chong CR, Badimon JJ, Kelly DP, de Boer RA, Westenbrink BD. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;77(13):1660–1669. [DOI] [PubMed] [Google Scholar]
- 138.Youm YH, Grant RW, McCabe LR, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ang QY, Alexander M, Newman JC, et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell. 2020;181(6):1263–1275.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cheng CW, Biton M, Haber AL, et al. Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell. 2019;178(5):1115–1131.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Newman JC, Verdin E. Beta-hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Newman JC, Covarrubias AJ, Zhao M, et al. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 2017;26(3):547–557.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nielsen R, Møller N, Gormsen LC, et al. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139(18):2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Selvaraj S, Hu R, Vidula MK, et al. Acute echocardiographic effects of exogenous ketone administration in healthy participants. J Am Soc Echocardiogr. 2022;35(3):305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kaeberlein M, Powers RW 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193–1196. [DOI] [PubMed] [Google Scholar]
- 146.Kennedy BK, Lamming DW. The mechanistic target of rapamycin: the grand conductor of metabolism and aging. Cell Metab. 2016;23(6): 990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Miller RA, Harrison DE, Astle CM, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13(3): 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Strong R, Miller RA, Bogue M, et al. Rapamycin-mediated mouse lifespan extension: Late-life dosage regimes with sex-specific effects. Aging Cell. 2020;19(11):e13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Urfer SR, Kaeberlein TL, Mailheau S, et al. A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience. 2017;39(2): 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. [DOI] [PubMed] [Google Scholar]
- 152.Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–1842. [DOI] [PubMed] [Google Scholar]
- 153.Vallurupalli M, MacFadyen JG, Glynn RJ, et al. Effects of interleukin-1beta inhibition on incident anemia: exploratory analyses from a randomized trial. Ann Intern Med. 2020;172(8):523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Everett BM, Cornel JH, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289–1299. [DOI] [PubMed] [Google Scholar]
- 155.Deftereos SG, Beerkens FJ, Shah B, et al. Colchicine in cardiovascular disease: in-depth review. Circulation. 2022;145(1):61–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saavedra JM. Angiotensin Receptor blockers are not just for hypertension anymore. Physiology (Bethesda). 2021;36(3):160–173. [DOI] [PubMed] [Google Scholar]
- 157.Ridker PM, Everett BM, Pradhan A, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. 2019;380(8):752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Saul D, Kosinsky RL, Atkinson EJ, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun. 2022;13(1):4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Translational Geroscience Network. Accessed June 8, 2023. https://www.gerosciencenetwork.org/
- 160.Justice J, Miller JD, Newman JC, et al. Frameworks for proof-of-concept clinical trials of interventions that target fundamental aging processes. J Gerontol A Biol Sci Med Sci. 2016;71(11): 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Barzilai N, Cuervo AM, Austad S. Aging as a biological target for prevention and therapy. JAMA. 2018;320(13):1321–1322. [DOI] [PubMed] [Google Scholar]
- 162.Crimmins EM. Social hallmarks of aging: suggestions for geroscience research. Ageing Res Rev. 2020;63:101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dwyer-Lindgren L, Bertozzi-Villa A, Stubbs RW, et al. Inequalities in life expectancy among US counties, 1980 to 2014: temporal trends and key drivers. JAMA Intern Med. 2017;177(7):1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Noren Hooten N, Pacheco NL, Smith JT, Evans MK. The accelerated aging phenotype: the role of race and social determinants of health on aging. Ageing Res Rev. 2022;73:101536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Scott AJ. The longevity economy. Lancet Healthy Longev. 2021;2(12):e828–e835. [DOI] [PubMed] [Google Scholar]
- 166.Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr. 2008;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cruz-Jentoft AJ, Landi F, Schneider SM, et al. Prevalence of and interventions for sarcopenia in ageing adults: a systematic review: report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing. 2014;43(6): 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.